
Converting the covalent organic framework linkage from hydrazone to thiadiazole toward blue light-powered selective conversion of organic sulfides†
Yuexin
Wang‡
a,
Ji-Long
Shi‡
b,
Xiaoyun
Dong
a,
Fulin
Zhang
a and
Xianjun
Lang
 *a
*a
aHubei Key Lab on Organic and Polymeric Optoelectronic Materials, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China. E-mail: xianjunlang@whu.edu.cn
bHubei Provincial Engineering Research Center of Racing Horse Detection and Application Transformation, Wuhan Business University, Wuhan 430056, China
First published on 13th August 2024
Abstract
Covalent organic frameworks (COFs) are composed of various organic linkers with dynamic covalent bonds as linkages, which, in part, establish functionality. Nevertheless, these covalent bonds are usually not robust enough under reaction conditions and, therefore, can be converted into irreversible ones because the application of COFs hinges on the characteristics of these linkages. In this work, TFPT-COF, a triazine-based COF with a hydrazone linkage, is constructed. Subsequently, the hydrazone linkage is converted to a thiadiazole linkage through thionation and oxidative cyclization at 115 °C, converting TFPT-COF to TDA-COF with a robust and irreversible thiadiazole linkage. The band gap is narrowed, and the electronic structure is altered from TFPT-COF to TDA-COF based on density functional theory calculations. Besides, experimental results suggest that TDA-COF possesses broadened light absorption and improved optoelectronic properties compared to TFPT-COF. Converting TFPT-COF to TDA-COF significantly shifts the photocatalytic activity for the selective conversion of thioanisoles to sulfoxides with oxygen (O2). TDA-COF drives the blue light-powered conversion of thioanisoles to sulfoxides with O2, whereas TFPT-COF shows almost no activity. TDA-COF is especially accessible for converting various sulfides to sulfoxides with high selectivities, while maintaining high photocatalytic activity and stability over six cycles. This work demonstrates that converting the dynamic linkages of COFs to irreversible ones contributes positively to photocatalytic activities.
1. Introduction
Covalent organic frameworks (COFs) have flourished as promising materials with distinct properties such as high stability, well-defined crystallinity, porous networks, and predictable topologies.1–5 Owing to sufficient flexibility and tunability in building units, the diverse functions of COFs have been exploited for various practical applications,6 such as adsorption,7,8 optoelectronics,9,10 catalysis,11 separation,12,13 ionic conduction,14,15 biomedicine,16 and energy conversion and storage.17,18 Notably, sustainable and environmentally benign visible light photocatalysis over COFs contributes to photodynamic therapy, wastewater purification, and chemical conversion.19–22 COFs are composed of various functional linkers connected by relatively strong and covalent linkages that determine the properties and functionalities of COFs. Generally, the linkages of COFs play a critical role in defining stability and crystallinity. Both are prerequisites for practical application as a photocatalyst.23–25Hitherto, COFs with imine linkages have been elegantly designed26,27 and generally utilized in photocatalysis.28,29 The reversibility of the imine linkage makes it easy to construct COFs with high crystallinity. Nevertheless, the intrinsic high polarization of imine linkages leads to interrupted π-conjugation, restricting the visible light absorption and optoelectronic property. Additionally, the reversibility of these imine linkages, in turn, compromises the chemical stability of COFs, resulting in the limitation of photocatalysis under prolonged light irradiation. To overcome the issues associated with imine, β-ketoenamine,30,31 azine,32,33 and hydrazone34,35 linkages for COFs have been investigated for photocatalysis. Among these, the hydrazone linkage, constructed from an aldehyde and a substituted acyl hydrazine, possess more heteroatomic sites and higher hydrolytic stability.36 Notably, the electronegativity and robust hydrogen-bonding interactions inherent in hydrazone linkages have been incorporated into COFs, enhancing their photocatalytic activities.37 Consequently, COFs with hydrazone linkages have been reported as photocatalysts for H2 evolution,35 CO2 reduction,38 H2O2 production,39 and selective organic conversions.40
TFPT-COF, a triazine-based COF with a hydrazone linkage, is the first COF to be adopted in photocatalysis.41 Notably, TFPT-COF cannot drive blue light-powered photocatalytic organic conversions. Likewise, the intrinsic non-conjugation in the hydrazone linkage hinders efficient charge carrier separation and migration over COFs.42 Intriguingly, the versatility of the chemical motif–NH–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– makes hydrazone linkages highly promising for conversion. Besides, based on the dual electrophilicity and nucleophilicity of carbon atoms,43 hydrazone has been widely exploited in multiple syntheses. Therefore, the chemical conversion of hydrazone linkages is accessible to break the limitation of intrinsic non-conjugation toward synthesizing diverse COFs. In addition, converting a hydrazone linkage into an aromatic heterocycle further contributes to the stability of the COF. Notably, the two N atoms in the hydrazone linkage facilitate the conversion in a COF, allowing the incorporation of thiadiazole to further enhance photocatalytic activities.
CH– makes hydrazone linkages highly promising for conversion. Besides, based on the dual electrophilicity and nucleophilicity of carbon atoms,43 hydrazone has been widely exploited in multiple syntheses. Therefore, the chemical conversion of hydrazone linkages is accessible to break the limitation of intrinsic non-conjugation toward synthesizing diverse COFs. In addition, converting a hydrazone linkage into an aromatic heterocycle further contributes to the stability of the COF. Notably, the two N atoms in the hydrazone linkage facilitate the conversion in a COF, allowing the incorporation of thiadiazole to further enhance photocatalytic activities.
Herein, through converting the hydrazone linkage of TFPT-COF to a thiadiazole linkage with Lawesson's reagent as a mild and convenient thionating agent, TDA-COF can be afforded. The electronic structure and band gap of TDA-COF are altered by the fully conjugated planar motif and unique optical property of thiadiazole. Thus, TDA-COF possesses a higher tendency of charge generation and separation and a lower impedance than TFPT-COF. Besides, TFPT-COF and TDA-COF are explored for the blue light-powered selective conversion of organic sulfides with oxygen (O2), in which TDA-COF shows superior photocatalytic activity with satisfactory selectivity. Notably, both singlet oxygen (1O2) and superoxide (O2˙−) as the reactive oxygen species (ROS) significantly contribute to the blue light-powered selective conversion of thioanisoles over TDA-COF. The generations of 1O2 and O2˙− are assigned to energy and electron transfer pathways, respectively. This work highlights that converting the linkages of COFs provides an access to enhanced photocatalysis.
2. Results and discussion
TFPT-COF was assembled through the condensation reaction between 1,3,5-tris-(4-formyl-phenyl)triazine (TFPT) and 2,5-diethoxyterephthalohydrazide (DETH) in the presence of acetic acid.41 TDA-COF was synthesized by converting the hydrazone linkage of TFPT-COF into a thiadiazole linkage through thionation and oxidative cyclization.44 This linkage conversion utilized Lawesson's reagent to enable thionation and oxidative cyclization at a temperature of 115 °C. The schematic representations of TFPT-COF and TDA-COF are discernibly illustrated (Fig. 1).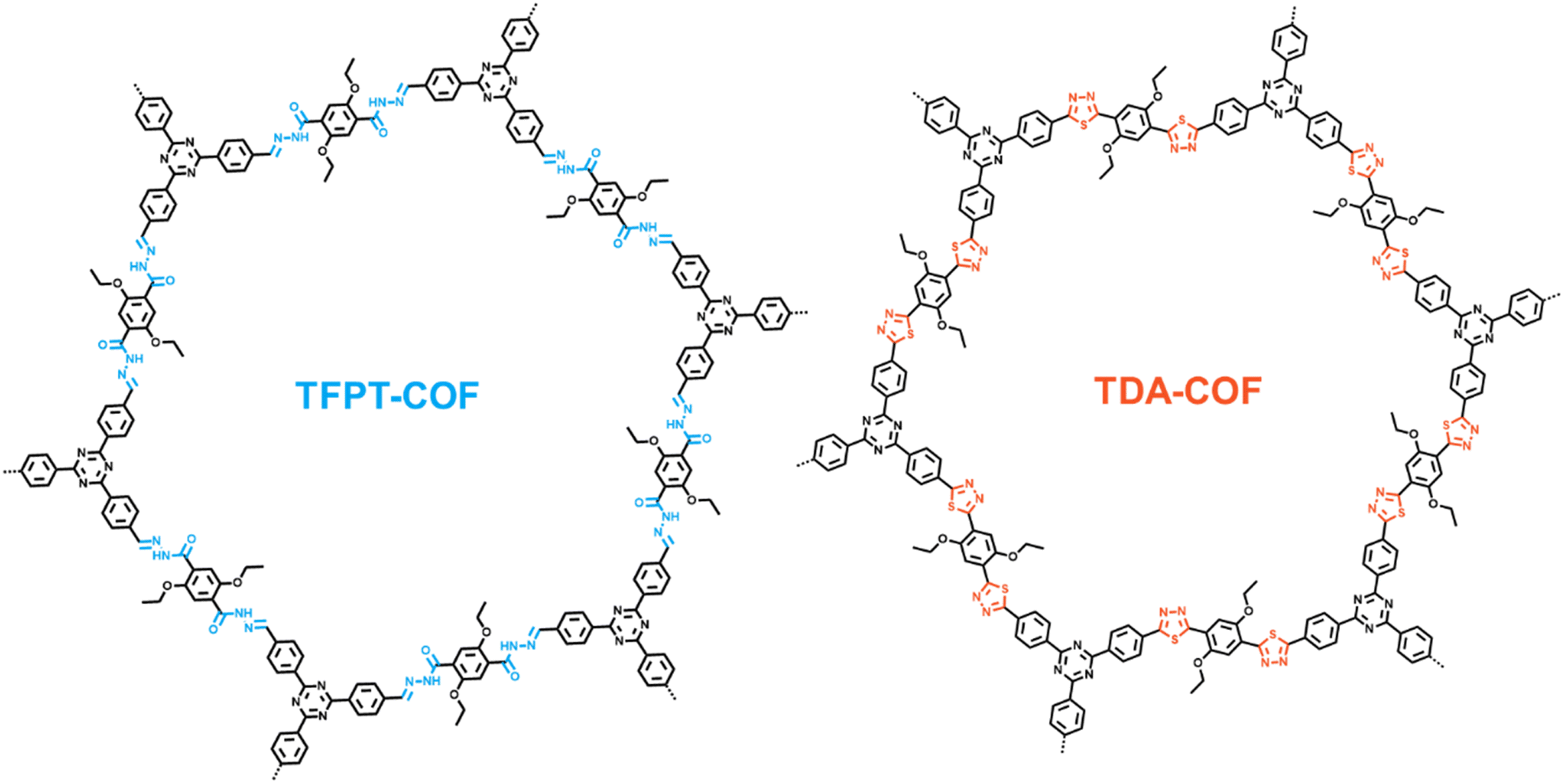 | ||
| Fig. 1 The schematic representations of TFPT-COF and TDA-COF. | ||
The electronic structures of TFPT-COF and TDA-COF were explored through density functional theory (DFT) calculations. The density of states (DOS) of their fragments are depicted in Fig. 2a and b. It can be found that the electronic structure of TFPT-COF was altered after converting the linkage from hydrazone to thiadiazole.45 In detail, both the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) of TFPT-COF are primarily located on the acylhydrazone group (Fig. 2a, fragment 1). Besides, the contribution of triazine (Fig. 2a, fragment 2) concentrates on LUMO+2. Diethoxybenzene (Fig. 2a, fragment 3) also makes a moderate contribution to the LUMO and a major contribution to HOMO−2. After converting the linkage from hydrazone to thiadiazole, the π-conjugation was enhanced, resulting in a narrower HOMO–LUMO gap of TDA-COF than that of TFPT-COF. The LUMO of TDA-COF incorporates contributions from thiadiazole and diethoxybenzene. Similar to TFPT-COF, the contribution of triazine (Fig. 2b, fragment 2) concentrates on LUMO+2 in TDA-COF. Different from TFPT-COF, the contribution of diethoxybenzene (Fig. 2b, fragment 3) is also located on the HOMO after the incorporation of thiadiazole (Fig. 2b, fragment 1) in TDA-COF. Indeed, diethoxybenzene makes a major contribution to the HOMO. It reveals that converting the linkage from hydrazone to thiadiazole altered the electronic structure obviously and consequently narrowed the band gap of the COF.46
 | ||
| Fig. 2 The DOS of TFPT-COF (a) and TDA-COF (b), and local molecular orbital distribution of the corresponding fragment. The UV-visible DRS (c) and Tauc plots (d) of TFPT-COF and TDA-COF. | ||
UV-visible DRS (diffuse reflectance spectra) were recorded to assess the optical property of TFPF-COF and TDA-COF. As depicted in Fig. 2c, converting TFPT-COF into TDA-COF extended the light absorption range from 450 to 600 nm derived from the UV-visible DRS. The Tauc plots of TFPT-COF and TDA-COF were obtained (Fig. 2d). Furthermore, the optical band gaps were determined to be 2.89 eV for TFPT-COF and 2.53 eV for TDA-COF, respectively. Converting the COF linkage from hydrazone to thiadiazole provides an access to an extended light absorption range and a narrower optical band gap.
The chemical structures of linkages and corresponding 13C solid-state nuclear magnetic resonance (NMR) spectra of TFPT-COF and TDA-COF were compared (Fig. 3a and b). After converting the COF linkage from hydrazone to thiadiazole, two new peaks appeared at 162.2 and 161.0 ppm in TDA-COF, assigned to thiadiazole carbons. Correspondingly, the disappearance of the peaks at 159.4 ppm (CO) and 150.2 ppm (CN) in TFPT-COF was observed, suggesting that hydrazone linkages were converted into thiadiazole linkages.
 | ||
| Fig. 3 The chemical structures of linkages (a), 13C CP-MAS NMR spectra (b), and FTIR spectra (c) of TFPT-COF and TDA-COF. | ||
The construction of TFPT-COF and TDA-COF was further verified using Fourier-transform infrared (FTIR) spectra (Fig. 3c and S1†). The disappearance of aldehyde carbonyl stretching vibration at 1710 cm−1 in the FTIR spectra of TFPT illustrated the complete condensation of TFPT and DETH. Besides, the peaks at 1663 and 1640 cm−1 in the FTIR spectra of TFPT-COF and TDA-COF, respectively, corresponded to the carbon atom of the CN bonds. The new peak at 653 cm−1 was attributed to the C–S bond, corresponding to the thiadiazole linkages.47 Moreover, the characteristic peak of the triazine group at 804 cm−1 was present in TFPT-COF and TDA-COF.
Powder X-ray diffraction (PXRD) was carried out to demonstrate the crystallinity of TFPT-COF and TDA-COF (Fig. 4a and b). The excellent crystallinity of TFPT-COF and TDA-COF was verified by a prominent peak of the (100) facet at 2.42° and 2.36°, respectively. Compared with the simulated results, TFPT-COF and TDA-COF were assigned to AA stacking. It revealed that converting the linkage from hydrazone to thiadiazole did not change the stackings of COFs. Besides, the COFs still possessed good crystallinity after converting the linkage, which was attested by incisive peaks in the PXRD pattern of TDA-COF. Notably, converting linkages leads to a degree of deformation of principal pores, which was attested by a slight change in the relative intensity of the peak of the (200) facet.48
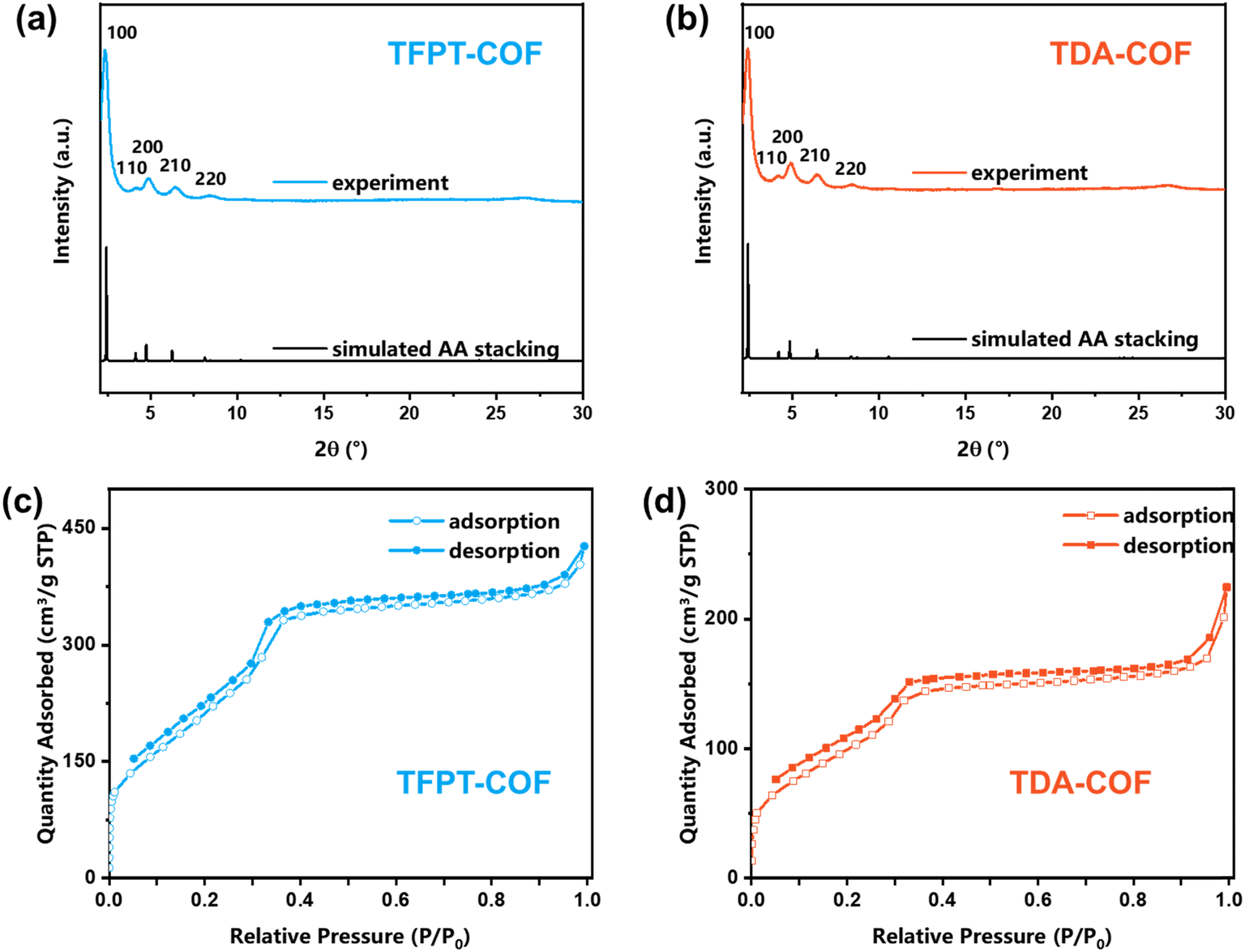 | ||
| Fig. 4 The PXRD patterns (a and b) and the N2 sorption isotherms (c and d) of TFPT-COF and TDA-COF. | ||
The porosity endows COFs with great adsorption capacity for various applications,49 which can be evaluated using N2 sorption isotherms at 77 K (Fig. 4c and d). A characteristic slope at P/P0 = 0.05–0.3 in the isotherms indicated the mesoporosity of TFPT-COF and TDA-COF, which was described as type-IV shape. The specific surface areas of TFPT-COF and TDA-COF were determined to be 996 and 402 m2 g−1, respectively. The reduction in specific surface areas was ascribed to the reduction of crystallinity and the increase of the intrinsic framework mass. Moreover, the pore size distributions of TFPT-COF and TDA-COF were evaluated using DFT fitting isotherm plots (Fig. S2†). It is revealed that the pore sizes of TFPT-COF and TDA-COF are distributed at about 3.2 nm based on their similar frameworks.
Field-emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM) were used to characterize the morphologies of TFPT-COF and TDA-COF. As shown in Fig. 5 and S3,† both TFPT-COF and TDA-COF exhibited clearly cross-linked rodlike structures. After converting TFPT-COF to TDA-COF through linkage conversion, the rod-like structure of TDA-COF appeared with a slightly smoother surface and a smaller diameter than that of TFPT-COF. Besides, the elemental mappings were explored using energy dispersive spectroscopy (EDS), which suggested that C and N elements were uniformly distributed in TFPT-COF and TDA-COF. Notably, S element only appeared in the elemental mapping of TDA-COF with a uniform distribution, revealing the successful conversion of TFPT-COF to TDA-COF. Besides, the elemental analysis (EA) results showed the content of S element before and after linkage conversion (Table S1†), further suggesting the construction of thiadiazole linkages in TDA-COF.
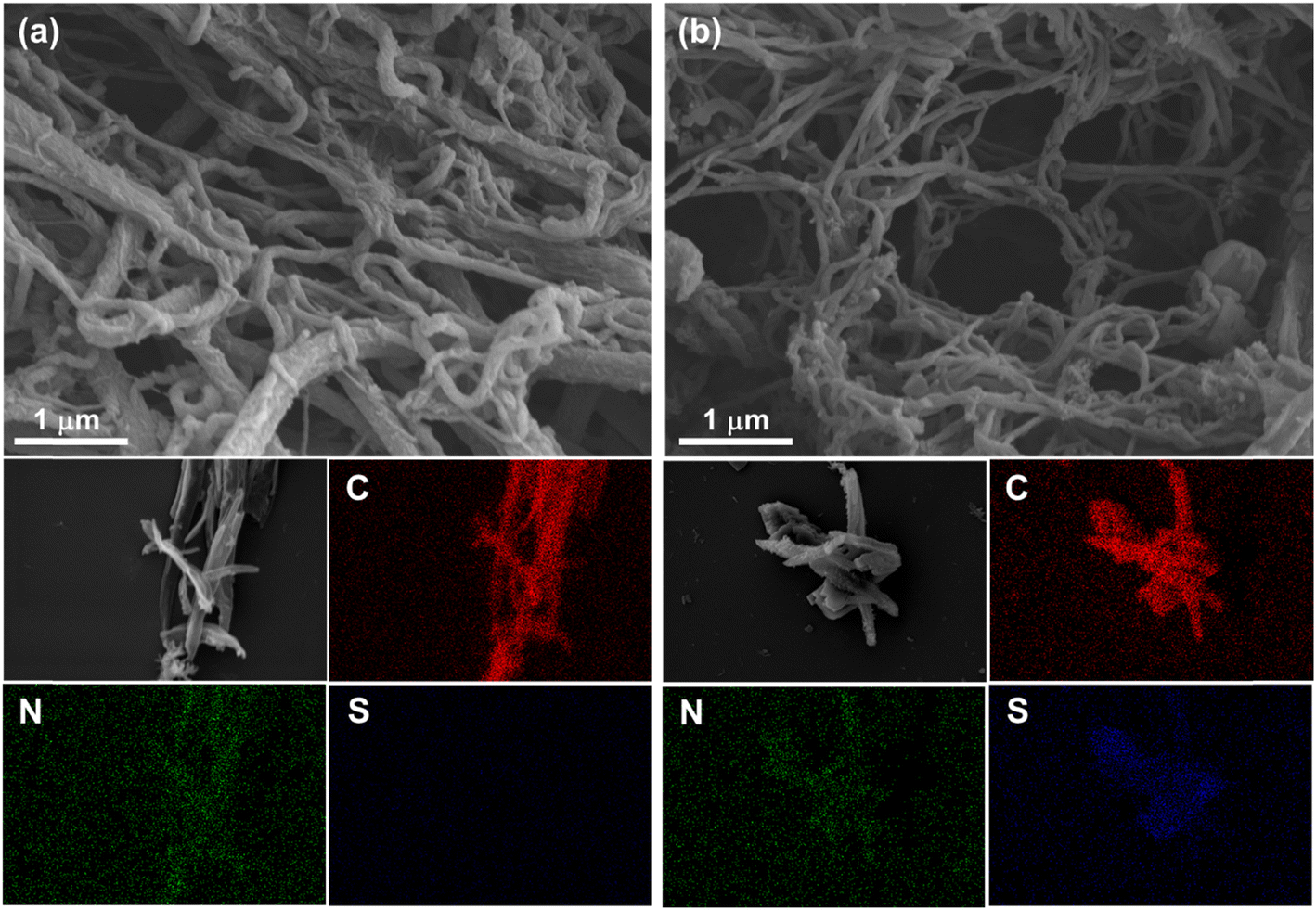 | ||
| Fig. 5 The SEM images and corresponding elemental mappings of TFPT-COF (a) and TDA-COF (b). | ||
The Mott–Schottky plots were obtained to assess the LUMO potentials of COFs (Fig. 6a and b). The LUMO potentials of TFPT-COF and TDA-COF were −0.88 and −1.22 V (vs. Ag/AgCl), respectively.50 In contrast, the LUMO potentials of the two COFs were more negative than the reduction potential of O2 (O2/O2˙− = −0.48 V vs. Ag/AgCl), suggesting that TFPT-COF and TDA-COF facilitated the electron transfer to O2 to afford O2˙−. Notably, the n-type semiconductor character of both TFPT-COF and TDA-COF was verified through positive Mott–Schottky plot slopes.
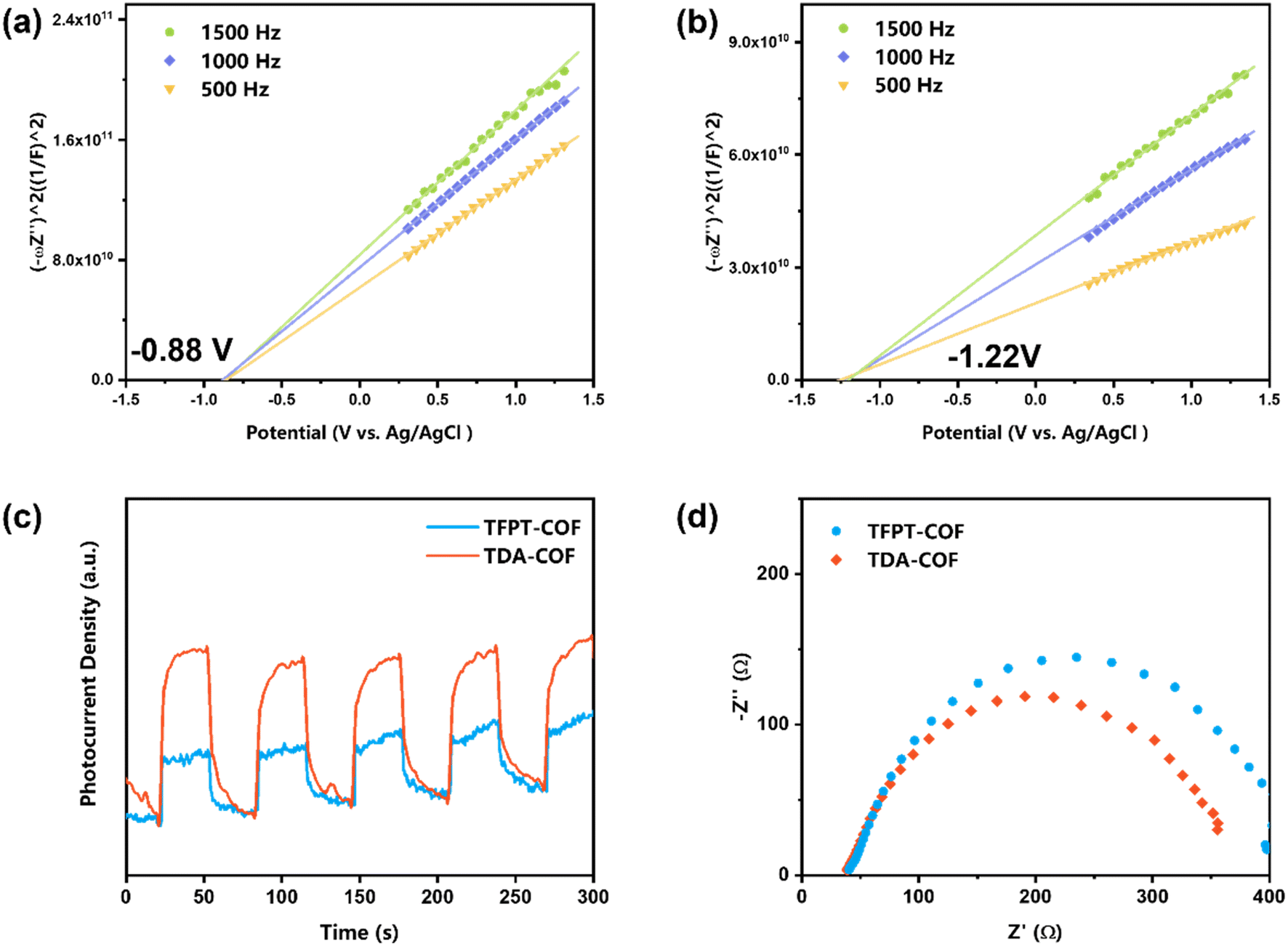 | ||
| Fig. 6 The Mott–Schottky plots of TFPT-COF (a) and TDA-COF (b). The transient photocurrent densities (c) and the EIS (d) of TFPT-COF and TDA-COF. | ||
Furthermore, to investigate the optoelectronic properties of TFPT-COF and TDA-COF, electrochemical impedance spectroscopy (EIS) and transient photocurrent measurements were performed (Fig. 6c and d). Notably, the photocurrent density of TDA-COF was much higher than that of TFPT-COF, which is indicative of superior charge separation efficiency of TDA-COF. Besides, compared with TFPT-COF, the EIS results attested to the lower charge transfer resistance of TDA-COF according to the smaller Nyquist arc radius. The electrochemical measurements revealed that converting the COF linkage from hydrazone to thiadiazole enhanced the optoelectronic properties patently.
The features mentioned above indicate that converting the COF linkage from hydrazone to thiadiazole ameliorates the optoelectronic properties and expands the light absorption range. Thus, these characteristics, including the capacity for the reduction of O2, make TDA-COF a promising candidate for photocatalytic oxidation with O2. Many organic conversions can be realized through photocatalysis as green and energy-efficient alternatives.51,52 The selective conversion of sulfides is an accessible method to obtain sulfoxides and has crucial applications in the pharmaceutical and agricultural industries.53–57 Herein, the selective conversion of thioanisoles to sulfoxides under blue light irradiation was exploited to evaluate the photocatalytic activities of TFPT-COF and TDA-COF (Fig. 7). When employing TDA-COF as a photocatalyst for selective conversion, thioanisoles can be converted into the corresponding sulfoxides with satisfactory selectivity. However, whether with electron-withdrawing or electron-donating para-substituent groups, thioanisole could not be converted over TFPT-COF under the same conditions. The peak wavelength (λp) of the blue light-emitting diode (LED) was determined to be 460 nm (Fig. S4†). However, the broad optical gap of TFPT-COF implied negligible response to the blue LED irradiation. There was almost no photocatalytic activity for the blue light-powered selective conversion of thioanisole over TFPT-COF. Additionally, TFPT and DETH showed no photocatalytic activity for the selective conversion of thioanisole (Table S2†). Thus, it can be concluded that converting the COF linkage from hydrazone to thiadiazole contributes positively to photocatalytic activities.
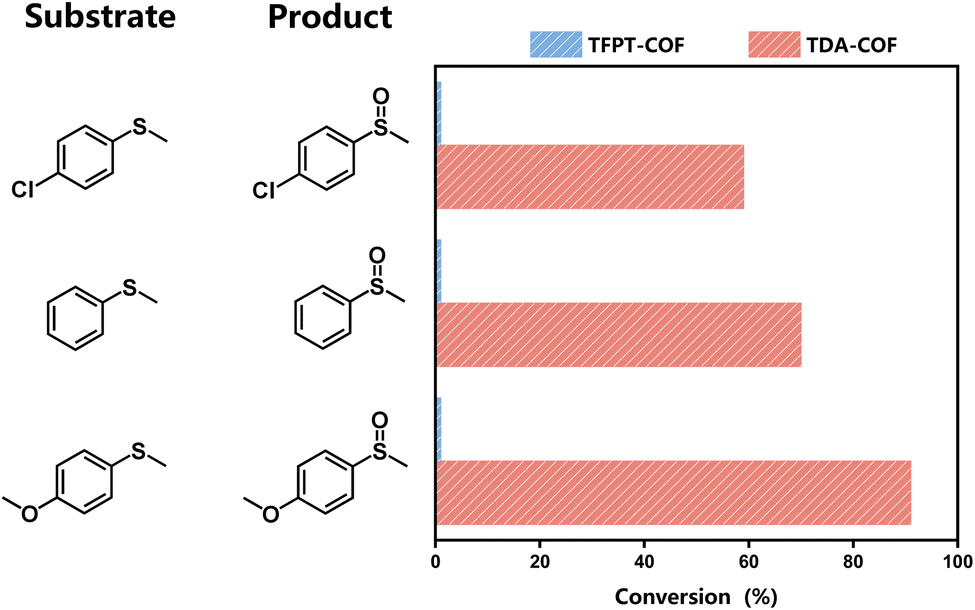 | ||
| Fig. 7 The comparison of TFPT-COF and TDA-COF for the blue light-powered selective conversion of thioanisole with O2. Reaction conditions: thioanisole (0.3 mmol), CH3OH (1 mL), photocatalyst (4 mg), O2 (1 atm), blue LED irradiation (460 ± 10 nm), and 15 min. | ||
The light source is crucial for the photocatalytic conversion of thioanisole, so it is essential to investigate the influence of λp of LEDs (Fig. S5†). Corresponding well with the UV-vis DRS (Fig. 2), there was almost no conversion of thioanisole under red, yellow, or green light irradiation. In contrast, blue light irradiation prominently improved the conversion of thioanisole. Besides, violet light irradiation further accelerated the conversion of thioanisole. Owing to the suitable conversion (69%) of thioanisole within 15 min, blue LEDs were selected as the desired light source.
The stability and reusability of TDA-COF were confirmed by recycling experiments (Fig. 8a), which is a prerequisite for practical application as a photocatalyst.51,58 Throughout 6 cycles, the blue light-powered conversion of thioanisole remained consistent in activity and selectivity, suggesting that the thiadiazole linkage endows TDA-COF with enough stability for photocatalytic conversion. Significantly, the recycled TDA-COF preserved the high crystallinity of TDA-COF (Fig. S6†). Besides, the similar rod-like structure observed in the SEM image of the recycled TDA-COF further confirmed the stability (Fig. S7†). Meanwhile, the kinetic experiment of the blue light-powered selective conversion of thioanisole over TDA-COF was conducted. As depicted in Fig. 8b, the concentration of thioanisole nearly retained a linear relationship with reaction time (t), corresponding well with a zero-order reaction. Besides, the reaction of blue light-powered selective conversion of thioanisole over TDA-COF was completed less than 25 min, emphasizing the outstanding activity.
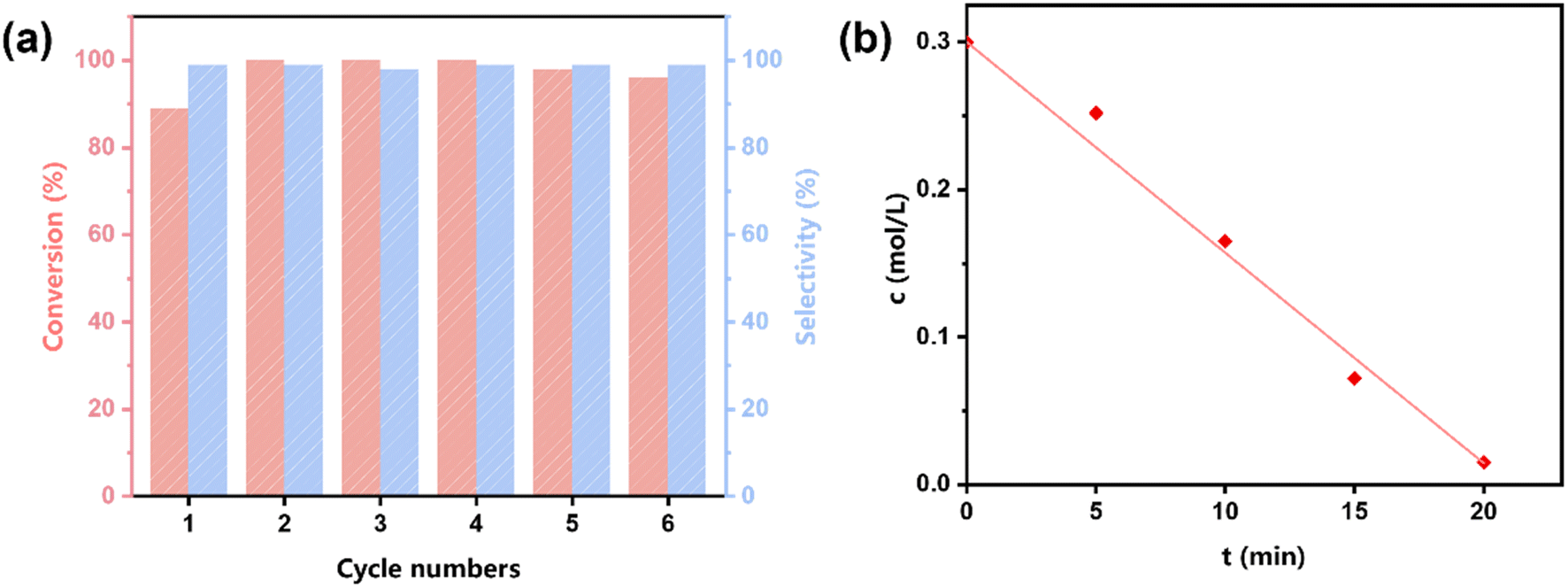 | ||
| Fig. 8 The recycling result (a) and kinetic curve (b) of TDA-COF for the blue light-powered selective conversion of thioanisole with O2. Reaction conditions: thioanisole (0.3 mmol), CH3OH (1 mL), O2 (1 atm), TDA-COF (4 mg), blue LED irradiation (460 ± 10 nm) and 20 min. | ||
Identifying ROS is crucial to understanding the reaction mechanism.59–64 Thus, the ROS involved in the blue light-powered selective conversion of thioanisole over TDA-COF were investigated through quenching experiments (Fig. 9a). Adding KI as the h+ (hole) scavenger prevented the conversion of thioanisole, emphasizing the significance of the photo-induced h+. Besides, O2 as an oxidizing agent is crucial for this reaction, which was attested by the inhibitory effect of replacing O2 with N2. The capture of O2˙− or 1O2 hampered the reaction markedly, suggesting that the ROS in the conversion of thioanisole were O2˙− and 1O2. By prolonging the lifetime of 1O2, CD3OD improved the conversion of thioanisole, further illustrating that 1O2 as the ROS contributes to the reaction. TDA-COF is fundamentally an organic material. However, the large π-conjugation of the 2D COF ensures the stability of TDA-COF under the attack of ROS.65 Thus, organic sulfides, rather than TDA-COF, are more inclined to be converted by ROS. The importance of photo-induced electron (e−) was investigated by adding AgNO3 as a scavenger of e−, further suppressing the conversion of thioanisole.
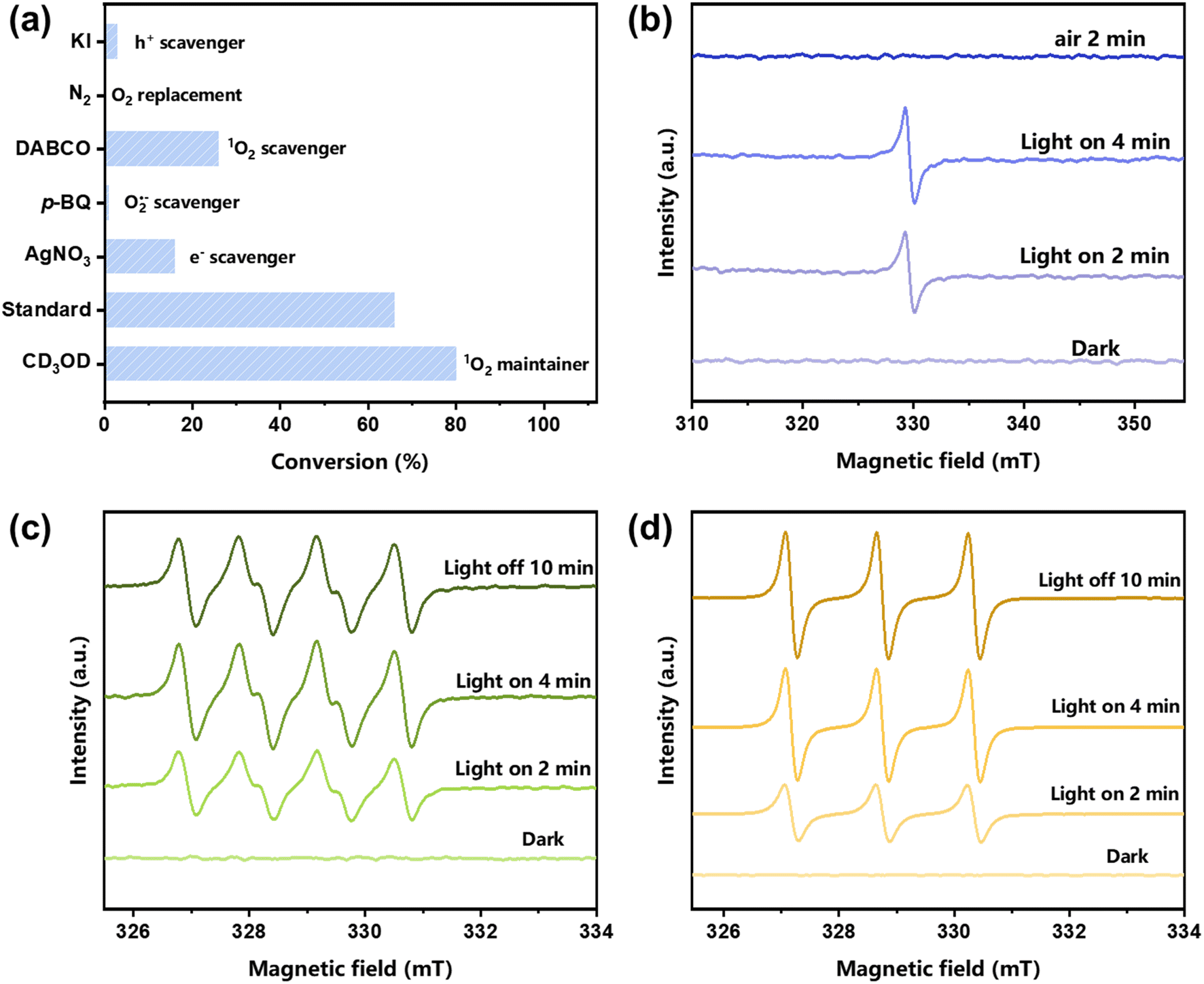 | ||
| Fig. 9 (a) The quenching experiments of TDA-COF for the blue light-powered selective conversion of thioanisole. Reaction conditions: thioanisole (0.3 mmol), CH3OH (1 mL), TDA-COF (4 mg), O2 (1 atm), blue LED irradiation (460 ± 10 nm), and 15 min. The EPR spectra of e− (b), O2˙− trapped with DMPO (c), and 1O2 trapped with 4-oxo-TMP (d). | ||
The mechanism of the photocatalytic selective conversion of thioanisole was further elucidated according to the electron paramagnetic resonance (EPR) spectra. In Fig. 9b, the intensity of the e− signal increased along with visible light irradiation time but diminished upon exposure to aerial O2. It can be concluded that TDA-COF generated e− under visible light irradiation. Subsequently, e− was transferred to O2. This transfer aligns adequately with the more negative LUMO potential of TDZ-COF. Besides, the visible light irradiation enhanced the signals of O2˙− and 1O2 obviously, and the intensity of the signal was gradually reinforced with irradiation time (Fig. 9c and d). The results further revealed that O2˙− and 1O2 as the ROS significantly contribute to the conversion of thioanisole. In like manner, the existence of both O2˙− and 1O2 as the ROS has been identified for the visible light-powered conversion of thioanisoles.66,67
Based on the meticulous studies, a plausible reaction mechanism for the blue light-powered selective conversion of thioanisole over TDA-COF is proposed in Fig. 10. There are two pathways in this photocatalytic system. Initially, TDA-COF is activated by blue light, leading to the production of e−–h+ pairs. For the energy transfer pathway, the energy transfer from the recombination of e−–h+ pairs into O2 generates 1O2, which combines with thioanisole to furnish persulfoxide. For the electron transfer pathway, thioanisole is attacked by h+, affording an S-central free cation radical. The O2˙− is formed by the combination of O2 and e− and consequently integrates with the cation radical to generate persulfoxide. Both energy and electron transfer pathways utilize a protic solvent, CH3OH, to assist in the formation of sulfoxide from persulfoxide.
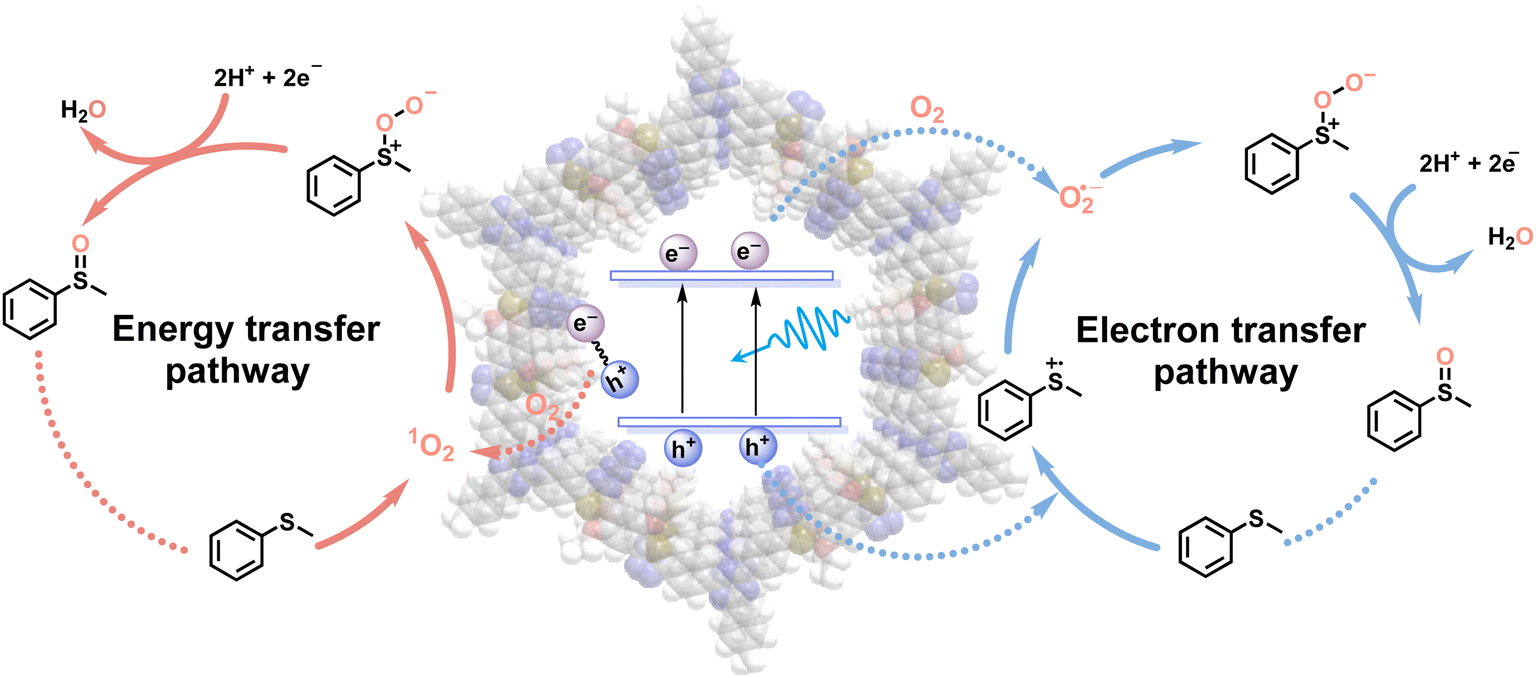 | ||
| Fig. 10 A plausible mechanism of TDA-COF photocatalysis for the blue light-powered selective conversion of thioanisole. | ||
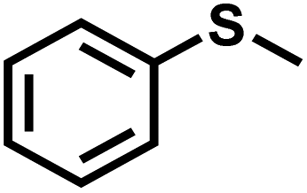
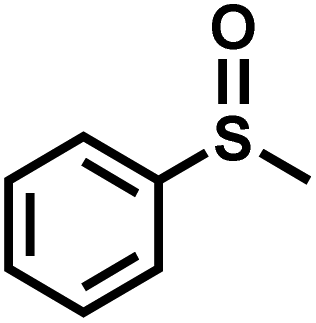
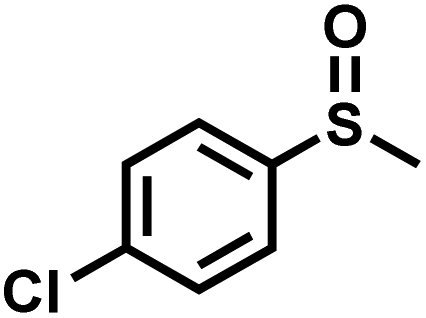
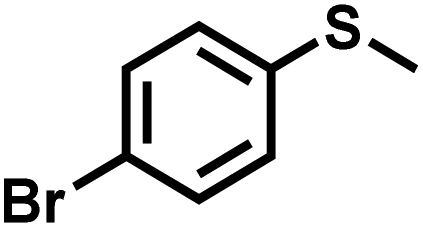
The substrate scope was explored to assess the universality of TDA-COF for the blue light-powered selective conversion of organic sulfides (Table 1). Compared with thioanisole, introducing p-F substituent to thioanisole enhanced the conversion slightly (Table 1, entries 1 and 2). However, introducing other halogen substituents inhibited the conversion and the relative activity of conversion improved as electronegativity decreased (Table 1, entries 3–5). Thereinto, the suppression effect of the Cl substituent on the conversion of thioanisoles followed o-Cl > m-Cl > p-Cl (Table 1, entries 3, 6 and 7). Besides, replacing the p-H of thioanisole with an electron-donating substituent group led to more effective conversions obviously (Table 1, entries 8 and 9). Steric effects made a great contribution to the conversion of thioanisoles, following the order p-OMe > o-OMe > m-OMe substituent (Table 1, entries 9–11). With the para-substituent of the benzene ring replaced by –NO2, p-nitrothioanisole was converted into the corresponding sulfoxide with more reaction time and relatively inferior selectivity (Table 1, entry 12). Notably, comparable conversions of linear and cyclic aliphatic sulfides needed less reaction time and achieved satisfactory selectivity (Table 1, entries 13 and 14). The methyl group of thioanisole could be replaced with an ethyl or phenyl group. In contrast, the replacement with phenyl resulted in less efficient conversion but superior selectivity (Table 1, entries 15 and 16). Moreover, 92% conversion of methyl naphthyl sulfide was completed in 24 min (Table 1, entry 17).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | t (min) | Conv. (%) | Sel. (%) |
| a Reaction conditions: organic sulfide (0.3 mmol), CH3OH (1 mL), TDA-COF (4 mg), O2 (0.1 MPa), and blue LED irradiation (460 ± 10 nm). | |||||
| 1 |
|
|
21 | 94 | 99 |
| 2 |

|

|
20 | 92 | 99 |
| 3 |

|
|
23 | 90 | 99 |
| 4 |
|

|
22 | 90 | 98 |
| 5 |
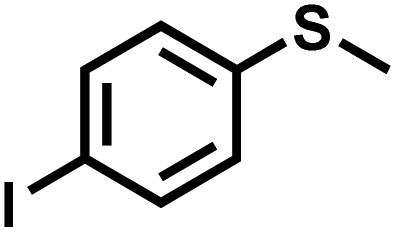
|
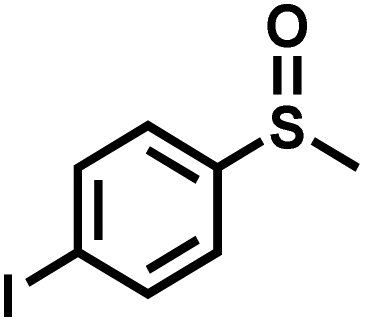
|
21 | 90 | 98 |
| 6 |
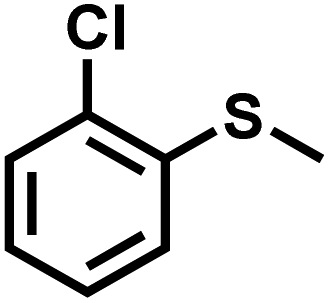
|

|
43 | 90 | 99 |
| 7 |
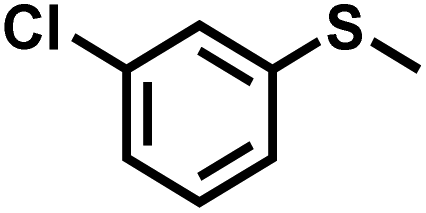
|

|
30 | 90 | 99 |
| 8 |

|

|
15 | 94 | 99 |
| 9 |

|

|
15 | 91 | 97 |
| 10 |

|
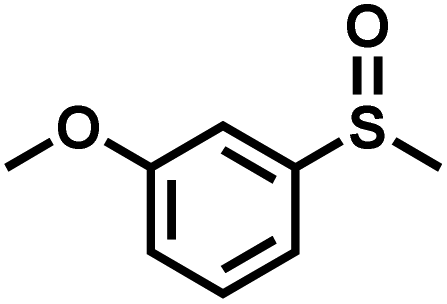
|
26 | 92 | 99 |
| 11 |

|
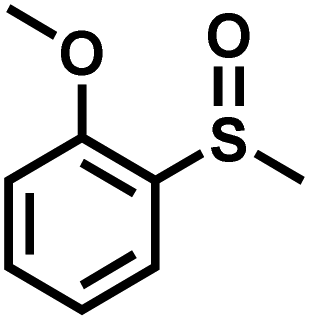
|
16 | 90 | 98 |
| 12 |

|

|
225 | 83 | 92 |
| 13 |

|

|
14 | 94 | 99 |
| 14 |

|
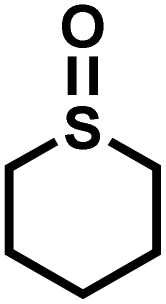
|
13 | 92 | 99 |
| 15 |

|

|
22 | 91 | 93 |
| 16 |

|
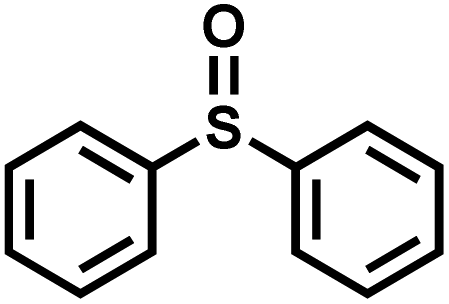
|
165 | 90 | 99 |
| 17 |

|

|
24 | 92 | 99 |

3. Conclusions
In summary, TFPT-COF as a triazine-based COF with a hydrazone linkage was synthesized through the condensation of an aldehyde, TFPT, and a substituted acyl hydrazine, DETH. Converting the hydrazone linkage of TFPT-COF through thionation and oxidative cyclization, TDA-COF with a thiadiazole linkage was constructed at 115 °C. The electronic structure and band gap of the COF were regulated by the fully conjugated planar and five-membered heterocycle of thiadiazole. Thus, the light absorption range of TDA-COF was broader than that of TFPT-COF. Meanwhile, owing to a relatively mild converting temperature, TDA-COF maintained excellent crystallinity and similar morphology. Notably, benefiting from an altered electronic structure, the optoelectronic properties of TDA-COF were superior to those of TFPT-COF. Therefore, TDA-COF showed higher photocatalytic activity for the conversion of organic sulfides than TFPT-COF. Recycle tests verified the outstanding stability of TDA-COF for the blue light-powered selective conversion of organic sulfides. This work reveals that converting the dynamic linkages of COFs is an emerging trend in achieving exceptional visible light photocatalysis.Data availability
Data are available upon request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22372124 and 22072108). The numerical calculations were done on the supercomputing system at the Supercomputing Center of Wuhan University. We also acknowledge the Core Facility of Wuhan University and the Center for Electron Microscopy at Wuhan University for materials characterization.References
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- H. S. Xu, Y. Luo, P. Z. See, X. Li, Z. X. Chen, Y. Zhou, X. X. Zhao, K. Leng, I. H. Park, R. L. Li, C. B. Liu, F. Z. Chen, S. B. Xi, J. L. Sun and K. P. Loh, Angew. Chem., Int. Ed., 2020, 59, 11527–11532 CrossRef CAS PubMed.
- C. Qian, X. Li, W. L. Teo, Q. Gao and W. Wei, Adv. Funct. Mater., 2024, 34, 2313905 CrossRef CAS.
- Z. Miao, G. Y. Liu, Y. M. Cui, Z. Y. Liu, J. H. Li, F. W. Han, Y. Liu, X. X. Sun, X. F. Gong, Y. F. Zhai, Y. L. Zhao and Y. F. Zeng, Angew. Chem., Int. Ed., 2019, 58, 4906–4910 CrossRef CAS PubMed.
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS PubMed.
- N. Huang, P. Wang and D. L. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- F. X. Duan, X. T. Liu, D. Qu, B. Li and L. X. Wu, CCS Chem., 2021, 3, 2676–2687 CrossRef CAS.
- M. H. Liu, Q. Xu and G. F. Zeng, Angew. Chem., Int. Ed., 2024, 63, e202404886 CrossRef CAS PubMed.
- S. S. Tao and D. L. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202408296 CrossRef CAS PubMed.
- Y. Liang, M. Xia, Y. X. Zhao, D. Wang, Y. P. Li, Z. Y. Sui, J. X. Xiao and Q. Chen, J. Colloid Interface Sci., 2022, 608, 652–661 CrossRef CAS PubMed.
- Y. Y. Qian and H. L. Jiang, Acc. Chem. Res., 2024, 57, 1214–1226 CrossRef CAS PubMed.
- S. S. Yuan, X. Li, J. Y. Zhu, G. Zhang, P. Van Puyvelde and B. Van der Bruggen, Chem. Soc. Rev., 2019, 48, 2665–2681 RSC.
- S. Kumar, M. A. Abdulhamid, A. D. Dinga Wonanke, M. A. Addicoat and G. Szekely, Nanoscale, 2022, 14, 2475–2481 RSC.
- Z. Xie, B. Wang, Z. F. Yang, X. Yang, X. Yu, G. L. Xing, Y. H. Zhang and L. Chen, Angew. Chem., Int. Ed., 2019, 58, 15742–15746 CrossRef CAS PubMed.
- Y. Cao, M. D. Wang, H. J. Wang, C. Y. Han, F. S. Pan and J. Sun, Adv. Energy Mater., 2022, 12, 2200057 CrossRef CAS.
- T. Li, D. W. Wang, M. Meng, X. Y. Guo, L. Lin, Z. Y. Yang, Z. Li, L. W. Xiang, C. Liu, J. Chen, X. Pang, K. Hao, H. Y. Tian and X. S. Chen, Chem. Eng. J., 2024, 492, 152163 CrossRef CAS.
- T. He and Y. L. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202303086 CrossRef CAS PubMed.
- C. R. DeBlase, K. E. Silberstein, T. T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- H. J. He, X. Fang, D. Zhai, W. Zhou, Y. M. Li, W. L. Zhao, C. C. Liu, Z. Li and W. Q. Deng, Chem.–Eur. J., 2021, 27, 14390–14395 CrossRef CAS PubMed.
- B. C. Luo, Y. B. Zhang, Y. Chen and J. Q. Huo, Mater. Adv., 2022, 3, 4699–4706 RSC.
- C. X. Zhao, C. H. Han, X. F. Yang and J. S. Xu, Green Chem., 2022, 24, 4728–4741 RSC.
- G. E. Fu, R. Ma, S. Q. Xu, T. Z. Xu, S. X. Li, Y. X. Zhao, Y. Jing, X. B. Li and T. Zhang, Mater. Today Energy, 2024, 40, 101477 CrossRef CAS.
- J. Y. Yue, L. P. Song, Z. X. Pan, P. Yang, Y. Ma, Q. Xu and B. Tang, ACS Catal., 2024, 14, 4728–4737 CrossRef CAS.
- Q. Sun, S. D. Jiang, H. Y. Niu, Y. Li, X. D. Wu, Y. L. Shi and Y. Q. Cai, J. Mater. Chem. A, 2024, 12, 5254–5260 RSC.
- W. K. Qin, C. H. Tung and L. Z. Wu, J. Mater. Chem. A, 2023, 11, 12521–12538 RSC.
- C. Qian, L. L. Feng, W. L. Teo, J. W. Liu, W. Zhou, D. D. Wang and Y. L. Zhao, Nat. Rev. Chem, 2022, 6, 881–898 CrossRef CAS PubMed.
- G. X. Jiang, W. W. Zou, Z. Y. Ou, W. F. Zhang, Z. X. Liang and L. Du, Chem.–Eur. J., 2023, 29, e202203610 CrossRef CAS PubMed.
- Z. W. Zhang, J. Jia, Y. F. Zhi, S. Ma and X. M. Liu, Chem. Soc. Rev., 2022, 51, 2444–2490 RSC.
- H. Chen, H. S. Jena, X. Feng, K. Leus and P. Van Der Voort, Angew. Chem., Int. Ed., 2022, 61, e202204938 CrossRef CAS PubMed.
- X. Y. Guan, Y. Y. Qian, X. Y. Zhang and H. L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202306135 CrossRef PubMed.
- Y. H. Liu, X. Y. Chu, Y. X. Jiang, W. Han, Y. Wang, L. H. Shao, G. L. Zhang and F. M. Zhang, Adv. Funct. Mater., 2024, 34, 2316546 CrossRef CAS.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- X. Y. Dong, F. L. Zhang, F. W. Huang and X. J. Lang, Appl. Catal., B, 2022, 318, 121875 CrossRef CAS.
- Q. Q. Tang, Y. Y. Gu, J. Ning, Y. K. Yan, L. Shi, M. S. Zhou, H. T. Wei, X. H. Ren, X. H. Li, J. X. Wang, C. Tang, L. Hao and J. H. Ye, Chem. Eng. J., 2023, 470, 144106 CrossRef CAS.
- R. F. Chen, Y. Wang, Y. Ma, A. Mal, X. Y. Gao, L. Gao, L. J. Qiao, X. B. Li, L. Z. Wu and C. Wang, Nat. Commun., 2021, 12, 1354 CrossRef CAS PubMed.
- H. F. Zhuang, C. Guo, J. L. Huang, L. W. Wang, Z. X. Zheng, H. N. Wang, Y. F. Chen and Y. Q. Lan, Angew. Chem., Int. Ed., 2024, 63, e202404941 CrossRef CAS PubMed.
- Y. J. He, Y. Zhao, X. F. Wang, Z. Y. Liu, Y. Yu and L. Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202307160 CrossRef PubMed.
- S. Q. You, J. Zhou, M. M. Chen, C. Y. Sun, X. J. Qi, A. Yousaf, X. L. Wang and Z. M. Su, J. Catal., 2020, 392, 49–55 CrossRef CAS.
- G. D. Pan, X. S. Hou, Z. Y. Liu, C. K. Yang, J. L. Long, G. C. Huang, J. H. Bi, Y. Yu and L. Y. Li, ACS Catal., 2022, 12, 14911–14917 CrossRef CAS.
- S. J. Wu, Y. F. Zhang, H. M. Ding, X. Li and X. J. Lang, J. Colloid Interface Sci., 2022, 610, 446–454 CrossRef CAS PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- X. Li, Q. Hou, W. Huang, H. S. Xu, X. W. Wang, W. Yu, R. L. Li, K. Zhang, L. Wang, Z. X. Chen, K. Y. Xie and K. P. Loh, ACS Energy Lett., 2020, 5, 3498–3506 CrossRef CAS.
- Y. Wang, S. X. Guo, L. J. Yu, W. Zhang, Z. C. Wang, Y. R. Chi and J. Wu, Chin. Chem. Lett., 2024, 35, 108207 CrossRef CAS.
- S. L. Yang, Z. Chen, L. Zou and R. Cao, Adv. Sci., 2023, 10, 2304697 CrossRef CAS PubMed.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- K. Merkel, J. Greiner and F. Ortmann, Sci. Rep., 2023, 13, 1685 CrossRef CAS PubMed.
- Y. H. Hou, F. Y. Liu, B. Q. Zhang and M. P. Tong, Environ. Sci. Technol., 2022, 56, 16303–16314 CrossRef CAS PubMed.
- Y. X. Wang, F. L. Zhang, F. W. Huang, X. Y. Dong, B. Zeng, X. K. Gu and X. J. Lang, Appl. Catal., B, 2024, 354, 124103 CrossRef CAS.
- X. Y. Xu, X. Y. Wu, K. Xu, H. Xu, H. Z. Chen and N. Huang, Nat. Commun., 2023, 14, 3360 CrossRef CAS PubMed.
- Y. J. Zou, S. Abednatanzi, P. G. Derakhshandeh, S. Mazzanti, C. M. Schusslbauer, D. Cruz, P. Van der Voort, J. W. Shi, M. Antonietti, D. M. Guldi and A. Savateev, Nat. Commun., 2022, 13, 2171 CrossRef CAS PubMed.
- C. H. Han, R. J. Qi, R. L. Sun, K. C. Fan, B. Johannessen, D. C. Qi, S. W. Cao and J. S. Xu, Appl. Catal., B, 2023, 320, 121954 CrossRef CAS.
- M. Q. Yang, C. F. Tan, W. H. Lu, K. Y. Zeng and G. W. Ho, Adv. Funct. Mater., 2020, 30, 2004460 CrossRef CAS.
- V. Nosek and J. Mísek, Angew. Chem., Int. Ed., 2018, 57, 9849–9852 CrossRef CAS PubMed.
- H. X. Zhao, F. L. Zhang, X. Y. Dong and X. J. Lang, J. Mater. Chem. A, 2023, 11, 18115–18125 RSC.
- F. W. Huang, Y. X. Wang, X. Y. Dong and X. J. Lang, J. Mater. Chem. A, 2024, 12, 7036–7046 RSC.
- M. A. Hoque, J. B. Gerken and S. S. Stahl, Science, 2024, 383, 173–178 CrossRef CAS PubMed.
- S. Li, L. Dai, L. Li, A. W. Dong, J. N. Li, X. J. Meng, B. Wang and P. F. Li, J. Mater. Chem. A, 2022, 10, 13325–13332 RSC.
- T. Yang, D. Zhang, A. G. Kong, Y. Y. Zou, L. Yuan, C. Liu, S. J. Luo, G. F. Wei and C. Z. Yu, Angew. Chem., Int. Ed., 2024, 63, e202404077 CrossRef CAS PubMed.
- Y. Liu, B. Zhang, D. P. Yan and X. Xiang, Green Chem., 2024, 26, 2505–2524 RSC.
- J. J. Zhang, J. H. Zhang, J. N. Shen, D. M. Li, J. L. Long, W. X. Dai, X. X. Wang and Z. Z. Zhang, ACS Catal., 2024, 14, 3855–3866 CrossRef CAS.
- K. K. Niu, T. X. Luan, J. Cui, H. Liu, L. B. Xing and P. Z. Li, ACS Catal., 2024, 14, 2631–2641 CrossRef CAS.
- Z. J. Gu, J. J. Wang, Z. Shan, M. M. Wu, T. T. Liu, L. Song, G. X. Wang, X. H. Ju, J. Su and G. Zhang, J. Mater. Chem. A, 2022, 10, 17624–17632 RSC.
- Q. Q. Hu, K. J. Liu, J. W. Ye, L. Ming, J. S. Xu and S. W. Cao, Appl. Surf. Sci., 2023, 623, 157012 CrossRef CAS.
- B. W. Liu, J. J. Cai, J. J. Zhang, H. Y. Tan, B. Cheng and J. S. Xu, Chin. J. Catal., 2023, 51, 204–215 CrossRef CAS.
- J. L. Li, Q. Lei, X. L. Dong, C. L. Chen, X. L. Liu, Z. Y. Zhang, F. Shui, M. Yi, B. Y. Li and X. H. Bu, Chem. Mater., 2023, 35, 4120–4127 CrossRef CAS.
- Z. P. Xie, W. B. Wang, X. T. Ke, X. Cai, X. Chen, S. B. Wang, W. Lin and X. C. Wang, Appl. Catal., B, 2023, 325, 122312 CrossRef CAS.
- Y. Z. Peng, G. C. Guo, S. Guo, L. H. Kong, T. B. Lu and Z. M. Zhang, Angew. Chem., Int. Ed., 2021, 60, 22062–22069 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04548c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |