
Divergent synthesis of indole-fused polycycles via Rh(II)-catalyzed intramolecular [3 + 2] cycloaddition and C–H functionalization of indolyltriazoles†
Yong-Sheng
Zhang
,
Xiang-Ying
Tang
* and
Min
Shi
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: siocxiangying@mail.sioc.ac.cn; mshi@mail.sioc.ac.cn
First published on 21st September 2015
Abstract
Rh(II)-catalyzed divergent synthesis of polycyclic indolines and azepino[4,5-b]indoles through intramolecular [3 + 2] cycloaddition and C–H functionalization of indoles with N-sulfonyl 1,2,3-triazoles is described. The reaction pathways are controlled by the substituent type of indole.
Indole derivatives present a key structural motif in many natural products and medicinal molecules, which exhibit a wide range of promising biological activities.1 In particular, indole-fused N-heterocycles, such as indoline2 and azepino[4,5-b]indole3 derivatives, are most attractive due to their wide existence in a number of natural products and pharmaceutical reagents. Thus, many synthetic methods have been developed to construct these compounds in recent years.4 Because a sequential reaction to synthesize such a complex and useful motif is of great importance, we herein disclose a divergent synthesis of polycyclic indolines and azepino[4,5-b]indoles from readily available indolyltriazoles. The reaction pathways are switchable according to different substituents at the indole N1 position: if the nitrogen is protected, the reaction goes through a formal [3 + 2] cycloaddition to yield polycyclic indolines 2, while for the non-protected indole substrate, the reaction delivers azepino[4,5-b]indoles 3via C–H functionalization.
N-Sulfonyl-1,2,3-triazoles, which can be simply prepared from terminal alkynes by copper-catalyzed 1,3-dipolar cycloaddition with N-sulfonyl azides, have recently attracted much attention.5 As reported by Fokin, Gevorgyan, Murakami and Davies, N-sulfonyl triazoles, as precursors of α-imino metal carbenes, can be effectively decomposed in the presence of a suitable metal catalyst6 and undergo various interesting and useful transformations, such as cyclopropanation,7 transannulation,8 C–H bond insertion,9 X–H (X = heteroatoms) bond insertions10 and other novel reactions based on the inherent properties of metal carbenes.11 Previously, we9b also developed an intramolecular annulation of 1-sulfonyl-1,2,3-triazoles with pyrroles and indoles to construct indole fused azepine derivatives (Scheme 1a). To continue our research interest in indole chemistry, we envisaged that 4-methyl-N-(2-(1-methyl-1H-indol-3-yl)ethyl)-N-((1-tosyl-1H-1,2,3-triazol-4-yl)methyl)benzenesulfonamide 1a could either undergo intramolecular [3 + 2] cycloaddition/ring expansion or C–H functionalization in the presence of a dirhodium complex (Scheme 1b). To our delight, indoline derivatives 2 were obtained after treatment of 1 (R3 is not H) with the rhodium catalyst. Moreover, for non-protected substrates (R3 = H), the reaction gave the desired azepine derivatives 4 after C–H functionalization. The structures of 2a and 4a were unambiguously determined by X-ray diffraction.12
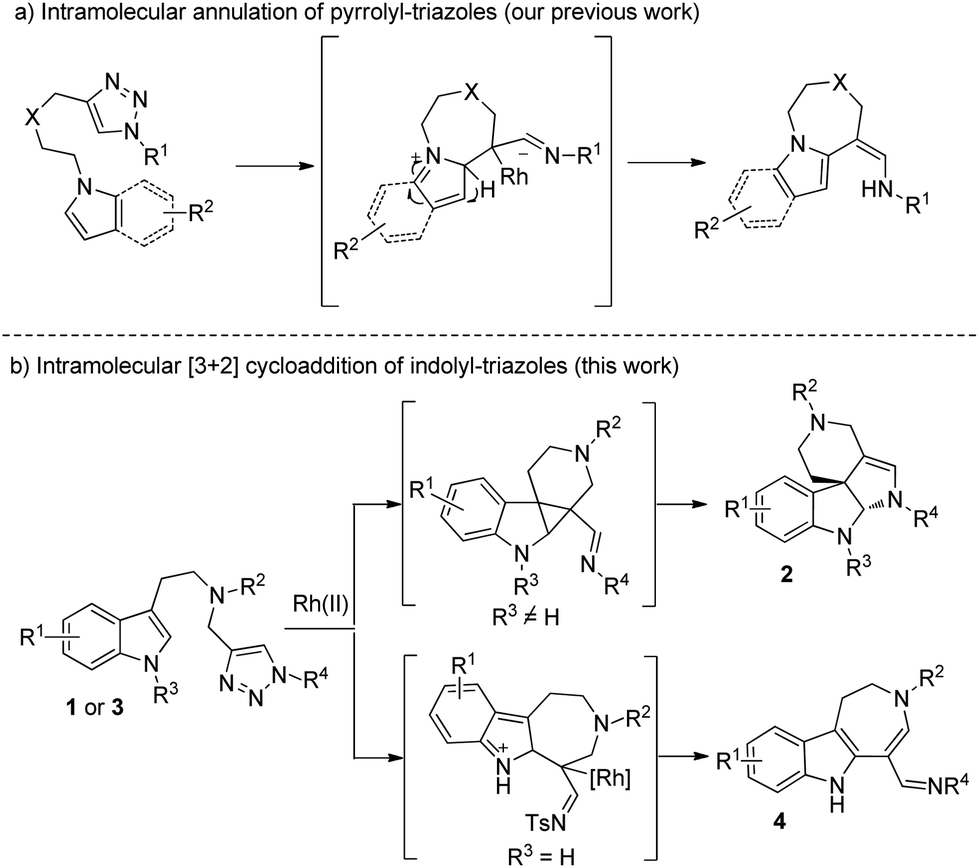 | ||
| Scheme 1 Previous work and this work. | ||
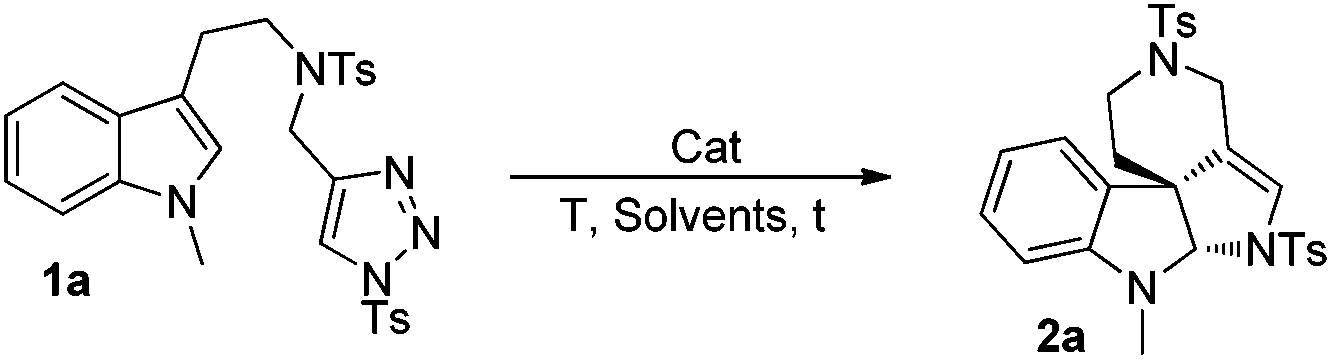
To optimize the reaction conditions, we started to screen different Rh(II) complexes, temperature as well as solvents, using 1a as the model substrate. As can be seen from Table 1, when the reaction was catalyzed by Rh2(Piv)4 (2 mol%) in DCM (dichloromethane) for 3 h at different temperatures, the corresponding product 2a was obtained in 21–30% yields (Table 1, entries 1–3). On changing the solvent to DCE and performing the reaction at 80 °C, the yield of 2a was only 8%, and by further increasing the reaction temperature to 110 °C, the reaction was very complex with no desired product detected (Table 1, entries 4–5). Other achiral dirhodium tetracarboxylates were also investigated, and all of these catalysts turned out to be of poor efficiency to this reaction (Table 1, entries 6–9). Then, we tried several chiral dirhodium complexes, which were proved to give better results than achiral catalysts in some reported cases.11a,8h To our delight, when Rh2(S-PTTL)4 was employed as a catalyst, the yield of 2a increased to 75% (Table 1, entry 10). Other chiral rhodium catalysts such as Rh2(S-NTTL), Rh2(S-DOSP)4 and Rh2(S-TBSP)4 were found less effective than Rh2(S-PTTL)4, affording the desired product 2a in 28–71% yields, respectively (Table 1, entries 11–13). Next, solvent effects were tested and it was found that using a mixed solvent of cyclohexane and DCM (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) would benefit the formation of 2a (77%) (Table 1, entries 14 and 15). The reaction did not take place without a catalyst under otherwise identical standard reaction conditions (Table 1, entry 16). In addition, the reaction of 1a could also run at a gram scale and the corresponding product of 2a was obtained in 67% yield (Table 1, entry 15).
:1) would benefit the formation of 2a (77%) (Table 1, entries 14 and 15). The reaction did not take place without a catalyst under otherwise identical standard reaction conditions (Table 1, entry 16). In addition, the reaction of 1a could also run at a gram scale and the corresponding product of 2a was obtained in 67% yield (Table 1, entry 15).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Cat. | T (°C) | Solvent | Time | Yieldb (%) |
| 2a | |||||
a Reaction conditions: 1a (0.2 mmol), and Rh cat. (2 mol%) were stirred in 2 ml of solvent.
b Yields of isolated product.
c The solvent was 6 ml.
d Tested by 1H NMR.
e 2.0 mmol scale, 1.1 g of 1a (67% yield).
f When the reaction was conducted without a catalyst, no reaction took place.
|
|||||
| 1 | Rh2(Piv)4 | 60 | DCM | 3 h | 26 |
| 2 | Rh2(Piv)4 | Rt | DCM | 24 h | 21 |
| 3 | Rh2(Piv)4 | 80 | DCM | 3 h | 30 |
| 4 | Rh2(Piv)4 | 80 | DCE | 3 h | 8d |
| 5 | Rh2(Piv)4 | 110 | DCE | 3 h | Traced |
| 6 | Rh2(Oct)4 | 80 | DCM | 3 h | 41 |
| 7 | Rh2(Oct)4 | 120 | DCE | 3 h | Traced |
| 8 | Rh2(esp)2 | 80 | DCM | 3 h | 35 |
| 9 | Rh2(OAc)4 | 80 | DCM | 3 h | 13d |
| 10 | Rh2(S-PTTL)4 | 75 | DCM | 3 h | 75 |
| 11 | Rh2(S-NTTL)4 | 75 | DCM | 3 h | 71 |
| 12 | Rh2S-DOSP)4 | 75 | DCM | 3 h | 28 |
| 13 | Rh2S-TBSP)4 | 75 | DCM | 3 h | 48 |
| 14c | Rh2(S-PTTL)4 | 75 | Cyclohexane | 3 h | 65 |
| 15 | Rh 2 (S-PTTL) 4 | 75 |
Cyclohexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :DCM = 5:1 :DCM = 5:1
|
3 h | 77 (67) |
| 16f | — | 75 | DCM | 3 h | — |
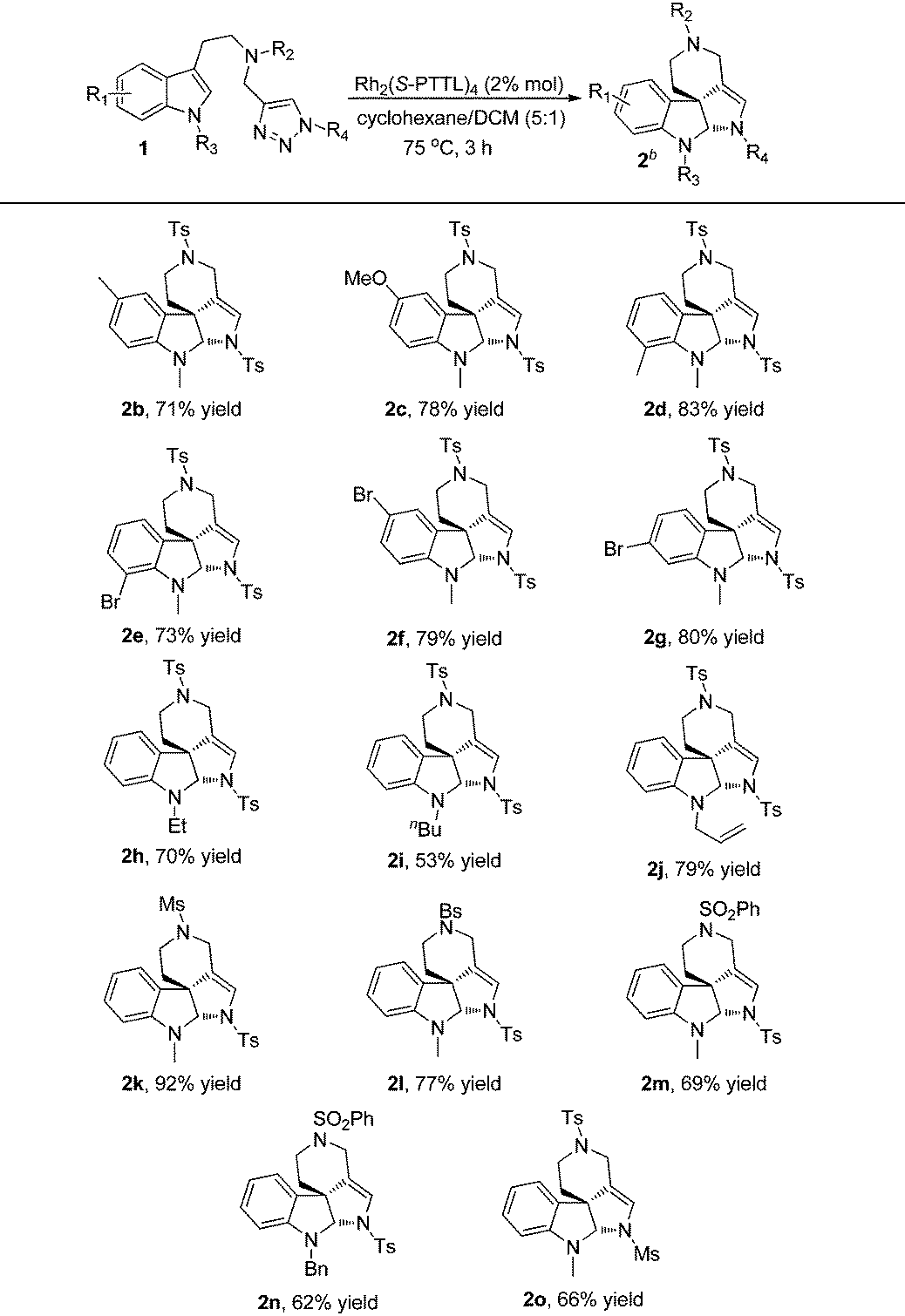
With the optimized reaction conditions in hand, we next investigated the scope and limitations of this reaction and the results are summarized in Table 2. As for substrates 1b–1g, the reactions proceeded smoothly to afford the corresponding products 2b–2g in good yields, and the electronic properties of the substituents on indole didn't have significant influence on the reaction outcomes. When indole was protected by ethyl, n-butyl or allyl groups (R3), the corresponding products 2h–2j were obtained in 53–79% yields. Other sulfonyl substituents (R2) were also well tolerated and the desired products 2k–2n were delivered in 62–92% yields. Moreover, changing the protecting group of triazole (R4) instead of Ts, such as Ms, the reaction also went on well to give 2o in 66% yield.
As a further study of the cycloaddition reaction, we next conducted a one-pot synthesis of the products 2 from terminal alkyne (Scheme 2). On treatment of alkynes (0.2 mmol) with TsN3 (0.2 mmol) in the presence of CuTc (0.016 mmol) in DCM (2.0 mL) at rt under Ar, a triazole intermediate was formed, then Rh2(S-PTTL)4 was added under Ar and the reaction was heated for 3 h at 75 °C. After completion, the reaction mixture was directly subjected to flash column chromatography to give the products 2a, 2b, 2e and 2m in moderate yields.
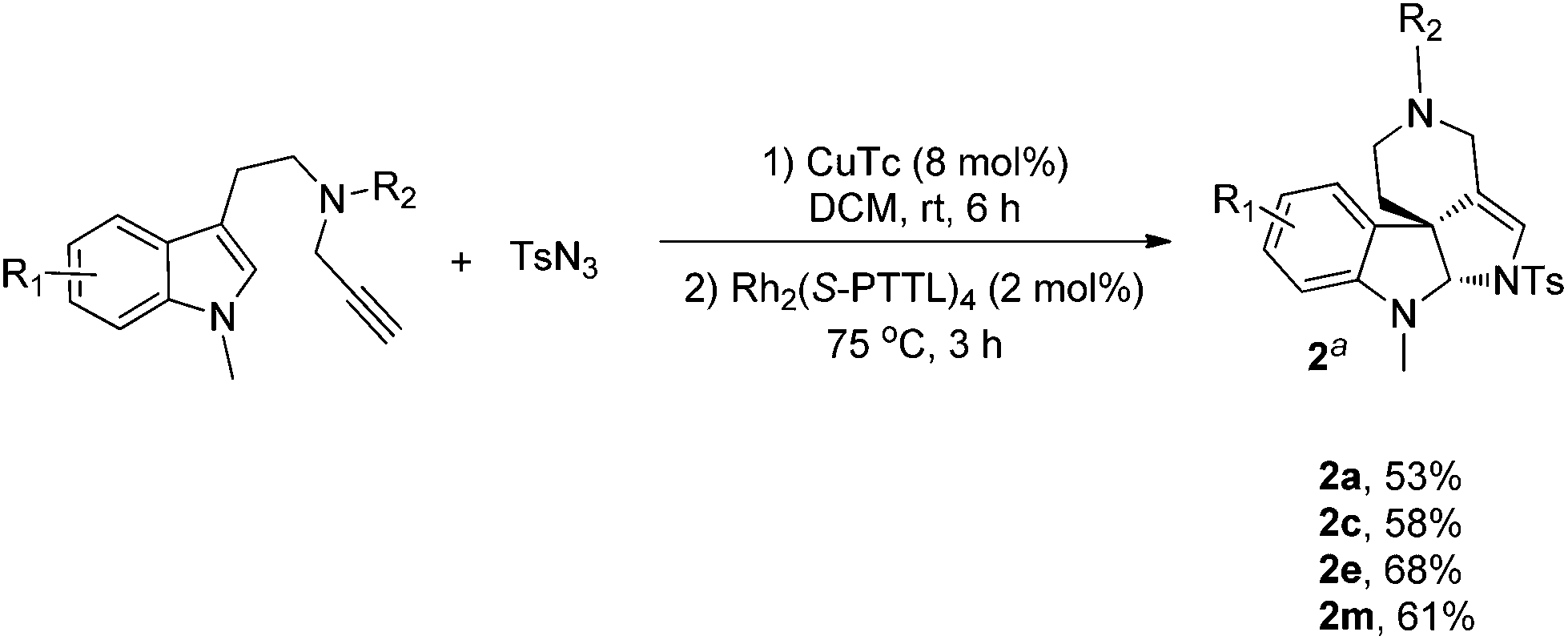 | ||
| Scheme 2 One-pot synthesis of polycyclic pyrroloindolines. Reaction conditions: (1) alkyne (0.2 mmol), TsN3 (0.2 mmol) and CuTc (8 mol%) were stirred in 2 mL of DCM at rt for 6 h. (2) Rh(II) (2 mol%) was added and the reaction mixture was heated at 75 °C for 3 h. aIsolated yield. | ||
To extend the substrate scope, we also examined other types of indolyltriazoles. As can be seen from Scheme 3, when oxygen tethered tryptopholtriazole 1p was treated with Rh2(S-PTTL)4 in DCM at 75 °C for 3 h, the desired spiro derivative 2p was obtained in 45% yield as well as the acrolein imine byproduct 2p′ derived from β-H elimination in 40% yield. However, when substrate 1q with a gem-diester linker was treated under the standard reaction conditions, the reaction became very complex and no desired product was observed as tested by 1H NMR of the crude reaction mixture. Interestingly, when indoly-triazole 3a with a free NH group (R3 = H) was employed as the substrate, the reaction gave azepine derivative 4a in 47% yield upon heating in DCM at 75 °C for 3 h when 1.0 eq. of Cu(OAc)2 was added to the reaction mixture. In comparison, the reaction gave both 4a and 2a in 30% and 18% yields without Cu(OAc)2, indicating that the copper salt plays an important role in controlling the reaction selectivity.
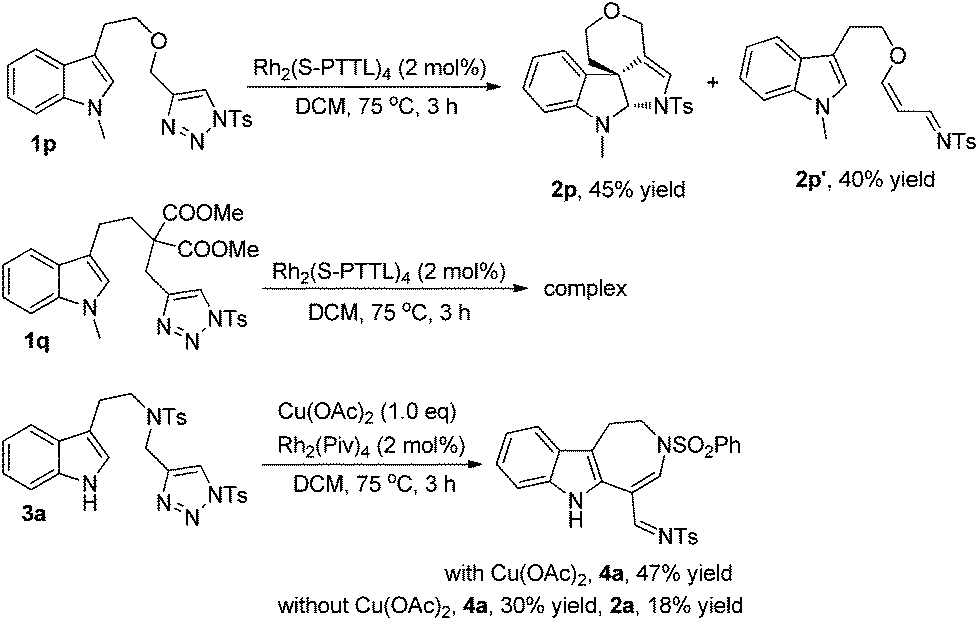 | ||
| Scheme 3 Further substrate scope study. | ||
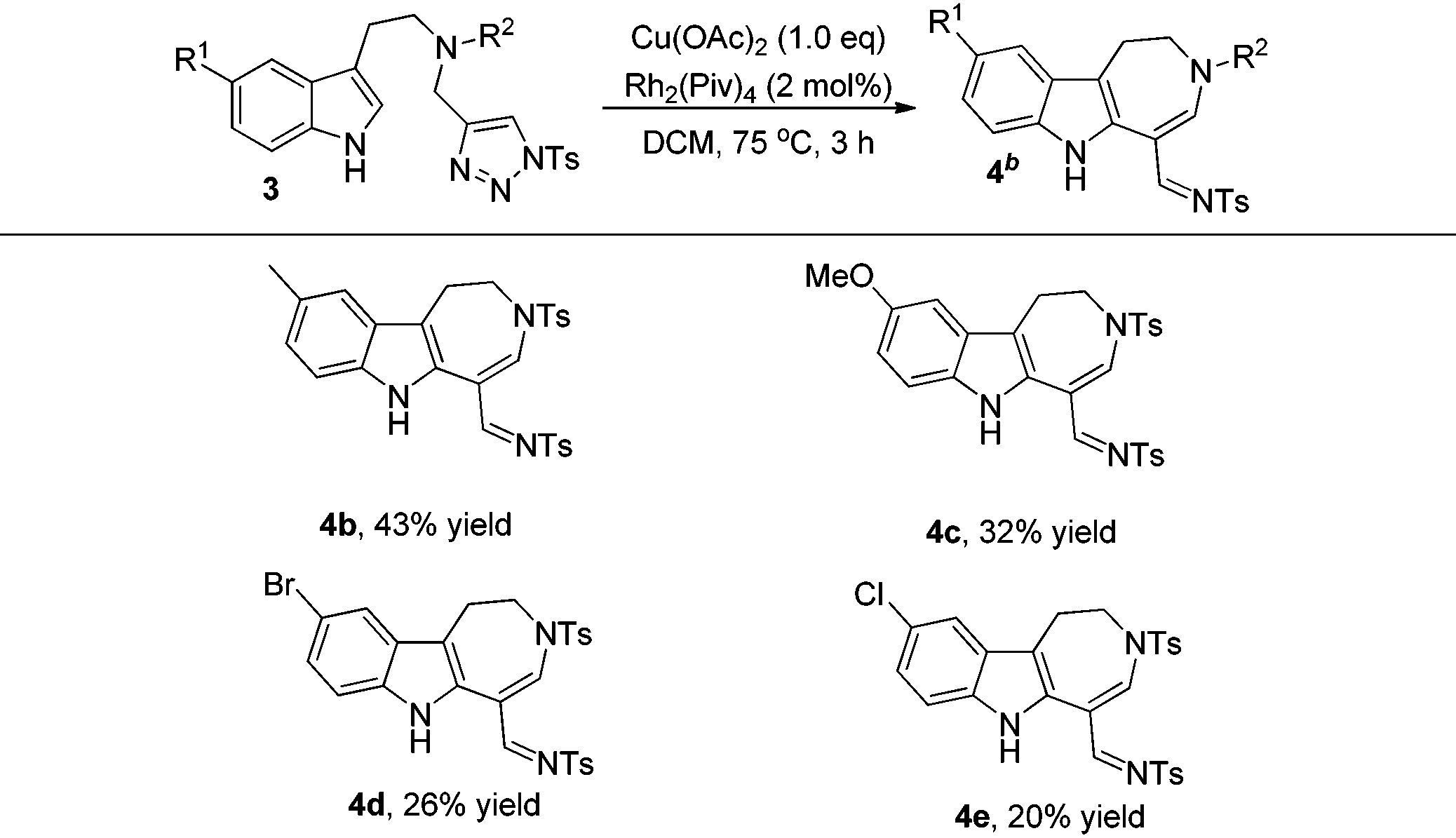
The formation of 4a stimulated our interest to further investigate the scope and limitations of this reaction. After screening the reaction conditions, it was found that using 2 mol% Rh2(Piv)4 and 1.0 eq. Cu(OAc)2 as additives, the reaction gave the best results (for more information, please see Table S1 in the ESI†). As can be seen from Table 3, the corresponding azepine derivatives 4b–4e could be obtained in 20–43% yields. The relatively low yield of the reaction might be due to the instability of the products.13
A plausible mechanism is outlined in Scheme 4. Initially, denitrogenation of 1 in the presence of a Rh(II) complex gives an azavinyl carbine intermediate A. According to Davies's report,7f if the indole substrate is protected by an alkyl group, then the cyclopropanation of the indole double bond by rhodium carbene takes place to yield intermediate B, which then undergoes ring expansion to give intermediate D. After ring closure, the final product 2 is obtained. On the other hand, if R3 is a proton, a Friedel–Crafts reaction occurs, giving product 4 instead via intermediate C. Intramolecular H bonding may exist between the indole N–H and the imine group, which could stabilize intermediate C, therefore, the formation of 4 is more favored than 2. When Cu(OAc)2 is added to the reaction system, the interaction between copper and imine is even more stronger than the H bonding to stabilize intermediate C, giving higher selectivity.
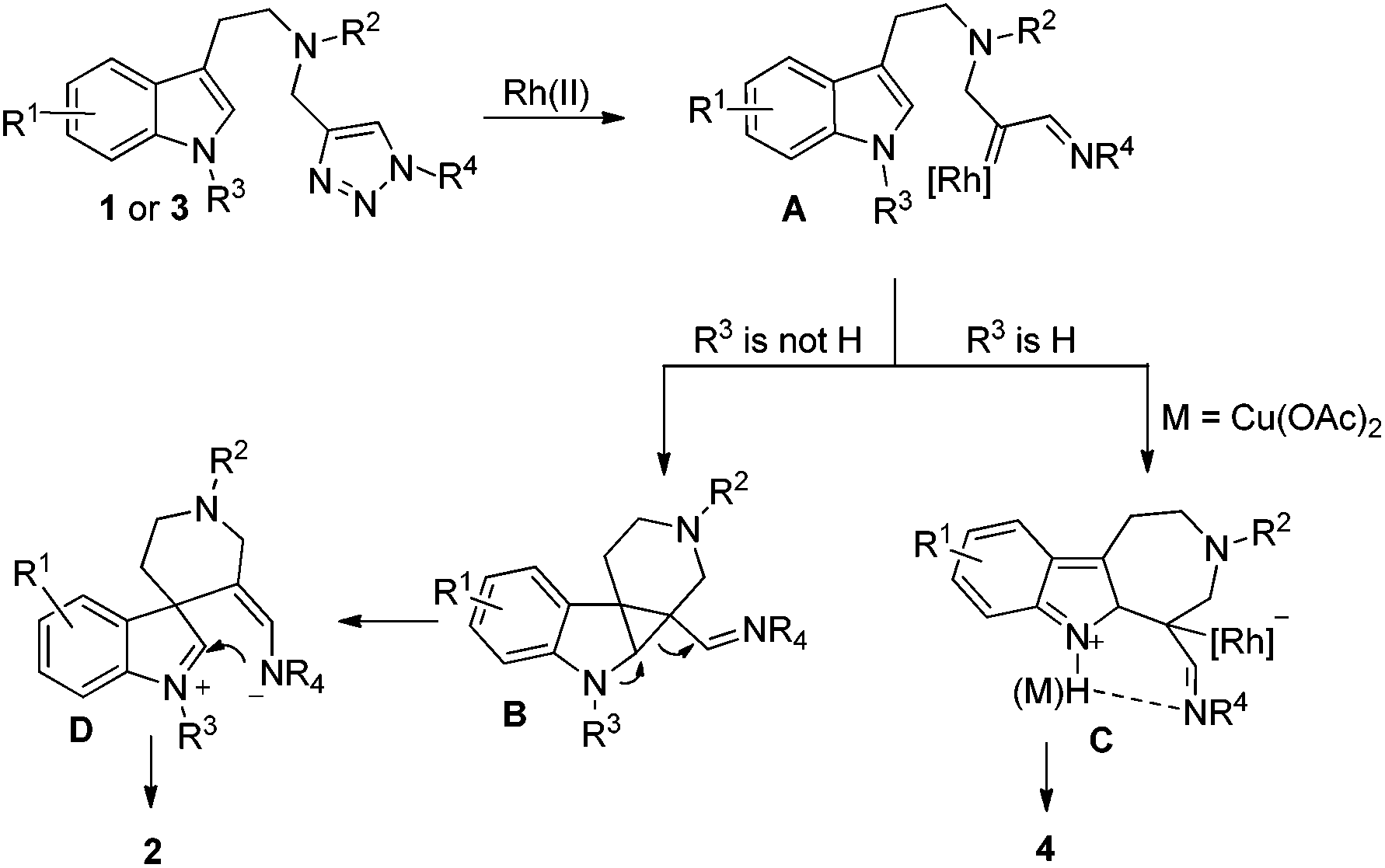 | ||
| Scheme 4 A proposed mechanism. | ||
In summary, we have developed a novel and effective method to synthesize a series of polycyclic pyrroloindolines and azepino[4,5-b]indoles via rhodium(II) catalyzed intramolecular [3 + 2] cycloaddition or C–H functionalization of indolyltriazoles. The reaction pathways are dependent on the substituents at the indole N1 position: when R3 is an alkyl group, the reaction delivers pyrroloindolines, while non-protected substrates result in azepino[4,5-b]indoles. Further investigations to extend the substrate scope as well as to examine the mechanistic details more extensively are currently underway in our laboratory.
We are grateful to the National Basic Research Program of China (973)-2015CB856603, and the National Natural Science Foundation of China (20472096, 21372241, 21361140350, 20672127, 21421091, 21302203, 21102166, 21372250 and 20732008).
Notes and references
- (a) R. J. Sundberg, The Chemistry of Indoles, Academic Press, New York, 1970 Search PubMed; (b) Alkaloids Chemical and Biological Perspectives, ed. S. W. Pelletier, Wiley-Interscience, New York, 1983, vol. 4, p. 211 Search PubMed; (c) R. J. Sundberg, Indoles, Academic Press, San Diego, 1996 Search PubMed; (d) J. P. Michael, Nat. Prod. Rep., 1998, 15, 571 RSC; (e) D. J. Faulkner, Nat. Prod. Rep., 1999, 16, 155–198 RSC; (f) M. Lounasmaa and A. Tolvanen, Nat. Prod. Rep., 2000, 17, 175 RSC; (g) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS PubMed; (h) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed.
- (a) R. Robinson, Experientia, 1946, 2, 28 CrossRef CAS; (b) S. M. Verbitski, C. L. Mayne, R. A. Davis, G. P. Concepcion and C. M. Ireland, J. Org. Chem., 2002, 67, 7124 CrossRef CAS PubMed; (c) R. Jadulco, R. A. Edrada, R. Ebel, A. Berg, K. Schaumann, V. Wray, K. Steube and P. Proksch, J. Nat. Prod., 2004, 67, 78 CrossRef CAS PubMed.
- R. M. Shaheen, D. W. Davis, W. Liu, B. K. Zebrowski, M. R. Wilson, C. D. Bucana, D. J. McConkey, G. McMahon and L. M. Ellis, Cancer Res., 1999, 59, 5412 CAS.
- For synthesis of indoline, see: (a) K. M. Depew, S. P. Mardsen, D. Zatorska, A. Zatorska, W. G. Bornmann and S. J. Danishefsky, J. Am. Chem. Soc., 1999, 121, 11953 CrossRef CAS; (b) V. R. Espejo, X. B. Li and J. D. Rainier, J. Am. Chem. Soc., 2010, 132, 8282 CrossRef CAS PubMed; (c) A. Steven and L. E. Overman, Angew. Chem., Int. Ed., 2007, 46, 5488 CrossRef CAS PubMed; (d) L. E. Overman and D. V. Paone, J. Am. Chem. Soc., 2001, 123, 9465 CrossRef CAS; (e) J. F. Austin, S. G. Kim, C. J. Sinz, W. J. Xiao and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5482 CrossRef CAS PubMed; (f) S. Ma, X. Han, S. Krishnan, S. C. Virgil and B. M. Stoltz, Angew. Chem., Int. Ed., 2009, 48, 8037 CrossRef CAS PubMed; (g) D. Crich and A. Banerjee, Acc. Chem. Res., 2007, 40, 151 CrossRef CAS PubMed; (h) J. Kim and M. Movassaghi, Chem. Soc. Rev., 2009, 38, 3035 RSC; (i) P. Ruiz-Sanchis, S. A. Savina, F. Albericio and M. Alvarez, Chem. – Eur. J., 2011, 17, 1388 CrossRef CAS PubMed. For synthesis of indole fused azepine, see: (j) C. Ferrer and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 1105 CrossRef CAS PubMed; (k) J. T. Lundquist, D. C. Harnish, C. Y. Kim, J. F. Mehlmann, R. J. Unwalla, K. M. Phipps, M. L. Crawley, T. Commons, D. M. Green, W.-X. Xu, W. T. Hum, J. E. Eta, I. Feingold, V. Patel, M. J. Evans, K.-D. Lai, L. B. Marcucci, P. E. Mahaney and J. E. Wrobel, J. Med. Chem., 2010, 53, 1774 CrossRef CAS PubMed; (l) X. Zhong, Y. Li, J. Zhang, W.-X. Zhang, S.-X. Wang and F.-S. Han, Chem. Commun., 2014, 50, 11181 RSC.
- (a) J. Raushel and V. V. Fokin, Org. Lett., 2010, 12, 4952 CrossRef CAS PubMed; (b) Y. Liu, X. Wang, J. Xu, Q. Zhang, Y. Zhao and Y. Hu, Tetrahedron, 2011, 67, 6294 CrossRef CAS PubMed; (c) E. J. Yoo, M. Ahlquist, S. H. Kim, I. Bae, V. V. Fokin, K. B. Sharpless and S. Chang, Angew. Chem., Int. Ed., 2007, 46, 1730 CrossRef CAS PubMed.
- For leading reviews, see: (a) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862 CrossRef CAS PubMed; (b) A. V. Gulevich and V. Gevorgyan, Angew. Chem., Int. Ed., 2013, 52, 1371 CrossRef CAS PubMed; (c) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151 RSC. For pioneering examples, see: (d) T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972 CrossRef CAS PubMed; (e) T. Miura, M. Yamauchi and M. Murakami, Chem. Commun., 2009, 1470 RSC; (f) J. S. Alford and H. M. L. Davies, Org. Lett., 2012, 14, 6020 CrossRef CAS PubMed.
- (a) S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034 CrossRef CAS PubMed; (b) N. P. Grimster, L. Zhang and V. V. Fokin, J. Am. Chem. Soc., 2010, 132, 2510 CrossRef CAS PubMed; (c) J. C. Culhane and V. V. Fokin, Org. Lett., 2011, 13, 4578 CrossRef CAS PubMed; (d) M. Zibinsky and V. V. Fokin, Org. Lett., 2011, 13, 4870 CrossRef CAS PubMed; (e) B. T. Parr and H. M. L. Davies, Angew. Chem., Int. Ed., 2013, 52, 10044 CrossRef CAS PubMed; (f) J. E. Spangler and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 6802 CrossRef CAS PubMed; (g) H. Shang, Y. Wang, Y. Tian, J. Feng and Y. Tang, Angew. Chem., Int. Ed., 2014, 53, 5662 CrossRef CAS PubMed; (h) E. E. Schultz, V. N. G. Lindsay and R. Sarpong, Angew. Chem., Int. Ed., 2014, 53, 9904 CrossRef CAS PubMed; (i) J. S. Alford and H. M. L. Davies, J. Am. Chem. Soc., 2014, 136, 10266 CrossRef CAS PubMed.
- (a) S. W. Kwok, L. Zhang, N. P. Grimster and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53, 3452 CrossRef CAS PubMed; (b) M. Zibinsky and V. V. Fokin, Angew. Chem., Int. Ed., 2013, 52, 1507 CrossRef CAS PubMed; (c) T. Miura, T. Tanaka, K. Hiraga, S. G. Stewart and M. Murakami, J. Am. Chem. Soc., 2013, 135, 13652 CrossRef CAS PubMed; (d) B. Chattopadhyay and V. Gevorgyan, Org. Lett., 2011, 13, 3746 CrossRef CAS PubMed; (e) Y. Shi and V. Gevorgyan, Org. Lett., 2013, 15, 5394 CrossRef CAS PubMed; (f) E. E. Schultz and R. Sarpong, J. Am. Chem. Soc., 2013, 135, 4696 CrossRef CAS PubMed; (g) S. Chuprakov, S. W. Kwok and V. V. Fokin, J. Am. Chem. Soc., 2013, 135, 4652 CrossRef CAS PubMed; (h) B. T. Parr, S. A. Green and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 4716 CrossRef CAS PubMed; (i) Y. Xing, G. Sheng, J. Wang, P. Lu and Y. Wang, Org. Lett., 2014, 16, 1244 CrossRef CAS PubMed; (j) X. Ma, S. Pan, H. Wang and W. Chen, Org. Lett., 2014, 16, 4554 CrossRef CAS PubMed; (k) D. J. Lee, H. S. Han, J. Shin and E. J. Yoo, J. Am. Chem. Soc., 2014, 136, 11606 CrossRef CAS PubMed.
- (a) S. Chuprakov, J. A. Malik, M. Zibinsky and V. V. Fokin, J. Am. Chem. Soc., 2011, 133, 10352 CrossRef CAS PubMed; (b) J.-M. Yang, C.-Z. Zhu, X.-Y. Tang and M. Shi, Angew. Chem., Int. Ed., 2014, 53, 5142 CAS; (c) D. Yadagiri and P. Anbarasan, Org. Lett., 2014, 16, 2510 CrossRef CAS PubMed.
- (a) T. Miura, T. Biyajima, T. Fujii and M. Murakami, J. Am. Chem. Soc., 2012, 134, 194 CrossRef CAS PubMed; (b) T. Miura, T. Tanaka, T. Biyajima, A. Yada and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 3883 CrossRef CAS PubMed; (c) S. Chuprakov, B. T. Worrell, N. Selander, R. K. Sit and V. V. Fokin, J. Am. Chem. Soc., 2014, 136, 195 CrossRef CAS PubMed.
- (a) N. Selander, B. T. Worrell, S. Chuprakov, S. Velaparthi and V. V. Fokin, J. Am. Chem. Soc., 2012, 134, 14670 CrossRef CAS PubMed; (b) N. Selander and V. V. Fokin, J. Am. Chem. Soc., 2012, 134, 2477 CrossRef CAS PubMed; (c) T. Miura, Y. Funakoshi, M. Morimoto, T. Biyajima and M. Murakami, J. Am. Chem. Soc., 2012, 134, 17440 CrossRef CAS PubMed; (d) R. Liu, M. Zhang, G. Winston-McPherson and W. Tang, Chem. Commun., 2013, 49, 4376 RSC; (e) T. Miura, T. Tanaka, A. Yada and M. Murakami, Chem. Lett., 2013, 42, 1308 CrossRef CAS; (f) J. S. Alford, J. E. Spangler and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 11712 CrossRef CAS PubMed; (g) K. Chen, Z.-Z. Zhu, Y.-S. Zhang, X.-Y. Tang and M. Shi, Angew. Chem., Int. Ed., 2014, 53, 6645 CrossRef CAS PubMed; (h) T. Miura, Y. Funakoshi, T. Tanaka and M. Murakami, Org. Lett., 2014, 16, 2760 CrossRef CAS PubMed; (i) T. Miura, T. Nakamuro, K. Hiraga and M. Murakami, Chem. Commun., 2014, 50, 10474 RSC; (j) Y.-Z. Zhao, H.-B. Yang, X.-Y. Tang and M. Shi, Chem. – Eur. J., 2015, 21, 3562 CrossRef CAS PubMed; (k) Y. Wang, X. Lei and Y. Tang, Chem. Commun., 2015, 51, 4507 RSC.
- The crystal data of 2a and 4a have been deposited in CCDC with numbers 1018373 and 1018374.
- The deprotection of Bn or allyl groups of 2n and 2j under various conditions turned out to be unsuccessful, the formation of a complex product mixture indicated that the corresponding products might be unstable. For more details, please see the ESI.†
.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. CCDC 1018373 and 1018374. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00216h |
| This journal is © the Partner Organisations 2015 |