
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA coiled-coil strategy for the directional display of multiple proteins on the surface of iron oxide nanoparticles
Brandon J. Hilla,
Yaolin Xub,
Jennifer Sherwoodb,
Andrew D. Raddatzb,
Yonghyun Kim b,
Yuping Baob and
Carol Duffy*a
b,
Yuping Baob and
Carol Duffy*a
aDepartment of Biological Sciences, University of Alabama, Tuscaloosa, AL 35487, USA. E-mail: cduffy3@ua.edu
bDepartment of Chemical and Biological Engineering, University of Alabama, Tuscaloosa, AL 35487, USA
First published on 20th February 2017
Abstract
Current methods for attachment of proteins to inorganic nanoparticle surfaces utilize strategies that can destroy the tertiary structure of large proteins and/or do not allow for directional attachment, thereby diminishing functionality. We report a novel approach for the conjugation of proteins to iron oxide nanoparticles (IONPs) through coiled-coil interactions. With this strategy, coiled-coiled interactions drive attachment of the desired protein, fused to a K-coil peptide, and E-coil peptides linked to the IONP surface via a nanoparticle attachment peptide (NAP). Importantly, this method allows for directional attachment of proteins without the use of either chemical conjugation or extreme pH solutions and, thus, allows maintenance of correct protein structure with functional orientation relative to the IONP surface. In our studies, monomeric red fluorescent protein (mRFP) and enhanced green fluorescent protein (EGFP) were used as model proteins to demonstrate the coiled-coil approach can be utilized to attach large proteins to IONP surfaces while maintaining complex tertiary structures. These proof-of-concept studies showed EGFP- and mRFP-K-coil fusion proteins interacted specifically with E-coil-coated nanoparticles and multiple different proteins could be displayed on individual IONPs. We did not observe fluorescence after conjugation of fluorescent proteins to either E-coil-coated IONPs or dopamine-coated IONPs, which was likely due to quenching or FRET activity induced by the iron oxide core. However, substituting an E-coil-bound chromatography resin for E-coil-coated IONPs allowed the preservation of protein fluorescence following coiled-coil attachment of K-coil-fused mRFP, demonstrating the coiled-coil attachment strategy provided maintenance of the mRFP tertiary structure required for fluorescence. The coiled-coil nanoparticle attachment strategy developed herein should have broad applications in nanomedicine.
Introduction
Iron oxide nanoparticles (IONPs) have been studied considerably for use in biomedical applications such as magnetic delivery, magnetic resonance imaging (MRI) contrast enhancement, hypothermia, and targeted drug delivery.1–4 With these goals in mind, much attention has been directed towards developing more effective conjugation strategies for the generation of multifunctional nanoparticles (NPs). An ideal strategy would (1) utilize a mechanism that allows stable attachment of multiple large biological molecules (enzymes, antibodies, receptor ligands, drugs) to the NP surface, (2) preserve the structure and function of the attached molecules, and (3) control the orientation of the attached molecules relative to the nanoparticle surface to promote downstream functions such as cell-specific targeting via receptor binding. Despite recent advances in nanotechnology, development of such a nanoparticle platform continues to remain a major challenge due to limitations associated with NP surface functionalization. The limitations are influenced by a combination of bio/physiochemical properties (size, shape, structure, and surface chemistry) shared by both the NP and the desired biological molecules.5Current technologies for the attachment of biological molecules to nanoparticle surfaces generally fall into the following categories. Covalent chemical attachment mechanisms include 1-ethyl-3-(3 dimethylaminopropyl) carbodimide hydrochloride (EDC)-mediated conjugation of amines to carboxyls and N-hydroxysuccinimide (NHS)-based conjugation to amines and carboxylic groups.6,7 Catechol conjugation involves the interaction of catechol groups on nanoparticle surfaces with biological molecules through Michael addition and/or Schiff base reactions.8,9 Biotin–streptavidin conjugation involves the interaction of biotin-labeled biological molecules with streptavidin-coated nanoparticles.10,11 Common challenges observed with these strategies include the requirement of specific reaction conditions that can disrupt the structure and function of the attached biological ligands. Although biotin–streptavidin conjugations can be conducted under near physiological conditions, decreased specificity due to background interference is a concern with this attachment strategy due to the high occurrence of biotin in biological materials. In addition, while biotin–streptavidin conjugation allows for the directional attachment of biological molecules to the particle surface, controlled directional attachment is not possible with EDC–NHS or catechol conjugation strategies in which attachment is spatially random.
Here we present a novel conjugation strategy that utilizes coiled-coils to overcome existing challenges associated with controlling directionality and maintaining functionality of proteins during attachment to magnetic IONP surfaces. The alpha-helical coiled-coil is a universal oligomerization domain.12,13 The individual peptides that comprise the coiled-coil each contain a number of heptad repeats (repeated sequence of seven amino acids). Complementary nonpolar residues lie at the interface of the two helices away from the solvent and stabilize the coiled-coil through hydrophobic and van der Waals interactions.14 Residues that flank the hydrophobic interface are often charged and promote electrostatic interactions between the helices.15,16 Formation of heterodimeric coiled-coils is promoted when the residues flanking the hydrophobic interface on the individual helices are of opposite charges. Our strategy involves the coiled-coil interaction between a glutamate coil (E-coil) linked to the NP surface via a nanoparticle attachment linker and a lysine coil (K-coil) partner genetically fused to the desired display protein. This attachment strategy provides for strong and specific binding of proteins to NP surfaces while offering the following advances over current technologies:
(1) Nanoparticle attachment occurs under physiological conditions and via a mechanism that does not damage the structure or function of the attached protein.
(2) The orientation of conjugated proteins relative to the NP surface can be controlled by genetically fusing the K-coil in a region of the protein opposite the domain required for downstream functionality.
(3) The mechanism allows for the attachment of multiple different proteins, peptides, or coil-fusion molecules to the same nanoparticle.
(4) The attachment mechanism is easily adaptable to most proteins.17–21
We envision applications of this strategy in imaging/diagnostics and therapeutics for diseases such as cancer. More importantly, by varying the types of nanoparticles (size, shape, chemical composition), one can widely expand and vary the applications for our strategy in medicine, technology, and basic research.
Materials and methods
Generation of recombinant expression constructs
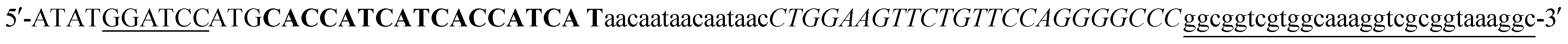 (BamHI site underlined, 6xHis in bold, 5xAsn in lower case, HRV3Cpcs in italics, NAP underlined lower case) and the reverse primer sequence was
(BamHI site underlined, 6xHis in bold, 5xAsn in lower case, HRV3Cpcs in italics, NAP underlined lower case) and the reverse primer sequence was  (HindIII site underlined, stop codon in bold). Following agarose gel extraction using the QIAquick Gel extraction kit (Qiagen, Germantown, MD), the purified PCR product and plasmid pET30a (+)-collapse (in which the upstream His-tag was previously removed via deletion of the NdeI-EcoRV sequences) were digested with BamHI and HindIII (New England Biolabs, Ipswich, MA). Following fragment purification, a ligation reaction was carried out overnight at 16 °C using T4 ligase (Promega, Madison, WI). E. coli DH10B competent cells (New England Biolabs, Ipswich, MA) were transformed with the ligation reaction, plated on LB agar plates containing 50 mg ml−1 kanamycin (Kan), and incubated overnight at 37 °C. Transformants were screened by PCR and sequenced to confirm correct sequence and orientation of the insert. E. coli BL21-CodonPlus (DE3)-RP competent cells (Agilent Technologies, Santa Clara, CA) were transformed with a clone of the recombinant plasmid, named pCD256, for overexpression of the 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil fusion protein.
(HindIII site underlined, stop codon in bold). Following agarose gel extraction using the QIAquick Gel extraction kit (Qiagen, Germantown, MD), the purified PCR product and plasmid pET30a (+)-collapse (in which the upstream His-tag was previously removed via deletion of the NdeI-EcoRV sequences) were digested with BamHI and HindIII (New England Biolabs, Ipswich, MA). Following fragment purification, a ligation reaction was carried out overnight at 16 °C using T4 ligase (Promega, Madison, WI). E. coli DH10B competent cells (New England Biolabs, Ipswich, MA) were transformed with the ligation reaction, plated on LB agar plates containing 50 mg ml−1 kanamycin (Kan), and incubated overnight at 37 °C. Transformants were screened by PCR and sequenced to confirm correct sequence and orientation of the insert. E. coli BL21-CodonPlus (DE3)-RP competent cells (Agilent Technologies, Santa Clara, CA) were transformed with a clone of the recombinant plasmid, named pCD256, for overexpression of the 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil fusion protein.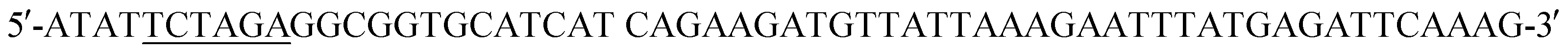 (XbaI site underlined) and the reverse primer
(XbaI site underlined) and the reverse primer  (SalI site underlined). Following purification using the QIAquick Gel Extraction Kit (Qiagen), the PCR product and pIDT-BamHI-GG-XbaI-GGGG-SalI-K-coil-HindIII were digested with XbaI and SalI (New England Biolabs, Ipswich, MA). Digested fragments were gel purified and ligated overnight at 16 °C using T4 ligase (Promega). E. coli DH10B competent cells (New England Biolabs) were transformed with the ligation reaction, plated on LB agar plates containing 50 mg ml−1 Kan, and incubated overnight at 37 °C. Transformants were screened by PCR and sequenced to confirm correct sequence and orientation of the desired clone, named pCD245. Next, the fragment of pCD245 containing mRFP and K-coil coding sequences was removed via digestion with BamHI and HindIII and subcloned into pH-MBP-3C22 cut with BamHI and HindIII. The multiple cloning site of pH-MBP-3C is downstream of a sequence encoding an N-terminal 6x His-tag fused to a maltose binding protein (MBP) gene and followed by a HRV 3C protease cleavage site. Thus, the recombinant plasmid encodes 6xHis followed by MBP, HRV3Cpcs, mRFP and eight K-coil heptad repeats (K-coil). E. coli DH10B competent cells were transformed with the ligation reaction, plated on LB agar plates containing 200 mg ml−1 ampicillin (Amp), and incubated overnight at 37 °C. Transformants were screened by PCR and sequenced to confirm correct sequence and orientation of the insert. E. coli BL21-CodonPlus (DE3)-RP competent cells (Agilent Technologies) were transformed with a clone of the desired recombinant plasmid, named pCD248, for overexpression of the 6xHis-MBP-HRV3Cpcs-mRFP-K-coil fusion protein.
(SalI site underlined). Following purification using the QIAquick Gel Extraction Kit (Qiagen), the PCR product and pIDT-BamHI-GG-XbaI-GGGG-SalI-K-coil-HindIII were digested with XbaI and SalI (New England Biolabs, Ipswich, MA). Digested fragments were gel purified and ligated overnight at 16 °C using T4 ligase (Promega). E. coli DH10B competent cells (New England Biolabs) were transformed with the ligation reaction, plated on LB agar plates containing 50 mg ml−1 Kan, and incubated overnight at 37 °C. Transformants were screened by PCR and sequenced to confirm correct sequence and orientation of the desired clone, named pCD245. Next, the fragment of pCD245 containing mRFP and K-coil coding sequences was removed via digestion with BamHI and HindIII and subcloned into pH-MBP-3C22 cut with BamHI and HindIII. The multiple cloning site of pH-MBP-3C is downstream of a sequence encoding an N-terminal 6x His-tag fused to a maltose binding protein (MBP) gene and followed by a HRV 3C protease cleavage site. Thus, the recombinant plasmid encodes 6xHis followed by MBP, HRV3Cpcs, mRFP and eight K-coil heptad repeats (K-coil). E. coli DH10B competent cells were transformed with the ligation reaction, plated on LB agar plates containing 200 mg ml−1 ampicillin (Amp), and incubated overnight at 37 °C. Transformants were screened by PCR and sequenced to confirm correct sequence and orientation of the insert. E. coli BL21-CodonPlus (DE3)-RP competent cells (Agilent Technologies) were transformed with a clone of the desired recombinant plasmid, named pCD248, for overexpression of the 6xHis-MBP-HRV3Cpcs-mRFP-K-coil fusion protein.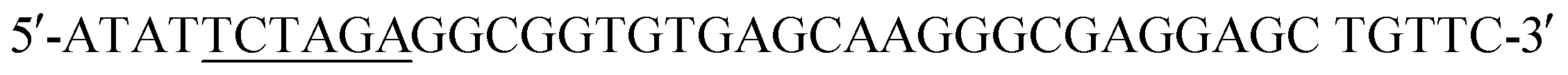 (XbaI site underlined) and the reverse primer
(XbaI site underlined) and the reverse primer 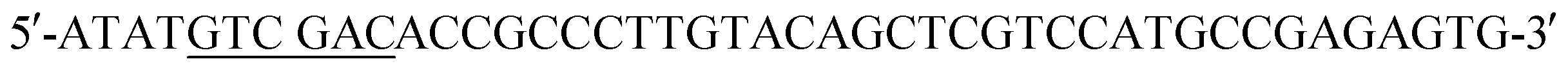 (SalI site underlined). Following gel extraction, the purified PCR product was cloned into pCD248 cut with XbaI and SalI, replacing the sequence encoding mRFP with that encoding EGFP. Ligation, transformation, clone screening and sequencing was performed as described above. E. coli BL21-CodonPlus (DE3)-RP competent cells (Agilent Technologies) were transformed with the recombinant plasmid, named pCD255, for overexpression of the 6xHis-MBP-HRV3Cpcs-EGFP-K-coil fusion protein.
(SalI site underlined). Following gel extraction, the purified PCR product was cloned into pCD248 cut with XbaI and SalI, replacing the sequence encoding mRFP with that encoding EGFP. Ligation, transformation, clone screening and sequencing was performed as described above. E. coli BL21-CodonPlus (DE3)-RP competent cells (Agilent Technologies) were transformed with the recombinant plasmid, named pCD255, for overexpression of the 6xHis-MBP-HRV3Cpcs-EGFP-K-coil fusion protein.Expression and purification of the 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil fusion protein
E. coli BL21-CodonPlus (DE3)-RP competent cells (Agilent Technologies) transformed with pCD256 were cultured in 1 l LB medium supplemented with 50 μg ml−1 Kan and 30 μg ml−1 chloramphenicol (Cm) at 37 °C with shaking (180 rpm). When the culture OD600 reached 0.6–0.7, IPTG (TEKNOVA, Hollister, CA) was added to a final concentration of 1 mM to induce protein expression. Cells were grown for an additional 4 h under the same conditions, then harvested at 9290 × g for 5 min at 4 °C. The supernatant was removed and the remaining pellet was frozen at −80 °C until protein purification.The frozen cell pellet was thawed on ice and resuspended in buffer 1 (25 mM NaP (pH 7.5), 140 mM NaCl) supplemented with 0.25 mg ml−1 lysozyme (Sigma Aldrich, St. Louis, MO) and 1 Sigma EDTA-free protease inhibitor tablet (Sigma Aldrich). The resuspended cells were incubated at 37 °C for 15 min with shaking (130 rpm), followed by cooling on ice for 5–10 min, and then sonicated briefly with a probe sonicator. The total cell lysate was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 860 × g for 20 min at 4 °C to separate soluble and insoluble cellular components. The soluble fraction was decanted and directly loaded onto 500 μl (1 ml bead slurry) Talon cobalt resin (Thermo Scientific, Rockford, IL) that had been pre-equilibrated in 10 ml buffer 1. Binding of the 6xHis-HRV3Cpcs-NAP-GRS-E-coil fusion protein to the resin took place via rocking at 4 °C for 12 h. The resin was washed in buffer 1 and then again in buffer 2 (25 mM NaP (pH 7.5), 500 mM NaCl). Each wash entailed rocking for 10 min at 4 °C followed by centrifugation and removal of the supernatant. The 6xHis-HRV3Cpcs-NAP-GRS-E-coil protein immobilized on the cobalt resin was mixed with 1 ml 1X HRV 3C protease cleavage buffer and 100 μl HRV 3C protease enzyme (EMD Chemicals, San Diego, CA). Protease cleavage took place via rotation for 24 h at 4 °C. Gravity flow through a disposable 5 ml polypropylene column was then used to recover the eluate-containing purified NAP-GRS-E-coil. E-coil remaining in the void volume was recovered by gravity flow of 200 μl of 1X HRV 3C buffer through the column. The eluate was dialyzed against 1.5 l dialysis buffer 1 (25 mM Tris (pH 7.5), 75 mM NaCl) for 20–21 h, then against two changes of 1.5 l dialysis buffer 2 (10 mM Tris (pH 7.5), 35 mM NaCl) for 5 h per change. Lastly, the eluate was dialyzed against two changes of 1.5 l dialysis buffer 3 (10 mM ammonium bicarbonate) for 4–5 hours per change. Purified NAP-GRS-E-coil was stored at −80 °C followed by lyophilization using a LABCONCO FreeZone 4.5 Liter Benchtop Freeze Dry System (Model 77500).
860 × g for 20 min at 4 °C to separate soluble and insoluble cellular components. The soluble fraction was decanted and directly loaded onto 500 μl (1 ml bead slurry) Talon cobalt resin (Thermo Scientific, Rockford, IL) that had been pre-equilibrated in 10 ml buffer 1. Binding of the 6xHis-HRV3Cpcs-NAP-GRS-E-coil fusion protein to the resin took place via rocking at 4 °C for 12 h. The resin was washed in buffer 1 and then again in buffer 2 (25 mM NaP (pH 7.5), 500 mM NaCl). Each wash entailed rocking for 10 min at 4 °C followed by centrifugation and removal of the supernatant. The 6xHis-HRV3Cpcs-NAP-GRS-E-coil protein immobilized on the cobalt resin was mixed with 1 ml 1X HRV 3C protease cleavage buffer and 100 μl HRV 3C protease enzyme (EMD Chemicals, San Diego, CA). Protease cleavage took place via rotation for 24 h at 4 °C. Gravity flow through a disposable 5 ml polypropylene column was then used to recover the eluate-containing purified NAP-GRS-E-coil. E-coil remaining in the void volume was recovered by gravity flow of 200 μl of 1X HRV 3C buffer through the column. The eluate was dialyzed against 1.5 l dialysis buffer 1 (25 mM Tris (pH 7.5), 75 mM NaCl) for 20–21 h, then against two changes of 1.5 l dialysis buffer 2 (10 mM Tris (pH 7.5), 35 mM NaCl) for 5 h per change. Lastly, the eluate was dialyzed against two changes of 1.5 l dialysis buffer 3 (10 mM ammonium bicarbonate) for 4–5 hours per change. Purified NAP-GRS-E-coil was stored at −80 °C followed by lyophilization using a LABCONCO FreeZone 4.5 Liter Benchtop Freeze Dry System (Model 77500).
