
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMononuclear metal complex catalysts on supports: foundations in organometallic and surface chemistry and insights into structure, reactivity, and catalysis
Bruce C.
Gates

Department of Chemical Engineering, University of California, Davis, California 95616, USA. E-mail: bcgates@ucdavis.edu
First published on 20th September 2024
Abstract
Catalysts that consist of isolated metal atoms bonded to solid supports have drawn wide attention by researchers, with recent work emphasizing noble metals on metal oxide and zeolite supports. Progress has been facilitated by methods for atomic-scale imaging the metals and spectroscopic characterization of the supported structures and the nature of metal–support bonding, even with catalysts in the working state. Because of the intrinsic heterogeneity of support surface sites for bonding of metals and the tendency of noble metal cations on supports to be reduced and aggregated, it is challenging to determine structures of individual metal complexes among the mixtures that may be present and to determine structures of catalytically active species and reactive intermediates. A central premise of this perspective is that synthesis of supported metal complexes that have nearly uniform structures—on supports such as dealuminated HY zeolite, chosen to have relatively uniform surfaces—is a key to fundamental understanding, facilitating progress toward determining the roles of the ligands on the metals, which include the supports and reactive intermediates in catalysis. Characterization of relatively uniform and well-defined samples nonetheless requires multiple spectroscopic, microscopic, and theory-based techniques used in concert and still leaves open many questions about the nature of reactive intermediates and catalytic reaction mechanisms.
| Bruce C. Gates Bruce Gates is Distinguished Professor Emeritus of Chemical Engineering at the University of California, Davis. His recent research has been focused on investigations of catalysts synthesized to be structurally well defined, including metal complexes and metal clusters supported on zeotype materials, MOFs, and metal oxides. The group collaborates widely, striving for fundamental understanding of catalyst structure and function by investigating catalysts with high-resolution electron microscopy and spectroscopies applied to functioning catalysts, bolstered by electronic structure calculations. |
Organometallic chemistry and catalysis
Organometallic chemistry emerged as an essential discipline in chemistry largely on the strength of its importance in catalysis.1,2 Advances in organometallic catalysis have been recognized with multiple Nobel Prizes. The catalysts are mononuclear metal complexes—many in Group 8—used in solution. These have for decades been essential in chemical technology, with examples of catalysts that have long histories of research, development, and industrial application. These include the rhodium and iridium complexes used for hydrogenation and hydroformylation of alkenes and for methanol carbonylation.1–3 Newer examples are chiral catalysts used for the manufacture of pharmaceuticals. The discovery processes leading to new catalysts have for the most part involved experimental testing of new metal–ligand combinations.Organometallic chemistry has advanced beyond mononuclear metal complexes to include many compounds with metal–metal bonds (metal clusters), but these have not led to notable successes in technology—largely because of the stability limitations of these compounds and the loss of reactivity that results when they are made stable by envelopment in ligand sheaths.
For many years, the family of organometallic catalysts has included metal complexes and clusters bonded to solid surfaces (supports), which are ligands that may help to stabilize the catalytic species. These supported catalysts and have drawn wide attention from researchers recently, but with barely any large-scale applications emerging from the recent work.
Processes involving homogeneous organometallic catalysis, even when they are economical and practiced on a large scale, are generally challenged by the costs of separating the catalysts from products for efficient recycle and sometimes by the need for expensive corrosion-resistant materials such as stainless steels for reactors and associated equipment. Further, only a small minority of organometallic catalysts are stable enough at high temperatures to allow separation from products by distillation, which would usually be the economically preferred separation method if the stability criteria could be met. The Wacker ethene oxidation3 is an exception that illustrates the benefit of catalyst stability—the palladium salt catalyst is stable enough at high temperatures to allow a gas–liquid separation as the organic products are distilled from the reactor (with the exothermic oxidation reaction providing the energy for continuously vaporizing the reactor contents).
Alternatively, the separations challenge is sometimes met with processes that involve two separate phases—with reactants and products predominantly in one fluid phase and the catalyst in the other—which can be either a solid or a liquid. In “biphasic” liquid–liquid catalysis, illustrated by industrial propene hydroformylation,4 an aqueous phase contains most of the catalyst (which incorporates hydrophilic ligands to make the metal complex soluble), and an organic liquid phase contains most of the reactants and products. The reactant mixture, consisting of drops of one phase in a continuum of the other, is vigorously stirred so that the drops are small and intimately mixed with the continuous phase, affording a high interfacial surface area that facilitates transport of reactant and product molecules between the phases. The two liquid phases are separated from each other downstream of the reactor in a device called a settler—a tank that functions like a separatory funnel, in which the phases split into two layers. The aqueous liquid containing the catalyst is recycled to the reactor, and the organic liquid containing the product is sent to a purification device such as a distillation column; unconverted reactants may be recycled to the reactor.
Catalysis on solid surfaces
In contrast to this liquid–liquid biphasic catalysis, the two separate phases are more typically a gas and a solid, with the catalyst being part of the solid surface. The catalyst may be an organometallic species that is a molecular analogue bonded to the solid surface, which is a ligand. A decisive advantage of a such supported catalysts is that they are readily and cheaply separated from fluid-phase products. Extensive research has been done with supported organometallic catalysts, and there are industrial applications, illustrated by the Chiyoda-UOP process for methanol carbonylation catalyzed by rhodium complexes anchored to an anion exchange resin.5 Another example, performed on an enormous scale for decades, is ethene polymerization catalyzed by mononuclear chromium complexes on porous, high-area silica; but in this process, the product (polymer) may not be separated from the catalyst—rather, the catalyst is swallowed up by the polymer and becomes a minor impurity.6,7In most large-scale catalytic processes, solid catalysts, such as metal nanoparticles dispersed on supports,8 are chosen over liquids because they offer the advantages of (a) reactivity combined with stability at high temperatures, (b) lack of corrosion, and (c), most important, ease of separation from fluid-phase products. Thus, the fluid stream consisting of reactants and the products formed in the reactor flows through a tube containing catalyst particles (a fixed-bed reactor) and then to product purification devices. The catalyst particles are porous, with a high internal surface area (e.g., hundreds of square meters per gram), and they incorporate high densities of surface catalytic sites that are accessible to reactants via transport (diffusion) through the catalyst pores. The catalyst stays in place in the reactor, so that virtually no cost is associated with separating the products from the catalyst.
In most large-scale processes involving fixed-bed reactors, the catalyst is stable enough to be used at high temperatures—which explains why most practical catalysts consist of robust solids such as metals, metal oxides, or metal sulfides. In contrast, when the catalyst is a molecular analogue bonded to a support, it is often not robust or stable—and, if it is not, to be useful in practice it must offer compensating economic advantages, such as high selectivity (which might be associated with a simple, essentially molecular, structure). We delve into the chemistry of such supported catalysts in the following paragraphs. In a number of processes for the manufacture of pharmaceuticals, the product is so valuable and so urgently needed for rapid entry into the marketplace that a metal complex catalyst in solution is used only a single time in a batch reactor—it is sacrificed rather than being separated and recycled to the reactor. When the metal is as expensive as palladium or rhodium, which are commonly used for pharma manufacture,9 this sacrificial operation is shockingly costly—but it is nonetheless viable because the rush to market of newly discovered and approved drugs forces the issue. Stable supported palladium and rhodium catalysts that are similar chemically to the soluble catalysts that are used in pharma manufacture would be extremely valuable technologically; they are grand-challenge research targets.
Catalysis terminology
Catalysis that takes place in a single fluid phase is called homogeneous catalysis. When the catalyst is a solid and the reactants are present in a gas or liquid phase, then the term “heterogeneous catalysis” is used. These conventional terms, “homogeneous catalysis” and “heterogeneous catalysis,” simply refer to the number of phases that are present in the reactor: one, or more than one, respectively. Homogeneous catalysis takes place in a single fluid phase that is usually liquid and includes an organic solvent, chosen to be compatible with the organic ligands on the catalytic metal. The temperatures applied in heterogeneous catalysis are usually much higher than those applied in homogeneous catalysis. Low temperatures predominate in homogeneous catalysis because of the catalyst stability limitations. Further, even if a soluble catalyst is stable at high temperatures, a high pressure would be needed to keep the reactants in the liquid state, and high pressures require expensive equipment such as thick-walled reactors. The terminology of catalysis has evolved in ways that have sometimes led to deviations from the established standards, and to some misunderstandings. A publication by Robert L. Burwell that appeared in 1976 (ref. 10) provides standard usage of terms in catalysis. Nonetheless, the term “heterogeneous catalysis” has led many authors to refer to solid catalysts as “heterogeneous catalysts,” contrary to Burwell's guidance. This misstatement is significant because the term “heterogeneous” used to describe a solid catalyst has an important meaning, which Burwell stated: solid surfaces are almost all nonuniform in structure (and often in composition), and it is common that minority surface species are responsible for the catalysis. Thus, solid catalyst surfaces are heterogeneous, and their heterogeneity is one of their fundamental characteristics. This point is implicit in Hugh S. Taylor's early recognition of “active sites”11 (or “active centers”), catalytic sites that are often a minority of the sites on a surface. The heterogeneity of surface sites is a vexing challenge to the understanding of surface catalysis, a point that is central to this perspective.Supported catalysts and supported molecular catalysts
Many industrial catalysts are dispersed on the surfaces of porous solid supports (also called carriers) so that they are readily accessible to reactants. Typical supported species are metal nanoparticles, made to be so small that a substantial fraction of their atoms are exposed at a surface—allowing efficient use of the catalytic materials. The supports are typically inexpensive materials that make up more than 99% of the catalyst. The supports are porous, usually having high internal surface areas (>100 m2 g−1), and the catalytic material is distributed over the internal surface. The supports are typically robust materials such as metal oxides and zeolites; carbons, including functionalized carbons, are also commonly used. Widely applied supports such as transition aluminas (e.g., γ-Al2O3) can be manufactured to have various particle sizes, surface areas, and pore size distributions, which affect the accessibility of the internal catalytic species to reactants. Many catalysts on supports exemplified by metal oxides are treated periodically to remove carbonaceous deposits (coke) that accumulate as side products during operation; the typical treatment involves exposure to oxygen in air to burn off these deposits. Carbon and related supports (including metal–organic frameworks, MOFs) do not survive such treatments, and we barely consider them further here.Catalytic metals are almost always used as nanoparticles that expose a large fraction of their atoms on a surface.8 In a limiting case, as these nanoparticles become smaller and smaller, the catalytic material is dispersed maximally—atomically—on the support surface. Such catalysts are the central focus of this perspective.
Support surface chemistry and installation of reactive groups
The surface chemistry underlying heterogeneous catalysis developed strongly in the 1950s and 1960s as researchers identified and quantified functional groups on silicas, aluminas, titanias, and other high-area porous materials that are common catalyst supports. Infrared (IR) spectroscopy was essential to the advancement of this science, as summarized in early reviews.12,13 As mentioned in these reviews, researchers went beyond just analyzing the surfaces of the materials and took the next step of doing synthesis to link various groups to them. The anchored groups include organics (e.g., alkoxides), halides, boron-containing species, and metal-containing species, such as those containing aluminum.Mononuclear metal complex catalysts on supports
In view of this history, it is no surprise that researchers thought to extend the family of supported reagents and catalysts to include mononuclear metal complexes. Some of the pioneering work has conceptual links to Ziegler–Natta catalysis for alkene polymerization; according to early interpretations of the catalytic properties of the Ziegler polymerization catalyst α-TiCl3, titanium ions at the surfaces of TiCl3 crystals where chloride ions are missing are regarded as if they were reactive (coordinatively unsaturated) mononuclear metal complexes. This simplified picture reflects the Cossee–Arlman interpretation.14–16 It is an important concept in catalysis science, because it provides an essentially molecular interpretation of surface catalysis.The Cossee–Arlman interpretation is related to Taylor's hypothesis of active centers, but it goes beyond it, extending it to organometallic chemistry and identification of the surface catalytic sites (which Taylor realistically regarded as not often identifiable on typical catalyst surfaces).
In the history of heterogeneous catalysis, some decades ago there were intense debates about whether the catalytic properties of solids depend fundamentally on the bulk properties of the solid (e.g., semiconducting properties) or, instead, on “molecular” properties of groups on the solid surface. Now it is evident that both viewpoints have validity: on the one hand, consider catalysis on a surface whereby electron transport through a conductive solid between complementary groups or reactive intermediates on the surface plays a role in the catalysis; and, on the other hand, consider catalysis by organometallic groups that are bonded to supports that are ligands. The former idea provides conceptual links between conventional heterogeneous catalysis (“thermal catalysis”) and electrocatalysis,17 a topic of intense practical interest today, motivated by the availability of low-cost electrons generated in solar cells.
This essay is focused on essentially “molecular” catalysts on support surfaces. In retrospect, it might seem as though a logical next step beyond the Cossee–Arlman interpretation of surface polymerization catalysis would have been to bond a molecular metal complex with known alkene polymerization activity onto a support. And indeed this advance was reported in 1973 by Ballard,18 who anchored Zr(allyl)4 to porous silica—presumably by reaction of this precursor (in solution) with two neighboring OH groups on the silica surface to release two mols of propene per mol of zirconium and anchor the complex by two Si–O–Zr bonds. Similar surface synthesis chemistry was carried out by Ballard with Zr(benzyl)4 and with chromium compounds. Ballard's supported metal complexes were found to catalyze alkene polymerization.
Thus, the support came to be regarded as a ligand (sometimes called a macroligand), and, although these early catalysts were not characterized in depth, it was recognized that the supported metal complexes themselves acted as polymerization catalysts comparable to zirconium and chromium complexes in solution, and to Ziegler catalysts.
Exemplary early work on such supported catalysts was done by Burwell and Brenner,19 who synthesized mononuclear molybdenum carbonyls on γ-Al2O3. That work, among others, was influential, because it illustrated the application of quantitative physical chemical methods for characterization of the supported species, linking compositions of the metal complexes to catalytic activities.
These examples are part of the foundation of what has grown into a large body of work involving organic, inorganic, and organometallic species bonded to alumina, silica, and other inorganic solids. Silica surfaces present an array of groups including hydroxyl groups (silanol groups) and oxygen ions (in siloxanes), and silica has long been recognized as a “pegboard” for anchoring reactive groups. A rich chemistry has emerged of species regarded as molecular analogues on silica surfaces; many are organometallics with varied and intriguing catalytic properties. An extensive literature of catalysis by a wide range of organometallics on silica has been published by J.-M. Basset and coworkers, as reviewed recently.20,21 These authors recognized that silica served as a ligand and that it presented a variety of structures at its surface that can bond to anchored metals in various ways—and they realized that the surface species were fluxional. From this foundation, they discovered numerous new catalysts, often anticipating their properties from the chemistry of the analogous metal complexes in solution.
The class of mononuclear metal complexes on supports has expanded enormously, especially in the preceding decade, and it now includes many supports and, on them, many metals with a wide variety of ligands. The field is generating a burgeoning literature, much of it directed to the discovery of new and improved catalysts, with only little focus on elucidation of the fundamental chemistry (Serna22 recently provided citation data showing the rapid growth of the field). Much of the recent emphasis is on group-8 metals on metal oxide, zeolite, carbon, and MOF supports.
A surge in research on complexes of noble metals on supports began when it became feasible to image individual heavy metal atoms on supports that consist of light atoms—by scanning transmission electron microscopy (STEM). Some of the early reports23–26 that show images of isolated metal atoms on the supports (precisely made and evidently in the absence of metal clusters or nanoparticles) are summarized in Table 1. The reports summarized in this table have been influential because the authors used STEM in combination with complementary spectroscopic methods, as well as catalyst performance measurements, to characterize their catalysts.
| Metal on support | Ligands on metal in initial state identified by EXAFS spectroscopy | Support (surface area, m2 g−1) | Metal loading, wt% | Results from HAADF STEM imaging | Results from IR spectroscopy | Results from IR spectroscopy of working catalyst | Metal oxidation state indicated by XANES | Results from EXAFS spectroscopy of working catalyst | Catalytic reaction (temperature, °C) | Results from in operando XAS | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Palladium | 4 O atoms of support; no evidence of others | Mesoporous Al2O3 (350) | <0.03 | Isolated Pd atoms without clusters, only at this low palladium loading-electron beam caused cluster formation | None | None | Pd(I) | None | Oxidation of crotyl alcohol and of cinnamyl alcohol to form corresponding aldehydes (60) | Evidence of catalyst stability, but data not shown | Extremely low metal loadings required to synthesize sample with exclusively mononuclear palladium species; EXAFS data challenging to obtain for such low loading; oxidizing atmosphere inferred to stabilize cationic, site-isolated metal | 23 |
| Iridium | 2 O atoms of support + 2 ethene ligands, the latter confirmed by IR spectroscopy | MgO powder, surface area (ca. 100) | 1.0 | Isolated Ir atoms without clusters; density of iridium atoms on nearly planar surfaces about 1/3 of that determined by overall metal loading; electron beam caused cluster formation. Evidence from images that iridium cluster formation had started after 2 h of catalysis in flow reactor | Evidence of ethene ligands; evidence of CO ligands replacing ethene when sample exposed to CO (indicating iridium gem-dicarbonyl) and confirming mono-nuclearity | Evidence of mono-nuclear iridium complexes without evidence of clusters | Ir(I) | Ir bonded to 2 O atoms of support and to hydrocarbon ligands, consistent with 2 ethene ligands per Ir atom; no evidence of iridium clusters | Ethene hydrogenation (25) | Evidence of mononuclear metal complexes with coordination not distinguishable from that of precursor | Under reducing conditions metal is readily reduced and converted into clusters | 24 and 25 |
| Platinum | FeOx (ca. 290) | 0.17 | Isolated platinum atoms without clusters; density close to that expected from overall metal loading; electron beam caused cluster formation | Carbonyl band evident after sample exposure to CO, suggesting cationic Pt | Pt with low positive charge | CO oxidation; CO oxidation in presence of H2 (preferential oxidation) | None | None | Oxidizing atmosphere inferred to stabilize cationic, site-isolated metal; authors reported modeling of surface species with DFT, but with support surface not matching catalyst support | 26 |
The focus on group 8 metals reflects their widespread importance in practical catalysis—both homogeneous and heterogeneous. But the latter applications almost all involve metal nanoparticles8 (on the commonly used supports mentioned above), and the reactivities of these zerovalent metals for the most part are markedly different from those of mononuclear complexes of those metals, because the metals in the complexes typically are positively charged.8
The following perspective addresses metal complexes on supports, with a focus on group-8 metals, and the primary goal is to assess progress and challenges in fundamental understanding and to place the subject in the context of organometallic chemistry, surface chemistry, and catalysis.
Metal–support bonding and limitations of catalyst stability
Because the supports used most commonly in practical catalysis include metal oxides and zeolites, metals that are highly dispersed on them are almost always anchored through metal–oxygen bonds. In contrast, metal–oxygen bonds (and oxygen-containing ligands) are relatively rare among the molecular metal complexes that have been investigated as catalysts in solution. Ligands in metal complexes used most commonly in homogeneous catalysis are instead those that afford metal–carbon, metal–phosphorus, and metal–nitrogen bonds. Thus, in terms of the ligands, there is a disconnect between the most common supported metal complexes and the metal complexes most relevant in homogeneous catalysis.Early work with catalysts that consist of metal complexes on supports includes many contributions from organometallic chemists, and, not surprisingly, many of the early examples involved ligands comparable to those that are important in homogeneous catalysis—exemplified by phosphines, comparable, for example, to the phosphines in rhodium complexes used for alkene hydrogenation and hydroformylation.
Sometimes ligands such as phosphines anchored to organic polymer and silica supports and bonded to the metals were replaced during catalysis by reactant-derived ligands that afforded catalytic activity, although the switches were typically not identified in early work.27,28 Such ligand exchanges are now recognized to occur commonly, whether the reactants are gases or liquids.
A discouraging turning point in the emerging field of supported metal complexes as catalysts appeared in 1977 when Lang et al.28 reported flow-reactor experiments characterizing hydroformylation of 1-hexene in the liquid phase in contact with a supported rhodium catalyst on a phosphine-functionalized polymer support. The data demonstrated ligand exchanges involving breaking of metal–support-ligand bonds that led to unlinking of the metals from the support—the metals that leaked off the support flowed downstream with the products of the catalytic reaction, demonstrating the lack of practical applicability of the catalyst—taking away a putative essential benefit of placing the catalyst on a support—that is, the elimination of an expensive product–catalyst separation step.
Nonetheless, there are examples showing that when the metal is linked to the support by sufficiently strong bonds, the stability may be sufficient—as illustrated by the aforementioned Chiyoda-UOP process5 for carbonylation of methanol in a polar solution (containing water) in contact with a catalyst that is a rhodium complex bonded to the anion exchange resin support; the catalyst is evidently stable enough to be used commercially.
Lack of uniformity of supported metal complexes
Recurrent statements in the recent literature of supported metal complexes refer to syntheses that are “atomically precise” and to supported species that are “uniform”—all the same or nearly all the same. This terminology can be traced back to early literature. For example, in a 1988 review, John Meurig Thomas29 emphasized the benefits of fundamental understanding accruing from investigations of solid catalysts having (bulk) crystalline structures; he emphasized zeolites, which are aluminosilicates with pore geometries that are determined by the crystal structures. The zeolites are much more nearly uniform than most practical catalytic materials, although recent high-resolution images now make it evident that some of these materials are far from uniform.30Extending his perspective about uniform catalyst supports, Thomas in a 2014 review31 wrote about “single-site heterogeneous catalysts” (SSHCs), stating that “…it is prudent to recall that SSHCs are, inter alia, those in which the active centres are energetically equivalent and spatially isolated from one another, and are uniformly distributed over an internal and accessible surface which is exceptionally large.” These points have gained traction in the recent literature, and many authors have gone beyond Thomas's statement and misrepresented the supported species as not just energetically equivalent but structurally equivalent. Thus, it has been common for authors to infer on the basis of the mononuclearity of the metal species on supports that they are equivalent in structure and in reactivity. The limitations of this viewpoint and evidence of the heterogeneity of supported metal complexes are developed further below.
Complexes of oxophilic metals on supports
Work on oxophilic metals such as rhenium and tungsten supported on alumina has led to practical catalysts for alkene metathesis that may be essentially mononuclear complexes, and extensive work has been done on supported catalysts for alkene polymerization; these are beyond the scope of this perspective. Because of their practical importance, the aforementioned silica-supported mononuclear chromium catalysts used for ethene polymerization have generated an enormous literature and made clear the challenge of unraveling the catalytic chemistry—the ligands in the catalytic reaction intermediates are challenging to identify. The complexity may be associated in part with the variety of groups on the silica surface (oxygen ions and hydroxyl groups in various configurations), which are fluxional, and the presence of difficult-to-detect minority metal complexes formed from reactants. In other words, the heterogeneity of the support surface contributes to the challenge of elucidating the chemistry.Complexes of noble metals on supports and their tendency to be reduced and aggregated
Through the years of investigation of metal complexes on supports, much of the emphasis, especially recently, has been on noble metals, primarily rhodium, iridium, palladium, platinum, and gold, often chosen because of their importance in practical catalysis as well as the availability of STEM for imaging the heavy metal atoms on supports consisting of light atoms. When these metals are present as mononuclear complexes on metal oxides and zeolites, they are often stable as cationic complexes under oxidative conditions. Such complexes catalyze oxidation reactions, including CO oxidation, which has been frequently chosen as a test reaction, one that offers the benefits of a simple product distribution and small reactant molecules with informative spectroscopic signatures. This reaction is important in practice because it takes place on an enormous scale in the clean-up of vehicle exhausts.However, most applications of supported noble metal catalysts involve metals in a more conventional state—as nanoparticles on supports. These are applied, for example, in numerous hydrocarbon conversion processes in the petroleum refining and petrochemical conversion industries, with the reactions taking place under reducing conditions; further, the metals in vehicle exhaust emission control catalysts are also predominantly present as zerovalent nanoparticles.
Noble metals in mononuclear complexes on metal oxide and zeolite supports are readily reduced in the presence of H2 and other reducing agents—so that the metals migrate and aggregate into zerovalent nanoparticles on the supports. In some recent research, the conversion of atomically dispersed noble metals on supports into reduced, aggregated metals has taken place to such a small degree that it has been missed, with the catalytic species being misidentified as mononuclear metal complexes. We return to this topic below when addressing the complexity of species that may be present in catalysts that nominally consist of mononuclear metal complexes.
A related point is that catalysts consisting of supported metal nanoparticles are often treated in oxidizing atmospheres to burn off carbonaceous deposits that form during hydrocarbon conversion catalysis (and other reactions of organic compounds)—and the treatments may take place at high temperatures and lead to oxidative fragmentation of the nanoparticles, converting them into mononuclear metal species; we delve into this issue below.
Metals on metals: dilute-alloy nanoparticles
The supports for metals include not just the conventional metal oxides, zeolites, etc., but also zerovalent metals themselves—and the zerovalent metals are commonly present as nanoparticles on the conventional supports. When a noble catalytic metal such as platinum is present in only a low loading on a less active coinage metal such as copper, it may be isolated—present in an alloy so dilute that the catalytic metal is surrounded only by atoms of the other metal—then it is called a single-atom alloy. Catalysts in this class offer unique properties, associated with the lack of neighboring noble metal centers and reactivities that are markedly different from those of the noble metal alone or the coinage metal alone.32These supported bimetallic catalysts have been postulated to consist of site-isolated noble metals on the surfaces of supported nanoparticles of coinage metals, but recent results include X-ray absorption spectra and IR spectra implying that the stable structures, depending on the reactive environment, may include the noble metal (e.g., platinum) within the bulk of the coinage metal (e.g., silver) in nanoparticles supported on Al2O3, and not on the surfaces of coinage metal nanoparticles.33
And matters may be still more complex: during catalysis of hydrogenation of unsaturated aldehydes to give unsaturated alcohols catalyzed by supported platinum–copper nanoparticles, the platinum in the nanoparticles has been found by extended X-ray absorption fine structure (EXAFS) spectroscopy to be present predominantly at the interface with the support and not on a metallic surface surrounded only by the copper.34
There is more to learn about the structures of supported dilute alloys and how to control their structures and reactivities and how they depend on reactive environments; this topic is beyond the scope of this perspective.
Methods for elucidating structure and reactivity of supported mononuclear metal complexes
Notwithstanding the complications mentioned above, substantial understanding of mononuclear noble metal complexes on metal oxide and zeolite supports has emerged. Much of the understanding pertains to chemistry taking place under conditions so mild that the structure and bonding can be characterized and changes in structure and bonding can be tracked. The most helpful characterization methods include atomic-resolution imaging and IR spectroscopy, EXAFS spectroscopy, and X-ray absorption near edge structure (XANES) spectroscopy.Methods for imaging individual heavy metal atoms on supports have become widely accessible in the preceding few years, and their availability has helped to create the recent deluge of research on supported mononuclear noble metal complexes.35 Thus, advances in high-resolution transmission electron microscopy (TEM), and especially aberration-corrected STEM, have helped accelerate research on supported metal complex catalysts, especially those incorporating heavy metals (such as iridium, platinum, and gold) on supports consisting of low-atomic-number atoms (such as zeolites and MgO). Many publications about these catalysts have appeared in catalysis journals—and also in general science and general chemistry and materials science journals, all leading to a broadening awareness of catalysis science and technology.
This energizing of the field of supported metal complex catalysts has led to the popular use of terms such as “atomically dispersed supported metal catalyst” and, more commonly, “single-atom catalyst.” The former is often accurate, but it is not sufficient to distinguish supported mononuclear from polynuclear metal complexes when all the metal atoms are accessible to reactants. The latter term may have encouraged authors to deemphasize or overlook the roles of the ligands, which are central to the chemistry and include the supports.
A related term is the aforementioned “single-atom alloy,” which describes some dilute alloys. In these materials, the metal is often present as nanoparticles, and the complexities mentioned above may arise as the highly dispersed metal atoms may be bonded within bulk structures and also to the support.
The following paragraphs address recent work on the fundamental chemistry of supported mononuclear noble metal complexes and their role in catalysis. The focus is on metals on metal oxide and zeolite supports—with this emphasis chosen in large part because the metals and supports may be stable when carbonaceous deposits that form on them during catalysis are removed by high-temperature combustion (carbon and MOF supports are less practical supports because they do not withstand such regeneration treatments).
Identifying ligands on metals and sites for metal bonding on high-area supports and evidence of site heterogeneity
Extensive data have been reported characterizing metal complexes on high-area metal oxides and zeolites, with structures determined primarily by IR and X-ray absorption spectroscopies and atomic-scale imaging of sample surfaces. Typical results reflect the heterogeneity of the support surfaces, with mixtures of supported species being present and poorly resolved. Some relatively uniform supported species have been formed on zeotype materials, with the broadness of νCO bands characterizing carbonyls of the metals typically being markedly less for these species than for comparable species on metal oxides.Thus, some of the clearest evidence of the supports as ligands has emerged from IR spectra of metal carbonyls on these supports. For example, mononuclear rhodium carbonyls on zeolite HY were shown by the spectra to be rhodium gem-dicarbonyls, with the symmetries shown by the spectra (compared with those of pure-compound analogues having fully determined structures) indicating two ligands besides CO bonded to the metal—typically two oxygen atoms of the support. EXAFS spectra were found to be consistent with the inferred structures, showing metal–oxygen coordination numbers of about two and typical metal–oxygen bonding distances.
For example, isostructural rhodium and iridium complexes on zeolite HY were characterized by IR and EXAFS spectroscopies when various ligands were present on the metals (CO, ethene, and hydride).36–39 The spectra identify the ligands and provide evidence of their influence on catalyst performance (e.g., for ethene hydrogenation and dimerization and H–D exchange in the reaction of H2 with D2), showing, for example, that CO is generally a reaction inhibitor.36 However, a limitation of such data is that they fall short of determining reaction intermediates, which are often present in amounts too low to determine. Another limitation is that such data do not determine the locations of the metals on the supports. These and related points are addressed in more depth below, with details about the characterization methods and the chemistry of the supported metal complexes.
Sites for bonding metals on single-crystal metal oxide surfaces
Surface bonding sites for metals such as gold and platinum, among numerous others, have been determined in experiments performed with structurally well-defined supports—single crystals of metal oxides, including TiO2, MgO, and Fe3O4. The experiments have typically been performed under ultrahigh-vacuum (UHV) conditions.In contrast to the methods that have been used most commonly to image metal atoms on surfaces of high-area supports and to determine metal oxidation states,40,41 those used to characterize single-crystal samples include imaging by scanning tunneling microscopy (STM) and characterization by spectroscopic methods including X-ray photoelectron spectroscopy to provide evidence of the metal oxidation states.42
The single-crystal metal oxides mentioned above are typical in presenting numerous potential bonding sites for metals, and the structural complexity makes determination of the chemistry challenging.42,43 Metals have been found to be coordinated, for example, to neighboring oxygen ions (bidentate sites), among others, and to occupy not just various sites on the surfaces, but also sites within the bulk materials, with the surface site preferences sometimes being influenced by defects in the bulk support below the binding site for the metal.42,43 Noble metals have been observed to be readily reduced and to aggregate into clusters. Only a few results have been reported characterizing reactivities of the metals on these well-defined supports, most commonly in experiments with CO as a reactant, providing evidence of metal carbonyls. Both monocarbonyls and dicarbonyls have been observed.
Assessment of a broad literature by the group of Diebold and Parkinson42,43 led to the inference that bonding environments for the metals on single-crystal metal oxides tend to be comparable to those in molecular metal complexes, at least when CO is a ligand. This observation, combined with the tendency of the metal atoms on the surface to distort the support structure to accommodate the adsorbates, led these authors to infer that atoms adsorbed on supports (undercoordinated under UHV conditions) should be viewed as analogous to metals bonded to ligands in metal complexes, with the observations in a broad sense being transferrable to numerous species on numerous supports. This insight is in line with extensive results characterizing metal complexes on high-area metal oxide and zeolite supports, as summarized in this perspective, and bolstering the broader conclusions stated here. The images and spectra characterizing single-crystal metal oxides and metal complexes on them are important in providing foundational information and determining precise structures of surface species that are not attainable with intrinsically nonuniform, high-area supports such as TiO2 and MgO powders.
Sites for bonding metals on single-crystal metal surfaces
STM images of single crystals of copper incorporating palladium atoms on their surfaces point to atomically dispersed palladium, and this was characterized by IR spectra of CO on the isolated palladium atoms, which were found to be stable enough to allow the characterization that indicated the near uniformity of the surroundings of the noble metal atoms.32 The results led to descriptions of the single-crystal materials as single-atom alloys.In this context, recall the aforementioned data obtained with supported bimetallic nanoparticles incorporating platinum (with silver or copper) which have led to a nuanced view of the structures of the bimetallics dispersed on metal oxide supports and the role of the support—these data point to the importance of noble metal atoms in the bulk of the particles and at the metal–support interface, raising questions about how to determine when the term “single-atom alloy” is descriptive of supported bimetallic nanoparticles.
The challenges of linking results obtained with single-crystal samples under UHV conditions with results characterizing conventional supported metal catalysts are substantial and seem to provide fruitful opportunities for future work.
Early examples of characterization of atomically dispersed noble metals on supports with STEM complemented by spectroscopy demonstrating isolation of individual metal atoms
Interest among researchers in supported mononuclear metal complex catalysts gained momentum when it became apparent that imaging by aberration-corrected STEM could demonstrate the presence of isolated metal atoms on supports in the absence of clusters or nanoparticles of the metals. Aberration-corrected high-angle annular dark field (HAADF) STEM became available in a number of laboratories and triggered a flood of research activity that continues to this day. Several early reports of images of noble metal catalysts dispersed atomically on metal oxide supports were complemented with spectroscopic data and catalyst performance data, as summarized in Table 1, which provides some details of the characterizations and comments about their limitations. The combinations of characterization methods illustrated in this early work have largely become the norm, and they are illustrated with specifics below.Identifying various support surface functional groups and sites for bonding of atomically dispersed metals
Metal oxide and zeolite supports typically present a variety of bonding sites for metal complexes, including sites with various surroundings, both on single-crystal surfaces and more complex surfaces. The sites incorporate various atoms and functional groups in various surroundings, exemplified by oxygen ions and hydroxyl groups with various coordinations, defect sites of various structures, and sites that are below the surface, including sites within the bulk (which are considered in a separate section below).Experimental evidence of structure and bonding at the metal–support interface
Evidence of bonding of isolated metal atoms on zeolite and metal oxide supports has been reported for years, with quantitative data determined by EXAFS spectroscopy. When the metals are present as mononuclear complexes or few-atom clusters, metal–support distances of about 2.1 Å are common.44 This is a bonding distance between positively charged metal atoms and oxygen anions, observed often, both for numerous molecular metal complexes in solution or in crystalline form and supported analogues.Only little structural evidence of metal–support interfaces has emerged from experimental methods other than EXAFS spectroscopy, however. An exception is illustrated by STEM images of gold atoms in zeolite NaY; the images, combined with the known crystal structure of the defect-free zeolite, allowed the authors45 to determine the sites of the gold atoms within that crystal structure (Fig. 1). The images show gold atoms in more than one crystallographic location, and the gold atoms shifted between locations (and changed oxidation state) when the catalyst was used for CO oxidation.45
 | ||
| Fig. 1 STEM image of gold atoms in NaY zeolite; the images, which demonstrate the porous structure of the zeolite (without defects) and the locations of the gold atoms, were used to infer two crystallographically different bonding sites for the gold cations in the catalyst; another site was occupied initially, after adsorption (physisorption) of the precursor Au(CH3)2(acac). Exposure to CO + O2 led to movement of the gold complex to other sites and to changes in the Au–O coordination; images showed that the bonding positions changed during CO oxidation catalysis, and complementary XANES data indicated the reduction of the gold from approximately Au(III) initially to Au(I) during catalysis.45 Reproduced from ref. 45 with permission from John Wiley and Sons, Copyright 2012. | ||
Theory for identifying support bonding sites and supported metal complexes
Density functional theory (DFT) has been used to represent many metal complexes on supports; many of the recent publications in this field include DFT results. The representations have mostly been based on the assumption of a single metal–support bonding mode, without accounting for multiple bonding sites or strong experimental evidence of the site structure. Thus, applications of theory have not yet advanced sufficiently to account for the structural complexity of support surface compositions and structures, and the surroundings of metals on supports.A recent critical assessment by Di Liberto and Pacchioni46 provides an evaluation of the role of electronic structure calculations in representing supported metal complexes and their reactivities. The authors emphasized that for the results of simulations to be reliable, realistic, and experimentally verifiable, they must take account of the complexity of the supported species. Major limitations thus include the lack of precise knowledge of the local coordination environment of the metal on the support in the various plausible structures—because even small changes in the surroundings of the metal can result in large changes in its reactivity. Further, realistic modeling needs to account for changes in the environment of the metal during reaction.
Most of the reported calculations of atomically dispersed supported metals are based on approximations that are good (but imperfect) for extended metals, but that have serious limitations when applied to structures with localized electronic states, exemplified by transition metal atoms in a surrounding matrix.46 According to Di Liberto and Paccioni,46 the calculated binding energy of an adsorbate on a reactive site can change by 0.5 eV or more when the exchange–correlation functional used in the computations is changed, with even qualitative effects on the computed results.
There are many detailed computational results characterizing supported species (including metal complexes) on a variety of idealized supports, but most of the reported results in this category have been presented without critical statements about the limitations of the simplified structure and bonding that were assumed. Further, the available results often do not account realistically for the ligands on the metal, which typically are not uniform from one species to another on a given support; in many reports, ligands besides the support are not even included.
Notwithstanding these limitations, theory is helpful and expected to play an essential role in advancing the science of supported metal complexes. It is valuable, for example, in clarifying the complexity of the samples, as illustrated by the work of Xia et al.,47 who investigated platinum complexes on ceria. They used computational chemistry to demonstrate that the reactive sites of stable supported metal complexes may often include one structure that is much more stable than other local-minimum structures—and this most stable structure may be responsible for only little of the catalytic activity, with that activity instead reflecting the properties of structures that may be orders of magnitude less common than the predominant structure. Results such as these alert researchers to the limitations of spectroscopic evidence of the supported species, even those measured during catalysis, as they may have little direct relevance to the catalytic reaction mechanism.
In summary, the complexity of the typical supported mononuclear metal complex is still beyond the reach of realistic computational chemistry to account for structure, bonding, and reactivity, and computational chemistry is not yet sufficient to predict the structures of catalysts with appealing new properties, although it may provide valuable leads and suggest candidate experiments.
Theory in concert with experimental EXAFS spectroscopy for identifying support bonding sites and supported metal complexes
We posit that some of the major benefits of theory in the next steps of advancing the science of supported mononuclear metal complexes may result from closer linking of theory with experiment. For example, DFT computations have been incorporated into the fitting of EXAFS data (by the QuantEXAFS method), with the results determining structural models for platinum on MgO (ref. 48) and palladium on MgO.49 The strategy was used to combine results of conventional EXAFS analysis, STEM, DFT calculations, automated EXAFS analysis, FEFF-XANES spectroscopy, and IR spectroscopy, all used in concert to characterize the isolated metal atoms on high-area MgO and to identify the structures most consistent with experiment.This approach is appropriate to the characterization of samples that have sufficient structural uniformity, that is, to metal complexes on supports that are not too far from uniform and present not too many different sites for bonding of the metals; thus, supports that are highly crystalline, such as MgO and zeolites, are among the most tractable, whereas structurally more complex supports such as CeO2 are less so.
The QuantEXAFS approach led to the identification of MgO-supported platinum, determining the most populated (thermodynamically stable) sites for these metals, as follows: Results were obtained for MgO-supported platinum made from K2PtCl4; the sample was calcined at 700 °C, and STEM images and IR and EXAFS data all indicated mononuclear platinum on the support. The QuantEXAFS analysis relied on DFT calculations (with the PBESol functional, although others led to similar results) to create a comprehensive library of all the plausible cationic platinum on crystalline MgO structures, considering three MgO facets (terrace sites: [100], Mg vacancy terrace sites: [100]Mg-vac, and step sites: [310]); various oxygen adsorbates; various types of vacancies (Ovac, Mgvac); and subsurface platinum locations. The platinum sites are denoted by the MgO facet, the platinum atom location, and adsorbates or vacancies (Fig. 2). Thus, [100]Mg-vac/sub1 refers to a 100 facet with the platinum atom in the first subsurface site neighboring a magnesium vacancy, for example. These structures were used to determine the relative stabilities of platinum atoms in the various sites. The calculations for the stoichiometric [100] and [310] surfaces show that the surface platinum ([100]/sub0/*O2) and leading step edge ([310]/pos1/*O2) sites are favored under the conditions of the experiments (temperature, 573 °C; O2 partial pressure, 1 bar). A comparison of the relative stabilities of platinum atoms at the surface and in subsurface layers of the various MgO facets shows that, for the stoichiometric [100] facet, platinum sites in the first and second subsurface layers are less favorable energetically than those at the MgO surface. However, although surface platinum sites are favored for the stoichiometric [100] surface, incorporation of a magnesium vacancy changes the relative stabilities, so that platinum substitution in the first subsurface layer ([100]Mg-vac/sub1) is energetically more favorable than that in the surface layer or the second subsurface layer. The EXAFS analysis of all of the aforementioned DFT-optimized structures (47 of them) was done including all the relevant scattering paths for each DFT-optimized structure.48
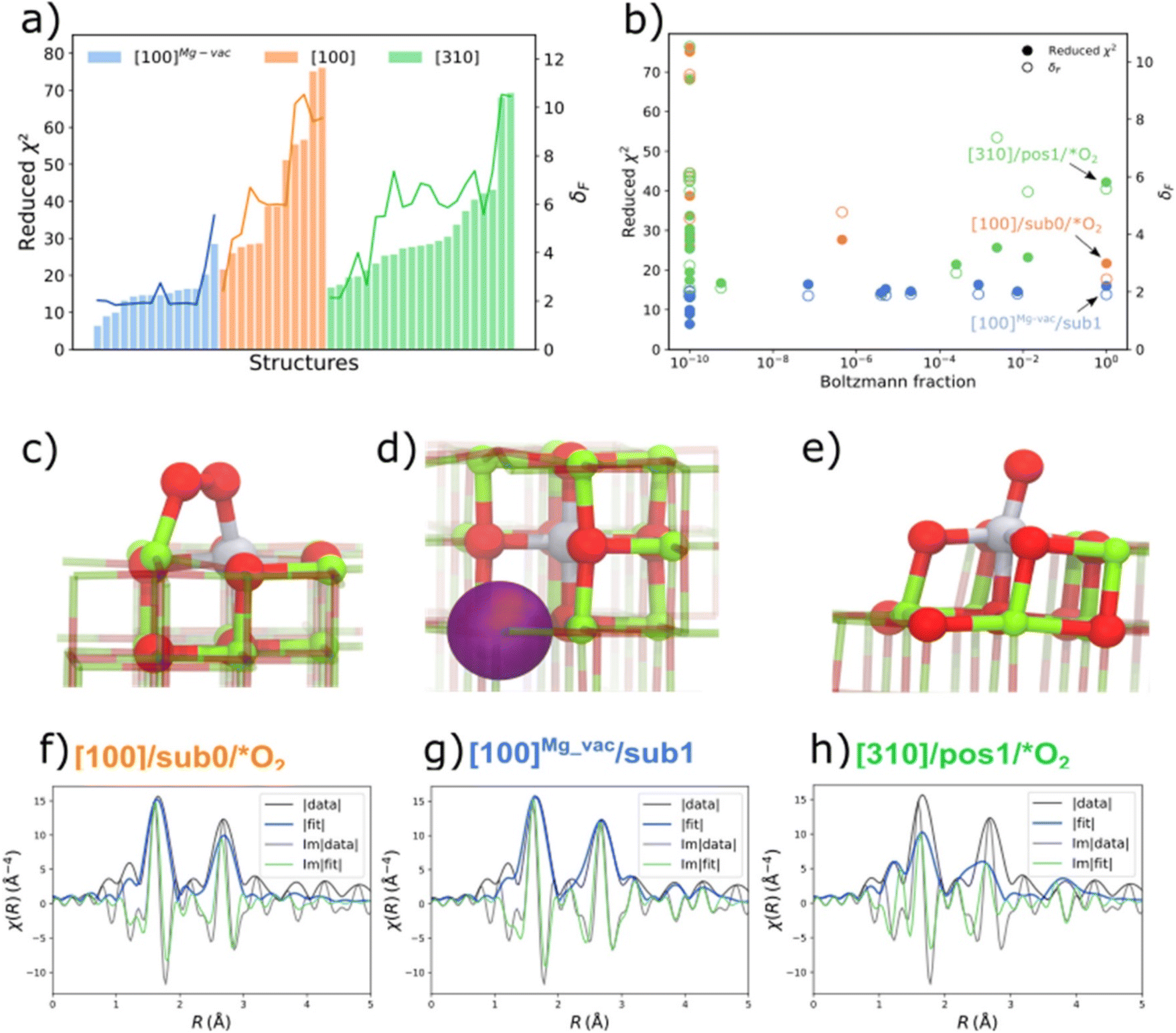 | ||
| Fig. 2 (a) Reduced-χ2 and Fréchet distance (δF) of EXAFS analysis for all the DFT-optimized structures of MgO-supported platinum that had been calcined at 700 °C; (b) Boltzmann fraction calculations for all facets. The DFT-optimized geometries of the most stable (c) [100]/sub0/*O2 (d) [100]Mg-vac/sub1, and (e) [310]/pos1/*O2 structures (with the terminology illustrated by the structures) are shown with the corresponding EXAFS fits in (f–h), representing the magnitude (fit: blue; experiment: black) and imaginary portions (fit: green; experiment: black) of the Fourier transforms of the EXAFS data. A k-range of 2.2–12.5 Å−1 and the R-range of 1.0–5.0 Å were used in the fitting (k is the wave vector; R is the interatomic distance). Note the good agreement between the data and the fit shown in (g). Colors: magnesium (green); oxygen (red); platinum (gray). The purple sphere in (d) represents the subsurface magnesium vacancy.48 Reproduced from ref. 48 with permission from the American Chemical Society, Copyright 2021. | ||
The results (Fig. 2) show, for example, that the stoichiometric [100] facet presents sites for platinum in the first subsurface layer that are 1.3 eV less favorable energetically than those at the MgO surface, and those for platinum in the second layer are 2.1 ev less favorable energetically. Although surface platinum sites were found to be preferred for the stoichiometric [100] surface, the presence of a magnesium vacancy changes the relative stabilities: then, platinum substitution in the first subsurface layer is energetically more favorable than that in the surface layer or the second subsurface layer (Fig. 2).
Thus, the results indicate that the supported mononuclear platinum is best represented by the structure (d) in Fig. 2, with the subsurface platinum cation next to a magnesium vacancy. We return to this sample in a following section to consider its catalytic properties.
A closely related investigation performed with the same strategy for MgO-supported palladium49 again took account of all the scattering paths of the plausible structures determined by DFT calculations for well-defined mononuclear palladium structures on the crystalline support. The candidate models in the database again included all the structures that were optimized by using the PBESol functional, and three representative MgO facets (terrace sites, [100], [100]Mg-vac (with a Mg vacancy), and step sites, [310]). Various surface and subsurface layer locations of palladium atoms in the MgO lattice were accounted for, as were adsorbates (O2 and O) and possible Mg and O vacancies. The results characterizing the sample calcined at the higher temperature, Pd/MgO700, show that the palladium was present predominantly as cations in the first MgO subsurface layer; each was located next to a magnesium vacancy in the [100] MgO facet. The corresponding Pd–O and Pd–Mg coordination numbers were found to be CNPd–O = 6 and CNPd–Mg = 11. However, the analysis carried out using the database of all the structures mentioned characterizing Pd/MgO500 showed that no single structure provided a satisfactory fit of the data. Thus, to go a step further and address the challenge of representing the heterogeneity of this sample, the method was adapted to simultaneously fit the scattering paths obtained for two distinct DFT-optimized structures in the sample. In addition to the second-layer (subsurface) site that accounts for the Pd/MgO700 sample, an alternative palladium site was identified where the O2-bound palladium cation is stabilized at the [310] step. The results show that Pd/MgO500 is well represented as a 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :63 mixture of the two structures, and it is significant in representing a first step toward accounting quantitatively for the heterogeneity of a support harboring supported mononuclear metal complexes.
:63 mixture of the two structures, and it is significant in representing a first step toward accounting quantitatively for the heterogeneity of a support harboring supported mononuclear metal complexes.
Challenges of elucidating catalytic intermediates
An essential general point about fundamental understanding of catalysts is the following: in molecular (homogeneous) catalysis, it is sometimes possible—with extensive work—to resolve the elementary reactions (steps) and thereby to elucidate a catalytic cycle. Resolving the steps requires isolation of intermediates in a cycle and determining their structures, most convincingly by using X-ray diffraction crystallography of isolated, crystallized intermediates, perhaps combined with spectroscopy. If only some of the essential species in a catalytic cycle can be identified experimentally, then theory can be used to fill in the gaps, to predict the full cycle. There are now a number of examples of molecular organometallic catalysis with elucidated cycles, although this depth of understanding of catalysis is exceptional, because of the complexity of the chemistry and the challenge of elucidating the intermediates.1–3A related general point is that comparable chemistry of catalysis on surfaces is still essentially unattainable. The best one can do is usually to identify some surface species spectroscopically—but the methods usually fall short because (a) the species that are identified may not be part of a catalytic cycle (they may be red herrings); (b) the intermediates that are part of the cycle may be present in concentrations too low to detect; and (c) even when catalytic intermediates can be identified spectroscopically, definitive structure-determination methods such as X-ray diffraction crystallography do not work for surface species. The complexity is amplified by the heterogeneity of the support surfaces and the attendant nonuniformity of the species on them.
Minority surface species are often kinetically significant in catalysis, and, in principle, theory may be valuable for identifying them, but the heterogeneity of most support surfaces makes matters highly challenging, and far-reaching advances are needed for progress toward accounting for the heterogeneity of the reactive surface intermediates.
Striving for uniform metal complexes on supports to facilitate fundamental understanding
Numerous authors have written cautionary statements stating the limitations of the assumption that mononuclear metals on supports are uniform,39,50–52—in other words, they have emphasized the importance of recognizing support surface heterogeneity. But some authors have considered it to be fruitful to begin investigations with syntheses intended to make samples that have a minimal degree of nonuniformity and to try to extract maximal fundamental understanding from investigations of such samples. A core thesis of this essay is that this approach has been helpful and may continue to be helpful in advancing the science of supported metal complex catalysts.Nearly uniform catalytic species offer the following prospective advantages: they would allow
(a) Accurate, essentially molecular interpretations of structure and reactivity;
(b) Prediction and design of families of catalysts having various controlled and systematically varied ligand environments; and
(c) Accurate modeling of the surface species with computational chemistry representing single species—thus providing a foundation for starting to elucidate reaction mechanisms.
These prospects have motivated a number of researchers to take up the synthesis challenge, and some examples are mentioned below. A corollary is that data are needed to demonstrate whether the surface metal complexes are nearly uniform and to assess the degree of deviation from this limiting case. We next address some synthetic approaches and methods of assessing the degree of uniformity of the supported metal species.
Approaches to synthesizing nearly uniform supported metal complexes
The following approaches have been applied in attempts to synthesize supported mononuclear metal complexes with simple, well-defined structures:39(1) Use supports that are crystalline and largely defect-free that present sparse and nearly uniform bonding sites for metals; because of their intrinsic complexity, most single-crystal metal oxides are not especially good candidates, and well-synthesized zeotype materials seem to be the best candidates among high-area supports, especially zeolites that are dealuminated and provide sparse bonding sites associated with aluminum in the framework (where proton-donor sites are located). MOFs have drawn significant attention as supports, but these crystalline high-area supports have defects and do not seem yet to be available with simple enough, nearly ideal structures to provide the opportunities that some well-made zeolites provide (more attention should be paid to imperfections in MOF structures).
(2) Treat supports, even those that are not crystalline and those that incorporate various surface sites (including defect sites), to minimize the number of different kinds of sites on their surfaces—and use these sites selectively to make the supported catalysts. A characterization challenge with this approach is to determine where and how the metals are bonded when the metal loadings are low and the characterization methods lack sufficient sensitivity.
(3) Make samples with low metal loadings, attempting to anchor the metals selectively to only the most reactive support surface sites—with the premise that these, in prospect, might be nearly unique sites and occupied preferentially by the metals in the synthesis. Again, a challenge is to determine where and how the metals are bonded, and the samples with the lowest metal loadings challenge the characterization methods.
Experimental criteria for assessing the degree of uniformity of metal complexes on supports
The methods for assessing the uniformity of the supported species include the following;39 none alone is sufficient; they are most effective when used in combination:(1) Image the metal atoms and determine whether they are all atomically dispersed. If they are not, the goal of uniformity has not been met. Many images may be needed for a critical check, as supports present various faces and zones for potential metal bonding (and it is a rarity when reports provide many images for checking). Further, in electron microscopy, attention needs to be paid to changes in the sample caused by the electron beam; supports such as zeolites and MOFs are especially susceptible to beam damage. STM is valuable but limited to nearly planar samples such as single crystals.
(2) If the metals are all atomically dispersed according to the available images, image their surroundings—and, if possible, check for uniformity. Such imaging is challenging, especially in view of the heterogeneity of most supports, but the goal is realistic with some single-crystal or highly uniform crystalline samples such as zeotypes that, if synthesized well, may have nearly uniform pore structures, as shown by images (e.g., Fig. 1)—but detailed scrutiny with the most advanced imaging methods has shown that some zeolites have many structural details different from those of the idealized (perfectly crystalline) materials.30
(3) Probe the metal atoms on the supports with small-molecule adsorbates and record spectra that provide evidence of the degree of uniformity. IR spectroscopy is especially useful, with CO as the probe; sharp νCO bands indicate bonding to nearly uniform sites, and NO may be a useful probe as well. But one needs to be aware of the possibility that the probes are non-innocent—that they react with the supported species and change their ligand environments, metal oxidation states, and/or structures, including bonding to the support and metal nuclearity. This point is illustrated below with examples.
(4) Modify the supported species by reaction with gas-phase reactants (probe molecules), rather than liquid-phase reactants, to simplify matters, and record spectra to track changes in the structures and the ligands on the metal. If the spectra include isosbestic points, then the data imply that one species is transformed to one other species—evidence implying the presence of uniform species before and after the transformation (but this method may not probe all the structures).
(5) Treat the sample to change the metal nuclearity (along with the ligands) and record spectra; again, if the spectra include isosbestic points, infer that one species is transformed to one other species—indirect evidence of uniform species before and after the transformation. It is helpful if the transformation is reversible. This point is illustrated below by example.
(6) Use nuclear magnetic resonance (NMR) spectroscopy (e.g., 13C NMR spectroscopy) to check for dynamic uniformity of the environment of the metal centers.53 There are surprisingly few reports of this approach.
(7) Use poisons to deactivate various fractions of the metal sites and determine whether there is a linear dependence of catalytic activity (turnover frequency, TOF) on the number of poison molecules per metal site—if there is, infer that they are catalytically equivalent and, by inference, structurally equivalent. There are surprisingly few data available to check for site uniformity of supported metal complexes39 with this approach, although it has been traditional in catalysis research for many years.
(8) Use temperature-programmed desorption, expecting that a sharp desorption profile, for example, of an adsorbed probe molecule such as CO, indicates that these adsorbates are widely separated from each other, for example, indicating a lack of dipole–dipole interactions between neighboring adsorbed CO molecules. Doing such experiments in concert with IR spectroscopy characterizing the adsorbed and desorbing species adds value to the data.54
(9) Use EXAFS spectroscopy to determine models of the structure of the supported species. This approach is most appropriate when the support is highly crystalline and presents only a limited number of kinds of sites for metal bonding. Samples with highly heterogeneous surfaces will have so many possible structures that it will be unrealistic to distinguish them with EXAFS spectroscopy. EXAFS spectra of supported metal complexes have often been overinterpreted, and recent work provides a cautionary set of examples to illustrate prudent data analysis.55
(10) Use various loadings of metal in attempts to populate the more reactive bonding sites preferentially (as suggested above)—and do this to different degrees to prepare samples with a range of metal loadings—and then characterize them with imaging, EXAFS spectroscopy, IR spectroscopy with adsorbed probe molecules, and other methods, with the goal of distinguishing the metals in separate kinds of sites—and searching for a limiting case of only one kind of site—and then determine whether the catalytic activity is independent of the metal loading.
To reemphasize a core thesis of this perspective: striving for uniform supported mononuclear metal complexes is a worthy goal that provides a path forward for advancing the understanding of supported mononuclear metal complexes and for tailoring their properties. It is significant, however, that only little work has been reported with the approaches stated above, and only few reports have provided critical assessments of the degree of uniformity of the supported species.
However, among the characterization methods mentioned above, there is a notable exception: many researchers have used gas-phase CO as a probe of supported metals and presented data that determine the narrowness of the νCO bands, which provide a basis for assessing the degree of uniformity of the metal species.51 These experiments are relatively straightforward and have long been popular in the catalysis community, and thus there is a substantial literature of their application. But there is a surprising lack of comments in the literature about the narrowness of the νCO bands and comparisons with those of related materials. Some details of the method and data are summarized in examples below.
Examples illustrating structure, reactivity, and catalytic performance of supported noble metal complexes
Evidence contrasting O and OH groups for bonding to metal complexes on metal oxides
There has been only little research done with the goal of distinguishing O from OH groups as bonding sites for metal complexes on metal oxides or zeolites. An exceptional investigation was reported for rhenium carbonyls on MgO, with the support samples treated to give widely different densities of OH groups on the surface. Relying on IR spectroscopy and evidence of the symmetries of the supported metal complexes, the authors56 determined that samples prepared with varying densities of support surface OH groups incorporated rhenium carbonyl mixtures well approximated as Re(CO)3{OMg}3 and Re(CO)3{HOMg}3 (where the braces denote surface groups), and the spectra are similar to those of molecular analogues. The latter was found to be predominant on the more highly hydroxylated support and vice versa. The support as a tridentate ligand implies that the rhenium was bonded at MgO sites such as corner sites.56Carbonyl ligands on the metals provide insights into structure and uniformity of supported metal complexes
Metal carbonyls on supports have been shown to provide uniquely valuable insights into the structures of supported metal complexes, with evidence of symmetry and structure of the supported species, bonding at the metal–support interface, and the degree of uniformity of the species. The advantages derive from the richness of information provided by IR spectra in the νCO region (including the high intensities of the bands and the information about symmetry provided by the spectra); the characteristics of multiple scattering paths representing linear M–C–O moieties in EXAFS spectra (M is the metal); and the availability of numerous molecular analogues for comparison of spectra (and metal–ligand bond distances) of surface species with those of analogous compounds having known crystal structures.These points are illustrated with rhodium gem-dicarbonyls on zeolite supports. Groundbreaking work by Miessner57,58 reporting IR spectra showed that transition metal carbonyl complexes with well-defined structures could be formed on dealuminated Y zeolite samples, as made evident by narrow νCO bands characterizing the metal carbonyls. It is advantageous to use zeolites with low aluminum contents, evidently simplifying the structures of the supported species by limiting the bonding sites to those in the zeolite structure near sparse Al sites (where reactive OH groups are initially present). Dealuminated HY zeolite has been used by researchers following on Miessner's work, as summarized below; although this zeolite typically contains amorphous impurities resulting from the dealumination, these may be sufficiently sparse and lacking in reactivity in the syntheses of the supported metal complexes to have a minimal influence on the bonding of the metal complexes at sites initially being OH groups (near aluminum sites).
Work with rhodium gem-dicarbonyls on this support,59 made by reaction with the precursor Rh(C2H4)2(acac) (acac is acetylacetonato) and replacement of the C2H4 ligands with CO, determined EXAFS spectra complementing IR spectra and DFT computations providing insights into the structures of the supported metal complexes. The results show that this precursor reacted with support hydroxyl groups and led to bonding of positively charged rhodium at support oxygen atoms. The IR spectra of the rhodium carbonyls provide essential evidence of the symmetry of the metal complexes, with comparisons with pure-compound standards showing the presence of rhodium gem-dicarbonyls, with the metal bonded to two non-carbonyl ligands, inferred to be support surface sites. EXAFS data confirmed the bonding of rhodium to the support, determining the sites to be pairs of O atoms in the zeolite, with Rh–O bonding distances being about 2.1 Å (a value that is quite general for mononuclear group-8 metal complexes on metal oxide and zeolite supports—and analogous compounds). The EXAFS data also provide evidence of multiple scattering, indicating linear Rh–C–O moieties (Fig. 3).
 | ||
| Fig. 3 Location of rhodium dicarbonyl complex at a four-ring of dealuminated HY zeolite. The atoms included in the isolated DFT cluster model are shown as circles, with the rhodium atom in the upper center shown with two carbonyl ligands. Below that, in the four-ring, are three silicon atoms and one aluminum atom.59 The dangling bonds of the cluster model are capped by hydrogen atoms. Reproduced from ref. 59 with permission from the American Chemical Society. Copyright 2000. | ||
These supported species may be sufficiently close to uniform (as indicated by νCO spectra; see below) to engender some confidence in the DFT results, which are in agreement, within error, with the IR and EXAFS data.59 This rhodium carbonyl is one of the structurally best-defined supported metal complexes and illustrates the advantages of dealuminated zeolite HY as a support when a goal is to simplify the structures bonded to the support.
As data characterizing this sample illustrate, IR spectra of supported metal carbonyl complexes provide easily measured criteria determining the degree of uniformity of the supported species. Complexes of metals on zeolites may have especially narrow νCO bands, with full width at half maximum (fwhm) values sometimes being about 5 cm−1 for metals on dealuminated HY zeolite (compared with a value of about 4 cm−1 for a pure-compound analogue, Ir(CO)2(acac)51). For comparison, values characterizing analogous metal carbonyls on metal oxides are typically 20–30 cm−1, although samples made with extremely low metal loadings in attempts to confine the metals to a single kind of site (e.g., platinum on titania) have been found to have fwhm values between 5 and 10 cm−1.41
Another benefit of CO ligands as probes of metal complexes on supports is that the νCO values are sensitive to the non-carbonyl ligands, that is, the structures of the surface sites where the metal is bonded, as illustrated for rhodium carbonyls on various TiO2 sites having the same symmetry but crystallographically different surroundings.60
Rhodium complexes on isostructural supports: evidence confirming the supports are ligands
A related investigation was performed with the primary goal of comparing two isostructural supports as ligands for a metal complex, with rhodium dicarbonyls and rhodium diethene complexes on dealuminated zeolite HY and, alternatively, on the isostructural silicoaluminophosphate SAPO-37.61 EXAFS and IR data led to structural models of the rhodium diethene complexes showing that they were isostructural (Scheme 1); an equivalent conclusion was determined for the respective rhodium dicarbonyls. | ||
| Scheme 1 Structural models of rhodium diethene complex on SAPO-37 (top)61 and rhodium diethene complex on DAY zeolite (bottom)60 determined by EXAFS spectroscopy. Reproduced from ref. 61 with permission from the American Chemical Society, Copyright 2021. | ||
IR spectra of the samples that had been exposed to CO show that the rhodium diethene species on the SAPO reacted to give rhodium gem-dicarbonyls, characterized by νCO bands at 2115 and 2052 cm−1 (with fwhm values of 7 and 10 cm−1, a few wavenumbers more than the νCO bands characterizing the zeolite-supported analogue). Correspondingly, the Rh–O and Rh–C distances determined by EXAFS spectroscopy are also nearly the same for the two carbonylated samples on these zeotype supports—with the differences being too small to distinguish them from each other within the error in the data (the equivalent statement pertains to the diethene complexes, Scheme 1).61
The zeolite- and SAPO-37-supported rhodium complexes are evidently the most closely related pairs in any reported family of supported metal complexes, but they are not the same, because the supports—ligands—are different, and the reactivities were correspondingly found to be different. Catalyst performance data61 were determined in flow-reactor experiments with each of the rhodium diethene complexes being the catalyst precursor; the data show that the complexes on SAPO-37 and on DAY zeolite are approximately the same in activity, as measured by the rate of product formation at near steady state (TOF) in ethene conversion in the presence of H2 at a molar H2 to ethene ratio of 1:1, but the product distributions associated with the two catalysts were found to differ substantially, with the rhodium complexes on SAPO-37 being more selective for hydrogenation than the zeolite-supported complexes, with butenes formed only in trace amounts on the former but as the majority product on the latter. This difference is consistent with the inference that the different electron-donor properties and/or the different surroundings of the catalytic groups affected the catalytic performance substantially. The catalytic data are a stronger indicator than the spectra of the differences between the two samples, and we suggest that this conclusion may have some generality.
The authors61 postulated that the different catalytic selectivities may be consistent with results of Vummaleti et al.,62 who used DFT to investigate the mechanism of butene formation on a zeolite-supported rhodium complex, inferring a mechanism proceeding through a metallacycle intermediate, whereby a Rh–zeolite oxygen bond is broken to create an open bonding site for a reactant. The data are consistent with the suggestion that the properties of the support as a ligand affect the reactivities of the rhodium species, favoring the Vummaleti oligomerization mechanism on zeolite-supported rhodium over that on the SAPO-supported rhodium.61
Tracking reactions of ligands on supported mononuclear metal complexes and determining kinetics
Nearly uniform supported metal complexes provide opportunities for fundamental characterization of the reactivities of the ligands, and this point was illustrated by Hoffman et al.63 for the reactions of dealuminated HY zeolite-supported rhodium complexes incorporating ethene and carbonyl ligands. Those authors used quick EXAFS spectroscopy to show the C2H4-for-CO ligand exchange is reversible. The high acquisition speeds of commonly available IR spectrometers allow observations of such ligand exchanges, but the typical IR cell designs are not those of an ideal reactor, and the less-than-well-defined flow patterns do not allow for determination of the intrinsic kinetics of the ligand exchanges. By using a once-through plug-flow capillary tube as the cell/flow reactor for EXAFS measurements, Hoffman et al.63 determined intrinsic kinetics of the slower of the two exchanges (with ethene replacing CO), although the reverse reaction was too fast to characterize with their equipment at the synchrotron. The characteristic time for the ligand exchange was of the order of 10 s at 25 °C, with the process being approximately first order in the reactant ethene. Data such as these may help to unravel and quantify some of the fundamental chemistry of supported mononuclear metal complexes, but faster measurements will be needed to characterize elementary reactions in all but the slowest catalytic reactions (and the widely used spectroscopic methods are usually not sensitive enough to even detect reactive intermediates in catalysis).When characterizing ligand exchanges, it is helpful to check for isosbestic points in families of spectra recorded during the transformations; both IR and XANES provide valuable information about whether one species is being transformed into one other species.64
Influences of ligands on catalysis by supported rhodium and iridium complexes
Samples supported on dealuminated HY zeolite initially consisting of M(C2H4)2, M(CO)(C2H4), and M(CO)2 (with M being rhodium in some experiments and iridium in others) were tested as catalysts for ethene conversion in the presence of H2 and for H–D exchange in the reaction of H2 with D2.36 The data determine the roles of systematically varied ligands in isostructural supported metal complexes, showing that the strongly bonded CO ligand is a strong reaction inhibitor, with the M(CO)2 species being catalytically inactive under the mild conditions (e.g., 25 °C) chosen to assure that the metals remained as mononuclear complexes (the data show that the CO ligands were not replaced at a measurable rate under the catalytic reaction conditions applied when ligands other that CO were present on the metals).36Thus, after the M(CO)2 species were brought in contact with ethene and H2, under more forcing conditions that led to replacement of one of the CO ligands to give M(CO)(C2H4) species, catalysis was observed. These supported metal complexes were active for both ethene hydrogenation and the H–D exchange reaction. When no CO ligand was present, each metal complex was then more active. Thus, the overall activity at room temperature followed the trend M(C2H4)2 > M(CO)(C2H4) > M(CO)2, demonstrating the inhibition by the CO ligand (these species are catalyst precursors rather than catalysts, because the ligands on the metals change over time in the flow reactor as a result of reactions with the ethene and H2 in the reactant stream to form the steady-state catalysts). IR spectra of the working catalysts show how the catalytic selectivities depend on the ligand composition (Fig. 4).
 | ||
| Fig. 4 Dependence of turnover frequency for dimerization on the intensities of two IR bands (arbitrary units) for reaction catalyzed by sample initially in form of Rh(CO)(C2H4) supported on dealuminated HY zeolite; reaction conditions: flowing H2 + C2H4 in 4:1 molar ratio at 298 K and 1 bar. The data represent the Rh(CO)(C2H4) band at 2056 cm−1 (●) and the band at 2063 cm−1 suggested to represent an intermediate in the dimerization (▲).36 Reproduced from ref. 36 with permission from the American Chemical Society, Copyright 2015. | ||
An advantage of the CO ligand on the catalytic metal complexes is that it facilitates IR identification of the predominant species present during catalysis; the frequency of the C–O stretch provides evidence of the electron density on the metal centers in the predominant species. The data demonstrate the strong influence of the various ligands on the catalytic properties and provide a clear contrast between rhodium and iridium when the predominant supported species are isostructural; the two metals are substantially different from each other in their catalytic properties (but data such as these are not sufficient to explain the differences).
Support heterogeneity influences structure and catalytic properties of iridium complexes on MgO
To address the issues of support heterogeneity, samples were made from Ir(C2H4)2(acac) on high-area MgO powder, with various iridium loadings chosen to populate various support surface sites to different degrees.64 Characterization of the samples with IR, XANES, and EXAFS spectroscopies, STEM, and DFT modelling led to the conclusion that samples with populations of iridium species with varied metal–support structures were synthesized. The samples thus contained mixtures of supported species—and some structures predominated in some of the samples, as determined by the images and summarized in Table 2.| MgO surface location indicated by images | Number of Ir atoms observed for sample containing 0.1 wt% Ir | Number of Ir atoms observed for sample containing 0.05 wt% Ir | Number of Ir atoms observed for sample containing 0.01 wt% Ir |
|---|---|---|---|
| (100) Face terrace | 3 | 4 | 7 |
| (100) Face edge | 1 + 4 within 1 nm | 3 | 9 + 6 within 1 nm |
| (100) Face corner | 0 | 0 | 0 |
| (100) Face defect | 0 | 1 | 3 |
| (111) Face terrace | 13 | 4 | 1 |
| (111) Face edge | 6 | 0 | 0 |
| (111) Face corner | 0 | 0 | 0 |
| (111) Face defect | 2 | 0 | 0 |
| (110) Face terrace | 7 | — | 0 |
| (110) Face edge | 2 | — | 0 |
| (110) Face corner | 0 | — | 0 |
| (110) Face defect | 4 | — | 1 |
These data demonstrate the heterogeneity of the surface sites on the MgO support.64 EXAFS data show that Ir(C2H4)2(acac) reacted at multiple sites that hosted mononuclear iridium complexes, which the spectra show to have been present initially as Ir(C2H2)2. When the iridium loading was low (≤0.05 wt%), almost all of the supported species were located near MgO crystal edges, as shown by STEM images (Table 2), with each iridium atom bonded to about three support oxygen atoms, as shown by EXAFS data. When the loadings were higher, the mixture of iridium complexes included more that were bonded through 2 Ir–O bonds, as shown by EXAFS data. The values represent averages for the heterogeneous samples and are approximate, but the data characterizing ethene hydrogenation catalysis show that the sample with the lowest iridium loading come closest to having unique metal species, and these are the least active catalytically—presumably because they have more Ir–O bonds than the others and fewer sites for bonding of reactants to the iridium.
When the catalysts were exposed to high temperatures in H2, the iridium sintered, and the iridium in the sample with most of the iridium bonded to three oxygen atoms of the support sintered less rapidly than the iridium in the samples with iridium bonded to fewer oxygen atoms, indicating a stabilization of the catalysts by stronger bonding of the metal to the support.64 The less active catalysts are the more stable ones, and the data thus indicate a tradeoff between catalytic activity and stability.
Stabilizing supported metal complexes by confinement in nests on supports
Progress toward stabilizing noble metals on supports was made by isolating them in metal oxide nests on supports.65 Platinum salt precursors were selectively anchored to nano-sized nests (shown by STEM images to be about 2 nm in average diameter) of cerium oxide isolated on silica, and not to the silica, as the negatively charged precursor PtCl62− was attracted to the positively charged cerium oxide nests and repelled by the silica that had a negative charge under the conditions of the aqueous synthesis.The platinum loading was chosen so that less than one platinum atom was present per nest, on average. The platinum atoms in the supported complexes were shown by STEM imaging and IR and EXAFS spectroscopies not to migrate from the nests onto the silica support or to be transported between nests, even under harsh oxidative or reductive conditions. In contrast, when the platinum complexes were present on silica without the confining nests, the metal sintered readily under reducing conditions. The strategy of stabilizing the metal complexes within nests on support surfaces could be of practical value for a wide range of supported metal complex catalysts.65
Reduction and aggregation of metals in supported noble metal complexes to form supported metal clusters and nanoparticles
Noble metal complexes are readily reduced, and, on supports, they have a strong tendency to migrate and form clusters/particles of metal dispersed on the support. A number of examples of catalyst characterization by spectroscopy have provided real-time evidence of the conversion of supported metal complexes into clusters, with attendant changes in catalytic properties.Sometimes, however, the clusters have barely formed and remained undetected and not recognized as the species responsible for the observed catalysis, which has instead been misattributed to the mononuclear metal complexes from which they formed. Numerous examples illustrate this point, as summarized elsewhere.66
An example illustrating an increase in catalytic activity associated with metal cluster formation from mononuclear metal complexes was reported for gold on ceria, which was used to catalyze CO oxidation in a flow reactor that was an X-ray absorption spectroscopy (XAS) cell.67 Data characterizing the catalyst in action (Fig. 5) show how the gold was reduced and aggregated during catalysis, with the catalyst becoming much more active as the gold clusters formed.67
 | ||
| Fig. 5 XAS data showing the evolution of a ceria-supported gold complex catalyst into supported gold clusters, with the catalytic activity increasing during the transformation. The XANES data shown in green demonstrate the decreasing amount of cationic gold in the sample, and the EXAFS data shown in red show the cluster formation; the data shown in black indicate the decrease in Au–O bonds as Au–Au bonds formed.67 Reproduced from ref. 67 with permission from the American Chemical Society, Copyright 2009. | ||
In contrast to these results for CO oxidation, EXAFS data characterizing gold on MgO show that for ethene hydrogenation gold clusters/nanoparticles lack catalytic activity, whereas mononuclear gold complexes are active.68,69 The data (Fig. 6) demonstrate the dependence of catalytic activity at 80 °C and atmospheric pressure on the average Au–Au coordination number for a family of catalysts, show that the clusters lack activity.
 | ||
| Fig. 6 Catalytic activity for ethene hydrogenation in a flow reactor that was an EXAFS cell. Samples initially incorporating mononuclear gold complexes were treated in helium under various conditions to convert fractions of the gold into clusters/nanoparticles. The catalyst consisting of mononuclear gold complexes (with an undetectable Au–Au scattering path) is the most active, and the activity decreased as increasing fractions of the gold were reduced and aggregated. The Au–O coordination number characterizing the isolated gold complex was approximately two, indicating that the support was a bidentate ligand.68 Reproduced from ref. 68 with permission from John Wiley and Sons, Copyright 2003. | ||
Reversible formation of supported rhodium clusters from supported rhodium complexes
As shown by the preceding examples, noble metal complexes on supports are readily reduced to form clusters/nanoparticles of metal. The reduction and aggregation of rhodium initially present as the aforementioned rhodium diethene complexes on dealuminated HY zeolite were characterized by IR and EXAFS spectroscopies. The data show that the rhodium complex, when exposed to flowing H2 for 1.5 h at atmospheric pressure and room temperature, was converted into extremely small rhodium clusters, consisting of only several atoms each, on average, as shown by EXAFS spectroscopy.69 The cluster formation was reversed when the sample was exposed to ethene, which caused oxidative fragmentation. The cluster formation/breakup processes were characterized by EXAFS spectroscopy during catalysis of ethene conversion in a once-through plug-flow reactor at atmospheric pressure and 30 °C with feeds containing various C2H4:H2 ratios. In ethene-rich mixtures (C2H4:H2 molar ratio = 4), the catalysts were found to be active and highly selective for ethene dimerization; ethene hydrogenation was a minor side reaction. When the feed composition to the reactor was switched from 4:1 to 1:4, the clusters formed and the catalyst became selective for ethene hydrogenation, with negligible dimerization. This process was reversible, as the rhodium complexes were reduced to form clusters in excess H2 and these were oxidatively fragmented in excess ethene. The chemistry is shown schematically in Fig. 7. The reversibility of the conversions and the spectra suggest that it is a good approximation to represent the rhodium complexes as unique species, bonded at equivalent sites on the zeolite support, consistent with the results presented above characterizing the mononuclear species.
 | ||
| Fig. 7 Schematic representation of rhodium species during reversible cluster formation from mononuclear rhodium complex on dealuminated HY zeolite during ethene conversion catalysis in the presence of excess H2 and cluster breakup (oxidative fragmentation by ethene) in the presence of excess ethene.69 Reproduced from ref. 69 with permission from the American Chemical Society, Copyright 2011. | ||
Generality of reversible formation of noble metal clusters and oxidative fragmentation of clusters on supports
The oxidative fragmentation of supported noble metal clusters and nanoparticles on supports is, we infer, quite general. It provides convenient routes to the synthesis of supported platinum complexes (by “atom trapping”) on ceria.70 Similar chemistry of platinum complexes and clusters takes place in the zeolite chabazite (with the process being reversible)71 and in the zeolite HZSM-5 (ref. 72) (with the process being reversible and involving platinum gem-dicarbonyls), among others. In these examples, the oxidizing agent triggering oxidative fragmentation of clusters/nanoparticles is O2; therefore, such chemistry is expected to take place under conditions of burnoff of carbonaceous deposits in catalysts such as zeolite-supported platinum used in hydrocarbon conversion processes, and likely occurs in vehicle exhaust emission catalysts under oxidative conditions.Migration of metal complexes and their fragments on supports
The evidence of oxidative fragmentation of supported metal clusters/nanoparticles (and the reverse) imply transport of the fragments, which could be mononuclear species themselves and could, in prospect, include larger species such as clusters. Many authors have recognized the importance of such transport, which could take place in a gas phase or on the support surface, but there has been barely any work characterizing the transport. However, the group of Ortalan73 reported time-resolved STEM images of mononuclear iridium species migrating on MgO, demonstrating transport on the surface and finding that iridium atoms moved between neighboring oxygen atoms of the support surface. The results suggest that breaking and forming of Ir–O bonds account for essential steps in the migration.Real-time EXAFS data showing the formation of clusters approximated as tetrairidium from mononuclear iridium species on MgO were reported by Uzun and Gates,74 who also observed the reverse process, with the respective reducing and oxidizing agents being H2 and ethene, respectively. The transport was likely being related to what was observed by Ortalan et al.,73 but the available data leave open many questions about the ligands on the metals and details of the chemistry as it depends on the reactive atmosphere. The open questions seem to present a fruitful opportunity for further research; understanding of the transport phenomena seems to require understanding the surface chemistry.
Loss of accessibility by trapping of metals within supports
The aforementioned oxidative fragmentation chemistry has typically been carried out under forcing (calcination) conditions—burnoff of carbonaceous deposits in practice takes place at high temperatures. The literature of the oxidative fragmentation processes mentioned just above raises questions about the nature and surroundings of the metal species formed by oxidative fragmentation on metal oxide supports. The work mentioned above with QuantEXAFS pointed to metals in support layers just below the surface, and it had been suggested earlier that atomically dispersed metals in metal oxide supports could be present in the bulk of the material75 (consistent with a large literature of “doped” materials consisting of various metals in various metal oxide hosts76).Recent work has shown that high-temperature oxidation caused the transport of platinum cations into the interior of a high-area MgO support, where they are inaccessible (or barely accessible) to reactants;40 details follow:
Platinum on MgO samples made from a platinum salt precursor, described above, were characterized by STEM imaging and QuantEXAFS complementing EXAFS, XANES, and IR spectroscopies. The analysis showed that the Pt–O and Pt–Mg coordination numbers changed significantly as the calcination temperature increased from 400 to 700 °C, as the location of platinum changed from a bonding site in the first MgO layer to a site in the second MgO layer. The oxidation state of platinum in these subsurface sites, determined from DFT calculations using Bader charge analysis, was found to be almost the same as that of Pt4+ in H2Pt(OH)6, whereas the oxidation state of platinum that was observed to have formed in the first MgO layer (before the high-temperature oxidation) was between those of Pt2+ and Pt4+. XANES data support these conclusions, showing that the white line intensity increased as the oxidation temperature increased from 400 to 700 °C, and high-energy-resolution fluorescence detection XANES data and FEFF-simulated XANES spectra calculated from the DFT-optimized geometries of the supported species indicated by QuantEXAFS provided confirmation.
The expectation of different surroundings of the platinum in the samples before and after high-temperature oxidation was confirmed by IR spectra of the samples exposed to CO.40 CO was adsorbed at room temperature on the samples exposing platinum at the MgO surface, as shown by IR spectra. In contrast, the IR spectra gave no evidence of room-temperature CO adsorption on the subsurface platinum. Thus, the IR experiments show that the highly coordinated Pt4+ species in the second MgO layer were less accessible to reactants than those in the first layer. The two samples with differently coordinated cationic platinum were compared as catalysts for CO oxidation in light-off experiments at atmospheric pressure. CO oxidation activity was first observed at about 120 °C for the catalyst incorporating surface platinum, but a much higher temperature, 180 °C, was required for the onset of catalysis with the sample incorporating the buried platinum.
IR spectra of the used catalysts show that the platinum on the support surface had sintered, implying its lack of stability; the data imply that the catalytically active species were primarily metallic platinum.40 In contrast, the IR data characterizing platinum in the subsurface sites remained unchanged after catalysis. The less accessible subsurface platinum atoms were thus much less active catalytically than those in the first MgO layer but also much more resistant to sintering. The stability came at the cost of catalytic activity associated with the limited accessibility of the buried platinum.
Platinum has also been observed to be present in the interior of titania supports, driven into the interior under high-temperature oxidation conditions, with the location dependent on the titania particle morphology.41 The subsurface locations of the platinum cations were determined in experiments in which the surface was etched away with argon ions, and confirming data were obtained with X-ray photoelectron spectroscopy and STEM.
The evidence of platinum located in subsurface locations in more than one kind of metal oxide support raises questions about the fate of noble metals in supported catalysts that are subjected to high-temperature oxidizing conditions, for example, in the burnoff of carbonaceous deposits; metals may become inaccessible to reactants and taken out of action by such treatments. The results also raise questions about the effectiveness of formation of supported mononuclear metal complex catalysts by oxidative fragmentation of metal clusters/nanoparticles: the “atom-trapping” method70 might lead to dispersion of metal not just onto the support, where it is accessible and active, but also into the support, where it is inaccessible.
Roles of surface sites interacting with supported metal complexes
The catalytic roles of metal complexes on supports are sometimes complicated by interactions of the catalytic metals with neighboring metal sites. Serna22 recently provided a review addressing this point. Examples illustrating the role of oxophilic metals on supports influencing catalyst by noble metal complexes have been reported for alkene hydroformylation77 (with a benefit in activity provided by the rhenium sites neighboring rhodium sites). Numerous catalysts in this class have been made from molecular bimetallic clusters incorporating one noble metal and one oxophilic metal.78Prospects for large-scale applications of atomically dispersed noble metal catalysts on supports remain largely unrealized, with one exception
Although some authors have expressed optimism that mononuclear metal complex catalysts are ripe for industrial application, there is a lack of such applications, with a notable exception—the application of supported mononuclear gold for acetylene hydrochlorination to make vinyl chloride monomer, as described by Hutchings, who presented summaries of the catalyst invention and development.79 The large-scale application reflects many years of work and early insights into the chemistry of cationic gold.79,80 EXAFS and XANES characterizing working carbon-supported catalysts combined with STEM imaging and DFT results led to the identification of Au(I) as the predominant form of the gold in the catalytically active material, stabilized on the carbon support, with chloride playing a role in the stabilization, likely as a ligand bonded to Au(I).80 Mixtures of mononuclear species were observed spectroscopically and with STEM of used catalysts. Sulfur-containing ligands added to the catalyst were found to improve its performance, and the data imply that both sulfur-containing ligands and chloride are bonded to gold in the working catalyst, providing further evidence of the importance of sites neighboring the noble metal cations.80 The catalyst is reported to be stable in long-term operation, but, understandably, practical information such as rates of the catalytic reaction and of catalyst deactivation and replacement are missing from the literature.A basis for optimism that further catalytic applications of supported mononuclear metal complexes may be found is that many new structures of have been discovered (with others likely to follow), combined with the principal practical advantage of the atomically dispersed catalysts—the efficient use of the expensive metal atoms, which may all be accessible to reactants. In prospect, any of these metal complexes could be placed on supports to be accessible to reactants and engaged in catalysis. A recent report of a dilute alloy catalyst, rhodium atoms in a single-crystal copper surface (copper (111)) and supported bimetallic nanoparticles includes encouraging evidence of catalysis of propane dehydrogenation, for example.81
But, to find practical applications, the catalysts need to produced reliably and economically on a large scale and be stable and regenerable. Part of what is missing from the literature is evidence of catalyst stability and regenerability, and a realization that industrial catalysts are found to meet processing needs, and not the reverse. Almost without exception, intriguing new catalysts fail to find practical application, notwithstanding many statements in the literature that a newly reported catalyst “outperforms” those already reported; only rarely are wide ranges of processing variables such as temperature, space velocity, time on stream, and pressure investigated. Supported noble metal catalysts that are applied successfully on a large scale in the conventional form of supported nanoparticles will not generally be good catalysts for the same reactions when the metal dispersion is taken to the limit of atomic dispersion: then, at least on a conventional metal oxide or zeolite support, the metal becomes cationic and has catalytic properties markedly different from those of the zerovalent metal.
Suggested directions for future research: catalyst synthesis, characterization, and performance testing
Research on supported metal complex catalysts is proceeding vigorously, and better characterization methods and more precise syntheses can be expected to help carry it forward. Any further large-scale applications would also provide a boost.Characterization methods that continue to develop include STEM; double aberration correction has recently been used to improve image quality,41 and more fine-grained images are emerging, such as those characterizing zeolites.30 More applications of these techniques seem sure to emerge, including measurements of time-resolved images to track changes in catalyst structure.
The technology for XAS at synchrotrons is also improving, with facilities emerging for better tracking of catalyst performance under more realistic reaction conditions. Synchrotron measurements providing evidence of metal oxidation states with high sensitivity are emerging from work with high-energy-resolution fluorescence-detection XANES, and the attendant theory, is having a positive influence.82 X-ray photoelectron spectroscopy measurements at synchrotrons at pressures as high as several mbar are also helping to move the characterization science forward. Better equipment is emerging for investigating catalysts in the working state, including simultaneous determination of IR and X-ray absorption spectra and experimentation at higher temperatures and pressures and for longer times on stream.
Understanding of solid catalysts synthesized to have relatively simple, uniform structures is expected to move the field of heterogeneous catalysis forward broadly, and precisely constructed catalysts consisting of mononuclear metal complexes and well-chosen supports might provide new reactions that combine catalyst stability with the high selectivities that are associated with industrial homogeneous catalysis.
It may be fruitful to investigate supported metal complex catalysts confined within small pores, such as those of MOFs, where conditions akin to those of homogeneous catalysis might pertain, with liquid-like environments in the confined spaces. These conditions might be substantially different from those encountered with sparsely populated metal complexes on surfaces of supports such as metal oxides, perhaps affecting the fundamental chemistry, such as rates of elementary reactions exemplified by ligand exchanges and breaking of metal–support bonds.
Data availability
No primary research results, software, or code have been included, and no new data were generated or analyzed as part of this review.Conflicts of interest
The author declares no conflict of interest.References
- J. P. Collman, L. S. Hegedus, J. R. Norton and R. G. Finke, Principles and Applications of Organotransition Metal Chemistry, University Science Books, Mill Valley, CA, 1987 Search PubMed.
- J. Hartwig, Organotransition Metal Chemistry: from Bonding to Catalysis, University Science Books, Mill Valley, CA, 2010 Search PubMed.
- G. W. Parshall and S. D. Ittel, Homogeneous Catalysis, Wiley-Interscience, New York, 1994 Search PubMed.
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225 CrossRef CAS.
- N. Yoneda, T. Minami, J. Weiszmann and B. Spehlmann, Stud. Surf. Sci. Catal., 1999, 121, 93–98 CrossRef CAS.
- M. P. McDaniel, Adv. Catal., 2010, 53, 123–606 CAS.
- K. C. Jayaratne, T. H. Cymbaluk and M. D. Jensen, ACS Catal., 2018, 8, 602–614 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- A. Hyer, D. Gregory, K. Kay, Q. Le, J. Turnage, F. Gupton and J. K. Ferri, Adv. Synth. Catal., 2024, 366, 357–389 CrossRef CAS.
- R. L. Burwell, Pure Appl. Chem., 1976, 46, 71–90 Search PubMed.
- H. S. Taylor, Proc. Roy. Soc., 1925, 108, 105–111 CAS.
- H. P. Boehm, Adv. Catal., 1966, 16, 179–274 CAS.
- H. P. Boehm and H. Knözinger, in Catalysis Science and Technology, ed. J. R. Anderson and M. Boudart, Springer, Berlin, 1983, vol. 4, pp. 39–207 Search PubMed.
- P. Cossee, J. Catal., 1964, 3, 80–88 CrossRef CAS.
- E. J. Arlman, J. Catal., 1964, 3, 89–98 CrossRef CAS.
- E. J. Arlman and P. Cossee, J. Catal., 1964, 3, 99–104 CrossRef CAS.
- G. V. Fortunato, E. Pizutilo, I. Katsounaros, D. Göhl, R. J. Lewis, K. J. J. Mayrhofer, G. J. Hutchings, S. J. Freakley and M. Ledendecker, Nat. Commun., 2022, 13, 1973 CrossRef CAS PubMed.
- D. G. H. Ballard, Adv. Catal., 1973, 23, 263–325 CAS.
- R. L. Burwell Jr and A. Brenner, J. Mol. Catal., 1975, 76(1), 77–84 Search PubMed.
- M. K. Samantaray, S. K. Mishra, A. Saidi and J.-M. Basset, J. Organomet. Chem., 2021, 945, 121864 CrossRef CAS.
- M. Samantaray, V. D'Elia, E. Pump, L. Falivene, M. Harb, S. Ould Chikh, L. Cavallo and J.-M. Basset, Chem. Rev., 2020, 120, 734–813 CrossRef CAS PubMed.
- P. Serna, Chem. Eng. J., 2024, 496, 153840 CrossRef CAS.
- S. F. J. Haskett, R. M. Brydson, M. H. Gaas, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef PubMed.
- A. Uzun, V. Ortalan, N. D. Browning and B. C. Gates, Chem. Commun., 2009, 4657–4659 RSC.
- A. Uzun, V. Ortalan, N. D. Browning and B. C. Gates, J. Catal., 2010, 269, 318–328 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- M. S. Jarrell and B. C. Gates, J. Catal., 1975, 40, 255–267 CrossRef CAS.
- W. H. Lang, A. T. Jurewicz, W. O. Haag, D. D. Whitehurst and L. D. Rollmann, J. Organomet. Chem., 1977, 134, 85–94 CrossRef CAS.
- J. M. Thomas, Angew. Chem., Int. Ed., 1988, 27, 1673–1691 CrossRef.
- H. Zhang, G. Li, J. Zhang, D. Zhang, Z. Chen, X. Liu, P. Guo, Y. Zhu, C. Chen, L. Liu, X. Guo and Y. Han, Science, 2023, 380, 633–638 CrossRef CAS PubMed.
- J. M. Thomas, Phys. Chem. Chem. Phys., 2014, 16, 7647–7661 RSC.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. Sykes, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS PubMed.
- J. Finzel and P. Christopher, Top. Catal., 2022, 65, 1587–1603 CrossRef CAS.
- T. Han, Y. Li, T. Wu, D. Motta Meira, S. Xiang, Y. Cao, I. Lee, X.-G. Zhou, D.-e. Jiang, A. I. Frenkel and F. Zaera, ACS Catal., 2024, 14, 7157–7165 CrossRef CAS PubMed.
- P. Tieu, X. Yan, M. Xu, P. Christopher and X. Pan, Small, 2021, 17, 2006482 CrossRef CAS PubMed.
- C. Martinez-Macias, P. Serna and B. C. Gates, ACS Catal., 2015, 5, 5647–5656 CrossRef CAS.
- K. Khivantev, A. Vityuk, H. A. Aleksandrov, G. N. Vayssilov, O. S. Alexeev and M. D. Amiridis, J. Chem. Phys., 2021, 154, 184706 CrossRef PubMed.
- K. Khivantev, M. A. Derewinski and J. Szanyi, Microporous Mesoporous Mater., 2023, 358, 112378 CrossRef.
- M. Babucci, A. Guntida and B. C. Gates, Chem. Rev., 2020, 120, 11956–11985 CrossRef CAS PubMed.
- Y. Chen, R. Rana, Z. Huang, F. D. Vila, T. Sours, J. E. Perez-Aguilar, X. Zhao, J. Hong, A. S. Hoffman, X. Li, C. Shang, T. Blum, J. Zeng, M. Chi, M. Salmeron, C. X. Kronawitter, S. R. Bare, A. R. Kulkarni and B. C. Gates, J. Phys. Chem. Lett., 2022, 13, 3896–3903 CrossRef CAS PubMed.
- W. Zang, J. Lee, P. Tieu, X. Yan, G. W. Graham, I. C. Tran, P. Wang, P. Christopher and X. Pan, Nat. Commun., 2024, 15, 998 CrossRef CAS PubMed.
- J. Hulva, M. Meier, R. Bliem, Z. Jakub, F. Kraushofer, M. Schmid, U. Diebold, C. Franchini and G. S. Parkinson, Science, 2021, 371, 375–379 CrossRef CAS PubMed.
- F. Kraushofer and G. S. Parkinson, Chem. Rev., 2022, 122, 14911–14939 CrossRef CAS PubMed.
- O. Alexeev and B. C. Gates, Top. Catal., 2000, 10, 273–293 CrossRef CAS.
- J. Lu, C. Aydin, N. D. Browning and B. C. Gates, Angew. Chem., Int. Ed., 2012, 51, 5842–5846 CrossRef CAS PubMed.
- G. Di Liberto and G. Pacchioni, Adv. Mater., 2023, 35, 2307150 CrossRef CAS PubMed.
- Z. Xia, Y. Yin, J. Li and H. Xiao, Chem. Sci., 2023, 14, 2631–2639 RSC.
- Y. Chen, R. Rana, T. Sours, F. D. Vila, S. Cao, T. Blum, J. Hong, A. S. Hoffman, C.-Y. Fang, Z. Huang, C. Shang, C. Wang, J. Zeng, M. Chi, C. X. Kronawitter, S. R. Bare, B. C. Gates and A. R. Kulkarni, J. Am. Chem. Soc., 2021, 143, 20144–20156 CrossRef CAS PubMed.
- Y. Chen, R. Rana, Y. Zhang, A. S. Hoffman, Z. Huang, B. Yang, F. D. Vila, J. E. Perez-Aguilar, J. Hong, X. Li, J. Zeng, M. Chi, C. X. Kronawitter, H. Wang, S. R. Bare, A. R. Kulkarni and B. C. Gates, Chem. Sci., 2024, 15, 6454–6464 RSC.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS PubMed.
- A. S. Hoffman, C.-Y. Fang and B. C. Gates, J. Phys. Chem. Lett., 2016, 7, 3854–3860 CrossRef CAS PubMed.
- P. Christopher, ACS Energy Lett., 2019, 4, 2249–2250 CrossRef CAS.
- J. O. Ehresmann, P. W. Kletnieks, A. Liang, V. A. Bhirud, O. P. Bagatchenko, E. J. Lee, M. Klaric, B. C. Gates and J. F. Haw, Angew. Chem., Int. Ed., 2006, 45, 574–576 CrossRef CAS PubMed.
- J. Resasco and P. Christopher, J. Phys. Chem. Lett., 2020, 11, 1014–10123 CrossRef PubMed.
- J. Finzel, K. M. Sanroman Gutierrez, A. S. Hoffman, J. Resasco, P. Christopher and S. R. Bare, ACS Catal., 2023, 13, 6462–6473 CrossRef CAS.
- C. J. Papile and B. C. Gates, Langmuir, 1992, 8, 74–79 CrossRef CAS.
- H. Miessner, I. Burkhardt, D. Gutschick, A. Zecchina, C. Morterra and G. Spoto, J. Chem. Soc., Faraday Trans., 1989, 85, 2113–2126 RSC.
- H. Landmesser and H. Miessner, J. Phys. Chem., 1991, 95, 10544–10546 CrossRef CAS.
- J. F. Goellner, B. C. Gates, G. N. Vayssilov and N. Rösch, J. Am. Chem. Soc., 2000, 122, 8056–8066 CrossRef CAS.
- H. V. Thang, G. Pacchioni, L. DeRita and P. Christopher, J. Catal., 2018, 367, 104–114 CrossRef CAS.
- J. E. Perez-Aguilar, C.-Y. Chen, J. T. Hughes, C.-Y. Fang and B. C. Gates, J. Am. Chem. Soc., 2020, 142, 11474–11485 CrossRef CAS PubMed.
- S. V. C. Vummaleti, N. Kuriakose, S. Dinda, Y. Wu, A. Geneste and N. Rösch, Catal. Sci. Technol., 2019, 9, 2781–2793 RSC.
- A. S. Hoffman, O. Müller, J. Hong, G. A. Canning, C.-Y. Fang, J. E. Perez-Aguilar, B. C. Gates and S. R. Bare, J. Phys. Chem. Lett., 2023, 14, 4591–4599 CrossRef CAS PubMed.
- A. S. Hoffman, L. M. Debefve, S. Zhang, J. E. Perez-Aguilar, E. T. Conley, K. R. Justl, I. Arslan, D. A. Dixon and B. C. Gates, ACS Catal., 2018, 8, 3489–3498 CrossRef CAS.
- X. Li, X. Isidro Pereira-Hernández, Y. Chen, J. Xu, J. Zhao, C.-W. Pao, C.-Y. Fang, J. Zeng, Y. Wang, B. C. Gates and J. Liu, Nature, 2022, 611, 284–288 CrossRef CAS PubMed.
- S. Das, U. Anjum, K. H. Lim, Q. He, A. S. Hoffman, S. R. Bare, S. M. Kozlov, B. C. Gates and S. Kawi, Small, 2023, 19, 2207272 CrossRef CAS PubMed.
- V. Aguilar-Guerrero, R. J. Lobo-Lapidus and B. C. Gates, J. Phys. Chem. C, 2009, 113, 3259–3269 CrossRef CAS.
- J. Guzman and B. C. Gates, Angew. Chem., Int. Ed., 2003, 42, 690–693 CrossRef CAS PubMed.
- P. Serna and B. C. Gates, J. Am. Chem. Soc., 2011, 133, 4714–4717 CrossRef CAS PubMed.
- J. Jones, H. Xiong, A. T. Delariva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- M. Moliner, J. E. Gabay, C. E. Kliewer, R. T. Carr, J. Guzman, G. L. Casty, P. Serna and A. Corma, J. Am. Chem. Soc., 2016, 138, 15743–15750 CrossRef CAS PubMed.
- K. Yalcin, R. Kumar, E. Zuidema, A. R. Kulkarni, J. Ciston, K. C. Bustillo, P. Ercius, A. Katz, B. C. Gates, C. X. Kronawitter and R. C. Runnebaum, ACS Catal., 2024, 14, 4999–5005 CrossRef CAS PubMed.
- C. W. Han, H. Idi, A. Uzun, L. A. Curtiss, N. D. Browning, B. C. Gates and V. Ortalan, J. Phys. Chem. Lett., 2015, 6, 4675–4679 CrossRef CAS PubMed.
- A. Uzun and B. C. Gates, Angew. Chem., Int. Ed., 2008, 120, 9385–9388 CrossRef.
- B. C. Gates, M. Flytzani-Stephanopoulos, D. A. Dixon and B. C. Gates, Catal. Sci. Technol., 2017, 7, 4259–4275 RSC.
- E. W. McFarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS PubMed.
- I. Ro, J. Qi, S. Lee, M. Xu, X. Yan, Z. Xie, G. Zakem, A. Morales, J. G. Chen, X. Pan, D. G. Vlachos, S. Caratzoulas and P. Christopher, Nature, 2022, 609, 287–292 CrossRef CAS PubMed.
- O. S. Alexeev, G. W. Graham, M. Shelef and B. C. Gates, J. Catal., 2000, 190, 157–172 CrossRef CAS.
- G. J. Hutchings, J. Catal., 2024, 432, 115392 CrossRef CAS.
- G. Malta, S. A. Kondrat, S. J. Freakley, C. J. Davies, L. Lu, S. Dawson, A. Thetford, E. K. Gibson, D. J. Morgan, W. Jones, S. P. Wells, P. Johnston, C. R. A. Catlow, C. J. Kiely and G. J. Hutchings, Science, 2017, 355, 1399–1402 CrossRef CAS PubMed.
- R. T. Hannagan, G. Giannakakis, R. Réocreux, J. Schumann, J. Finzel, Y. Wang, A. Michaelides, P. Deshlahra, P. Christopher, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Science, 2021, 372, 1444–1447 CrossRef CAS.
- A. S. Hoffman, D. Sokaras, S. Zhang, L. M. Debefve, C.-Y. Fang, A. Gallo, T. Kroll, D. A. Dixon, S. R. Bare and B. C. Gates, Chem.–Eur. J., 2017, 23, 14760–14768 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |