
Ultrasensitive electrochemical detection of UO22+ based on DNAzyme and isothermal enzyme-free amplification†
Wen Yuna,
Jiaolai Jiang‡
a,
Dingzhou Caia,
Xiaofang Wanga,
Ge Sang*a,
Junsheng Liao*a,
Tiecheng Lub and
Kangping Yanb
aScience and Technology on Surface Physics and Chemistry Laboratory, Jiangyou 621908, Sichuan, China. E-mail: yunwenyunwen@126.com; jshliao711@163.com
bSichuan University, Chengdu 610065, China
First published on 18th December 2015
Abstract
A novel strategy for ultrasensitive detection of UO22+ was proposed by using a UO22+-specific DNAzyme and two free labeled DNA hairpins. The principle and detection processes of this electrochemical biosensor for amplification detection of UO22+ were described. The detection conditions of the biosensor, such as the concentration of hairpins, pH, and the incubation time of HCR, were optimized. Under optimal conditions, this biosensor demonstrated a high sensitivity of UO22+ detection with the dynamic concentration range spanning from 0.05 to 4 nM and a detection limit of 20 pM at the 3Sblank level, providing a promising strategy for on site and real-time monitoring of uranium.
Introduction
Designing metal ion sensors which are comparable to the most sensitive and selective analytical instrument has always been a great challenge for a long time. Many metal ions are similar in charge, ionic radius and other properties, making them difficult to detect at ultra-low concentration with no interference by other metal ions.1–3 Because of its high toxicity and radioactivity, uranium has attracted much attention in the past half-century. Since uranium has been significantly enriched and widely used in nuclear energy generation and nuclear weapons, human beings have a high chance of being exposed to the circumstance of uranium, which can cause long-term health damage.4–6 Assessment of contamination condition of uranium and monitoring of the environmental remediation requires a simple and portable method for onsite and real-time detection of uranium. At present, many techniques have been developed for routine uranium detection, including X-ray fluorescence spectroscopy,7 inductively coupled plasma atomic emission spectroscopy (ICP-AES),8 high-performance liquid chromatography (HPLC),9 and mass spectrometry (MS).10 Most instrument-based methods with high sensitivity and selectivity are high cost, requiring expensive instrument and tedious pretreatment with poor portability, making them only conduct in well equipped analytical laboratories. Developing a simple, convenient and ultrasensitive method for on-site determination of uranium has become increasingly necessary and desirable.11 Although some chemical and biological sensors for uranyl ion (UO22+), the most stable chemical form of uranium in water, have been developed.12 Most of them cannot match instrument-based methods in sensitivity or selectivity.13–17DNAzymes are catalytic DNA sequences derived via in vitro selection from a large DNA library.18 Most DNAzymes require metal ion cofactors for structure and function, and many of them show high binding affinity and specificity.19 This high specificity has been used to convert these DNAzymes into highly selective biosensors for metal ions.20–22 Recently, Lu and co-worker reported a catalytic beacon sensor for uranyl based on an in vitro-selected UO22+-specific DNAzyme with high sensitivity.12 Furthermore, uranyl specific DNAzyme and gold nanoparticles were also reported by them for uranium sensors in extracellular and intracellular environment.23,24 Recently, DNAzyme as a specific recognization element has been customized for uranyl detection with various signaling transduction strategies, including resonance light scattering, colorimetry, and fluorescence, but few of them combined an amplification strategy for ultrasensitive detection of UO22+.25–27
In order to realize ultrasensitive detection, DNA amplification strategies such as polymerase chain reaction (PCR),27 rolling circle amplification (RCA), strand-displacement amplification (SDA)29 and hybridization chain reaction (HCR)30 were widely used. HCR was first introduced by Dirks and Pierce.31 The outstanding advantage of HCR is an isothermal and enzyme-free condition. In addition, it successfully avoided the disadvantages of enzyme-based amplification protocol such as complexity of the experimental system and increased detection cost which limited its application. These advantages make HCR a fascinating strategy in DNA-based sensing applications. So far, although a lot of papers have been reported by HCR for signal amplification in detection of DNA,32 protein33 and cell,34 few paper has tried to apply this promising technique for metal ion detection.
Herein, we proposed a novel strategy for detection of UO22+ by using a UO22+-specific DNAzyme and two free labeled DNA hairpins (Scheme 1). This electrochemical biosensor realized the ultrasensitive detection of UO22+ by combing the high specificity of DNAzyme and the advantages of HCR, isothermal and enzyme-free condition. The UO22+-specific DNAzyme consisted of a enzyme strand DNA (E-DNA) and a substrate strand DNA (S-DNA). The DNAzyme was formed and immobilized on the surface of gold electrode in advance. In the presence of UO22+, the cleaved strand DNA produced by the DNAzyme catalytic cleavage of S-DNA triggered the hybridization chain reaction between two alternating hairpin DNA structures to form a nicked double-helix DNA polymers on the electrode surface. Then, methylene blue (MB) which could be intercalated into the minor groove of the long dsDNA polymers, was introduced to produce electrochemical signal.35 In the absence of UO22+, the S-DNA of the DNAzymes cannot be cleaved, the whole DNAzymes cannot initiate the HCR between the hairpins (H1 and H2), and thus they were still in closed form. The concentration of UO22+ could be indirectly determined with ultra-sensitivity by monitoring the change of the electrochemical signal.
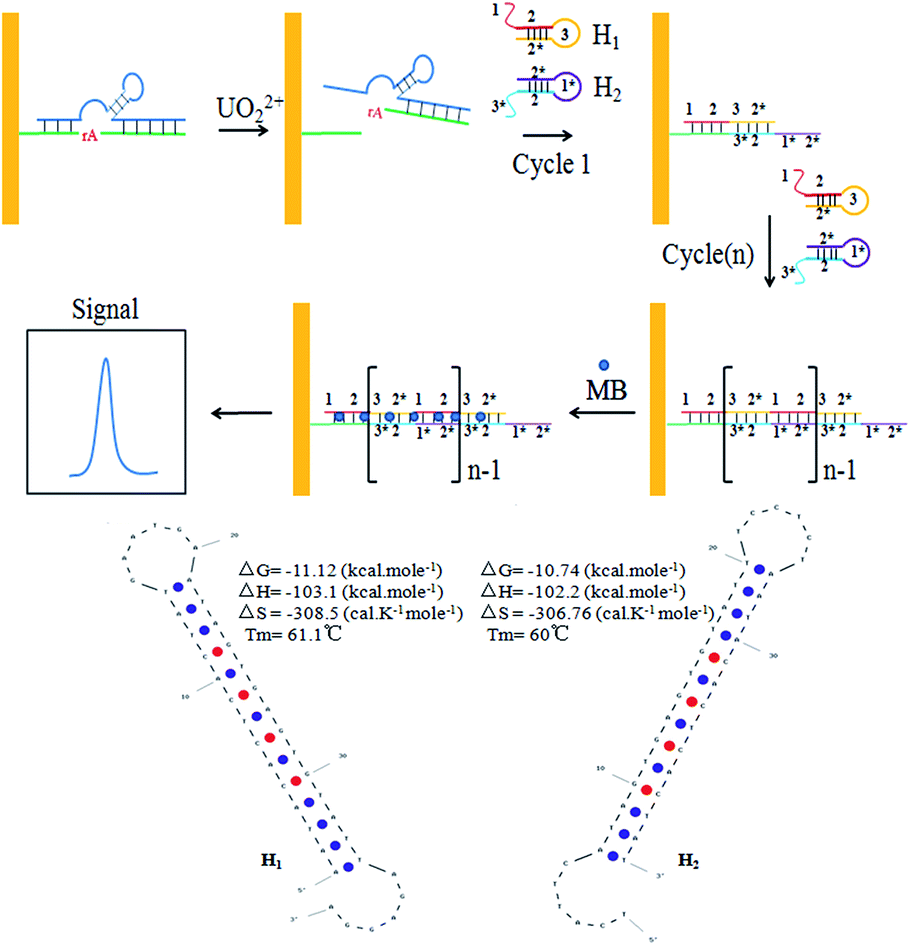 | ||
| Scheme 1 Schematic illustration of the sensing principle for UO22+ detection. The ideal structure and parameters of H1 and H2 were exhibited at the bottom. | ||
Experimental
Materials and chemicals
Uranyl nitrate was purchased from the China National Nuclear Corporation (Lanzhou, China), dissolved in water to make a 10 mM stock solution, then diluting to the final concentration before use. Oligonucleotides were synthesized and purified with HPLC by Sangon Biotechnology Co. Ltd. (Shanghai, China). The sequences of these oligonucleotides were shown in Table S1 in the ESI.† 2-(N-Morpholino) ethanesulfonic acid (MES) (pH 5.5) and phosphate buffered saline (PBS) (pH 7.2) were purchased from Beijing Leagene Biotech. CO, Ltd. (Beijing, China). K3Fe(CN)6 and K4Fe(CN)6 were purchased from Chengdu Kelong Chemical Reagents Factory (Chengdu, China). Methylene blue and 6-mercaptohexanol (MCH) were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals were analytical grade. All solutions were prepared with ultrapure water with resistivity of 18.2 MΩ cm.Apparatus
Electrochemical measurements were performed a CHI660D electrochemical analyzer using a traditional three-electrode system. The gold electrode was used as working electrode; a platinum wire and Ag/AgCl electrode were used as counter electrode and reference electrode respectively. Square wave voltammetry (SWV) measurements were performed in 10 mM PBS solution (pH 7.2) and scanned from −0.40 to 0.00 V with amplitude of 50 mV by SWV scanning. The EIS experiments were measured in 1 mM [Fe(CN)6]3−/4− solution containing 0.5 M KCl with the frequency range from 0.1 Hz to 10 kHz.Sensor preparation and DNAzyme cleavage
50 μL of 100 μM thiol modified S-DNA and equal amount of E-DNA were mixed in 900 μL of 10 mM MES buffer solution containing with 300 mM NaCl (pH 5.5) in microcentrifuge tube. After vortex, sample was heated up to 90 °C and cooled down to room temperature in 2 hours.The gold electrode (3 mm in diameter) was firstly polished by 0.05 μm alumina powder. Then it was thoroughly ultrasonicated in water, absolutely ethanol for 3 min, respectively. Following that, the electrode was electrochemically cleaned in 0.5 M H2SO4 solution via potential cycling between −0.20 and 1.25 V, until a stable background has been reached. After drying with nitrogen, the gold electrode was immersed into the above hybridized DNAzyme solution for 3 h, followed by 2 h treatment with 1 mM MCH to remove the nonspecific DNA adsorption and obtain a well aligned DNAzyme modified surface. The modified electrode was thoroughly rinsed with PBS buffer for the further use. Following that, an appropriate amount of UO22+ or the sample solution were added, and reacted for 10 min at room temperature.
Hybridization chain reaction
After the DNAzyme based cleavage reaction, the sensor was washed and incubated with a mixture of 100 nM hairpin1 (H1) and 100 nM hairpin2 (H2) in HCR reaction buffer (50 mM PBS with 0.5 M NaCl, pH 7.2) at 37 °C for 2 h. H1 and H2 were heated up to 90 °C for 5 min and cool slowly down to room temperature in 2 h prior to use to form hairpin structure.MB intercalation and voltammetric transduction
As an organic dye, MB has been used as an electron transfer mediator for the different interaction between ssDNA and dsDNA.36 The voltammetric signals of MB reflect the extent of the hybrid formation. MB could intercalate into base pairs of dsDNA by immersing the electrode into the stirring 10 mM PBS buffer solution (pH 7.2) containing 20 μM MB for 10 min. After the intercalation of MB, the electrode was rinsed for 10 s to remove the nonspecifically bounding of MB. Subsequently, the electrode was transferred into the 10 mM PBS buffer solution (pH 7.2) and scanned from −0.40 to 0.00 V by SWV scanning with an amplitude of 25 mV, step potential of 4 mV and frequency of 15 Hz.Results and discussion
The principle of electrochemical sensor for amplification detection of UO22+
In the present study, a electrochemical sensor was proposed for UO22+ ultrasensitive detection based on the highly specific, metal-induced activation of DNAzyme and two free labeled DNA hairpins. The S-DNA was firstly annealed with E-DNA to form the DNAzyme structure. Then the thiol group functionalized DNAzyme structure was immobilized onto the gold electrode surface by Au–S interaction, followed by blocking with MCH. The backfilling MCH monolayer has been proved to be necessary for efficient formation of active DNAzyme complex on gold surface.37 In the presence of UO22+, the DNAzyme was activated, causing a cleaved strand DNA of S-DNA to release. The hairpins (H1 and H2) can stably co-exist in solution by the 14 base pairs stem without the cleaved strand DNA. The melting temperature of H1 and H2 are 61.1 °C and 60 °C respectively (calculated by Oligo Analyzer 3.1), shown at the bottom of Scheme 1, which is much higher than the reaction temperature (37 °C). As shown in Scheme 1, when the cleaved strand DNA was appeared, it would hybridize with a toehold mediated strand (1–2) on H1 and open the hairpin structure through an unbiased strand-displacement interaction. The newly released sticky sequence of H1 (3–2*) could serve as a new toehold to unfold the hairpin H2. The released sticky sequence of H2 (1*–2*) was identical in sequence to the effective part of cleaved strand DNA. In this manner, the cleaved strand DNA could specifically trigger the chain reaction between H1 and H2 to form a long nicked dsDNA. Hence, a small amount of the cleaved strand DNA could initiate the HCR and generated a long dsDNA for the intercalation of numerous MB, leading to a significantly increase in the corresponding electrochemical signal intensity. By monitoring the change of the electrochemical signal, the concentration of UO22+ could be indirectly determined with ultra-sensitivity.Electrochemical study of UO22+ biosensor fabrication process
The feasibility of the proposed UO22+ biosensor was firstly investigated. The typical electrochemical responses of methylene blue at different experimental stages were shown in Fig. 1. Only a teeny reduction peak was observed at bare electrode, probably owning to the non-specific protein absorption of MB (Fig. 1 curve a). The peak current increased after the DNAzyme structure modifying process due to the intercalation of MB into the double-helix structure of DNAzyme (Fig. 1 curve b). After the UO22+ was introduced, the electrochemical responses obviously decreased, probably owning to the cleavage of DNAzyme in the presence of UO22+, which reduced the intercalation of MB (Fig. 1 curve c). In the condition of only H1, the H1 was linked to the electrode surface by partly complementary hybridization, and the double-helix part formed by the cleaved strand with H1 accumulated the MB, inducing an evident increased signal (Fig. 1 curve d). In the condition of H1 and H2, a remarkably signal enhancement was observed for the result of HCR amplification (Fig. 1 curve e). The appreciable signal increased significantly comparing with the condition without HCR amplification, revealing an efficient and excellent signal amplification capability. This phenomenon was consistent with the fact that the S-DNA of DNAzyme can be cleaved in the presence of UO22+, and the cleaved strand DNA on the surface can initiate the HCR and propagate the hybridization between H1 and H2 to form the long nicked double-helix DNA.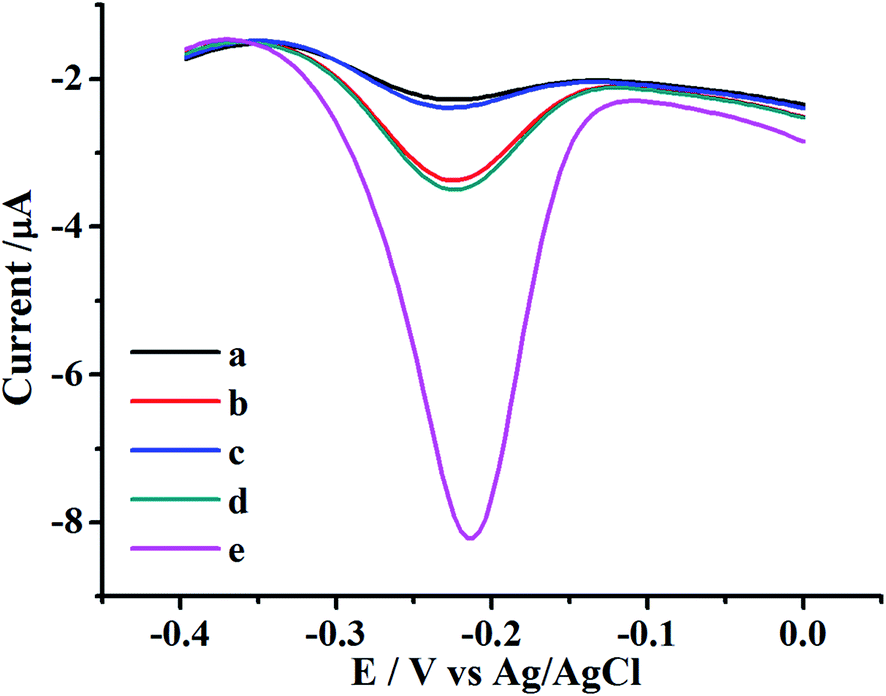 | ||
| Fig. 1 SWV plots of the proposed UO22+ electrochemical biosensor in the 10 mM PBS buffer solution (pH 7.2) from −0.40 to 0.00 V with an amplitude of 25 mV by SWV scanning. The curves were the cases for different states of electrode after incubation with MB: (a) the bare gold electrode, (b) modified by 5 μM DNAzyme, (c) reaction with 6 nM UO22+, (d) treated with 100 nM H1 only for 2 h, (e) treated with 100 nM H1 and 100 nM H2 for 2 h. | ||
Electrochemical characterization of the UO22+ biosensor
The DNAzyme-based biosensor fabrication process was also characterized by electrochemical impedance spectroscopy (EIS). As shown in inset of Fig. 2, the typical electrochemical interface was shown as an electrical circuit. The equivalent circuit was composed by the solution resistance (Rs), charge-transfer resistance (Rct), constant phase element (CPE), Warburg impedance (Zw), and diffusion resistance (Rw). In the Nyquist plot of impedance spectroscopy, the diameter of the semicircle indicates the Rct. As shown in Fig. 2, the EIS plot of the bare electrode only had a small semicircular domain (curve a), indicating a fast charge-transfer process on the bare electrode surface. After the DNAzyme was immobilized on the electrode surface, the semicircle on EIS increased significantly, owing to the negatively charged phosphate backbone of DNAzyme which could prevent the diffusion of [Fe(CN)6]3−/4− towards to the electrode surface (curve b).38 The resistance decreased dramatically after incubation with UO22+, which attributed to the catalytic cleavage of S-DNA of DNAzyme, reducing negative charges of phosphate backbone to facilitate the electron transfer (curve c). Subsequently, the hairpin H1 was introduced to hybridize with electrode. The resistance increased again (cure d), showing that the H1 was successfully opened and hybridized on the remnant of S-DNA. However, a large semicircle domain was observed in the presence of H1 and H2 simultaneously for HCR (curve e), resulting a high resistance of the electrode interface causing by the forming the long nicked double-helix that obstruct the electron transfer of the electrochemical probe.39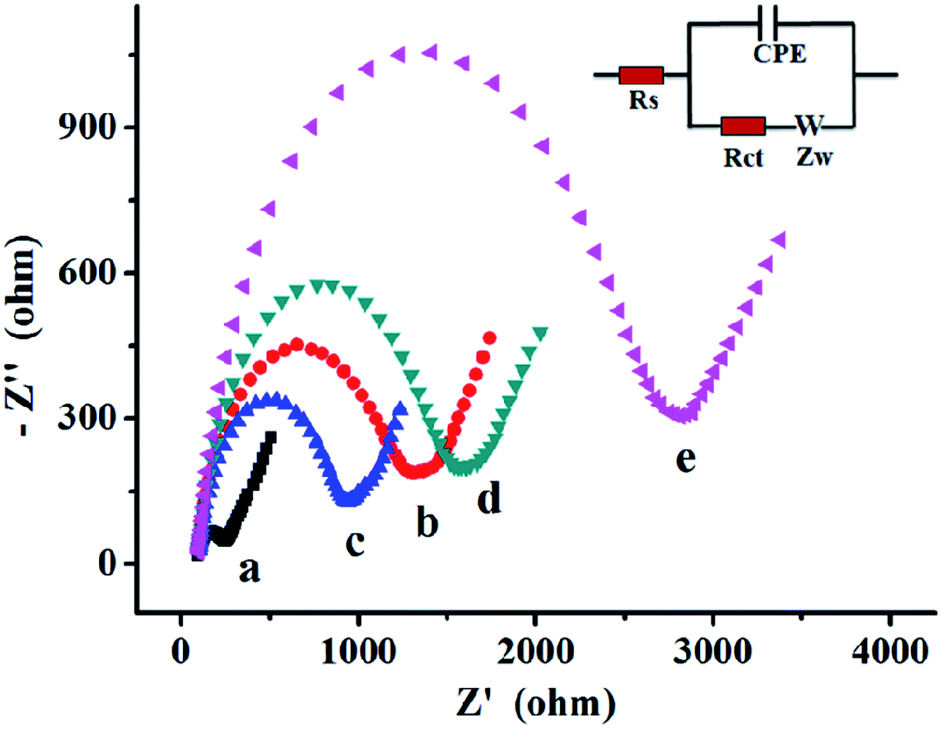 | ||
| Fig. 2 EIS plots in 1 mM [Fe(CN)6]3−/4− solution containing 0.5 M KCl at (a) bare gold electrode, (b) DNAzyme modified electrode with MCH, (c) incubation with 6 nM UO22+, (d) hybridization with 100 nM H1 for 2 h, (e) hybridization with 100 nM H1 and H2 for 2 h. | ||
Uranyl ion-dependent activities of DNAzyme
It was important to study the enzyme activity as a function of UO22+ concentration, owing to the highly effective activity of DNAzyme in detection of uranium.12 To determine the apparent dissociation constant (Kd) for uranium binding, the rate of signal change with UO22+ concentration was calculated and plotted in Fig. 3. The rate increased with UO22+ concentration and reached a platform at 1500 nM. The dissociation constant of uranium binding was 370 nM by fitting the curve. This Kd value was higher than that obtained from fluorescence assays,12 suggesting weaker affinity measured using electrochemical methods. This discrepancy could be explained for two reasons. Firstly, the higher enzyme concentration was used in our method compared with the fluorescence method where the enzyme concentration was only 50 nM. The excess amount of enzyme might bind with the UO22+ and thus decreased the effective concentration of the free uranyl ion.40 Secondly, the DNAzyme which was immobilized on the surface of gold surface has been proved less active than its free in solution.37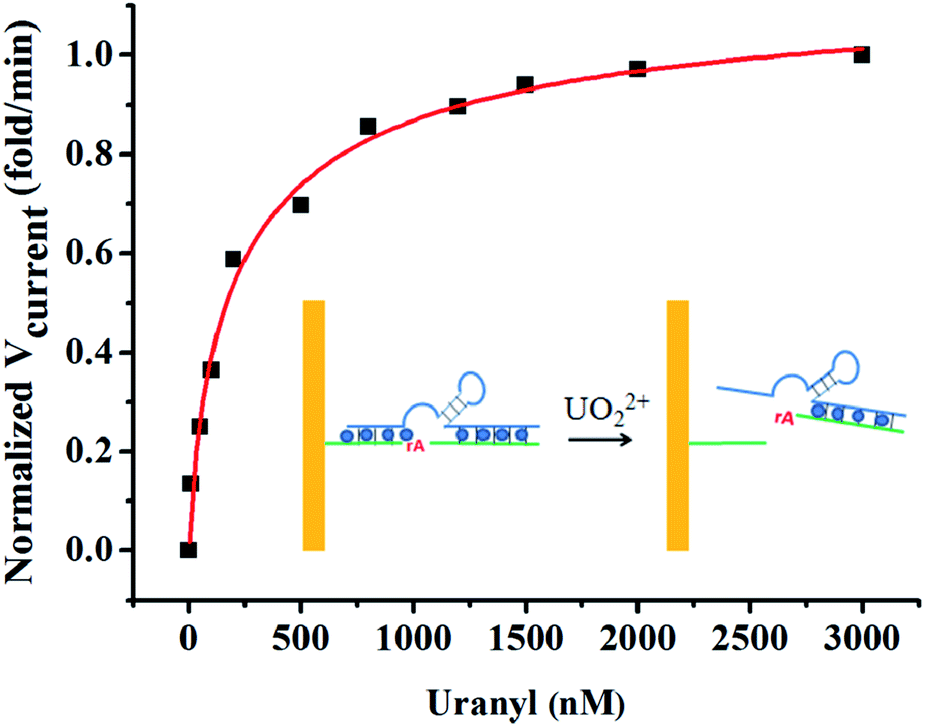 | ||
| Fig. 3 Plot of the initial rate of signal change vs. UO22+ concentration in 10 mM MES buffer solution containing 300 mM NaCl (pH 5.5). Inset: the schematic illustration of reaction process. | ||
The Kd of DNAzyme for UO22+ was small compared with other DNAzymes for divalent metal ions: Kd for Pb2+, Mg2+, Ca2+, and Mn2+ were 5 μM, 180 mM, 60 mM, and 2.6 mM, respectively.40 The small Kd of DNAzyme for UO22+ indicated the strong binding affinity and high sensitivity for the biosensor.
Optimization of detection conditions
To achieve the best performance of this biosensor, the assay conditions such as both the concentration of H1 and H2, pH, and the incubation time of HCR were optimized in this work. Because the same concentration of H1 and H2 was used for HCR amplification, both these concentration were optimized together. The result for optimization of the concentration of H1 and H2 was shown in Fig. S1A.† The Δcurrent (current peak value deduct the background value) increased gradually with the concentration of hairpins. It could be found when the concentration was 100 nM, a better electrochemical signal were obtained than other concentrations. When the concentration was higher, the signal did not show an obvious increase. The higher hairpins concentration increased the length of the nicked dsDNA for more MB intercalation, but it is also restricted to the distant-sensitive electrochemical reaction.39The incubation time of HCR also greatly influenced the performance of the biosensor. The incubation time were optimized, and the result was shown in Fig. S1B.† The Δcurrent increased gradually with the augment of the assembling time and reached a platform after 2 h. Then the Δcurrent did not increase obviously as the HCR time extended to 3 h. Therefore, 2 h was chosen as the optimum HCR time.
It has been reported the pH is a crucial parameter for the DNAzyme cleavage reaction, the DNAzyme activity is greatly affected by pH of solution. Therefore, the pH of MES (DNAzyme cleavage reaction buffer) was optimized here. Fig. S1C† illustrated that the Δcurrent increased with the rise of pH and reached a peak in MES buffer of pH 5.5, which was consistent with those reports.23,40 When the pH was higher than 5.5, the Δcurrent value decreased dramatically, indicating the activity of DNAzyme was highly pH dependent. That is because the UO22+ hydrolyzes and exists in the form of UO2(OH)+ when the pH is higher than 5.5, which can not initiate the DNAzyme cleavage reaction. Finally, MES buffer of pH 5.5 was selected for the cleavage reaction process.
Analytical performance of the biosensor for UO22+
Under optimum conditions, the analytical performance of the sensor was studied by varying the UO22+ concentration. Fig. 4 showed the signal responses with different concentrations of UO22+, and the signal response showed a good linear correlation to the UO22+ concentration from 0.05 to 4 nM. The dynamic region here only covered about two orders, this is same with other DNAzyme based UO22+ detection reports.23,25,42 The linear calibration equation was I = −0.606 − 0.647C (I is the Δcurrent (μA) and C is the concentration of UO22+ (nM)) and the correlation coefficient R2 = 0.994. The detection limit was calculated to be 20 pM by three times standard deviation of the blank (10 replicates). The detection limit was much lower than the maximum contamination level of UO22+ defined by the U.S. Environmental Protection Agency (EPA) with 130 nM (http://water.epa.gov/drink/contaminants/index.cfm), the proposed sensor could completely meet the requirement for monitoring UO22+.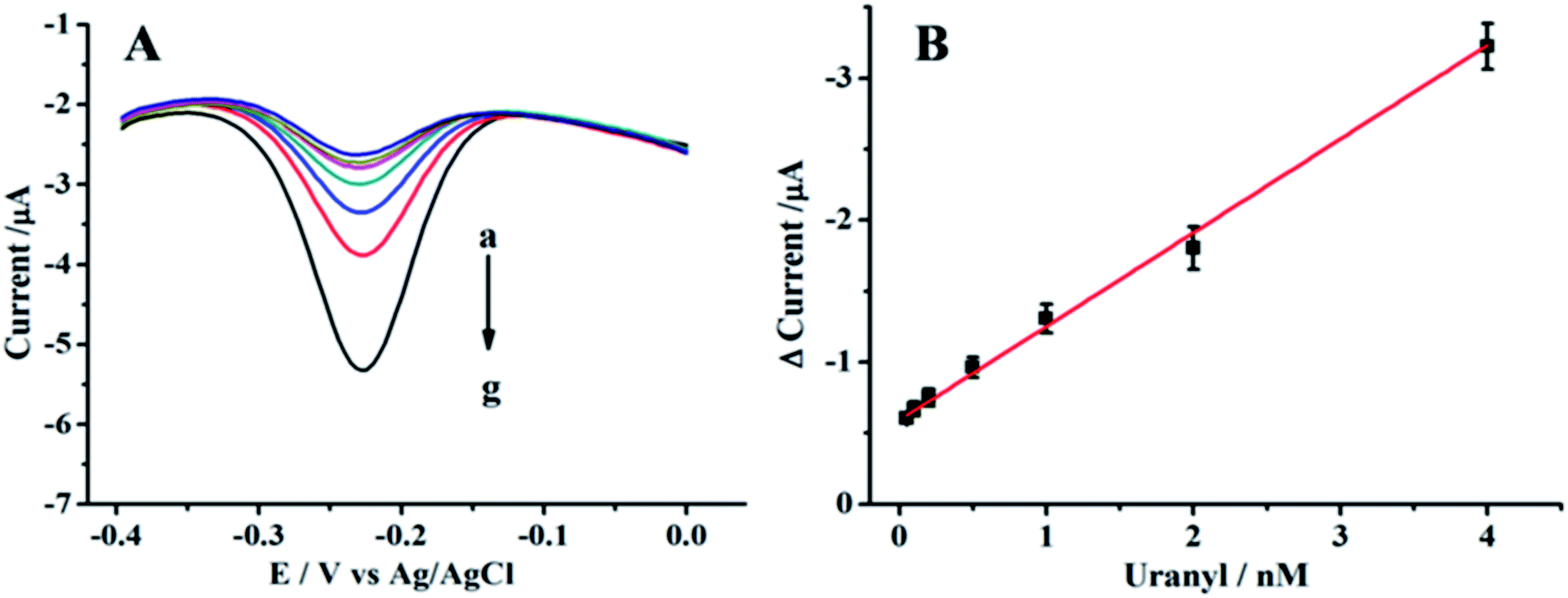 | ||
| Fig. 4 (A) SWVs for the different concentrations of UO22+ with the proposed method in 10 mM PBS (pH 7.2): (a) 0.05 nM, (b) 0.1 nM, (c) 0.2 nM, (d) 0.5 nM, (e) 1 nM, (f) 2 nM, (g) 4 nM. (B) The calibration plot for concentration of UO22+ vs. Δcurrent intensity in the range from 0.05 nM to 4 nM. | ||
We have compared the analytical performance of our proposed biosensor with other methodologies for UO22+ detection which have been reported including SERS, colorimetry, wireless magnetoelastic sensor, and resonance light scattering. As shown in Table 1, the LOD of our method was at least 2 orders of magnitude lower than those without HCR amplification. This can be attributed to two reasons: firstly, the strong binding affinity between DNAzyme with UO22+ may offer relatively high sensitivity for detection;40 secondly, the high amplification efficiency of HCR increases the sensitivity.
| Detection method | Detection limit | Linear range | Ref. |
|---|---|---|---|
| Electrochemistry | 5.2 × 10−6 M | 1 × 10−5 to 1 × 10−1 M | 13 |
| Surface enhanced Raman scattering | 1 × 10−8 M | — | 42 |
| DNA-based electrochemistry | 3 × 10−8 M | — | 16 |
| DNAzyme-based colorimetry | 1 × 10−9 M | 5 × 10−8 to 5 × 10−7 M | 23 |
| DNAzyme-based wireless magnetoelastic sensor | 2.87 × 10−9 M | 9.57 × 10−9 to 1.2 × 10−7 M | 43 |
| DNAzyme-based resonance light scatter | 4.09 × 10−9 M | 1.36 × 10−8 to 1.5 × 10−7 M | 25 |
| DNAzyme with HCR amplification | 2 × 10−11 M | 5 × 10−11 to 4 × 10−9 M | This work |
Reproducibility, selectivity of the proposed biosensor
Reproducibility of the proposed method was evaluated by relative standard deviation of six repetitive measurements for 0.05 and 1 nM UO22+. The RSD for the intra-assay and the inter-assay were valued by using six parallel prepared biosensors, the RSD of 1 nM were 10.6% and 11.5%, and the RSD of 1 nM were 6.3% and 4.8%, respectively. All the results of RSD were comparable with previous articles. These results indicated the satisfactory repeatability and reproducibility for the UO22+ biosensor.Under the optimal condition, the influence of other metal ions was evaluated. As shown in Fig. S2,† the signal intensities of 100 times concentration of other metal ions such as Mg2+, Ca2+, Zn2+, Pb2+, Cu2+, Fe2+, Co2+, Sn2+ were much lower than that of UO22+. This indicated that the proposed method had good selectivity for UO22+ detection and could distinguish UO22+ in complex samples from other metal ions. These results also proved that selectivity of the proposed method was comparable with other UO22+ sensor.23,26
Analysis of UO22+ in river water
To investigate the validity of the proposed biosensor to river water samples. River water samples were filtered for the detection procedure. River water samples were filtered with a 0.45 micron filter and then adjusted pH to 5.5 using 1 M HCl for the detection procedure. For a spiked sample, a known amount of UO22+ was added into the river water sample. Three spiked samples with the UO22+ concentration of 0.05 nM, 1 nM and 4 nM were measured. The results were listed in Table S2.† The recoveries and RSD were range from 94.6% to 108.0% and 2.75% to 4.37%, respectively, which clearly demonstrated the biosensor had potential for analytical application in real samples.Conclusions
In summary, a novel and ultrasensitive electrochemical detection method for uranyl based on UO22+-specific DNAzyme and HCR amplification was developed. This strategy combined the high specificity of DNAzyme and the advantages of hybridization chain reaction (HCR) in isothermal and enzyme-free conditions, which greatly improved the sensitivity. To our best knowledge, this is the first time to employ HCR amplification and DNAzyme into uranyl detection. Importantly, this strategy also provided a promising potential for the detection of other DNAzyme based metal ions and aptamer based small molecules. Moreover, due to the unique merit of electrochemistry, such as low cost, ease of use and potential of miniaturization, commercial portable biosensor could be developed for on site and real-time detection for uranium contamination assessment and the environmental remediation monitoring.Acknowledgements
This work was financially supported by Radiochemical Discipline 909 Funds by China Academy of Engineering Physics (Item No. xk909-2), the National Natural Science Foundation of China (Grant No. 21401174), China Academy of Engineering Physics (Item no. 2013B0302047) and Science and Technology on Surface Physics and Chemistry Laboratory (TP201302-1).Notes and references
- P. Chen, B. Greenberg, S. Taghavi, C. Romano, D. van der Lelie and C. He, Angew. Chem., Int. Ed., 2005, 117, 2775–2779 CrossRef.
- Y. Hitomi, C. E. Outten and T. V. O'Halloran, J. Am. Chem. Soc., 2001, 123, 8614–8615 CrossRef CAS PubMed.
- E. M. Nolan and S. J. Lippard, J. Am. Chem. Soc., 2003, 125, 14270–14271 CrossRef CAS PubMed.
- A. Jones, B. Miller, S. Walker, J. Anderson, A. Colvin, P. Hutchison and C. Soutar, Environ. Res., 2007, 104, 216–223 CrossRef CAS PubMed.
- M. Rožmarić, A. G. Ivšić and Ž. Grahek, Talanta, 2009, 80, 352–362 CrossRef PubMed.
- P. Zoriy, P. Ostapczuk, H. Dederichs, J. Höbig, R. Lennartz and M. Zoriy, J. Environ. Radioact., 2010, 101, 414–420 CrossRef CAS PubMed.
- D. Rathore, Talanta, 2008, 77, 9–20 CrossRef CAS PubMed.
- A. Dejeant, L. Bourva, R. Sia, L. Galoisy, G. Calas, V. Phrommavanh and M. Descostes, J. Environ. Radioact., 2014, 137, 105–112 CrossRef CAS PubMed.
- P. Jaison, V. M. Telmore, P. Kumar and S. K. Aggarwal, J. Chromatogr. Sci., 2011, 49, 657–664 CAS.
- M. Xu, S. Frelon, O. Simon, R. Lobinski and S. Mounicou, Talanta, 2014, 128, 187–195 CrossRef CAS PubMed.
- J. Mehta, B. V. Dorst, E. Rouah-Martin, W. Herrebout, M. L. Scippo, R. Blust and J. Robbens, J. Biotechnol., 2011, 155, 361–369 CrossRef CAS PubMed.
- J. Liu, A. K. Brown, X. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2056–2061 CrossRef CAS PubMed.
- I. H. Badr, W. Zidan and Z. Akl, Talanta, 2014, 118, 147–155 CrossRef CAS PubMed.
- R. Banerjee, Y. Katsenovich, L. Lagos, M. Senn, M. Naja, V. Balsamo, K. H. Pannell and C. Z. Li, Electrochim. Acta, 2010, 55, 7897–7902 CrossRef CAS.
- A. Becker, H. Tobias and D. Mandler, Anal. Chem., 2009, 81, 8627–8631 CrossRef CAS PubMed.
- M. Jarczewska, R. Ziółkowski, Ł. Górski and E. Malinowska, Bioelectrochemistry, 2014, 96, 1–6 CrossRef CAS PubMed.
- X. Shu, Y. Wang, S. Zhang, L. Huang, S. Wang and D. Hua, Talanta, 2015, 131, 198–204 CrossRef CAS PubMed.
- N. Carmi, S. R. Balkhi and R. R. Breaker, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2233–2237 CrossRef CAS.
- B. Cuenoud and J. W. Szostak, Nature, 1995, 375, 611–614 CrossRef CAS PubMed.
- M. Liu, H. Zhao, S. Chen, H. Yu, Y. Zhang and X. Quan, Biosens. Bioelectron., 2011, 26, 4111–4116 CrossRef CAS PubMed.
- G. Wang, L. Chen, Y. Zhu, X. He, G. Xu and X. Zhang, Analyst, 2014, 139, 5297–5303 RSC.
- X. H. Zhao, R. M. Kong, X. B. Zhang, H. M. Meng, W. N. Liu, W. Tan, G. L. Shen and R. Q. Yu, Anal. Chem., 2011, 83, 5062–5066 CrossRef CAS PubMed.
- J. H. Lee, Z. Wang, J. Liu and Y. Lu, J. Am. Chem. Soc., 2008, 130, 14217–14226 CrossRef CAS PubMed.
- P. Wu, K. Hwang, T. Lan and Y. Lu, J. Am. Chem. Soc., 2013, 135, 5254–5257 CrossRef CAS PubMed.
- B. Zhou, L. F. Shi, Y. S. Wang, H. X. Yang, J. H. Xue, L. Liu, Y. S. Wang, J. C. Yin and J. C. Wang, Spectrochim. Acta, Part A, 2013, 110, 419–424 CrossRef CAS PubMed.
- Y. Luo, Y. Zhang, L. Xu, L. Wang, G. Wen, A. Liang and Z. Jiang, Analyst, 2012, 137, 1866–1871 RSC.
- R. K. Saiki, S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich and N. Arnheim, Science, 1985, 230, 1350–1354 CAS.
- G. Nallur, C. Luo, L. Fang, S. Cooley, V. Dave, J. Lambert, K. Kukanskis, S. Kingsmore, R. Lasken and B. Schweitzer, Nucleic Acids Res., 2001, 29, e118 CrossRef CAS PubMed.
- Y. Liao, R. Huang, Z. Ma, Y. Wu, X. Zhou and D. Xing, Anal. Chem., 2014, 86, 4596–4604 CrossRef CAS PubMed.
- Y. Yu, Z. Chen, W. Jian, D. Sun, B. Zhang, X. Li and M. Yao, Biosens. Bioelectron., 2015, 64, 566–571 CrossRef CAS PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed.
- Y. Qian, C. Wang and F. Gao, Biosens. Bioelectron., 2015, 63, 425–431 CrossRef CAS PubMed.
- L. Bai, Y. Chai, R. Yuan, Y. Yuan, S. Xie and L. Jiang, Biosens. Bioelectron., 2013, 50, 325–330 CrossRef CAS PubMed.
- G. Zhou, M. Lin, P. Song, X. Chen, J. Chao, L. Wang, Q. Huang, W. Huang, C. Fan and X. Zuo, Anal. Chem., 2014, 86, 7843–7848 CrossRef CAS PubMed.
- R. Rohs, H. Sklenar, R. Lavery and B. Röder, J. Am. Chem. Soc., 2000, 122, 2860–2866 CrossRef CAS.
- K. Kerman, D. Ozkan, P. Kara, B. Meric, J. J. Gooding and M. Ozsoz, Anal. Chim. Acta, 2002, 462, 39–47 CrossRef CAS.
- D. P. Wernette, C. Mead, P. W. Bohn and Y. Lu, Langmuir, 2007, 23, 9513–9521 CrossRef CAS PubMed.
- S. Grützke, S. Abdali, W. Schuhmann and M. Gebala, Electrochem. Commun., 2012, 19, 59–62 CrossRef.
- W. Ren, Z. F. Gao, N. B. Li and H. Q. Luo, Biosens. Bioelectron., 2015, 63, 153–158 CrossRef CAS PubMed.
- A. K. Brown, J. Liu, Y. He and Y. Lu, ChemBioChem, 2009, 10, 486–492 CrossRef CAS PubMed.
- A. Liang, Y. Zhang, Y. Fan, C. Chen, G. Wen, Q. Liu, C. Kang and Z. Jiang, Nanoscale, 2011, 3, 3178–3184 RSC.
- S. Dutta, C. Ray, S. Sarkar, M. Pradhan, Y. Negishi and T. Pal, ACS Appl. Mater. Interfaces, 2013, 5, 8724–8732 CAS.
- J. C. Yin, Y. S. Wang, B. Zhou, X. L. Xiao, J. H. Xue, J. C. Wang, Y. S. Wang and Q. M. Qian, Sens. Actuators, B, 2013, 188, 147–155 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra22773a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |