Ultrasonic spraying of Ce(Mn,Fe)O2 nanocatalysts onto a perovskite surface for highly efficient electrochemical CO2 reduction†
Received
28th August 2024
, Accepted 13th November 2024
First published on 25th November 2024
Abstract
Solid oxide electrolysis cells (SOECs) are promising devices for application in electrochemical CO2 reduction towards achieving a carbon-neutral society. However, the low durability of Ni-based electrodes during CO2 electrolysis hinders their commercial viability. Here, a fuel electrode with a nano-convex structure, i.e., (La0.75Sr0.25)0.97Cr0.5Mn0.5O3@Ce0.6Mn0.3Fe0.1O2 (LSCM@nano-CMF), is designed with an all-ceramic phase to enhance the electrochemical activity by following a simple and scalable approach. Ultrasonic spraying enables one-step formation of uniform nano-electrodes, contrasting with the tedious, consumable, and typically hired multi-step infiltration process. The excellent performance (3.89 A cm−2 at 1.5 V in the CO2 electrolysis at 850 °C) attributed to the CMF nanocatalyst with abundant oxygen vacancies and the unique perovskite/fluorite interface in a regulated structure, accelerating CO2 adsorption and displaying the synergistic catalytic effect of the dual phases. Additionally, the durability and coking tolerance of the LSCM@nano-CMF fuel electrode are demonstrated for 180 h, with a high faradaic efficiency of nearly 92%. This work provides insights for using SOECs for large-scale applications in CO2 reduction.
Broader context
Achieving a carbon-neutral society requires innovative CO2 reduction technologies and preparing for the era of space exploration, such as Mars, on which CO2 can be converted into O2. Solid oxide electrolysis cells (SOECs) are highly efficient in converting CO2 into O2, fuels, and chemicals. However, the commonly used Ni-based fuel electrode suffers from inherent redox instability during direct CO2 conditions. This work develops a novel all-ceramic fuel electrode with a nano-convex structure, enhancing activity and durability via a unique processing technique. Unlike the typical multi-step process, ultrasonic spraying offers a one-step method to create uniform nanostructured electrodes. This improves performance and simplifies manufacturing, making large-scale CO2 reduction feasible. The LSCM@nano-CMF electrode demonstrated excellent performance in both the H2 fuel cell mode (1.35 W cm−2) and CO2 electrolysis mode (3.89 A cm−2 at 1.5 V) at 850 °C and stability (180 h with 92% faradaic efficiency), highlighting its potential to advance CO2 reduction and contribute to global carbon neutrality.
|
1. Introduction
Amidst the ongoing expansion of energy-intensive industrial infrastructure, particularly within the cement and steel sectors, the resultant CO2 emissions from these industries are becoming an increasing concern.1,2 To achieve a sustainable society, the CO2 capture, utilization, and storage (CCUS) strategies are generally considered crucial for mitigating CO2 emission from fossil-fuel-driven power and industrial plants.3–5 Among them, the electrochemical reduction of CO2 to CO not only converts CO2 but also leads to the production of valuable fuels and chemicals using CO in a sustainable manner.6–10 Currently, electrochemical CO2 reduction powered by renewable electricity mainly utilizes aqueous systems, such as those used in alkaline electrolysis and photocatalysis, because these systems can be handled easily at room temperature and utilize existing industrial-scale applications.11–13 Nonetheless, the catalytic activity tends to be inadequate primarily due to the extremely stable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of CO2 (particularly at lower temperatures), low solubility of CO2, and side reaction with OH− in liquid electrolytes. As a result, the reaction kinetics is sluggish in these systems.14,15 On the other hand, solid oxide electrolysis cells (SOECs) are drawing considerable attention because of their high faradaic and energy conversion efficiencies stemming from the high operating temperatures (>800 °C), leading to favorable thermodynamics/kinetics of gaseous CO2 reduction.16–18 This SOEC has notably been proven in NASA's MOXIE program, showcasing its potential for sustainable CO2 management and utilization, enabling the transformation and recycling of CO2 in the future.19,20 In the SOECs, the conventional Ni-based electrode is widely used as the fuel electrode responsible for CO2 reduction owing to its significant catalytic activity. However, Ni-based electrodes are susceptible to intrinsic redox instability and carbon coking via the Boudouard reaction (2CO ↔ CO2 + C), hampering their application in CO2 electrolysis.21 Hence, mixed ionic and electronic conductors (MIECs), containing perovskite oxides and fluorite oxides, have been demonstrated to exhibit robust structural stability and adequate conductivity against CO/CO2 conditions.22–25 Nevertheless, the insufficient chemical adsorption/dissociation of nonpolar CO2 on MIECs constrains CO2 reduction at high temperatures compared to conventional Ni-based electrodes.23,26 Therefore, creating CO2-active sites in fuel electrodes is important for enhancing the performance of CO2 electrolysis. One efficient strategy is the incorporation of stable doped ceria materials to provide abundant oxygen vacancies as active sites.27,28 Moreover, the doped ceria exhibits MIEC properties resulting from Ce4+/Ce3+ redox coupling, thereby augmenting its surface activity in the fuel electrode.29 Comprehensively, doped ceria is exceptional at inhibiting carbon coking by facilitating the formation of carbonate species,30,31
O bond of CO2 (particularly at lower temperatures), low solubility of CO2, and side reaction with OH− in liquid electrolytes. As a result, the reaction kinetics is sluggish in these systems.14,15 On the other hand, solid oxide electrolysis cells (SOECs) are drawing considerable attention because of their high faradaic and energy conversion efficiencies stemming from the high operating temperatures (>800 °C), leading to favorable thermodynamics/kinetics of gaseous CO2 reduction.16–18 This SOEC has notably been proven in NASA's MOXIE program, showcasing its potential for sustainable CO2 management and utilization, enabling the transformation and recycling of CO2 in the future.19,20 In the SOECs, the conventional Ni-based electrode is widely used as the fuel electrode responsible for CO2 reduction owing to its significant catalytic activity. However, Ni-based electrodes are susceptible to intrinsic redox instability and carbon coking via the Boudouard reaction (2CO ↔ CO2 + C), hampering their application in CO2 electrolysis.21 Hence, mixed ionic and electronic conductors (MIECs), containing perovskite oxides and fluorite oxides, have been demonstrated to exhibit robust structural stability and adequate conductivity against CO/CO2 conditions.22–25 Nevertheless, the insufficient chemical adsorption/dissociation of nonpolar CO2 on MIECs constrains CO2 reduction at high temperatures compared to conventional Ni-based electrodes.23,26 Therefore, creating CO2-active sites in fuel electrodes is important for enhancing the performance of CO2 electrolysis. One efficient strategy is the incorporation of stable doped ceria materials to provide abundant oxygen vacancies as active sites.27,28 Moreover, the doped ceria exhibits MIEC properties resulting from Ce4+/Ce3+ redox coupling, thereby augmenting its surface activity in the fuel electrode.29 Comprehensively, doped ceria is exceptional at inhibiting carbon coking by facilitating the formation of carbonate species,30,31| |  | (1) |
| | | CO2,g + e− + O×O,s ↔ (CO3)− | (2) |
where  and O×O,s represent the surface oxygen vacancy and lattice oxygen, respectively. CO2,ads and (CO3)− denote the carbonate species. Recently, our group demonstrated that Ce(Mn,Fe)O2 (CMF) particles could function as an excellent catalyst when introduced into La(Sr)Cr(Mn)O3 (LSCM) via a simple mixing process to form the non-coking dual-phase electrode (LSCM/CMF).32 This CMF catalyst provided abundant oxygen vacancies on the surface of the LSCM electrode, accelerating CO2 adsorption and resulting in the subsequent dissociation of carbonate intermediates in CO2 reduction, thereby enhancing the electrochemical performance. However, an optimal design (including catalyst introduction) is still needed for the dual-structure electrodes. Building upon previous research, in this study, we aimed at designing the structure of the CMF nanocatalyst to further enhance their electrocatalytic activity for CO2 electrolysis. Two easily accessible approaches are often used to introduce nanocatalysts: the wet process of infiltration and the simple mixing process.33,34 However, both processes involve repetitive steps and high time/energy consumption. In particular, because electrochemical reactions are strongly affected by microstructural characteristics of the electrocatalyst, fine control is needed over structural factors like catalyst distribution and particle size. An efficient and simple process should be developed to ensure uniform catalyst distribution within the electrode. Furthermore, we applied the encapsulation technique of ultrasonic spray (ETUS) to incorporate and coat CMF nanocatalyst particles over LSCM, resulting in a highly uniform distribution of CMF. This spraying process was highly efficient because it formed the LSCM electrode with an incorporated CMF catalyst (LSCM@nano-CMF) in a single step without further processing. We demonstrated CO2 electrolysis on the LSCM@nano-CMF electrode with a faradaic efficiency (FE) of 92% at 800 °C. This all-ceramic electrode showed excellent performance in CO2 electrolysis with a promising current density of 3.89 and 2.61 A cm−2 at 850 and 800 °C (applied at 1.5 V). This electrode was also found to be stable enough for 180-h operation at 800 °C.
and O×O,s represent the surface oxygen vacancy and lattice oxygen, respectively. CO2,ads and (CO3)− denote the carbonate species. Recently, our group demonstrated that Ce(Mn,Fe)O2 (CMF) particles could function as an excellent catalyst when introduced into La(Sr)Cr(Mn)O3 (LSCM) via a simple mixing process to form the non-coking dual-phase electrode (LSCM/CMF).32 This CMF catalyst provided abundant oxygen vacancies on the surface of the LSCM electrode, accelerating CO2 adsorption and resulting in the subsequent dissociation of carbonate intermediates in CO2 reduction, thereby enhancing the electrochemical performance. However, an optimal design (including catalyst introduction) is still needed for the dual-structure electrodes. Building upon previous research, in this study, we aimed at designing the structure of the CMF nanocatalyst to further enhance their electrocatalytic activity for CO2 electrolysis. Two easily accessible approaches are often used to introduce nanocatalysts: the wet process of infiltration and the simple mixing process.33,34 However, both processes involve repetitive steps and high time/energy consumption. In particular, because electrochemical reactions are strongly affected by microstructural characteristics of the electrocatalyst, fine control is needed over structural factors like catalyst distribution and particle size. An efficient and simple process should be developed to ensure uniform catalyst distribution within the electrode. Furthermore, we applied the encapsulation technique of ultrasonic spray (ETUS) to incorporate and coat CMF nanocatalyst particles over LSCM, resulting in a highly uniform distribution of CMF. This spraying process was highly efficient because it formed the LSCM electrode with an incorporated CMF catalyst (LSCM@nano-CMF) in a single step without further processing. We demonstrated CO2 electrolysis on the LSCM@nano-CMF electrode with a faradaic efficiency (FE) of 92% at 800 °C. This all-ceramic electrode showed excellent performance in CO2 electrolysis with a promising current density of 3.89 and 2.61 A cm−2 at 850 and 800 °C (applied at 1.5 V). This electrode was also found to be stable enough for 180-h operation at 800 °C.
2. Results and discussion
2.1. Highly uniform CMF catalyst produced using ETUS
The main determinants of electrode performance are the particle size and distribution of the catalyst. Therefore, preparing SOEC with a stable and uniformly distributed active catalyst is important for high-performance CO2 electrolysis. Herein, we applied the encapsulation technique of ultrasonic spray (ETUS) to fabricate electrodes with high catalytic activity in a simple and scalable manner (Fig. 1a). This technology utilizes ultrasonic spraying to uniformly introduce a nanocatalyst into all spaces within the electrode and achieve superior electrochemical performance. The cross-sectional scanning electron microscopy (SEM) images of the LSCM@nano-CMF electrode show that the catalyst is favorably distributed over the entire electrode area, a feature that is hard to achieve using the conventional infiltration process (Fig. 1b and c and top views in Fig. S1, ESI†). Fig. 1d and e show that the CMF catalyst with a size of ∼40 nm is uniformly decorated on the LSCM surface. Therefore, this single sintering-and-coating process produced SOECs with a nano-convex-structured LSCM@nano-CMF electrode.
 |
| | Fig. 1 Fabrication of highly uniform CMF catalyst using ultrasonic spray: (a) schematic of the ultrasonic spray coating process at La0.8Sr0.2Ga0.8Mg0.2O3 (LSGM) based cells coated with La0.4Ce0.6O2 (LDC) layer. (b)–(e) Cross-sectional SEM images of LSCM@nano-CMF electrode fabricated by ultrasonic spray. High-resolution TEM analysis of (f) FIB-milled LSCM@nano-CMF, (g) enlarged CMF catalyst, and (h) and (i) interface between LSCM and CMF. (j) Scanning TEM-EDS profiles of CMF catalyst on LSCM surface. | |
Transmission electron microscopy (TEM) analysis was used to investigate the structure and interface of LSCM@nano-CMF fabricated by ETUS. The TEM image (Fig. 1f–i) also shows the cross section of the CMF nanocatalyst on the LSCM electrode, except for the area treated with focused ion beam (FIB) milling. Fig. 1g shows the lattice plane of the CMF phase with an interplanar distance of 0.309 nm, which is assigned to the (111) plane of the cubic fluorite structure. This result, which is consistent with the X-ray diffraction (XRD) observations (Fig. S2, ESI†), indicates the successful formation of CMF on the LSCM electrode. The LSCM@nano-CMF is coherently connected at the unique perovskite/fluorite interface (Fig. 1h). As shown in Fig. 1i, the (200) plane of CMF exhibits a coherent connection in the same direction with the (110) plane of LSCM at the interface, leading to interfacial interaction. Additionally, the lattice spacings of 0.27 and 0.274 nm correspond to the (110) planes of LSCM and (200) planes of CMF, respectively. This mismatch of spacings at the interface, which causes lattice strain, is expected to enhance electrochemical reactions during cell operation, such as creating active sites or accelerating ion transfer.35–37 Moreover, energy-dispersive X-ray spectroscopy (EDS) using scanning TEM images reaffirmed that the CMF phase is clearly located on the LSCM surface, with obvious segregation of specific elements (Fig. 1j). On the general surface, the CMF is uniformly distributed across the LSCM surface because the liquid encapsulated droplets cover the entire LSCM surface during the ultrasonic spraying process. Therefore, we believe that the CMF might uniformly exist in a film-like manner across the entire LSCM surface. Additionally, after the heat treatment process, the CMF takes on a convex, dot-like form as shown in Fig. 1e and j, indicating the formation of a dramatic nano-convex-structured LSCM@nano-CMF electrode.
2.2. Effect of CMF concentration on LSCM@nano-CMF fabricated by ETUS
The cell performance depends on the electrochemical reactions, which in turn are determined by surface reactions on the electrode fabricated by ETUS. Therefore, we try to identify the optimal CMF concentration for the ETUS process. Fig. 2a shows the SEM images of different LSCM@nano-CMF electrodes fabricated using 0.1 mL of spray solution with different CMF concentrations. The CMF nanoparticles were readily observed on the surface at ≥6.25 wt% CMF (denoted as LSCM@nano-CMF 6.25%, 12.5%, and 25%). Generally, the coverage by nanoparticles increases upon increasing the mass concentration of the CMF solution. Notably, in LSCM@nano-CMF 25% the nanoparticles covered almost the entire LSCM surface, and this change is expected to influence the electrochemical behavior. Fig. 2b shows a comparison of the CO2 electrolysis performances of LSCM@nano-CMF fuel electrodes with different CMF concentrations, all measured at 850 °C under 50% CO2/50% CO conditions. LSCM@nano-CMF 12.5% demonstrates a remarkable current density of 3.01 A cm−2 at 850 °C and 1.5 V. Furthermore, this current density is ∼126% higher than that of the cell using an LSCM electrode (1.33 A cm−2) at the same temperature. Another intriguing aspect is that the performance does not increase monotonically with the CMF concentration. For example, increasing the CMF concentration from 6.25 wt% to 25 wt% actually reduced the current density at 850 °C and 1.5 V from 1.79 to 1.48 A cm−2, indicating a change in the overall catalyst activity in the electrode. Fig. 2c and Fig. S3 (ESI†) display the electrochemical impedance spectroscopy (EIS) profiles of SOECs with different LSCM@nano-CMF fuel electrodes collected at 850 °C under open-circuit voltage (OCV) conditions. The ohmic resistance (Ro) is 0.20, 0.15, 0.11, and 0.16 Ω cm2 for LSCM, LSCM@nano-CMF 6.25%, 12.5%, and 25%, respectively, first decreasing from 0 to 12.5% and then increasing at 25%. These results suggest that the unique LSCM/CMF fabricated by ultrasonic spray enhances interfacial contacts within the electrode resulting from the unique interface of LSCM and CMF, as confirmed by TEM analysis. The increase in Ro at 25% could be attributed to an excess of CMF, which has a lower conductivity than LSCM.38,39 The polarization resistance (Rp), which represents that of the electrochemical reaction, showed a trend similar to that of Ro and cell performance, reaching the lowest value of 0.09 Ω cm2 in LSCM@nano-CMF 12.5%. To obtain more detailed insight into the electrode reaction, the EIS data were analyzed using a distribution of relaxation time (DRT) approach (Fig. 2d).40 Each plot contains three distinct peaks, indicating that there are three rate-determining steps in the electrochemical reaction. The peaks denoted as P1, P2, and P3 have high, medium, and low frequencies, respectively. Based on the literature, we assigned these peaks to the following processes: P1 is associated with O2− transport at the electrode/electrolyte interface, while P2 and P3 are related to surface exchange process and the gaseous reaction of the CO2 adsorption/dissociation process, respectively.35,41,42 The estimated values for these three processes are summarized in Table 1 and Fig. S4 (ESI†). The variation in DRT among these fuel electrodes shows that the P3 process dominates the electrochemical reaction when CMF nanocatalyst is introduced. This implies that the oxygen vacancy-rich CMF accelerates the adsorption/dissociation process of CO2 molecules on the electrode surface.32,43 However, at a higher content of surface CMF (≥25%), the ability to elicit a catalytic effect diminishes. Accordingly, identifying the optimal nano-convex structure is crucial for boosting the synergistic catalytic effect of LSCM and CMF on the electrode surface. The surface exchange process (P2) also exhibits a trend similar to that of P3, increasing with the CMF concentration from 0% to 12.5% and then decreasing at 25%. This might be explained in a manner consistent with the aforementioned observations. Even in the P1 process, the nano-convex structure is advantageous for O2− transport at the electrode/electrolyte interface. However, the only difference is that at a high CMF concentration (25%) the resistance is lower than that of the pristine LSCM electrode, probably due to two coexisting ion diffusion pathways of (i) conventional internal diffusion and (ii) diffusion by connected CMF, resulting in the transport of O2−. The schematic shown in Fig. 2e well illustrates the specific electrochemical processes of the LSCM@nano-CMF fuel electrodes with different CMF concentrations. By comprehensively understanding the various electrochemical perspectives, we identified LSCM@nano-CMF 12.5% as the ideal nano-convex structure and used it in the subsequent investigations.
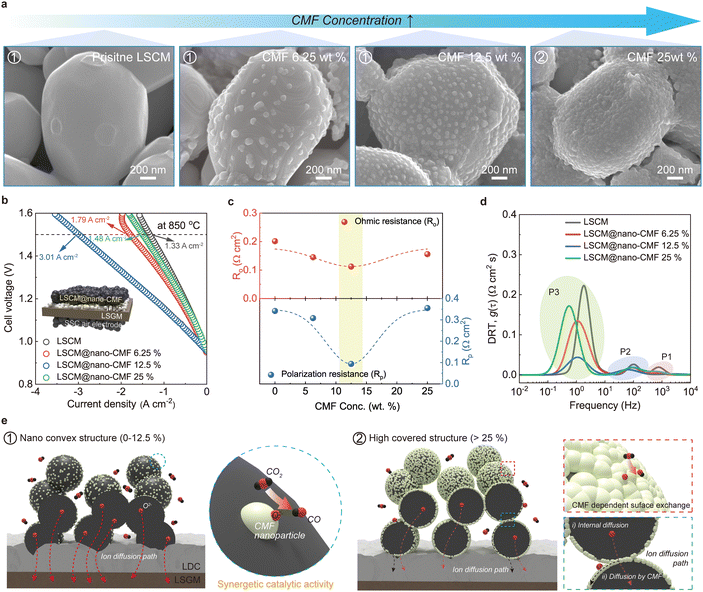 |
| | Fig. 2 Optimization and properties of the LSCM@nano-CMF fuel electrode: (a) SEM images of LSCM@nano-CMF electrodes fabricated by ETUS with different concentrations of CMF. (b) I–V curves of the CO2 electrolysis cell with different LSCM@nano-CMF electrodes at 850 °C under 50% CO2/50% CO conditions. Inset: Schematic illustration of the SOEC configuration. (c) Comparison of Ro and Rp for different LSCM@nano-CMF electrodes based on EIS data at 850 °C. (d) DRT plots for different LSCM@nano-CMF electrodes at 850 °C. (e) Schematic of electrochemical behaviors in different LSCM@nano-CMF electrodes. These electrodes have a nano-convex structure at 0–12.5% CMF and a highly covered structure at ≥25% CMF. | |
Table 1 Resistances at P1, P2, and P3 of the LSCM@nano-CMF fuel electrodes with different CMF contents during CO2 electrolysis at 850 °C
| Fuel electrode |
R
p (Ω cm2) |
R
P1 (Ω cm2) |
R
P2 (Ω cm2) |
R
P3 (Ω cm2) |
| LSCM@nano-CMF 25 |
0.355 |
0.011 |
0.030 |
0.314 |
| LSCM@nano-CMF 12.5 |
0.094 |
0.004 |
0.018 |
0.072 |
| LSCM@nano-CMF 6.25 |
0.308 |
0.008 |
0.028 |
0.269 |
| LSCM |
0.342 |
0.013 |
0.031 |
0.298 |
2.3. Electrochemical performance of the optimized LSCM@nano-CMF fuel electrode
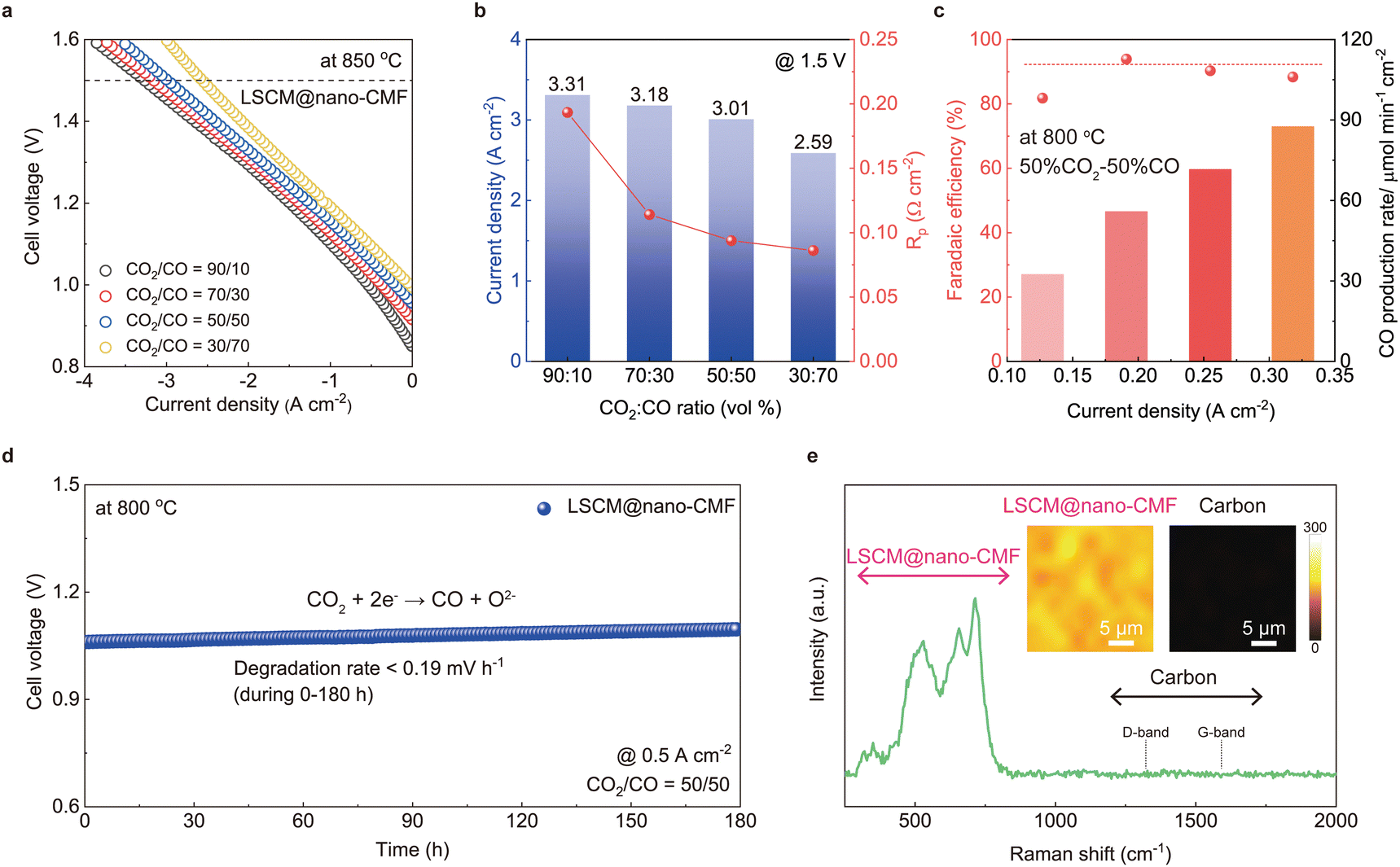
To further demonstrate the capability of the LSCM@nano-CMF fuel electrode fabricated by ultrasonic spray, we compared two SOECs with the optimal LSCM@nano-CMF fuel electrode (Fig. 3a and Table 2) and an electrode of identical composition (12.5% CMF) prepared by a conventional ball-milling process (denoted as the LSCM/CMF composite electrode). From Fig. 3b, the all-ceramic LSCM@nano-CMF electrode cell operated in both the H2 fuel cell mode (1.35 W cm−2) and CO2 electrolysis mode (3.01 A cm−2 at 1.5 V) at 850 °C. In contrast, the LSCM/CMF electrode cell showed a lower CO2 electrolysis performance of 2.56 A cm−2 at 1.5 V (Fig. 3c). The reason is that the nanocatalyst in the LSCM@nano-CMF electrode created additional active sites along with the distinctive perovskite/fluorite interface, while the LSCM/CMF electrode is a randomly mixed without forming a specific interface (Fig. S5, ESI†). Additionally, the Rp values obtained from EIS data are displayed in Fig. 3d. Across all temperatures, LSCM@nano-CMF has lower Rp due to nanocatalyst introduction. The resistance of P3, which is due to molecular CO2 adsorption/dissociation, decreases during CO2 electrolysis in both cells and more significant in the LSCM@nano-CMF cell. This is attributed to the elongation of active sites by the nanocatalyst (Fig. 3e). Additionally, LSCM@nano-CMF demonstrates an enhanced capability for the amount of CO2 adsorption at a temperature above 600 °C in the CO2-temperature programmed desorption (CO2-TPD) because the CO2-TPD intensity curves show that the peak area of LSCM@nano-CMF is the largest, following the trend LSCM@nano-CMF > LSCM/CMF > LSCM (Fig. S7, ESI†). Furthermore, the LSCM@nano-CMF shows a small peak position signal at temperatures between 700 and 600 °C, suggesting the relatively higher adsorption strength of CO2. This superior CO2 adsorption is attributed to the presence of abundant oxygen vacancies in the uniformly distributed nano CMF catalyst compared to the LSCM and LSCM/CMF electrodes.44–46 These oxygen vacancies provide active sites for CO2 adsorption at elevated temperatures. On the other hand, in the P2 process, significant changes were observed only in the nano-convex-structured LSCM@nano-CMF that has an increasing resistance. This increase of resistance in the P2 process, which corresponds to a surface exchange reaction under bias conditions, can be considered in two ways. A pessimistic interpretation is an increase in resistance due to degradation of the electrode surface, such as carbon deposition during electrochemical reactions in CO2 electrolysis. However, the typical degradation mechanisms in electrochemical reactions imply an increase in the surface CO2 adsorption/desorption capability, which disagrees with our observations of the P3 process. Therefore, further investigation on the stability of CO2 electrolysis was carried out and described below. The most reliable interpretation is that the nano-dot structure of CMF, along with the distinctive perovskite/fluorite interface, enhances the catalytic activity on the electrode surface. In other words, the superior CO2 adsorption capability of CMF under bias conditions leads to an increase in surface activity, such as molecule spillover, thereby inducing the synergistic catalytic effect between LSCM and CMF in the LSCM@nano-CMF fuel electrode.23,47 We also evaluated CO2 electrolysis using the LSCM@nano-CMF fuel electrode at 850 °C in various CO2/CO mixtures, and the results are shown in Fig. 4a and b. As shown in Table S1 (ESI†), the OCVs are 0.995, 0.956, 0.915, and 0.850 V for CO2/CO compositions of 30/70, 50/50, 70/30, and 90/10, respectively. The current densities for CO2 electrolysis at 1.5 V are 3.31, 3.18, and 2.59 A cm−2 for the CO2/CO conditions of 90/10, 70/30, and 30/70, respectively, showcasing the superior electrochemical performance. Additionally, the current densities for CO2 electrolysis at 1.2V are 1.49, 1.39, 1.27, and 1.07 A cm−2 at 90/10, 70/30, 50/50, and 30/70 CO2/CO compositions, respectively. In similar systems utilizing LSCM-GDC or LSCM-YSZ composite electrodes, as described by Yue and Irvine,48,49 the I–V curves for varying CO2/CO ratios (from 90/10 to 30/70) shift in parallel, driven by the differences in OCV values. This suggests that higher concentrations of CO2 are essential for achieving optimal electrolysis performance.32
 |
| | Fig. 3 Electrochemical properties of the nano-convex LSCM@nano-CMF electrode: (a) cross-sectional SEM image of CO2 electrolysis cell with LSCM@nano-CMF fuel electrode. (b) Electrochemical performance of the CO2 electrolysis cell with LSCM@nano-CMF fuel electrode. (c) I–V curves of LSCM@nano-CMF, LSCM/CMF composite, and LSCM cells at 850 °C. (d) Temperature dependence of Rp for the LSCM@nano-CMF, LSCM/CMF, and LSCM electrodes. Inset: EIS data at 850 °C. The electrochemical properties of (e) DRT plots and (f) variation of DRT plots for LSCM@nano-CMF and LSCM/CMF composite electrodes under an applied bias at 850 °C. | |
Table 2 Electrochemical performance profiles of LSCM@nano-CMF, LSCM/CMF, and LSCM fuel electrodes during CO2 electrolysis at 850 °C
| Fuel electrode |
OCV (V) |
Current density (A cm−2@1.5 V) |
R
o (Ω cm2) |
R
p (Ω cm2) |
| LSCM@nano-CMF |
0.956 |
3.01 |
0.11 |
0.09 |
| LSCM/CMF |
0.952 |
2.56 |
0.16 |
0.14 |
| LSCM |
0.950 |
1.33 |
0.20 |
0.34 |
 |
| | Fig. 4 CO2 electrolysis performance and durability of SOEC: (a) I–V curves of CO2 electrolysis using the LSCM@nano-CMF electrode under different CO2/CO conditions. (b) Cell performance and Rp under an applied voltage of 1.5 V and different CO2/CO conditions. (c) Faradaic efficiency and (d) stability of CO2 electrolysis using the LSCM@nano-CMF fuel electrode at 800 °C. (e) Raman spectrum of the LSCM@nano-CMF fuel electrode after stability test of CO2 electrolysis. Inset: Raman mapping. | |
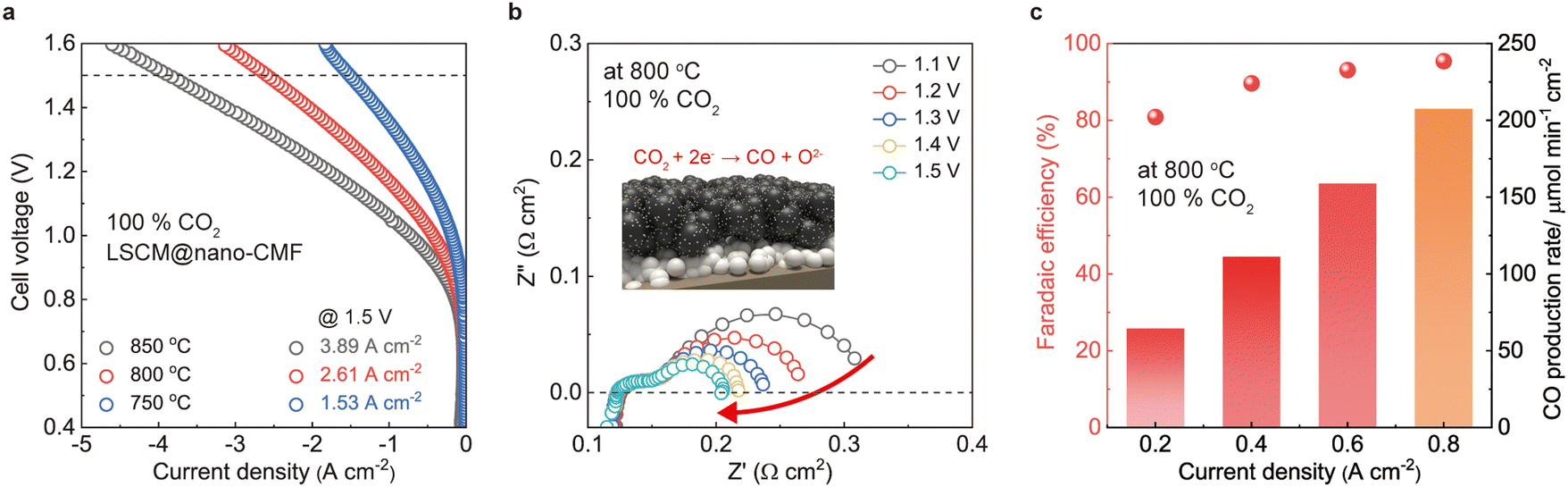
The FEs of the LSCM@nano-CMF fuel electrode were also measured at different current densities using Faraday's law (Fig. 4c). At most current densities, the estimated FE is ∼92%, demonstrating remarkable electrochemical CO2 reduction on the LSCM@nano-CMF fuel electrode at 800 °C. We then tested the SOEC stability for CO2 reduction at 800 °C (Fig. 4d). At a current density of 0.5 A cm−2, the SOEC with LSCM@nano-CMF fuel electrode operated continuously in CO2 electrolysis mode for 180 h, achieving a degradation rate of ≤0.19 mV h−1. As shown in Fig. S9 (ESI†), the degradation rate is lower than that of both LSCM and LSCM/CMF, demonstrating the superior stability of LSCM@nano-CMF, which could be attributed to its perovskite/fluorite interface. Fig. 4e shows the Raman spectra after the stability test in CO2 electrolysis, and no carbon was deposited on the LSCM@nano-CMF fuel electrode. The mapping area of the electrode is depicted in Fig. S10 (ESI†). Apart from the Raman spectrum of LSCM@nano-CMF (400–750 cm−1), there were no peaks due to the D band (1359 cm−1) or G band (1581 cm−1) of carbon. Moreover, Raman mapping of LSCM@nano-CMF did not detect any carbon signals,34 confirming the absence of carbon coking on the electrode during CO2 electrolysis operation. To compare this CO2 electrolysis with the most recently developed ones,48,49 we investigated CO2 electrolysis using the LSCM@nano-CMF fuel electrode in pure CO2. The LSCM@nano-CMF fuel electrode achieved remarkably high current densities of 3.89, 2.61, and 1.53 A cm−2 under an applied voltage of 1.5 V at 850, 800, and 750 °C, respectively (Fig. 5a). These electrochemical performances of LSCM@nano-CMF outperform various LSGM-based cells in the literature (Table 3 and Fig. S11, ESI†).50–57Fig. 5b shows the decreasing Rp of the EIS data under bias, indicating the favorable operation of the LSCN@nano-CMF fuel electrode on pure CO2 at 800 °C. Moreover, the FEs of the LSCM@nano-CMF fuel electrode were also calculated in pure CO2. In Fig. 5c, the estimated FE of over 92% demonstrates the superiority of the LSCM@nano-CMF fuel electrode in electrochemical CO2 reduction in pure CO2 at 800 °C. Thus, this active and durable LSCM@nano-CMF fabricated by ultrasonic spray is a reasonable fuel electrode for catalyzing CO2 electrolysis in SOECs.
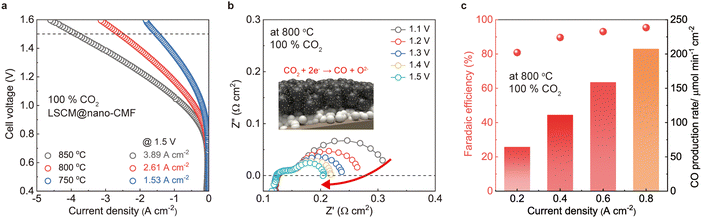 |
| | Fig. 5 Extremely high-CO2 electrolysis performance of SOEC: (a) I–V curves of CO2 electrolysis using the LSCM@nano-CMF electrode under pure CO2. (b) EIS data of LSCM@nano-CMF fuel electrode at 800 °C under bias conditions. (c) Faradaic efficiency of CO2 electrolysis using LSCM@nano-CMF fuel electrode at 800 °C under pure CO2. | |
Table 3 Comparisons of current densities obtained during CO2 electrolysis between various fuel electrodes
| Cell configuration |
Current density at 1.5 V (A cm−2) |
Ref. |
| Fuel electrode |
Electrolyte |
Air electrode |
850 °C |
800 °C |
750 °C |
| LSCM@nano-CMF |
LDC/LSGM |
SSC |
3.89 |
2.61 |
1.53 |
This work |
| H-LSCFP |
LDC/LSGM |
LSCF–GDC |
3.7 |
2.2 |
1.37 |
50
|
| HE-PSCFMMN |
LSGM |
LSCF |
— |
1.95 |
— |
51
|
| SFMMc–SDC |
LSGM |
LSCF–SDC |
1.80 |
1.35 |
0.90 |
52
|
| Fe@PSFM |
LDC/LSGM |
LSCF–GDC |
0.85 |
0.7 |
0.58 |
53
|
| SF1.5M-R |
LDC/LSGM |
LSCF–SDC |
— |
1.5 |
0.9 |
54
|
| F-SFM |
LDC/LSGM |
LSCF–SDC |
— |
1.34 |
0.96 |
55
|
| LSFT |
LSGM |
LSFT |
— |
1.57 |
0.84 |
56
|
| PBSFG |
LSGM |
PBSFG |
0.83 |
0.44 |
0.2 |
57
|
3. Conclusions
In summary, LSCM electrodes incorporating oxygen vacancy-rich CMF nanocatalysts were successfully fabricated using an encapsulation technique of ultrasonic spray (ETUS). Unlike conventional processes such as infiltration, our technique formed this CMF catalyst in situ without additional steps and achieve a highly uniform nanocatalyst distribution within the electrode. Additionally, we provided guidelines for the design of electrochemically optimal nano-convex structures, such as the expanding the active sites and the unique interface of perovskite/fluorite formed by encapsulation. At 850 °C, the SOECs developed in this study showed outstanding performance in both the H2 fuel cell mode (1.35 W cm−2) and CO2 electrolysis mode (3.89 A cm−2 at 1.5 V), due to enhanced electrode surface activity facilitated by a better chemisorption/dissociation process of gas molecules such as CO2 on the CMF catalyst. For CO2 electrolysis, the SOEC achieved a high FE of 92% and good durability of 180 h at 800 °C without any carbon coking. These results further highlight the feasibility of applying ETUS to produce SOECs for large-scale CO2 reduction applications.
Author contributions
T. H. S. conceived the main idea and guided the work. S. W. L. and T. H. S. designed all the experiments for characterization. S. W. L. and T. H. N. fabricated the LSCM@nano-CMF composite electrode materials and SOEC single cells and their performance; S. W. L., S. H. L., and T. H. S. analyzed all measured results. S. W. L. and T. H. S. performed the reproducible experiments and wrote the manuscript. T. I., J. T. S. I., and T. H. S. guided and participated in building the hypothesis and main discussion. All authors were involved in the preparation of the manuscript.
Data availability
Data are available upon request from the authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Nano/Material Technology Development Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT (NRF-2021M3H4A3A02086499). This research was also supported by the Ceramic Strategic Technology R&D program through the Korea Institute of Ceramic Engineering & Technology (KICET) (grant NTIS no. 1415187241).
References
- T. Lei, D. Wang, X. Yu, S. Ma, W. Zhao, C. Cui, J. Meng, S. Tao and D. Guan, Nature, 2023, 622, 514–520 CrossRef CAS.
- P. Fennell, J. Driver, C. Bataill and S. J. Davis, Nature, 2022, 603, 574–577 CrossRef CAS PubMed.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. M. Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS.
- M. T. Dvorak, K. C. Armour, D. M. W. Frierson, C. Proistosescu, M. B. Baker and C. J. Smith, Nat. Clim. Change, 2022, 12, 547–552 CrossRef.
- J.-P. Gattuso, A. Magnan, R. Billé, W. W. L. Cheung, E. L. Howes, F. Joos, D. Allemand, L. Bopp, S. R. Cooley, C. M. Eakin, O. Hoegh-Guldberg, R. P. Kelly, H.-O. Portner, A. D. Rogers, J. M. Baxter, D. Laffoley, D. Osborn, A. Rankovic, J. Rochette, U. R. Sumaila, S. Treyer and C. Turley, Science, 2015, 249, aac4722 CrossRef PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chen, C. Hahn, J. K. Norskov, T. F. Jaramilo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- S. H. Jensen, C. Graves, M. Mogensen, C. Wendel, R. Braun, G. Hughes, Z. Gao and S. A. Barnett, Energy Environ. Sci., 2015, 8, 2471 RSC.
- L. Chen, F. Chen and C. Xia, Energy Environ. Sci., 2014, 7, 4018–4022 RSC.
- A. Hauch, R. Kungas, P. Blennow, A. B. Hansen, J. B. Hansen, B. V. Mathiesen and M. B. Mogensen, Science, 2020, 370, aba6118 CrossRef PubMed.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511 RSC.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 CrossRef CAS.
- Y. Song, X. Zhang, K. Xie, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1902033 CrossRef CAS PubMed.
- J. A. Rabinowitz and M. W. Kanon, Nat. Commun., 2020, 11, 5231 CrossRef CAS.
- S. D. Ebbesen, S. H. Jensen, A. Hauch and M. B. Mogensen, Chem. Rev., 2014, 114, 10697–10734 CrossRef CAS PubMed.
- T. H. Shin, J.-H. Myung, M. Verbraeken, G. Kim and J. T. S. Irvine, Faraday Discuss., 2015, 182, 227–239 RSC.
- S. D. Ebbesen and M. B. Mogensen, J. Power Sources, 2009, 193, 349–358 CrossRef CAS.
- J. A. Hoffman, M. H. Hecht, D. Rapp, J. J. Hartvigsen, J. G. Soohoo, A. M. Aboobaker, J. B. Mcclean, A. M. Liu, E. D. Hinterman, M. Nasr, S. Hariharan, K. J. Horn, F. E. Meyen, H. Okkels, P. Steen, S. Elangovan, C. R. Graves, P. Khopkar, M. B. Madsen, G. E. Voecks, P. H. Smith, T. L. Skafte, K. R. Araghi and D. J. Eisenman, Sci. Adv., 2022, 8, eabp8636 CrossRef CAS PubMed.
- M. Hecht, J. Hoffman, D. Rapp, J. Mcclean, J. Soohoo, R. Schaefer, A. Aboobaker, J. Mellstrom, J. Hartvigsen, E. Hinterman, G. Voecks, A. Liu, M. Nasr, J. Lewis, J. Johnson, C. Guernsey, J. Swoboda, C. Eckert, C. Alcalde, M. Poirier, P. Khopkar, S. Elangovan, M. Madsen, P. Smith, C. Graves, G. Sanders, K. Araghi, M. Juarez, D. Larson, J. Agui, A. Burns, K. Lackner, R. Nielsen, T. Pike, B. Tata, K. Wilson, T. Brown, T. Disarro, R. Morris, R. Schaefer, R. Steinkraus, R. Surampudi, T. Werne and A. Ponce, Space Sci. Rev., 2021, 217, 1–76 CrossRef.
- E. P. Murray, T. Tsai and S. A. Barnett, Nature, 1999, 400, 649–651 CrossRef CAS.
- S. Sengodan, S. Choi, A. Jun, T. H. Shin, Y.-W. Ju, H. Y. Jeong, J. Shin, J. T. S. Irvine and G. Kim, Nat. Mater., 2015, 14, 205–209 CrossRef CAS.
- T. Tan, Z. Wang, M. Qin, W. Zhong, J. Hu, C. Yang and M. Liu, Adv. Funct. Mater., 2022, 32, 2202878 CrossRef CAS.
- T. H. Shin, S. Ida and T. Ishihara, Chem. Soc., 2011, 133, 19399–19407 CrossRef CAS PubMed.
- J. T. S. Irvine, D. Neagu, M. C. Verberaeken, C. Charzichristodoulou, C. Graves and M. B. Mogensen, Nat. Energy, 2016, 1, 1–13 Search PubMed.
- X. Yang, K. Sun, M. Ma, C. Xu, R. Ren, J. Qiao, Z. Wang, S. Zhen, R. Hou and W. Sun, Appl. Catal., B, 2020, 272, 118968 CrossRef CAS.
- Z. A. Feng, F. E. Gabaly, X. Ye, Z.-X. Shen and W. C. Chueh, Nat. Commun., 2014, 5, 4374 CrossRef CAS.
- R. Schmitt, A. Nenning, O. Kraynis, R. Korobko, A. I. Frenkel, I. Lubomirsky, S. M. Haile and J. L. M. Rupp, Chem. Soc. Rev., 2020, 49, 554–592 RSC.
- M. Mogensen, N. M. Sammes and G. A. Tompsett, Solid State Ion, 2000, 129, 63–94 CrossRef CAS.
- T. L. Skafte, Z. Guan, M. L. Machala, C. B. Gopal, M. Monti, L. Martinez, E. Stamate, S. Sanna, J. A. G. Torres, E. J. Crumlin, M. Garcia-Melchor, M. Bajdich, W. C. Chueh and C. Graves, Nat. Energy, 2019, 4, 846–855 CrossRef CAS.
- Z. A. Feng, M. L. Machala and W. C. Chueh, Phys. Chem. Chem. Phys., 2015, 17, 12273–12281 RSC.
- S. Lee, M. Kim, K. T. Lee, J. T. S. Irvine and T. H. Shin, Adv. Energy Mater., 2021, 11, 2100339 CrossRef CAS.
- D. Ding, X. Li, S. Y. Lai, K. Gerdes and M. Liu, Energy Environ. Sci., 2014, 7, 552–575 RSC.
- J. M. Vohs and R. J. Gorte, Adv. Mater., 2009, 21, 943–956 CrossRef CAS.
- F. He, M. Hou, F. Zhu, D. Liu, H. Zhang, F. Yu, Y. Shou, Y. Ding, M. Liu and Y. Chen, Adv. Energy Mater., 2022, 12, 2202175 CrossRef CAS.
- Z. Du, L. Shen, Y. Gong, M. Zhang, J. Zhang, J. Feng, K. Li, K. Swierczek and H. Zhao, Adv. Funct. Mater., 2024, 34, 2310790 CrossRef CAS.
- S. W. Lee, J. W. Park, Y. W. Ju and T. H. Shin, Small Struct., 2024, 5, 2300292 CrossRef CAS.
- S. Tao and J. T. S. Irvine, J. Electrochem. Soc., 2004, 151, A252–A259 CrossRef CAS.
- T. H. Shin, P. Vanalabhpatana and T. Ishihara, J. Electrochem. Soc., 2010, 157, B1896–B1901 CrossRef CAS.
- T. H. Wan, M. Saccoccio, C. Chen and F. Cuicci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- X. Xi, Y. Fan, J. Zhang, J.-L. Luo and X.-Z. Fu, J. Mater. Chem. A, 2022, 10, 2509–2518 RSC.
- S. W. Lee, T. H. Nam, M. Kim, S. Lee, K. H. Lee, J. H. Park and T. H. Shin, Inorg. Chem. Front., 2023, 10, 3536 RSC.
- X. Ding, Z. Gao, D. Diong, X. Zhao, H. Hou, S. Zhang and G. Yuan, Appl. Catal., B, 2019, 243, 546–555 CrossRef CAS.
- X. Sun, Y. Ye, M. Zhou, H. Chen, Y. Li, P. Chen, D. Dong, Y. Ling and M. Khan, J. Mater. Chem. A, 2022, 10, 2327–2335 RSC.
- H. Lv, L. Lin, X. Zhang, D. Gao, Y. Song, Y. Zhou, Q. Liu, G. Wang and X. Bao, J. Mater. Chem. A, 2019, 7, 11967–11975 RSC.
- W. Qi, Y. Gan, D. Yin, Z. Li, G. Wu, K. Xie and Y. Wu, J. Mater. Chem. A, 2014, 2, 6904–6915 RSC.
- H. Kang, L. Zhu, S. Li, S. Yu, Y. Niu, B. Zhang, W. Chu, X. Liu, S. Perathoner, G. Centi and Y. Liu, Nat. Catal., 2023, 6, 1062–1072 CrossRef CAS.
- X. Yue and J. T. S. Irvine, J. Electrochem. Soc., 2012, 158, F442–F448 CrossRef.
- X. Yue and J. T. S. Irvine, J. Mater. Chem. A, 2017, 5, 7081–7090 RSC.
- A. Akhmadjonov, K. T. Bae and K. T. Lee, Nano-Micro Lett., 2024, 16, 93 CrossRef CAS PubMed.
- Z. Wang, T. Tan, K. Du, Q. Zhang, M. Liu and C. Yang, Adv. Mater., 2024, 36, 2312119 CrossRef CAS PubMed.
- Y. Jiang, Y. Yang, C. Xia and H. J. M. Bouwmeester, J. Mater. Chem. A, 2019, 7, 22939–22949 RSC.
- K. Zhang, Y. Zhao, W. He, P. Zhao, D. Zhang, T. He, Y. Wang and T. Liu, Energy Technol., 2020, 8, 2000539 CrossRef CAS.
- X. Xi, J. Liu, W. Luo, Y. Fan, J. Zhang, J. L. Luo and X. Z. Fu, Adv. Energy Mater., 2021, 11, 2102845 CrossRef CAS.
- Y. Li, Y. Li, Y. Wan, Y. Wan, Y. Xie, J. Zhu, H. Pan, X. Zhang and C. Xia, Adv. Energy Mater., 2019, 9, 1803156 CrossRef.
- Y. Hou, L. Wang, L. Bian, Y. Wang and K. Chou, ACS Appl. Mater. Interfaces, 2021, 13, 22381–22390 CrossRef CAS.
- H. J. Lee, S. J. Son, S. K. Kim, S. Choi, M. K. Kim, T. H. Shin and J. H. Joo, J. Power Sources, 2024, 595, 234032 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Tae Heon
Nam
a,
Seok Hee
Lee
a,
Tatsumi
Ishihara
ab,
Tae Heon
Nam
a,
Seok Hee
Lee
a,
Tatsumi
Ishihara

 and O×O,s represent the surface oxygen vacancy and lattice oxygen, respectively. CO2,ads and (CO3)− denote the carbonate species. Recently, our group demonstrated that Ce(Mn,Fe)O2 (CMF) particles could function as an excellent catalyst when introduced into La(Sr)Cr(Mn)O3 (LSCM) via a simple mixing process to form the non-coking dual-phase electrode (LSCM/CMF).32 This CMF catalyst provided abundant oxygen vacancies on the surface of the LSCM electrode, accelerating CO2 adsorption and resulting in the subsequent dissociation of carbonate intermediates in CO2 reduction, thereby enhancing the electrochemical performance. However, an optimal design (including catalyst introduction) is still needed for the dual-structure electrodes. Building upon previous research, in this study, we aimed at designing the structure of the CMF nanocatalyst to further enhance their electrocatalytic activity for CO2 electrolysis. Two easily accessible approaches are often used to introduce nanocatalysts: the wet process of infiltration and the simple mixing process.33,34 However, both processes involve repetitive steps and high time/energy consumption. In particular, because electrochemical reactions are strongly affected by microstructural characteristics of the electrocatalyst, fine control is needed over structural factors like catalyst distribution and particle size. An efficient and simple process should be developed to ensure uniform catalyst distribution within the electrode. Furthermore, we applied the encapsulation technique of ultrasonic spray (ETUS) to incorporate and coat CMF nanocatalyst particles over LSCM, resulting in a highly uniform distribution of CMF. This spraying process was highly efficient because it formed the LSCM electrode with an incorporated CMF catalyst (LSCM@nano-CMF) in a single step without further processing. We demonstrated CO2 electrolysis on the LSCM@nano-CMF electrode with a faradaic efficiency (FE) of 92% at 800 °C. This all-ceramic electrode showed excellent performance in CO2 electrolysis with a promising current density of 3.89 and 2.61 A cm−2 at 850 and 800 °C (applied at 1.5 V). This electrode was also found to be stable enough for 180-h operation at 800 °C.
and O×O,s represent the surface oxygen vacancy and lattice oxygen, respectively. CO2,ads and (CO3)− denote the carbonate species. Recently, our group demonstrated that Ce(Mn,Fe)O2 (CMF) particles could function as an excellent catalyst when introduced into La(Sr)Cr(Mn)O3 (LSCM) via a simple mixing process to form the non-coking dual-phase electrode (LSCM/CMF).32 This CMF catalyst provided abundant oxygen vacancies on the surface of the LSCM electrode, accelerating CO2 adsorption and resulting in the subsequent dissociation of carbonate intermediates in CO2 reduction, thereby enhancing the electrochemical performance. However, an optimal design (including catalyst introduction) is still needed for the dual-structure electrodes. Building upon previous research, in this study, we aimed at designing the structure of the CMF nanocatalyst to further enhance their electrocatalytic activity for CO2 electrolysis. Two easily accessible approaches are often used to introduce nanocatalysts: the wet process of infiltration and the simple mixing process.33,34 However, both processes involve repetitive steps and high time/energy consumption. In particular, because electrochemical reactions are strongly affected by microstructural characteristics of the electrocatalyst, fine control is needed over structural factors like catalyst distribution and particle size. An efficient and simple process should be developed to ensure uniform catalyst distribution within the electrode. Furthermore, we applied the encapsulation technique of ultrasonic spray (ETUS) to incorporate and coat CMF nanocatalyst particles over LSCM, resulting in a highly uniform distribution of CMF. This spraying process was highly efficient because it formed the LSCM electrode with an incorporated CMF catalyst (LSCM@nano-CMF) in a single step without further processing. We demonstrated CO2 electrolysis on the LSCM@nano-CMF electrode with a faradaic efficiency (FE) of 92% at 800 °C. This all-ceramic electrode showed excellent performance in CO2 electrolysis with a promising current density of 3.89 and 2.61 A cm−2 at 850 and 800 °C (applied at 1.5 V). This electrode was also found to be stable enough for 180-h operation at 800 °C.