
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous flow synthesis enabling reaction discovery
Antonella Ilenia
Alfano†
 a,
Jorge
García-Lacuna†
a,
Oliver M.
Griffiths
b,
Steven V.
Ley
*b and
Marcus
Baumann
*a
a,
Jorge
García-Lacuna†
a,
Oliver M.
Griffiths
b,
Steven V.
Ley
*b and
Marcus
Baumann
*a
aSchool of Chemistry, University College Dublin, Science Centre South, Belfield, Dublin 4, Ireland. E-mail: marcus.baumann@ucd.ie
bYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW, Cambridge, UK. E-mail: svl1000@cam.ac.uk
First published on 28th February 2024
Abstract
This article defines the role that continuous flow chemistry can have in new reaction discovery, thereby creating molecular assembly opportunities beyond our current capabilities. Most notably the focus is based upon photochemical, electrochemical and temperature sensitive processes where continuous flow methods and machine assisted processing can have significant impact on chemical reactivity patterns. These flow chemical platforms are ideally placed to exploit future innovation in data acquisition, feed-back and control through artificial intelligence (AI) and machine learning (ML) techniques.
Introduction
Synthetic chemistry exploits at its core numerous selective and reliable methods for the precise manipulation of complex molecular architectures. Countless transformations have been discovered and optimised over the last 150 years that now form the foundation of modern synthetic chemistry. Based on this rich pedigree, chemists can today avail of a diverse toolbox for forging new bonds, new stereo centres, or even entire molecular scaffolds with high precision and selectivity. While many classical bond forming reactions (i.e., C–C, C–O, C–N, C–X bonds) can be traced back to serendipitous discoveries involving simple chemical building blocks, more sophisticated transformations have their origins in total synthesis campaigns where the observation of unexpected reactivity patterns enabled the development of more specific reaction modes.1 As topics such as asymmetric catalysis, chemical sustainability and process selectivity are critical for today's chemists in both academia and industry,2 the rate at which fundamentally new reactions are reported appears to have slowed down. This trend is however countered by significant developments in the modern era, for example photo-3 and electrocatalysis4 as well as the application of existing biocatalysts5 and the creation of non-natural variants6 that provide for far superior means towards the synthesis of complex chemical entities.The use of enabling methods, including reaction automation,7 high-throughput technologies,8 machine learning (ML)9 and artificial intelligence (AI)10 are already contributing to the development of not only more efficient chemical reactions but also the discovery of new transformations. Continuous flow chemistry thereby plays a central role as it facilitates the application of various enabling technologies whilst providing for safer and miniaturised equipment platforms.11 This leads to further advantages such as the safe use of high temperatures and pressures, straightforward scalability, as well as benefits arising from spatiotemporal processing and flash chemistry.12 Unsurprisingly, this has led to many new reactions and reactivity patterns being discovered that exploit the salient features of miniaturised flow set-ups. In this Perspective we will highlight the most significant examples of flow-based reaction discoveries as categorised by photochemical, electrochemical, and high/low temperature processes which will include important industrial contributions. The value of AI and ML for future efforts in this field will be discussed and it is hoped that this will both inform, and guide synthetic chemists interested in technology-based reaction discovery.
Discovery of photochemical reactions
In the context of photochemistry, the use of continuous flow processing is commonly favoured over batch-mode operation due to its advantages such as uniform irradiation, increased photon transfer and scalability.13 Therefore, continuous flow photochemistry emerged as an efficient alternative for the development of scalable protocols using either bespoke or commercial equipment to produce multi-gram quantities of products in hours instead of days. For this reason, synthetic chemists have made significant efforts to adapt classical photochemical methodologies to realise efficient and economical flow processes.14 However, the unique conditions that can be harnessed through flow have also been exploited to discover new reactivities.One representative example was reported by the Jamison group where the ability of continuous flow set-ups to effectively handle gases such as CO2 as well as photochemistry for rapidly screening reaction conditions was exploited. Superior gas–liquid mixing and better irradiation of the reaction mixture allowed for two new reactions to be uncovered. In the first case, a new amino acid synthesis was reported in which tertiary amines and CO2 gas are activated under UV irradiation (Scheme 1a).15 In this process single-electron activation of CO2 using p-terphenyl as catalyst takes place which is facilitated by the efficient gas–liquid mixing. More than 20 examples of this distinctive benzylic photocarboxylation reaction are reported with moderate to excellent yields.
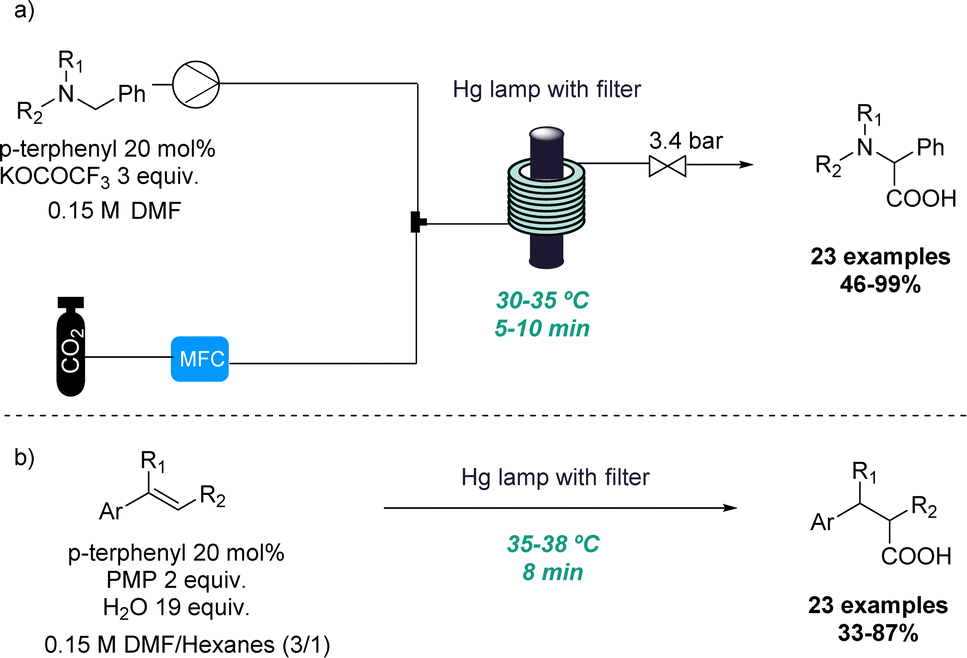 | ||
| Scheme 1 Photocarboxylation reactions in flow mode. | ||
Secondly, the same group explored this approach for the efficient β-selective hydrocarboxylation of styrenes in a metal-free process (Scheme 1b). This photocarboxylation protocol reports good selectivity to give the unusual anti-Markovnikov product for a wide range of styrenes.16 The authors provide a comparison of this flow approach to a batch alternative in which CO2 gas had to be introduced into the solution continuously. This resulted in longer reaction times and lower selectivity and yield. Finally, the flow approach is demonstrated for more than 23 examples including variably substituted styrenes with moderate to excellent yields and high selectivity towards the monocarboxylated product.
In a similar fashion, the combination of gases and photochemistry allowed Noël and coworkers to achieve the impressive photochemical activation of light alkanes.17 This approach exploited the pressurisation of the gaseous reagents to favour their photochemical reaction in the liquid phase (Scheme 2a). Therefore, light alkanes such methane, ethane, propane and isobutane were successfully functionalised using decatungstate-catalysed C(sp3)–H activation reactions under the intensified reaction conditions.
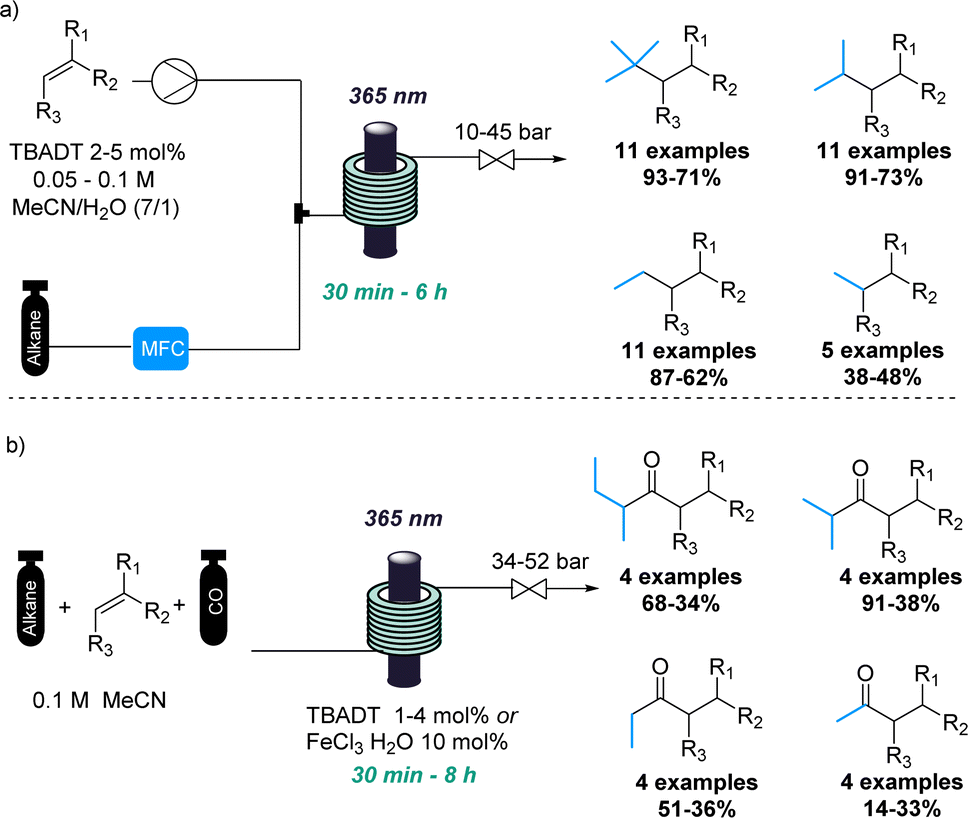 | ||
| Scheme 2 Flow intensification activating light alkanes via photocatalysis. | ||
Additionally, the same authors achieved the photocatalytic carbonylation of diverse alkanes following a similar C(sp3)–H activation approach.18 In this case, flow technology facilitated the safe handling of toxic carbon monoxide gas at elevated pressures (Scheme 2b). Although variable yields were obtained in some cases, this transformation involving light alkanes, carbon monoxide and photocatalysis would have been very difficult to achieve using traditional batch techniques. Recently, a flow set up was also used for the tungsten-catalysed C–H amination of light alkanes with nitroarenes.19 This new reaction was developed in batch, however, a flow approach was used to enhance mass transfer and increase overall safety of the process.
Carbon monoxide has also been exploited in flow-based photochemical carbonylations by the Polyzos group. The inherent hazards of this gas in combination with the high pressures required were easily solved by using a tube-in-tube flow reactor system.20 Therefore, the authors have applied this strategy in different rationally designed photochemical carbonylation processes. This culminated in the successful carbonylative hydroacylation of styrenes with alkyl halides providing more than 40 examples with mostly good to excellent yields.21 (Scheme 3). The precise addition of carbon monoxide combined with a short residence time thereby provided for good chemoselectivity and excellent functional group tolerance. Furthermore, a novel carbonylative amidation process of aryl and alkyl halides was discovered involving a highly reducing second excited state of the iridium catalyst.22 This transformation was demonstrated by accessing a large number of amide products including natural products and complex examples. Previously, the same authors had also reported a photocatalytic radical carbonylation to access 2,3-benzofurans exploiting allyloxy-tethered benzene diazonium salts in the presence of pressurised carbon monoxide gas.23
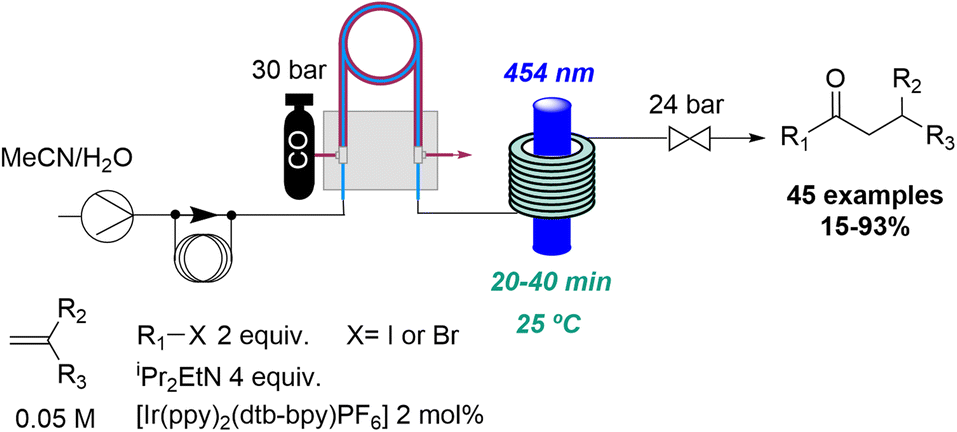 | ||
| Scheme 3 Carbonylative hydroacylation of styrenes exploiting photochemical flow conditions. | ||
The handling of insoluble substrates, reagents, or catalysts is a widely recognized challenge in continuous flow processes. For this reason, Barham and coworkers prepared an analogue of 9,10-dicyanoanthracene as a more soluble and potent oxidative photocatalyst.24 This was then explored in a new N–CH3 oxidation reaction of trialkylamines to give N-formamides in the presence of O2. After a short optimisation in a recirculation flow system, the authors opted for a continuous flow set-up to benefit from improved gas–liquid mixing and irradiation of the reaction mixture. The underlying mechanism is noteworthy as it involves a change from photoinduced SET to energy transfer, followed by a downstream HAT step. A total of 18 new compounds were synthesised in moderate to good yields, showing good functional group tolerance and opportunities for late-stage functionalisation. In the field of C–H activation reactions, Ackermann and coworkers coupled manganese-catalysis with flow photochemistry.25 Here flow was again critical over batch-mode processing to avail from better irradiation that provided for higher yields, and better catalyst performance. Ultimately, a powerful manganese-catalyzed C–H arylation of (hetero)arenes was developed affording more than 30 compounds in good to excellent yields.
The examples highlighted above showcase scenarios where the discovery of new reactions is enabled by the safe and effective use of gases at elevated pressures in combination with photocatalysis in flow mode. Through miniaturisation flow chemistry thereby provides for excellent photon transfer and micro-mixing of gaseous reactants under intensified conditions. An alternative approach towards discovering new photochemical reactions is based on the direct excitation of suitably conjugated substrates with UV light using flow reactor set-ups. This can be exploited to generate short-lived reactive intermediates that trigger new reaction cascades and thus generate new types of products.
This approach was recently demonstrated in the context of discovering a new photochemical cascade in flow as reported by Baumann and coworkers.26 Exploiting alkyne bearing chalcones thereby generated a transient carbene that was trapped by a nearby aryl ring system to afford isolable cycloheptatrienes and ultimately, complex cyclobutenes via a sequence of pericyclic reactions (Scheme 4).
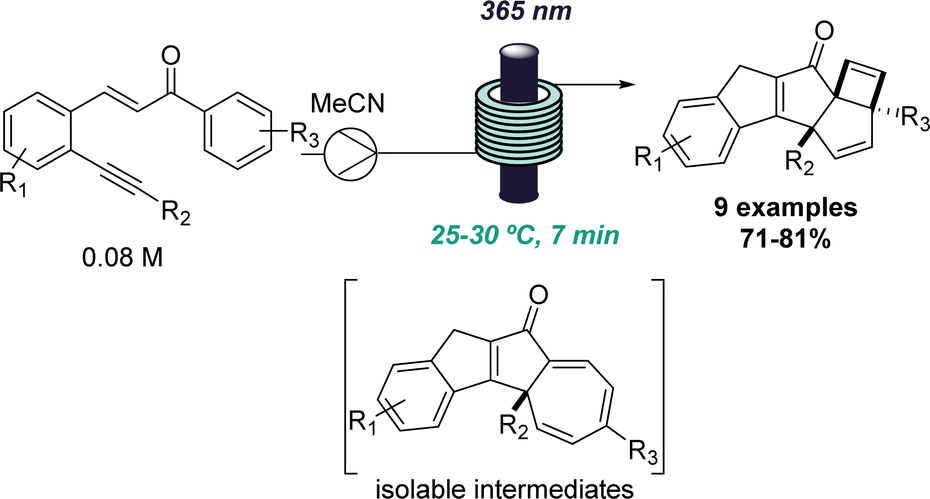 | ||
| Scheme 4 Photochemical reaction cascade discovered in flow. | ||
The uniform irradiation of the substrate solution with a high-power LED (365 nm) in combination with short residence times also allowed for the isolation of late-stage intermediates in support of the proposed mechanism. Subsequently, continuous flow processing facilitated the expansion of the substrate scope to identify a highly regioselective entry into highly substituted polycyclic products that shows how flow-based reaction discovery can be merged with exploring new chemical space.27
The same group also reported another intriguing photochemical rearrangement process. In this case, highly reactive ketenimines were obtained from trisubstituted isoxazoles upon short exposure to UV-C light (Scheme 5).28 This finding emanated from a previous publication where isoxazoles were rearranged to form oxazoles via keto azirine intermediates.29 However, a different reactivity was observed by altering the substitution pattern of the isoxazoles. A set of isolable ketenimines was generated in good yields and reacted in situ with hydrazines to give functionalised trisubstituted pyrazoles. This example demonstrates the power of flow processing for the serendipitous discovery of new reactions that enable the preparation of unusual intermediates as well as valuable heterocycles via a telescoped approach.
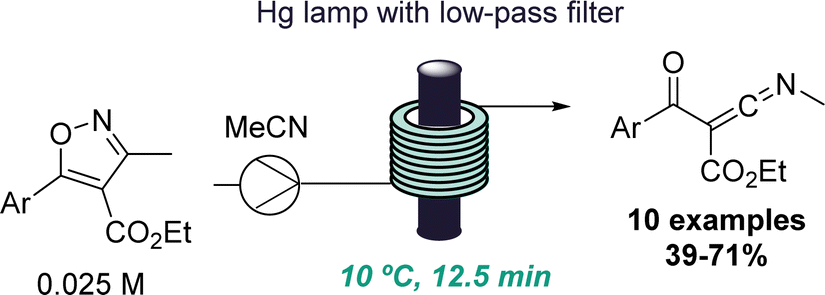 | ||
| Scheme 5 Skeletal rearrangements of isoxazoles discovered in flow mode. | ||
The value of flow photochemistry for accessing highly reactive intermediates has also been exploited in the context of generating benzynes from stable triazine precursors. Here, the benzyne precursor was found to undergo an unusual amide substitution under photochemical conditions if secondary amines were present.30 Instead of the predicted insertion of the benzyne into the N–H bond of the amine, a new triazine species was formed in a high-yielding process. This unexpected reactivity enabled the development of a new triazine synthesis affording these underutilised compounds in moderate to good yields (Scheme 6). In this case, flow photochemistry was harnessed for more uniform irradiation which is especially important when handling highly reactive species.
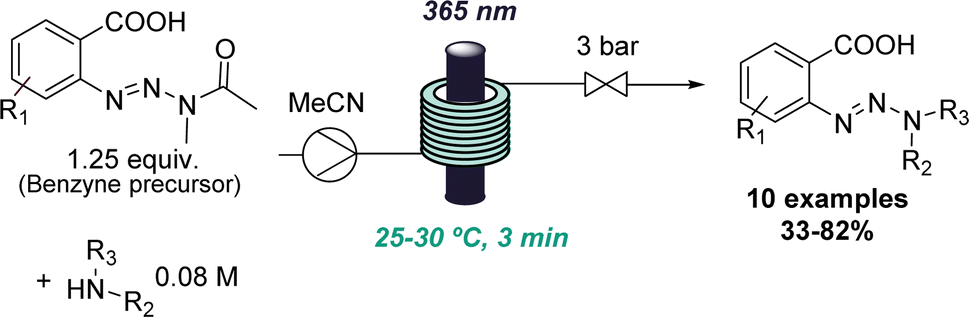 | ||
| Scheme 6 Photochemical benzyne formation as a starting point for discovering a new route to triazines. | ||
Additionally, flow photochemistry has recently gained traction as a powerful tool for the rational design of new procedures, based on known photochemical reactions that benefit from greener conditions, accelerated reaction screening, and better irradiation in flow to generate various target compounds. One representative example is the synthesis of α-aminoamide derivatives by Aleman and collaborators.31 Based on known radical addition concepts, a new synthesis of the desired α-aminoamides was developed whereby flow provided vast improvements compared to similar batch processes including the possibility of telescoping the hydrolysis of the amine. Furthermore, Rueping and coworkers identified flow conditions to develop both C–H functionalisation and cross-dehydrogenative coupling reactions using rose Bengal as a photocatalyst under irradiation with green LEDs.32 Although these transformations were not conceptually novel, utilizing flow approaches enabled the use of green light and a benign photocatalyst which improves the green credentials of this chemistry. Consequently, good yields for 10 different substrates were obtained albeit with relatively long reactions times (ca. 20 h in flow compared to 3 days in batch). In a similar fashion, Fang and Tranmer reported different flow photocyclization reactions that avoided the use of metals and multi-step sequences. The effective use of flow chemistry enabled the desired transformation and provided rapid access to a range of different phenanthridinones,33 thieno[3,2-c]quinolin-4(5H)-ones, and benzo[h]-1,6-naphthyridine-5(6H)-ones34 with potential biological interest.
In relation to industry-led advances towards new reactions involving flow photochemistry, the main trend is the adaptation of known procedures, but using greener, readily scalable, and highly efficient methods. A representative example of this is Jansen's effort to develop new photochemical metal-catalysed cross coupling methods. In most cases, these studies start with a batch exploration with the aim of transferring the conditions to a future flow process where the final optimisation takes place. In this context, a visible light induced nickel-catalysed Negishi reaction was reported.35 Apart from all the previously mentioned advantages of flow photochemistry over batch, the authors highlight the use of freshly prepared organozinc derivatives and only 2 mol% of nickel catalyst needed. Consequently, more than 20 products were generated in moderate to excellent yields, and a scale-up operating for more than 8 hours was also demonstrated (Scheme 7). Moreover, it was possible to expand the substrate scope to historically unsuccessful substrates for this transformation.36 A palladium-catalysed procedure accelerated by blue light was disclosed in this case, further expanding the scope to 31 more compounds.
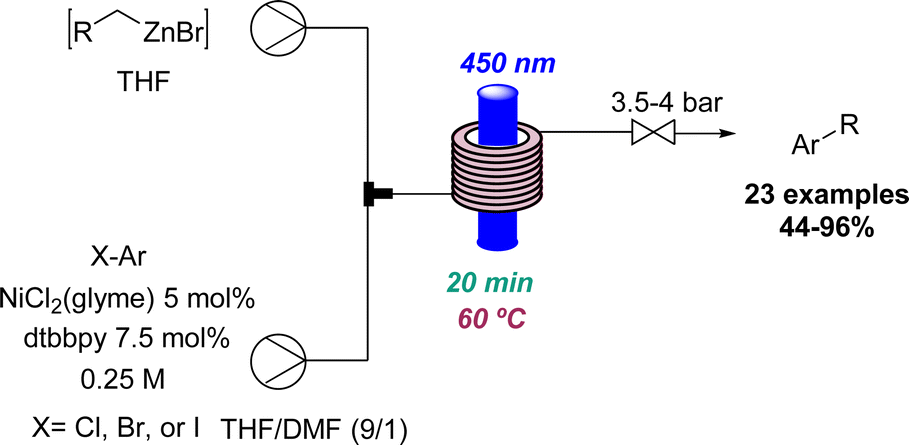 | ||
| Scheme 7 Ni-catalysed photochemical Negishi cross coupling reactions in flow mode. | ||
Subsequently, a visible light promoted iron-catalysed Kumada–Corriu coupling was reported in a collaboration between Janssen and the Noël group.37 The use of flow processing provided a rapid and scalable approach to access to a wide range of compounds (up to 27 with good to excellent yields) with good functional group tolerance and including electron-rich aryl chlorides which usually require more forcing conditions when using other protocols.
Additional applications that highlight the value of flow photochemistry for the identification of more sustainable conditions include photoredox C(sp2)–C(sp3) cross-coupling reactions developed through collaborative efforts between the Ley group and a team from Novartis.38 Here flow photochemistry was used to overcome issues associated with potassium trifluoroborate salts and the limited scalability of analogous batch methods. This work demonstrates the efficient cross coupling using visible light and readily accessible boron pinacol esters as substrates for more than 30 substrates with good to excellent yields (Scheme 8).
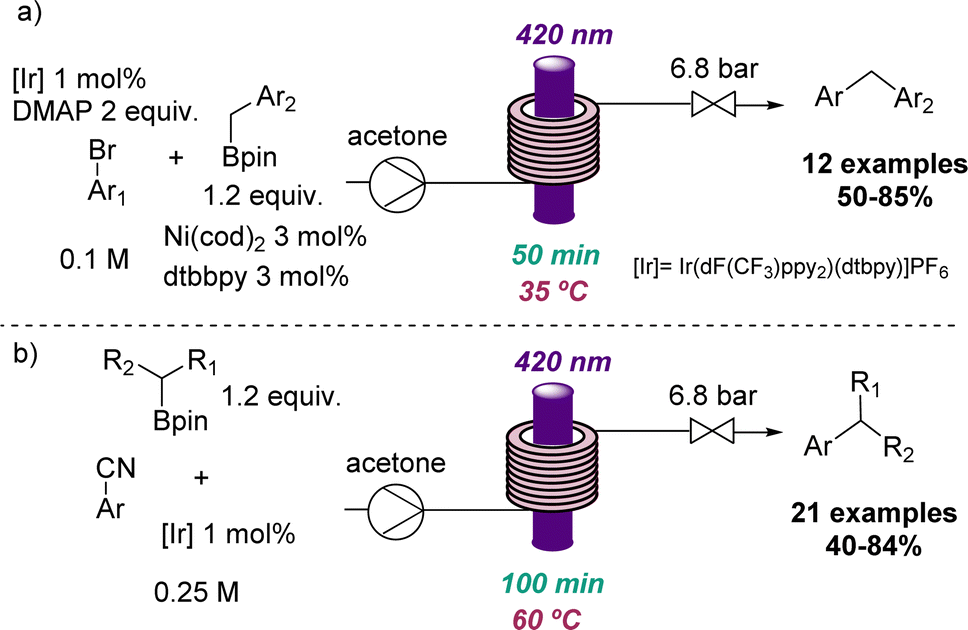 | ||
| Scheme 8 Photochemical C(sp2)–C(sp3) cross-coupling reactions in flow. | ||
A team of researchers from Janssen and the Kappe group reported the flow synthesis of arylhydrazines via a nickel-catalysed photoredox coupling between aryl halides and Boc-protected hydrazine.39 After preliminary batch experiments, most of the screening and optimisation was performed in continuous flow mode which included a DoE study. Ten different substrates were demonstrated including two scale-ups showing process robustness and avoiding issues with nickel precipitation.
A light induced C–H arylation of arenes using diazo anhydrides prepared in situ was recently reported through a collaboration of between Lilly and the Kappe group.40 The required diazo anhydrides were generated from anilines through a new photochemical pathway that does not require the intermediacy of diazonium salts. Although the exploration started in batch, the authors quickly adopted flow processing to avail of better irradiation and consequently minimise side product formation. A set of 17 different compounds was then synthesised in moderate to good yields.
A final case study demonstrating the value of academia-industry collaborations concerns aerobic photochemical C(sp3)–H oxidations. A team from the Noël group and Abbvie reported this transformation using molecular oxygen and decatungstate photocatalysis.41 After a short exploration in batch, the authors exploited flow chemistry to improve both mixing and irradiation and thus realise better yields. This resulted in a large scope of substrates being converted into related ketones through efficient flow processing which would not be achievable in batch mode.
Electrochemistry
Driven by the increasing demand for sustainable synthesis methods, researchers worldwide are focusing on the development of electrochemical reactions. This is as electrons can be considered as traceless reagents which leads to significant advantages over the use of stoichiometric and potentially toxic reductants and oxidants. Electrochemical flow cells overcome several issues associated with analogous batch set-ups, whereby the large ratio of electrode surface to reactor volume, efficient mass transfer and easy scalability stand out as key features. The small inter-electrode distance furthermore results in a reduction or even elimination in the use of supporting electrolytes needed in the reaction mixture, thus enabling an improvement in terms of sustainability and material costs. Furthermore, the reaction selectivity can be easily tuned by changing the nature of the electrode material and the applied potential.42As the following case studies demonstrate, the role of flow electrochemistry in the discovery of new reactions has become notable in recent years.
An intramolecular dehydrogenative C–S coupling using a modular flow electrolysis cell was highlighted by Xu and co-workers in 2019 to synthetise 1,4-benzoxathiins and 1,4-benzothiazines. Despite several studies performed in batch on the C–S coupling for the synthesis of benzothiazoles, oxidative desulfurisation reactions still represent an unsolved challenge for fused six-membered heterocycles. By conducting the electrolysis under acid conditions using a flow cell, a new pathway was discovered, and no traces of desulfurised amides were observed, which contrasts with the batch reaction. The reactions proceeded with evolution of hydrogen gas, overcoming the need for oxidising reagents and proton acceptors. The reaction robustness was demonstrated by preparing a library of 30 compounds, affording yields ranging between 48 and 81% (Scheme 9).43
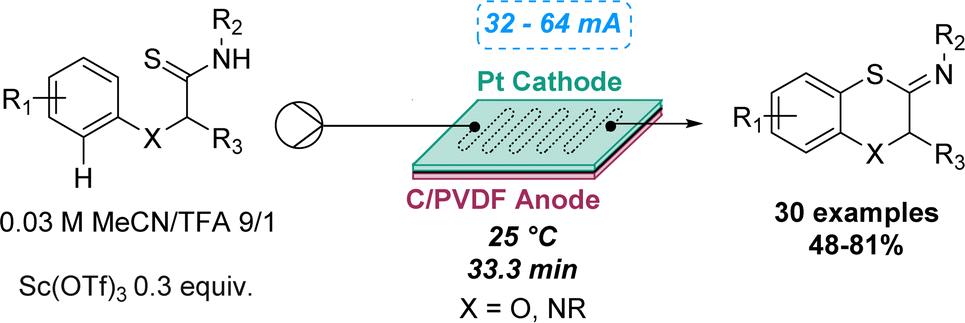 | ||
| Scheme 9 Electrochemical flow synthesis of 1,4-benzoxathiins. | ||
In the same year, Ackermann and co-workers developed one of the first electrocatalytic C–H activation in flow, whereby the active rhodium(III) carboxylate catalyst was generated in situ on the anode. Metalla-electrocatalysis represents an effective route towards more sustainable synthesis and the flow platform increases the robustness of this method in terms of functional group tolerance, chemoselectivity, regioselectivity and scale-up. Several alkynes underwent flow-assisted annulation reactions with a variety of C–H/N–H functionalisations in an inter- or intramolecular manner, resulting in a library of 26 compounds in moderate to excellent yields. This flow platform led to the discovery a new catalytic reactivity by employing electricity in a dual role of terminal oxidant and promotor for the reductive elimination in the overall rhodium-catalysed C–N bond formation (Scheme 10).44
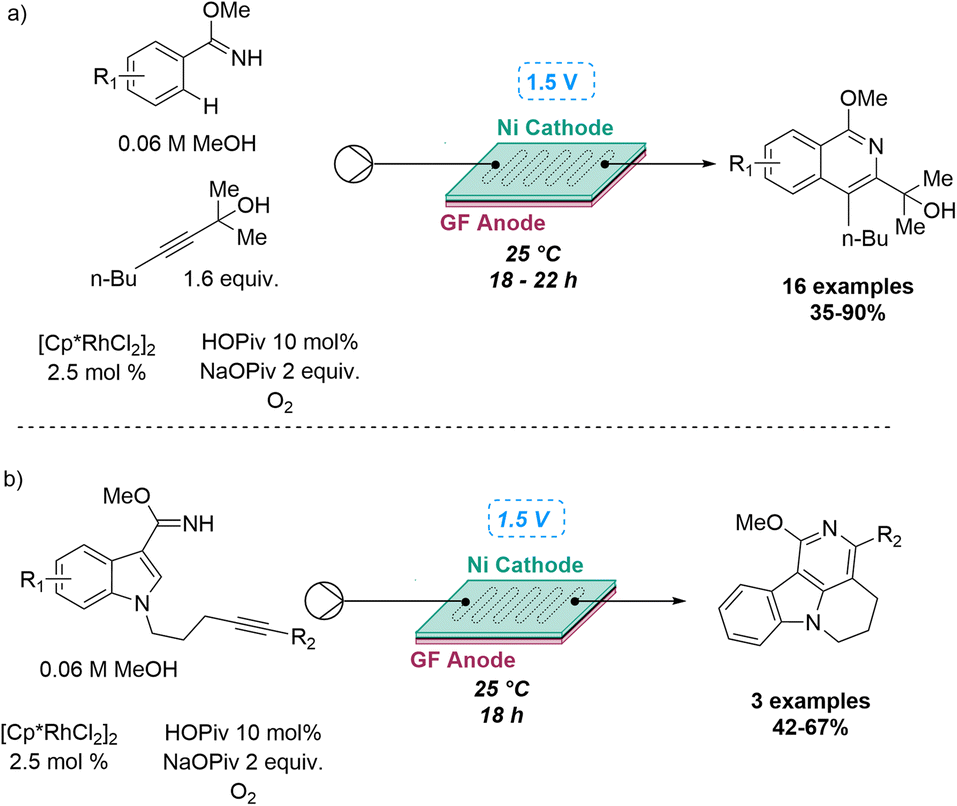 | ||
| Scheme 10 Electrochemical flow synthesis via Rh-catalysis. | ||
A similar electrochemically C–H functionalisation process was also explored by the Xu group for two different reactions. This included a flow-based electrochemical C–H phosphorylation of arenes to prepare target aryl phosphorus compounds. In previous batch reactions of this type, the substrate scope was limited to electron-rich arenes or azoles, due to over-oxidation issues of the intermediate radical cation and the moderate reactivity of the neutral P-radical formed. Flow chemistry enabled the investigation of new reactivity for anodically generated P-radical cations with the subsequent trapping by arenes in a process that did not require a catalyst or an external oxidant. By using H2O as an additive, 44 aryl phosphorus compounds were synthetised, exploring both electron-rich and electron-deficient arenes, natural products and synthetic bioactive compounds, with yields ranging between 26 and 94%.45
Similarly, an electrochemical C–H hydroxylation of arenes was developed in flow by the same group in 2022, that did not require catalysts or oxidants. Previous reactions of this type suffered from a limited substrate scope, overoxidation issues and low reactivity of the arene radical cation intermediate. By using an undivided flow cell under acidic conditions, electron-rich, neutral and electron-deficient arenes were used, accessing a large number of products in good to excellent yields (Scheme 11).46
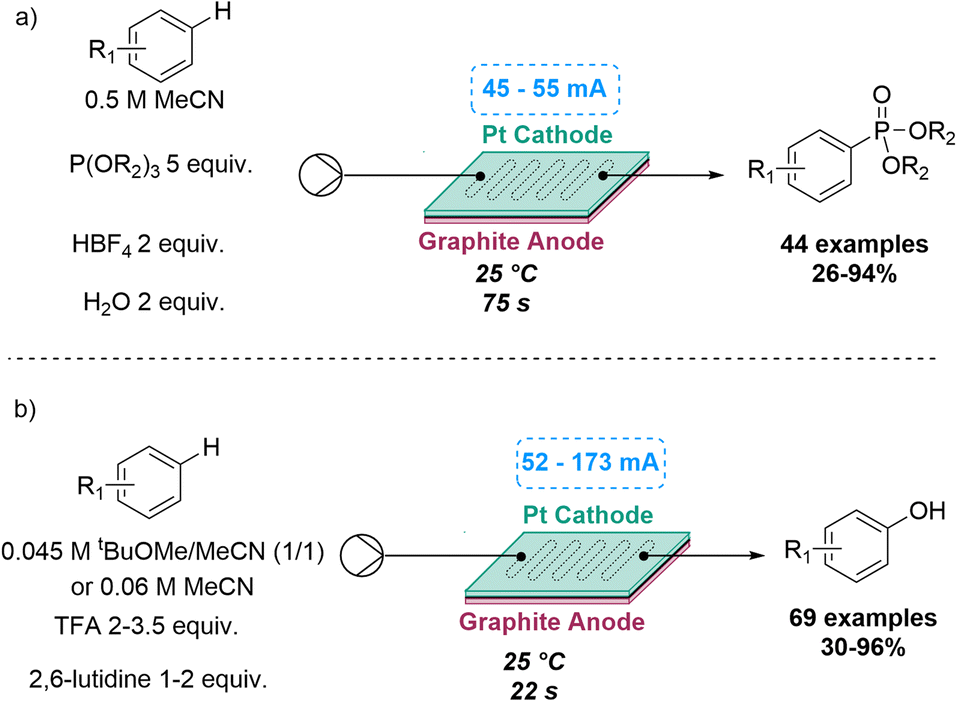 | ||
| Scheme 11 Flow-based C–H phosphorylation and hydroxylation reactions exploiting electrochemistry. | ||
Electrochemistry can also be employed as an alternative pathway in the oxidative decarboxylative ether formation through Hofer–Moest-type reactions, as described by Brown and Linclau in 2020.47 This methodology was applied to cubane carboxylic acids as challenging substrates for their functionalisation under previously reported batch conditions. Carrying out this reaction in continuous flow mode was essential to work under electrolyte-free conditions. By using a commercially available flow cell and no supporting electrolyte, a library of 14 compounds could be prepared in moderate yields, exploring different alcohols as nucleophiles as well as functional groups on the cubane moiety.
