
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNear-infrared photocatalysis with cyanines: synthesis, applications and perspectives
Nicolas
Sellet†
 ,
Johanna
Frey†
,
Morgan
Cormier
* and
Jean-Philippe
Goddard
*
,
Johanna
Frey†
,
Morgan
Cormier
* and
Jean-Philippe
Goddard
*
Laboratoire d'Innovation Moléculaire et Applications (LIMA), UMR 7042, Université de Haute-Alsace (UHA), Université de Strasbourg, CNRS, Mulhouse, 68100, France. E-mail: morgan.cormier@uha.fr; jean-philippe.goddard@uha.fr
First published on 1st May 2024
Abstract
Cyanines are organic dyes bearing two aza-heterocycles linked by a polymethine chain. Excited states, fluorescence, redox activity, and energy transfer are interesting properties of cyanines which have been used by chemists. Moreover, they are easily accessible and highly tunable. For all these reasons, cyanines are often selected for applications like fluorescent probes, phototherapy and photovoltaics. However, considering cyanines as photocatalysts is a new field of investigation and has been sparsely reported in the literature. This field of research has been launched on the basis of near-infrared light photocatalysis. With a deeper NIR light penetration, the irradiation is compatible with biological tissues. Due to the longer wavelengths that are involved, the safety of the operator can be guaranteed. In this perspective review, the photophysical/redox properties of cyanines are reported as well as their preparations and applications in modern synthetic approaches. Finally, recent examples of cyanine-based NIR-photocatalysis are discussed including photopolymerization and organic synthesis.
Introduction
Photocatalysis is a powerful method to promote transformations induced by light as a source of renewable energy.1 To do so, a photocatalyst is required to achieve the first activation step of the reaction. Mechanistically, the excited photocatalyst can promote either a single electron transfer (SET) or an energy transfer (EnT) to a substrate. Various applications have been developed based on this concept including water splitting,2 organic synthesis,3 photopolymerization,4 and life-science.5 Until 2019, most of the photocatalysts were developed in the UV-visible range6 but these wavelengths present some drawbacks. First, the irradiation under UV-visible light can be energetic enough to provide side-reactions and selectivity issues especially when a polyfunctionalized molecule is considered.7 Secondly, light penetration required for scale-up procedures or photopolymerization is higher with a longer light wavelength like near infrared (NIR) light.8 Finally, the NIR-light is biocompatible since the biological tissue does not absorb NIR-photons resulting in a non-damageable irradiation (Fig. 1a).9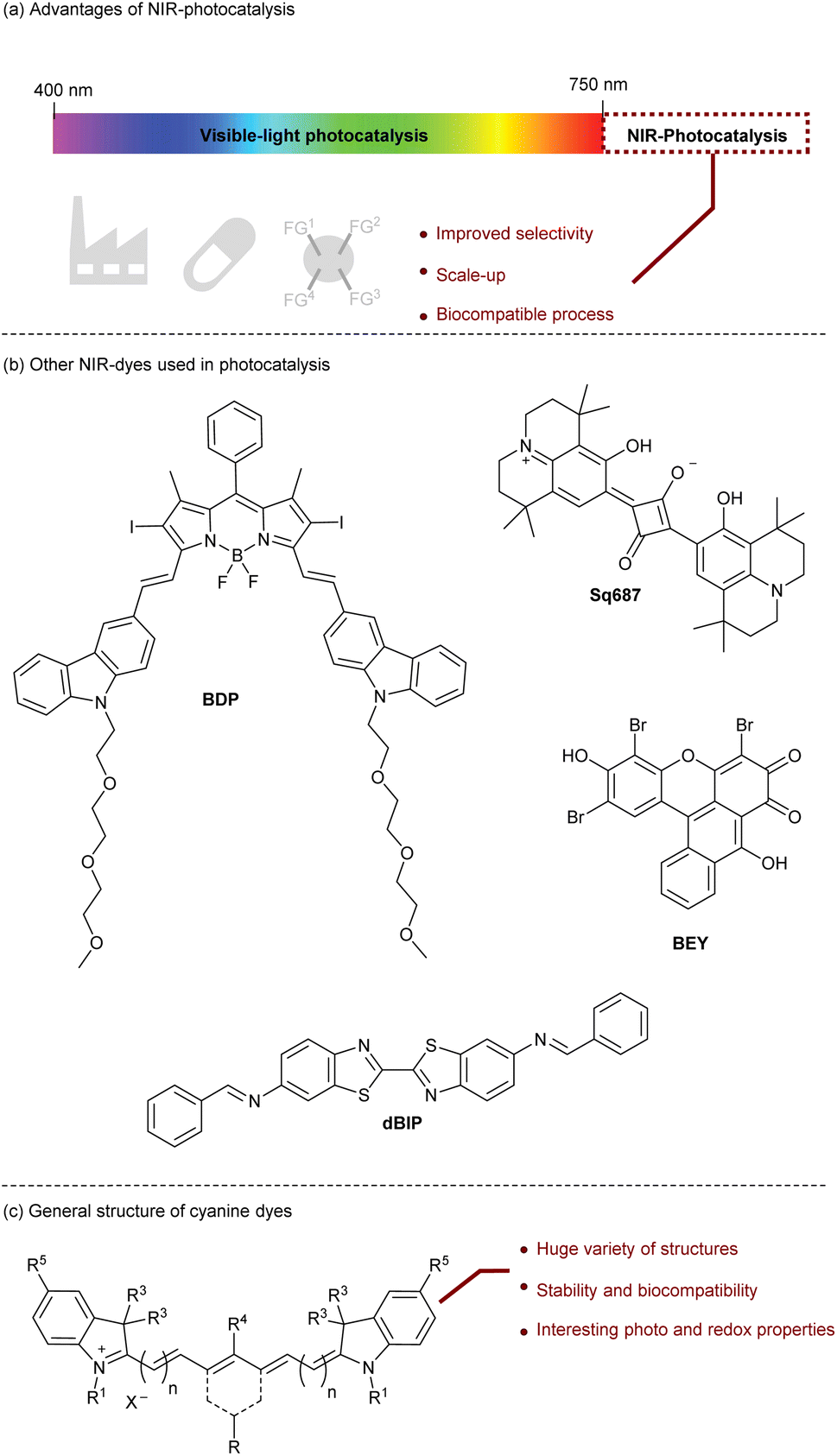 | ||
| Fig. 1 Context. | ||
Two main families of NIR-photocatalysts have been developed.10 Organometallic complexes such as Os-complexes,11 Zn-phthalocyanines,12 and Ru-polypyridyl13 offer a large panel of photophysical and redox properties that can be tuned by modifying the ligands. However, their preparation and sensitivity are usually the limiting factors that prevent some potential applications. Additionally, some organometallic complexes (aluminum 2,3-naphthalocyanines,14 metal-phthalocyanines,15,16 bacteriochlorophyll a17) have been employed as photocatalysts to trigger radical photopolymerization in an efficient manner. The second family of NIR-photocatalysts is based on organic structures (Fig. 1b). In this case, redox potentials are currently rather limited, but such catalysts are accessible, stable, and non-toxic. For instance, the squaraine family drew the attention of chemists thanks to their ability to promote SET, they have been used to initiate photopolymerization by the reduction of an iodonium salt18 but also to promote oxidation of amines in organic processes with Sq687.19 Recently, other organic dyes such as bridged Eosin Y (BEY),20 BODIPY (BDP)21 and dibenzothiazole (dBIP)22 have also been considered and applied for organic synthesis. The huge variety of complementary NIR-organic dyes opens new perspectives to reach interesting redox potentials and photophysical properties that can be applied in molecular and macro-molecular chemistry.
To complete this collection of NIR-organic photocatalysts, cyanine derivatives are probably the most valuable candidates. Cyanines are formed by a polymethine chain with odd carbons surrounded by two heterocycles that offer an unlimited number of structures by changing and functionalizing these two parts (Fig. 1c). In this perspective, we will present the recent advances in the development of cyanine photocatalysts, a field of research that has been developed recently and showing a great potential.
Cyanines: properties and non-catalytic applications
The cyanine family is huge and can be classified according to the chemical structures of the compounds. Among them we can cite closed chain cyanines, streptocyanines and merocyanines. They are all different in their terminal parts which can be a heterocyclic ring (symmetric or non-symmetric), a non-heterocyclic ring, an amino or carbonyl group for example. Cyanines can also be classified according to the number of methine groups present: monomethine, trimethine, pentamethine, heptamethine.23 Their absorption properties are clearly influenced by the size of the polymethine chain. Indeed, adding an extra conjugated carbon–carbon double bond provides a red-shift of about 100 nm in the absorption spectrum. For applications in the near-infrared domain, heptamethine cyanines are the most relevant dyes and this perspective review will be focused on this type of scaffold. Furthermore, other important photophysical properties are tunable thanks to modifications of specific parts of the cyanines (Fig. 2a).24 The synthesis and modification of cyanines have undergone intensive investigations, empowering chemists to design a wide range of structures. This will be further explained in the dedicated section of this review. Adding cyclohexyl (or cyclopentyl) to the polymethine chain brings rigidification of the structure and prevents photoisomerization and photodegradation which slightly improve the quantum yield of fluorescence (ϕ). The photostability is also enhanced by attaching sterically congested groups in various parts of the cyanine. For instance, in indole-based cyanines, the addition of a dimethyl group at position 3 of the indolenium ring is common and efficient for this purpose. The photostability of cyanines is a crucial parameter that should be considered for their promising applications. Usually, the degradation is due to dioxygen reactive species25 (1O2 or superoxide anion) but recently it was also demonstrated that cyanines can undergo iterative phototruncations of the polymethine chain.26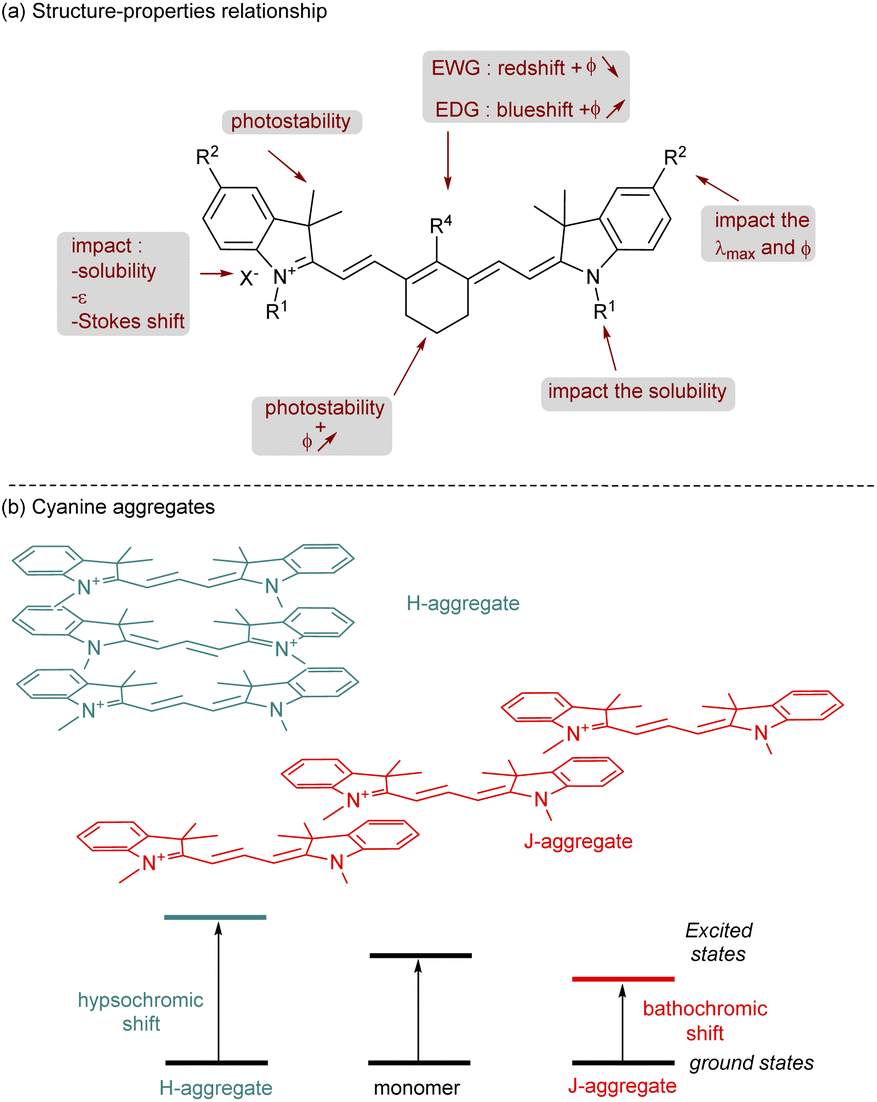 | ||
| Fig. 2 Photophysical properties of cyanines. | ||
Then, photophysical properties are highly customizable by changing the heterocycle or by adding a functional group on it (modification of R2). Since the conjugation on the π-system is modified the value of the λmax and quantum yield of fluorescence is affected. Playing with functional groups at the meso-position (R4) will also change these properties. If R4 is an electron-withdrawing group (EWG) the absorption is shifted to the red part of the spectrum and the quantum yield of the fluorescence is decreasing, the opposite results are obtained with electron-donating groups (EDG). The nature of the counter anion is also important, classically halide, perchlorate, hexafluorophosphate, nitrate or tetrafluoroborate are selected. The counter anion affects the charge distribution which impacts the ε and the Stokes shift. Moreover, it plays a crucial role in cyanine solubility.27
This parameter is also tunable through the modification of R1 attached to the nitrogen of the heterocycle. Chemists were mainly focused on the solubility of cyanines in water for various applications and to do so, adding a sulfonate or a carboxylate is a determining factor to reach this goal. This modification is also crucial to avoid aggregate formation in solution (especially in water). Indeed, in solution cyanines are able to form, by self-association, supramolecular structures known as H-aggregates or J-aggregates (Fig. 2b).28 An H-aggregate is obtained when the structure is parallel to form a sandwich-type arrangement. The head-to-tail organization of these dyes is called the J-aggregate. The H-aggregate involves a hypsochromic shift in absorption whereas the corresponding J-aggregate gives a bathochromic shift and a narrow Stokes shift. Quantum yields are substantially impacted by the formation of aggregates, and most of the time H-aggregates are unable to reach high values of ϕ. Except for specific applications with J-aggregates,29 usually these conditions are chosen to inhibit the formation of aggregates.
In addition to their photophysical properties, cyanines possess interesting redox properties from their ground states and excited states. However, the structure–redox property relationship is less commented in the literature.30 It was proved that extending the polymethine chain increases the Eox1/2 and decreases the Ered1/2. The effect of donor–acceptor properties of functional groups attached to the cyanine is important but not often regular. Nonetheless, the impact of counter-anions on redox properties is less pronounced.
Thanks to these remarkable properties, heptamethine cyanines have been used for many applications (Fig. 3). Their fluorescence properties and biocompatibility are currently used for daily analysis as biosensors (pH, ROS, thiols, H2O2…), for bioimaging or even for drug delivery by photocaging.31 For all these applications, a modification of the meso position is required to attach a functional group used for the biomolecule recognition.
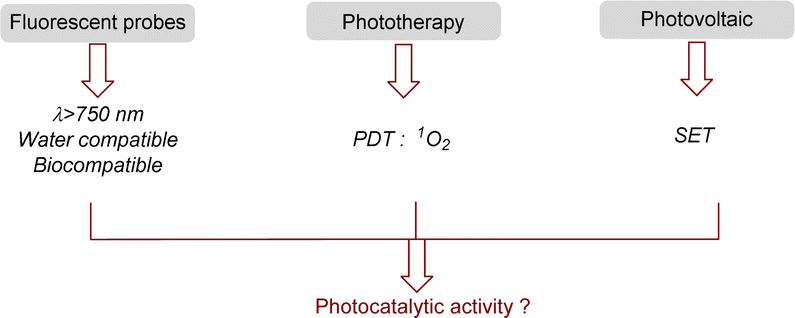 | ||
| Fig. 3 Main applications of NIR-cyanine dyes. | ||
In addition to the fluorescence, cyanines present other interesting features in the excited state that are useful for various applications. They are able to convert light energy into thermal energy through a non-radiative process, this property is applied to photothermal therapy (PTT).32 Even more interestingly, cyanines can promote energy transfer (EnT) from their triplet state to photosensitize 3O2 into 1O2. This photosensitizer character is applied for photodynamic therapy (PDT).33 To reach the best quantum yields of 1O2, simple modifications of the cyanine can be done to favor inter-system crossing (ISC) such as the introduction of heavy atoms (iodine,34 selenium35), anthracene36 or paramagnetic scaffolds.37 Cyanines are also well-known for their applications in solar-cells as photosensitizers.38 Their ability to transfer an electron via a SET process from their exited state is useful in Dye-Sensitized Solar Cells (DSSCs) but this property could also be employed in photocatalytic reactions.
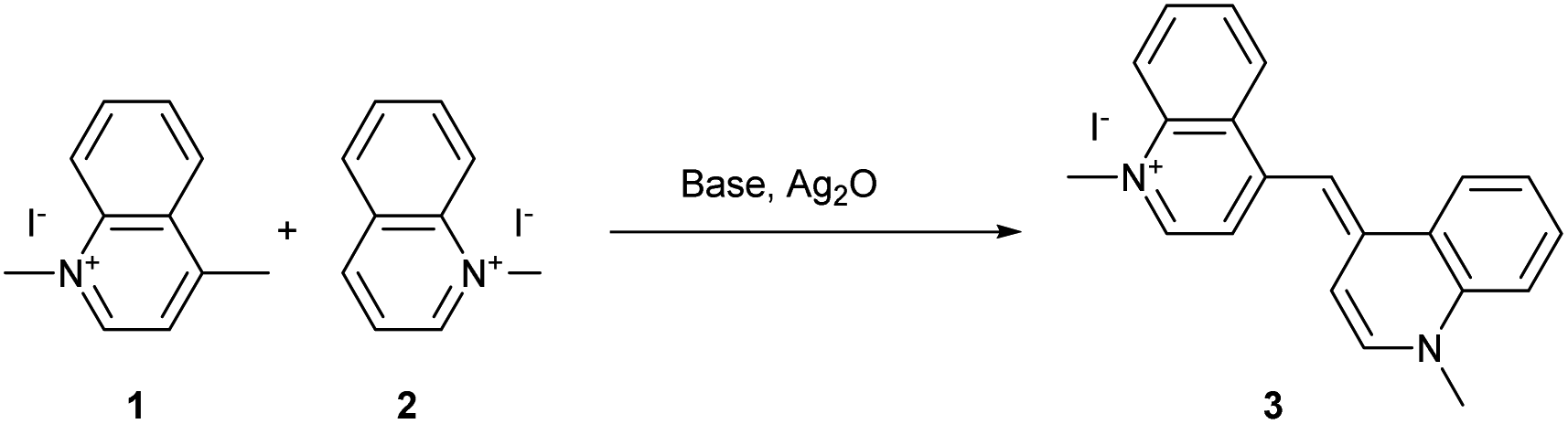 | ||
| Scheme 1 First cyanine dye synthesis described by Greville Williams. | ||
During the 19th century, newly discovered dyes were often quickly applied in the textile industry as dyeing agents. Unfortunately, the first cyanine 3 was not useful for the dyeing industry due to its low photostability.40 To improve this parameter, other cyanines have been developed through different synthetic routes. For decades, different trimethine, tetramethine, and pentamethine cyanine dyes were synthesized.41 In this section dedicated to cyanine synthesis, heptamethine cyanine dyes (seven carbons on the central chain) will be highlighted according to their absorption in the near infrared window and their application in photocatalysis. The first classical synthesis of heptamethine dyes involves two key substrates: a quaternary indolenium 8 and a modified Schiff base 10 (Scheme 2).
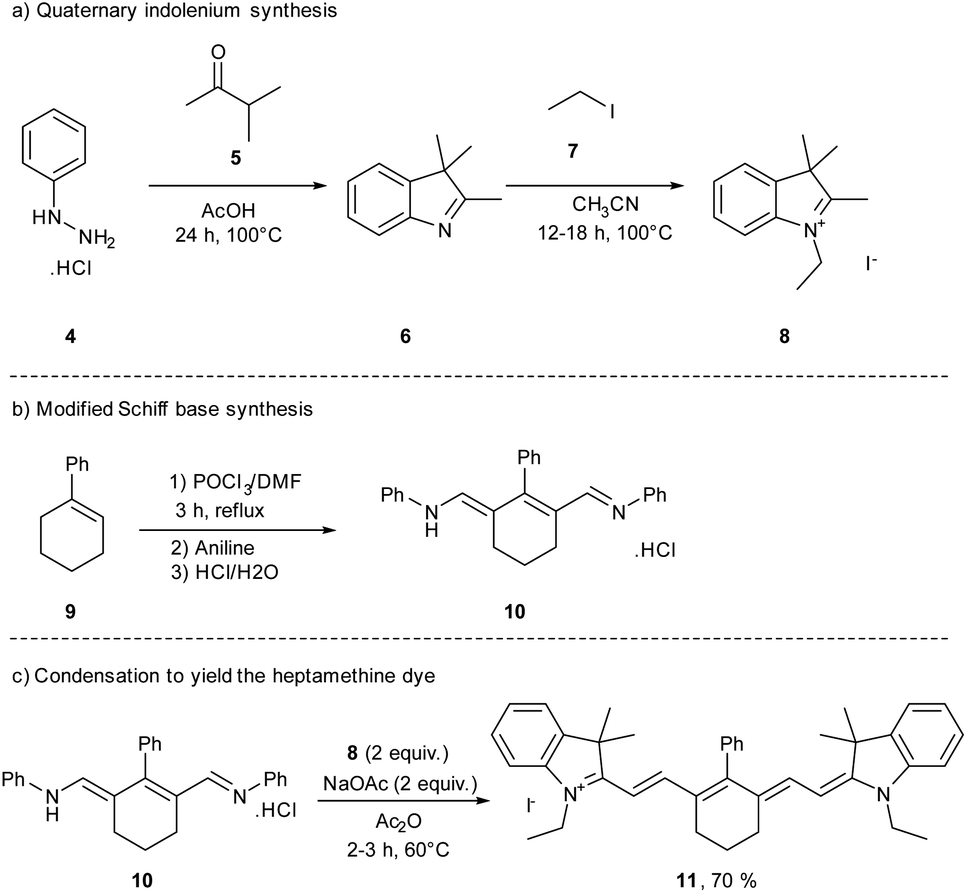 | ||
| Scheme 2 Classical synthesis route toward heptamethine dyes. | ||
To obtain the quaternary indolenium moiety 8, the strategy starts with a Fischer indole type synthesis from the phenylhydrazine hydrochlorate salt 4 and a ketone. The 3-methylbutan-2-one 5 is often used to improve the stability of the cyanine by increasing the steric hindrance. Then, the alkyl iodide 7 is substituted by the indole 6 to quaternize the indolenine nitrogen and afford the indolenium 8. The modified Schiff base 10 was obtained through a Vilsmeier–Haack formylation, starting from a cyclohexenyl derivative 9. At the end, a condensation between 8 and 10, with a ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, under basic conditions, gives the corresponding cyanine dye with 70% yield for the reported example. This approach is reasonably general and by modifications of different building blocks several cyanines can be prepared in moderate to excellent yields.
:1, under basic conditions, gives the corresponding cyanine dye with 70% yield for the reported example. This approach is reasonably general and by modifications of different building blocks several cyanines can be prepared in moderate to excellent yields.
Even more interesting, by modification of the key intermediates, unsymmetrical cyanine dyes have been obtained.42 Indeed, this synthesis route was applied with two different quaternary indolenium moieties condensed on a Schiff base derivative to obtain unsymmetrical dyes. This strategy was pushed further with a microwave assisted solid-phase synthesis, in which a resin-bound hemicyanine 12 is prepared by addition under microwave conditions of indolenium into a grafted Schiff base. Then 12 is released after reaction with a quaternary ammonium salt 13 to afford the unsymmetrical cyanine 14 in 86% yield (Scheme 3).43
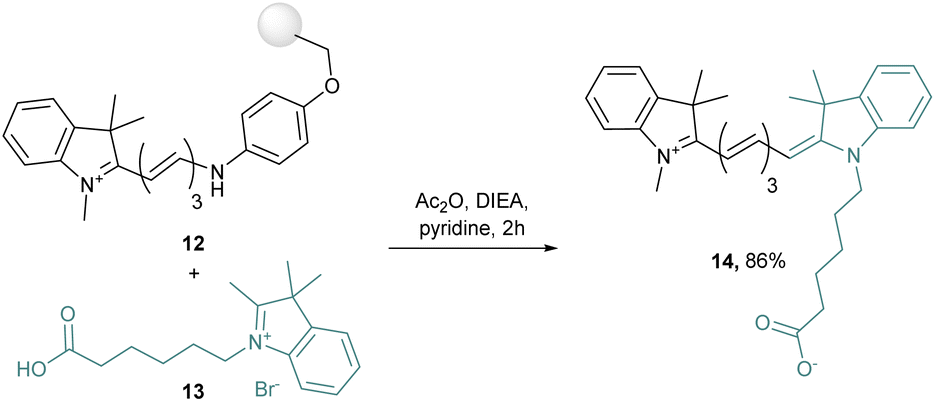 | ||
| Scheme 3 Heterogeneous unsymmetrical cyanine synthesis. | ||
This strategy has been extended to many different cyanines and takes advantage of the solid-support since only one purification in the final step is required.
Another route of heptamethine synthesis was then developed by the group of Klàn in 2019 (ref. 44) using a modification of the classical Zincke reaction.45 The authors reported the use of an open form of the pyridinium salt 15 (also called the Zincke salt) obtained by substitution of two equivalents of the p-bromoaniline 16. The resulting intermediate 17 was then reacted with quaternary indolenium moieties 18 under mild conditions giving the heptamethine dyes with good yields (up to 90%) with high purity after simple precipitation in ether (Scheme 4a).
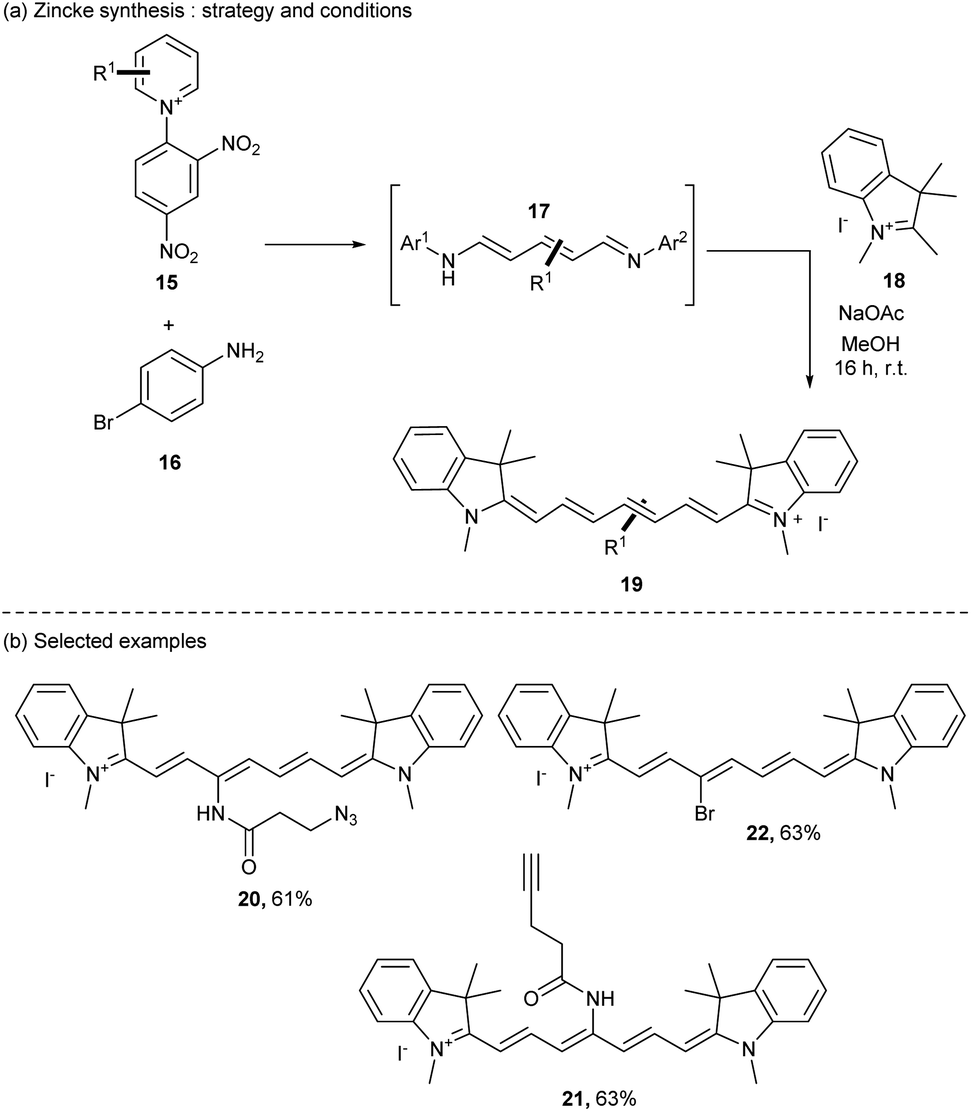 | ||
| Scheme 4 The Zincke route strategy. | ||
This strategy is versatile and offers a large diversification of the heptamethine dyes. For instance, cyanine dyes bearing interesting functional groups on the heptamethine scaffold like azide (20), terminal alkyne (21), and bromide (22) were prepared in 61%, 63% and 63% yields respectively (Scheme 4b). This strategy opens the door to direct modifications of the conjugated chain of cyanines to tune their properties or even to attach a linker which is useful for many applications. A new update has been achieved in the pyridinium strategy to prepare cyanines with a huge diversity of structures using pyridinium benzoxazole (PyBox). This pyridinium is well tolerant to various functional groups offering original substituted cyanines.46
Recently, a novel synthesis of cyanines was published by the group of Mo and the strategy is based on the one-pot preparation of a Stenhouse salt intermediate from the corresponding furfural 23 (Scheme 5).47 The Stenhouse salt 24 is generated in situ by the addition of aniline into the furfural derivatives. This intermediate is placed under reflux and basic conditions with quaternary indolenium 25 to yield the symmetrical cyanines 25 which contain various functional groups in the polymethine chain.
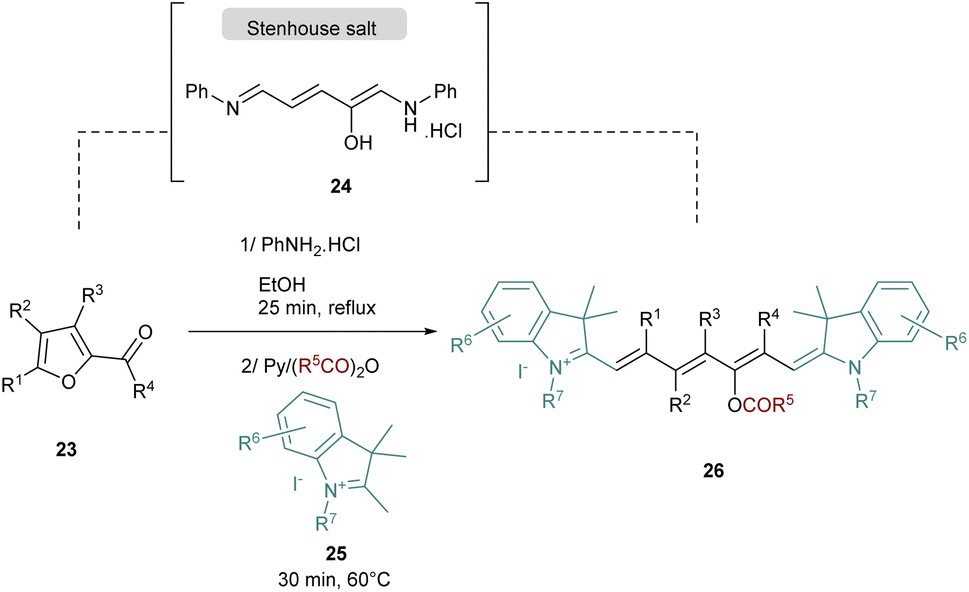 | ||
| Scheme 5 One-pot strategy to yield cyanine dyes via Stenhouse salt. | ||
It should be noted that this strategy requires the evaporation of the ethanol after the Stenhouse salt formation to avoid the cyclization and the formation of the corresponding cyclopentenone. The synthesis of unsymmetrical cyanine dyes following this strategy was also reported after a slight modification of the procedure. The key intermediate 27 to obtain unsymmetrical dyes is a “half-cyanine” bearing a furan moiety which is stable at room temperature and easy to isolate (Scheme 6). The furan containing precursor 27 is then opened using zinc chloride and p-anisidine. The corresponding intermediate in this reaction is a half Stenhouse salt which is directly involved in the condensation with a second different quaternary indolenium 28. This strategy allows access to unsymmetrical heptamethine cyanines 29 with a shorter reaction time and with a high degree of functionalization.
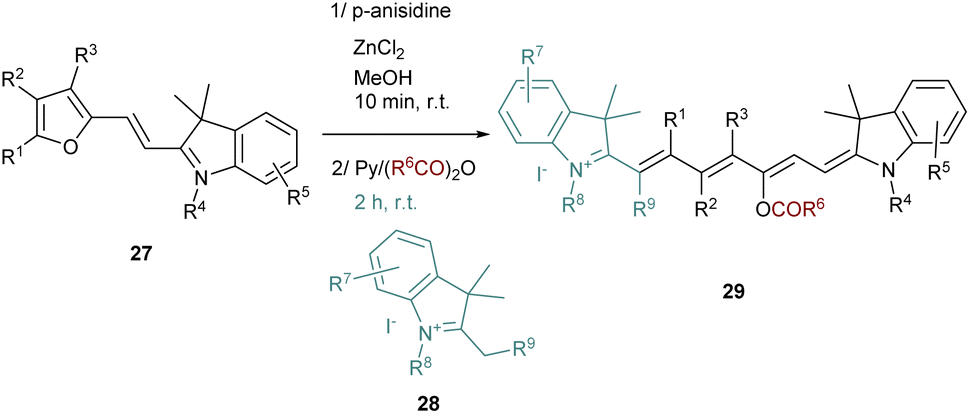 | ||
| Scheme 6 Unsymmetrical dyes obtained through the one-pot strategy. | ||
The meso position is crucial to tune the properties of cyanines, and several methods have been investigated to modify it. For instance, Suzuki–Miyaura coupling reactions were described to be efficient in this specific position for different halogenated cyanines (Scheme 7).48 Thus, chlorocyanines 30 react in water with the boronic acid 31 in the presence of a catalytic amount of Pd(Ph3)4 to reach the arylated cyanine 32. This meso-chlorine atom substitution creates a new robust C–C bond under environment-friendly aqueous conditions with good yields (68–83%). The cross-coupling strategy is rather general and other conditions are compatible such as Heck, Sonogashira or Stille cross-coupling reactions.49
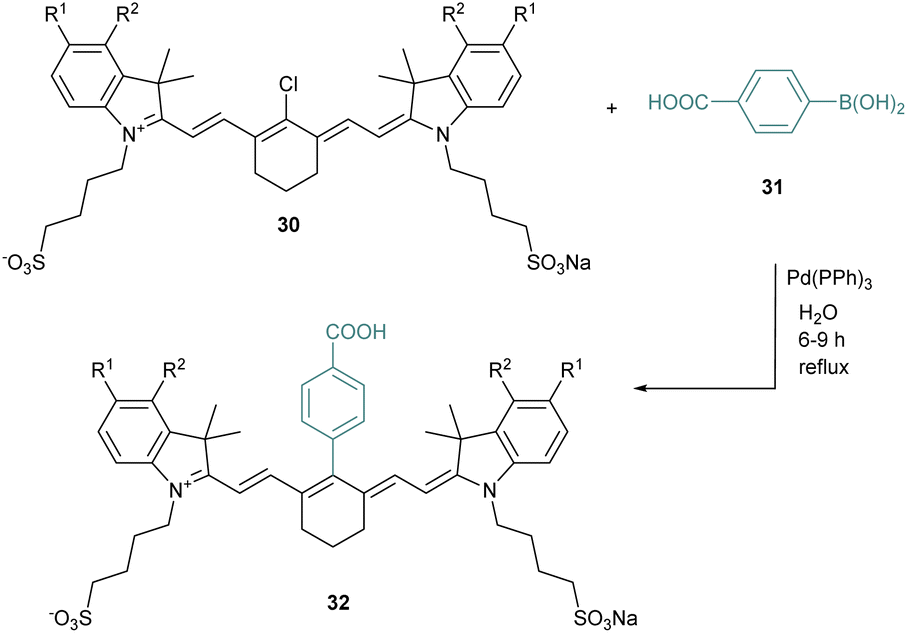 | ||
| Scheme 7 Suzuki–Miyaura coupling on heptamethine dyes. | ||
The halogenated heptamethine at the meso position can also be substituted by nucleophiles as proposed originally by the group of Strekowski for the introduction of alcoholates and thiolates (Scheme 8).50 They reported that the substitution would probably follow an SRN1 (substitution radical nucleophilic unimolecular) pathway which was proved by EPR experiments. This strategy was then applied to various nucleophiles51 including alkyl amines,52 azides,53 selenols54 or acetylacetone (acac).55 This approach is versatile and generally provides an interesting tool to quickly tune the properties of cyanine derivatives.
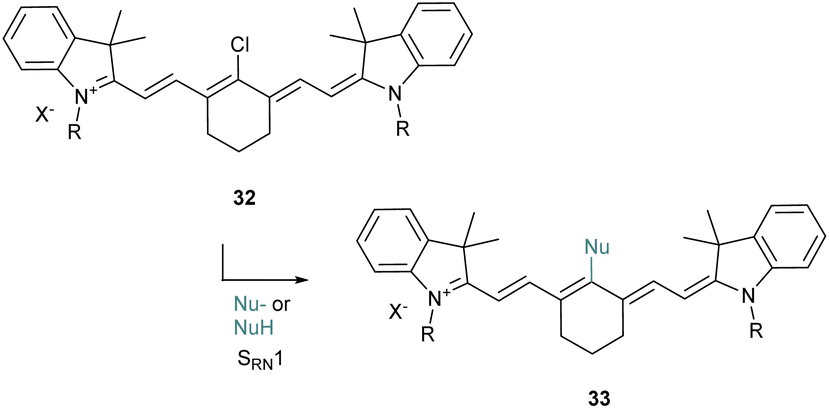 | ||
| Scheme 8 SRN1 pathway for heptamethine diversification. | ||
Cyanines as NIR-photocatalysts
Among the NIR-photosensitizers,58,59 cyanines have been intensively employed but, originally, they were used for photopolymerization with visible light. In this case, the radical polymerization occurs through the oxidation of the borate salt which is generally the counter anion of the cyanine (Scheme 9a).60 In the excited state the cyanine can oxidize the borate through a Single Electron Transfer (SET) to produce the first radical species able to initiate the polymerization. This system was also extended to the reduction of 2,4,6-tris(chlorodifluoromethyl)-1,3,5-triazine 34 in a three-component system with borate to achieve under visible light conditions the first step of the radical photopolymerization (Scheme 9b).61
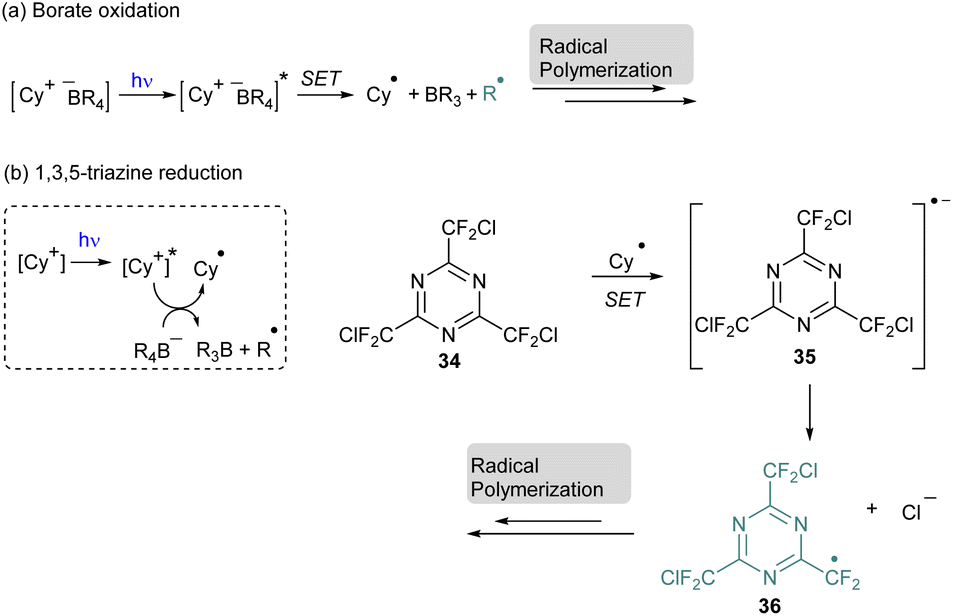 | ||
| Scheme 9 Visible-light photopolymerization with cyanine: (a) borate oxidation; (b) 1,3,5 triazine reduction. | ||
Then, with the development of NIR sources of irradiation, these interesting tools were adapted to be used in this corresponding window. The polymerization triggered by NIR-cyanine dyes proceeds through different pathways. The initiation step can occur via a SET process from the excited state of the cyanine to generate a radical species (Scheme 10a).62,59a–f In this case iodonium salts are the most employed radical initiators. A specific design of this cationic species has been achieved and usually the iodonium 37 bearing a p-tertbutyl group is commonly used. The effect of the counteranion is also very important especially to solve solubility issues. After the first step, the reduction of the iodonium salt, the aryl radical species can initiate radical polymerization. Additionally, this photocatalytic system is compatible with phosphines, this additive prevents polymerization inhibition of acrylates due to oxygen. Another possible pathway is to use the intermediate radical species derived from the cyanine as a source of proton. In this case, after the first SET with an iodonium salt the cyanine intermediate undergoes decomposition to provide a proton able to trigger a cationic polymerization. The release of the proton was monitored by UV-vis analyses with rhodamine B. Cyanines are also known to produce heat through a non-radiative process from their excited state. This phenomenon increases the reaction media temperature and can activate a thermal initiator, which in turn can enable the polymerization (Scheme 10b). For instance, cyanines in combination with peroxides result in the photothermal radical polymerization of acrylates or methacrylate.63 Apart from peroxide, thermal initiators, such as alkoxyamine or azo derivatives, are also efficient for this purpose. Recently, the photothermal process has been applied to iodonium salts in combination with electron donating anilines. In this case a Charge Transfer Complex (CTC) is formed in situ, which allows the formation of an aryl radical and a subsequent free-radical polymerization after photothermal activation.59g For all of them, the temperature increase is tunable and depends on the concentration of the dye and the light power. It is often discussed that a photoredox and a photothermal process occur simultaneously which reinforced the efficiency of cyanines as photosensitizers. For instance, a combination of these two processes allows the formation of interpenetrating polymer networks (IPNs).59e,f
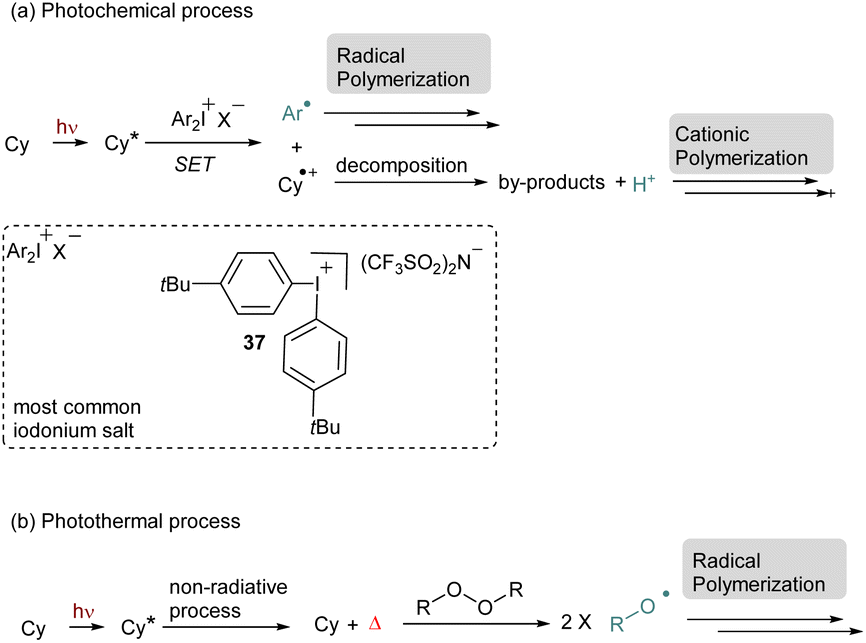 | ||
| Scheme 10 General mechanism of NIR-polymerization with cyanines. | ||
The photoinitiation driven by cyanines and NIR-light has proved to be compatible with classical monomers used in regular UV-vis photopolymerization (Scheme 11a). For the free-radical polymerization, acrylates, such as TMPTA (trimethylolpropane triacrylate) or TPGDA (tri(propylene glycol) diacrylate) and methacrylates like HPMA (hydroxypropyl methacrylate) or UDMA (urethane dimethacrylate) are often considered for this application. However, epoxides (ERL-4211, Epikote 828) or oxetanes are mainly employed in cationic polymerization.
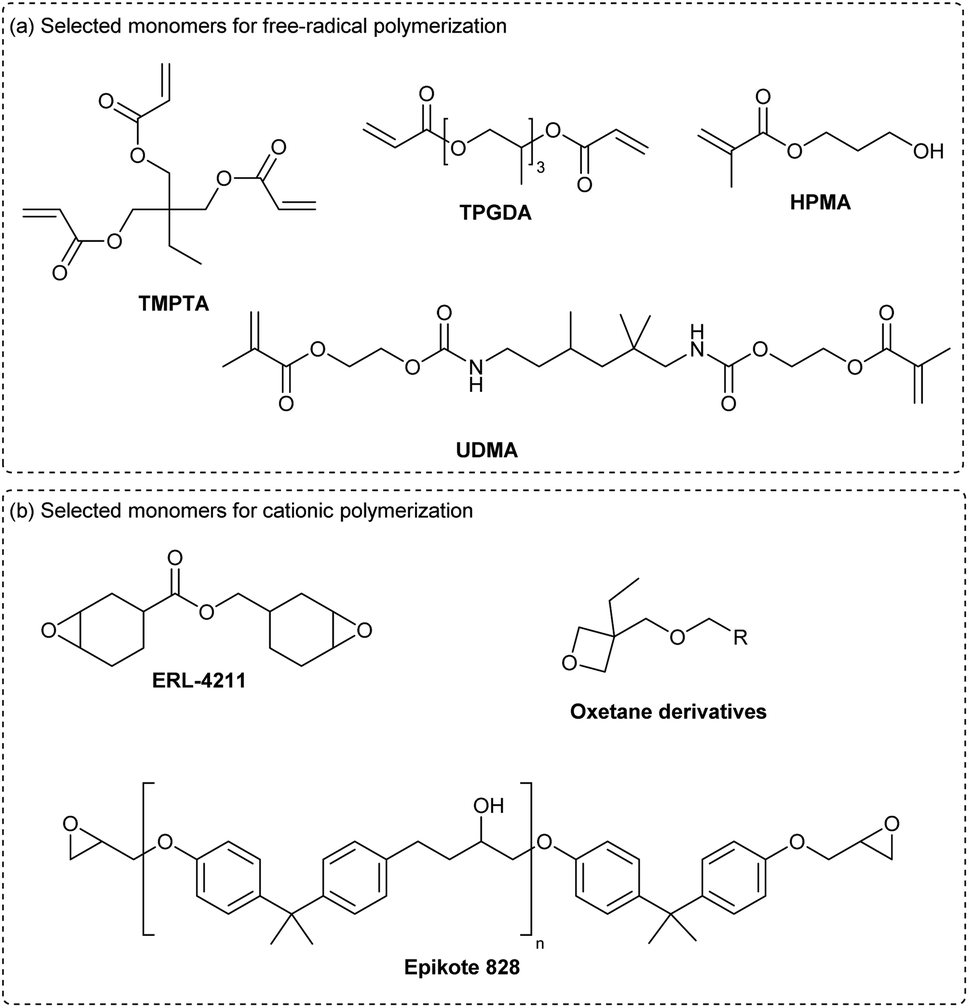 | ||
| Scheme 11 Selected examples of monomers compatible with cyanine-based NIR photopolymerization. | ||
Among the huge number of accessible cyanines available, photopolymerization is often achieved by commercial ones. The group of Strehmel has provided many photophysical and redox data for the most efficient cyanines.59d Interestingly, the structures of the best photosensitizers are relatively different. A selection of cyanines that are efficient for radical and cationic polymerization are presented below: Cy791, Cy746, Cy794, Cy854 (Fig. 4).
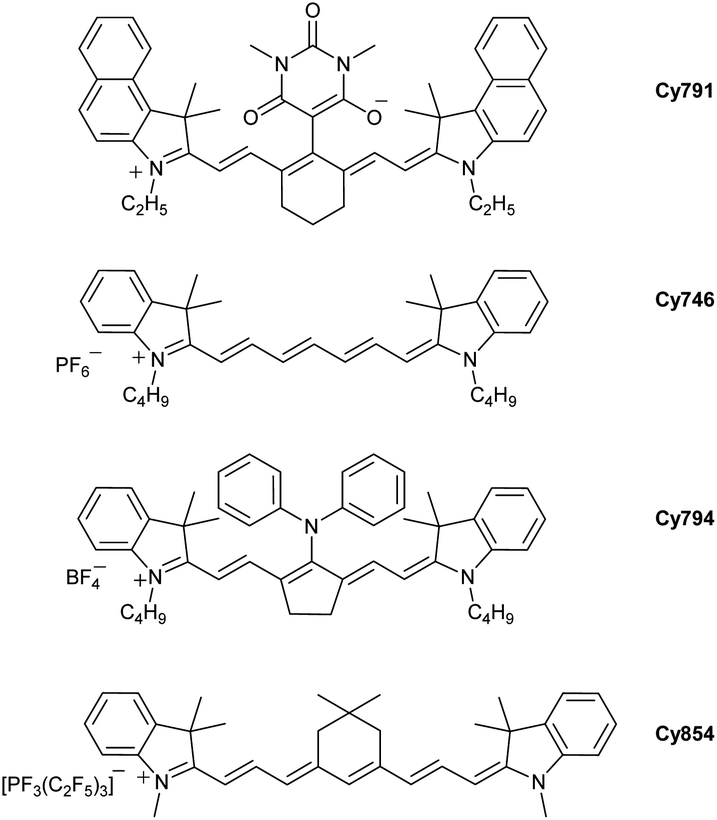 | ||
| Fig. 4 Examples of cyanines used as photosensitizers. | ||
Still in the context of polymer chemistry, Strehmel and Yagci reported a cyanine-based NIR-photoinduced Copper-catalyzed Azide–Alkyne Cycloaddition click reaction (CuAAC).64 This approach has been employed to prepare bloc copolymers such as polystyrene-β-poly(ε-caprolactone) (PS-β-PCL) 40 (Scheme 11a). Mechanistically, CuAAC requires a Cu(I) catalyst to achieve the cycloaddition under mild conditions. However, Cu(I) salts are sensitive to air and several solutions have been used to reduce stable Cu(II)-complexes in situ to Cu(I)-complexes. The cyanine Cy791 bearing a barbiturate group in the meso position was found to be efficient for NIR-photoreduction of CuBr2 to promote the CuAAC (Scheme 11b).65 In order to regenerate the initial state of the cyanine after the first SET, N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) is added in the solution as a co-reductant (Scheme 12).
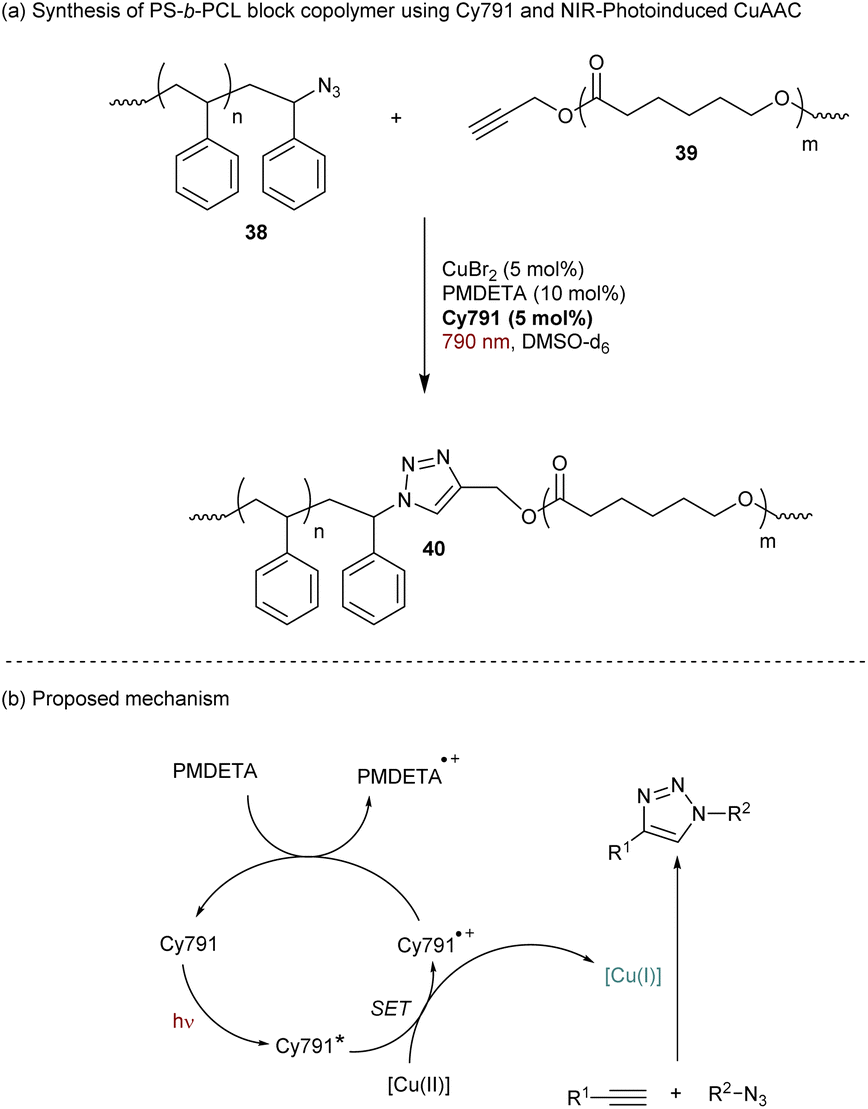 | ||
| Scheme 12 Copper-catalyzed alkyne–azide cycloaddition under NIR light using Cy791. | ||
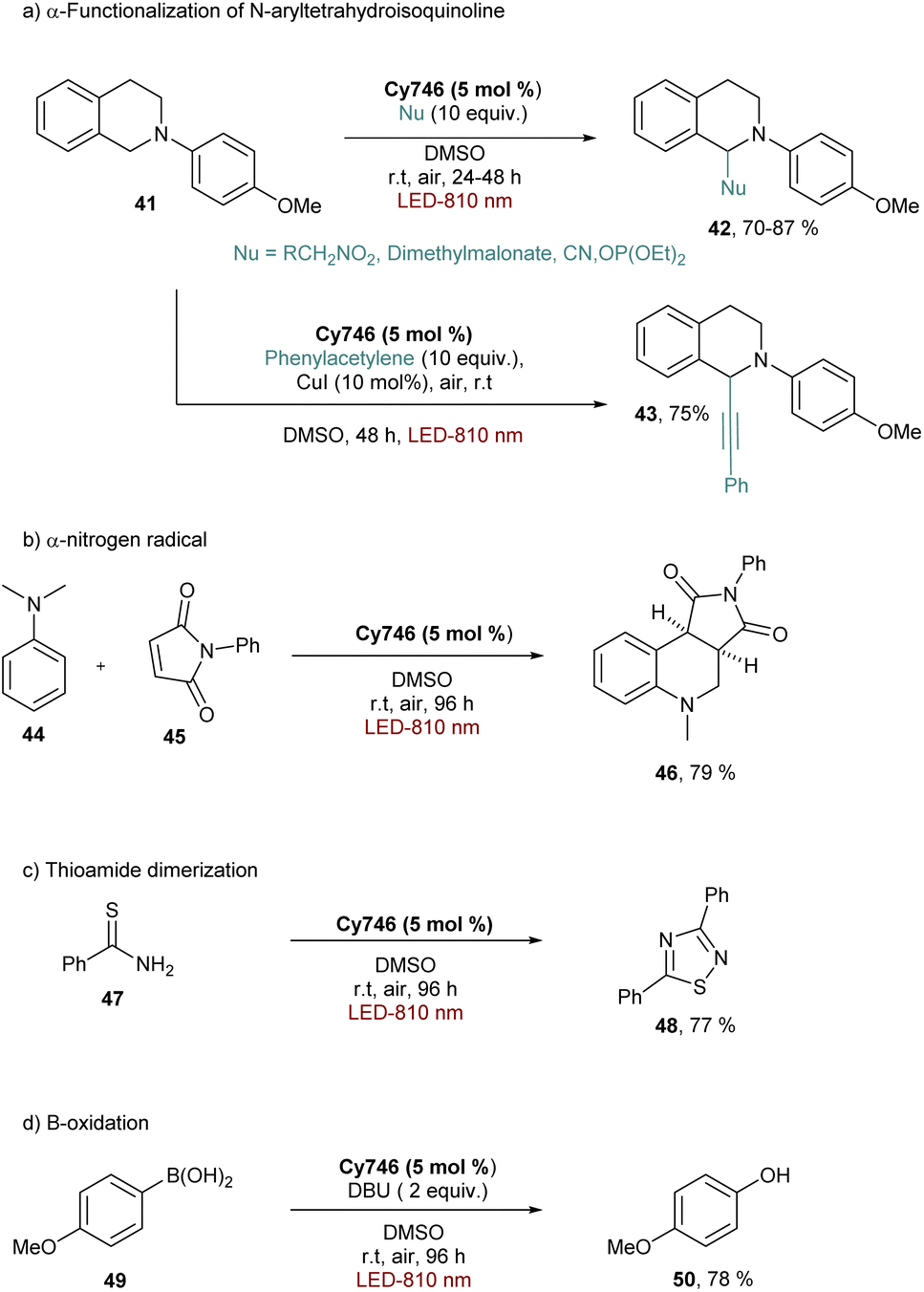 | ||
| Scheme 13 Oxidation performed with Cy746, the first generation of cyanine-based NIR-photocatalysts. | ||
Reduction of substrates can also take place, as exemplified by the reduction of the Umemoto salt to form the trifluoromethylated alkene 52 (Scheme 14). In this case, the mechanism involves a SET between the cyanine and the sulfonium to provide the trifluoromethyl radical which is trapped by a radical acceptor. The reaction is compatible in water media since adding H2O as a co-solvent and nucleophile provided the hydroxytrifluoromethylated adduct 54.
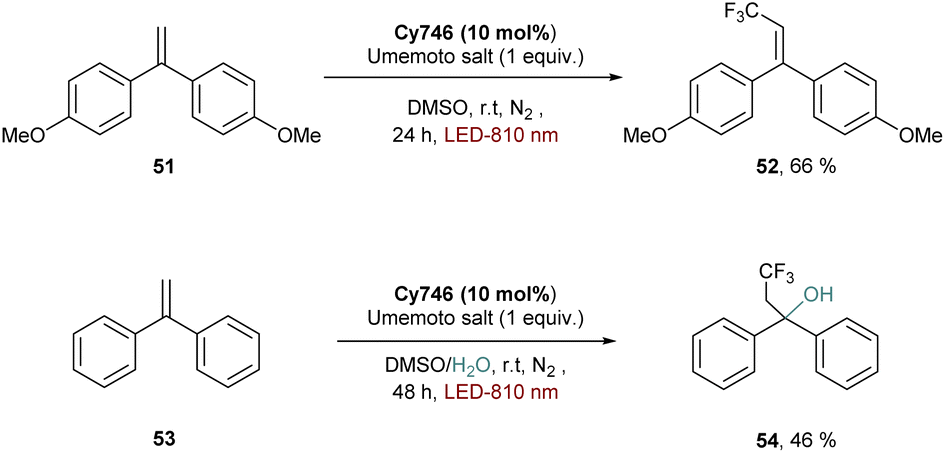 | ||
| Scheme 14 Reduction of the Umemoto salt with Cy746. | ||
Cy746 proved its versatility and synthetic utility, however, one major issue is the slow kinetics of the mentioned reactions. To improve this aspect, a second generation of cyanine dyes was described.68 A range of cyanines were synthesized and fully characterized with the estimation of their redox potentials in the excited state. They were tested in the aza-Henry and the trifluoromethylation reactions. Concerning the oxidation of tetrahydroisoquinolines, a secondary amino group on the meso position of the heptamethine chain was able to accelerate kinetics of different reactions (Scheme 15).
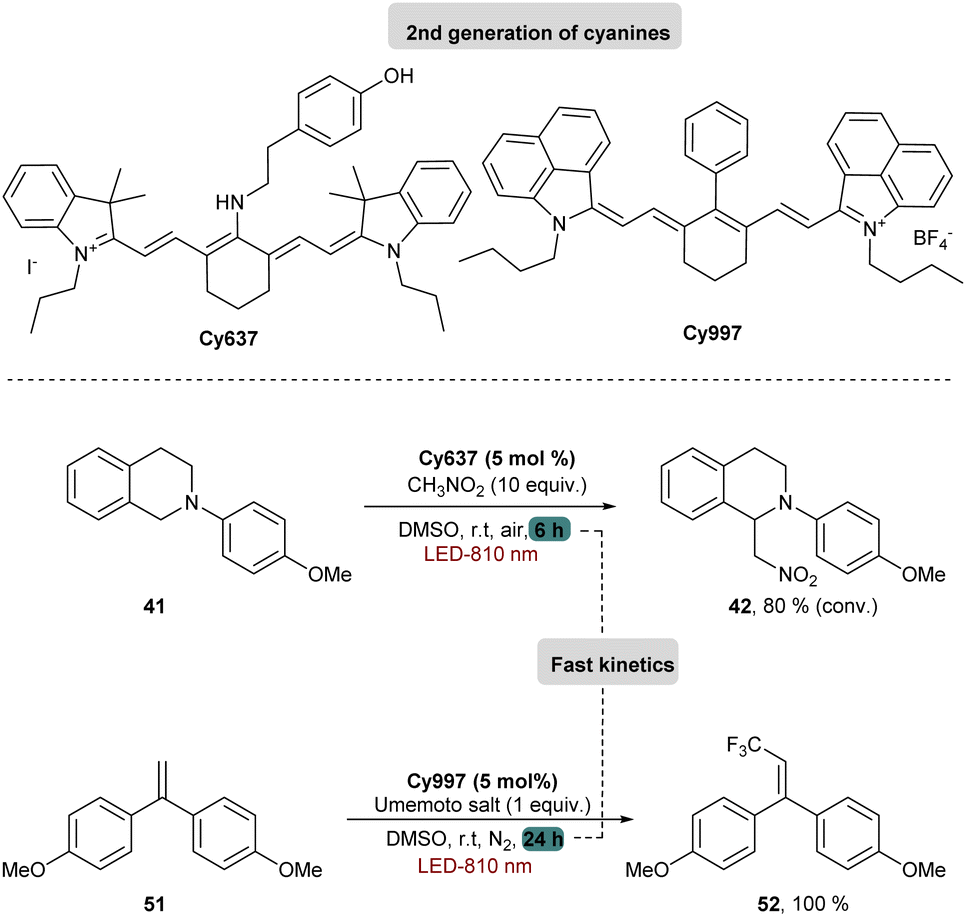 | ||
| Scheme 15 Improvement of kinetic rates with the second generation of cyanines. | ||
These cyanines, which have the lowest 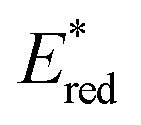 values (0.68–075 V), allow conversion rates greater than 50% after only 6 h of reaction. The best result was obtained with Cy637 in converting 41 in 80% after only 6 h compared to the 24 h needed with Cy746. However, comparing all cyanines did not allow demonstration of the obvious correlation between their redox potential in the excited state and the rate of the aza-Henry reaction. This can again suggest competitive mechanisms between SET and EnT.
values (0.68–075 V), allow conversion rates greater than 50% after only 6 h of reaction. The best result was obtained with Cy637 in converting 41 in 80% after only 6 h compared to the 24 h needed with Cy746. However, comparing all cyanines did not allow demonstration of the obvious correlation between their redox potential in the excited state and the rate of the aza-Henry reaction. This can again suggest competitive mechanisms between SET and EnT.
Looking at the trifluoromethylation kinetics of 51, a plateau is generally reached after 4 h of reaction, suggesting a possible degradation of the photocatalyst by non-selective trifluoromethylation of the cyanine. The best results were obtained with Cy997 (Scheme 15) which was among the fastest at the beginning of the reaction, and more importantly was the only one to achieve full conversion after 24 h. Its stability was a key parameter for efficient and rapid conversion, despite the low potential of this cyanine (−0.38 V). Cy997 also proved its efficiency in giving the trifluoromethylated product under irradiation at lower energy 940 nm (with 83% yield instead of 100% at 810 nm).
The ability of cyanines to process via an EnT mechanism was also explored for organic transformations. Indeed, Cy746 can also perform the photosensitization of oxygen to generate 1O2 and oxidize the furfural 55 in 45% yield, the tetraphenylcyclopentadienone 57 (81% yield) and the 9,10-dimethylanthracene 59 to form the endoperoxide 60 quantitatively (Scheme 16).61,62Cy746 is highly adaptable and can also promote the photosensitization of the vinyl azide 61 to obtain, after formation of a nitrene in situ, the disubstituted pyrrole 62 in 68% isolated yield.
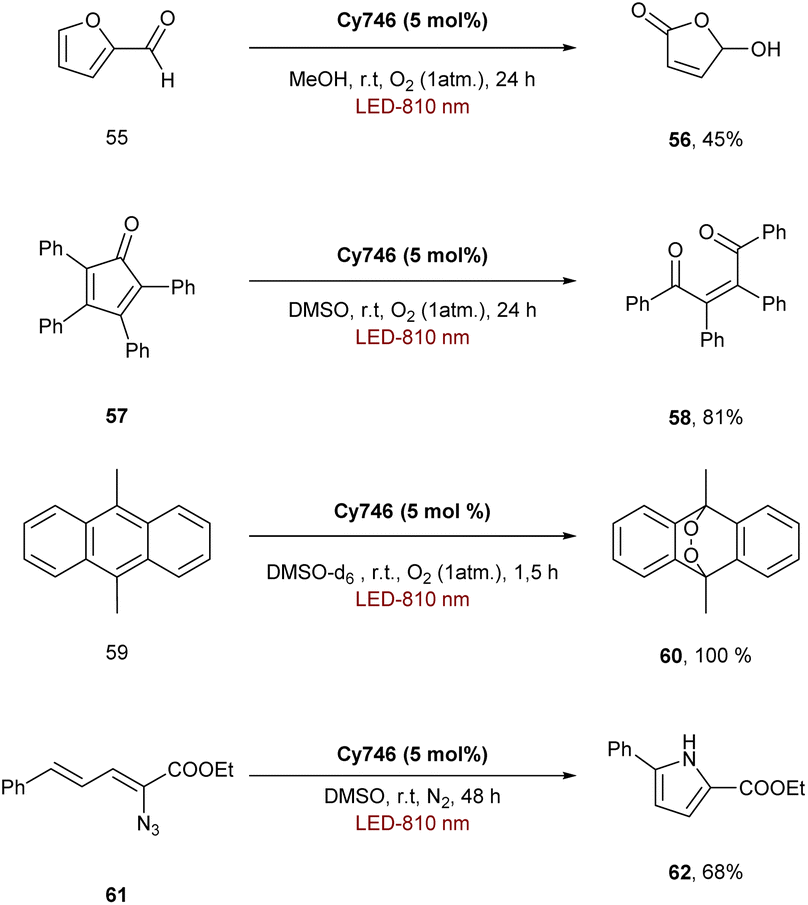 | ||
| Scheme 16 Photosensitization transformations performed with Cy746. | ||
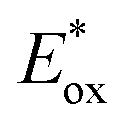 value (−1.19 V vs. SCE) which is still far from that of Sq637 (
value (−1.19 V vs. SCE) which is still far from that of Sq637 (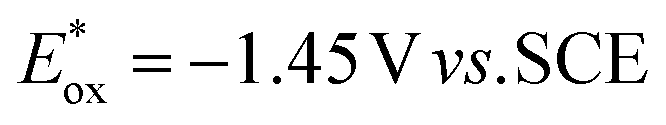 ). For the other cyanines,
). For the other cyanines, 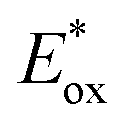 is between −0.80 and −1.00 V vs. SCE, consistent with other PCat in the same range of wavelengths of absorption (BEY and MB). Surprisingly, Cy997, while producing optimal results in the Umemoto salt reduction, exhibits the least negative potential in the excited state (
is between −0.80 and −1.00 V vs. SCE, consistent with other PCat in the same range of wavelengths of absorption (BEY and MB). Surprisingly, Cy997, while producing optimal results in the Umemoto salt reduction, exhibits the least negative potential in the excited state (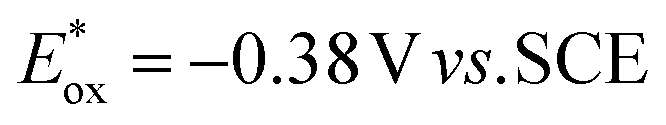 ). This outcome confirms that the redox potential alone cannot be regarded as the unique parameter in optimizing the photocatalyst. In this case, the stability of sensitive NIR-dyes is also very crucial. Interestingly, the
). This outcome confirms that the redox potential alone cannot be regarded as the unique parameter in optimizing the photocatalyst. In this case, the stability of sensitive NIR-dyes is also very crucial. Interestingly, the 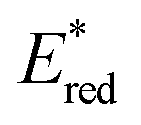 seems to be less impacted by the cyanine structure since very similar values have been determined (0.70 to 0.8 V) for these selected cyanines. Except for Cy854 (not determined in the literature), the current cyanine photocatalysts present limited
seems to be less impacted by the cyanine structure since very similar values have been determined (0.70 to 0.8 V) for these selected cyanines. Except for Cy854 (not determined in the literature), the current cyanine photocatalysts present limited 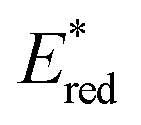 compared to other organic NIR-PCat. Indeed, Sq637, BEY and MB seem to be more powerful oxidants in the excited state (
compared to other organic NIR-PCat. Indeed, Sq637, BEY and MB seem to be more powerful oxidants in the excited state (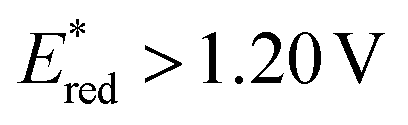 ), providing a perspective of amelioration for the design of new cyanine-based photocatalysts.
), providing a perspective of amelioration for the design of new cyanine-based photocatalysts.
| PCat | λ max (nm) | E red/ox vs. SCEb (V) |
|
||
|---|---|---|---|---|---|
| E ox | E red |
|
|
||
a In DMSO.
b
E
red/ox was measured by cyclic voltammetry (CV) with respect to KCl (3 M)/Ag/AgCl reference electrode and corrected vs. SCE. The solvent was DMSO.
c
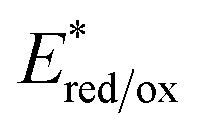 was calculated according to the Rehm–Weller equation.
d In acetonitrile.
e
E
red/ox was measured by cyclic voltammetry (CV) with respect to an Ag/AgCl reference electrode in CH3CN.
f In MeCN/H2O (4:1).
g
E
red/ox was measured by cyclic voltammetry (CV) with respect to Ag/AgCl and referenced to SCE in DCM. was calculated according to the Rehm–Weller equation.
d In acetonitrile.
e
E
red/ox was measured by cyclic voltammetry (CV) with respect to an Ag/AgCl reference electrode in CH3CN.
f In MeCN/H2O (4:1).
g
E
red/ox was measured by cyclic voltammetry (CV) with respect to Ag/AgCl and referenced to SCE in DCM.
|
|||||
| Cy637 68 | 637 | 0.38 | −0.85 | −1.19 | 0.72 |
| Cy746 68 | 746 | 0.61 | −0.68 | −0.88 | 0.80 |
| Cy791 68 | 791 | 0.41 | −0.93 | −1.00 | 0.48 |
| Cy794 68 | 794 | 0.58 | −0.64 | −0.80 | 0.74 |
| Cy854 59d | 854d | 0.46e | −0.33e | n.d | n.d |
| Cy997 68 | 997 | 0.68 | −0.27 | −0.38 | 0.79 |
| Sq687 19 | 687 | 0.23 | −0.43 | −1.45 | 1.22 |
| BEY 20 | 731 | 1.16 | −0.34 | −0.81 | 1.63 |
| Methylene blue (MB)3d | 664f | 1.13g | −0.30g | −0.68 | 1.60 |
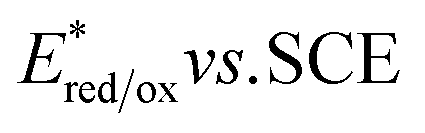
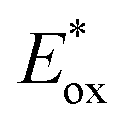
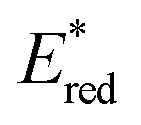
Conclusions and perspectives
In conclusion, NIR-photocatalysis is only in its launching sequence. Currently, the main applications of NIR-photocatalysts are devoted to radical and cationic photopolymerization which combine SET and photothermal processes. The application in organic synthesis is promising as it was demonstrated with a first generation of cyanine (Cy746) in oxidation, reduction and photosensitization. Then an updated and optimized version was reported with Cy637 and Cy997 which provides efficient and fast reactions in the oxidation of tetrahydroisoquinolines and reduction of the Umemoto salt.Due to the benefits of the NIR photocatalysis compared to visible light and the potential applications of this field of research, the photochemistry driven by cyanines will play a key role in the future. However, to enrich the toolbox of cyanine photocatalysts, there are still some questions that need to be solved. First, the rationalization of the structure–photocatalytic activity relationship is required to accelerate the design of this family of photocatalysts. This is particularly interesting, to reach higher 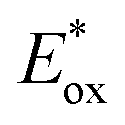 which is currently limited to 0.80 V for most of the cyanines. Concerning the energy transfer (EnT), the application is currently mainly restricted to 1O2 generation and the design of cyanines with high triplet energy would open the door to a new class of reactions triggered by NIR-light. Currently the NIR-light used either for the SET or EnT process is around 800 nm, and the highest wavelength irradiation has been achieved at 940 nm for the reduction of the Umemoto reagent with Cy997. Would it be possible to shift the light irradiation to very low energetic wavelengths (>1000 nm)? Finally, the stability of cyanines is still a challenge to achieve a more efficient process. Although some modifications on cyanines have been done to protect the heptamethine chain, these conjugated molecules remain sensitive to radical species generated in the photocatalytic process. Thus, the ideal cyanine-based photocatalyst will combine all these parameters in one structure and the future developments will tend towards its design.
which is currently limited to 0.80 V for most of the cyanines. Concerning the energy transfer (EnT), the application is currently mainly restricted to 1O2 generation and the design of cyanines with high triplet energy would open the door to a new class of reactions triggered by NIR-light. Currently the NIR-light used either for the SET or EnT process is around 800 nm, and the highest wavelength irradiation has been achieved at 940 nm for the reduction of the Umemoto reagent with Cy997. Would it be possible to shift the light irradiation to very low energetic wavelengths (>1000 nm)? Finally, the stability of cyanines is still a challenge to achieve a more efficient process. Although some modifications on cyanines have been done to protect the heptamethine chain, these conjugated molecules remain sensitive to radical species generated in the photocatalytic process. Thus, the ideal cyanine-based photocatalyst will combine all these parameters in one structure and the future developments will tend towards its design.
Author contributions
N. S, J. F, M. C. and J.-P. G. conceptualized, wrote and edited the perspective.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Université de Haute-Alsace (UHA) for providing facilities and funding.Notes and references
- R. C. McAtee, E. J. McClainand and C. R. J. Stephenson, Trends Chem., 2019, 1, 111 CrossRef CAS.
- (a) S. Nishioka, F. E. Osterloh, X. Wang, T. E. Mallouk and K. Maeda, Nat. Rev. Methods Primers, 2023, 3, 42 CrossRef CAS; (b) K. Villa, J.-R. Galán-Mascarós, N. López and E. Palomares, Sustainable Energy Fuels, 2021, 5, 4560 RSC; (c) K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655 CrossRef CAS.
- (a) M. S. Kwon and Y. Lee, Eur. J. Org Chem., 2020, 6028 Search PubMed; (b) J.-P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924 CrossRef CAS PubMed; (c) M. H. Shaw, J. Twiltonand and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (e) R. A. Angnes, Z. Li, C. R. D. Correia and G. B. Hammond, Org. Biomol. Chem., 2015, 13, 9152 RSC; (f) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- (a) V. Ferraro, C. R. Adam, A. Vranic and S. Bräse, Adv. Funct. Mater., 2024, 34, 2302157 CrossRef CAS; (b) C. Wu, N. Corrigan, C.-H. Lim, W. Liu, G. Miyake and C. Boyer, Chem. Rev., 2022, 122, 5476 CrossRef CAS; (c) N. Zivic, M. Bouzrati-Zerelli, A. Kermagoret, F. Dumur, J.-P. Fouassier, D. Gigmes and J. Lalevée, ChemCatChem, 2016, 8, 1617 CrossRef CAS.
- L. Candish, K. D. Collins, G. C. Cook, J. J Douglas, A. Gómez-Suárez, A. Jolit and S. Keess, Chem. Rev., 2022, 122, 2907 CrossRef CAS PubMed.
- (a) D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343 CrossRef PubMed; (b) M. Freitag, N. Möller, A. Rühling, C. A. Strassert, B. J. Ravoo and F. Glorius, ChemPhotoChem, 2019, 3, 24 CrossRef CAS.
- H. Bartling, A. Eisenhofer, B. König and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 11860 CrossRef CAS.
- E. B. Corcoran, J. P. McMullen, F. Lévesque, M. K. Wismer and J. R. Naber, Angew. Chem., Int. Ed., 2020, 59, 11964 CrossRef CAS.
- (a) B. F. Buksh, S. D. Knutson, J. V. Oakley, N. B. Bissonnette, D. G. Oblinsky, M. P. Schwoerer, C. P. Seath, J. B. Geri, F. P. Rodriguez-Rivera, D. L. Parker, G. D. Scholes, A. Ploss and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144(14), 6154 CrossRef CAS PubMed; (b) S. Mallidi, S. Anbil, A.-L. Bulin, G. Obaid, M. Ichikawa and T. Hasan, Theranostics, 2016, 6, 2458 CrossRef CAS PubMed; (c) A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710 CrossRef CAS; (d) K. Szacilowski, W. Macyk, A. Drzewiecka-Matuszek, M. Brindell and G. Stochel, Chem. Rev., 2005, 105, 2647 CrossRef CAS.
- N. Sellet, M. Cormier and J.-P. Goddard, Org. Chem. Front., 2021, 8, 6783 RSC.
- (a) D. C. Cabanero, J. A. Nguyen, C. S. Cazin, S. P. Nolan and T. Rovis, ACS Catal., 2023, 13, 4384 CrossRef CAS; (b) N. Eng Soon Tay, K. Ah Ryu, J. L. Weber, A. K. Olow, D. C. Cabanero, D. R. Reichman, R. C. Oslund, O. O. Fadeyl and T. Rovis, Nat. Chem., 2023, 15, 101 CrossRef PubMed; (c) S. L. Goldschmid, N. Eng Soon Tay, C. L. Joe, B. C. Lainhart, T. C. Sherwood, E. M. Simmons, M. Sezen-Edmonds and T. Rovis, J. Am. Chem. Soc., 2022, 144, 22409 CrossRef CAS; (d) S. L. Goldschmid, E. Bednarova, L. R. Beck, K. Xie, N. E. S. Tay, B. D. Ravetz, J. Li and C. L. Joe, Synlett, 2022, 33, 247 CrossRef CAS; (e) I. M. Ogbu, D. M. Bassani, F. Robert and Y. Landais, Chem. Commun., 2022, 58, 8802 RSC; (f) B. D. Ravetz, N. E. S. Tay, C. L. Joe, M. Sezen-Edmonds, M. A. Schmidt, Y. Tan, J. M. Janey, M. D. Eastgate and T. Rovis, ACS Cent. Sci., 2020, 6, 2053 CrossRef CAS.
- (a) C. Grundke, R. C. Silva, W. R. Kitzmann, K. Heinze, K. T. de Oliveira and T. Opatz, J. Org. Chem., 2022, 87, 5630 CrossRef CAS; (b) Y. Katsurayama, Y. Ikabata, H. Maeda, M. Segi, H. Nakai and T. Furuyama, Chem.–Eur. J., 2022, 28, e202103223 CrossRef CAS PubMed.
- G. Han, G. Li, J. Huang, C. Han, C. Turro and Y. Sun, Nat. Commun., 2022, 13, 2288 CrossRef CAS.
- C. Ng, Z. Wu, T. Zhang, A. Phong Dang, Y. Yao, A. R. J. Nelson, S. W. Prescott, A. Postma, G. Moad, C. J. Hawker and C. Boyer, Macromolecules, 2023, 56, 7898 CrossRef.
- J. Sun, S. Ren, H. Zhao, S. Zhang, X. Xu, L. Zhang and E. Cheng, ACS Macro Lett., 2023, 12, 165 CrossRef CAS PubMed.
- N. Corrigan, J. Xu and C. Boyer, Macromolecules, 2016, 49, 3274 CrossRef CAS.
- S. Shanmugam, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55, 1036 CrossRef CAS.
- A. Bonardi, F. Bonardi, G. Noirbent, F. Dumur, C. Dietlin, D. Gigmes, J.-P. Fouassier and J. Lalevée, Polym. Chem., 2019, 10, 6505 RSC.
- N. Sellet, M. Sebbat, M. Elhabiri, M. Cormier and J.-P. Goddard, Chem. Commun., 2022, 58, 13759 RSC.
- M. Tanioka, A. Kuromiya, R. Ueda, T. Obata, A. Muranaka, M. Uchiyama and S. Kamino, Chem. Commun., 2022, 58, 7825 RSC.
- L. Zeng, Z. Wang, T. Zhang and C. Duan, Molecules, 2022, 27, 4047 CrossRef CAS.
- B. K. Kundu, G. Han and Y. Sun, J. Am. Chem. Soc., 2023, 145, 3535 CrossRef CAS.
- G. S. Gopika, P. M. Hari Prasad, A. G. Lekshmi, S. Lekshmyapriya, S. Sreesaila, C. Arunima, M. S. Kumar, A. Anil, A. Sreekumar and Z. S. Pillai, Mater. Today: Proc., 2021, 46, 3102 CAS.
- (a) K. Colas, S. Doloczki, M. Posada Urrutia and C. Dyrager, Eur. J. Org Chem., 2021, 15, 2133 CrossRef; (b) L. Feng, W. Chen, X. Ma, S. H. Liu and J. Yin, Org. Biomol. Chem., 2020, 18, 9385 RSC.
- C. Schwechheimer, F. Rönicke, U. Shepers and H.-A. Wagenknecht, Chem. Sci., 2018, 9, 6587 RSC.
- S. S. Matikonda, D. A. Helmerich, M. Meub, G. Beliu, P. Kollmannsberger, A. Greer, M. Sauer and M. J. Schnermann, ACS Cent. Sci., 2021, 7, 1144 CrossRef CAS.
- J. N. Gayton, S. Autry, R. C. Fortenberry, N. I. Hammer and J. H. Delcamp, Molecules, 2018, 23, 3051 CrossRef PubMed.
- J. L. Bricks, Y. L. Slominskii, I. D. Panas and A. P. Demchenko, Methods Appl. Fluoresc., 2018, 6, 012001 CrossRef.
- C. Sun, B. Li, M. Zhao, S. Wang, Z. Lei, L. Lu, H. Zhang, L. Feng, C. Dou, D. Yin, H. Xu, Y. Chen and F. Zhang, J. Am. Chem. Soc., 2019, 141, 19221 CrossRef CAS PubMed.
- (a) A. V. Kulinich, N. A. Derevyanko, A. A. Ishchenko, N. B. Gusyak, I. M. Kobasa, P. P. Romanczyk and S. S. Kurek, Dyes Pigm., 2019, 161, 24 CrossRef CAS; (b) J. R. Lenhard and B. R. Hein, J. Phys. Chem., 1996, 100, 17287 CrossRef CAS; (c) C. Chen, B. Zhou, D. Lu and G. Xu, J. Photochem. Photobiol., A, 1995, 89, 25 CrossRef CAS; (d) J. R. Lenhard and A. D. Cameron, J. Phys. Chem., 1993, 97, 4916 CrossRef CAS; (e) T. Tani, J. Electrochem. Soc., 1973, 120, 254 CrossRef CAS.
- (a) H. Li, H. Kim, F. Xu, J. Han, Q. Yao, J. Wang, K. Pu, X. Peng and J. Yoon, Chem. Soc. Rev., 2022, 51, 1795 RSC; (b) N. G. Medeiros, C. A. Braga, V. S. Camara, R. C. Duarte and F. S. Rodembusch, Asian J. Org. Chem., 2022, 11, e202200095 CrossRef CAS; (c) P. J. Choi, T. I.-H. Park, E. Cooper, M. Dragunow, W. A. Denny and J. Jose, Bioconjugate Chem., 2020, 31, 1724 CrossRef CAS PubMed; (d) A. P. Gorka, R. R. Nani and M. J. Schnermann, Acc. Chem. Res., 2018, 51, 3226 CrossRef CAS PubMed; (e) W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Chem. Rev., 2016, 116, 7768 CrossRef CAS PubMed; (f) A. P. Gorka, R. R. Nani and M. J. Schnermann, Org. Biomol. Chem., 2015, 13, 7584 RSC.
- G. Della Pelle, A. D. Lopez, M. S. Fiol and N. Kostevsek, Int. J. Mol. Sci., 2021, 22, 6914 CrossRef CAS.
- K. Bilici, S. Cetin, E. Celikbas, H. Y. Acar and S. Kolemen, Front. Chem., 2021, 9, 707876 CrossRef CAS PubMed.
- S. M. Usama, S. Thavornpradit and K. Burgess, ACS Appl. Bio Mater., 2018, 1, 1195 CrossRef CAS.
- J. Sun, E. Feng, Y. Shao, F. Lv, Y. Wu, J. Tian, H. Sun and F. Song, ChemBioChem, 2022, 23, e202200421 CrossRef CAS.
- X. Zhao, Q. Yao, S. Long, W. Chi, Y. Yang, D. Tan, X. Liu, H. Huang, W. Sun, J. Du, J. Fan and X. Peng, J. Am. Chem. Soc., 2021, 143, 12345 CrossRef CAS.
- L. Jiao, F. Song, J. Cui and X. Peng, Chem. Commun., 2018, 54, 9198 RSC.
- (a) E. N. Bifari, P. Almeida and R. M. El-Shishtawy, Mater. Today: Proc., 2023, 36, 101337 CAS; (b) W. Ghann, H. Kang, E. Emerson, J. Oh, T. Chavez-Gil, F. Nesbitt, R. Williams and J. Uddin, Inorg. Chim. Acta, 2017, 467, 123 CrossRef CAS.
- C. H. Greville Williams, Earth Environ. Sci. Trans. R. Soc. Edinburgh, 1857, 21, 377 Search PubMed.
- F. M. Hamer, Q. Rev., Chem. Soc., 1950, 4, 327 RSC.
- (a) M. Panigrahi, S. Dash, S. Patel and B. K. Mishra, Tetrahedron, 2012, 68, 781 CrossRef CAS; (b) D. S. Pisoni, L. Todeschini, A. C. A. Borges, C. L. Petzhold, F. S. Rdoembusch and L. F. Campo, J. Org. Chem., 2014, 79, 5511 CrossRef CAS PubMed; (c) G. S. Gopika, P. M. Hari Prasad, A. G. Lekshmi, S. Lekshmypriya, S. Sreesaila, C. Arunima, M. S. Kumar, A. Anil, A. Sreekumar and Z. S. Pillai, Mater. Today: Proc., 2021, 46, 3102 CAS.
- N. G. Medeiros, C. A. Braga, V. S. Câmara, R. C. Duarte and F. S. Rodembusch, Asian J. Org. Chem., 2022, 11, e202200095 CrossRef CAS.
- (a) M. Lopalco, E. N. Koini, J. K. Cho and M. Bradley, Org. Biomol. Chem., 2009, 7, 856 RSC; (b) S. J. Mason, J. L. Hake, J. Nairne, W. J. Cummins and S. Balasubramanian, J. Org. Chem., 2005, 70, 2939 CrossRef CAS PubMed.
- L. Stackova, P. Stacko and P. Klàn, J. Am. Chem. Soc., 2019, 141, 7155 CrossRef CAS PubMed.
- (a) For review see: C. D. Vanderwal, J. Org. Chem., 2011, 76, 9555 CrossRef CAS PubMed; (b) S. Kunugi, T. Okubo and N. Ise, J. Am. Chem. Soc., 1976, 98, 2282 CrossRef CAS; (c) E. N. Marvell, G. Caple and I. Shahidi, J. Am. Chem. Soc., 1970, 92, 5641 CrossRef CAS; (d) E. N. Marvell and I. Shahidi, J. Am. Chem. Soc., 1970, 92, 5646 CrossRef CAS; (e) W. König and J. Prakt, Chem, 1904, 69, 105 Search PubMed.
- S. M. Usama, S. C. Marker, D.-H. Li, D. R. Caldwell, M. Stroet, N. L. Patel, A. G. Tebo, S. Hernot, J. D. Kalen and M. Schnermann, J. Am. Chem. Soc., 2023, 145, 14647 CrossRef CAS PubMed.
- Z. Hao, L. Hu, X. Wang, Y. Liu and S. Mo, Org. Lett., 2023, 25, 1078 CrossRef CAS PubMed.
- H. Lee, J. C. Mason and S. Achilefu, J. Org. Chem., 2006, 71, 7862 CrossRef CAS PubMed.
- H.-H. Johannes, W. Grahn, A. Reisner and P. G. Jones, Tetrahedron Lett., 1995, 36, 7225 CrossRef CAS.
- L. Strekowski, M. Lipowska and G. Patonay, J. Org. Chem., 1992, 57, 4578 CrossRef CAS.
- R. M. Exner, F. Cortezon-Tamarit and S. I. Pascu, Angew. Chem., Int. Ed., 2021, 60, 6230 CrossRef CAS PubMed.
- R. Gray, D. Walton, J. Bickerton, P. Richard and J. Heptinstall, Dyes Pigm., 1998, 38, 97 CrossRef CAS.
- R. J. Mellamby, J. I. Scott, I. Mair, A. Fernandez, L. Saul, J. Arlt, M. Moral and M. Vandrell, Chem. Sci., 2018, 9, 7261 RSC.
- K. Xu, H. Chen, J. Tian, B. Ding, Y. Xie, M. Qiang and B. Tang, Chem. Commun., 2011, 47, 9468 RSC.
- K. Mitra, C. E. Lyons and M. C. T. Hartman, Angew. Chem., Int. Ed., 2018, 57, 10263 CrossRef CAS PubMed.
- G. Moad and D. H. Solomon, The Chemistry of Radical Polymerization, Elsevier, 2006 Search PubMed.
- (a) C. Nacci, M. Schnied, D. Civita, E. Magnano, S. Nappini, I. Pis and L. Grill, J. Phys. Chem. C, 2021, 125, 22554 CrossRef CAS; (b) M. Kaur and A. K. Srivastava, J. Macromol. Sci., Part C: Polym. Rev., 2006, 42, 481 CrossRef.
- For review: B. Strehmel, C. Schmitz, C. Kütahya, Y. Pang, A. Drewitz and H. Mustroph, Beilstein J. Org. Chem., 2020, 16, 415 CrossRef CAS PubMed.
- (a) X. He, Y. Shao, Y. Pang, J. Wang, M. Lui, Y. Xin and Y. Zou, Polym. Chem., 2023, 14, 1543 RSC; (b) Q. Wang, S. Popov, V. Strehmel, J. S. Gutmann and B. Strehmel, Polym. Chem., 2023, 14, 116 RSC; (c) H. Mokbel, G. Noirbent, D. Gigmes, F. Dumur and J. Lalevée, Beilstein J. Org. Chem., 2021, 17, 2067 CrossRef CAS PubMed; (d) Q. Wang, S. Popov, A. Feilen, V. Strehmel and B. Strehmel, Angew. Chem., Int. Ed., 2021, 60, 26855 CrossRef CAS PubMed; (e) Y. Pang, A. Shiraishi, D. Keil, S. Popov, V. Strehmel, H. Jiao, J. S. Gutmann, Y. Zou and B. Strehmel, Angew. Chem., Int. Ed., 2021, 60, 1465 CrossRef CAS PubMed; (f) Y. Pang, H. Jiao, Y. Zou and B. Strehmel, Polym. Chem., 2021, 12, 5752 RSC; (g) A. Caron, F. Dumur and J. Lalevée, J. Polym. Sci., 2020, 58, 2134 CrossRef CAS.
- (a) B. Jedrzejewska, M. Pietrzak and Z. Rafinski, Polymer, 2011, 52, 2110 CrossRef CAS; (b) S. Chatterjee, P. Gottschalk, P. D. Davis and G. B. Schuster, J. Am. Chem. Soc., 1988, 110, 2326 CrossRef CAS.
- J. Kabatc, M. Zasada and J. Paczkowski, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3626 CrossRef CAS.
- A. H. Bonardi, F. Dumur, T. M. Grant, G. Noirbent, D. Gigmes, B. H. Lessard, J.-P. Fouassier and J. Lalevée, Macromolecules, 2018, 51, 1314 CrossRef CAS.
- A. H. Bonardi, F. Bonardi, F. Morlet-Savary, C. Dietlin, G. Noirbent, T. M. Grant, J.-P. Fouassier, F. Dumur, B. H. Lessard, F. Gigmes and J. Lalevée, Macromolecules, 2018, 51, 8808 CrossRef CAS.
- C. Kütahya, Y. Yagci and B. Strehemel, ChemPhotoChem, 2019, 3, 1180 CrossRef.
- C. Kütahya, C. Schmitz, V. Strehmel, Y. Yagci and B. Strehemel, Angew. Chem., Int. Ed., 2018, 57, 7898 CrossRef.
- A. R. Obah Kosso, N. Sellet, A. Baralle, M. Cormier and J.-P. Goddard, Chem. Sci., 2021, 12, 6964 RSC.
- Discussion about SET vs. EnT mechanisms for photocatalytic oxidation of amine: Y. Pan, S. Wang, C. W. Kee, E. Dubuisson, Y. Yang, K. P. Loh and C.-H. Tan, Green Chem., 2011, 13, 3341 RSC.
- N. Sellet, L. Clement-Comoy, M. Elhabiri, M. Cormier and J.-P. Goddard, Chem.–Eur. J., 2023, 29, e202302353 CrossRef CAS PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |