
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational analysis of R–X oxidative addition to Pd nanoparticles†
Mikhail V.
Polynski
 *a,
Yulia S.
Vlasova
bc,
Yaroslav V.
Solovev
d,
Sergey M.
Kozlov
a and
Valentine P.
Ananikov
*bc
*a,
Yulia S.
Vlasova
bc,
Yaroslav V.
Solovev
d,
Sergey M.
Kozlov
a and
Valentine P.
Ananikov
*bc
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: polynskimikhail@gmail.com; mvp@nus.edu.sg
bFaculty of Chemistry, Moscow State University, Leninskiye Gory 1-3, Moscow 119991, Russia
cZelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky Prospect 47, Moscow 119991, Russia. E-mail: val@ioc.ac.ru
dM. M. Shemyakin and Yu. A. Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Miklukho-Maklaya 16/10, Moscow 117997, Russia
First published on 24th May 2024
Abstract
Oxidative addition (OA) is a necessary step in mechanisms of widely used synthetic methodologies such as the Heck reaction, cross-coupling reactions, and the Buchwald–Hartwig amination. This study pioneers the exploration of OA of aryl halide to palladium nanoparticles (NPs), a process previously unaddressed in contrast to the activity of well-studied Pd(0) complexes. Employing DFT modeling and semi-empirical metadynamics simulations, the oxidative addition of phenyl bromide to Pd nanoparticles was investigated in detail. Energy profiles of oxidative addition to Pd NPs were analyzed and compared to those involving Pd(0) complexes forming under both ligand-stabilized (phosphines) and ligandless (amine base) conditions. Metadynamics simulations highlighted the edges of the (1 1 1) facets of Pd NPs as the key element of oxidative addition activity. We demonstrate that OA to Pd NPs is not only kinetically facile at ambient temperatures but also thermodynamically favorable. This finding accentuates the necessity of incorporating OA to Pd NPs in future investigations, thus providing a more realistic view of the involved catalytic mechanisms. These results enhance the understanding of aryl halide (cross-)coupling reactions, reinforcing the concept of a catalytic “cocktail”. This concept posits dynamic interconversions between diverse active and inactive centers, collectively affecting the outcome of the reaction. High activity of Pd NPs in direct C–X activation paves the way for novel approaches in catalysis, potentially enhancing the field and offering new catalytic pathways to consider.
1. Introduction
Facile synthesis of functionalized organic molecules is of paramount importance for the development of personalized medicine, fine chemical synthesis, advancements in drug design, and innovative materials, among many other areas. Within this conceptual demand, activation of the carbon–halogen (C–X) bond in organic halides (R–X) with subsequent transfer and functionalization of the organic group (R) is the basis for many well-recognized synthetic approaches known for their cost-efficiency, wide scope, and general applicability.1–3 Activation of the C–X bond occurs through oxidative addition (OA), initiating a series of transformations in catalyst active centers.Developing synthetic methodologies involving OA as an essential step in the reaction mechanism uncovered dynamic catalyst interconversions as a common phenomenon.4 Dynamic catalyst interconversions are now at the forefront of catalysis as the key concept to developing a new generation of synthetic technologies and addressing sustainability problems.4–7 This dynamic behavior was exceptionally evident in one of the cornerstone classes of organic reactions, Pd-catalyzed (cross-)couplings.8,9 Previous studies have shown that a “cocktail” of dynamically interconverting Pd species can easily form under catalytic conditions, including metal complexes, halides, and nanoparticles as catalytically active centers or pre-activated Pd reservoirs, with their activity varying tremendously in some cases.10–17 The dynamic phenomena occur under homogeneous and heterogeneous catalysis conditions.7 It has been demonstrated that oxidative addition of aryl halides plays a central role in the formation of “cocktail”-type systems.17,18
OA to Pd(0) metal complexes was investigated in experimental, DFT, and combined experimental/theoretical studies.19–28 However, the high level of reaction mechanism complexity still drastically challenges our understanding of OA, even at the level of molecular complexes.29
Pd NPs have shown remarkable activity in cross-coupling reactions as versatile and practical catalysts. In many studies, the activity was associated with Pd leached into the solution, not with the surface reactions on Pd NPs.9,30–34 At the same time, Pd nanoparticles are often formed spontaneously during cross-coupling reactions.35–39
While OA plays a key role in Pd leaching,17 its mechanism on the nanoparticle surface remains unexplored since previous computational and experimental–computational studies focused on energies of Pd detachment from the NP surface or cross-coupling reactions on molecular subnanoclusters.40–45 In the present study, for the first time, we investigated the oxidative addition of an organic halide to the Pd surface at the nano-scale. The studied process is relevant to C–C and C–heteroatom bond formation in applied fine organic synthesis involving nanocatalysts. Comparison with regular Pd complexes, including various ligands, highlighted a critical difference in C–X bond activation at the molecular and nano-scale levels.
2. Results
We conducted metadynamics (MTD) modeling of oxidative addition (OA) to truncated octahedral Pd79 and Pd140 nanocrystallites, along with a smaller Pd55 nanocrystallite, using the GFN1-xTB Hamiltonian (Pd79 and Pd55: Fig. 1; Pd140: Fig. S2†). We observed OAs in almost all Pd55 and Pd79 systems within tens of picoseconds (Fig. S1a† and 1a, respectively). A representative case of the PhBr OA to Pd79 is depicted in Fig. 1a(1). PhBr dissociation occurred at the edge of the nanoparticle. Subsequently, the Br atom migrated to the (1 0 0) facet and remained bonded to it. Concurrently, the Ph group exhibited active migration along the edge between two (1 1 1) facets and the adjacent (1 0 0) facet, as illustrated in the snapshot structures at the top of Fig. 1a.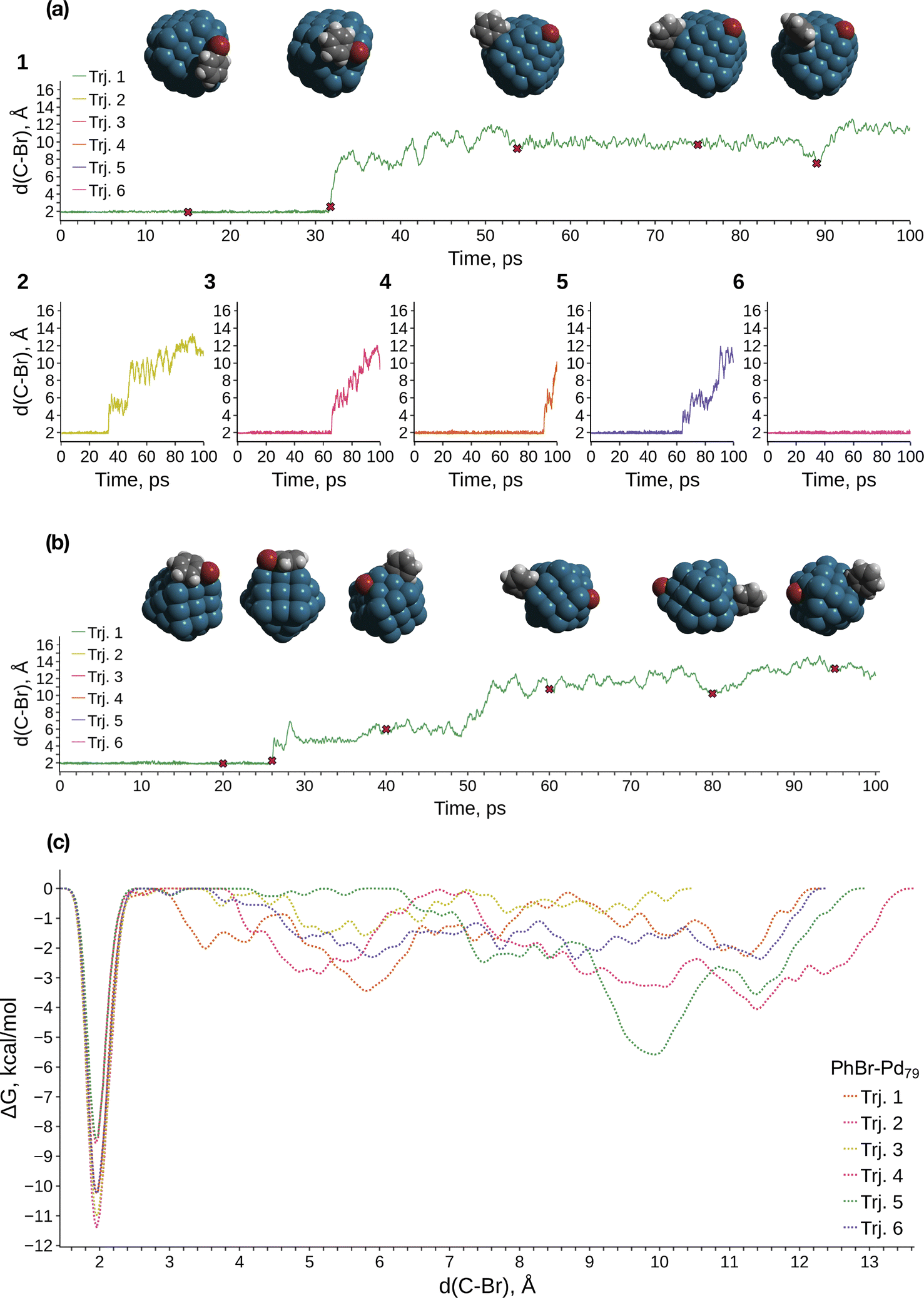 | ||
| Fig. 1 (a) Collective variable (C–Br distance) evolution during the metadynamics simulations of the Pd79 system; (b) selected case of the collective variable evolution in the Pd55 system; (c) free energy profiles obtained from the metadynamics runs of PhBr OA to Pd79. The color scheme is as follows: Pd – dark cerulean; Br – dark red; C – grey; H – white. | ||
The OA mechanism and post-OA system evolution were similar across all cases examined. The majority of OA events occurred at nanoparticle edges. In a few cases involving Pd55, the OA occurred at the vertex of the nanoparticle, which is also a site comprised of a Pd atom with a low coordination number (see details below). Only one OA to Pd140 was observed within 100 ps of sampling due to the increased configuration space size; this reaction also occurred on the edge (Fig. S2†). In the Pd55 and Pd140 systems, Ph migrated along the NP edges similarly to the case of Pd79 throughout all MTD simulations (Fig. 1a, b and S2a†). After the OA, Br similarly remained tightly bound to one of the (1 0 0) facets of Pd140. In contrast, some mobility of the adsorbed Br was observed on Pd55, with Br migrating along the (1 0 0) facet, which is larger in Pd55 than in Pd79 and Pd140 (Fig. 1b).
Fig. 1c presents the free energy profiles (FEPs) obtained from individual MTD runs involving Pd79. The movement of the dissociated Ph along the edge corresponds to a series of shallow minima on the right side of the obtained FEPs. Although these basins are deeper than the “thermal energy”, RT, at 298.15 K (0.6 kcal mol−1), Ph migration occurs relatively easily. The corresponding FEP minima are significantly higher than the lowest sharp minimum at approximately 1.9 Å, which corresponds to the undissociated state of PhBr. According to the obtained FEP, the OA activation free energy does not exceed approximately 11 kcal mol−1, and OA is kinetically feasible at ambient temperature. OA can lead to the accumulation of tightly bound Br atoms on the Pd surface, with a preference for the (1 0 0) surface. This is accompanied by the chemisorption of Ph groups, which migrate easily near low-coordinate Pd atom sites such as edges, steps, vertices, etc.
MTD sampling of dissociation trajectories allowed us to identify transition states relevant to modeled nanoparticles. Upon examining the MTD trajectories, we determined two groups of OA processes in the Pd79 system occurring at the edge of (1 1 1) facets, with variations in the positioning of the Ph group (Fig. 2a). In the first group, the Ph group was situated on the (1 1 1) facet with the Ph–Br bond nearly perpendicular to the edge (Pd79 fac–ed), whereas in the second group, the Ph moiety was positioned such that the Ph–Br bond nearly collinearized with the edge (Pd79 ed–ed). The OA of PhBr to Pd55 proceeded via two distinct pathways: in the first, OA occurred on the edge (labeled Pd55 ed in Fig. 2a), and in the second, the vertex Pd atom acted as the reactive center (labeled Pd55 ver). The TS found during the MTD sampling of the PhBr–Pd140 interaction (Pd140 ed in Fig. 2a) was fully similar to that in the Pd79 system.
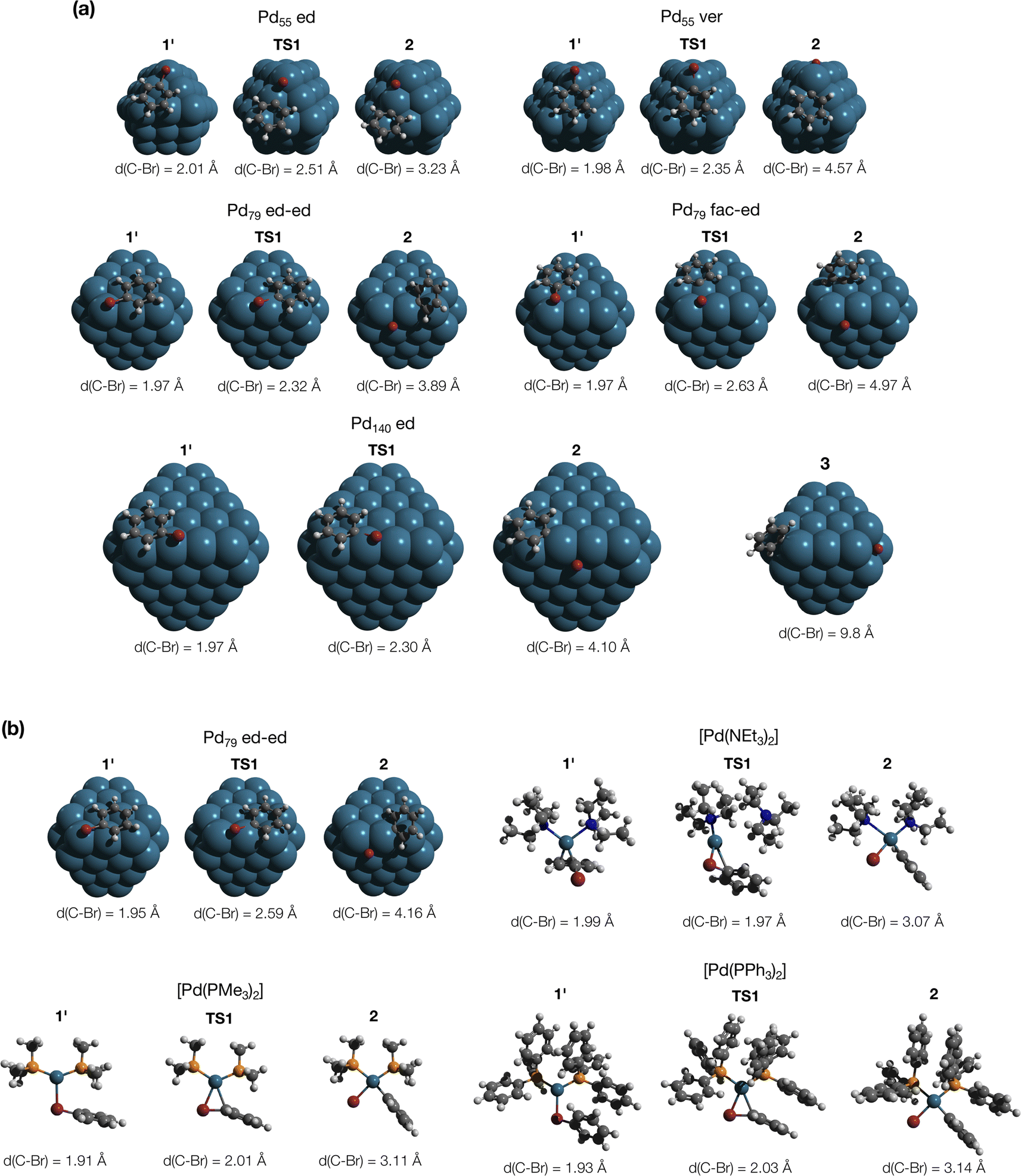 | ||
| Fig. 2 (a) Optimized structures of the OA transition states along with pre-reaction and post-reaction states in the PhBr interactions with Pd55, Pd79, and Pd140 nanoparticles, derived from metadynamics simulations. Intermediate 3, identified during the MTD sampling, is discussed in the text. (b) Optimized structures of the intermediates in the pathways involving Pd79 (facet active site) and [PdL2] (L = NEt3, PMe3, and PPh3), elucidated in nudged elastic band calculations. The color scheme is as follows: Pd – dark cerulean; Br – dark red; P – orange; N – blue; C – grey; H – white. Intermediate and transition states are numbered according to the scheme in Fig. 3. All optimizations were performed using DFT (see Section S1† for details). | ||
In addition to the OA pathways identified in MTD simulations, we employed the DyNEB method46 to explore several alternative active centers. Firstly, we examined OA to Pd–phosphine complexes: the electron-donating PMe3 and the widely used relatively weakly electron-donating PPh3. Secondly, we included [Pd(NEt3)2] into the consideration to model a nitrogen base added in some coupling reactions conducted under “ligandless” conditions (the Heck reaction and others). Lastly, using the DyNEB method, we investigated a hypothetical pathway in which the reaction occurred on a facet of the Pd79 nanoparticle (labeled Pd55 ed in Fig. 2b). Although the latter pathway was not observed in the MTD simulations, it can be used a valuable reference to enhance our understanding of the effects of nanostructuring on OA to Pd catalysts.
The focal point of this study is the computed free energy profiles of OA to Pd79 and molecular complexes depicted in Fig. 3. The corresponding numerical values are given in Table S1.† The process begins with the formation of the pre-OA complex 1′. Pd79 was selected to compare the adsorption affinity of PhBr towards Pd nanoparticles vs. the thermodynamic effect of 1 → 1′ in the case of molecular complexes [PdL2]. The adsorption of PhBr onto Pd79 is markedly exergonic, with ΔG1→1′ being lower than −46.9 kcal mol−1. This result indicates a probable intensive coverage of Pd NP precatalysts by the aryl halide during coupling reactions.
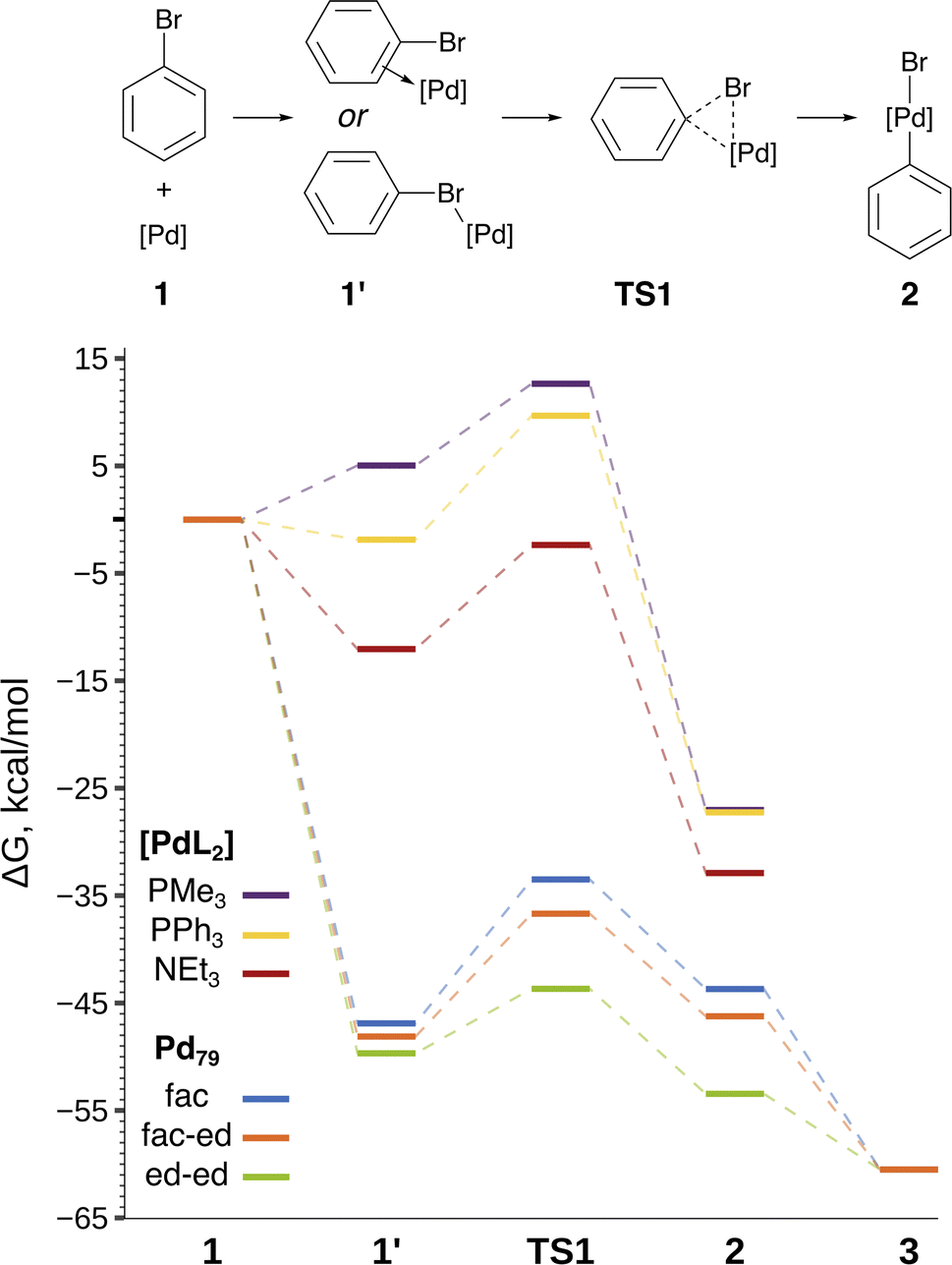 | ||
| Fig. 3 Reaction scheme and the free energy profile. Optimized structures of the intermediates are shown in Fig. 2. See Table S1† for numerical data. | ||
The transition 1 → 1′ is endergonic for [Pd(PMe3)2] (ΔG1→1′ = 5.0 kcal mol−1) and exergonic for [Pd(NEt3)2] and [Pd(PPh3)2], with ΔG1→1′ being equal to −12.0 and −1.9 kcal mol−1. In the case of phosphine ligands, PhBr in 1′ binds to [PdL2] only through the Br atom, and there is considerable translational and rotational entropy loss. For 1′ with L = NEt3, a strong Pd–π bond forms between the aryl halide and the [PdL2] moiety, effectively counterbalancing the entropic loss.
The comparison of the DFT-calculated activation energies of oxidative addition is given in Table S1,† and the corresponding plot is given in Fig. 3. Since the transition 1 → 1′ was exergonic in the cases of Pd79, [Pd(NEt3)2], and [Pd(PPh3)2], we regarded 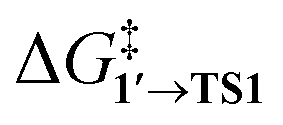 as the OA activation energy for these species. For [Pd(PMe3)2], however,
as the OA activation energy for these species. For [Pd(PMe3)2], however, 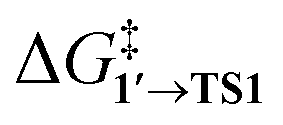 was considered as the activation energy instead of
was considered as the activation energy instead of 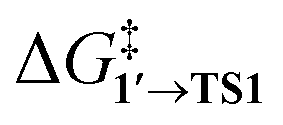 due to the positive value of ΔG1→1′.
due to the positive value of ΔG1→1′.
In all the molecular complexes, classic three-center Br–Pd–C transition states were identified (Fig. 2), characterized by imaginary modes representing the migration of Ph to Pd and the Ph–Br bond cleavage. The activation barriers of the OA to [Pd(PMe3)2] and [Pd(PPh3)2] did not surpass 12.6 kcal mol−1, aligning well with the typically facile OA to complexes of Pd(0) with two non-overly sterically hindered phosphine ligands. The OA of PhBr to [Pd(PPh3)2] proceeded with 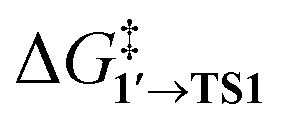 equal to 9.7 kcal mol−1. It is important to note the detachment of the second NEt3 from the Pd center in the transition state, whereas both the initial and final states, 1′ and 2, featured NEt3 attached to Pd.
equal to 9.7 kcal mol−1. It is important to note the detachment of the second NEt3 from the Pd center in the transition state, whereas both the initial and final states, 1′ and 2, featured NEt3 attached to Pd.
The calculated activation energies of PhBr OA to Pd79 are comparable to or lower than those involving molecular Pd complexes, with 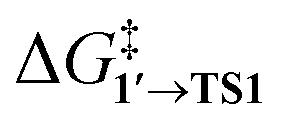 being equal to 13.4, 11.4, and 6.0 kcal mol−1 for Pd79 fac, Pd79 fac–ed, and Pd79 ed–ed, respectively. Evidently, the edge is a highly reactive site with the activation barrier for the Pd79 ed–ed transition state well below those observed with the [PdL2] complexes. Furthermore, the formation of the pre-reaction state 1′ in pathway Pd79 ed–ed is also the most exergonic among these three model reaction channels. Therefore, we may expect the preferable reactivity via the ed–ed channel. It should also be noted that the (1 1 1) facet exhibited the lowest activity in the model OA process, indicating that oxidative addition preferentially occurs at the edges of Pd nanoparticles.
being equal to 13.4, 11.4, and 6.0 kcal mol−1 for Pd79 fac, Pd79 fac–ed, and Pd79 ed–ed, respectively. Evidently, the edge is a highly reactive site with the activation barrier for the Pd79 ed–ed transition state well below those observed with the [PdL2] complexes. Furthermore, the formation of the pre-reaction state 1′ in pathway Pd79 ed–ed is also the most exergonic among these three model reaction channels. Therefore, we may expect the preferable reactivity via the ed–ed channel. It should also be noted that the (1 1 1) facet exhibited the lowest activity in the model OA process, indicating that oxidative addition preferentially occurs at the edges of Pd nanoparticles.
Fig. 3 and the last columns of Table S1† show that the transitions 1′ → 2 and 1 → 2 with all the molecular complexes are highly exergonic. In the case of Pd79, the notable exergonicity of 1 → 2 is primarily due to the substantial exergonicity of PhBr adsorption (1 → 1′). The ΔG1′→2 values for the Pd79 fac–ed and fac channels are positive (1.9 and 3.2 kcal mol−1, respectively), while ΔG1′→2 for ed–ed is negative, being −3.8 kcal mol−1. However, state 2 in the Pd79 fac–ed and ed–ed channels does not correspond to the structures of the OA products observed in the MTD runs (see Fig. 1a). In particular, we can see that most structures of the post-OA complexes at the top of Fig. 1a have Br and Ph chemisorbed on two adjacent (1 0 0) facets.
Such a structure resulting from the facile migration of Br and Ph to the (1 0 0) facets is depicted in Fig. 1a as 3. The values of ΔG1′→2 are negative for all three reaction channels. This suggests that the accumulation of chemisorbed Ph and Br (the products of 1′ → 3) can be thermodynamically favorable when higher surface energy (1 0 0) facets are available on the NP surface along with edges between (1 1 1) facets. The availability of (1 0 0) surfaces was previously associated with a higher propensity for Pd leaching and activity in Suzuki cross-coupling.34 This final product 3 differs significantly from species 2 in molecular complexes; in the NP system, the Ph and Br groups are separated, whereas in the molecular complexes, they remain bonded to a single Pd center.
By comparing 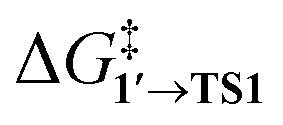 and ΔG1′→2 in Pd55, Pd79, and Pd140 systems, we can assess the effect of the nanoparticle size on the kinetic and thermodynamic feasibility of PhBr oxidative addition. The lowest
and ΔG1′→2 in Pd55, Pd79, and Pd140 systems, we can assess the effect of the nanoparticle size on the kinetic and thermodynamic feasibility of PhBr oxidative addition. The lowest 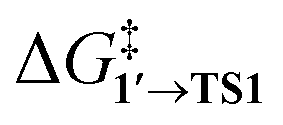 values for each NP are presented in Table 1, while Table S1† contains all the computed values. The
values for each NP are presented in Table 1, while Table S1† contains all the computed values. The 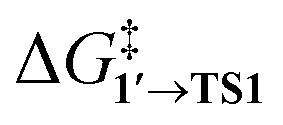 values indicate that the activity of the edge sites increases with the increase of the nanoparticle size. The non-monotonous behavior of ΔG1′→2 with the increase of the nanoparticle's size is also evident, while all ΔG1′→2 in Table 1 are negative. Given that the post-OA migration of Ph was observed in all MTD simulations (see Fig. 1, S1 and S2†), and that process 1′ → 3 was highly exergonic in the Pd79 system, we may expect the high propensity of Pd NPs to undergo the OA of PhBr when there is a sufficient surface density of edge sites and presence of (1 0 0) surfaces. At the same time, the fraction of edge atoms is smaller in larger nanoparticles; hence, the density of active sites decreases with the increase in the size.
values indicate that the activity of the edge sites increases with the increase of the nanoparticle size. The non-monotonous behavior of ΔG1′→2 with the increase of the nanoparticle's size is also evident, while all ΔG1′→2 in Table 1 are negative. Given that the post-OA migration of Ph was observed in all MTD simulations (see Fig. 1, S1 and S2†), and that process 1′ → 3 was highly exergonic in the Pd79 system, we may expect the high propensity of Pd NPs to undergo the OA of PhBr when there is a sufficient surface density of edge sites and presence of (1 0 0) surfaces. At the same time, the fraction of edge atoms is smaller in larger nanoparticles; hence, the density of active sites decreases with the increase in the size.
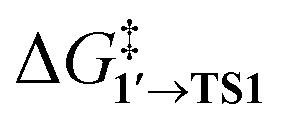 for each NP
for each NP
| System |
|
ΔG1′→2 |
|---|---|---|
| Pd55 ed | 7.5 | −6.0 |
| Pd79 ed–ed | 6.0 | −3.8 |
| Pd140 ed | 3.9 | −9.7 |
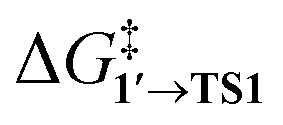
Although previous experimental studies associated OA of PhBr with the leaching of Pd into the solution,11,18,47 within the context of this study, we can only hypothesize that Ph and Br on the surface may kinetically facilitate this process. Notably, our earlier analysis, which considered only thermodynamic factors, indicated that OA leads to negative (favorable) formation energies of molecular forms (metal complexes) of leached Pd in solution.17 Additionally, the spontaneous formation of Pd NPs is a common phenomenon in coupling reactions where the (pre)catalyst is initially introduced as a metal complex. Therefore, OA to Pd NPs should be recognized as a crucial mechanistic step. OA to Pd NPs is as kinetically feasible as the OA to Pd(0) complexes, reinforcing the concept of the catalytic “cocktail”, in which a variety of potentially active and inactive interconverting centers contribute to the overall reaction outcome.
3. Discussion and conclusions
The conclusions of this study are schematically depicted in Fig. 4, which highlights the interplay of the elucidated transformations in the (cross-)coupling catalysis of reactions involving aryl halides. In our DFT modeling, we analyzed the activity of the nanoparticles ranging from ∼1.08 (Pd55) to ∼1.56 nm (Pd140). We focused primarily on the edges between their (1 1 1) facets that exhibit the highest thermodynamic stability;48 however, we recognize that other centers containing low-valent Pd atoms (e.g., various vertices and kinks) could also be active in OA.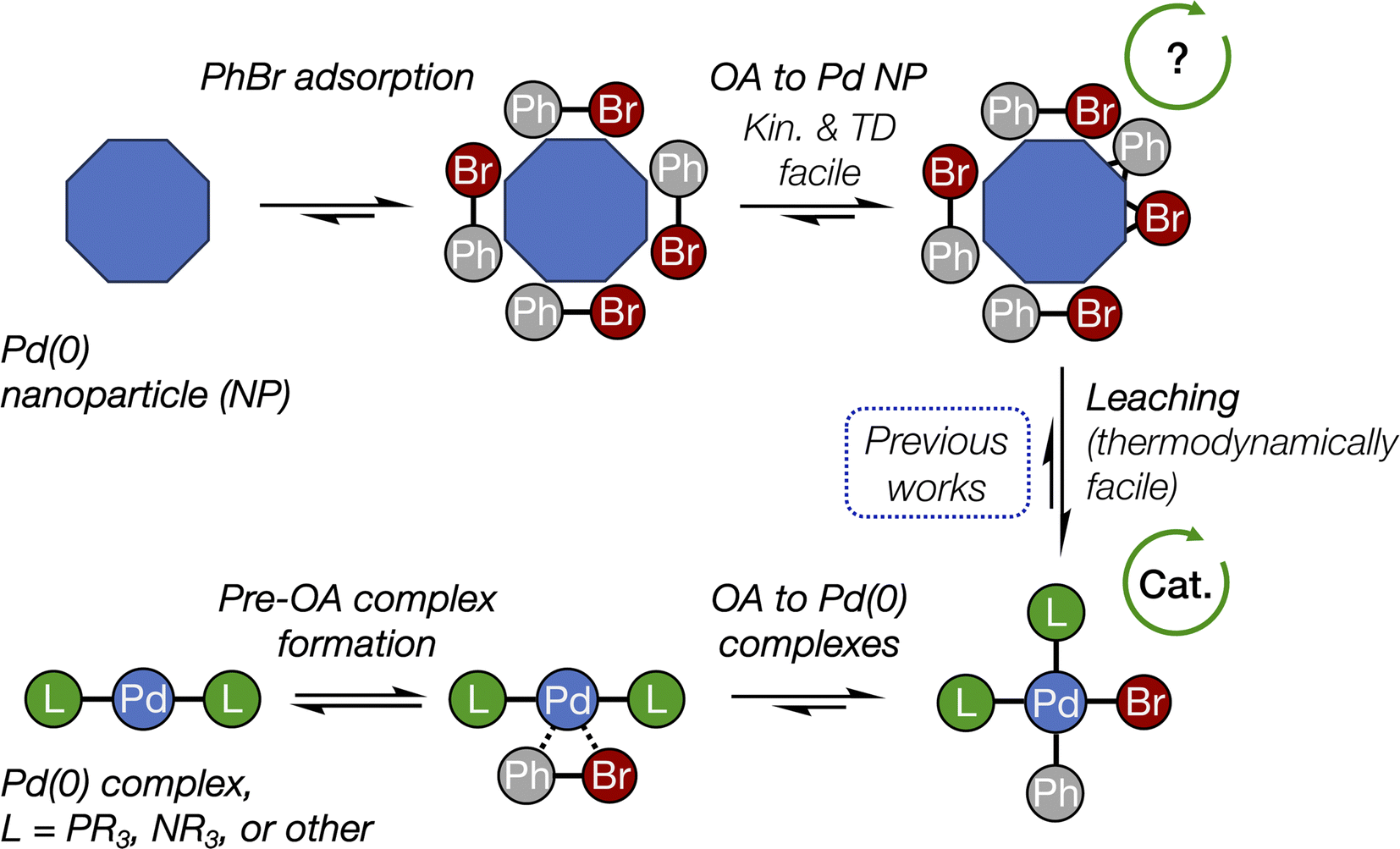 | ||
| Fig. 4 The interplay of oxidative addition in catalyst interconversions in the course of (cross-)couplings with Pd(0) nanoparticles and molecular complexes. See Fig. 2 for more detailed structures and Fig. 3 for reaction (activation) free energies. The arrowed circles represent (possible) participation in the catalytic cycle (see text for discussion). | ||
Pd nanoparticles featuring a sufficient surface concentration of low-coordinated atoms, such as facet edges, and atoms on higher-surface energy facets, such as (1 0 0), can undergo oxidative addition of aryl halides. The presented DFT modeling suggests that OA to Pd nanoparticles is well comparable to OA to Pd(0) complexes by its activation barrier and also being thermodynamically favorable. (1 1 1) terraces on Pd nanoparticles also showed relatively high activity, though lower than that of the considered edge sites. OA to all nanoparticles was both kinetically facile at 25 °C and thermodynamically favorable.
For the first time, the present study revealed the critical difference between the OA involving monometallic centers, i.e., molecular complexes, and metal nanoparticles. Classic OA to a monometallic Pd center proceeds as reversible coordination of Ph–Br followed by three-center interaction and breakage of C–Br bond with both Ph and Br groups remaining in close proximity. In contrast, OA to Pd NPs involves practically irreversible trapping of Ph–Br on the metal surface followed by facile C–Br bond breakage and separation of Ph and Br from each other via multicentered interactions on the surface.
The debate49 about the occurrence of a (cross-)coupling process at the nanoparticle surface continues in recent literature, with works reporting the reaction at the metal surface,50,51 exclusively in solution,33,52 or in both modes, depending on the reaction conditions.53 Here, we show that the OA stage alone would not hamper (cross-)coupling at the nanoparticle surface, according to its calculated reaction (activation) free energies that are comparable with those related to Pd(0) complexes. Another important point is that the type of the ligand, Pd ligation state, type of the organic substrate, and even the solvent can strongly affect the mechanism, kinetics, and selectivity of OA to metal complexes.23,28,29,54–61 Therefore, the relative activity of Pd complexes and nanoparticles in OA depends on the choice of reference ligand. The rate of the OA to Pd complexes bearing some designer ligands may surpass that to Pd NPs. In addition, mass transfer effects may be at play in the case of aryl halides OA to Pd NPs. A definitive answer to the question of the feasibility of truly heterogeneous (cross-)coupling on Pd(0) surface requires further investigation, particularly regarding the surface activity of Pd NPs in other coupling steps, such as transmetalation and reductive elimination.
The discussions in the present study highlight many significant phenomena related to coupling reactions involving Pd nanoparticles that require further elucidation. This finding emphasized that our understanding of oxidative addition is still evolving and incomplete.
4. Computational details
GFN1-xTB62 was utilized for MTD simulations and thermochemical correction calculations in ASE.63 The GBSA solvation model parameterized for GFN1-xTB was employed to account for solvent effects.64 MTD simulations were conducted using DFTB+ 22.2 (ref. 65) and PLUMED 2.8.2.66,67 DFT calculations were carried out using the revPBE functional68 with the D3(BJ) correction69,70 in VASP 6.3.2.71 Core electron density was modeled using PAW.72 The single-point Hessian (SPH) method73 implemented in the xtb program64 was used to perform thermochemical calculations. Additional computational details are provided in the ESI.† ChatGPT 4 was used for initial text proofreading.Data availability
The data supporting this article have been uploaded as part of the ESI.†Author contributions
Mikhail V. Polynski: conceptualization; formal analysis; investigation; methodology; supervision; writing − original draft; writing − review & editing. Yulia S. Vlasova: formal analysis; investigation; writing − original draft. Yaroslav V. Solovev: formal analysis; investigation; visualization. Sergey M. Kozlov: resources; writing − original draft; writing − review & editing. Valentine P. Ananikov: conceptualization; supervision; writing − original draft, writing − review & editing.Conflicts of interest
There are no conflicts of interest to declare.References
- C. Torborg and M. Beller, Recent Applications of Palladium-Catalyzed Coupling Reactions in the Pharmaceutical, Agrochemical, and Fine Chemical Industries, Adv. Synth. Catal., 2009, 351(18), 3027–3043, DOI:10.1002/adsc.200900587.
- J. Magano and J. R. Dunetz, Large-Scale Applications of Transition Metal-Catalyzed Couplings for the Synthesis of Pharmaceuticals, Chem. Rev., 2011, 111(3), 2177–2250, DOI:10.1021/cr100346g.
- J. P. Corbet and G. Mignani, Selected Patented Cross-Coupling Reaction Technologies, Chem. Rev., 2006, 106(7), 2651–2710, DOI:10.1021/cr0505268.
- A. S. Galushko, A. S. Kashin, D. B. Eremin, M. V. Polynski, E. O. Pentsak, V. M. Chernyshev and V. P. Ananikov, Introduction to Dynamic Catalysis and the Interface Between Molecular and Heterogeneous Catalysts, in Nanoparticles in Catalysis: Advances in Synthesis and Applications, ed. K. Philippot and A. Roucoux, WILEY-VCH GmbH, 2021, pp. 13–42 Search PubMed.
- S. Chavez, B. Werghi, K. M. Sanroman Gutierrez, R. Chen, S. Lall and M. Cargnello, Studying, Promoting, Exploiting, and Predicting Catalyst Dynamics: The Next Frontier in Heterogeneous Catalysis, J. Phys. Chem. C, 2023, 127(5), 2127–2146, DOI:10.1021/ACS.JPCC.2C06519.
- A. J. Martín, S. Mitchell, C. Mondelli, S. Jaydev and J. Pérez-Ramírez, Unifying Views on Catalyst Deactivation, Nat. Catal., 2022, 5(10), 854–866, DOI:10.1038/s41929-022-00842-y.
- A. S. Galushko and V. P. Ananikov, 4D Catalysis Concept Enabled by Multilevel Data Collection and Machine Learning Analysis, ACS Catal., 2023, 14, 161–175, DOI:10.1021/ACSCATAL.3C03889.
- D. B. Eremin and V. P. Ananikov, Understanding Active Species in Catalytic Transformations: From Molecular Catalysis to Nanoparticles, Leaching, “Cocktails” of Catalysts and Dynamic Systems, Coord. Chem. Rev., 2017, 346, 2–19, DOI:10.1016/j.ccr.2016.12.021.
- A. M. Trzeciak and J. J. Ziółkowski, Monomolecular Nanosized and Heterogenized Palladium Catalysts for the Heck Reaction, Coord. Chem. Rev., 2007, 251(9–10), 1281–1293, DOI:10.1016/J.CCR.2006.11.013.
- J. G. de Vries, When Does Catalysis with Transition Metal Complexes Turn into Catalysis by Nanoparticles?, in Selective Nanocatalysts and Nanoscience, ed. A. Zecchina, S. Bordiga and E. Groppo, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011, pp. 73–103, DOI:10.1002/9783527635689.ch3.
- C. Deraedt and D. Astruc, “Homeopathic” Palladium Nanoparticle Catalysis of Cross Carbon–Carbon Coupling Reactions, Acc. Chem. Res., 2014, 47(2), 494–503, DOI:10.1021/ar400168s.
- C. G. Baumann, S. De Ornellas, J. P. Reeds, T. E. Storr, T. J. Williams and I. J. S. Fairlamb, Formation and Propagation of Well-Defined Pd Nanoparticles (PdNPs) during C–H Bond Functionalization of Heteroarenes: Are Nanoparticles a Moribund Form of Pd or an Active Catalytic Species?, Tetrahedron, 2014, 70(36), 6174–6187, DOI:10.1016/J.TET.2014.06.002.
- M. Górna, M. S. Szulmanowicz, A. Gniewek, W. Tylus and A. M. Trzeciak, Recyclable Pd(0)-Pd(II) Composites Formed from Pd(II) Dimers with NHC Ligands under Suzuki–Miyaura Conditions, J. Organomet. Chem., 2015, 785, 92–99, DOI:10.1016/J.JORGANCHEM.2015.03.009.
- B. P. Carrow and J. F. Hartwig, Ligandless, Anionic, Arylpalladium Halide Intermediates in the Heck Reaction, J. Am. Chem. Soc., 2010, 132(1), 79–81, DOI:10.1021/ja909306f.
- C. C. C. Johansson Seechurn, T. Sperger, T. G. Scrase, F. Schoenebeck and T. J. Colacot, Understanding the Unusual Reduction Mechanism of Pd(II) to Pd(I): Uncovering Hidden Species and Implications in Catalytic Cross-Coupling Reactions, J. Am. Chem. Soc., 2017, 139(14), 5194–5200, DOI:10.1021/jacs.7b01110.
- N. W. J. Scott, M. J. Ford, C. Schotes, R. R. Parker, A. C. Whitwood and I. J. S. Fairlamb, The Ubiquitous Cross-Coupling Catalyst System ‘Pd(OAc)2’/2PPh3 Forms a Unique Dinuclear PdI Complex: An Important Entry Point into Catalytically Competent Cyclic Pd3 Clusters, Chem. Sci., 2019, 10(34), 7898–7906, 10.1039/C9SC01847F.
- M. V. Polynski and V. P. Ananikov, Modeling Key Pathways Proposed for the Formation and Evolution of “Cocktail”-Type Systems in Pd-Catalyzed Reactions Involving ArX Reagents, ACS Catal., 2019, 9(5), 3991–4005, DOI:10.1021/acscatal.9b00207.
- A. S. Galushko, D. O. Prima, J. V. Burykina and V. P. Ananikov, Comparative Study of Aryl Halides in Pd-Mediated Reactions: Key Factors beyond the Oxidative Addition Step, Inorg. Chem. Front., 2021, 8(3), 620–635, 10.1039/D0QI01133A.
- C. Amatore and A. Jutand, Anionic Pd(0) and Pd(II) Intermediates in Palladium-Catalyzed Heck and Cross-Coupling Reactions, Acc. Chem. Res., 2000, 33(5), 314–321, DOI:10.1021/ar980063a.
- Z. Li, Y. Fu, Q.-X. Guo and L. Liu, Theoretical Study on Monoligated Pd-Catalyzed Cross-Coupling Reactions of Aryl Chlorides and Bromides, Organometallics, 2008, 27(16), 4043–4049, DOI:10.1021/om701065f.
- F. Niroomand Hosseini, A. Ariafard, M. Rashidi, G. Azimi and S. M. Nabavizadeh, Density Functional Studies of Influences of Ni Triad Metals and Solvents on Oxidative Addition of MeI to [M(CH3)2(NH3)2] Complexes and C–C Reductive Elimination from [M(CH3)3(NH3)2I] Complexes, J. Organomet. Chem., 2011, 696(21), 3351–3358, DOI:10.1016/J.JORGANCHEM.2011.07.024.
- A. B. González-Pérez, R. Álvarez, O. N. Faza, Á. R. de Lera and J. M. Aurrecoechea, DFT-Based Insights into Pd–Zn Cooperative Effects in Oxidative Addition and Reductive Elimination Processes Relevant to Negishi Cross-Couplings, Organometallics, 2012, 31(5), 2053–2058, DOI:10.1021/om300024p.
- K. Vikse, T. Naka, J. S. McIndoe, M. Besora and F. Maseras, Oxidative Additions of Aryl Halides to Palladium Proceed through the Monoligated Complex, ChemCatChem, 2013, 5(12), 3604–3609, DOI:10.1002/cctc.201300723.
- Y. Yang, K. Niedermann, C. Han and S. L. Buchwald, Highly Selective Palladium-Catalyzed Cross-Coupling of Secondary Alkylzinc Reagents with Heteroaryl Halides, Org. Lett., 2014, 16(17), 4638–4641, DOI:10.1021/ol502230p.
- B. U. W. Maes, S. Verbeeck, T. Verhelst, A. Ekomié, N. Von Wolff, G. Lefèvre, E. A. Mitchell and A. Jutand, Oxidative Addition of Haloheteroarenes to Palladium(0): Concerted versus SN</Inf>Ar-Type Mechanism, Chem.–Eur. J., 2015, 21(21), 7858–7865, DOI:10.1002/chem.201406210.
- B. Noverges Pedro, M. Medio-Simón and A. Jutand, Influence of the Ligand of Palladium(0) Complexes on the Rate of the Oxidative Addition of Aryl and Activated Alkyl Bromides: Csp2−Br versus Csp3−Br Reactivity and Selectivity, ChemCatChem, 2017, 9(12), 2136–2144, DOI:10.1002/cctc.201700041.
- M. Kolter and K. Koszinowski, Second Comes First: Switching Elementary Steps in Palladium-Catalyzed Cross-Coupling Reactions, Chem.–Eur. J., 2020, 26(53), 12212–12218, DOI:10.1002/CHEM.202001041.
- T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Computational Studies of Synthetically Relevant Homogeneous Organometallic Catalysis Involving Ni, Pd, Ir, and Rh: An Overview of Commonly Employed DFT Methods and Mechanistic Insights, Chem. Rev., 2015, 115(17), 9532–9586, DOI:10.1021/acs.chemrev.5b00163.
- J. Rio, H. Liang, M. E. L. Perrin, L. A. Perego, L. Grimaud and P. A. Payard, We Already Know Everything about Oxidative Addition to Pd(0): Do We?, ACS Catal., 2023, 13(17), 11399–11421, DOI:10.1021/ACSCATAL.3C01943.
- A. Biffis, M. Zecca and M. Basato, Metallic Palladium in the Heck Reaction: Active Catalyst or Convenient Precursor?, Eur. J. Inorg. Chem., 2001, 2001(5), 1131–1133, DOI:10.1002/1099-0682(200105)2001:5<1131::AID-EJIC1131>3.0.CO;2-3.
- A. Leyva-Pérez, J. Oliver-Meseguer, P. Rubio-Marqués and A. Corma, Water-Stabilized Three- and Four-Atom Palladium Clusters as Highly Active Catalytic Species in Ligand-Free C—C Cross-Coupling Reactions, Angew. Chem., Int. Ed., 2013, 52(44), 11554–11559, DOI:10.1002/anie.201303188.
- M. Al-Amin, T. Honma, N. Hoshiya, S. Shuto and M. Arisawa, Ligand-Free Buchwald-Hartwig Aromatic Aminations of Aryl Halides Catalyzed by Low-Leaching and Highly Recyclable Sulfur-Modified Gold-Supported Palladium Material, Adv. Synth. Catal., 2012, 354(6), 1061–1068, DOI:10.1002/adsc.201100761.
- G. Collins, M. Schmidt, C. O'Dwyer, G. McGlacken and J. D. Holmes, Enhanced Catalytic Activity of High-Index Faceted Palladium Nanoparticles in Suzuki–Miyaura Coupling Due to Efficient Leaching Mechanism, ACS Catal., 2014, 4(9), 3105–3111, DOI:10.1021/cs5008014.
- G. Collins, M. Schmidt, C. O'Dwyer, J. D. Holmes and G. P. McGlacken, The Origin of Shape Sensitivity in Palladium-Catalyzed Suzuki-Miyaura Cross Coupling Reactions, Angew. Chem., Int. Ed., 2014, 53(16), 4142–4145, DOI:10.1002/anie.201400483.
- R. H. Crabtree, Deactivation in Homogeneous Transition Metal Catalysis: Causes, Avoidance, and Cure, Chem. Rev., 2015, 115(1), 127–150, DOI:10.1021/cr5004375.
- A. S. Sigeev, A. S. Peregudov, A. V. Cheprakov and I. P. Beletskaya, The Palladium Slow-Release Pre-Catalysts and Nanoparticles in the “Phosphine-Free” Mizoroki–Heck and Suzuki–Miyaura Reactions, Adv. Synth. Catal., 2015, 357(2–3), 417–429, DOI:10.1002/adsc.201400562.
- A. V. Astakhov, O. V. Khazipov, A. Y. Chernenko, D. V. Pasyukov, A. S. Kashin, E. G. Gordeev, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, A New Mode of Operation of Pd-NHC Systems Studied in a Catalytic Mizoroki-Heck Reaction, Organometallics, 2017, 36(10), 1981–1992, DOI:10.1021/acs.organomet.7b00184.
- D. Canseco-Gonzalez, A. Gniewek, M. Szulmanowicz, H. Müller-Bunz, A. M. Trzeciak and M. Albrecht, PEPPSI-Type Palladium Complexes Containing Basic 1,2,3-Triazolylidene Ligands and Their Role in Suzuki-Miyaura Catalysis, Chem.–Eur. J., 2012, 18(19), 6055–6062, DOI:10.1002/chem.201103719.
- V. M. Chernyshev, O. V. Khazipov, M. A. Shevchenko, A. Y. Chernenko, A. V. Astakhov, D. B. Eremin, D. V. Pasyukov, A. S. Kashin and V. P. Ananikov, Revealing the Unusual Role of Bases in Activation/Deactivation of Catalytic Systems: O–NHC Coupling in M/NHC Catalysis, Chem. Sci., 2018, 9(25), 5564–5577, 10.1039/C8SC01353E.
- H. Ramezani-Dakhel, P. A. Mirau, R. R. Naik, M. R. Knecht and H. Heinz, Stability, Surface Features, and Atom Leaching of Palladium Nanoparticles: Toward Prediction of Catalytic Functionality, Phys. Chem. Chem. Phys., 2013, 15(15), 5488, 10.1039/c3cp00135k.
- B. D. Briggs, N. M. Bedford, S. Seifert, H. Koerner, H. Ramezani-Dakhel, H. Heinz, R. R. Naik, A. I. Frenkel, M. R. Knecht and M. R. Knecht, Atomic-Scale Identification of Pd Leaching in Nanoparticle Catalyzed C–C Coupling: Effects of Particle Surface Disorder, Chem. Sci., 2015, 6(11), 6413–6419, 10.1039/C5SC01424G.
- Y. Yang, A. C. Reber, S. E. Gilliland, C. E. Castano, B. F. Gupton and S. N. Khanna, More than Just a Support: Graphene as a Solid-State Ligand for Palladium-Catalyzed Cross-Coupling Reactions, J. Catal., 2018, 360, 20–26, DOI:10.1016/j.jcat.2018.01.027.
- C.-R. Chang, Z.-J. Zhao, K. Köhler, A. Genest, J. Li and N. Rösch, Theoretical Study on the Leaching of Palladium in a CO Atmosphere, Catal. Sci. Technol., 2012, 2(11), 2238, 10.1039/c2cy20441j.
- E. E. Zvereva, S. A. Katsyuba, P. J. Dyson and A. V. Aleksandrov, Leaching from Palladium Nanoparticles in an Ionic Liquid Leads to the Formation of Ionic Monometallic Species, J. Phys. Chem. Lett., 2017, 8(15), 3452–3456, DOI:10.1021/acs.jpclett.7b01212.
- K. S. S. V. Prasad Reddy and P. A. Deshpande, DFT Reveals the Support Effects in Pd Nanoclusters over Defect-Ridden Graphene for the Oxidative Addition of Bromobenzene, Mol. Catal., 2022, 521, 112205, DOI:10.1016/j.mcat.2022.112205.
- P. Lindgren, G. Kastlunger and A. A. Peterson, Scaled and Dynamic Optimizations of Nudged Elastic Bands, J. Chem. Theory Comput., 2019, 15(11), 5787–5793, DOI:10.1021/acs.jctc.9b00633.
- A. M. Trzeciak and A. W. Augustyniak, The Role of Palladium Nanoparticles in Catalytic C–C Cross-Coupling Reactions, Coord. Chem. Rev., 2019, 384, 1–20, DOI:10.1016/j.ccr.2019.01.008.
- C. Popa, T. Zhu, I. Tranca, P. Kaghazchi, T. Jacob, E. J. M. Hensen, P. Kaghazchi, O. Balmes, R. van Rijn, J. N. Andersen, K. Deppert, H. Bluhm, Z. Liu, M. E. Grass, M. Hävecker and E. Lundgren, Structure of Palladium Nanoparticles under Oxidative Conditions, Phys. Chem. Chem. Phys., 2015, 17(3), 2268–2273, 10.1039/C4CP01761G.
- A. Bej, K. Ghosh, A. Sarkar and D. W. Knight, Palladium Nanoparticles in the Catalysis of Coupling Reactions, RSC Adv., 2016, 6(14), 11446–11453, 10.1039/C5RA26304B.
- M. Kim, Y. Kim, J. W. Hong, S. Ahn, W. Y. Kim and S. W. Han, The Facet-Dependent Enhanced Catalytic Activity of Pd Nanocrystals, Chem. Commun., 2014, 50(67), 9454–9457, 10.1039/c4cc02494j.
- Y. Zhao, L. Du, H. Li, W. Xie and J. Chen, Is the Suzuki-Miyaura Cross-Coupling Reaction in the Presence of Pd Nanoparticles Heterogeneously or Homogeneously Catalyzed? An Interfacial Surface-Enhanced Raman Spectroscopy Study, J. Phys. Chem. Lett., 2019, 10(6), 1286–1291, DOI:10.1021/ACS.JPCLETT.9B00351.
- B. Sun, L. Ning and H. C. Zeng, Confirmation of Suzuki-Miyaura Cross-Coupling Reaction Mechanism through Synthetic Architecture of Nanocatalysts, J. Am. Chem. Soc., 2020, 142(32), 13823–13832, DOI:10.1021/JACS.0C04804.
- N. W. J. Scott, M. J. Ford, N. Jeddi, A. Eyles, L. Simon, A. C. Whitwood, T. Tanner, C. E. Willans and I. J. S. Fairlamb, A Dichotomy in Cross-Coupling Site Selectivity in a Dihalogenated Heteroarene: Influence of Mononuclear Pd, Pd Clusters, and Pd Nanoparticles-The Case for Exploiting Pd Catalyst Speciation, J. Am. Chem. Soc., 2021, 143(25), 9682–9693, DOI:10.1021/JACS.1C05294.
- F. Barrios-Landeros, B. P. Carrow and J. F. Hartwig, Effect of Ligand Steric Properties and Halide Identity on the Mechanism for Oxidative Addition of Haloarenes to Trialkylphosphine Pd(0) Complexes, J. Am. Chem. Soc., 2009, 131(23), 8141–8154, DOI:10.1021/JA900798S.
- B. Noverges Pedro, M. Medio-Simón and A. Jutand, Influence of the Ligand of Palladium(0) Complexes on the Rate of the Oxidative Addition of Aryl and Activated Alkyl Bromides: Csp2−Br versus Csp3−Br Reactivity and Selectivity, ChemCatChem, 2017, 9(12), 2136–2144, DOI:10.1002/CCTC.201700041.
- E. K. Elias, S. M. Rehbein and S. R. Neufeldt, Solvent Coordination to Palladium Can Invert the Selectivity of Oxidative Addition, Chem. Sci., 2022, 13(6), 1618–1628, 10.1039/D1SC05862B.
- S. Roland, P. Mangeney and A. Jutand, Reactivity of Pd(0)(NHC)2 (NHC: N-Heterocyclic Carbene) in Oxidative Addition with Aryl Halides in Heck Reactions, Synlett, 2006, 2006(18), 3088–3094, DOI:10.1055/S-2006-951519.
- J. Lu, S. Donnecke, I. Paci and D. C. Leitch, A Reactivity Model for Oxidative Addition to Palladium Enables Quantitative Predictions for Catalytic Cross-Coupling Reactions, Chem. Sci., 2022, 13(12), 3477–3488, 10.1039/D2SC00174H.
- F. Proutiere, E. Lyngvi, M. Aufiero, I. A. Sanhueza and F. Schoenebeck, Combining the Reactivity Properties of PCy3 and PtBu3 into a Single Ligand, P(iPr)(tBu)2. Reaction via Mono- or Bisphosphine Palladium(0) Centers and Palladium(i) Dimer Formation, Organometallics, 2014, 33(23), 6879–6884, DOI:10.1021/OM5009605.
- J. P. Norman, N. G. Larson and S. R. Neufeldt, Different Oxidative Addition Mechanisms for 12- and 14-Electron Palladium(0) Explain Ligand-Controlled Divergent Site Selectivity, ACS Catal., 2022, 12(15), 8822–8828, DOI:10.1021/ACSCATAL.2C01698.
- C. L. McMullin, N. Fey and J. N. Harvey, Computed Ligand Effects on the Oxidative Addition of Phenyl Halides to Phosphine Supported Palladium(0) Catalysts, Dalton Trans., 2014, 43(36), 13545–13556, 10.1039/c4dt01758g.
- S. Grimme, C. Bannwarth and P. Shushkov, A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All Spd-Block Elements (Z = 1–86), J. Chem. Theory Comput., 2017, 13(5), 1989–2009, DOI:10.1021/acs.jctc.7b00118.
- A. Hjorth Larsen, J. JØrgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. SchiØtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, The Atomic Simulation Environment—a Python Library for Working with Atoms, J. Phys.: Condens. Matter, 2017, 29(27), 273002, DOI:10.1088/1361-648X/AA680E.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Extended Tight-Binding Quantum Chemistry Methods, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11(2), e1493, DOI:10.1002/WCMS.1493.
- B. Hourahine, B. Aradi, V. Blum, F. Bonafé, A. Buccheri, C. Camacho, C. Cevallos, M. Y. Deshaye, T. Dumitric, A. Dominguez, S. Ehlert, M. Elstner, T. Van Der Heide, J. Hermann, S. Irle, J. J. Kranz, C. Köhler, T. Kowalczyk, T. Kubař, I. S. Lee, V. Lutsker, R. J. Maurer, S. K. Min, I. Mitchell, C. Negre, T. A. Niehaus, A. M. N. Niklasson, A. J. Page, A. Pecchia, G. Penazzi, M. P. Persson, J. Řezáč, C. G. Sánchez, M. Sternberg, M. Stöhr, F. Stuckenberg, A. Tkatchenko, V. W. Z. Yu and T. Frauenheim, DFTB+, a Software Package for Efficient Approximate Density Functional Theory Based Atomistic Simulations, J. Chem. Phys., 2020, 152(12), 124101, DOI:10.1063/1.5143190.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, PLUMED 2: New Feathers for an Old Bird, Comput. Phys. Commun., 2014, 185(2), 604–613, DOI:10.1016/J.CPC.2013.09.018.
- M. Bonomi, G. Bussi, C. Camilloni, G. A. Tribello, P. Banáš, A. Barducci, M. Bernetti, P. G. Bolhuis, S. Bottaro, D. Branduardi, R. Capelli, P. Carloni, M. Ceriotti, A. Cesari, H. Chen, W. Chen, F. Colizzi, S. De, M. De La Pierre, D. Donadio, V. Drobot, B. Ensing, A. L. Ferguson, M. Filizola, J. S. Fraser, H. Fu, P. Gasparotto, F. L. Gervasio, F. Giberti, A. Gil-Ley, T. Giorgino, G. T. Heller, G. M. Hocky, M. Iannuzzi, M. Invernizzi, K. E. Jelfs, A. Jussupow, E. Kirilin, A. Laio, V. Limongelli, K. Lindorff-Larsen, T. Löhr, F. Marinelli, L. Martin-Samos, M. Masetti, R. Meyer, A. Michaelides, C. Molteni, T. Morishita, M. Nava, C. Paissoni, E. Papaleo, M. Parrinello, J. Pfaendtner, P. Piaggi, G. M. Piccini, A. Pietropaolo, F. Pietrucci, S. Pipolo, D. Provasi, D. Quigley, P. Raiteri, S. Raniolo, J. Rydzewski, M. Salvalaglio, G. C. Sosso, V. Spiwok, J. Šponer, D. W. H. Swenson, P. Tiwary, O. Valsson, M. Vendruscolo, G. A. Voth and A. White, Promoting Transparency and Reproducibility in Enhanced Molecular Simulations, Nat. Methods, 2019, 16(8), 670–673, DOI:10.1038/s41592-019-0506-8.
- Y. Zhang and W. Yang, Comment on “Generalized Gradient Approximation Made Simple.”, Phys. Rev. Lett., 1998, 80(4), 890, DOI:10.1103/PhysRevLett.80.890.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104, DOI:10.1063/1.3382344.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32(7), 1456–1465, DOI:10.1002/jcc.21759.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775, DOI:10.1103/PhysRevB.59.1758.
- S. Spicher and S. Grimme, Single-Point Hessian Calculations for Improved Vibrational Frequencies and Rigid-Rotor-Harmonic-Oscillator Thermodynamics, J. Chem. Theory Comput., 2021, 17(3), 1701–1714, DOI:10.1021/ACS.JCTC.0C01306.
Footnote |
| † Electronic supplementary information (ESI) available: The PDF file includes computational details, parameter selection procedures, and supporting tables and figures. All (free) energy values for PES stationary points (TS′ and minima) are included in the supporting .xlsx table. The supporting ZIP archive includes all structures of the PES stationary points. See DOI: https://doi.org/10.1039/d4sc00628c |
| This journal is © The Royal Society of Chemistry 2024 |