
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-active chemical chaperones exhibiting promiscuous binding promote oxidative protein folding under condensed sub-millimolar conditions†
Koki
Suzuki‡
a,
Ryoya
Nojiri‡
a,
Motonori
Matsusaki
b,
Takuya
Mabuchi
 cd,
Shingo
Kanemura
c,
Kotone
Ishii
c,
Hiroyuki
Kumeta
e,
Masaki
Okumura
*c,
Tomohide
Saio
*b and
Takahiro
Muraoka
*af
cd,
Shingo
Kanemura
c,
Kotone
Ishii
c,
Hiroyuki
Kumeta
e,
Masaki
Okumura
*c,
Tomohide
Saio
*b and
Takahiro
Muraoka
*af
aDepartment of Applied Chemistry, Graduate School of Engineering, Tokyo University of Agriculture and Technology, Koganei, Tokyo 184-8588, Japan. E-mail: muraoka@go.tuat.ac.jp
bDivision of Molecular Life Science, Institute of Advanced Medical Sciences, Tokushima University, Tokushima 770-8503, Japan. E-mail: saio@tokushima-u.ac.jp
cFrontier Research Institute for Interdisciplinary Sciences, Tohoku University, Sendai, Miyagi 980-8578, Japan. E-mail: okmasaki@tohoku.ac.jp
dInstitute of Fluid Science, Tohoku University, Sendai, Miyagi 980-8577, Japan
eFaculty of Advanced Life Science, Hokkaido University, Sapporo, Hokkaido 060-0810, Japan
fKanagawa Institute of Industrial Science and Technology (KISTEC), Kanagawa 243-0435, Japan
First published on 29th July 2024
Abstract
Proteins form native structures through folding processes, many of which proceed through intramolecular hydrophobic effect, hydrogen bond and disulfide-bond formation. In vivo, protein aggregation is prevented even in the highly condensed milieu of a cell through folding mediated by molecular chaperones and oxidative enzymes. Chemical approaches to date have not replicated such exquisite mediation. Oxidoreductases efficiently promote folding by the cooperative effects of oxidative reactivity for disulfide-bond formation in the client unfolded protein and chaperone activity to mitigate aggregation. Conventional synthetic folding promotors mimic the redox-reactivity of thiol/disulfide units but do not address client-recognition units for inhibiting aggregation. Herein, we report thiol/disulfide compounds containing client-recognition units, which act as synthetic oxidoreductase-mimics. For example, compound βCDWSH/SS bears a thiol/disulfide unit at the wide rim of β-cyclodextrin as a client recognition unit. βCDWSH/SS shows promiscuous binding to client proteins, mitigates protein aggregation, and accelerates disulfide-bond formation. In contrast, positioning a thiol/disulfide unit at the narrow rim of β-cyclodextrin promotes folding less effectively through preferential interactions at specific residues, resulting in aggregation. The combination of promiscuous client-binding and redox reactivity is effective for the design of synthetic folding promoters. βCDWSH/SS accelerates oxidative protein folding at highly condensed sub-millimolar protein concentrations.
Introduction
Chemical reactions under highly condensed conditions often result in the formation of undesired kinetic byproducts due to multiple competitive factors. The promotion of condensed-phase reactions can increase the synthetic yield of drugs and materials, but currently there are few methodologies for controlling the reactions of concentrated polypeptides due to their high propensities to aggregate in aqueous media. Protein folding is a biochemical pathway followed by each unique polypeptide sequence to construct the native structure of the folded protein. Many of these pathways proceed through cooperative intramolecular hydrophobic effect, and hydrogen bond and disulfide (SS) bond formation (Fig. 1a).1 Under condensed conditions, protein folding competes with non-specific intermolecular interactions and SS bonding between multiple polypeptide chains to afford misfolded and aggregated proteins (Fig. 1a, Route A).2 Over 20 members of the protein disulfide isomerase (PDI) family of oxidoreductases recognize nascent and unfolded reduced proteins such as proinsulin and immunoglobulin G and promote folding by redox reactions, thereby facilitating SS-bond formation. The chaperone activity of PDIs prevents protein aggregation in a cell (Fig. 1a, Route B).3 The interior of a cell is a typical condensed medium. Bioinspired and enzyme-mimetic approaches could aid in the design of synthetic methodologies to regulate and promote condensed-phase polypeptide reactions.4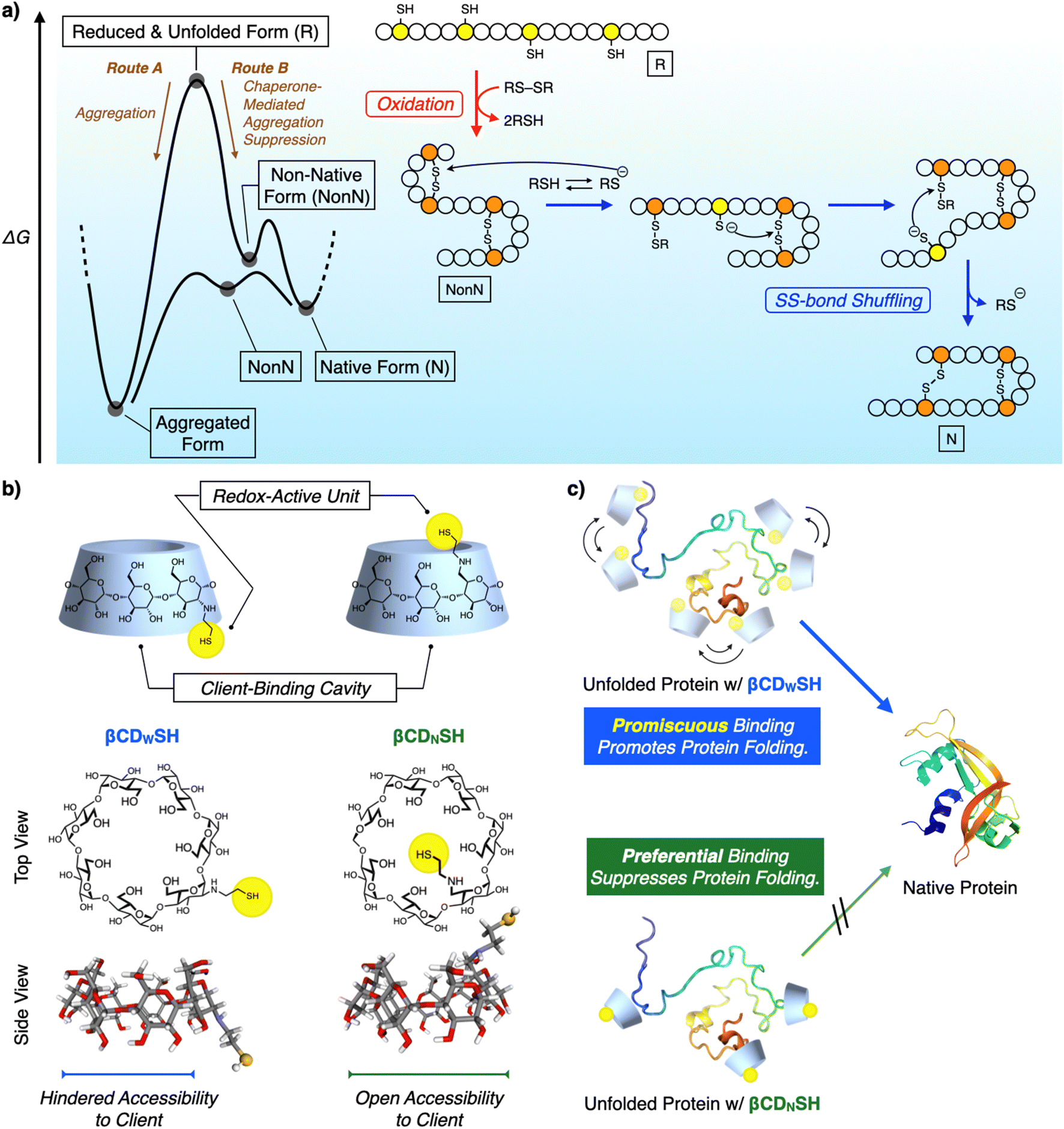 | ||
| Fig. 1 (a) Schematic illustration of the oxidative folding of a polypeptide chain to form two disulfide bonds. Yellow and orange circles represent cysteine residues that are in the thiol/thiolate form and disulfide form, respectively. Open circles represent amino acid residues other than cysteine. Red arrow step: oxidation reaction from a reduced and unfolded form (R) to a non-native form (NonN). Blue arrow steps: disulfide bond isomerization from NonN to N by the nucleophilic attack of thiolates. (b) Molecular structures of βCDWSH and βCDNSH. Schematic images showing the redox-active units and client-binding cavities, and the molecular models calculated using DFT B3LYP 6-31G*. (c) The concept of (blue) the promiscuous binding property of βCDWSH and (green) the preferential binding property of βCDNSH with an unfolded protein. The promiscuous binding property of βCDWSH is advantageous for promoting protein folding compared to the preferential binding property of βCDNSH. | ||
Enzymes are conventionally considered to be specific catalysts that recognize preferential substrates to provide single products.5 The well-defined structures of enzyme–substrate complexes have afforded important clues helpful for the design of synthetic mimics of enzyme recognition domains and active sites, a useful approach for developing drugs and catalysts.6 In contrast to such preferential recognition systems, enzymes that act as chaperones exhibit promiscuous binding.7 Oxidoreductases recognize unstructured proteins, and an active center facilitates SS-bond formation in such unstructured proteins (called clients).3 The dynamic binding of promiscuous enzymes with a range of client unfolded polypeptides can address the conflicting demands for protein folding: namely, maintaining conformational mobility of the client polypeptide chains, allowing the formation of intramolecular covalent and non-covalent bonds, while simultaneously blocking intermolecular contacts. Such client-binding properties significantly influence the functionalities of folding catalysts. PDI and ERp46 are representative oxidoreductases belonging to the PDI family. Both have thioredoxin-like domains as their active centers, but ERp46 promotes faster SS-bond formation in the client protein than does PDI.3c,8 The characteristic binding properties of the two enzymes are different, such as the promiscuous client-binding of ERp46 and the preferential recognition of a client's specific local structure by PDI. The different binding behavior of PDI and ERp46 likely leads to functional switches in their enzymatic activity, such as oxidative SS-bond formation and their anti-aggregation activity when acting as chaperones.9 Binding kinetics contribute to the functional characteristics of molecular chaperones such as SecB and trigger factor (TF). The higher kon (client-binding rate constant) value for SecB than for TF enhances the holdase activity of SecB, while the moderate kon value for TF allows it to exhibit both foldase and holdase activities, depending on the conditions affecting aggregation suppression.10 Such structure–activity analyses suggest the effectiveness of combining promiscuous recognition and redox activity to promote folding and suppress the aggregation of unfolded proteins.
Synthetic mimics of oxidoreductases have been developed to promote polypeptide folding into native structures. In conventional approaches, the mimic focuses on the enzymes' redox active centers for forming and rearranging the SS bonds of the client polypeptides. However, this approach often does not incorporate the binding units that prevent aggregation.11 Protein folding using these approaches is generally conducted at micromolar concentrations, i.e., dilute conditions to suppress the aggregation of unfolded and misfolded proteins. In contrast, here we report synthetic mimics of oxidoreductases that exhibit redox activity and recognize client unfolded polypeptides in a preferential and promiscuous manner (Fig. 1b and c). The enzyme-mimic with promiscuous binding ability significantly promoted folding and inhibited aggregation compared to the mimic exhibiting preferential binding. The former mimic promoted protein folding under condensed conditions, even at sub-millimolar concentrations.
Results and discussion
β-Cyclodextrin (βCD) is a representative synthetic receptor that binds with hydrophobic guest molecules via a concave pocket.12 βCD also interacts with the side chains of amino acids.13 A thiol group was conjugated with βCD to introduce redox activity, and two structural isomers, βCDNSH and βCDWSH, bearing a thiol group at different positions were designed to investigate the steric effect of the thiol group on promoting protein folding and suppressing aggregation (Fig. 1b). The thiol group of βCDWSH is located at the wide rim of βCD, while that of βCDNSH is attached at the narrow rim of the concave structure. These geometrical differences in substituent placement would likely influence the ability of the βCD unit to access the client polypeptide, thus affecting the binding dynamics of the synthetic oxidoreductase-mimics.Biochemical studies on oxidoreductases show that adding disulfide and thiol compounds to unfolded reduced proteins facilitates their folding. Disulfides act as an oxidant to prompt SS-bond formation of the client polypeptide, whereas thiols react with SS bonds through nucleophilic attack, aiding recombination between the native cysteine pairs and driving folding towards the native structure (Fig. 1a). βCDNSH and βCDWSH were synthesized from the corresponding mono-tosylated βCDs (ESI†). Oxidizability and nucleophilicity were assessed from the redox potential E°′ and pKa of βCDNSH and βCDWSH (Fig. S1–S3†). In the pKa analysis, βCDNSH and βCDWSH showed characteristic sigmoidal curves with two inflexion points, which is likely assigned to the deprotonation processes of the thiol and ammonium groups (Fig. S2†). Based on the deprotonation process at the lower pH condition, pKa values of the thiol groups of βCDNSH and βCDWSH were evaluated at 4.68 ± 0.20 and 6.69 ± 0.94, respectively, which were significantly lower than that of reduced glutathione (GSH, pKa 9.17) used as a standard.14 These pKa values indicate that most percentages of βCDNSH and βCDWSH adopt the thiolate forms that possess nucleophilicity at the experimental conditions of the protein folding assays in this study (pH 7.5), and the lower pKa value of the thiol group of βCDNSH suggests higher nucleophilicity than that of βCDWSH. E°′ values of the disulfide forms of βCDNSH and βCDWSH were comparable with each other as well as that of oxidized glutathione (GSSG),11c indicating the oxidizability of the three compounds in the oxidized forms are comparable among one another.
The binding properties of βCDWSH and βCDNSH were investigated through molecular dynamics simulations. Three systems were constructed, each comprising one unfolded bovine pancreatic trypsin inhibitor (BPTI) molecule and five molecules of βCD, βCDWSH or βCDNSH in 300 mM NaCl solution. BPTI was used as a model to compare the simulation results with experimental assays. Each system underwent a 100 ns NPT run at 300 K and 1 atm. The time-averaged fraction of contacts between βCD and each BPTI residue revealed an inhomogeneous distribution of contacts for βCD and βCDNSH with the BPTI residues compared to βCDWSH (Fig. 2 and S4, ESI Movies 1–3†). Specifically, βCDNSH exhibited higher contacts with specific residues, including F4–P8, I19–Y23, and L29–V34. These observed contact distribution trends for each type of βCD align reasonably well with our nuclear magnetic resonance (NMR) measurement results, as discussed in a later section. Furthermore, in comparison to βCD, both βCDWSH and βCDNSH showed fewer contacts (reported as percentages) on the side with the SH group—i.e., on the wide side for βCDWSH and the narrow side for βCDNSH, likely due to steric hindrance by the SH group (Fig. S4 and S5†). These results suggest that specific contacts between βCD with unfolded BPTI residues are associated with contacts with the wide side of βCD. Steric hindrance by the SH group on the wide side can lead to promiscuous contacts with all residues (i.e., βCDWSH), while hindrance on the narrow side enhances the specificity of the wide side of βCD (i.e., βCDNSH).
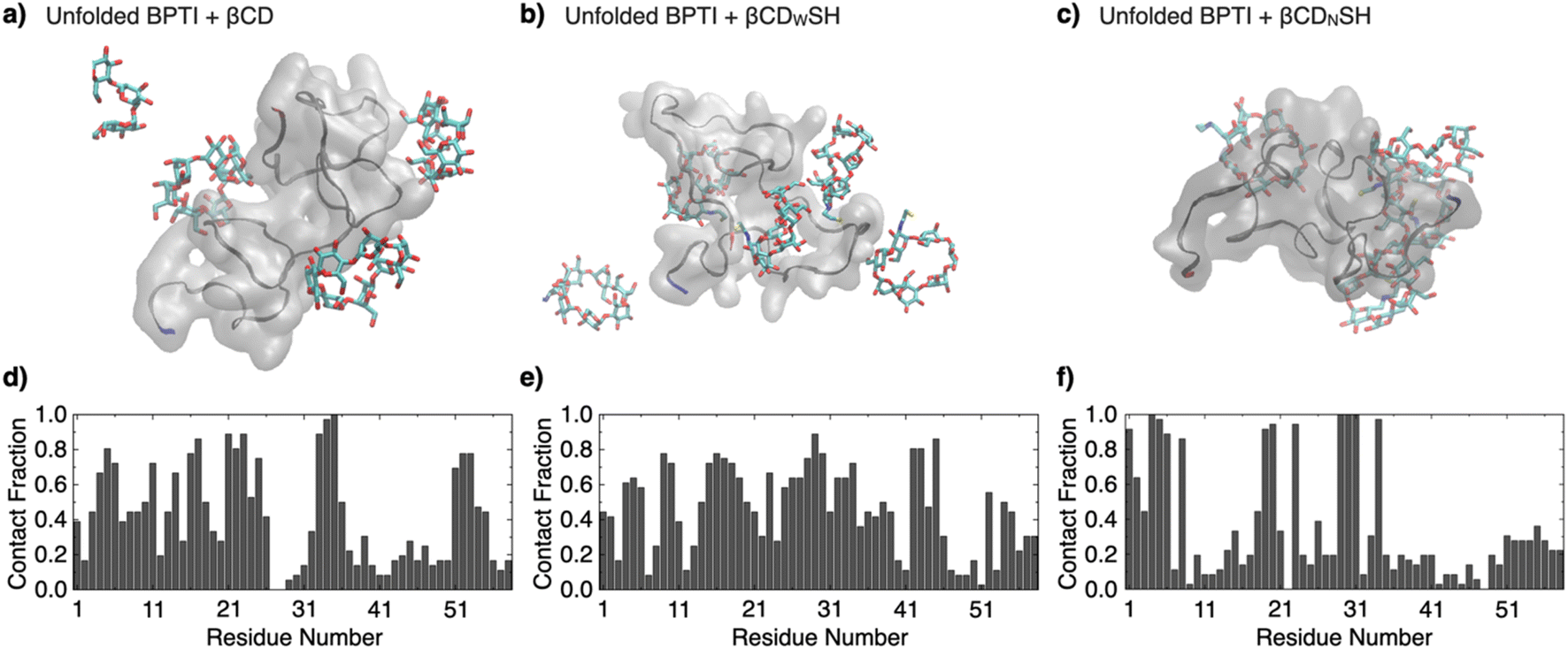 | ||
| Fig. 2 Molecular dynamics simulations over 100 ns of one unfolded BPTI molecule in the presence of five molecules of (a and d) βCD, (b and e) βCDWSH, and (c and f) βCDNSH in 300 mM NaCl aq. (a–c) Representative snapshots. The N-terminus and C-terminus of BPTI are colored in blue and red, respectively. (d–f) Time-averaged fraction of contacts between the additives (βCD, βCDWSH, or βCDNSH, respectively) and each BPTI residue. See original movies in the ESI Movies (Movie 1: βCD, Movie 2: βCDWSH, Movie 3: βCDNSH†). | ||
The ability of βCDNSH and βCDWSH to promote folding was investigated using BPTI. Native (N) BPTI contains three disulfide bonds, between Cys5–Cys55, Cys14–Cys38 and Cys30–Cys51, and was used as a representative model protein for the following folding study. The entire folding pathway of BPTI was elucidated by Weissman and Kim.15 The folding of reduced (R) BPTI proceeds through the quasi-native intermediates N′ and N*, which adopt native-like structures with two SS bonds prior to the formation of N (Fig. 3a). The folding assays were conducted using 30 μM R-BPTI in the presence of 90 μM disulfide compounds and 450 μM thiol compounds, with essentially one equivalent of disulfide compound being added to the reduced-form of BPTI. The chosen ratio of disulfide and thiol compounds was based on a previous study.16 Reverse-phase high-performance liquid chromatography (RP-HPLC) analysis of the oxidative folding of reduced BPTI in the presence of GSH and GSSG showed a gradual decrease in the R-fraction over a 60 min incubation period (Fig. 3b and e) whereas the N-fraction of BPTI emerged during the first 10 min of incubation to a final yield of 15% after 60 min incubation (Fig. 3f). Interestingly, the presence of βCDWSH and its oxidized form βCDWSS resulted in the rapid folding of reduced BPTI. The R-fraction decreased after 10 min incubation, and the N-fraction emerged after 5 min incubation (Fig. 3c). Sixty minutes incubation yielded 43% of the N-form (Fig. 3f). In sharp contrast, the addition of βCDNSH and βCDNSS to reduced BPTI improved folding only to a degree comparable to that obtained using the GSH/GSSG system, and the yield of N-form after 60 min incubation decreased (to 7%, Fig. 3f).
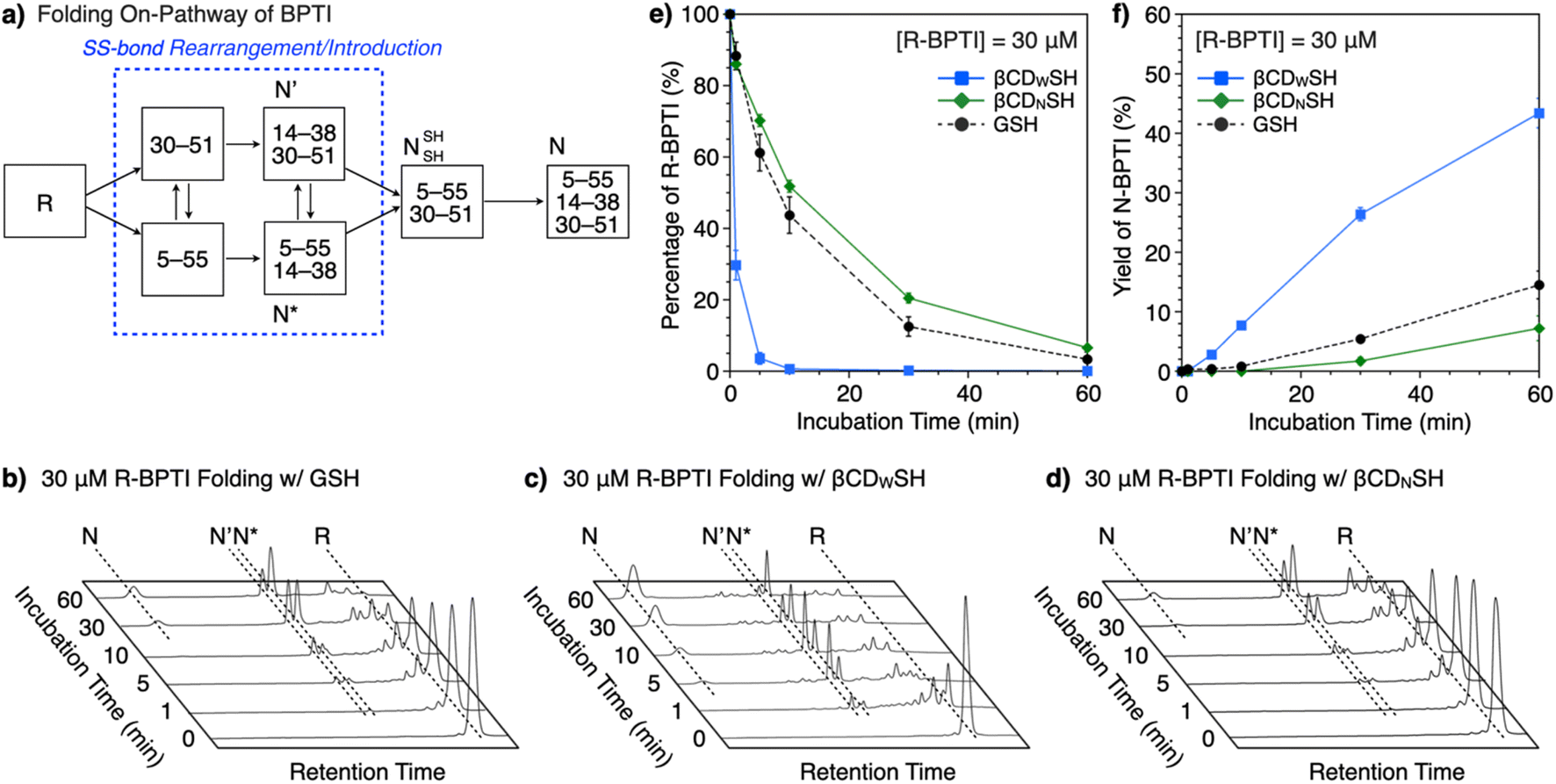 | ||
| Fig. 3 Time course of BPTI oxidative folding as analyzed by RP-HPLC. (a) Folding pathway of BPTI. Time-course reverse-phase HPLC analyses of oxidative folding of BPTI (30 μM) in the presence of (b) GSH and GSSG, (c) βCDWSH and βCDWSS, and (d) βCDNSH and βCDNSS, in the retention time range of 16 to 45 min. N and R denote the native and reduced forms of BPTI, respectively. N′ and N* are the folding intermediates indicated in (a). [Thiols] = 450 μM, [disulfides] = 90 μM. Eluent buffers: water (containing 0.05% TFA) and CH3CN (containing 0.05% TFA) with a linear gradient; flow rate: 1.0 mL min−1; detection wavelength: 229 nm; temperature: 50 °C. Time course plots of (e) the percentage of R-BPTI and (f) the yield of N-BPTI in the presence of (black circles) GSH and GSSG, (blue squares) βCDWSH and βCDWSS, and (green diamonds) βCDNSH and βCDNSS. Error bars indicate the means ± SEM of three independent experiments. | ||
NMR spectroscopic studies were conducted to investigate the mechanism promoting folding and the binding properties of the cyclodextrin-conjugated thiols with unfolded BPTI. The interactions between unfolded BPTI and the cyclodextrin-conjugated thiols were visualized using an 15N-labelled BPTI mutant in which all the cysteine residues were replaced with serine (15N BPTI All-Ser) to provide a model of the R-form. 1H–15N correlation spectra in the amide region were obtained following the addition of βCD using the selective optimized flip angle short transient (SOFAST) technique coupled to heteronuclear multiple quantum correlation (HMQC) experiments.17 Significant chemical shift perturbations to specific signals of 15N BPTI All-Ser were observed (Fig. 4a and S6†). The bar graph showing the chemical shift differences (Δδ) for each residue after adding βCD indicates that the resonances of some specific residues, including Y10, G12, Y35, and G37, shifted markedly, while most other residues showed much smaller chemical shift changes (Fig. 4d) consistent with significant changes in Δδ at the resonances of Y10, G12, Y35, and G37 upon the addition of βCDNSH (Fig. 4c and f). Interestingly, the addition of βCDWSH triggered smaller changes in Δδ relative to that resulting from the addition of βCD or βCDNSH, but the resonances of most residues showed Δδ changes (Fig. 4b and e and S7†). These results suggest that, while βCD or βCDNSH preferentially interacted with several specific residues with high selectivity and affinity, βCDWSH interacted promiscuously with many residues in 15N BPTI All-Ser (Fig. 1c). Furthermore, the resonance of I19 showed a larger Δδ change upon the addition of βCDWSH than upon the addition of βCDNSH, and the signal assigned to A48 shifted in opposite directions upon the addition of βCDWSH or βCDNSH. These changing spectral profiles suggest that βCDWSH interacts with the unfolded protein not only in a weak and indiscriminate manner, but also via a different geometry than does βCDNSH.
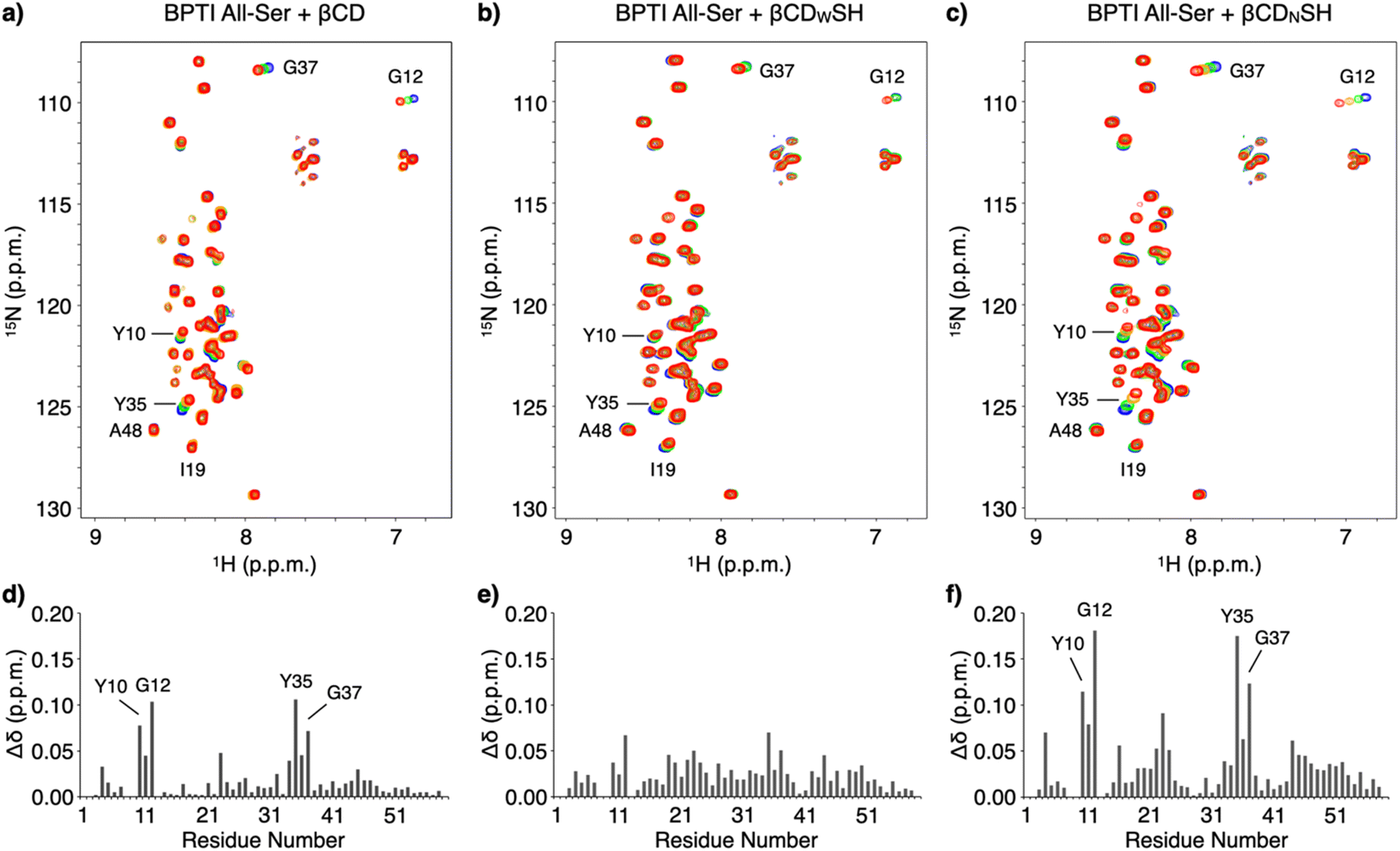 | ||
| Fig. 4 1H–15N correlation SOFAST-HMQC spectra of 15N BPTI All-Ser upon titration with (a) βCD, (b) βCDWSH, and (c) βCDNSH. [15N BPTI All-Ser] = 100 μM, 50 mM HEPES (pH 7.0), 10 v/v% D2O, 10 °C, 500 MHz. Concentration of additives: 0 mM (blue), 1 mM (green), 3 mM (orange), and 5 mM (red). Chemical shift changes for 15N BPTI All-Ser upon the addition of 5 mM (d) βCD, (e) βCDWSH, and (f) βCDNSH. | ||
To directly monitor the interaction at the side chain of an amino acid residue with βCDs, we synthesized a peptide (BPTI7-13) consisting of the same amino acid sequence at 7–13 residues of BPTI. We plotted Δδ of the signal corresponding to Tyr10 of BPTI7-13 because the resonance of Tyr10 showed relatively larger shift of Δδ than the ones of other residues. As expected, titration with 0, 1, 3, and 5 mM of βCDNSH induced the largest Δδ and reached plateau, while the addition of βCDWSH elicited only small change (Fig. S8 and S9†). These analyses suggest that βCDNSH interacts with unfolded BPTI not only with the higher selectivity at some specific residues but also with the higher affinity than βCDWSH. It is likely that the property of the high selectivity and affinity with the unfolded protein endows βCDNSH with the preferential client-binding character, while the promiscuous character of βCDWSH should be emerged by the lower selectivity and affinity property with the amino acid residues.
It would be of importance to discuss relationships between the BPTI primary structure and putative βCD binding sites such as Tyr and Phe (Fig. S10a and b†). The analysis indicates relatively larger Δδ at the signals of Tyr than those of Phe as an overall trend. Among the Tyr residues, the order of Δδ is as follows: (largest) Tyr10 > Tyr35 > Tyr23 > Tyr21 (smallest). Interestingly, the hydrophobic score analysis of each amino acid residue corresponds well with this order (Fig. S10c†). Namely, the Tyr residue with lower hydrophobic score showed larger Δδ, and vice versa, suggesting that βCD and βCDNSH interacts more effectively with the Tyr residues possessing relatively higher hydrophilicity. It is likely that the Tyr residue in a relatively hydrophilic region can expose out to the aqueous phase more favorably, which should be advantageous to the access of βCD and βCDNSH. Meanwhile, the Tyr residue in the hydrophobic region might be buried inside of the polypeptide chain, which would inhibit the complexation with βCD and βCDNSH.
Based on the interactions of βCDWSH and βCDNSH with unfolded proteins, we studied their aggregation suppression properties. Unfolded and reduced BPTI precipitates at 150 μM, a highly condensed condition (Fig. 5a) and protein aggregation was apparently unaffected by βCD (Fig. 5b). Interestingly, the βCDWSH/βCDWSS and βCDNSH/βCDNSS systems acted oppositely to each other. The addition of βCDNSH and βCDNSS caused unfolded BPTI to aggregate, whereas little protein aggregation was observed in the presence of βCDWSH or βCDWSS (Fig. 5c and d). The ability of the βCDWSH/βCDWSS system to suppress aggregation likely promotes folding of the reduced proteins under condensed conditions. We assayed the folding of unfolded and reduced 150 μM BPTI upon addition of the cyclodextrin-conjugated thiols and disulfides ([thiols] = 1000 μM, [disulfides] = 450 μM as one equivalent for the full oxidation of R-BPTI). The concentration of the thiol compounds was limited to 1000 μM due to their poor solubility. In the presence of βCDWSH and βCDWSS, the fraction corresponding to R-BPTI rapidly decreased during a 10 min incubation period, during which time a fraction assigned to N-BPTI became evident (Fig. 5e). Extending the incubation time to 300 min provided a fraction area of N-BPTI corresponding to a 45% yield (Fig. 5g). In contrast, the βCDNSH/βCDNSS or GSH/GSSG system only slowly decreased the fraction area of R-BPTI, resulting in only 23% and 27% yields of N-BPTI, respectively (Fig. 5f and g, S11, and S12†).
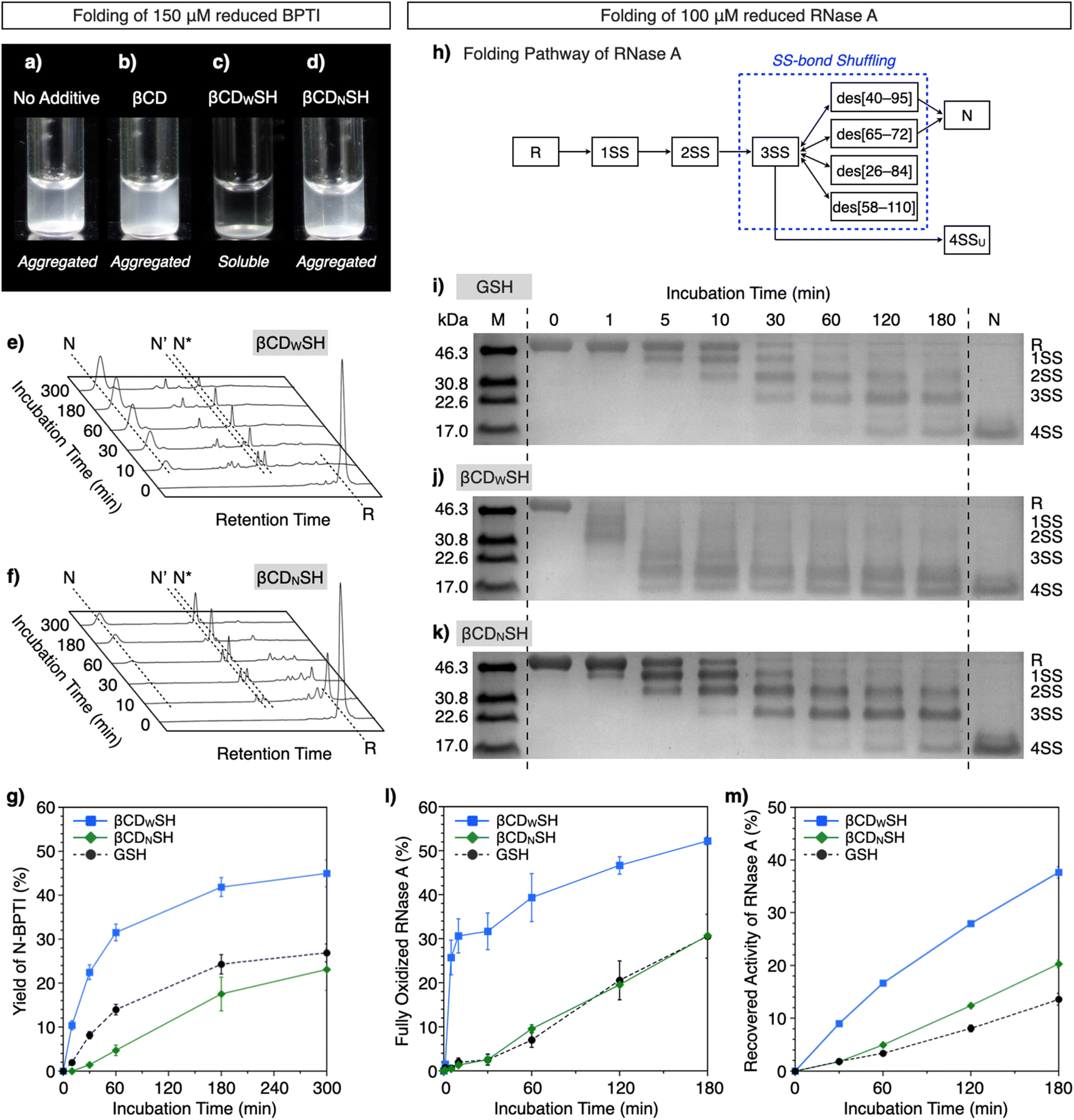 | ||
| Fig. 5 BPTI and RNase A folding under condensed conditions ([BPTI] = 150 μM, [RNase A] = 100 μM). (a–d) Photographs of solutions of unfolded and reduced BPTI (150 μM) (a) without additives, and in the presence of (b) βCD, (c) βCDWSH/βCDWSS and (d) βCDNSH/βCDNSS ([additives] = 1000 μM). (e and f) Time-course reverse-phase HPLC analyses of the oxidative folding of BPTI (150 μM) in the presence of (e) βCDWSH and βCDWSS and (f) βCDNSH and βCDNSS in the retention time range of 16 to 45 min. N and R represent the native and reduced forms of BPTI, respectively. N′ and N* are the folding intermediates. The fraction of N and N′ in (e) corresponds to complexes between BPTI and βCDWSH based on MALDI-TOF MS analysis (Fig. S13†). Eluent buffers: water (containing 0.05% TFA) and CH3CN (containing 0.05% TFA) with a linear gradient; flow rate: 1.0 mL min−1; detection wavelength: 229 nm; temperature: 50 °C. (g) Time course plots of the yields of N-BPTI in the presence of (black circles) GSH and GSSG, (blue squares) βCDWSH and βCDWSS, and (green diamonds) βCDNSH and βCDNSS. (h) Folding pathway of RNase A. (i–k) SDS-PAGE gel images monitoring the oxidation of RNase A (100 μM) in the presence of (i) GSH and GSSG, (j) βCDWSH and βCDWSS, and (k) βCDNSH and βCDNSS in buffer (50 mM Tris–HCl, 300 mM NaCl, pH 7.5) at 30 °C. The folding reactions were quenched with malPEG-2000 after 1, 5, 10, 30, 60, 120, and 180 min of incubation. The leftmost and rightmost lanes contain molecular weight markers (M) and native RNase A (N), respectively. Time-course changes of (l) formation of fully oxidized RNase A (4SS forms) quantified by SDS-PAGE analyses and (m) recovered enzymatic activity of RNase A in the presence of GSH and GSSG, βCDWSH and βCDWSS, and βCDNSH and βCDNSS in buffer (50 mM Tris–HCl, 300 mM NaCl, pH 7.5) at 30 °C. Activity was evaluated by spectroscopic monitoring of the hydrolysis of cCMP to 3′-CMP at 30 °C. Error bars indicate the means ± SEM of three independent experiments. | ||
The ability of βCDNSH and βCDWSH to promote folding was further investigated utilizing ribonuclease (RNase) A. Native RNase A contains four disulfide bonds, between Cys26–Cys84, Cys40–Cys95, Cys58–Cys110, and Cys65–Cys72. The folding of R-RNase A to the N-form proceeds through stepwise oxidations of 1SS, 2SS, and 3SS intermediates containing one, two, and three SS-bonds, respectively (Fig. 5h).18 As a side reaction, the 3SS intermediate can generate 4SSU, a non-native form of fully oxidized RNase A containing four SS-bonds. SS-bond formation of R-RNase A at a high concentration was investigated in the presence of the chemical additives ([RNase A] = 100 μM, [thiol] = 800 μM, [disulfide] = 400 μM). The progress of oxidation involving SS-bond formation was monitored by adding malPEG-2000 (average Mn = 2000) to the reaction medium at selected time points (0, 1, 5, 10, 30, 60, 120, and 180 min) to quench the reaction. The maleimide moiety of malPEG-2000 reacts with the cysteine thiol groups of RNase A irreversibly, increasing its mass; therefore, RNase A bearing more thiol groups increases the mass of the protein correspondingly more. RNase A conjugated with different numbers of malPEG-2000 were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) to quantitatively monitor the oxidation reactions. In the presence of GSH and GSSG, the band corresponding to fully oxidized RNase A containing N and 4SSU appeared after 60 min incubation, and the percentage of N/4SSU-RNase A increased to 31% after 180 min incubation (Fig. 5i and l). Interestingly, the βCDWSH/βCDWSS system accelerated the oxidation of RNase A more effectively and rapidly than did the GSH/GSSG system, while the efficiency of the βCDNSH/βCDNSS system was very similar to that of the GSH/GSSG system (Fig. 5j–l). Accordingly, the βCDWSH/βCDWSS system supported the most rapid recovery of RNase A enzymatic activity, used to evaluate the formation of the N-form. After 180 min incubation, the βCDWSH/βCDWSS system afforded a 38 ± 1.1% yield of N-RNase A. Under otherwise identical conditions, the βCDNSH/βCDNSS and GSH/GSSG systems provided yields of N-form of up to 20 ± 0.7% and 14 ± 1.1%, respectively (Fig. 5m).
As described above, βCDWSS and βCDNSS have similar oxidizability (E°′), and their E°′ values are also comparable to that of GSH/GSSG (Table 1). Based on the pKa values, the nucleophilicity of βCDWSH and βCDNSH are higher than that of GSH, and particularly, the thiol group of βCDNSH should have higher nucleophilicity than βCDWSH. The higher nucleophilicity of the thiol group is generally advantageous for prompting SS-bond rearrangement. Nevertheless, the βCDWSH/βCDWSS system facilitated oxidative SS-bond formation of reduced BPTI and RNase A more efficiently than did the βCDNSH/βCDNSS system. Furthermore, folding to the native forms of BPTI and RNase A proceeded faster with the βCDWSH/βCDWSS system than with the other systems, suggesting effective cooperation between the formation and rearrangement of the client protein's SS-bonds (Fig. 1a). The higher reactivities of the βCDWSH/βCDWSS system for protein folding are likely due to the system's binding properties with the unfolded client protein, increasing the reaction rate at the thiol and disulfide unit of the cyclodextrin additive with the protein cysteine residues. Attaching the thiol group at the wide rim of βCD imparts a promiscuous and global binding property with unfolded proteins, likely due to steric hindrance at the client-binding pocket, as visualized by the MD simulation and supported by NMR measurements. Since the βCDNSH/βCDNSS system, which binds with unfolded proteins tightly at specific domains, only weakly promoted protein folding and caused aggregation, it is likely that the promiscuous binding property of βCDWSH/βCDWSS is advantageous for balancing polypeptide conformational mobility for forming intramolecular SS-bonds and non-covalent bonds required for folding, and for inhibiting polypeptide intermolecular contacts, thereby suppressing aggregation under condensed conditions. We suggest that the promiscuous binding behavior of βCDWSH is analogous to the client-recognition behavior of ERp46, while the more preferential recognition character of βCDNSH corresponds to that of PDI, which binds to specific local regions of the client protein. Interestingly, βCDNSH and PDI recognize several common sites in unfolded BPTI, such as the residues around G12 and G37, despite their large differences in size and chemical structure. Furthermore, consistent with the trend observed between ERp46 and PDI in the initial phase of the folding process, the ERp46-mimetic βCDWSH supported faster SS-bond formation than the PDI-mimetic βCDNSH.9 As observed in the BPTI folding process catalyzed by ERp46, off-pathway intermediate species were also observed in the presence of βCDWSH.8b,9 These analogies between biological enzymes and the described chemical mimics demonstrate that the binding characteristics of chemical chaperones can be controlled by steric effects at the client-recognition site to deter aggregation. The bio-inspired design described here, combining a redox-active center and client-recognition unit with a tendency for promiscuous binding, is an effective approach for promoting the correct folding of error-prone and aggregation-prone polypeptide folding under condensed conditions.
In this study, one equivalent of disulfide compounds relative to the client proteins was added to the reaction solutions, indicating that the reaction systems are non-catalytic. Meanwhile, the native system of the oxidative protein folding is catalytic, in which the reduced forms of oxidoreductases were re-oxidized in a multi-step electron transfer reactions from O2, an oxidation source, to H2O.3c Installing such regeneration process of the oxidants should enable development of bio-mimetic catalytic reaction systems of the oxidative protein folding, which is an important subject in this research field to further enhance the production efficiency as high as that of the cellular system.
There is an increasing number of reports describing a link between disulfide chemistry and the pathogenesis of diseases that involve misfolded proteins.19 Over 20 members of the PDI family, which are disulfide catalysts, play key roles in maintaining protein homeostasis by both catalyzing oxidative folding and by chaperoning aggregation-prone clients, thereby likely decreasing the risk of misfolding-induced pathologies. The dysfunction of some disulfide catalysts thus may result in pathologies such as amyotrophic lateral sclerosis, Alzheimer's disease, Parkinson's disease, type 2 diabetes, and intellectual disability. There are few examples of the development of chemical chaperones to treat misfolding-related diseases,20 and new approaches using thiol compounds in place of PDI family proteins are attractive for this purpose.
Conclusions
In this study, we developed a redox-active promiscuous binder, βCDWSH, which bears a client-recognition unit and a redox-active center. βCDWSH promotes oxidative protein folding at sub-millimolar concentrations under condensed conditions. The design of a sterically hindered client-binding pocket in βCD coupled with a redox-active group satisfies the conflicting demands for protein folding: to enhance the frequency of cysteine residue reactions, to maintain conformational mobility of the polypeptide chain, and to block intermolecular protein aggregation. The molecular design of redox-active chemical chaperones with promiscuous binding properties represents an important breakthrough for promoting protein folding under highly condensed conditions and is promising for producing functional and pharmaceutical proteins. This chemical approach can also find medical applications for the prevention of misfolding diseases caused by protein denaturation and aggregation.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
T. Mu. conceived the idea of this study. K. S., R. N., T. Ma., S. K., K. I. and H. K. performed the experiments. M. O., T. S., and T. Mu. supervised the conduct of this study. M. O., T. S. and T. M. wrote the manuscript. All authors critically reviewed and revised the manuscript draft and approved the final version for submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ms Atsuko Hishiyama, Ms Miki Suda, and Mr Takaaki Toyoda for their help in conducting experiments. This work was supported by a research grant from JSPS Grant-in-Aid for Transformative Research Areas (JP21H05096 to TMu and TMa, JP21H05095 to MO, JP21H05094 to TS), JSPS Fund for the Promotion of Joint International Research (JP23KK0105 to MM, MO and TMu), JST CREST (JPMJCR19S4 to TMu), JST FOREST (JPMJFR2122 to TMu, JPMJFR201F to MO, JPMJFR204W to TS, JPMJFR212H to TMa), KISTEC (to TMu), Takeda Science Foundation (TMu), and Asahi Glass Foundation (TMu), G-7 Scholarship Foundation (TMu).Notes and references
- (a) C. B. Anfinsen, Principles that govern the folding of protein chains, Science, 1973, 181, 223–230 CrossRef CAS PubMed; (b) M. J. Gething and J. Sambrook, Protein folding in the cell, Nature, 1992, 355, 33–45 CrossRef CAS PubMed; (c) T. E. Creighton, Protein folding coupled to disulphide bond formation, Biol. Chem., 1997, 378, 731–744 CAS; (d) C. M. Dobson, Protein folding and misfolding, Nature, 2003, 426, 884–890 CrossRef CAS PubMed; (e) F. Baneyx and M. Mujacic, Recombinant protein folding and misfolding in Escherichia coli, Nat. Biotechnol., 2004, 22, 1399–1408 CrossRef CAS PubMed; (f) J. L. Arolas, F. X. Aviles, J.-Y. Chang and S. Ventura, Folding of small disulfide-rich proteins/clarifying the puzzle, Trends Biochem. Sci., 2006, 31, 292–301 CrossRef CAS PubMed; (g) S. J. Kim, J. S. Yoon, H. Shishido, Z. Yang, L. A. Rooney, J. M. Barral and W. R. Skach, Translational tuning optimizes nascent protein folding in cells, Science, 2015, 348, 444–448 CrossRef CAS PubMed.
- (a) O. B. Ptitsyn, V. E. Bychkova and V. N. Uversky, Kinetic and equilibrium folding intermediates, Philos. Trans. R. Soc., B, 1995, 348, 35–41 CrossRef CAS PubMed; (b) J. L. Sohl, S. S. Jaswal and D. A. Agard, Unfolded conformations of α-lytic protease are more stable than its native state, Nature, 1998, 395, 817–819 CrossRef CAS PubMed; (c) P. T. Lansbury Jr, Evolution of amyloid: What normal protein folding may tell us about fibrillogenesis and disease, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3342–3344 CrossRef CAS PubMed; (d) J. Ren, Y. Bi, J. R. Sowers, C. Hetz and Y. Zhang, Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases, Nat. Rev. Cardiol., 2021, 18, 499–521 CrossRef PubMed.
- (a) J. S. Weissman and P. S. Kim, Efficient catalysis of disulphide bond rearrangements by protein disulphide isomerase, Nature, 1993, 365, 185–188 CrossRef CAS PubMed; (b) B. van den Berg, E. W. Chung, C. V. Robinson, P. L. Mateo and C. M. Dobson, The oxidative refolding of hen lysozyme and its catalysis by protein disulfide isomerase, EMBO J., 1999, 18, 4794–4803 CrossRef CAS PubMed; (c) M. Okumura, H. Kadokura and K. Inaba, Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum, Free Radical Biol. Med., 2015, 83, 314–322 CrossRef CAS PubMed; (d) R. A. Lumb and N. J. Bulleid, Is protein disulfide isomerase a redox-dependent molecular chaperone?, EMBO J., 2002, 21, 6763–6770 CrossRef CAS PubMed; (e) M. Okumura, H. Kadokura, S. Hashimoto, K. Yutani, S. Kanemura, T. Hikima, Y. Hidaka, L. Ito, K. Shiba, S. Masui, D. Imai, S. Imaoka, H. Yamaguchi and K. Inaba, Inhibition of the functional interplay between endoplasmic reticulum (ER) oxidoreduclin-1α (Ero1α) and protein-disulfide isomerase (PDI) by the endocrine disruptor bisphenol A, J. Biol. Chem., 2014, 289, 27004–27018 CrossRef CAS PubMed; (f) M. Okumura, K. Noi, S. Kanemura, M. Kinoshita, T. Saio, Y. Inoue, T. Hikima, S. Akiyama, T. Ogura and K. Inaba, Dynamic assembly of protein disulfide isomerase in catalysis of oxidative folding, Nat. Chem. Biol., 2019, 15, 499–509 CrossRef CAS PubMed; (g) M. Okumura, K. Noi and K. Inaba, Visualization of structural dynamics of protein disulfide isomerase enzymes in catalysis of oxidative folding and reductive unfolding, Curr. Opin. Struct. Biol., 2021, 66, 49–57 CrossRef CAS PubMed; (h) M. Chinnaraj, R. Flaumenhaft and N. Pozzi, Reduction of protein disulfide isomerase results in open conformations and stimulates dynamic exchange between structural ensembles, J. Biol. Chem., 2022, 298, 102217 CrossRef CAS PubMed.
- T. Muraoka, M. Okumura and T. Saio, Enzymatic and synthetic regulation of polypeptide folding, Chem. Sci., 2024, 15, 2282–2299 RSC.
- E. Rajakumara, S. Abhishek, K. Nitin, D. Saniya, P. Bajaj, U. Schwaneberg and M. D. Davari, Structure and cooperativity in substrate–enzyme interactions: Perspectives on enzyme engineering and inhibitor design, ACS Chem. Biol., 2022, 17, 266–280 CrossRef CAS PubMed.
- J.-M. Choi, S.-S. Han and H.-S. Kim, Industrial applications of enzyme biocatalysis: Current status and future aspects, Biotechnol. Adv., 2015, 33, 1443–1454 CrossRef CAS PubMed.
- (a) R. Rosenzweig, A. Sekhar, J. Nagesh and L. E. Kay, Promiscuous binding by Hsp70 results in conformational heterogeneity and fuzzy chaperone-substrate ensembles, eLife, 2017, 6, e28030 CrossRef PubMed; (b) L. He and S. Hiller, Common patterns in chaperone interactions with a native client protein, Angew. Chem., Int. Ed., 2018, 57, 5921–5924 CrossRef CAS PubMed.
- (a) J. C. Edman, L. Ellis, R. W. Blacher, R. A. Roth and W. J. Rutter, Sequence of protein disulphide isomerase and implications of its relationship to thioredoxin, Nature, 1985, 317, 267–270 CrossRef CAS PubMed; (b) Y. Sato, R. Kojima, M. Okumura, M. Hagiwara, S. Masui, K. Maegawa, M. Saiki, T. Horibe, M. Suzuki and K. Inaba, Synergistic cooperation of PDI family members in peroxiredoxin 4-driven oxidative protein folding, Sci. Rep., 2013, 3, 2456 CrossRef PubMed; (c) R. Kojima, M. Okumura, S. Masui, S. Kanemura, M. Inoue, M. Saiki, H. Yamaguchi, T. Hikima, M. Suzuki, S. Akiyama and K. Inaba, Radically different thioredoxin domain arrangement of ERp46, an efficient disulfide bond introducer of the mammalian PDI family, Structure, 2014, 22, 431–443 CrossRef CAS PubMed.
- T. Saio, K. Ishii, M. Matsusaki, H. Kumeta, S. Kanemura and M. Okumura, bioRxiv, 2024, preprint, DOI:10.1101/2024.03.04.583432.
- (a) T. Saio, X. Guan, P. Rossi, A. Economou and C. G. Kalodimos, Structural basis for protein antiaggregation activity of the trigger factor chaperone, Science, 2014, 344, 1250494 CrossRef PubMed; (b) C. Huang, P. Rossi, T. Saio and C. G. Kalodimos, Structural basis for the antifolding activity of a molecular chaperone, Nature, 2016, 537, 202–206 CrossRef CAS PubMed.
- (a) E. Welker, M. Narayan, W. J. Wedemeyer and H. A. Scheraga, Structural determinants of oxidative folding in proteins, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2312–2316 CrossRef CAS PubMed; (b) J. D. Gough, R. H. Williams, A. E. Donofrio and W. J. Lees, Folding disulfide-containing proteins faster with an aromatic thiol, J. Am. Chem. Soc., 2002, 124, 3885–3892 CrossRef CAS PubMed; (c) J. Beld, K. J. Woycechowsky and D. Hilvert, Selenoglutathione: Efficient oxidative protein folding by a diselenide, Biochemistry, 2007, 46, 5382–5390 CrossRef CAS PubMed; (d) W. J. Lees, Small-molecule catalysts of oxidative protein folding, Curr. Opin. Chem. Biol., 2008, 12, 740–745 CrossRef CAS PubMed; (e) M. Okumura, M. Saiki, H. Yamaguchi and Y. Hidaka, Acceleration of disulfide-coupled protein folding using glutathione derivatives, FEBS J., 2011, 278, 1137–1144 CrossRef CAS PubMed; (f) A. S. Patel and W. J. Lees, Oxidative folding of lysozyme with aromatic dithiols, and aliphatic and aromatic monothiols, Bioorg. Med. Chem., 2012, 20, 1020–1028 CrossRef CAS PubMed; (g) J. C. Lukesh III, M. J. Palte and R. T. Raines, A potent, versatile disulfide-reducing agent from aspartic acid, J. Am. Chem. Soc., 2012, 134, 4057–4059 CrossRef PubMed; (h) P. S. Reddy and N. Metanis, Small molecule diselenide additives for in vitro oxidative protein folding, Chem. Commun., 2016, 52, 3336–3339 RSC; (i) K. Arai, H. Ueno, Y. Asano, G. Chakrabarty, S. Shimodaira, G. Mugesh and M. Iwaoka, Protein folding in the presence of water-soluble Cyclic diselenides with novel oxidoreductase and isomerase activities, ChemBioChem, 2018, 19, 207–211 CrossRef CAS PubMed; (j) S. Okada, M. Matsusaki, K. Arai, Y. Hidaka, K. Inaba, M. Okumura and T. Muraoka, Coupling effects of thiol and urea-type groups for promotion of oxidative protein folding, Chem. Commun., 2019, 55, 759–762 RSC; (k) S. Tsukagoshi, R. Mikami and K. Arai, Basic amino acid conjugates of 1,2-diselenan-4-amine with protein disulfide isomerase-like functions as a manipulator of protein quality control, Chem.–Asian J., 2020, 15, 2646–2656 CrossRef CAS PubMed; (l) S. Okada, Y. Matsumoto, R. Takahashi, K. Arai, S. Kanemura, M. Okumura and T. Muraoka, Semi-enzymatic acceleration of oxidative protein folding by N-methylated heteroaromatic thiols, Chem. Sci., 2023, 14, 7630–7636 RSC.
- G. Crini, Review: A history of cyclodextrins, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS PubMed.
- (a) J. M. Alexander, J. L. Clark, T. J. Brett and J. J. Stezowski, Chiral discrimination in cyclodextrin complexes of amino acid derivatives/β-cyclodextrin/N-acetyl-L-phenylalanine and N-acetyl-D-phenylalanine complexes, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5115–5120 CrossRef CAS PubMed; (b) M. S. Semrau, G. Giachin, S. Covaceuszach, A. Cassetta, N. Demitri, P. Storici and G. Lolli, Molecular architecture of the glycogen-committed PP1/PTG holoenzyme, Nat. Commun., 2022, 13, 6199 CrossRef CAS PubMed.
- S.-S. Tang and G.-G. Chang, Nucleophilic aromatic substitution of glutathione and 1-chloro-2,4-dinitro-benzene in reverse micelles. A model system to assess the transition-state stabilization in glutathione transferase catalyzed conjugation, J. Org. Chem., 1995, 60, 6183–6185 CrossRef CAS.
- J. S. Weissman and P. S. Kim, Reexamination of the folding of BPTI: Predominance of native intermediates, Science, 1991, 253, 1386–1393 CrossRef CAS PubMed.
- M. M. Lyles and H. F. Gilbert, Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: Dependence of the rate on the composition of the redox buffer, Biochemistry, 1991, 30, 613–619 CrossRef CAS PubMed.
- P. Schanda and B. Brutscher, Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds, J. Am. Chem. Soc., 2005, 127, 8014–8015 CrossRef CAS PubMed.
- (a) A. Bierzynski, P. S. Kim and R. L. Baldwin, A salt bridge stabilizes the helix formed by isolated C-peptide of RNase A, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 2470–2474 CrossRef CAS PubMed; (b) D. M. Rothwarf and H. A. Scheraga, Regeneration of bovine pancreatic ribonuclease A. 2. Kinetics of regeneration, Biochemistry, 1993, 32, 2680–2689 CrossRef CAS PubMed; (c) M. Iwaoka, D. Juminaga and H. A. Scheraga, Regeneration of three-disulfide mutants of bovine pancreatic ribonuclease A missing the 65-72 disulfide bond: Characterization of a minor folding pathway of ribonuclease A and kinetic roles of Cys65 and Cys72, Biochemistry, 1998, 37, 4490–4501 CrossRef CAS PubMed; (d) M. Narayan, E. Welker, W. J. Wedemeyer and H. A. Scheraga, Oxidative folding of proteins, Acc. Chem. Res., 2000, 33, 805–812 CrossRef CAS PubMed.
- (a) T. Uehara, T. Nakamura, D. Yao, Z. Q. Shi, Z. Gu, Y. Ma, E. Masliah, Y. Nomura and S. A. Lipton, S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration, Nature, 2006, 441, 513–517 CrossRef CAS PubMed; (b) D. Eletto, D. Eletto, D. Dersh, T. Gidalevitz and Y. Argon, Protein disulfide isomerase A6 controls the decay of IRE1α signaling via disulfide-dependent association, Mol. Cell, 2014, 53, 562–576 CrossRef CAS PubMed; (c) O. B. Oka, M. van Lith, J. Rudolf, W. Tungkum, M. A. Pringle and N. J. Bulleid, ERp18 regulates activation of ATF6α during unfolded protein response, EMBO J., 2019, 38, e100990 CrossRef PubMed; (d) M. Matsusaki, S. Kanemura, M. Kinoshita, Y.-H. Lee, K. Inaba and M. Okumura, The protein disulfide isomerase family: from proteostasis to pathogenesis, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864, 129338 CrossRef CAS PubMed; (e) D. B. Medinas, P. Rozas and C. Hetz, Critical roles of protein disulfide isomerases in balancing proteostasis in the nervous system, J. Biol. Chem., 2022, 298, 102087 CrossRef CAS PubMed.
- (a) C. R. Brown, L. Q. Hong-Brown, J. Biwersi, A. S. Verkman and W. J. Welch, Chemical chaperones correct the mutant phenotype of the ΔF508 cystic fibrosis transmembrane conductance regulator protein, Cell Stress Chaperones, 1996, 1, 117–125 CrossRef CAS PubMed; (b) T. Okiyoneda, G. Veit, J. F. Dekkers, M. Bagdany, N. Soya, H. Xu, A. Roldan, A. S. Verkman, M. Kurth, A. Simon, T. Hegedus, J. M. Beekman and G. L. Lukacs, Mechanism-based corrector combination restores ΔF508-CFTR folding and function, Nat. Chem. Biol., 2013, 9, 444–454 CrossRef CAS PubMed; (c) T. Kuramochi, Y. Yamashita, K. Arai, S. Kanemura, T. Muraoka and M. Okumura, Boosting the enzymatic activity of CxxC motif-containing PDI family members, Chem. Commun., 2024, 60, 6134–6137 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02123a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |