
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompelling DNA intercalation through ‘anion–anion’ anti-coulombic interactions: boron cluster self-vehicles as promising anticancer agents†
Laura
Gutiérrez-Gálvez
a,
Tania
García-Mendiola
 ab,
Encarnación
Lorenzo
abc,
Miquel
Nuez-Martinez
d,
Carmen
Ocal
d,
Shunya
Yan
d,
Francesc
Teixidor
d,
Teresa
Pinheiro
ef,
Fernanda
Marques
fg and
Clara
Viñas
*d
ab,
Encarnación
Lorenzo
abc,
Miquel
Nuez-Martinez
d,
Carmen
Ocal
d,
Shunya
Yan
d,
Francesc
Teixidor
d,
Teresa
Pinheiro
ef,
Fernanda
Marques
fg and
Clara
Viñas
*d
aDepartamento de Química Analítica y Análisis Instrumental, Universidad Autónoma de Madrid, 28049, Madrid, Spain
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049, Madrid, Spain
cIMDEA-Nanociencia, Ciudad Universitaria de Cantoblanco, 28049, Madrid, Spain
dInstitut de Ciència de Materials de Barcelona (C.S.I.C.) Campus U.A.B, 08193 Bellaterra, Barcelona, Spain. E-mail: clara@icmab.es
eiBB – Instituto de Bioengenharia e Biociências, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais 1, 1049-001 Lisboa, Portugal
fDepartamento de Engenharia e Ciências Nucleares, Instituto Superior Técnico, Universidade de Lisboa, Estrada Nacional 10, 2695-066 Bobadela LRS, Portugal
gC2TN – Centro de Ciências e Tecnologias Nucleares, Instituto Superior Técnico, Universidade de Lisboa, Estrada Nacional 10, 2695-066 Bobadela LRS, Portugal
First published on 7th August 2024
Abstract
Anticancer drugs inhibit DNA replication by intercalating between DNA base pairs, forming covalent bonds with nucleotide bases, or binding to the DNA groove. To develop safer drugs, novel molecular structures with alternative binding mechanisms are essential. Stable boron hydrides offer a promising alternative for cancer therapy, opening up additional options like boron neutron capture therapy based on 10B and thermal neutron beams or proton boron fusion therapy using 11B and proton beams. These therapies are more efficient when the boron compound is ideally located inside cancer cells, particularly in the nucleus. Current cancer treatments often utilize small, polycyclic, aromatic, planar molecules that intercalate between ds-DNA base pairs, requiring only a spacing of approximately 0.34 nm. In this paper, we demonstrate another type of intercalation. Notably, [3,3′-Fe(1,2-C2B9H11)2]−, ([o-FESAN]−), a compact 3D molecule measuring 1.1 nm × 0.6 nm, can as well intercalate by strong non-bonding interactions preferentially with guanine. Unlike known intercalators, which are positive or neutral, [o-FESAN]− is a negative species and when an [o-FESAN]− molecule approaches the negatively charged DNA phosphate chain an anion–anion interaction consistently anti-electrostatic via Ccluster–H⋯O–P bonds occurs. Then, when more molecules approach, an elongated outstandingly self-assembled structure of [o-FESAN]−–[o-FESAN]− forms moving anions towards the interthread region to interact with base pairs and form aggregates of four [o-FESAN]− anions per base pair. These aggregates, in this environment, are generated by Ccluster–H⋯O–C, N–H⋯H–B and Ccluster–H⋯H–B interactions. The ferrabis(dicarbollide) boron-rich small molecules not only effectively penetrate the nucleus but also intercalate with ds-DNA, making them promising for cancer treatment. This amphiphilic anionic molecule, used as a carrier-free drug, can enhance radiotherapy in a multimodal perspective, providing healthcare professionals with improved tools for cancer treatment. This work demonstrates these findings with a plethora of techniques.
Introduction
Amid historical focus on carbon-based drugs, the current landscape of therapeutic approaches urges diversification of methodologies as, in particular, exploring drugs that incorporate boron. This strategic shift opens avenues for addressing complex medical challenges. Despite achievements made, ongoing discussions between medicinal and pharmaceutical chemists emphasize the need for new drugs to enhance clinical efficacy against persistent challenges such as cancer, inflammatory conditions, and antibiotic resistance.The unique characteristics of boron-based systems stem from the distinctive traits of boron atoms, such as electron deficiency and the capacity to substitute carbon atoms, as well as the properties of boron compounds, including hydrophobicity, lipophilicity, and versatile stereochemistry. Consequently, the distinctive properties exhibited by boron hydride clusters underscore their significant relevance and profound influence on the metabolism and biodistribution of molecules that incorporate them. These key features include (i) exceptional metabolic stability attributed to their inorganic structure, which confers substantial metabolic stability, evading recognition by conventional enzymes associated with biodegradation;1–4 (ii) amphiphilic character5–8 (i.e., good solubility in water as well as in non-aqueous solvents) that can be explored to shuttle these small molecules between immiscible solvents1,9 and cross membranes;10–15 and (iii) unusual intermolecular interactions as boron clusters engage in atypical intermolecular interactions, such as dihydrogen bonds and bridging interactions, in addition to conventional hydrogen bonds.16–20 These interactions, formed with biological targets (carbohydrates,21 amino acids,22 proteins23–26 and DNA27,28), contribute to their distinctive pharmacological profile. The potential of the non-covalent dihydrogen bond interactions due to their physicochemical properties (especially their amphiphilic character) can be observed in practical applications in the field of materials science and bioorganic chemistry.29–33
Consequently, these systems (boranes, carboranes and metallacarboranes) emerge as compelling alternatives to conventional organic frameworks in the design of novel drugs. An aspect rarely considered is that voids or unfilled spaces in proteins, enzymes, and DNA are volumes with spherical or elongated ellipsoidal shape. In these cavities, molecules with appropriate dimensions, a robust 3D shape and a high likelihood of anchoring would fit or alternatively interact better than molecules with a 2D base. For this reason, boron hydride clusters, with their 3D bulky characteristic and multiple possibilities of forming hydrogen and dihydrogen bonds with receptors in the cavity, should be significant alternatives to be considered for achieving perfect molecular docking. Their resistance to biodegradation,1–4 improved cellular membrane permeability,10–15 and unique intermolecular interactions22,27,28 make them promising candidates for developing drugs with enhanced residence times in living organisms.34–40
One of the most prominent metallacarboranes is the metallabis(dicarbollide) anion, denoted as [M(C2B9H11)2]−, with the ortho-cobaltabis(dicarbollide) (abbreviated as [o-COSAN]−),41–45 being the most well-known variant. This anion, with an empirical formula of [3,3′-Co(1,2-C2B9H11)2]−, boasts dimensions of approximately 1.1 nm × 0.6 nm.5,46 This compound that ranks high, in terms of chemical and thermal stabilities (withstanding strong acid, moderate base, high temperatures and intense radiation),1 exhibits distinctive 3D global aromaticity.47 Its negative charge is differentially distributed across the 45 atoms within the icosahedral cluster.17,48–50 Such a distribution imparts amphiphilic properties, leading to the formation of aggregates (vesicles and micelles) in low-concentration aqueous solutions.5–8,51,52
Based on experimental and theoretical demonstrations of the 3D global aromaticity displayed by both neutral and anionic boron clusters,47 coupled with the pivotal roles that aromatic compounds play in various fields such as biochemistry, drug delivery, nanotechnology, and biological markers, a new frontier has emerged.
DNA is considered a primary target for several anticancer medications,53 which induce apoptosis by damaging DNA.54–58 Unfortunately, these drugs often present significant side effects. Enhancing the DNA-cleaving capabilities of these drugs in a non-toxic manner would involve lower doses and reduce side effects, absolutely improving chemotherapy effectiveness.59 A captivating concept revolves around DNA-binding ligands, which work by crosslinking with specific DNA base sequences through interactions with the DNA microstructure disrupting DNA repair mechanisms, causing DNA damage, and triggering apoptosis in cancer cells.60,61 More efficient DNA-binding drugs are those that have improved binding affinity and specificity toward the target DNA.62–64
Studies on the paramagnetic ortho-ferrabis(dicarbollide), with an empirical formula of [3,3′-Fe(1,2-C2B9H11)2]− and abbreviated as [o-FESAN]− (Fig. 1a), remain relatively unexplored.65–69 Earlier, we investigated the Mössbauer effect for radiotherapy using 57Fe-ferrabis(dicarbollide),65 and the documented cell-killing effects from γ-rays and protons (PBFR) with Na[o-FESAN] in U87 glioblastoma cells.70 Our current study deeply investigates the interactions at the molecular level as well as structural and conformational changes associated with DNA binding of the small (1.01 nm × 0.56 nm),46 anionic, and purely inorganic [o-FESAN]− for potential development of more effective drugs. As such, it will be very important to ascertain the binding affinity and specificity toward the DNA, to know if the small anionic [o-FESAN]− molecule somehow inserts into the ds-DNA structure of the cancer cells in order to evaluate damage to DNA during the irradiation.
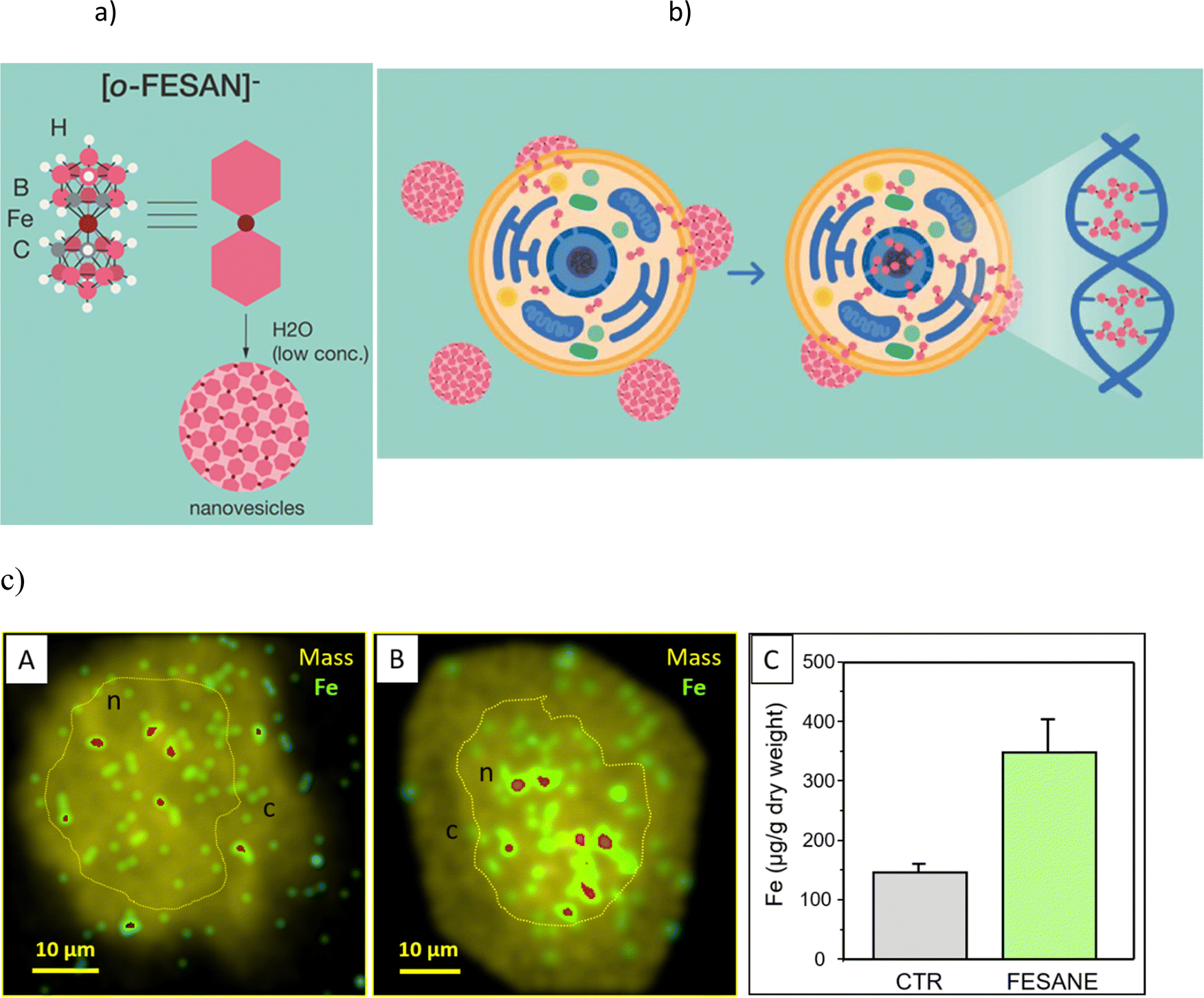 | ||
| Fig. 1 (a) Schematic representation of the small anionic ferrabis(dicarbollide) molecule [3,3′-Fe(1,2-C2B9H11)2]−, abbreviated as [o-FESAN]−, and [o-FESAN]− nanovesicles formed in aqueous low concentration of its Na+ salt. Circles in grey correspond to Ccluster–H vertices, the light pink to B, the white to H and the dark red to Fe. (b) Cartoon illustrating how the [o-FESAN]− nanovesicles in the culture media interact and cross the nuclear membrane to release [o-FESAN]− into the cytosol. Magnification displays a simplified representation of [o-FESAN]− clusters incorporated to ds-DNA. (c) Nuclear microscopy images of Fe content distribution (green to red stand for low to high intensity) in U87 glioblastoma cells overlapped to mass density (mass): (A) control; (B) incubated for 24 h with 50 μM Na[o-FESAN]; showing a preferential accumulation of Fe in the nucleus region. The high-density area corresponding to nucleus (n) is delineated by the yellow dotted line; (c) cytoplasm; (C) quantitative uptake of Na[o-FESAN] (average Fe concentration in N>5 single cells). | ||
Discussion
Lipid membranes are notorious for their ability to maintain ionic concentration gradients, acting as efficient barriers to ions. The uptake and cellular distribution of compounds are pivotal factors that significantly influence the effectiveness of drug treatments. There is experimental evidence that certain nanometric metallabis(dicarbollide) anions are able to spontaneously cross lipid bilayers in spite of the high Born free energy estimated by these ions.10,11 In a recent study employing density functional theory (DFT) calculations along with implicit solvent and atomistic molecular dynamics (MD) simulations revealed that the cisoid [o-COSAN]− rotamer, which proves to be the most stable in water, aligns with the anticipated architecture for a classical surfactant.8 This configuration locates, respectively, the hydrophobic apolar tail at the Ccluster–H vertices and the hydrophilic polar head at the B–H vertices providing interesting self-assembly properties in water.7 Building upon this understanding, we have identified the mechanism responsible for this effect through detailed MD simulations, which involve the formation of temporary elongated self-assembled structures inside the bilayer that help anions to overcome the free energy barrier posed by the membrane.13A schematic representation (Fig. 1b) shows the translocation process of monolayer vesicles of small anionic metallabis(dicarbollide) molecules across the cytoplasmic and nuclear membranes.
In fact, our previous studies on the stability of Na[o-FESAN] in cellular medium showed that the metallabis(dicarbollide) did not suffer degradation.65 The visualization of iron (57Fe) distribution in U87 glioblastoma cells, as a signature of Na[o-FESAN] intracellular trafficking was also studied by micro-PIXE (Fig. 1c).70 An increased concentration of Fe was noted towards the cell center, presumably the nucleus (Fig. 1c, images B), in comparison to untreated U87 glioblastoma controls that exhibited a uniform distribution of Fe (Fig. 1c, images A). Consistent with expectations, treated cells displayed a notably high concentration of iron (Fig. 1c, graph C) without cytotoxic effect. These findings validate the effectiveness of [o-FESAN]− uptake after crossing cell and nuclear membranes, offering a quantitative assessment of the extent to which compounds are uptaken by the cells as well as their subcellular localization.
The above uptake studies conducted with the U87 glioblastoma cells, demonstrating an elevated concentration of Na[o-FESAN] within the nucleus, are in accordance with theoretical studies by Rabin and Szleifer.71 These theoretical insights underscore the pivotal roles played by charge and hydrophobicity in facilitating the translocation of model particles through the nuclear pore complex.71 This alignment between theory and practice further substantiates our results. The subcellular distribution of Fe indicates the nucleus as a cellular target and suggests a potential connection to the interaction between small anionic [o-FESAN]− molecules and DNA of the nucleus.
The suggested interaction of Na[o-FESAN] with double-stranded calf thymus DNA (CT-dsDNA) was first studied by dynamic light scattering (DLS). In this study, the initial concentration solution of Na[o-FESAN] was fixed (1.0 mM in water) and the concentration of CT-dsDNA increased from 0 to 900 μM. As depicted in Fig. 2, the diameter of [o-FESAN]− aggregates exhibits a linear increase in particle size, ranging from approximately 39 nm to 62 nm upon the introduction of CT-dsDNA (<160 μM). This is succeeded by a plateau that is observed up to 380 μM concentration. Subsequently, a secondary linear increase is noted up to 610 μM, succeeded by another plateau extending at least until 900 μM. The modulation of the Na[o-FESAN] aggregate size with CT-dsDNA content provides evidence of the interaction between dsDNA and the anionic ferrabis(dicarbollide) molecules.
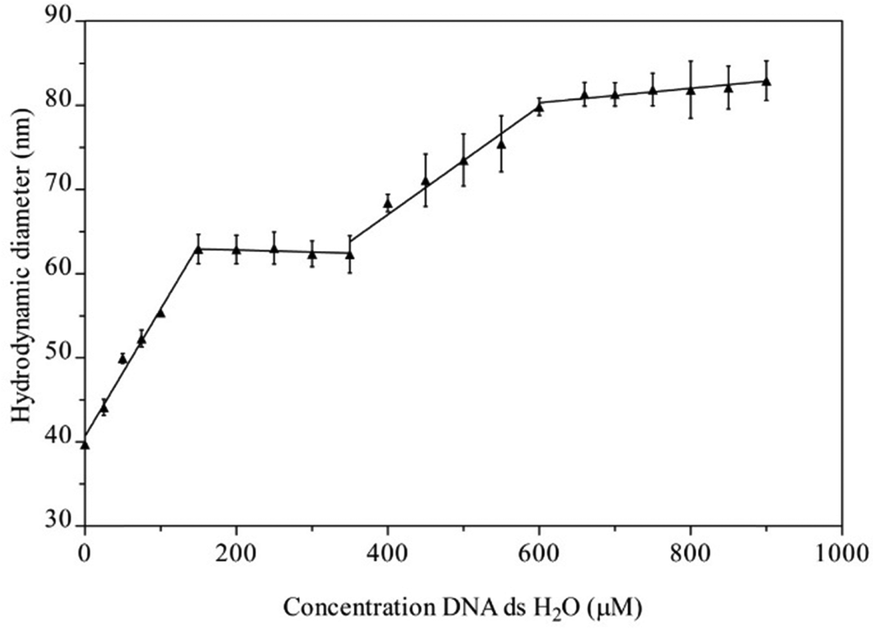 | ||
| Fig. 2 Graphical representation of the hydrodynamic diameter (nm) of Na[o-FESAN] aggregates in aqueous 1 mM solution measured by Dynamic Light Scattering (DLS) vs. concentration of CT-dsDNA in water, in the range from 0 to 900 μM. | ||
To unambiguously decipher the interaction mode, intercalative or ionic, between the anionic small [o-FESAN]− molecules and dsDNA, we drew up a procedure to prepare the nanohybrid CT-dsDNA/Na[o-FESAN]. This involves saturating the backbone of the CT-dsDNA with the small anionic [o-FESAN]− molecules. As illustrated in Fig. 3, the nanohybrid CT-dsDNA/Na[o-FESAN] was obtained by incubating Na[o-FESAN] solution with CT-dsDNA solution for 72 hours at room temperature as described in the Experimental section. Following isolation, the coexistence of both components in the CT-dsDNA/Na[o-FESAN] nanohybrid was confirmed through FT-IR, Raman, and X-ray photoelectron (XPS) spectroscopies and electrochemical studies.
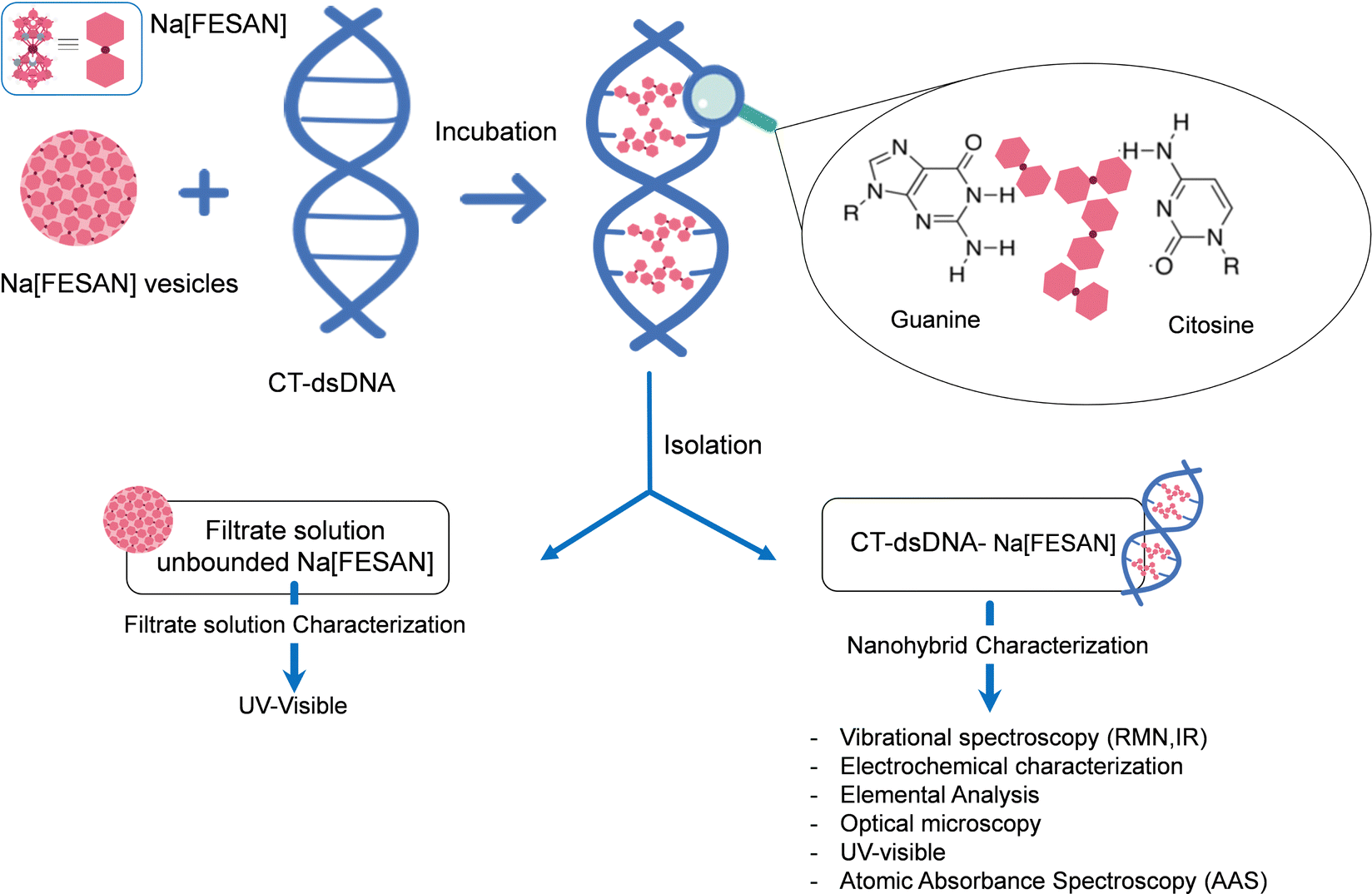 | ||
| Fig. 3 Schematic representation of the preparation, isolation and characterization of the nanohybrid CT-dsDNA-Na[o-FESAN]; emphasizing the amplification of intercalation between amphiphilic small [o-FESAN]− molecules and the double strand of DNA molecules with the double strand of DNA. | ||
Taking advantage of the fact that metallabis(dicarbollides) display a strong and characteristic ν(B–H) frequency in the infrared range 2600–2500 cm−1 in which no other frequencies of organic compounds appear,72 the IR (Fig. 4a) and Raman (Fig. 4b) spectra of the nanohybrids CT-dsDNA/Na[o-FESAN] (orange line) were run to confirm the presence of the ferrabis(dicarbollide) clusters. The corresponding spectra of Na[o-FESAN] and CT-dsDNA were also run as references (grey and blue lines, respectively). As can be seen in Fig. 4a, the presence of metallacarborane clusters in the nanohybrid is proven, on the one hand, by the strong absorption at 2527 cm−1 due to the B–H stretching frequency dominating the IR spectra in both samples, CT-dsDNA/Na[o-FESAN] (orange line) and the Na[o-FESAN] reference (grey line). On the other hand, the IR spectrum of the CT-dsDNA/Na[o-FESAN] nanohybrid contains the characteristic stretching bands of the CT-dsDNA reference (blue line) corresponding to the phosphate backbone at 1219 and 1089 cm−1, which are ascribed to asymmetric νas(POO–) and symmetric νs(POO–) stretch of coordinated phosphinate group73,74 as well as the bands at 976 cm−1 from C–C stretching backbone and at 1057, 881, 799 and 787 cm−1 from the DNA sugar–phosphate vibration.75,76
 | ||
| Fig. 4 (a) Infrared spectra of Na[o-FESAN] (grey line), CT-dsDNA (blue line) and CT-dsDNA-Na[o-FESAN] nanohybrid (orange line). (b) Raman spectra of Na[o-FESAN] (grey line), CT-dsDNA (blue line) and CT-dsDNA-Na[o-FESAN] (orange line). | ||
The most useful part of the Raman spectra (Fig. 4b) is in the 1000–3500 cm−1 region. The vibrational peaks associated with cage B–H vibrations (around 2500 cm−1) and Ccluster–H stretching bands (3030 cm−1) are remarkable in the CT-dsDNA/Na[o-FESAN] nanohybrid as in the Na[o-FESAN] reference spectra. Additionally, the phosphate backbone characteristic between 500–1000 cm−1 is present in the spectra of CT-dsDNA and CT-dsDNA/Na[o-FESAN].
The oxidation state of each element in the compounds was analyzed using XPS (ESI,† Fig. S1). The presence of [o-FESAN]− and CT-dsDNA in the nanohybrid is unambiguosly supported by spectra of the dry CT-dsDNA/Na[o-FESAN] sample displaying the two spin orbit Fe 2p peaks at the bonding energy (BE) position characteristic of Fe3+,77 as well as the expected for B 1s, N 1s and P 2p. Long survey as well as short BE energy range spectra at the B, Fe, N, P, and Na regions are shown in the figure for the three samples.
Next, we also studied the electrochemical behaviour of the nanohybrid (3.0 mM CT-dsDNA/Na[o-FESAN] solution in 0.1 M PB pH 7.0) on a gold screen printed electrode (AuSPE), (ESI,†, Fig. S2) and compared to that of the Na[o-FESAN] salt. The nanohybrid shows a reversible redox peak adscribed to the Fe3+/Fe2+ couple at a formal potential (E°) of −0.289 V vs. Ag with an ΔE of 86 mV, which is higher than the expected value (60 mV) for a freely diffusing one-electron reversible redox process. Na[o-FESAN] salt shows the same redox couple of the oxidation/reduction of Fe3+/Fe2+ at an E° value of −0.283 V with a ΔE of 74 mV. The nanohybrid exhibited a slight shift towards a negative potential, accompanied by an observed increase in ΔE. This suggests constraints on both diffusion and charge transfer kinetics of the anionic small [o-FESAN]− molecules after binding to DNA, a notion further supported by the decrease in peak current.
All the aforementioned measurements align consistently with the successful preparation of the new nanohybrid biomaterial, CT-dsDNA/Na[o-FESAN], composed of small amphiphilic anionic [o-FESAN]− units and CT-dsDNA. Indeed, the UV-visible spectrum of CT-dsDNA/Na[o-FESAN] in aqueous solution (ESI,† Fig. S3) exhibits a band around 270–280 nm, characteristic of Na[o-FESAN], confirming its presence in the new nanohybrid biomaterial. Moreover, considering the molar absorptivity of Na[o-FESAN] calculated from the slope of the calibration plot (ESI,† Fig. S3a and inset), the estimated concentration of Na[o-FESAN] in the nanohybrid (ESI,† Fig. S3c), was 14 mM. This value closely resembles that obtained from the final filtrate solution (15 mM) (refer to the ESI,† Fig. S3b), which contains the excess Na[o-FESAN] unbound to CT-dsDNA, as described in the Experimental section. As demonstrated later, this concentration of Na[o-FESAN] in the hybrid aligns with the findings from atomic absorption spectroscopy (AAS) studies.
At this point, to ascertain the ratio between the anionic [o-FESAN]− and the CT-dsDNA in the novel nanohybrid, we employ combined results from elemental analysis, electrochemical titration and AAS.
The elemental analysis of the two dry samples gave C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H:N ratios of 2.65:4.18:1 and 6.71:22.5:1 for the CT-dsDNA reference and CT-dsDNA/Na[o-FESAN] nanohybrid, respectively (ESI,† Table S1), i.e., approximately four extra C atoms and eighteen H per N atom in the nanohybrid. Since the empirical molecular formula of [o-FESAN]− is FeC4B18H22, the results point to the presence of one molecule of [o-FESAN]− per N atom in the reference CT-dsDNA in the new nanohybrid. These results are consistent with a ratio of four [o-FESAN]− anions per DNA base pair (see the ESI†).
:H:N ratios of 2.65:4.18:1 and 6.71:22.5:1 for the CT-dsDNA reference and CT-dsDNA/Na[o-FESAN] nanohybrid, respectively (ESI,† Table S1), i.e., approximately four extra C atoms and eighteen H per N atom in the nanohybrid. Since the empirical molecular formula of [o-FESAN]− is FeC4B18H22, the results point to the presence of one molecule of [o-FESAN]− per N atom in the reference CT-dsDNA in the new nanohybrid. These results are consistent with a ratio of four [o-FESAN]− anions per DNA base pair (see the ESI†).
To validate the estimated ratio of [o-FESAN]− anions per base pair in the DNA within the nanohybrid, we conducted an electrochemical titration and cyclic voltammetry (CV) studies. For this purpose, CT-dsDNA was kept at a fixed concentration of 2.0 mM while the Na[o-FESAN] concentration was varied from 0 to 20.0 mM (same conditions as in the nanohybrid preparation) and both the CV and differential pulse voltammogram (DPV) were recorded after each Na[o-FESAN] addition. As expected, no electrochemical signal is observed in the absence of Na[o-FESAN] because DNA is electrochemically inactive in the potential range studied. However, upon addition of increasing amounts of Na[o-FESAN], a pair of redox peaks appear (Fig. 5a), which we ascribe to the metal center (Fe) of Na[o-FESAN]. In addition, a shift in the formal potential (E°) of Na[o-FESAN] as well as an increase in the peak current are observed. The potential shifts are quite proportional to the concentration of Na[o-FESAN] up to 8.0 mM and then level off (Fig. 5b). From this value, a [o-FESAN]−/[CT-dsDNA] ratio of 4 is calculated, in excellent agreement to the one obtained by the elemental analysis. DPV of Na[o-FESAN] at the anodic or cathodic scan corroborates these results (ESI,† Fig. S4).
 | ||
| Fig. 5 (a) Cyclic voltammograms of 2.0 mM CT-dsDNA (red line) and increasing concentrations of Na[o-FESAN] (colored lines) from 1.0 to 20.0 mM in 0.1 M PB pH 7.0 solution at AuSPE. Scan rate is 100 mV s−1. (b) Representation of the potential (E°) vs. Na[o-FESAN] concentration. | ||
AAS studies were carried out to determine the concentration of Fe in the CT-dsDNA/Na[o-FESAN] nanohybrid. First, the atomic absorbance of aqueous solutions containing different concentrations of Na[o-FESAN] was measured to obtain the calibration plot (ESI,† Fig. S5). The absorption of the nanohybrid solution (dilution 1:125) was measured under the same conditions. By interpolation of this value in the calibration plot, an estimated value of 14.9 mM of Na[o-FESAN] for the nanohybrid CT-dsDNA/Na[o-FESAN] was obtained, in parallel with those extracted above from the UV-vis studies.
To get insight on the material distribution within the nanohybrid, optical microscopy and atomic force microscopy (AFM) measurements were performed (Fig. 6 and 7). Glass slides, each containing CT-dsDNA/Na[o-FESAN] nanohybrid, Na[o-FESAN] and CT-dsDNA, respectively, were meticulously prepared according to the experimental procedure and examined under an optical microscope. Samples obtained by drop casting the hybrid CT-dsDNA/Na[o-FESAN] solution on a Si substrate (see the Experimental section) were used for AFM. As can be seen, the CT-dsDNA appears as flower-like dendritic shapes in both optical and AFM micrographs (top panels in Fig. 6), remarkably different to those observed for Na[o-FESAN] and CT-dsDNA/Na[o-FESAN] nanohybrid. Na[o-FESAN] appears as large rods of about 8 μm wide randomly distributed onto the glass surface (Fig. 7, image a). Outstandingly, CT-dsDNA/Na[o-FESAN] nanohybrids show a completely different landscape. As can be seen in the micrography (Fig. 6, image b), a non-crystalline matrix contains spherical inclusions of 5–10 μm diameter formed by crystaline thin rods. According to the size of these structures observed by DLS is consistent with CT-dsDNA chains surrounding Na[o-FESAN] vesicles. This interpretation is further supported by the AFM data of the nanohybrid material (Fig. 7).
 | ||
| Fig. 6 Top left: Optical microscopy image of a glassy slide modified with ds-DNA (50 × objective lens) and top right: AFM topography for a 4 mM solution of ds-DNA drop casted on a Si substrate. Bottom left: Na[o-FESAN] dry solution (20 × objective lens) and bottom right: CT-dsDNA/Na[o-FESAN] nanohybrid (100 × objective lens). | ||
 | ||
| Fig. 7 AFM images of the hybrid CT-dsDNA/Na[o-FESAN] (1:100) deposited by drop casting on a Si substrate. Topographic images in (a) and (b) show the coexistence of round shaped structures and crystalline needles. The image in (b) corresponds to the region marked by a red square in (a). Images in (c) and (e) are the magnified topography taken on each of the observed structures (round-like and needles, respectively) while those in (d) and (f) are the simultaneously measured lateral force (forward) images. Total z ranges from black to white are 106 nm (a), 65 nm in (b) and (c) and 32 nm in (e), respectively. | ||
As seen by AFM, the morphology of the drop casted hybrid CT-dsDNA/Na[o-FESAN] solution shows a series of round shaped structures (5 μm in diameter and about 10 μm apart), which are distributed over the entire surface coexisting with some isolated thin rods or needles and a surrounding discontinuous network, likely the result of on-surface drop drying, that reaches up to 50 nm in height (Fig. 7, images a and b). A magnified region containing one of the round structures is also shown (Fig. 7, image c). Close inspection reveals that two distinct parts are distinguished: a central region (∼10 nm thick) coupled to and enclosed by a non-uniform ring of material that matches with the discontinuous network covering the rest of the surface. The isolated needles, randomly oriented, are extremely flat and exhibit very thin layers, separated by steps ∼1.2 nm in height (Fig. 7, image e and ESI,† Fig. S6), evidencing their crystalline nature. Based on crystallographic data, the measured step height is compatible with the inter-planar distance in Na[o-FESAN] single crystals, as reported.78 The presence of diverse structural elements on the surface prompts us to investigate the potential phase separation or segregation of the two constituents, [o-FESAN]− and CT-dsDNA, within the nanohybrid by friction force microscopy (FFM)79,80 (see details in the ESI,†). As local differences in the frictional properties of a surface permit visualizing regions of different chemical nature on samples of heterogeneous composition, the spatial redistribution of the drop casted material is unambiguously determined. Remarkably, the FFM analysis indicates that the central part of the round structures (Fig. 7, images c and d) and the needles (Fig. 7, images e and f) have the same composition, which is attributed to [o-FESAN]−. On the other hand, the irregular rim at the perimeter of the structures has the same frictional contrast (same composition) as the extended disrupted network (Fig. 7, image a and ESI,† Fig. S7) to which we attribute to a majority content of CT-dsDNA polymeric chains forming the nanohybrid matrix seen by optical microscopy (Fig. 6b). Unfortunately, these AFM techniques cannot detect to which extent small [o-FESAN]− molecules are incorporated to dsDNA.
In an effort to gain a deeper understanding of the intercalative interaction mode between the anionic small [o-FESAN]− molecules and dsDNA, we subjected the new nanohybrid CT-dsDNA/Na[o-FESAN] to analysis using electrospray ionization (ESI-MS) and matrix-assisted laser desorption/ionization (MALDI-TOF-MS) mass spectroscopies.
Over the past decades, mass spectrometry (MS) has revolutionized the analytical chemistry field because it does not only report on the exact mass of a given compound, but also generates an ion fragments spectrum of it, which helps on determining the structure of an unknown molecule.64,81 ESI-MS and MALDI-TOF-MS are among the most employed analytical techniques used in mass spectroscopy nowadays in the biopharmaceutical industry for the detection, quantification and structural elucidation of macromolecules in the fields of biomarkers and metabolomics.82–85 Because of the differences in sample introduction and ionization methods, variations in their respective mass spectra are expected, thus their complementary use can contribute new data or confirm and strengthen them, as it has been the case proving the [o-FESAN]− strong interaction with DNA.
We have used ESI-MS as an effective technique to study the noncovalent interaction of CT-dsDNA with the small anionic [o-FESAN]− molecules. In the negative ion mode, only the molecular peak (M) of the [o-FESAN]− at 321.0 with isotopic distribution is observed (Fig. 8b) while the positive ion mode displays peaks also with isotopic distribution at 473.4 (M + guanine), 580.5 (M + guanine + cytosine), 709.5 (M + guanine + cytosine + thymine), and 778.5 (M + 3 guanine) (Fig. 8a and ESI,† Fig. S8). It should be emphasized that CT-dsDNA, which was run as reference, does not ionize under the same ESI-MS conditions.
 | ||
| Fig. 8 (a) Magnification of the ESI-MS spectrum of the CT-dsDNA/Na[o-FESAN] nanohybrid at the positive mode in the range 400–650 m/z with isotopic (+1) distribution; Fig. S8 at the ESI,† displays the full spectrum in the range 0–1200 m/z. (b) ESI-MS spectrum of the CT-dsDNA/Na[o-FESAN] nanohybrid at the negative mode with isotopic (−1) distribution in the range 270–470 m/z; only molecular peak of the [o-FESAN]− at 321.0. (c) MALDI-TOF-MS of the nanohybrid CT-dsDNA/Na[o-FESAN] using DHB (2,5-dihydroxybenzoic acid) matrix at the anode with 30% attenuation. | ||
The MALDI-TOF-MS of the CT-dsDNA and the CT-dsDNA/Na[o-FESAN] nanohybrid samples (diluted 100 times in H2O and acetone) was run at the positive and negative ionization modes in either the presence or absence of matrix (2,5-dihydroxybenzoic acid, DHB) (Fig. 8c and Fig. S9–S13, Table S2 in the ESI†). The positive ionization mode did not present the isotopic distribution typical of the boron clusters while the negative ionization mode provided peaks with isotropic distribution (z = 1) at 437.284 (M + 116.634 (cytosine)), 473.440 (M + 152.78 (guanine)), 624.611(M + 304.05 (two guanines)), 776.76 (M + 304.05 (three guanines)), being M = 320.66, the molecular weight of the small anionic [o-FESAN]− molecule. It should be noted that the peak corresponding to the [o-FESAN]− anion does not appear in the spectrum of the CT-dsDNA/Na[o-FESAN] nanohybrid, which fully supports that there are no free anionic [o-FESAN]− clusters but that all molecules are strongly bonded to the CT-dsDNA biopolymer. Both techniques ESI† and MALDI-TOF albeit different in their ionization and sample introduction ways have corroborated the strong non-bonding interactions of [o-FESAN]− anions with the pair bases.
All characterization techniques described so far demonstrate the successful formation of the corresponding nanohybrid in which the 3D aromatic [o-FESAN]− inserts as an intercalating agent into the dsDNA structure. The mass spectra results aligned with our previous observed increase of 4.9 ± 0.2 °C in the melting temperature of CT-dsDNA are indicative of the presence of [o-FESAN]−.66 This observation, consistent with an interaction between the anion and DNA, also suggests an intercalative interaction.
In summary, our findings confirm a robust interaction between Na[o-FESAN] and dsDNA, enabling the formation of the CT-dsDNA/Na[o-FESAN] nanohybrid that accommodates four [o-FESAN]− anions per base pair within a confined space. The binding between ds-DNA and [o-FESAN]− does not occur through alkylation, as seen with mechlorethamine and mitomycin C, nor through intercalation between the ds-DNA base pairs, as observed with doxorubicin. Although [o-FESAN]− is aromatic, it is three-dimensional and large, making it unable to fit between the ds-DNA base pairs to accommodate these 3D anions. [o-FESAN]− interacts with guanine and cytosine through weak dihydrogen bonds rather than covalent binding. Crucially, Na[o-FESAN] shows no DNA damage. The notable distinction lies in the charge, as typical DNA-binding agents are cations62,86,87 whereas [o-FESAN]− clusters are anions. Then, how can four anions be placed in a confined space if Coulomb's law predicts that anions minimize electron repulsion by increasing distances88 and preventing aggregation? The answer must be in the anti-electrostatic [o-FESAN]−–[o-FESAN]−, “anion–anion” formation.78
But, how do [o-FESAN]− anions engage with the bases of dsDNA and partake in intercalated interactions, especially considering the presence of external phosphate anions along DNA strands? In simpler terms, how do [o-FESAN]− anions in solution navigate around the repulsion posed by phosphate anions? The explanation is in the initial anti-electrostatic interaction between [o-FESAN]− anions of the solution and the external phosphate anions of the nucleotide dsDNA backbone, forming an anti-coulombic “anion–anion” interaction via Ccluster–H⋯O–P or Ccluster–H⋯OH hydrogen bonds (Fig. 9, step 1). Then, when additional [o-FESAN]− molecules approach, a cooperative effect among them facilitates the formation of temporary elongated self-assembled structures of [o-FESAN]−–[o-FESAN]−. This process counteracts the repulsion between anions by fostering the creation of numerous Ccluster–H⋯H–B and/or B–H⋯H–B dihydrogen bonds (depicted in Fig. 9, step 2 and 3). Accordingly, [o-FESAN]− approaches the innermost part of the dsDNA where the base pairs are situated. Finally, the formation of the CT-dsDNA/Na[o-FESAN] nanohybrid is facilitated by hydrogen bonding (Ccluster–H⋯O) and/or dihydrogen bonding (B–H⋯H–N) interactions between the nucleoside groups and the four [o-FESAN]− aggregates (refer to Fig. 9, step 4). This interaction pattern is showcased by its ability to interact with amino groups present in antibiotics,89 amino acids22 and proteins.23–26 It is worth noting that the proposed pathway outlined in Fig. 9 parallels a recently identified mechanism uncovered through atomistic molecular dynamics simulations, elucidating how metallabis(dicarbollide) anions can cross phospholipid bilayer membranes spontaneously.13
 | ||
| Fig. 9 Proposed pathway elucidating how [o-FESAN]− anions in solution maneuver around the repulsion exerted by external phosphate anions lining DNA strands to interact with the bases of dsDNA and engage in intercalated interactions. | ||
In the above mentioned simulations, it was demonstrated that while a single metallacarborane molecule cannot traverse a lipid membrane, a cooperative effect facilitated by other identical molecules enables the metallacarborane to reach the opposite side of the membrane. These simulations further revealed the mechanism behind this phenomenon, involving the creation of temporary elongated self-assembled structures within the membrane that help anions to overcome the free energy barrier posed by the membrane.13
To substantiate our proposed pathway, we utilized the powerful measurement technique of XPS that not only determine the presence of elements and their chemical states but also permit identifying their bonding partners. We analyzed the binding energy (BE) ranges of specific core levels of B, N, P, and Na for the CT-dsDNA/Na[o-FESAN] nanohybrid, as well as for [o-FESAN]− and dsDNA, to assess whether the electronic states of these elements were affected by the new chemical environment of the nanohybrid respect to those on the pure materials.
The BE of Na 1s in the nanohybrid increases with respect to that in the pristine dsDNA (Fig. 10a), aligning with Na in Na[o-FESAN], indicating that at least most of Na+ is no longer surrounded by the external phosphate anions of the nucleotide dsDNA backbone. Can the lack of Na-phosphate interaction as revealed by XPS help us demonstrating the interaction between [o-FESAN]− and the dsDNA? To address this question, we conducted an analysis of the binding energies of P 2p and B 1s, following peak deconvolution (see details in Table S3, ESI†). As depicted in Fig. 10b, the intensity of the P 2p peak at 131.5 eV, corresponding to phosphate surrounded by Na+ in dsDNA, significantly diminishes in the nanohybrid. This decrease is accompanied by the emergence of a new peak at a higher binding energy of 138.05 eV (ΔBE = 3.95 eV compared to the main peak of the hybrid and 6.55 eV compared to that of the dsDNA reference). Furthermore, the B 1s peak of the CT-dsDNA/Na[o-FESAN] nanohybrid exhibits a highest peak at 188.7 eV, which correlates with the [o-FESAN]− small molecule (anion–anion aggregates) but, a new peak at a lower binding energy of 185.9 eV (ΔBE = −3 eV compared to pure Na[o-FESAN]) is found, which is correlated to the energy shift observed in the new P 2p peak (Fig. 10c). Although it is known that the absolute magnitude of the measured shift may notably vary due to factors such as specific atom electronegativity and/or screening effects, the opposite sign of the energy shifts for each element (B and P) is an accurate indicator of the mutual interaction. The N 1s XPS deconvolved spectrum of dsDNA exhibits a two-component structure (Fig. 10d), which is characteristic of dsDNA nucleobases (in particular, the similar weight of both components is typical from guanine and cytosine), while the CT-dsDNA/Na[o-FESAN] nanohybrid exhibits only one at the higher energy region (Fig. 10d), reflecting an important involvement of these nucleobases in the nanohybrid formation. It is worth noting that the higher energy peak in ds-DNA90 and amino acids91 is attributed to amino N sites that connect with single bonds related to the NH2 group. The absence of the peak at lower binding energy is consistent with the presence of [o-FESAN]− interacting with the N–H groups of the dsDNA nucleobases through N–H⋯H–B dihydrogen bonds.
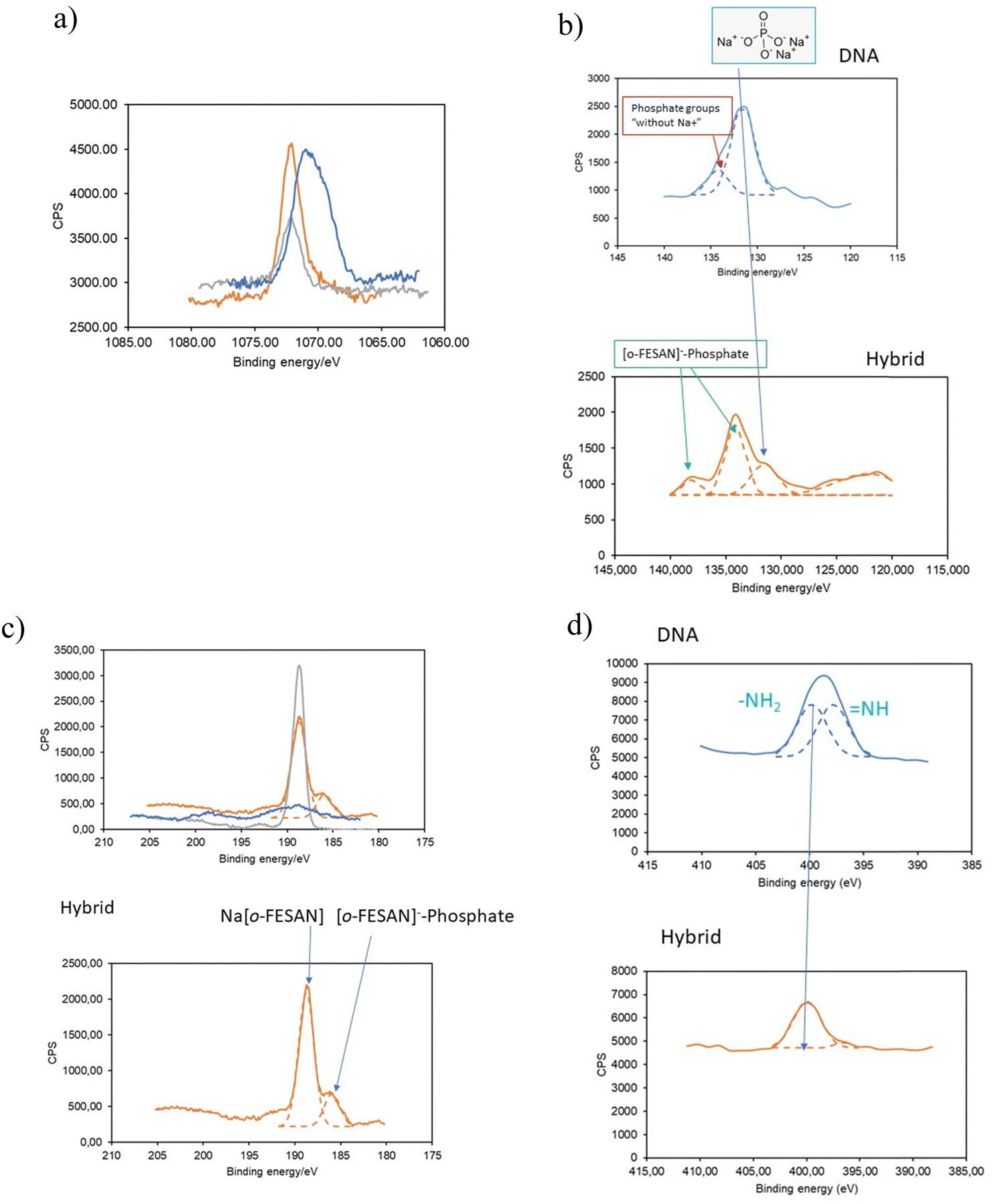 | ||
| Fig. 10 XPS short range spectra for the following core levels: (a) Na 1s, (b) P 2p, (c) B 1s and (d) N 1s. Nanohybrid CT-dsDNA/Na[o-FESAN] (orange), Na[o-FESAN] (grey) and, CT-dsDNA (blue). The adscription of peaks by arrows is used as a guide to the explanation provided in the text. | ||
Summarizing, the analysis of the Na 1s, P 2p, B 1s and N 1s peaks observed in the CT-dsDNA/Na[o-FESAN] nanohybrid supports the presence of anion–anion anti-Coulombic interaction between the [o-FESAN]− and phosphate as well as the [o-FESAN]− interactions mostly with guanine and cytosine nucleobases of the ds-DNA. These findings agree with the found peaks of fragmentation in the ESI† and MALDI-TOF mass spectra and with the reported interactions between metallabis(dicarbollide) anions and N–H groups of amino acids22 and proteins.23–26
It is important to note that the metallabis(dicarbollide) anions ([o-COSAN]− and [o-FESAN]−) exhibit intriguing self-assembly properties in water. Moreover, the [o-FESAN]− anion has been experimentally observed to form anti-electrostatic “anion–anion” aggregates.78 Thus, [o-FESAN]− ability to cross the phosphate anionic dsDNA backbone is again a manifestation of the rich self-assembly behavior of these amphiphilic nano-ions that have a bridging character between traditional anions and small molecules.
Our discoveries hold significance not only in advancing fundamental scientific understanding but also in their potential implications for groundbreaking biomedical applications. Metallacarboranes emerge as promising candidates for cutting-edge drug development, with conceivable applications in cancer therapy and the fight against bacterial infections, among others. In this regard, alternative therapeutic approaches such as boron neutron capture therapy (BNCT), proposed by Locher already in 1936,92 emerge as an opportunity to treat high-grade cancers that are resistant to chemotherapy and conventional radiotherapy.40,93–97 Only two 10B-enriched drugs, 4-dihydroxyborylphenylalanine (BPA) and the sodium salt of mercaptoundecahydro-closo-dodecaborate (BSH) have received FDA approval for clinical studies in BNCT.98 Recently, on March 25, 2020, Stella Pharma corporation developed Steboronine®, marking it as the first boron drug specifically designed for clinical use to be utilized in BNCT.99 The development of boron delivery agents for BNCT is currently a major area of research.100–106 The BNCT therapy occurs when cells accumulating 10B are irradiated with thermal neutrons to produce 4He (α particles) and 7Li.36–40,93–97,107 Since the path lengths of the particles are approximately 9–10 μm, similar to the dimensions of a single cell, 10B-containing cells (approximately 109 atoms of 10B per cell) are selectively destroyed by BNCT, which results in a therapeutic effect. The generated radiation destroys malignant cells containing the boron compound. For an efficient BNCT, highly boron-enriched molecules with an adequate vector might allow a selective delivery of a consistent amount of boron (>20 μg of 10B per g of tumour)108,109 to the tumor. Furthermore, in silico studies employing Monte Carlo simulations110 demonstrate that the BNCT treatment is more efficient if the boron compound is located in the cell nucleus as compared to a uniform distribution throughout the entire cell. Our studies have demonstrated that in uptake experiments conducted with U87 glioblastoma cells,70 the subcellular distribution of Fe indicates the nucleus as a primary target within the cell. These findings are consistent with a robust intercalative interaction between small anionic [o-FESAN]− molecules and guanine and citosine regions of dsDNA, indicative of a preference akin to that observed with many intercalators.62,111
Crucially, the [o-FESAN]− anions, boron-rich small molecules, not only penetrate the nucleus effectively but also intercalate with dsDNA, making them promising contenders for cancer treatment such as boron neutron capture therapy (BNCT) and proton boron fusion therapy (PBFT). Our ongoing research in this area, conducted within our laboratories, is nearing completion and will be published imminently.
Conclusions
The urgent need and challenge in cancer therapy research involve crafting multifunctional small molecules that offer additional therapeutic benefits. These molecules aim to enhance anticancer efficacy, thus enabling reduced dosage and mitigated side effects for the advancement of effective chemotherapy and improved safety. Having in mind this demand, we have previously studied amphiphilic small molecules for multimodal radiation therapy.70 One of these molecules is a purely inorganic amphiphilic water soluble anionic small molecule, the sodium salt of ferrabis(dicarbollide), [3,3′-Fe(1,2-C2B9H11)2]− herein abbreviated as Na[o-FESAN].It is very important to ascertain the damage of ds-DNA during irradiation therapies to know in advance if the small anionic [o-FESAN]− molecule inserts into the ds-DNA structure of the cancer cells and bonds to the DNA as an intercalating agent. Therefore, to unambiguously decipher the interaction mode between the anionic small [o-FESAN]− molecules and ds-DNA, we drew up a plan to prepare the nanohybrid CT-dsDNA/Na[o-FESAN]. This nanohybrid was synthesized, purified, and characterized by elemental analysis and ESI-MS, MALDI-TOF-MS, XPS, FTIR, UV-vis, CV and AAS spectroscopies, which support the presence of [o-FESAN]− and CT-dsDNA in the nanohybrid. Furthermore, optical microscopy and AFM displayed the images of the nanohybrid morphology, which being different for the two components matches the nanohybrid characterization results. Elemental analysis indicated a ratio of four small anionic molecules/pair base. ESI-MS displays a peak at 580.5 with isotopic distribution corresponding to the monocationic species guanine⋯[o-FESAN]−⋯citosine and, very important, is consistent with the intercalative interaction between the small anionic [o-FESAN]− molecule and the pair base guanine⋯citosine of the CT-dsDNA. The MALDI-TOF-MS displays fragmentation peaks corresponding to the CT-dsDNA plus the mass of one or two small anionic [o-FESAN]− molecules. The positioning of the four [o-FESAN]− anions within the interthread region of base pairs arises from the formation of anti-electrostatic [o-FESAN]−–[o-FESAN]− “anion–anion” interactions, strategically minimizing Coulombic electron repulsion. This arrangement is facilitated by the establishment of numerous Ccluster–H⋯H and B–H⋯H–B dihydrogen bonds.
Our findings are relevant not only from a “fundamental” point of view of basic science but also may have an impact for clinical applications. Based on the reported results, metallacarboranes emerge as highly promising candidates for cutting-edge drug development, particularly for cancer radiation therapies such as BNCT and PBFT. This potential stems from the ability of small anionic [o-FESAN]− molecules to effectively insert as intercalating agents into the dsDNA structure, with four anions per base pair and thus enhancing radiation induced DNA damage.
Drawing from the reported findings, Na[o-FESAN] also offers unique possibilities to improve and optimize radiotherapy in a multimodal perspective, providing healthcare professionals with stronger and well-suited tools in the treatment of cancer. This small amphiphilic anionic [o-FESAN]− molecule employed as a carrier-free drug can certainly deliver more with less in the fight against cancer. Results obtained are quite encouraging, which leads to expect that this or some derivatives or new small molecule drugs for cancer treatment with similar dsDNA binding characteristics will gain significant development in the near future.
Experimental section
The NaCl was purchased from Sigma–Aldrich, whereas the cationic exchanging resin used (Amberlite IR120, H form) was purchased from Acros Organics and the hydrochloric acid (37%) was purchased from Carlo Erba Reagents. Solvents used were from Carlo Erba SDS and purified by distillation from sodium and benzophenone under a nitrogen atmosphere before used.
Preparation, isolation and characterization of CT-dsDNA/Na[o-FESAN]
To evaluate the maximum content of Na[o-FESAN] that saturates CT-dsDNA, the first step was the nanohybrid CT-dsDNA/Na[o-FESAN] preparation.The nanohybrid CT-dsDNA/Na[o-FESAN] was synthesized by incubating 0.50 mL of a 50 mM Na[o-FESAN] solution with 0.5 mL of a 4.0 mM CT-dsDNA solution for 72 hours at room temperature. The resulting solution, containing 25 mM Na[o-FESAN] and 2.0 mM CT-dsDNA, was transferred to a filter unit (Amicon Ultracentrifugal 100 K) and adjusted to a final volume of 4.0 mL with water. Subsequently, the solution was centrifuged using a Hettich 320R centrifuge at 120000 rpm for 30 minutes. Then, the supernatant containing the nanohybrid CT-dsDNA/Na[o-FESAN] was washed with water.
Evaluation of the content of Na[o-FESAN] that saturates CT-dsDNA to produce the nanohybrid CT-dsDNA/Na[o-FESAN]
The concentration of Na[o-FESAN] unbonded to the nanohybrid CT-dsDNA/Na[o-FESAN] was determined by UV spectroscopy (ESI,† Fig. S1) from the rinsed water solutions using the absorption band of Na[o-FESAN] at 271 nm. Previously, the calibration plot absorption of Na[o-FESAN] in aqueous solution and its molar absorptivity in the UV at 271 nm were meassured. Considering the amount (μmol) of Na[o-FESAN] added initially and the amount (μmol) of the unbounded calculated, the amount of Na[o-FESAN] bound to the CT-dsDNA was calculated to be 3 μmol (Fig. 3).Equipment for nanohybrid characterization
:19.00 kV (Uis1), ion source 2:16.59 kV (Uis2)]. The software flexControl and flexAnalysis from Bruker were used for the data treatment of acquisition and analysis, respectively. The matrix was 2,5-dihydroxybenzoic acid (DHB) at a concentration of 10 mg mL−1 in acetonitrile and 0.1% TFA (trifluoracetic acid) in a volume ratio of 1:2. The MALDI-TOF-MS samples were prepared by mixing the matrix and the sample at ratio 1:1. In the absence of matrix: MALDI-TOF/TOF mass spectrometer (ABSciex 4800 TOF TOF) in the range 200–1000 m/z at the reflector negative and positive mode was used to run the MALDI-TOF-MS spectra in the absence of matrix of the samples (nanohybrid CT-dsDNA/Na[o-FESAN] and the pristine CT-dsDNA). Then, 5 μL of 4 mM CT-dsDNA solution were deposited in the irradiation cavity. The CT-dsDNA/Na[o-FESAN] nanohybrid samples were prepared in two different solvents, water, or acetone, by diluting the sample 100 times and depositing 5 μL of this solution in the irradiation cavity. After the solutions were dried, MALDI-TOF-MS spectra were recorded using a MALDI-TOF/TOF 4800 AB Sciex spectrometer with a laser power in the range of 5100–5200 W cm−2. ESI-MS spectra were recorded using an esquire 6000 ESI† ion Trap LC/MS (Bruker Daltonics) equipped with an electrospray ion source. The instrument was operated either in the positive ESI(+) or negative ESI(−) ion mode. Samples (nanohybrid CT-dsDNA/Na[o-FESAN] and the pristine CT-dsDNA) were introduced into the mass spectrometer ion source directly using a HPLC autosampler with a 1 μL sample. The mobile phase flow (100 μL min−1 of 50:50 v/v CH3CN/H2O) was delivered by a 1200 Series HPLC pump (Agilent). Nitrogen was employed as both a drying and nebulizing gas. Association of the substrate were often observed, the latter appeared from reactions with traces of cations, even in HPLC-grade solvent. Optical microscopy studies: optical microscope images were carried out using an Olympus microscope with a 50×, 20× or 100× objective lens. The samples were prepared by drop casting (i) 50 μL of aqueous solutions of 25 μM Na[o-FESAN], (ii) 50 μL of 2.0 mM CT-dsDNA and (iii) 50 μL of CT-dsDNA/Na[o-FESAN] prepared as described in the Experimental section, respectively, on a glass slide and let to dry at 4 °C. The images of Na[o-FESAN] and CT-dsDNA were taken as references. Atomic absorption spectroscopy (AAS): NovAA 350 atomic absorption spectrophotometer was used for atomic absorbance meassurements. Different concentrations of Na[o-FESAN] aqueous standard solutions were prepared and atomic absorption was measured using an iron cathod lamp (248.3 nm) to obtain the calibration plot. Later, the diluted solutions of CT-dsDNA/Na[o-FESAN] sample were measured to estimate Fe concentration present in the complex, and subsequently in the nanohybrids. Dynamic light scattering (DLS): the hydrodynamic diameters of CT-dsDNA/Na[o-FESAN] nanohybrid was measured by DLS (Malvern Zetasizer Nano ZS) equipped with a He/Ne 633 nm laser, starting with 1 mM solution of Na[o-FESAN] in milliQ water versus increasing concentrations of CT-dsDNA in water in the range of 0 to 900 μM. Measurements were performed for each sample in triplicate at ambient temperature and each with 13–17 scans depending on the sample. X-ray photoelectron spectroscopy (XPS): the XPS measurements were performed using a Specs Phoibos 150 hemispherical analyzer (SPECS GmbH, Berlin, Germany) at room temperature, in a base pressure of 5 × 10−10 mbar using monochromatic Al K-α radiation (1486.74 eV) as excitation source operated at 300 W. The energy resolution as measured by the FWHM of the Ag 3d5/2 peak for a sputtered silver foil was 0.62 eV. The spectra were calibrated in respect to the C 1s at 284.8 eV, main component present in all compounds. Atomic force microscopy (AFM): the AFM measurements working in contact mode and amplitude modulation (AM-AFM) operation (Cypher ES, Oxford instruments) were performed under ambient conditions to investigate the surface morphology of the samples. The lateral distribution of the materials on the surface was analyzed by either phase-shift imaging or lateral force imaging (see details in the ESI†), signals that are highly sensitive to the local chemical termination and/or composition of the surface. For the AFM experiments, the different molecular solutions were prepared and deposited at room temperature by drop casting on clean Si substrates with their native oxide. For contact mode, silicon nitride tips and cantilevers with nominal spring constant of k = 0.02 or 0.03 N m−1 (Bruker) were used, whereas for dynamic operation, cantilevers with a nominal spring constant of k = 3 N m−1 (BudgetSensors) were employed. Force versus distance curves were regularly obtained during imaging acquisition to check the tip conditions through measurement of the adhesion force. All data were analyzed using WxSM free software.116
Author contributions
Laura Gutiérrez-Gálvez and Tania García-Mendiola (T. G. M.) carried out the nanohybrid preparation and some characterization (Raman, UV-vis, electrochemistry, AAS, optical microscopy). Tania García-Mendiola and Encarnación Lorenzo (E. L.) supervised nanohybrid preparation, the Raman, UV-vis, electrochemistry, AAS, optical microscopy characterization. Miquel Nuez-Martinez performed and analysed FT-IR and magnetic studies on breast cancer cells as well as DLS, elemental analysis, ESI-MS, MALDI-TOF-MS and XPS of nanohybrids characterization. Carmen Ocal (C. O.) and Shunya Yan performed the AFM experiments and analysed the data. Francesc Teixidor provided starting boron clusters and participated in chemical discussions. Teresa Pinheiro (T. P.) performed Proton irradiation studies. Fernanda Marques (F. M.) conceptualization, coordination of the biological studies. Clara Viñas (C. V.) conceptualization, coordination of the chemical studies. T. G. M., E. L., C. O., T. P., F. M. and C. V. writing original draft preparation. All authors writing – review and editing manuscript.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors received support from the Spanish Ministerio de Economía y Competitividad (PID2020-116728RB-I00, PID2022-136802NB-I00, RED2022-134120-T and TED2021-129738B-I00), the Generalitat de Catalunya (2017SGR1720). L. Gutiérrez-Gálvez was supported by FPU19/06309 grant from the Spanish Ministry of Universities. S. Y. was supported by the China Scholarship Council (CSC) under Grant No. 202006990034.References
- J. Plesek, Chem. Rev., 1992, 92, 269–278 CrossRef CAS.
- M. Tarrés, E. Canetta, E. Paul, J. Forbes, K. Azzouni, C. Viñas, F. Teixidor and A. J. Harwood, Sci. Rep., 2015, 5, 7804 CrossRef PubMed.
- I. Fuentes, T. García-Mendiola, S. Sato, M. Pita, H. Nakamura, E. Lorenzo, F. Teixidor, F. Marques and C. Viñas, Chem. – Eur. J., 2018, 24, 17239–17254 CrossRef CAS PubMed.
- K. Fink and M. Uchman, Coord. Chem. Rev., 2021, 431, 213684 CrossRef CAS.
- P. Bauduin, S. Prevost, P. Farràs, F. Teixidor, O. Diat and T. Zemb, Angew. Chem., Int. Ed., 2011, 50, 5298–5300 CrossRef CAS PubMed.
- M. Uchman, V. Ďorďovič, Z. Tošner and P. Matějíček, Angew. Chem., Int. Ed., 2015, 54, 14113–14117 CrossRef CAS PubMed.
- D. C. Malaspina, C. Viñas, F. Teixidor and J. Faraudo, Angew. Chem., Int. Ed., 2020, 59, 3088–3092 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Angew. Chem., Int. Ed., 2018, 57, 13968–13981 CrossRef CAS PubMed.
- B. Grüner, J. Rais, P. Selucky and M. Lucaníková, in Boron science: new technologies and applications, ed.N. S. Hosmane, CRC Press-Taylor & Francis group, 2012, pp. 463–490 Search PubMed.
- C. Verdiá-Báguena, A. Alcaraz, V. M. Aguilella, A. M. Cioran, S. Tachikawa, H. Nakamura, F. Teixidor and C. Viñas, Chem. Commun., 2014, 50, 6700–6703 RSC.
- M. Tarrés, E. Canetta, C. Viñas, F. Teixidor and A. J. Harwood, Chem. Commun., 2014, 50, 3370–3372 RSC.
- L. M. Alvero-Gonzalez, D. Aurora Perini, M. Queralt-Martín, A. Perálvarez-Marín, C. Viñas and A. Alcaraz, Bioelectrochemistry, 2023, 154, 108527 CrossRef CAS PubMed.
- D. C. Malaspina, F. Teixidor, C. Viñas and J. Faraudo, Phys. Chem. Chem. Phys., 2023, 25, 27942–27948 RSC.
- T. I. Rokitskaya, I. D. Kosenko, I. B. Sivaev, Y. N. Antonenko and V. I. Bregadze, Phys. Chem. Chem. Phys., 2017, 19, 25122–25128 RSC.
- T. I. Rokitskaya, L. S. Khailova, A. V. Makarenkov, V. A. Ol'shevskaya, V. N. Kalinin and Y. N. Antonenko, Phys. Chem. Chem. Phys., 2016, 18(24), 16476–16482 RSC.
- M. A. Fox and A. K. Hughes, Coord. Chem. Rev., 2004, 248(5–6), 457–476 CrossRef CAS.
- P. Farràs, E. J. Juárez-Pérez, M. Lepšík, R. Luque, R. Núñez and F. Teixidor, Chem. Soc. Rev., 2012, 41, 3445 RSC.
- X. Zhang and H. Yan, Coord. Chem. Rev., 2019, 378, 466–482 CrossRef CAS.
- S. P. Fisher, A. W. Tomich, S. O. Lovera, J. F. Kleinsasser, J. Guo, M. J. Asay, H. M. Nelson and V. Lavallo, Chem. Rev., 2019, 119, 8262–8290 CrossRef CAS PubMed.
- L. Pazderová, E. Z. Tüzün, D. Bavol, M. Litecká, L. Fojt and B. Grűner, Molecules, 2023, 28, 6971 CrossRef PubMed.
- J. F. Valliant, K. J. Guenther, A. S. King, P. Morel, P. Schaffer, O. O. Sogbein and K. A. Stephenson, Coord. Chem. Rev., 2002, 232, 173–230 CrossRef CAS.
- A.-I. Stoica, C. Viñas and F. Teixidor, Chem. Commun., 2009, 4988–4990 RSC.
- I. Fuentes, J. Pujols, C. Viñas, S. Ventura and F. Teixidor, Chem. – Eur. J., 2019, 25, 12820–12829 CrossRef CAS PubMed.
- T. M. Goszczyński, K. Fink, K. Kowalski, Z. J. Leśnikowski and J. Boratyński, Sci. Rep., 2017, 7, 9800 CrossRef PubMed.
- P. Cígler, M. Kožíšek, P. Řezáčová, J. Brynda, Z. Otwinowski, J. Pokorná, J. Plešek, B. Grüner, L. Dolečková-Marešová, M. Máša, J. Sedláček, J. Bodem, H.-G. Kräusslich, V. Král and J. Konvalinka, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15394–15399 CrossRef PubMed.
- J. Fanfrlík, J. Brynda, J. Řezáč, P. Hobza and M. Lepšík, J. Phys. Chem. B, 2008, 112, 15094–15102 CrossRef PubMed.
- A. B. Olejniczak, J. Plešek, O. Křiž and Z. J. Lesnikowski, Angew. Chem., Int. Ed., 2003, 42(46), 5740–5743 CrossRef CAS PubMed.
- M. Nuez-Martínez, L. Pedrosa, I. Martinez-Rovira, I. Yousef, D. Diao, F. Teixidor, E. Stanzani, F. Martínez-Soler, A. Tortosa, À. Sierra, J. J. Gonzalez and C. Viñas, Int. J. Mol. Sci., 2021, 22, 9937 CrossRef PubMed.
- X. Chen, J.-C. Zhao and S. G. Shore, Acc. Chem. Res., 2013, 46, 2666–2675 CrossRef CAS PubMed.
- N. V. Belkova, L. M. Epstein, O. A. Filippov and E. S. Shubina, Chem. Rev., 2016, 116, 8545–8587 CrossRef CAS PubMed.
- J. Echeverría, G. Aullón and S. Alvarez, Dalton Trans., 2017, 46, 2844–2854 RSC.
- S. J. Grabowski, J. Phys. Org. Chem., 2013, 26, 452–459 CrossRef CAS.
- I. Alkorta, J. Elguero, M. Solimannejad and S. J. Grabowski, J. Phys. Chem. A, 2011, 115, 201–210 CrossRef CAS PubMed.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W.-Y. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917 RSC.
- A. Marfavi, P. Kavianpour and L. M. Rendina, Nat. Rev. Chem., 2022, 6, 486–504 CrossRef PubMed.
- A. M. Hughes, Expert Rev. Mol. Med., 2022, 24, e14 CrossRef CAS PubMed.
- N. S. Hosmane, J. A. Maguire, Y. Zhu and M. Takagaki, Boron and Gadolinium Neutron Capture Therapy for Cancer Treatment, World Scientific, 2012 Search PubMed.
- P. Stockmann, M. Gozzi, R. Kuhnert, M. B. Sárosi and E. Hey-Hawkins, Chem. Soc. Rev., 2019, 48, 3497–3512 RSC.
- T. D. Marforio, A. Carboni and M. Calvaresi, Cancers, 2023, 15, 4944 CrossRef CAS PubMed.
- R. J. Grams, W. L. Santos, I. R. Scorei, A. Abad-García, C. A. Rosenblum, A. Bita, H. Cerecetto, C. Viñas and M. A. Soriano-Ursúa, Chem. Rev., 2024, 124, 2441–2511 CrossRef CAS PubMed.
- R. A. Wiesboeck and M. Frederick Hawthorne, J. Am. Chem. Soc., 1964, 86, 1642–1643 CrossRef CAS.
- M. F. Hawthorne, D. C. Young, P. M. Garrett, D. A. Owen, S. G. Schwerin, F. N. Tebbe and P. A. Wegner, J. Am. Chem. Soc., 1968, 90, 862–868 CrossRef CAS.
- R. N. Grimes, Carboranes, Elsevier Inc., 3rd edn, 2016 Search PubMed.
- I. B. Sivaev and V. I. Bregadze, Collect. Czech. Chem. Commun., 1999, 64, 783–805 CrossRef CAS.
- O. N. Kazheva, G. G. Alexandrov, A. V. Kravchenko, I. B. Sivaev, I. D. Kosenko, I. A. Lobanova, M. Kajňaková, L. I. Buravov, V. I. Bregadze, A. Feher, V. A. Starodub and O. A. Dyachenko, Inorg. Chem. Commun., 2012, 15, 106–108 CrossRef CAS.
- I. Bennour, M. N. Ramos, M. Nuez-Martínez, J. A. M. Xavier, A. B. Buades, R. Sillanpää, F. Teixidor, D. Choquesillo-Lazarte, I. Romero, M. Martinez-Medina and C. Viñas, Dalton Trans., 2022, 51, 7188–7209 RSC.
- J. Poater, C. Viñas, I. Bennour, S. Escayola, M. Solà and F. Teixidor, J. Am. Chem. Soc., 2020, 142, 9396–9407 CrossRef CAS PubMed.
- C. Masalles, J. Llop, C. Viñas and F. Teixidor, Adv. Mater., 2002, 14, 826–829 CrossRef CAS.
- M. Uchman, A. I. Abrikosov, M. Lepšík, M. Lund and P. Matějíček, Adv. Theory Simul., 2018, 1, 1870003 CrossRef.
- Ž. Medoš, B. Hleli, Z. Tošner, P. Ogrin, T. Urbič, K. Kogej, M. Bešter-Rogač and P. Matějíček, J. Phys. Chem. C, 2022, 126(12), 5735–5742 CrossRef.
- A. Zaulet, F. Teixidor, P. Bauduin, O. Diat, P. Hirva, A. Ofori and C. Viñas, J. Organomet. Chem., 2018, 865, 214–225 CrossRef CAS.
- A. M. A. Abdelgawwad, J. A. M. Xavier, D. Roca-Sanjuán, C. Viñas, F. Teixidor and A. Francés-Monerris, Angew. Chem., Int. Ed., 2021, 60, 25753–25757 CrossRef CAS PubMed.
- L. H. Hurley, Nat. Rev. Cancer, 2002, 2, 188–200 CrossRef CAS PubMed.
- S. Tada-Oikawa, S. Oikawa, M. Kawanishi, M. Yamada and S. Kawanishi, FEBS Lett., 1999, 442, 65–69 CrossRef CAS PubMed.
- H. Mizutani, S. Tada-Oikawa, Y. Hiraku, S. Oikawa, M. Kojima and S. Kawanishi, J. Biol. Chem., 2002, 277, 30684–30689 CrossRef CAS PubMed.
- S. H. Kaufmann and W. C. Earnshaw, Exp. Cell Res., 2000, 256, 42–49 CrossRef CAS PubMed.
- C. J. Norbury and B. Zhivotovsky, Oncogene, 2004, 23, 2797–2808 CrossRef CAS PubMed.
- T. Hishita, S. Tada-Oikawa, K. Tohyama, Y. Miura, T. Nishihara, Y. Tohyama, Y. Yoshida, T. Uchiyama and S. Kawanishi, Cancer Res., 2001, 61, 2878–2884 CAS.
- S. Kawanishi and Y. Hiraku, Curr. Med. Chem.: Anti-Cancer Agents, 2004, 4, 415–419 CrossRef CAS PubMed.
- S. Kawanishi, S. Oikawa, M. Kawanishi, H. Sugiyama, I. Saito, L. Strekowski and W. D. Wilson, Biochemistry, 2000, 39, 13210–13215 CrossRef CAS PubMed.
- C. Hélène, Curr. Opin. Biotechnol, 1993, 4, 29–36 CrossRef PubMed.
- K. X. Wan, T. Shibue and M. L. Gross, J. Am. Chem. Soc., 2000, 122, 300–307 CrossRef CAS.
- K. X. Wan, M. L. Gross and T. Shibue, J. Am. Soc. Mass Spectrom., 2000, 11, 450–457 CrossRef CAS PubMed.
- E. Largy, A. König, A. Ghosh, D. Ghosh, S. Benabou, F. Rosu and V. Gabelica, Chem. Rev., 2022, 122, 7720–7839 CrossRef CAS PubMed.
- A. B. Buades, L. C. J. Pereira, B. J. C. Vieira, A. C. Cerdeira, J. C. Waerenborgh, T. Pinheiro, A. P. A. Matos, C. G. Pinto, J. F. Guerreiro, F. Mendes, S. Valic, F. Teixidor, C. Viñas and F. Marques, Inorg. Chem. Front., 2022, 9, 1490–1503 RSC.
- T. García-Mendiola, V. Bayon-Pizarro, A. Zaulet, I. Fuentes, F. Pariente, F. Teixidor, C. Viñas and E. Lorenzo, Chem. Sci., 2016, 7, 5786–5797 RSC.
- K. Bednarska-Szczepaniak, K. Dziedzic-Kocurek, E. Przelazły, J. Stanek and Z. J. Leśnikowski, Chem. Commun., 2022, 58(3), 391–394 RSC.
- D. Kaniowski, J. Suwara, K. Ebenryter-Olbińska, A. Jakóbik-Kolon and B. Nawrot, Int. J. Mol. Sci., 2022, 23, 14793 CrossRef CAS PubMed.
- A. B. Buades, V. S. Arderiu, L. Maxwell, M. Amoza, D. Choquesillo-Lazarte, N. Aliaga-Alcalde, C. Viñas, F. Teixidor and E. Ruiz, Chem. Commun., 2019, 55(26), 3825–3828 RSC.
- M. Nuez-Martínez, M. Queralt-Martín, A. Muñoz-Juan, V. M. Aguilella, A. Laromaine, F. Teixidor, C. Viñas, C. G. Pinto, T. Pinheiro, J. F. Guerreiro, F. Mendes, C. Roma-Rodrigues, P. V. Baptista, A. R. Fernandes, S. Valic and F. Marques, J. Mater. Chem. B, 2022, 10, 9794–9815 RSC.
- M. Tagliazucchi, O. Peleg, M. Kröger, Y. Rabin and I. Szleifer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3363–3368 CrossRef CAS PubMed.
- L. A. Leites, Chem. Rev., 1992, 92(2), 279–323 CrossRef CAS.
- R. Pothiraja, M. Sathiyendiran, R. J. Butcher and R. Murugavel, Inorg. Chem., 2005, 44, 6314–6323 CrossRef CAS PubMed.
- R. Pothiraja, S. Shanmugan, M. G. Walawalkar, M. Nethaji, R. J. Butcher and R. Murugavel, Eur. J. Inorg. Chem., 2008, 1834–1845 CrossRef CAS.
- M. Banyay, M. Sarkar and A. Gräslund, Biophys. Chem., 2003, 104, 477–488 CrossRef CAS PubMed.
- M. Banyay, J. Sandbrink, R. Strömberg and A. Gräslund, Biochem. Biophys. Res. Commun., 2004, 324, 634–639 CrossRef CAS PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy. A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, ed.J. Chastain, Physical Electronics, Inc., 1995 Search PubMed.
- D. Tu, J. Li, F. Sun, H. Yan, J. Poater and M. Solà, J. Am. Chem. Soc., 2022, 1(11), 2047–2057 Search PubMed.
- R. W. Carpick and M. Salmeron, Chem. Rev., 1997, 97, 1163–1191 CrossRef CAS PubMed.
- M. Salmeron, S. Kopta, E. Barrena and C. Ocal, Atomic Scale Origin of Adhesion and Friction. In Fundamentals of Tribology and Bridging the Gap Between the Macro- and Micro/Nanoscales. NATO Science Series, ed.B. Bhushan, Springer, 2001, pp. 41–52 Search PubMed.
- C. Prakash, C. L. Shaffer and A. Nedderman, Mass Spectrom. Rev., 2007, 26, 340–369 CrossRef CAS PubMed.
- A. Pandey and M. Mann, Nature, 2000, 405, 837–846 CrossRef CAS PubMed.
- C. Jurinke, P. Oeth and D. van den Boom, Mol. Biotechnol., 2004, 26, 147–164 CrossRef CAS PubMed.
- R. Zenobi and R. Knochenmuss, Mass Spectrom. Rev., 1998, 17, 337–366 CrossRef CAS.
- S. A. Hofstadler and R. H. Griffey, Chem. Rev., 2001, 101, 377–390 CrossRef CAS PubMed.
- J. N. Lisgarten, M. Coll, J. Portugal, C. W. Wright and J. Aymami, Nat. Struct. Biol., 2002, 9(1), 57–60 CrossRef PubMed.
- S. Kawanishi, Y. Hiraku, M. Murata and S. Oikawa, Free Radic. Biol. Med., 2002, 32, 822–832 CrossRef CAS PubMed.
- C. A. Coulomb, Mém. Acad. R. Sci., 1788, 567–577 Search PubMed.
- A.-I. Stoica, C. Viñas and F. Teixidor, Chem. Commun., 2008, 6492–6494 RSC.
- S. Ptasińska, A. Stypczyńska, T. Nixon, N. J. Mason, D. V. Klyachko and L. Sanche, J. Chem. Phys., 2008, 129, 065102 CrossRef PubMed.
- Y. Zubavichus, O. Fuchs, L. Weinhardt, C. Heske, E. Umbach, J. D. Denlinger and M. Grunze, Radiat. Res., 2004, 161, 346–358 CrossRef CAS PubMed.
- G. L. Locher, Am. J. Roentgenol. Radium Ther., 1936, 36, 1–13 CAS.
- A. H. Soloway, W. Tjarks, B. A. Barnum, F.-G. Rong, R. F. Barth, I. M. Codogni and J. G. Wilson, Chem. Rev., 1998, 98, 2389–2390 CrossRef CAS PubMed.
- H. Cerecetto and M. Couto, Medicinal chemistry of boron-bearing compounds for BNCT-glioma treatment: Current challenges and perspectives. Glioma Contemporary Diagnostic and Therapeutic Approaches, ed.I. Omerhodzic and K. Arnautovic, IntechOpen Ltd, 2018 Search PubMed.
- J. Xu, J. Wang and Q. Wei, Chin. Sci. Bull., 2021, 67, 1479–1489 CrossRef.
- X. Cheng, F. Li and L. Liang, Curr. Oncol., 2022, 29(10), 7868–7886 CrossRef PubMed.
- Z. Xie and T. B. Marder, Chem, 2023, 9, 3015–3029 CAS.
- T. D. Malouff, D. S. Seneviratne, D. K. Ebner, W. C. Stross, M. R. Waddle, D. M. Trifiletti and S. Krishnan, Front Oncol., 2021, 11, 601820 CrossRef CAS PubMed.
- STELLA PHARMA. Available online: https://stella-pharma.co.jp/en/blog/1292/accessed on May 9th 2024.
- M. Couto, I. Mastandrea, M. Cabrera, P. Cabral, F. Teixidor, H. Cerecetto and C. Viñas, Chem. – Eur. J., 2017, 33(39), 9233–9238 CrossRef PubMed.
- J. Wang, L. Chen, J. Ye, Z. Li, H. Jiang, H. Yan, M. Y. Stogniy, I. B. Sivaev, V. I. Bregadze and X. Wang, Biomacromolecules, 2017, 18, 1466–1472 CrossRef CAS PubMed.
- M. Couto, M. F. García, C. Alamón, M. Cabrera, P. Cabral, A. Merlino, F. Teixidor, H. Cerecetto and C. Viñas, Chem. – Eur. J., 2018, 24(13), 3122–3126 CrossRef CAS PubMed.
- M. Couto, C. Alamon, S. Nievas, M. Perona, M. A. Dagrosa, F. Teixidor, P. Cabral, C. Viñas and H. Cerecetto, Chem. – Eur. J., 2020, 26(63), 14335–14340 CrossRef CAS PubMed.
- J. Li, Q. Sun, C. Lu, H. Xiao, Z. Guo, D. Duan, Z. Zhang, T. Liu and Z. Liu, Nat. Commun., 2022, 13, 2143–2154 CrossRef CAS PubMed.
- W. Ma, Y. Wang, Y. Xue, M. Wang, C. Lu, W. Guo, Y.-H. Liu, D. Shu, G. Shao, Q. Xu, D. Tu and H. Yan, Chem. Sci., 2024, 15, 4019–4030 RSC.
- R. F. Barth, N. Gupta and S. Kawabata, Cancer Commun., 2024 DOI:10.1002/cac2.12582.
- R. F. Barth, Mg. H. Vicente, O. K. Harling, W. Kiger, K. J. Riley, P. J. Binns, F. M. Wagner, M. Suzuki, T. Aihara, I. Kato and S. Kawabata, Radiat. Oncol., 2012, 7, 1–21 CrossRef PubMed.
- R. F. Barth, J. Neurooncol., 2003, 62, 1–5 Search PubMed.
- A. F. F. da Silva, R. S. G. R. Seixas, A. M. S. Silva, J. Coimbra, A. C. Fernandes, J. P. Santos, A. Matos, J. Rino, I. Santos and F. Marques, Org. Biomol. Chem., 2014, 12, 5201–5211 RSC.
- D. Gabel, S. Foster and R. G. Fairchild, Radiat. Res., 1987, 111, 14–25 CrossRef CAS PubMed.
- B. H. Geierstanger and D. E. Wemmer, Annu. Rev. Biophys. Biomol. Struct., 1995, 24, 463–493 CrossRef CAS PubMed.
- C. Viñas, J. Pedrajas, J. Bertran, F. Teixidor, R. Kivekäs and R. Sillanpää, Inorg. Chem., 1997, 36, 2482–2486 CrossRef PubMed.
- M. F. Hawthorne and T. D. Andrews, J. Chem. Soc. Chem. Commun., 1965, 443–444 RSC.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–218 CrossRef CAS.
- P. Doty and S. A. Rice, Biochim. Biophys. Acta, 1955, 16, 446–448 CrossRef CAS PubMed.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S4 display the characterization of the nanohybrid by XPS, CV, UV-vis, DPV (anodic and cathodic), and AAS. Friction force microscopy procedure and the corresponding studies (Fig. S6 and S7). ESI-MS and MALDI-TOF-MS spectra (Fig. S8–S14 and Table S2). Table S1 contains the results and studies of elemental analysis. Table S2 displays the anodic peaks of the MALDI-TOF-MS spectra. Table S3 analyses the deconvoluted XPS spectra. See DOI: https://doi.org/10.1039/d4tb01177e |
| This journal is © The Royal Society of Chemistry 2024 |