Samples collected during the various stages of protein purification were examined via SDS-PAGE. Because purified NAP-GRS-E-coil protein runs higher than expected on denaturing SDS-PAGE, matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometry was performed using a Bruker Daltonic Ultraflex MALDI TOF/TOF mass spectrometer (Mass Spec Facility, University of Alabama) to verify the purified NAP-GRS-E-coil peptide was the correct size (5.8 kDa).
Expression and purification of the K-coil fusion proteins
For expression of mRFP- and EGFP-K-coil fusion proteins, E. coli BL21-CodonPlus (DE3)-RP competent cells (Agilent Technologies) transformed with the recombinant plasmids pCD248 or pCD255, respectively, were cultured at 30 °C in 1 l LB medium supplemented with 30 μg ml−1 of carbenicillin (Cellgro, Manassas, VA) and 30 μg ml−1 of Cm (Fisher Scientific, Fair Lawn, NJ). When the culture OD600 reached 0.3, IPTG was added to a final concentration of 0.3 mM to induce protein expression and cells were grown for an additional 8 h at 25 °C. Cells were then harvested at 9290 × g for 5 min at 4 °C. The supernatant was removed and the cell pellet was frozen at −80 °C until protein purification.The frozen cell pellet was resuspended in buffer 1 supplemented with 20 mg ml−1 egg white lysozyme (Sigma Aldrich) as well as one Sigma Fast EDTA-free protease inhibitor tablet (Sigma Aldrich). Resuspended cells were incubated at 37 °C for 15 min, followed by cooling on ice for 5 min, and then sonicated briefly with a probe sonicator. The cell lysate was centrifuged at 17000 × g for 45 min at 4 °C. The soluble fraction was decanted and mixed with 100 μl (200 μl slurry) Talon cobalt resin (Thermo Scientific) that had been pre-equilibrated in 10 ml buffer 1. Binding of the K-coil fusion proteins to the resin took place via rocking 30 min at 4 °C. The resin was then washed twice in buffer 1, three times in buffer 3 (12 mM NaP (pH 7.2–7.5) 1.14 M NaCl), and once in buffer 1. Each wash consisted of gentle rocking at 4 °C for 10 min followed by centrifugation and removal of the supernatant. The K-coil fusion proteins immobilized on the cobalt resin were mixed with 500 μl 1X HRV 3C protease cleavage buffer and 25 μl HRV 3C protease enzyme (EMD Chemicals). Protease cleavage took place via rocking for 15 h at 4 °C. The eluate was dialyzed against 1.5 l dialysis buffer 1 for 20 h, then against 1.5 l dialysis buffer 2 for 8 h.
Samples collected during the various stages of protein purification were examined via SDS-PAGE. To confirm mRFP- and EGFP-K-coil fusion proteins were properly folded, fluorescence intensity was measured using a Cary Eclipse fluorescence spectrophotometer and photos of samples excited with a standard UV exposure box were taken with a digital camera.
Iron oxide nanoparticle ligand exchange
IONP synthesis and ligand exchange were performed according to the method of Xu et al.23 Specifically, 200 μl IONP stock solution (1 mg ml−1) was mixed with 2 mg lyophilized NAP-GRS-E-coil in 9.8 ml DMSO. The molar ratio of NAP-GRS-E-coil molecules to IONP NAP binding sites was roughly 5:1. The suspension was briefly sonicated to disperse NPs and mixed for 48 h at room temperature using a shaking incubator. NAP-GRS-E-coil-coated NPs (E-coil-NPs) were then magnetically collected and re-dispersed in 1 ml sterile water (pH = 7.0) for characterization. The ligand exchange process for preparation of polyacrylic acid-coated NPs (PAA-NPs) was performed using the method described previously.23 Specifically, 1 ml stock solution (5 mg ml−1) of IONPs was mixed with 90.5 mg of 5000 MW PAA in 49 ml DMSO. The molar ratio of the PAA ligand to NP Fe surface atoms was roughly 5:1. After 48 h of mixing at room temperature, the IONPs were magnetically collected and re-dispersed in water (pH = 7.0) at a concentration of 1 mg ml−1 for characterization.
Characterization of E-coil-coated iron oxide nanoparticles
The morphology and size of E-coil-IONPs were studied using a Hitachi 7860 transmission electron microscope. The hydrodynamic size, measured via dynamic light scattering (DLS), and surface charge, measured via zeta potential, of E-coil-IONPs and E-coil-IONPs conjugated to mRFP-K-coil were measured in water or dialysis buffer 2 using a Zetasizer Nano ZS system (Malvern Instruments, Worcestershire, UK).Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting
Protein samples were mixed with 2X SDS-PAGE sample buffer (New England Biolabs) and incubated for 5 min at 100 °C prior to separation by 12% or 15% denaturing SDS-PAGE. Gels were stained with Coomassie Brilliant Blue (Bio-Rad, Hercules, CA) to visualize proteins. For immunoblotting, proteins were transferred to a nitrocellulose membrane (GE Water and Process Technologies, Pittsburg, PA) using a wet electroblotting system (Bio-Rad) then blocked for several hours with 5% (w/v) non-fat dry milk in PBS-T (0.1% Tween-20 in phosphate buffered saline). Membranes were then washed three times with PBS-T prior to rocking overnight at 4 °C with primary antibodies diluted in 1% (w/v) bovine serum albumin in PBS-T. Primary antibodies used include rabbit polyclonal anti-RFP (diluted 1:5000, Rockland 600-401-379) and goat polyclonal anti-GFP (diluted 1:1000, Rockland 600-101-215). Following three 10 min washes in PBS-T, membranes were rocked for 2 h at 4 °C with horseradish peroxidase-conjugated secondary antibodies diluted in 1% (w/v) bovine serum albumin in PBS-T. Secondary antibodies used include donkey anti-rabbit IgG (diluted 1:5000, GE Healthcare NA934V) and rabbit anti-goat IgG (diluted 1:5000, Rockland Immunochemicals 605-4302). Following three 10 min washes in PBS-T, membranes were developed with either the Pierce ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL) or Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare, Little Chalfont Buckinghamshire, UK) followed by exposure to autoradiography film (Amersham Hyperfilm ECL, GE Healthcare, Little Chalfont Buckinghamshire, UK).
Specificity of mRFP-K-coil conjugation to E-coil-coated IONPs
To assess the specificity of protein attachment to IONPs via the E/K coiled-coil interaction, 1 mg of PAA-IONPs and 1 mg E-coil-IONPs were mixed in separate reactions with 0.6 mg mRFP-K-coil in a total volume of 4 ml IONP buffer (0.6 mM Tris (pH 7.5), 3.5 mM NaCl) for a mRFP-K-coil:IONP molar ratio of 60:1. The reactions were rocked overnight at 4 °C. Conjugated IONPs were magnetically separated and washed with IONP buffer to remove unbound proteins. IONPs were then resuspended in 1 ml IONP buffer and 100 μl was removed and mixed with 100 μl of 2X SDS-PAGE sample buffer. Samples were boiled for 5 min at 100 °C, resolved by 12% SDS-PAGE and subjected to immunoblotting using an anti-RFP polyclonal antibody.
Fluorescence of mRFP-K-coil conjugated to E-coil-coated resin
The 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil fusion protein was expressed and bound to the Talon cobalt resin as described above. Approximately 200 μg purified mRFP-K-coil was mixed, in separate reactions, with 100 μl 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil-bound resin and 100 μl of pre-equilibrated unbound resin in 900 μl total volumes. The mixtures were rocked at 4 °C for 18.5 h then washed 3 × 10 min at 4 °C in 10 ml buffer 1. Following centrifugation and removal of the supernatant, resin samples were resuspended in 200 μl buffer 1 and 150 μl was immediately transferred to a 96-well plate (BRANDTECH, Scientific, INC, Wertheim, Germany). Fluorescence intensity was measured using a Synergy H1 Hybrid Multi-Mode Microplate Reader (Biotech Winooski, VT) at fixed excitation and emission wavelengths of 550 nm and 620 nm, respectively. Capture of RFP-K-coil on unbound and 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil-bound resin was confirmed by immunoblotting using an anti-RFP antibody (Rockland Immunochemicals) following separation of the above samples by 12% SDS-PAGE.Characterization of mRFP-K-coil-conjugated dopamine-coated IONPs
To examine the potential quenching of mRFP by IONPs, 0.5 mg of activated, dopamine-coated IONPs was mixed with 85 mg of purified mRFP-K-coil in a total volume of 650 μl buffer 4 (10 mM Tris (pH 7.5), 10 mM NaCl) by rocking at 4 °C for 4 h. After centrifugation at 4 °C for 10 min at 21000 × g and two washes with buffer 4 to remove unbound mRFP-K-coil, the conjugated IONPs were dispersed in 1 ml buffer 4. The fluorescence of mRFP-K-coil conjugated to dopamine-coated IONPs was measured using a Cary Eclipse fluorescence spectrophotometer. The hydrodynamic size and surface charge of unconjugated and mRFP-K-coil-conjugated dopamine-coated IONPs were evaluated in water using a Zetasizer Nano ZS (Malvern Instruments). The NPs were then pelleted by centrifugation at 16000 × g for 5 minutes, resuspended in 100 μl SDS-PAGE sample buffer and analyzed via immunoblotting with an anti-RFP antibody (Rockland Immunochemicals).
Co-immunoprecipitation
To determine whether multiple different proteins could be directionally attached via coiled-coils to a single IONP, we performed co-immunoprecipitation experiments using mRFP-K-coil and EGFP-K-coil as model proteins. IONPs were coated with purified NAP-GRS-E-coil as described above. E-coil-IONPs (1.5 mg) were mixed with mRFP-K-coil (0.3 mg) and EGFP-K-coil (0.3 mg) in a total volume of 4 ml buffer 5 (0.6 mM Tris (pH 7.5), 3.5 mM NaCl) for a mRFP-K-coil:EGFP-K-coil:E-coil-IONP molar ratio of 30:30:1. The mixture was rocked overnight at 4 °C. The NPs were then magnetically separated, washed twice with buffer 5, resuspended in 1 ml of the same buffer, and incubated overnight at 4 °C with 10 μl polyclonal anti-GFP antibody. The immunocomplex was captured by adding pre-washed protein G-coated sepharose (100 μl) followed by gentle rocking for 4 hours at 4 °C. The sepharose resin was collected by pulse centrifugation and washed four times in 800 μl ice-cold sterile PBS containing 0.05% Tween-20. The sepharose resin was boiled in 60 μl 2X SDS loading buffer for 5 min to dissociate and denature immunocomplexes, then pelleted by centrifugation. The supernatants were resolved by 12% SDS-PAGE and subjected to immunoblotting using antibodies specific to mRFP and EGFP. Purified mRFP-K-coil and EGFP-K-coil were run as controls for specificity of the anti-RFP and anti-GFP antibodies. E-coil-IONPs (1.13 mg) were mixed with mRFP-K-coil (0.2 mg) and EGFP-K-coil (0.2 mg) in the absence of the anti-GFP antibody to ensure the conjugated IONPs did not bind the protein G-coated sepharose directly. E-coil-IONPs (0.75 mg) were also mixed with mRFP-K-coil (0.3 mg) in the absence of EGFP-K-coil, then immunoprecipitated with the anti-GFP antibody to ensure the immunocomplex did not form in the absence of EGFP-K-coil.
Flow cytometry
E-coil-IONPs (200 μg) diluted in 150 μl dialysis buffer 2 were briefly sonicated with a probe sonicator, mixed with 130 μg purified mRFP-K-coil in a total volume of 800 μl, and rotated at 4 °C for 20.5 hours. Unbound mRFP-K-coil was removed via dialysis against dialysis buffer 2 for 20 h in a Float-A-Lyzer G2 Dialysis Device (100 MWCO) (Spectrum Labs, Rancho Dominquez, CA). Approximately 100 μl E-coil-IONPs conjugated to mRFP-K-coil were removed from the dialysis device and diluted in 400 μl dialysis buffer 2 for detection by flow cytometry. Unconjugated E-coil-IONPs diluted in dialysis buffer 2 were used as a negative control. Flow cytometry analyses were performed in an S3e Cell Sorter equipped with 488 nm/561 nm dual lasers and ProSort Software (BIO-RAD Laboratories, Hercules, CA) by counting 10000 total events per run per sample, followed by data analysis using FlowJo software (v.10.2, FlowJo LLC, Ashland, OR).
Results and discussion
Expression, purification, and IONP conjugation of the E-coil fusion protein
Many of the current NP bioconjugation strategies require chemical conditions that can destroy the tertiary structure of larger proteins and/or do not allow for directional attachment critical for downstream function. In this study, E/K coiled-coil fusion domains were examined for the ability to facilitate controlled, directional attachment of proteins to IONPs under physiological conditions. For proof-of-concept purposes, we utilized monomeric red fluorescent protein (mRFP) and enhanced green fluorescent protein (EGFP) for IONP coiled-coil conjugation. Because the fluorescence of these proteins is inherent to and dependent upon their tertiary structure, we reasoned retention of their inherent fluorescence could be used as a proxy for retention of protein tertiary structure. We first designed an expression construct that encoded a six-histidine tag (6xHis) genetically fused to a five-asparagine linker (5xAsn), an HRV 3C protease cleavage site (HRV3Cpcs), a nanoparticle attachment peptide (NAP), a glycine-rich spacer (GRS), and a glutamate coil (E-coil) (Fig. 1A). The 6xHis tag facilitated protein purification via affinity chromatography with a cobalt resin. The HRV3Cpcs provided a site at which the fusion protein could be cleaved from the purification resin. The E-coil consisted of a 5× repeat of the amino acid heptad sequence valine (V), serine (S), alanine (A), leucine (L), glutamate (E), glutamine (Q), glutamate (E). The NAP upstream of the E-coil is comprised of arginine, lysine, glycine and proline residues; the arginine and lysine residues conjugate the fusion protein to IONPs through coordination bonds while proline residues promote outward orientation of the downstream E-coil.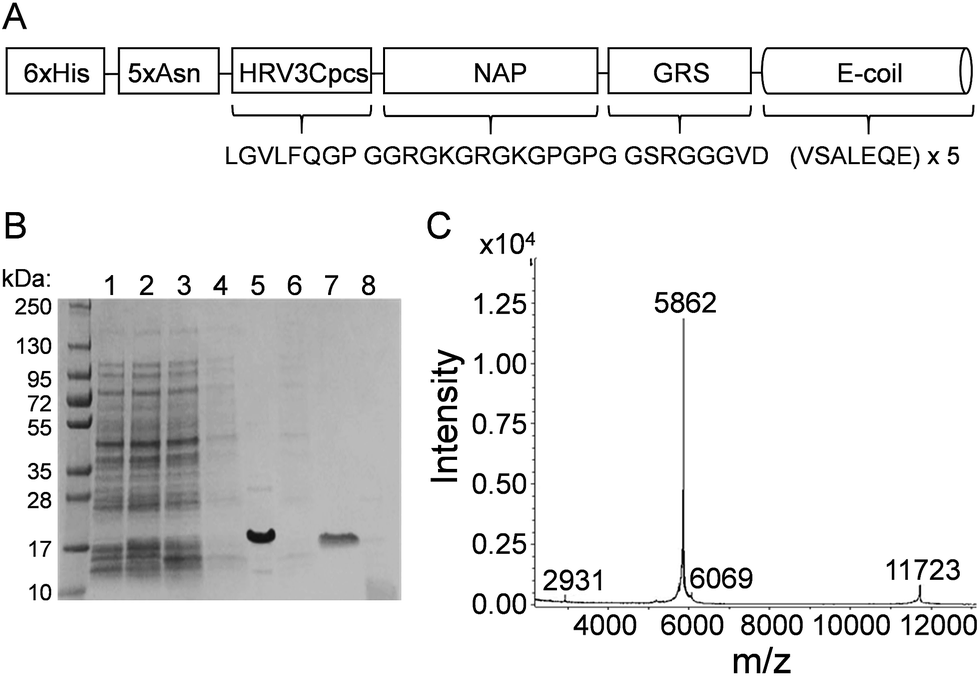 | ||
| Fig. 1 E-coil fusion protein design and purification. (A) Schematic of the 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil fusion protein. Following HRV 3C protease cleavage, the NAP-GRS-E-coil fusion protein was linked to IONPs via the NAP for coiled-coil NP conjugation studies. (B) Coomassie Brilliant Blue-stained SDS-PAGE gel showing the expression and purification of the NAP-GRS-E-coil fusion protein. A molecular weight marker is shown in the left-hand lane. Lane 1, uninduced cell lysate; 2, cell lysate following IPTG induction prior to freezing pellet; 3, total cell lysate following treatment with lysozyme and sonication; 4, soluble lysate following centrifugation; 5, 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil fusion protein bound to cobalt resin; 6, resin flow-through; 7, purified NAP-GRS-E-coil fusion protein post HRV 3C protease cleavage; 8, cobalt resin post HRB 3C protease cleavage. (C) Mass spectrum of purified NAP-GRS-E-coil fusion protein. | ||
The 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil was expressed in E. coli BL21-CodonPlus (DE3)-RP cells and purified via affinity chromatography followed by HRV 3C cleavage. Protein samples collected at various stages of the purification process were separated by SDS-PAGE and visualized by Coomassie Brilliant Blue staining (Fig. 1B). The expression-purification protocol developed provided highly pure protein (Fig. 1B, lane 7) at a yield of approximately 1 mg per liter culture. Following HRV 3C cleavage from the purification resin the expected molecular weight of the NAP-GRS-E-coil fusion protein was 5861.3 daltons, but migrated via SDS-PAGE more slowly than expected. However, with some proteins SDS-PAGE does not provide an accurate estimate of molecular weight. Therefore, we performed MALDI-TOF mass spectrometry post-dialysis (Fig. 1C). According to the spectrum, the NAP-GRS-E-coil fusion protein was highly pure and the expected molecular weight.
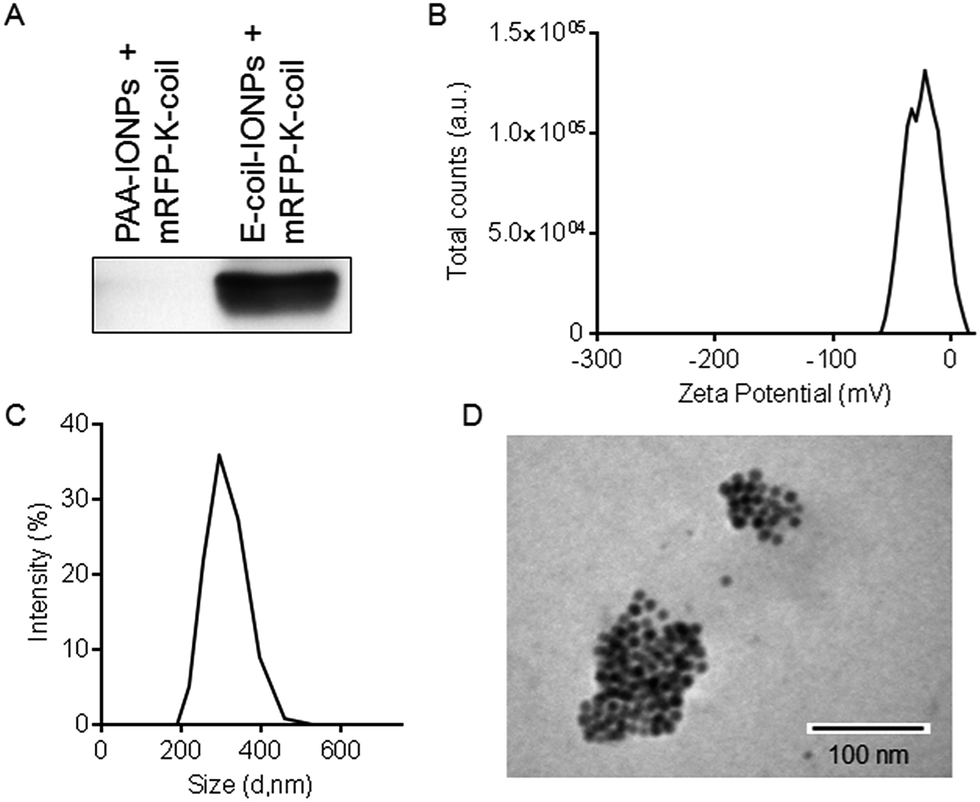
Following purification, the NAP-GRS-E-coil fusion protein (hereafter referred to as E-coil) was conjugated to IONPs produced as previously described.23 IONPs generated with this protocol are typically 12 nm in diameter. NP conjugation via the NAP fusion protein domain was performed using a previously reported ligand exchange protocol.23 To promote efficient interaction with the K-coil fusion partner, E-coil-coated IONPs (E-coil-IONPs) must be well-dispersed in solution with minimal to no aggregation. Dynamic light scattering (DLS) analysis was performed with E-coil-IONPs to characterize their size and dispersion in water. The E-coil-IONPs were larger than expected (220 nm) indicating formation of aggregates which was confirmed via transmission electron microscopy (Fig. 2A and C). The possibility of electrostatic interactions attributing to such rapid agglomeration is unlikely as the negative surface charge of the E-coil-IONPs should aid dispersion through repulsion. Such agglomeration could be attributed to the inherent magnetic properties of the iron oxide nanoparticles used in these studies and/or to insufficient sonication prior to DLS measurements. This is important considering DLS is extremely sensitive towards the presence of small aggregates in solution. To determine electrical charge properties and stability of E-coil-IONPs in water, zeta-potential measurements were performed using polyacrylic-acid-coated IONPs (PAA-IONPs) as a control. As expected, a net negative surface charge was observed following E-coil conjugation due to the presence of the glutamate residues within the E-coil heptad repeat. IONPs are generally considered stable if their zeta potential is >30 mV or <−30 mV. An absolute value of −44.3 mV was observed for the E-coil-IONPs, indicating high stability in solution (Fig. 2B).
 | ||
| Fig. 2 Characterization of E-coil-IONPs. (A) Dynamic light scattering (DLS) measurements of E-coil-IONPs and PAA-IONPs. (B) Zeta potential measurements of E-coil-IONPs and PAA-IONPs. (C) Transmission electron micrograph of E-coil-IONPs. | ||
Expression and purification of mRFP and EGFP K-coil fusion proteins
The mRFP-K-coil and EGFP-K-coil expression constructs included coding sequences for a 6xHis, maltose binding protein (MBP), HRV3Cpcs, mRFP or EGFP, and a K-coil (Fig. 3A). The K-coil consisted of an 8× repeat of the amino acid heptad sequence valine (V), serine (S), alanine (A), leucine (L), lysine (K), glutamine (Q), lysine (K). K-coil fusion proteins were expressed in E. coli BL21-CodonPlus (DE3)-RP cells and purified to homogeneity via affinity chromatography using a cobalt resin. Following cleavage from the resin with HRV 3C protease, the predicted molecular weights for mRFP-K-coil and EGFP-K-coil were 32.47 kDa and 33.95 kDa respectively. Both proteins migrated more quickly than expected with SDS-PAGE (Fig. 3B). This observation was consistent with a previous study that described the susceptibility of K-coils to E. coli proteases.21 Despite some apparent degradation, a sufficient number of K-coil heptad repeats for coiled-coil conjugation remained intact. The fluorescence of mRFP and EGFP is inherent to their tertiary structure. To ensure the purified mRFP-K-coil and EGFP-K-coil fusion proteins were correctly folded, we measured their fluorescence emission intensity and examined their brightness under ultraviolet light (Fig. 3C). Both proteins emitted maximum fluorescence at their correct, respective wavelengths (mRFPmars: 602 nm; EGFP: 509 nm).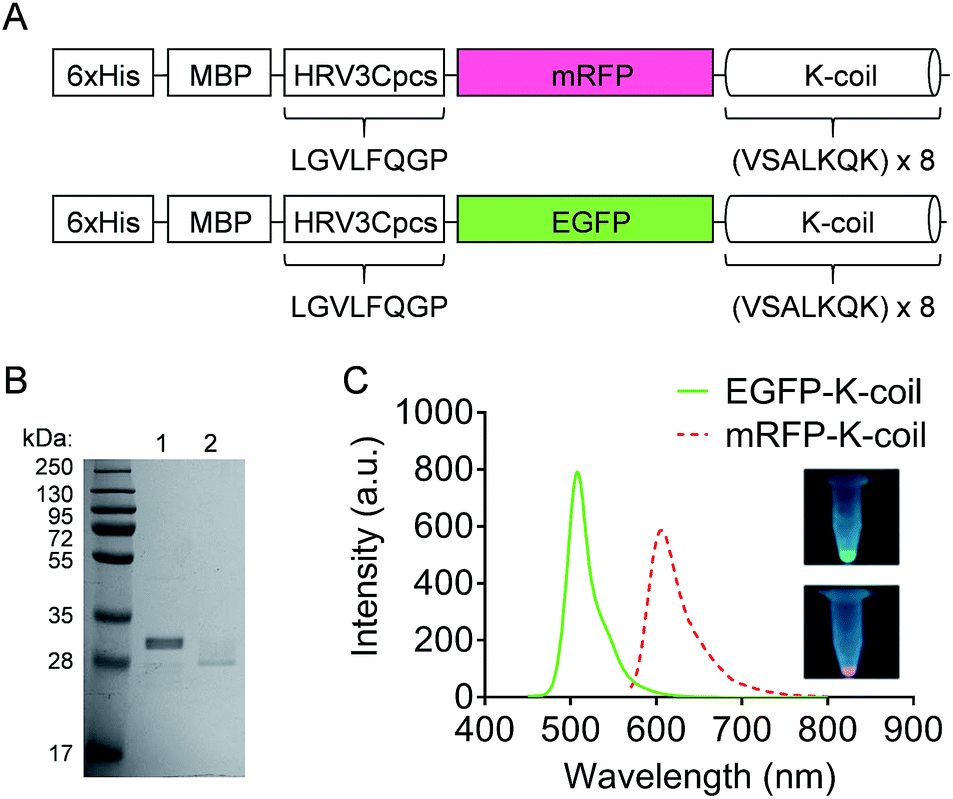 | ||
| Fig. 3 K-coil fusion protein design and purification. (A) Schematic of the 6xHis-MBP-HRV3Cpcs-mRFP-K-coil and 6xHis-MBP-HRV3Cpcs-EGFP-K-coil fusion proteins. Following HRV 3C protease cleavage, the mRFP-K-coil and EGFP-K-coil fusion proteins were used in coiled-coil IONP conjugation studies. (B) Coomassie Brilliant Blue-stained SDS-PAGE gel showing purification of mRFP-K-coil and EGFP-K-coil fusion proteins. A molecular weight marker is shown in the left-hand lane. Lane 1, purified mRFP-K-coil post HRV 3C protease cleavage and dialysis; 2, purified EGFP-K-coil post HRV 3C protease cleavage and dialysis. (C) Fluorescence intensity of EGFP-K-coil (green line) and mRFP-K-coil (pink dashed line) measured by fluorescence spectrophotometry. Insets show digital photographs of purified EGFP-K-coil (top) and purified mRFP-K-coil illuminated with a UV light box. | ||
Specificity of the coiled-coil interaction between mRFP-K-coil and E-coil-IONPs
The nonpolar leucine and valine residues are positioned in E- and K-coils in a complementary manner at the interface of the two helices, forming a hydrophobic core that contributes greatly to the stability of the coiled-coil.14 Electrostatic interactions between the lysine residues of the K-coil and the glutamate residues of the E-coil also contribute to coiled-coil stability through salt bridge formation.24 The number of heptad repeats is a contributing factor in the strength of coiled-coil interactions, with a minimum of three repeats required for stable coiled-coil formation.25 The strength of the E/K coiled-coil interaction should provide a measure of specificity for the linking of proteins to NPs by withstanding the washing stages of the conjugation process.To assess the specificity of IONP protein conjugation via E/K coiled-coils, purified mRFP-K-coil was mixed with either E-coil-IONPs or PAA-IONPs at molar ratio of 60:1. Because PAA mimics the acidity of the E-coil and, therefore, could potentially interact with the K-coil, it served as an excellent control to test the specificity of coiled-coil NP conjugation. Following extensive washing and magnetic separation, proteins bound to the IONPs were separated by SDS-PAGE and immunoblotted with an antibody specific to mRFP (Fig. 4A). We found the mRFP-K-coil interacts with E-coil-IONPs, but not with PAA-IONPs, demonstrating the specificity of the coiled-coil interaction. Particle surface charge and size were also measured following mRFP-K-coil conjugation to E-coil-IONPs. The zeta potential of −44.3 mV measured with E-coil-IONPs (Fig. 2B) shifted to −21.8 mV following conjugation of RFP-K-coil (Fig. 4B). We also observed a shift in NP size from 220 nm for E-coil-IONPs (Fig. 2A) to 295 nm following attachment of mRFP-K-coil (Fig. 4C). The decrease in the zeta potential and increase in diameter is indicative of the increasing ionic strength (35 mM NaCl) of dialysis buffer 2 used during characterization of mRFP-K-coil-conjugated E-coil-NPs which may have caused some particle destabilization and aggregation. The magnetic separation process utilized in these studies may also have contributed to the size increase by enriching for aggregates that were more responsive to the magnet. A TEM image of E-coil-IONPs conjugated to mRFP-K-coil is shown in Fig. 4D.
 | ||
| Fig. 4 Specificity of E/K coiled-coil IONP conjugation. (A) Immunoblot of PAA-IONPs + mRFP-K-coil (lane 1) E-coil-IONPs + mRFP-K-coil using an antibody specific to RFP. (B) Transmission electron micrograph of mRFP-K-coil-conjugated E-coil-IONPs. (C) Dynamic light scattering (DLS) measurements of mRFP-K-coil-conjugated E-coil-IONPs. (D) Zeta potential measurements of mRFP-K-coil-conjugated E-coil-IONPs. | ||
Fluorescence of mRFP-K-coil is maintained upon conjugation to an E-coil-coated resin, but is quenched by IONPs
One of the primary objectives of this study was the development of a NP bioconjugation strategy for large proteins that retained their tertiary fold/structure. As mentioned above, mRFP and EGFP were utilized as proof-of-concept proteins with the idea that retention of their inherent fluorescence could be used as a proxy for retention of tertiary structure. However, upon conjugation of mRFP-K-coil to E-coil-IONPs, fluorescence was not detectable via fluorescence spectrophotometry (data not shown). Several studies have shown IONP iron oxide cores can induce quenching of a variety of fluorophores.11,26,27 To ensure the absence of fluorescence was not a result of misfolding or degradation of the mRFP-K-coil fusion protein upon coiled-coil conjugation, we examined the fluorescence of mRFP-K-coil upon coiled-coil conjugation to a 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil-coated cobalt resin. Following affinity purification of the 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil fusion protein via interactions between the 6xHis tag and the cobalt resin, the bound resin was mixed with purified mRFP-K-coil, washed thoroughly, and examined for fluorescence intensity. Controls included unbound resin, 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil-coated resin, unbound mRFP-K-coil, and mRFP-K-coil mixed with equilibrated cobalt resin not bound to the 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil fusion protein. We observed no significant difference in the relative fluorescence units (RFU) per μg input mRFP-K-coil between free (unbound) mRFP-K-coil and mRFP-K-coil conjugated to the 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil-coated resin (p = 0.47), indicating coiled-coil conjugation does not promote protein unfolding/degradation (Fig. 5A). Although some mRFP-K-coil was retained on the unbound resin, its fluorescence was significantly decreased compared to mRFP-K-coil retained on 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil-coated resin (p ≤ 0.0001) and was not significantly different than that observed with unbound resin or 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil-coated resin (p = 0.410 and p = 0.432, respectively), indicating non-specific binding played a very minor role in the retention of mRFP-K-coil on the 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil-coated resin. Immunoblot analyses of these samples using an antibody specific to RFP confirmed the specificity of interaction between mRFP-K-coil and the 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil-coated resin (Fig. 5B). These results, together with those shown in Fig. 4A, demonstrate coiled-coils allow for highly specific conjugation while retaining protein structure.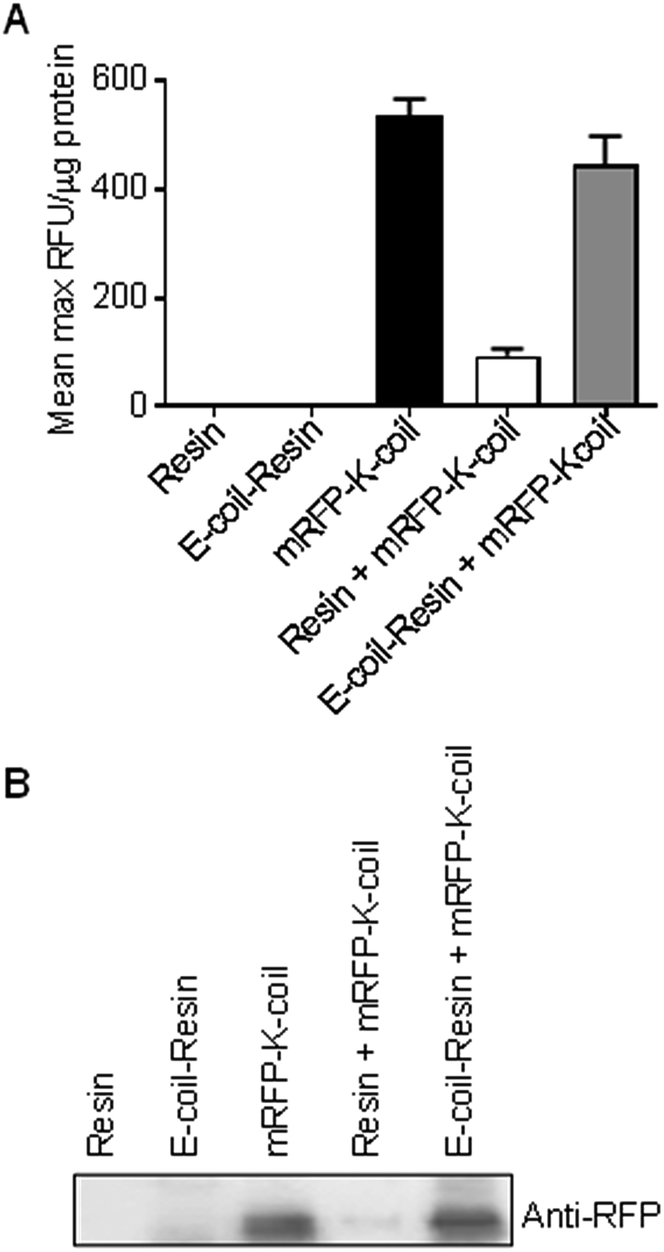 | ||
| Fig. 5 mRFP-K-coil fluorescence retention following coiled-coil conjugation to an E-coil-coated cobalt resin. (A) Mean maximum relative fluorescence per microgram mRFP-K-coil. Three measurements for each group were made via fluorescence spectrophotometry and analyzed by one-way ANOVA with Boniferroni's multiple comparisons test. Error bars represent the SEM. (B) Immunoblot analysis of cobalt resin conjugation of mRFP-K-coil using an antibody specific to RFP. Lane 1, cobalt resin; 2, 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil-coated cobalt resin; 3, purified mRFP-K-coil; 4, cobalt resin + mRFP-K-coil; 5, mRFP-K-coil conjugated to 6xHis-5xAsn-HRV3Cpcs-NAP-GRS-E-coil-coated cobalt resin. | ||
We next asked whether the lack of fluorescence upon conjugation of mRFP-K-coil to E-coil-IONPs was a result of a limited number of fluorescent proteins being conjugated to the particle surface via E/K coiled-coil interactions. Several reports have shown the successful conjugation of proteins to dopamine modified nanoparticles.28–30 For the below studies, a catechol reaction was performed between dopamine-coated IONPs and mRFP-K-coil according to the method of Xu et al.29 The catechol groups on the surface of IONPs can be easily oxidized into dopamine quinone at pH > 8.5, forming a very thin polydopamine surface layer with many catechol groups available for protein conjugation upon activation of thiol or amino groups.29 Although dopamine functionalization of IONPs does not provide for directional attachment, it does afford for the attachment of a large number of protein molecules without affecting their tertiary structure. Thus, utilizing this conjugation strategy provided a mechanism to determine if the lack of fluorescence observed with mRFP-K-coil bound to E-coil-IONPs was the result of quenching or inefficient conjugation. No fluorescence was observed by spectrophotometry for mRFP-K-coil conjugated to dopamine-coated IONPs (Fig. 6A). Fluorescence was observed for the mRFP-K-coil control indicating the input protein was folded correctly (Fig. 6A). The dopamine-conjugated mRFP-IONPs were characterized to ensure conjugation had occurred. Following SDS-PAGE of dopamine-conjugated mRFP-IONPs, mRFP-K-coil was detected via immunoblot using an antibody specific to RFP (Fig. 6B). The conjugation of mRFP-K-coil to the active dopamine surface yielded a hydrodynamic size increase from 43.8 nm to 50 nm (Fig. 6C). Furthermore, a shift from −39.5 mV to −22.2 mV was observed for the zeta potential (Fig. 6D). The above data indicate the lack of mRFP-K-coil fluorescence observed following coiled-coil conjugation to E-coil-IONPs is likely due, at least in part, to quenching of the fluorophore by the iron oxide core.
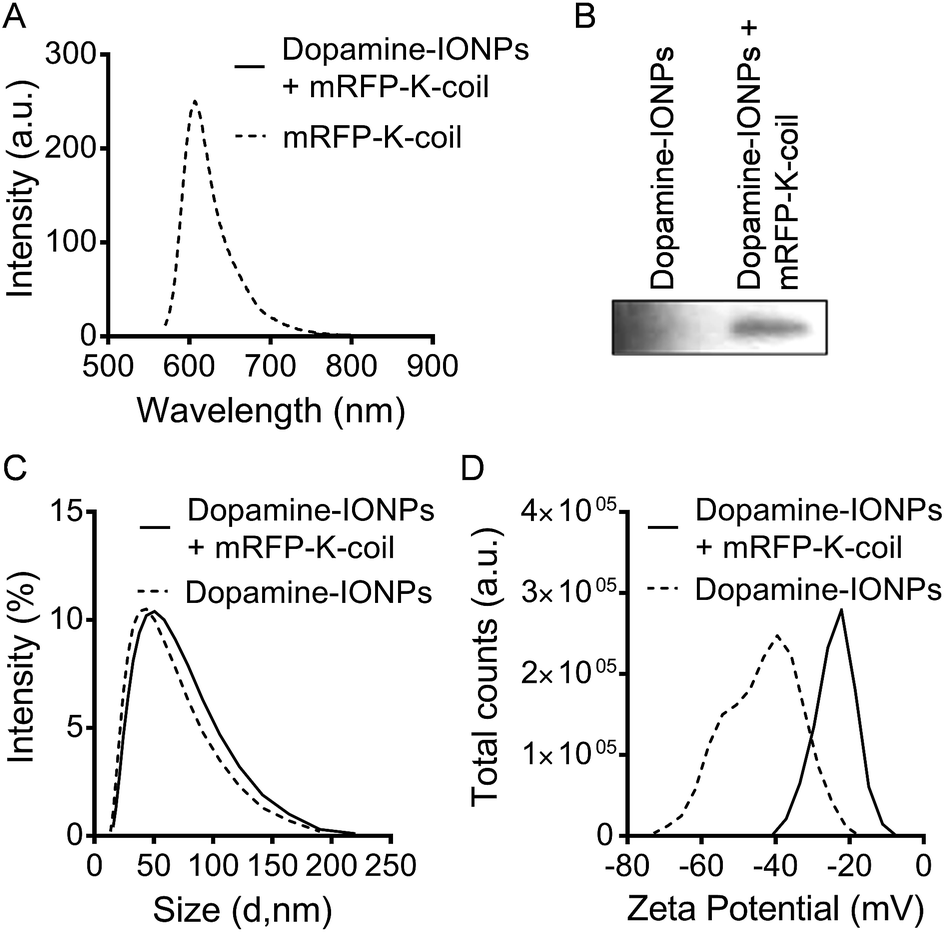 | ||
| Fig. 6 Characterization of mRFP-K-coil-conjugated dopamine-coated IONPs. (A) Fluorescence intensity of purified mRFP-K-coil and mRFP-K-coil-conjugated dopamine-coated IONPs measured by fluorescence spectrophotometry. (B) Immunoblot analysis of dopamine-coated IONPs (lane 1) and mRFP-K-coil-conjugated dopamine-coated IONPs (lane 2) using an antibody specific to RFP. (C) Dynamic light scattering (DLS) measurements of dopamine-coated IONPs and mRFP-K-coil-conjugated dopamine-coated IONPs. (D) Zeta-potential measurements of dopamine-coated IONPs and mRFP-K-coil-conjugated dopamine-coated IONPs. | ||
Fluorescence detection in the above studies was performed via spectrophotometry. To determine if a more sensitive method could detect mRFP-K-coil fluorescence following coiled-coil conjugation to E-coil-IONPs, flow cytometry was performed. Fig. 7A and B show representative scatter plots of forward scatter (FSC) and mRFP fluorescence (FL2 channel) of the E-coil-IONPs (control) and E-coil-IONPs conjugated to mRFP-K-coil (10000 events each). Although three different populations were observed, the boxed area refers to the gated population which represented 25% of the total population. According to the overlay histogram representative of the gated population in the FL2 channel (Fig. 7C), no discernible difference was observed for fluorescence between E-coil-IONPs (dotted line) and E-coil-IONPs conjugated to mRFP-K-coil (solid line). However, the FSC histogram shows there was a shift in the size for E-coil-IONPs conjugated to mRFP-K-coil compared to unconjugated E-coil-IONPs (Fig. 7D). For a more quantitative analysis, relative mean fluorescence intensity (MFI) of mRFP-K-coil-conjugate E-coil-IONPs (FL2) and size (FSC) was normalized to the control group (E-coil-IONPs). The mean fluorescence of mRFP-K-coil-conjugated E-coil-IONPs increased a modest 8% (Fig. 7E) whereas the forward scatter (size) mean intensity increased 86% compared to E-coil-IONPs (Fig. 7F). These observations indicate the mRFP-K-coil fluorescence signal was quenched dramatically upon conjugation to E-coil-IONPs. Moreover, the difference in FSC observed for the E-coil-IONPs compared to mRFP-K-coil-conjugated E-coil-IONPs is consistent with the percent size increase predicted for mRFP-K-coil conjugation, based on the DLS data presented in Fig. 2A and 4C.
 | ||
| Fig. 7 Flow cytometry analysis of mRFP-K-coil-conjugated E-coil-IONPs. (A and B) Representative scatter plot of forward scatter (FSC; size) and mRFP-K-coil fluorescence (FL2) of E-coil-IONPs (A) and mRFP-K-coil-conjugated E-coil-IONPs (B); 10000 events each. The boxed areas refer to the gated populations which were ∼25% of the total populations. (C and D) Representative plots of the gated populations in the FL2 (fluorescence) channel (C) and the FSC (size) channel (D). Dotted line: E-coil-IONPs; solid line: mRFP-K-coil-conjugated E-coil-IONPs. (E) Relative mean fluorescence intensity (MFI) of mRFP-K-coil-conjugated E-coil-IONPs normalized to E-coil-IONPs (mean ± CV). (F) Relative size of mRFP-K-coil-conjugated E-coil-IONPs normalized to E-coil-IONPs (mean ± CV). | ||
Multiple, different proteins can be conjugated to NP surfaces via E/K coiled-coil interactions
The generation of a NP platform in which different proteins could be attached to the same NP would allow for multiple functions (e.g. cell-specific targeting and endosome escape), and would have numerous biomedical applications. We hypothesized the coiled-coil conjugation mechanism would allow for the attachment of multiple different proteins, peptides, or coil-fusion molecules on the same NP. To test this idea, both mRFP-K-coil and EGFP-K-coil were mixed with E-coil-IONPs to allow coiled-coil conjugation as described above. Due to the lack of fluorescence observed with mRFP-K-coil-conjugated E-coil-IONPs, we used co-immunoprecipitation (Co-IP) analyses to determine whether individual IONPs were conjugated to both mRFP-K-coil and EGFP-K-coil. Fig. 8A shows a schematic diagram illustrating how Co-IP can be used to examine the conjugation of different proteins on NPs. An antibody specific to the EGFP “bait” protein was used to immunoprecipitate EGFP-K-coil-conjugated IONPs following binding of the antibody Fc domain to a protein G-coated sepharose resin. Once the EGFP-K-coil-conjugated IONPs were separated, they were examined for the co-conjugation of mRFP-K-coil via immunoblotting using an antibody specific to RFP. Lanes 1 and 2 of Fig. 8B shows the anti-GFP and anti-RFP antibodies utilized in these studies were not cross-reactive. Lane 3 shows EGFP-K-coil and mRFP-K-coil could be detected in the double conjugation mixture. Importantly, lane 4 shows EGFP-K-coil-conjugated E-coil-IONPs immunoprecipitated with an anti-GFP antibody were also conjugated to mRFP-K-coil. Critical controls for this type of experiment are shown in lanes 5 and 6. Non-specific binding of mRFP- and EGFP-K-coil proteins to the G protein-coated sepharose resin was examined by performing the above experiment in the absence of anti-EGFP antibody. Although a low level of non-specific binding did occur (Fig. 8B, lane 5), it is clear the EGFP and mRFP signals observed in lane 4 chiefly result from the formation of a specific immunocomplex. As an additional control for antibody specificity, E-coil-IONPs conjugated to mRFP-K-coil, but not EGFP-K-coil, were immunoprecipitated with an anti-GFP antibody. This control (lane 6) shows the immunoprecipitates examined in lane 4 were not the product of non-specific immunoprecipitation. The above data shows the coiled-coil conjugation strategy provides for the attachment of multiple different proteins to individual IONPs. | ||
| Fig. 8 Co-immunoprecipitation analysis of mRFP-K-coil and EGFP-K-coil dual conjugation to E-coil-IONPs. (A) Illustration of co-immunoprecipitation of E-coil-IONPs conjugated to both mRFP-K-coil and EGFP-K-coil using an antibody specific to GFP. (B) Immunoblot analysis of the co-immunoprecipitation experiment using antibodies specific to either GFP (top) or RFP (bottom). Lane 1, purified mRFP-K-coil fusion protein; 2, purified EGFP-K-coil fusion protein; 3, E-coil-IONPs mixed with mRFP-K-coil and EGFP-K-coil; 4, immunoprecipitation of E-coil-IONPs mixed with mRFP-K-coil and EGFP-K-coil using an antibody specific to GFP; 5, protein G sepharose precipitation of E-coil-IONPs mixed with mRFP-K-coil and EGFP-K-coil in the absence of GFP antibody; 6, immunoprecipitation of E-coil-IONPs mixed with mRFP-K-coil using an antibody specific to GFP. | ||
Conclusions
In this paper, we describe the development of a novel and facile platform for the display of multiple proteins on nanoparticles under physiological conditions. Unlike current conjugation mechanisms, this method circumvents the use of harsh chemical conditions that often destroy the structure and function of biological molecules. To show proof-of-concept, mRFP and EGFP were used as model proteins for conjugation to IONPs via E/K coiled-coil interactions. This mechanism utilizes direct linkage of E-coils to NPs through a NP attachment peptide followed by coiled-coil conjugation of K-coil fusion proteins. The rationally designed NP attachment peptide utilizes arginine and lysine residues to conjugate the E-coil to IONPs through coordination bonds and proline residues to promote outward orientation of the E-coil relative to the NP surface. Genetic fusion of the K-coil to the mRFP and EGFP model proteins provided strong and specific NP conjugation via coiled-coil interactions and did not disrupt their fluorescent biological activity, providing promise for the application of this technique to other proteins. Thus far, we have shown specificity of attachment as well as attachment of multiple different proteins to E-coil-IONPs using the coiled-coil conjugation strategy. Our findings provide a valuable mechanism to expand the use and functionality of inorganic nanoparticles in biomedical applications.Acknowledgements
We thank Dr Paul LeBlanc for advice and helpful discussions. We also thank The University of Alabama Department of Biological Sciences Optical Analysis Facility and Dr Kim Lackey, Optical Analysis Facility Coordinator. B. J. H. was supported as a Future Faculty Graduate Student on a 2010 Howard Hughes Medical Institute Precollege and Undergraduate Science Education Program Grant to The University of Alabama. This work was funded by Department of Education Graduate Assistance in Areas of National Need (GAANN) Grant No. P200A100109, National Science Foundation (NSF) Grant No. 1342388 (Y. K.), NSF Grant No. DMR 1149931 (Y. B.), and University of Alabama institutional support to C. D.References
- C. Nazli, G. S. Demirer, Y. Yar, H. Y. Acar and S. Kizilel, Colloids Surf., B, 2014, 122, 674–683 CrossRef CAS PubMed
.
- J. Giri, A. Ray, S. Dasgupta, D. Datta and D. Bahadur, Bull. Mater. Sci., 2003, 13, 387–399 CAS
- Y. X. Wang, Quant. Imag. Med. Surg., 2011, 1, 35–40 Search PubMed
- H. Ittrich, K. Peldschus, N. Raabe, M. Kaul and G. Adam, ROFO-Fortschr. Rontg., 2013, 185, 1149–1166 CrossRef CAS PubMed
- S. Hassan and A. V. Singh, J. Nanosci. Nanotechnol., 2014, 14, 402–414 CrossRef CAS PubMed
- F. Shamsipour, A. H. Zarnani, R. Ghods, M. Chamankhah, F. Forouzesh, S. Vafaei, A. A. Bayat, M. M. Akhondi, M. Ali Oghabian and M. Jeddi-Tehrani, Avicenna J. Med. Biotechnol., 2009, 1, 27–31 CAS
- L. Liu, X. Zhang, Y. Zhang, Y. Pu, L. Yin, M. Tang and H. Liu, J. Nanosci. Nanotechnol., 2013, 13, 8137–8143 CrossRef CAS PubMed
- K. Ni, X. Zhou, L. Zhao, H. Wang, Y. Ren and D. Wei, PLoS One, 2012, 7, e41101 CAS
- A. K. Yuen, G. A. Hutton, A. F. Masters and T. Maschmeyer, Dalton Trans., 2012, 41, 2545–2559 RSC
- S. Sun, M. Ma, N. Qiu, X. Huang, Z. Cai, Q. Huang and X. Hu, Colloids Surf., B, 2011, 88, 246–253 CrossRef CAS PubMed
- N. Hirata, K. Tanabe, A. Narita, K. Tanaka, K. Naka, Y. Chujo and S. Nishimoto, Bioorg. Med. Chem., 2009, 17, 3775–3781 CrossRef CAS PubMed
- B. Berger, D. B. Wilson, E. Wolf, T. Tonchev, M. Milla and P. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 8259–8263 CrossRef CAS
- E. Wolf, P. S. Kim and B. Berger, Protein Sci., 1997, 6, 1179–1189 CrossRef CAS PubMed
- R. S. Hodges, Curr. Biol., 1992, 2, 122–124 CrossRef CAS PubMed
- R. S. Hodges, J. Sodek, L. B. Smillie and L. Jurasek, Cold Spring Harbor Symp. Quant. Biol., 1973, 37, 299–310 CrossRef
- A. D. McLachlan and M. Stewart, J. Mol. Biol., 1975, 98, 293–304 CrossRef CAS PubMed
- R. A. Hanna, B. E. Garcia-Diaz and P. L. Davies, FEBS Lett., 2007, 581, 2894–2898 CrossRef CAS PubMed
- C. Boucher, J. C. Ruiz, M. Thibault, M. D. Buschmann, M. R. Wertheimer, M. Jolicoeur, Y. Durocher and G. De Crescenzo, Biomaterials, 2010, 31, 7021–7031 CrossRef CAS PubMed
- A. Demolliens, C. Boucher, Y. Durocher, M. Jolicoeur, M. D. Buschmann and G. De Crescenzo, Bioconjugate Chem., 2008, 19, 1849–1854 CrossRef CAS PubMed
- B. Steinmann, A. Christmann, T. Heiseler, J. Fritz and H. Kolmar, Appl. Environ. Microbiol., 2010, 76, 5563–5569 CrossRef CAS PubMed
- Q. Lian, S. J. Szarka, K. K. Ng and S. L. Wong, J. Biol. Chem., 2003, 278, 26677–26686 CrossRef CAS PubMed
- A. Alexandrov, K. Dutta and S. M. Pascal, BioTechniques, 2001, 30, 1194–1198 CAS
- Y. Xu, Y. Qin, S. Palchoudhury and Y. Bao, Langmuir, 2011, 27, 8990–8997 CrossRef CAS PubMed
- E. K. O'Shea, J. D. Klemm, P. S. Kim and T. Alber, Science, 1991, 254, 539–544 Search PubMed
- J. Y. Su, R. S. Hodges and C. M. Kay, Biochemistry, 1994, 33, 15501–15510 CrossRef CAS PubMed
- A. Manciulea, A. Baker and J. R. Lead, Chemosphere, 2009, 76, 1023–1027 CrossRef CAS PubMed
- E. M. de Oliveira, F. L. Coelho, M. L. Zanini, R. M. Papaleo and L. F. Campo, ChemPhysChem, 2016, 17, 1–6 CrossRef PubMed
- Y. Xu, S. Palchoudhury, Y. Qin, T. Macher and Y. Bao, Langmuir, 2012, 28, 8767–8772 CrossRef CAS PubMed
- Y. Xu, D. C. Baiu, J. A. Sherwood, M. R. McElreath, Y. Qin, K. H. Lackey, M. Otto and Y. Bao, J. Mater. Chem. B, 2014, 2, 6198–6206 RSC
- K. Turcheniuk, A. V. Tarasevych, V. P. Kukhar, R. Boukherroub and S. Szunerits, Nanoscale, 2013, 5, 10729–10752 RSC
| This journal is © The Royal Society of Chemistry 2017 |