
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetrathiafulvalene-based covalent organic frameworks as high-voltage organic cathodes for lithium batteries†
Gonçalo
Valente
a,
Raquel
Dantas
 a,
Pedro
Ferreira
a,
Rebecca
Grieco
b,
Nagaraj
Patil
b,
Ana
Guillem-Navajas
c,
David
Rodríguez-San Miguel
cd,
Félix
Zamora
ce,
Roman
Guntermann
f,
Thomas
Bein
f,
João
Rocha
a,
M. Helena
Braga
gh,
Karol
Strutyński
a,
Manuel
Melle-Franco
*a,
Rebeca
Marcilla
*b and
Manuel
Souto
*ai
a,
Pedro
Ferreira
a,
Rebecca
Grieco
b,
Nagaraj
Patil
b,
Ana
Guillem-Navajas
c,
David
Rodríguez-San Miguel
cd,
Félix
Zamora
ce,
Roman
Guntermann
f,
Thomas
Bein
f,
João
Rocha
a,
M. Helena
Braga
gh,
Karol
Strutyński
a,
Manuel
Melle-Franco
*a,
Rebeca
Marcilla
*b and
Manuel
Souto
*ai
aDepartment of Chemistry, CICECO-Aveiro Institute of Materials, University of Aveiro, Aveiro, 3810-393, Portugal
bElectrochemical Processes Unit, IMDEA Energy Institute, Avda. Ramón de La Sagra 3, 28935 Móstoles, Spain
cDepartamento de Química Inorgánica, Universidad Autónoma de Madrid, Madrid, 28049, Spain
dInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
eCondensed Matter Physics Center (IFIMAC), Universidad Autónoma de Madrid, 28049 Madrid, Spain
fDepartment of Chemistry and Center for NanoScience (CeNS), Ludwig-Maximilians-Universität München, Butenandtstraße 5-13 (E), 81377 Munich, Germany
gEngineering Physics Department, Engineering Faculty, University of Porto, 4200-465 Porto, Portugal
hMatER – Materials for Energy Research Laboratory, Engineering Faculty, University of Porto, Portugal
iCiQUS, Centro Singular de Investigación en Química Bioloxica e Materiais Moleculares, Departamento de Química-Física, Universidade de Santiago de Compostela, 15782, Santiago de Compostela, Spain. E-mail: manuel.souto.salom@usc.es
First published on 9th August 2024
Abstract
Redox-active covalent organic frameworks (COFs) are promising electrode materials for metal-ion batteries owing to their tunable electrochemical properties, adjustable structure, and resource availability. Herein, we report a series of two-dimensional tetrathiafulvalene (TTF)-based COFs incorporating different organic linkers between the electroactive moieties. These COFs were investigated as p-type organic cathode materials for lithium-organic batteries. The electrical conductivity of both neutral and doped TTF-COFs was measured using a van der Pauw setup, and their electronic structures were investigated through quantum-chemical calculations. Binder-free buckypaper TTF-based electrodes were prepared and systematically tested as organic cathodes in lithium half-cells. The results revealed high average discharge potentials (∼3.6 V vs. Li/Li+) and consistent cycling stability (80% capacity retention after 400 cycles at 2C) for the three TTF-COF electrodes. In addition, the specific capacity, rate capability, and kinetics varied depending on the structure of the framework. Our results highlight the potential of TTF-COFs as high-voltage organic cathodes for metal-ion batteries and emphasize the importance of molecular design in optimizing their electrochemical performance.
Introduction
Covalent organic frameworks (COFs) are crystalline porous polymers composed of molecular organic building blocks linked by strong covalent bonds.1–4 The structure and porosity of COFs are predetermined by the connectivity and size of the organic linkers, which are of great interest in the pre-design of new materials with specific functionalities and applications. For example, the electrical and optical properties of layered two-dimensional (2D) COFs can be fine-tuned by careful selection of electroactive organic building blocks and linkages.5 Such control over the structure and properties of COFs has attracted considerable attention in their design and synthesis, particularly in expanding their use to a wider range of applications. Therefore, these materials are interesting for a wide variety of applications such as gas storage and separation,6,7 catalysis,8 (opto)electronics,9,10 photo/electrocatalysis,11,12 and energy storage and conversion.13,14In recent years, COFs have emerged as promising organic electrode materials (OEMs) for lithium-ion batteries and post-lithium battery technologies.11,13–15 Some advantages over conventional OEMs include their insolubility in electrolytes, permanent porosity that enhances ion diffusion, and the ability to incorporate numerous redox-active centres within the framework. Moreover, their crystalline nature allows their structure and properties to be predicted through computational modelling, providing meaningful insights into the structure–property relationship, enabling researchers to rationally design COFs with targeted functionalities, and accelerating the development of highly efficient and purpose-specific materials. To date, most COF electrodes have used n-type building blocks,15,16 which typically result in high specific capacities but moderate potentials (<3 V vs. Li/Li+). Conversely, redox-active COFs built up from p-type moieties have been much less explored, even though they may show much higher working potentials (>3 V vs. Li/Li+). For example, COFs based on TEMPO radical,17 phenoxazine,18 or dibenzopentalene19 moieties have achieved potentials exceeding 3.5 V vs. Li/Li+. In this case, the p-type building blocks are oxidized to the cation state and combine with anions from the electrolyte such as PF6− to compensate the positive charges.15 Despite their promise as high-potential organic cathodes, p-type COFs are limited in variety compared to n-type COFs.
Tetrathiafulvalene (TTF) is an electroactive molecule widely recognised in the field of organic electronics for its tuneable redox and electron-donor properties, as well as its role as a building block in developing organic conductors.5,20 TTF can be reversibly oxidized to TTF˙+ and TTF2+, making it appealing for energy storage applications. For example, TTF has been used as a redox mediator in Li–O2 batteries,21 and some TTF-based extended molecules22 and conventional linear polymers23–25 have been investigated as p-type electrodes for lithium-organic batteries, where they exhibit a relatively high average voltage. Although some studies have reported the use of TTF COFs for other applications,26–32 to our knowledge, detailed studies into the electrochemical performance of TTF-based COFs used as p-type cathodes in lithium batteries are still lacking.
Herein, we report the synthesis and characterization of a series of three imine-linked 2D TTF-COFs, investigated as p-type cathodes for lithium-organic batteries. These three TTF-COFs share a similar topology but incorporate different organic spacers (phenyl, diphenyl, 4,7-diphenyl-benzothiadiazole) between the redox-active TTF building blocks (Scheme 1). This design allowed us to assess how various linkers affect the electrical and electrochemical properties of the COFs. The electronic conductivities of neutral and iodine- and F4TCNQ-doped TTF-COFs were measured as pressed pellets using a van der Pauw setup. Additionally, quantum chemical calculations were performed to get further insights into the electronic structure of the TTF-COFs. Self-supported binder-free buckypaper electrodes using the TTF-COFs as active materials were prepared and investigated as p-type cathodes in lithium-organic half-cells showing high average potentials (∼3.6 V vs. Li/Li+) and tuneable electrochemical performance depending on the spacers used between the TTF moieties.
 | ||
| Scheme 1 Schematic representation of the three 2D TTF-COFs. | ||
Results and discussion
Synthesis and characterisation of TTF-COFs
The synthesis of TTF-Ph-COF was previously reported,26–28 whereas TTF-(Ph)2-COF and TTF-BT-COF were obtained by optimizing the synthetic parameters. In general, TTF-COFs were synthesized under solvothermal conditions by the condensation reaction of tetrathiafulvalene–tetrabenzaldehyde and the corresponding diamine derivatives in mixtures of mesitylene, dioxane, and acetic acid (6 M) at 120 °C for different times (see ESI† for Experimental details). After cooling, the resulting precipitates were washed with anhydrous THF to remove unreacted precursors, and the dark maroon powders were activated using supercritical CO2 (see ESI†). Fourier-transform infrared (FTIR) and 13C solid-state cross-polarization magic-angle spinning (CPMAS) NMR spectroscopies confirmed the formation of the imine bonds. FTIR spectra of TTF-COFs exhibited characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching bands around 1620 cm−1, along with a reduction in the intensity of the carbonyl band from residual aldehyde groups (Fig. S1†). 13C CPMAS NMR spectra of TTF-COFs showed signals around 153–157 ppm attributed to the formation of the new CN bond (Fig. S2†).26,28 Thermal gravimetric analysis (TGA) demonstrated that TTF-COFs are thermally stable up to 370 °C (Fig. S3†). Scanning electron microscopy (SEM) was used to study the morphology of TTF-COF powders, revealing spherical particles of 1–2 μm (Fig. S4–S6†).
N stretching bands around 1620 cm−1, along with a reduction in the intensity of the carbonyl band from residual aldehyde groups (Fig. S1†). 13C CPMAS NMR spectra of TTF-COFs showed signals around 153–157 ppm attributed to the formation of the new CN bond (Fig. S2†).26,28 Thermal gravimetric analysis (TGA) demonstrated that TTF-COFs are thermally stable up to 370 °C (Fig. S3†). Scanning electron microscopy (SEM) was used to study the morphology of TTF-COF powders, revealing spherical particles of 1–2 μm (Fig. S4–S6†).
Structural determination
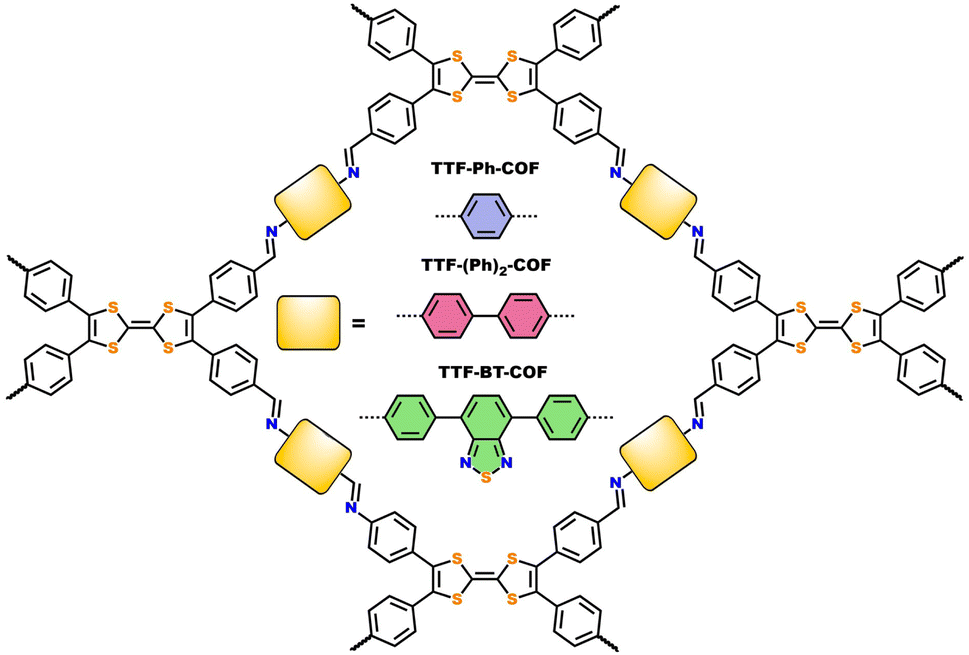
Powder X-ray diffraction (PXRD) measurements were employed to assess the crystallinity of the synthesized TTF-COFs (Fig. 1). PXRD of TTF-Ph-COF showed an intense reflection at 2θ = 3.8°, accompanied by a weak peak at 7.6°, which are attributed to the (100), and (200) reflections, respectively.26–28 The PXRD pattern of TTF-(Ph)2-COF revealed peaks at lower angles (2θ = 3.4° and 6.6°), while TTF-BT-COF displayed an intense diffraction peak at 2.8° and a weaker peak at 5.6°. To confirm the structure of TTF-COFs and determine their framework topology, computational models were built to explore possible conformations and align them with the observed PXRD patterns. The structural models were simulated using AA or AB stacking modes (Fig. S7–S10†). The theoretical PXRD patterns using the AA stacking mode were found to be comparable to the experimental ones (Fig. S11–S13†). The unit cells of these models underwent Pawley refinement to achieve a close match with the experimental PXRD results, showing minimal discrepancies in the fitting (Fig. 1).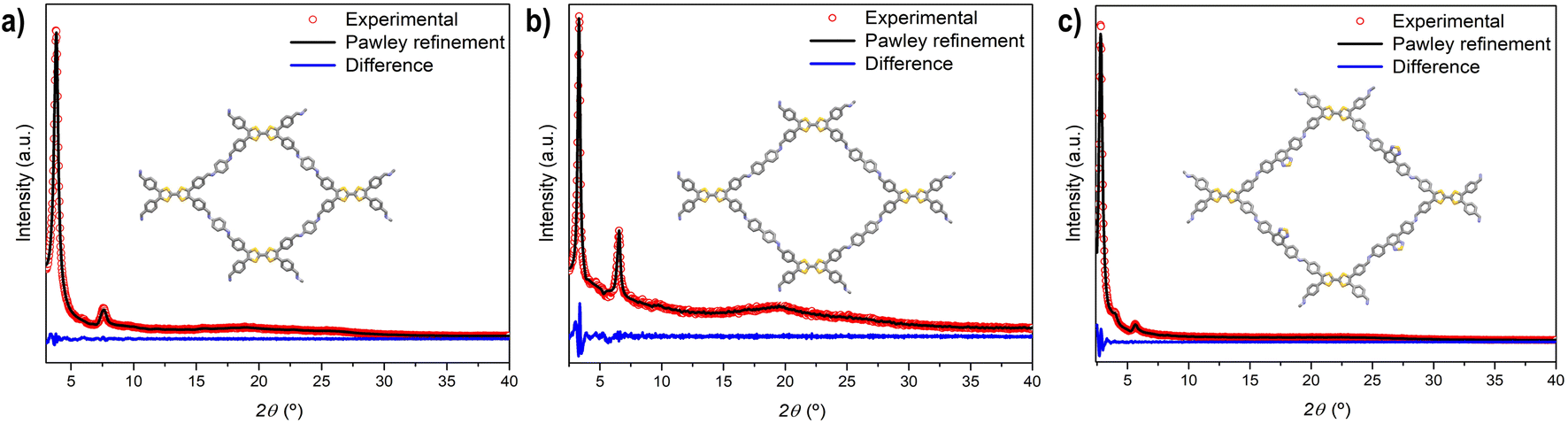 | ||
| Fig. 1 PXRD patterns and Pawley refinement (AA stacking mode) of (a) TTF-Ph-COF, (b) TTF-(Ph)2-COF and (c) TTF-BT-COF. | ||
Gas sorption measurements
Nitrogen adsorption–desorption isotherms of activated TTF-COFs were performed at 77 K (Fig. 2a–c) and a BETSI analysis33 of the results was performed to obtain the Brunauer–Emmett–Teller (BET) surface area. This analysis yielded a surface area of 1148 m2 g−1 for TTF-Ph-COF, in agreement with previous findings,26–28 and 1278 m2 g−1 for TTF-(Ph)2-COF (Fig. S14–S15†). Additionally, the pore width distribution was determined using the non-local density functional theory (NLDFT) method (Fig. 2d). As expected, an increase in the length of the diamine used in each TTF-COF led to a corresponding increase in pore size. Thus, the experimental measurements show that TTF-Ph-COF and TTF-(Ph)2-COF have a pore width of about 1.6 nm and 1.9 nm, respectively. These values are in good agreement with the computed results from PoreBlazer v4.0,34 which calculated values of 1.49 nm and 1.82 nm for TTF-Ph-COF and TTF-(Ph)2-COF, respectively (Table S4†). For TTF-BT-COF, measuring the porosity was proved to be more challenging due to its larger pore size and thus more fragile structure. To prevent the collapse of the structure, a delicate activation process was necessary (see ESI† for details). By performing a solvent exchange with a low surface tension solvent such as perfluorohexane35 before the activation with supercritical CO2, it was possible to maintain the integrity of the COF structure. This approach yielded a BET surface area of 487 m2 g−1 (Fig. 2c and S16†) and a pore width distribution centred at 2.2 nm (Fig. 2d), which agrees with the calculated value of 2.12 nm (Table S4†). The BET value is similar to a recently reported pyrene-BT COF analogue.36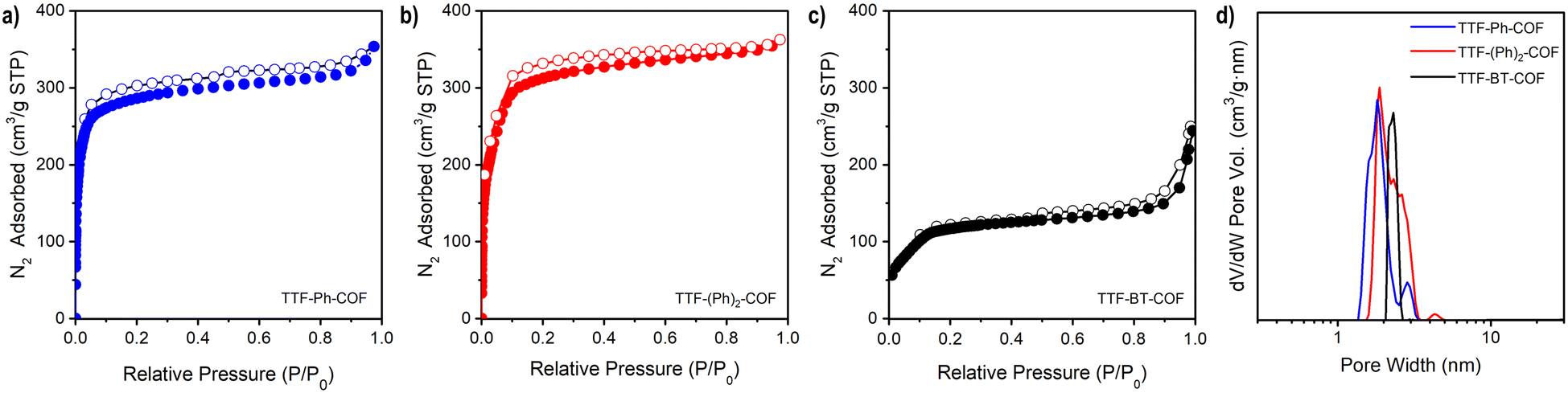 | ||
| Fig. 2 N2 adsorption (filled symbols) and desorption (empty symbols) isotherms of (a) TTF-Ph-COF, (b) TTF-(Ph)2-COF, (c) TTF-BT-COF. (d) Pore width distributions of TTF-Ph-COF (blue), TTF-(Ph)2-COF (red) and TTF-BT-COF (black). | ||
Electrical conductivity and chemical doping
Electrical conductivities of neutral TTF-COF bulk materials as pressed pellets at room temperature were measured using a van der Pauw four-probe setup (Experimental details in the ESI†). Similar conductivity values (∼10−8 S cm−1) were obtained for the 3 TTF-COFs (Table 1), indicating that porosity does not have a significant impact on their charge transport. In the case of TTF-Ph-COF and TTF-(Ph)2-COF, the conductivities of TTF-COFs were increased up to 3 and 1 orders of magnitude upon chemical doping with iodine (I2) or 2,3,5,6-tetrafluoro-tetracyanoquinodimethane (F4TCNQ), respectively (Table 1). These dopants are commonly employed for chemical doping of COFs and the observed increase in conductivity is attributed to the increment in the number of free charge carriers.37 To prepare the doped materials, bulk TTF-COFs were either immersed in a solution of F4TCNQ in acetonitrile or exposed to iodine vapor (see ESI†). The room-temperature conductivities of both pristine and iodine-doped TTF-Ph-COF are in line with those previously reported in pressed pellets form using a two-probe configuration.26 It is worth noting that, across all samples, higher conductivities were achieved with iodine doping. These results are in contrast with some Wurster-type COFs, where the highest conductivities were observed with F4TCNQ doping.38| COF system | RT conductivity (S cm−1) | E g (eV) |
|---|---|---|
| TTF-Ph-COF, pristine | 2.66 × 10−8 | 1.79 |
| TTF-Ph-COF/I2 | 8.50 × 10−5 | 1.57 |
| TTF-Ph-COF/F4TCNQ (1 eq.) | 1.18 × 10−7 | 1.52 |
| TTF-(Ph)2-COF, pristine | 1.95 × 10−8 | 1.84 |
| TTF-(Ph)2-COF/I2 | 6.98 × 10−5 | 1.49 |
| TTF-(Ph)2-COF/F4TCNQ (1 eq.) | 1.65 × 10−7 | 1.52 |
| TTF-BT-COF, pristine | 3.43 × 10−8 | 1.82 |
| TTF-BT-COF/I2 | 3.98 × 10−7 | 1.68 |
| TTF-BT-COF/F4TCNQ (1 eq.) | 9.72 × 10−8 | 1.50 |
The doped TTF-COFs were further characterized by different spectroscopic techniques. After doping, the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching IR band of F4TCNQ shifted to lower frequencies, consistent with the degree of charge transfer between the F4TCNQ acceptor and TTF donor moieties (Fig. S17†).39 Additionally, the formation of TTF˙+ radical cation species in the doped TTF-COFs was confirmed by EPR spectroscopy, which showed a significant increase in the intensity of the signal at g = 2.005 upon doping with either iodine or F4TCNQ (Fig. S18–S20†). The uniform distribution of the dopants was verified by energy-dispersive X-ray spectroscopy (EDS) (Fig. S21 and S22†). The optical band gaps of the neutral and doped TTF-COFs were determined by linearly fitting the absorption onsets in the Tauc plots of the Kubelka–Munk-transformed data (Fig. 3 and S23, S24†). All neutral TTF-COFs showed optical band gaps of approximately 1.8 eV (Table 1), consistent with theoretical band gap predictions (see below) and corresponding to similar orders of magnitude in conductivity. Conversely, the doped TTF-COFs showed lower optical band gaps, consistent with the highest measured conductivities.
N stretching IR band of F4TCNQ shifted to lower frequencies, consistent with the degree of charge transfer between the F4TCNQ acceptor and TTF donor moieties (Fig. S17†).39 Additionally, the formation of TTF˙+ radical cation species in the doped TTF-COFs was confirmed by EPR spectroscopy, which showed a significant increase in the intensity of the signal at g = 2.005 upon doping with either iodine or F4TCNQ (Fig. S18–S20†). The uniform distribution of the dopants was verified by energy-dispersive X-ray spectroscopy (EDS) (Fig. S21 and S22†). The optical band gaps of the neutral and doped TTF-COFs were determined by linearly fitting the absorption onsets in the Tauc plots of the Kubelka–Munk-transformed data (Fig. 3 and S23, S24†). All neutral TTF-COFs showed optical band gaps of approximately 1.8 eV (Table 1), consistent with theoretical band gap predictions (see below) and corresponding to similar orders of magnitude in conductivity. Conversely, the doped TTF-COFs showed lower optical band gaps, consistent with the highest measured conductivities.
 | ||
| Fig. 3 Normalized Tauc plot of the Kubelka–Munk-transformed data for TTF-Ph-COF, TTF-(Ph)2-COF and TTF-BT-COF. Dashed lines depict linear fits to the absorption onsets. | ||
Electronic structure of TTF-COFs
Theoretical models were used to elucidate the electronic structure of TTF-COFs (see ESI† for details). Band structures and band gaps were obtained using the B3LYP functional and shown in Table S5 and Fig. S25.† The theoretical band gaps were found to be similar (2.07, 2.15, and 1.94 eV) for the monolayers of TTF-Ph-COF, TTF-(Ph)2-COF and TTF-BT-COF, respectively, which are in close agreement with the experimental values (Table 1). It is worth noting that the tested conformations (AA and AB) exhibited considerable variability in band gap values. In all cases, the highest occupied crystal orbitals (HOCO), responsible for two distinct bands below the Fermi level, were localized on the TTF moieties, whereas the lowest unoccupied crystal orbitals (LUCO) were localized on the imine linkages and the BT units in the case of TTF-BT-COF (Fig. 4, S26 and S27†). Furthermore, it was found that the eigenvalue of the lowest unoccupied orbital of the iodine molecule lies within the gap of the investigated TTF-COFs. This confirms the theoretical prediction that doping arises from the spontaneous formation of electron holes in TTF moieties in contact with iodine molecules for all COFs under study (Fig. S28†).Electrochemical performance of TTF-COFs
The electrochemical properties of TTF-COFs were first assessed by solid-state cyclic voltammetry (CV) of the COFs deposited on a glassy carbon working electrode performed at room temperature using 0.1 M TBAPF6/CH3CN as electrolyte (Fig. S29†). CVs of the three TTF-COFs show two reversible redox processes assigned to the radical cation (∼0.55 V vs. Agwire) and dication (∼0.90 V vs. Agwire) states at similar potentials.26 The potentials are similar for all three COFs and consistent with the computational calculations that revealed similar HOCO energy values. Furthermore, the average computed potential for the three TTF-COFs (0.5 V vs. Ag) matches remarkably well with the measured potentials (see ESI†). When applying more negative potentials, a reduction process (−0.90 V vs. Agwire) was also observed in the case of TTF-BT-COF (Fig. S30†). This could be attributed to the reduction of the BT unit to the radical anion state, appearing at similar potentials as in other BT-based COFs.36,40–43 In addition, this matches our computer modelling results, which predict a first reduction at −1.4 V vs. Ag localized in the BT units, Fig. 4b (see ESI†). Therefore, TTF-BT-COF could be a potential candidate to be explored as a redox-bipolar active material due to the combination of n-type BT and p-type TTF building blocks (Fig. S31†).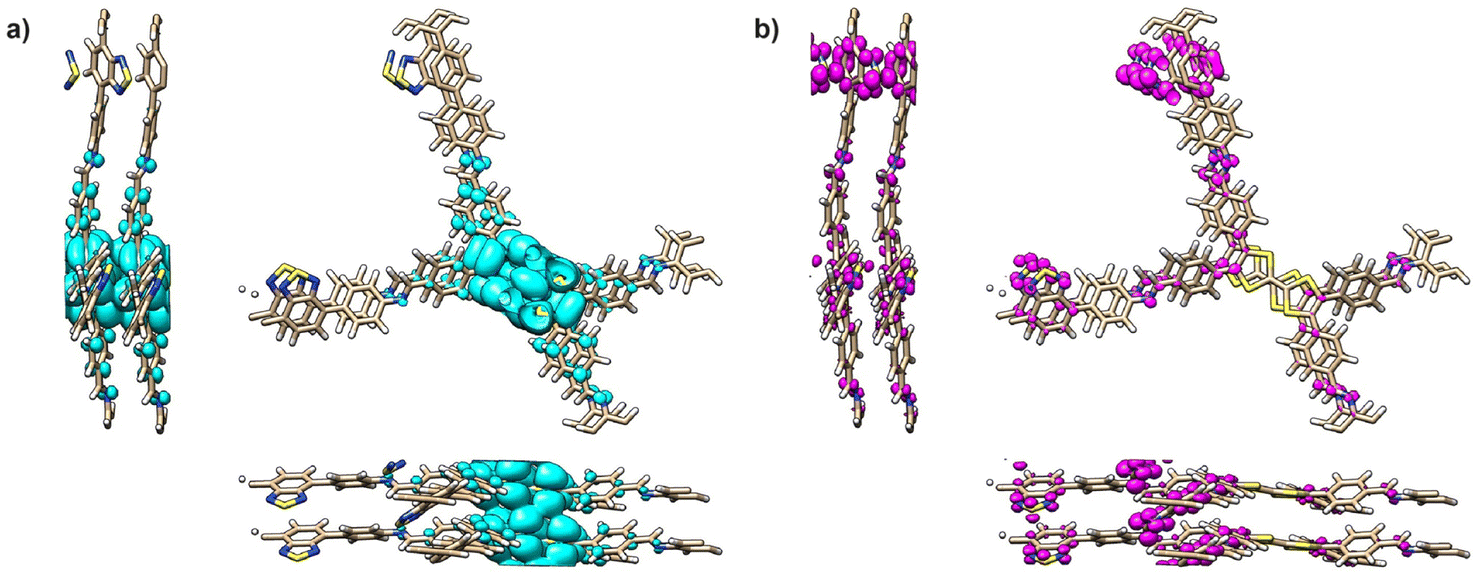 | ||
| Fig. 4 Integrated electronic density of TTF-BT-COF for (a) the two highest occupied bands and (b) the two lowest unoccupied bands. | ||
Binder- and metal current collector-free self-standing buckypaper electrodes were fabricated to evaluate TTF-COFs as potential cathodes for lithium-organic batteries. This electrode preparation method has been used with other redox-active conjugated microporous polymers,44 but unexplored with COFs. The self-supported buckypaper electrodes comprised a blend of COF as active material, single-walled carbon nanotubes (SWCNTs) and reduced graphene oxide (rGO) in a weight ratio of 50/35/15. The electrodes were tested in a coin-type Li-ion half-cell configuration, using lithium metal as both the reference and counter electrodes. The electrolyte used was 1 M lithium hexafluorophosphate (LiPF6) in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) with a volume ratio of 3/7 (EC/DMC). The electrodes were fabricated with a target mass loading of active material (TTF-COFs) of 1 mg cm−2. As a commonly used procedure for organic batteries, specific capacities were calculated based on the amount of the active material.
Fig. 5a shows the CVs at 0.1 mV s−1 for the three p-type TTF-COFs, exhibiting two pairs of redox processes associated with the TTF moieties in the potential window of 2.5–4.2 V vs. Li/Li+. These p-type TTF-COF electrodes may undergo two reversible redox processes in this potential window. During charging, the TTF moiety undergoes first oxidation to the radical cation state and subsequently to the dication state, during which it loses two electrons that are compensated by one and two PF6− counterions from the electrolyte, respectively (Fig. 5b). The first set of peaks centred at 3.41, 3.42, and 3.47 V vs. Li/Li+ for TTF-Ph-COF, TTF-(Ph)2-COF, and TTF-BT-COF, respectively, corresponds to the redox transformation of the TTF moieties between the neutral and the radical cation states. A less pronounced second peak pair centred at 3.75, 3.80, and 3.81 V vs. Li/Li+ for TTF-Ph-COF, TTF-(Ph)2-COF, and TTF-BT-COF, respectively, is related to the redox transformation between the radical cation and dication states.
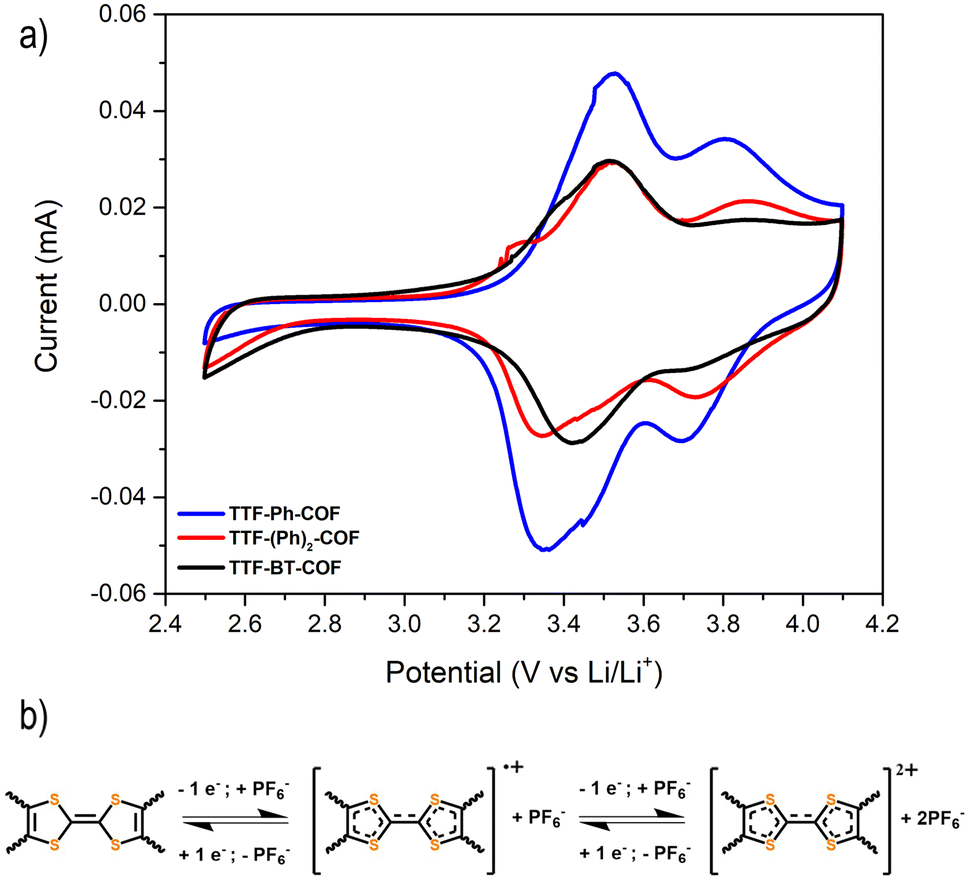 | ||
| Fig. 5 (a) Cyclic voltammetry of TTF-Ph-COF, TTF-(Ph)2-COF, and TTF-BT-COF using 1 M LiPF6 in EC/DMC (3/7 v/v) at a scan rate of 0.5 mV s−1. (b) Redox mechanism of the p-type TTF moiety in TTF-COFs. | ||
The TTF-BT-COF, which combines the p-type properties of TTF and n-type properties of the BT unit, could be considered a potential redox-bipolar COF. In this case, when the battery is discharged, the BT building block could be further reduced to the radical anion state, with one Li+ compensating for the negative charge. Galvanostatic charge–discharge (GCD) experiments were first performed for TTF-BT-COF using different potential windows to preliminary assess the BT moieties redox activity. However, differential capacity analysis (dQ/dV) shows that when decreasing the lower potential cut-off limit, the signals associated with the TTF units gradually decrease in intensity, eventually leading to the irreversible oxidation of TTF without traces of BT redox activity (Fig. S32†). This behaviour can be related to the large voltage separation between the redox processes associated with the p-type TTF and n-type BT building blocks and/or incompatibility of BT in the tested carbonate electrolyte.16 This makes it unsuitable to exploit both electroactive units within the same potential window in this specific electrolyte, despite the redox-bipolar nature of the COF.
Therefore, unless specifically stated, the remaining electrochemical studies have always focused on the potential window of 2.5–4.2 V. In this potential window, the theoretical specific capacities of TTF-Ph-COF, TTF-(Ph)2-COF and TTF-BT-COF were calculated to be 70, 59 and 45 mA h g−1, respectively. These values were obtained considering only two-electron redox processes involving the p-type TTF moieties. GCD experiments were performed to evaluate the rate capability and long-term cyclic stability of the three TTF-COF electrodes. The rate capability tests for the TTF-COF electrodes were conducted within the potential window of 2.5–4.2 V and using current rates (C-rates) from 1C up to 120C, as shown in Fig. 6. TTF-Ph-COF, TTF-(Ph)2-COF and TTF-BT-COF revealed reasonable gravimetric capacity of 57, 41, and 49 mA h g−1 at 1C. Moreover, Ph-based COFs exhibited better rate performance than the BT-COF. For instance, at a C-rate of 10C, the capacity retention was 64%, 53% and 41% for TTF-Ph-COF, TTF-(Ph)2-COF and TTF-BT-COF, respectively, relative to the capacity observed at 1C (Fig. 6c).
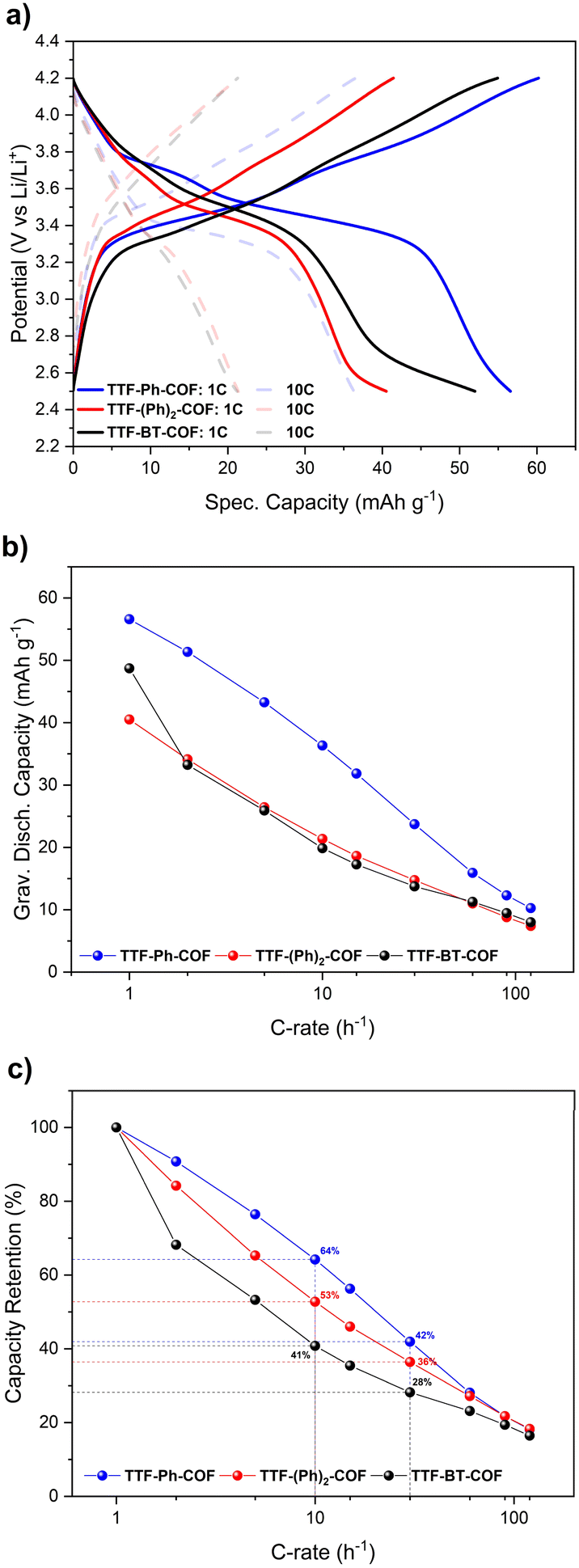 | ||
| Fig. 6 Galvanostatic charge–discharge rate capability experiments of TTF-Ph-COF, TTF-(Ph)2-COF, and TTF-BT-COF, using a potential window of 2.5–4.2 V. (a) Galvanostatic profiles at different current rates. (b) Gravimetric specific capacity versus C-rate. (c) Capacity retention as a function of C-rate. | ||
We made a preliminary attempt to understand the rate performance differences among these COFs by CV, electrochemical impedance spectroscopy (EIS) and galvanostatic intermittent titration technique (GITT) experiments. CVs at different scan rates from 0.1 to 1 mV s−1 (Fig. S33†), enabled a detailed kinetics study of the redox processes. This analysis involved determining the cathodic and anodic b-values for each redox processes (see Fig. S34†) and deconvoluting bulk and surface charge storage contributions using Dunn's method (see Fig. S35†). In general, b-value for TTF-BT-COF was lower than TTF-Ph-COF, TTF-(Ph)2-COF for the first redox peak, and additionally, found to be lower for the second redox process (Table S6†). Moreover, all three TTF-COFs have similar capacitive/diffusion contributions (around 65/35%), with slightly higher capacitive contribution for TTF-(Ph)2-COF (Fig. S35d†).
To complete the electrochemical study, we performed a comprehensive EIS analysis (Fig. 7 and S36–S40†). During this analysis, equivalent series and charge transfer resistances, phase angles and characteristic times were measured (see ESI†).45TTF-Ph-COF and TTF-(Ph)2-COF consistently showed better performance in terms of lower resistances (Fig. S38†), higher phase angles (Fig. S39†) and characteristic times (Fig. S40†) compared to the TTF-BT-COF. The equivalent series resistances (Rs) did not show significant variation at different state-of-charge and depth-of-discharges during different charging and discharging potentials, respectively (Fig. S38†). Additionally, the Rs charge-transfer resistances (RCT) values were found to be lower for TTF-Ph-COF and TTF-(Ph)2-COF compared to TTF-BT-COF. Therefore, Ph-based COFs are expected to provide better capacitance performance in line with higher capacity utilization. The same trend was observed in the diffusivity values of PF6−, which were obtained using the GITT technique (Fig. 7c and S41†). The better diffusivity values could be explained by the higher porosity, i.e. higher BET surface area (Fig. 2) of TTF-Ph-COF and TTF-(Ph)2-COF, justifying the poor rate performance for the TTF-BT-COF. These results demonstrate that, in addition to selecting the appropriate electroactive building blocks, it is necessary to optimize the COF pore size to improve ion diffusion.15,46 However, increasing pore size may be inefficient if the density of redox sites decreases significantly, so the right balance between pore size and specific capacity should be considered when designing new redox-active COF electrodes for rechargeable batteries.15
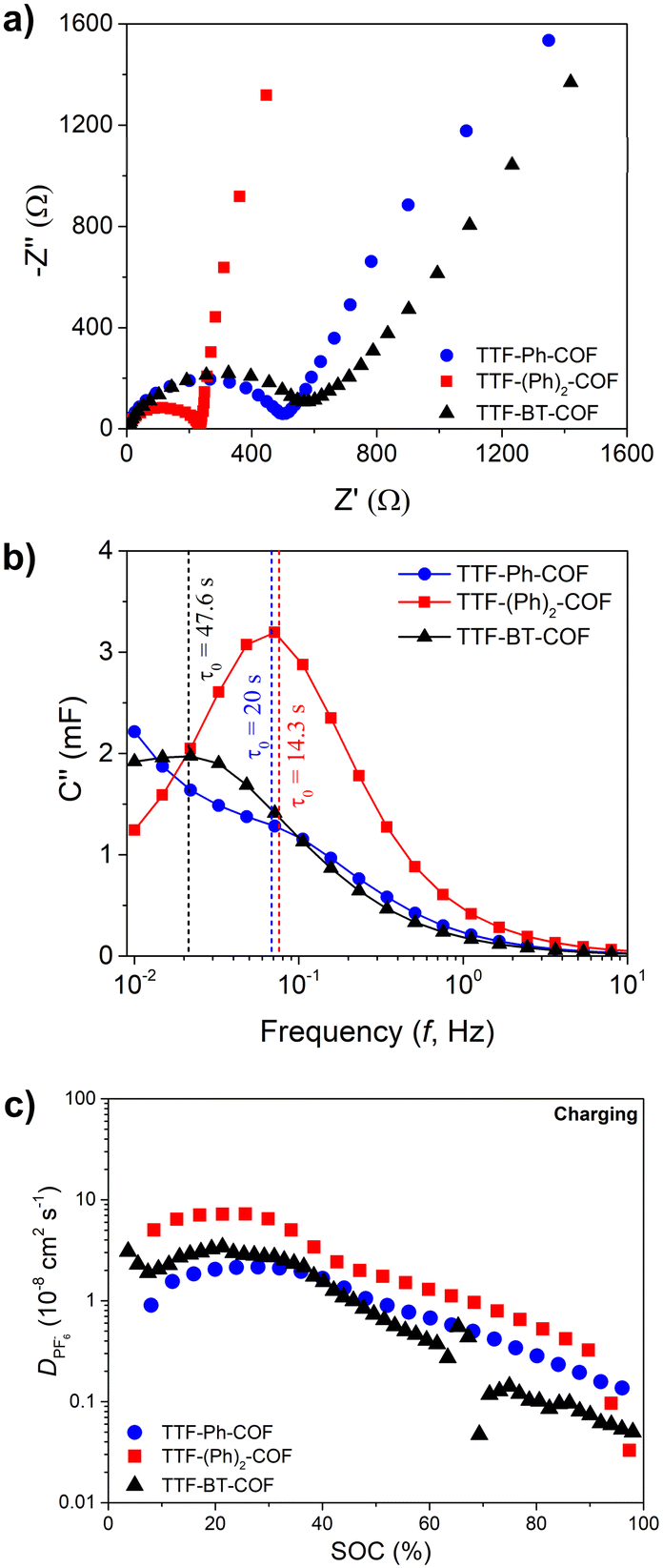 | ||
| Fig. 7 (a) Electrochemical impedance spectroscopy (EIS) results at 3.7 V (vs. Li/Li+) upon charging, (b) imaginary capacitance analysis and characteristic time, and (c) diffusion coefficients of the PF6− anion (charging) for TTF-Ph-COF (blue), TTF-(Ph)2-COF (red) and TTF-BT-COF (black). | ||
To further evaluate the performance of TTF-COF electrodes, the cycling stability of the three COFs was assessed by conducting GCD experiments at 2C rate, as shown in Fig. 8. The capacity retentions were calculated to be 83%, 84% and 76% for TTF-Ph-COF, TTF-(Ph)2-COF and TTF-BT-COF, respectively, after 400 cycles at 2C, showing relatively good cycling stability. The low coulombic efficiency during the first cycles can be due to parasitic reactions from the electrolyte at high potentials. Differential capacity analysis (dQ/dV) of the three TTF-COF electrodes (Fig. 8c) indicated that the first redox process is relatively more stable than the second one. To explore this behavior more thoroughly, additional electrochemical tests were performed on the TTF-Ph-COF, restricting the potential window to encompass only the first redox process (Fig. S42†). Evidently, the one-electron redox process exhibited better cycle stability, with no capacity decay observed over 500 cycles and quantitative CEs. In contrast, capacity retention was 85% over the same number of cycles with lower CEs when considering both redox processes. Interestingly, the comparative rate capability experiments also revealed better rate performance when limiting the potential window to the first redox process (Fig. S43†). The results indicate that the reversibility of the second redox process at high C-rates can be significantly compromised because such a redox process is too slow. This hypothesis is partially corroborated by the observed lower potential peak separation (Fig. 8c) and generally higher b-value (Table S6†) for the first redox process over the second, and higher anion diffusivity during charging, below 50% SOC (Fig. 7c). This diminished reversibility at high C-rates may be attributed to conformational changes that occur during the second oxidation of TTF radical cation moieties to the dication state, which normally adopts a twisted conformation.47 Volume expansion and increased charge repulsion in the dication state could also explain this behavior.
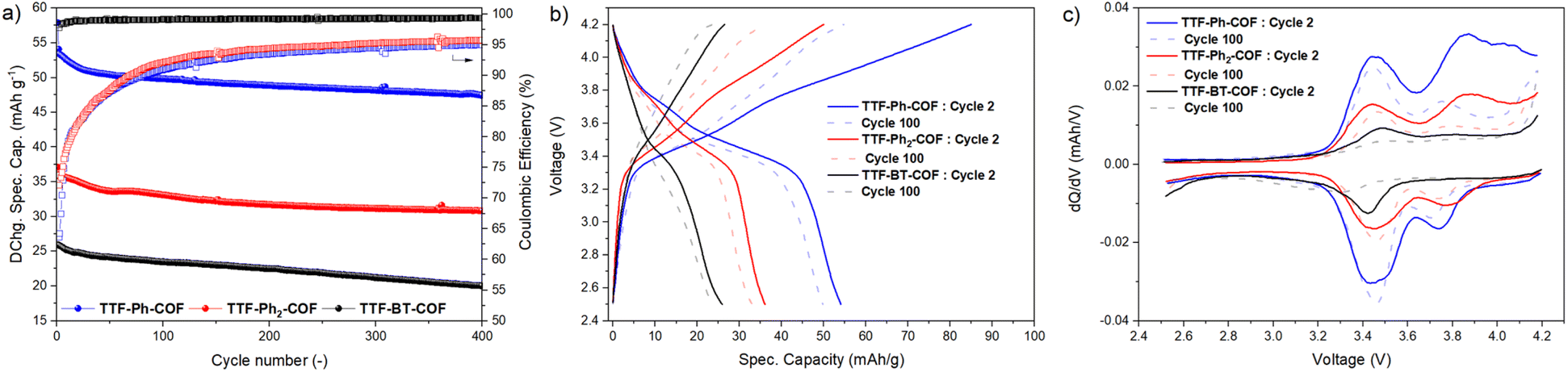 | ||
| Fig. 8 Galvanostatic charge–discharge (GCD) long cycling experiments. (a) Cyclic stability and coulombic efficiency (square symbols) of TTF-Ph-COF, TTF-(Ph)2-COF, and TTF-BT-COF during long-term cycling at 2C, using a potential window of 2.5–4.2 V. (b) Galvanostatic profiles of the 2nd and 100th cycles. (c) Differential capacity analysis (dQ/dV) of the 2nd and 100th cycles. | ||
Conclusions
In conclusion, the comprehensive study of TTF-COFs as p-type cathode materials has demonstrated their potential as high-voltage organic cathodes in lithium batteries. The series of TTF-COFs explored, comprising different organic linkers, exhibited significant effects on electrochemical properties and performance metrics. Structural analyses confirmed the crystalline nature of the COFs, with different linkers influencing the framework porosity and electronic structure. All three TTF-COFs show a high average discharge potential (∼3.6 V), being one of the highest for COF-based cathodes in lithium batteries,15 and good cycling stability (∼80% capacity retention after 400 cycles at 2C). Although TTF-BT-COF may have a redox-bipolar character, the n-type BT units could not be exploited which considerably decreases its specific capacity. The cathodes based on TTF-Ph-COF and TTF-(Ph)2-COF showed faster kinetics, lower resistance, and facile diffusion than BT-TTF-COF. Overall, TTF-Ph-COF showed the best electrochemical performance in terms of higher specific capacity, capacity retention, and faster kinetics. Our results show that increasing the pore size can be counterproductive if it does not have a significant effect on the kinetics and ion diffusion. Future research will focus on the incorporation of extended TTF derivatives with improved electrochemical performance as well as on the design of redox-bipolar COFs combining n- and p-type building blocks synergistically.Data availability
Data for this article, including PXRD, N2 isotherms, diffuse reflectance, IR, solid-state NMR, EPR, electrochemical raw data will be available at Zenodo.org repository (https://doi.org/10.5281/zenodo.13305166) as a source data files.Author contributions
G. V. and P. F. synthesized and characterized the materials. A. G, D. R.-S. and F. Z. performed the gas sorption measurements. R. G. and T. B. performed the electrical conductivity measurements. R. D., R. G., N. P. and R. M. performed the electrochemical studies. K. S. and M. M.-F. performed the computational modelling. M. S. designed and supervised the whole project. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Research Council (ERC) under the European Union’s Horizon Europe Framework Programme (ERC-2021-Starting Grant, grant agreement no. 101039748-ELECTROCOFS). This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020, UIDP/50011/2020, and LA/P/0006/2020, financed by national funds through the FCT/MEC (PIDDAC). The NMR spectrometers are part of the National NMR Network (PTNMR) and are partially supported by Infrastructure Project No. 022161 (co-financed by FEDER through COMPETE 2020 and POCI, PORL, and FCT through PIDDAC). We also thank FCT for funding the PTDC/QUI-ELT/2593/2021 and 2022.07534.CEECIND. G. V. and P. F. are grateful to FCT for their PhD grants (2020.08520.BD and UI/BD/151049/2021, respectively). This study was also funded by the PRR-Plano de Recuperação e Resiliência and by the NextGenerationEU funds at the University of Aveiro through the scope of the Agenda for Business Innovation “New Generation Storage” (project no. 58 with the application C644936001–00000045). This work has received financial support from the Xunta de Galicia (Centro de investigación do Sistema universitario de Galicia accreditation 2023–2027, ED431G 2023/03), the Oportunius program (Gain) and the European Union (European Regional Development Fund – ERDF). F. Z. acknowledges the support from the Spanish Ministry of Science, Innovation, and Universities through the “María de Maeztu” Programme for Units of Excellence in R&D (CEX2023-001316-M) and the financial support to the projects PDC2022-133498-I00, TED2021-129886B-C42, PID2022-138908NB-C31. Gratitude is also extended to the “(MAD2D-CM)-UAM” project funded by Comunidad de Madrid, the Recovery, Transformation, and Resilience Plan, NextGenerationEU from the European Union, and the European Innovation Council under grant Agreement 101047081 (EVA). T. B. acknowledges support from the SolTech network funded by the Free State of Bavaria, and funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2089/1 – 390776260. Authors also thank the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant agreement (Grant No. 860403) and Spanish Government; MCIN/AEI/10.13039/501100011033/FEDER “A way of making Europe” (PID2021-124974OB-C21 and IJC2020-043076-I-I) for the funding.Notes and references
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, aal1585 CrossRef PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. Feng, A. Thomas and D. Jiang, Nat. Rev. Methods Primers, 2023, 3, 1 CrossRef CAS.
- M. Souto, K. Strutyński, M. Melle-Franco and J. Rocha, Chem.–Eur. J., 2020, 26, 10912–10935 CrossRef CAS PubMed.
- H. Fan, A. Mundstock, A. Feldhoff, A. Knebel, J. Gu, H. Meng and J. Caro, J. Am. Chem. Soc., 2018, 140, 10094–10098 CrossRef CAS PubMed.
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- J. Guo and D. Jiang, ACS Cent. Sci., 2020, 6, 869–879 CrossRef CAS PubMed.
- M. D. Allendorf, R. Dong, X. Feng, S. Kaskel, D. Matoga and V. Stavila, Chem. Rev., 2020, 120, 8581–8640 CrossRef CAS PubMed.
- N. Keller and T. Bein, Chem. Soc. Rev., 2021, 50, 1813–1845 RSC.
- X. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, C. Schlesiger, S. Praetz, J. Schmidt and A. Thomas, J. Am. Chem. Soc., 2019, 141, 6623–6630 CrossRef CAS PubMed.
- K. Prakash, B. Mishra, D. D. Díaz, C. M. Nagaraja and P. Pachfule, J. Mater. Chem. A, 2023, 11, 14489–14538 RSC.
- T. Sun, J. Xie, W. Guo, D. S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1–23 Search PubMed.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- R. Dantas, C. Ribeiro and M. Souto, Chem. Commun., 2024, 60, 138–149 RSC.
- O. Lužanin, R. Dantas, R. Dominko, J. Bitenc and M. Souto, J. Mater. Chem. A, 2023, 11, 21553–21560 RSC.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- Z. Meng, Y. Zhang, M. Dong, Y. Zhang, F. Cui, T.-P. Loh, Y. Jin, W. Zhang, H. Yang and Y. Du, J. Mater. Chem. A, 2021, 9, 10661–10665 RSC.
- J. Sprachmann, T. Wachsmuth, M. Bhosale, D. Burmeister, G. J. Smales, M. Schmidt, Z. Kochovski, N. Grabicki, R. Wessling, E. J. W. List-Kratochvil, B. Esser and O. Dumele, J. Am. Chem. Soc., 2023, 145, 2840–2851 CrossRef CAS PubMed.
- N. Martín, Chem. Commun., 2013, 49, 7025–7027 RSC.
- Y. Chen, S. A. Freunberger, Z. Peng, O. Fontaine and P. G. Bruce, Nat. Chem., 2013, 5, 489–494 CrossRef CAS PubMed.
- M. Kato, K. Senoo, M. Yao and Y. Misaki, J. Mater. Chem. A, 2014, 2, 6747–6754 RSC.
- B. Häupler, R. Burges, C. Friebe, T. Janoschka, D. Schmidt, A. Wild and U. S. Schubert, Macromol. Rapid Commun., 2014, 35, 1367–1371 CrossRef PubMed.
- A. Yoshimura, K. Hemmi, H. Moriwaki, R. Sakakibara, H. Kimura, Y. Aso, N. Kinoshita, R. Suizu, T. Shirahata, M. Yao, H. Yorimitsu, K. Awaga and Y. Misaki, ACS Appl. Mater. Interfaces, 2022, 14, 35978–35984 CrossRef CAS PubMed.
- A. Yoshimura, M. Yoshinouchi, K. Hemmi, Y. Aso, R. Utsumi, T. Shirahata, M. Yao and Y. Misaki, New J. Chem., 2023, 47, 11760–11764 RSC.
- H. Ding, Y. Li, H. Hu, Y. Sun, J. Wang, C. Wang, C. Wang, G. Zhang, B. Wang, W. Xu and D. Zhang, Chem.–Eur. J., 2014, 20, 14614–14618 CrossRef CAS PubMed.
- S. Jin, T. Sakurai, T. Kowalczyk, S. Dalapati, F. Xu, H. Wei, X. Chen, J. Gao, S. Seki, S. Irle and D. Jiang, Chem.–Eur. J., 2014, 20, 14608–14613 CrossRef CAS PubMed.
- S. L. Cai, Y. B. Zhang, A. B. Pun, B. He, J. Yang, F. M. Toma, I. D. Sharp, O. M. Yaghi, J. Fan, S. R. Zheng, W. G. Zhang and Y. Liu, Chem. Sci., 2014, 5, 4693–4700 RSC.
- H. Li, J. Chang, S. Li, X. Guan, D. Li, C. Li, L. Tang, M. Xue, Y. Yan, V. Valtchev, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2019, 141, 13324–13329 CrossRef CAS PubMed.
- Q. Wu, R. K. Xie, M. J. Mao, G. L. Chai, J. D. Yi, S. S. Zhao, Y. B. Huang and R. Cao, ACS Appl. Mater. Interfaces, 2020, 1005–1012 CAS.
- J. Chang, H. Li, J. Zhao, X. Guan, C. Li, G. Yu, V. Valtchev, Y. Yan, S. Qiu and Q. Fang, Chem. Sci., 2021, 12, 8452–8457 RSC.
- Y. Li, T. Wei, C. Liu, Z. Zhang, L. Wu, M. Ding, S. Yuan, J. Zhu and J. Zuo, Chem.–Eur. J., 2023, 29, e202301048 CrossRef CAS PubMed.
- J. W. M. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider, R. Ameloot, J. Marreiros, C. Ania, D. Azevedo, E. Vilarrasa-Garcia, B. F. Santos, X. Bu, Z. Chang, H. Bunzen, N. R. Champness, S. L. Griffin, B. Chen, R. Lin, B. Coasne, S. Cohen, J. C. Moreton, Y. J. Colón, L. Chen, R. Clowes, F. Coudert, Y. Cui, B. Hou, D. M. D'Alessandro, P. W. Doheny, M. Dincă, C. Sun, C. Doonan, M. T. Huxley, J. D. Evans, P. Falcaro, R. Ricco, O. Farha, K. B. Idrees, T. Islamoglu, P. Feng, H. Yang, R. S. Forgan, D. Bara, S. Furukawa, E. Sanchez, J. Gascon, S. Telalović, S. K. Ghosh, S. Mukherjee, M. R. Hill, M. M. Sadiq, P. Horcajada, P. Salcedo-Abraira, K. Kaneko, R. Kukobat, J. Kenvin, S. Keskin, S. Kitagawa, K. Otake, R. P. Lively, S. J. A. DeWitt, P. Llewellyn, B. V. Lotsch, S. T. Emmerling, A. M. Pütz, C. Martí-Gastaldo, N. M. Padial, J. García-Martínez, N. Linares, D. Maspoch, J. A. Suárez del Pino, P. Moghadam, R. Oktavian, R. E. Morris, P. S. Wheatley, J. Navarro, C. Petit, D. Danaci, M. J. Rosseinsky, A. P. Katsoulidis, M. Schröder, X. Han, S. Yang, C. Serre, G. Mouchaham, D. S. Sholl, R. Thyagarajan, D. Siderius, R. Q. Snurr, R. B. Goncalves, S. Telfer, S. J. Lee, V. P. Ting, J. L. Rowlandson, T. Uemura, T. Iiyuka, M. A. van der Veen, D. Rega, V. Van Speybroeck, S. M. J. Rogge, A. Lamaire, K. S. Walton, L. W. Bingel, S. Wuttke, J. Andreo, O. Yaghi, B. Zhang, C. T. Yavuz, T. S. Nguyen, F. Zamora, C. Montoro, H. Zhou, A. Kirchon and D. Fairen-Jimenez, Adv. Mater., 2022, 34, 2201502 CrossRef CAS PubMed.
- L. Sarkisov, R. Bueno-Perez, M. Sutharson and D. Fairen-Jimenez, Chem. Mater., 2020, 32, 9849–9867 CrossRef CAS.
- D. Zhu, J.-J. Zhang, X. Wu, Q. Yan, F. Liu, Y. Zhu, X. Gao, M. M. Rahman, B. I. Yakobson, P. M. Ajayan and R. Verduzco, Chem. Sci., 2022, 13, 9655–9667 RSC.
- S. Wang, Q. Sun, W. Chen, Y. Tang, B. Aguila, Y. Pan, A. Zheng, Z. Yang, L. Wojtas, S. Ma and F.-S. Xiao, Matter, 2020, 2, 416–427 CrossRef CAS.
- M. Souto and D. F. Perepichka, J. Mater. Chem. C, 2021, 9, 10668–10676 RSC.
- J. M. Rotter, R. Guntermann, M. Auth, A. Mähringer, A. Sperlich, V. Dyakonov, D. D. Medina and T. Bein, Chem. Sci., 2020, 11, 12843–12853 RSC.
- J. S. Chappell, A. N. Bloch, W. A. Bryden, M. Maxfield, T. O. Poehler and D. O. Cowan, J. Am. Chem. Soc., 1981, 103, 2442–2443 CrossRef CAS.
- L. Chen, C. R. Bridges, G. Gao, T. Baumgartner and X. He, ACS Appl. Energy Mater., 2019, 2, 7315–7320 CrossRef CAS.
- G. B. Wang, S. Li, C. X. Yan, Q. Q. Lin, F. C. Zhu, Y. Geng and Y. Bin Dong, Chem. Commun., 2020, 56, 12612–12615 RSC.
- W. Chen, Z. Yang, Z. Xie, Y. Li, X. Yu, F. Lu and L. Chen, J. Mater. Chem. A, 2019, 7, 998–1004 RSC.
- J. N. Chang, Q. Li, J. W. Shi, M. Zhang, L. Zhang, S. Li, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202218868 CrossRef CAS PubMed.
- A. Molina, N. Patil, E. Ventosa, M. Liras, J. Palma and R. Marcilla, ACS Energy Lett., 2020, 5, 2945–2953 CrossRef CAS.
- N. Patil, A. Aqil, F. Ouhib, S. Admassie, O. Inganäs, C. Jérôme and C. Detrembleur, Adv. Mater., 2017, 29, 1703373 CrossRef PubMed.
- M. Wu, Y. Zhao, R. Zhao, J. Zhu, J. Liu, Y. Zhang, C. Li, Y. Ma, H. Zhang and Y. Chen, Adv. Funct. Mater., 2022, 32, 1–8 Search PubMed.
- H. V. Schröder and C. A. Schalley, Beilstein J. Org. Chem., 2018, 14, 2163–2185 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General methods and materials, synthesis and characterization of TTF-COFs, computational modelling, additional electrochemical characterisation. See DOI: https://doi.org/10.1039/d4ta04576a |
| This journal is © The Royal Society of Chemistry 2024 |