Furthermore, flow electrochemistry has been applied with great effect to aza-Wacker cyclisation reactions generating different N-heterocycles, as reported by Xu and co-workers in 2021. This approach led to an increase in the electrode surface area, facilitating the mass transfer in the alkene cyclisation.48 This catalyst-, reagent- and additive-free method tolerated a wide range of substrates resulting in a library of 48 compounds in high yields. Notably, the same method under batch conditions gave inferior results and a supporting electrolyte was needed to increase the conductivity. In addition, flow electrochemistry was also applied to the synthesis of spiro-indolinines via the dearomative arylation of indoles. Although optimisation and discovery were carried out in batch, continuous flow was investigated to improve scalability.49
Reaction discovery via thermal processes
Historically, the use of high temperatures has allowed for the discovery of new reactions as energy barriers can be overcome easily whilst the increased rate of reactions means that thermodynamically favoured products can be accessed more quickly.In the context of flow chemistry, improved heat transfer resulting from high surface area to volume ratios is one of the main advantages over batch processing.11 This does not only lead to process intensification and improved safety through reactor miniaturisation, but also newly discovered thermal reactions that would not be viable in analogous batch set-ups. In addition, the ability to exploit backpressure regulators and thus work above the atmospheric boiling point of common organic solvents, and the intrinsic scalability of continuous flow systems make this the method of choice for many industrial processes. Being able to control the exothermicity of a reaction often leads to better selectivity towards a given product, providing routes to otherwise inaccessible compounds. Some examples where high temperature conditions have been used to explore new reactivities in a safe and controlled manner are discussed below.
Two representative examples illustrating the effect of flow methods at elevated temperature to promote a desired reactivity were reported by Ackermann and coworkers. Here, different Mn-catalysed C–H arylations were achieved after exploring the effect of temperature, solvent, catalysts, and additives in continuous flow mode. As shown in Scheme 12 this rendered a set of over 20 indoles and related structures via chemoselective hydroarylation reactions.50 Afterwards, the reaction was expanded to include C–H arylation reactions of azines with a different catalytic system.51 In both cases, the authors performed a batch comparison with similar and sometimes improved yields. However, the reaction discovery and the initial optimisation were executed in flow mode due to its intrinsic advantages of working at high temperatures and rapid screening of reaction conditions.
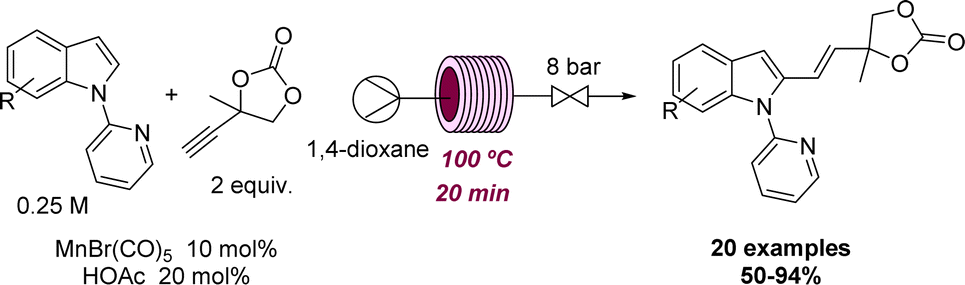 | ||
| Scheme 12 Development of a flow-based C–H alkynylation reaction. | ||
A recent effort by the Ley group led to the discovery of a new method for the preparation of homoallylic alcohols in flow mode.52 This process operates at 60 °C and was effectively used to generate a set of 15 products that were prepared from TMS-diazomethane, E-vinyl boronic acids and aldehydes in good to excellent yields (Scheme 13). After the initial flow discovery, the method was expanded to allow batch processing in view of wider uptake.
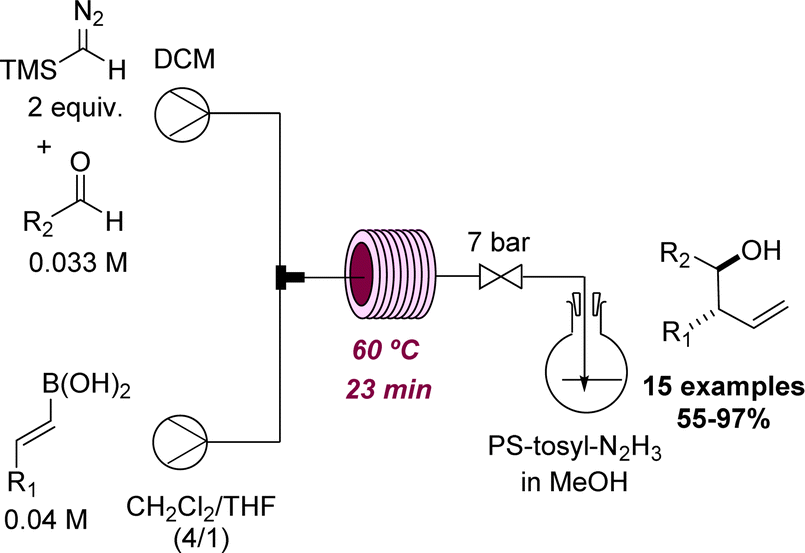 | ||
| Scheme 13 Synthesis of homoallyl alcohols in flow mode. | ||
A related flow process exploiting in situ generated diazo alkanes via the MnO2-mediated oxidation of hydrazones was reported by a team of researchers from the Ley group and Pfizer.53 This provided a convenient access to reactive diazo species that were then coupled with boronic acids at ambient temperature. Flow chemistry thus served as a discovery tool for these important sp3–sp2 cross coupling reactions based on diazo species that could be expanded to generate various cyclopropane products.54
For similar reasons as discussed above in the context of photochemistry, working with gases in continuous flow is usually preferred over batch because of the better mixing achieved and reduced safety concerns arising from reactor miniaturisation. The formation of cyclic carbonates from epoxides and CO2 is a well-known reaction, however, Jamison and coworkers recently achieved this transformation using a novel strategy. The epoxide is thereby activated by electrophilic bromine with CO2 in the presence of DMF.55 The initial exploration was done using batch set-ups, but the final optimisation used a flow set-up allowing the reaction time to be reduced to 30 minutes with full conversion of the substrate. A set of 12 different cyclic carbonates was synthesised following this procedure in good to excellent yields.
Additionally, a new Rh-catalysed hydroaminomethylation reaction was discovered by Abolhasani and coworkers.56 Based on preliminary batch procedures and a thorough optimisation in flow, a more general and scalable procedure was developed, which allowed the use of hindered amines and highlighted the possibility to generate the intermediary enamine species in preference to the reduced amine which shows a new hydroaminovinylation pathway.
Supercritical carbon dioxide (scCO2) is often employed in industry as a versatile and green solvent. However, the extreme conditions applied can lead to unexpected reactivities. In a related study George and Poliakoff employed γ-Al2O3 as a heterogeneous catalyst for the direct amination of alcohols in scCO2. Unexpectedly, this study found that cyclic ureas and urethanes can be accessed by incorporation of CO2 under more forcing conditions (Scheme 14).57 This study is related to earlier work by the authors reporting the thermal reaction between tetrahydrofurans, dimethylcarbonate, and anilines in the presence of γ-Al2O3 which led to a variety of new products based on the opening of the tetrahydrofuran ring system. Here the use of flow reactors allowed their use towards self-optimising and potentially automated processes.58
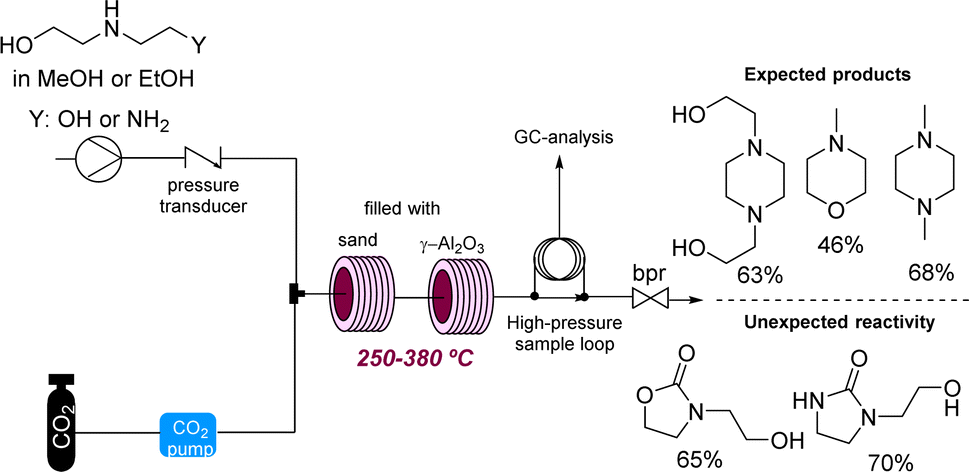 | ||
| Scheme 14 Discovery of new reactivities under thermal conditions. | ||
Another unexpected outcome in relation to a well-known transformation was reported through a collaboration between the Baumann group and Almac.59 When using quaternary proline derivatives as substrates for Curtius rearrangements unsaturated pyrrolidines and ring-opened ketones were obtained as main products. This new reactivity can be explained via an interrupted Curtius rearrangement occurring at elevated temperatures of 100 °C. Although this unknown reactivity was first observed under batch conditions, flow processing was preferred during the following optimisation study in view of reaction safety, substrate scope, and scalability of the process.
Flow technology has also been exploited to access thermodynamically unfavourable compounds under more forcing conditions. For instance, Pérez Castells and coworkers achieved the successful Pauson–Khand reaction of vinyl ethers by using high temperatures and pressures in continuous flow.60 Moreover, it was possible to detect products not previously reported via cobalt catalysis. Another example can be seen in the successful synthesis of 1,4-benzothiazines which was achieved by Oh and collaborators.61 Unlike under batch conditions, the use of flow processing allowed to control the ambivalent reactivity of the (E)-β-vinyl ketone substrate towards the desired benzothiazine targets (Scheme 15).
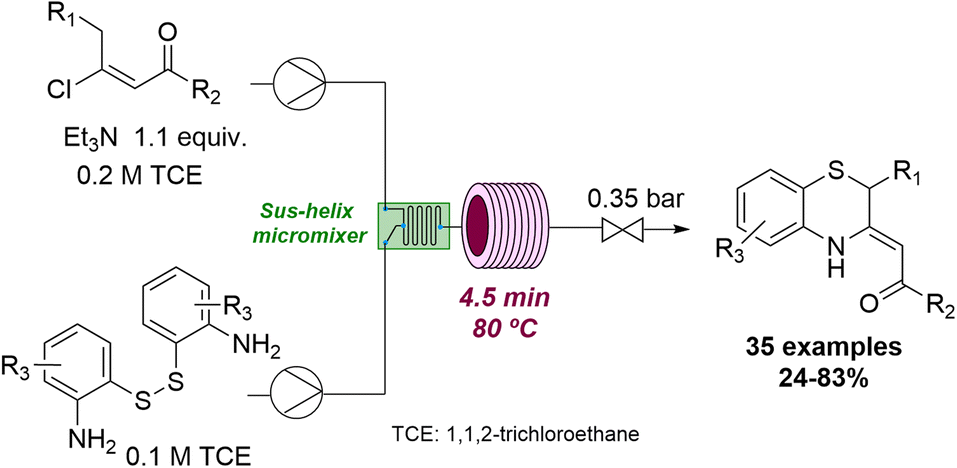 | ||
| Scheme 15 Flow synthesis of benzothiazines from (E)-β-vinyl ketones. | ||
The generation and use of highly reactive species at elevated temperatures can lead to various unexpected flow reactions (Scheme 16). Examples include a recently published synthesis of substituted benzo[d][1,3]dioxoles from peroxides and amides as reported by Gnanaprakasam and coworkers.62
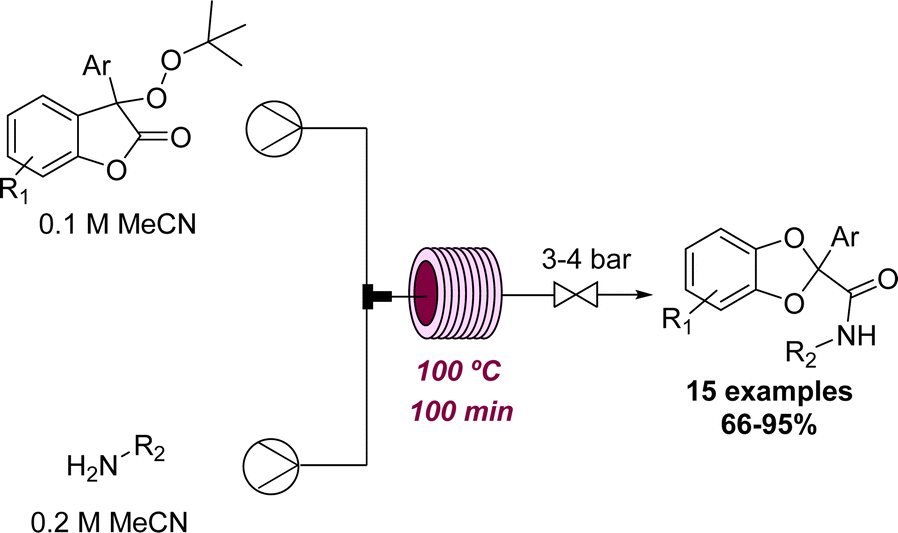 | ||
| Scheme 16 Thermal rearrangement of peroxides in flow. | ||
Similarly, the Kirschning group combined flow processing with inductive heating methods leading to the oxidative conversion of cyclic ketones into ring-expanded esters as well as hydrocarbons in the presence of H2O2/HNO3.63 The Jamison group reported a flow-based access to diimide from N,O-bistrifluoroacetyl hydroxylamine and free hydroxylamine to affect the saturation of various alkenes at elevated temperatures in flow mode.64 In analogy, Kappe and coworkers have developed a related method to generate diimide in situ for metal- and hydrogen-free transfer hydrogenations.65 Hydrazine hydrate and molecular oxygen were thereby exploited in a continuous flow process at 100 °C. This flow approach compared favourably to a related batch method that was characterised by low selectivity and productivity.
Lithiation reactions
Lithiation reactions are amongst the most versatile synthetic methods based on the metalation of suitable organic precursors that enable a variety of subsequent transformations. Despite the value of these reactions in academia and industry, lithiations are very challenging due to the associated exothermicity when generating these high-energy intermediates. Further challenges arise when scaling these processes as insufficient mixing can lead to side reactions that may only be controlled under cryogenic conditions. Continuous flow processing is thus an invaluable tool for lithiations as the improved heat and mass transfer mitigates these problems. Spatiotemporal control furthermore allows chemists to execute highly selective lithiations that are not possible in batch mode which has resulted in the coining of the term flash chemistry describing these unique flow-based lithiation processes.66 In analogy to lithiation reactions, similar advantages have been reported for analogous magnesiation reactions67 as well as the generation and use of organo-sodium and -potassium species.68The last ten years have seen a steady increase in flow-based examples of effective and innovative lithiation reactions. An early example exploring new reactivity by taking advantages of precise control of temperature and residence time was published by the Ley group in 2014 (Scheme 17).69 This study reports the lithiation of dibromomethane in flow mode which is challenging in batch due to the decomposition of this intermediate. The lithiated dibromomethyl species was thereby added to several esters affording the resulting ketone products in high yields. Notably, for substrates bearing acidic α-protons this flow approach allowed for new reactivity patterns as competitive enolisation reactions could be avoided.
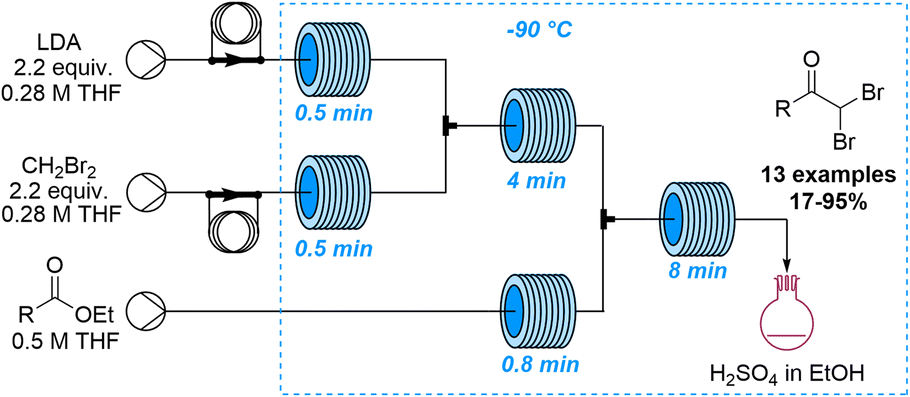 | ||
| Scheme 17 Generation and use of lithiated dibromomethyl species in flow. | ||
Yoshida and Kim reported on the selective lithiation of isocyanate-substituted aryl halides using phenyllithium as the organometallic reagent.70 Preliminary batch experiments showed that the undesired reaction at the isothiocyanate site cannot be prevented. However, the precise control of residence time in microflow set-ups allowed the lithiation of m- and p-iodophenyl isothiocyanate with phenyllithium to selectively occur at −40 °C (tRes = 14 ms). The resulting aryl lithium species can then be trapped with various electrophiles and further reactions on the isocyanate with external nucleophiles can be achieved via a telescoped flow process in only six seconds. This process was demonstrated for 20 compounds that were generated in high yields (Scheme 18).
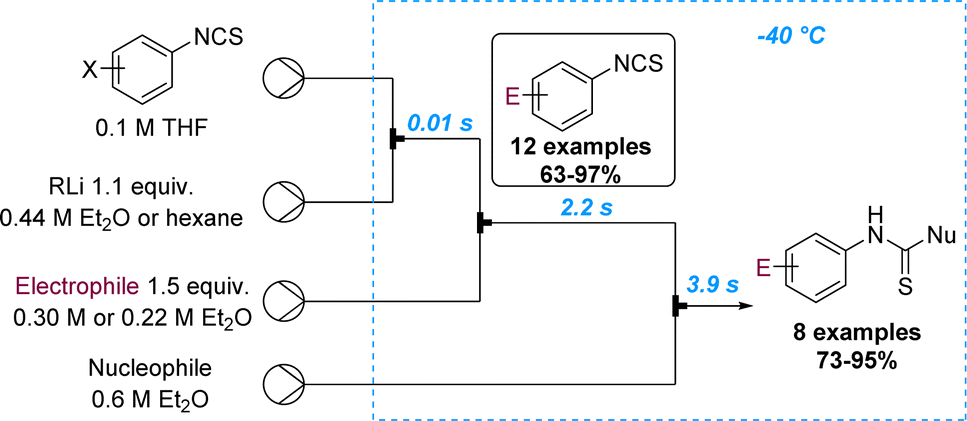 | ||
| Scheme 18 Lithiation of m- and p-iodophenyl isothiocyanate with phenyllithium. | ||
Another powerful example of flash chemistry was published by Kim, Yoshida and co-workers in 2018 (Scheme 19).71 In this flow protocol ortho-lithiated aryl benzyl ethers are generated in situ from tBuLi at ambient temperature. The superb reaction control of the resulting microreactor allowed to either trap this aryl lithium species immediately with an electrophile (tRes < 1 second) or promote its isomerisation to a benzylic anion over a period of 6 seconds prior to trapping. Lastly, longer reaction times (1 h at 50 °C in batch) resulted in a [1,2]-Wittig rearrangement providing tertiary alcohols after protonation. A collection of 18 different products exploiting this flow-based approach rendered these targets in high selectivity and excellent chemical yields.
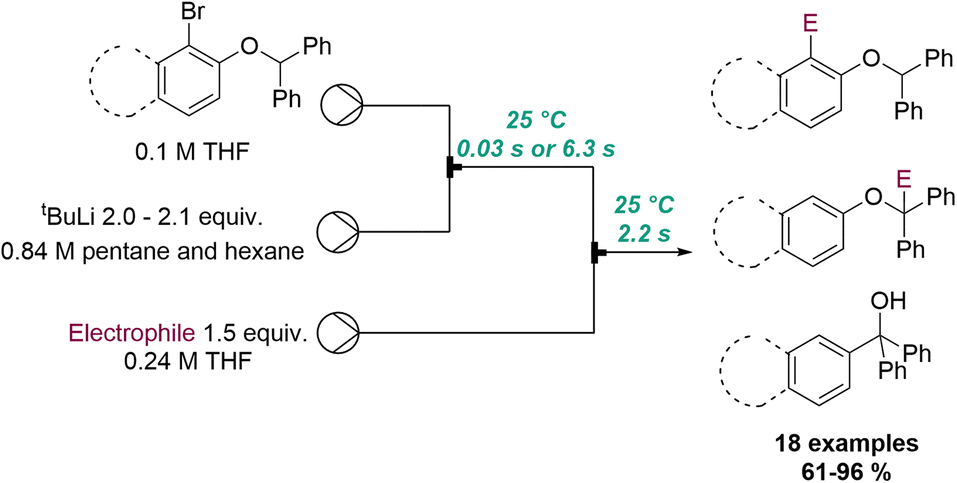 | ||
| Scheme 19 Flow protocols exploiting ortho-lithiated aryl benzyl ethers. | ||
The importance of precise control of residence time in the millisecond range was also showcased by the same authors exploiting the cis–trans isomerisation of α-anionic stilbenes (Scheme 20).72 In batch mode the rapid isomerisation from cis to the more stable trans isomer cannot be prevented. However, flow processing allowed to immediately quench the α-lithiated cis-stilbene within 55 ms. Alternatively, by increasing the residence time to 94 s, full isomerisation to the more stable trans-stilbene isomer was observed, highlighting the benefits of flow chemistry as tool for inaccessible reactivity in batch. A library of 24 examples with yields ranging between 54 and 99% was described, exploring several electrophiles.
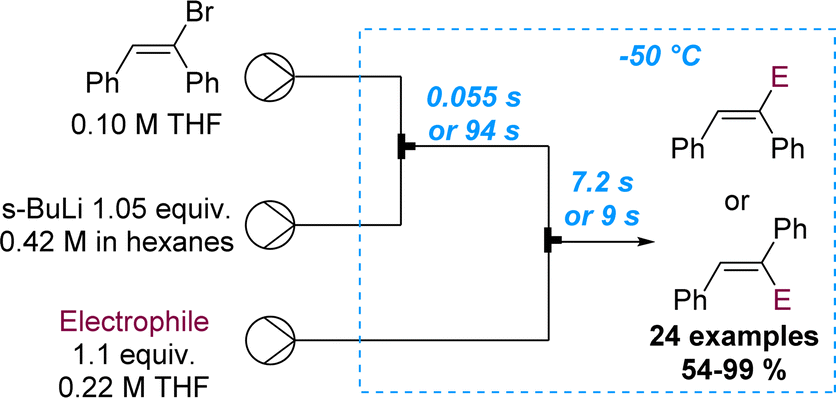 | ||
| Scheme 20 Regioselective flow synthesis of α-functionalised stilbenes via precise residence time control. | ||
Further applications of flow-based lithiation protocols were reported by Barker in 2019 for the α-lithiation of 1,3,4-oxadiazoles at room temperature.73 Improved mixing and heat transfer in flow mode allowed for the selective generation of this species that is not possible in batch due to inevitable ring fragmentation and related side reactions.
Early studies74 describing the addition of dichloromethyl lithium to carbonyl compounds and a subsequent homologation through a Meinwald-type epoxide-aldehyde isomerisation process inspired Luisi and co-workers to exploit flow processing to investigate this transformation. This work demonstrated how flow chemistry effectively tames the unstable lithium dihalocarbenoid species en route to generating α-chloro aldehydes bearing a quaternary center. Through precisely controlled residence times in the millisecond range relatively high temperatures of −20 °C can be exploited in flow mode which is not a viable option in batch.75 The same group reported the C3- and C2- functionalisation of constrained aza-heterocycles through the generation of highly reactive lithium species in flow.76 Preliminary experiments were performed in batch, however, flow processing was quickly identified for its superiority as it guaranteed better reaction control that was crucial for the generation and use of unstable organolithium species, resulting in a library of N-Boc azetidines with good yields. In addition, Luisi and co-workers showcased how flow processing can be exploited for the exploration of new chemical space for strained heterocyclic targets (Scheme 21).77
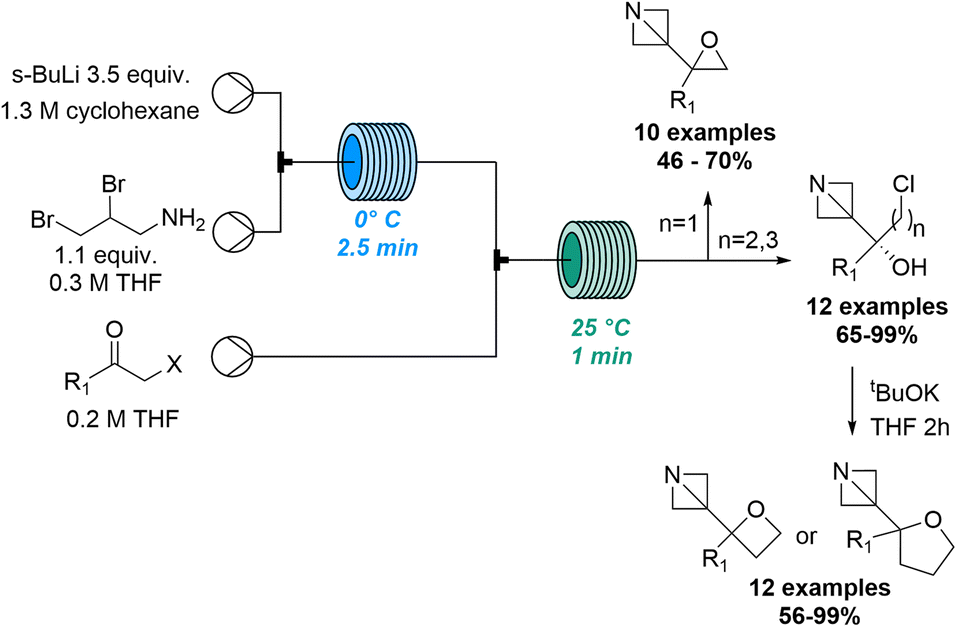 | ||
| Scheme 21 Exploration of new chemical space via lithiated azabicyclo[1.1.0]butanes. | ||
Building on prior studies by the Kappe and Luisi groups78 this innovative approach thereby describes a robust strategy to synthetise series of complex 1-azabicyclo[1.1.0]butanes (ABB) which are highly strained C(sp3)-rich compounds. Using sBuLi in combination with a residence time of 2.5 minutes the ABB-Li species was generated at 0 °C in flow mode. This was then trapped in a second reactor coil with different haloalkyl ketones to produce a variety of target compounds. A library of 34 products was accessed in high yields and excellent chemoselectivity and allowed the concomitant formation of oxiranes, oxetanes and tetrahydrofurans adjacent to the azetidine core.
In the same year, Yorimitsu and co-workers highlighted the use of flow chemistry in the reductive dimerisation of styrenes for the generation of reactive 1,4-dilithium species. By taking advantage of microreactor technology, the unwanted polymerisation of these highly reactive intermediates could be avoided allowing for immediate trapping reactions with several electrophiles.79
The regioselective deprotonation of dihalopyridines using LDA was studied by Legros and co-workers and showcased divergent functionalisation reactions in flow mode. Aided by superb control over residence time and temperature it was possible to either trap the lithiated intermediate with electrophiles at −60 °C or trigger a halogen dance process at −20 °C (Scheme 22).80 Analogous batch reactions were flawed due to the rapid degradation of the lithiated pyridine species resulting in low selectivity and yields. This study thus demonstrates the value of flow processing in divergent functionalisation reactions of pyridines affording 28 target compounds in good yields.
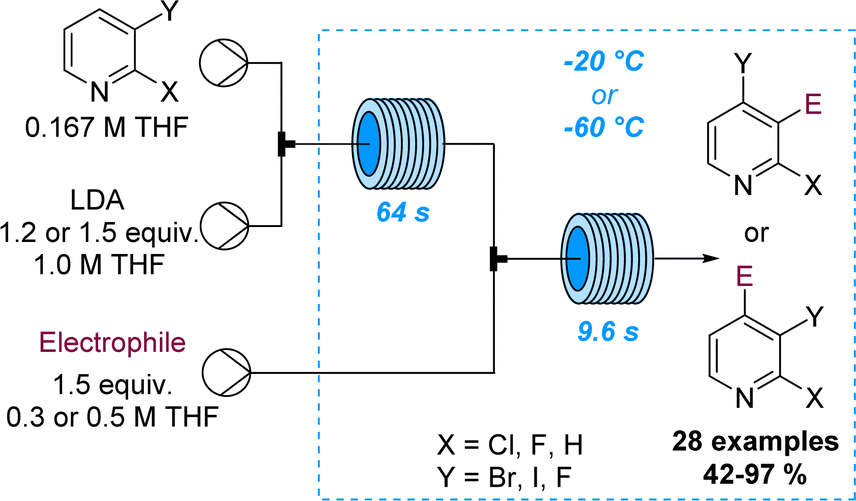 | ||
| Scheme 22 Product divergency in flow-based lithiations of dihalogenated pyridine scaffolds. | ||
Whilst the above examples highlight strategies based on flow-based organolithium species and their reaction with electrophiles, recent efforts also exploit transmetalation reactions. As demonstrated by Kim and co-workers, flow processing can thereby improve the chemoselectivity for the direct C–H metalation of tetrahydrofuran (THF).81 In batch mode the generation of the α-anionic THF species will lead to fragmentation giving ethylene gas and a lithium enolate species, however, due to the precise control over residence time and temperature in a microfluidic system flow processing can prevent this competing pathway. This demonstrated that α-methylated THF can be rapidly prepared at −50 °C in situ using Schlosser's base (KOtBu/sBuLi) prior to transmetalation via cupration and borylation reactions.
In 2020, Jamison and co-workers discovered the rapid conversion of α,β-unsaturated aldehydes into related nitriles when studying flow-based aziridination reactions (Scheme 23).82 Continuous flow chemistry was initially exploited for the rapid on-demand generation of chloramine from sodium hypochlorite and ammonium species. Chloramine was to be used as an electrophile in a telescoped procedure that avoided handling of this hazardous species. However, when employing chloramine for the aziridination of unsaturated aldehydes in flow, the serendipitous formation of unsaturated nitriles was observed instead. Upon further optimisation a set of nitrile products was obtained in a flow process within 5 minutes of residence time and yields up to 71%.
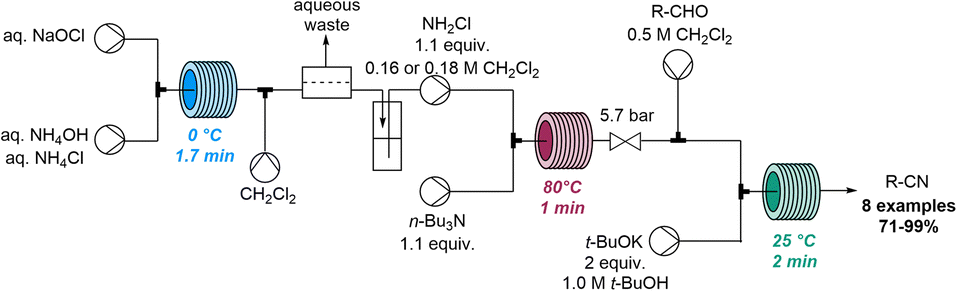 | ||
| Scheme 23 Flow-based formation of unsaturated nitriles via in situ generated chloramine. | ||
Jamison and co-workers also disclosed a method for the deuterio difluoromethylation of aldehydes using gaseous chlorodifluoromethane (ClCF2H, Scheme 24).83 Flow chemistry thereby guaranteed control of gas stoichiometry and allowed to overcome challenges relating to the use of this species in pressurised gas–liquid reactions. Specifically, singlet difluorocarbene (:CF2) was generated through deprotonation of ClCF2H with LiOtBu as base and subsequent α-elimination. Upon trapping with triphenylphosphine the resulting ylide (Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2) underwent a Wittig reaction with aldehydes to generate a difluorinated oxaphosphetane as key intermediate. Quenching this species with deuterated water rendered the deuterio difluoromethylation products under alkaline conditions, or gem-difluoroalkenes upon heating.
CF2) underwent a Wittig reaction with aldehydes to generate a difluorinated oxaphosphetane as key intermediate. Quenching this species with deuterated water rendered the deuterio difluoromethylation products under alkaline conditions, or gem-difluoroalkenes upon heating.
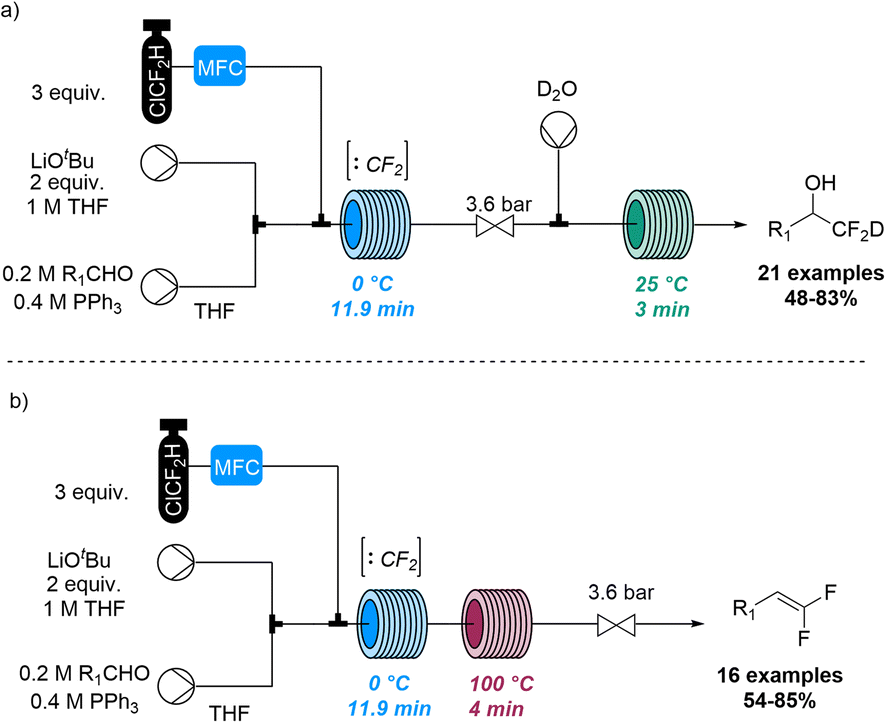 | ||
| Scheme 24 Deuterio difluoromethylation of aldehydes exploiting in situ generated difluorocarbene. | ||
The importance of AI and ML
The above sections provide a summary of representative studies whereby flow processing was instrumental in the discovery of new reactions and reactivity patterns. These exploited light, electricity, or thermal means to trigger the desired reactions and the use of flow principles was critical in controlling vital reaction parameters such as residence time, mixing and heat transfer. The discovery of these processes was sometimes inspired by prior observations in batch mode leading to rationally developed methods, whilst in some cases serendipity led to the resulting transformations. In the future, artificial intelligence (AI) and machine learning (ML) methods along with associated robotic systems can be expected to have an increasing role in the assembly of society's functional molecules. The machine assisted benefits that arise by adoption of flow chemistry techniques play especially well into this rapidly evolving agenda.On the other hand, the data field available to AI/ML for synthesis currently is very limited and not yet standardised and slows progress in the area. Nevertheless, there are obvious areas where AI/ML impacts, for example in synthesis route design,84 optimisation,84a–d scale-up85 and automation.84e,85 More innovation, however, will likely arise through their application in new high-throughput reaction discovery platforms, self-optimisation protocols and function linked synthesis. The serendipitous nature of new reaction discovery and the disconnected leaps of faith common to innovative chemical progress provide a very challenging but exciting environment for the future incorporation of AI.
Also, going forward, ethical issues in the use of these methods will need careful monitoring and control, whereby bespoke flow reactor systems will again likely serve the community well in this regard.
Conclusions
As demonstrated in this Perspective flow chemistry has started to impact on the discovery of new reactions and reactivity patterns allowing for the subsequent exploration of new synthetic methodology. Unsurprisingly, most discoveries were reported in fields that lay dormant for some time (i.e., photochemistry, electrochemistry, temperature sensitive reactions) due to the absence of mild and effective processing technologies and the improved safety envelope offered by miniaturised flow set-ups. Further discoveries in these and many other areas are expected to emerge in the near future, especially when combinations of new synthesis modalities are exploited. Future efforts will also embrace reaction automation, inline analysis, AI and ML to a larger extent, and it is likely that dedicated robotic flow platforms will be conceived in both industry and academia to generate large numbers of new discoveries going forward. Fostering collaborative links between scientists of different backgrounds paired with an open mind towards new discoveries and their subsequent exploration will be crucial assets in these exciting endeavours. Undoubtably, these developments highlight how flow chemistry does not merely serve chemists to translate challenging reactions from batch to flow, or develop continuous processes ready for large scale synthesis, but that it enables the discovery of new and highly efficient chemical reactions.Author contributions
M. B. and S. V. L. conceived the topic of the perspective. All authors collected and curated relevant case studies. A. I. A. and J. G.-L. drafted the original manuscript and created the schemes. All authors reviewed and edited the manuscript. M. B. and S. V. L. acquired the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. G. acknowledges the Fundación Ramón Areces for a postdoctoral fellowship (BEVP33P01S12222). We wish to thank Science Foundation Ireland for supporting our research program through grants 12/RC2275_P2, 19/IFA/7420 and 20/FFP-P/8712. SVL was supported by the American Chemical Society through receipt of the Arthur C. Cope Award.Notes and references
- (a) R. Herges, Tetrahedron Comput. Methodol., 1988, 1, 15–25 CrossRef CAS; (b) C. Lavigne, G. Gomes, R. Pollice and A. Aspuru-Guzik, Chem. Sci., 2022, 13, 13857–13871 RSC; (c) W. Bort, I. I. Baskin, T. Gimadiev, A. Mukanov, R. Nugmanov, P. Sidorov, G. Marcou, D. Horvath, O. Klimchuk, T. Madzhidov and A. Varnek, Sci. Rep., 2021, 11, 3178 CrossRef CAS PubMed.
- (a) E. S. Beach, Z. Cui and P. T. Anastas, Energy Environ. Sci., 2009, 2, 1038–1049 RSC; (b) C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS; (c) A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615–4628 CrossRef CAS PubMed; (d) S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2022, 122(3), 3637–3710 CrossRef CAS PubMed.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (b) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed; (c) M. D. Kärkäs, J. A. Porco Jr and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (b) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed; (c) C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS.
- (a) C. K. Prier and F. H. Arnold, J. Am. Chem. Soc., 2015, 137, 13992–14006 CrossRef CAS; (b) E. Alfonzo, A. Das and F. H. Arnold, Curr. Opin. Green Sustainable Chem., 2022, 38, 100701 CrossRef CAS; (c) B. A. Sandoval and T. K Hyster, Curr. Opin. Chem. Biol., 2020, 55, 45–51 CrossRef CAS.
- (a) Y. Shen, J. E. Borowski, M. A. Hardy, R. Sarpong, A. G. Doyle and T. Cernak, Nat. Rev., 2021, 1, 23 CAS; (b) E. Farrant, ACS Med. Chem. Lett., 2020, 11, 1506–1513 CrossRef CAS; (c) A. Gioiello, A. Piccinno, A. M. Lozza and B. Cerra, J. Med. Chem., 2020, 63, 6624–6647 CrossRef CAS PubMed.
- S. Berritt, M. Christensen, M. J. Johansson, S. W. Krska, S. G. Newman, J. Sampson, E. M. Simmons, Y. Wang and N. A. Strotman, Perspectives on the Past, Present, and Future of High-Throughput Experimentation, in ACS Symposium Series, ch. 1, vol. 1419, pp. 3–9, ISBN13: 9780841297579 Search PubMed.
- (a) K. Jorner, A. Tomberg, C. Bauer, C. Sköld and P.-O. Norrby, Nat. Rev. Chem., 2021, 5, 240–255 CrossRef CAS PubMed; (b) H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2018, 4, 1465–1476 CrossRef CAS; (c) J. M. Granda, L. Donina, V. Dragone, D.-L. Long and L. Cronin, Nature, 2018, 559, 377–381 CrossRef CAS PubMed.
- (a) A. F. de Almeida, R. Moreira and T. Rodrigues, Nat. Rev. Chem., 2019, 3, 589–604 CrossRef; (b) F. Peiretti and J. M. Brunel, ACS Omega, 2018, 3, 13263–13266 CrossRef CAS.
- L. Capaldo, Z. Wen and T. Noël, Chem. Sci., 2023, 14, 4230–4247 RSC.
- J.-i. Yoshida, Y. Takahashia and A. Nagaki, Chem. Commun., 2013, 49, 9896–9904 RSC.
- For recent reviews in flow photochemistry see: (a) F. Politano and G. Oksdath-Mansilla, Org. Process Res. Dev., 2018, 22, 1045–1062 CrossRef CAS; (b) C. Sambiagio and T. Noël, Trends Chem., 2020, 2, 92–106 CrossRef CAS; (c) J. D. Williams and C. O. Kappe, Curr. Opin. Green Sustainable Chem., 2020, 25, 100351 CrossRef; (d) L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS.
- (a) K. Donnelly and M. Baumann, J. Flow Chem., 2021, 11, 223–241 CrossRef CAS; (b) C. Xiouras, K. Kuijpers, D. Fanfair, M. Dorbec and B. Gielen, Curr. Opin. Chem. Eng., 2023, 41, 100920 CrossRef.
- H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2017, 9, 453–456 CrossRef CAS.
- H. Seo, A. Liu and T. F. Jamison, J. Am. Chem. Soc., 2017, 139, 13969–13972 CrossRef CAS PubMed.
- G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed.
- F. Raymenants, T. M. Masson, J. Sanjosé-Orduna and T. Noël, Angew. Chem., Int. Ed., 2023, 62, e202308563 CrossRef CAS PubMed.
- Q. Wang, S. Ni, X. Wang, Y. Wang and Y. Pan, Sci. China: Chem., 2022, 65, 678–685 CrossRef CAS.
- M. Brzozowski, M. O’Brien, S. V. Ley and A. Polyzos, Acc. Chem. Res., 2015, 48, 349–362 CrossRef CAS.
- J. A. Forni, V. H. Gandhi and A. Polyzos, ACS Catal., 2022, 12, 10018–10027 CrossRef CAS.
- J. A. Forni, N. Micic, T. U. Connell, G. Weragoda and A. Polyzos, Angew. Chem., Int. Ed., 2020, 59, 18646–18654 CrossRef CAS PubMed.
- N. Micic and A. Polyzos, Org. Lett., 2018, 20, 4663–4666 CrossRef CAS PubMed.
- M. J. P. Mandigma, J. Žurauskas, C. I. MacGregor, L. J. Edwards, A. Shahin, L. d'Heureuse, P. Yip, D. J. S. Birch, T. Gruber, J. Heilmann, M. P. John and J. P. Barham, Chem. Sci., 2022, 13, 1912–1924 RSC.
- Y.-F. Liang, R. Steinbock, L. Yang and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 10625–10629 Search PubMed.
- M. Di Filippo, C. Trujillo, G. Sánchez-Sanz, A. S. Batsanov and M. Baumann, Chem. Sci., 2021, 12, 9895–9901 RSC.
- M. Di Filippo and M. Baumann, Org. Biomol. Chem., 2023, 21, 2930–2934 RSC.
- C. Bracken and M. Baumann, Org. Lett., 2023, 25, 6593–6597 CrossRef CAS.
- C. Bracken and M. Baumann, J. Org. Chem., 2020, 85, 2607–2617 CrossRef CAS.
- J. García-Lacuna and M. Baumann, Adv. Synth. Catal., 2023, 365, 2628–2635 CrossRef.
- R. I. Rodríguez, M. Sicignano, M. J. García, R. G. Enríquez, S. Cabrera and J. Alemán, Green Chem., 2022, 24, 6613–6618 RSC.
- M. Rueping, C. Vila and T. Bootwicha, ACS Catal., 2013, 3, 1676–1680 CrossRef CAS.
- Y. Fang and G. K. Tranmer, Med. Chem. Commun., 2016, 7, 720–724 RSC.
- Y. Fang and G. K. Tranmer, Org. Biomol. Chem., 2016, 14, 10799–10803 RSC.
- I. Abdiaj, A. Fontana, M. V. Gomez, A. de la Hoz and J. Alcázar, Angew. Chem., Int. Ed., 2018, 57, 8473–8477 CrossRef CAS.
- I. Abdiaj, L. Huck, J. M. Mateo, A. de la Hoz, M. V. Gomez, A. Díaz-Ortiz and J. Alcázar, Angew. Chem., Int. Ed., 2018, 57, 13231–13236 CrossRef CAS PubMed.
- X.-J. Wei, I. Abdiaj, C. Sambiagio, C. Li, E. Zysman-Colman, J. Alcázar and T. Noël, Angew. Chem., Int. Ed., 2019, 58, 13030–13034 CrossRef CAS PubMed.
- F. Lima, M. A. Kabeshov, D. N. Tran, C. Battilocchio, J. Sedelmeier, G. Sedelmeier, B. Schenkel and S. V. Ley, Angew. Chem., Int. Ed., 2016, 55, 14085–14089 Search PubMed.
- A. Mata, D. N. Tran, U. Weigl, J. D. Williams and C. O. Kappe, Chem. Commun., 2020, 56, 14621–14624 RSC.
- D. Cantillo, C. Mateos, J. A. Rincon, O. de Frutos and C. O. Kappe, Chem.–Eur. J., 2015, 21, 12894–12898 CrossRef CAS.
- G. Laudadio, S. Govaerts, Y. Wang, D. Ravelli, H. F. Koolman, M. Fagnoni, S. W. Djuric and T. Noël, Angew. Chem., Int. Ed., 2018, 57, 4078–4082 CrossRef CAS PubMed.
- For recent reviews on flow electrochemistry see: (a) M. Elsherbini and T. Wirth, Acc. Chem. Res., 2019, 52, 3287–3296 CrossRef CAS PubMed; (b) T. Noël, Y. Cao and G. Laudadio, Acc. Chem. Res., 2019, 52, 2858–2869 CrossRef PubMed; (c) N. Tanbouza, T. Ollevier and K. Lam, iScience, 2020, 23, 101720 CrossRef CAS PubMed.
- C. Huang, X.-Y. Qian and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 6650–6653 CrossRef CAS.
- W.-J. Kong, L. H. Finger, A. M. Messinis, R. Kuniyil, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2019, 141, 17198–17206 CrossRef CAS PubMed.
- H. Long, C. Huang, Y.-T. Zheng, Z.-Y. Li, L.-H. Jie, J. Song, S. Zhu and H.-C. Xu, Nat. Commun., 2021, 12, 6629 CrossRef CAS PubMed.
- H. Long, T.-S. Chen, J. Song, S. Zhu and H.-C. Xu, Nat. Commun., 2022, 13, 3945 CrossRef CAS.
- D. E. Collin, A. A. Folgueiras-Amador, D. Pletcher, M. E. Light, B. Linclau and R. C. D. Brown, Chem.–Eur. J., 2020, 26, 374–378 CrossRef CAS.
- C. Huang, Z.-Y. Li, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2021, 60, 11237–11241 CrossRef CAS.
- W. Zhao, Y. Lu, Y. Qiao, X. Yin, C. Liu, Z. Fang, J. Zhu and K. Guo, Org. Lett., 2023, 25, 7451–7456 CrossRef CAS PubMed.
- H. Wang, F. Pesciaioli, J. C. A. Oliveira, S. Warratz and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 15063–15067 CrossRef CAS.
- C. Zhu, J. C. A. Oliveira, Z. Shen, H. Huang and L. Ackermann, ACS Catal., 2018, 8, 4402–4407 CrossRef CAS.
- J.-S. Poh, S.-H. Lau, I. G. Dykes, D. N. Tran, C. Battilocchio and S. V. Ley, Chem. Sci., 2016, 7, 6803–6807 RSC.
- D. N. Tran, C. Battilocchio, S.-B. Lou, J. M. Hawkins and S. V. Ley, Chem. Sci., 2015, 6, 1120–1125 RSC.
- N. M. Roda, D. N. Tran, C. Battilocchio, R. Labes, R. J. Ingham, J. M. Hawkins and S. V. Ley, Org. Biomol. Chem., 2015, 13, 2550–2554 RSC.
- J. A. Kozak, J. Wu, X. Su, F. Simeon, T. A. Hatton and T. F. Jamison, J. Am. Chem. Soc., 2013, 135, 18497–18501 CrossRef CAS.
- M. Y. S. Ibrahim and M. Abolhasani, Nat. Commun., 2022, 13, 2441 CrossRef CAS.
- E. S. Streng, D. S. Lee, M. W. George and M. Poliakoff, Beilstein J. Org. Chem., 2017, 13, 329–337 CrossRef CAS PubMed.
- Z. Amara, E. S. Streng, R. A. Skilton, J. Jin, M. W. George and M. Poliakoff, Eur. J. Org Chem., 2015, 2015, 6141–6145 CrossRef CAS.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, J. Org. Chem., 2021, 86, 14199–14206 CrossRef CAS PubMed.
- J. García-Lacuna, M. Alonso, G. Domínguez and J. Pérez Castells, RSC Adv., 2022, 12, 7313–7317 RSC.
- S. Borra, S. Chae, H. Y. Kim and K. Oh, Org. Lett., 2022, 24, 5287–5292 Search PubMed.
- A. S. Ubale, M. A. Shaikh, N. Mohanta and B. Gnanaprakasam, Adv. Synth. Catal., 2023, 365, 3094–3100 CrossRef CAS.
- A. Seemann, J. Panten and A. Kirschning, J. Org. Chem., 2021, 86, 13924–13933 Search PubMed.
- A. S. Kleinke and T. F. Jamison, Org. Lett., 2013, 15, 710–713 CrossRef CAS PubMed.
- B. Pieber, S. T. Martinez, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2013, 52, 10241–10244 CrossRef CAS.
- J. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896–9904 RSC.
- M. A. Ganiek, M. R. Becker, M. Ketels and P. Knochel, Org. Lett., 2016, 18, 828–831 CrossRef CAS PubMed.
- J. H. Harenberg, N. Weidmann, A. J. Wiegand, C. A. Hoefer, R. R. Annapureddy and P. Knochel, Angew. Chem., Int. Ed., 2021, 60, 14296–14301 CrossRef CAS PubMed.
- J. Hartwig, J. B. Metternich, N. Nikbin, A. Kirschning and S. V. Ley, Org. Biomol. Chem., 2014, 12, 3611–3615 RSC.
- H.-J. T. Lee, D. Torii, Y. Jeon, J. Yoshida and H. Kim, Synlett, 2020, 31, 1899–1902 CrossRef CAS.
- H.-J. Lee, H. Kim, J. Yoshida and D.-P. Kim, Chem. Commun., 2018, 54, 547–550 RSC.
- H.-J. Lee, Y. Yonekura, N. Kim, J. Yoshida and H. Kim, Org. Lett., 2021, 23, 2904–2910 CrossRef CAS PubMed.
- J. Y. F. Wong, J. M. Tobin, F. Vilela and G. Barker, Chem.–Eur. J., 2019, 25, 12439–12445 CrossRef CAS PubMed.
- J. Grosser and W. Werner, Chem. Ber., 1973, 106, 2610–2619 CrossRef.
- P. Musci, M. Colella, A. Sivo, G. Romanazzi, R. Luisi and L. Degennaro, Org. Lett., 2020, 22, 3623–3627 CrossRef CAS.
- M. Colella, P. Musci, D. Cannillo, M. Spennacchio, A. Aramini, L. Degennaro and R. Luisi, J. Org. Chem., 2021, 86, 13943–13954 CrossRef CAS.
- P. Musci, M. Colella, M. Andresini, A. Aramini, L. Degennaro and R. Luisi, Chem. Commun., 2022, 58, 6356–6359 RSC.
- P. Musci, T. von Keutz, F. Belaj, L. Degennaro, D. Cantillo, C. O. Kappe and R. Luisi, Angew. Chem., Int. Ed., 2021, 60, 6395–6399 CrossRef CAS PubMed.
- Y. Jiang and H. Yorimitsu, JACS Au, 2022, 2, 2514–2521 CrossRef CAS.
- T. Brégent, M. V. Ivanova, T. Poisson, P. Jubault and J. Legros, Chem.–Eur. J., 2022, 28, e202202286 CrossRef PubMed.
- D. Kim, H.-J. Lee, Y. Shimizu, J. Yoshida and H. Kim, Nat. Synth., 2022, 1, 558–564 CrossRef.
- K. E. Danahy, E. D. Styduhar, A. M. Fodness, L. M. Heckman and T. F. Jamison, Org. Lett., 2020, 22, 8392–8395 CrossRef CAS PubMed.
- W. C. Fu and T. F. Jamison, Angew. Chem., Int. Ed., 2020, 59, 13885–13890 CrossRef CAS PubMed.
- (a) A. F. de Almeida, R. Moreira and T. Rodrigues, Nat. Rev. Chem., 2019, 3, 589–604 CrossRef; (b) T. J. Struble, J. C. Alvarez, S. P. Brown, M. Chytil, J. Cisar, R. L. DesJarlais, O. Engkvist, S. A. Frank, D. R. Greve, D. J. Griffin, X. Hou, J. W. Johannes, C. Kreatsoulas, B. Lahue, M. Mathea, G. Mogk, C. A. Nicolaou, A. D. Palmer, D. J. Price, R. I. Robinson, S. Salentin, L. Xing, T. Jaakkola, W. H. Green, R. Barzilay, C. W. Coley and K. F. Jensen, J. Med. Chem., 2020, 63, 8667–8682 CrossRef CAS PubMed; (c) A. Thakkar, S. Johansson, K. Jorner, D. Buttar, J. L. Reymond and O. Engkvist, React. Chem. Eng., 2021, 6, 27–51 RSC; (d) R. S. K. Vijayan, J. Kihlberg, J. B. Cross and V. Poongavanam, Drug Discovery Today, 2022, 27, 967–984 CrossRef CAS PubMed; (e) M. Lu, J. Yin, Q. Zhu, G. Lin, M. Mou, F. Liu, Z. Pan, N. You, X. Lian, F. Li, H. Zhang, L. Zheng, W. Zhang, H. Zhang, Z. Shen, Z. Gu, H. Li and F. Zhu, Engineering, 2024 DOI:10.1016/j.eng.2023.01.014.
- D. J. Griffin, C. W. Coley, S. A. Frank, J. M. Hawkins and K. F. Jensen, Org. Process Res. Dev., 2023, 27, 1868–1879 CrossRef CAS.
- P. L. Nichols, Prog. Med. Chem., 2021, 60, 191–272 Search PubMed.
Footnote |
| † These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |