
Gram-scale production of vertically aligned holey graphene nanosheet arrays derived from a renewable biomass precursor via a facile hydrothermal/salt-assisted pyrolysis method for aqueous high-performance redox supercapacitors†
Qincheng
Yang‡
a,
Shuaibing
Liu‡
a,
Qianglin
Li
*b,
Ling
Wu
 *c,
Binghua
Zhou
d,
Zhipeng
Wang
d,
Zheng-Hong
Huang
e,
Hao
Yang
f and
Ming-Xi
Wang
*a
*c,
Binghua
Zhou
d,
Zhipeng
Wang
d,
Zheng-Hong
Huang
e,
Hao
Yang
f and
Ming-Xi
Wang
*a
aKey Laboratory of Biomass-based Materials for Environment and Energy in Petroleum & Chemical Industries, School of Chemical and Environmental Engineering, Wuhan Institute of Technology, Wuhan 430205, China. E-mail: Wangmx14@wit.edu.cn
bDepartment of Material and Environmental Engineering, Chengdu Technological University, Chengdu, 611730, China. E-mail: lqlxp1010@163.com; Tel: +86-27-87195680
cHubei Province Key Laboratory of Coal Conversion and New Carbon Materials, School of Chemistry and Chemical Engineering, Wuhan University of Science and Technology, Wuhan 430081, China. E-mail: wuling2018@wust.edu.cn
dInstitute of Adv. Mater., Jiangxi Normal University, 99 Ziyang Avenue, Nanchang 330022, China
eLab of Adv. Mater., School of Materials Science and Engineering, Tsinghua University, Beijing 100084, China
fKey Laboratory for Green Chemical Process of Ministry of Education, School of Environmental Ecology and Biological Engineering, Wuhan Institute of Technology, Wuhan, 430205, China
First published on 17th August 2024
Abstract
Herein, we developed a facile method for the gram-scale production of high-quality vertically aligned graphene nanosheet arrays (VAGNAs) named as hydrothermal/salt-assisted pyrolysis (HSP) for constructing high-performance redox supercapacitors. VAGNAs were fabricated on a large scale using HSP from low-cost, green and renewable biomass instead of fossil gas precursors; no expensive instrument or extra substrate was required. The key fabrication process parameters were screened and optimized, including the pretreatment method, selected salts and pyrolysis temperature. The as-fabricated VAGNAs exhibit outstanding properties, including high graphitization degree, thin nanosheets, good hydrophilicity and a hierarchically porous structure. The as-fabricated VAGNAs were employed as electrodes to construct redox supercapacitors. The VAGNA electrodes possess an ultrahigh specific capacitance of 3148 F g−1 in 1.0 M KOH electrolyte with the addition of 0.10 M K3Fe(CN)6. The assembled symmetrical redox supercapacitor based on VAGNA-L-1000 delivers a high specific capacitance of 92.8 F g−1 at 1 A g−1 under a wide operation potential window of 2.0 V and high energy density of 36.7 W h kg−1 at a power density of 712 W kg−1. The present study highlights the large-scale synthesis of high-value added nanocarbons derived from renewable biomass for developing high-performance supercapacitors with high energy density in an economical way.
 Ling Wu | Ling Wu received her PhD in October 2017 in applied chemistry at Tokyo University, Japan. Later, she worked as a distinguished professor through the “Chutian Scholar Program” in Hubei Province at the School of Chemistry and Chemical Engineering in Wuhan University of Science and Technology. Her research interests include the development of sustainable materials from biomass, their applications in nanocatalysis, water pollution control, energy storage and conversion, and the mechanism of catalysis and energy storage. |
1. Introduction
Supercapacitors (SCs) exhibit significant advantages as energy storage devices, such as high specific power capability, rapid charge/discharge processes and long lifetimes.1,2 However, SCs often suffer from their relatively low energy density, which is determined by the energy storage mechanism and restricts their further application as independent devices.3,4 In particular, the energy storage in electrical double-layer capacitors (EDLCs) occurs primarily via electrostatic charge accumulation at the electrode/electrolyte interface; thus, their energy densities essentially depend on the specific surface area (SSA) of active materials. Thus, an ideal electrode material should have a high SSA to achieve enhanced capacitance together with good conductivity to ensure efficient ion and electron transport for charge storage.5 Along this line, porous carbon materials such as activated carbon, carbon nanofibers, carbon aerogels and graphene have been explored as SC electrode materials owing to their large SSA, high electrical conductivity and chemical stability.6–8 Among these carbon materials, graphene is recognized as a promising electrode material because of its excellent conductivity and theoretically high SSA.9 Nevertheless, the practical performance of graphene-based SCs is not satisfactory because of the following reasons: (1) the inevitable π–π aggregation and restacking of graphene sheets lead to a low ion-accessible surface area; (2) the randomly oriented and parallel graphene structure results in a limited accessible surface area and transport pathways for electrolyte ions.10,11Recently, some studies have demonstrated that electrode materials with three-dimensional (3D) structures exhibit markedly enhanced supercapacitor performance because their inherent non-stacking structure can provide a large SSA favoring ion diffusion and storage. Vertically arranged graphene nanosheet arrays (VAGNAs) as a type of emerging 3D graphene have attracted great interest from researchers owing to their unique nanostructures, excellent properties and wide range of potential applications.12 Structurally, VAGNAs are mainly composed of vertically independent few-layer (1–10-layer) graphene-containing carbon nanosheets (CNSs) with an interlayer spacing of about 0.34 nm.13 Compared with horizontal graphene, VAGNAs have higher active open-edge densities, larger surface-to-volume ratios, specific orientations, and nonstacking properties. Due to the vertically aligned few-layer graphene structure, in addition to the basic properties of graphene, VAGNAs have other advantages: (1) the array structure prevents the aggregation of graphene nanosheets and confers structural stability; (2) the vertical graphene nanosheets ensure efficient electrical conductivity of the graphene/electrolyte interface; (3) a large number of sharp and exposed graphene edges facilitate electrode reaction kinetics and mass transfer; (4) VAGNAs can be easily and uniformly modified with other active components, making it an ideal electrode material in the field of supercapacitors.14–17 However, the vast majority of VAGNAs reported so far have been prepared by various chemical vapor deposition (CVD) methods, such as plasma-enhanced chemical vapor deposition (PECVD),18 atmospheric high voltage PECVD (AHV-PECVD),19 magnetron sputtering and thermal hot wire chemical vapor deposition (HWCVD),20 and microwave plasma-enhanced chemical vapor deposition (MPCVD).21 These CVD-based growth methods always involve a series of complex steps, starting from the deposition of a thin layer of carbon atoms on the substrate, and the structure and properties of the grown VAGNAs are affected by a number of factors, such as the substrate, the composition of the gas, and the applied power and pressure.22 These methods also suffer from other disadvantages, such as expensive instrumentation, unsustainable carbon sources (CH4/H2, C2H2/H2, etc.), and required substrate, time-consuming processes, and low efficiency. These drawbacks severely restrict the large-scale production of VAGNAs and its practical applications.
Bio-renewable materials (biomass waste and its derivatives) have become alternative feedstocks for various carbon materials due to their renewable, green, abundant, and inexpensive features.23 Wang et al.24,25 prepared VAGNAs using rice husk and polyimide as precursors via the MPCVD method, which demonstrated that biomass can be converted to VAGNAs through a “bottom-up” strategy. Liu et al.26 prepared VAGNAs by hydrothermal carbonization (HTC)-potassium hydroxide (KOH) activation using spruce bark biowaste as a precursor. Compared with the CVD-based method, the HTC-KOH method does not require any expensive and sophisticated instruments, substrates or catalysts; thus, large-scale production is feasible. On the other hand, the HTC-KOH method has some disadvantages due to the use of a strong base, KOH, as an activator. KOH is known to be corrosive and destroys tube furnaces during high-temperature pyrolysis, while the yield of VAGNAs remains low (8.9% at 900 °C). Therefore, the development of a facile, reproducible and green VAGNA strategy from renewable precursors is still urgently needed. In recent years, salt-assisted pyrolysis has been used to prepare a variety of heteroatom-doped carbons, such as oxygen–nitrogen co-doped carbons27 and lignin-derived Fe, N, P, and S co-doped porous carbons.28 Salt-assisted pyrolysis has been shown to be an effective strategy for designing carbon materials, but there are no reports on the preparation of VAGNA using this method.
To achieve ideal energy density, efficient carbon-based electrode materials alone are not enough as they are limited by the storage mechanism. The energy density of the currently reported non-functionalized VAGNAs-based SCs was less than 10 W h kg−1,29,30 which is far from the practical requirement. Besides electrode materials, the electrolyte also plays a crucial role in the supercapacitor performance. According to the equation of E = 0.5CΔV2, the energy density (E) can be improved by increasing the capacitance (C, F g−1) and voltage (V). An effective strategy is to add active additives into basic electrolytes (KOH, H2SO4, etc.) to construct the redox electrolyte, which not only increases the capacitance, but also widens the potential window.31 K3Fe(CN)6 is an ideal redox-active additive6,32,33 due to its high activity and the reversibility of the redox ion pair (Fe(CN)63−/Fe(CN)64−) produced during the reduction/oxidation process. So far, there is no literature available on a VAGNA-based supercapacitor with redox electrolyte.
Herein, the gram-scale production of high-quality VAGNA from a variety of renewable biomasses was successfully performed via a two-step strategy consisting of a hydrothermal step, followed by a salt-assisted pyrolysis step (HSP). The as-fabricated VAGNA was used as electrode materials to assemble high-energy-density supercapacitors with the redox electrolyte. The method uses a variety of biomass (including bark, shells and pollen) that can be used as a carbon source, and uses a variety of salts (K2CO3, CH3COOK, Na2CO3, etc.) for the production of VAGNAs via the HSP method without the need for any substrate or catalyst. The preparation process is green and facilitates the large-scale production of high-quality VAGNAs from renewable precursors. K3Fe(CN)6 was employed as a redox-active additive, which was added to KOH to form the redox electrolyte. The electrochemical performance of the as-fabricated VAGNA-based supercapacitors was then investigated. The specific capacitance of the optimized electrode material can reach 3148 F g−1 in 1.0 M KOH + 0.10 M K3Fe(CN)6 electrolyte. The assembled VAGNA-based symmetrical redox supercapacitor delivered a high energy density of 36.7 W h Kg−1 under a power density of 712 W Kg−1 with a large potential window of 2 V, which is superior to the electrochemical performance reported for the VAGNA-based supercapacitor.
2. Experimental section
2.1 Biomass raw materials and chemicals
The renewable precursors, including spruce bark, grapefruit bark, tamarind fruit shell, pecan shell, lotus pollen and rape pollen, were collected from Hubei province, P. R. China. Before use, the raw biomass was firstly rinsed with deionized (DI) water, and dried in an oven at 105 °C overnight. In each experiment, the weight of the cleaned raw biomass was around 5 g. The chemical reagents, K2CO3, CH3COOK, Na2CO3, Li2CO3, KCl, K3PO4 and K3Fe(CN)6 were purchased from Sinopharm Chemical Reagent Co., Ltd, and all reagents were of analytical grade and used without further purification.2.2 Fabrication of VAGNAs
The biomass-derived gram-scale VAGNAs were fabricated via a facile and green hydrothermal procedure, followed by salt-assisted pyrolysis (HSP method), shown in Fig. 1. In a typical fabrication experiment, the cleaned and dried raw biomass was first ground into a powder, and dispersed into 60 mL DI water under stirring. The resulting suspension was transferred into a 100 mL Teflon hydrothermal reactor, which was heated to 180 °C and maintained at this temperature for 12 hours, followed by cooling down naturally to room temperature. The hydrothermal products were washed with DI water to neutral condition, and dried at 105 °C overnight. Salts were then added to the hydrothermal products with a weight ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and mixed by grinding in an agate crucible. Six kinds of salts were examined in fabricating the VAGNAs, including K2CO3, CH3COOK, Na2CO3, Li2CO3, KCl and K3PO4. After grinding and mixing, the grinding blender with salt was dried again at 80 °C before pyrolysis. The dried mixture was annealed at the desired temperature (800–1000 °C) for 2 hours under N2 or Ar flow in a tube furnace, and the heating rate was controlled at 5 °C min−1 from room temperature to the set temperature. During the salt-assisted pyrolysis process at higher temperature under an inert atmosphere, the hydrothermal biochar was carbonized and activated. The resulting black powder was immersed in diluted HCl acid several times and washed with DI water until pH = 7, followed by drying at 105 °C for 12 hours. Finally, the as-obtained VAGNAs derived from spruce bark (S), grapefruit bark (G), tamarind fruit shell (T), pecan shell (P), lotus pollen (L) and rape pollen (R) at different temperatures were denoted as VAGNA-S/G/T/P/L/R-t, where t indicates the pyrolysis temperature. The yield of VAGNA is ca. 20% relative to the hydrothermal treated samples.
:1, and mixed by grinding in an agate crucible. Six kinds of salts were examined in fabricating the VAGNAs, including K2CO3, CH3COOK, Na2CO3, Li2CO3, KCl and K3PO4. After grinding and mixing, the grinding blender with salt was dried again at 80 °C before pyrolysis. The dried mixture was annealed at the desired temperature (800–1000 °C) for 2 hours under N2 or Ar flow in a tube furnace, and the heating rate was controlled at 5 °C min−1 from room temperature to the set temperature. During the salt-assisted pyrolysis process at higher temperature under an inert atmosphere, the hydrothermal biochar was carbonized and activated. The resulting black powder was immersed in diluted HCl acid several times and washed with DI water until pH = 7, followed by drying at 105 °C for 12 hours. Finally, the as-obtained VAGNAs derived from spruce bark (S), grapefruit bark (G), tamarind fruit shell (T), pecan shell (P), lotus pollen (L) and rape pollen (R) at different temperatures were denoted as VAGNA-S/G/T/P/L/R-t, where t indicates the pyrolysis temperature. The yield of VAGNA is ca. 20% relative to the hydrothermal treated samples.
 | ||
| Fig. 1 Schematic of the fabrication process for the gram-scale production of the vertically aligned graphene nanosheet array (VAGNA) from multiple renewable biomasses via the hydrothermal/salt-assisted pyrolysis (HSP) method. | ||
2.3 Electrode fabrication and its electrochemical testing
The prepared VAGNA was used as the SC electrode material. VAGNA (80 wt%), acetylene black (10 wt%) and polytetrafluoroethylene (PTFE, 10 wt%) were mixed to form a slurry and coated on 1 cm2 of nickel foam, which was then dried in a vacuum oven at 80 °C for 12 h. Finally, a nickel foam loaded with the electrode material was pressed at 10 MPa to obtain the electrodes, and the active electrode material (VAGNA) weighed 3–4 mg. Different electrolyte concentrations of K3Fe(CN)6 were added into 1 M KOH and 1 M KOH. Symmetric capacitors were assembled using two identical VAGNA-based electrodes with the above electrolytes. All electrochemical measurements were made at an electrochemical workstation (CHI760E). In the three-electrode system, the prepared VAGNA electrode was the working electrode, and the Pt and Hg/HgO electrodes were used as the counting and reference electrodes, respectively. The electrochemical performance of the electrode was evaluated by cyclic voltammetry (CV) and constant current charge/discharge (GCD) tests at different scan rates. The same tests were performed in a two-electrode system to evaluate the performance of the assembled capacitor. Electrochemical impedance spectroscopy (EIS) measurements were performed in the frequency range of 0.01–100 kHz, and used to study the resistance and ion transport behavior of the supercapacitor. The specific capacitances of a single VAGNA electrode (Cm, F g−1) and VAGNA-based supercapacitor (Ccell, F g−1) were calculated from the discharge curve of the GCD curve, according to eqn (1) and (2), and the energy density (E, W h kg−1) and power density (P, W kg−1) of the VAGNA-based supercapacitors were calculated based on eqn (3) and (4) as follows: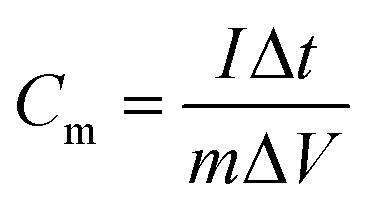 | (1) |
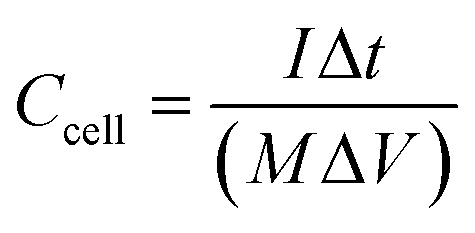 | (2) |
 | (3) |
 | (4) |
2.4 Material characterization
Field emission scanning electron microscopy (FESEM, Gemini SEM 300), transmission electron microscopy (TEM, JEOL JEM-2100), and atomic force microscopy (AFM, NanoMan VS microscope, tapping mode) were employed to investigate the microstructure and surface morphology of the as-obtained VAGNAs. An automatic contact angle measuring instrument (CA, Dataphysics OCA20, Germany) was used to measure the contact angle of the materials for evaluating the wettability of the carbon materials. Raman spectra were recorded on a Renishaw inVia Raman spectrometer using a 532 nm laser beam in the wavelength range from 100 to 3500 cm−1. The crystalline structures of the VAGNAs were examined by X-ray diffractometer (Rigaku SmartLab SE, Japan) at a scan rate of 5° min−1. The nitrogen adsorption/desorption isotherm profiles at 77 K were acquired on an ASAP 2460 gas adsorption instrument, and the specific surface area (SSA) and pore size distributions (PSDs) of the carbon materials were calculated based on the Brunauer–Emmett–Teller (BET) equation and nonlocal density functional theory (NLDFT) model, respectively.3. Results and discussion
3.1 VAGNAs derived from multiple biomasses under optimized conditions
To fabricate VAGNAs by the HSP method, we chose six kinds of biomass as the carbon source, including spruce bark (S), grapefruit bark (G), tamarind fruit shell (T), pecan shell (P), lotus pollen (L) and rape pollen (R). We found that VAGNAs can be successfully fabricated from all of the biomasses by this HSP method under optimized conditions, demonstrating its universality for various biomasses. The optimal salt-assisted pyrolysis temperature is 1000 °C, and the most suitable salt is K2CO3. The as-fabricated VAGNAs derived from these chosen biomasses were designated as VAGNA-S-1000, VAGNA-G-1000, VAGNA-T-1000, VAGNA-P-1000, VAGNA-L-1000 and VAGNA-R-1000, respectively. The morphology, microstructure and surface wettability of these VAGNAs were characterized.The top-view FESEM images of the as-obtained products are shown in Fig. 2. Obviously, all of the pyrolysis products from different biomasses obtained via this HSP method show the typical characteristics of VAGNAs. These images show that the two-dimensional (2D) carbon nanosheets stand almost vertically, and an open structure with large and detached channels can be distinctly observed. In addition, the detached nanosheets possess extended edges, and they partly connect with each other, forming a continuously interconnected porous network. The porous honeycomb structure consists of carbon nanosheets with a thickness of 1–10 nm, which is far thinner than the VAGNAs prepared by PECVD.34 It should also be noted that there are some curvatures and wrinkles in these VAGNAs due to their thin, large sheets. These sheets will transfer from a 2D to 3D structure to reduce the surface energy under the released stress during the thermal shrinkage process.35,36
 | ||
| Fig. 2 FE-SEM images of VAGNAs from the pyrolysis of different biomasses with K2CO3 at 1000 °C: (a) VAGNA-S-1000, (b) VAGNA-G-1000, (c) VAGNA-T-1000, (d) VAGNA-P-1000, (e) VAGNA-L-1000 and (f) VAGNA-R-1000. | ||
Upon closer inspection of these biomass-derived VAGNAs by HSP method, there is a certain degree of differences in their morphologies. As can be seen, the VAGNAs prepared from barks (Fig. 2a and b) display the highest vertical alignment with regular edges and large space among the carbon nanosheets. Meanwhile, those from pollens (Fig. 2e and f) are observed to have lower vertical alignment and highest density of sheets, and the VAGNAs from shells (Fig. 2c and d) show medial regularity and vertical alignment. The increase in the disorder and curvature of VAGNAs originate from the diversity of the intrinsic structure and complexity of the chemical composition of these biomass precursors. Generally speaking, barks have a denser structure than shell and pollen,37,38 which will affect the VAGNAs growth during the salt-assisted pyrolysis process. As shown in Fig. S1,† the lotus pollen was almost entirely transformed into VAGNAs, whereas part of the bark and shell were converted into VAGNAs. As a result, VAGNAs can be observed easily in the whole range for lotus pollen-derived VAGNAs, while it was observed locally in the spruce bark-derived VAGNAs. These results indicated that pollen is a relatively ideal precursor for preparing VAGNAs compared with the other two precursors under the same fabrication conditions.
The morphology and microstructure of lotus pollen-derived VAGNAs were also investigated by TEM and high-resolution TEM (HRTEM) technique. The TEM images in Fig. 3a and b display the honeycomb-like and discontinuous lattice fringes of carbon, which is consistent with the SEM observations. Based on the matrix, the maximal height of the vertical nanosheet can reach several microns. The HRTEM image in Fig. 3c clearly shows the lattice information of the as-prepared VAGNA, which consisted of 2–4 layers of graphene nanosheet with smooth, exposed and highly extended edges. According to the Bragg equation: 2dsinθ = nλ, the calculated interlayer spacing between the layers was 0.343 nm, which is very close to the graphite layer spacing (0.334 nm). These indicated that the as-prepared VAGNA was highly graphitized. The selected electron diffraction (SAED) pattern that is displayed presents a typical ring feature of (002) and (100) crystal planes of graphene (Fig. 3d), which arises from the few layers of scrolled/fold graphene sheets,26,39 confirming the formation of the graphitic structure and curved shape. The few-layer nature of the as-fabricated VAGNAs was further confirmed by atomic force microscopy (AFM) and height analysis. Fig. 3e shows a typical AFM topographic image of VAGNA-L-1000. It can be observed that the VAGNA had a smooth surface, and the nanoflake dimensions indicated that they averaged several nm in size. Fig. 3f displays the height profile, corresponding to the solid line in Fig. 3e. The height and root mean square roughness (RMS) value of VAGNA were 2.850 nm and 0.905 nm, respectively, which were consistent with the results observed by TEM.
 | ||
| Fig. 3 (a) TEM images, (b) enlarged image of (a) and (c) HRTEM images and (d) SAED images of VAGNA-L-1000, (e) AFM images and (f) the corresponding cross-section profiles. | ||
Raman spectroscopy, a fast and effective technique to identify the carbon materials while offering structural information, was employed to characterize the VAGNAs. The Raman spectra of VAGNA-L-1000, VAGNA-T-1000 and VAGNA-S-1000 are presented in Fig. 4a. Two pronounced bands located at 2694 cm−1 (2D-band) and 1577 cm−1 (G-band), and a very weak band at 1342 cm−1 (D-band) are found in all of the Raman spectra of VAGNAs. The D-band is correlated with the defects and disorders in carbon materials,40 while the G-band stems from the in-plane sp2 phonon vibration.41 The intensity ratio of the D and G bands (ID/IG) is generally used to evaluate the degree of crystallization or defect density of carbon materials.42,43 The 2D band is the second order D band corresponding to the two phonon lattice vibrations, which is a characteristic peak of the graphene structure.44,45 The number of graphene layers can be inferred from the ratio of the 2D/G band intensities (I2D/IG), as well as the shape and position of these bands.46 A bigger I2D/IG value and sharper 2D band indicate that there are fewer layers. As seen from Fig. 4a, the G and 2D bands are both sharper with a narrow full-width at half-maximum (FWHM), and their intensities are much larger than that of the D band for all three samples. As listed in Table 1, the calculated ID/IG values are 0.18, 0.10 and 0.55 for VAGNA-L-1000, VAGNA-S-1000 and VAGNA-T-1000, and the corresponding I2D/IG values are 1.66, 1.65 and 0.89, respectively. These results indicated that these VAGNAs were composed of graphene sheets with several layers with high graphitization, which agree well with the TEM observations. By contrast, the VAGNAs prepared by PECVD method were observed to have a more random arrangement and thicker layers in Fig. S2.† Thus, it can be said that the HSP method is effective in fabricating VAGNAs with few defects and high purity. These indicated that the as-developed HSP method can easily produce good-crystallinity biomass-derived graphene materials.
 | ||
| Fig. 4 (a) Raman spectra, (b) nitrogen adsorption–desorption isotherms and (c) pore size distributions (PSDs) of VAGNA-L-1000, VAGNA-S-1000 and VAGNA-T-1000. | ||
| VAGNAs | S BET (m2 g−1) | V total (cm3 g−1) | V micro (cm3 g−1) | V meso (cm3 g−1) | V micro/Vtotal (%) | I D/IG | I 2D/IG |
|---|---|---|---|---|---|---|---|
| VAGNA-L-1000 | 1997 | 1.28 | 0.61 | 0.67 | 47.76 | 0.18 | 1.66 |
| VAGNA-S-1000 | 1088 | 1.12 | 0.25 | 0.87 | 22.32 | 0.10 | 1.65 |
| VAGNA-T-1000 | 883 | 0.69 | 0.23 | 0.46 | 33.33 | 0.55 | 0.89 |
Generally, VAGNAs prepared from PECVD have a specific surface area of about 1000 m2 g−1,16 while the VAGNAs obtained from the as-developed HSP method have a much bigger specific surface area and pore volume, which were evaluated by N2 adsorption/desorption measurements. The N2 adsorption/desorption isotherms of all VAGNAs in Fig. 4b exhibit a combined I/IV type isotherm,47 which has a sharp adsorption knee at low relative pressure (P/P0 < 0.01) and a distinct hysteresis loop in the P/P0 range from 0.4 to 1.0. This kind of isotherm indicated that the measured samples consisted of both micropores and mesopores, because the sharp adsorption and hysteresis loop resulted from the micropore filling and capillary condensation in mesopores.48
The PSDs shown in Fig. 4c confirms the presence of micropores and mesorpores in all of the VAGNAs. The calculated SSA and pore volume are listed in Table 1. It can be observed that the SSA of VAGNA-L-1000 can reach 1997 m2 g−1, which is almost twice that of VAGNAs prepared by PECVD, and the total pore volume is up to 1.28 cm3 g−1. The SSA of the VAGNA-S-1000 and VAGNA-T-1000 samples are 1088 and 883 m2 g−1, respectively. This demonstrated that the VAGNAs prepared via HSP method have developed a hierarchical pore structure, which is ascribed to the pore-forming capability of the salt (K2CO3, Na2CO3, etc.) beyond its facilitation of the vertical alignment of the carbon sheet. The differences of the SSA and pore volume among the VAGNAs originate from the diverse structure of the raw biomass. Comparatively speaking, pollen is an ideal biomass raw material for preparing VAGNAs with a developed pore structure.
The surface wettability is a critical property of carbon materials that will influence their applications.49 To assess the wetting behavior of the VAGNAs, the water contact angles (WCAs) were measured. In the WCAs tests, the sample was first pressed into a sheet under 10 MPa, and the contact angle between the sheet and DI water were detected. Fig. 5 shows the WCAs of the typical VAGNAs by HSP method and the control sample by PECVD method. It is clear that the as-fabricated VAGNAs by HSP method show much better hydrophilicity than the control sample. As seen from Fig. 5, the instantaneous contact angles measured upon immediate water droplet formation are 68.2°, 52.6° and 73.8° for VAGNA-S-1000, VAGNA-T-1000 and VAGNA-L-1000, respectively, which are much lower than that of the control sample. Moreover, the contact angles of the as-fabricated VAGNAs decreased rapidly with the time elapsing, and the droplets completely disappeared within 2 seconds. In contrast, the contact angle of the control sample hardly decreased over time, and the contact angle almost remained constant even when the time was extended to 2 minutes. The excellent wettability of the as-fabricated VAGNAs with water is attributed to the hydrothermal pre-treatment and the salt-assisted pyrolysis process.
 | ||
| Fig. 5 Wettability measurements: dynamic contact angle for the water droplet on (a–a′′) VAGNA-S-1000, (b–b′′) VAGNA-T-1000, (c–c′′) VAGNA-L-1000, and (d–d′′) the control sample-VAGNA via the PECVD method. | ||
3.2 Screening and optimization of the fabrication process conditions
According to the fabrication process of VAGNAs shown in Fig. 1, the process involves the pretreatment of raw biomass and the salt-assisted pyrolysis at a certain temperature. It is found that the quality of the final products mainly depends on the pretreatment method, salt used, pyrolysis temperature and other parameters. To obtain high-quality VAGNAs, we screened and optimized these process conditions, such as adopting several pretreatment methods, selecting different salts, and adjusting the pyrolysis temperature. Their effects on the final VAGNAs were investigated in detail. | ||
| Fig. 6 FE-SEM images of lotus pollen-derived carbons (VAGNA) from pretreated precursors with different methods. (a and a′): hydrothermal treatment, (b) NaOH soaking, (c) H2SO4 soaking, (d) ammonia soaking and (e) without pretreatment. | ||
The above results demonstrated that the hydrothermal pretreatment played a crucial role in the formation of nanosheets from raw biomass. During the mild hydrothermal process, part of lignin, and most of the semicellulose and protein are transformed into soluble organic compounds,50,51 while the crystalline cellulose is partially carbonized.52 Subsequently, they will be converted into carbon sheets in the following pyrolysis step at higher temperature in the presence of salt. On the contrary, under the soaking process with strong alkali (KOH) or acid (H2SO4) and ammonia, some of the components will react with the alkali or acid.53,54 The carbon skeleton will be inevitably damaged in the etching process of these agents, resulting in the failure of the formation of carbon sheets. This indicates the importance of selecting a suitable pretreatment method for the formation of versatile carbon materials with a desirable microstructure. The mechanism for the formation of VAGNAs will be further discussed later.
Characterization of the morphology and structure verifies that salt plays a vital role in the formation of the final products by pyrolysis of biomass. Fig. 7 shows the FE-SEM images of lotus pollen-derived carbons undergoing hydrothermal treatment and pyrolysis at 1000 °C in the presence of different salts. As seen in Fig. 7a, the samples prepared by pyrolyzing the mixture of lotus pollen and K2CO3 show an ideal carbon sheet structure with a smooth surface, and the continuous 2D carbon nanosheets are aligned vertically to form a unique 3D architecture. Interestingly, the samples prepared in the existence of CH3COOK also show a similar structure of VAGNAs prepared in the presence of K2CO3 (Fig. 7b), in which nanosheets with the thickness of about tens of nanometers were clearly observed. Relatively, when lotus pollen was pyrolyzed with Na2CO3 (Fig. 7c), neither nanosheet were formed nor were they aligned vertically. Only irregular and thick lamellar structures were observed with a disordered arrangement. In sharp contrast with the K2CO3, CH3COOK and Na2CO3-assisted pyrolysis samples, the samples prepared in the presence of Li2CO3, KCl and K3PO4 (Fig. 7d–f) show totally different structure from VAGNAs. There were hardly any sheets or fragments to be found in these samples. Instead, there were some blocks with particles on the surface.
 | ||
| Fig. 7 FE-SEM images of lotus pollen-derived carbons via HSP method using different salts at 1000 °C: (a) K2CO3, (b) CH3COOK, (c) Na2CO3, (d) Li2CO3, (e) KCl and (f) K3PO4. | ||
The morphologies fully confirm that the cation and anion ions of the salt both synergistically determine the final microstructure of the products. In view of the formation of VAGNAs, the priority obeys the following order: K2CO3 ≥ CH3COOK > Na2CO3 ≫ Li2CO3 > KCl > K3PO4. The outstanding performance of K2CO3 and CH3COOK in fabricating VAGNAs results from the excellent catalytic graphitization and intercalation capability of the potassium ion and the released gas (CO2) from the anion ions during the reaction with the carbon matrix. It is recognized that K2CO3 can be used as an activating agent to prepare porous carbons. During the salt-assisted pyrolysis process under an inert atmosphere, K2CO3 reacts with carbon via the following reactions indicated by eqn (5)–(8):
| K2CO3 + 2C → 2K + 3CO | (5) |
| K2CO3 → K2O + CO2 | (6) |
| K2O + 2C → 2K + CO | (7) |
| CO2 + C → 2CO | (8) |
According to reactions (5) and (7), metallic element (K) will be generated,55 which is the key to the formation of VAGNAs, due to the catalytic graphitization and intercalation role of K. Generally speaking, biomass-derived carbon belongs to hard carbon, which is difficult to convert into ordered carbon even at a high temperature of 2500 °C.56 Having said that, the biomass-derived carbons by HSP method in this study have a remarkably high graphitization, as confirmed by the above Raman spectra and morphological characterizations. This is because the generated metal (K) acts as a catalyst, facilitating the graphitization of the biomass-derived carbon. Specifically, the metal (K) can react with the highly active sp3 hybrid carbon to yield double bonded carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, sp2) at higher temperature,57 thus promoting the formation of the graphite structure. Moreover, the intercalated metal element (K) can break the interlayer van der Waals bonds without breaking the covalent bonds within the layers, which increases the graphene lattice spacing to form a thinner graphene sheet. Simultaneously, a climb–glide type dislocation motion occurs in the internal lattice of graphene, which increases the ductility and plasticity of graphene.58,59 On the other hand, the released gases (CO and CO2) from reactions (5)–(8) are beneficial to the continuous upward peeling effect, leading to the vertical displacement of the graphene sheet.60 Finally, VAGNAs were successfully obtained.
C, sp2) at higher temperature,57 thus promoting the formation of the graphite structure. Moreover, the intercalated metal element (K) can break the interlayer van der Waals bonds without breaking the covalent bonds within the layers, which increases the graphene lattice spacing to form a thinner graphene sheet. Simultaneously, a climb–glide type dislocation motion occurs in the internal lattice of graphene, which increases the ductility and plasticity of graphene.58,59 On the other hand, the released gases (CO and CO2) from reactions (5)–(8) are beneficial to the continuous upward peeling effect, leading to the vertical displacement of the graphene sheet.60 Finally, VAGNAs were successfully obtained.
Compared to K2CO3, CH3COOK has the same cation ion (K+) and an analogous anion ion (CH3COO−). Similar reactions as those indicated by eqn (5)–(8) will occur during the salt-assisted pyrolysis process. Thus, nearly identical VAGNAs can be obtained when CH3COOK is employed. As for Na2CO3, it just begins to decompose at 1000 °C.61 The similar reactions (6) and (7) will not occur at this temperature, and Na+ has a weaker intercalation capability into the graphene layer. Thus, only thick lamellar structures were obtained under this condition. For Li2CO3, although the reaction between it and C (Li2CO3 + CLi2O + 2CO) can occur, Li2O is so stable that the lithium metal (Li) will not be formed by carbothermic reduction under the inert gas condition.62 Thus, the intercalation stripping is weak and no sheets are formed. When KCl or K3PO4 was used as activator, blocks were observed with some pores or particles on the surface. This is because neither metal (K) nor gas can be generated. K3PO4 has a slight etching effect, in that phosphate crystallization can inhibit the etching of molten salt.63 Another biomass (Spruce bark) was chosen as the precursor to examine the influence of salt. Nearly identical fine products were also achieved and are shown in Fig. S3,† which further confirmed the role of salt. The priority also obeyed the following order: K2CO3 > Na2CO3 ≫ Li2CO3 > KCl > K3PO4. Even so, it does not mean that salts including Li2CO3, KCl and K3PO4 are not appropriate for the fabrication of VAGNAs, as VAGNAs also can be fabricated by adjusting the pretreatment method and/or temperature.
As seen in the FE-SEM images (Fig. 8), the morphology of the lotus pollen-derived carbons changed significantly with the increase of the pyrolysis temperature. The carbon materials synthesized at 800 °C show a pristine spherical structure (Fig. 8a). The higher magnification images further confirm that the structure has a very smooth surface with few detachable particles (Fig. 8a′). When the pyrolysis temperature was 850 °C, the spherical structure was still maintained in the material (Fig. 8b). However, the surface showed irregular pits and some nanosheets can be observed (Fig. 8b′), which demonstrated that the graphene sheets began to form at this temperature. Nevertheless, the formed nanosheets were disconnected and thin with a lower height. As the pyrolysis temperature further increased up to 900 °C, more dense and continuous carbon sheets were observed, although they were not vertically aligned (Fig. 8c and c′). Fig. 8d and d′ shows the VAGNA-L-1000 prepared at 1000 °C. A fluffy 3D interconnection framework consisting of smooth carbon nanosheets was formed, and the nanosheets were much bigger than those at lower temperatures. In order to further validate the synergistic influences of the pretreatment, salt and temperature, the raw biomass (LP) was directly pyrolyzed without hydrothermal pretreatment at higher temperature (1100 °C) in the presence of K2CO3. Unfortunately, no VAGNA can be formed, as shown in Fig. S4.† This confirmed that the hydrothermal pretreatment was indispensable for the formation of the ideal VAGNA, and merely raising the temperature could not produce VAGNA.
 | ||
| Fig. 8 Low- and high-magnification FE-SEM images of hydrothermal-treated lotus pollen-derived VAGNAs pyrolyzed with K2CO3 at different temperatures: (a and a′) 800 °C, (b and b′) 850 °C, (c and c′) 900 °C and (d and d′) 1000 °C. | ||
To gain more insight on the effect of the pyrolysis temperature on the final products, another precursor (spruce bark) was also pyrolyzed with the aid of K2CO3 at various temperatures (850, 900 and 1000 °C), and the FE-SEM images are displayed in Fig. S5.† It is clear that the morphologies of the samples show a similar evolution trend with the increase of pyrolysis temperature. With higher temperature, the formation of VAGNAs becomes easier. This may be ascribed to the temperature-dependent reaction of salt and the intercalation capability of the metal (K). K2CO3 begins to decompose at 850 °C when reaction (2) occurs to yield metal (K),64 so carbon sheets are just starting to be generated, which in turn confirms the key role of the metal in the formation of VAGNAs. Moreover, the intercalation capability of metal (K) increases with the rise of temperature. Thus, VAGNAs were successfully obtained at higher temperature (1000 °C). Notably, carbon sheets began to form at 850 °C in the lotus pollen-derived carbons. Meanwhile, the sheets just began to form until 900 °C for the spruce bark-derived carbons, and they only formed in local regions such as the crevices and breakages in the matrix. This variation was caused by the difference of the natural structure of the precursor. Fig. S6† shows the FE-SEM images of the hydrothermal products of spruce bark and lotus pollen. It is seen that spruce bark displays a large and dense strip-like structure, while lotus pollen displays a small and loose ellipsoidal structure, which determine the difficulty of the formation of VAGNAs.
aman spectra and XRD patterns were employed to investigate the phase evolution of the VAGNAs with the pyrolysis temperature. As displayed in Fig. 9a, the sample (AC-L-800) prepared at 800 °C only shows the two common bands for the carbon materials, including the broad and strong D band at 1327 cm−1 and weak G band at 1582 cm−1.65–67 Meanwhile, the typical 2D band belonging to graphene is completely invisible, suggesting that no graphene sheet has been formed at 800 °C. When the pyrolysis temperature rose to 850, 900 and 1000 °C, the D band became increasingly weaker. Meanwhile, the G band became increasingly stronger. In addition, the typical 2D band started to emerge at 850 °C and grew sharper with the increase of the pyrolysis temperature. As seen from Table 2, the calculated ID/IG value decreased from 1.84 to 0.18, whereas the I2D/IG increased from 0.42 to 1.66. These results confirmed that carbon sheets were formed from 850 °C and the degree of graphitization was also enhanced. Thus, ideal VAGNAs with good crystallinity and thin sheets were obtained at 1000 °C. Fig. 9b shows the XRD patterns of these samples. It is clear that no sharp peaks can be found in the AC-L-800, indicative of its amorphous nature. The characteristic graphitic peaks oriented around 2θ = 26.0° and 43.2°, which correspond to the (002) and (100) planes, respectively,68 can be distinctly observed in the XRD patterns of the as-prepared VAGNAs with increasing pyrolysis temperature. Especially, the peak of the (002) plane became very sharp in VAGNA-L-1000, indicating the existence of the graphene sheet.69
 | ||
| Fig. 9 (a) Raman spectra, (b) XRD patterns, (c) nitrogen adsorption/desorption isotherms and (d) pore size distributions (PSDs) curves of lotus pollen-derived carbons fabricated at different temperatures. | ||
| Samples | SSABET (m2 g−1) | SSAmicro (m2 g−1) | SSAmeso (m2 g−1) | V total (cm3 g−1) | V micro (cm3 g−1) | V meso (cm3 g−1) | V micro/Vtotal (%) | I D/IG | I 2D/IG |
|---|---|---|---|---|---|---|---|---|---|
| AC-L-800 | 1856 | 1560 | 296 | 0.70 | 0.67 | 0.03 | 95.71 | 2.29 | — |
| VAGNA-L-850 | 1980 | 1288 | 692 | 0.90 | 0.69 | 0.21 | 76.67 | 1.84 | 0.42 |
| VAGNA-L-900 | 2250 | 1756 | 494 | 1.09 | 0.90 | 0.19 | 82.57 | 1.09 | 1.08 |
| VAGNA-L-1000 | 1997 | 1055 | 942 | 1.28 | 0.61 | 0.67 | 47.66 | 0.18 | 1.66 |
The variation of the pore structure for the as-prepared VAGNAs at different pyrolysis temperatures was also characterized by nitrogen adsorption/desorption measurements. The isotherms, PSDs plots and the corresponding textural parameters are displayed in Fig. 9c, d and Table 2. It is seen that AC-L-800 exhibits the typical type-I isotherm with a very sharp adsorption knee at low relative pressure and standard plateau at higher pressure, suggesting that its porosity is mainly composed of micropores.70 The calculated SSA of AC-L-800 is 1856 m2 g−1, in which the micropore SSA (SSAmicro) is 1560 m2 g−1. Furthermore, the micropore volume accounts for 95.71% of the total volume, verifying the predominance of micropores. When the temperature rose to 850 and 900 °C, small hysteresis loops beginning at P/P0 = 0.4 appeared in the isotherms of VAGNAL-850 and VAGNAL-900, representing the presence of a small number of mesopores.71 The PSDs curve in Fig. 9d clearly confirms the existence of mesopores, and the micropore volume declines to 76.67% and 82.57% (Table 2). As the pyrolysis temperature further increased to 1000 °C, the hysteresis loops became obvious and bigger, while the total adsorption amount did not increase significantly. This indicated that more mesopores were generated by enlarging the micropores. The total SSA of VAGNA-L-1000 is only 1997 m2 g−1, even is less than that of VAGNA-L-900 with 2250 m2 g−1. The change tendency of pores with pyrolysis temperature was close to the previous study, in which spruce bark was used as a carbon source with KOH as the activator.26 Taking into account the morphology change, microcrystalline and pore structure evolution, 1000 °C is the optimal temperature.
3.3 Electrochemical performance of VAGNAs
 | ||
| Fig. 10 Electrochemical performance of VAGNA-L-1000 in 1 M KOH electrolyte: (a) CV curves at different scanning rates, (b) GCD curves at different current densities, (c) variation of the magnification curve and Coulomb efficiency curve with current density. (d) The cycling performance of VAGNA-L-1000 at a current density of 10 A g−1. | ||
| [Fe(CN)6]3− + e → [Fe(CN)6]4− | (9) |
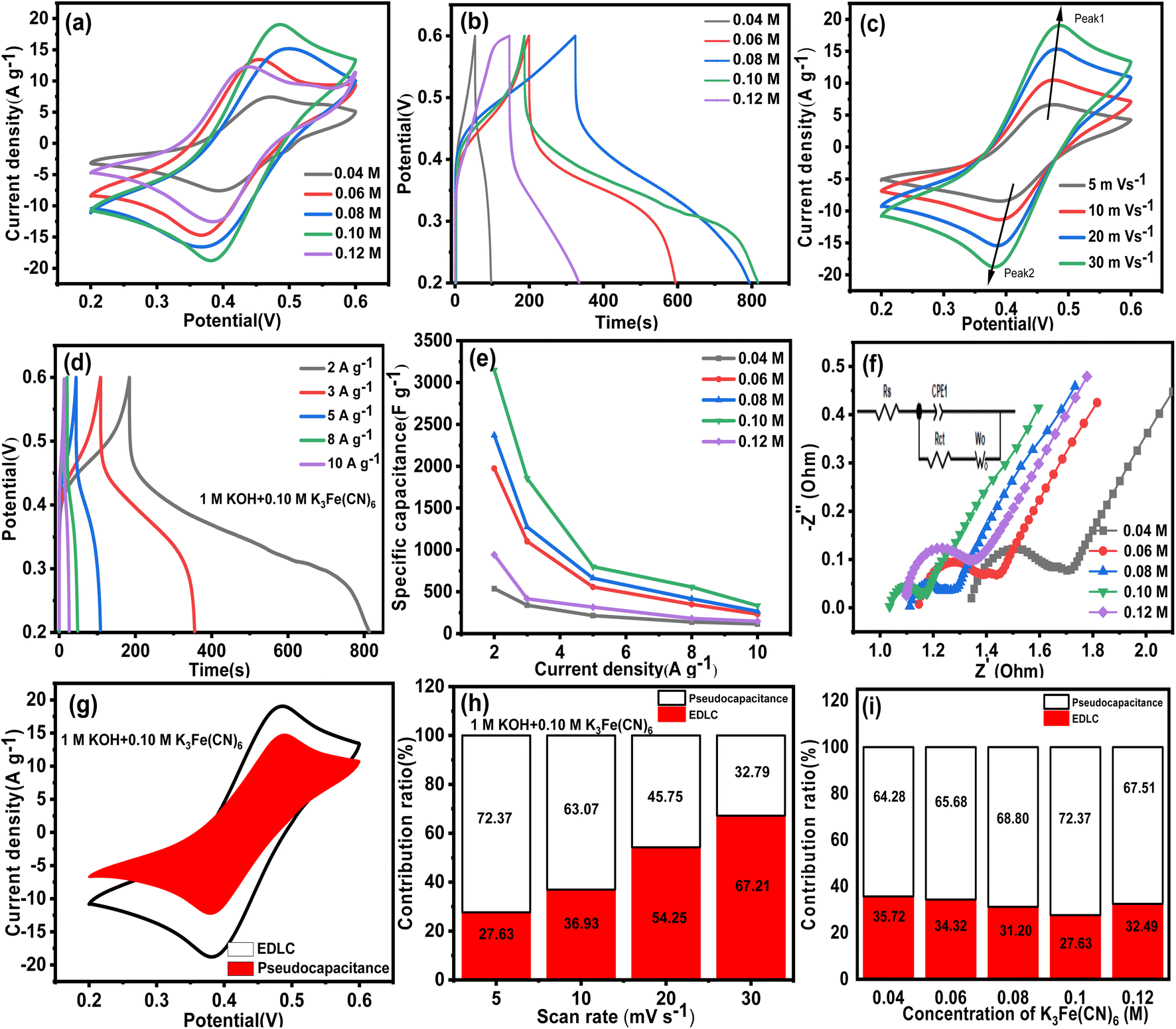 | ||
| Fig. 11 Electrochemical properties of VAGNA-L-1000 in 1 M KOH electrolyte with different concentrations of K3Fe(CN)6: (a) CV curves at a scan rate of 30 mV s−1, (b) GCD curve at a current density of 2 A g−1, (c) CV at different scan rates in 1 M KOH + 0.10 M K3Fe(CN)6, (d) GCD curves at different current densities in 1 M KOH + 0.10 M K3Fe(CN)6, (e) specific capacitances in 1 M KOH + K3Fe(CN) (0.04 M–0.12 M). (f) Electrochemical impedance spectroscopy (EIS) Nyquist plots for the VAGNA-L-1000 electrode in 1 M KOH solution with K3Fe(CN)6 (inset shows the equivalent circuit). (g) Capacitance contribution ratio diagram at a scan rate of 30 mV s−1 in 1 M KOH + 0.10 M K3Fe(CN)6. (h) Capacitance contribution ratio in 1 M KOH + 0.10 M K3Fe(CN)6 at different scan rates. (i) Capacitance contribution ratio diagram in 1 M KOH with the addition of different concentrations of K3Fe(CN)6 at a scan rate of 5 mV s−1. | ||
The observed oxidation peak is correlated with the charging process of [Fe(CN)6]4− to [Fe(CN)6]3−, while the reduction peak is attributed to the reverse process. It is proved that the [Fe(CN)6]3−/[Fe(CN)6]4− redox couple distinctly offers a pseudocapacitance contribution to the VAGNA-based electrode, but VAGNA in traditional electrolytes (KOH solution) has dominant electric double-layer capacitance.
Fig. 11c shows the CV curves in 1 M KOH + 0.10 M K3Fe(CN)6 under different scan rates from 5 to 30 mV s−1. With the increase of the scan rate, the oxidation peak shifted slightly to the positive potential, while the reduction peak gradually shifted to the negative potential. This is due to the polarization effect and internal resistance.73 Meanwhile, the intensities of the redox peaks increased with increasing scan rate, which is indicative of the higher electrochemical activity and better charge/discharge reversibility. Moreover, the shape of the CV curves showed minimal change as the scan rate increased from 5 to 30 mV s−1, which demonstrated that the VAGNA-K3Fe(CN)6 supercapacitors were excellent electron conductors with a small equivalent series resistance. Fig. 11d shows the GCD curves at various current densities in the electrolyte of 1 M KOH + 0.10 M K3Fe(CN)6 aqueous solutions. The charge/discharge curves are nonlinear, and the discharge time is much longer than the charge time. Furthermore, the calculated Coulomb efficiency reached high values of 341.6%, 227.1%, 143.9%, 133.6% and 101.5% for the current densities of 2, 3, 5, 8 and 10 A g−1, respectively. The greater Coulomb efficiency above 100% was also found in the redox supercapacitors,32,74,75 which can be interpreted by the initial addition of the redox additives (e.g., Fe(CN)63−). Based on eqn (5), in the charging process, Fe(CN)64− loses an electron and is oxidized completely into Fe(CN)63−. By contrast, in the discharging process, not all Fe(CN)63− gain electrons. Thus, only part of Fe(CN)63− will be reduced into Fe(CN)64−. Accordingly, the released charges contain both discharging and non-discharging Fe(CN)63−, which can be much more than the reserved charges in the charging process. Thus, the coulombic efficiency will be larger than 100%.
The specific capacitance was calculated according to the discharge branch, and the calculated values are depicted in Fig. 11e. As the concentration of K3Fe(CN)6 is 0.04, 0.06, 0.08, 0.10 and 0.12 M, the specific capacitances calculated at the current density of 2 A g−1 are 537, 1974, 2370, 3148, 942 F g−1, respectively. Compared with the specific capacitance (102.2 F g−1) in pristine 1 M KOH at the same current density, the capacitance increased significantly with the addition of redox additives into aqueous KOH electrolytes. When the concentration is 0.10 M, the maximal increase amplitude is about 30 times. In addition, the high capacitance can be maintained at higher current density, and the specific capacitances are 3148, 1854, 798.8, 558, and 333 F g−1 at 2, 3, 5, 8, and 10 A g−1, respectively. The resulting capacitance at a higher concentration of K3Fe(CN)6 is actually comparable with those of nitrogen-doped VAGNA films and RuO2-decorating VAGNA structures.34 To better understand the advantage of VAGNA in energy storage, the electrochemical performance of a commercial activated carbon (AC) electrode was also investigated in the same electrochemical system. Fig. S7† shows the electrochemical results of the AC-based electrode in a three-electrode system including the CV, GCD and EIS measurements. The CV curves were similar with those of VAGNA-L-1000 either in the pristine 1 M KOH or 1 M KOH + K3Fe(CN)6. The calculated specific capacitance in 1 M KOH was only 52.9 F g−1 at the current density of 1 A g−1, which was in accordance with the reported values in the previous study.34 In the 1 M KOH + 0.10 M K3Fe(CN)6 redox electrolyte, the specific capacitance was 625.5 F g−1 at 2 A g−1. It decreased to 90 F g−1 at 10 A g−1, which was far less than that of VAGNA.
The charge transfer resistance and reaction kinetics were also examined by EIS. Fig. 11f represents the EIS Nyquist for the VAGNA-L-1000 electrode in 1 M KOH containing various concentrations of K3Fe(CN)6. All the Nyquist plots exhibit a semicircle in the low frequency region and a straight line in the high frequency region. Furthermore, their positions and shapes show minimal change, indicating that the resistance should be ascribed to the redox additives and affected by the concentration. By analysing these Nyquist plots (Fig. 11f), the values of the solution resistance (Rs), the electrolyte and electrode interface of charge transfer resistance (Rct) and equivalent series resistance (ESR) can be obtained and are listed in Table 3. The Rs includes the intrinsic resistance of the electrode material, ionic resistance of the electrolyte, and the contact resistance between the electrode and current collector,76 and the ESR is the sum of Rs and Rct.77 Notably, the Rct values in every concentrations are very small for the redox electrolyte and VAGNA-based electrode. This is attributed to the formation of Fe(CN)63−/Fe(CN)64− redox pairs, and the unique structure of VAGNA with 3D vertically aligned graphene nanosheet being beneficial to the charge transfer. As the concentration of the redox additive increases, the ESR values firstly decreases and then increases, and the lowest ESR value is 1.13 Ω when 0.10 M K3Fe(CN)6 was added in 1 M KOH aqueous solution. Taking all of the above together, the low concentration of K3Fe(CN)6 resulted in a low specific capacitance due to a lower contribution of the redox faradaic reaction from K3Fe(CN)6/K4Fe(CN)6. By contrast, a high concentration of redox species can theoretically lead to a high specific capacitance. However, this will produce greater resistance, deteriorating the rate capacity and chemical stability. Consequently, it is crucial to select an appropriate concentration of redox species to assemble redox supercapacitors with satisfactory electrochemical performance, such as high capacitance, energy density, coulombic efficiency and long cycle stability.
| K3Fe(CN)6 conc. (M) | 0.04 | 0.06 | 0.08 | 0.10 | 0.12 |
|---|---|---|---|---|---|
| R s (Ω) | 1.36 | 1.16 | 1.13 | 1.06 | 1.10 |
| R ct (Ω) | 0.26 | 0.18 | 0.09 | 0.07 | 0.25 |
| ESR (Ω) | 1.62 | 1.34 | 1.22 | 1.13 | 1.35 |
To further elucidate the kinetic process and capacitance contribution of the VAGNA-based electrode in the redox electrolyte, the capacitance from EDLC and pseudocapacitance was evaluated according to Dunn's formula:78i = abv, i = k1v + k2v1/2. The total current response i is the sum of the surface-controlled k1v and diffusion-controlled k2v1/2 processes.78,79 In general, the b value ranges from 0.5 to 1.0, indicating that the electrode material possesses both diffusion-controlled and pseudocapacitive characteristics.6 The b value can be obtained from the slope of the line by plotting the logarithm of the measured peak current (logI) and scan rate (logV) (logi = blogv + loga). As displayed in Fig. S8,† the calculated values of b1 (anode peak) and b2 (cathode peak) were 0.5236 and 0.5875, respectively. Both are close to 0.50, which means that the kinetic process is a slow diffusion-controlled process, and the capacitance of VAGNA in 1 M KOH + 0.10 M K3Fe(CN)6 electrolyte is contributed by EDLC and pseudocapacitance.
The capacitance contribution from EDLC and pseudocapacitance was further evaluated. Fig. 11g shows the distribution diagram of capacitance at a scan rate of 30 mV s−1. It can be observed that most of the capacitance resulted from the pseudocapacitance at a higher concentration of redox additives. The quantitative results under different scan rates and various concentrations of K3Fe(CN)6 are plotted in Fig. 11h and i. The ratio of pseudocapacitance decreases with the increase of the scan rate, and the values are 72.37, 63.07, 45.75 and 32.79% at a scan rate of 5, 10, 20 and 30 mV s−1, respectively. Apparently, the pseudocapacitance accounted for an overwhelming percentage of the total capacitance at lower scan rates, and also contributed a certain percentage at higher scan rates because the redox reaction requires some time. These indicate that the pseudocapacitance from the redox reaction of K3Fe(CN)6/K4Fe(CN)6 plays a key role in enhancing the electrochemical performance. From Fig. 11i, the ratio of the pseudocapacitance at 5 mV s−1 is 64.28, 65.68, 68.80, 72.37 and 67.51%, corresponding to the concentrations of K3Fe(CN)6 of 0.04, 0.06, 0.08, 0.10 and 0.12 M, respectively. The ratio of pseudocapacitance also firstly increased, and then decreased with the increasing concentration of the redox additives, which is consistent with the trend of the ESR value. Therefore, 0.10 M is a proper concentration of K3Fe(CN)6 in this electrode–electrolyte system, and was chosen as the target concentration for further investigation.
Aside from the concentration of the redox additive, the concentration of the supporting electrolyte also has an important effect on the electrochemical performance of the redox supercapacitor. Thus, different concentrations of KOH (0.30, 1.0 and 3.0 M) were used as supporting electrolytes to construct the redox electrolyte, in which the concentration of K3Fe(CN)6 was fixed at 0.10 M. The electrochemical performance of the VAGNA-L-1000-based electrode in the prepared redox electrolytes was thoroughly investigated. Fig. 12a–c shows the CV curves of VAGNA-L-1000 in xM KOH + 0.10 M K3Fe(CN)6 (x = 0.30, 1.0 and 3.0). It is noticeable that the shape, area of CV covering, and positions of redox peak pairs are greatly affected by the concentration of KOH. When the concentration of KOH is 1.0 M, the shapes of the CV curves under all scan rates were basically symmetrical. Furthermore, a small distortion occurred with increasing scan rate from 5 to 30 mV s−1. Moreover, the positions of the redox peaks were relatively fixed at around 0.37 V and 0.45 V. In contrast, when the KOH concentration was 0.30 M or 3.0 M, the shape of the CV curves was not symmetrical and obvious distortions occurred under different scan rates. The positions of the redox peaks shifted significantly, and even the redox peaks were inconspicuous. This demonstrated that the redox electrolyte using 0.30 or 3.0 M KOH as a supporting electrolyte was not stable, leading to poor electrochemical performance. Fig. 12d compares the CV curves at all concentrations of KOH at a scan rate of 30 mV s−1. It can be found that the CV curve in 1.0 M KOH had the largest area, while the CV curve in 0.30 M KOH had the smallest area. In addition, the position and intensity of the redox peaks varied significantly. The standard potential for the Fe(CN)63−/Fe(CN)64− pairs is 0.37 V.80 The activity of Fe(CN)63−/Fe(CN)64− will gradually increase with increasing pH, resulting in better electrochemical performance. Nevertheless, under strongly alkaline condition, the actual potential of the redox electron pair will increase. This is due to the increase of the ionic strength and potential deviation,81 resulting in a deterioration of the electrochemical activity and energy storage performance.
 | ||
| Fig. 12 Electrochemical performances of the VAGNA-L-1000-based electrode in different concentrations of KOH added with 0.10 M K3Fe(CN)6 electrolyte: CV curves of (a) 0.3 M, (b) 1.0 M, (c) 3.0 M KOH at different scan rates. (d) CV curve comparison at a scan rate of 30 mV s−1. (e) GCD curves of different concentrations of KOH at a current density of 5 A g−1. (f) Specific capacitance at different current densities. (g) EIS Nyquist plots (inset shows the equivalent circuit). (h) Histogram of the EDLC contribution ratio at different scan rates for all electrochemical systems. | ||
Fig. 12e displays the GCD curves at a current density of 5 A g−1, and the specific capacitances at different current densities are illustrated in Fig. 12f. The discharge time in 1.0 M KOH + 0.10 M K3Fe(CN)6 is greater than those in 0.30 and 3.0 M KOH as the supporting electrolyte, indicating the optimal specific capacitance among them. The calculated specific capacitance at 2 A g−1 is 413.5 F g−1 and 1605 F g−1 for 0.30 M KOH and 3.0 M KOH + 0.10 M K3Fe(CN)6 redox electrolytes, respectively, which are far below than that (3148 F g−1) in 1.0 M KOH + 0.10 M K3Fe(CN)6. At other current densities, the specific capacitance in 1.0 M KOH + 0.10 M K3Fe(CN)6 is always greater than those of others. Fig. 12g shows the Nyquist plots in the three systems with different KOH concentrations. When the concentration of KOH was 1.0 M, the smallest semicircle was measured compared with the other concentrations. The calculated Rs values are 1.30, 1.06 and 0.98 Ω, corresponding to the KOH concentration of 0.30, 1.0 and 3.0 M, respectively, while the Rct values are 0.28, 0.07 and 0.16 Ω. The smallest ESR is 1.13 Ω measured in 1.0 M KOH + 0.10 M K3Fe(CN)6. KOH has an activating effect on K3Fe(CN)6, leading to the enhancement of the electrochemical performance. The low concentration of KOH only improves the electrochemical performance to a small extent due to the limited activation effect on the redox electrolyte, while the high concentration of KOH can activate the redox electrolyte performance more thoroughly. However, it will affect the peak potential shift, resulting in an insufficient redox reaction and ultimately lowering the electrochemical performance. Therefore, the appropriate concentration of KOH is essential for the selection of redox electrolytes involving K3Fe(CN)6. Fig. 12h depicts the histogram of the capacitance contribution in the three redox electrolytes. Under all of the scan rates, the highest ratio of pseudocapacitance was obtained in 1.0 M KOH + 0.10 M K3Fe(CN)6, demonstrating that the maximal pseudocapacitance can be acquired in a moderate concentration (1.0 M) of KOH as the supporting electrolyte.
From the above results, the electrochemical performance of VAGNA-L-1000-based electrode was greatly enhanced in KOH + K3Fe(CN)6 redox electrolyte with proper concentrations. This is mainly attributed to the addition of the redox-active additive (K3Fe(CN)6), which has the capability to produce both an electric double layer and a surface redox activity due to the redox activity provided by its own “electron shuttling effect”.82 According to eqn (5), both reactions move forward (using it as the cathode) upon discharging. [Fe(CN)6]4− is generated by the reduction of [Fe(CN)6]3− on the anode and flows into the bank.83 At the same time, the oxidized species [Fe(CN)6]3− circulates back to the cell regenerate [Fe(CN)6]4−, starting a new round of reactions. During this process, K+ ions are transported from the counter electrode and are stored in VAGNA. Upon charging, the above redox reactions are reversible. Moreover, the Density-functional-theory (DFT) calculations on the redox system containing K3Fe(CN)6 show that the [Fe(CN)6]4− presence increases the Fermi energy level, and significantly reduces the work function of the whole energy system.84 The reversible reaction between [Fe(CN)6]3−/[Fe(CN)6]4− and VAGNA transmits an electron/charge, and the energy is then reversibly stored in the electrode materials, thus significantly improving the electrochemical performance.
For testing the performance in practical applications, a two-electrode symmetrical supercapacitor device was then assembled, employing two identical VAGNA-L-1000 s as the capacitor electrodes. The electrochemical performance of the as-assembled device was evaluated in the redox electrolyte of 1.0 M KOH + 0.10 M K3Fe(CN)6 using CV and GCD. CV measurements were performed at different scan rates (5, 10, 20, 30, 50 and 100 mV s−1) with an operation voltage of 2.0 V. As observed from Fig. 13a, the CV curves exhibited a pair of prominently well-defined peaks, similar shapes as those measured in the three-electrode system, which doubtlessly resulted from the redox reaction of [Fe(CN)6]3−/[Fe(CN)6]4−. With increasing scan rate, a similar increase in the curve areas occurred as those in the three-electrode system. To calculate the specific capacitance at a constant current, GCD measurements were also carried out. Fig. 13b shows the GCD profiles of the VAGNA-based redox supercapacitor at current densities of 1, 2, 3, 5, 8 and 10 A g−1. An evident charging/discharging plateau occurs at a voltage of 0 V, relating to the redox reaction of [Fe(CN)6]3−/[Fe(CN)6]4−, which greatly increases the electrochemical storage/release capacity. As shown in Fig. 13c, specific capacitances of 92.8, 67.7, 55.8, 47.5, 34.8, and 30 F g−1 were achieved at current densities from 2 to 10 A g−1.
 | ||
| Fig. 13 Electrochemical performances of the VAGNA-L-1000-based symmetrical supercapacitor device: (a) CV curves at different scan rates. (b) GCD profiles at different current densities. (c) Specific capacitance at different current densities. (d) EIS Nyquist plots (inset: zoomed-in Nyquist plots under a high-frequency region). (e) Cycling stability tests at a current density of 10 A g−1 and (f) Ragone plots for the VAGNA electrodes-derived redox supercapacitor in comparison with other reported supercapacitors. | ||
It is interesting that the potential window is up to 2.0 V. This potential window is higher than the previously reported value of 1.0 V using VG@Ni films55 and 1.6 V using MnO2/rGO85 as electrodes with the same KOH + K3Fe(CN)6 redox electrolyte. The higher potential window is mainly attributed to the addition of the redox additive and the electrode materials. K3Fe(CN)6 has a redox potential near the window of H2O decomposition, and the kinetics of the redox mediator is faster than that of water electrolysis as an electrolyte additive, thus reducing the activity of water electrolysis.86,87 Additionally, the decomposition voltage between the Fe(CN)63−/Fe(CN)64− redox pairs is close to that between water and electrolyte. The high solvation energy of the Fe(CN)63−/Fe(CN)64− oxidation pairs leads to the tendency of the energy in initiating the decomposition of conventional electrolytes to drive the redox reaction of the redox pairs, which protects the solvent water from decomposition and widens the operating voltage window.88,89 Besides the active additive, the unique structure of VAGNA also plays a key role in widening the potential window. This is due to the rapid ion/electron transfer originating from the 3D structure with ultrathin nanosheet. The electrocatalytic behavior of VAGNA towards Fe(CN)63−/Fe(CN)64− indicates the dominant contribution of graphene in the electron transfer kinetics. Furthermore, it is recognized that the capacitive behavior of VAGNAs is mainly derived from the surface graphene layers90 because ion adsorption/desorption generally occurs at/near the surface of the electrode during the charge/discharge process for both the EDLC and pseudocapacitor. The as-fabricated VAGNA has an ultrathin graphene nanosheet and aligns vertically with enough accessible surface area, which is beneficial to the electrochemical process, leading to the excellent electrochemical performance.
The facilitated Ion accessibility of the as-fabricated VAGNA-based electrode was verified by EIS study. Fig. 13d exhibits the EIS Nyquist of the device based on the as-fabricated VAGNA-L-1000 and the redox electrolyte of 1.0 M KOH + 0.10 M K3Fe(CN)6. The steeper slope in the low-frequency region for VAGNA indicates a high ion diffusion nature, and the small semicircle in the high-frequency region suggests low resistance. The calculated ESR value is just 0.88 Ω, which is less than that measured in the three-electrode system, indicative of the outstanding electric conductivity of VAGNA. The long-term cycling stability of the VAGNA-L-1000-based device was further examined using the GCD test at the current density of 10 A g−1 and is shown in Fig. 13e. The device still retained 84.6% of its initial specific capacitance after 2500 cycles. As described above, the VAGNA-based redox supercapacitors have multiple merits, such as ultrahigh specific capacitance, excellent rate capacity and cycling stability. In addition, the energy density (Ecell, W h kg−1) and power density (P, W kg−1) are key parameters for evaluating the supercapacitors. The Ragone plots of the as-fabricated VAGNA-based redox supercapacitor are illustrated in Fig. 13f, and compared with those of other supercapacitors. The energy densities of the assembled device are 36.7, 27.7, 22.6, 19.5, 13.6 and 11.8 W h kg−1 at power densities of 712, 1472, 2187, 3702, 5628 and 7080 W kg−1, respectively, which are much higher than those of other reported VAGNAs or carbon-based supercapacitors.91–98
4. Conclusion
A novel method was developed for the gram-scale fabrication of high-quality vertically aligned graphene nanosheets (VAGNAs) from multifarious kinds of biomasses. This method consists of a hydrothermal pretreatment step, followed by a salt-assisted pyrolysis (HSP) step, under an inert atmosphere at higher temperature. The key fabrication process parameters were screened and optimized, such as the pretreatment method, selected salts and pyrolysis temperature. It was found that the hydrothermal treatment of the biomass precursor was the optimal pretreatment for obtaining VAGNAs. Both cation and anion ions of the salt synergistically determine the structure of the final products. K2CO3 and CH3COOK are ideal salts for fabricating VAGNAs in the HSP process. Moreover, the pyrolysis temperature has an obvious effect on the final products, with 1000 °C being an appropriate temperature for obtaining VAGNAs. Compared with the well-established CVD methods for fabricating VAGNAs, which require expensive instruments and unsustainable gas (CH4/H2, C2H2/H2, etc.) as the carbon source and additional substrates, the as-developed HSP method in this study has obvious advantages, such as low-cost, green chemistry, universality and high-efficiency. VAGNAs can be fabricated on a large scale by this HSP method from low-cost, green and renewable biomass instead of fossil gas precursors. Furthermore, no expensive instrument or extra substrate is required.In view of these advantages, we designed a novel redox supercapacitor with the as-fabricated VAGNA electrodes in KOH electrolyte with the redox additive of K3Fe(CN)6. The VAGNA electrodes possess an ultrahigh specific capacitance of 3148 F g−1 in the 1.0 M KOH electrolyte with the addition of 0.10 M K3Fe(CN)6. The assembled symmetrical redox supercapacitor based on VAGNA-L-1000 delivers a high specific capacitance of 92.8 F g−1 at 1 A g−1 under a wide operation potential window of 2.0 V, and high energy density of 36.7 W h kg−1 at the power density of 712 W kg−1. These outstanding electrochemical performances were mainly attributed to the unique 3D structure of the as-fabricated VAGNA materials, high electronic conductivity of the VAGNA-based electrode, faradaic properties of the redox electrolyte and the synergistic effects between the VAGNA and redox electrolyte. The as-fabricated VAGNAs exhibit outstanding properties, including high graphitization degree, thin nanosheet and good hydrophilicity, as well as a developed hierarchically porous structure, rendering them as ideal electrode materials. This study highlights the large-scale synthesis of high-value nanomaterials derived from biomass for developing high-performance supercapacitors with high energy density in an economical way.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the Fund of National Natural Science Foundation of China (No. 52063016, No. 52372048), Science and Technology Project Founded by the Education Department of Hubei Province (No. D20211102) and the Innovation Project of Key Laboratory of Novel Biomass-based Environmental and Energy Materials in Petroleum and Chemical Industry (22BEEA05).References
- Y. Tong, J. Yang, J. Li, Z. Cong, L. Wei, M. Liu, S. Zhai, K. Wang and Q. An, J. Mater. Chem. A, 2023, 11, 1061–1082 RSC.
- Z. Bi, Q. Kong, Y. Cao, G. Sun, F. Su, X. Wei, X. Li, A. Ahmad, L. Xie and C.-M. Chen, J. Mater. Chem. A, 2019, 7, 16028–16045 RSC.
- K. A. Owusu, L. Qu, J. Li, Z. Wang, K. Zhao, C. Yang, K. M. Hercule, C. Lin, C. Shi, Q. Wei, L. Zhou and L. Mai, Nat. Commun., 2017, 8, 14264 CrossRef.
- J. Lin, H. Jia, Y. Cai, S. Chen, H. Liang, X. Wang, F. Zhang, J. Qi, J. Cao, J. Feng and W.-d. Fei, J. Mater. Chem. A, 2018, 6, 908–917 RSC.
- J. Mao, J. Iocozzia, J. Huang, K. Meng, Y. Lai and Z. Lin, Energy Environ. Sci., 2018, 11, 772–799 RSC.
- Y. Mei, S. Liu, L. Wu, B. Zhou, Z. Wang, Z.-H. Huang, Y. Zhu and M.-X. Wang, J. Energy Storage, 2024, 77, 109878 CrossRef.
- D. He, Y. Gao, Y. Yao, L. Wu, J. Zhang, Z.-H. Huang and M.-X. Wang, Front. Chem., 2020, 8, 719 CrossRef CAS PubMed.
- J. Yin, W. Zhang, N. A. Alhebshi, N. Salah and H. N. Alshareef, Small Methods, 2020, 4, 1900853 CrossRef CAS.
- X. Zhang, L. Hou, A. Ciesielski and P. Samorì, Adv. Energy Mater., 2016, 6, 1600671 CrossRef.
- K. Chen, S. Song, F. Liu and D. Xue, Chem. Soc. Rev., 2015, 44, 6230–6257 RSC.
- X. Yang, C. Cheng, Y. Wang, L. Qiu and D. Li, Science, 2013, 341, 534–537 CrossRef CAS.
- Z. Zhang, C. Lee and W. Zhang, Adv. Energy Mater., 2017, 7, 1700678 CrossRef.
- Z. Bo, Y. Yang, J. Chen, K. Yu, J. Yan and K. Cen, Nanoscale, 2013, 5, 5180–5204 RSC.
- Z. Xu, W. Deng and X. Wang, Electrochem. Energy Rev., 2021, 4, 269–335 CrossRef CAS.
- E. Anghel, B. Adiaconita, I. Demetrescu and A. Avram, Coatings, 2023, 13, 761 CrossRef CAS.
- W. Zheng, X. Zhao and W. Fu, ACS Appl. Mater. Interfaces, 2021, 13, 9561–9579 CrossRef CAS.
- F. Zhou, J. Shan, L. Cui, Y. Qi, J. Hu, Y. Zhang and Z. Liu, Adv. Funct. Mater., 2022, 32, 2202026 CrossRef CAS.
- Z. Hu, Z. Li, Z. Xia, T. Jiang, G. Wang, J. Sun, P. Sun, C. Yan and L. Zhang, Energy Storage Mater., 2019, 22, 29–39 CrossRef.
- Z. Bo, S. Cui, K. Yu, G. Lu, S. Mao and J. Chen, Rev. Sci. Instrum., 2011, 82, 086116 CrossRef.
- J. Deng, S. Wu, Y. Yang, R. Zheng and G. Cheng, Nucl. Instrum. Methods Phys. Res., Sect. B, 2013, 307, 177–180 CrossRef CAS.
- Y. Wu, P. Qiao, T. Chong and Z. Shen, Adv. Mater., 2002, 14, 64–67 CrossRef CAS.
- H. Shi, W. Wu, F. Wei and Q. Chen, FlatChem, 2021, 30, 100306 CrossRef CAS.
- X. Tang, D. Liu, Y. Wang, L. Cui, A. Ignaszak, Y. Yu and J. Zhang, Prog. Mater. Sci., 2021, 118, 100770 CrossRef CAS.
- Z. Wang, M. Shoji, K. Baba, T. Ito and H. Ogata, Carbon, 2014, 67, 326–335 CrossRef CAS.
- Z. Wang, H. Ogata, S. Morimoto, M. Fujishige, K. Takeuchi, Y. Hashimoto and M. Endo, Carbon, 2014, 72, 421–424 CrossRef CAS.
- Z. Sun, M. Zheng, H. Hu, H. Dong, Y. Liang, Y. Xiao, B. Lei and Y. Liu, Chem. Eng. J., 2018, 336, 550–561 CrossRef CAS.
- D. Yan, Y. Wu, R. Kitaura and K. Awaga, J. Mater. Chem. A, 2019, 7, 26829–26837 RSC.
- P. Li, H. Wang, W. Fan, M. Huang, J. Shi, Z. Shi and S. Liu, Chem. Eng. J., 2021, 421, 129704 CrossRef CAS.
- J. Xia, F. Chen, J. Li and N. Tao, Nat. Nanotechnol., 2009, 4, 505–509 CrossRef CAS PubMed.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- N. Yang, S. Yu, W. Zhang, H.-M. Cheng, P. Simon and X. Jiang, Adv. Mater., 2022, 34, 2202380 CrossRef CAS.
- C. Zhao, W. Zheng, X. Wang, H. Zhang, X. Cui and H. Wang, Sci. Rep., 2013, 3, 2986 CrossRef.
- C. Wei, R. Zhang, X. Zheng, Q. Ru, Q. Chen, C. Cui, G. Li and D. Zhang, Inorg. Chem. Front., 2018, 5, 3126–3134 RSC.
- T. M. Dinh, A. Achour, S. Vizireanu, G. Dinescu, L. Nistor, K. Armstrong, D. Guay and D. Pech, Nano Energy, 2014, 10, 288–294 CrossRef CAS.
- M. Wang, M. Huang, D. Luo, Y. Li, M. Choe, W. K. Seong, M. Kim, S. Jin, M. Wang, S. Chatterjee, Y. Kwon, Z. Lee and R. S. Ruoff, Nature, 2021, 596, 519–524 CrossRef CAS PubMed.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60–63 CrossRef CAS.
- D. Liu, T. Zhang, X. Cheng, B. Wang, Y. Guo, Z. Liu, H. Jiang and Y. Lu, Small, 2022, 18, e2200037 CrossRef PubMed.
- B. Basak, R. Kumar, A. V. S. L. S. Bharadwaj, T. H. Kim, J. R. Kim, M. Jang, S.-E. Oh, H.-S. Roh and B.-H. Jeon, Bioresour. Technol., 2023, 369, 128413 CrossRef CAS PubMed.
- H. Wang, K. Sun, F. Tao, D. J. Stacchiola and Y. H. Hu, Angew. Chem., 2013, 125, 9380–9384 CrossRef.
- D. He, Y. Gao, Z. Wang, Y. Yao, L. Wu, J. Zhang, Z.-H. Huang and M.-X. Wang, J. Colloid Interface Sci., 2021, 581, 238–250 CrossRef CAS PubMed.
- Z. Wang, Y. Li, J. Liu, G. Tian, G. Liu, M. Wang, H. Ogata, W. Gong, A. K. Vipin, G. J. Hong Melvin, J. Ortiz-Medina, S. Morimoto, Y. Hashimoto, M. Terrones and M. Endo, Carbon, 2021, 172, 26–30 CrossRef CAS.
- M. Qi, Y. Zhang, Q. Li, L. Wu, B. Zhou, Z. Wang, Z. Huang and M. Wang, Chem. Eng. J., 2023, 474, 145823 CrossRef CAS.
- Z. Zhu, X. Yang, X. Ye, Q. Li, J. Wang, L. Wu, Z.-H. Huang and M.-X. Wang, Sep. Purif. Technol., 2024, 127456 CrossRef CAS.
- G. Li, S. Wang, J. Zeng and J. Yu, Carbon, 2021, 171, 111–118 CrossRef CAS.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- D. He, L. Wu, Z. Wang, Z.-H. Huang and M.-X. Wang, Carbon, 2021, 184, 340–345 CrossRef CAS.
- C. Li, D. He, Z. Huang and M. Wang, J. Electrochem. Soc., 2018, 165, A3334–A3341 CrossRef CAS.
- M. Wang, D. He, M. Zhu, L. Wu, Z. Wang, Z. Huang and H. Yang, J. Power Sources, 2023, 560, 232703 CrossRef CAS.
- H. Wang, Z. Xu, A. Kohandehghan, Z. Li, K. Cui, X. Tan, T. J. Stephenson, C. K. King’ondu, C. M. B. Holt, B. C. Olsen, J. K. Tak, D. Harfield, A. O. Anyia and D. Mitlin, ACS Nano, 2013, 7, 5131–5141 CrossRef CAS PubMed.
- R. Wang, Z. Lin, S. Meng, S. Liu, Z. Zhao, C. Wang and Q. Yin, Energy, 2022, 259, 125063 CrossRef CAS.
- Y. Gong, L. Xie, C. Chen, J. Liu, M. Antonietti and Y. Wang, Prog. Mater. Sci., 2023, 132, 101048 CrossRef CAS.
- J. H. Bang and K. S. Suslick, Adv. Mater., 2010, 22, 1039–1059 CrossRef CAS.
- Q. Xu, M. Zhao, Z. Yu, J. Yin, G. Li, M. Zhen and Q. Zhang, Ind. Crop. Prod., 2017, 109, 220–226 CrossRef CAS.
- J. i. Hayashi, T. Horikawa, I. Takeda, K. Muroyama and F. N. Ani, Carbon, 2002, 40, 2381–2386 CrossRef CAS.
- E. Irisarri, A. Ponrouch and M. R. Palacin, J. Electrochem. Soc., 2015, 162, A2476–A2482 CrossRef CAS.
- Y. Xi, D. Yang, X. Qiu, H. Wang, J. Huang and Q. Li, Ind. Crop. Prod., 2018, 124, 747–754 CrossRef CAS.
- M. M. M. Ahmed, T. Imae, J. P. Hill, Y. Yamauchi, K. Ariga and L. K. Shrestha, Colloids Surf., A, 2018, 538, 127–132 CrossRef CAS.
- W. Zhou, J. Wang, B. Lin, Z. Wang, J. Li and Z.-F. Huang, Carbon, 2019, 153, 242–256 CrossRef CAS.
- J. Zhou, Z. Lin, H. Ren, X. Duan, I. Shakir, Y. Huang and X. Duan, Adv. Mater., 2021, 33, e2004557 CrossRef PubMed.
- J.-W. Kim and H.-G. Lee, Metall. Mater. Trans. B, 2001, 32, 17–24 CrossRef.
- D. W. McKee, Fuel, 1983, 62, 170–175 CrossRef CAS.
- S. Huang, J. Wang, X. Wei, Y. Zhou, L. Wang and J. Zhang, RSC Adv., 2019, 9, 18767–18775 RSC.
- M. Sevilla and A. B. Fuertes, ChemSusChem, 2016, 9, 1880–1888 CrossRef CAS PubMed.
- X. Ye, L. Wu, M. Zhu, Z. Wang, Z.-H. Huang and M.-X. Wang, Sep. Purif. Technol., 2022, 300, 121899 CrossRef CAS.
- M. Xu, L. Wu, M. Zhu, Z. Wang, Z.-H. Huang and M.-X. Wang, Carbon, 2022, 193, 242–257 CrossRef CAS.
- D. He, L. Wu, Y. Yao, J. Zhang, Z.-H. Huang and M.-X. Wang, Appl. Surf. Sci., 2020, 507, 145108 CrossRef CAS.
- Y. Zhang, M. Zhu, Q. Wei and M. Wang, Atmosphere, 2022, 13, 2074 CrossRef CAS.
- X. Wang, Y. Zhang, C. Zhi, X. Wang, D. Tang, Y. Xu, Q. Weng, X. Jiang, M. Mitome, D. Golberg and Y. Bando, Nat. Commun., 2013, 4, 2905 CrossRef PubMed.
- Y. Gao, D. He, L. Wu, Z. Wang, Y. Yao, Z.-H. Huang, H. Yang and M.-X. Wang, Chem. Eng. J., 2021, 420, 127411 CrossRef CAS.
- Q. Wei, L. Wu, M. Zhu, Z. Wang, Z.-H. Huang and M.-X. Wang, iScience, 2023, 26, 106155 CrossRef CAS PubMed.
- J. F. Gao, J. F. Hou and L. B. Kong, Electrochim. Acta, 2023, 439, 141692 CrossRef CAS.
- X. Chen, Y. Li, M. He, B. Zhou, D. Cheng, S. Guo, K. Xu, C. Yuan, M. Wang and H. Ogata, J. Power Sources, 2023, 575, 233183 CrossRef CAS.
- Y. Chen, Y. Ma, J. Huang and H. Xu, Electrochim. Acta, 2022, 414, 140215 CrossRef CAS.
- T. M. Patil, A. H. Patil, M. Krishnaiah, D. Mishra, A. Kumar, N. Kumar, V. D. Chavan, D.-K. Kim, S. B. Patil and S. B. Sadale, J. Energy Storage, 2023, 63, 106945 CrossRef.
- P. K. Stys, B. R. Ransom and S. G. Waxman, Brain Res., 1991, 546, 18–32 CrossRef CAS PubMed.
- L. Suárez, V. Barranco and T. A. Centeno, J. Colloid Interface Sci., 2021, 588, 705–712 CrossRef PubMed.
- Y. Sun, D. Xu, Z. He, Z. Zhang, L. Fan and S. Wang, J. Mater. Chem. A, 2023, 11, 20011–20020 RSC.
- X. Wei, B. Qiu, H. Tian, Y. Lv, W. Zhang, Q. Qin, Z. Liu and F. Wei, Appl. Surf. Sci., 2023, 615, 156280 CrossRef CAS.
- P. A. Rock, J. Phys. Chem., 2002, 70, 576–580 CrossRef.
- V. P. Shilov and A. V. Gogolev, Russ. J. Gen. Chem., 2009, 79, 1773–1777 CrossRef CAS.
- L. Mai, A. Minhas-Khan, X. Tian, K. M. Hercule, Y. Zhao, X. Lin and X. Xu, Nat. Commun., 2013, 4, 2923 CrossRef PubMed.
- Y. Chen, M. Zhou, Y. Xia, X. Wang, Y. Liu, Y. Yao, H. Zhang, Y. Li, S. Lu, W. Qin, X. Wu and Q. Wang, Joule, 2019, 3, 2255–2267 CrossRef CAS.
- S. Sun, D. Rao, T. Zhai, Q. Liu, H. Huang, B. Liu, H. Zhang, L. Xue and H. Xia, Adv. Mater., 2020, 32, 2005344 CrossRef CAS.
- N. Kumar, V. Gajraj, R. Rameshbabu, R. V. Mangalaraja, N. C. Joshi and N. Priyadarshi, Mater. Charact., 2022, 189, 377–387 Search PubMed.
- Q. Gou, S. Zhao, J. Wang, M. Li and J. Xue, Nano-Micro Lett., 2020, 12, 98 CrossRef CAS PubMed.
- S.-E. Chun, B. Evanko, X. Wang, D. Vonlanthen, X. Ji, G. D. Stucky and S. W. Boettcher, Nat. Commun., 2015, 6, 7818 CrossRef CAS PubMed.
- M. Maher, S. Hassan, K. Shoueir, B. Yousif and M. E. A. Abo-Elsoud, J. Mater. Res. Technol., 2021, 11, 1232–1244 CrossRef CAS.
- J. Y. Hwang, M. Li, M. F. El-Kady and R. B. Kaner, Adv. Funct. Mater., 2017, 27, 1605745 CrossRef.
- E. Irisarri, A. Ponrouch and M. R. Palacin, J. Electrochem. Soc., 2015, 162, A2476–A2482 CrossRef CAS.
- C. Zhang, Z. Peng, J. Lin, Y. Zhu, G. Ruan, C.-C. Hwang, W. Lu, R. H. Hauge and J. M. Tour, ACS Nano, 2013, 7, 5151–5159 CrossRef CAS.
- H. Shen, H. Li, M. Li, C. Li, L. Qian, L. Su and B. Yang, Electrochim. Acta, 2018, 283, 410–418 CrossRef CAS.
- H. Jiang, X. Cai, Y. Qian, C. Zhang, L. Zhou, W. Liu, B. Li, L. Lai and W. Huang, J. Mater. Chem. A, 2017, 5, 23727–23736 RSC.
- W. Yan, Z. Meng, M. Zou, H. Miao, F. Ma, R. Yu, W. Qiu, X. Y. Liu and N. Lin, Chem. Eng. J., 2020, 381, 122547 CrossRef CAS.
- J. Wang, Y. Huang, Y. Gao, J. Dai and X. Sun, J. Energy Storage, 2022, 51, 104581 CrossRef.
- D. Xuan, J. Liu, D. Wang, Z. Lu, Q. Liu, Y. Liu, S. Li and Z. Zheng, Energy Fuels, 2020, 35, 796–805 CrossRef.
- F. Miao, C. Shao, X. Li, K. Wang and Y. Liu, J. Mater. Chem. A, 2016, 4, 4180–4187 RSC.
- X. Wang, W. Li, Y. Xue, J. Liu, Y. Yue, C. Zhou, K. Zhu, D. Cao, Y. Chen and G. Wang, J. Energy Storage, 2022, 50, 104220 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01328j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |