CeNiO3 perovskite nanoparticles synthesized using gelatin as a chelating agent for CO2 dry reforming of methane†
Received
3rd June 2024
, Accepted 22nd October 2024
First published on 30th October 2024
Abstract
CeNiO3 perovskite nanoparticles were synthesized using gelatin as a chelating agent to catalyze the CO2 dry reforming reaction. The optimization of gelatin concentration affects the formation of Ni coordinated on the B-sites of CeNiO3. CeNiO3 shows catalytic stability for 60 h with ∼50% CO2/CH4 conversion and a H2/CO ratio of ∼0.8 when obtained using low concentrations of gelatin (0.05 g, 0.1 g). The exsolution of Ni nanoparticles from CeNiO3 structures occurs during reduction treatment, expanding the crystal lattices of CeO2. A high gelatin concentration (0.2 g) reduced Ni mobility and restricted Ni–Ce contacts. The uncoordinated Ni in CeNiO3 exposes a high surface area (10.12 m2 g−1) for CH4 dissociation, consequently increasing the H2/CO ratio to ∼1.5 with 78% CH4 and 53% CO2 conversion. In situ DRIFTS analysis showed that CH4 readily dissociates in the absence of CO2, but CO2 completes the reaction cycles by removing the carbon as CO gas.
Sustainability spotlight
The exponential rise in energy demand is responsible for the depletion of natural fossil fuels and the rapid increase in Earth's temperature. The capture and utilization of CO2 pave the way for a carbon-free industry. Dry reforming of methane (DRM) efficiently uses two greenhouse gases, carbon dioxide and methane, to produce synthesis gas (CO + H2) as feedstock in the Fischer–Tropsch reaction. This study aims to utilize gelatin as a chelating agent for synthesizing CeNiO3 perovskite to enhance stability for CO2 dry reforming. The H2/CO ratio relies on Ni coordination that can be controlled by variations in gelatin concentration. We emphasize the route to achieve SDG 7 (affordable and clean energy) and SDG 13 (climate action) of the UN sustainable development goals.
|
Introduction
The exponential rise of fossil fuel usage to sustain global energy demand is responsible for the depletion of natural fossil fuels and the rapid increase in CO2 levels in the atmosphere.1 The Paris Agreement of 2015 aimed to reduce greenhouse gas emissions to limit global temperature rise to 1.5 °C.2,3 The agreement encouraged the energy sector to adopt more sustainable and greener sources,4 paving a path for carbon-neutral energy. Dry reforming of methane (DRM) uses two greenhouse gases, carbon dioxide and methane, to produce synthesis gas (CO + H2) as feedstock in the Fischer–Tropsch reaction.5 The ideal H2/CO ratio is one, but side reactions such as reverse water gas shift (RWGS) and Boudouard reactions cause variations in the H2/CO ratio.6| | 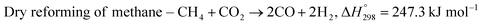 | (1) |
| | 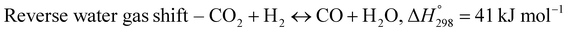 | (2) |
| | 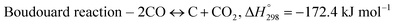 | (3) |
The main challenge in dry reforming is the highly endothermic reaction for co-activation of the C–O bond in CO2 and the C–H bond in CH4. Due to thermodynamic constraints, many developments have been made for high-temperature DRM (>700 °C),7 causing thermal degradation of catalysts and carbon deposition.8,9 Designing stable catalysts with carbon-resistant properties can enhance catalyst stability.10 Metal oxides with stable structures, such as pyrochlore, perovskites, and mixed oxides, have been employed to improve structural stability.11 Ce-based catalysts exhibited high performance in DRM due to the oxygen mobility in ceria that inhibits carbon deposition.12 The general formula of ABO3 perovskite consists of metallic cations with different ionic radii occupying the A and B sites of the cubic structure, with the small metal occupying the B site. Under a reducing environment such as in dry reforming reactions, Ni on the B-sites of CeNiO3 perovskite exsolves to form highly dispersed Ni nanoparticles.13 The multivalency of perovskite from rare earth elements offers excellent redox properties and oxygen vacancies. Perovskite comprises 60% oxygen atoms that can facilitate carbon gasification.14
The synthesis of CeNiO3 that requires high calcination temperatures up to 800 °C produced low surface area perovskites. CeNiO3 produced using auto-combustion of glycine and metallic nitrate precursors showed CO2 conversion reduced from 65% to 55% in 7 h at 700 °C.12,15 High surface area perovskites were synthesized by modification of sol–gel synthesis with an organic template that can reach 30–40 m2 g−1.16 Nanocasting methods with hard templates such as SBA-15 and MCM-41 increased the surface area to 100 m2 g−1.17 Since most catalytic applications of perovskite for dry reforming of methane rely on the exsolution of isolated Ni nanoparticles, having a high surface area CeNiO3 will enhance Ni dispersion and reduce the nanoparticle size. Most studies focused on partially substituting CeNiO3 perovskite with other metals such as Zr and Y to enhance lattice oxygen capacity.18 CeNiO3 substituted with La produced using citric acid as a chelating agent reduced Ni aggregation to enhance catalytic stability.19
Employing a chelating agent in the synthesis mixtures formed a stable and uniform gel between the metal precursors.20 The optimization of chelating agents stabilized the octahedral Ni bonds for transformation into stable ionic structures. In dry reforming of methane, the reaction under a continuous reducing environment exsolves Ni to form highly dispersed nanoparticles. Metal exsolution in perovskites is initialized from the surfaces and/or grain boundaries,21 which are enriched with oxygen vacancies and A-site deficiencies.22 Despite the multitude of benefits of CeNiO3, the agglomerated crystals reduce the available surface for Ni exsolution. Gelatin has been utilized as an organic template and chelating agent for metal ions for synthesizing porous materials. As a chelating agent, gelatin forms a strong bond with metals via interaction with the amino and carbonyl groups.23 The stable hydrophilic colloid protects the sol during sol–gel synthesis, controlling the growth of nanoparticles.24 The chelating ability of gelatin arises from the combination of high molecular weight protein and amine and carboxyl group functionality. Therefore, this study aims to investigate the effect of gelatin as a chelating agent on the catalytic performance of CeNiO3 perovskites in a dry reforming reaction. The correlation between crystal structures and catalytic properties was observed upon variation in gelatin concentration.
Results and discussion
XRD analysis
The XRD analysis of as-synthesized CeNiO3 perovskite with different concentrations of gelatin was conducted on the catalysts after calcination at 900 °C and after a 24 h dry reforming reaction at 600 °C (Fig. 1a). The peaks at 2θ = 28.7°, 33.3°, 37.3°, 43.3°, 47.7°, and 56.5° confirm the formation of a crystalline CeNiO3 perovskite structure.12,25–27 The diffraction peaks of the cubic phase NiO can be observed at 2θ = 37.3° and 43.3°. Close observation of the range 2θ = 27.5–30° reveals the shift of the (111) peak from 28.6° at 0.05 g of gelatin to 28.7° at 0.1 g of gelatin (Fig. 1b). However, increasing gelatin to 0.2 g shifted the (111) peak to 2θ = 28.6°. The calculated unit cell parameters in Table 1 further indicate the reduction of lattice parameters from 5.389 Å to 5.379 Å with increasing gelatin concentration from 0.05 g to 0.1 g. The lattice contraction of the CeO2 (111) plane implies a different degree of Ni incorporation in the B-site of perovskite.
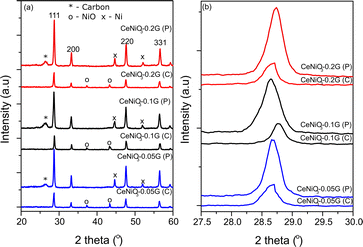 |
| | Fig. 1 XRD of CeNiO3 perovskite with (a) different concentrations of gelatin after calcination (C) and after the reaction (P); (b) changes in the high intense diffraction peak with different concentrations of gelatin before and after the reaction. | |
Table 1 Crystallite size and unit cell parameters of CeNiO3 perovskites with different concentrations of gelatina
| Amount of gelatin |
D (nm) |
a (Å) |
V (Å3) |
|
C: calcined; P: post-reaction.
|
| 0.05 g (C) |
29 |
5.389 |
156.581 |
| 0.05 g (P) |
29 |
5.397 |
157.224 |
| 0.10 g (C) |
34 |
5.379 |
155.624 |
| 0.10 g (P) |
24 |
5.401 |
157.546 |
| 0.20 g (C) |
32 |
5.388 |
156.421 |
| 0.20 g (P) |
28 |
5.383 |
155.942 |
The variation in contraction of CeO2 lattice spacing suggests that the inclusion of Ni in the CeNiO3 perovskite structure relies on gelatin concentration. There is also a possibility of the formation of a CeNiO3 + CeO2 + NiO mixture. However, such formation is difficult to prove by XRD analysis. Nevertheless, the shift of the CeO2 peak to high 2θ, while the NiO peak remains at similar 2θ = 37.3° and 43.3°, suggests the inclusion of Ni in the CeO2 lattice, forming CeNiO3 perovskite.
XRD analysis of CeNiO3 after 24 h of dry reforming of methane (DRM) shows the absence of the NiO peak due to reduction to Ni metal at 2θ = 44.5° and 52.1°. By comparing the XRD of synthesized CeNiO3-0.05G and after the DRM reaction, there is a slight shift of the (111) peak toward a lower angle, as shown in Fig. 1b. The shift is more apparent in CeNiO3-0.1G. The exsolution of NiO to the surface of CeO2 under reduction causes Ni nanoparticle formation and CeO2 lattice expansion. However, for CeNiO3-0.2G, the (111) peaks do not shift to a lower 2θ, which might indicate that NiO formed as nanoparticles deposited on CeO2, rather than CeNiO3. A new peak also appeared at 2θ = 26.2°, which shows carbon formation on the catalysts after a 24 h reaction (Fig. 1).
Table 1 summarizes the crystallite size, unit cell parameters (a), and volume of CeNiO3 catalysts before and after a 24 h catalytic reaction. The crystallite sizes of CeNiO3 perovskites synthesized at different gelatin concentrations are quite similar at approximately ∼29–34 nm. The unit cell parameters of CeNiO3 synthesized using different gelatin concentrations were determined to understand the incorporation of Ni in B sites. At 0.05 g of gelatin, CeNiO3 exhibited a unit cell parameter of 5.389 Å, slightly larger than that of CeNiO3 obtained using 0.1 g gelatin at 5.379 Å. However, CeNiO3 obtained using 0.2 g gelatin showed a unit cell parameter of 5.388 Å. The decrease in unit cell parameters indicates the incorporation of Ni2+, which has a smaller ionic radius, into CeO2.28 Using 0.1 g gelatin enhanced Ni incorporation into CeNiO3 perovskite, causing significant lattice contraction. High lattice strain in perovskite implies a weak metal–oxygen bond in BO6 octahedra, which is susceptible to oxygen vacancy formation.29 The increase of lattice volume (V) after a 24 h catalytic reaction suggested the expansion of the CeO2 lattice following the exsolution of Ni nanoparticles. A high degree of lattice expansion was observed on CeNiO3, obtained using 0.05 g and 0.1 g of gelatin, in which the lattice volume of CeNiO3-0.05G enhanced by 0.4% and that of CeNiO3-0.1G enhanced by 1.21% after a 24 h reaction. However, CeO2-0.2G exhibited different behaviors, exhibiting lattice volume reduction by 0.03%. The observation implied that at high gelatin concentration, NiO was deposited as nanoparticles on CeO2. Following long catalytic reduction under a reducing environment, Ni was doped onto CeO2, causing lattice contraction.30
Raman analysis
Fig. 2 shows the recorded Raman spectra from 2000 cm−1 to 200 cm−1. CeNiO3 perovskite shows a highly intense peak at ∼460 cm−1, a characteristic peak of cubic fluorite structure CeO2.31 This sharp peak is related to the F2g symmetric stretching of oxygen atoms around the cerium ions.32 The variation of gelatin concentrations from 0.05 g to 0.2 g reduced the intensity of the F2g peak. The peak also broadens and is red-shifted at 0.2 g gelatin concentration, implying a decrease in CeO2 crystallite size. The strain due to the high curvature of nanoparticles and surface defects also red-shifted the peak.33–35 A small peak at 225 cm−1 only appeared in ceria when doping with ionic metals with different ionic radii,36 attributed to the second-order transverse acoustic (2TA) mode. The peaks at 595 cm−1 are ascribed to defect-induced (D) mode, presumably due to the presence of oxygen vacancies or cation substitution in the lattices.36,37 The oxygen vacancies produced at the redox site in ceria-based materials play a prime role in removing the deposited carbon during the dry reforming reaction. The ratio between the intensity of defect induced (D) mode and the strong F2g mode (ID/IF2g) can be used for the estimation of oxygen vacancies in ceria-based catalysts.38,39 The concentration of oxygen vacancies in CeNiO3 synthesized using 0.05 g, 0.1 g and 0.2 g gelatin was 0.85, 0.88 and 0.90, respectively (Table 2).
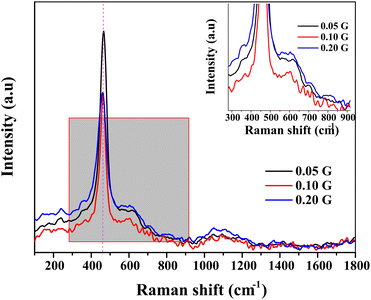 |
| | Fig. 2 Raman spectra of CeNiO3 perovskite with varied concentrations of gelatin. | |
Table 2 Physicochemical properties of CeNiO3 perovskites synthesized using different concentrations of gelatin
| Catalysts |
I
D/IF2ga |
Temp (°C) |
H2b (mmol gcat−1) |
Ni dispersionc (%), (μmol g−1) |
Ni surface areac (m2 g−1) |
CO2 emissiond (mmol gcat−1) |
Carbone (%) |
| P1 |
P2 |
P3 |
|
Calculated from Raman spectroscopy.
Calculated from H2-TPR analysis.
Calculated from CO chemisorption.
Calculated from TPO analysis.
Calculated from CHNS–O analysis.
|
| CeNiO3 0.05G |
0.85 |
393 |
531 |
802 |
44.48 |
0.58, 19.64 |
3.84 |
1.54 |
58.8 |
| CeNiO3 0.10G |
0.88 |
394 |
534 |
805 |
44.79 |
0.24, 8.01 |
1.57 |
1.43 |
56.7 |
| CeNiO3 0.20G |
0.90 |
435 |
545 |
833 |
55.08 |
1.52, 51.76 |
10.12 |
0.90 |
42.4 |
Temperature programmed reduction (TPR)
Temperature programmed reduction (TPR) examines the metal–support interaction and reducibility of the catalysts. Fig. 3 shows the TPR profiles of CeNiO3 perovskite synthesized at different concentrations of gelatin. Three reduction peaks appeared for the TPR profile of CeNiO3 synthesized using 0.05 g of gelatin. The broad reduction peaks at 393 °C correspond to Ni2+ to Ni0 reduction.12 A small peak at 530 °C was due to the reduction of surface ceria, while a high temperature reduction peak at 802 °C corresponds to the reduction of bulk oxygen in ceria.40 CeNiO3 perovskite synthesized using 0.1 g of gelatin shows similar TPR profiles to CeNiO3 (0.05 g). However, for CeNiO3 synthesized with 0.2 g gelatin, the reduction peaks of NiO shift to 435 °C with a small shoulder peak at 395 °C. The reduction peak for surface CeO2 also shifts to 545 °C, while the peak ascribed to the reducibility of lattice oxygen species appeared to shift to 833 °C. Increasing gelatin concentration resulted in enhanced interfacial interaction between NiO and CeO2 based on the high temperature reduction. CeNiO3 with 0.05 g and 0.10 g gelatin also shows similar reduction peaks and hydrogen consumption at ∼44 mmol gcat−1 while 0.20 g of gelatin shows a slight enhancement to 55 mmol gcat−1 (Table 2). The higher hydrogen consumption implies a high oxidation ability of CeNiO3-0.2G. CO chemisorption on reduced CeNiO3 provides information on Ni dispersion and the surface area. The results suggest that increasing gelatin concentration to 0.2 g significantly enhanced Ni dispersion by 1.52% and the Ni surface area by 10.12 m2 g−1. There is a possibility that at high gelatin concentration, some of the NiO were not incorporated into the CeNiO3 perovskite lattice but were instead impregnated on the surface of CeO2 as nanoparticles, as implied by XRD analysis.
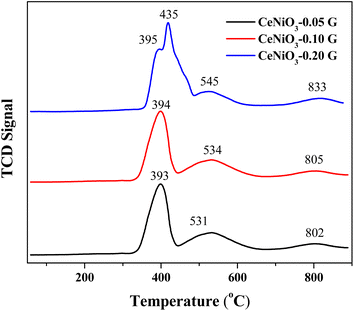 |
| | Fig. 3 H2-TPR profiles of CeNiO3 perovskites obtained using different concentrations of gelatin. | |
Temperature programmed oxidation
Temperature programmed oxidation (TPO) has been performed to determine the carbonaceous species on 24 h post-reaction catalysts. The TPO profiles of the fresh CeNiO3 catalyst show negligible oxidation up to 800 °C (ESI, Fig. S1†). The TPO profiles of all spent catalysts confirm that the oxidation of graphitic carbon occurred at 600–700 °C (Fig. 4).41,42 No peak was associated with the amorphous carbon oxidation at 250–400 °C,43 suggesting efficient oxidation of amorphous carbon to CO during the reaction. As indicated by Raman analysis, oxygen vacancies facilitate the oxidation of carbonaceous species.44 However, since the reaction was performed at 600 °C, the temperature is too low to oxidize graphitic carbon. The intensity of the oxidation peak at ∼600 °C decreases when using CeNiO3 synthesized using a high concentration of gelatin. CO2 emissions were reduced from 1.54 mmol gcat−1 to 0.90 mmol gcat−1 when using CeNiO3 with the gelatin concentration increasing from 0.05 g to 0.2 g. The results were further supported by CHNS–O analysis, which is summarized in Table 2.
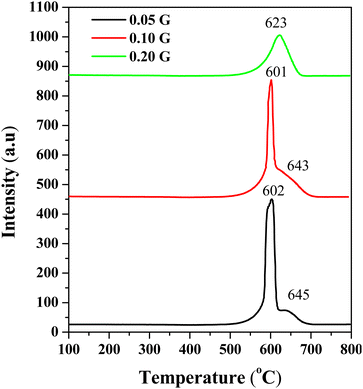 |
| | Fig. 4 TPO profiles of substituted CeNiO3 perovskites synthesized using different concentrations of gelatin after a 24 h dry reforming reaction at 600 °C. | |
N2 adsorption/desorption analysis
The textural properties of CeNiO3 perovskite were analyzed using N2 adsorption–desorption analysis (ESI, Fig. S2†). According to the IUPAC porosity classification, CeNiO3 perovskites synthesized using 0.05 g and 0.1 g gelatin show type IV isotherms,45 indicating the formation of mesoporous structures. The isotherm also exhibits a hysteresis loop H3 due to the capillary condensation occurring in mesoporous structures. However, CeNiO3 obtained using 0.2 g gelatin shows a very slow nitrogen uptake up to P/Po = 0.9, resembling a type III nonporous material. All the catalysts show strong adsorption at high relative pressure, indicating the presence of large pores. Large pores are beneficial for the diffusion of adsorbate gas. There is a trend in the surface area with adding more gelatin. CeNiO3 perovskite with 0.05 g concentration of gelatin has a surface area of 17.65 m2 g−1. At 0.1 g of gelatin, the surface area reduced to 13.30 m2 g−1, while increasing the gelatin concentration to 0.20 g significantly reduced the surface area of CeNiO3 to 11.48 m2 g−1 (Table 3).
Table 3 Textural properties of CeNiO3 perovskites
| Catalysts |
BET surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Pore diameter (nm) |
| CeNiO3-0.05G calcined |
17.65 |
0.055 |
10.4 |
| Post-reaction |
74.18 |
0.19 |
8.5 |
| CeNiO3-0.10G calcined |
13.30 |
0.044 |
11.3 |
| Post-reaction |
58.72 |
0.18 |
9.98 |
| CeNiO3-0.20G calcined |
11.48 |
0.072 |
21.05 |
| Post-reaction |
35.45 |
0.1 |
11.03 |
N2 analysis was also conducted on the catalysts recovered from a 24 h dry reforming reaction at 600 °C. There is clear evidence of changes in the N2 isotherm with a higher N2 uptake compared to the fresh catalysts (ESI, Fig. S2†). All CeNiO3 catalysts displayed a larger hysteresis loop at P/Po = 0.4 to 0.9, indicating the formation of mesoporous structures. The surface area enhances to 35.45 m2 g−1 for CeNiO3-0.2G, 58.72 m2 g−1 for CeNiO3-0.1G and 74.18 m2 g−1 for CeNiO3-0.05G. The surface areas of CeNiO3 catalysts were enhanced due to the formation of porous carbon as the by-product of carbon deposition on the catalysts during long catalytic reactions.
HR-TEM analysis
High resolution transmission electron microscopy (HR-TEM) was employed to study the morphology of CeNiO3-0.1G after calcination and a 24 h DRM reaction at 600 °C (Fig. 5a). The average estimated particle size of calcined CeNiO3 perovskite was 43.7 nm (Fig. 5g). The selected area electron diffraction pattern (SAED) shows (111), (200), (222), (400) and (220) lattice planes, which confirm the formation of CeNiO3 perovskite (Fig. 5b). The HR-TEM image after 24 hours of the DRM reaction shows the exsolution of Ni from CeNiO3 and the formation of carbon nanotubes (Fig. 5c). The estimated particle size of CeNiO3 perovskite after DRM was 71.5 nm, slightly larger than the calcined catalysts due to particle sintering (Fig. 5h). The SAED pattern shows the enlargement of (220) and (111) lattice planes, implying CeO2 phase formation after the reduction of CeNiO3 perovskite (Fig. 5d). By comparing the TEM images of calcined and post-reaction CeNiO3, Ni nanoparticles were observed as large nanoparticles at ∼8 nm and small nanoparticles at ∼1 nm (Fig. 5f). The results show non-uniform exsolution of Ni from CeNiO3 after a 24 h reaction. Fig. 5e shows the formation of carbon nanotubes on post-reaction catalysts with a lattice diameter of 0.34 nm. The carbon nanotube diameter was estimated to be 45 nm. The carbon nanotube existed as crystalline carbon with no distinguishable amorphous carbon shell.
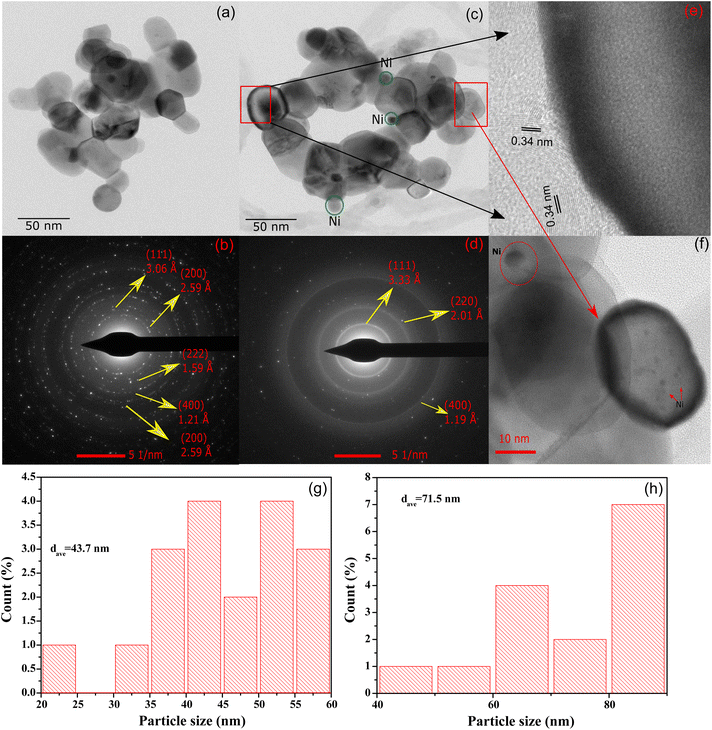 |
| | Fig. 5 HR-TEM images of (a) calcined CeNiO3-0.1G perovskite; (b) SAED pattern; (c) HR-TEM images of CeNiO3 after DRM; (d) SAED pattern; (e) carbon nanotube on post-reaction CeNiO3; (f) Ni nanoparticles on CeO2 after a 24 h reaction: (g) particle size distribution of CeNiO3-0.1G after calcination; and (h) particle size distribution of CeNiO3-0.1G after 24 h DRM reaction. | |
XPS analysis
Fig. 6 shows the XPS analysis of CeNiO3-0.1G after calcination at 900 °C and after a 24 h dry reforming reaction at 600 °C. The Ni 2p spectra of calcined CeNiO3 show peaks at 853.5 eV and 855.2 eV ascribed to Ni2+ of NiO and Ni3+ of Ni2O3.46–48 Ni exists as NiO, occupying the B-sites of CeNiO3 and the undercoordinated Ni defects. The uncoordinated Ni3+ species implies the presence of Ni2+ vacancies that may act as active sites for C–H dissociation. Post-reaction catalysts show binding energy shifts to 852.5 eV, implying NiO reduction to Ni metal after the dry reforming reaction.49 The second Ni 2p peak shifted to a higher binding energy of ∼856.5 eV, presumably due to the formation of NiOOH species.50 NiO was exsolved from the perovskite structure, forming Ni nanoparticles on CeO2 surfaces. Following CH4 decomposition, Ni was partially oxidized on the surface, forming NiO(OH) species. The changes in Ni 2p binding energy prove the changes in the Ni environment following 24 h of reactions.
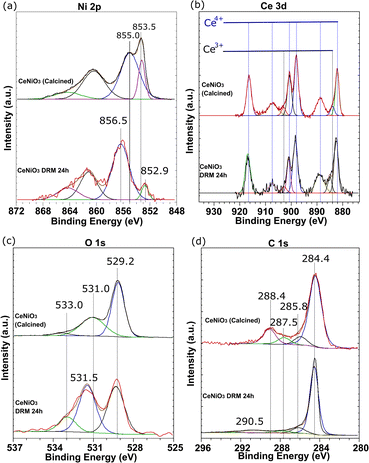 |
| | Fig. 6 XPS analysis of (a) Ni 2p, (b) Ce 3d, (c) O 1s, and (d) C 1s of CeNiO3 (0.1 g gelatin) after calcination at 900 °C and the catalyst recovered from 24 h dry reforming at 600 °C. | |
The Ce 3d spectra confirm the presence of Ce3+ and Ce4+ in CeNiO3 before and after the catalytic reaction. The deconvoluted peaks divided the spectra into five pairs of 3d5/2 and 3d3/2 spin–orbit components, representing the electronic transition between Ce4+ and Ce3+. The high intensity peaks at 882.2 eV, 900.8 eV, 898.1 eV, and 916.2 eV and the medium intensity peaks at 888.8 eV and 906.9 eV are assigned to Ce4+. The low intensity peaks at 885.5 eV and 903.9 eV are assigned to Ce3+ cations.51 Increased concentrations of Ce3+ species are evident on the catalyst after a 24 h reaction. The ratios between Ce4+ and Ce3+ were determined based on the sums of the integrated peak areas of each species. The intensities of Ce3+ peaks increase relative to the Ce4+ peak after a prolonged catalytic reaction. The oxidation of adsorbed C species involves the mobility of oxygen on CeO2. The transition of tetravalent Ce4+ to trivalent Ce3+ correlates with the formation of oxygen vacancies.52 A detailed analysis of surface oxygen species was conducted based on O 1s spectra. The O 1s spectra revealed a peak at 529.2 eV due to lattice oxygen species. The peak at 531.0 eV is assigned to the surface hydroxyl group and is used to imply the presence of defective oxygen. The peak at 533.0 eV is ascribed to oxygen bonded to carbon species. An increase in surface oxygen vacancies was observed on the post-reaction catalysts based on the enhanced surface hydroxyl group peak at 531.0 eV. The composition of oxygen from the carbonyl compound (O–C) was also enhanced to 13.73%. The percentage of O 1s species tabulated in Table 4 shows the enhanced concentration of O–C and O–H. The shift to higher BE, ascribed to the less electron-rich oxygen species in the adsorbed state, represents the surface oxygen vacancies generated following Ni exsolution. The results are in agreement with the increased Ce3+ peaks after catalytic reactions. The reducibility of Ce4+ to Ce3+ is responsible for fast oxygen vacancy production and promotes carbon oxidation to CO.
Table 4 XPS analysis of CeNiO3-0.1G after calcination and 24 h dry reforming of methane at 600 °C; the [Ce4+]/[Ce3+] atomic ratio, the binding energy (eV) for the C 1s components, and the O 1s surface composition
| Cat. |
[Ce4+]/[Ce3+] |
C 1s percentage (atom%) |
O 1s percentage (atom%) |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.8 eV) C (284.8 eV) |
C–C (285.8 eV) |
C–O (287.5 eV) |
CO (288.4 eV) |
COO– (290.5 eV) |
Ol (529.1 eV) |
OOH (531.03 eV) |
Ooc (533.31) |
| Calcined |
14.41 |
69.52 |
8.34 |
6.98 |
15.17 |
— |
57.73 |
38.01 |
4.19 |
| Post-reaction |
10.86 |
79.99 |
10.20 |
5.17 |
2.23 |
2.27 |
43.15 |
43.12 |
13.73 |
The nature of the C 1s peak of the fresh and post-reaction catalysts provides information on the carbon deposition after the reaction. The first peak at 284.4 eV indicates the sp2 CC bond of graphite carbon.53 Note that adventitious carbon appeared at 284.4 eV as the major C 1s peak on the fresh catalysts,54 together with the C–C peak at 285.8 eV, the CO peak at 287.5 eV and the CO peak at 288.4 eV. The calcined samples were dominated by graphitic CC carbon at 69%. Five carbon species were determined for the post-reaction catalyst, with an additional carboxyl or ester carbon COO– peak at 290.5 eV.55 The peak at 284.4 eV is ascribed to the graphitic sp2 CC carbon nanotube as evidenced by HRTEM analysis. Another small peak at 285.8 eV indicates C–C and C–H hydrocarbon.56 The third peak at 287.5 eV is assigned to the C–O bond, while the peak at 288.4 eV is assigned to the CO bond.57 It appears that graphite C–C carbon is the main composition with 76% concentration.
Catalytic performance
All CeNiO3 perovskites were reduced in situ before the reaction using 20 mL min−1 of hydrogen at 500 °C for 3 hours. Catalytic activity was investigated for a long-term reaction at 600 °C for 24 hours (Fig. 7). CeNiO3-0.05G displayed stable CH4 conversion at 50% throughout the 24 h catalytic reaction (Fig. 7a). Approximately similar CO2 conversion was observed at 48%, although the catalysts experienced an induction period within three hours of the reaction. CeNiO3 underwent exsolution to form Ni/CeO2 under hydrogen pre-treatment. The induction period during the catalytic reaction might suggest that the exsolution of Ni nanoparticles continuously occurred following the introduction of CH4/CO2 mixtures until they reached a steady state. The generation of oxygen vacancies from the Ce4+/Ce3+ transition enhanced CO2 adsorption and reduction to carbon monoxide. Consequently, the H2/CO ratios were gradually reduced to reach 0.85 at 24 h of reaction (Fig. 7d). A 60 h stability test was performed on CeNiO3 perovskite synthesized using 0.1 g of gelatin. The CeNiO3 showed a higher CH4 conversion than CO2 within five hours of the reaction (Fig. 7b). The CH4 conversion was gradually reduced to reach approximately similar CO2 conversion of 52%. The H2/CO ratio was initially high at ∼0.95 and then continued to decrease with time, reaching stability at 0.87 between 9 and 60 hours of the reaction. In contrast, the CO2 and CH4 conversion of CeNiO3 synthesized using 0.2 g of gelatin is stable for 24 h at 74% and 51%, respectively (Fig. 7c). Due to a higher conversion of CH4 than CO2, the ratio of H2/CO exceeds 1.0, reaching ∼1.6 throughout the 24 h reaction.
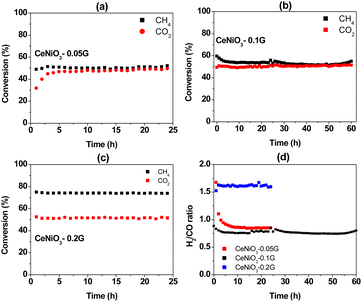 |
| | Fig. 7 Catalytic activity for a 24 h dry reforming reaction at 600 °C using CeNiO3 perovskite: CH4 and CO2 conversion using (a) CeNiO3-0.05 G catalyst; (b) CeNiO3-0.1G catalyst; (c) CeNiO3-0.2G catalyst; and (d) H2/CO ratios. | |
The deviation of the H2/CO ratio from stoichiometry is due to side reaction reactions that take place, such as reverse water gas shift reaction (RWGS) (eqn (2)) and Boudouard reaction (eqn (3)). In CeNiO3 synthesized using 0.05 g and 0.1 g gelatin, the low H2/CO < 1 may be due to the reaction of hydrogen with CO2via a reverse water gas shift reaction. This step further enhances CO production, thus causing less hydrogen yield despite achieving approximately similar CO2 and CH4 conversion. However, for CeNiO3 obtained using 0.2 g gelatin, a high surface area of Ni caused the fast decomposition of CH4 into hydrogen gas. Interestingly, the catalysts were highly stable despite the formation of graphitic carbon, as evidenced by XRD, TEM, and TPR analysis. Based on N2 adsorption and TEM analysis, the graphitic carbon was identified as a highly porous carbon nanotube. Therefore, there is a possibility that the porous carbon allows diffusion of CO2/CH4 gases to reach active sites, thus preventing deactivation.
Based on the difference in the catalytic performance of CeNiO3 perovskite with 0.05 g and 0.20 g of gelatin, the catalysts were investigated at different reaction temperatures. The performance of CeNiO3 perovskite was studied at 400–600 °C under a constant flow of 20 mL min−1 mixed CO2/CH4/N2 gases. Fig. 8(a–c) shows the plot of methane conversion, carbon dioxide conversion and the H2/CO ratio at different temperatures. Initially, carbon dioxide conversion, methane conversion, and the H2/CO ratio are low at 400 °C with less than 10% conversion. However, CeNiO3 synthesized using 0.2 g gelatin concentration exhibited 9.22% CH4 conversion and 10.04% CO2 conversion to give 1.26 H2/CO ratios. The catalytic performance was slightly higher than that of CeNiO3 obtained at lower gelatin concentrations. There was an exponential rise in CH4 and CO2 conversion with increased temperatures. The highest 69.62% CH4 conversion with 49.05% carbon dioxide conversion was obtained on CeNiO3(0.20) to give a H2/CO ratio of 1.54. CeNiO3 synthesized at 0.2 g gelatin concentration shows a higher catalytic conversion to CH4 than CO2, resulting in a higher H2/CO ratio.
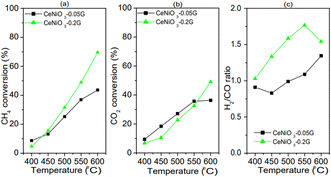 |
| | Fig. 8 Catalytic performance of CeNiO3 perovskites at different temperatures: (a) CH4 conversion; (b) CO2 conversion; and (c) H2/CO ratios from CeNiO3-0.05G, CeNiO3-0.1G and CeNiO3-0.2G. | |
In situ DRIFTS analysis
In situ DRIFTS analysis was performed to investigate intermediate species in the DRM reaction at different temperatures (Fig. 9). The catalyst was reduced with hydrogen under a constant flow of 20 mL min−1 at 500 °C before the DRM reaction. CH4 gas flowed to the CeNiO3-0.2G in the DRIFTS cell. The CH4 peak was identified by a strong absorbance band at 3007 cm−1 and 1298 cm−1, corresponding to the symmetric CH4 bond. The gradual growth of the 2838 cm−1 band implies the dissociation of CH4 on the Ni metal surface into  . The band was accompanied by the slow growth of the 1860 cm−1 peak assigned to the CO stretch. The results imply that CH4 is dissociated via multiple C–H dissociation steps on the metal surface, forming adsorbed carbon on Ni. The surface oxygen on CeO2 might react with adsorbed carbon at the Ni/CeO2 interface, forming adsorbed carbonate species. The suggestion was confirmed by the peak assigned to carbonate and bicarbonate in the 1742–1389 cm−1 region. The peak at 1742 cm−1 was also identified as a cerium oxycarbonate species (Ce2O2CO3).19 The peak at 2312 cm−1 implies the reaction of CH4 with surface oxygen on the CeO2, forming adsorbed –CO3 species. The Ni surface is selective to CH4 dissociation into atomic C. However, since CeO2 has high oxygen mobility, it is clear from DRIFTS analysis that the adsorbed carbon reacts with surface oxygen to form adsorbed carbonate. However, the adsorbed carbonate is strongly attached to the surface, based on the increased intensity of such species with prolonged holding temperatures at 600 °C.
. The band was accompanied by the slow growth of the 1860 cm−1 peak assigned to the CO stretch. The results imply that CH4 is dissociated via multiple C–H dissociation steps on the metal surface, forming adsorbed carbon on Ni. The surface oxygen on CeO2 might react with adsorbed carbon at the Ni/CeO2 interface, forming adsorbed carbonate species. The suggestion was confirmed by the peak assigned to carbonate and bicarbonate in the 1742–1389 cm−1 region. The peak at 1742 cm−1 was also identified as a cerium oxycarbonate species (Ce2O2CO3).19 The peak at 2312 cm−1 implies the reaction of CH4 with surface oxygen on the CeO2, forming adsorbed –CO3 species. The Ni surface is selective to CH4 dissociation into atomic C. However, since CeO2 has high oxygen mobility, it is clear from DRIFTS analysis that the adsorbed carbon reacts with surface oxygen to form adsorbed carbonate. However, the adsorbed carbonate is strongly attached to the surface, based on the increased intensity of such species with prolonged holding temperatures at 600 °C.
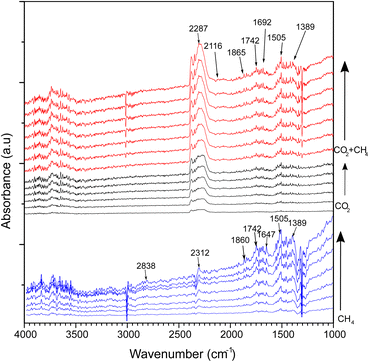 |
| | Fig. 9
In situ DRIFTS spectra of CeNiO3 perovskite (0.2 g gelatin) under continuous flows of CH4, CO2 and a mixture of CO2 and CH4 at 600 °C. | |
CO2 adsorption on the catalysts was carried out at 600 °C. Note that the CeNiO3 was treated again with a H2 flow at 500 °C before CO2 was introduced. The peak assigned to the absorbed *CO2 gas phase was observed between 2500 and 2250 cm−1. The small peak assigned to bidentate carbonate *bCO32− was observed at 1743 and 1646 cm−1. Additionally, the bidentate CO3 peak was also observed at 1040 cm−1. The peak at 1455 cm−1 refers to monodentate carbonate. The intensity of the bidentate carbonate band is higher than that of monodentate carbonate. The bicarbonate peak (HCO3−) was observed at 1425 cm−1, implying the reaction of CO2 with the surface OH− species.
Different FTIR bands appeared after introducing mixed methane and carbon dioxide gases. The broad band at 2313 cm−1 is assigned to gaseous carbon dioxide, while the bands at 1306 cm−1, 1336 cm−1 and 3010 cm−1 represent gaseous methane.19 A negative methane peak at 3010 cm−1 was observed with increasing time at 600 °C, implying CH4 decomposition during the DRM reaction. Monodentate carbonate and bidentate carbonate bands at 1455 cm−1 and 1510 cm−1 gained intensity in the presence of CH4. The bicarbonate absorption band was also observed at 1643 cm−1. A new species was observed at 2116 cm−1 assigned to CO formation. The peak at 1745 cm−1 was also identified as a cerium oxycarbonate species (Ce2O2CO3).19 The Ce2O2CO3 species play a vital role as intermediates for removing the deposited carbon from the active metal during DRM. The formed Ce2O3 under reducing condition generates Ce3+ that reacted with CO2 to form Ce2O2CO3 species. The presence of Ce3+ was observed in XPS analysis. The lower intensity of Ce2O2CO3 species in the CH4 + CO2 mixture compared to CH4 gas only further evidenced the removal of deposited carbon from CeNiO3. Another IR-band was observed at 2116 cm−1, representing carbon monoxide formation during the reaction. These C–O bands begin to appear following CH4 addition, which further supports the Ce2O2CO3 species as vital intermediates for the oxidation of the dissociated oxygen atom from CO2. Methane adsorption and dissociation occur at the surface of Ni active metal, while carbon dioxide adsorption and dissociation occur at the CeO2 support. Carbon dioxide is absorbed on the oxygen vacancies of CeO2 as bicarbonate species before undergoing oxygen dissociation to form Ce2O2CO3 species. The proposed mechanism for the reaction is given below.
| | | Ce2O3 + CO2 → Ce2O2CO3 | (6) |
| | | Ce2O2CO3 + C–Ni → 2CO + Ni* + CeO2 | (7) |
The difference in catalytic performance for CeNiO3 catalysts synthesized using different gelatin concentrations might give insights into the structural properties of catalysts. In dry reforming of methane, CH4 decomposed on the Ni via consecutive bond dissociation, producing adsorbed carbon species. CO2 is absorbed on the CeO2 surface, occupying surface oxygen vacancies that subsequently weaken the C–O bond. The dissociated oxygen migrated to the Ni/CeO2 interface for the oxidation of adsorbed C as CO, while the CO leaves the CeO2 surfaces. Strong CH4 dissociation potential caused the conversion to deviate from the stoichiometric CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH4 ratio. As suggested by characterization data, the high dispersion of Ni on the CeO2 surface, via either a high degree of NiO exsolution or the formation of uncoordinated Ni species, readily absorbs and dissociates CH4 into H2 gas. There is also a possibility that the generated H2 reacts with the CO2 reactant, producing CO through a reverse water gas shift reaction (eqn (2)). This step may be possible on CeNiO3-0.1 G and CeNiO3-0.05G because, despite similar CO2 and CH4 conversion, the H2/CO ratio was less than 1. In the synthesis of CeNiO3 perovskite, gelatin controlled the Ni2+ ion mobility in the cross-linked gel precursor. The Ni–gelatin complex inadvertently restricts the contact of Ni with the Ce2+ cation. At high gelatin concentrations, gelatin as a chelating agent might heavily saturate Ni2+. The slow mobility of Ni2+ leads to the formation of uncoordinated Ni species, which causes deposition as NiO nanoparticles. The formation of uncoordinated NiO species alongside the main phase CeNiO3 perovskite leads to a strong CH4 dissociation site. Consequently, CeNiO3-0.2G, obtained at high gelatin concentration, produced H2/CO ratios >1.
:CH4 ratio. As suggested by characterization data, the high dispersion of Ni on the CeO2 surface, via either a high degree of NiO exsolution or the formation of uncoordinated Ni species, readily absorbs and dissociates CH4 into H2 gas. There is also a possibility that the generated H2 reacts with the CO2 reactant, producing CO through a reverse water gas shift reaction (eqn (2)). This step may be possible on CeNiO3-0.1 G and CeNiO3-0.05G because, despite similar CO2 and CH4 conversion, the H2/CO ratio was less than 1. In the synthesis of CeNiO3 perovskite, gelatin controlled the Ni2+ ion mobility in the cross-linked gel precursor. The Ni–gelatin complex inadvertently restricts the contact of Ni with the Ce2+ cation. At high gelatin concentrations, gelatin as a chelating agent might heavily saturate Ni2+. The slow mobility of Ni2+ leads to the formation of uncoordinated Ni species, which causes deposition as NiO nanoparticles. The formation of uncoordinated NiO species alongside the main phase CeNiO3 perovskite leads to a strong CH4 dissociation site. Consequently, CeNiO3-0.2G, obtained at high gelatin concentration, produced H2/CO ratios >1.
Conclusion
Using gelatin at different concentrations to produce CeNiO3 perovskite nanoparticles for dry reforming of methane significantly affects the stoichiometric H2/CO formation. The preference for CH4 decomposition on Ni metal surfaces was observed on CeNiO3-0.2G due to the increase of the Ni surface area to 10 m2 g−1. Stoichiometric CH4 and CO2 conversion were achieved on CeNiO3 obtained using low gelatin concentration (0.05 g and 0.1 g), suggesting that the exsolution of Ni from CeNiO3 prevents fast methane decomposition. The expansion of the CeO2 unit cells after the reaction suggests the exsolution of Ni from CeNiO3 perovskite. Gelatin, as amine-based chelating agent, coordinates with Ni ions from the lone pair electrons on the nitrogen, forming octahedral binding. An optimized chelating agent amount is essential for forming stable metal ion bonds. Gelatin reduced Ni mobility in the cross-linked gel precursor, restricting the contact between the cations. Therefore, gelatin facilitates uniformity in the particle size distribution and forms smaller particles. However, at higher gelatin concentrations, the reduced Ni mobility may restrict the self-assembly of CeNiO3 perovskite. CeNiO3 perovskite shows catalytic stability at 600 °C with a H2/CO ratio of ∼8.5 when produced using low gelatin amounts (0.05 g and 0.1 g).
Experimental
Materials
Cerium nitrate hexahydrate (Ce(NO3)3·6H2O, Merck, >99%), nickel nitrate hexahydrate (Ni(NO3)2·6H2O, Merck, >99%), gelatin (bovine skin type B, Sigma Aldrich) and distilled water were directly used without further purification.
Catalyst preparation
The synthesis of CeNiO3 was conducted based on previous studies with slight modifications.23 The amount of gelatin was varied to obtain the weight ratios of gelatin to the final product of 1:0.01 (0.05 g gelatin), 1:0.03 (0.1 g gelatin) and 1:0.06 (0.2 g gelatin). Gelatin was used as a chelating agent for Ni to synthesize CeNiO3 perovskites.58 Gelatin was dissolved in distilled water (30 g) and stirred at 50 °C for 30 min to obtain a clear solution. Equimolar solutions of metal nitrates such as Ce(NO3)2·6H2O and Ni(NO3)2·6H2O were added to the solution and stirred at 70 °C. The solution temperature was raised to 90 °C using a hot plate and stirring until the solution turned into a gel. The gel was heated in a muffle furnace with double calcination. Firstly, the gel was calcined at 350 °C for 2 hours to obtain a dried powder. This powder was ground and calcined at 900 °C for 4 hours at a ramping rate of 5 °C min−1. The purpose of double calcination is to remove the impurities to form a high purity product.
Characterization
The properties of the perovskite were investigated using a Shimadzu XRD-7000 with Cu Kα 1.5418 Å radiation. The scan rate was 5° min−1 in the 2θ range of 10°–90° and the crystal structure was analyzed using X'Pert High Score Plus software. The lattice parameters are calculated using eqn (8),| | 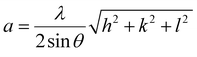 | (8) |
where λ is the Cu Kα wavelength, θ is the Bragg angle, and hkl is Miller's index.
H2-TPR analyses were performed using a ChemBET Pulsar TPR/TPD equipped with a thermal conductivity detector. H2-TPR analyses involve the activation of the catalyst with H2 at a high temperature of 900 °C with a ramping rate of 10 °C min−1. A very small amount of samples, i.e. 20 mg, was placed in a U-shaped tube. The textural properties of perovskites were measured using N2-physisorption using a Micromeritics ASAP2020 instrument at 77 K. The Brunauer–Emmett–Teller (BET) method was employed to determine the surface area of the catalyst, while the pore diameter of the catalyst was determined by applying the Barrett–Joyner–Halenda (BJH) method using nitrogen isotherms. X-ray photoelectron spectroscopy (XPS) analysis was performed using a Kratos Axis Ultra-DLD XPS spectrometer with a monochromatic Al Kα source (75–150 W) and an analyzer pass energy of 160 eV for survey scans or 40 eV for detailed scans. High-resolution transmission electron microscopy (Tecnai G2 20S) determined the nanostructure and surface morphology. Raman spectra were recorded using a Scanner 5.0 Raman system assembled at National Taiwan University with a 532 nm wavelength laser.
Catalytic dry reforming reaction
The catalytic activity of CeNiO3 was determined in a tubular fixed bed quartz reactor with a 4 mm internal diameter and 40 cm length. CeNiO3 was reduced in situ with pure hydrogen at 20 mL min−1 at 500 °C for 3 hours before the reaction. The H2 gas was switched to a mixed gas containing 25% CO2 and 25% CH4 in nitrogen at 20 mL min−1 after the furnace reached room temperature. The flow rates of gases were controlled using calibrated mass flow controllers (MFCs). Online gas chromatography (GC), which is equipped with thermal conductivity (TCD) and flame ionizing (FID) detectors, was used for the analysis of the reaction mixture.
The conversion of the reactants was calculated using the following formula:
| | 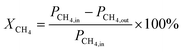 | (9) |
| | 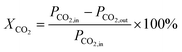 | (10) |
where
XCH4 represent the % conversion of CH
4,
XCO2 represents the % conversion of CO
2,
PCH4,in and
PCH4,out are the peak areas of CH
4 before and after the reaction, and
PCO2,in and
PCO2,out are the peak areas of CO
2 before and after the reaction.
Conclusions
Using gelatin at different concentrations to produce CeNiO3 perovskite nanoparticles for dry reforming of methane has a significant effect on stoichiometric H2/CO formation. The preference for CH4 decomposition on Ni metal surfaces was observed on CeNiO3-0.2G due to the increase of the Ni surface area. Stoichiometric CH4 and CO2 conversion was achieved on CeNiO3 obtained using low gelatin concentration (0.05 g and 0.1 g), which suggests that control exsolution of Ni from CeNiO3 prevents fast methane decomposition. The shift of the CeO2 (111) plane to lower 2θ after the reaction suggests the exsolution of Ni from CeNiO3-0.05G perovskite. Meanwhile, CeNiO3-0.2G shows a similar peak position, suggesting that NiO was deposited on CeO2 at high gelatin concentration, instead of being incorporated into CeNiO3 perovskite. Gelatin as an amine based chelating agent coordinates with Ni ions from the lone pair of electrons on the nitrogen, forming octahedral binding. An optimized chelating agent amount is essential to form stable bonds of metal ions. Gelatin reduced Ni mobility in the cross-linked gel precursor, restricting the contact between the Ni and Ce cations. Therefore, gelatin facilitates uniformity in the particle size distribution and forms smaller particles. However, at higher gelatin concentrations, the reduced Ni mobility may restrict the self-assembly of CeNiO3 perovskite. CeNiO3 perovskite shows catalytic stability at 600 °C with a H2/CO ratio close of 0.85 when produced using low gelatin amounts (0.05 g and 0.1 g).
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the UBD UGRS scholarship for Usman Zahid and the UBD FIC grant UBD/RSCH/1.9/FICBF(b)/2023/020 for funding this research.
References
- T. Abbasi, M. Premalatha and S. A. Abbasi, Renewable Sustainable Energy Rev., 2011, 15, 891–894 CrossRef.
- C.-F. Schleussner, J. Rogelj, M. Schaeffer, T. Lissner, R. Licker, E. M. Fischer, R. Knutti, A. Levermann, K. Frieler and W. Hare, Nat. Clim. Change, 2016, 6, 827–835 CrossRef.
- J. Rogelj, M. Den Elzen, N. Höhne, T. Fransen, H. Fekete, H. Winkler, R. Schaeffer, F. Sha, K. Riahi and M. Meinshausen, Nature, 2016, 534, 631–639 CrossRef CAS PubMed.
- J. Hansen, R. Ruedy, M. Sato and K. Lo, Rev. Geophys., 2010, 48, RG4004 CrossRef.
- J. Szczygieł, K. Chojnacka, D. Skrzypczak, G. Izydorczyk, K. Moustakas and M. Kułażyński, J. Environ. Manage., 2023, 325, 116463 CrossRef PubMed.
- F. G. M. de Medeiros, F. W. B. Lopes and B. Rego de Vasconcelos, Catalysts, 2022, 12, 363 CrossRef CAS.
- B. C. Ekeoma, M. Yusuf, K. Johari and B. Abdullah, Int. J. Hydrogen Energy, 2022, 41596–41620 CrossRef CAS.
- E. Le Saché and T. R. Reina, Prog. Energy Combust. Sci., 2022, 89, 100970 CrossRef.
- T. Ban, X.-Y. Yu, H.-Z. Kang, H.-X. Zhang, X. Gao, Z.-Q. Huang and C.-R. Chang, J. Catal., 2022, 408, 206–215 CrossRef CAS.
- Z. Li, Z. Wang and S. Kawi, ChemCatChem, 2019, 11, 202–224 CrossRef CAS.
- S. Bhattar, M. A. Abedin, S. Kanitkar and J. J. Spivey, Catal. Today, 2021, 365, 2–23 CrossRef CAS.
- N. Ahmad, R. Wahab, S. Manoharadas, B. F. Alrayes, M. Alam and F. A. Alharthi, Molecules, 2022, 27, 356 CrossRef CAS PubMed.
- G. Valderrama, A. Kiennemann, C. U. de Navarro and M. R. Goldwasser, Appl. Catal., A, 2018, 565, 26–33 CrossRef CAS.
- M. Tyunina, J. Levoska, O. Pacherova, T. Kocourek and A. Dejneka, J. Mater. Chem. C, 2022, 10, 6770–6777 RSC.
- N. Ahmad, F. Alharthi, M. Alam, R. Wahab, S. Manoharadas and B. Alrayes, Energies, 2021, 14, 2928 CrossRef CAS.
- D. Navas, S. Fuentes, A. Castro-Alvarez and E. Chavez-Angel, Gels, 2021, 7, 275 CrossRef CAS PubMed.
- Z. Mhlwatika, R. Meijboom and N. Bingwa, Inorg. Chem. Commun., 2021, 133, 108962 CrossRef CAS.
- A. E. Abasaeed, M. L. Sofiu, K. Acharya, A. I. Osman, A. H. Fakeeha, R. L. AL-Otaibi, A. A. Ibrahim, A. S. Al-Awadi, H. Bayahia, S. A. Al-Zahrani, R. Kumar and A. S. Al-Fatesh, Energy Sci. Eng., 2023, 11, 1436–1450 CrossRef CAS.
- Z. Du, C. Petru, X. Yang, F. Chen, S. Fang, F. Pan, Y. Gang, H.-C. Zhou, Y. H. Hu and Y. Li, J. CO2 Util., 2023, 67, 102317 CrossRef CAS.
- B. G. Kim, W. Jang, Y. J. Park, J. H. Kang, J. H. Seo and D. H. Wang, J. Phys. Chem. C, 2020, 124, 25184–25195 CrossRef CAS.
- M. Awais, S. Kundu, D. Zhang, V. Yeddu, M. R. Kokaba, Y. Ahmed, W. Zhou, S. Dayneko, F. Tan and M. I. Saidaminov, Cell Rep. Phys. Sci., 2023, 4, 101634 CrossRef CAS.
- Q. A. Islam, S. Paydar, N. Akbar, B. Zhu and Y. Wu, J. Power Sources, 2021, 492, 229626 CrossRef CAS.
- N. Azad, H. Arabi, S. R. Ghorbani and A. Davodi, J. Sol-Gel Sci. Technol., 2018, 88, 465–473 CrossRef CAS.
- G. Monros, J. Cards, M. A. Tena, P. Escribano, J. Badenes and E. Cordoncillo, J. Mater. Chem., 1995, 5, 85–90 RSC.
- F. Dehghani, S. Ayatollahi, S. Bahadorikhalili and M. Esmaeilpour, Pet. Chem., 2020, 60, 731–743 CrossRef CAS.
- M. Harikrishnan, A. J. C. Mary and A. C. Bose, Electrochim. Acta, 2020, 362, 137095 CrossRef CAS.
- J. Chen, Z. He, G. Li, T. An, H. Shi and Y. Li, Appl. Catal., B, 2017, 209, 146–154 CrossRef CAS.
- S. Mahammadunnisa, P. M. K. Reddy, N. Lingaiah and C. Subrahmanyam, Catal. Sci. Technol., 2013, 3, 730–736 RSC.
- H. Chen, C. Lim, M. Zhou, Z. He, X. Sun, X. Li, Y. Ye, T. Tan, H. Zhang, C. Yang, J. W. Han and Y. Chen, Advanced Science, 2021, 8, 2102713 CrossRef CAS PubMed.
- Z. Yang, D. Zheng, X. Yue, K. Wang, Y. Hou, W. Dai and X. Fu, Appl. Surf. Sci., 2023, 615, 156311 CrossRef CAS.
- J. Kojčinović, D. Tatar, S. Šarić, C. B. Pravda, A. Mavrič, I. Arčon, Z. Jagličić, M. Mellin, M. Einert, A. Altomare, R. Caliandro, Á. Kukovecz, J. P. Hofmann and I. Djerdj, Dalton Trans., 2024, 53, 2082–2097 RSC.
- S. Loridant, Catal. Today, 2021, 373, 98–111 CrossRef CAS.
- J. E. Spanier, R. D. Robinson, F. Zhang, S.-W. Chan and I. P. Herman, J. Appl. Phys., 2001, 64, 245407 Search PubMed.
- I. Kosacki, T. Suzuki, H. U. Anderson and P. Colomban, Solid State Ionics, 2002, 149, 99–105 CrossRef CAS.
- C. C. Yang and S. Li, J. Phys. Chem. B, 2008, 112, 14193–14197 CrossRef CAS PubMed.
- Z. Popović, Z. Dohčević-Mitrović, N. Paunović and M. Radović, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 014302 CrossRef.
- J. R. McBride, K. C. Hass, B. D. Poindexter and W. H. Weber, J. Appl. Phys., 1994, 76, 2435–2441 CrossRef CAS.
- Y. Li, Z. Wei, F. Gao, L. Kovarik, R. A. L. Baylon, C. H. F. Peden and Y. Wang, ACS Catal., 2015, 5, 3006–3012 CrossRef CAS.
- T. Taniguchi, T. Watanabe, N. Sugiyama, A. K. Subramani, H. Wagata, N. Matsushita and M. Yoshimura, J. Phys. Chem. C, 2009, 113, 19789–19793 CrossRef CAS.
- D. B. Long, B. T. Hau, N. T. T. Van, N. P. Anh, T. G.-T. Ho, H. C. Anh, P. H. Phuong, C. D. Huy, N. Tri and L. C. Loc, J. Sci.: Adv. Mater. Devices, 2023, 8, 100610 CAS.
- B. M. Al-Swai, N. Osman, M. S. Alnarabiji, A. A. Adesina and B. Abdullah, Ind. Eng. Chem. Res., 2018, 58, 539–552 CrossRef.
- B. Kitiyanan, W. E. Alvarez, J. H. Harwell and D. E. Resasco, Chem. Phys. Lett., 2000, 317, 497–503 CrossRef CAS.
- Y. Feng, G. Zhou, G. Wang, M. Qu and Z. Yu, Chem. Phys. Lett., 2003, 375, 645–648 CrossRef CAS.
- O. Omoregbe, H. T. Danh, S. Z. Abidin, H. D. Setiabudi, B. Abdullah, K. B. Vu and D.-V. N. Vo, Procedia Eng., 2016, 148, 1388–1395 CrossRef CAS.
- N. Tri, N. P. Anh, T. D. Huy, D. B. Long, H. C. Anh, P. H. Phuong, N. T. T. Van, T.-T. Nguyen and L. C. Loc, J. Sci.: Adv. Mater. Devices, 2023, 8, 100529 CAS.
- M. B. Islam, M. Yanagida, Y. Shirai, Y. Nabetani and K. Miyano, ACS Omega, 2017, 2, 2291–2299 CrossRef CAS PubMed.
- D. S. Mann, S.-N. Kwon, P. Patil and S.-I. Na, Nano Energy, 2023, 106, 108062 CrossRef CAS.
- C. C. Boyd, R. C. Shallcross, T. Moot, R. Kerner, L. Bertoluzzi, A. Onno, S. Kavadiya, C. Chosy, E. J. Wolf, J. Werner, J. A. Raiford, C. de Paula, A. F. Palmstrom, Z. J. Yu, J. J. Berry, S. F. Bent, Z. C. Holman, J. M. Luther, E. L. Ratcliff, N. R. Armstrong and M. D. McGehee, Joule, 2020, 4, 1759–1775 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- X. Wang, B. Zhang, W. Zhang, M. Yu, L. Cui, X. Cao and J. Liu, Sci. Rep., 2017, 7, 1584 CrossRef PubMed.
- E. Y. Konysheva and S. M. Francis, Appl. Surf. Sci., 2013, 268, 278–287 CrossRef CAS.
- F. Rao, Y. An, G. Zhu, S. Gong, L. Zhu, H. Lu, X. Shi, Y. Huang, F. Zhang and M. Hojamberdiev, Sep. Purif. Technol., 2024, 333, 125951 CrossRef CAS.
- I. Bertóti, M. Mohai and K. László, Carbon, 2015, 84, 185–196 CrossRef.
- G. Greczynski and L. Hultman, Sci. Rep., 2021, 11, 11195 CrossRef CAS PubMed.
- X. Chen, X. Wang and D. Fang, Fullerenes, Nanotubes Carbon Nanostruct., 2020, 28, 1048–1058 CrossRef CAS.
- A. Manakhov, P. Kiryukhantsev-Korneev, M. Michlíček, E. Permyakova, E. Dvořáková, J. Polčák, Z. Popov, M. Visotin and D. V. Shtansky, Appl. Surf. Sci., 2018, 435, 1220–1227 CrossRef CAS.
- Y. Liu, H. Yan, X. Chen, X. Feng and C. Yang, J. Anal. Appl. Pyrolysis, 2019, 141, 104629 CrossRef CAS.
-
F. S. Oliveira, P. M. Pimentel, D. M. d. A. Melo, R. M. P. B. Oliveira, M. A. d. F. Melo and O. R. Bagnato, Synthesis of Sm_1_-_xSr_xNiO"3 systems for application in dry reforming reaction, PTECH 2009: 7 International latin-american conference on powder technology, Brazil, 2009 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c and
Hasliza
Bahruji
c and
Hasliza
Bahruji











 . The band was accompanied by the slow growth of the 1860 cm−1 peak assigned to the C
. The band was accompanied by the slow growth of the 1860 cm−1 peak assigned to the C


