
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photodynamic therapy targeting VCAM-1-expressing human umbilical vein endothelial cells using a PpIX–VCAM-1 binding peptide–quantum dot conjugate
Huijuan Yin ab,
Xiafei Shib,
Hong Wangb,
Wendong Jinb,
Yingxin Lib and
Ying Fu*a
ab,
Xiafei Shib,
Hong Wangb,
Wendong Jinb,
Yingxin Lib and
Ying Fu*a
aSection of Cellular Biophysics, Department of Applied Physics, Royal Institute of Technology, Science for Life Laboratory, SE-171 21 Solna, Sweden. E-mail: fu@kth.se; Tel: +46 737650507
bLaser Medicine Laboratory, Institute of Biomedical Engineering, Chinese Academy of Medical Sciences, Peking Union Medical College, Tianjin 300192, China
First published on 30th October 2017
Abstract
With increasing knowledge of the relevance of vascular cell adhesion molecule 1 (VCAM-1) for tumor growth, metastasis, angiogenesis, and related processes, it has become an attractive anti-tumor strategy to target VCAM-1 expression on the tumor vasculature. We designed a new targeted nanodrug, denoted PVQ, based on a photosensitizer (for the photodynamic effect), VCAM-1 target and quantum dot (QD) carrier, using conjugated water-dispersible colloidal CdSe–CdS/ZnS QDs, protoporphyrin IX (PpIX) photosensitizers, and VCAM-1 binding peptides. Its targeting ability and photodynamic therapy (PDT) efficiency against VCAM-1 expression in human umbilical vein endothelial cells (HUVECs) were then investigated. Conjugates of QD–VCAM-1 binding peptide (VQ), PpIX–VCAM-1 binding peptide (PV), and PVQ prepared using amide coupling were verified by agarose gel electrophoresis, Fourier transform infrared spectroscopy, and fluorescence spectrometry. VCAM-1 expression in HUVECs was induced by TNF-α treatment. PVQ conjugates were co-cultured with VCAM-1 expressing (VCAM-1(+)) and non-expressing (VCAM-1(−)) HUVECs, and target imaging, ROS generation, cell death, and apoptosis were analyzed using confocal fluorescence microscopy. VCAM-1 target imaging could not distinguish between VCAM-1(+) and VCAM-1(−) HUVECs after only 6 h of incubation; however it could distinguish between the cells after incubation for 24 h. After incubation for ca. 30 min, PVQ generated a significantly higher yield of ROS (3.6 fold) in VCAM-1(+) HUVECs compared with VCAM-1(−) cells, during 10 min of irradiation at a wavelength of 405 nm, and this was followed by a second rise in ROS at 30 min after irradiation. Moreover, cell destruction was observed clearly in VCAM-1(+) cells treated with PVQ and almost all cells became round after 30 min of irradiation at 405 nm. PVQ-induced PDT effects caused a significant apoptosis (onset and late apoptosis) in VCAM-1(+) HUVECs at 6 h after PDT treatment. In conclusion, PVQ shows a great potential for targeted PDT in cancer therapy.
Introduction
Since the late 1980s, when the first insight was provided into endothelial leukocyte adhesion, the relevance of vascular cell adhesion molecule 1 (VCAM-1) for tumor cell adhesion and metastasis has been investigated.1,2 Banks first exhibited augmented soluble VCAM-1 (sVCAM-1) levels in the serum of 110 cancer patients.3 This was the starting point for extensive investigations aiming to correlate sVCAM-1 with cancer progression and metastasis. Most of the studies revealed that augmented sVCAM-1 is a type of biomarker for overall survival and post-operative recurrence.4–6 Besides the utilization of VCAM-1 as a biomarker for cancer progression, the blocking of endothelial VCAM-1 adhesive functions appears to interfere with cancer disease. Direct blocking of VCAM-1 by small molecules or antibodies has not been reported yet due to its complexity. Endothelial VCAM-1 has attracted much attention recently, as a potential target for drug carriers such as liposomes or nanoparticles.7–9 These nanoparticle approaches have mainly been devised for cancer diagnosis using upregulated VCAM-1. Currently, a direct VCAM-1 blockade for cancer therapy is not available.Quantum dots (QDs) have two distinctive properties which allow them to play an increasingly important role in drug carrier systems. One is their unique optical properties of bright and stable fluorescence, which can be used to locate and identify target tissue; and the other is their versatile encapsulation and surface functionalization which offer improved water solubility, biocompatibility, and targeting.10 As nanoparticles, QDs also show the macromolecular enhanced permeability and retention effect (EPR) in solid tumors.11 To design a VCAM-1 blockade for cancer therapy, we selected QDs as the carrier, a VCAM-1 binding peptide to enhance targeting, and the photosensitizer (PS) protoporphyrin IX (PpIX) as a drug which causes the photodynamic effect when irradiated by light, because irradiation provides another targeting control.
Photodynamic therapy (PDT) is widely used for treating tumors on the surface of the skin or in body cavities, including tumors of the skin, head, and neck areas, as well as solid tumors that can be accessed with relative ease, such as tumors of the esophagus, lungs, larynx, and uterine cervix.12–14 PDT has also been developed recently for application to non-solid tumors such as leukaemia,15 and for AIDS16 and infectious disease.17 This therapeutic approach involves the administration of a tumor-localizing PS, by intravenous injection or external application, and the subsequent targeted activation of this PS by exposure to light of a specific wavelength.18 The selectivity of PDT is achieved from the somewhat preferential localization of the PS in the target tissue and from the irradiation of a specified area. Provided that the PS is not toxic, only the irradiated areas will be affected, even if the PS does bind to normal tissues. This rationale works well if light can be directed specifically to the lesion as in dermatological and ophthalmological applications. However, for applications of PDT in complex anatomical sites, such as the abdominal or thoracic cavities, confined irradiation is hardly possible so that targeted PS delivery becomes a necessity. Three strategies are under study and development: (1) passive targeting of the PS (due to greater proliferative rates of neoplastic cells compared with normal cells); (2) target modulation to increase PS accumulation (exogenous 5-aminolevulinic acid); and (3) active targeting of PS conjugates (saccharide, peptide, protein, etc.).19 However, most PSs target tumors rather than vessels.
In this paper, we design an active targeting conjugate comprising protoporphyrin IX (PpIX; PS for the photodynamic effect), a VCAM-1 target, and a QD carrier, for vascular endothelial cells, in order to explore the possibility of a universal targeted drug for advanced solid tumors which rely on the role of VCAM-1 for tumor metastasis.
Materials and methods
Reagents
EGM-2 MV BulletKit (CC-3202 = CC-3156 + CC-4147) was purchased from Lonza; TNF recombinant human protein (TNF-α, PHC3015), penicillin–streptomycin–glutamine (100×, 10378016), Gibco™ amphotericin B (15290018) and DPBS, no calcium, no magnesium (14190250) were purchased from Thermofisher; VCAM-1 binding peptide (VHPKQHRGGSKGC) and 5-carboxyfluorescein (5-FAM)-labelled VCAM-1 binding peptide (VHPKQHRGGS(K-5-FAM)GC) were custom made by Innovagen, Sweden; Cell Proliferation Kit I (MTT, 11465007001 ROCHE), Annexin V-FITC Apoptosis Detection Kit (APOAF-50T ST), and protoporphyrin IX (PpIX, P8293-1G) were obtained from Sigma-Aldrich; NucBlue® Live ReadyProbes® Reagent (Hoechst 33342, Cat R37605) and di(acetoxymethyl ester) (6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate) (H2DCFDA, C2938) were obtained from Thermofisher.Water-dispersible 3-mercaptopropionic acid (3MPA)-coated CdSe–CdS/ZnS core–shell QDs were synthesized in-house using a high-temperature organic synthesis method (the details can be found in our previous paper20). There were three major steps: (1) the formation of the CdSe core: cadmium oxide (CdO), tetradecylphosphonic acid (TDPA), octadecylamine (ODA) and trioctylphosphine oxide (TOPO) were mixed and heated to 330 °C for clear melting, and then cooled to 260 °C for Se-TOP (selenium-trioctylphosphine) addition. The desired CdSe cores were formed by controlling the reaction time. (2) Core–shell QDs: ODA and ODE (1-octadecene) were melted at 100 °C as the reaction solution, in which CdSe cores from the previous step were covered with Cd-, S-, and Zn-precursors sequentially at 230 °C. The size of the QDs was regulated by controlling the time for growth of each layer and the number of layers. The size of the QDs determined the fluorescence peak wavelength. All the procedures were performed under nitrogen flow. (3) Surface ligand exchange: core–shell QDs from step 2 were dispersed in chloroform, mixed with an equal volume of water containing 3-MPA, and stirred for 2 h, resulting in water-soluble carboxyl-coated QDs. These QDs consisted of a CdSe core, a CdS shell of 2 monolayers, another shell of 1 monolayer Cd0.5Zn0.5S, 1.5 monolayers ZnS, and a coating of 3-MPA surface ligands. They had a fluorescence peak at about 580 nm at room temperature. They were dispersed in deionized water (pH = 7.2) at a concentration of 10 μM.
There are other reported QD fabrication and conversion methods21 with differences in growth times and temperatures; however, a key difference is that our QDs have a core–shell structure that avoids possible leakage of heavy metal in the QD cores. This may contribute significantly to the fact that free 3MPA–QDs showed no toxicity to Calu-3 cells.22
Synthesis of conjugates
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VCAM-1 binding peptide:QD was 50:50:1. The concentration of PpIX in the PVQ conjugates was 50 μM.:50.:1. The concentration of PpIX in the PV conjugates was 50 μM.
:VCAM-1 binding peptide:QD was 50:50:1. The concentration of PpIX in the PVQ conjugates was 50 μM.:50.:1. The concentration of PpIX in the PV conjugates was 50 μM.Un-conjugated peptides were removed using Amicon Ultra-4 filters (100 kDa cut-off). Final conjugates were dissolved in 1 mL of PBS.
Characterizations of the conjugates
The conjugates were characterized by agarose gel electrophoresis, Fourier transform infrared spectroscopy (FTIR) and fluorescence spectrometry.Agarose gel electrophoresis was performed to check the conjugations between PpIX, QD and VCAM-1 peptide. 20 μL samples were added to loading wells in 0.5% agarose gels. The gels were run at a voltage of 100 V for 0.5 h.
HUVEC culture and VCAM-1 expression
Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza and cultured in EGM-2 medium (EGM-2MV Basal Medium + EGM-2MV SingleQuots) complemented with 1% penicillin/streptomycin/glutamine, and 0.1% amphotericin B. Cells of passage 2–5 were used in the experiments.VCAM-1 expression in HUVECs was induced by TNF-α treatment. In detail, HUVECs at 8 × 105 mL−1 were incubated for 24 h, then changed to a medium with 20 ng mL−1 TNF-α (1:5000 dilution) and incubated for a further 24 h. VCAM-1 expression was assessed by the fluorescence of 5-FAM contained in the labelled 5FAM–VCAM-1 peptide using a LSM780 confocal microscope (Carl Zeiss, Jena, Germany). In detail, after TNF-α treatment, 50 μL of 200 μM 5FAM–VCAM-1 peptide was added into the cells and co-cultured for 1 h. The cells were then rinsed three times to remove free peptides and studied using fluorescence imaging.
Target imaging of VCAM-1 expression
HUVECs were seeded onto glass-bottomed Petri dishes and incubated for 24 h for adherence and a further 24 h with TNF-α for VCAM-1 expression. Then the cells were treated with PVQ (final concentration of 1 μM), VQ (1 μM) and PV (10 μM) for 20 min. After incubation, the cells were rinsed three times with PBS and resupplied with growth medium EGM-2. Then the cells were placed on the sample stage of the L780 confocal laser scanning microscope and the images were acquired using Ex 488 nm/Em 510–540 nm for 5-FAM conjugated to the VCAM-1 binding peptide, Ex 405 nm/Em 560–600 nm for the QDs, and Ex 405 nm/Em 600–650 nm for PpIX.Killing effects of PDT using conjugates
HUVECs were seeded onto glass-bottomed 96-well plates and cultured for 24 h at 37 °C. Then the cells were treated either with 20 ng mL−1 TNF-α for VCAM-1 expression or normal medium for another 24 h. After that the cells were treated with the conjugates separately in different groups with 4 wells per group. There were five groups: PV–PDT, VQ-PDT, PVQ–PDT, light control, and blank control. The concentration of PVQ/VQ was 80–400 nM, and the concentration of PV was 0.8–4 μM. The incubation time with the conjugates was 30 min. The light-emitting diode (LED) light source at 630 nm was designed and developed in-house, with an optical power density of 40 mW cm−2. For an irradiation time of 60 s, the energy density was calculated to be 2.4 J cm−2.At a time point 24 h after PDT treatment, the killing effects on the cells were assessed by MTT assay. MTT at a concentration of 5 mg mL−1 was added to the cells at the volume ratio of 1:10, and then cultured for 4 h. The same volume of solvent (10% SDS in 0.01 M HCl) was added to dissolve the formazan crystals overnight in a humidified atmosphere. An ELISA reader was used to detect absorbance at 570 nm.
ROS detection
HUVECs were seeded onto glass-bottomed Petri dishes and incubated for 24 h for adherence and then 24 h with TNF-α for VCAM-1 expression. They were treated with PV (final concentration of 10 μM), PVQ (1 μM) and VQ (1 μM). At the same time, the cells were dyed with H2DCFDA (10 μM). After 30 min of incubation, the cells were rinsed three times in PBS and resupplied with EGM-2 growth medium. Then the cells were placed under the LSM 780 microscope for ROS detection. The imaging was divided into two steps. In the first step, the cells were irradiated with a 405 nm laser while ROS signals (the fluorescence of DCF (2′,7′-di-chlorofluorescein) which was converted by H2DCFDA in the presence of ROS) were detected with Ex 488 nm/Em 525 nm for 10 min at time intervals of 2 s. In the second step, the 405 nm laser was switched off and the ROS signals were detected for a further 30 min at time intervals of 3 min using a z-stack and time series scanning mode.Apoptosis detection
HUVECs were seeded onto four glass-bottomed Petri dishes, incubated for 24 h for adherence and another 24 h for VCAM-1 expression with TNF-α (or without TNF-α in the case of the negative control). Then the dishes were treated with PVQ (final concentration of 1 μM) or PV (10 μM) for 20 min. After rinsing with PBS three times, the cells were irradiated with a 630 nm LED for 2–4 min at 28.6 mW cm−2.Apoptosis was detected at 2 h and 6 h. After incubation for 2 h or 6 h, the cells were stained with Annexin V-FITC, PI and Hoechst 33342 for 10 min at room temperature and images were obtained across the irradiation area with ex 405 nm/em 461–497 nm for Hoechst 33342, ex 488 nm/em 495–546 nm for FITC, and ex 561 nm/em 588–642 nm for PI, using the LSM780 confocal laser scanning microscope.
Results
Conjugations of QD, PpIX, and VCAM-1 binding peptides
The 3MPA-coated QDs possess carboxylic acid groups on their surfaces for conjugation with amine-terminated peptides, and PpIX contains two carboxylic acid groups which are also suitable for conjugation to amine-terminated peptides. The VCAM-1 binding peptide (VHPKQHRGGSKGC) used in these experiments contains 5 amine groups.24 Our conjugations were designed as shown Fig. 1.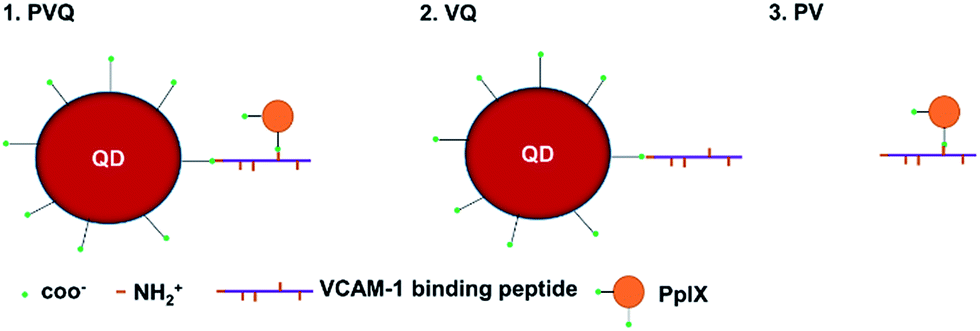 | ||
| Fig. 1 The structures of the conjugates. PVQ: PpIX and 3MPA–QDs were simultaneously conjugated to VCAM-1 binding peptides by amino linkage. VQ: 3MPA–QDs and VCAM-1 binding peptides were conjugated by condensation reaction. PV: PpIX and VCAM-1 binding peptides were conjugated by condensation reaction. | ||
The conjugations of PpIX and QD with VCAM-1 binding peptides were first verified by electrophoresis experiments. Fig. 2A shows different samples in the gel after 0.5 h electrophoresis under a voltage of 100 V. The green fluorescence came from 5FAM (ca. 523 nm), the red came from PpIX (ca. 637 nm), and the yellow came from the QDs (ca. 584 nm). The QDs and PpIX migrated towards the anode since their carboxyl groups were de-protonated. 5FAM-labelled VCAM-1 binding peptides (V–peptide–FAM) migrated towards the cathode, since the amino acids of the peptides were either neutral (V, P, Q, G, C) or positively charged (H, K, R). PV, VQ, and PVQ all moved towards the anode, although their electrophoretic mobility differed due to their different sizes/masses. Moreover, we noticed size/mass-dependent retentions in the agarose gel (the movements were more apparent inside the loading wells; the dashed line is the “−” cathode side of the loading wells and PV/VQ/PVQ conjugates were all located at the “+” anode side of the loading wells).
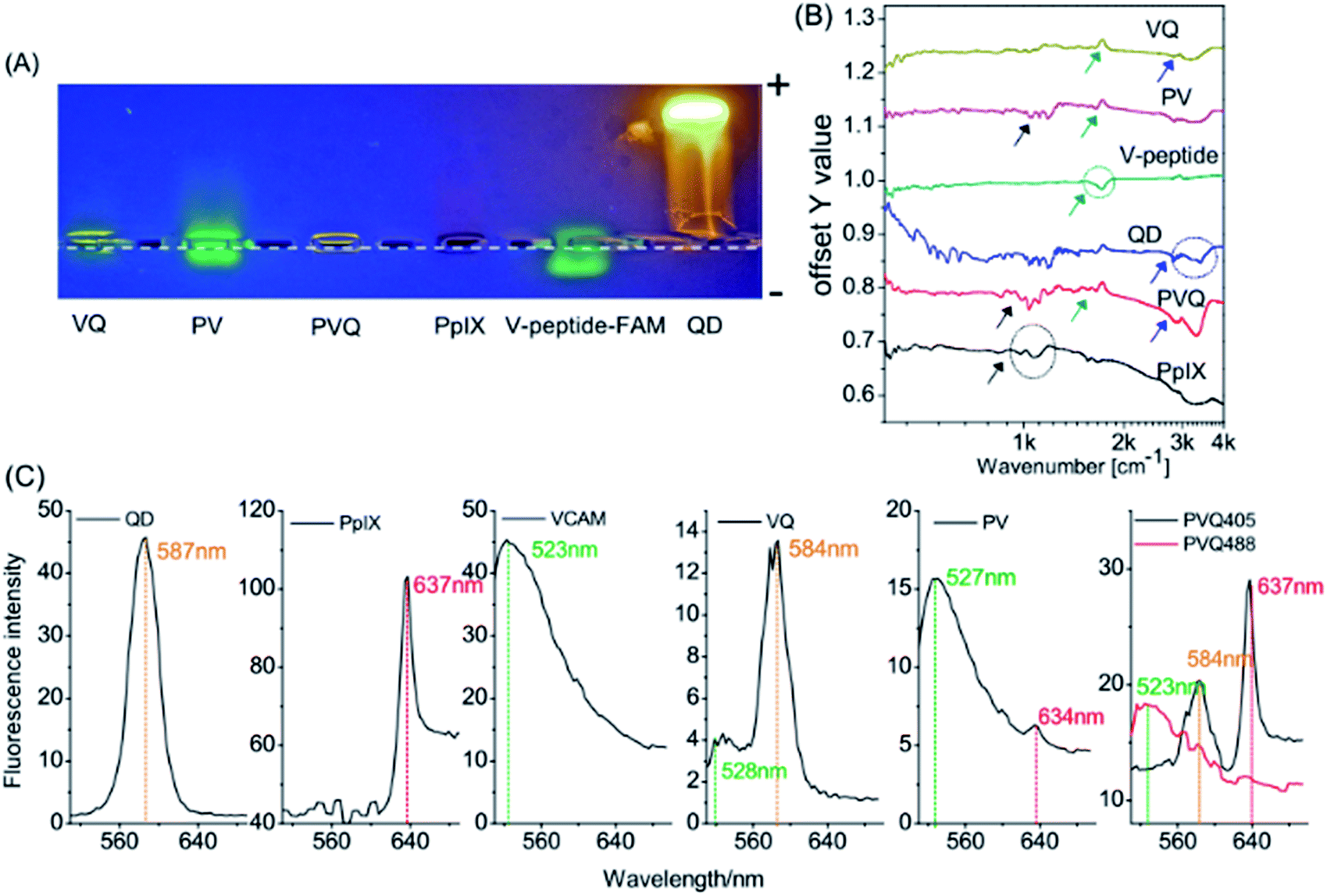 | ||
| Fig. 2 Characterizations of the conjugates. (A) Electrophoresis assay; (B) FTIR; (C) emission spectra. | ||
FTIR was used to identify functional groups in the conjugates. We know that each molecule has its own characteristic infrared “fingerprint” spectrum which will change when its structure changes. Fig. 2B shows clearly the characteristic features of the V-peptide (green ring and arrow), PpIX (black ring and arrow) and QDs (blue ring and arrow). The spectra at the same sites (characteristic peaks) showed marked change when the components became part of VQ, PV, and PVQ (see the arrows in the corresponding colours).
We also measured the emission spectra of the conjugates. Fig. 2C shows the fluorescence spectra of many regions of interest (ROIs) in the lambda-mode micrographs, and confirmed the formation of the conjugates (the spectral resolution was 3 nm). The peak wavelengths of the QDs (587 nm), PpIX (637 nm) and FAM–VCAM (523 nm) displayed no significant modifications (less than 5 nm) in the conjugates of VQ, PV and PVQ. The fluorophore did not change when QD, PpIX and FAM–VCAM were linked by carbodiimide-based chemistry. Note that the fluorescence came from the product conjugates rather than from the reactants because the free small molecules PpIX and FAM–VCAM were removed by ultracentrifugation from the conjugates.
VCAM-1 expression, and conjugate targets
We used the conjugated PVQ and VQ in co-cultures with VCAM-1(+)/(−) HUVECs to identify the VCAM-1 target of the conjugates. After co-culture for 6 h we did not find any significant differences between the binding abilities of the conjugates to VCAM-1 in the VCAM-1(+) and VCAM-1(−) HUVECs from the fluorescent signals, but after 24 h, specific targeting between PVQ and VCAM-1(+) HUVECs appeared significant (see Fig. 3A). In order to identify the early interaction between PVQ and VCAM-1, we observed the HUVECs which had been co-cultured with PVQ for 20 min using L780 confocal laser scanning microscopy in time-lapse imaging mode for 30 min with time intervals of 5 min. The 405 nm laser of the microscope acted not only as a light source for the imaging, but also as the light source to generate the PDT effects. As shown in Fig. 3B, the morphological structures of all VCAM-1(+) cells looked normal at 5 min, while the cells became round during the imaging process and gradually detached from the cell culture dish, clearly implying that the PDT effects of PVQ induced by the 405 nm laser were killing the cells. By contrast, the cells without VCAM-1 expression stayed normal. This indicated that PVQ could attach to the cell membrane non-specifically, while it bound closely to VCAM-1 molecules on VCAM-1(+) HUVECs. The close, specific binding between PVQ and the VCAM-1 molecules resulted in the observed PDT effects since PpIX exerts its PDT effects only within nanometer range. The conjugate of VCAM-1 binding peptide, PpIX and QD, i.e., PVQ, and its specific binding with the VCAM-1 molecule in VCAM-1(+) HUVECs are the determining factors in the observed PDT effects.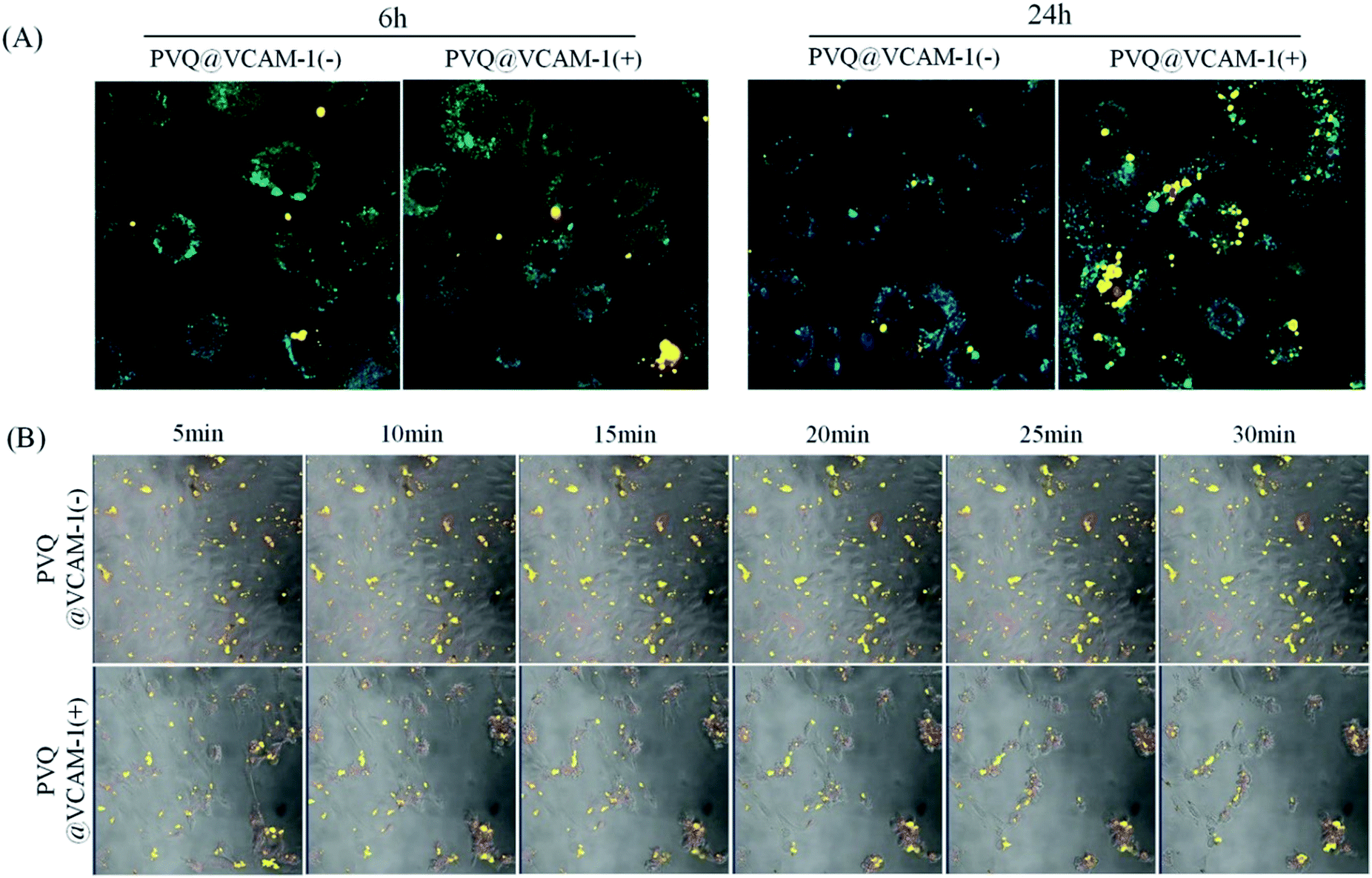 | ||
| Fig. 3 The targeted binding between VCAM-1 and PVQ conjugates. (A) Fluorescence images of PVQ in HUVECs. 1 μM PVQ was added to VCAM-1(+)/(−) HUVECs for 6 h or 24 h, and imaged in lambda mode (objective 40×); (B) time lapse images showing PDT effects of PVQ on VCAM-1(+)/(−) cells; time intervals of 5 min, 20× magnification. | ||
MTT analysis of PDT effect
To quantify the PDT effect with the targeted conjugates, we compared the effects of PDT on VCAM-1(+)/(−) HUVECs by MTT assay. As seen in Fig. 4, PVQ and PV both caused around 40% cell death and VQ caused about 20% in both VCAM-1(+) and VCAM-1(−) HUVECs. The lethal effects analyzed using MTT displayed no significant differences between VCAM-1(+) and VCAM-1(−) HUVECs (P > 0.05) using LED irradiation. We believe that MTT was not a proper analytical tool since the PVQ–PDT treated cells were not totally dead, even if they became round and detached from the culture dish; see more below in the apoptosis analysis.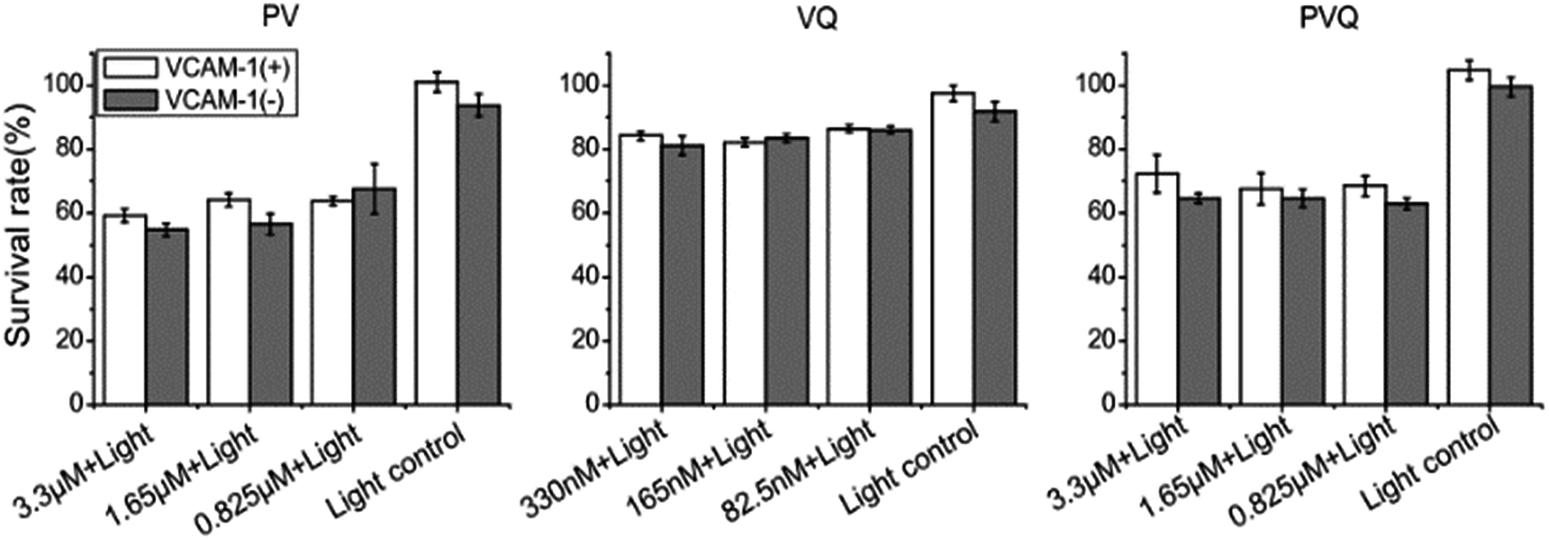 | ||
| Fig. 4 MTT analysis of the PDT effect induced by conjugates on VCAM-1(+)/(−) HUVECs. | ||
ROS produced by PDT with PVQ
Reactive oxygen species (ROS) are the main effectors of PDT. To analyze directly the photodynamic effect of the conjugates, the products of ROS were detected. Firstly, we detected ROS products when exciting the conjugates using the 405 nm laser from the microscope, due to the short lifetime of ROS. As shown in Fig. 5A, when the cells were illuminated at 405 nm, the DCF signal was simultaneously collected for ROS detection (Ex 488 nm/Em 525 nm) over 10 min; we then switched off the 405 nm laser, and continued to collect the DCF signal for a further 30 min. ROS in VCAM-1(+) cells treated with PVQ increased continuously during the irradiation at 405 nm, and reached its peak at the time point of switching off the irradiation. After irradiation, ROS decreased initially. However, a second ascending peak of ROS appeared 20 min after stopping irradiation, which indicated the occurrence of a secondary reaction induced by the primary ROS. In the VCAM-1(−) cells treated with PVQ, ROS remained at low levels during and after 405 nm illumination. PV could not affect ROS production in either the cells expressing VCAM-1 or those not. In right-hand upper panels of Fig. 5A show the total ROS signal modulations (i.e., after subtraction of the values at t = 0) were averaged by the numbers of cells (ca. 90 cells in PVQ@VCAM-1(+)/(−) and PV@VCAM-1(−) samples; only 40 cells in the PV@VCAM-1(+) sample). To understand the distribution of ROS along the z axis, we plotted fire plots from the same cells along the z axis (Fig. 5A right-hand lower panels). The brightest ROS signal in PVQ@VCAM-1(+) was obtained close to the upper planes of z axis because the cells had become round as a result of the abundant ROS effects. However, the ROS signals in the other samples showed no changes along the z orientation or along the time axis.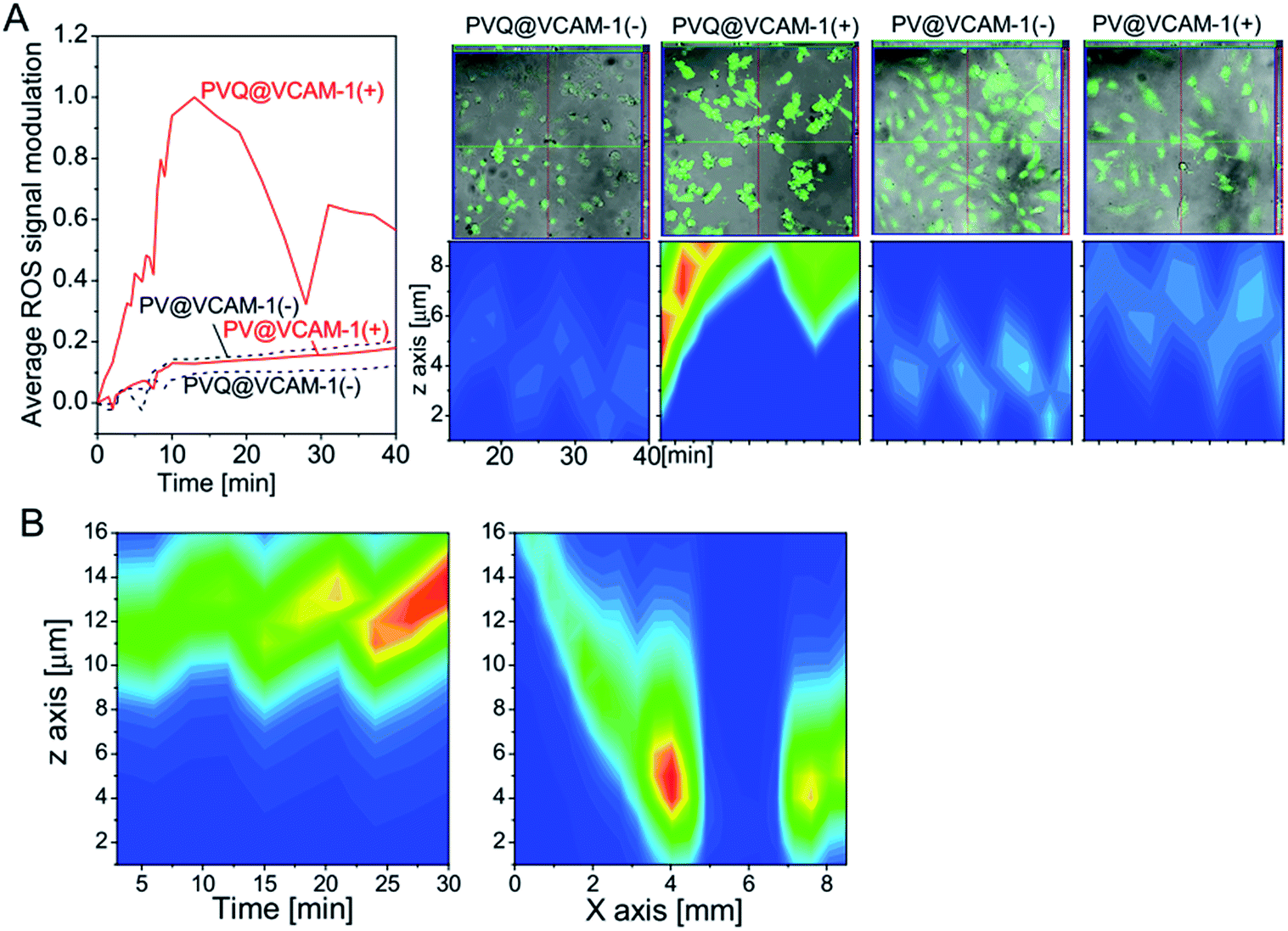 | ||
| Fig. 5 Temporal–spatial distributions of ROS products induced by conjugates. (A) (Left) The temporal distribution of ROS induced by conjugates with 405 nm laser irradiation; (Right) the distribution of ROS along the z axis. (B) The temporal–spatial distribution of ROS induced by PVQ with LED irradiation at 630 nm. | ||
We also analyzed the temporal–spatial distribution of ROS products induced by PVQ with 630 nm LED irradiation in VCAM-1(+) cells. In this experiment, the ROS signals were collected firstly after LED irradiation from one view for 30 min with time intervals of 3 min, and then covering 20 views along the center line of the light spot at the time point of 33 min. As shown in Fig. 5B, ROS products increased continuously over the time period, similar to the results seen for the point irradiation with the laser of the microscope (Fig. 5A). Interestingly, ROS was totally absent at the center of the light spot in 2 views of 850 μm where the light energy was the weakest due to the array formation of the LED. It decayed linearly from the center to the border of the LED array, thus confirming the photosensitizing effect of PVQ.
Apoptosis induced by PDT with PVQ
We used laser confocal microscopy rather than flow cytometry (FCM) to measure the apoptosis of HUVECs treated with PVQ–PDT because the QD signal overlaps with that of propidium iodide (PI), which could produce false-positive results when using FCM. To obtain statistical significance, we ran tile scans for large observation areas (2560 × 512 pixels and 5120 × 512 pixels) to cover a large number of cells, and ran in z-stack mode (δz = 2 μm for total 20 μm thickness) to cover the whole cell. Fig. 6A–E shows the VCAM-1(+) cells 6 h after treatment with PVQ–PDT: (A) is the merged image of bright-field, blue (461–497 nm, Hoechst 33342) and red (588–642 nm, both PI and QD) channels. Here only a part (200 × 200 pixels) of the 2560 × 512 pixel tile image is presented. The blue and red channels are separately shown in Fig. 6B and C. Note that the red channel also contained the QD signal (peak 580 nm). However, QDs were distributed only outside the nuclei, so we generated a mask (D) from (B) to exclude signals outside of the nuclei in (C), resulting in (E), which was the real PI (late-apoptosis) signal.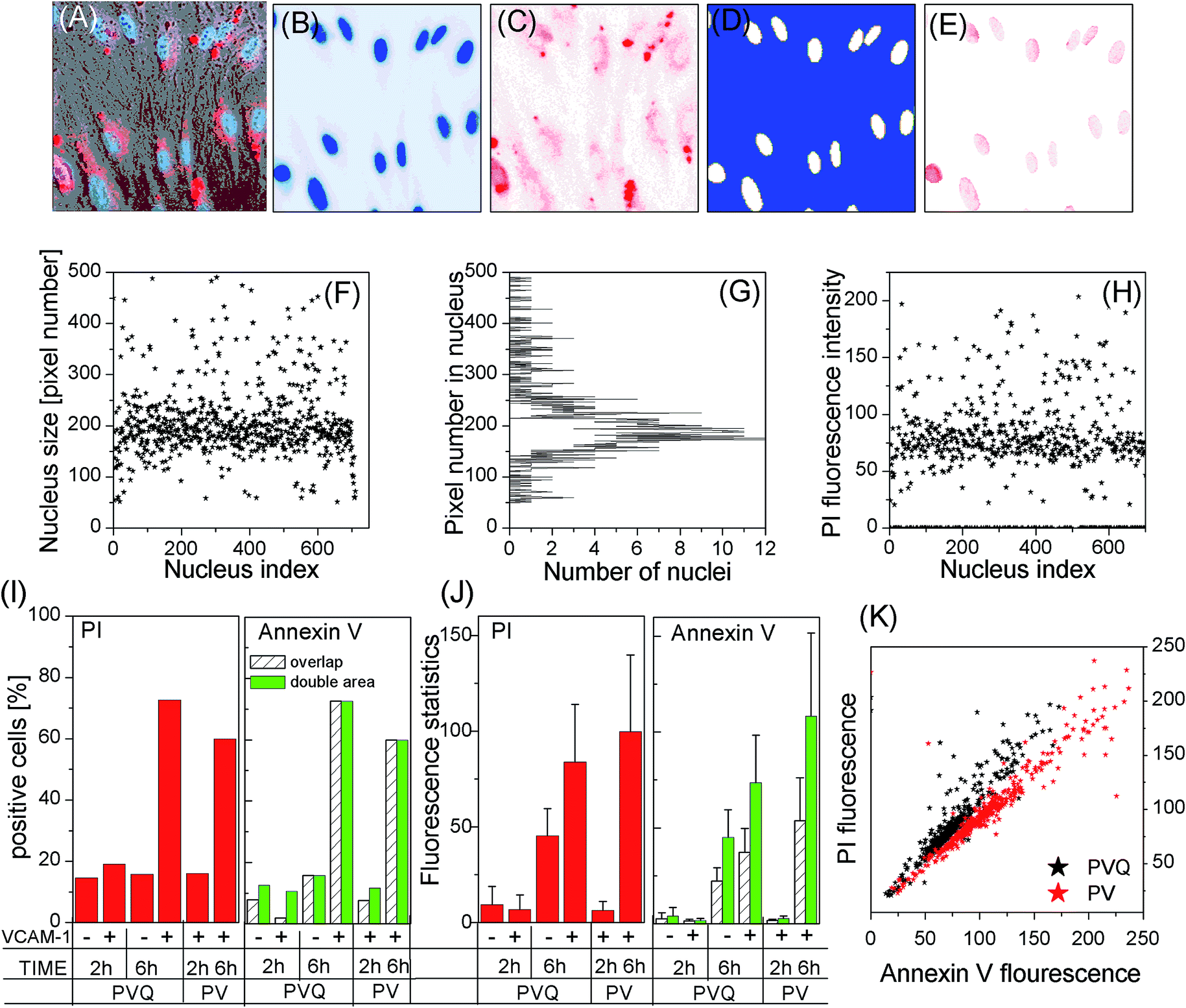 | ||
| Fig. 6 Apoptosis induced by PDT from the conjugates. (A–H) method to quantify PI signal; (I) the percentage of PI and Annexin-positive cells in different cell groups; (J) PI/Annexin signal intensities in PI/Annexin-positive cells; (K) the correlation of PI and Annexin V in PVQ and PV-induced PDT. | ||
To study PDT-induced apoptosis at the individual cell level, we first calculated the number of pixels (pixel size) of each individual nucleus (white areas) in (D), which resulted in (F) and (G) showing that a large number of nuclei were in the size range of 150 to 210 pixels, see (G). We picked up only those nuclei whose pixel numbers exceeded 50 (excluding small-size nuclei in the grey area in (G) which are either on the edge of the observation view, or are noise), resulting in the pixel size of each nucleus (F).
We then calculated the total PI fluorescence signal intensities inside these nuclei, and the data are presented in (H). Here we see clearly that the majority of cells, 419 nuclei out of a total 710, had a PI fluorescence intensity around 75. However there were many cells which did not express late apoptosis at all. We counted the number of nuclei which had PI signals then divided it by the total number of nuclei in the observation view, which is defined as the PI expression ratio in (I). The mean value and standard deviations (SDs) of PI fluorescence intensities of all PI expressed nuclei were calculated and are presented in (J) (in line with common practice, the 10 most-highly expressed nuclei were excluded from statistical analysis). (I) and (J) display the data of the four PVQ samples plus two PV samples. Note that the numbers in the bars in (I) are the numbers of cells in the samples.
Fig. 6I shows that the numbers of cells displaying late apoptosis were only significant in VCAM-1(+) samples 6 h after PVQ–PDT, while (J) implies that the level of late apoptosis in apoptosis-expressing cells depended kinetically on the lapsed time after PDT treatment.
Annexin V-FITC is known to bind to cell membranes when early apoptosis occurs. It is however difficult to calculate its fluorescence intensity per cell because of the irregular cell geometries. We first calculated only Annexin fluorescence signals inside nuclei, denoted as the “overlap” scheme. Next we calculated the center-of-mass of each nucleus, and then the Annexin fluorescence signal inside a circle having a double area as the nuclear pixel size centered at the center-of-mass of the nucleus (denoted as the “double-area” scheme). The results are presented in the right of (I) and (J). For the 2 h samples, we observed differences in Annexin V binding ratios (the ratio is large for the “double-area” scheme), while the ratios remained the same for both 6 h samples. The mean values and SDs of the “double-area” scheme were almost twice the values of “overlap”. Moreover, (K) shows that the PI and Annexin V expressions of individual cells were clearly correlated. All these data suggest that the early apoptosis results were similar to the results for late apoptosis, i.e., the VCAM-1(+) cells displayed the highest degree of apoptosis 6 h after PVQ–PDT.
Discussion
In this study, we designed a nanoparticle drug, denoted PVQ, to target tumor vessels, using QDs as the drug carrier, PpIX as the drug with PDT effect, and the VCAM-1 binding peptide for targeting; i.e., PpIX–VCAM-1 peptide–QD.We conjugated water-dispersible colloidal CdSe–CdS/ZnS QDs with VCAM-1 binding peptide and PpIX. Two principal results were obtained: we observed enhanced PDT effects and specific tumor targeting via vascular VCAM-1. As shown in Fig. 5A, the efficiency of ROS generation from PVQ excited by a 405 nm laser was significantly higher than that from PV (the conjugate containing only PpIX and VCAM-1 binding peptide) at the same concentration of PpIX. The apoptosis test gave similar results as shown in Fig. 6I and J. After irradiation with a 630 nm LED, PVQ caused a higher PI positive rate and Annexin V positive rate compared with PV at both 2 h and 6 h. This implies that PpIX within the PVQ conjugate has a higher PDT effect than it displays in the form of small molecules such as PV. We believe that the local high PpIX concentration of PVQ enhanced the PDT effect, generated ROS, and destroyed the target cells, without fluorescence resonance energy transfer (FRET). Tsay et al. reported similar conjugates of PS–peptide–QD.25 They verified that for the PS–peptide–QD conjugates, generation of singlet oxygen could be achieved via indirect excitation through FRET from the nanocrystals to the PS, or by direct excitation of the PS. They also found that the higher singlet oxygen quantum yields were achieved by direct excitation using 532 nm excitation wavelengths rather than by indirect excitation via FRET. Li et al. reported somewhat different results however.26 They used a 532 nm laser, which was at the absorption region of QDs but not at the absorption region of sulfonated aluminium phthalocyanines (AlPcSs), to excite the AlPcS-QD conjugates that they had synthesized and which were taken up by human nasopharyngeal carcinoma cells. They found that the AlPcS-QD conjugates underwent FRET in cells, with an efficiency of around 80%, and destroyed most cancer cells. Tsay and Li gave inconsistent conclusions about FRET, so in this paper, we did not excite the FRET effect between the QDs and PS, and the enhanced PDT can only result from the aggregated PpIX.
Tumor targeting was our primary aim in this study. VCAM-1 expression is upregulated at the periphery of a tumor. Gosk et al. found that VCAM-1 increasingly targeted immunoliposomes (ILs) accumulated in tumor vessels from 30 min to 24 h in mice bearing human Colo 677 xenograft tumors, while control ILs accumulated in non-affected organs, mainly the liver and spleen.27 A target to VCAM-1 can destroy the blood vessels around the tumor through PDT, and cut off the nutrition support of the tumor and the pathway of tumor metastasis. Zhan et al. found that VCAM-1 expression was markedly increased after PDT with hematoporphyrin monomethyl ether (HMME) treatment of Wistar rats bearing C6 glioma, but when they used VCAM-1 monoclonal antibodies to treat the C6 glioma-bearing rats following PDT, VCAM-1 expression was significantly reduced.28 Therefore a PS designed to target VCAM-1 will function as a universal PS for most tumors. In our study, we used a binding peptide with 13 amino acids to target VCAM-1, and used its amine groups to link QDs and PpIX via carbodiimide based chemistry. Our experiments using fluorescence imaging (see Fig. 3) were clearly designed to differentiate VCAM-1 targeting by PVQ between VCAM-1(+) and VCAM-1(−) HUVECs after 24 h incubation. Moreover, we found that the excitation laser of the fluorescence microscope induced significant morphological changes in VCAM-1(+) cells incubated with PVQ for 30 min – the cells became round and cell–cell junctions broke – while the VCAM-1(−) cells stayed unchanged. This implies clearly the existence of specific target binding between VCAM-1 and PVQ with binding distances shorter than hydrophobic-group binding between PVQ and the cell membrane. The result was further verified by the ROS and apoptosis experiments. It can thus be concluded that our PVQ displays specific targeting to VCAM-1 and that the PVQ conjugates have a great potential in cancer therapy.
Conclusions
We have shown that water-stable, PpIX–VCAM-1 binding peptide-coated QD (PVQ) conjugates can be synthesized without any deterioration in the photophysical properties of either the QDs or the photosensitizers. Furthermore, PVQ conjugates specifically target VCAM-1 expressing HUVECs, efficiently generate ROS, and cause distinct cell destruction, while these PDT effects do not apply to normal HUVECs. PVQ-induced PDT generates efficient ROS which continue to rise under irradiation and have a secondary burst 20 min after the irradiation is stopped; this causes significant apoptosis (onset and late apoptosis) in VCAM-1-expressing HUVECs 6 h after PDT treatment. Future work will focus on demonstrating targeting, imaging, and PDT in live animals.Conflicts of interest
There are no conflicts to declare.Notes and references
- U. Prabhakar, H. Maeda, R. K. Jain, E. M. Sevick-Muraca, W. Zamboni, O. C. Farokhzad, S. T. Barry, A. Gabizon, P. Grodzinski and D. C. Blakey, Cancer Res., 2013, 73, 2412–2417 CrossRef CAS PubMed.
- M. Schlesinger and G. Bendas, Int. J. Cancer, 2015, 136, 2504–2514 CrossRef CAS PubMed.
- R. E. Banks, A. J. Gearing, I. K. Hemingway, D. R. Norfolk, T. J. Perren and P. J. Selby, Br. J. Cancer, 1993, 68, 122–124 CrossRef CAS PubMed.
- Y. Yamada, T. Arao, K. Matsumoto, V. Gupta, W. Tan, J. Fedynyshyn, T. E. Nakajima, Y. Shimada, T. Hamaguchi, K. Kato, H. Taniguchi, Y. Saito, T. Matsuda, Y. Moriya, T. Akasu, S. Fujita, S. Yamamoto and K. Nishio, Cancer Sci., 2010, 101, 1886–1890 CrossRef CAS PubMed.
- V. Dymicka-Piekarska, K. Guzinska-Ustymowicz, A. Kuklinski and H. Kemona, Thromb. Res., 2012, 129, e47–50 CrossRef CAS PubMed.
- Y. Liu, M. D. Starr, A. Bulusu, H. Pang, N. S. Wong, W. Honeycutt, A. Amara, H. I. Hurwitz and A. B. Nixon, Cancer Med., 2013, 2, 234–242 CrossRef CAS PubMed.
- D. I. Kang, S. Lee, J. T. Lee, B. J. Sung, J. Y. Yoon, J. K. Kim, J. Chung and S. J. Lim, J. Microencapsulation, 2011, 28, 220–227 CrossRef CAS PubMed.
- L. Rouleau, R. Berti, V. W. Ng, C. Matteau-Pelletier, T. Lam, P. Saboural, A. K. Kakkar, F. Lesage, E. Rheaume and J. C. Tardif, Contrast Media Mol. Imaging, 2013, 8, 27–39 CrossRef CAS PubMed.
- H. Yang, F. Zhao, Y. Li, M. Xu, L. Li, C. Wu, H. Miyoshi and Y. Liu, Int. J. Nanomed., 2013, 8, 1897–1906 Search PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- Y. Nakamura, A. Mochida, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2016, 27, 2225–2238 CrossRef CAS PubMed.
- C. A. Robertson, D. H. Evans and H. Abrahamse, J. Photochem. Photobiol., B, 2009, 96, 1–8 CrossRef CAS PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- A. E. O'Connor, W. M. Gallagher and A. T. Byrne, Photochem. Photobiol., 2009, 85, 1053–1074 CrossRef PubMed.
- H. Yin, X. Ye, Y. Li, Q. Niu, C. Wang and W. Ma, J. Photochem. Photobiol., B, 2015, 153, 13–19 CrossRef CAS PubMed.
- H. Yin, Y. Li, Y. Zheng, X. Ye, L. Zheng, C. Li and Z. Xue, Lasers Med. Sci., 2012, 27, 943–950 CrossRef PubMed.
- X. J. Fu, Y. Fang and M. Yao, BioMed Res. Int., 2013, 2013, 159157 Search PubMed.
- J. M. Dabrowski and L. G. Arnaut, Photochem. Photobiol. Sci., 2015, 14, 1765–1780 CAS.
- N. Solban, I. Rizvi and T. Hasan, Lasers Surg. Med., 2006, 38, 522–531 CrossRef PubMed.
- Z. Ning, M. Molnar, Y. Chen, P. Friberg, L. Gan, H. Agren and Y. Fu, Phys. Chem. Chem. Phys., 2011, 13, 5848–5854 RSC.
- J. Yan, M. J. Uddin, T. J. Dickens, D. E. Daramola and O. I. Okoli, Adv. Mater. Interfaces, 2014, 1, 1400075 CrossRef.
- H. Yin, J. M. Fontana, J. Solandt, J. I. Jussi, H. Xu, H. Brismar and Y. Fu, Int. J. Nanomed., 2017, 12, 2781–2792 CrossRef PubMed.
- S. Singh, A. Chakraborty, V. Singh, A. Molla, S. Hussain, M. K. Singh and P. Das, Phys. Chem. Chem. Phys., 2015, 17, 5973–5981 RSC.
- Y. Chen, M. Molnar, L. Li, P. Friberg, L. M. Gan, H. Brismar and Y. Fu, PLoS One, 2013, 8, e83805 Search PubMed.
- J. M. Tsay, M. Trzoss, L. Shi, X. Kong, M. Selke, M. E. Jung and S. Weiss, J. Am. Chem. Soc., 2007, 129, 6865–6871 CrossRef CAS PubMed.
- L. Li, J. F. Zhao, N. Won, H. Jin, S. Kim and J. Y. Chen, Nanoscale Res. Lett., 2012, 7, 386 CrossRef PubMed.
- S. Gosk, T. Moos, C. Gottstein and G. Bendas, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 854–863 CrossRef CAS PubMed.
- Q. Zhan, W. Yue and H. Shaoshan, Braz. J. Med. Biol. Res., 2011, 44, 489–490 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |