
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances of metal active sites in photocatalytic CO2 reduction
Wa
Gao
 *a,
Haonan
Li
a,
Jianqiang
Hu
f,
Yong
Yang
*e,
Yujie
Xiong
h,
Jinhua
Ye
g,
Zhigang
Zou
cd and
Yong
Zhou
*bcd
*a,
Haonan
Li
a,
Jianqiang
Hu
f,
Yong
Yang
*e,
Yujie
Xiong
h,
Jinhua
Ye
g,
Zhigang
Zou
cd and
Yong
Zhou
*bcd
aSchool of Physical Science and Technology, Tiangong University, Tianjin 300387, P. R. China
bSchool of Chemical and Environmental Engineering, Anhui Polytechnic University, Wuhu 241000, P. R. China
cSchool of Physics, Jiangsu Key Laboratory of Nanotechnology, Eco-materials and Renewable Energy Research Center (ERERC), National Laboratory of Solid State Microstructures, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210093, P. R. China
dSchool of Science and Engineering, The Chinese University of Hongkong (Shenzhen), Shenzhen, Guangdong 518172, P. R. China
eKey Laboratory of Soft Chemistry and Functional Materials (MOE), Nanjing University of Science and Technology, Nanjing 210094, P. R. China
fJiangxi Normal Univ., Inst. Adv. Mat. IAM, Coll. Chem. & Chem. Engn., Nanchang 330022, P. R. China
gNational Institute for Materials Science (NIMS), International Center Materials Nanoarchitecture MANA, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
hSchool of Chemistry and Materials Science, University of Science and Technology of China, Hefei 230036, Anhui, P. R. China
First published on 15th August 2024
Abstract
Photocatalytic CO2 reduction captures solar energy to convert CO2 into hydrocarbon fuels, thus shifting the dependence on rapidly depleting fossil fuels. Among the various proposed photocatalysts, systems containing metal active sites (MASs) possess obvious advantages, such as effective photogenerated carrier separation, suitable adsorption and activation of intermediates, and achievable C–C coupling to generate multi-carbon (C2+) products. The present review aims to summarize the typical photocatalytic materials with MAS, highlighting the critical role of different formulations of MAS in CO2 photoreduction, especially for C2+ product generation. State-of-the-art progress in the characterization and theoretical calculations for MAS-containing photocatalysts is also emphasized. Finally, the challenges and prospects of catalytic systems involving MAS for solar-driven CO2 conversion are outlined, providing inspiration for the future design of materials for efficient photocatalytic energy conversion.
1 Introduction
Over-reliance on fossil fuels as energy carriers leads to excessive CO2 emissions from combustion, which in turn causes climate change, environmental damage and energy crisis.1–3 Shifting from fossil fuels to renewable energy sources to achieve carbon neutrality is an important step towards sustainable development. Therefore, there is an urgent need to find a suitable, effective, and green strategy to facilitate the conversion of CO2 and access renewable energy through sustained technological innovation.CO2 capture and conversion by artificial photosynthesis can simultaneously address the rising global CO2 emissions and produce hydrocarbon fuels to accomplish carbon neutrality.4–6 It is proving to be an elegant and promising solution, attracting the interest of scientists and making great strides in the efficiency of CO2 conversion.7–11 Specifically, the photocatalytic reduction of CO2 achieves a sustainable alternative to conventional fossil fuels, which is carried out in relatively mild conditions-room temperature and pressure, driving the conversion of CO2 to hydrocarbons directly without additional energy supply or harmful substance release. Photocatalytic CO2 reduction consists of three main processes:12 the catalyst generates electron–hole pairs under sunlight;13–15 carriers separate and migrate to the surface;16 CO2 is activated and converted at the surface active sites, involving C–O bond breaking, C–H bond formation, and C–C coupling.17,18 In fact, the CO2 molecule possesses a rather stable structure with high dissociation energy to cleave the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (750 kJ mol−1), which poses a challenge for the activation and conversion of CO2.19 In addition, solar-driven CO2 reduction involves complex multi-step conversions that, depending on the number of electrons and protons transferred, produces CO,20–22 CH3OH,23–25 CH4,26,27 C2H4,28–30 C2H6,31,32 C2H5OH,33etc. Due to the diversity of products of CO2 photoreduction, achieving highly selective CO2 conversion to generate specific target products is extremely challenging.
O bond (750 kJ mol−1), which poses a challenge for the activation and conversion of CO2.19 In addition, solar-driven CO2 reduction involves complex multi-step conversions that, depending on the number of electrons and protons transferred, produces CO,20–22 CH3OH,23–25 CH4,26,27 C2H4,28–30 C2H6,31,32 C2H5OH,33etc. Due to the diversity of products of CO2 photoreduction, achieving highly selective CO2 conversion to generate specific target products is extremely challenging.
Various strategies have been employed to improve the efficiency and selectivity of CO2 photoreduction, including enhancing light absorption,34,35 promoting carrier separation,36 constructing active sites,37 and promoting C–C coupling.38 According to the current findings, MAS possesses significant advantages in improving the performance of photocatalytic reduction of CO2 and promoting C–C coupling to obtain C2+ products.39–41 This review mainly introduces the research progress of MAS in driving photocatalytic CO2 conversion (Fig. 1). Photocatalytic systems containing MAS including metal oxides, metal sulfides, layered double hydroxides (LDHs), metal organic framework (MOF), covalent organic frameworks (COF), single atom catalysts, and metal complexes are summarized. The achievement of MAS with different formulations in photocatalytic CO2 conversion is systemically explored while mechanisms that drive performance improvement are discussed in detail. The development of characterization techniques and theoretical calculation to determine the structure and function of MAS in CO2 photoreduction is also highlighted. Finally, the difficulties, challenges, and novel viewpoints of feasible solutions are addressed regarding the design of advanced photocatalysts containing MAS for efficient solar energy conversion.
 | ||
| Fig. 1 Overview of the metal active sites in photocatalytic CO2 reductions. | ||
2 Photocatalytic systems with MAS
Micro-nanostructures containing MAS, including metal oxides, metal sulfides, LDHs, MOF, COF, single atom catalysts, and metal complexes are widely used in photocatalytic CO2 reduction. According to the process and mechanism of CO2 photoreduction, the catalytic systems with high light absorption capacity, sufficient carrier separation, and abundant active sites show obvious advantages in the yield and selectivity of CO2 conversion.2.1 Metal oxides
In the complex and diverse photocatalytic systems, metal oxides have become the most representative and well-established photocatalysts. A large number of strategies aimed at modulating the band gap, facilitating carrier separation, and establishing active sites have been proposed and intensively investigated in metal oxides.42–44 TiO2, as a representative metal oxide, has become a photocatalyst of wide interest due to its low toxicity, high stability and economic advantages. Defect engineering, heterostructure construction, heteroatom doping, and surface modulation have been proposed to regulate the carrier migration, CO2 adsorption and activation of TiO2-based catalysts. The synergistic effect of Ru and oxygen vacancies (Vo) on the photocatalytic performance of TiO2 (Ru–TiO2−x) was systematically investigated.45 It was found that Ru–TiO2−x exhibited high activity for the photoreduction of CO2 to CH4. The introduced Vo in Ru–TiO2−x provided important contributions to promote photogenerated carrier separation and facilitate CO2 conversion. The Ru species effectively captured photogenerated electrons, which inhibited further photogenerated carrier recombination.Photocatalytic CO2 reduction for the generation of C2+ products was achieved in metal–oxide systems. The vacancy-rich TiO2 with Cu single atoms loading produced C3H8, which involved an overall 20 e− reduction and two sequential C–C coupling processes.46 The Cu–Ti–VO unit in the Ti0.91O2 matrix was formed through the modulation of the electronic coupling interaction between the Cu atoms and adjacent Ti atoms by Vo. This unique unit lowered the energy levels of the key *CHOCO and *CH2OCOCO intermediates, thereby tuning the C1–C1 and C1–C2 couplings to thermodynamically favourable exothermic processes (Fig. 2a). Moreover, the metastable hexagonal WO3 (h-WO3), offering a suitable bandgap, exhibited efficient photoreduction of CO2 to C2H4.47 The blue color of the metastable state of WO3 was attributed to the reduction of W6+ to W5+ or W4+ with the increase of the reduced state, which indicated the existence of Vo. The surface Vo enhanced the light absorption capability and promoted the photogenerated carrier separation. More importantly, the W–S–W sites formed by the S atom, which replaced oxygen atoms and bridged the adjacent W atoms, benefited the adsorption of *CH2 intermediates and promoted the C–C coupling to generate C2H4 (Fig. 2b).
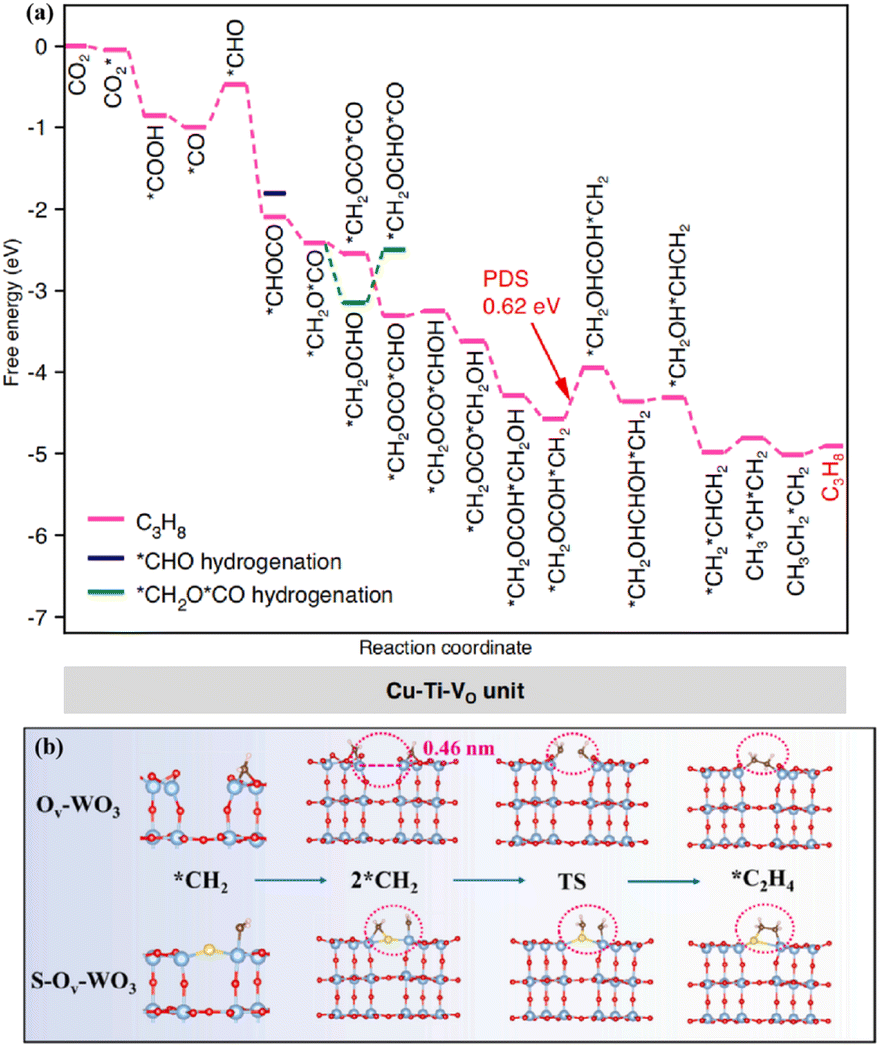 | ||
| Fig. 2 (a) Gibbs free energy diagrams of CO2 reduction on the Cu–Ti–VO unit. This figure has been reproduced from ref. 46 with permission from Springer Nature, copyright 2023. (b) The C–C coupling process of Ov–WO3 and S–Ov–WO3. This figure has been reproduced from ref. 47 with permission from Elsevier, copyright 2023. | ||
Based on the above analysis, the MAS constructed in the metal oxide system is structurally stable, successfully achieving efficient photocatalytic CO2 conversion with C2+ generation. In fact, MAS in metal oxides often synergizes with defect and doping atoms to achieve performance enhancement. Therefore, MAS introduced in metal oxides usually needs multi-step experimental treatment processes.
2.2 Transition metal sulfides/phosphorus sulfides
Various metal chalcogenides, such as ZnS, CdS, MnS, CdSe, ZnIn2S4 (ZIS), and CuIn5S8, have exhibited progressive photocatalytic performances. Their excellent properties, including their low cost, wide range of light absorption, and high carrier mobility, have made them one of the most promising materials for CO2 conversion.The reduction potential of ZnS is −1.04 V vs. NHE, which is relatively negative and particularly suitable for the photocatalytic reduction reaction.48 However, the wide band gap (3.6–3.8 eV), fast recombination of the charge carriers, and low selectivity toward CO2 reduction competing with the hydrogen evolution reaction (HER) limit the efficiency of ZnS. Cu-doped ZnS, containing abundant sphalerite and wurtzite phase (S–W) junctions, enhanced the photocatalytic activity with high CO selectivity.49 The S–W phase junction containing abundant sulfur vacancies (Vs) effectively facilitated the separation of charge carriers and the localization of photoelectrons for surface catalysis. The addition of Cu increased the electron density at the Fermi energy level, promoted the reactivity of the Cu sites, and enhanced the bonding of the catalyst surface to the *CO intermediate, thereby improving the CO selectivity. Furthermore, the dual functional ZnS coupled with g-C3N4 simultaneously enhanced the surface and bulk carrier separation of g-C3N4 for the highly selective reduction of CO2 to CH4.50 ZnS acted as a cocatalyst to capture the photogenerated electrons of g-C3N4. It contributed a polarization electric field, which was created inside the ZnS nanoparticle along the direction of spontaneous polarization, to prompt the migration and separation of photogenerated charges from bulk to surface.
ZIS, belonging to the AB2X4 group of ternary compounds, has been used in photocatalysis with visible light absorption.53 The metallic ZnIn2S4, which is rich in indium vacancies (VIn), exhibited full-spectrum responsiveness for high CO2 photoreduction efficiency.51 The VIn defect state captured the excited hole (h+) and increased the minority carrier diffusion length, resulting in a large number of carriers moving to the surface and participating in the conversion of CO2. Theoretical calculations revealed that VIn lowered the energy barrier of the rate-limiting step (the formation of COOH*, Fig. 3a and b), leading to the high rate of CO evolution. In addition, anchoring single Au atoms to ultrathin ZIS (Au1/ZIS) nanosheets with Vs resulted in the Au1–S2 low coordination structure, which enabled exceptional photocatalytic CO2-to-CH4 conversion.52 The introduction of Au single atoms enhanced the efficiency of carrier separation and transfer. Moreover, the low-coordinated single Au atom significantly enhanced CO2 activation, lowered the energy barrier for *CO protonation, and stabilized the *CH3 intermediate, leading to the selective generation of CH4 by CO2 photoreduction.
 | ||
| Fig. 3 Free energy diagrams for the reduction of CO2 to CO over the (001) facets of VIn-rich-ZIS (a) and VIn-poor-ZIS (b). These figures have been reproduced from ref. 51 with permission from American Chemical Society, copyright 2022. Free energy diagrams of photocatalytic CO2 to CH4 for Au1–S2/ZIS (c). This figure has been reproduced from ref. 52 with permission from Wiley-VCH, copyright 2022. | ||
Transition metal phosphorous trichalcogenides have been well-explored in the photoreduction of CO2 to valuable fuels. The introduction of Vs in AgInP2S6 regulated the CO2 photoreduction reaction pathway to steer the dominant generation of C2H4.54 The VS led the charge accumulation on the Ag atoms near VS, which effectively captured the forming *CO. This phenomenon enriched the catalyst surface with key reaction intermediates and promoted C–C coupling to generate C2 species with low binding energy barriers. In addition, the tandem synergistic effect of the charge-enriched Cu–In dual site, which was confined on the lateral edge of the CuInP2S6 monolayer, became the main reason for the efficient conversion and high selectivity of C2H4.55 In the presence of light, the limbic In site of the CuInP2S6 monolayer converted CO2 mainly to *CO, which was transferred to the neighbouring Cu site for the subsequent C–C coupling reaction to C2H4.
By reason of the foregoing, MAS in metal chalcogenides shows unique advantages in the generation of C2+ products. However, due to the limitation of the stability of the metal sulfide, the stability of MAS in them may be lacking. Therefore, in photocatalytic applications, attention should be paid to improving the stability of MAS in metal sulfides.
2.3 LDHs
LDHs are typically layered materials consisting of laminates and interlaminar anions with a high degree of flexible tunability in terms of morphology and ionic composition.57 The adjustment of the metal cation or anion in LDH can easily regulate the electronic structure and influence the CO2 conversion process. For example, the selectivity of the photocatalytic reaction could be precisely tuned by the composition of metal cations in the ZnM-LDH photocatalysts (M = Ti4+, Fe3+, Co3+, Ga3+, Al3+).56 Specifically, the main reduction product of ZnTi-LDH was CH4. ZnFe-LDH and ZnCo-LDH generated H2 by water splitting, and ZnGa-LDH and ZnAl-LDH produced CO (Fig. 4a and b). Experimental characterization and theoretical calculations revealed that the d-band center of the M3+ or M4+ cations affected the adsorption strength of CO2: cations with the d-band centers close to the Fermi level adsorbed CO2 strongly, benefiting CH4 or CO formation. Conversely, the deviation of the d-band centre from the Fermi level led to poor CO2 adsorption and hence H2 production. In addition, the strategies involving morphology modulation, heterostructure construction, doping, and defect introduction have a crucial effect on the CO2 conversion process of LDH. The S-scheme heterojunction of g-C3N4/CoCo-LDH preserved the water oxidation ability of CoCo-LDH and redox ability of g-C3N4.58 The atomic-level interface chemical bond (Co–N2 bond) of g-C3N4/CoCo-LDH realized the high-speed transfer of electrons. The addition of CoCo-LDH benefited the CO2 adsorption, reduced the energy barrier of the key intermediate *COOH, and promoted water decomposition. | ||
| Fig. 4 (a) Scheme showing CO2 and H2O photoreduction on the different ZnM-LDH photocatalyst (carbon: dark gray; oxygen: red; hydrogen: white). DFT calculations of the elementary steps of (b) H2 evolution, (c) CO2 reduction to CO, and (d) CO2 reduction to CH4 involving a methanol intermediate over ZnTi-LDH, ZnAl-LDH, and ZnFe-LDH. These figures have been reproduced from ref. 56 with permission from Elsevier, copyright 2020. | ||
Obviously, MAS can take advantage of the two-dimensional (2D) structure, as well as the highly flexible adjustability in the morphology and ionic composition of LDH. However, the synergies of different metals and their roles in C2+ generation remain to be explored.
2.4 MOF
One of the crystalline porous materials, MOF, which possesses diversified metal ions/clusters and organic linkers, as well as atomically precise and tailorable structures, provides the natural MAS for catalysis.60 Zirconium-based MOF (PCN-222) hybridized with cellulose acetate (CA@PCN-222) increased the activity of CO2 photoreduction to formate compared with pristine PCN-222 by regulating the atomic interface structure of the MAS (Fig. 5a).59 The valence band (VB) across the Fermi energy levels resulted in higher charge-transfer kinetics of CA@PCN-222 relative to PCN-222. The interfacial electron transfer from CA to PCN-222 led to the redistribution of the Zr d-orbital electrons. Moreover, the ZrIV-cluster unit in CA@PCN-222 was turned into a ZrIII-cluster active site after accepting two electrons from two adjacent tetrakis(4-carboxyphenyl)-porphyrin ligands under visible-light irradiation, which selectively reduced CO2 to HCOO− in a two-electron process (Fig. 5b). | ||
| Fig. 5 (a) Schematic atomic interface model of CA@PCN-222. (b) Proposed mechanism for the CO2 photoreduction reaction over CA@PCN-222. These figures have been reproduced from ref. 59 with permission from Wiley-VCH, copyright 2023. | ||
The modulation of the coordination environment around the MAS in MOFs helps to reveal the relationship between structure and activity during CO2 photoreduction. In UiO-type MOFs bearing bipyridine linkers, the number of coordinated N atoms around a single Co site was tuned to provide UiO-Co-Nx (x = 2, 3 and 4) for photocatalytic CO2 reduction.61 UiO-Co-N3 exhibited superior activity to the other counterparts, which was mainly attributable to the difference in the number of coordinating N atoms around the Co site. Particularly, UiO-Co-N3 endowed the lowest energy barriers of the rate-determining step (CO2 → COOH*) and the desorption of CO* among all UiO-Co-Nx samples, accounting for the optimized CO2 photoreduction activity.
The MAS in MOF enables liquid C2+ generation. The specific NH2–Cu–NH2 triple atom site was constructed by incorporating Cu sites into the connected nodes of defective UiO-66-NH2, realizing photocatalytic CO2-to-acetone conversion.62 Specifically, one of the N active sites on the NH2–Cu–NH2 adsorbed CO2 and converted CO2 to form *CH3 (CO2 → COOH* → CO* → *CH3). Meanwhile, the Cu site activated its adsorbed CO2 to CO*. *CH3 and *CO underwent the first C–C coupling on CuN2O2 to generate *CO–*CH3. At the same time, another N site on the CuN2O2 fragment activated the adsorbed CO2 into CO*, which underwent a second C–C coupling process with the *COCH3 intermediate to generate the crucial *COCOCH3. Due to the synergistic interaction between the Cu site and the N site, the C2 intermediate on the CuN2O2 ultimately generated C3.
In summary, the MAS in MOF connected with organic ligands exhibits good dispersion and high utilization, which can make full use of the porous property of MOF to optimize the photocatalytic CO2 reduction performance. However, its synthesis process involves complex chemical reactions and utilizes expensive organic ligands, making practical production applications difficult.
2.5 COF
COF is a class of organic polymers linked by reversible covalent bonds, which possesses periodical structures, well-defined porosity, an extended π-conjugated framework, and pore structures for mass transfer. An adequate MAS of COF could be constructed by incorporating metal atoms into the skeletons or pore channels by coordination interaction.63 2D COF ultrathin nanobelts coordinated with single Cu–O/N sites (Cu2+ anchored by both O and N) exhibited high CO selectivity of 94% under visible-light-driven CO2 reduction.64 Crucially, the Cu–O/N sites benefited the electron transfer from COF upon light irradiation, and served as the active sites for the highly selective reduction of CO2 to CO. Two types of cobalt Schiff base COF composites, Co-2,3-DHTA-COF with Co–O4 sites and Co-TP-COF with Co-O3N sites, realized the CO2 photoreduction into CO (Fig. 6).65 The supereminent photocatalytic performance of Co-COF with Co-O4 sites was mainly attributed to the high CO2 adsorption capacity, low charge-transfer resistance, strong separation of electrons and holes, and the lower energy barrier in the ligand exchange process between Co-2,3-DHTA-COF and CO2. For hydrocarbon product generation, single-atom MoN2 sites were introduced into COF (Mo-COF) to realize the photoreduction of CO2 to CH4 and C2H4 under visible light.66 The MoN2 sites contributed to the efficient separation efficiency of photogenerated electrons and holes, enhanced the adsorption and activation of CO2 and CO, and reduced the energy barriers for the formation of hydrocarbon intermediates, thus leading to the production of high value-added hydrocarbons over Mo-COF. | ||
| Fig. 6 Gibbs energy profiles of the CO2RR-to-CO reaction over Co-2,3-DHTA-COF (A, blue line) and Co-TP-COF (B, orange line). This figure has been reproduced from ref. 65 with permission from American Chemical Society, copyright 2023. | ||
Particularly, MAS can utilize the π-conjugated framework in COF to promote the photogenerated carrier separation and enhance the photocatalytic CO2 reduction. However, MAS needs to be grafted onto the COF structure with fine regulation. In addition, the construction of multiple MAS in COF and the study of their catalytic mechanism still need to be continuously explored.
2.6 Single-atom catalysts
Single-atom catalysis with MAS uniformly and individually loaded on a supporting material provides a research platform for structure–performance correlation at the atomic level. Atomically dispersed MAS improves the atomic utilization, modulates the charge distribution of the catalyst, and affects the adsorption and conversion of reactants and intermediates, ultimately influencing the product efficiency and selectivity of the photocatalytic CO2 reduction.Loading rare-earth La single atoms on carbon nitride (O/La–CN) constructed the active centers of La–N charge-transfer bridges for photocatalytic CO2 reduction.67 Electronic state changes induced by the hybridization of the 4f and 5d orbitals of La single atoms and the p–d orbitals of La–N atoms established charge-transfer channels for La–N bridges to promote carrier separation. Bader charge and differential charge distributions suggested that electrons were transferred through La atoms into connected N atoms, and eventually through C atoms to O atoms. The O/La–CN strengthened CO2 adsorption, endowed the high capacity for CO2 uptake, and reduced the activation energy barrier for COOH* formation. Moreover, the desorption of CO from the surface of O/La–CN required the lowest energy (0.2 eV) compared with the dissociation reaction (COH*, 2.96 eV and C* + H2O, 4.59 eV) or hydrogenation (HCO*, 1.53 eV), contributing CO formation with high selectivity (Fig. 7a). Cu loading on BiOBr nanosheets (Cu1@BiOBr) established a strong built-in electric field with isolated Cu sites that acted as electron traps to promote charge transfer and stabilize charge carriers.68 The high selectivity of methanol within this photocatalytic system could be ascribed to the energy-favorable hydrogenation of the *CO intermediate into *CHO (Fig. 7b). Furthermore, the unfavorable adsorption of CH3OH on Cu1@BiOBr relative to H2O prevented methanol from being oxidized by photogenerated holes.
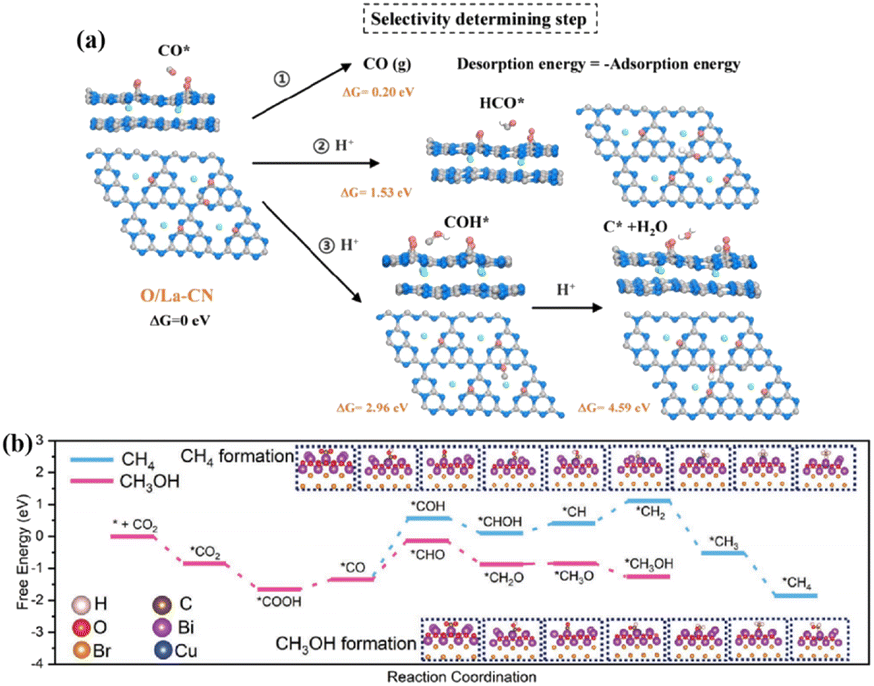 | ||
| Fig. 7 (a) Calculated free energy for the photocatalytic CO2 reduction reaction of the selectivity-determining step. This figure has been reproduced from ref. 67 with permission from American Chemical Society, copyright 2020. (b) The DFT-calculated free energy diagram for the hydrogenation of CO2 to CH3OH and CH4 over Cu1@BiOBr. This figure has been reproduced from ref. 68 with permission from Wiley-VCH, copyright 2023. | ||
In addition to single MAS loading, dual MAS incorporation has been explored to drive CO2 photoreduction with diverse functions. The dual MAS of cobalt (Co) and ruthenium (Ru) supported on a conjugated porous carbon nitride polymer exhibited efficient photocatalytic CO2 reduction.69In situ characterizations and theoretical calculations revealed that the active Co sites facilitated dynamic charge transfer, along with Ru working as adsorption sites for CO2 photoactivation. In detail, the atomic Co facilitated dynamic electron transfer from the carbon nitride polymer to the atomic Ru site, resulting in the COOH* intermediate being effectively stabilized by the charged-rich Ru site. Moreover, the smaller Gibbs free energy of CO formation than that of the protonation of CO* to CHO* resulted in high CO selectivity. Similarly, Ru and Cu single atoms simultaneously incorporated into polymeric carbon nitride (poly-CN) exhibited higher selectivity (95%) for CH4 production than that decorated with Ru or Cu individually.70 The atomically dispersed Ru–N4 and Cu–N3 sites tuned the electronic structure of poly-CN, and were identified as active centers. During CO2 photoreduction, the Ru single atom was essential for proton production, while the Cu single atom played a dominant role in the reduction process. Due to the thermodynamically more favorable conversion of *CO to *CHO, the coexistence of the Ru and Cu single atoms resulted in the efficient conversion of CO2 to CH4.
Remarkably, the MAS in single-atom catalysts exhibits high metal atom utilization and achieves synergistic effects of multiple MASs. However, the metal is easily aggregated to form particles during the construction process. Thus, a delicate experimental design is required to successfully construct MAS in single-atom form.
2.7 Metal complexes
The realization of CO2 photoreduction by transition metal complexes with tunable and definable chemical structures is accompanied by the activation and immobilisation of neutral CO2 molecules at the metal centre.71 By installing a 2-phenol, 2-amino or 2-mercapto to a tripodal skeleton, the Co-based homogeneous catalysts (named [CoN3O]ClO4, [CoN4]ClO4 and [CoN3S]ClO4, respectively) possessed different coordination microenvironments for CO2 reduction.72 The optimal [CoN3O]ClO4 photocatalyst had a maximum turnover number (TON) of 5652 in photocatalytic CO2 reduction among [CoN4]ClO4 and [CoN3S]ClO4. The strong electronegativity of the oxygen atom in the ligand endowed the Co(II) catalytic center with a low reduction potential and more stable *COOH intermediate, thus substantially promoting the CO2-to-CO conversion. The dinuclear heterometallic [CuNiL2] showed the highest CO2-to-CO conversion relative to dinuclear homometallic [Ni2L2(CH3OH)2] and mononuclear [NiL1(CH3OH)].73 A theoretical calculation revealed that the introduction of inactive CuII in CuNi-L2 promoted the transportation of photo-generated electrons to the coupled active NiII site. This allowed NiII with an O4 coordination environment in [CuNiL2] to express strong reducing ability, which significantly accelerated the photocatalytic CO2-to-CO conversion.Metal complexes could drive C–C coupling to generate C2+ products. The synergistic dual sites of rhenium(I) bipyridine fac-[ReI(bpy)(CO)3Cl] (Re-bpy) and copper porphyrinic triazine framework [PTF(Cu)] working in tandem strategy achieved the photocatalytic conversion of CO2 to C2H4 (Fig. 8).74 The CO generated at the Re-bpy sites was adsorbed by the nearby Cu single sites in PTF(Cu), followed by a synergistic C–C coupling process to ultimately produce C2H4. Critically, Rebpy-*CO could enter the porous PTF(Cu) and move to the nearby Cu–*CO, leading to practicable free energy for the CO–CO coupling between Cu–*CO and Re-bpy-*CO (Re-bpy-*CO-*CO-PTF(Cu)). However, PTF(Cu) catalysts alone produced CO under similar conditions. This was caused by the large distance between the adjacent Cu centers in PTF(Cu) hindering C2+ formation via the coupling of Cu–*CO–*CO–Cu.
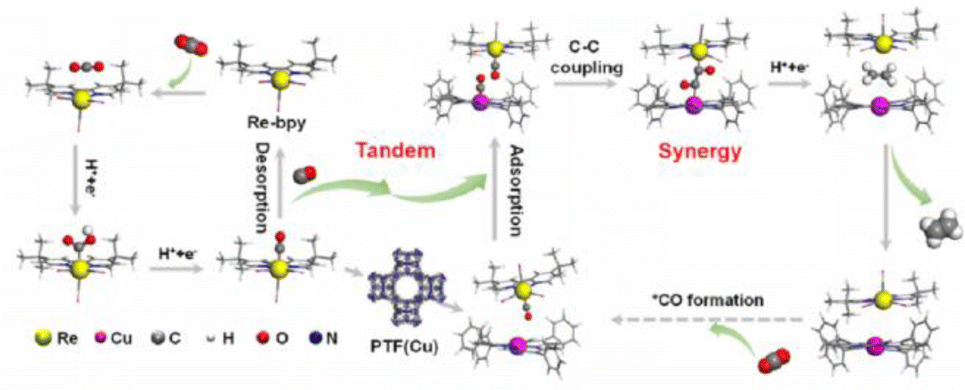 | ||
| Fig. 8 Tandem catalysis mechanism of Re-bpy/PTF(Cu). This figure has been reproduced from ref. 74 with permission from American Chemical Society, copyright 2023. | ||
As mentioned above, the alteration of the MAS coordination environment can be achieved by modulating the skeleton in metal complexes, which in turn regulates the photocatalytic CO2 reduction performance. It is conducive to revealing the in-depth reaction mechanism. However, the typical problems in photocatalysis, such as the high separation cost and low chemical stability, still limit their practical applications.75
3 Different formulations of MAS in the photocatalytic CO2 reduction
Incorporation of MAS can regulate the transport of photogenerated carriers, increase the local electron density of the adsorption sites, affect the adsorption strength of reactants and intermediates, and ultimately adjust the barrier and selectivity of the reaction. In this section, the role of different formulations of MAS in photocatalytic CO2 reduction is described in detail (Table 1).| Catalyst | Reactive sites | Light source | Reaction condition | Performance (μmol g−1 h−1) | Ref. | |
|---|---|---|---|---|---|---|
| Single MAS | Cu-doped ZnS containing abundant S–W junctions | Cu | 300 W Xe lamp | 18 mL solution containing chloroform + 2 mL 2-propanol + CO2 | CO: 68.9 (99.9%) | 49 |
| VIn-rich-ZIS | VIn | 300 W Xe lamp AM 1.5 cut-off filter | 45 mL acetonitrile (MeCN) + 5 mL trolamine (TEOA) + CO2 | CO: 298 | 51 | |
| ZnM-LDH (M = Ti4+, Ga3+, Al3+) | M | 300 W Xe lamp | 0.1 mL H2O + 0.08 MPa CO2 | CH4: 1.2 (68% M = Ti), CO: 1.3 (90% M = Al), CO: 1.6 (78% M = Ga) | 56 | |
| CA@PCN-222 | ZrIII-cluster | 300 W Xe lamp (800 nm > λ > 400 nm) | 60 mL mixed solution (MeCN/TEOA = 30/1) + CO2 | HCOO−: 281.6 | 59 | |
| UiO-Co-N3 | Co–N3 | 300 W Xe lamp (λ ≥ 420 nm) | 20 mL MeCN + 30 mg [Ru(bpy)3]Cl2·6H2O + 0.025 M 1,3-dimethyl-1,3-dihydro-2-phenyl-2H-benzimidazole (BIH) + CO2 | CO: 179.3 (99.3%) | 61 | |
| Incorporating Cu sites into the connected nodes of defective UiO-66-NH2 | NH2-Cu-NH2 | 300 W Xe lamp (λ > 420 nm) | 2 mL H2O + 6 mL N,N-dimethylformamide (DMF) + CO2 | CH3COCH3: 70.9 (97%) | 62 | |
| 2,2′-Bipyridine-based COF bearing non-noble single Cu sites | Cu2+ sites | 300 W Xe lamp (λ > 420 nm) | 10 mL DMF or H2O + 1 mL triethylamine + CO2 | CH4: 17.5; CO: 1.6 (H2O), CO: 10.2 (100%; DMF), CO: 22.5 (66.4%; DMF/H2O = 50%) | 63 | |
| Cu-COF | Cu–O/N sites | 300 W Xe lamp (λ > 420 nm) | 12 mL mixed solution + 600 torr CO2, MeCN/H2O/TEOA (8 mL/2 mL/2 mL) | CO: 206 (94%) | 64 | |
| Co-2,3-DHTA-COF | Co–O4 sites | 300 W Xe lamp (λ > 420 nm) | 46 mL mixed solution + 10 mg [Ru(bpy)3]Cl2·6H2O + 1 atm CO2, MeCN/H2O/TEOA (32 mL/8 mL/6 mL) | CO: 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (95.7%) 000 (95.7%) |
65 | |
| Mo-COF | MoN2 sites | 300 W Xe lamp (λ ≥ 420 nm) | 5 mL H2O + 1 mL CO2 | CO: 6.19, C2H4: 3.57, CH4: 1.08 | 66 | |
| O/La–CN | La–N charge-transfer bridge | Xe lamp | 12 mL mixed solution + 1 atm CO2MeCN/H2O/TEOA (6 mL/4 mL/2 mL) | CO: 92 (80.3%) | 67 | |
| Cu1@BiOBr | Isolated Cu sites | 300 W Xe lamp | 5 mL H2O + 4 MPa CO2 | CH3OH: 627.66 (86.6%) | 68 | |
| [CoN3O]ClO4 | Co(II) catalytic center | 300 W Xe lamp (λ > 420 nm) | 5 mL mixed solution + [Ru(bpy)3]Cl2·6H2O + BIH + CO2 MeCN/H2O (4 mL/1 mL) | CO: 88.14% ± 2.09% | 72 | |
| CuNi-L2 | Active NiII site | LED light (λ > 420 nm) | 25 mL mixed solution + [Ru(bpy)3]Cl2·6H2O (0.85 mmol L−1)+ Triisopropanolamine (TIPA 0.8 mol L−1) + CO2, MeCN/H2O (20 mL/5 mL) | CO: (93.5%) | 73 | |
| Cu single atoms on the nitrogen-doped carbon anchored on TiO2 | Cu SAs | 300 W Xe lamp | H2O vapor + CO2 | CO: 65.8 | 77 | |
| 2Re-In2O3 | Odef | 300 W Xe lamp | 100 mL H2O + 1.01 bar CO2 | CH3OH: 265.6 (100%) | 78 | |
| Cu1/TiO2 | Cu1–O2+1 | Xe lamp | 20 mL H2O + TEOA + CO2 | C2H4: 60.4 (75.2%) | 79 | |
| MIL-88B-NS40 | Fe–N coordinated sites/uncoordinated S | 300 W Xe lamp (λ > 420 nm) | 2 mL H2O + CO2 | C2H4: 17.7 | 80 | |
| B-Ni1/WO2.72 | Ni site | 300 W Xe lamp (λ > 420 nm) | 6 mL mixed solution + 15 mg [Ru(bpy)3]Cl2·6H2O + CO2, MeCN/H2O/TEOA (3 mL/2 mL/1 mL) | CO: 80500 (98.7%) |
82 | |
| Isolated Mn atoms over the multi-edged TiO2 nano-pompons | Mn atoms | 300 W Xe lamp (λ > 400 nm) | 6 mL mixed solution + 10 mg [Ru(bpy)3]Cl2·6H2O + 1 atm CO2 MeCN/H2O/TEOA (3 mL/2 mL/1 mL) | CO: 80510, H2: 23070 |
83 | |
| In-bonded N-atom (Inδ+-N4) in the (002) crystal planes of g-C3N4 | Inδ+-N4 | 300 W Xe lamp | 400 μL H2O + CO2 | CO: 398.87 | 84 | |
| CuACs/PCN | Cu–N4 sites doped P | 300 W Xe lamp | 5 mL TEOA + [Ru(bpy)3]Cl2·6H2O + 45 mL H2O + CO2 | C2H4: 10.17 (53.2%), CH4: 9.55 | 85 | |
| 3Er-ZnIn2S4 | Er | 300 W Xe lamp (λ ≥ 420 nm) | TEOA + 100 mL H2O + 80 kPa CO2 | CH4: 6.68 (>90%) | 86 | |
| Cu1N3@PCN | Different coordination structures Cu-atom | 300 W Xe lamp (λ > 420 nm) | 1 mL H2O + CO2 | CO: 49.8 | 87 | |
| Ag1@PCNT | Ag–N3, Ag single atoms | 300 W Xe lamp AM 1.5 cut-off filter | 3 mL H2O + 70 kPa CO2 | CO: 160 (>94%) | 88 | |
| Ultra-high Pr loading CN | Pr1–N4O2 sites | 300 W Xe lamp | 1.0 mL H2O + 4.0 mL CO2 | CH3OH: 511.1 | 89 | |
| MCOF-Ti6Cu3 | Cu cluster | Xe lamp AM 1.5 cut-off filter | 30 mL H2O + CO2 | HCOOH: 169.8 | 90 | |
| Dual MAS | Mn1Co1/CN | Two compatible active centers of Mn and Co | 300 W Xe lamp (λ > 400 nm) | 2 mL H2O + 80 kPa CO2 | CO: 47 | 17 |
| Cu–Ti–VO/Ti0.91O2-SL | Cu–Ti–VO unit | 300 W Xe lamp | 12.5 mL MeCN + 2.5 mL H2O + 1 atm CO2 | C2H4: 7.6, C3H8: 13.8, CO: 18.6 | 46 | |
| CuInP2S6 monolayer | Cu–In dual site | 300 W Xe lamp AM 1.5 cut-off filter | 0.4 mL H2O + CO2 | C2H4: 20.89 (56.4%), CO: 8.59, CH4: 6.22 | 55 | |
| V into NiAl-LDH | V; lower-valence Ni | 300 W Xe lamp (λ > 400 nm) | 10 mL mixed solution + 0.005 mmol [Ru(bpy)3]Cl2·6H2O + 1.8 bar CO2 MeCN/H2O/TEOA (6 mL/2 mL/2 mL) | CH4: 217 (78.9%) | 57 | |
| Dispersed Co–Ru bimetal into conjugated porous carbon nitride polymer | Co sites; Ru sites | 300 W Xe lamp | 500 μL H2O + 70–80 kPa CO2 | CO: 27.3 | 69 | |
| Ru Cu single atom incorporated into polymeric carbon nitride | Ru–N4 sites, Cu–N3 sites | 300 W Xe lamp | 9 mL NaHCO3 (3 M) + 1 mL TEOA + CO2 | CH4: 154 (95%), CO: 2, H2: 6 | 70 | |
| Re-bpy/PTF(Cu) | Re-bpy sites; Cu single sites in PTF(Cu) | 300 W Xe lamp (1100 nm > λ > 400 nm) | 5 mL mixed solution + 1 atm CO2 MeCN/TEOA (2.5 mL/2.5 mL) | C2H4: 73.2 | 74 | |
| Cu–N2–V | Cu+/Cu2+ | 300 W Xe lamp | 21 mL DMF + 3 mL H2O + 0.8 MPa CO2 | C2H5OH: 69.8 | 81 | |
| LaNi-Phen/COF-5 | Optically active (La site) catalytically active (Ni site) | 300 W Xe lamp | 10 mM BIH + 48 mL MeCN + 2 mL H2O + 80 kPa CO2 | CO: 605.8 (98.2%) | 91 | |
| CuIn5S8 | Cu–In dual sites | 300 W Xe lamp (λ > 400 nm) | 2 mL H2O + 0.1 atm CO2 | CH4: 8.7 | 92 | |
| Ag2Cu2O3 | Cu–Ag Lewis acid–base dual sites | 300 W Xe lamp | 5 mLH2O or 5 mL 0.2% methanol aqueous solution was injected + CO2 | CH4: 3.6 | 93 | |
| In/TiO2-Vo | Vo-regulated In–Ti dual sites | Xe lamp (320 nm < λ < 780 nm) | H2O + CO2 | CH4: 35.49 (91.3%) | 94 | |
| Au/Co dual single atom loaded CdS NPs | Au/Co DSA | 300 W Xe lamp (λ > 400 nm) | CO2 | CO: 64.1 μmol g−1, CH4: 7.7 μmol g−1 | 95 | |
| Vo-rich Zn2GeO4 nanobelts | Asymmetric Zn–O–Ge sites | Xe lamp light | 12 mL H2O + 0.03 vol% CO2/Ar gas mixture | CH3COOH: 12.7 (29.95%) | 96 | |
| Au–CeO2 | Au–O–Ce sites | 300 W Xe lamp | 5 mL H2O + 80 kPa CO2 | CO: 11.07, C2H6: 11.07 (65.3%) | 97 | |
| InCu/PCN | In–Cu dual sites | 300 W Xe lamp | 24 mL DMF/H2O (12.5 vol% H2O) + 0.8 MPa CO2 | C2H5OH: 28.5 (92%) | 98 | |
| Bi2WO6 nanosheets with Vo anchoring Au and Cu dual single atoms | Au–Cu dual-single-atom sites; Vo | 300 W Xe lamp (780 nm > λ > 320 nm) | 1.0 mL H2O + (CO2-1%, N2-99%) | Total amount of production (CO, CH4, C2H4, C3H6): 83.9 | 99 | |
| 2% Pd–Cu/TiO2 | Pd–Cu; Vo | 300 W Xe lamp | 15 mL H2O + 0.5 MPa CO2 | CH3OH: 71.84 (91.71) | 100 | |
| Single atoms of Ni/Co loaded on TiO2 | Ni Co single atoms | 300 W Xe lamp | 50 mL H2O (0.1 M Na2SO3 + 0.2 M CsOH) + CO2 | CH3COOH: 22.6, 71% | 101 | |
| Incorporated the redox-active Co2+/Ni2+ centers into the chemically stable layered lead iodide hybrids | Co2+/Ni2+–O–Pb bimetallic catalytic sites | 300 W Xe lamp AM 1.5 G filter | 1 mL H2O + CO2 | C2H5OH: 24.9–31.4, 89.5–93.6% | 102 | |
| g-C3N4/UiO-66(Zr/Ce) | N–Zr/Ce–O | 300 W Xe lamp | 15 mL MeCN + 15 mL H2O + 101 kPa CO2 | CH3OH: 54.71, C2H5OH: 38.10 | 103 | |
| Cu-CuTCPP/g-C3N4 | Cu1+δ | 300 W Xe lamp (800 nm > λ > 360 nm) | 400 μL H2O + 200 μL TEA + 0.1 MPa CO2 | CO: 12.3, CH4: 11.6, C2H6: 18.5 | 104 | |
| MAS combined with vacancies | P and Cu dual sites anchored on graphitic carbon nitride | P–N and Cu–N4 dual sites | 300 W Xe lamp | 3 mL TEOA + H2O + 0.65 atm CO2 | C2H6: 616.6 | 31 |
| 1% Ru–TiO2−x | Ru; Vo | 300 W Xe lamp | 0.14 g NaHCO3 + N2 + H2O + H2SO4 (2 M) | CH4: 31.63 (90.93%) | 45 | |
| A series of metastable WO3 photocatalysts with the coexistence of S doping and Vo | W–S–W bridge; Vo | 300 W Xe lamp | 0.4 mL H2O + CO2 | C2H4: 224.278, 87.6% | 47 | |
| Au atoms to ultrathin ZnIn2S4 nanosheets with Vs | Au1–S2 | 300 W Xe lamp (λ > 420 nm) | 5 mL mixed solution + 10 mg [Ru(bpy)3]Cl2·6H2O + 1 atm CO2 MeCN/H2O/TEOA (3 mL/1 mL/1 mL) | CH4: 275 (77%) | 52 | |
| Vs-AgInP2S6 single atomic layer | Ag atoms; Vs | 300 W Xe lamp | 0.4 mL H2O + CO2 | C2H4: 44.3 (73%) | 54 | |
| Mn dopants and Vo were engineered in Zn2GeO4 nanorods | Mn dopants and Vo | 300 W Xe lamp | 1 mL H2O + CO2 | CO: 40.02 (82.9%) | 105 | |
| Cu1/N2CV-CN | Cu single atoms; N2C vacancies | 300 W Xe lamp AM 1.5 cut-off filter | 200 μL H2O + 105 kPa CO2 | CO: 11.12 | 106 | |
| CuGaS2/Ga2S3 containing Vs | Cu–Ga metallic bond | 300 W Xe lamp (λ > 420 nm) | 3 mL H2O + TEOA + 70 kPa CO2 | C2H4: 335.67 (93.87%) | 107 | |
| Cu1.95S1−x | Dual Cu and Vs | 300 W Xe lamp AM 1.5 cut-off filter | 1 atm CO2 | CH4: 12.42 | 108 | |
| MAS synergizing with nanoparticles | CA/Ni1CuNP N–C | Cu nanoparticles; Ni single-atom sites; asymmetric Ni–N4 sites | 300 W Xe lamp (λ > 420 nm) | 95 mL H2O + 5 mL TEOA + 100 kPa CO2 | CH4: 35.245, CO: 32.067, C2H6: 25.328 | 109 |
| Co1Ag(1+n)-PCN | Co–N6–P bonds, Ag–N2C2 SA sites, Ag NPs | 300 W Xe lamp | 6 mL CH3CN + 4 mL H2O + 80 kPa CO2 | CO: 46.82 (70.1%) | 110 | |
| Cu single atoms and nanoclusters supported on defective TiO2 | Cu SAs, Cu NCs | 300 W Xe lamp | 100 μL H2O + CO2 | CH4: 19.63 (98%) | 111 | |
| single Pd atoms and Pd nanoparticles on graphitic carbon nitride | Pd1 sites; PdNPs sites | Xe lamp | 10 mL H2O + 1 atm CO2 | CH4: 20.3 (97.8%) | 112 | |
| MAS within heterostructure | g-C3N4/CoCo-LDH | Co–N2 bonds | 300 W Xe lamp | 3 mL H2O + 1 atm CO2 | CO: 71.39 | 58 |
| CuO/Cu2V2O7 | Two metal atomic sites of Cu and V | 300 W Xe lamp | 20 mL TEOA + 80 mL H2O + 1.01 bar CO2 | CO: 118, C2H4: 29.57 | 113 | |
| 2D/2D Ni-doped CsPbBr3/Bi3O4Br Z-scheme heterojunction | Ni2+ | 300 W Xe lamp | 2 mL H2O + 101 kPa CO2 | CO: 96.89 (98.2%) | 114 | |
| Co-TCPP/Bi3O4Br | Bi–O bridge bond; Co atoms | 300 W Xe lamp | 50 mL H2O + 0.08 MPa CO2 | CO: 71.3 | 115 | |
| Cu-SAEB | N–Cu1–S single-atom electron bridge | 300 W Xe lamp | 0.1 mL H2O + CO2 | CO: 236.0, O2: 120.1 | 116 | |
| In2O3/CdSe-diethylenetriamine | In–O–Cd bonds; Vo | 300 W Xe lamp | 120 mg NaHCO3 + 0.5 mL H2SO4 | CO: 70.08, CH4: 27.92 | 117 | |
| 2D/0D g-C3N4/Cu2SnS3 | Cu–C and Cu–N dual chemical bond; Cu sites | 100 W Xe lamp AM 1.5 filter | H2O + CO2 | CO: 18.2 | 118 | |
| NiAl–Fe-TCPP | Fe; Ni | 300 W Xe lamp | 0.005 mmol [Ru(byp)3]Cl2·6H2O + 10 mL mixed solution + 1.8 bar CO2MeCN/H2O/TEOA (6 mL/2 mL/2 mL) | C2H4: 2470 | 119 | |
| r-In2O3/InP | O–In–P polarized sites | 300 W Xe lamp (λ > 420 nm) | 100 mL H2O + 80 kPa CO2 | CH3COOH: 9.67 (96.1%) | 120 | |
| CdS:Dy3+/g-C3N4 | Dy3+ single atom | 300 W Xe lamp (λ > 400 nm) | 4 mL H2O + CO2 | CH4: 8.06 | 121 | |
| MAS as cocatalysts | Bi NCs/Bi2O3 | Bi0 of Bi NCs | 300 W Xe lamp (λ > 420 nm) | 1 mL H2O + 0.08 MPa CO2 | CH4: 7.45 (94.8%) | 122 |
| CsCuCl3/Cu NCs | Cu NCs | 150 W Xe lamp AM 1.5 cut-off filter | 4 mL ethyl acetate (EA) + 1 mL propan-2-ol (IPA) + CO2 | CH4: 7.2 (92.7%) | 123 | |
| Ultrathin Bi12O17Cl2 nanosheets | Vo; Bi clusters | 300 W Xe lamp | 50 mL H2O + 0.08 MPa CO2 | CO: 64.3 | 124 | |
| Bi–BiOCl plasmonic nanoparticles decorated TiO2 nanosheets | Bi | 300 W Xe lamp AM 1.5 cut-off filter | 5 mL 0.2 M NaHCO3 + CO2 | CH3OH: 235.78 (≈90%) | 125 | |
| Au/TiO2/W18O49 | Au–O–Ti, W–O–Ti | 300 W Xe lamp (λ > 420 nm) | 0.1 mL H2O + CO2 | CH4: 35.55, CO: 2.57, CH4 (93.3%) | 126 | |
| PtAg-2/HNb3O8 | Pt–Ag alloy (7:43 Pt–Ag molar ratio) |
300 W Xe lamp (320 nm < λ < 780 nm) | 2 mL H2O + 0.09 MPa CO2 | CH4: 93.6 (74.3%) | 127 | |
| PtCu/TiO2 | Pt/Cu alloy (0.4:0.6) |
300 W Xe lamp (300 nm < λ < 400 nm) | Water vapor + CO2 | CH4: 100% | 128 | |
| Cu–Ag/TiO2 | Cu–Ag alloy (0.8:0.2) |
300 W Xe lamp AM 1.5 cut-off filter | 150 mL H2O + 90 kPa CO2 | C2H4: 1110.6 | 129 | |
| AuIr with InGaN nanowires on silicon | Au3Ir1 alloy (0.44:0.56) |
300 W Xe lamp | 30 mL H2O + 2 atm CO2 | C2H6: 58800, CH4: 125400, H2: 735600, CO: 127800 |
130 | |
| Au/TZO | Au nanoparticles | 300 W Xe lamp AM 1.5 G filter | 20 mL mixed solution + 1 atm CO2 MeCN/H2O/TEOA (16 mL/2 mL/2 mL) | H2: 271.6, CO: 260.6, C2H4: 6.80, C2H6: 4.05 | 131 | |
| Bi nanoparticles grown on the Bi2MoO6 with Vo | Bi nanoparticles Vo | Xe lamp | 0.42 g NaHCO3 + 30 mL H2O + CO2 | C2H5OH: 17.93 (92%) | 132 |
3.1 Single MAS
The incorporation of individual MAS into the catalyst backbone from an atomic level perspective maximizes atomic utilisation efficiency and optimizes the performance of photocatalytic CO2 conversion. The bipyridine-based polyimide polymer (Bpy-PDI) anchoring single Ni site achieved diluted CO2 photoreduction with high generation activity for CO2-to-CO in 0.1 atm CO2 pressure.76 The obviously larger adsorption energy of CO2 for the Ni site (1.44 eV) compared to that of Bpy-PDI (0.19 eV) indicated a much stronger adsorption affinity of the Ni site towards CO2. Moreover, the free energy of COOH* generation (the rate-determining step) on the Ni sites (−0.07 eV) was obviously smaller than that of Bpy-PDI (1.69 eV), revealing the contribution of the dispersed Ni atom toward the energy barrier decline. Cu single atoms on the nitrogen-doped carbon anchored on TiO2 with the anatase–rutile mixed phase exhibited 100% CO selectivity in the photocatalytic CO2 reduction.77 Cu single atoms became the adsorption and activation sites for CO2, where the strong hybridization of the Cu 3d and CO2-O 2p orbitals facilitated the transfer of electrons from the Cu single atoms to CO2, effectively optimizing the rate-limiting step (CO2* → COOH*). Moreover, single MAS also improved the selectivity of the liquid product of the photocatalytic CO2 reduction. The atomically dispersed rhenium (Re) in In2O3 changed the product from CO on pure In2O3 to CH3OH on 2Re-In2O3.78 DFT calculations disclosed that the Re site promoted the H2O dissociation to form sufficient H atoms for CO2 reduction. Critically, CO preferred to be hydrogenated into CHO instead of desorption due to the strong binding between CO and Re1-In2O3(111). Through multi-step hydrogenation, CHO was eventually converted to CH3OH with high selectivity.Photocatalytic CO2 conversion mainly involves CO bond cleavage and C–H bond formation, accompanied by the challenging C–C coupling toward the generation of C2+ products. The MAS could reduce electrostatic repulsion between the C1 intermediates, promote C–C coupling, and thus lead to the highly selective formation of the C2+ product. The photogenerated electron transition from TiO2 to atomically dispersed Cu atoms rearranged the energy levels of the Cu 3d orbitals.79 Consequently, the initial four-coordinated Cu1–O4 was distorted into a Cu1–O2+1 structure (twofold normal Cu–O coordination and one stretched Cu–O coordination), which could be reversibly recovered after removing the synergistic light field. The photoinduced metastable intermediate of Cu1–O2+1 delivered an C2H4 yield rate of 60.4 μmol gcat−1 h−1. The energy barrier of the first protonation step from CO2 to *COOH showed an obvious decline over Cu1–O2+1, indicating the stronger activation capability of the metastable asymmetrical structure to reactants than that of Cu1–O4. *CO molecules adsorbed more strongly on the Cu1–O2+1 (−1.49 eV) surface than on the Cu1–O4 (−0.44 eV) surface, leading to subsequent hydrogenation rather than desorption. It was noteworthy that, in contrast to Cu1–O4, Cu1–O2+1 promoted C–C coupling and further formation of C–C bonds, thus facilitating the generation of C2H4. Furthermore, N,S-codoped Fe-MOF MIL-88B with a well-defined bipyramidal hexagonal prism shape was designed.80 The synergistic effect of the Fe–N coordinated sites and reasonable defects from uncoordinated S increased the electron density disorder around Fe, accelerated the migration of photogenerated carriers, benefited electron storage, and effectively promoted the formation of C–C coupling intermediates for C2H4. For liquid products, the carbon nitride-supported Cu single-atom catalyst with a defective low-coordination Cu–N2 motif (Cu–N2–V) exhibited superior photocatalytic activity for CO2 reduction to ethanol relative to Cu–N3 and Cu–N4.81 In particular, Cu in Cu–N2–V existed in both Cu+ and Cu2+ valence states. On the one hand, the Cu+ sites benefited CO2 activation. On the other hand, the coexistence of Cu+/Cu2+ sites contributed to the strong adsorption of *CO and subsequent *CO–*CO dimerization. Finally, ethanol was ultimately produced from *CO–*CO, which underwent a series of hydrogenation processes.
The d-band center tuning strategy influences the carrier transportation and the adsorption of the reactant and intermediate to promote photocatalytic CO2 conversion, which can be achieved by incorporating MAS. Bulk doping of single Ni atoms in WO2.72 (B-Ni1/WO2.72) displayed superior solar-driven CO2 reduction performance to surface anchoring of single Ni atoms on WO2.72 (S-Ni1/WO2.72).82 The introduction of Ni atoms led to an upward shift of the d-band center of W atoms in the WO2.72 host structure due to the overlapping orbital hybridization. Critically, the d-band center of W in B-Ni1/WO2.72 shifted upward to a greater extent relative to S-Ni1/WO2.72. Therefore, the Ni atoms in B-Ni1/WO2.72 exhibited stronger electronic interactions with the WO2.72 host, facilitating the formation of charge-transfer channels that enabled the rapid transfer of photogenerated electrons to the surface Ni atoms. Moreover, the free energies of *CO2, *COOH, and *CO intermediates of B-Ni1/WO2.72 were integrally decreased compared to those on S-Ni1/WO2.72, benefiting the conversion of CO2 to CO. Analogously, the decoration of isolated Mn atoms over the multi-edged TiO2 nano-pompons shifted the d-band center upwards and pushed the antibonding orbital closer to the Fermi level, thus facilitating CO2 adsorption.83 The Mn site acted as an active center for CO2 activation, and significantly reduced the formation energy barriers of *COOH to accelerate the decisive step of the reaction.
Besides in-plane regulation, MAS can realize interlayer-spacing adjustment, which may significantly influence the carrier transport of the bulk-catalyst, thereby affecting CO2 photoreduction. Single-atom In-bonded N-atom (Inδ+-N4) in the (002) crystal planes of g-C3N4 reduced the (002) interplanar spacing of g-C3N4, benefiting the separation of bulk carriers.84 More charges were transferred to the adsorbed CO2 molecule from the Inδ+-N4 active center (0.116 eV) compared to the bare C3N4 site (0.006 eV), which was favorable for CO2 adsorption. Moreover, the Inδ+-N4-led CO2 hydrogenation to *COOH was downhill by −0.114 eV and evidently surpassed the uphill step by 2.09 eV on the bare C3N4, which optimized the reaction path.
3.2 Dual MAS
The precise construction of photocatalysts with dual MAS to achieve the simultaneous fostering of light absorption and catalytic activity is a formidable challenge. The bifunctional LaNi sites within COF were synthesized by electrostatically driven self-assembly approach.91 The La–Ni dual MAS accelerated the dynamic behavior of the photogenerated charge carriers, comprising photoelectron transfer from La-Phen to the COF-5 colloid and subsequent electron injection into Ni-Phen for the CO2 reduction process. In addition, the bimetallic LaNi coordination facilitated the generation of abundant *COOH groups, and effectively decreased the activation barrier of CO2 transformation. Moreover, the CO desorption was thermodynamically preferred over CHO* formation, with an energy barrier of 0.72 eV versus 1.02 eV, resulting in high CO selectivity.Precise control of the formation of Metal1⋯CO⋯Metal2 (M1⋯CO⋯M2) intermediates at the photocatalyst interface is one of the critical steps in the formation of hydrocarbons. This is due to the fact that the energy required for simultaneous cleavage of the M1⋯O and M2⋯C bonds is much greater than that required for C–O bond breaking (Fig. 9a).92 Ag2Cu2O3 nanowires with abundant Cu–Ag Lewis acid–base dual sites on the preferentially exposed (110) surface were utilized as a model catalyst to achieve 100% selectivity in the photogeneration of CH4 from CO2.93 The Cu⋯Ag Lewis acid–base dual sites on Ag2Cu2O3(110) regulated the M1⋯CO⋯M2 intermediate formation, converting CO2 into hydrocarbons. The rate-determining step with a corresponding Gibbs free energy (ΔG(CHO*)) of 0.75 eV led to greater feasibility in CO* forming CHO* than desorbing from the catalyst surface (1.16 eV, Fig. 9b). Similarly, the highly stable Cu–C–O–In intermediate at the Cu–In dual sites of the sulfur-deficient CuIn5S8 converted the endoergic protonation step to an exoergic reaction process, changing the reaction pathway to form CH4 instead of CO.92 Furthermore, the VO-regulated In–Ti dual sites enabled the formation of a stable adsorption conformation of the In–C–O–Ti intermediate, leading to the highly selective reduction of CO2 to CH4.94
 | ||
| Fig. 9 (a) CO2 photoreduction into fuels such as CH4 and CO through the use of dual-metal-site catalytic systems (M represents the metal site, ‘H+ + e−’ refers to the proton coupled electron transfer process and ‘−H2O’ means the desorption of H2O molecules after the intermediates react with the proton–electron pair). This figure has been reproduced from ref. 92 with permission from Springer Nature, copyright 2019. (b) The rate-determined step of photoreduction of CO2 into CH4 over Ag2Cu2O3 nanowires. This figure has been reproduced from ref. 93 with permission from Wiley-VCH, copyright 2023. | ||
Catalysts containing dual MAS regulates the electron distribution by creating asymmetric atomic configurations, which significantly affects the photocatalytic performance, especially for multi-electron CO2 reduction. The mechanism of reverse electron transfer over Au and Co bimetallic atom catalysts was reported.95 Electrons were delocalized from Au and accumulated around the Co atoms, resulting in the electron-rich Co atoms adsorbing/activating CO2 molecules readily, which significantly promoted photocatalytic CO2 reduction. In this regard, the Au/Co double single-atom loaded CdS increased the yield of CO and CH4 by nearly 2800% and 700%, respectively, compared to CdS alone. In addition, the disparate electron distributions and valence states of two distinct metal atoms could endow significantly different charge distributions of the neighboring C1 intermediates to effectively suppress the electrostatic repulsion. The asymmetric Zn–O–Ge triatomic sites in the Vo-rich Zn2GeO4 nanobelts induced distinct charge distributions of neighboring C1 intermediates, which facilitated the C–C coupling with a high CO2-to-CH3COOH conversion rate of 29.95%.96 Under similar mechanisms, incorporating redox-active Co2+/Ni2+ cations (TM) into layered lead iodide hybrids (TJU-39(Pb)) achieved efficient photocatalytic CO2-to-C2H5OH conversion (yield of 24.9–31.4 μmol g−1 h−1, selectivity exceeding 90%).102 It was experimentally demonstrated that the interlayer TMs were delocalized to the lead iodide layers to construct TM–O–Pd sites with substantial asymmetric charge distribution, which reduced the reaction barrier for C–C coupling. Specifically, the two-electron reduction of CO2 to CO* occurred simultaneously at the charge-enriched Pb2+ sites and the Ni2+ site, which subsequently underwent C–C coupling to form OC–CO* intermediates. The OC–CO* intermediate coupled multiple e−/H+ pairs, combining the dehydration process to finally yield C2H5OH.
From an overall reaction perspective, the CO2 photoreduction contains oxidation and reduction half-reactions, involving multiple proton-coupled electron transfer processes. The MAS construction not only enhances CO2 reduction, but also influences the corresponding oxidation half-reaction. The existence of an internal electric field pointing from the Ti cluster to the Cu cluster in MCOF-Ti6Cu3 facilitated carrier separation, leading to electron and hole transfer to the Cu cluster and Ti cluster, respectively.90 Consequentially, electrons reaching the Cu cluster drove the reductive reactions, and the oxidative reaction proceeded at the Ti cluster. Analogously, two compatible active centers of Mn and Co were loaded onto carbon nitride (Mn1Co1/CN).17 The active center of Mn promoted H2O oxidation by accumulating photogenerated holes, along with Co-facilitated CO2 activation by increasing the bond length and bond angle of CO2 molecules.
3.3 MAS combined with vacancies
Vacancies exhibit great influence on the properties of MAS. Vo-containing Co3O4 hollow nanoparticles loaded on a macroporous N-doped carbon framework realized the photoreduction of low-concentration CO2 to CO.133 Vo distorted the nearby Co–O bonds, broke the local structural symmetry of the Co–O–Co sites, and thus caused an asymmetric distribution of charge density compared with pristine Co3O4. This asymmetrical active site with a polarized electronic structure endowed a stronger electrostatic interaction between the CO2 molecule and Vo, which switched the CO2 configuration from a single-site linear model to a multiple-sites bending one, therefore facilitating CO2 activation. Moreover, the hydrogenation of adsorbed *CO2 proceeded smoothly on the Vo-Co3O4 surface, and the adsorption capacity of *COOH was obviously stronger than that of Co3O4. Mn dopants and Vo were engineered in Zn2GeO4 nanorods (denoted as Mn-ZGO-Vo), which exhibited prominent photocatalytic CO2 reduction performance.105 The introduction of Vo in Mn-ZGO-Vo caused the charge accumulation on the Mn atoms near Vo, certainly favoring the electron-rich Mn sites for CO2 molecule capture. The lattice strain and ligand effects caused by Mn doping and Vo engineering elevated the d-band centers, endowing less filling of the antibonding states and hence stronger binding strength of CO2. Moreover, the formation of COOH* on the Mn-ZGO-Vo slab changed from endothermic to exothermic, and the rate-limiting step was tuned to CO* formation with a lowest energy barrier among ZGO and Mn-ZGO. Precisely constructed Cu single-atom centers and two-coordinated N vacancies as dual active sites on carbon nitride (Cu1/N2CV-CN) achieved a high carbon-based selectivity of 98.50% for CO production.106 The atomic Cu active sites promoted the chemisorption of CO2 by changing the electronic structure of the surface. The N2C vacancy became the active site for the adsorption of H2O, which accelerated the dissociation of H2O by reducing the dissociation energy barrier of H2O from 2.59 eV to 1.68 eV. Moreover, the decrease in the rate-limiting step (the formation of COOH* species) by the dual active sites in Cu1/N2CV-CN led to a decent CO production rate of 11.12 μmol g−1 h−1.The geometry of MAS regulated by vacancies ultimately affects the catalytic performance to generate C2+. Ultra-thin CuGaS2/Ga2S3 containing Vs realized the selectivity of C2H4, reaching ≈93.87% with the yield of ≈335.67 μmol g−1 h−1.107 Vs induced a highly delocalized electron distribution in the original region of S atoms, forming a Cu–Ga metallic bond. The upshift of the d-band center of Cu–Ga ions induced by Vs could not only enforce the adsorption ability for the intermediates of *CHOH*CO to initiate C–C coupling, but also accumulate electrons to drive the kinetic process of photocatalytic CO2 reduction.
3.4 MAS synergizing with nanoparticles
The synergistic effect of MAS and nanoparticle accelerates the complex activation and hydrogenation process of photocatalysis. Introducing such a complex structure for simultaneous CO2 reduction and H2O dissociation is still a formidable challenge, and its underlying mechanism remains to be elucidated. The nitrogen-doped carbon anchoring with Cu nanoparticles and Ni single-atom sites (Ni1CuNP N–C) were hybridized with CoAl-LDH (CA) for photocatalytic CO2 reduction (CA/Ni1CuNP N–C).109 The photogenerated electrons of CoAl-LDH were transferred to N–C in the presence of the internal potential, and eventually accumulated on dispersed Ni single-atom sites. Meanwhile, the loading of Cu nanoparticles could significantly facilitate this charge transfer. The Ni center showed a high charge density, which facilitated reactant/intermediate adsorption and promoted catalytic reactions with multi-electron participation. Furthermore, the charge density around the electron-rich center Ni–N4 showed an asymmetric distribution in the presence of Cu nanoparticles. This asymmetric active site was favorable for coupling adjacent *CO, thus promoting the generation of C2 products. A cooperative catalyst containing Ag nanoparticles with adjacent atomic cobalt–silver dual-metal sites on P-doped carbon nitride (Co1Ag(1+n)-PCN) was reported for photocatalytic CO2 reduction.110 The asymmetric Co–N6–P sites coupled with Ag–N2C2 sites could serve as the charge transfer channel to facilitate the migration of electrons to the surface reaction sites, while Ag nanoparticles acted as the electron acceptor to enrich and separate photogenerated electrons. Moreover, the synergistic function of Ag nanoparticles with adjacent atomically dispersed Ag–N2C2 and Co–N6–P single-atom sites promoted the adsorption of CO2 molecules onto the photocatalyst surface, and facilitated the formation of CO2* and COOH*.3.5 MAS within a heterostructure
The construction of heterogeneous structures enhances light absorption and provides sufficient reaction sites.44 In addition, the heterostructure can realize the spatial separation of the reduction and oxidation reaction sites, improving the utilization rate of the photogenerated carrier and preserving high-energy electrons and holes. MAS is conducive to the tight connection of heterogeneous structures and promotes the directional carrier transportation. The presence of Vo exposed Bi atoms on the Bi3O4Br surface, which formed an interfacial Bi–O bond by grafting the terminal O of the cobalt porphyrin (Co-TCPP).115 The Bi–O bond, as a charge-transfer bridge, promoted the extraction and transfer of photogenerated electrons from the external [Bi3O4] layers to Co-TCPP. This contributed an effective space charge separation and the slow recombination process on the μs–ms time scale of CoTCPP. MoS2 decorated with Cu species (Cu1/MS) was coated on the surface of MIL-125-NH2 (MIL), which constructed a N–Cu1–S single-atom electron bridge (denoted as Cu-SAEB).116 The Cu-SAEB achieved a Z-scheme charge-transfer mode between Cu1/MS and MIL, thus prolonging the lifetimes of carriers with a strong redox potential. Benefiting from the strengthened contact interface of Cu-SAEB, the highly active and stable CO2 reduction performance was achieved, with CO and O2 formation rates of 236.0 and 120.1 μmol g−1 h−1, respectively. Similarly, an In–O–Cd bond-modulated S-scheme heterojunction of In2O3/CdSe-diethylenetriamine accelerated the photogenerated electron transfer.117 Combined with the electron capture effect of Vo in In2O3, the In–Vo–In–O–Cd structural units at the interface led to the extraction of electrons and the rapid transfer to the surface-active sites, improving the electronic coupling of CO2.The interface of the heterostructure could natively provide MAS for photocatalytic reactions. For example, the unique Cu–C and Cu–N dual chemical bond at the interface of the fabricated g-C3N4/Cu2SnS3 nanocomposite led to a superior CO formation rate from CO2 photoreduction.118 The Z-scheme carrier conduction was highly activated due to the interfacial nitrogen vacancies via the Cu–C and Cu–N bonding for CO2 reduction on the conduction band (CB) of Cu2SnS3 and water oxidation on the VB of g-C3N4. The more negative overall free energy of Cu sites relative to Sn and S implied that the CO2 conversion reaction occurred mainly via effective dual bonds between Cu sites and the C and N elements in g-C3N4.118
Heterogeneous structures enriched with MAS can generate C2+ products. A novel supramolecular assembly of NiAl–Fe-TCPP obtained by intercalating iron porphyrin (Fe-TCPP) into NiAl-layered double hydroxide (NiAl-LDH) exhibited superior catalytic performance of CO2 photoreduction to derive C2H4 with high selectivity up to 93.4%.119 The strong host–guest interaction between LDH and Fe-TCPP led to the electron transfer from NiAl-LDH to Fe-TCPP. According to the ex/in situ XAS, the valence of Fe decreased along with the increase of Ni valence compared with that of Fe-TCPP and LDH, respectively. The low valence of Fe in NiAl–Fe-TCPP facilitated the CO* hydrogenation and coupling with CHO* to form COCHO*. Particularly, the CO2 activation to CO was accomplished on NiAl-LDH, and then spilled to Fe-TCPP and coupled to generate COCHO*, which was further hydrogenated to produce C2H4. The O–In–P polarized sites at the r-In2O3/InP interface promoted C–C coupling with a productivity of 96.7 μmol g−1 and selectivity >96% for CO2 photoreduction to CH3COOH.120 The interfacial In atoms accumulated electrons and transferred electrons to CO2, thus promoting CO2 adsorption and activation. The quite different carbon Bader charges of OCCO* on r-In2O3/InP caused by the formation of rich O–In–P reactive sites at the interface alleviated dipole–dipole repulsion and promoted C–C coupling (Fig. 10a–c). Ultimately, the C–C coupling energy barrier of r-In2O3/InP (0.5461 eV) was lower than that of pure In2O3 (0.9446 eV), suggesting that the formation of heterostructures by modifying In2O3 with P favored the formation of C2+ products (Fig. 10d).
 | ||
| Fig. 10 Calculated electron density difference diagrams and the Bader charge values of (a) r-In2O3/InP, (b) In2O3, and (c) p-In2O3/InP with OCCO* absorbed. (d) Free energy diagrams for the reduction of CO2 to CH3COOH over r-In2O3/InP, In2O3, and p-In2O3/InP. These figures have been reproduced from ref. 120 with permission from American Chemical Society, copyright 2023. | ||
3.6 MAS as cocatalysts
The cocatalyst plays a crucial role in semiconductor-based photocatalysis, which is conducive to the migration of electrons or holes, and improves the utilization efficiency of the photogenerated carrier. Moreover, cocatalysts can reduce the activation energy and overpotential of the CO2 conversion to speed up the surface reaction.44 MAS used as cocatalysts can effectively improve photocatalytic performance. Bi nanoclusters (NCs) were prepared in situ on Bi2O3 through dehalogenation of Bi3O4Br by regulating the pH value in the hydrothermal process.122 The construction of Bi sites on the basis of Bi2O3 could promote photogenerated charge transfer through the Schottky junction. Moreover, the change of the valence state of Bi regulated the type of active sites from O of the original Bi2O3 to Bi0 of the Bi NCs in Bi NCs/Bi2O3, thus regulating the path of CO2 photoreduction. Thermodynamically, the *CO intermediate on the Bi site tended to hydrogenate to *CHO species, accompanied by a subsequent multi-step hydrogenation process that selectively generated CH4 on Bi NCs/Bi2O3. The *CO desorption to form CO over Bi2O3 was spontaneous and exothermic, whereas the hydrogenation of *CO to form the *CHO species required overcoming a large energy barrier (2.29 eV), leading to CO being the only reduction product of Bi2O3. Similarly, the CsCuCl3/Cu heterojunction synthesized via a simple acid-etching solution process exhibited high CH4 selectivity.123 The accumulating of charges at the contact interface between CsCuCl3 and Cu nanocrystals formed a Schottky contact, facilitating the extraction of photoelectrons from CsCuCl3 to Cu nanocrystals, thereby realizing the effective separation of photogenerated electron–hole pairs. Moreover, the Cu nanocrystals accelerated the photocatalytic CO2 reduction toward CH4via manipulating the adsorption and activation of CO2 and stabilizing the reaction intermediates.The surface plasmon resonance of MAS plays a crucial role in the collection and conversion of solar energy by the strong local fields. The coexistences of Vo and Bi clusters generated in situ on ultrathin Bi12O17Cl2 nanosheets contributed to the high efficiency of the CO2-to-CO conversion.124 The Bi clusters exhibited a plasmon effect that extended the light absorption and enabled more sunlight harvest. Furthermore, Bi clusters acted as hole trapping centres in synergy with Vo as electron trapping sites, leading to the spatial separation of photogenerated electron–hole pairs. As another example, the attachment of non-noble plasmonic Bi particles with BiOCl shells to self-assembled TiO2 nanosheets created a transformative hybrid plasmonic nanostructure. Bi stabilized by the BiOCl shell generated robust localized surface plasmon resonances, inducing a local field enhancement of 7–9 times, thus enabling the efficient and selective CO2-to-methanol conversion at the TiO2–BiOCl heterointerfaces.125 In addition, it is worth mentioning that the localized surface plasmon resonance effect of Bi nanoparticles could significantly improve the rate and selectivity of the C2H5OH generation.132 The high localized electron density and abundance of hot electrons in the active site drove the multi-electron reduction reaction, favoring the generation of C2+ products.
Plasmon resonance-mediated photocatalysis on precious metal surfaces (mainly Ag and Au) shows great potential for solar energy harvesting and conversion. For instance, plasmonic Au mediated the S-scheme charge transfer, and generated additional energetic hot electrons and holes to inject to the CB of ZnIn2S4 and VB of CuS, respectively, enabling a more thorough separation of carriers for CO2 reduction and H2O oxidation.134 Moreover, a new idea of plasmonic active “hot spot”-confined photocatalysis was proposed to improve the photocatalytic CO2 conversion. Specifically, tiny gaps (<10 nm) between the plasma nanostructures promoted plasma coupling between the nanostructures, resulting in the formation of classical “hot spots”. The Au/TiO2/W18O49 sandwich-like substructures with the short distance (<10 nm) between Au and adjacent W18O49 induced an intense plasmon-coupling to form the active “hot spots” in the substructures.126 These active “hot spots” could gather the incident light to enhance “hot electron” generation and migration, and capture protons and *CO through the dual-hetero-active-sites (Au–O–Ti and W–O–Ti) at the Au/TiO2/W18O49 interface, thus accelerating the protonation of *CO intermediates to derive CH4.
Alloys as MAS enhance light absorption, inhibit charge recombination, and adsorb and activate reactants, thereby improving the selectivity of specific products. The PtAg alloy over HNb3O8 nanosheets trapped the electrons and improved the charge separation efficiency, mitigating the kinetical challenge of the eight-electron transfer process for CH4.127 In addition, the PtAg alloy acted as synergistic sites to reduce CO2 to CO intermediates at the Ag site, which were then spilled over or sequentially adsorbed at neighboring Pt sites for further hydrogenation to CH4. As another example, PtCu alloys with appropriate Pt/Cu ratios were deposited onto TiO2 nanocrystals, which significantly improved CH4 production with 100% selectivity.128 The PtCu alloy effectively facilitated the separation/transfer of photogenerated charges, benefited the adsorption of CO2, and promoted the formation and activation of intermediates (CO2−, *COOH, *CO, and *CHO). The synergistic effect of PtCu lowered the activation energy barriers of *CO2 and *CHO, inhibited the desorption of *CO, and ultimately optimized the efficiency and selectivity of CH4.
The alloy MAS promotes the C–C coupling and desorption of *C2 intermediates from the catalyst surface. Loading Cu–Ag alloy sub-nanoclusters (ASNCs) on TiO2 for CO2 photoreduction produced C2H4 with a record-high formation rate (1110.6 ± 82.5 μmol g−1 h−1).129 The interaction between Cu and Ag in the Cu–Ag ASNCs promoted the C–C coupling of CH2* at the Cu active site, which led to the spontaneous formation of *C2H4 from CH2* on Cu–Ag alloy/TiO2. In addition, the desorption energy of *C2H4 in the Cu–Ag alloy/TiO2 was lower than that in Ag/TiO2. This suggested that the interaction between Cu and Ag promoted the desorption of *C2H4, and thus the selective and efficient production of C2H4. Ag, in conjunction with Ir, achieved C–C coupling by inserting CO2 into –CH3, facilitating the formation of C2+ during CO2 photoreduction.130 Specifically, the assembly of AuIr with InGaN nanowires on silicon achieved a C2H6 activity of 58.8 mmol g−1 h−1 with a turnover number of 54595 over 60 h. The Ir sites in Au–Ir alloys increased CO2 reduction activity by lowering the reaction energy of key elementary steps (for example, CO2 to *COOH on pure Au and *CO to *CHO on Au3Ir1 alloy) and steered the selectivity from the dominant HER to C–C coupling. The insertion of CO2 into *CH3 toward *CH3COO exhibited the lowest reaction energy compared with other C–C coupling forms (CH + *CH → *C2H2, *CH2 + *CH2 → *C2H4, *CH3 + *CH3 → C2H6(g) and CO2 insertion into *CH3 to *CH3COO), revealing the possible mechanism of C–C coupling for C2H6 synthesis.
4 Identification the role of MAS in photocatalysis
With the growing development in nanoscale characterization, a variety of techniques have emerged to assess the role played by MAS in photocatalysis. Specifically, microscopic visual characterization combined with spectroscopic measurements can help to reveal the structure and the coordination environments of the sample, along with the in situ observation of intermediary transformations to profoundly reveal the reaction mechanisms. In addition, theoretical calculations simulating the role of MAS in photocatalysis reveal the physical and chemical properties exhibited by MAS during CO2 conversion.1354.1 High-resolution aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM)
In the structural characterization of single-atom catalysts, high-resolution AC-HAADF-STEM demonstrates paramount importance in elucidating the distribution of the MAS and its geometry. Isolated Co single-atom-modified 2D monolayer ZIS nanosheet composites (Co-sZIS) possessed dual active centers of Co and Zn.136 AC-HAADF-STEM was used to characterize the dispersion and configuration of the atoms involved, and to assist in determining the atomic structure of Co-sZIS. Fig. 11a clearly shows the distribution of isolated Co atoms in sZIS, with the yellow circles marking the atomic spots that were brighter compared to the surrounding support. Fig. 11b and c reflects the fact that the corresponding atoms exhibited different contrasts, with one of the largest peaks corresponding to the overlapping intensity spectra of an In atom and the individual Co atom. In addition, AC-HAADF-STEM was used to further examine the detailed distribution state of the MAS. The bright spots in Fig. 11d and e confirmed the isolated dispersion of Ni or Fe atoms on the carbon skeleton of the Ni–N–C and Fe–N–C single-atom catalysts, respectively, and no atom pairs were observed. In contrast, the AC-HAADF-STEM images of the (Ni, Fe) dual-single-atom catalysts (Fig. 11f) clearly showed most of the bright spots appearing in pairs, marked by yellow rectangles, confirming the formation of Ni/Fe dual-atom pairs with an average distance of about 4.1 Å.137 | ||
| Fig. 11 (a) Aberration-corrected HAADF STEM image of the Co-sZIS; single-atomic Co sites are highlighted by yellow circles. (b) Magnified view of the white box in (a) (inset is the simulated atom distribution of Co-sZIS, and the green, gray, and red spheres represent the Zn, In, and Co atoms, respectively). (c) Intensity profile corresponding to the dark cyan arrow in (b). These figures have been reproduced from ref. 136 with permission from American Chemical Society, copyright 2023. Aberration-corrected HAADF-STEM images of (d) Ni–N–C, (e) Fe–N–C, and (f) (Ni, Fe)–N–C. These figures have been reproduced from ref. 137 with permission from Wiley-VCH, copyright 2023. | ||
4.2 X-ray photoelectron spectroscopy (XPS)
XPS is considered to be an effective method to analyze the surface properties of the catalysts, reflecting the chemical bonding composition and the migration paths of carriers based on peak shifts. Porous covalent triazine frameworks (CTFs) were combined with CdS nanorods to obtain CdS@CTF-HUST-1 heterojunction photocatalysts with core–shell structures for CO2-to-CO conversion.138 Essentially, the full XPS spectra showed the coexistence of Cd, S, C and N elements in the CdS@CTF-HUST-1 with an optimal CdS content, referred to as Cd(0.9)@CTF. In situ XPS was conducive to revealing S-scheme charge transfer mechanisms of CdS@CTF-HUST-1. As shown in Fig. 12, the Cd 3d and S 2p peaks of the CdS(0.9)@CTF sample showed a positive displacement with respect to CdS, while the C 1s and N 1s peaks of CdS(0.9)@CTF exhibited a negative shift relative to CTF-HUST-1 under dark conditions. Such shifts indicated the interfacial transfer pathway of electrons from CdS to CTF-HUST-1. Under light illumination, the binding energies of Cd 3d and S 2p of the CdS(0.9)@CTF were negatively shifted compared to those in darkness, while the binding energies of C 1s and N 1s underwent positive displacement. These peak shifts confirmed that the internal electric field could drive the transfer of photogenerated electrons from CTF-HUST-1 to CdS under light irradiation. Furthermore, in situ irradiated XPS validated the Z-scheme charge transfer path of Ni-doped CsPbBr3/Bi3O4Br with an internal electric field directing from Ni-doped CsPbBr3 to Bi3O4Br.114 The binding energies of Cs, Pb and Br of CsPbBr3 were all positively shifted upon Ni doping, suggesting that the introduced Ni sites interacted with the surrounding atoms to reduce the electron density around the Cs, Pb and Br atoms. After composing the heterostructure, the Cs 3d and Pb 4f of Ni-doped CsPbBr3/Bi3O4Br were shifted to higher binding energy by 0.3 eV with respect to Ni-doped CsPbBr3, whereas Bi 4f and O 1s underwent a 0.2 eV shift to lower binding energy with respect to Bi3O4Br. In addition, the distance between the Br 3d5/2 and Br 3d3/2 peaks increased, indicating an enhanced spin–orbit splitting function of Br 3d. These results revealed changes in the charge loss and accumulation states of Ni-doped CsPbBr3 and Bi3O4Br in the heterostructures, demonstrating the strong interfacial interaction between Bi3O4Br and Ni-doped CsPbBr3. Upon illumination, Cs 3d and Pb 4f moved toward lower binding energies relative to the characteristic peaks in the dark environment, while Bi 4f and O 1s exhibited significant positive shifts. These results suggested that the light-induced electrons follow a Z-scheme path from Bi3O4Br to Ni-doped CsPbBr3. | ||
| Fig. 12 High-resolution XPS spectra of (a) Cd 3d, (b) S 2p, (c) C 1s, and (d) N 1s of CdS, CTF-HUST-1, and CdS(0.9)@CTF samples. The in situ XPS measurement was performed under light irradiation. These figures have been reproduced from ref. 138 with permission from Wiley-VCH, copyright 2023. | ||
4.3 X-ray absorption fine structure (XAFS)
Based on synchrotron radiation, XAFS has become an important technique to reveal the electronic structure, coordination number and bonding environment of MAS, and is widely used to study MAS-containing catalytic systems. Specifically, the technique is usually classified into X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS).139Cu–N4 sites-anchored phosphorus-modulated carbon nitride (CuACs/PCN) regulated the intermediate energy levels to achieve C2H4 formation during CO2 photoreduction.85 The Cu K-edge XANES of CuACs/PCN was between Cu2O and CuO (Fig. 13a), indicating that the oxidation state of Cu is between +1 and +2. In the EXAFS spectrum (Fig. 13b), CuACs/PCN had a prominent peak near 1.5 Å, corresponding to the scattering of the Cu–N bond in the first coordination shell. The absence of the Cu–Cu coordination (2.2 Å) indicated that the Cu sites were atomically dispersed. From the fitting results (Fig. 13c), Cu atoms in CuACs/PCN were coordinated by four N atoms at a distance of 1.97 Å. The wavelet transform contour plot of CuACs/PCN exhibited a radial distance of 1.5 Å (Y-axis), which was significantly different from that of the Cu foil. These results strongly demonstrated that Cu atoms were dispersed with Cu–N4 coordination in CuACs/PCN. In addition, XAFS was applied to study the coordination environment of MAS in Co1Ag(1+n)PCN.110 In the Ag K-edge XANES spectra, the near-edge absorption peak of Co1Ag(1+n)PCN was located above Ag2O, indicating that the valence state of Ag was near +1 (Fig. 13e). Two main peaks at 1.5 and 2.4 Å in the Fourier-transformed Ag k2-weighted EXAFS spectra (Fig. 13f) corresponded to the first shell layer of Ag–N and the second shell layer of Ag–C, respectively. The presence of Ag–Ag coordination (2.6 Å) indicated that Ag existed in the form of single atoms and nanoparticles. The wavelet transform Ag K-edge EXAFS of Co1Ag(1+n)PCN showed a maximum peak around 4.7 Å, which was different from that of the Ag foil and Ag2O, attributed to Ag–N/C coordination. Specifically, the EXAFS fitting of the shell coordination elucidated that the Ag atom was coordinated to 2.3 N atoms and 1.7C atoms, referred as the Ag–N2C2 site. Based on a similar analytical procedure, the existence of a Co–N6–P configuration of the Co single atom in Co1Ag(1+n)PCN was also revealed.
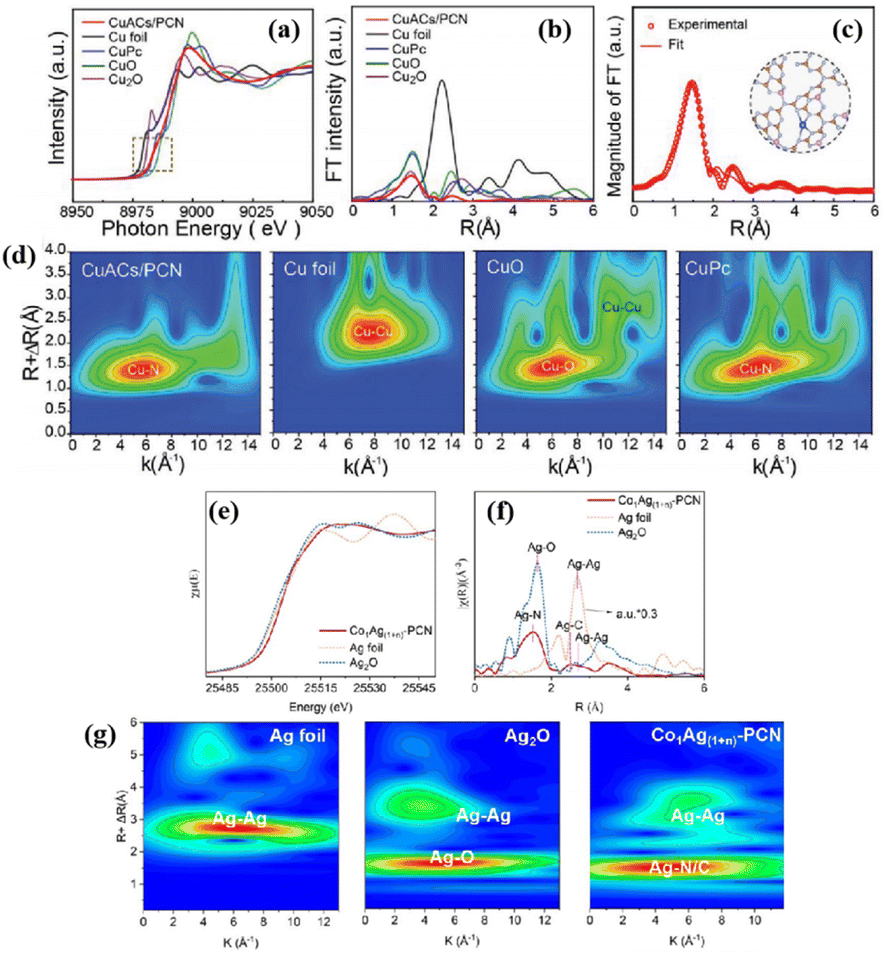 | ||
| Fig. 13 (a) XANES and (b) EXAFS spectra of the Cu K-edge of CuACs/PCN, Cu foil, Cu2O, CuO, and CuPc. (c) The EXAFS fitting curves of CuACs/PCN at R-space. (d) WT EXAFS of the Cu K-edge. These figures have been reproduced from ref. 85 with permission from Wiley-VCH, copyright 2022. Ag K-edge (e) XANES, (f) EXAFS, and (g) WT-EXAFS spectra of Co1Ag(1+n)-PCN. These figures have been reproduced from ref. 110 with permission from American Chemical Society, copyright 2023. | ||
4.4 Diffuse reflectance infrared Fourier-transformed spectroscopy (DRIFTS)
DRIFTS can be used to deduce the structure of the compound, reflecting the type of functional groups contained in the substance. Characterization of intermediates generated on the catalyst surface using in situ DRIFTS provides support for the reaction pathway revelation and MAS identification. In situ DRIFTS was used to probe the reaction intermediates in the CO2 photoreduction of Er-doped ZnIn2S4.86 Under dark conditions, Er-doped ZnIn2S4 exhibited distinct absorption peaks corresponding to monodentate carbonate (m-CO3) and bidentate carbonate (b-CO3), indicating the effective adsorption and activation of CO2 and H2O. Under light conditions, the peaks of *CHO (962 cm−1), *OCH3 (1043 cm−1) and *COOH (1530 cm−1) appeared in Er-ZnIn2S4, which were the key intermediates for the subsequent generation of CH4. Both Er-ZnIn2S4 and pure ZnIn2S4 showed absorption peaks for the *CH2 group at 2856 and 2935 cm−1, respectively, reflecting their tendency to generate CH4. Compared to pure ZnIn2S4, the Er-ZnIn2S4 exhibited stronger b-CO3 dissipation with stronger CO2 reduction capacity. The spectrum of Er-ZnIn2S4 had additional *OCH2 peaks between 1210–1240 cm−1, reflecting different CO2 reduction pathways between Er-ZnIn2S4 and ZnIn2S4 (Er-ZnIn2S4: CO2 → *COOH → *CO → *CHO → *OCH2 → *OCH3 → CH4; ZnIn2S4: CO2 → *COOH → *CO → *CHO → *CH2O → *CH → *CH2 → *CH3 → CH4).In situ DRIFTS identifies key intermediates of the C2+ product, contributing to the inference of C–C coupling processes. In this regard, crucial C2 intermediates including COCO* (1374 and 1486 cm−1) and COCOH* (1233 and 1574 cm−1) were observed on the Au–CeO2 nanocomposite, providing strong evidence that the Au–O–Ce sites drove the generation of C2H6 from the C–C coupling.97 In addition, the peaks at 1367 and 1485 cm−1 appearing in the In2O3/InP heterojunction were related to OCCO*, a key intermediate in the generation of CH3COOH.120 The peak at 1332 cm−1 belonged to the C–H vibration of the hydrogenated intermediate after C–C coupling, and the peak at 1433 cm−1 belonged to the COO stretching vibration of CH3COOH, further reflecting the generation of C2+.
4.5 Raman
Raman spectroscopy identifies chemical bonds and functional groups. In situ Raman spectra can reveal the structural evolution of catalysts during catalytic reactions. For Vo-rich Zn2GeO4, CO2 adsorption on Ge and Zn atoms gradually shifted the peak positions of the Zn–O–Ge stretching vibrations (∼730 cm−1) and Zn–O–Ge bending vibrations (∼785 cm−1) to higher wave numbers.96 The Raman peak blue shift revealed the transfer of the electron cloud from the lattice oxygen to the metal atoms. The Zn–O–Ge stretching and Zn–O–Ge bending vibration peaks were again shifted to higher frequencies during CO2 photoreduction, indicating that the electron cloud in the O 2p orbitals of the lattice oxygen was further transferred to the nearby Zn 4s or Ge 4s empty orbitals. This process provided electrons for the reduction of adsorbed CO2 molecules and reaction intermediates. In situ Raman enables the real-time monitoring of the intermediate generation and conversion processes on the catalyst surface, revealing the reaction mechanism. In the in situ Raman spectrum, the peak before 500 cm−1 was related to the structural properties of r-In2O3/InP.120 With increasing light duration, the peak at 887 cm−1 was designated as C–C–O stretching, reflecting the critical C–C coupling process for CH3COOH generation. The C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching vibration of CO* (2909 cm−1) further validated that the CO* coupling produced CH3COOH.
O stretching vibration of CO* (2909 cm−1) further validated that the CO* coupling produced CH3COOH.
4.6 Electrochemical analysis
Electrochemical analysis is widely used to reveal the kinetic processes of the migration and separation of photogenerated carriers in photocatalysis. Among them, transient photocurrent response and electrochemical impedance measurement (EIS) are the most representative and widely used methods.140–144 For example, the Cu–N4 sites-anchored phosphorus-modulated carbon nitride (CuACs/PCN) exhibited the strongest photocurrent response relative to carbon nitride (CN), CuACs-anchored carbon nitride (CuACs/CN), and CuACs/PCNs with different P doping contents, reflecting its superior interfacial charge separation efficiency.85 Moreover, EIS also demonstrated the lowest resistance of CuACs/PCN, thus confirming the efficient separation of photoexcited electron–hole pairs.As another crucial feature, electrochemical analysis contributes to reveal the change of activation and reduction energy of CO2 conversion. Kang's research group examined the Tafel plots of pure Cu, Cu/PVP and Cu/rGO/PVP/Nafion composite electrodes to estimate the reaction overpotentials of the samples.145 The overpotentials of Cu/PVP (6.16 kJ mol−1) and Cu/rGO/PVP/Nafion (9.00 kJ mol−1) electrodes were significantly lower than those of the pure Cu electrode, revealing a decrease in the CO2 reduction energy.
In addition, the Mott–Schottky measurement can be used to specify the semiconductor type and determine the valence band maximum (VBM) and conduction band minimum (CBM) potentials. For example, according to the results of the Mott–Schottky test, the Zr/Ti bimetallic oxide solid solution integrated with Au nanoparticles (Au/TZO) exhibited the characteristics of n-type semiconductors.131 As the CBM of Au/TZO was estimated as −0.80 V with respect to a normal hydrogen electrode (NHE), the photocatalytic conversion of CO2 to CO (–0.53 vs. NHE), CH4 (–0.24 vs. NHE), C2H4 (–0.34 vs. NHE) and C2H6 (–0.27 vs. NHE) was thermodynamically feasible over Au/TZO. It is also worth mentioning that the electrochemical active surface area (ECSA) can be used to reveal the number of catalytically active sites. A larger ECSA indicates more active sites, which is more favorable for photocatalytic CO2 conversion.146
4.7 Theoretical calculations
Theoretical calculations can be used to obtain the atomic and electronic structures of the MAS, the adsorption strength of the intermediates, and the optimal reaction path.Electronic property analysis can infer underlying factors affecting the catalytic activity. The d-band centers of Cu in Cu1N3@P(1)CN, Cu1N3@CN, and Cu1P3@PCN were determined to be −1.24 eV, −1.46 eV, and 11.60 eV, respectively, by partial density of states analysis, which depended on the local coordination environment of Cu.87 As the d-band center of Cu approached the Fermi energy level, the catalytic activity for the COOH* formation became higher. It is worth mentioning that the overlap between Cu 3d and P 3p in Cu1N3@P(1)CN was more pronounced than the overlap of Cu 3d with C 2p at the same corner position in Cu1N3@CN. This reflected the more pronounced orbital and electronic interactions of Cu and P in Cu1N3@P(1)CN than that of Cu and C in Cu1N3@CN, which moved the d-band center of Cu1N3@PCN closer to the Fermi energy level. Charge density difference analysis further demonstrated that the electronic interaction between Cu and COOH* in Cu1N3@P(1)CN was the strongest relative to Cu1N3@CN and Cu1P3@PCN, prompting a large number of electrons to be transferred from the Cu center to COOH* in Cu1N3@P(1)CN, benefiting CO2 activation.
Theoretical calculations provide insight into the mechanism of the photocatalytic CO2 reduction reaction. From the free energy analysis, the introduction of Ag single atoms on hollow porous polygonal C3N4 nanotubes (Ag1@PCNT) significantly enhanced the adsorption of *CO2 and weakened the adsorption of *H, suggesting that Ag1@PCNT was more favorable for CO2 conversion than for hydrogen production.88 The COOH* formation energy (the rate-limiting step) of Ag1@PCNT was lower than that of C3N4 nanotubes. This suggests that the strong interaction of the Ag–N3 coordination with *COOH stabilized the *COOH intermediate and reduced the barrier for CO2 photoreduction (Fig. 14a and b). In addition, the generation of CO molecules by desorption of *CO groups on the surface of Ag1@PCNT had a smaller Gibbs free energy than the protonation of *CO into *CHO, which contributed to the highly selective generation of CO.
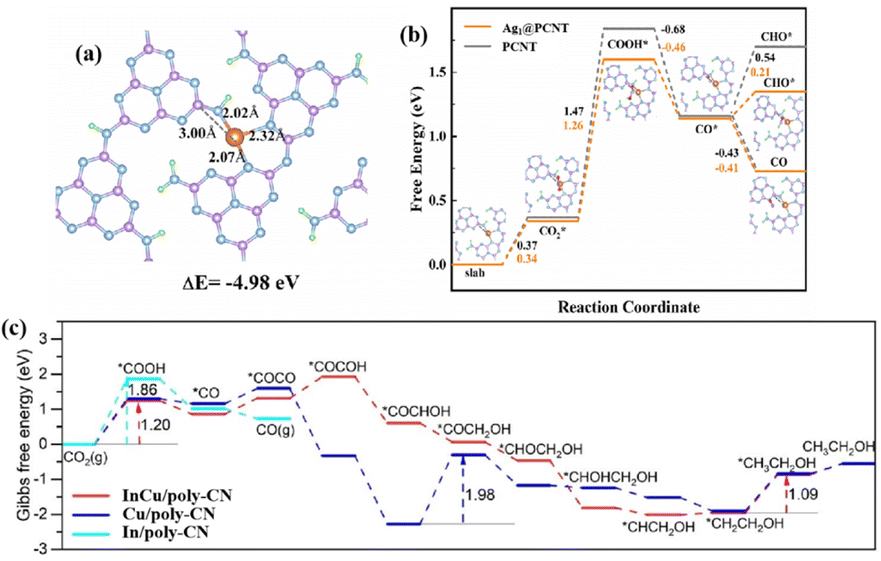 | ||
| Fig. 14 (a) Structure of Ag1@PCNT. (b) Gibbs free energies of the CO2 photoreduction pathways by DFT calculations over the Ag1@PCNT (Ag sites) and PCNT surfaces. Atom key: N (blue), C (purple), H (green), and Ag (orange). These figures have been reproduced from ref. 88 with permission from Wiley-VCH, copyright 2023. Gibbs free energy diagrams and CO2-to-ethanol photoreduction pathways on InCu/poly-CN, Cu/poly-CN, and In/poly-CN (c). This figure has been reproduced from ref. 98 with permission from Wiley-VCH, copyright 2022. | ||
Theoretical calculations can reveal the specific processes involved in the generation of C2+ products, especially for C–C coupling. Calculation of the Gibbs free energy diagrams for CO2 photoreduction on In and/or Cu-anchored poly-CN (InCu/poly-CN, Cu/poly-CN, and In/poly-CN) (Fig. 14c) revealed that the formation of *COOH was a rate-determining step on InCu/poly-CN (ΔG = +1.20 eV) and In/poly-CN (ΔG = +1.86 eV), while the generation of *COCH2OH was a rate-determining step on Cu/poly-CN (ΔG = +1.98 eV).98 On InCu/poly-CN, *CO underwent a dimerization reaction (ΔG = 0.45 eV) and a subsequent hydrogenation–electron addition process to ultimately generate CH3CH2OH. In the Cu/poly-CN system, *CO was also coupled to form *COCO, but its Gibbs free energy was higher than that of the InCu/poly-CN. Furthermore, the process from *COCHOH to *COCH2OH in Cu/poly-CN was endothermic with the highest gain (ΔG = +1.98 eV), resulting in lower CH3CH2OH yields for Cu/poly-CN relative to InCu/poly-CN. However, the *CO intermediates on In/poly-CN preferred desorption as CO products (exothermic) rather than C–C coupling, resulting in a poor CH3CH2OH yield on In/poly-CN.
5 Conclusion and perspectives
The photocatalysts containing MAS have been proved as efficient systems for converting incident solar energy into chemical fuel. In this review, typical photocatalytic materials containing MAS have been briefly introduced with clear examples. Then, the different roles of MAS in various forms in the photocatalytic CO2 reduction from the perspective of principles and applications are discussed in detail, greatly accelerating the surface charge separation/transfer, and enhancing the adsorption and activation of CO2. Finally, advanced characterization techniques and theoretical calculation for revealing the function played by MAS in CO2 photoreduction are presented. Although considerable progress has been made in MAS-containing photocatalytic systems, there are still great challenges in practical applications that need to be further explored.(1) The complexity of the preparation process and the difficulty of achieving uniform high-volume production are the major drawbacks of MAS-containing catalysts. In addition, the exposed MAS are susceptible to oxidation, resulting in performance degradation. Therefore, effective technologies should be continuously developed for the large-scale production of MAS-containing photocatalysts with stable and controllable surface states. Elaborate design strategies to improve the catalyst stability (e.g., surface encapsulation and ligand grafting) need to be further explored. Continuous optimization of existing synthesis methods is an effective and viable solution.
(2) Although photocatalysts containing MAS have shown great potential in artificial photosynthesis, their performance is still unsatisfactory for practical production. Particularly, photocatalytic CO2 conversion involves a variety of intermediates and products, making it difficult to regulate the selectivity of the target product. For photocatalytic systems with MAS, the interactions of reactants and intermediates with the MAS affect the activation and conversion of CO2, as well as C–C coupling. Defect design, doping, construction of heterostructures, and introduction of co-catalysts have become widely used strategies to enhance the CO2 photoreduction performance. However, there is still a need to continuously explore effective and novel strategies to improve the performance for real-world production, which remains elusive at the present stage.
(3) Accurate characterization and in-depth understanding of the mechanism of MAS in photocatalysis still face great challenges. It is crucial to carefully reveal the coordination environments of the MAS and its changes during the photocatalytic reaction. Specifically, the working mechanism of MAS in photocatalysis can be explored in-depth by advanced in situ characterisation techniques and accurate theoretical calculations, which guide the rational design and regulation of photocatalysts. Therefore, there is a need for the continuous development of highly sophisticated characterisation techniques, and theoretical computational models close to the actual reaction processes.
(4) In the face of the explosion of research data, the traditional research method of relying on pre-investigation to identify scientific problems has become overwhelming. This approach is costly, time-consuming, and increasingly difficult to meet the research needs of modern materials science. Nowadays, machine learning is changing the traditional research paradigm and bringing new opportunities for the development of related technologies and industrial upgrading. Combining machine learning techniques with the design of photocatalytic systems to rapidly mine effective information from a large amount of data is conducive to the construction of novel materials and the excavation of reaction mechanisms. Specifically, machine learning techniques are becoming a powerful tool for scientific research in the design and construction of photocatalysts, as well as in the prediction of physicochemical properties and mechanisms.
In summary, through continuous exploration and in-depth research, it is found that photocatalytic CO2 reduction technology is expected to realize the establishment of a resource-saving and environmentally-friendly energy system, and improve human life in the near future.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
All authors participated in this review work. W. Gao, Y. Yang, and Y. Zhou conceived the structure of this perspective. W. Gao, H. N. Li, and J. Q. Hu prepared the draft. Y. J. Xiong, J. H. Ye, and Z. G. Zou reviewed and refined the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the support of the National Key R&D Program of China (2018YFE0208500 and 2021YFA1500700), the NSF of China (21972065 and 22202152), the NSF of Jiangsu Province (No. BK20220006), the Hefei National Laboratory for Physical Sciences at the Microscale (KF2020006), the Program for Guangdong Introducing Innovative and Entrepreneurial Team (2019ZL08L101) and the University Development Fund (UDF01001159).References
- C. Y. Feng, Z. P. Wu, K. W. Huang, J. H. Ye and H. B. Zhang, Adv. Mater., 2022, 34, 202200180 Search PubMed.
- E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim, H. Kim and S. I. In, Energy Environ. Sci., 2022, 15, 880–937 RSC.
- H. W. Lin, S. N. Luo, H. B. Zhang and J. H. Ye, Joule, 2022, 6, 294–314 CrossRef CAS.
- R. Ham, C. J. Nielsen, S. Pullen and J. N. H. Reek, Chem. Rev., 2023, 123, 5225–5261 CrossRef CAS PubMed.
- X. J. Liu, T. Q. Chen, Y. H. Xue, J. C. Fan, S. L. Shen, M. Hossain, M. A. Amin, L. K. Pan, X. T. Xu and Y. Yamauchi, Coord. Chem. Rev., 2022, 459, 214440 CrossRef CAS.
- B. C. Qiu, M. M. Du, Y. X. Ma, Q. H. Zhu, M. Y. Xing and J. L. Zhang, Energy Environ. Sci., 2021, 14, 5260–5288 RSC.
- K. H. Chen, X. W. Wang, Q. Y. Li, Y. N. Feng, F. F. Chen and Y. Yu, Chem. Eng. J., 2021, 418, 129476 CrossRef CAS.
- L. Cheng, D. N. Zhang, Y. L. Liao, J. J. Fan and Q. J. Xiang, Chin. J. Catal., 2021, 42, 131–140 CrossRef CAS.
- J. W. Jiang, X. F. Wang, Q. J. Xu, Z. Y. Mei, L. Y. Duan and H. Guo, Appl. Catal., B, 2022, 316, 121679 CrossRef CAS.
- G. H. Li, Y. Y. Sun, Q. M. Zhang, Z. Gao, W. Sun and X. X. Zhou, Chem. Eng. J., 2021, 410, 128397 CrossRef CAS.
- J. X. Liang, H. Yu, J. J. Shi, B. Li, L. X. Wu and M. Wang, Adv. Mater., 2023, 35, 202209814 Search PubMed.
- W. B. Jiang, H. Y. Loh, B. Q. L. Low, H. J. Zhu, J. X. Low, J. Z. X. Heng, K. Y. Tang, Z. B. Li, X. J. Loh, E. Y. Ye and Y. J. Xiong, Appl. Catal., B, 2023, 321, 122079 CrossRef CAS.
- S. M. Mao, J. W. Shi, G. T. Sun, D. D. Ma, C. He, Z. X. Pu, K. L. Song and Y. H. Cheng, Appl. Catal., B, 2021, 282, 119550 CrossRef CAS.
- Z. Y. Dong, L. Zhang, J. Gong and Q. Zhao, Chem. Eng. J., 2021, 403, 126383 CrossRef CAS.
- S. K. Yin, L. L. Sun, Y. J. Zhou, X. Li, J. Z. Li, X. H. Song, P. W. Huo, H. Q. Wang and Y. S. Yan, Chem. Eng. J., 2021, 406, 126776 CrossRef CAS.
- J. Zhang, L. T. Li, M. Du, Y. Cui, Y. H. Li, W. Yan, H. J. Huang, X. A. Li and X. B. Zhu, Small, 2023, 19, 202300402 Search PubMed.
- H. H. Ou, S. B. Ning, P. Zhu, S. H. Chen, A. Han, Q. Kang, Z. F. Hu, J. H. Ye, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2022, 61, 202206579 CrossRef.
- J. P. Sheng, Y. He, J. Y. Li, C. W. Yuan, H. W. Huang, S. Y. Wang, Y. J. Sun, Z. M. Wang and F. Dong, ACS Nano, 2020, 14, 13103–13114 CrossRef CAS PubMed.
- N. Podrojková, V. Sans, A. Orinak and R. Orinaková, ChemCatChem, 2020, 12, 1802–1825 CrossRef.
- J. Wang, W. B. Zhu, F. Y. Meng, G. Y. Bai, Q. F. Zhang and X. W. Lan, ACS Catal., 2023, 13, 4316–4329 CrossRef CAS.
- F. F. Chen, L. H. Zhou, C. Peng, D. T. Zhang, L. Y. Li, D. F. Xue and Y. Yu, Appl. Catal., B, 2023, 331, 122689 CrossRef CAS.
- J. Di, C. Chen, C. Zhu, P. Song, M. L. Duan, J. Xiong, R. Long, M. Z. Xu, L. X. Kang, S. S. Guo, S. M. Chen, H. L. Chen, Z. Chi, Y. X. Weng, H. M. Li, L. Song, M. H. Wu, Q. Y. Yan, S. Z. Li and Z. Liu, Nano Energy, 2021, 79, 105429 CrossRef CAS.
- J. Li, B. J. Huang, Q. Guo, S. Guo, Z. K. Peng, J. Liu, Q. Y. Tian, Y. P. Yang, Q. Xu, Z. Y. Liu and B. Liu, Appl. Catal., B, 2021, 284, 119733 CrossRef CAS.
- H. W. Guo, S. P. Wan, Y. A. Wang, W. H. Ma, Q. Zhong and J. Ding, Chem. Eng. J., 2021, 412, 128646 CrossRef CAS.
- J. Li, W. F. Pan, Q. Y. Liu, Z. Q. Chen, Z. J. Chen, X. Z. Feng and H. Chen, J. Am. Chem. Soc., 2021, 143, 6551–6559 CrossRef CAS PubMed.
- Y. Y. Wang, H. L. Huang, Z. Z. Zhang, C. Wang, Y. Y. Yang, Q. Li and D. S. Xu, Appl. Catal., B, 2021, 282, 119570 CrossRef CAS.
- L. Cheng, P. Zhang, Q. Y. Wen, J. J. Fan and Q. J. Xiang, Chin. J. Catal., 2022, 43, 451–460 CrossRef CAS.
- Q. J. Wu, J. Liang, Y. B. Huang and R. Cao, Acc. Chem. Res., 2022, 55, 2978–2997 CrossRef CAS.
- F. Guo, R. X. Li, S. Z. Yang, X. Y. Zhang, H. J. Yu, J. J. Urban and W. Y. Sun, Angew. Chem., Int. Ed., 2023, 62, 202216232 CrossRef.
- W. Wang, C. Y. Deng, S. J. Xie, Y. F. Li, W. Y. Zhang, H. Sheng, C. C. Chen and J. C. Zhao, J. Am. Chem. Soc., 2021, 143, 2984–2993 CrossRef CAS.
- G. Wang, Z. Chen, T. Wang, D. S. Wang and J. J. Mao, Angew. Chem., Int. Ed., 2022, 61, 202210789 CrossRef.
- X. H. Yang, X. W. Lan, Y. Z. Zhang, H. S. Li and G. Y. Bai, Appl. Catal., B, 2023, 325, 122393 CrossRef CAS.
- H. Yu, C. Sun, Y. Xuan, K. Zhang and K. Chang, Chem. Eng. J., 2022, 430, 132940 CrossRef CAS.
- S. P. Wan, M. Ou, Q. Zhong and X. M. Wang, Chem. Eng. J., 2019, 358, 1287–1295 CrossRef CAS.
- S. L. Wang, M. Xu, T. Y. Peng, C. X. Zhang, T. Li, I. Hussain, J. Y. Wang and B. E. Tan, Nat. Commun., 2019, 10, 08651 Search PubMed.
- G. Wang, R. Huang, J. W. Zhang, J. J. Mao, D. S. Wang and Y. D. Li, Adv. Mater., 2021, 33, 202105904 Search PubMed.
- X. L. Zhuang, S. T. Zhang, Y. J. Tang, F. Yu, Z. M. Li and H. Pang, Coord. Chem. Rev., 2023, 490, 215208 CrossRef CAS.
- T. Wang, L. Chen, C. Chen, M. T. Huang, Y. J. Huang, S. J. Liu and B. X. Li, ACS Nano, 2022, 16, 2306–2318 CrossRef CAS.
- P. Y. Li, L. Liu, W. J. An, H. Wang, H. X. Guo, Y. H. Liang and W. Q. Cui, Appl. Catal., B, 2020, 266, 118618 CrossRef CAS.
- Y. J. Liang, X. Wu, X. Y. Liu, C. H. Li and S. W. Liu, Appl. Catal., B, 2022, 304, 120978 CrossRef CAS.
- S. J. Xie, W. C. Ma, X. J. Wu, H. K. Zhang, Q. H. Zhang, Y. D. Wang and Y. Wang, Energy Environ. Sci., 2021, 14, 37–89 RSC.
- Y. N. Teja and M. Sakar, Small, 2023, 19, 202303980 CrossRef.
- W. G. Tu, Y. Zhou, Q. Liu, Z. P. Tian, J. Gao, X. Y. Chen, H. T. Zhang, J. G. Liu and Z. G. Zou, Adv. Funct. Mater., 2012, 22, 1215–1221 CrossRef CAS.
- X. H. Yan, J. J. Zhang, G. Z. Hao, W. Jiang and J. Di, Small, 2024, 20, 202306742 Search PubMed.
- Y. M. Zhou, Q. X. Zhang, X. L. Shi, Q. Song, C. J. Zhou and D. L. Jiang, J. Colloid Interface Sci., 2022, 608, 2809–2819 CrossRef CAS PubMed.
- Y. Shen, C. J. Ren, L. R. Zheng, X. Y. Xu, R. Long, W. Q. Zhang, Y. Yang, Y. C. Zhang, Y. F. Yao, H. Q. Chi, J. L. Wang, Q. Shen, Y. J. Xiong, Z. G. Zou and Y. Zhou, Nat. Commun., 2023, 14, 1117 CrossRef CAS PubMed.
- L. J. Xiong, Y. J. Hu, Y. Wang, W. Dong, X. Y. Zhang, K. Zhang, T. Y. Wang, J. Y. Shen and Y. Yang, Appl. Catal., B, 2024, 340, 123263 CrossRef CAS.
- H. Pang, X. G. Meng, H. Song, W. Zhou, G. L. Yang, H. W. Zhang, Y. Izumi, T. Takei, W. Jewasuwan, N. Fukata and J. H. Ye, Appl. Catal., B, 2019, 244, 1013–1020 CrossRef CAS.
- X. D. Zhang, D. Kim and L. Y. S. Lee, ACS Appl. Energy Mater., 2021, 4, 2586–2592 CrossRef CAS.
- C. Hu, H. Y. Sun, X. M. Jia, H. L. Lin, J. Cao and S. F. Chen, ChemPhotoChem, 2022, 6, 202200150 CrossRef.
- Y. Q. He, C. L. Chen, Y. X. Liu, Y. L. Yang, C. G. Li, Z. Shi, Y. Han and S. H. Feng, Nano Lett., 2022, 22, 4970–4978 CrossRef CAS.
- S. Si, H. Shou, Y. Mao, X. Bao, G. Zhai, K. Song, Z. Wang, P. Wang, Y. Liu, Z. Zheng, Y. Dai, L. Song, B. Huang and H. Cheng, Angew. Chem., Int. Ed., 2022, 61, e202209446 CrossRef CAS PubMed.
- Y. Q. He, H. Rao, K. P. Song, J. X. Li, Y. Yu, Y. Lou, C. G. Li, Y. Han, Z. Shi and S. H. Feng, Adv. Funct. Mater., 2019, 29, 201905153 Search PubMed.
- W. Gao, S. Li, H. C. He, X. N. Li, Z. X. Cheng, Y. Yang, J. L. Wang, Q. Shen, X. Y. Wang, Y. J. Xiong, Y. Zhou and Z. G. Zou, Nat. Commun., 2021, 12, 4747 CrossRef CAS PubMed.
- W. Gao, L. Shi, W. Hou, C. Ding, Q. Liu, R. Long, H. Chi, Y. Zhang, X. Xu, X. Ma, Z. Tang, Y. Yang, X. Wang, Q. Shen, Y. Xiong, J. Wang, Z. Zou and Y. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202317852 CrossRef CAS.
- X. Y. Xiong, Y. F. Zhao, R. Shi, W. J. Yin, Y. X. Zhao, G. I. N. Waterhouse and T. R. Zhang, Sci. Bull., 2020, 65, 987–994 CrossRef CAS PubMed.
- S. Yu, L. Tan, S. Bai, C. J. Ning, G. H. Liu, H. J. Wang, B. Liu, Y. F. Zhao and Y. F. Song, Small, 2022, 18, 202202334 Search PubMed.
- Y. L. Li, Q. Zhao, X. J. Liu, Y. Liu, Y. J. Hao, X. J. Wang, X. Y. Liu, D. Hildebrandt, F. Y. Li and F. T. Li, Small Struct., 2023, 4, 202300177 Search PubMed.
- Z. H. Wei, S. J. Song, H. F. Gu, Y. Q. Li, Q. Sun, N. Ding, H. Tang, L. R. Zheng, S. H. Liu, Z. X. Li, W. X. Chen, S. H. Li and S. P. Pang, Adv. Sci., 2023, 10, 202303206 Search PubMed.
- K. Sun, Y. Qian and H. L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202217565 CrossRef CAS.
- J. X. Wang, K. Sun, D. A. Wang, X. W. Niu, Z. Y. Lin, S. Y. Wang, W. J. Yang, J. R. Huang and H. L. Jiang, ACS Catal., 2023, 13, 8760–8769 CrossRef CAS.
- M. R. Zhang, D. Zhang, X. Jing, B. J. Xu and C. Y. Duan, Angew. Chem., Int. Ed., 2024, 63, 202402755 CrossRef.
- Y. Z. Zhang, L. L. Cao, G. Y. Bai and X. W. Lan, Small, 2023, 19, 202300035 Search PubMed.
- W. G. Tu, Y. Q. Yang, C. P. Chen, T. H. Zhou, T. H. Li, H. J. Wang, S. Y. Wu, Y. Zhou, D. O'Hare, Z. G. Zou and R. Xu, Small Struct., 2023, 4, 202200233 Search PubMed.
- Q. Zhang, S. Q. Gao, Y. Y. Guo, H. Y. Wang, J. S. Wei, X. F. Su, H. C. Zhang, Z. M. Liu and J. J. Wang, Nat. Commun., 2023, 14, 1147 CrossRef CAS.
- M. P. Kou, W. Liu, Y. Y. Wang, J. D. Huang, Y. L. Chen, Y. Zhou, Y. Chen, M. Z. Ma, K. Lei, H. Q. Xie, P. K. Wong and L. Q. Ye, Appl. Catal., B, 2021, 291, 120146 CrossRef CAS.
- P. Chen, B. Lei, X. A. Dong, H. Wang, J. P. Sheng, W. Cui, J. Y. Li, Y. J. Sun, Z. M. Wang and F. Dong, ACS Nano, 2020, 14, 15841–15852 CrossRef CAS.
- K. Wang, M. Cheng, F. J. Xia, N. Cao, F. X. Zhang, W. K. Ni, X. Y. Yue, K. P. Yan, Y. He, Y. Shi, W. X. Dai and P. F. Xie, Small, 2023, 19, 202207581 Search PubMed.
- L. Cheng, X. Y. Yue, L. X. Wang, D. N. Zhang, P. Zhang, J. J. Fan and Q. J. Xiang, Adv. Mater., 2021, 33, 202105135 Search PubMed.
- L. Zeng, J. W. Chen, L. X. Zhong, W. L. Zhen, Y. Y. Tay, S. Z. Li, Y. G. Wang, L. M. Huang and C. Xue, Appl. Catal., B, 2022, 307, 121154 CrossRef CAS.
- W. Xia and F. Wang, Mol. Catal., 2023, 535, 112884 CrossRef CAS.
- Y. C. Wang, J. H. Zhang, W. Yang, W. X. Tao, K. Y. Tao, J. H. Deng, W. J. Shi, D. C. Zhong and T. B. Lu, Chin. J. Chem., 2023, 41, 3305–3310 CrossRef CAS.
- L. Yuan, L. Zhang, X. X. Li, J. Liu, J. J. Liu, L. Z. Dong, D. S. Li, S. L. Li and Y. Q. Lan, Chin. Chem. Lett., 2023, 34, 039 Search PubMed.
- R. Xu, D. H. Si, S. S. Zhao, Q. J. Wu, X. S. Wang, T. F. Liu, H. Zhao, R. Cao and Y. B. Huang, J. Am. Chem. Soc., 2023, 145, 8261–8270 CrossRef CAS.
- J. Q. Ning, W. Chen, Q. Niu, L. Y. Li and Y. Yu, ChemSusChem, 2024 DOI:10.1002/cssc.202301963.
- S. J. Liang, X. H. Zhong, Z. Q. Zhong, H. Deng and W. Y. Wong, Appl. Catal., B, 2023, 337, 122958 CrossRef CAS.
- H. B. Yin, F. Dong, D. S. Wang and J. H. Li, ACS Catal., 2022, 12, 14096–14105 CrossRef CAS.
- C. Y. Shen, X. Y. Meng, R. Zou, K. H. Sun, Q. L. Wu, Y. X. Pan and C. J. Liu, Angew. Chem., Int. Ed., 2024, 63, 202402369 CrossRef.
- Q. Y. Wang, Y. D. Zhang, M. X. Lin, H. W. Wang, Y. Bai, C. Y. Liu, J. L. Lu, Q. Q. Luo, G. M. Wang, H. L. Jiang, T. Yao and X. S. Zheng, Adv. Energy Mater., 2023, 13, 202302692 Search PubMed.
- F. Guo, R. X. Li, S. Yang, X. Y. Zhang, H. Yu, J. J. Urban and W. Y. Sun, Angew. Chem., Int. Ed., 2023, 62, 202216232 CrossRef.
- H. Shi, Y. Liang, J. Hou, H. Wang, Z. Jia, J. Wu, F. Song, H. Yang and X. Guo, Angew. Chem., Int. Ed., 2024, 63, e202404884 CrossRef CAS.
- Y. Y. Mao, M. H. Zhang, S. H. Si, G. Y. Zhai, X. L. Bao, K. P. Song, L. R. Zheng, Y. Y. Liu, Z. Y. Wang, Z. K. Zheng, P. Wang, Y. Dai, H. F. Cheng and B. B. Huang, ACS Catal., 2023, 13, 8362–8371 CrossRef CAS.
- C. Y. Feng, T. T. Bo, P. Maity, S. W. Zuo, W. Zhou, K. W. Huang, O. F. Mohammed and H. B. Zhang, Adv. Funct. Mater., 2023, 34, 202309761 Search PubMed.
- C. Ding, X. X. Lu, B. Tao, L. Q. Yang, X. Y. Xu, L. Q. Tang, H. Q. Chi, Y. Yang, D. M. Meira, L. Wang, X. Zhu, S. Li, Y. Zhou and Z. G. Zou, Adv. Funct. Mater., 2023, 33, 202302824 Search PubMed.
- W. K. Xie, K. J. Li, X. H. Liu, X. Zhang and H. W. Huang, Adv. Mater., 2023, 35, 202208132 Search PubMed.
- F. H. Zhou, Y. L. Zhang, J. Wu, W. Yang, X. Fang, T. Jia, Y. Ling, P. He, Q. Z. Liu and J. Lin, Appl. Catal., B, 2024, 341, 123347 CrossRef CAS.
- X. Sun, L. Sun, G. Li, Y. Tuo, C. Ye, J. Yang, J. Low, X. Yu, J. H. Bitter, Y. Lei, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, 202207677 CrossRef.
- S. Hu, P. Z. Qiao, X. L. Yi, Y. M. Lei, H. L. Hu, J. H. Ye and D. F. Wang, Angew. Chem., Int. Ed., 2023, 62, 202304585 CrossRef.
- M. Z. Ma, Z. Huang, L. A. Li, W. D. Zhang, R. Guo, R. Y. Zhang, W. Fa, C. Q. Han, Y. H. Cao, S. Yu and Y. Zhou, Appl. Catal., B, 2023, 330, 122626 CrossRef CAS.
- J. Zhou, J. Li, L. Kan, L. Zhang, Q. Huang, Y. Yan, Y. F. Chen, J. Liu, S. L. Li and Y. Q. Lan, Nat. Commun., 2022, 13, 32449 Search PubMed.
- M. Zhou, Z. Q. Wang, A. H. Mei, Z. F. Yang, W. Chen, S. Y. Ou, S. Y. Wang, K. Q. Chen, P. Reiss, K. Qi, J. Y. Ma and Y. L. Liu, Nat. Commun., 2023, 14, 2473 CrossRef CAS.
- X. D. Li, Y. F. Sun, J. Q. Xu, Y. J. Shao, J. Wu, X. L. Xu, Y. Pan, H. X. Ju, J. F. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- S. M. Deng, R. H. Wang, X. Z. Feng, R. J. Zheng, S. K. Gong, X. H. Chen, Y. Z. Shangguan, L. L. Deng, H. Tang, H. Dai, L. L. Duan, C. Y. Liu, Y. Pan and H. Chen, Angew. Chem., Int. Ed., 2023, 62, 202309625 CrossRef.
- C. Chen, L. Chen, Y. G. Hu, K. Yan, T. Wang, Y. J. Huang, C. Gao, J. J. Mao, S. J. Liu and B. X. Li, J. Energy Chem., 2023, 86, 599–608 CrossRef CAS.
- Y. Z. Zhang, B. Johannessen, P. Zhang, J. L. Gong, J. R. Ran and S. Z. Qiao, Adv. Mater., 2023, 35, 202306923 Search PubMed.
- J. C. Zhu, W. W. Shao, X. D. Li, X. C. Jiao, J. F. Zhu, Y. F. Sun and Y. Xie, J. Am. Chem. Soc., 2021, 143, 18233–18241 CrossRef CAS.
- J. X. Ji, R. R. Li, H. Zhang, Y. N. Duan, Q. Liu, H. Z. Wang and Z. R. Shen, Appl. Catal., B, 2023, 321, 122020 CrossRef CAS.
- H. Shi, H. Wang, Y. Zhou, J. Li, P. Zhai, X. Li, G. G. Gurzadyan, J. Hou, H. Yang and X. Guo, Angew. Chem., Int. Ed., 2022, 61, 202208904 CrossRef PubMed.
- B. Liu, M. Cheng, C. Zhang, Y. Si, J. Zhou, Y. Ren, J. Guan, L. Duan, M. Liu, D. Jing and N. Li, Appl. Catal., B, 2024, 357, 124263 CrossRef CAS.
- X. F. Shang, G. J. Li, R. A. Wang, T. Xie, J. Ding and Q. Zhong, Chem. Eng. J., 2023, 456, 140805 CrossRef CAS.
- G. R. Jia, M. Z. Sun, Y. Wang, Y. B. Shi, L. Z. Zhang, X. Q. Cui, B. L. Huang and J. C. Yu, Adv. Funct. Mater., 2022, 32, 202206817 Search PubMed.
- J. L. Yin, D. Y. Li, C. Sun, Y. L. Jiang, Y. K. Li and H. H. Fei, Adv. Mater., 2024, 36, 2403651 CrossRef CAS PubMed.
- W. W. Wang, S. J. Song, P. Wang, M. He, Z. Fang, X. L. Yuan, H. Li, C. Y. Li, X. Wang, Y. C. Wei, W. Y. Song, H. Xu and Z. X. Li, ACS Catal., 2023, 13, 4597–4610 CrossRef CAS.
- S. J. Xie, Y. F. Li, B. Sheng, W. Y. Zhang, W. Wang, C. C. Chen, J. K. Li, H. Sheng and J. C. Zhao, Appl. Catal., B, 2022, 310, 121320 CrossRef CAS.
- Z. F. Ma, X. Liu, X. S. Wang, Z. G. Luo, W. R. Li, Y. H. Nie, L. Pei, Q. A. Mao, X. Wen and J. S. Zhong, Chem. Eng. J., 2023, 468, 143569 CrossRef CAS.
- Y. Y. Duan, Y. Wang, W. X. Zhang, J. W. Zhang, C. G. Ban, D. M. Yu, K. Zhou, J. J. Tang, X. Zhang, X. D. Han, L. Y. Gan, X. P. Tao and X. Y. Zhou, Adv. Funct. Mater., 2023, 33, 202301729 Search PubMed.
- J. Y. Wang, C. Yang, L. Mao, X. Y. Cai, Z. K. Geng, H. Y. Zhang, J. Y. Zhang, X. Tan, J. H. Ye and T. Yu, Adv. Funct. Mater., 2023, 33, 202213901 Search PubMed.
- X. Shi, W. D. Dai, X. A. Dong, Q. Ren, J. P. Sheng and F. Dong, Appl. Catal., B, 2023, 339, 123147 CrossRef CAS.
- W. G. Pan, C. F. Li, Z. R. Zhang, T. Wu and R. T. Guo, Appl. Catal., B, 2024, 343, 123492 CrossRef CAS.
- A. X. Deng, E. Zhao, Q. Li, Y. Sun, Y. Z. Liu, S. G. Yang, H. He, Y. Xu, W. Zhao, H. O. Song, Z. Xu and Z. P. Chen, ACS Nano, 2023, 17, 11869–11881 CrossRef CAS.
- M. H. Zhang, Y. Y. Mao, X. L. Bao, P. Wang, Y. Y. Liu, Z. K. Zheng, H. F. Cheng, Y. Dai, Z. Y. Wang and B. B. Huang, ACS Catal., 2024, 14, 5275–5285 CrossRef CAS.
- P. G. Liu, Z. X. Huang, X. P. Gao, X. Hong, J. F. Zhu, G. M. Wang, Y. E. Wu, J. Zeng and X. S. Zheng, Adv. Mater., 2022, 34, 202200057 Search PubMed.
- X. H. Li, F. H. Li, S. L. Tong, Y. J. Cao, Y. W. Jiang, Z. M. Wang, W. Lu, J. Wu, T. Zhou, J. Lin and Y. S. Liu, J. Alloys Compd., 2024, 984, 173986 CrossRef CAS.
- X. T. Wang, Z. Z. Wang, Y. Li, J. T. Wang and G. K. Zhang, Appl. Catal., B, 2022, 319, 121895 CrossRef CAS.
- Y. Zhang, F. Y. Guo, K. K. Wang, J. Di, B. K. Min, H. Y. Zhu, H. L. Chen, Y. X. Weng, J. Y. Dai, Y. B. She, J. X. Xia and H. M. Li, Chem. Eng. J., 2023, 465, 142663 CrossRef CAS.
- G. Wang, Y. Wu, Z. Li, Z. Lou, Q. Chen, Y. Li, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2023, 62, e202218460 CrossRef CAS PubMed.
- Z. W. Zhao, Z. L. Wang, J. F. Zhang, C. F. Shao, K. Dai, K. Fan and C. H. Liang, Adv. Funct. Mater., 2023, 33, 202214470 Search PubMed.
- H. A. E. Omr, R. Putikam, S. P. Feng, M. C. Lin and H. Lee, Appl. Catal., B, 2023, 339, 123103 CrossRef CAS.
- C. J. Ning, J. R. Yang, S. Bai, G. B. Chen, G. H. Liu, T. Y. Shen, L. R. Zheng, S. M. Xu, X. G. Kong, B. Liu, Y. F. Zhao and Y. F. Song, Adv. Funct. Mater., 2023, 33, 202300365 Search PubMed.
- S. Q. Gong, Y. L. Niu, X. Liu, C. Xu, C. C. Chen, T. J. Meyer and Z. F. Chen, ACS Nano, 2023, 17, 4922–4932 CrossRef CAS PubMed.
- Y. Zhao, Z. D. Han, G. Y. Gao, W. Y. Zhang, Y. Qu, H. Y. Zhu, P. F. Zhu and G. F. Wang, Adv. Funct. Mater., 2021, 31, 202104976 Search PubMed.
- W. L. Dai, P. Wang, J. F. Long, Y. Xu, M. Zhang, L. X. Yang, J. P. Zou, X. B. Luo and S. L. Luo, ACS Catal., 2023, 13, 2513–2522 CrossRef CAS.
- H. B. Zhao, J. N. Huang, Q. Qin, H. Y. Chen and D. B. Kuang, Small, 2023, 19, 202302022 Search PubMed.
- M. L. Guan, N. Lu, X. Zhang, Q. W. Wang, J. Bao, G. Y. Chen, H. Yu, H. M. Li, J. X. Xia and X. Z. Gong, Carbon Energy, 2023, 6, e420 CrossRef.
- H. J. Lu, N. Uddin, Z. H. Sun, Z. B. Chen, Z. Mahfoud, Y. L. Wu, A. A. Wibowo, Z. C. Su, X. M. Yin, C. S. Tang, X. Z. Liao, S. P. Ringer, X. S. Zhao, A. T. S. Wee, M. Bosman and Z. Y. Yin, Nano Energy, 2023, 115, 108684 CrossRef CAS.
- X. Y. Jiang, J. D. Huang, Z. H. Bi, W. J. Ni, G. Gurzadyan, Y. A. Zhu and Z. Y. Zhang, Adv. Mater., 2022, 34, 202109330 Search PubMed.
- D. Li, C. J. Zhou, X. L. Shi, Q. Zhang, Q. Song, Y. M. Zhou and D. L. Jiang, Appl. Surf. Sci., 2022, 598, 153843 CrossRef CAS.
- J. Y. Wang, Y. Z. Li, J. T. Zhao, Z. Xiong, Y. C. Zhao and J. Y. Zhang, Catal. Sci. Technol., 2022, 12, 3454–3463 RSC.
- Y. Y. Yu, Y. He, P. Yan, S. Y. Wang and F. Dong, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, 2307320120 CrossRef.
- B. W. Zhou, Y. J. Ma, P. F. Ou, Z. W. Ye, X. Y. Li, S. Vanka, T. Ma, H. D. Sun, P. Wang, P. Zhou, J. K. Cooper, Y. X. Xiao, I. A. Navid, J. Pan, J. Song and Z. T. Mi, Nat. Catal., 2023, 6, 987–995 CrossRef CAS.
- N. Y. Huang, B. Li, D. J. Wu, Z. Y. Chen, B. Shao, D. Chen, Y. T. Zheng, W. J. Wang, C. Z. Yang, M. Gu, L. Li and Q. Xu, Angew. Chem., Int. Ed., 2024, 63, e202319177 CrossRef CAS PubMed.
- D. W. Zhao, Y. M. Xuan, K. Zhang and X. L. Liu, ChemSusChem, 2021, 14, 3293–3302 CrossRef CAS.
- W. Lyu, Y. Liu, J. Y. Zhou, D. T. Chen, X. Zhao, R. Q. Fang, F. L. Wang and Y. W. Li, Angew. Chem., Int. Ed., 2023, 62, 202310733 CrossRef.
- Y. Zhang, H. L. Shi, S. Y. Zhao, Z. L. Chen, Y. Y. Zheng, G. M. Tu, S. X. Zhong, Y. L. Zhao and S. Bai, Small, 2024, 20, 202304050 Search PubMed.
- Y. X. Miao, Y. X. Zhao, S. Zhang, R. Shi and T. R. Zhang, Adv. Mater., 2022, 34, 202200868 Search PubMed.
- C. L. Tan, M. Y. Qi, Z. R. Tang and Y. J. Xu, ACS Catal., 2023, 13, 8317–8329 CrossRef CAS.
- S. P. Mo, X. Y. Zhao, S. D. Li, L. L. Huang, X. Zhao, Q. M. Ren, M. Y. Zhang, R. S. Peng, Y. A. Zhang, X. B. Zhou, Y. M. Fan, Q. L. Xie, Y. B. Guo, D. Q. Ye and Y. F. Chen, Angew. Chem., Int. Ed., 2023, 62, 202313868 CrossRef PubMed.
- G. P. Zhang, X. X. Li, D. Y. Chen, N. J. Li, Q. F. Xu, H. Li and J. M. Lu, Adv. Funct. Mater., 2023, 33, 202308553 Search PubMed.
- R. Z. Chen, S. H. Chen, L. Q. Wang and D. S. Wang, Adv. Mater., 2023, 36, 202304713 Search PubMed.
- M. Jung, C. W. Kim, S. Y. Kim, A. U. Pawar, D. K. Lee and Y. S. Kang, Adv. Energy Mater., 2022, 12, 202202160 Search PubMed.
- C. W. Kim, Y. S. Son, M. J. Kang, D. Y. Kim and Y. S. Kang, Adv. Energy Mater., 2016, 6, 201501754 Search PubMed.
- C. W. Kim, S. J. Yeob, H. M. Cheng and Y. S. Kang, Energy Environ. Sci., 2015, 8, 3646–3653 RSC.
- A. U. Pawar, C. W. Kim, M. J. Kang and Y. S. Kang, Nano Energy, 2016, 20, 156–167 CrossRef CAS.
- C. W. Kim, A. U. Pawar, T. Hawari, N. H. Ahn, D. K. Lee, L. Yang, R. P. Sivasankaran, J. Tang, Z. B. Zhuo and Y. S. Kang, Sci. Rep., 2022, 12, 20045–20046 CrossRef.
- A. U. Pawar, U. Pal, J. Y. Zheng, C. W. Kim and Y. S. Kang, Appl. Catal., B, 2022, 303, 120921 CrossRef CAS.
- L. A. Li, Z. C. Chai, W. Jin, H. Sun, J. H. He, G. Q. Wu and W. W. Xia, J. Alloys Compd., 2023, 932, 167658 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |