
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA molecular view of single-atom catalysis toward carbon dioxide conversion
Xin
Shang
 ab,
Xiaofeng
Yang
a,
Guodong
Liu
a,
Tianyu
Zhang
*c and
Xiong
Su
*a
ab,
Xiaofeng
Yang
a,
Guodong
Liu
a,
Tianyu
Zhang
*c and
Xiong
Su
*a
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, No. 457 Zhongshan Road, Dalian 116023, China. E-mail: suxiong@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cCollege of Environmental Science and Engineering, Beijing Forestry University, Beijing 100083, China. E-mail: tzhang@bjfu.edu.cn
First published on 9th February 2024
Abstract
Carbon dioxide (CO2) conversion has attracted much interest recently owing to its importance in both scientific research and practical applications, but still faces a bottleneck in selectivity control and mechanism understanding owing to diversified active sites. Single-atom catalysts (SACs) featuring isolated and well-defined active centers are proved to not only exhibit unparalleled performances in various processes of CO2 conversion but also provide excellent research paradigms by circumventing the heterogeneity of active sites. Herein, we will not only critically review recent progress on the application of SACs in chemical CO2 conversion based on previous comprehension of general thermodynamics and kinetics, but also try to offer a multi-level understanding of SACs from a molecular point of view in terms of the central atom, coordination environment, support effect and synergy with other active centers. Meanwhile, crucial scientific issues of research methods will be also identified and highlighted, followed by a future outlook that is expected to present potential aspects of further developments.
 Xin Shang | Xin Shang received his B.S. degree from Tianjin University in 2016. Now, he is pursuing his PhD degree in the University of Chinese Academy of Sciences (UCAS), Dalian Institute of Chemical Physics, Chinese Academy of Sciences (DICP, CAS), supervised by Prof. Yanqiang Huang. His research interests include C1 chemistry and synthesis and application of molecular sieves and nanostructured materials. |
 Xiaofeng Yang | Xiaofeng Yang received his B.S. from Nanjing University (2003) and PhD degree in 2010 from the Dalian Institute of Chemical Physics under the supervision of Prof. Tao Zhang. After 2 years of postdoctoral research in Prof. Jun Li's group at Tsinghua University on theoretical and computational catalysis, he joined the DICP again and was promoted to professor in 2019. His research interests include theoretical and computational catalysis and nano- and single-atom catalysis. |
 Guodong Liu | Guodong Liu received his B.S degree in 1998 and PhD degree in 2019 from the Dalian University of Technology. He has been working in the Dalian Institute of Chemical Physics, Chinese Academy of Sciences (DICP, CAS), since 2019 and is in charge of developing heterogeneous catalysts on the industrial scale. His research interests include the synthesis of zeolite materials and their applications in aromatization of alkanes, alkylation of arenes, and epoxidation of alkenes. |
 Tianyu Zhang | Tianyu Zhang received his B.S. degree from the Dalian University of Technology and PhD degree from Southern Illinois University. He has been working in Beijing Forestry University as an assistant professor since 2021. His research interests include C1 molecular catalytic conversion and theoretical chemistry. |
 Xiong Su | Xiong Su received his B.S. degree from the Dalian University of Technology and PhD degree in industrial catalysis from the Dalian Institute of Chemical Physics (DICP), Chinese Academy of Sciences (CAS). He continued his academic career in the DICP as an assistant professor and was promoted to associate professor in 2017. His research interests include C1 chemistry and synthesis and application of molecular sieves and nanostructured materials. |
1. Introduction
The utilization of fossil fuels has greatly changed people's way of life and propelled the progress of human civilization. Now, however, it has become a double-edged sword damaging the climate and environment by the notorious greenhouse effect due to the substantial carbon dioxide (CO2) emission.1 The cumulative CO2 emissions have caused global mean temperature changes for more than a century (Fig. 1a), implying an imperative need to impose restrictions on carbon emissions. Therefore, a growing consensus has been established that realizing ‘carbon peak’ and ‘carbon neutrality’ is of great significance in seeking a balance between efficiency and quality, further contributing to the desired sustainable development.2,3 Besides applying renewable clean energy sources, such as solar, wind, wave, etc., as substitutes for conventional fossil resources, carbon capture, utilization and storage (CCUS) is expected to provide supplementary solutions to meet the challenges of mitigating climate change by recycling the released CO2 from the atmosphere.4–7 Indeed, artificial transformation of CO2, a nontoxic, widespread and renewable carbon source, into various value-added products might contribute to cleaner industrial production,8–10 although there is still controversy regarding the economic efficiency of its scale development11,12 (Fig. 1b), in which the high cost and low efficiency of practical processes are considered to be key barriers. In fact, not only the high energy input to break the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (803 kJ mol−1) but also the complicated reaction networks pose huge obstacles for the practical exploitation of CO2 conversion for chemical synthesis. Feasible catalytic processes and advanced devices are thus in great demand, for which the fabrication of effective catalysts is one of the core issues.
O bonds (803 kJ mol−1) but also the complicated reaction networks pose huge obstacles for the practical exploitation of CO2 conversion for chemical synthesis. Feasible catalytic processes and advanced devices are thus in great demand, for which the fabrication of effective catalysts is one of the core issues.
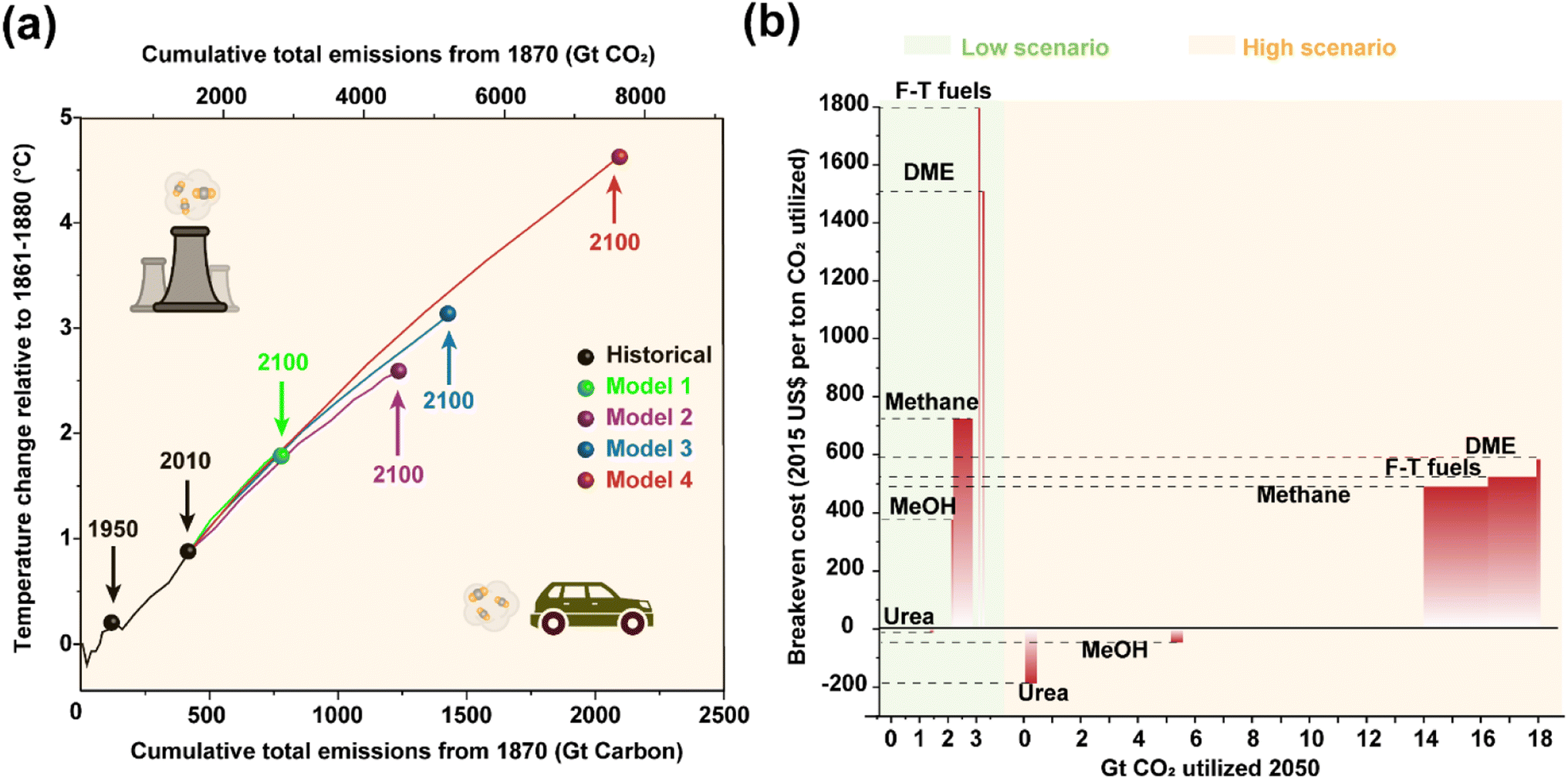 | ||
| Fig. 1 (a) Simulated global mean surface temperature increase as a function of the cumulative total global CO2 emissions as predicted by different models. Data are acquired from ref. 2 with permission from Springer Nature, copyright 2016. (b) Estimated CO2 utilization potential and breakeven cost of different sub-pathways in low and high scenarios. Data are acquired from ref. 12 with permission from Springer Nature, copyright 2019. | ||
Currently, catalysts play a vital role in most chemical syntheses by steering the reaction pathways towards lower activation energy barriers for the formation of specific intermediate species, thereby extensively increasing the rate of desired reactions. Homogeneous catalysts containing metal sites with well-defined coordination environments have been extensively used in CO2 conversion for the synthesis of various organic compounds,9,13–16 exhibiting a high turnover number (TON) and turnover frequency (TOF) due to the full contact between active centers and substrates in one phase. However, the difficulty of catalyst separation and inferior durability under harsh conditions still limit their practical application in large-scale industrial production. Heterogeneous catalysts, in contrast, are more easily separated and recycled,17 but only a small fraction of surficial metal sites can efficiently participate in the catalytic cycles, since the chemical interactions always occur on specific interfaces.18 Metal species with an uneven nature in traditional heterogeneous catalysts might even serve as troublemakers causing undesired side reactions or interfering in certain elementary steps, leading to deterioration of catalytic performance. Furthermore, the intrinsic complexity of CO2 transformation regarding the multiple-branch reaction pathways and corresponding challenges in selectivity control lead to ambiguous structure–activity relationships and labyrinthine mechanism understanding owing to the diversity of active sites. And this makes the rational design of effective catalysts a difficult task because of the absence of systematic theories.
To overcome these problems and merge the merits of homogeneous and heterogeneous catalysts, a series of catalysts featuring atomically dispersed metal atoms on the interface as sustained active sites, i.e., single-atom catalysts (SACs), have been proposed in recent decades18–20 (Fig. 2), and were first introduced by Zhang and his co-workers in 2011.21 And already before that, a few studies on catalysts with highly dispersed metal species have been conducted, disclosing that atoms in the isolated ionic or free state rather than ensembles might contribute to their major activity in various systems.22–29 With continuous advances in high-resolution characterization techniques and computational chemistry, more unique properties of SACs have been unveiled and are expected to inject new vigor into a broader frame of catalytic science.30–37
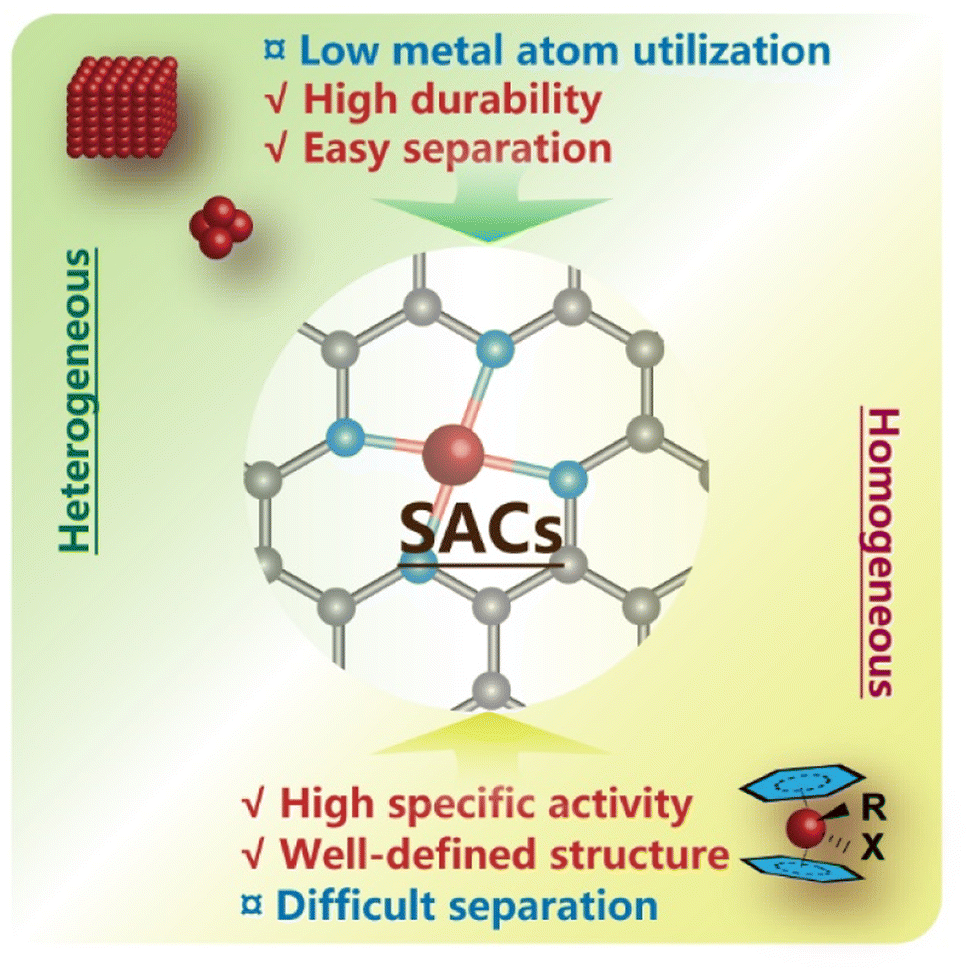 | ||
| Fig. 2 Single-atom catalysts (SACs) incorporate the advantageous features of homogeneous and heterogeneous catalysts. | ||
Recently, fruitful academic achievements involving SAC materials have been springing up toward expanding application fields,38–44 attracting worldwide interest for in-depth studies. In the meantime, the practical application of catalytic CO2 conversion is also expected to benefit from the SAC materials,45–48 demonstrating not only the boosted catalytic activity propelled by complete exposure of metal sites with nearly 100% atom utilization, but also controllable selectivity stemming from the distinct geometric and electronic properties. Compared with the conventional heterogeneous metal catalysts based on nanoparticles (NPs) and nanoclusters (NCs), the viable regulation of chemical microenvironments on the atomic scale via modulating the coordination states, spatial distances as well as the synergy between metal sites and supports might lead to a great difference in electronic states, orbital hybridization and energy bands. Additionally, the incorporation of other types of active sites can make SACs adaptable for extensive applications without tarnishing the advantages of single atomic sites. These factors further promote the activation of CO2 and stabilization of intermediate species with a corresponding match in energy and structure, offering an outstanding performance in multiple CO2 conversion reactions.49 More importantly, SACs with well-defined active sites, not only allow an exclusive selectivity, but also provide excellent paradigms for acquiring in-depth and comprehensive insights into catalysis. The studies on transformation of simple molecules such as CO2 exemplify the modeling approach to gain deep insight into the intrinsic thermodynamics and kinetics of typical chemical reactions assisted by a combination of experimental and theoretical aspects, which might also inspire in-depth research studies of other more complex catalytic systems and rational design of more advanced catalytic materials from an atomic-scale perspective.
In this review, we will first introduce the general understanding of typical chemical CO2 transformation. Then, we will concentrate on the recent progress on the application of SACs in chemical CO2 conversion based on various reaction routes and products and attempt to provide a comprehensive understanding of catalyst design, process optimization and reaction mechanisms (Fig. 3). Thanks to their well-defined geometric and electronic structures, we could further offer a molecular understanding of SACs in CO2 conversion with specific examples. The crucial scientific issues regarding SAC research and application will be also concluded and highlighted to gain in-depth insight. To inspire future research studies in such catalytic systems, we will also elucidate critical study methods and technological processes involving SACs for CO2 conversion. An outlook for future research will be demonstrated eventually based on the analysis of the current research direction, expected to present a potential prospect for further development of SAC application in CO2 conversion.
 | ||
| Fig. 3 An overview of SAC application in chemical CO2 conversion displayed in this review. | ||
2. General understanding of CO2 transformation
Before starting the discussion on the application of SACs in CO2 transformation, the general understanding in terms of thermodynamics and kinetics should be briefly introduced in advance. The typical CO2 transformation reactions involved not only the common reduction with hydrogen (H2) and water (H2O), but also other complicated synthesis processes with various heteroatomic and organic compounds, in which CO2 could also be partially or completely reduced following distinct reaction routes. Therefore, understanding the reduction reactions will be the focus in the following discussion especially regarding thermodynamics, as other processes will also be discussed in brief.2.1 Thermodynamics
Generally, the thermochemical reduction processes of CO2 involve the co-transformation of CO2 and H2 molecules. Accordingly, we calculated the free Gibbs energy change and enthalpy change dependent on temperature in typical hydrogenation routes toward various products (Fig. 4). The increasing free Gibbs energy change with increasing temperature for most routes apart from the CO2-to-CO reaction implies that lower temperature could be a more suitable condition for the synthesis of target products, which is also proved by the exothermic nature of most reactions. However, sufficiently higher operating temperature is always necessary in practical processes to ensure feasible efficiency, leading to inevitable side reactions due to their intersectional thermodynamic ranges. Therefore, the difficult selectivity control of CO2 hydrogenation makes the design of efficient catalysts a great challenge.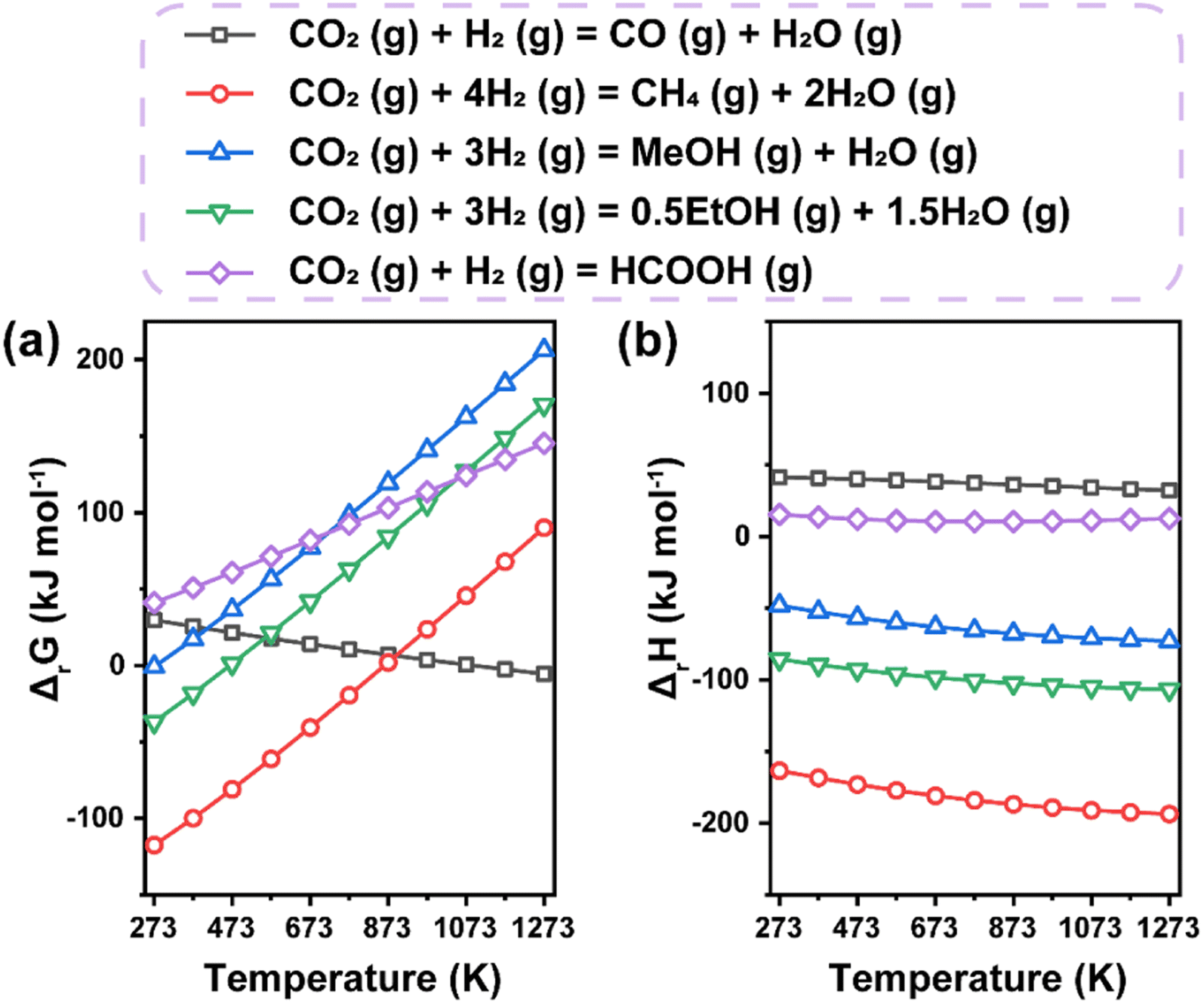 | ||
| Fig. 4 Thermodynamic analysis of main thermochemical reduction routes to various products. (a) Free Gibbs energy change. (b) Enthalpy change. Data are based on HSC chemistry 6.0 software. | ||
On the other hand, the electrochemical reduction of CO2 commonly involves the co-transformation of H2O molecules and operation at ambient temperature and pressure, in which H2O molecules provide protons and lose electrons to form O2, as CO2 molecules accept protons and electrons and are transformed into various reduction products. There have been also reports on high-temperature electrolysis of CO2 in solid-electrolyte cells for higher energy efficiency,50 in which CO and O2− are generated from CO2 molecules, and then O2− is transmitted to the anode surface through the electrolyte, losing electrons and forming O2. For a more comprehensive understanding, the equilibrium reduction potentials of different electrochemical routes are demonstrated in Fig. 5, since the free Gibbs energy change could be well correlated with redox potentials according to the equation ΔG = −nFE. Obviously, the redox potentials of most half reactions of electrochemical reduction processes are close to that of the hydrogen evolution reaction (HER, 2H+ + 2e− → H2), making the HER a parallel reaction competing with CO2 reduction by consuming the reactive electrons. Therefore, one of the core issues that the design of catalysts should take into account is to circumvent the influence of the thermodynamically inclined HER.
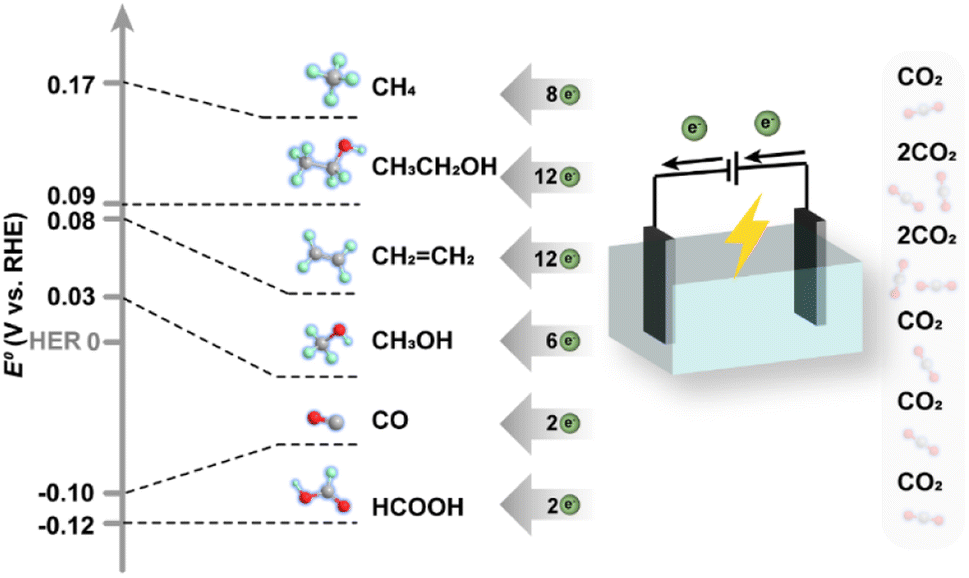 | ||
| Fig. 5 Equilibrium reduction potentials and electron transfer numbers for various products from CO2 reduction. | ||
The photochemical reduction processes, much more complicatedly, are limited by not only the thermodynamic barriers lying on the CO2 reaction pathways but also the energy transition for the light-to-electron procedures. Generally, the photoreduction of CO2 encompasses three pivotal procedures, which are consecutive light adsorption, charge separation and migration, and catalytic transformation45,51–54 (Fig. 6). For the facile formation and separation of photoexcited electron–hole pairs, semiconductor materials are commonly used as substrates due to their appropriate energy bandwidth. With the input of light in the specific wavelength range, electrons are excited from the valence band (VB) to the conduction band (CB), leaving the corresponding number of holes. Then, the electrons might participate in the reduction reactions, while the holes are responsible for the oxidation reactions. Quite similar to the cases in electrochemical processes, as the HER in the water photolysis process needs only two-electron transfer, CO2 reduction always needs more electrons and involves more complex pathways in the whole reaction. This suggests that it should be more urgent to accelerate the photo-induced electron separation and migration rather than focusing on the CO2 reduction kinetics. The challenges in both optoelectronic and catalytic processes still make the efficiency the Achilles' heel in limited practical application of the photocatalytic systems for CO2 to valuable products.45,51 Hence, so-called cocatalysts, certain kinds of modifiers and active components, have been introduced into the photocatalytic systems, playing a crucial role in enhancing charge separation and transfer and manipulating the activity and selectivity of CO2 reduction towards specific products.51,54
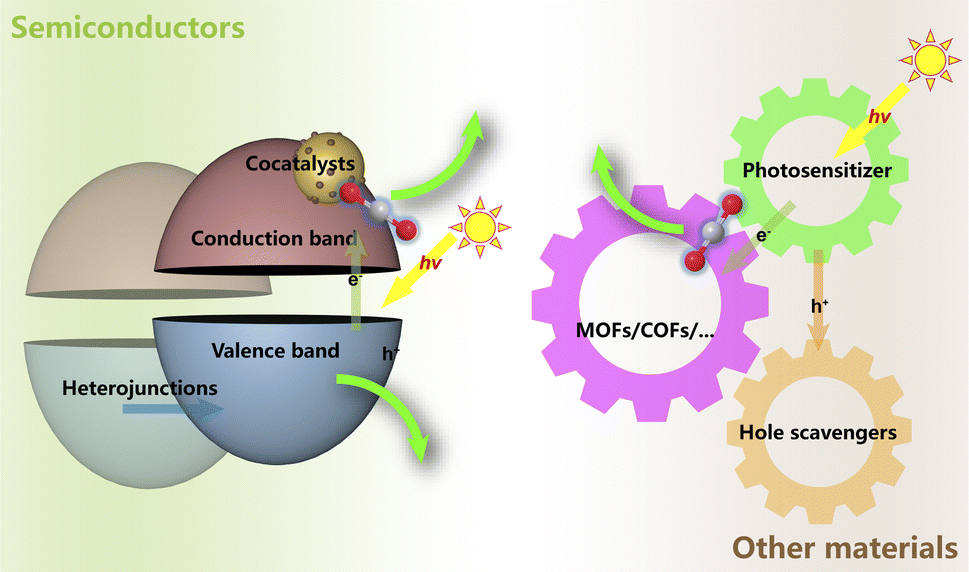 | ||
| Fig. 6 Typical photocatalytic systems for CO2 reduction. | ||
The co-transformation of CO2 with other molecules from simple C1 hydrocarbon methane (CO2 + CH4 → 2CO + 2H2, ΔG = 170 kJ mol−1, ΔH = 243 kJ mol−1, 298 K) to various heteroatomic compounds and multiple-atom organics renders the systematic analysis of thermodynamics an impossible task due to the diversified conditions and products. In these cases, reaction kinetics is always much more important for its role in determining the specific activity and selectivity toward certain products.
2.2 Kinetics
Prior to the chemical reactions, mass transfer, i.e., the diffusion of CO2 molecules and other reactants onto the reaction interface and the release of products from the active sites play a critical role in the kinetics of whole CO2 transformation process. This is largely determined by the chemical interaction that can only function effectively in a limited range of distance, although potential long-range influences still exist. Reaction equilibrium and the regeneration of active centers would also partially depend on a sufficient diffusion rate rather than only the effect of chemical forces. In fact, diffusion processes in most heterogeneous catalytic systems cannot be ignored, and researchers endeavor to design catalysts with higher dispersion and smaller size and develop advanced reactors with enhanced capability of mass transfer to alleviate the negative influence of insufficient diffusivity. In this way, in-depth research into the intrinsic reaction kinetics of CO2 transformation has a great significance. Herein, we will focus on the CO2 reaction kinetics of reduction reactions in a hydrogen atmosphere or under aqueous conditions because of the universality of research studies and the great attention to them (Fig. 7). The other reactions involving CO2 transformation kinetics would be discussed in conjunction with the introduction of specific catalysts.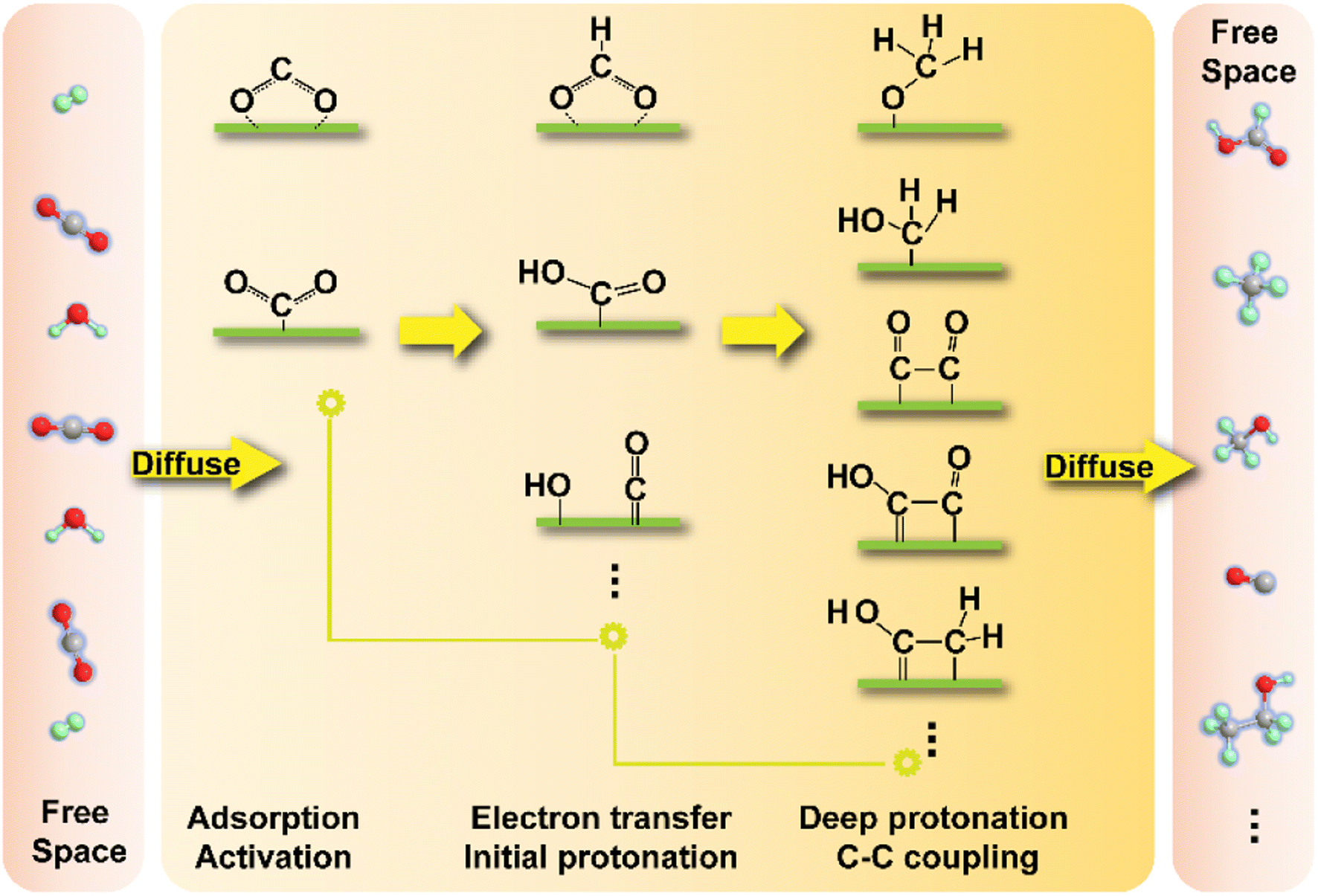 | ||
| Fig. 7 Schematic illustration of specific important kinetic procedures in typical CO2 reduction processes. In particular, we only list a part of the kinetic steps in this figure, and there could be other possible steps in various CO2 transformation processes. | ||
The first stage of reaction kinetics is the initial adsorption and activation of CO2 molecules, commonly involving the transfer of electrons between different reactants or the reactant and reactive interface. CO2 is a typical linear nonpolar molecule comprising two σ bonds and two delocalized π bonds between one carbon and two oxygen atoms. The chemical bonds between carbon and oxygen fall in between a conventional double bond and triple bond, showing higher bonding energy and relative chemical sluggishness. The electron cloud in delocalized bonds is mainly bound to two oxygen atoms and the higher electronegativity of oxygen leads to lower reactivity of electron pairs in such bonding orbitals. In contrast, the lone electron pairs in the oxygen atom are believed to be able to be involved in the chemical transformation, making the CO2 molecule an electron donor. On the other hand, the carbon atom in the CO2 molecule has electron deficiency and could be an electron acceptor. Therefore, the typical activation routes of a CO2 molecule include (i) input of electrons from catalysts to the carbon atom in the CO2 molecule, (ii) the carbon atom in the CO2 molecule takes away electrons from other electron-rich reactants, (iii) the CO2 molecule donates lone electron pairs to catalysts and (iv) the CO2 molecule donates lone electron pairs to certain electron-deficient reactants. Currently, most of the reduction processes follow the first two routes especially in the case of metal catalysis systems, while the latter two routes are more likely responsible for the initial activation of CO2 in some oxide catalysis and coordination chemistry systems for certain organic syntheses. To enhance the adsorption and activation, researchers have devoted their efforts to delicate design of catalysts, mainly through modifying the energy bands and electronic orbitals to match the features of the CO2 molecule for most thermochemical and electrochemical systems, and also through enhanced light-to-electron processes for photochemical systems.
After the initial activation, the transformation routes of CO2 might go through certain protonation and C–C coupling processes and experience stepwise evolution of various intermediates and transition states, forming a labyrinthine reaction network. This brings one of the greatest challenges in the control of product selectivity, for which catalysts have to subtly steer the reaction pathway toward specific targets. Only eliminating the side reaction pathway can ensure the efficiency of the target reaction, just like trimming the hedge and removing the weeds from crops. The different intermediate routes are highly dependent on the electronic properties of reactive interfaces that are closely related to the chemical interaction between guest molecules and substrates, as the stability or the Gibbs free energy of transition states could be influenced by the interaction, thereby determining the activation energy barriers from the initial to the secondary stages. In some cases, the spatial location of active sites and specific geometric properties could also have an effect on the stabilization of intermediates especially for those needing the synergy of different active components in multiple-step synthesis or with distinctive steric structures. Decided by the nature of active sites, including but not limited to *COOH, HCOO* and *COHO (* represents the adsorption site) moieties could be the key intermediates in a typical reduction reaction toward C1 products. They further go through protonation and electron transfer and are transformed into products with a higher H/C ratio. The kinetics of protonation steps in the thermochemical hydrogenation reaction and other multiple-molecule reactions (such as that with methane) might be also dependent on the supply of active H species stemming from activation and dissociation of H2 and other H-rich molecules. A balance between the activation of different reactants therefore should also play a vital role in the reaction kinetics.
The situations are even more complex for the synthesis of C2+ products, as there might be more than one kinetically controlled step, including the bonding between diversified active intermediates. Uneven reactive interfaces and non-uniform active centers in conventional heterogeneous catalysts further aggravate these issues and interfere not only in in-depth research studies on the intrinsic mechanisms of catalytic processes but also in the rational design of catalysts for selective synthesis of specific products with higher efficiency. Fortunately, SACs have provided opportunities for disentangling the labyrinthine system.
3 Single-atom catalysis in typical CO2 conversion routes
3.1 CO2 conversion toward C1 products
While conventional metal catalysts have realized plenty of catalytic processes at the expense of a harsh operating temperature and/or pressure as well as the high cost of precious metals, the utilization of SACs might become a powerful weapon to cope with the severe challenges in terms of energy cost and atom efficiency, and be expected to contribute to the higher selectivity of specific products due to their unique chemical properties. Herein, the thermochemical hydrogenation of CO2 into valuable products including CO, CH4, typical organic oxygenates, formic acid (HCOOH)/formate, methanol (MeOH) and ethanol (EtOH) by SACs will be discussed in the following section and critical scientific problems will be also summarized based on recent progress.
3.1.1.1 Carbon monoxide and methane. CO, an important platform chemical resource in industry for the production of a variety of valuable organic compounds, can be obtained via the well-known reverse water gas shift reaction (RWGS) from CO2 and H2. As a typical endothermic reaction (Fig. 4), RWGS would benefit from a relatively high operating temperature (usually above 673 K). Side reactions, especially methanation, however, might be also enhanced under such operating conditions, leading to a lowered CO yield and limited practical application. The competitive adsorption and activation of H2 and COx molecules might play a vital role in the eventual selectivity of methane and CO, which could be adjusted by using the distinct properties of SACs.
Kwak et al. studied the activity of palladium and ruthenium-based SACs in CO2 hydrogenation, taking support and particle size effects, respectively, into consideration.77,78 As for Pd-based catalysts, the temperature programmed surface reaction (TPSR) and scanning transmission electron microscopy (STEM) experiments were used to show the disparate reactivity patterns of Pd on different supports (Al2O3 and carbon nanotubes) in an atomic dispersion and the morphology of traditional 3D clusters, respectively, under CO2 hydrogenation. The morphology of Ru, on the other hand, was controlled by varying the loading on the same Al2O3 support, as also investigated by the STEM technique. In both studies, the shift of product distribution from CH4 to CO was found in both systems when SA sites were dominant, although the feature of supports also played a critical role. Matsubu et al. applied diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) with CO as the probe molecule to quantify the fraction of isolated Rh sites (Rhiso) and Rh sites (RhNP) on the surface of Rh NPs for a series of TiO2 supported rhodium SACs.79 Strong correlations were observed between the turnover frequency (TOF) of RWGS and the fraction of Rhiso, and also between the TOF of methanation and the fraction of RhNP sites (Fig. 8a–e). The changing activity in a long-term operation could also be rationalized by a reaction condition-induced disintegration of RhNP into Rhiso. This work provided a preliminary understanding of distinct catalytic behaviors between isolated atoms and nanoparticles for competing parallel reaction pathways. A similar dynamic change of SAC local structures in response to various redox conditions was also revealed by Tang et al. with more details through a combination of theoretical and experimental studies.80Operando studies showed that the single Rh atoms on TiO2 could adapt their structures to the reaction atmosphere, and the first-principles modeling showed that the substitutional structure for the Rh atom over Rh1@TiO2−2x was not catalytically active due to the strong adsorption energy of hydrogen on Rh that prevented CO2 adsorption, indicating that the active site for Rh SAC on TiO2 in the case of the RWGS reaction was the supported Rh1/TiO2−x site rather than the substitutional sites. This type of structural evolution could also be extended to various metals on supports for other redox reactions,81–84 for which deeper insights have been gained with the advances in operando characterization techniques and modeling calculation. Li et al., on the other hand, reported an alkali-promoted rhodium SAC for CO2 hydrogenation.85 The introduction of alkali ions (e.g., Na) enables efficient switching of the reaction products from nearly 100% CH4 to above 99% CO in a wide temperature range (513–713 K) with high activity and long-term stability at 573 K (Fig. 8f–h). The complete inhibition of deep hydrogenation of the CO intermediate products to CH4 could be attributed to the significantly higher apparent activation energy and facilitated CO desorption over the Na-doped SAC by performing control experiments of H2-D2 exchange and CO hydrogenation reactions. Moreover, the intermediate change from formate to carboxy using alkali ions was the key to favorable CO formation, as unveiled by in situ spectroscopy and theoretical calculations. The improved stability also benefited from the addition of alkali ions due to the reinforced electronic interaction between isolated Rh sites and ZrO2 supports. Notably, noble metal SAs could not only serve as active sites for the dissociation of CO2 to CO, but could also surprisingly promote the dynamic evolution of the support, which in turn enhances the catalytic performance under specific reaction conditions. Du et al. observed such a dynamic evolution driven by Pd SAs over a Pd1–FeOx catalyst during the RWGS reaction86 (Fig. 8i). The introduction of Pd atoms significantly changed the reduction and carburization behaviors of the FeOx support via inducing the formation of oxygen vacancies. The atomically dispersed Pd species enabled continuous H2 dissociation with a lower formation energy of oxygen vacancies, thereby endowing the FeOx support with a deeper reduction state, facilitating further carburization with the CO product. In contrast, Pd NPs tended to exhibit a strong metal–support interaction (SMSI) with the FeOx support as shown by the remarkable overlayers on the metal surface. Such a SMSI inhibited deep reduction and further carburization, resulting in an inferior activity compared with that of Pd1–FeOx, since iron carbide was believed to be a critical component for lowering the activation energy of CO2 conversion to CO.
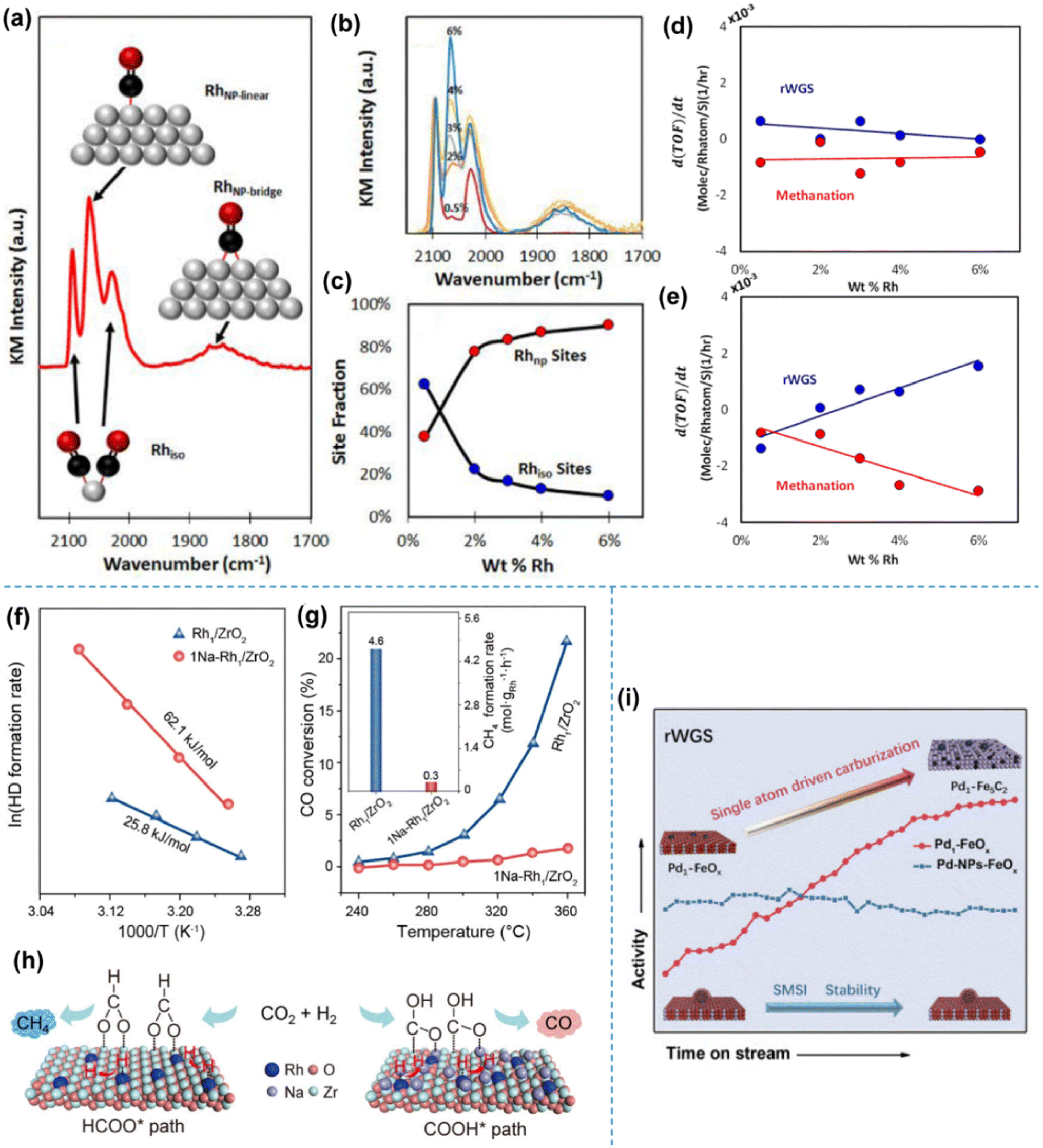 | ||
Fig. 8 (a) DRIFT spectrum obtained from a saturated layer of CO adsorbed at 300 K on 4% Rh/TiO2. Insets show ball-and-stick models of the assigned vibrational modes. (b) DRIFT spectra of CO at all five weight loadings of Rh/TiO2 catalysts. (c) Site fraction (%) of isolated (Rhiso) and nanoparticle-based Rh sites (RhNP). Rate of TOF change (d(TOF)/dt) for both reaction pathways as a function of wt% Rh measured at 473 K and a feed ratio of (d) 0.25 CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 and (e) 10 CO2:H2.79 (f) Arrhenius plots of Rh1/ZrO2 and 1Na–Rh1/ZrO2 in the H2-D2 exchange reaction. (g) Catalytic performance of Rh1/ZrO2 and 1Na–Rh1/ZrO2 in CO hydrogenation. (h) A schematic illustration of the regulation of Rh1–ZrO2 cooperativity by Na ions for selectivity tuning.85 (i) Schematic of boosting CO2 conversion to CO over Pd1–FeOxvia a SA driven carburization process.86 Reproduced from ref. 79, 85 and 86 with permission from American Chemical Society, Wiley and Elsevier, copyright 2015, 2022, 2022. :H2 and (e) 10 CO2:H2.79 (f) Arrhenius plots of Rh1/ZrO2 and 1Na–Rh1/ZrO2 in the H2-D2 exchange reaction. (g) Catalytic performance of Rh1/ZrO2 and 1Na–Rh1/ZrO2 in CO hydrogenation. (h) A schematic illustration of the regulation of Rh1–ZrO2 cooperativity by Na ions for selectivity tuning.85 (i) Schematic of boosting CO2 conversion to CO over Pd1–FeOxvia a SA driven carburization process.86 Reproduced from ref. 79, 85 and 86 with permission from American Chemical Society, Wiley and Elsevier, copyright 2015, 2022, 2022. | ||
In addition, SACs based on non-noble metals have been also applied for tuning the activity and selectivity in RWGS. Millet et al. prepared a series of Ni SACs with MgO as the support by a solid solution approach.87 Low-coordinated Ni sites on the surface of MgO predominated among the Ni species in the SACs, as predicted by hybrid-functional calculations. The isolated Ni atoms were active for the RWGS but were incapable of propelling deep hydrogenation, while Ni clusters might lead to the formation of CH4 in contrast. The substitution of Mg atoms by Ni atoms on the surface of oxide was found to not only reduce the strength of CO binding at low-coordinated sites but also promote H2 dissociation, so that the CO formation rates could show a positive correlation with the concentration of Ni on the surface. Liang et al. fabricated a cobalt-based SAC comprising isolated Co sites bonded to SBA-15 by the atomic layer deposition (ALD) technique, utilizing which 99% CO selectivity and near-equilibrium conversion of CO2 were maintained for a 500-hour RWGS reaction at 873 K.88 Co sites remained in a cationic state which was stabilized by strong Co–O–Si bonds, contributing to the H-assisted dissociation of CO2 that was distinct from the commonly reported mechanisms. Moreover, it was found that the CO products were generated along with the shifting of Co sites between the tetrahedral field and the octahedral field, by which the whole catalytic cycle was driven. A similar result was reported in a recent study by Li et al.89 The cobalt-based SAC synthesized by anchoring 4% Co atoms onto the nitrogen-doped carbon supports (Co–N–C) achieved nearly 100% CO selectivity and 52.4% CO2 conversion at 773 K, while the 20%-Co-loading catalyst with mostly Co NPs favored the formation of CH4. The H-assisted dissociation of CO2 was also proposed to be responsible for the excellent performance over the Co SAC via the COOH* intermediate, whereas direct dissociation took place on Co NPs. However, it might be quite different in a case where other types of active sites were introduced. In a study by Dostagir et al., the Co SA sites co-existed with oxygen vacancy sites over a ZrO2-supported Co SAC, catalyzing CO2 hydrogenation to produce CO with more than 95% selectivity.90Operando DRIFTS analysis showed that formate was the key intermediate, whose stabilization was achieved by the generation of oxygen vacancies derived from the synergy between Co and Zr. The subsequent decomposition of formate led to the formation of CO instead of methanol and methane by deep hydrogenation, since the adsorption of CO on active sites was less favorable than that of CO2. Furthermore, Jiang et al. reported a molybdenum-based SAC for CO2 hydrogenation to CO with a selectivity of almost 100%.91 Single Mo atoms with a MoN3 moiety supported by nitrogen-doped carbon enabled remarkable CO2 adsorption and facile CO desorption on the basis of the calculated transition state energy. And the formation of CO was considered through a direct dissociation path rather than a H-assisted path as mentioned above.
Synthesis of CH4 from thermochemical CO2 hydrogenation, namely the CO2 methanation (Sabatier) reaction, is another important pathway for CO2 chemical conversion. In contrast to RWGS, methanation is a highly exothermic process, suggesting an advantageous operating range at lower temperature regarding thermodynamics (Fig. 4). In terms of kinetics, nevertheless, higher temperature is always needed to ensure significant reaction efficiency. In general, the methanation reaction can either go through a direct hydrogenation step in the absence of the CO intermediate, or undergo an indirect step with CO as ‘bridge’ species. Within a certain temperature, RWGS is believed to be a parallel reaction to the methanation reaction over a variety of catalytic systems just as mentioned above. Therefore, it might be a good angle to inhibit RWGS or enhance the deep hydrogenation of CO for higher yield of CH4 from thermochemical CO2 hydrogenation.55,69,92 Indeed, the ostensibly different selectivities over various metal catalysts might be rationalized by the inherent kinetic differences, such as the activation and ad/de-sorption energy in each reaction pathway. With both theoretical and experimental insights, Chen et al. correlated the selectivity of CO2 hydrogenation with the distinct activation energy between the dissociation barrier and the desorption of metal carbonyls over supported metal catalysts.93 They concluded that decreasing the coordination number of metal sites to SA structures could contribute to the suppressed carbonyl dissociation with facile CO desorption, leading to increased CO formation, since the C–O bond scission of the main intermediates was considered as the rate-limiting step in CO2 hydrogenation (Fig. 9). Correspondingly, the design of methanation catalysts could also be inspired by this descriptor regarding activation energy.
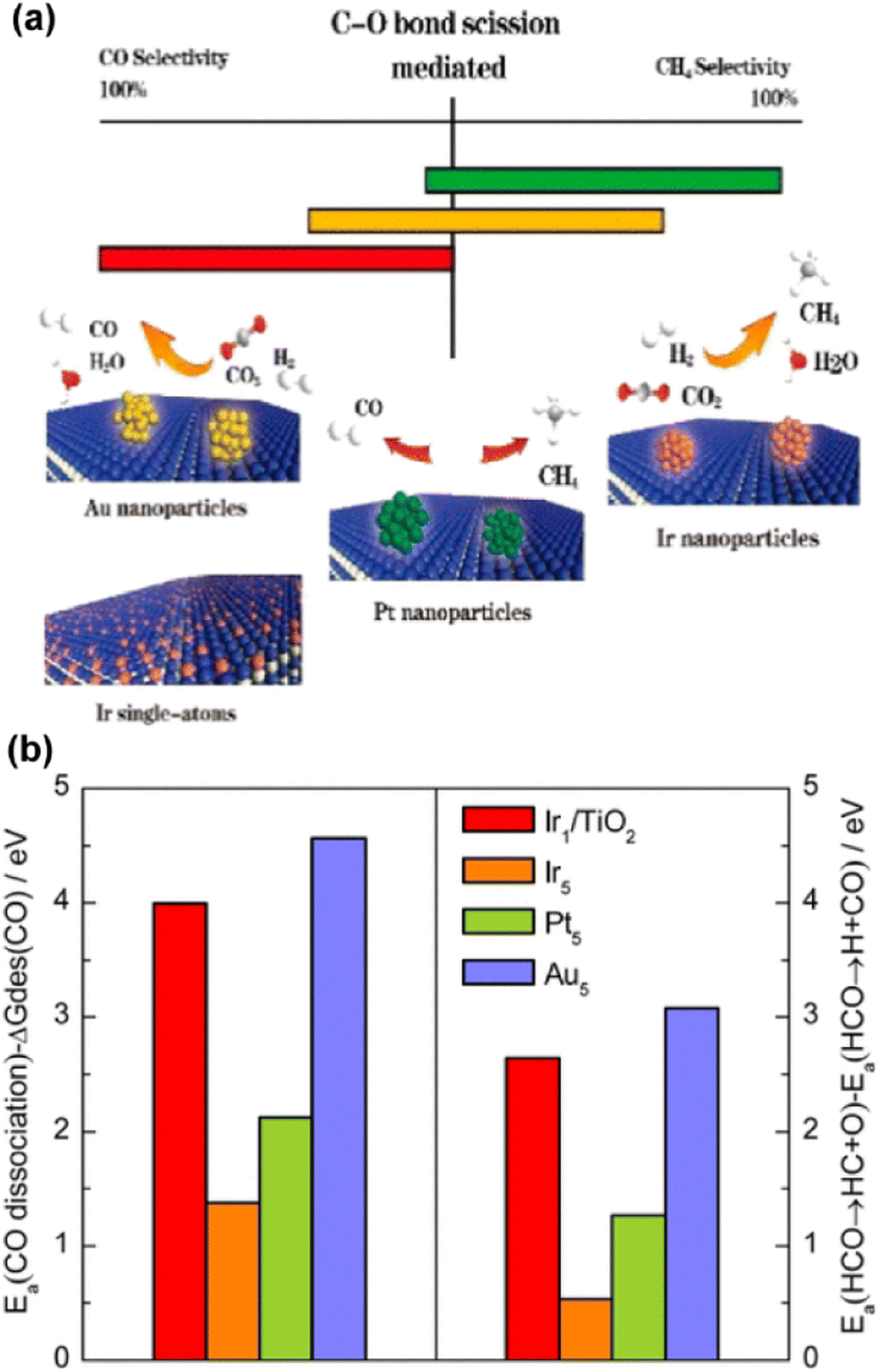 | ||
| Fig. 9 (a) Correlation between the C–O bond scission and the selectivity of CO2 hydrogenation. (b) (left) Difference between activation energies Ea for CO dissociation and desorption free energies of CO. (right) Difference in activation energies between HCO → HC + O and HCO → H + CO on PR1-Ir1/TiO2 (Ir1/TiO2) and stepped Ir (Ir5), Pt (Pt5), and Au (Au5) surfaces.93 Reproduced from ref. 93 with permission from American Chemical Society, copyright 2017. | ||
As reported by Kwak et al., the tendency of methanation in CO2 hydrogenation was enhanced with the increase in metal loading, in which the atomically dispersed metal centers were not the dominant species, suggesting that the SAC might not be suitable for methanation, although specific conditions would make some differences.77,78 A similar view was held by Guo et al.,418 as proposed in a study on low-temperature CO2 methanation over a CeO2-supported Ru SAC, nanoclusters (NCs), and NPs. The NCs with a size of 1.2 nm showed superior activity in comparison with the SAC and NPs, as well as 98–100% CH4 selectivity at 463 K. In situ measurements demonstrated that CH4 was formed dominantly via the CO route with metal carbonyls as the key intermediates, and Ce3+–OH sites and Ru sites located at the metal–support interfaces were active for CO2 dissociation and carbonyl hydrogenation, respectively. The strongest metal–support interaction and hydrogen spillover were present over the SAC and Ru NPs, respectively, leading to the corresponding suppression of metal carbonyl activation and the H2O removal on the support surfaces. In contrast, there was a balance between the two effects over NCs, which might be the key parameter to optimize their catalytic performance.
Recently, on the other hand, research studies have confirmed that the preferential synthesis of CH4 over SACs is also feasible in certain specific catalytic systems, breaking through the conventional understanding to some extent. In the work by Li et al., Rh SAs supported on ZrO2 without alkali promotor modification offered nearly 100% CH4 selectivity in CO2 hydrogenation.85 The formation of CH4 was through a HCOO* intermediate route, and facilitated by the uninhibited hydrogen activation with the help of Rh sites in the low valence state. This performance would be changed into a higher CO selectivity once alkali ions were introduced, since the Rh atomic sites were rendered into an electron deficient state with less H2 activation capability and weakened CO adsorption. By regulating the electronic state of single metal atoms via tuning the interaction between metals and supports, the catalytic properties of SACs might be significantly changed and the corresponding performance would be quite different. Fan et al. used porous hexagonal boron nitride (pBN) to immobilize Ru in the atomic dispersion and obtained an enhanced catalytic activity of 1.86 mmol CO2 per gcat per s and CH4 selectivity up to 93.5% for CO2 methanation at 623 K and 1.0 MPa.94 A vacuum filtration process allowed the Ru precursor to transform quickly into atomic Ru restricted onto the defects via B, N coordination. The Ru sites in a valence state reduced by interaction with B and N on the surface of supports remarkably lowered the maximum energy barrier from 1.56 to 1.07 eV of the consecutive hydrogenation process, contributing to the improved CH4 production rate at an atomic scale (Fig. 10). Thus, it should be possible to extend the practical application of SACs into methanation for higher metal atom utilization efficiency, in case that further efforts are devoted for this research aspect.
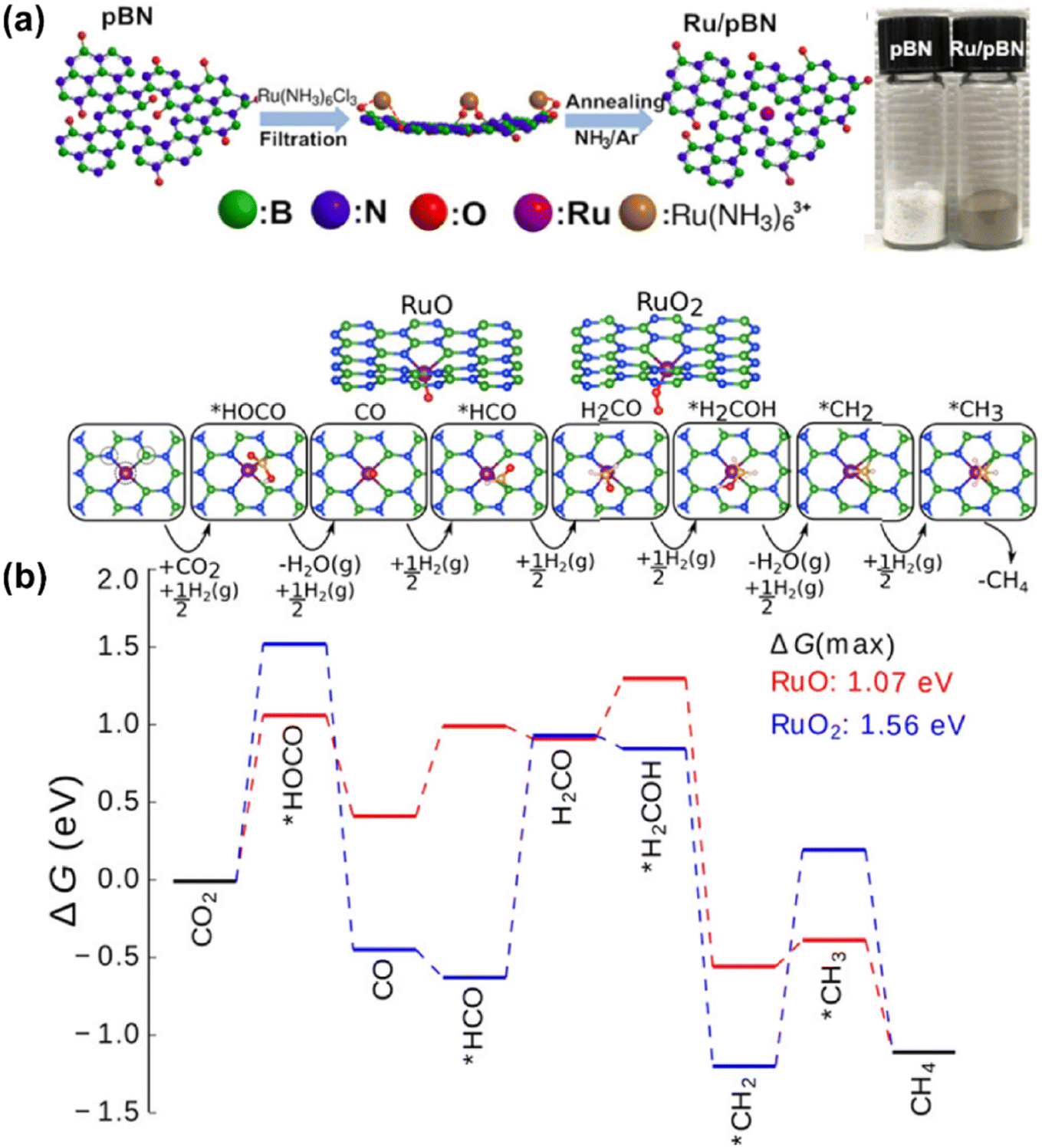 | ||
| Fig. 10 (a) Schematic illustration for fabricating Ru/pBN-xF assisted by vacuum filtration drying. (b) Reaction free-energy diagram for CO2 hydrogenation at 623 K.94 Reproduced from ref. 94 with permission from American Chemical Society, copyright 2019. | ||
Organic oxygenates such as formic acid (formate), methanol, ethanol and higher alcohols are a group of valuable compounds that are possible to be produced from CO2 hydrogenation.57,58,95,96 The controlled formation of organic oxygenates always needs an accurately regulated hydrogenation process to avoid either the generation of CO due to insufficient capability toward adsorption and hydrogenation of intermediates or the generation of CH4 due to excessive hydrogenation. Complicated procedures involving adsorption, reaction and desorption of various reactants, intermediates and products make it a challenging task to develop catalysts with high efficiency for directional catalytic hydrogenation of CO2 into organic oxygenates, in which SACs with specific structures could take advantage relying on their distinct advantages in not only high atom utilization but also their unique chemical properties.
3.1.1.2 Formic acid and formate. Formic acid is regarded as an important hydrogen storage medium, the synthesis of which might be of great significance in the promoted application of renewable and clean energy.96–98 Fruitful results have been achieved for CO2 hydrogenation to formic acid or formate via homogeneous metal complexes in basic media,99 while the development of heterogeneous catalysts is more and more appealing due to the practicality in mass production. At this moment, SACs are expected to offer a unique opportunity for the transition from homogeneous to heterogeneous systems.100 Shao et al. designed a porous organic polymer with aminopyridine functionalities to fabricate a stable Ir SAC for liquid-phase hydrogenation of CO2 to formate with an extremely high turnover number of up to 25
135, comparable with that of homogeneous systems (Fig. 11).101 The chemical structure of the Ir SA active site was identified to resemble that of a homogeneous mononuclear Ir pincer complex catalyst. Hence, the catalytic mechanism was supposed to be quite similar to that over a homogeneous Ir catalyst with this Ir-based SAC, i.e., (i) Ir SA sites encountered the CO2 reactant, (ii) the electron transfer from the metal hydride to CO2 imposed a bend of 135° of the O–C–O unit and an elongation from 1.18 to 1.23 Å of the corresponding C–O bond, with an exothermic binding energy of 0.14 eV, (iii) then this intermediate was readily transferred to HCOO– by the rotation of its C–O bond to allow binding at the Ir site, with an exothermic energy as high as 0.70 eV and a barrier of only 0.07 eV, (iv) the amide-H near the other dangling O-end of the HCOO– intermediate assisted the preliminary formation of HCOOH– by bonding to the O atom, which was induced by the formation of a six-member ring involving an Ir atom in the AP-POP framework, and (v) this initial HCOOH– promoted further H2 activation and dissociation on the site with the simultaneous transfer of H from the formic acid to the amide group (with a 1.00 eV barrier) to generate the final HCOOH product. The energy profile of the catalytic mechanism demonstrated the effectiveness of the Ir SAC and a quite similar catalytic mechanism that occurred during homogeneous catalysis, and thereby a concept of quasi-homogeneous catalysis was assigned to this system for the inherent associations between the homo- and hetero-catalytic processes. This result further suggested the great potential of SACs in CO2 conversion to formate. Indeed, quite a few attempts have been made for the fabrication of heterogeneous catalysts by mimicking the previous homogeneous systems via introducing metal–organic complexes into a rigid framework, such as CTFs, MOFs and other organic polymers, in which electron-donating functional groups comprising N, O, and P moieties were commonly used as building blocks to enhance the electron density at metal SAs.102–107 As a result, such SACs always delivered high TONs/TOFs, whereas the demand for enhancing the stability of metal SAs to overcome inferior recyclability is still urgent. For this, substantial efforts have been made on the structural engineering of the exquisitely designed catalysts. Moreover, there have been a few studies exploring the possibility of preparing materials without organic ligands. For instance, Mori et al. developed a stable and well-defined Ru SAC on the surface of a layered double hydroxide (LDH) for efficient selective hydrogenation of CO2 to formic acid under mild reaction conditions (2.0 MPa, 373 K).108 An active electron-rich Ru center was established by using triads of basic hydroxyl ligands at a particular location, with the electron-donating feature. A strong electronic metal–support interaction (EMSI) present between the Ru atoms and basic supports was evaluated by XPS spectroscopy and could be well correlated with the catalytic activity. By adjusting the type of metal ions and corresponding ratio in LDH, the CO2 adsorption capacity in the vicinity of the Ru center could be also tuned. The catalytic performance would be also improved with higher CO2 concentration on the surface. Wang et al. designed a solid micellar Ru SAC for efficient and stable water-free CO2 hydrogenation to formate under mild reaction conditions,109 directly obtaining concentrated formate solutions up to 4 mol L−1 in tertiary amines. The Ru SAC sites were stabilized by using the hexadecyl trimethyl ammonium (CTA+) surfactant in the pores of the MCM-41 support, forming a solid micelle structure during preparation. By DFT modelling, the reaction was found to proceed via heterolytic hydrogen splitting, forming metal hydride species and subsequent hydride transfer over low-coordinated Ru(III).
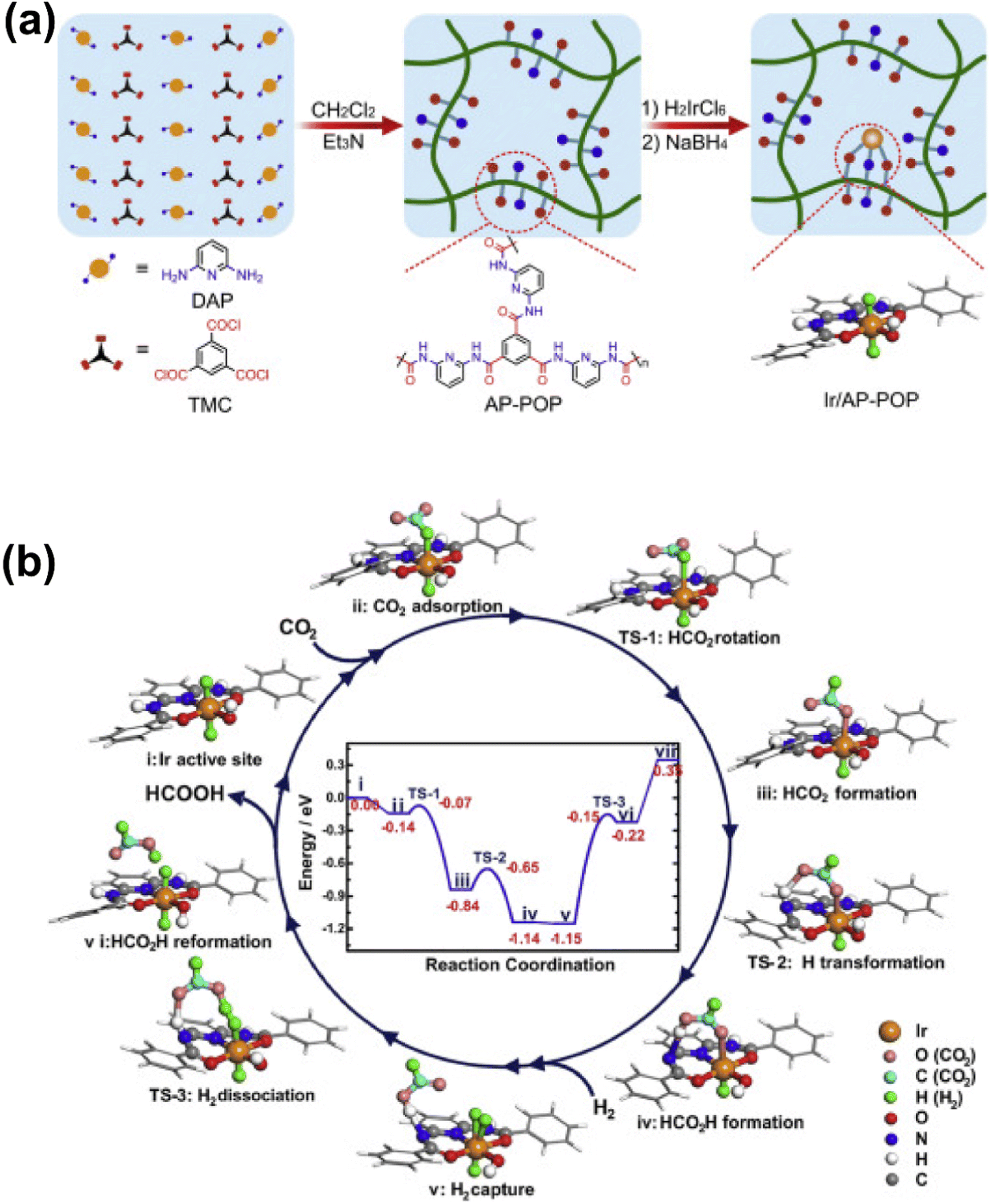 | ||
| Fig. 11 (a) Schematic illustration of the synthesis of the Ir/AP-POP SAC. (b) The proposed reaction mechanism for CO2 hydrogenation over the oppositely H bonded Ir SA active sites.101 Reproduced from ref. 101 with permission from Elsevier, copyright 2019. | ||
In addition to the investigations into the design of various active SACs based on the vital metal–support interaction, the interaction between active metal species has also been taken into account especially in the case where metal loading is high enough to reduce the distance between separated sites to some extent. Indeed, the synergistic effect of active metal sites has been reported and proposed to play a critical role in CO2 hydrogenation. Ren and co-workers reported their progress on the utilization of Pd SACs for CO2 hydrogenation to formate.110,111 One of their studies used a highly active dual single-Pd-atom catalyst with a microporous layered 2,6-pyridinedicarbonitrile-derived covalent triazine framework (2,6-DCP-CTF) as the support, efficiently converting CO2 to formate under ambient conditions with a turnover frequency (TOF) as high as 13.46 h−1.110 The microporous structures could enrich the concentration of reactants, while the dual Pd atoms within a nitrogen-rich environment exhibited a much lower barrier for this reaction than the Pd SA, and a rich nitrogen environment could further improve the intrinsic activity of Pd. A ternary synergetic effect was shown to be responsible for the outstanding performance, as confirmed by modelling calculations. They also developed a synergistic structure with Pd SAs and NPs on 2,6-DCP-CTF as an efficient active site for CO2 hydrogenation to formate.111 The Pd SAs were believed to be in charge of CO2 activation, while the Pd NPs were more likely to contribute to hydrogen dissociation based on theoretical calculations. Therefore, compared with the catalysts mainly comprising Pd SAs or NPs, this hybrid catalyst presented a better balance between the two key steps in CO2 hydrogenation, leading to the improved catalytic activity. By further optimizing the ratio of Pd SAs to Pd NPs, a formate formation rate of 3.66 mol molPd−1 h−1 could be obtained under ambient conditions (303 K, 0.1 MPa). Li et al. explored the activity of CO2 hydrogenation dependent on the interaction between two neighboring Pt monomers over a Pt/MoS2 SAC.112 As indicated, the majority of Pt monomers were in the isolated state over the 0.2% loading SAC, while the neighboring Pt monomers predominated in the Pt species over SAC with a high Pt loading up to 7.5% in contrast. As for the catalytic performance, the SAC with neighboring Pt monomers demonstrated a significantly lower reaction barrier for CO2 hydrogenation, and a stepwise reaction path via a formic acid intermediate rather than 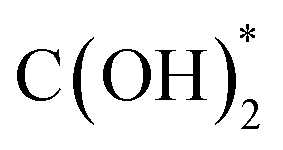 was demonstrated, leading to relatively high selectivity of formic acid. In contrast, the SAC with isolated Pt monomers favored a path with the absence of formic acid formation and higher activation energy (Fig. 12). These results clearly indicated the importance of the synergetic effect derived from the interaction between active metal sites.
was demonstrated, leading to relatively high selectivity of formic acid. In contrast, the SAC with isolated Pt monomers favored a path with the absence of formic acid formation and higher activation energy (Fig. 12). These results clearly indicated the importance of the synergetic effect derived from the interaction between active metal sites.
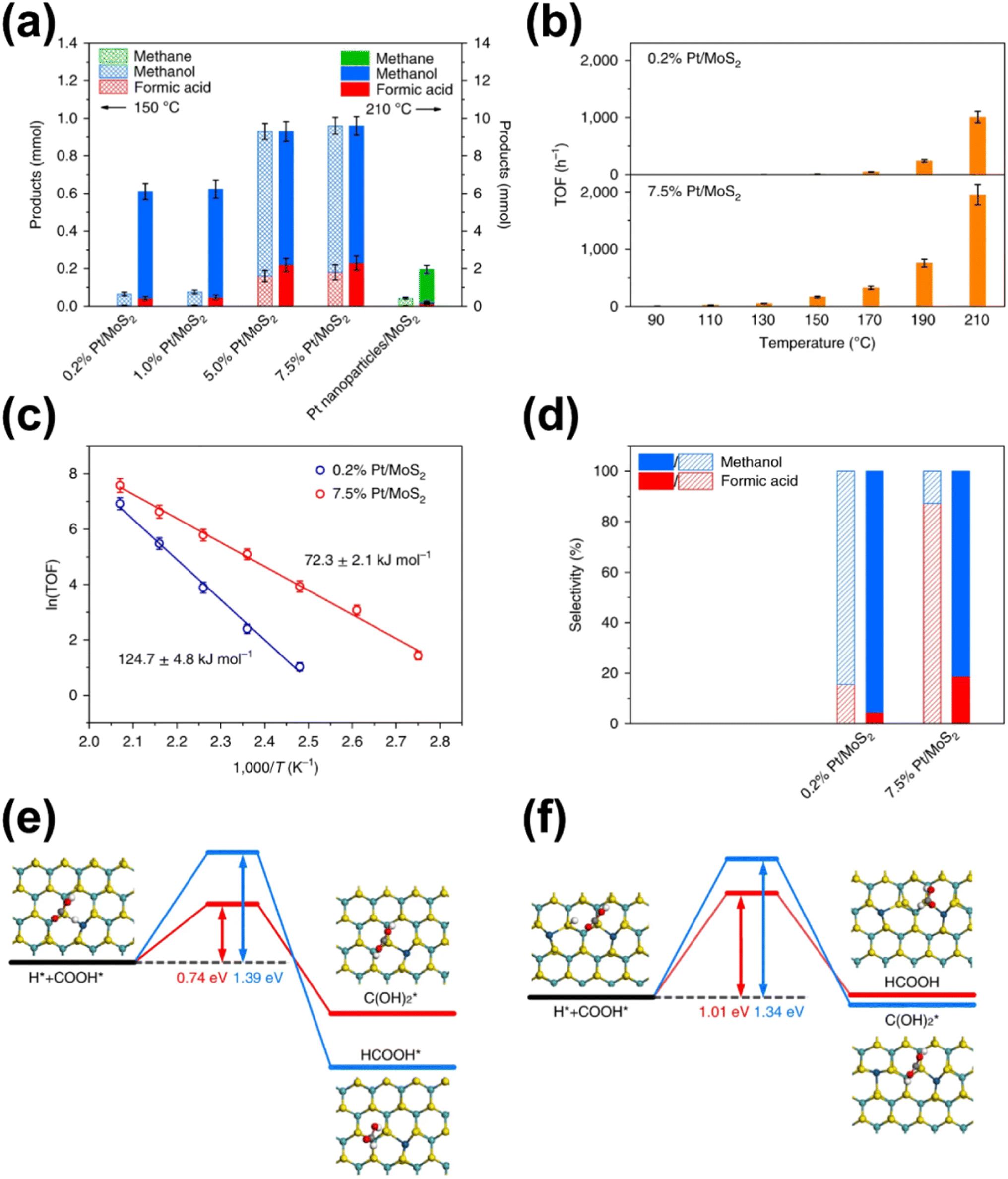 | ||
| Fig. 12 (a) Comparison of products obtained by using atomically dispersed Pt/MoS2 with different Pt loadings and Pt nanoparticles/MoS2. (b) Comparison of TOFs with 0.2% Pt/MoS2 and 7.5% Pt/MoS2 as the catalyst at different temperatures. (c) The Arrhenius plots of 0.2% Pt/MoS2 and 7.5% Pt/MoS2. (d) Comparison of selectivity of products obtained by using 0.2% Pt/MoS2 and 7.5% Pt/MoS2 as catalysts at different ratios of CO2 to H2 after 3 h. Steps for the addition of a H atom to COOH* over (e) Pt1/MoS2 and (f) Pt2iii/MoS2, respectively.112 Reproduced from ref. 112 with permission from Springer Nature, copyright 2018. | ||
3.1.1.3 Methanol. As one of the simplest liquid energy storage compounds and versatile C1 building blocks, methanol has been appealing in both chemical and energy sectors over the past few decades, whose production from CO2 hydrogenation has been considered to be a solution for excessive CO2 emission and contribute to ‘carbon neutrality’.57,113–116 The initial studies on the methanol synthesis from CO2 were originated from the conversion of syngas consisting of CO, CO2 and H2, which is mainly acquired through coal gasification, natural gas reforming and biomass transformation. The commercial production of methanol from CO2 is generally realized with the utilization of copper-based catalysts, particularly those based on copper, zinc oxide and alumina (Cu/ZnO/Al2O3).117,118 However, the efficiency of CO2 conversion is still severely limited by thermodynamics (Fig. 4) under the specific operation conditions (503–573 K and 50–75 bar) applied with the standard Cu/ZnO/Al2O3. Thus, great efforts have been devoted for the modification of conventional Cu-based catalysts, as well as designing new catalysts containing entirely distinct compositions such as noble metals and reducible metal oxides.57,63,119–121 Meanwhile, homogeneous hydrogenation of CO2 into methanol undergoes a renaissance due to its high activity at low temperature,122 which suggests the promising potential of SACs similar to that of homogeneous catalysts.
Recent studies have unveiled that SAs could play a critical role in Cu-based catalysts as either active components or promoters. Zhao et al. reported a Cu-based catalyst Cu/ZrO2 with isolated active copper sites for methanol synthesis from CO2 hydrogenation.123 A strong dependence of CO2 hydrogenation activity and selectivity on the nature of surface copper species was revealed. The undercoordinated cationic Cu1–O3 species, accumulating on the catalyst surface, were considered to form stable active sites and be in charge of high selectivity to MeOH (100%) during the catalysis process at 453 K. The pathway with HCOO* as the key intermediate was believed to be the most viable route for CO2 hydrogenation on the isolated Cu1–O3 active sites, as the generation of HCOO* was achieved through the reaction between activated CO2 and dissociative hydrogen formed over Cu SAs with the help of nearby oxygen atoms. In contrast, small copper NCs and NPs favored CO formation via RWGS, while larger particles exhibited little activity at low temperature (Fig. 13). Wu et al. used a Cu-based SAC (Cu/ZnO) to study the effect of introduced water content on the methanol synthesis from CO2 hydrogenation.124 They found that introduced water could act as a bridge between H atoms and CO2 or intermediates, facilitating the transformation of COOH* and CH2O*. The enhanced activity induced the generation of more water to react with CO via the water–gas shift reaction, resulting in an increase in methanol selectivity. With the introduction of optimal-content of water, a methanol selectivity of 99.1% could be obtained, while the CO2 conversion exhibited a volcano-type curve with a peak value of 4.9%. Yang et al. developed a Cu-based catalyst with atomically dispersed Zn on a ZrO2 support via a double-nozzle flame spray pyrolysis (DFSP) method.60 Dynamic structural evolution of Zn species analyzed by operando XAS showed that atomic Zn sites were formed under specific reaction conditions via the strong interaction between the highly dispersed ZnO clusters and ZrO2 support. The operando experiments as well as modeling calculations revealed that this unique Zn species promoted the selective formation of methoxy and subsequently methanol instead of decomposition to CO due to the stabilized formate intermediate and decreased hydrogen activation energy. As a result, a superior performance could be achieved over this SAC in comparison with that over the generally accepted active Cu–ZnO interface, while large and independent ZnO nanoparticles exhibited the poorest performance due to the absence of promotion of both hydrogen activation and intermediate stabilization. Furthermore, Wu et al. performed a systematic theoretical study on the mechanistic nature and catalytic pathways of Zn SAs and NCs on the surface of Zn/Cu alloy catalysts, in which surface oxidation was proved to make a great difference.125 The Zn SAs were more active than the Zn NCs initially with more difficulty in subsequent surface oxidation under specific reaction conditions. After catalyst surface oxidation during CO2 decomposition, more active species were switched to oxidized Zn NCs from Zn SAs. Further analyses show that this effect of surface oxidation could be attributed to not only the occupation of the strongest binding sites at the center of Zn NCs by O atoms, but also the higher positive charge and work function on the oxidized surface. The former made oxygenate intermediates adsorb on the Zn/Cu interface with moderate binding for further hydrogenation, while the latter directly promoted the hydrogenation of oxygenate intermediates.
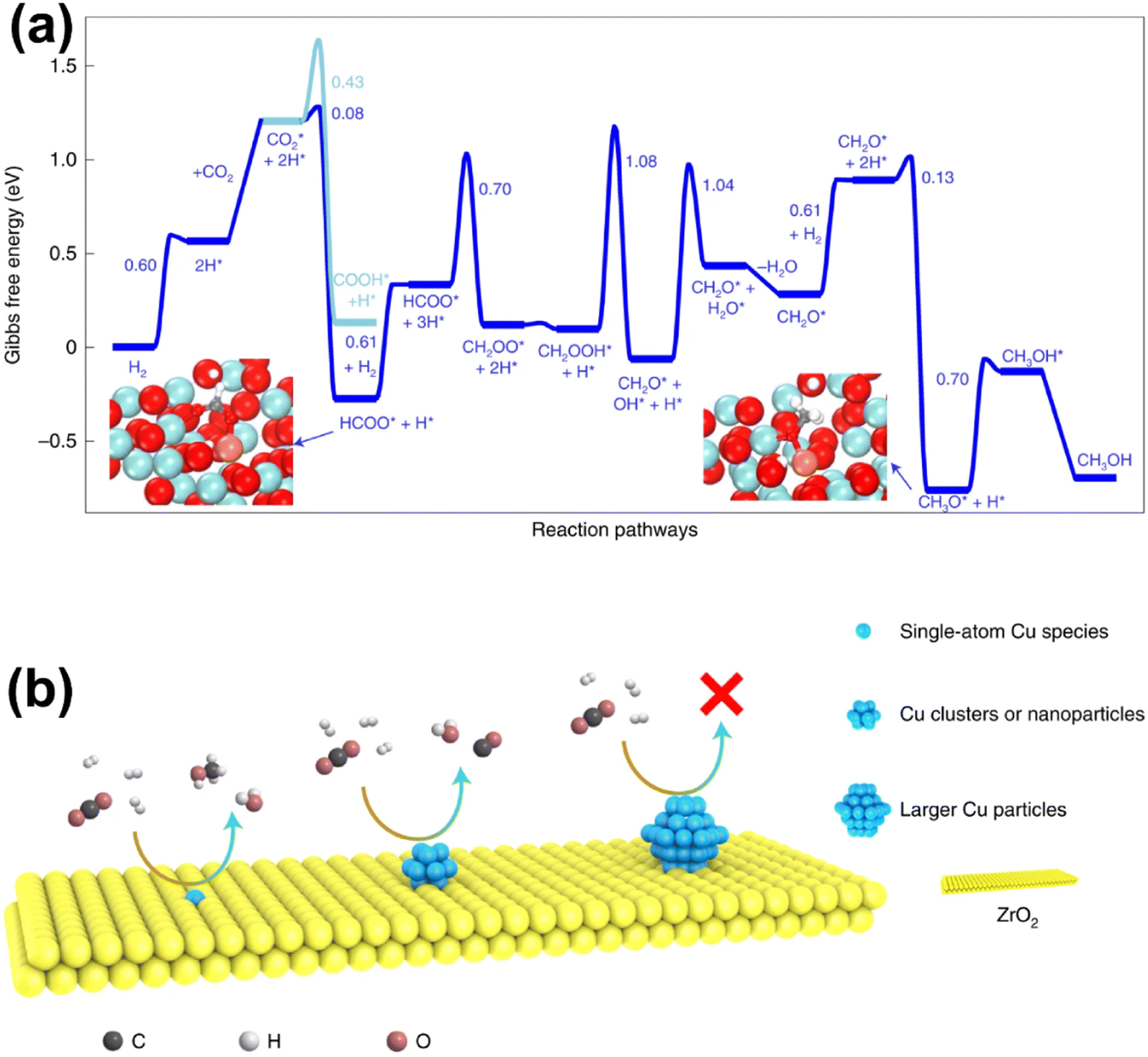 | ||
| Fig. 13 (a) Gibbs free-energy profile of CO2 hydrogenation to CH3OH/CO. (b) Schematic diagram for the CO2 hydrogenation reaction on different types of copper species.123 Reproduced from ref. 123 with permission from Springer Nature, copyright 2022. | ||
Apart from Cu-based catalysts, SACs based on noble metals for CO2 hydrogenation to methanol have also attracted considerable attention due to their superior hydrogen activation capability at relatively lower temperature. Chen et al. reported a Pt-based SAC comprising an ensemble of Pt SAs coordinated with oxygen atoms in MIL-101 (Pt1@MIL), with a turnover frequency (TOF) of 117 h−1 and a methanol selectivity of 90.3% in DMF at 3.2 MPa and 423 K for selective CO2 hydrogenation to methanol, which were much higher than that of Ptn@MIL with nanocrystals as the major Pt species.126 In contrast to Ptn@MIL with COOH* as the intermediate in CO2 hydrogenation, Pt1@MIL favored the pathway via HCOO* as the key intermediate, leading to a lower activation energy for the enhanced catalytic activity and corresponding higher selectivity toward methanol by suppressing the formation of CO. This research altered the reaction path with the help of a SAC to achieve enhanced performance, whereas some other studies reached similar goals by promoting the kinetics of the key reaction step during CO2 hydrogenation. Dostagir et al. used an In2O3 catalyst promoted by single Rh atoms to obtain a high methanol productivity of 1.0 gMeOH h−1 gcat−1 with intrinsic high selectivity over pure In2O3.120 Rh species were atomically dispersed in the In2O3 matrix and were stabilized by charge transfer from neighboring In atoms under specific reaction conditions. Rh SAs promoted the dissociation of H2 and consequent partial reduction of In2O3, leading to the formation of more oxygen vacancies on the catalyst surface, as the oxygen vacancies in In2O3 were considered to be the key active sites for the adsorption and activation of CO2. The generation of formate species, a key intermediate in In2O3-catalyzed CO2 hydrogenation, was also propelled by Rh SAs, as verified by DFT calculations regarding the activation barrier.
3.1.1.4 Summary. Despite suffering from relatively rigorous operation conditions, thermochemical hydrogenation processes still benefit from sophisticated equipment, high scalability and versatility. SACs with nearly 100% atomic utilization efficiency not only provide a promising prospect for wider application of noble metals in the effective conversion of CO2, but also steer the reaction pathway towards exclusive synthesis of valuable products. The distinct electronic and geometric properties derived from specific coordination and interaction with neighboring atoms offer highly active interfaces for activation of CO2 and stabilization of specific intermediates, thereby propelling the formation of C1 products. Furthermore, the studies on the production of C2+ products have also achieved inspiring results with the application of SACs, in which the specific features of SAs in combination with vacancy sites generated from defect engineering are incorporated for realizing C–C coupling in the systems. All these results indicate the potential of SACs in CO2 thermochemical hydrogenation (Fig. 14).
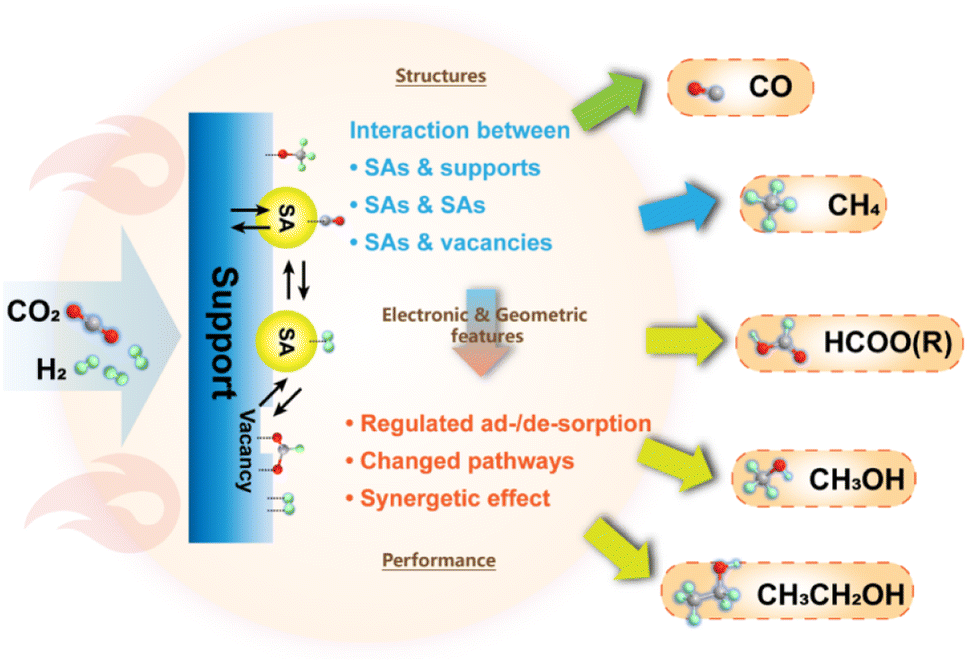 | ||
| Fig. 14 A brief summary of SAC applications and key scientific problems in thermochemical CO2 hydrogenation. | ||
3.1.2.1 Carbon monoxide. With the rapid development of electro-catalysis research over recent years, electrochemical CO2 reduction has been considered a feasible alternative approach for the production of various chemicals, especially CO which is synthesized traditionally by RWGS via a thermal reduction process under harsh conditions.129 As a reaction with a slightly negative equilibrium reduction potential of circa. −0.10 V versus the reversible hydrogen electrode (RHE) and involving two-electron transfer (Fig. 5), the production of CO might be one of the most fundamental and practical processes in electrochemical CO2 reduction because of its possibly higher selectivity and larger current densities. Over a long period, substantial effort has been devoted to exploring efficient catalysts for reduction of CO2 to CO. Noble metals like Au, Ag and Pd have been identified as useful catalysts with a high current efficiency and selectivity, but their high cost limits their practical application.137 To further improve the efficiency of CO2 reduction to CO and lower the cost to make it a more profitable process, ingenious catalyst design strategies have been developed. The application of SACs is one at the center of attention to overcome the existing complex problems regarding activity and selectivity compared to conventional bulk metal-based catalysts,138 and this even provides non-noble metals with performance exceeding that of noble metals.47,130,139
Given that nickel (Ni) is a relatively low-cost metal commonly used in reduction reactions,138,141 and inspired by the previous research studies that unveiled the potential of Ni-based molecular catalysts in CO2 transformation,142–144 plenty of studies have been devoted for the application of Ni-based SACs in electrochemical CO2 reduction. Yang and his co-workers (Fig. 15) fabricated a Ni-based SAC containing isolated, high-density, low-valent Ni anchored on a N-doped graphene matrix for CO2 reduction.140 Through the combination of operando X-ray absorption spectroscopy (XAS) and X-ray photoelectron spectroscopy (XPS) techniques, Ni(I) was identified as the active center, for which the structure evolution during the CO2 reduction was further illustrated. The active sites had an effect by the delocalization of the unpaired electron in the Ni 3dx2−y2 orbital, and a CO2δ− species was formed via spontaneous charge transfer from Ni(I) to the carbon 2p orbital in CO2. In this way, the energy barrier for electrochemical CO2 reduction was remarkably reduced, leading to a specific current of 350 A gcat−1 and turnover frequency of 14800 h−1 at a mild overpotential of 0.61 V with 97% faradaic efficiency (FE) for CO formation. Generally, the preparation methods of SACs have a great effect on the fine structure of active sites, which further determines the catalytic performance. Since the carbon-based supports are always essential for maintaining sufficient electron transfer capability, the kernel task for the fabrication of SACs is embedding the metal SAs into the carbon matrix, for which substantial efforts have been made. One of the recently proposed strategies is using metal organic framework (MOF) materials as precursors.145,146 The metal atoms with high density can be fixed into the MOF crystals preliminarily by the strong bonding between metals and the moiety of ligands. Then ligands should be partially removed in an appropriate way, so that the low coordination SAs can be well exposed to the reactant phase in a stable form. Another strategy is introducing specific dopants (e.g., nitrogen) in the preparation process or post-treatment to as-obtained carbon supports, since the lone pair electrons in doping elements provide an enhanced capability for chemical bonding to the metal atoms.147–149 Since the coordination of SAs on carbon supports with high loading always needs excess dopants, the formation of miscellaneous structures is always inevitable with non-uniform introduction of dopants onto the catalysts, resulting in some adverse effects on catalytic performance. Hence, Xi et al. synthesized carbon-supported Ni SACs via a joule heating strategy, ensuring the coordination of 80% of N dopants with metal atoms.150 It was plausible that the exclusion of the ineffective N species led to the superior performance of optimized Ni SACs in electrocatalytic CO2 reduction with a high FE above 92% for CO formation in the potential range of −0.7 to −1.9 V versus RHE, while the competing hydrogen evolution reaction (HER) was suppressed. Such a preparation strategy could be also facilely extended to the synthesis of various metal-based SACs. Moreover, not only do the metal atom sites have great influence on the eventual catalytic performance of SACs catalyst, but the features of the support could play an important role in many systems for CO2 electroreduction.132 He et al. constructed a Ni-based SAC comprising a porous carbon membrane with interconnected nanofibers and hierarchical pores, supporting abundant Ni SAs for CO2 reduction to CO.151 This membrane with high mechanical strength and well-dispersed Ni SAs combined gas-diffusion and catalyst layers into one configuration, which could establish a stable three-phase interface for CO2 electroreduction. Eventually, CO could be produced via a *COOH intermediated route with a 308.4 mA cm−2 partial current density and 88% FE over a 120-hour test. Moreover, the defects of certain supports could also exhibit a significant effect on the catalytic performance.152 In the work by Boppella et al.,152 the synergy between Ni–N4 and metal free defect sites over the N-doped carbon support was revealed in CO2 reduction. They investigated and compared the kinetic and thermodynamic behaviors of four possible active sites during CO2 transformation, which were Ni–N4-defects (defects as active centers), Ni–N4–C (C near pyrrolic N as active centers), Ni–N4–Ni (Ni as active centers) and Ni–N4–N (pyrrolic N as active centers), according to the free energy diagrams determined by DFT calculations. As a result, Ni–N4–N sites coupled with metal-free C near pyrrolic N sites were supposed to provide not only a lower energy barrier for the formation of COOH* intermediate species leading to promoted CO2 activation, but also an optimum CO* binding energy facilitating CO desorption, resulting in a higher intrinsic catalytic activity (CO FE of 94% at −0.6 V and CO partial current density of 59.6 mA cm−2 at −1 V). To gain a comprehensive understanding of reaction mechanisms regarding the structure of active sites and possible reaction kinetics over Ni-based SACs, advanced computational chemistry tools have been also used besides the great efforts made in experimental research. Hossain et al. used a new grand canonical potential kinetics (GCP-K) formulation, which could describe the electron transfer with proton or hydrogen transfer as a continuous process.153 Since the commonly accepted reaction route of electrochemical CO2 reduction to CO involves four consecutive steps: (a) linear CO2 binding to the catalyst surface (*), forming bent *CO2−via electron transfer; (b) hydrogen transfer from water which converts adsorbed *CO2− into the adsorbed *COOH intermediate; (c) a second hydrogen transfer from water which converts the adsorbed *COOH intermediate into bound *CO; and (d) bound *CO desorption from the catalyst surface as gaseous CO, leaving the surface empty (*) for another reaction cycle, the possible sites over Ni-SACs including Ni–N2C2, Ni–N3C1, and Ni–N4 were evaluated based on these procedures (Fig. 16). Distinct kinetics was exhibited over each kind of site, and the best overall performance was predicted to be present on Ni–N4 with a CO FE of nearly 100% and current density of 40 mA cm−2 at a potential of −1.05 V, while the other sites showed slightly inferior performance. Meanwhile, the XPS N, C 1s and Ni 2p binding energy (BE) shift and the CO vibrational frequencies for various sites were also predicted for helping in the identification of the active sites, and all these predicted values showed surprising agreement with the experimental results. This work further suggests that it should be a good modeling material for SAC to associate the theoretical analysis and experimental research. More recently, Li et al. studied the effect of acidic microenvironments on a Ni–N–C SAC for
CO2 electrolysis in a membrane electrode assembly (MEA) electrolyzer.154 To overcome the frequent severe carbon loss in common alkaline and neutral electrolytes, the authors developed an acidic membrane electrode assembly electrolyzer with a tailored local concentration ratio of H+ and K+. They found that the addition of both H+ and K+ favored the adsorption of CO2 and desorption of CO species energetically, despite simultaneously enhancing the adsorption of H* (competitive adsorption with CO2). As a result, they achieved an energy efficiency of 45% for CO production at pH 0.5 with CO2 loss reduced by 86% at 300 mA cm−2, as compared to that under alkaline conditions. Notably, they also realized a CO2–CO–C2+ process with 99% FE of C2+ products (46% FE of ethene) via a two-step CO2 electrolysis process by the tandem combination of such an acidic MEA electrolyzer and an alkaline MEA electrolyzer.
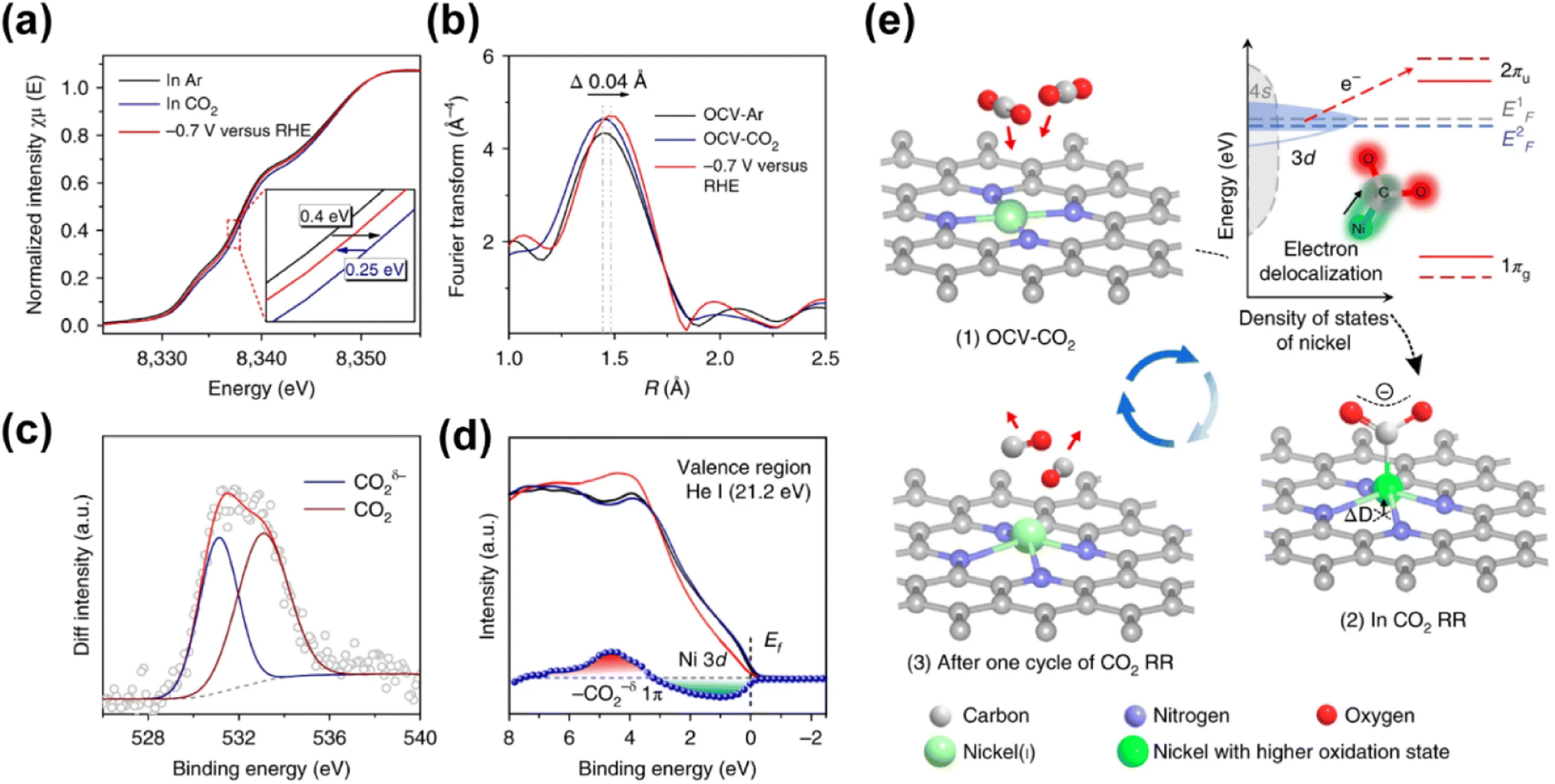 | ||
| Fig. 15 (a) Normalized operando Ni K-edge XANES spectra for A-Ni-NG at various biases (versus RHE) in 0.5 M KHCO3 aqueous solution at room temperature and 1 atm of Ar or CO2. The inset shows the enlarged Ni K-edge XANES spectra. (b) Fourier transform magnitudes of EXAFS spectra (without phase correction) of A-Ni-NG under open-circuit voltage bias in Ar (OCV-Ar) and CO2 (OCV-CO2), and at −0.7 V (versus RHE), in which an expanded Ni–N bond was detected. (c) Change in the XPS O 1s intensity induced by CO2 adsorption for A-Ni-NG. (d) Valence band spectra of A-Ni-NG before (black line) and after (red line) CO2 gas exposure, and after desorption of CO2 by thermal treatment at 773 K for 20 min in a vacuum (dark blue line). The blue line shows the variation in the valence band spectra of A-Ni-NG induced by CO2 adsorption. (e) Structural evolution of the active site in electrochemical CO2 reduction.140 Reproduced from ref. 140 with permission from Springer Nature, copyright 2018. | ||
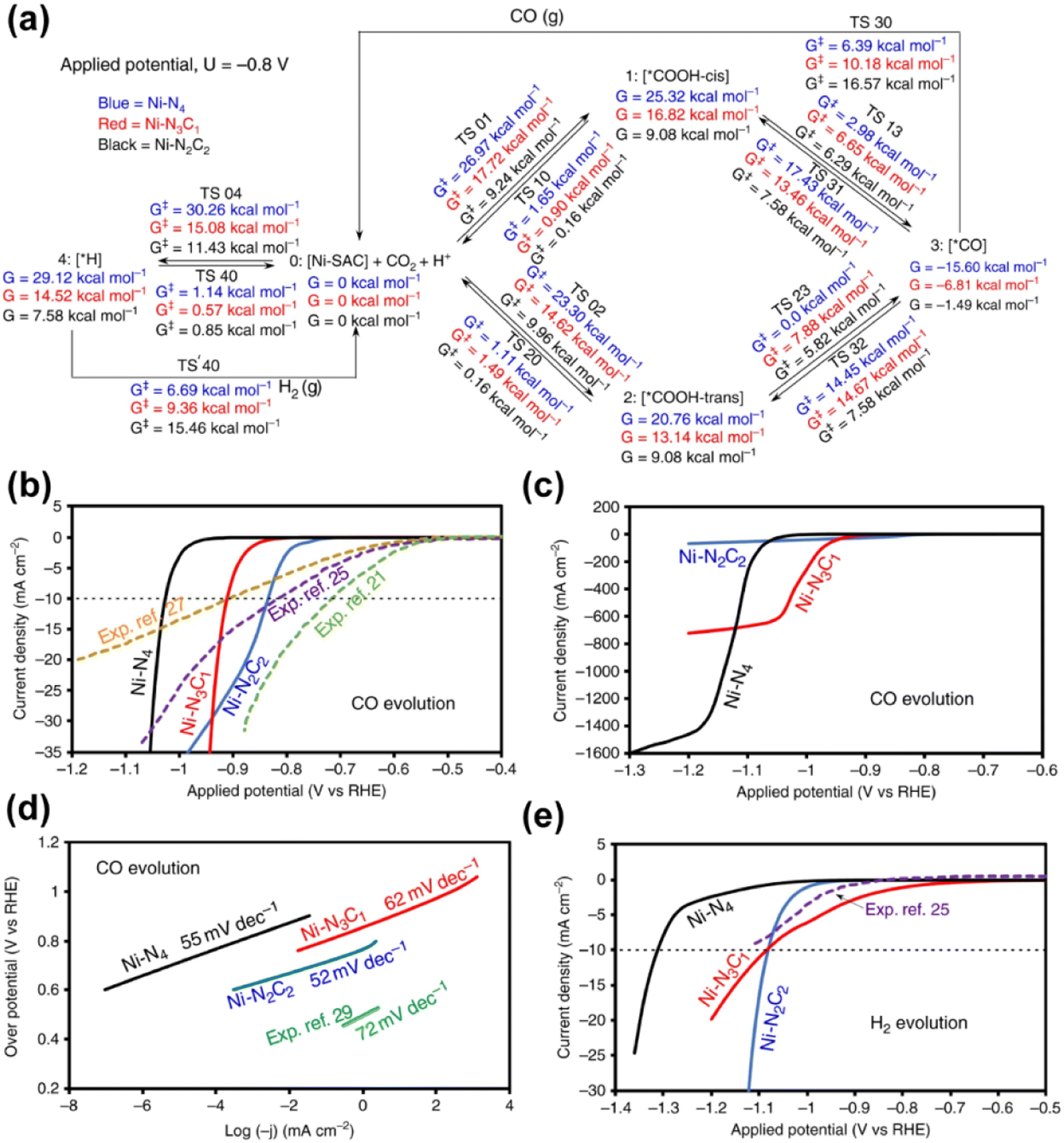 | ||
| Fig. 16 (a) Schematic representation of free energies at 298 K and pH 7 for −0.8 V applied potential. This summarizes all reaction intermediates (0–4) and transition state (TS) free energies involved in the reduction of CO2 on a nickel single site at −0.8 V constant applied potential. (b) Calculated partial current densities for CO evolution during CO2 reduction on Ni–N2C2, Ni–N3C1 and Ni–N4 along with experimental data for comparison (equivalent number of nickel atoms). (c) Large partial current densities for CO evolution during CO2 reduction on Ni–N2C2, Ni–N3C1 and Ni–N4. (d) Tafel slopes calculated from the I–V curves for CO evolution on Ni–N2C2, Ni–N3C1 and Ni–N4 during CO2 reduction, showing good agreement with the Tafel slope from the experiment. (e) Calculated partial H2 current densities as a function of applied potential during CO2 reduction on Ni–N2C2, Ni–N3C1 and Ni–N4 along with experimental data for comparison (equivalent number of nickel atoms), at pH 7 and 298 K.153 Reproduced from ref. 153 with permission from Springer Nature, copyright 2020. | ||
In addition to Ni-based SACs, SACs comprising iron species have been also used in electrochemical CO2 reduction. For example, Gu et al. reported an iron-based SAC that produced CO from CO2 reduction at an overpotential as low as 80 mV, and the partial current density could reach 94 mA cm−2 at an overpotential of 340 mV.139 Discrete Fe3+ ions, coordinated to pyrrolic N atoms of the N-doped carbon support, were identified as active sites by the operando XAS technique. During the electrocatalysis process, the active centers could maintain a +3 oxidation state through a possible electronic coupling to the conductive carbon support, driving faster CO2 adsorption and weaker CO absorption in comparison with conventional Fe2+ species. In this way, a comparable performance to that of noble metal catalysts was obtained. In order to further improve the activity of the Fe SAC, phosphorous SAs were introduced into N-doped carbon supported Fe SAs (Fe-SAC/NPC) for CO2 electroreduction to CO in an aqueous solution, as reported by Sun et al.155 Experimental analysis combined with DFT calculations suggested that P SAs mainly in the form of P–C bonds were located in high coordination shells, particularly the third coordination shell of the Fe center, leading to enhanced electronic localization of Fe. Thus, the Fe center could contribute more electrons to bond with the key *COOH intermediate, enabling the facile formation and stabilization of this intermediate on Fe with a decreased energy barrier. As a result, this SAC exhibited a CO FE of nearly 97% at an overpotential of 320 mV, comparable to state-of-the-art gold catalysts. Taking mass transfer into consideration, Zhang et al. prepared a hierarchical carbon foam confining Fe-based SAC with the assistance of a SiO2 template.156 The Fe SAs coordinated with four nitrogen atoms (FeN4) confined in a carbon matrix at the atomic-scale served as the active sites for electrocatalytic CO2 reduction to CO with an optimized CO FE of up to 94.9% at a moderate potential of −0.5 V vs. RHE. With the utilization of the special carbon support, the pore-enriched effect at the macro-scale favored the diffusion of substrate molecules, while the graphene nanosheets at the nano-scale enhanced charge transfer. 57Fe Mössbauer spectroscopy, involving the resonant and recoil-free emission and absorption of γ-rays by atomic nuclei, is useful to probe tiny changes in the energy levels of the atoms. Li et al. exploited this technique for operando monitoring of the dynamic behaviors of Fe SA moieties under specific reaction conditions (Fig. 17).157 By means of operando XAS and a quantum-chemical study, the in situ-generated four pyrrolic nitrogen atom-coordinated low-spin (LS) Fe(I) (LS FeIN4), featuring monovalent Fe(I, d7) with an unpaired electron in the dz2 orbital of a pseudo-D4h ligand field, had been identified as the reactive center for the conversion of CO2 to CO. The optimal binding strength of CO2 to the LS FeIN4 site, with strong orbital interactions between the singly occupied dz2 orbital of the Fe(I) site and the singly occupied π* orbital of the [COOH] fragment, was considered as the key factor for the excellent performance.
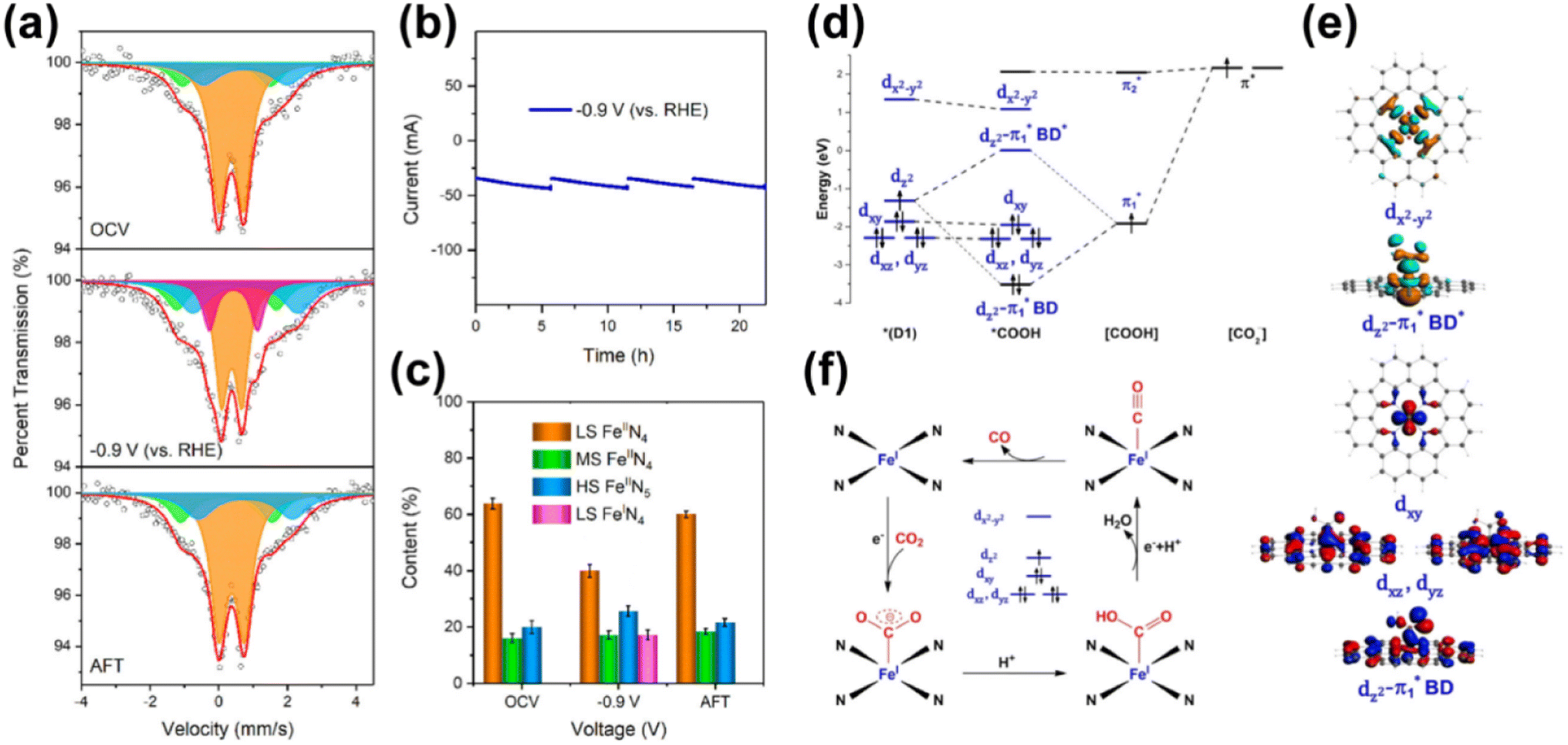 | ||
| Fig. 17 (a) Operando57Fe Mössbauer spectra of 57Fe-enriched Fe-NC-S recorded at OCV, −0.9 V (vs. RHE), and after the CRR. The orange, green, blue, and purple doublets could be assigned to LS Fe2+ in FeIIN4, MS Fe2+ in FeIIN4, HS Fe2+ in N–FeIIN4, and LS Fe2+ in FeIN4, respectively. (b) Current–time response of 57Fe-enriched Fe-NC-S for the CRR. (c) Content of different Fe moieties and reactive intermediates obtained from operando 57Fe Mössbauer measurements. (d) Schematic Kohn–Sham molecular orbital (MO) energy-level correlation diagram between CO2 and the active Fe site of D1 for the CO2 activation step. (e) Selected contour plots of MOs for the *COOH intermediate with the D1 model. (f) Proposed reaction pathway of the CRR over D1. The acronym CRR refers to the CO2 reduction reaction conducted in CO2-saturated 0.5 M KHCO3 solution.157 Reproduced from ref. 157 with permission from American Chemical Society, copyright 2021. | ||
Copper-based catalysts have been widely used in electrochemical reduction of CO2 to valuable chemicals, especially involving the C–C coupling process.158–164 When the size of Cu species is downsized to the atomic scale, distinct catalytic performances have been found, which makes the selective production of CO possible.165–167 In a pioneering research study by Li and his co-workers,166 a catalyst featuring two adjacent copper atoms, namely the ‘atom pair’, was reported to carry out the critical bimolecular step in CO2 reduction. With stable Cu01–Cu1x+ pair structures, this catalyst could readily adsorb H2O and CO2 over neighboring Cu1x+ and Cu01 sites, respectively. This adsorption configuration was verified by a combination of experiments and modeling calculations. It significantly reduced the activation energy of CO2, so that the performance could be promoted with a FECO of above 92% at relatively low potentials. Another strategy was proposed by Chen et al. via introducing other active component dispersed Cu clusters (Cux) into a Cu SA co-coordinated with a N and S anchored carbon matrix (Cu–S1N3) to develop a tandem catalyst (Cu–S1N3/Cux) for CO2 reduction to CO.167 In a typical reaction process, the Cu–S1N3 atomic interface offered an optimized binding energy for the key intermediate *COOH, while the adjacent Cux accelerated the protonation of *CO2− by enhancing the water dissociation and *H transfer to the Cu–S1N3 sites. In this way, the tandem catalyst exhibited much higher FECO (above 90%) compared with the catalysts with a single component (50–70%).
Zinc, as an earth-abundant metal, has shown some promising properties as an active component in CO2 electrochemical reduction.168 Recently, there have been also some achievements in terms of Zn-based SACs for the production of CO from CO2 reduction.169–171 Li et al. fabricated a nitrogen-stabilized SAC featuring low-valence zinc atoms (Znδ+–NC).169 Saturated four-coordinate (Zn–N4) and unsaturated three-coordinate (Zn–N3) sites coexisted, and the latter was in charge of Zn species at the low-valence state, as speculated from XPS, XAS, electron paramagnetic resonance (EPR), and DFT. Further calculations suggested that the unsaturated Zn–N3 could remarkably reduce the energy barrier for the formation and stabilization of the key COOH* intermediate thanks to the electron-rich environment of Zn (Fig. 18), leading to a high FECO of up to 95% at a current density of 1 A cm−2 in a flow cell. Furthermore, the preparation and corresponding properties of supports were also considered as key factors that made a great difference in some Zn-based SACs.170–172 Hao et al.170 succeeded in loading Zn SAs on the N-doped carbon nano-onions (ZnN/CNO) for CO2 electro-reduction to CO. A FE of CO up to 97% at −0.47 V (vs. RHE) could be obtained, beyond that of Zn SAs on a two-dimensional planar and porous graphene substrate with larger surface area. Systematical characterization studies and DFT calculations indicated that the superior performance of the CNO support could be attributed to the increased curvature, which could tune the surface charge and subsequent adsorption energies of key intermediates, and thereby improve the selectivity for CO generation. Coincidentally, Fang et al.171 also unveiled the effect of curvature on Zn SAs supported on curved N-doped carbon nanofibers (Zn SAs/N–C) by a facile noncovalent self-assembly approach. In this SAC, Zn atoms in Zn–N4 sites were identified as the actual active sites, while adjacent pyridine-N could synergistically decrease the free energy barrier for intermediate *COOH formation. In particular, the curvature of the catalyst made Zn 3d electrons in the Zn–N bonds return to the Zn atom, leading to an increase in electron density of Zn and accelerating CO2 electroreduction to CO. In addition to commonly reported Zn-based SACs with Zn–Nx sites as the sole active component, Zheng et al. develop a SAC with an isolated zinc site embedded in nitrogen, sulfur co-doped hierarchically porous carbon (Zn–NS–C) to realize CO2 electrochemical reduction to CO.173 Combined structural characterization and electrochemical experiments as well as theoretical calculations revealed the synergistic effects of auxiliary S and isolated Zn–N4 sites. The bicarbonate adsorption and dissociation were promoted by the adjacent S atoms, resulting in an increased proton transfer rate and boosted reaction kinetics of *CO2 protonation to form *COOH. In this way, this SAC provided enhanced activity of CO2 reduction with a high CO FE of 99% at an industrial-level current density of 200 mA cm−2 and a corresponding turnover frequency of 11419 h−1. More recently, Hu et al. reported a so-called ‘synergistic near- and long-range regulation’ strategy with the help of a modified Zn SAC.174 They embedded ZnN4 sites in hollow carbon supports with co-decorated sulfur and phosphorus in the first coordination shell and surrounding carbon matrix, respectively (ZnN4S1/P-HC). According to theoretical calculations, the S atom present in the axial position significantly distorted the planar coordination structure of the Zn SA site, thereby increasing the charge density of the active center, which could be further elevated by the redistribution of electrons stemming from the introduction of P atoms in the second coordination layer. Such a remarkable change in electronic state provided varied interactions between Zn SA sites and specific reaction intermediates, especially *COOH. An enhanced adsorption strength of *COOH was observed due to the increased overlap of Zn (3d) and *COOH (2p) orbitals, thus promoting the formation of *COOH with a lower energy barrier, then leading to the production of CO.
 | ||
| Fig. 18 (a) Calculated Gibbs free energy diagram for CO2-to-CO conversion using graphitic Zn–N4, Zn–N3 and Zn–N3–V models at U = 0 V and pH = 0. (b) DFT calculated geometry of graphitic Zn–N4, Zn–N3 and Zn–N3–V sites with key intermediate COOH*-adsorbed species. (c) Comparison of the active site of electron-rich Zn–N3 with that of Zn–N4.169 Reproduced from ref. 169 with permission from Wiley, copyright 2021. | ||
The application of SACs in CO2 electroreduction can be further extended to cobalt.175–178 Pan et al. developed a robust Co-based SAC with an atomically dispersed Co–N5 site anchored on polymer-derived hollow N-doped porous carbon spheres for CO2 electroreduction to CO.176 The hollow structure of the SAC was fabricated by a combination of pyrolysis and etching procedures with SiO2 as the template. Meanwhile, the metal atoms were embedded by coordination with N. On the basis of various experiments and modeling calculations, Co–N5 sites were disclosed to be the main active center for CO2 activation, the formation of the key intermediate COOH* and the desorption of the CO product. As a result, a high FE of CO of above 90% could be achieved over a wide potential range from −0.57 to −0.88 V with remarkable stability. Lin et al. constructed a Co-based catalyst based on covalent organic frameworks (COFs) comprising cobalt porphyrins for the aqueous electrochemical reduction of CO2 to CO.177 XAS data revealed that the chemical environment offered by the COF plays an important role in the electronic structure of the catalytic cobalt centers, thereby contributing to the high activity of catalysts and FE to CO. Han et al. established a Co-based catalyst featuring atomically dispersed active centers for electrocatalytic CO2 reduction,178 which was synthesized via an axial coordination assembly using tetra(4-pyridyl) porphyrin cobalt(II) as building blocks, resulting in an ultra-thin morphology. The well-defined structure enabled the precise identification of the active sites for d-orbital energy engineering. The promoted activity of this catalyst for electrocatalytic CO2 reduction originated from the elevated energy level of dz2 in Co 3d orbitals, leading to a remarkable FE of 96% to CO at an overpotential of 500 mV. A similar strategy was also reported by Shen et al.,179 in which an organic molecule porphyrin was used as the ligand to coordinate the Co atoms, and a performance dependent on the pH value was exhibited when the SAC was used in electrocatalytic CO2 reduction.
Apart from the metals mentioned above, other kinds of transition metals have been also explored regarding the design of SACs and their application in electrochemical CO2 reduction to CO. For instance, a cadmium (Cd)-based SAC was prepared by a unique gas-migration, trapping, and emitting strategy for CO2 reduction to CO.180 A Cd loading amount as high as 30.3 wt% could be achieved through the gas-migration and trapping processes, while the emitting process could readily modulate it from 30.3 to 1.4 wt%. At an optimum loading amount of 18.4 wt%, the maximum FE of 91.4% for CO was obtained at −0.728 V, following the proton-decoupled electron-transfer mechanism. The Cd–N4 structure was revealed to be the main active site for the reduction of the Gibbs free energy barrier in the rate-determining hydrogenation step of the key intermediate *COOH. Manganese (Mn) as the third transition metal after iron and titanium among all transition elements in natural abundance, has also been applied in the construction of SACs for electrochemical CO2 reduction to CO.181,182 Through a halogen and nitrogen dual-coordination strategy, a Mn-based SAC was successfully fabricated with graphene as the support for effective CO2 reduction to CO.182 The dual coordination involving chlorine, bromine and iodine all achieved enhanced performance by modifying the electronic structure of active Mn sites, and thus the free energy barrier for the rate-determining step of the formation of the intermediate was reduced. In contrast, the enhanced effect on Fe and Co SACs with the same dual coordination was not as significant as that on Mn, indicating a strong correlation with the nature of metal. Wang et al. also applied a Mn-based SAC supported on a carbon substrate for CO2 electrochemical reduction to CO.181 The difference was that oxygen instead of halogens was co-doped with N into the SAC. Isolated Mn–N4 moieties were considered to be the main active sites for CO2 reduction, while the epoxy groups in the second coordination spheres adjusted the electronic structure of the catalyst with reduced reaction energy barriers. The catalytic performance was enhanced accordingly, achieving a high CO FE of 94.5% with a CO current density of 13.7 mA cm−2, as well as a low overpotential of 0.44 V in an aqueous environment. In addition, a silver-based SAC was also prepared by thermal transformation of silver (Ag) NPs over a non-carbon substrate MnO2, as Zhang et al. reported.183 With simultaneous MnO2 reconstruction from the MnO2 (211) to (310) lattice plane, the Ag NPs drove the formation of Ag SAs confined firmly on the surface. Compared with the Ag NPs, the Ag SAC exhibited a much lower formation energy of the *COOH intermediate at −0.7 V, as well as a higher electronic density close to the Fermi level, leading to strong interactions between Ag SA sites and the *COOH intermediate. This difference contributed to the excellent performance of the Ag SAC with a CO FE of 95.7%, in contrast to that of the Ag NPs of less than 60%. Moreover, SACs based on rare earth metals such as yttrium (Y) and scandium (Sc) have been synthesized successfully on a carbon support (Y1/NC and Sc1/NC) and exhibited excellent catalytic activities for carbon dioxide reduction.184 Compared with common SACs featuring atomic transition metals supported on N-doped carbon supports in a N-coordinated form (M–Nx), the reported Y1/NC and Sc1/NC contained metal atoms with a large radius, which tended to be anchored to the large-sized carbon defects through six coordination bonds of N and C. This coordination further modulated the local electronic structure of Y/Sc sites, and surprisingly endowed the generally inactive Y- and Sc-based nanomaterials with considerable activity for room-temperature electrochemical reaction, suggesting the distinctive effect of SACs on catalytic performance.
On the basis of the well-known d band center theory, transition metals with a narrow d band have been widely used as active components in various catalysis systems, whereas main group metals had always been thought to be nearly inactive due to a broadened single resonance derived from the interaction of valence orbitals of adsorbates with the delocalized s/p-bands.185–189 However, there have been quite a few studies concentrating on the catalytic application of main group metals over recent years,190 where SACs also distinguish themselves for electrochemical CO2 reduction. For instance, Wang et al. reported an s-block magnesium-based SAC with atomically dispersed magnesium atoms embedded in graphitized C3N4 (Mg–C3N4) for electroreduction of CO2 to CO.191 Since the formation of the *COOH intermediate is assumed to be the rate determining step of the whole reaction process with transition metals as active sites, the desorption of *CO is always ignored although it has a higher energy barrier according to some modeling calculations. Indeed, the strong hybridization between localized d orbitals and CO would lead to limited CO desorption and further reaction cycles. In this case, the Mg SAs with a delocalized s orbital would provide a solution by weakening the CO adsorption (Fig. 19a). As a result, Mg–C3N4 demonstrated a high TOF of 18000 h−1 at a a CO FE above 90%, while the flow cell fabricated with Mg–C3N4 offered a large current density of −300 mA cm−2. More recently, Wang et al. developed an O doping strategy to localize electrons on p orbitals of a calcium SAC with active sites in asymmetric coordination (Ca–N3O, one O and three N coordinated atoms with one Ca).192 The asymmetric coordination of Ca–N3O improves *COOH formation, thereby enhancing the CO2 activation, in addition to the intrinsic virtue of the ability of main group metals to inhibit the competitive HER and CO poisoning induced by strong adsorption of CO species, leading to an excellent TOF of ∼15000 per hour in an H-cell and a large current density of −400 mA cm−2 with a CO FE of no less than 90% in a flow cell. Moreover, Zhang et al. inventively fabricated main group element gallium-based (Ga) SACs with a nitrogen, phosphorous and sulfur atomic coordination environment for electrochemical CO2 reduction.193 The Ga-N3S-PC structure in this SAC that was constructed via a polymer pyrolysis strategy surprisingly displayed specific fluxional properties in contrast to a typical Ga–N4–C structure with a rigid skeleton. Theoretical simulations further revealed the adaptive dynamic transition of Ga derived from a flexible three-dimensional structure, optimized the adsorption energy of the *COOH intermediate and renewed the active sites on time, leading to a CO FE of ∼92% at −0.3 V vs. RHE (Fig. 19b). Moreover, indium (In) that is from the same group as gallium, has been also applied in SAC-catalyzed electrochemical CO2 reduction to CO by Guo et al.194 They synthesized an atomic In-anchored N-doped carbon catalyst through pyrolysis of In-bearing MOFs, in which the In species in oxidized state predominated and coordinated with N. This SAC exhibited higher double-layer capacitance, larger CO2 adsorption capacity, and lower interfacial charge transfer resistance thanks to the highly dispersed active sites, resulting in a FE of CO of up to 97.2% and total current density of 39.4 mA cm−2 as well as a turnover frequency (TOF) of ∼40000 h−1. The specific In–N sites also contributed to CO selectivity by suppressing the formation of formate owing to the changed energy barrier, as revealed by theoretical calculations (Fig. 19c–f). Bismuth (Bi) and antimony (Sb) from the same main group have been also used to construct SACs for electrochemical CO2 reduction to CO.195–197 Zhang et al. prepared a Bi–N4 dominated catalyst by thermal decomposition of a Bi-based metal–organic framework (Bi-MOF) and dicyandiamide (DCD).196In situ environmental transmission electron microscopy (ETEM) analysis offered an opportunity to directly observe the reduction of a Bi-MOF into Bi NPs and subsequent redispersion of NPs into atomic species with the help of NH3 released from DCD decomposition. The obtained Bi SAC exhibited much higher FE of CO (97%) than that over Bi NP or NC dominated catalysts (Fig. 19g), to which the Bi–N4 sites made great contributions by accelerating the formation of the key intermediate COOH* with a lower free energy barrier. Wang et al., on the other hand, developed an atomically dispersed N, S co-coordinated bismuth SAC (Bi-SAs-NS/C) via a cation and anion simultaneous diffusion strategy for electrocatalytic CO2 reduction.197 The bonded Bi cation and S anion could be synchronously diffused into the N-doped carbon layer as a Bi2S3 compound. Bi and S were captured by the abundant N-rich vacancies and carbons on the support, respectively, at high temperature, leading to the formation of co-coordinated Bi sites (Bi–N3S/C). The replacement of one coordinated-N with less electronegative S in the Bi–N4C center was proved to be highly effective in reducing the energy barrier of intermediate formation in the rate-limiting step and enhancing the reaction kinetics. Thus, a high FE of CO of over 88% in a wide potential range with a maximum value of 98.3% at −0.8 V vs. RHE and a current density of 10.24 mA cm−2 could be achieved. Recently, Sb has also been exploited for electrochemical CO2 reduction to CO, in which the single atom Sb sites exhibited superior performance compared with bulk Sb or Sb2O3, even surpassing other reported SACs.195 In addition, SACs based on tin (Sn) were reported as well for efficient electrochemical CO2 reduction.198 Guo et al.198 prepared Sn SACs comprising atomically dispersed SnN3O1 active sites supported on a N-rich carbon matrix (Sn–NOC). The atomic arrangement of SnN3O1 reduced the activation energy for *COO and *COOH formation and significantly increased the energy barrier for HCOO* formation. Sn–NOC gave CO as the exclusive product, whereas HCOOH and H2 were the predominant products for the classic Sn–N4 configuration (Fig. 19h).
 | ||
| Fig. 19 (a) Mechanism of CO production on Mg–C3N4.191 (b) Schematic diagram of the CO2RR catalytic process on the fluxional Ga-N3S-PC catalyst.193 (c) The catalytic pathway on InN4/C based on the COOH* and CO* adsorption intermediates after optimization.194 Gibbs free-energy diagrams of (d) CO2-to-CO and (e) CO2-to-formate over different simulated models.194 (f) Difference in limiting potentials for CO2 reduction to CO and formate over different simulated models.194 (g) Performance of the Bi SA compared with NCs and NPs.196 (h) The calculated Gibbs free energy diagrams for CO2-to-CO over SnN3O1 and SnN4.198 Reproduced from ref. 191, 193, 194, 196 and 198 with permission from Wiley and American Chemical Society, copyright 2019, 2021, 2022. | ||
Apart from these SACs with only single atom active sites mentioned above, there have been also a few reports on catalysts involving dual atom sites or synergy between single atom sites and other types of active sites. Since these catalysts are also inspired by the theories of SACs, we include them in the following discussion for more a comprehensive understanding. For instance, Ni dual-atom sites and Pd dual-atom sites have been constructed and reported for electrocatalytic CO2 reduction to CO.199,200 In the former case, Ni dual-atom sites were revealed to be key to induce the adsorption of hydroxyl and the formation of electron-rich active centers, providing a moderate reaction kinetic barrier of *COOH formation and *CO desorption.199 As for the latter case, Pd dual-atom sites showed advantages in CO production owing to the moderate adsorption strength of CO* derived from the electron transfer between Pd atoms.200 Hetero-diatomic catalysts such as Ni–Fe, Ni–Cu and Ni–Zn have been also applied in electrocatalytic CO2 reduction to CO.201–203 For Ni–Cu202 (Fig. 20g–k) and Ni–Zn201 (Fig. 20a–c), heteronuclear coordination was considered to modify the d-states of the metal atoms, narrowing the gap between the d-band center of the Ni orbitals and the Fermi energy level to strengthen the electronic interaction at the reaction interface, resulting in a lower free energy barrier and a reduced activation energy for the formation of the key intermediate. The excellent performance of Ni–Fe,203 on the other hand, was attributed to the orbital coupling between the catalytic iron center and the adjacent nickel atom, resulting in an alteration in the orbital energy level, unique electronic states and a higher oxidation state of iron (Fig. 20d–f). The binding strength to *CO was weakened over Ni–Fe compared with Fe-SAC, playing a vital role in the enhanced efficiency for CO production. Furthermore, catalysts featuring the synergy between SAs and other active species have been studied for the electrocatalytic CO2 reduction to CO. Wang et al. applied a novel solid-state atomic replacement transformation strategy for constructing a catalyst containing both PtZn nanocrystals and Ni SA sites.204 Through the exchange between the Ni in the PtNi nanoalloy and the Zn in ZIF-8-derived Zn1 on nitrogen-doped carbon (Zn1–CN) at high temperature, (PtZn)n/Ni1–CN was obtained. This fabricated catalyst showed a synergistic effect combining PtZn nanocrystals and Ni SA sites on the CO2 reduction reaction with a lower energy barrier for CO2 protonation and CO desorption. The application of this unique strategy could be extended to other metal alloys such as PtPd. Li et al. tuned the CO selectivity of Cu by alloying a second metal Sb into Cu, forming an antimony-copper single-atom alloy catalyst (Sb1Cu) with isolated Sb–Cu interfaces.205 The atomic Sb–Cu interface promoted CO2 adsorption/activation and weakened the binding strength of CO*, suppressing the facile C–C coupling reactions over conventional Cu crystals, leading to enhanced CO selectivity with a FE of over 95%.
 | ||
| Fig. 20 (a) The free energy of each intermediate state on the metal atom sites in Zn–N4–C, Ni–N4–C, and ZnNi–N6–C. Projected DOS (PDOS) for Zn/Ni 3d orbitals and oxide 2p orbitals including COOH (b) and CO (c).201 Schematic illustration of orbital interactions between adsorbed CO (5σ and 2π*) and the 3d orbital (dz2, dxz/dyz) of the Fe site in the (d) Fe-SAC and (e) NiFe-DASC. (f) Calculated free energy diagrams for CO2RR and OER processes.203 (g) The catalytic pathway on NiN3–CuN3 based on the *COOH and *CO adsorption intermediates after optimization. (h) The density of states of Ni d-orbitals in NiN3–CuN3 and NiN3. (i–k) Gibbs free energy diagrams of CO2 conversion to CO over NiN3–CuN3 and NiN3 with free energy correction at pH = 3, 7, and 14.202 Reproduced from ref. 201–203 with permission from Wiley and Springer Nature, copyright 2021, 2023. | ||
3.1.2.2 Formic acid and formate. To synthesize formic acid and formate via an electrochemical route is another appealing subject in CO2 reduction due to the versatility of formic acid or formate in chemical production and energy industries. The process of CO2 conversion to HCOOH features two-electron transfer, identical to that of CO2 conversion to CO, with a slight lower reduction potential of −0.12 V versus RHE (Fig. 5). This indicates that the production of HCOOH might be another fundamental process involving the most preliminary procedures in CO2 conversion. However, practical application of CO2 reduction is still limited by the sluggish kinetics and significant side reactions such as the HER.190 Owing to the challenges in both increasing the reaction rate and steering reaction selectivity, it is urgent to develop catalysts with high efficiency and reliability. In addition to modifying the conventional nanostructure catalysts, SACs have been exemplified as an affordable solution to the above problems. For instance, Shang et al. designed an In SAC with Inδ+–N4 atomic interface active sites for electroreduction of CO2 to formate to overcome the low FE and poor durability over common In-based catalysts.206 Compared with the reference sample with dominant NPs, the SAC exhibited a lower free energy barrier for the formation of the HCOO* intermediate leading to higher activity, while the superior selectivity of formate could be rationalized based on the more positive difference between limiting potentials for CO2 reduction and H2 evolution. This strategy could further be extended to other main group elements, such as Sb.207 The positively charged Sbδ+–N4 (0 < δ < 3) active sites could endow the Sb SAC with a formate FE of 94.0% at −0.8 V versus RHE in a similar way as Inδ+–N4 did, transcending the performance over the catalyst with NPs. On the other hand, Xie and co-workers fabricated Mo and Sn SACs with N-doped graphene as a support, both of which demonstrated excellent performance for formate production.208,209 The reduction of CO2 to formate could not only benefit from the Mo SAs anchored on the N-doped graphene, which provided the substrate with a higher current density at relatively lower overpotential,208 but could also be favored by the positively charged Sn SA sites, which promoted CO2 activation and protonation via stabilizing CO2−* and HCOO−*.209 The N doping in graphene also facilitated the reaction by expediting the desorption of formate products via lowering the desorption energy and increasing the length of the bond between the metal and HCOO−* intermediate.209 Notably, to gain an in-depth understanding of CO2 electrochemical reduction reactions, Deng et al. developed a series of model Sn SACs with well-defined structures and employed a combination of operando characterization techniques for monitoring the variation in both SAC structures and surface key intermediate species.210 Particularly, 119Sn Mössbauer spectra were collected for more accurate analysis of chemical state evolution of the Sn moiety due to the specialty of Mössbauer with regard to the Sn element. Surprisingly, Sn(IV) was exclusively formed in the case where SAs were immobilized on a functionalized carbon nanotube (CNT–OH), while Sn(II) of different concentrations coexisted in other reference samples. Assisted by DFT simulations, the authors proposed a reasonable SA structure featuring Sn(IV)–N4 moieties axially coordinated with oxygen (O–Sn–N4) and correlated it with CO2 reduction performance. They further confirmed that surface-bound bidentate Sn carbonate species were formed from O–Sn–N4 sites in an initial stage and changed the electronic distribution for favorable formation and protonation of *OCHO species, thereby propelling the CO2 conversion into HCOOH (Fig. 21).
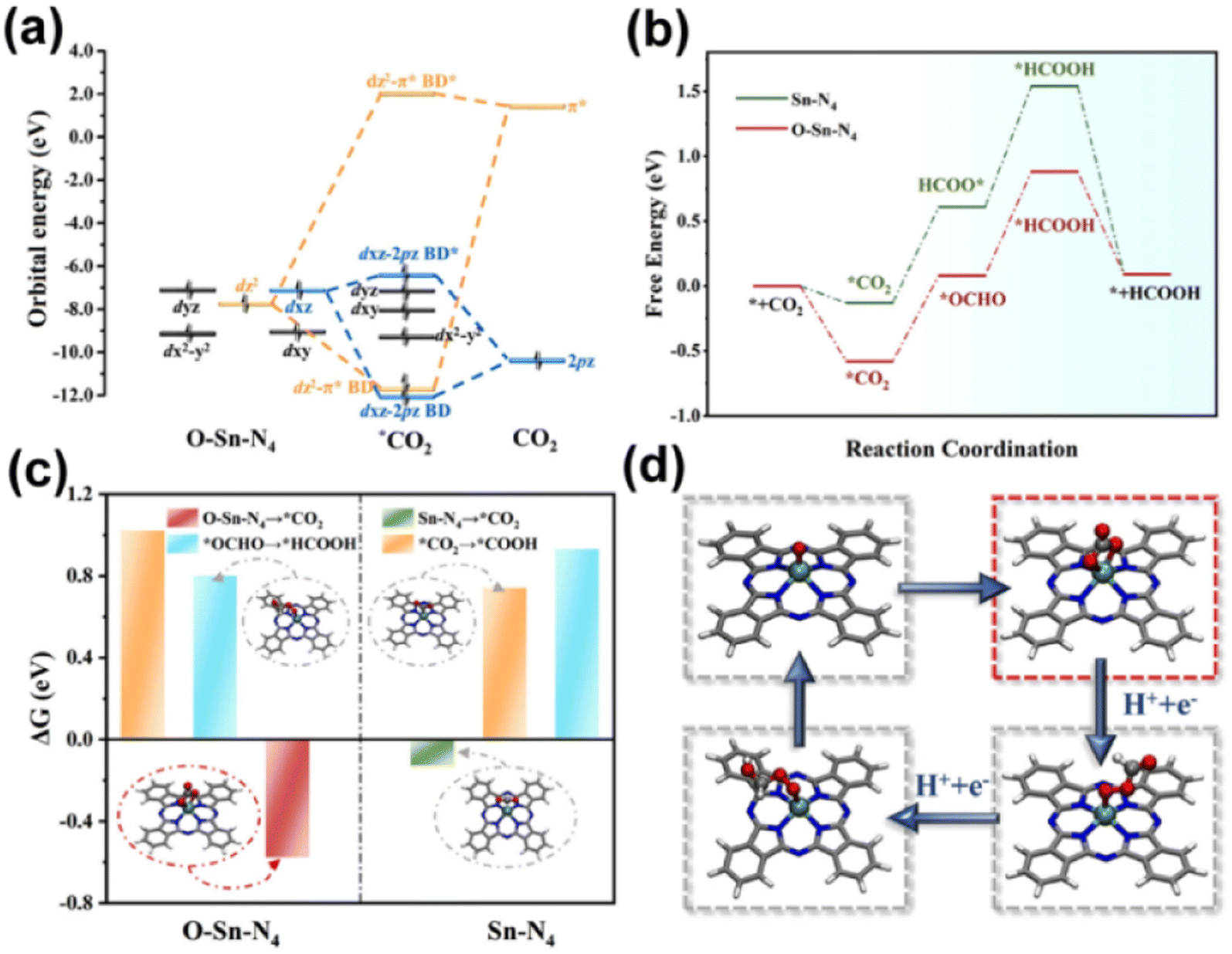 | ||
| Fig. 21 (a) Schematic Kohn–Sham molecular orbital (MO) energy-level correlation diagram between CO2 and the O–Sn–N4 site during the CO2 capture step (* + CO2 → *CO2). (b) Calculated Gibbs free energy diagrams for CO2 reduction to HCOOH over O–Sn–N4 and Sn–N4 sites. (c) Calculated ΔG for HCOOH and CO production over O–Sn–N4 and Sn–N4 sites. (d) Proposed reaction pathway for CO2 reduction over the O–Sn–N4 site.210 Reproduced from ref. 210 with permission from American Chemical Society, copyright 2023. | ||
Moreover, given that the applications of Cu-based catalysts are always limited by their poor selectivity towards specific products, Zheng et al. found an opportunity to inhibit the side reactions with the assistance of lead (Pb) SAs.211 Although the identification of true active sites was still ambiguous, it was plausible that exclusive formate production was promoted by the Cu sites of Pb1Cu via guiding the first protonation step towards a HCOO* path instead of a COOH* path. Such a promotion effect was also reported over a SnO2-based catalyst, in which the abundant oxygen vacancies (Ov) were triggered by the doping of Cu/Bi/Pt single atoms. These Ov held the key to stabilizing the oxidation state of Sn species and promoting the adsorption of the *OCHO reaction intermediate, resulting in a high formate FE of >80% and a cell energy efficiency of about 50–60% in a wide range of current densities up to 500 mA cm−2 in a commercial flow cell.212
3.1.2.3 Methane. CH4 is also an important light hydrocarbon resource, whose production from electrochemical CO2 reduction is attracting worldwide attention in both scientific research and practical industry. Nevertheless, the process of CO2 reduction to CH4 involves reaction paths of transferring as much as eight electrons and eight protons, indicating a much more complicated pathway with more possible parallel reactions in comparison with that of CO and HCOOH formation. Indeed, comprehensive understanding of the reaction pathways toward CH4 is still equivocal and highly dependent on the different catalytic systems. Copper (Cu) is known for its good performance in multi-electron transfer. SACs based on Cu have been designed for CO2 electrochemical reduction to CH4 correspondingly, whereas reasonable regulation of active sites is indispensable for remarkable efficiency. By anchoring Cu SAs on the unique platform graphdiyne, Shi et al. achieved a considerable CH4 FE of 81% via CO2 reduction.213 The authors believed that the platform graphdiyne not only stabilized Cu SAs against aggregation, but also favored the formation of Cu–C bonds, which was confirmed to play a vital role in the generation of the *OCHO intermediate rather than *COOH (Fig. 22). This shift of intermediate significantly inhibited the formation of CO and other side products by efficiently modulating the reaction pathway, leading to the preferential production of CH4. Further tailoring the structure of normal graphdiyne with specific electron-withdrawing/-donating groups, on the other hand, offered another good opportunity for the regulation of Cu SA sites.214 It was found that subtly introduced functional groups –F (fluorine) had a great effect on decreasing the pKa of the adsorbed water molecule, then forming a more acidic local environment that promoted the protonation of CO2 reduction intermediates for CH4 production. In contrast, other groups such as –H and –OMe (methoxy) exhibited inferior performance. This difference arose from the discrepancy of the Cu valence in various chemical environments, while a more positive charge state stemming from the electron-withdrawing effect provided an excellent CH4 FE of 72% from CO2 reduction. Furthermore, for the cases in the absence of carbon supports, other strategies have also been proposed to modify the Cu SA sites.215,216 Loading Cu SAs onto the metal oxide substrates with abundant Lewis acid sites was considered as an effective strategy to improve the activity of CO2 reduction to CH4.216 It was speculated that strong Lewis acid sites in the form of a metal center with positive charge could facilitate the activation of CO2 into *COOH and HCOO* intermediates with a lower energy barrier, while the selectivity was determined by using the formation energy in different pathways. This rationalized the superior performance of the Cu/Al2O3 SAC with substantial strong Lewis acid sites compared to that of the Cu/Cr2O3 SAC with weak acid sites. The effect of Ov was also highlighted in a Cu/CeO2 SAC for CO2 reduction to CH4,215 in which Cu SAs in the most stable structure with a low coordination number of 5 were well dispersed on CeO2 nanorods. With the assistance of Ov, the adsorption and activation of carbon dioxide molecules were both enhanced. An inhibited C–C coupling was also achieved, thanks to the isolated state of active Cu SA sites, leading to enhanced formation of CH4.
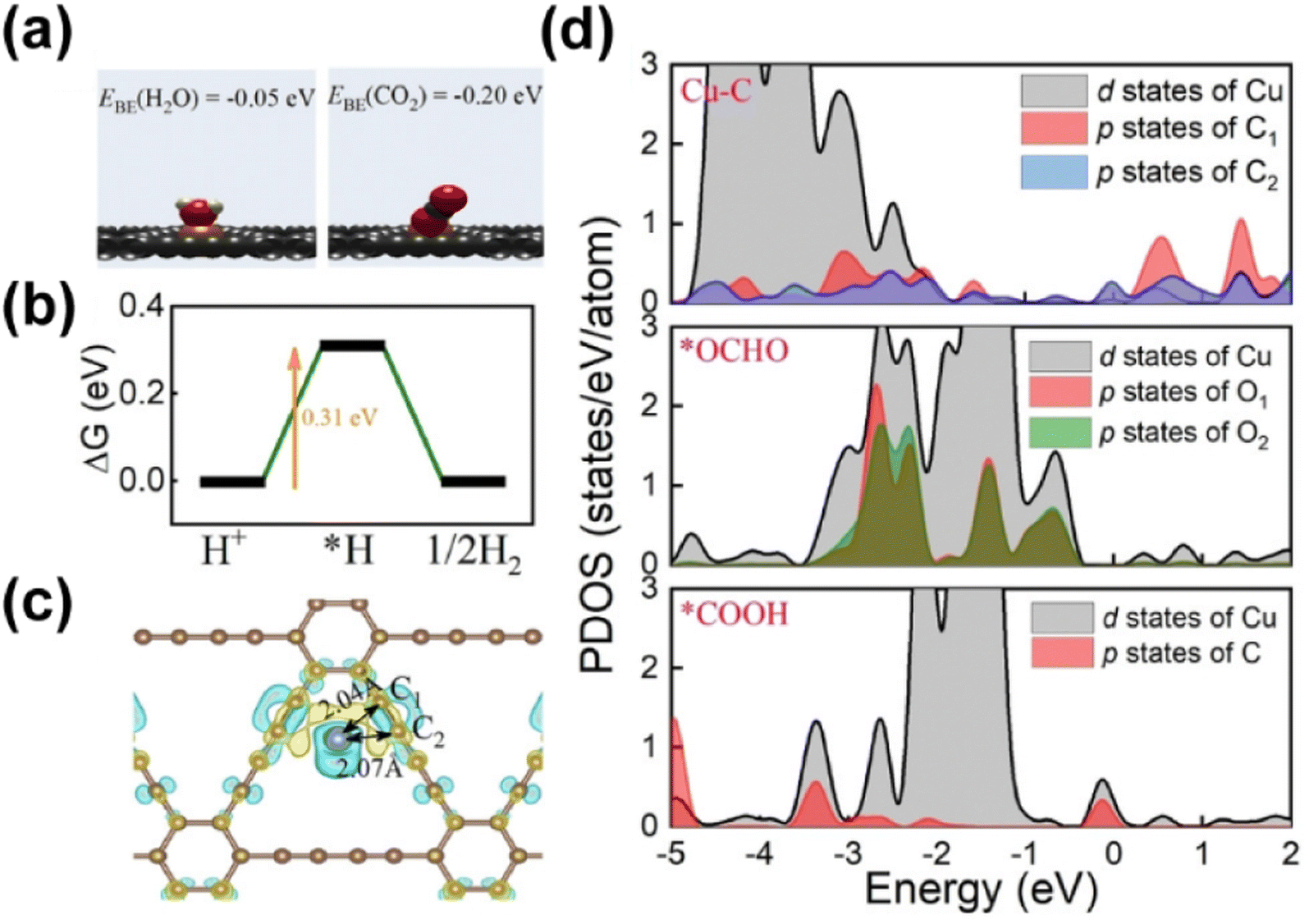 | ||
| Fig. 22 (a) The binding energy of CO2 and H2O on Cu SAs/GDY. Insers show the calculation models of adsorbed CO2 and H2O, respectively. (b) Free energy diagram for the HER. (c) The charge density difference (CDD) before and after the bonding of the Cu atom and GDY. (d) Projected density of states (PDOS) of the interface Cu–C atoms in Cu SAs/GDY, and the intermediates *OCHO and *COOH.213 Reproduced from ref. 213 with permission from Wiley, copyright 2022. | ||
Metals other than Cu have been also studied for CO2 electrochemical reduction to CH4. For instance, Han et al. developed a SAC with Zn SAs dispersed on microporous N-doped carbon.217 Zn–N4 was identified as the dominant active sites which preferred to bond to the O atoms of intermediates rather than C atoms. As a result, *OCHO was formed during the reduction process, suppressing the generation of CO and enhancing the production of CH4 accordingly. Ren et al. constructed a series of SACs with Fe SAs coordinated with boron (B) and C for promoting multi-electron reduction to CH4.218 By extensive theoretical calculations, the authors found that the introduction of B changed the adsorption strength of reaction intermediates by forming various FeBxCy structures, among which FeB2C exhibited the best performance. The adsorption energy of the O atom played a crucial role in the regulated pathway with the weakened binding strength of *OCHO and enhanced binding strength of *HCOOH on the surface, which reduced the energy barrier of *OCHO to *HCOOH and accelerated the reaction kinetics. This change in adsorption energy of the O atom was confirmed to be associated with the more negative d-band center and the optimal Fe atomic magnetic moment of FeB2C.
In addition, theoretical calculations further suggested some distinctive SACs that might take advantages in CO2 reduction to CH4.219 By using DFT calculations, Back et al.219 investigated the catalytic properties of TiC, TiN, and the corresponding single-atom catalysts for CO2 electrochemical reduction. Among various SACs with single transition-metal atoms inserted into the surface defect sites of TiC, iridium-doped TiC (Ir@d-TiC) was found to exhibit a remarkably low overpotential of −0.09 V, which is the lowest value among that of any catalyst reported in the literature used to selectively produce CH4 (−0.3–−1.0 V). This superior performance originated from a lacked sigma-type bonding interaction between *CO and Ir SA as compared to the cases of bare TiC or Ir(111), resulting in weakened *CO binding and the corresponding easier reduction in the limiting potential.
3.1.2.4 Methanol. Due to the wide applications of methanol in both chemical and energy industries, direct synthesis of methanol from electrochemical CO2 reduction has raised considerable concern, although it is still a great challenge to achieve such a process encompassing six-electron and six-proton transfer. Given that Cu is still the only metal that exhibits appreciable efficiency in CO2 reduction involving multiple electron transfer currently,158,220 pioneering studies on SACs for methanol production have been focused on retrofitting Cu-based catalysts. Yang et al. developed an effective strategy for massive production of SACs with Cu SAs decorated through-hole carbon nanofibers, denoted as Cu SAs/TCNFs, for utilization as a cathode for CO2 reduction.221 Benefitting from the specific structure of the carbon membrane and highly dispersed Cu–N4 sites, more active Cu SAs could effectively participate in the reaction. As such, the reduction process was promoted dynamically, resulting in a high current density of up to −93 mA cm−2. As for the selectivity, DFT calculations indicated that Cu–N4 sites possessed a relatively higher binding energy for the *CO intermediate, so that further reduction into methanol could become feasible rather than forming a gaseous CO product. As a result, nearly pure methanol in the liquid phase with 44% FE could be achieved over this Cu SAC. Inspired by the observation of the clearly different selectivities of methanol produced from CO2 reduction in aqueous and non-aqueous solutions, Kong et al. designed a cuprous cyanamide (Cu2NCN) crystal featuring isolated Cu(I) ions strongly conjugated with NCN2− (Fig. 23).222 They compared the dissociation enthalpy of O–C and Cu–O over typical Cu/Cu2O sites and Cu2NCN sites, respectively. It was found that ΔHCu–O > ΔHC–O in the former sites which propelled the formation of CH4 without a C–O bond, whereas ΔHCu–O < ΔHC–O in the latter case, offering a chance to retain the C–O bond for methanol formation. They rationalized this conclusion by using a modern hard–soft acid–base (HSAB) theory, where weakened bonding was always formed between softer acidic sites (delocalized sites) and hard bases such as *O and *OR (R = H or alkyl). By introducing suitable delocalized electron states into isolated ionic Cu species, their soft acidic nature might be enhanced with reduced Cu–O bond strength. Based on this strategy, the formation of methane was suppressed and 70% FE towards methanol was reached with a methanol partial current density of −92.3 mA cm−2 and a production rate of 0.160 μmol s−1 cm−2 in MEA-based cells. Furthermore, based on the investigations into the binding energy of *CO, Wu et al. developed a non-copper SAC with cobalt phthalocyanine immobilized on carbon nanotubes (CoPc/CNT), which could catalyze the CO2 reduction to methanol via a so-called domino process via CO as the intermediate.223 They found that Co–N4 possessed a similar binding energy to that of Cu, compared with too strong binding over Fe–N4 and too weak binding over Ni–N4. Intuitively, Co–N4 with a moderate binding energy mimicking Cu was expected to achieve a deep reduction just like that in the case of Cu. Indeed, individual CoPc dispersed by CNTs exhibited an appreciable methanol FE of up to 40%.
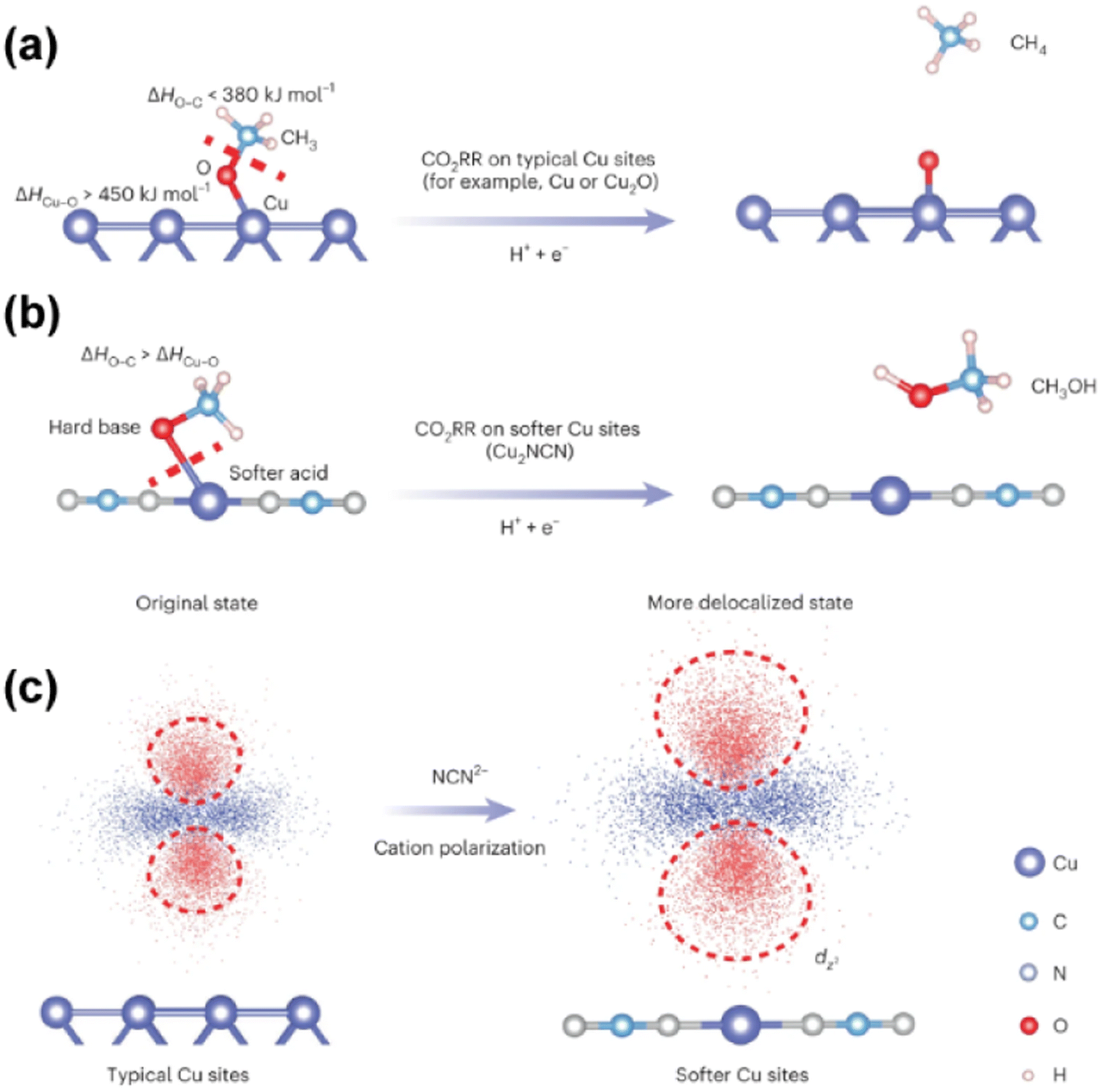 | ||
| Fig. 23 (a) Typical Cu catalytic sites (for example, Cu or Cu2O) display a relatively strong Cu–O bond (that is, the bond dissociation enthalpy, ΔHCu–O > 450 kJ mol−1) compared with the O–C bond (ΔHO–C in OCH3 is <380 kJ mol−1), thus leading to cleavage of the O–C bond to release *CH3 and then form CH4. (b) ΔHCu–O can be tuned to a lower value than ΔHO–C at a softer acid site (for example, Cu2NCN), enhancing the CO2-to-CH3OH selectivity. (c) Schematic illustration of the delocalized d-electron cloud induced by NCN2− in Cu2NCN.222 Reproduced from ref. 222 with permission from Wiley, copyright 2022. | ||
3.1.2.5 Summary. The direct electric energy input endows CO2 reduction with feasible operation under mild conditions. The application of SACs in CO2 electrochemical reduction not only pushes forward the practical utilization of noble metals, but also renders non-noble metals a suitable choice for preferential synthesis of specific products. Through modifying the chemical microenvironment of SACs via regulating parameters such as coordination (with heteroatoms like N, S, and O) and spatial distance (with other SA sites), appreciable performance regarding production of C1 chemicals is demonstrated. Notably, the common well-defined active sites of SACs make theoretical calculations always take advantage in the investigation of the structure and mechanisms, as the theoretical calculations would in turn contribute to the reasonable prediction and good design of advanced SACs.224
C1 products such as CO, HCOOH, CH3OH and CH4 are versatile platform chemicals in industry, whose production from photochemical CO2 reduction is expected to confront the current excessive emission issues. Given the fairly long history of studies on conventional non-carbon-based semiconductors, researchers have achieved fruitful results on enhancing the selective synthesis of C1 products by establishing SACs on the basis of previous studies. For instance, by adsorbing isolated Bi ions onto the surface of TiO2 2D nanosheets, electron transfer from Bi ions to the TiO2 substrate was presented by analyzing the density of states and corresponding charge density difference.230 Such a transfer induced a built-in electric field from the surface of TiO2 to the interior, and propelled the separation of electrons from photoexcited charge carriers and subsequent migration towards the surficial reaction region, thereby facilitating the reduction of CO2 to CH4 with an evolution rate of 4.1 μmol g−1 h−1, 5.6 times larger than that for blank TiO2. Embedding Cu SAs into anatase TiO2, on the other hand, took effect in a distinct way, which results in CH4 production with a 66-fold enhancement with respect to that of pristine TiO2.231 The introduction of Cu SAs first induced the formation of Ov by fortifying the surface reducibility of TiO2 with an exposed mid-gap O state above the Fermi level in Cu1/TiO2. The combination of Cu SAs and vacancies contributed to the stabilization of intermediates on the TiO2 surface. In contrast to Cu2/TiO2 with neighboring Cu atoms rather than the isolated state, in which electron localization was disturbed, Cu1/TiO2 with electron localization to the active Cu centers possessed strong Lewis basicity. This could further promote the electron transfer to the oxygen p state of CO2, breaking the symmetry of CO2 into two p states of each terminal oxygen atom and bending the C–O–C bond angle, favoring the chemisorption of CO2 molecules (Fig. 24). The effect of the Cu valence state might also play an important role in the performance of Cu/TiO2 SACs and has drawn attention in some other reports. Jiang et al. thought that Cu0 served as the active sites for CO2 reduction to CO over the Cu/TiO2 SAC, which could be formed through reduction of Cu2+ by photogenerated CB electrons of TiO2 under solar irradiation.232 However, Cu0 active sites suffered from gradual oxidation into Cuq+ by the generated O2, providing the Cu/TiO2 SAC with slowly decreasing activity, since Cuq+ was difficult to be reduced under the reaction conditions thermodynamically. In this case, the activity could be restored by opening the reactor for exposure to air or oxygen leading to complete oxidation to Cu2+. Then, the reactor should be resealed for the reduction of Cu2+ to Cu0 and the next reaction cycle. A similar dynamic evolution of Cu species over a Cu SAC with atomically dispersed Cu on mesoporous TiO2 was also observed by an in situ XAS technique.233 The difference was that the Cu0/Cu+ mixture was proposed to be more efficient for CH4 production based on a synergy with the meso-structure and enhanced charge carrier transfer. Further introducing other types of active components into the Cu/TiO2 SAC provided opportunities for boosted CO2 reduction. By using such a strategy, Yu et al. constructed an excellent photocatalyst where Cu SAs and Au–Cu alloy NPs coexisted on TiO2, achieving a record-high formation rate of 3578.9 μmol g−1 h−1 for CH4 and appreciable production of C2H4.234 It was found that there was a synergistic effect between Cu SAs and Au–Cu alloy NPs. Such an effect could enhance the adsorption and activation of CO2 and H2O and lower the overall activation energy barrier, particularly that of the rate-determining steps for CH4 and C2H4 formation.
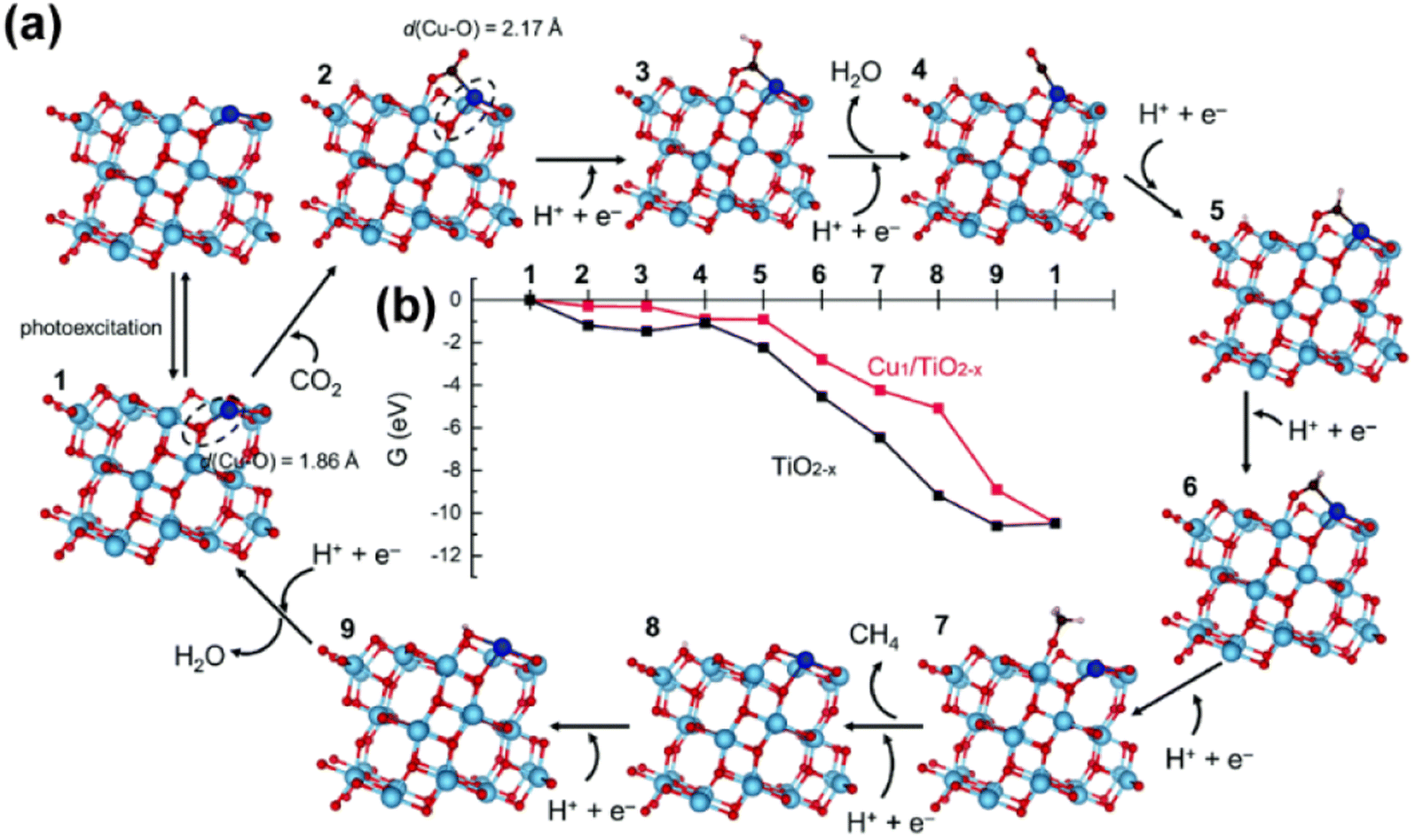 | ||
| Fig. 24 Photocatalytic CO2 reduction at Cu1/TiO2 with an oxygen vacancy. (a) CO2 reduction mechanism at Cu1/TiO2−x and (b) DFT energetics of CO2 reduction at Cu1/TiO2−x and TiO2−x. During the reduction process, the photoexcited electron is assumed to be transferred from the conduction band minimum of TiO2 (−0.25 V versus RHE).231 Reproduced from ref. 231 with permission from Royal Society of Chemistry, copyright 2022. | ||
Apart from TiO2, SACs based on other non-carbon-based substrates have been also extensively reported. One of them was based on a combination of Ni SAs and defect-rich zirconia.235 The Ni SAs were considered to have an effect on the kinetics of CO2 conversion to CO by lowering the energy barrier in a *COOH intermediated pathway. Besides, the H2 desorption as a competitive reaction consuming photo-generated electrons was also suppressed to a certain extent with the assistance of Ni SAs. Ding et al. compared the performances of a photocatalyst with Ag SAs supported on manganese oxide (Ag-HMO) and its counterpart with Ag NPs (Ag/HMO) for photocatalytic CO2 reduction.236 As a result, the Ag SAC exhibited a higher activity of CH4 production of up to 0.61 mol mol−1, which was 1.53 times as high as that of Ag NPs. This difference was rationalized by the promoted electron transfer from Ag to HMO, adsorption of visible light and the activation of CO2 over the SAC. A similar contrast was presented over the Pt–V2O5 SAC derived from vanadium-based polyoxometalates and Pt NPs supported on V2O5.237 Separated groups containing Pt–V and V in the precursor were converted into mono-dispersed Pt centers and surrounding vanadium oxide, respectively. The Pt SA sites were in the positive oxidation state, and tetra-coordination geometry assisted by four surface oxygen atoms of V2O5 favored the formation of the *COOH intermediate via a proton-coupled electron transfer process, followed by hydrogenation into *CO. The Pt SA site further stabilized *CO for its deep hydrogenation into CH4. The distinctive properties of Pt–O and tremendously enhanced atom utilization contributed to excellent activity that was 25 times higher than that of Pt-NP-V2O5. In some cases, significant dependence of activity on the distinct vacancies was shown on SACs with the same metal SAs. This correlation was unveiled over a CdS supported Au SAC by selectively creating Cd or S vacancies before anchoring Au atoms (Fig. 25).238 As for S vacancy dominated CdS, electrons tended to be localized at Au SA sites based on DFT calculations. In contrast, electrons were more likely to accumulate at the vacancy sites over CdS with abundant Cd vacancies. This difference in electron spatial arrangement made the adsorption type of CO2 change from physical adsorption to chemical adsorption. In contrast to catalysts without Au or with NCs, the strong hybridization of Au 5d and S 2p orbitals for the SA sites endowed CdS with superior efficiency of photo-electron transfer on the surface and a corresponding lower energy barrier for the formation of key intermediates during CO2 reduction. More recently, a silicon-based Co SAC was reported for the production of syngas with a highly tunable H/C ratio.239 A solid-state epitaxy method was used for the construction of such a SAC with Co SAs well dispersed on crystalline silicon, through a multistep variable-temperature annealing including the preliminary incorporation of Co atoms in the Si lattice and the following decomposition of the CoSi2 domain. With the addition of a particular photosensitizer and sacrificial agent, CO and H2 yields of 4.7 mol g(Co)−1 and 4.4 mol g(Co)−1 were achieved through visible-light-driven CO2 reduction, in which the H/C ratio could be tuned from 0.8 to 2 by modulating the Co loading amount.
 | ||
| Fig. 25 Schematic illustration of single Au atoms in CdS to promote CO2 photoreduction.238 Reproduced from ref. 238 with permission from Springer Nature, copyright 2021. | ||
Owing to their distinctive electronic energy band structures and fairly controllable physical properties such as porous structure and electrical and thermal conductivity, carbon-based semiconductors including various pure or heteroatomic carbon materials have been also widely studied and applied in photocatalysis.51,240,241 More importantly, carbon materials are considered as a renewable resource that is promising for sustainable utilization. Thus, there have been quite a few reports on SACs with carbon-based semiconductors as substrates. Graphitic carbon nitride (g-C3N4), as one of the most advanced carbon-based materials with excellent photochemical properties,242 has exhibited superior performance in CO2 reduction in collaboration with various metal SAs.243–256 Generally, SACs based on noble metals such as Pd, Pt, Au, and Ru played a crucial role in two aspects: the first is functioning as active sites for CO2 adsorption and activation as well as the stabilization of various intermediates; the second is manipulating the band structure of g-C3N4 for enhanced visible-light absorption (Fig. 26a–d).250–252 The former feature also offered chances for the modulation of the product selectivity by controlling the kinetic pathways involving the formation and transformation of key intermediates with varied free energy barriers, e.g., the dominant product was shifted from HCOOH over Pd SAs to CH4 over Pt SAs,252 while a higher yield of methanol was exhibited over Ru SAs.250 The latter feature, on the other hand, could afford higher efficiency of energy utilization in a practical process. In addition, non-noble metals such as Co, Ni, and Cu have also demonstrated outstanding performance in the SA state on a g-C3N4 substrate.245,253–255 In the above cases, the effect of N coordination in charge of the stabilization of SAs and local electron density of metal sites was mentioned more than one time. This effect was mainly derived from the interaction between the lone-pair electrons in the adjacent N atoms and d orbitals of metal SAs, imposing a positive oxidation state on SA sites in g-C3N4 that was slightly lower than that in oxides.252 This was an intuitive result according to the differences in electronegativity of N and O. SAs in the lower positive oxidation state were considered to be in favor of the adsorption and activation of reactants in some cases. However, in a niobium (Nb) SAC, carbon atoms were believed to coordinate with metal SAs exclusively in g-C3N4 with abundant N vacancies (Fig. 26e–h).243 In this case, polarized Nb–C bonds provided a high-speed channel for the directional transfer of photogenerated electrons and accordingly restrained the recombination, while the N vacancies were more likely to be responsible for the enhanced formation of charge carriers. Recently, it has been reported that SACs, particularly those that were based on rare earth metals such as lanthanum (La), dysprosium (Dy), and erbium (Er), could serve as a so-called electron bridge to assist charge transfer especially in heterojunction photocatalysts with a Z-scheme structure.244,247,248,257 To clarify, the Z-scheme structure was proposed for describing the photosynthesis process in natural bionts initially, and the concept was introduced into artificial photocatalysis systems for establishing excellent heterojunction photocatalysts with both enhanced electron transfer and sufficient redox potential for surface reactions.258–260 As a heterojunction was constructed to solve the dilemma of stronger light absorption (need a narrow gap between the CB and VB) or higher potential (need a wide gap between the CB and VB), a heterojunction with a Z-scheme structure featured a staggered band configuration with a medium for electron transfer from the lower CB in the oxidation semiconductor to the higher VB in the reduction semiconductor. In this way, both enhanced charge-separation efficiency and retained redox ability can be simultaneously achieved. Coincidentally, SAs of some rare earth metals with 4f levels between the VB of a certain semiconductor and CB of g-C3N4 were supposed to act as such a medium, as reported in recent studies.244,247,257 Specific rare earth metal SAs could also function as key active centers for CO2 activation, intermediate formation and products, e.g., CO desorption.248 In contrast, other transition metal SAs, like cobalt, were also claimed to have an effect on the prolonged lifetime besides engaging in the reduction reaction over Z-scheme heterojunctions.261
 | ||
| Fig. 26 (a) Optimized structure and plots of 3D differential charge densities, (b) band structures, and (c) optical absorption spectra of g-C3N4, Pd/g-C3N4, and Pt g−1-C3N4.252 (d) Possible schematic representation of the photocatalytic mechanism over the RuSA-mC3N4 surface under visible light irradiation.250 The WT-EXAFS plots of Nb-foil (e), CNNb0.06 (f), and Nb2O5 (g), respectively; free energy (h) of various possible modes of single-atom Nb using DFT calculations.243 Reproduced from ref. 240, 243, 250 and 252 with permission from American Chemical Society, Wiley and Elsevier, copyright 2016, 2020, 2023. | ||
The SAs could further be introduced into other functional carbon-based materials, such as N-doped CNTs,262 N-doped oxidized graphene,263 and N-doped carbon.264,265 Different supports provided not only distinct porous structures for mass transfer which imposed an enrichment or evacuation effect, but also specific chemical environments for the stabilization of SAs that were also endowed with regulated electronic properties. Notably, in some cases, N-doped carbon supports did not possess the attributes of a semiconductor with insufficient capability to harvest light; photosensitive reagents such as [Ru(bpy)3]Cl2·6H2O (bpy = 2,2′-bipyridine) should be added into reactors for the generation of photo-excited electrons, and a sacrificial hole scavenger such as triethanolamine (TEOA) should be also involved in the systems for the removal of holes. This suggested that carbon supports in contact with both SAs and photosensitive reagents also functioned as a bridge for electron transfer.263–265
Apart from widely used semiconductors, reticular materials represented by metal organic frameworks (MOFs) and covalent organic frameworks (COFs) featuring high specific area and an abundant porous structure, have been also exploited as photocatalysts for CO2 reduction recently.226,266–270 Thanks to the tunable structural and chemical properties, MOFs and COFs are also ideal choices for bearing metal SAs to further modify the catalytic performance. Resembling the circumstances in N-doped carbon substrates, most MOF and COF materials need auxiliary reagents to assist in light harvesting and hole scavenging as well. SAs embedded in MOFs and COFs are more likely to serve as active sites for CO2 activation, and thereby the reticular materials show advantages as they are endowed with a large active interface.266 Besides these general scenarios, some other factors might be equally important for optimizing the performance. Zhong et al. fabricated a 2,2′-bipyridine-based COF bearing single Ni sites (Ni-TpBpy) for exclusive selective production of CO from CO2 photoreduction.267 Under visible light irradiation, Ru(bpy)32+ was excited and transferred electrons for the subsequent reduction of coordinated CO2 molecules on Ni-TpBpy. H2 evolution could be well inhibited due to the higher affinity of CO2 species than that of H+. As CO2 was activated by Ni SAs, verified by the bent configuration of coordinated CO2 molecules, the significantly decreased free energy of the COF–Ni–CO2H intermediate could be ascribed to the formation of hydrogen bonds between COOH and the keto group of the TpBpy moiety (Fig. 27a). Thus, TpBpy not only functioned as a host for CO2 molecules and Ni SAs but also played a vital role in the enhanced activity and selectivity of the reduction of CO2 to CO. Given that the activation of reactants and transformation of intermediates always involve complex variations in chemical bonding between C–O, C–H and C–C during the whole process of CO2 reduction, the capability to adapt such variations might be the key to stabilize the intermediates for a lower energy barrier in a specific reaction pathway. Hence, inspired by the mechanisms of enzymes in organisms, Li et al. demonstrated a photocatalyst with flexible Cu–Ni dual-metal-site pairs (DMSPs) featuring dynamic self-adaptive behavior to match mutative C1 intermediates for CO2 photoreduction to CH4.268 The DMSPs were incorporated into a MOF to afford their respective SA forms in a flexible microenvironment. Based on DFT calculations, it was proposed that the distance between Cu and Ni SA pairs (dCu–Ni) with an original value of 4.312 Å, was adjusted to 4.454 Å to match the COOH* intermediate after one proton-coupled electron transfer process, followed by a further change to 4.356 Å after COOH* was converted into HCOOH*. Such accommodations provided key intermediates with a strong binding, thereby suppressing desorption, and leading to facilitated deep protonation to CH4 (Fig. 27b). Similar dependences of SA sites and intermediate species have been also reported in other systems, e.g., Cu SAs in a MOF (Cu/UiO-66-NH2) exhibited lower formation energy of COOH* and CHO* and capability for subsequent coupling between them, resulting in appreciable selectivity to methanol and ethanol;271 Mo SAs in a COF (Mo-TpBpy) favored CO adsorption, thereby enhancing the synthesis of CH4 and C2H4.269 Such dependences are also associated with distinct ligands, e.g., Co–O4 atomic sites in Co-2,3-DHTA-COF exhibited superior performance for CO production from CO2 photoreduction due to the lower energy barrier in the ligand exchange process between Co-2,3-DHTA-COF and CO2, compared with that of its counterpart with different ligands (Co-TP-COF).270
 | ||
| Fig. 27 (a) Proposed reaction mechanism for the photoconversion of CO2 into CO on Ni-TpBpy.267 (b) Implanting flexible and self-adaptive Cu/Ni DMSPs into MOF-808 for highly selective CO2 photoreduction to CH4.268 Reproduced from ref. 267 and 268 with permission from American Chemical Society and Springer Nature, copyright 2019, 2021. | ||
Recently, MXenes, a family of two-dimensional (2D) transition metal carbides, nitrides, and carbonitrides, have attracted burgeoning research interest for their application in photocatalysis due to their outstanding structural and electronic properties. However, the scarcity of active sites limits the exploitation of MXene-based photocatalysts, and therefore metal SAs have been incorporated into certain MXenes, providing a high-efficiency reaction interface for CO2 reduction.272 Co SAs immobilized on a Ti3C2Tx MXene by bonding with C and O atoms, were found to facilitate the formation of the *COOH intermediate with lower free energy in comparison with that of a blank MXene support, while the MXene support not only served to host the isolated Co sites, but also accepted electrons from the light absorber [Ru(bpy)3]3+ and transferred them to Co sites toward CO2 conversion.
3.1.3.1 Summary. Light-drive CO2 reduction might be a truly sustainable process which is expected to realize a net CO2 emission of nearly zero due to the utilization of a completely renewable energy source. SACs function not only as co-catalysts with incorporation into existing semiconductor materials but also as the sole catalyst in the presence of photosensitizers and sacrificial agents. In the former cases, SACs play a crucial role in both the activation of reactants and generation/transfer of photoexcited electrons via modulating the energy band of semiconductors. In the latter cases, MOFs and COFs with high specific area and abundant porous systems serve as common substrates for anchoring metal SAs, providing larger interfaces for the contact between photosensitizers and active sites, thereby enhancing the electron transfer and reaction kinetics.
 | ||
| Fig. 28 Top views and side views of transition-state geometries of CH4 activation and intermediate structures of the formed CH3 and H on Ni1 and Ru1 sites on CeO2. Top and side views of transition-state geometries of CH4 activation on Ni1 (a and b) and Ru1 (c and d) and intermediate geometries of CH3 and H formed on the Ni1 site (e and f) and Ru1 site (g and h). (i) Turnover frequency (TOF) of reforming CH4 with CO2 in terms of hydrogen production per Ni1 site of Ce0.95Ni0.05O2, per Ru1 site of Ce0.95 Ru0.05O2, and per Ni1 or Ru1 site for Ce0.95Ni0.025Ru0.025O2 at 773–833 K. In the calculation of TOFs, the number of catalytic sites for Ce0.95Ni0.025Ru0.025O2 is the total number of Ni and Ru atoms of the topmost surface layer of Ce0.95Ni0.025Ru0.025O2.273 Reproduced from ref. 273 with permission from American Chemical Society, copyright 2019. | ||
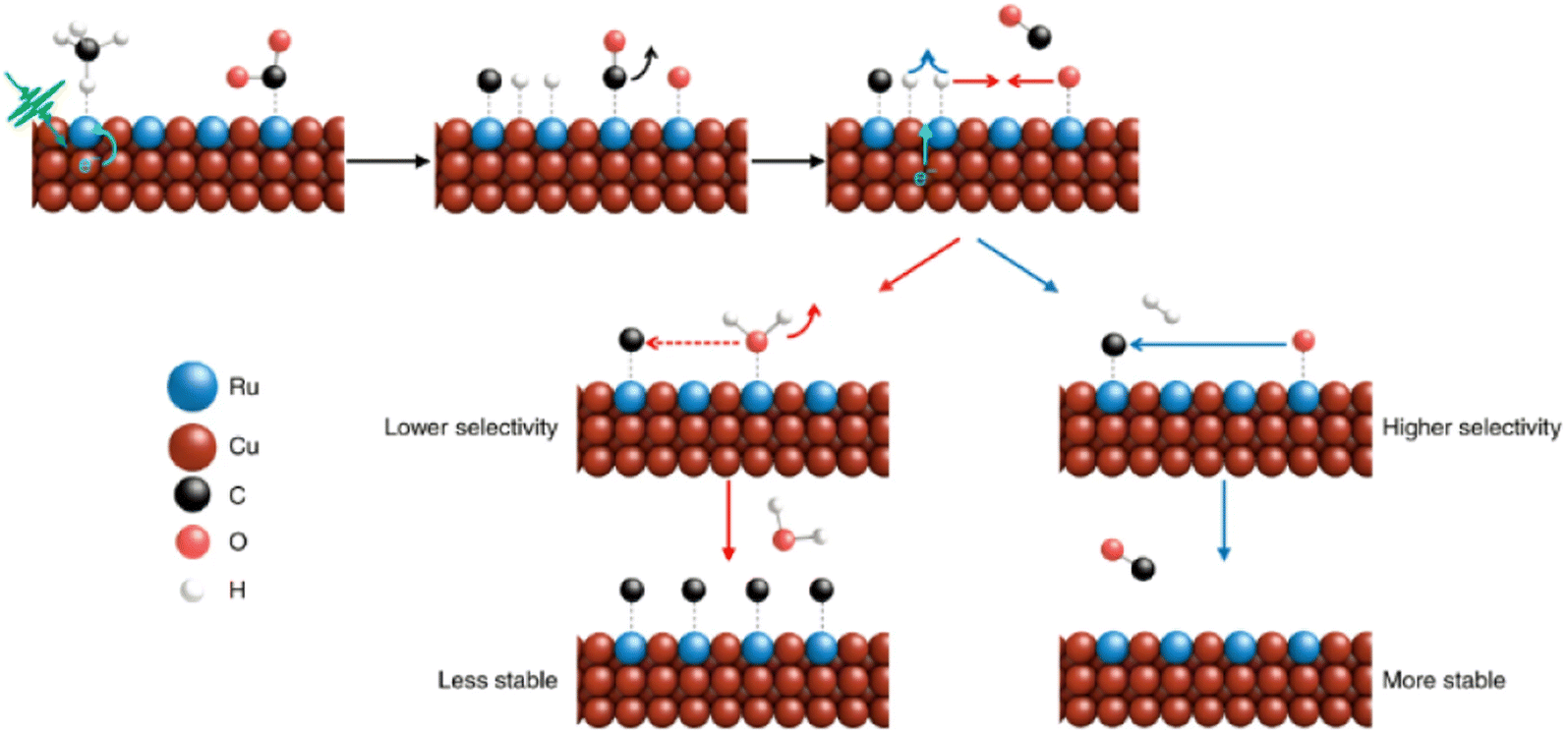 | ||
| Fig. 29 Schematics of enhanced selectivity and stability in photocatalysis via the electronic transition mechanism. The green pulse indicates light excitation. e− represents the hot carrier and the cyan arrow indicates transfer and surface excitation by hot carriers. Blue and red arrows indicate the reaction path of photocatalysis and thermocatalysis, respectively. The dashed arrow indicates a less probable reaction path.280 Reproduced from ref. 280 with permission from Springer Nature, copyright 2020. | ||
3.2 CO2 conversion toward C2+ products
A group of Pd-based SACs fabricated by Caparrós et al. with various supports, were applied in gas phase hydrogenation of CO2 into ethanol.282 A catalytic performance dependent on the specific interaction between Pd SAs and different inorganic oxide supports was found, which was attributed to the particular architecture established for C–C coupling. NP species were confirmed to be inactive for the formation of ethanol by increasing the reaction temperature to create a NP-dominated catalyst. It was an inspiring result that a SAC could work in a complicate hydrogenation process leading to the formation of ethanol. However, ethanol yield was limited by severe RWGS over this Pd SAC, and deep insights into the relevant reaction mechanism were still absent. Ye et al. studied the whole process of CO2 hydrogenation into ethanol in depth over an Ir-based SAC.59 They designed a bifunctional SAC (Ir1–In2O3) by anchoring monoatomic Ir onto the In2O3 carrier with abundant vacancy sites. On the one hand, a part of the feed CO2 was activated by a Lewis acid–base pair formed by the coupling between the isolated Ir atom and the adjacent oxygen vacancy on the surface of In2O3, followed by the generation of intermediate species of carbonyl (CO*) adsorbed on the Ir atom (Irδ+−CO*). On the other hand, another part of the feed CO2 was activated and transformed into methoxide species by oxygen vacancies (CH3O*–Ov). Cohesive corporation between the two types of active sites offered great opportunities for an effective C–C coupling reaction, resulting in a high selectivity for ethanol (>99%) with an excellent initial turnover frequency (481 h−1) for the hydrogenation of CO2 in the liquid phase (Fig. 30). In a subsequent study by the same group, 1D Mo2C nanowires with dominant (101) crystal surfaces were modified by the deposition of atomic Rh and K for CO2 hydrogenation to ethanol.283 In comparison with unmodified β-Mo2C that only converted CO2 to methanol, K0.2Rh0.2/β-Mo2C demonstrated an ethanol selectivity of 72.1% at 423 K. As the active carrier β-Mo2C was in charge of the formation of CH3O* intermediates, Rh SAs served as the active sites for activation of CO2 and stabilization of CO adsorbed species (CO*) in an electron-deficient state, favoring the subsequent CO* insertion. As such, the synergistic effect between these two functionalities led to highly specific controlled C–C coupling. The introduction of K had an effect by promoting the adsorption and activation of CO2, as well as regulating the activation of hydrogen, resulting in a more balanced performance of the two active centers and a further improved ethanol selectivity. Coincidentally, a similar SAC system containing such bifunctional components with Lewis acid–base pairs was reported by Zheng et al. They established a Rh-based SAC by embedding monoatomic Rh onto a Ti-doped CeO2 support (Rh1/CeTiOx).284 The formation of oxygen vacancies on the surface of reducible CeO2 was promoted with the introduction of Rh SAs and Ti. Meanwhile, the Rh SAs coupled with neighboring oxygen vacancies, existing in the form of Rh1–Ov Lewis acid–base pairs, played a vital role in the enhanced CO2 adsorption and activation. Different from the reaction pathway over Ir1–In2O3, CO2 was converted into 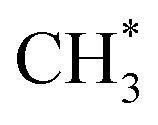 species via the HCOO* intermediate over Rh1–Ov active sites, and subsequently CO* generated from RWGS was inserted into Rh–CH3, leading to the formation of C–C bonds. Ethanol was formed and desorbed through the following hydrogenation eventually, completing a catalytic cycle (Fig. 30). In particular, the SAs ensured the high selectivity of ethanol by regulating the CO adsorption over metal sites, while NPs and NCs provided methanol-dominated products.
species via the HCOO* intermediate over Rh1–Ov active sites, and subsequently CO* generated from RWGS was inserted into Rh–CH3, leading to the formation of C–C bonds. Ethanol was formed and desorbed through the following hydrogenation eventually, completing a catalytic cycle (Fig. 30). In particular, the SAs ensured the high selectivity of ethanol by regulating the CO adsorption over metal sites, while NPs and NCs provided methanol-dominated products.
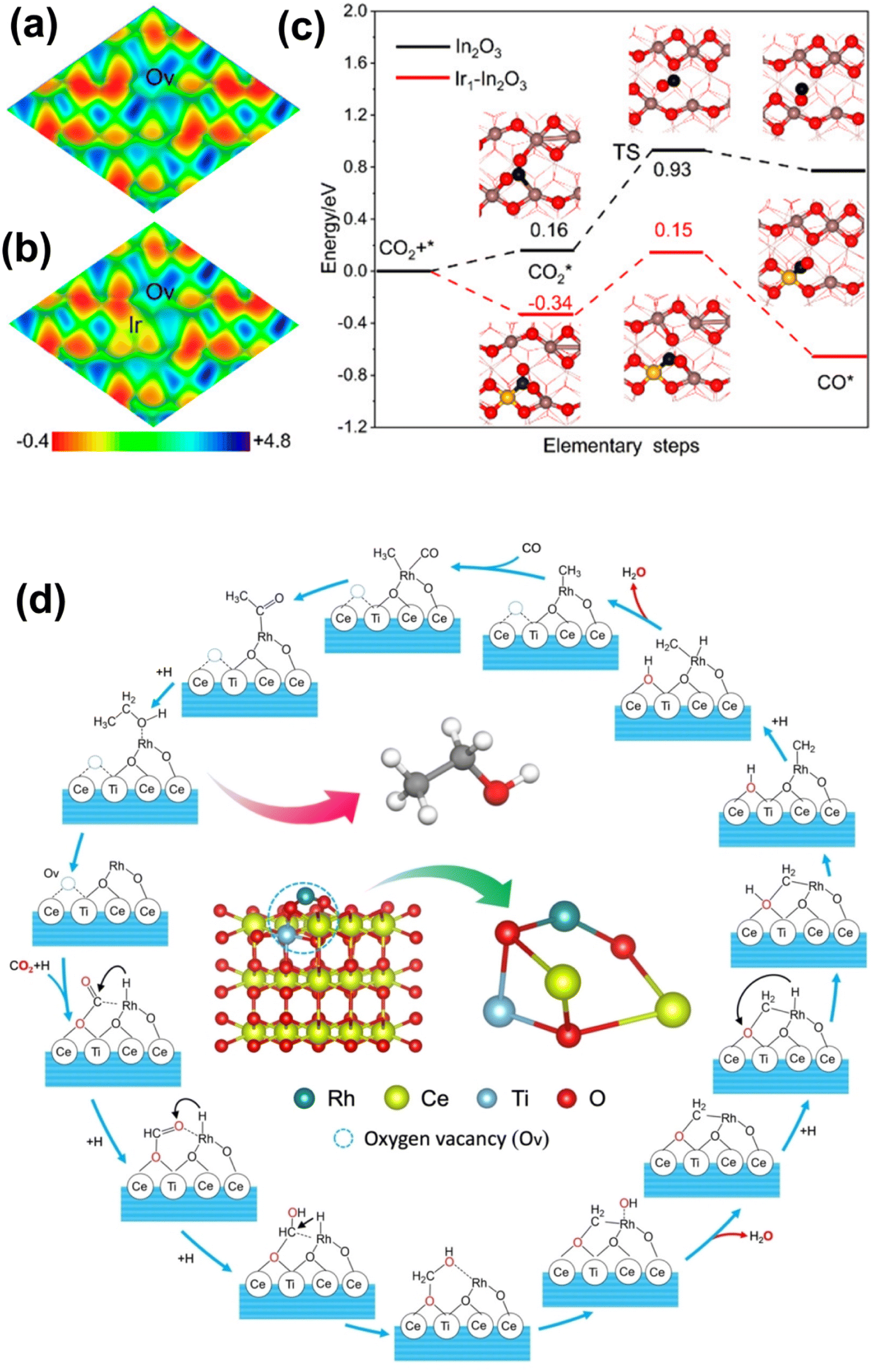 | ||
| Fig. 30 Electrostatic potential diagrams of the In2O3 (a) and Ir1–In2O3 catalyst surface (b). Free energy diagram for CO2 dissociation over the In2O3 and Ir1–In2O3 catalyst (c).59 (d) The illustrated catalytic cycle of ethanol formation from CO2 hydrogenation over the Rh1/CeTiOx catalyst. The inset figure displays the structure of the Rh1/CeTiOx catalyst.284 Reproduced from ref. 59 and 284 with permission from American Chemical Society and Wiley, copyright 2020 and 2022. | ||
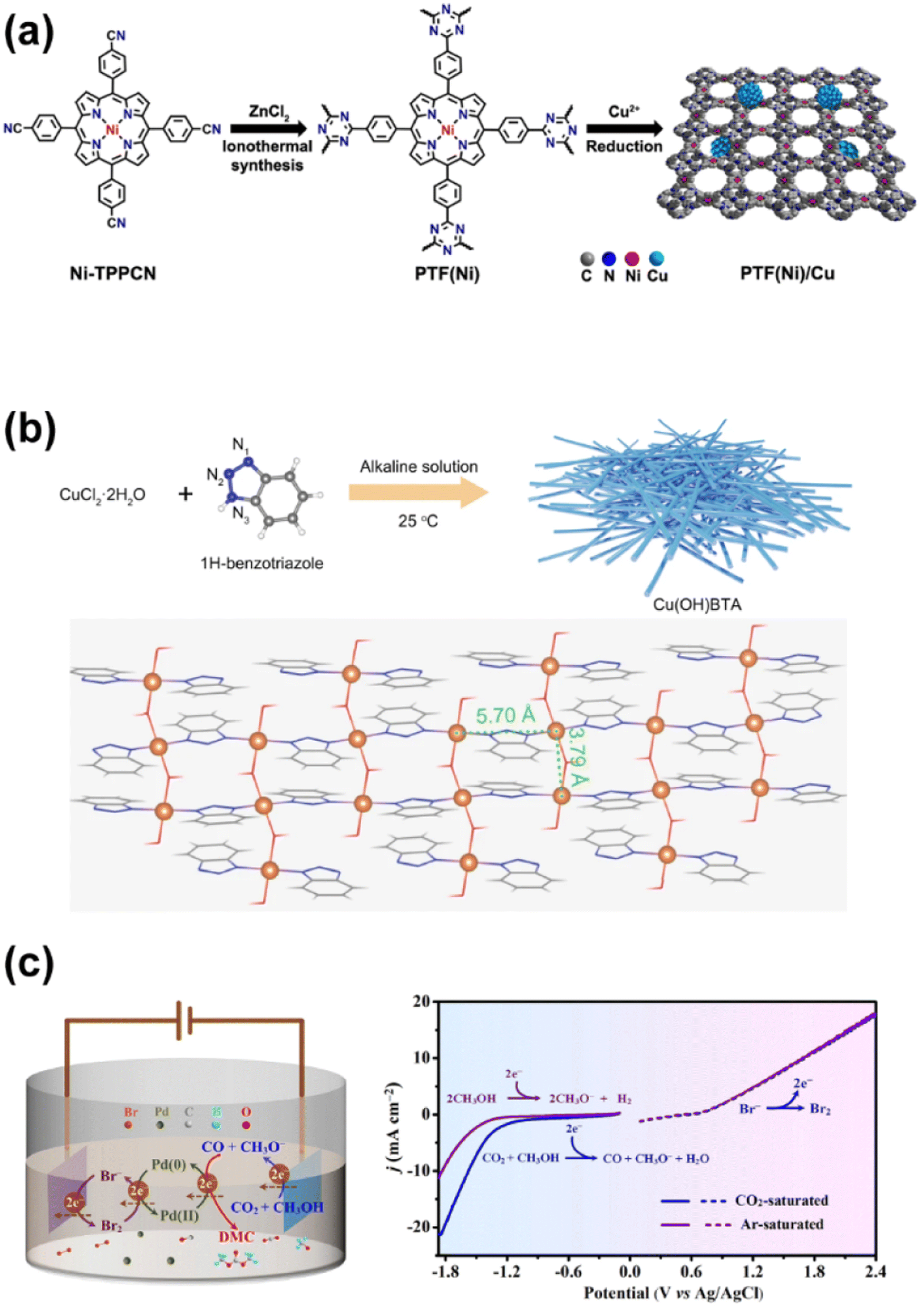 | ||
| Fig. 31 (a) Schematic illustration of the synthesis procedure and the wireframe model of Cu(OH)BTA.164 (b) Fabrication of PTF(Ni)/Cu with well dispersed copper sites from Ni-TPPCN. (c) Schematic for the convergent paired electrosynthesis of DMC from CO2.159 (b) LSV of Ni SAs/OMMNC for electrocatalytic CO2RRs in CO2-saturated 0.1 M KBr–CH3OH in H-type cells.291 Reproduced from ref. 159, 164 and 291 with permission from Wiley, Springer Nature and Royal Society of Chemistry, copyright 2021, 2023. | ||
Apart from the synthesis of C2+ products via C–C couplings, Li et al. reported a novel process for the production of dimethyl carbonate (DMC) through a so-called convergent paired electrosynthesis from CO2 by integrating the anodic Br2 evolution reaction and cathodic CO2 reduction reaction in a membrane-free single cell (Fig. 31c).291 Initially, they implemented a dual-channel SAC with Ni SAs asymmetrically coordinated with four planar nitrogen and one axial oxygen, which exhibited a lower energy barrier for CO2 activation to *COOH in comparison with the common Ni SAC featuring a symmetric structure with only planar four-nitrogen coordination. By virtue of this SAC, they obtained an exclusively high CO FE of 99% and a partial current density of 325 mA cm−2 at −0.6 V versus RHE. This reduction process (CO2 + 2e− + 2H+ → CO + H2O) was innovatively integrated with a Br2 evolution reaction (2Br− → Br2 + 2e−) in CO2-saturated 0.1 M KBr–CH3OH electrolyte, accompanied by a subsequent chemical oxidation of Pd0 to Pd2+ (Pd0 + Br2 → Pd2+ + 2Br−). Eventually, DMC was produced by the chemical oxidation of the formed CO and CH3O− with the synchronous reduction of Pd2+ to Pd0 (CO + 2CH3O− + Pd2+ → (CH3O)2CO + Pd0). As a result, a high FE of DMC of up to 80% could be achieved at room temperature.
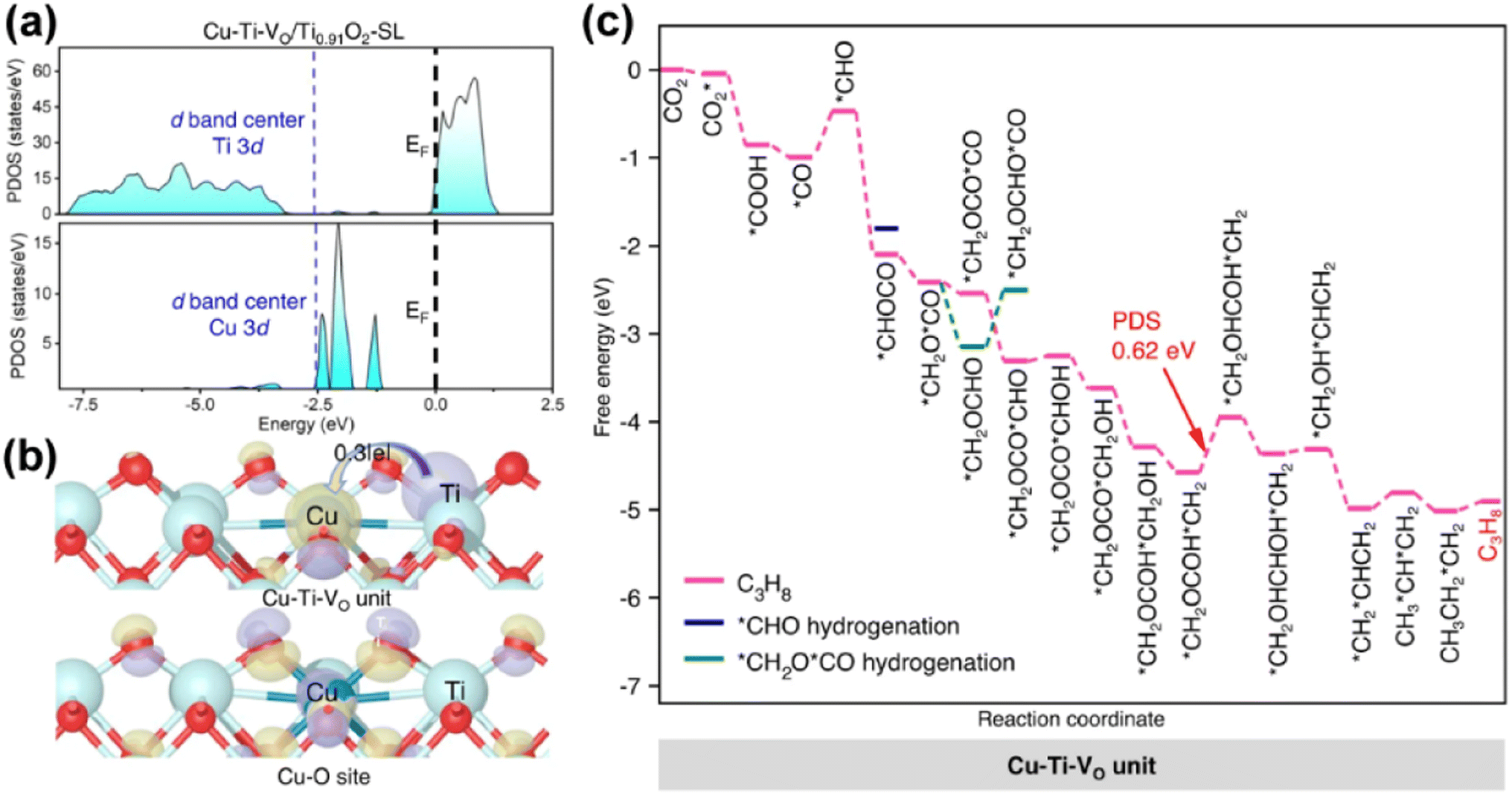 | ||
| Fig. 32 (a) PDOS and d-band centres of Cu 3d and Ti 3d orbitals. (b) Charge density differences of the Cu–O site and Cu–Ti–VO unit. (c) Gibbs free energy diagrams of CO2 reduction on the Cu-Ti-VO unit.295 Reproduced from ref. 295 with permission from Springer Nature, copyright 2023. | ||
Indeed, the above studies and other related research studies have addressed an effective strategy for the construction of superior SACs to realize preferential synthesis of complex products, that is, focusing on the intermediate steps, matching the electronic and geometric properties of SACs with key intermediates by accurately controlling microenvironments adjoining SAs and relying on the synergy between metal SAs and substrates with special properties. Certainly, a sufficient supply of electrons and protons should be ensured in the meantime. This strategy is quite consistent with that proposed in the thermal and electric process,59,159,284 further suggesting the universality of SAC design in CO2 reduction.
3.3 CO2 conversion toward other organic products
The above CO2 reduction reactions, no matter what the source of energy input is, show that nearly all carbon atoms of the products stem from CO2 conversion and these carbon atoms constitute the main frame of product molecules. Massive production of valuable carbon-based platform chemicals and fuels, as well as alleviation of carbon emission is expected to be achieved to some extent with the practical exploitation of these reactions, providing a viable opportunity for a sustainable industry. Besides, there have been also quite a few synthetic reactions encompassing not only CO2 conversion but also the transformation of other compounds (not including H2 or H2O) towards the production of various chemicals with skeletal structures not based on CO2-derived carbon atoms exclusively. A case in point is dry reforming reactions, which produces CO and H2via co-conversion of CO2 and CH4. Carbon atoms in CO are from both CO2 and CH4, as distinguished from other reactions of CO synthesis from CO2 reduction mentioned above, although the CO2 molecules also go through the procedures of reduction. Not just CH4, but other reactants such as alkenes, alkynes, arenes, and heteroatom-containing compounds can be incorporated in the CO2 conversion reactions as well, in which CO2 might be transformed into carbonyl, carboxyl, and alkyl with different degrees of protonation.9,10,14–16,296–303 On the basis of these reactions, the production of fine chemicals, drugs and some advanced organic materials might receive a good opportunity towards more environment-friendly operations with such a nontoxic carbon source. These processes are also expected to provide an opportunity for comprehensive utilization of CO2.Due to the increased complexity of feed reactants compared with that of CO2 reduction reactions, the thermodynamics and kinetics involved in these systems are more complicated correspondingly, which indicates that design of efficient catalysts and exploration into the mechanism are in great demand. Given that homogeneous catalysts have been extensively used in certain reactions,296,301 the applications of SACs possessing combined advantages of homogeneous and heterogeneous catalysts have been also reported for enhancing both activity and selectivity, as well as long-term stability under harsh conditions. Herein, we will focus on these synthetic reactions regarding the design strategies of SACs and relevant mechanism analysis for a more comprehensive understanding of SAC application in a broader spectrum of CO2 conversion.
As a C1 synthon, ubiquitous CO2 provides a green, non-toxic carbon source for the synthesis of a large variety of chemicals by pharmaceutical and organic engineering in terms of the functionalization of C–H and the formation of C–C and C–X (X denotes heteroatoms).14,296,300,304 For instance, in organic synthesis, CO2 has been extensively used for the carboxylation of alkenes, alkynes, and arenes.296 CO2 plus H2 could be also applied in carbonylation and alkylation reactions based on different reduction levels.14 Generally, homogeneous catalysts are commonly chosen to realize effective conversion with appreciable activity in these reactions. As heterogeneous systems with the applications of SACs have been burgeoning in formylation and carbonylation reactions with CO,305–307 the SACs are also expected to provide a glimmer of hope to the corresponding utilization of CO2, as CO can be selectively generated in situ from CO2 conversion via various routes. Fu et al. integrated cobalt SAs and CuPd NCs into a porphyrin-based MOF to construct composite photocatalysts (Cu1Pd2)z@PCN-222(Co) for coupling the CO2-to-CO photoconversion and Suzuki coupling reactions.308 Under visible light irradiation, excited porphyrin could concurrently transfer electrons to Co SAs and CuPd NCs. CO could be formed over Co SA sites, and then participate in the subsequent Suzuki reactions with various substrate molecules (Fig. 33a–c). The synergy effect between SA sites and NCs led to the photosynthesis of benzophenone with over 97% selectivity and 90% yield. Fenofibrate, a well-known blood cholesterol lowering drug, could also be obtained with a yield of 67.3% under mild conditions. Similar coupling strategies could be also carried out in separated reactors combining different inputs of energy, in which CO was a bridge to link the primary and secondary reactions.309,310 CO2 fixation to cyclic carbonates could be also realized over SACs.311,312 Jiang and his co-workers fabricated a class of novel hollow porous carbons (HPC) containing well dispersed dopants of nitrogen and Zn SAs through pyrolysis of template-directed hollow ZIF-8 spheres.312 The optimized SAC featuring an ultrahigh loading of (11.3 wt%) Zn and surrounding N active sites achieved efficient catalytic CO2 cycloaddition with epoxides via high-efficient photothermal conversion under light irradiation at ambient temperature. The enhanced photothermal effect was mainly contributed by the abundant ZnN4 sites with a specific coordination structure derived from the parent ZIF-8 precursor plus the hollow structure of the carbon support which could harvest light in a broad spectrum by using the multiple reflections within the cavity (Fig. 33e). Coincidentally, Wang et al. also reported a Zn SAC achieving a cycloaddition reaction of epoxides and CO2 with high yield (99%) and selectivity (98%) of propylene carbonate with a TOF of 2889 h−1.311 They anchored Zn on a nitrogen-doped graphene support (NG) with atomically dispersed [ZnN3.76±0.2] as active sites, as verified by XANES, EXAFS and XPS characterization studies. The active sites functioned by interacting with epoxide to polarize the C–O bond, followed by a ring-opening step via nucleophilic attack by the bromide anion. The subsequent nucleophilic attack of the intermediate at the CO2 molecule led to the formation of an alkyl carbonate anion and final cyclic carbonate via ring closing (Fig. 33f). During the whole process, the Zn SA sites were considered to have facilitated the activation of epoxides and stabilization of the intermediate significantly. Xu et al., on the other hand, exploited the strong electronic metal-support interaction between iridium single atoms and a WO3 support, promoting the CO2 cycloaddition of styrene oxide to styrene carbonate with 100% efficiency and high durability.313 This is another validation of SACs used for an effective cycloaddition reaction involving CO2 conversion and the synthesis of complex organic chemicals. Besides these mentioned reactions, more types of reactions were predicted to be realized promisingly over SACs based on the DFT calculations, such as conversion of methane and carbon dioxide to acetic acid.314
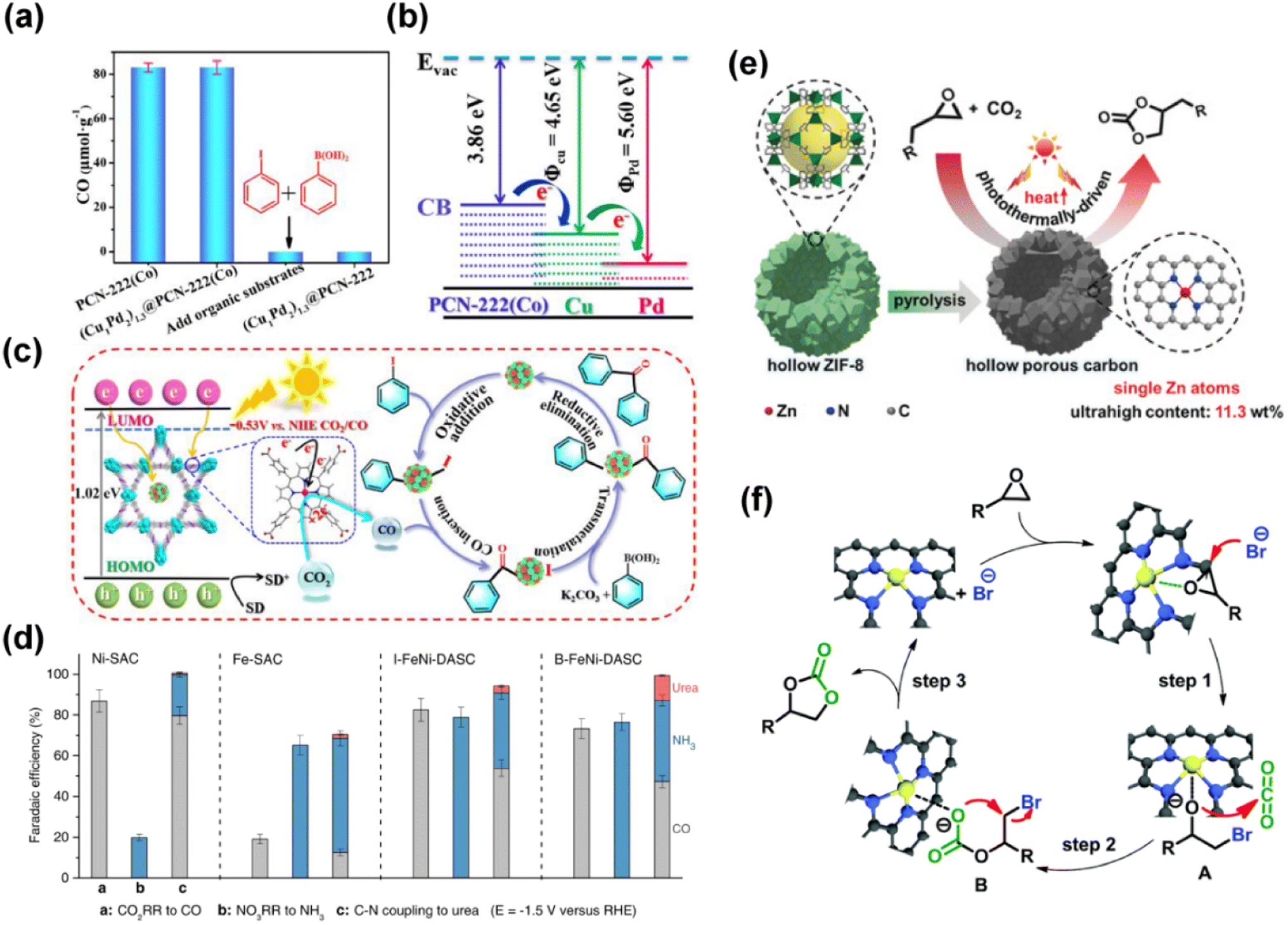 | ||
| Fig. 33 (a) Photocatalytic evolution of CO under irradiation with a 300 W xenon lamp (λ >420 nm) in a CO2-saturated solution of DMF/H2O (1:1, v/v, 5 mL). (b) Cu-mediated electron transfer process over (Cu1Pd2)1.3@PCN-222(Co). (c) Proposed mechanism for (Cu1Pd2)1.3@PCN-222(Co)-catalyzed carbonylation Suzuki coupling CO2 photoreduction.308 Reproduced from ref. 308 with permission from American Chemical Society, copyright 2021. (d) The product distributions of the CO2RR, the NO3RR and urea synthesis on Ni-SAC, Fe-SAC, I-FeNi-DASC, and B-FeNi-DASC at −1.4 V versus RHE.315 Reproduced from ref. 315 with permission from Springer Nature, copyright 2022. (e) Fabrication of the Zn SAC and its use in photothermal-driven CO2 cycloaddition.312 Reproduced from ref. 312 with permission from Wiley, copyright 2019. (f) Plausible atomic [ZnN] based reaction mechanism.311 Reproduced from ref. 311 with permission from Royal Society of Chemistry, copyright 2019. | ||
The production of urea, one of the most important nitrogen fertilizers, involving C–N formation between CO2 and NH3, has been operated at high pressure and temperature with large energy consumption conventionally (e.g., Haber–Bosch process). Electrocatalytic urea synthesis, on the other hand, could be conducted under mild conditions, and is expected to be a promising alternative to current industrial protocols. Wang and his co-workers reported a diatomic catalyst (DA) with bonded Fe–Ni pairs for electrochemical urea synthesis.315 As the capture and activation of CO2 were both suppressed over sole-metal SACs, the subsequent C–N coupling was hindered with a much higher formation free energy of the key intermediate *COOH. On introducing a second metal into the system, the transfer of CO to engage in subsequent coupling became another obstacle with a high energy barrier due to the large distance, although the problems in CO formation were assuaged. The bonded Fe–Ni pairs, in contrast, could serve as efficient sites for coordinated adsorption and activation of multiple reactants, thereby enhancing the crucial C–N coupling thermodynamically and kinetically (Fig. 33d). In a later report, they further investigated the application of a Cu-based SAC with Cu SAs decorated on a CeO2 support (denoted as Cu1–CeO2) in electrochemical urea synthesis from carbon dioxide and nitrate.316 They found a reconstitution of copper SAs (Cu1) to NCs (Cu4) during electrolysis through operando X-ray absorption spectra. These electrochemically reconstituted Cu4 clusters were identified as real active sites for electrocatalytic urea synthesis, since the favorable C–N coupling reactions and urea formation on Cu4 were validated by operando synchrotron-radiation Fourier transform infrared spectroscopy and theoretical calculations. Such transformations between NCs and SAs were even reversible by switching the applied potential to an open-circuit potential. A similar formation of C–N bonds by SACs was also reported in organic synthesis. Zhao et al. prepared a stabilized Pt SAC over ultrathin two-dimensional Ti3−xC2Ty MXene nanosheets (metal carbide with titanium vacancies; T: O, OH, F).317 The Pt precursor ions went through a simultaneous self-reduction stabilization process at room temperature when adsorbing on a support treated by HCl/LiF. With the simultaneous formation of Ti vacancies, the SAs hence exhibited strong metal–carbon bonds with the Ti3–xC2Ty support and were anchored onto the vacancy sites. Pt1/Ti3−xC2Ty afforded the efficient incorporation of CO2 in the formylation of amines. More importantly, compared to Pt NPs, the SAs possessing partial positive charges significantly favored the adsorption and activation of silane, CO2, and aniline energetically, leading to boosted catalytic performance.
3.4 Summary
Actually, SACs have been exploited in all kinds of systems involving CO2 conversion so far, suggesting their unparalleled potential and vitality. We provide a systematic summary of some representative cases of SAC application in different systems, especially various CO2 reduction reactions in Table 1.| Catalysts | Systemsa | Target products | Performances | Ref. | |
|---|---|---|---|---|---|
| Selectivity (%) | Activityb | ||||
| a T: thermochemical reduction; E: electrochemical reduction; P: photochemical reduction. b TON: Abbreviation of the turnover number; TOF: abbreviation of the turnover frequency. | |||||
| Rh1–ZrO2 | T | CH4 | ∼100 | — | 85 |
| Na–Rh1–ZrO2 | CO | ∼99 | 9.4 mol CO per gRh per h | ||
| Pd1-FeOx | T | CO | ∼98 | 42.0 mmol CO·per gcat·per h | 86 |
| Co/SBA-15 | T | CO | ∼99 | 304.6 mol CO per molCo per·h | 88 |
| Mo/NC | T | CH4 | ∼100 | Yield ∼46.3% | 91 |
| Ru/pBN | T | CH4 | ∼93.5 | 1.86 mmolCO2 gcat−1 s−1 | 94 |
| Ir/AP-POP | T | HCOOH | — | TON 25135 | 101 |
| Ru/LDH | T | HCOOH | — | TON 698 | 108 |
| Ru@MCM | T | HCOOH | — | TON ∼2000 | 109 |
| Pd/2,6-DCP-CTF | T | HCOOH | — | TOF 13.46 h−1 | 110 |
| Pt/MoS2 | T | CH3OH | ∼95 | TOF 162.5 h−1 at 150 °C | 112 |
| Cu/ZrO2 | T | CH3OH | ∼100 | TOF 1.37 h−1 | 123 |
| Cu/ZnO | T | CH3OH | 99.1 | Yield ∼4.9% | 124 |
| Pt1@MIL | T | CH3OH | 90.3 | TOF 117 h−1 | 126 |
| A-Ni-NG | E | CO | 97 | TOF 14800 h−1 | 140 |
| Ni-NC | E | CO | 92 | — | 150 |
| NiSA/PCFM | E | CO | 88 | Current density 308.4 mA cm−2 | 151 |
| Fe3+–N–C | E | CO | ∼90 | Current density 94 mA cm−2 | 139 |
| Fe-SAC/NPC | E | CO | ∼97 | — | 155 |
| Fe–N4/CF | E | CO | 94.9 | Current density ∼10 mA cm−2 | 156 |
| Cu–APC on Pd10Te3 nanowires | E | CO | 92 | Current density 18.74 mA cm−2 | 166 |
| Cu–S1N3/Cux | E | CO | > 90 | — | 167 |
| Znδ+-NC | E | CO | 95 | Current density 1 A cm−2 in flow cell | 169 |
| Zn–NS–C | E | CO | 99 | TOF 11419 h−1 | 173 |
| Co–N5/HNPCSs | E | CO | 99 | Current density 6.2 mA cm−2 | 176 |
| Cd-NC-t SAC | E | CO | 91.4 | Current density ∼5 mA cm−2 | 180 |
| Mn-MCs-(N,O) | E | CO | 94.5 | Current density 13.7 mA cm−2 | 181 |
| AgSA/MnO2 | E | CO | 95.7 | — | 183 |
| Mg–C3N4 | E | CO | >90 | TOF ∼18000 h−1 |
191 |
| Ca–N3O | E | CO | >90 | TOF ∼15000 h−1 |
192 |
| Ga-N3S-PC | E | CO | ∼92 | — | 193 |
| InN4/C | E | CO | 97.2 | TOF ∼40000 h−1 |
194 |
| Bi-SAs-NS/C | E | CO | 98.3 | Current density 10.24 mA cm−2 | 197 |
| Sn(IV)–N4/CNT–OH | E | HCOOH | 89.4 | Current density 74.8 mA cm−2 | 210 |
| Sbδ+–N4 (0 < δ < 3) | E | HCOOH | 94 | — | 207 |
| Cu SAs/GDY | E | CH4 | 81 | — | 213 |
| Cu SAs/TCNFs | E | CH3OH | 44 | Current density 93 mA cm−2 | 221 |
| CoPc/CNT | E | CH3OH | 40 | — | 223 |
| Cu2NCN | E | CH3OH | 70 | Current density 92.3 mA cm−2 | 222 |
| Cu1/TiO2 | P | CH4 (C2H6) | — | 1416.9 (64.2) ppm g−1 h−1 | 231 |
| Bi/TiO2 | P | CH4 | — | 4.1 μmol g−1 h−1 | 230 |
| Cu0.8Au0.2/TiO2 | P | CH4 (C2H4) | — | 3578.9 (369.8) μmol g−1 h−1 | 234 |
| Ag-HMO | P | CH4 | — | 0.61 mol mol−1 | 236 |
| Pt–V2O5 | P | CH4 | — | 247.6 μmol g−1 h−1 | 237 |
| CoSi2 | P | CO | — | 4.7 mol g(Co)−1 | 239 |
| Co-2,3-DHTA-COF | P | CO | 95.7 | 18000 μmol g−1 h−1 |
270 |
| Ni-TpBpy | P | CO | 96 | ∼4057 μmol g−1 (5 h) | 267 |
| Cu SAs/UiO-66-NH2 | P | CH3OH (CH3CH2OH) | — | 5.33 (4.22) μmol g−1 h−1 | 271 |
| RuSA-mC3N4 | P | CH3OH | — | 1500 μmol g−1 (6 h) | 250 |
| Co/g-C3N4 | P | CH3OH | — | 941.9 μmol g−1 (4 h) | 255 |
| Co1–C3N4@α-Fe2O3 | P | CO | > 99 | 14.9 μmol g−1 h−1 | 261 |
| InCu/PCN | P | CH3CH2OH | 92 | 28.5 μmol g−1 h−1 | 318 |
| Cu-Ti-VO/Ti0.91O2-SL | P | C3H8 (C2+) | 32.4 (50.2) | 13.8 μmol g−1 h−1 | 295 |
| SnS2/Sn1–O3G | E | CH3CH2OH | 82.5 | Current density 17.8 mA cm−2 | 290 |
| PTF(Ni)/Cu | E | C2H4 | 57.3 | — | 164 |
| 0.1Pd/Fe3O4 | T | CH3CH2OH | 97.5 | 413 mmolEtOH gPd−1 h−1 | 282 |
| Ir1–In2O3 | T | CH3CH2OH | > 99 | TOF 481 h−1 | 59 |
| Rh1/CeTiOx | T | CH3CH2OH | 99.1 | TOF 493.1 h−1 | 284 |
| K0.2Rh0.2/β-Mo2C | T | CH3CH2OH | 72.1 | 33.7 μmol g−1h−1 | 283 |
4. A molecular understanding of SACs in CO2 conversion
In the above sections, we have provided a comprehensive summary of different reaction routes for CO2 transformation and the outstanding performances of SACs in highly selective synthesis of various valuable products. However, the current stage of research mainly depends on case-by-case evaluation and empirical design. We must avoid pointless scientific research input on tediously screening catalysts and repetitively investigating mechanisms, or otherwise use excessive resources for future research studies. We truly expect a prospect that the construction and exploitation of optimized catalysts could be driven by rational design and get rid of the limitation of human resources.319 Nowadays, we could catch a glimpse of such a promising prospect thanks to the findings on SACs, of which well-arranged and well-defined structures regarding both electronic and geometric aspects shed light on the systematic analysis and study (Fig. 34). Furthermore, the transformation of CO2, a simple but important C1 molecule, should offer an excellent opportunity to dive into the molecular understanding into all levels of SACs in specific catalytic reactions. As such, the discussion in this section might guide the establishment of potential research paradigms with unambiguous structure–activity relationships and inspire in-depth exploration into the intrinsic reaction mechanisms.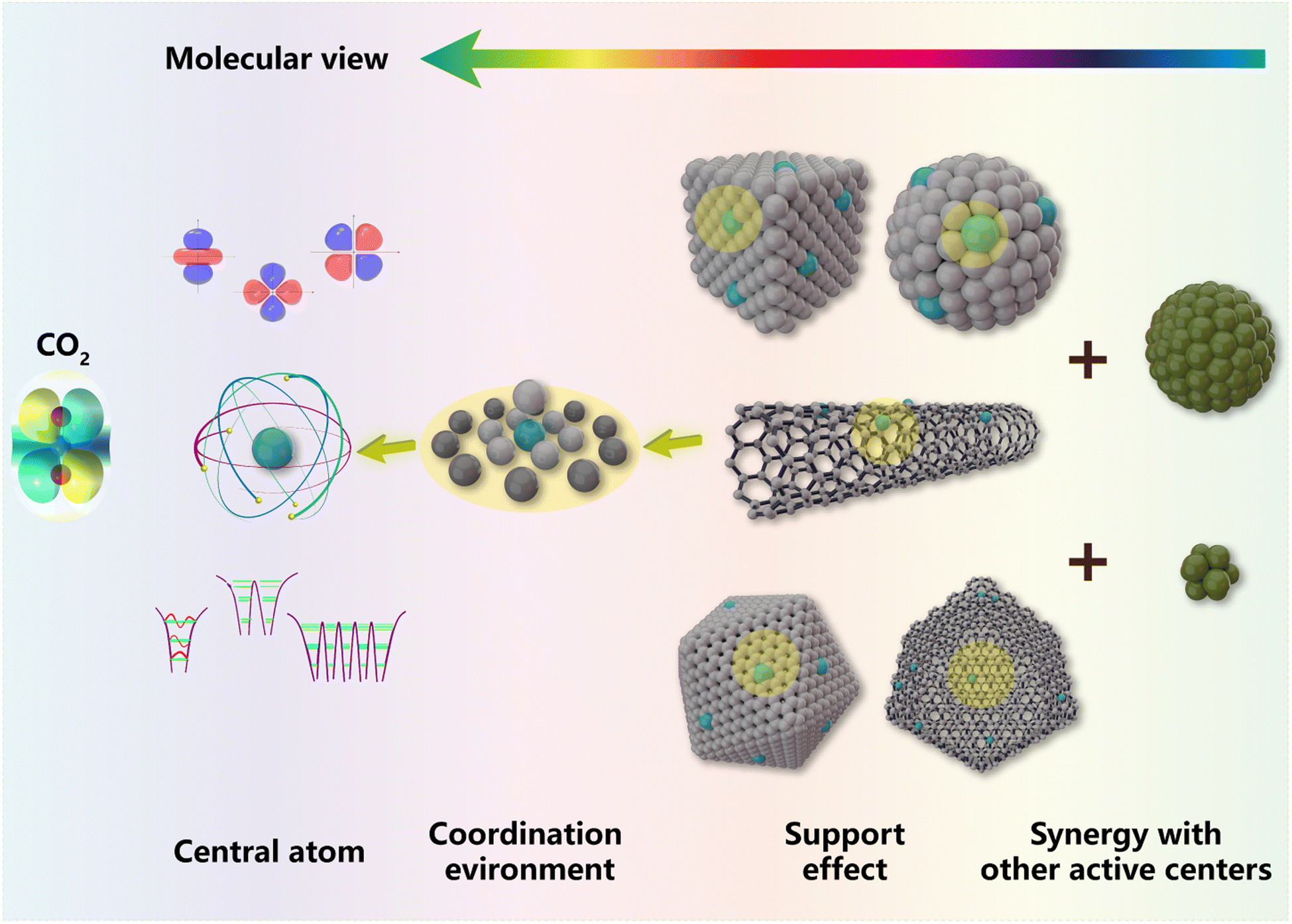 | ||
| Fig. 34 Schematic of multi-level understanding of SACs for CO2 transformation from a molecular point of view. | ||
4.1 Central atoms
Generally, central atoms endow the SACs with fundamental catalytic properties at the core and top level, especially for those CO2 reduction reactions, which just involve electron transfer and protonation procedures (Fig. 35). The intrinsic features of different types of central metal atoms have an effect on not only the primary interaction with neighboring atoms on the substrate but also the chemical adsorption of guest molecules including reactants and intermediates.320 The former effect partially affords the specific localized structures of active moieties, the dynamic evolution and even the durability of active sites under certain redox conditions. The latter effect consists of even more abundant connotations and drives the proposal of strategies for regulating the reaction pathway.321 It has influence on the practical reaction kinetics following the Sabatier principle, which has qualitatively pointed that there should be an optimum “bond strength” of reactants and transition states on the active centers, defining the best catalyst for a given reaction.322 Researchers further try hard to develop specific descriptors closely correlated with the intrinsic activity by mapping various parameters for quantitative implementation to open a path for predictable catalytic performance.323,324 d-band and other d-electron related models have been proposed as a popular theoretical basis for offering comprehension of the scaling relations and variations in catalytic performance with regard to the electronic structure of transition-metal sites.325 In addition, more specific descriptors might be more effective, which is decided by the catalytic systems.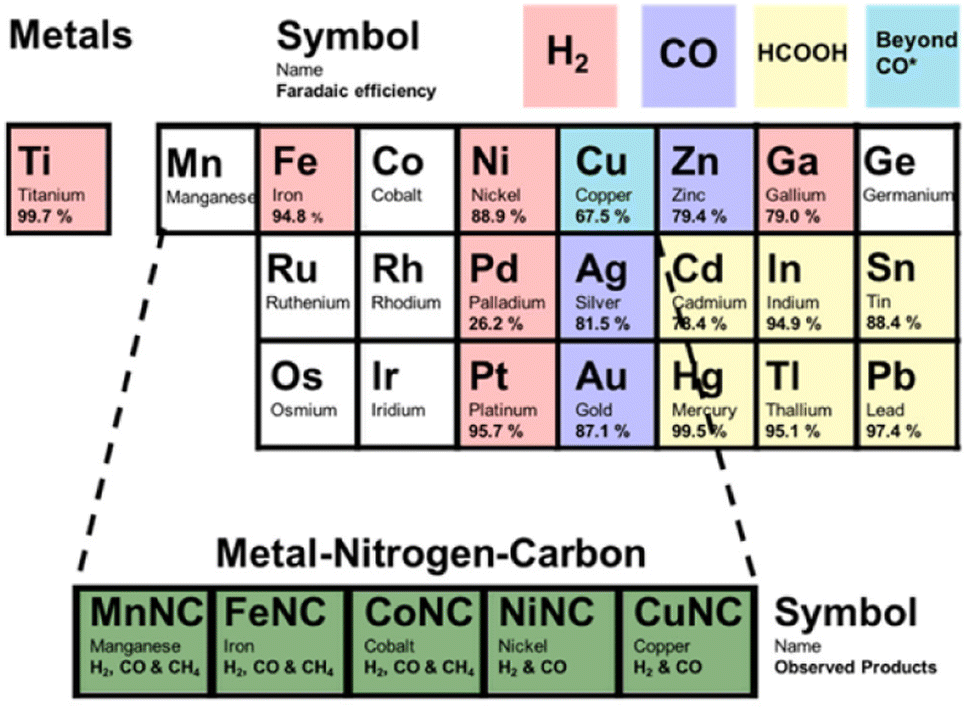 | ||
| Fig. 35 A colored periodic table with metal catalyst performance obtained in the CO2 electrochemical reduction reaction. Reproduced from ref. 326 with the permission from American Chemical Society, copyright 2019. | ||
For some electrochemical reactions over SACs, there has been primary progress recently.327,328 Taking commonly used nitrogen-doped carbon as an identical substrate, SACs based on various transition metals, such as Fe, Co, Ni, Cu, and Mn, have been applied in electrochemical CO2 reduction under consistent conditions (Fig. 36).326,329 Strasser et al.326 correlated the apparent performance of these M–N–C catalysts with specific energy descriptors via combined experimental and theoretical approaches. The catalytic reactivity trends and reaction pathway are divided into three potential regions deliberately with distinct kinetic features. By simply calculating the binding energy of possible intermediates *COOH and *CO, the potential onset could be correlated with the binding energy of *COOH in the low overpotential region, as the formation rate of CO correlates with the free energy of *CO in the medium overpotential region. Based on these relationships, the authors could infer that the rate-determining steps change from the first proton-coupled electron transfer into *COOH (CO2 + H+ + e− → *COOH) to the subsequent proton-coupled electron transfer into *CO (*COOH + H+ + e− → *CO + H2O). Even the higher CO FE of Ni at a certain current density at technologically relevant levels could be rationalized by using the gaps between the energy barriers of different types of metals regarding the HER. Adopting different substrates as platforms might lead to distinctly different descriptors.
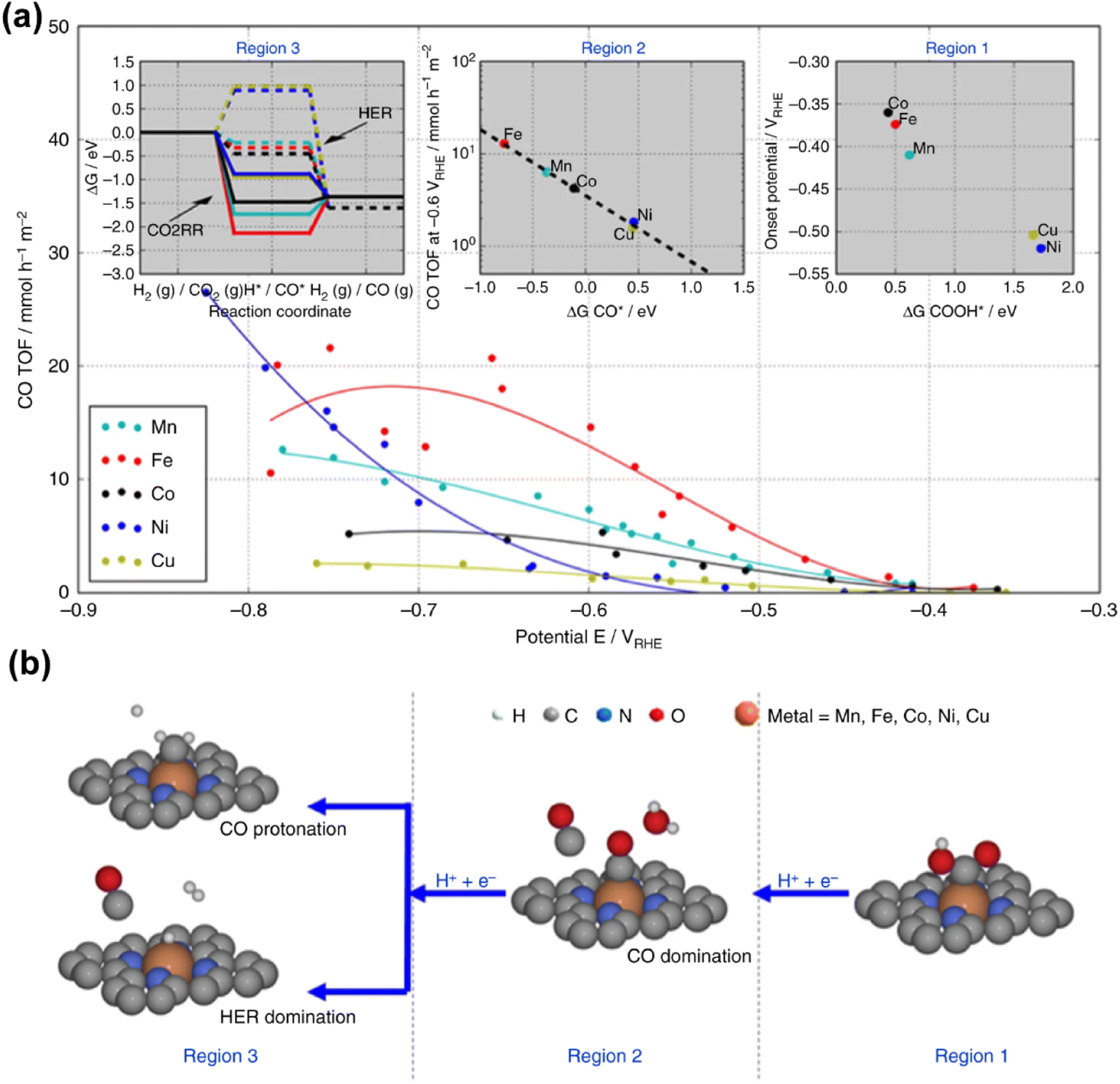 | ||
| Fig. 36 Experimental correlation with simulations. The catalytic reactivity trends (a) and reaction pathway (b) are divided into 3 potential regions with distinctly different rate-determining mechanistic features. Reproduced from ref. 329 with the permission from Springer Nature, copyright 2017. | ||
Chang et al. studied and compared the isolated transition metals (Fe, Co, Ni, Cu, and Zn) using a series of crystalline molecular catalysts, namely metal-coordinated phthalocyanines, to investigate the intrinsic role of the central metals with identical local coordination environments in the performance of CO2 electrochemical reduction.330 In their discussion, they found that the free energy difference of *HOCO and *CO adsorption, rather than any single energy parameter for different metals, was well-correlated with the partial current density of CO production, and exhibited linear scaling relations over a wide range of potentials (Fig. 37). Their findings not only unveil that the optimal binding mode of *HOCO and *CO controls the CO production for various metal-coordinated phthalocyanines, but also manifest again that the electronic nature of central atoms have a great effect on the intrinsic kinetics by regulating the evolution of key intermediates. And the descriptors for correlating the microscopic structure and energy parameters with apparent activity might highly depend on the specific catalytic systems. Such conclusions are not limited to conventional transition metals. As mentioned in Section 3.1, plenty of main group elements, such as Ga,193 In,194 Sn,198,210 Sb,207 Mg,191 Bi,196,197etc., could also take advantage in CO2 reduction reactions in the form of single atoms, although classical d band theory is not responsible for the explanation of their specific activity. The distinct variation in binding energy of key intermediates over SACs based on them stems from their inimitable electronic features, offering opportunities for designing innovative catalysts for efficient CO2 transformation. Furthermore, such research strategies on the basis of the investigation into the molecular-scale interaction between central atoms and specific intermediate species could be also generalized to other SAC systems with different substrates and coordination environments, which are operated under thermochemical and photochemical conditions.46,227,331
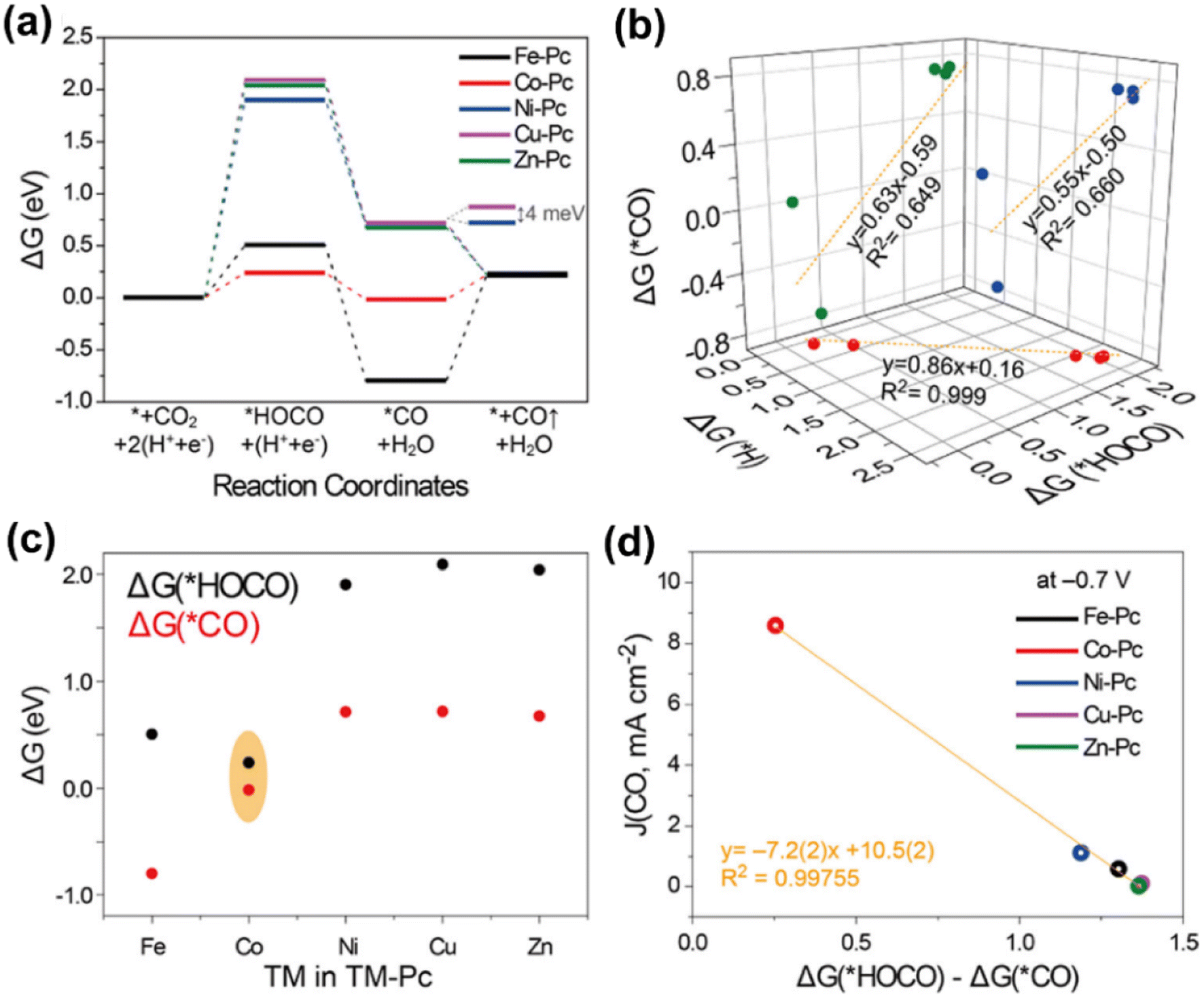 | ||
| Fig. 37 Correlation between experimental results and theoretical calculations. (a) Free energy diagram for CO2 reduction at E = 0 V. (b) Linear fitting between the free energy of reaction intermediates over different transition metals. (c) Free energy of *HOCO and *CO with different transition metals. (d) Plot of the free energy difference of *HOCO and *CO adsorption versus current density of CO production at −0.7 V. Reproduced from ref. 330 with the permission of American Chemical Society, copyright 2022. | ||
The above discussion of central atoms regards atomically dispersed active sites as ‘insular islands’ in the sea of substrates or ‘scattered stars’ in the night sky of supports. They seem to function in an independent and equivalent way. However, the variation of catalytic properties might be potentially caused by the interplay between the central atoms, particularly when they are within close proximity. This implies that the spatial distance of the central atoms might be one of the critical factors, which is decided by using the distribution diagrams of single atoms, resulting from the loading amount and fabrication strategies. The distance of central atoms could be shortened continuously by applying specific precursors and preparation methods, until two or several atoms bond with each other or locate at adjacent sites.332 The double-atom systems, so-called dual-atom or diatomic catalysts (DACs), have been burgeoning research subjects in recent years.35,333–335 Strictly speaking, the DACs do not belong to the SACs, as the metal dimers might function as single sites but show distinctly different properties in comparison with ordinary single atoms, i.e., they are ‘single’ dual atoms rather than ‘double’ single atoms. Such aggregations might further develop and comprise even more than two atoms until reaching the category of NCs. Generally, DACs also exhibit well-defined structures and have research methods similar to SACs. Therefore, we include the DACs in the discussion of SACs in this review and view them as the ultimate situation of shortening the distance between central atoms. Apart from the effect of spatial distance, different assemblies of single-atom sites (more than one type of central atom) might also remarkably affect the reaction kinetics and be exploited for enhanced performance of specific catalytic systems.
As mentioned in Section 3.1, neighboring Pt monomers not only render synergy to vary the reaction barrier, but also steer the reaction to go through a distinct pathway compared with isolated Pt atoms.112 In this way, the catalyst based on neighboring sites exhibits much higher low-temperature performance due to the reduced activation energy in a stepwise process of CO2 hydrogenation into formic acid and methanol. For the reforming reaction of methane with CO2, two sets of single-atom sites, Ru and Ni, are anchored on the same surface of the CeO2 support.273 On the one hand, they play different roles in the whole reaction: Ni is responsible for the CH4 conversion to CO and Ru is in charge of the dissociation of CO2 into CO. On the other hand, however, they also contribute to the sequential formation of H atoms and generation of H2 molecules in synergy: CH4 is activated on Ni to form H atoms and then H atoms are coupled to H2 over Ru sites.
There are more achievements emerging in electrochemical and photochemical circumstances,337,338 such as Co dual-atom,339 Ni–Cu,340 Mn–Co,341 Ni–Co,342 and those mentioned in Section 3.1, and Ni dual-atom,199 Pd dual-atom,200 Ni–Fe,203 Ni–Zn,201 and Ni–Sn,343 in which the central atoms could either be directly bonded, or bridged via non-metal atoms, or not be chemical bonded, yet remain close to each other. Relying on these catalysts featuring multiple-atom active sites, the production of a single C1 product is promoted to different extents. To overcome the intrinsic restriction of the competition between molecular CO2 activation and CO2 reduction product release, a heteronuclear Fe1–Mo1 dual-metal catalytic pair on ordered porous carbon has been designed and achieved a high catalytic performance for driving electrochemical CO2 reduction to CO336 (Fig. 38). Chemical adsorption of CO2 is proved on the Fe1–Mo1 catalytic pair through a bridge configuration, which prompts the bending of the CO2 molecule for activation of the subsequent protonation. The dynamic change from the bridge configuration of adsorbed CO2 species on hetero-diatomic Fe1–Mo1 centers to the linear configuration of CO on the Fe1 center disentangles the scaling relationship between the CO2 activation and the CO release, offering opportunities for regulating the reaction kinetics at the molecular scale in a promoted reaction pathway.
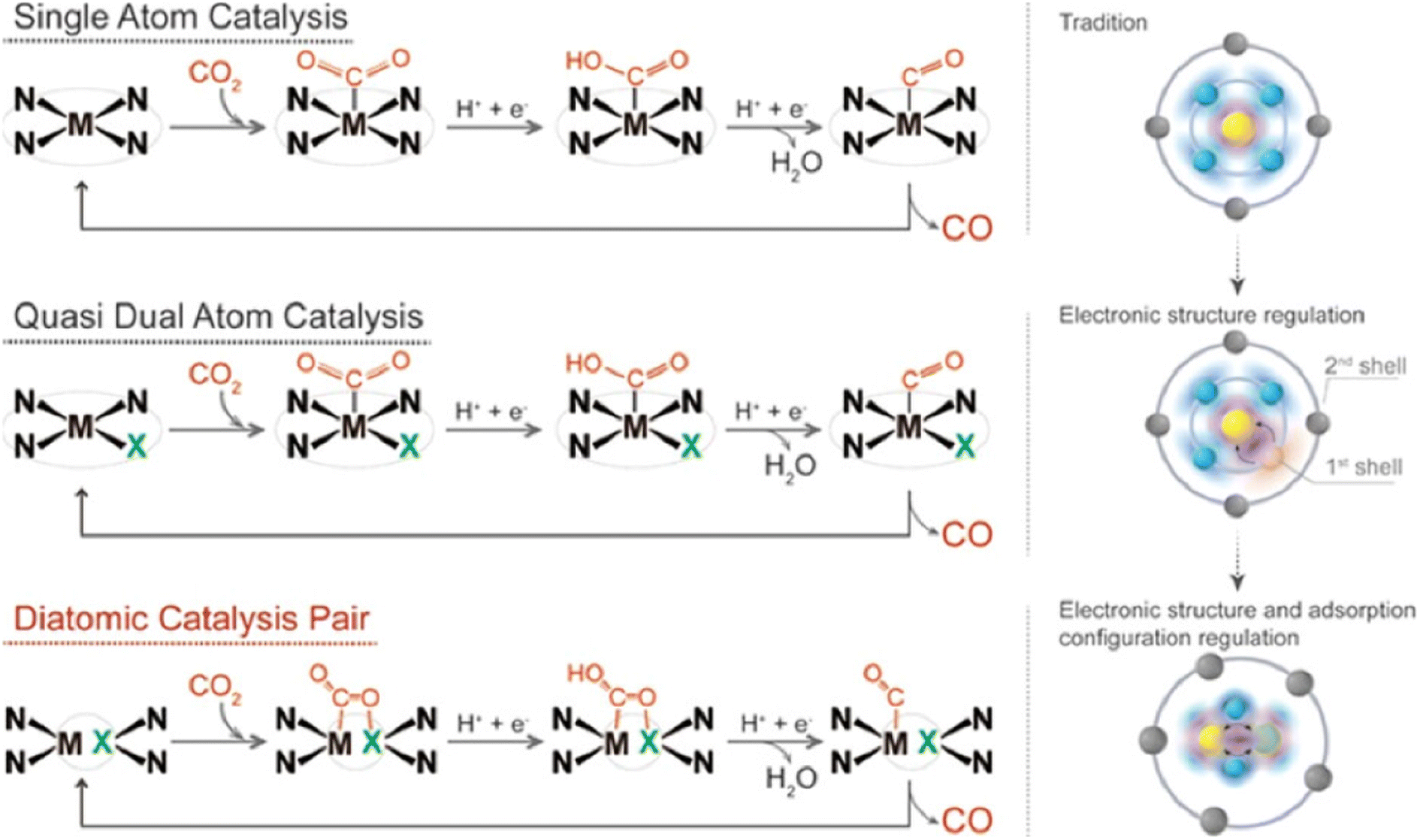 | ||
| Fig. 38 The design of a heteronuclear Fe1–Mo1 DAC and proposed mechanisms for CO2 electrochemical reduction. Reproduced from ref. 336 with the permission of American Chemical Society, copyright 2023. | ||
Besides improving the activity toward C1 products, adjacent central atoms are always considered as desired active centers for the synthesis of C2+ products by enhancing specific C–C coupling processes, which are believed to be difficult on conventional SACs.345,346 As mentioned in Section 3.1, the production of ethanol from CO2 hydrogenation could be realized by the dimers of Pd with nearly 100% selectivity.347 The Pd dual-atom structures not only promote the cleavage of C–O species in *CHxOH to form *CHx, but also favor the C–C coupling between *CHx and *CO. Such a synergistic effect could contribute to superior ethanol selectivity with an appreciable persistence in a continuous-flow fixed-bed reactor, as the formed H2O product could be enriched by a constructed hydrophobic SiO2-shell layer, which creates a quasi-H2O-solvent condition (Fig. 39).344 As for electrochemical reduction of CO2 toward ethanol, adjacent Cu single atom sites have been reported to promote C–C coupling with a higher efficiency compared with that of isolated sites (Fig. 40).348 Such structures are established by increasing the loading amount and corresponding density of Cu atoms anchored on the thin-walled nanotube-shaped nitrogen-doped carbon support. The well-defined Cu–N3 sites are maintained regardless of the extremely high loading, and create synergy between neighboring sites. As a result, the key intermediate species *CO, which are readily generated over single Cu–N3 sites, go through a coupling step in a much more thermodynamically favorable path with ΔG <0 according to the theoretical simulations. In contrast, the isolated sites afford a much higher ΔG value of *CO conversion to *COCO, suggesting the huge thermodynamic barrier for the coupling step, leading to impossible formation of the ethanol product.
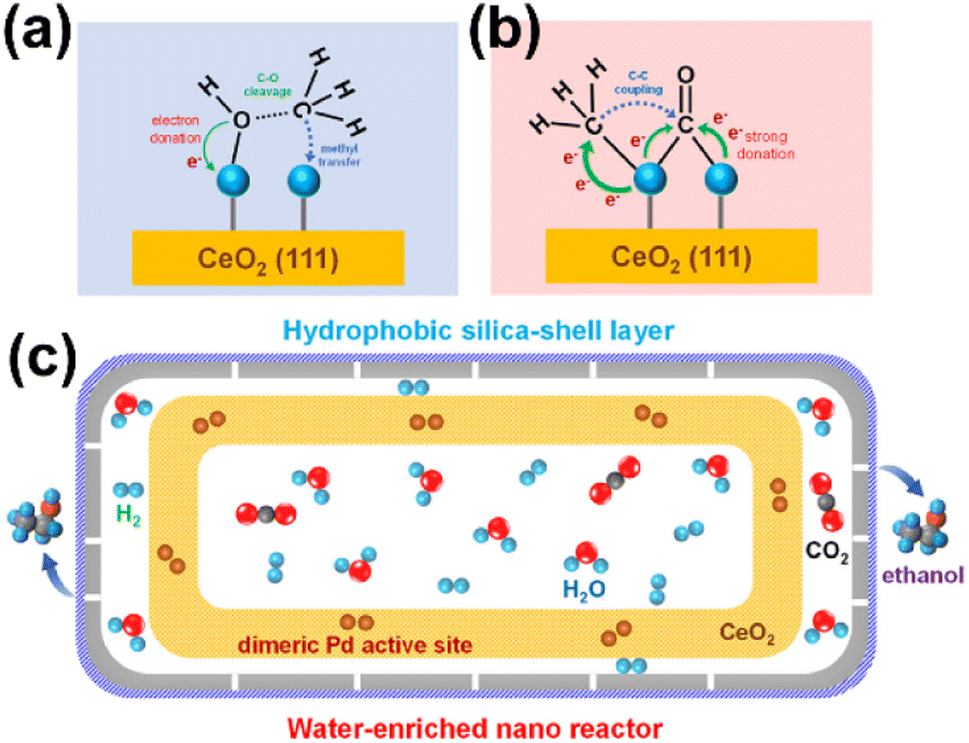 | ||
| Fig. 39 Proposed mechanism for C–O bond cleavage (a) and C–C coupling (b) over the dual-atom Pd sites. (c) Model presenting the in situ generated water enriched in a nanoreactor with a hydrophobic silica shell layer. Reproduced from ref. 344 with the permission of American Chemical Society, copyright 2023. | ||
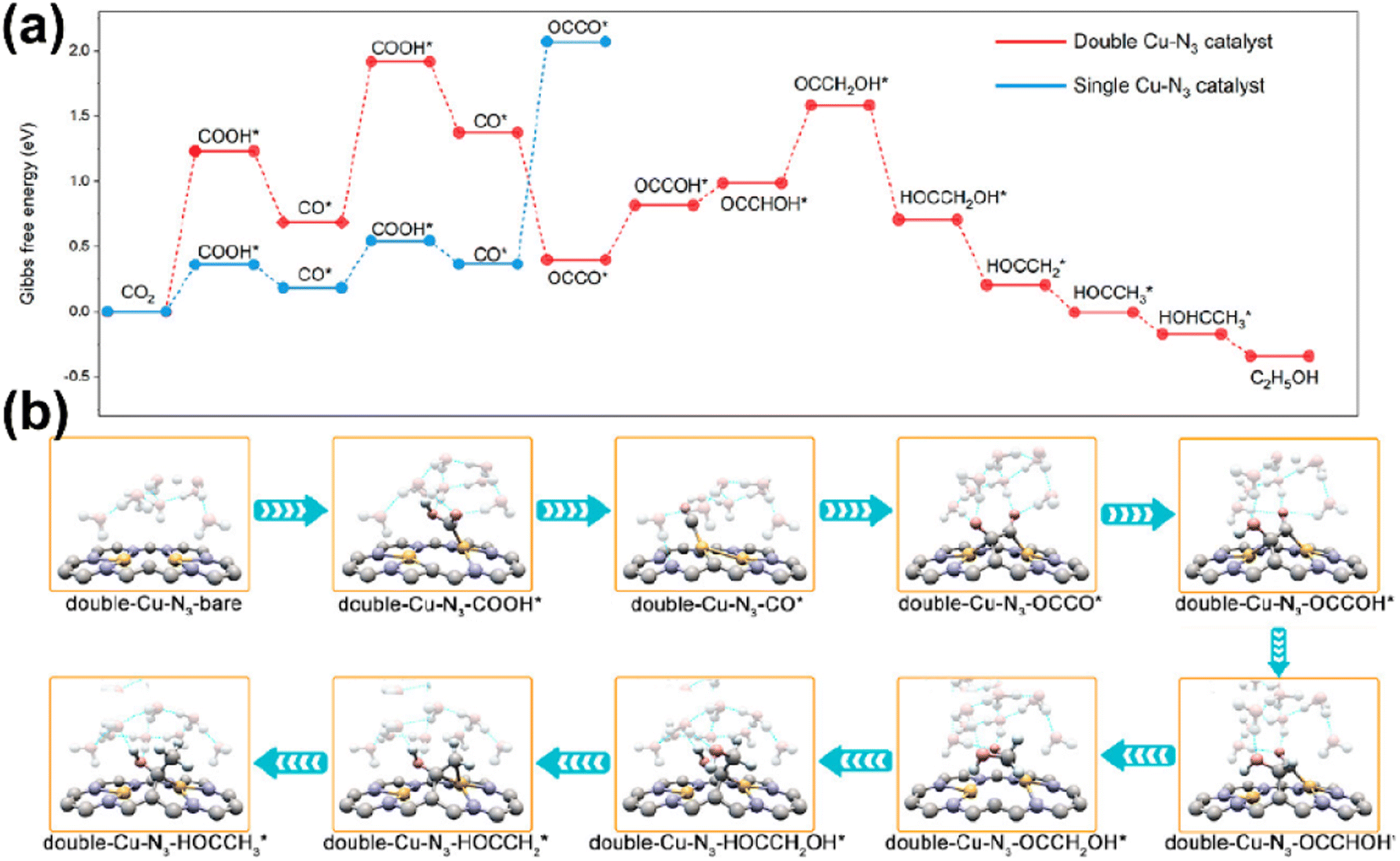 | ||
| Fig. 40 (a) Free energy diagrams calculated for the reduction of CO2 to CH3CH2OH on double Cu–N3 sites and single Cu–N3 sites. (b) Optimized structures in the pathways of CO2 reduction on the double Cu–N3 sites (gray: C of the catalyst; black: C of the adsorbate; red: O; orange: Cu; blue: N; white: H). Reproduced from ref. 348 with the permission of American Chemical Society, copyright 2022. | ||
In photochemical reduction, on the other hand, hetero-diatomic sites have been believed to be more efficient for the C–C coupling reactions.318,349 An In–Cu based photocatalyst could endow the CO2 photochemical conversion toward ethanol with a superior selectivity of 92%. The In–Cu heteroatomic sites are found to promote the adsorption of *CO intermediates and lower the energy barrier of C–C coupling.339 More specific discussion is provided by using a heteronuclear RuCu DAC, by which over 95% selectivity of acetate is achieved through CO2 photochemical reduction (Fig. 41).349 Intuitively, the intermediate atoms absorbed on the symmetrical homonuclear DAC sites (such as Cu dual-atom or Ru dual-atom active sites) would show identical electron scattering readily, which might lead to a strong repulsion between them and thus impede the subsequent C–C couplings. In contrast, possibly benefitting from the distinct electronegativity and gradient orbital coupling of the heteroatoms, the intermediates on heteronuclear DACs might exhibit an asymmetric charge distribution, which would deliver a mitigated repulsive effect and promote the molecular interactions of the formed intermediates, resulting in an augmentation of the collision probability of adjacent intermediates and the subsequent formation of the C2+ intermediates and products. This assumption has been manifested by the followed experimental results and theoretical simulations. Thorough investigations confirm that the strong gradient orbital coupling of Ru 4d–Cu 3d resonance leads to a remarkably weaker electrostatic repulsion of the two formed *CO intermediates on the Ru–Cu heteroatom with an asymmetric charge distribution. The adsorbed CO species are stabilized due to a decreasing energy splitting level, which stems from the strongly overlapped Ru/Cu-d and CO molecular orbitals and their facile split into bonding and antibonding orbitals. All these distinct features of electronic structures and chemical interaction contribute to a more favorable path toward C2+ products in terms of both thermodynamics and kinetics, especially the step of coupling between two side-to-side adsorbed CO.
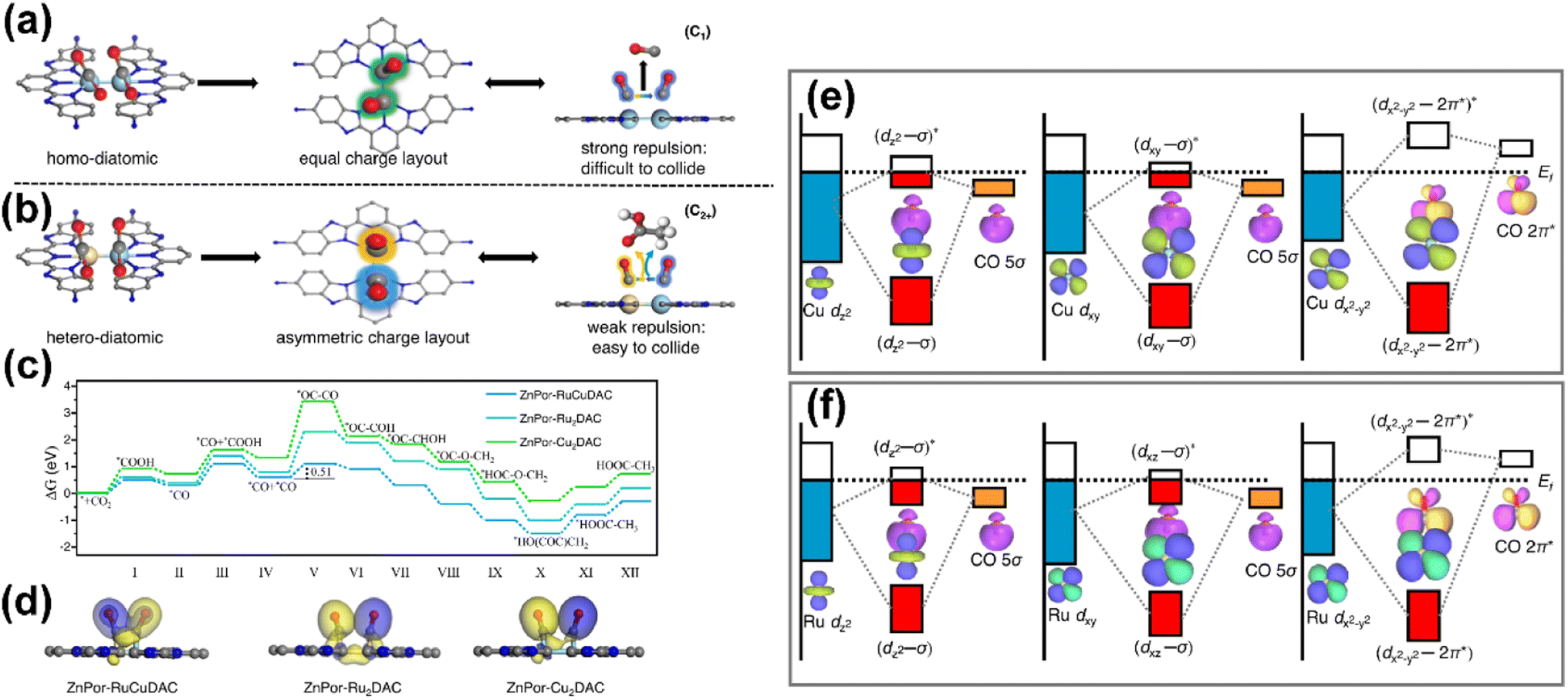 | ||
| Fig. 41 (a) and (b) Assumed photocatalytic CO2 reduction mechanism of C1 and C2+ product formation using homo- and hetero-diatomic catalytic sites, respectively. (c) Gibbs free energy diagrams of CO2 photoreduction to acetate of DACs based on RuCu, Ru2 and Cu2 active sites. (d) Differential charge density maps. Schematic illustration of the adsorbed CO (5σ, 2π*) orbital interactions with the (e) Cu 3d and (f) Ru 4d orbitals in a RuCu hetero-diatomic catalyst. Reproduced from ref. 349 with the permission from Springer Nature, copyright 2023. | ||
In summary, regarding central atoms, two critical factors should be taken into account: the element type that determines the fundamental electronic properties of active sites and the peer effect of identical or different atomically dispersed active sites, which is influenced by both the spatial distance and element type. On the basis of the binding of key intermediate species with the central atom, various descriptors have been proposed for clarifying the molecular-scale structure–activity relationship and gaining in-depth understanding of the intrinsic mechanisms. Either for the enhancement of C1 production or for the realization of C2+ synthesis, the cooperation between single atom sites, especially those in the neighboring location, has been exploited to break through the thermodynamic and kinetic obstacles on the reaction pathways, especially in terms of the stabilization of key intermediate species, subsequent C–C couplings and further transformation. In a word, the selection and engineering of central atoms will be continued for the rational design of SACs used for highly efficient CO2 transformation.
4.2 Coordination environments
The effects of coordination have been fully discussed in homogeneous catalytic systems, as the catalytic properties of central atoms could be readily adjusted by manipulating the properties of ligands, such as steric hindrance effects, pH values and some other electronic features.350–352 SACs, which can combine the merits of both heterogeneous and homogeneous catalysts, should benefit from not only the scaling relationship on the well-defined active sites, but also the adjustable performance by tailoring the local coordination environments,353 as many concepts of coordination chemistry in homogeneous systems could also be extended to SAC systems.354 Intuitively, we could envision a similarity between metal–ligand coordination in homogeneous catalysts and metal–support coordination in heterogeneous catalysts. The adsorption and activation of reactants and intermediates might either competitively or cooperatively co-exist with the interaction between central atoms and ligands, rendering distinctly controllable reaction pathways toward specific target products. The differences might be in the defect sites and standby sites of the support, where the dynamic evolution and the optimization of the coordination structure might take place for the metal-support and the metal–adsorbate interactions under redox conditions, as observed in some realistic catalytic systems.80,81,316,354–356 On the other hand, distinguished from the cases involving metal–metal interplay in the DACs and the cases involving the metal–support interaction in a much longer range, we just discuss the coordination environment in terms of the interplay between metal and non-metal atoms/groups in a relatively near-range level in this section.The effect of ligand atoms in the first coordination shell, which are nearest to the central atoms, has been studied in thermochemical CO2 hydrogenation reactions. As mentioned in section 3.1, Pt1@MIL provides an active center comprising a single Pt atom and its coordinated O atoms for CO2 hydrogenation.126 The metal–ligand cooperativity contributes to the dissociation of H2 and leads to the formation of hydroxyl groups, in which the hydroxy H atoms are added to CO2 to produce HCOO*. Subsequently, the HCOO* species could function as intermediates and go through stepwise hydrogenation, resulting in the preferential production of methanol. For the RWGS reaction, Co–N4 (ref. 89) and Mo–N3 (ref. 91) moieties have been found to afford excellent selectivity to CO. These distinct active centers efficiently steer the reaction pathway by modulating specific kinetic steps, which is *COOH conversion to *CO and the desorption of CO for Co–N4, and direct dissociation of CO2 to *CO. The valence and durability of certain metals could be also sustained with the assistance of coordination groups. Atomically dispersed Co2+, which is stabilized by the –O–Si–ligand, has exhibited nearly 100% selectivity of CO in RWGS.88 With the unique shift between the tetrahedral and the octahedral coordination structure, Co2+–O–Si bonded on an SBA-15 support drives the adsorption of CO2, the dissociation of H2, and the formation and desorption of CO.
For electroreduction of CO2 operated at a moderate temperature, a series of molecular catalysts which are fabricated by fixing metal complexes onto solid supports have been widely applied.357,358 Such molecular catalysts could be viewed either as a transition between homogeneous and heterogeneous catalysts, or as roughly defined SACs that have to combine with ligands to have an effect on the catalytic reactions.359,360 The well-defined isolated central metal atoms and corresponding ligand molecules are collectively responsible for the activation of reactants and the regulation of reaction pathways. For instance, phthalocyanine (Pc) ligands, commonly used in transition-metal-based SACs, are subjected to a one-electron reduction when coordinated to an iron single atom in a FePc SAC (Fig. 42). The environmental ligand functionalities, together with the adsorption and activation of the CO2 molecule, prevent the over-reduction of Fe species from bivalent to univalent and even the metallic state. The Pc-coordinated bivalent iron species in the high-spin state (HS Fe(II)Pc−), which are generated from in situ reduction of trivalent iron, are identified as efficient active centers with a modified d-band center and correspondingly stronger binding with CO2 molecules. In such a SAC, the ligand molecule has two types of effects on the ultimate reaction performance: (i) modify and maintain the electronic configuration of the central atoms; (ii) help in the formation of intermediate species as an efficient charge transfer channel. These effects have been also reported in other SACs for CO2 reduction, such as Cu–C,213 Ni–N,175,361 Fe–N,175 Fe–B,218 Zn–N,173 Co–N,362 In–N,206 Fe–P,363etc.
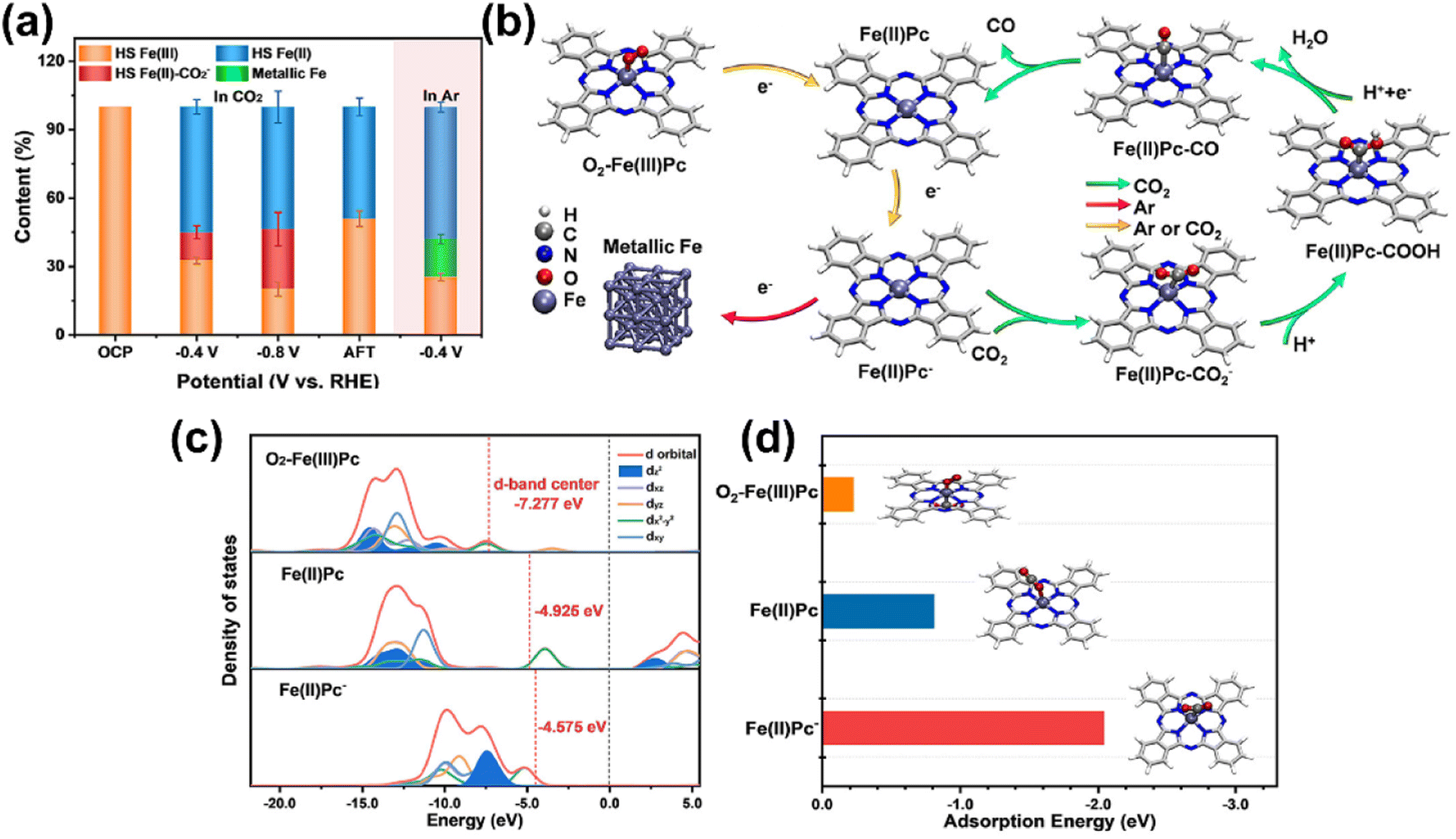 | ||
| Fig. 42 (a) Relative signal intensity of different Fe species obtained from the fitting of 57Fe Mössbauer measurements. (b) Proposed dynamic evolution routes of single-Fe-atom species during catalysis under CO2- and Ar-saturated conditions. (c) Density of states (DOS) for Fe-3d orbitals in O2–Fe(III)Pc, Fe(II)Pc, and Fe(II)Pc−. (d) CO2 adsorption energy over O2–Fe(III)Pc, Fe(II)Pc, and Fe(II)Pc−. (c) dz2 orbital of Fe(II)Pc and Fe(II)Pc− and the Kohn–Sham molecular orbital (MO) contours of the dz2–π* bonding orbital (BD) of Fe(II)Pc–CO2−, where the red and blue regions are positive and negative orbital phases whose isovalue is 0.05. Reproduced from ref. 357 with the permission of American Chemical Society, copyright 2023. | ||
Further doping of heteroatoms, such as O, S, P, Cl, etc., into pristine SACs, or changing the coordination number might also have potentially positive influences on the reaction kinetics and apparent performances. For instance, main-group metals, which could intrinsically overcome the too strong adsorption of *H and *CO usually occurring over traditional d-block metals, have attracted more and more attention in the construction of SACs for CO2 electrochemical reduction. Nevertheless, they always suffer from the delocalized p-orbitals and accordingly insufficient capability of CO2 activation, as the localization of electrons is widely accepted as an effective strategy for promoted CO2 activation.190 Wang et al. developed a Ca-based SAC featuring localized electrons on p-orbitals through an asymmetric coordination by O (Ca–N3O) doping (Fig. 43).192 According to the theoretical analysis, such an asymmetric coordination creates a larger charge shift from active centers toward *COOH and a larger shift of the p-band center in the projected density of states (PDOS) in comparison with that of Ca–N4. This always suggests the increasing ability for stabilizing the *COOH and a lower energy barrier from *CO2 to *COOH. In addition, Ca–N3O still retains an intrinsically higher energy barrier for the HER and easier desorption of *CO compared with the representative transition metal site Fe–N4. Therefore, a reinforced performance of CO2 conversion to CO could be achieved by Ca–N3O. A similar result has also be reported in a Cu-based SAC, in which the asymmetric atomic interface of Cu single atoms (Cu–N3O) affords enhanced performance of CO2 electrochemical reduction toward CO.364
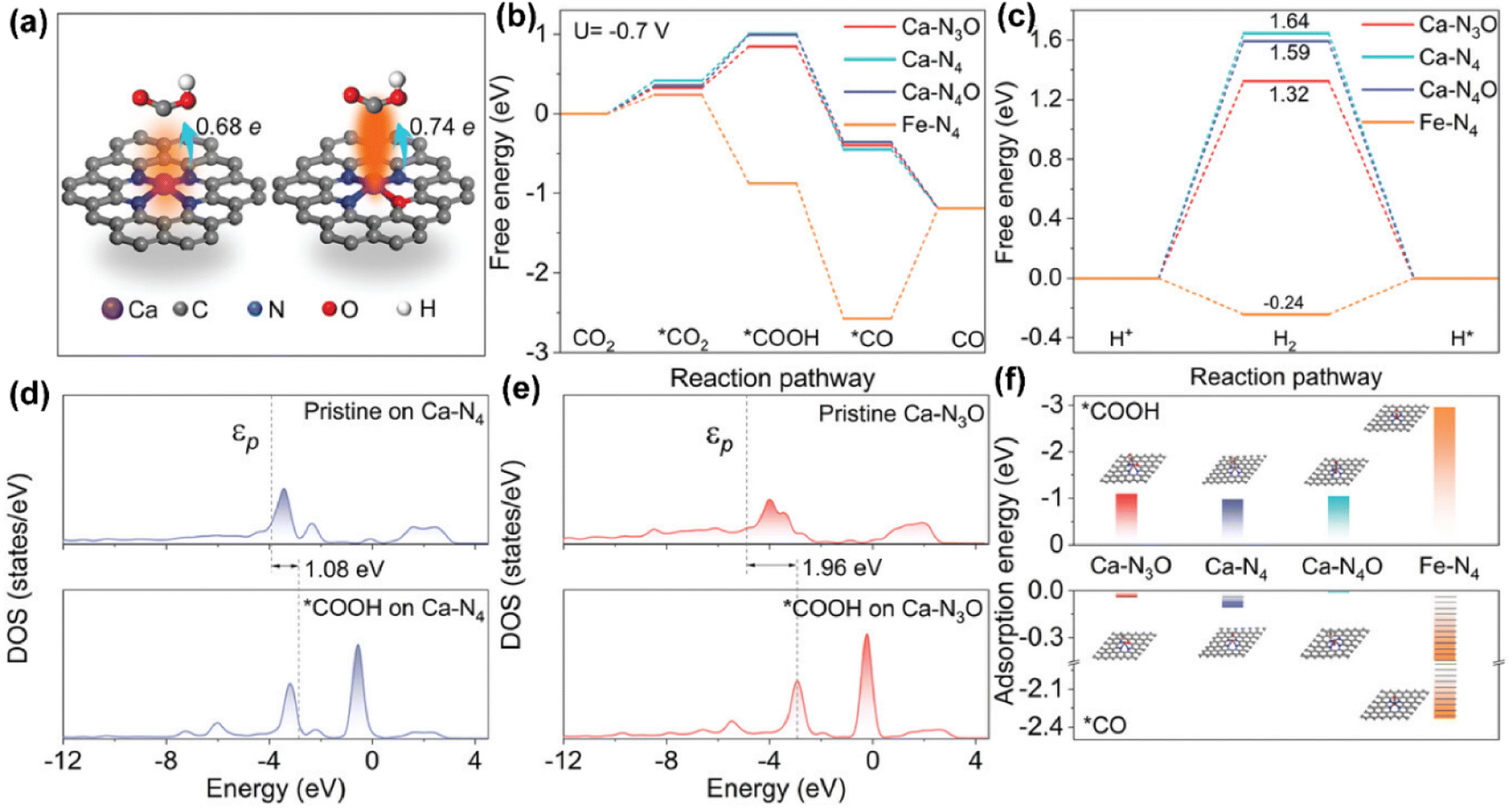 | ||
| Fig. 43 (a) Comparison of the schematic diagram and Mulliken charge analysis for *COOH adsorbed on Ca–N3O and Ca–N4 orbitals. Free energy diagram for (b) CO2-to-CO conversion and (c) hydrogen evolution. PDOS and p-band center (εp) before and after *COOH adsorption on (d) Ca–N4 and (e) Ca–N3O. (f) Adsorption energy of *COOH and *CO on catalysts. Reproduced from ref. 192 with the permission of Wiley, copyright 2023. | ||
To realize localization of electrons, Zhang et al. fabricated a low-coordinate Ni SAC featuring Ni–N3 sites365 (Fig. 44). The change in the coordination number from the traditionally reported four to three in this work is carried out by retrofitting a Zn-based MOF derived material with Zn–N3 sites. Such a Ni–N3 active center delivers a much higher ability to stabilize the *COOH intermediate and thus contributes to a lower energy barrier for *CO2 conversion to *COOH, which is the rate-determining step of the whole reaction. We should notice that the strategy of constructing an asymmetric configuration of the coordination environment of central atoms might be universal for the activation and transformation of such a symmetric nonpolar molecule CO2, through either heteroatom doping198,366–369 or coordination number changing.146,176,370–373 Although possibly following different mechanisms, this strategy has also been reported in more generalized CO2 reaction systems,349,374 and might be extended to the activation of other nonpolar molecules.375
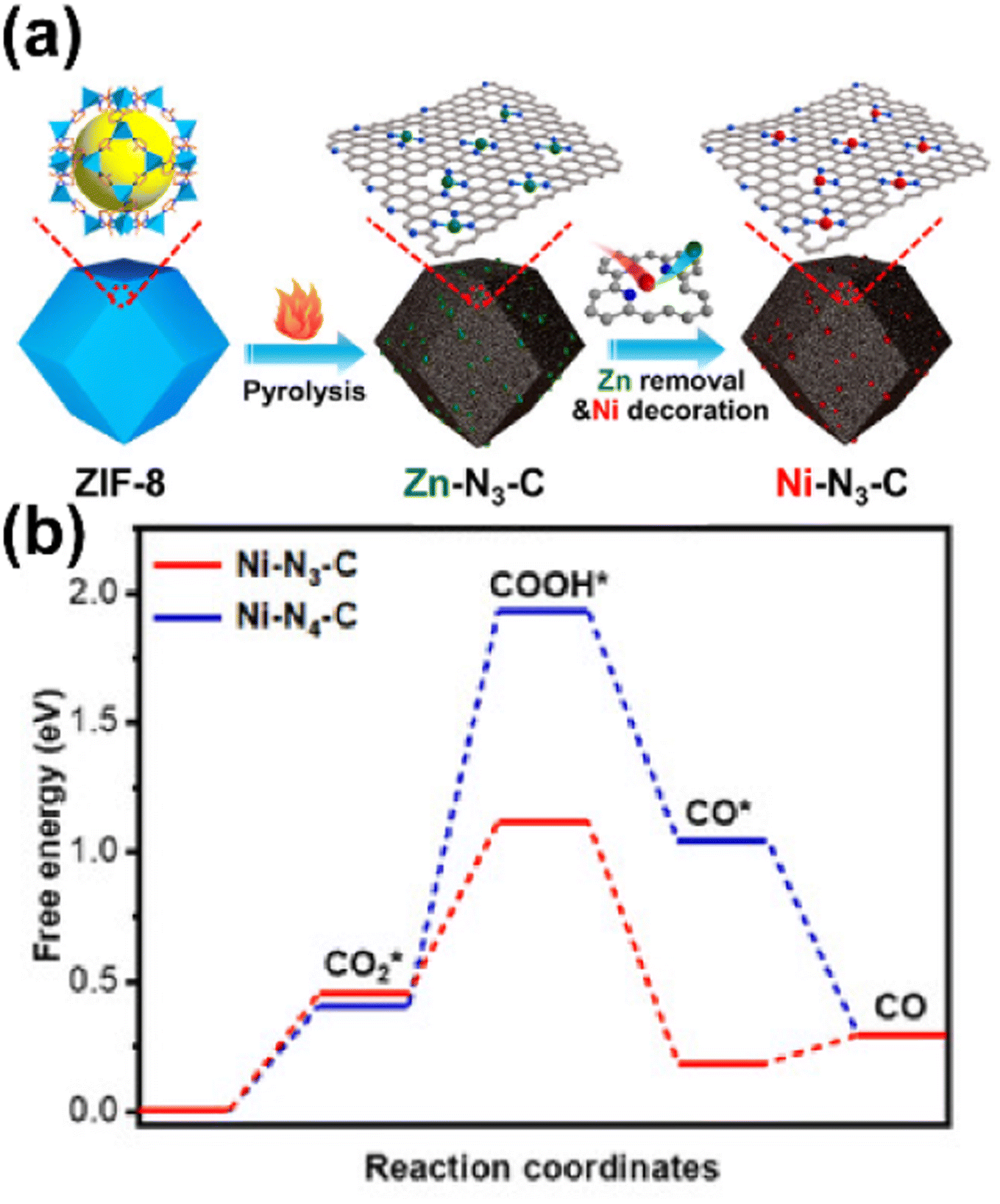 | ||
| Fig. 44 (a) Fabrication of low-coordination single-atom Ni electrocatalysts. (b) Reaction paths and free energy diagrams of CO2 reduction to CO for Ni–N4–C and Ni–N3–C. Reproduced from ref. 365 with the permission of Wiley, copyright 2021. | ||
As for photochemical transformation of CO2, metal–ligand assemblages might also play a vital role in the charge transfer process, which is possibly the rate-controlling step in a stepwise evolution from light to electron then to chemicals. In these catalysts, single atom sites with their adjacent ligand atoms or groups could function as not only an electron bridge for the promoted charge transfer, but also the active centers for CO2 activation. As mentioned in Section 3.1, the rare-earth metal single atoms La,248 Er247,257 and Dy244 in different coordination environments all exhibit dual functions of CO2 molecular activation and charge transfer, in which the 4f levels of these metals and the interplay between their orbitals and the orbitals of neighboring coordination atoms both make a critical difference. Wang et al. developed a Cu single-atom-based electron bridge, N–Cu–S, in a Z-scheme photocatalyst for the production of CO and O2 from CO2 reduction.376 Such an atomic structure achieves a great enhancement of the interfacial charge-transport process, thereby favoring the efficient catalytic transformation of CO2 even in the absence of sacrificial agents. In contrast, impeded charge transfer is exhibited over the heterojunction material without any Cu species, and inferior promotion effect is presented by Cu NPs (Fig. 45).
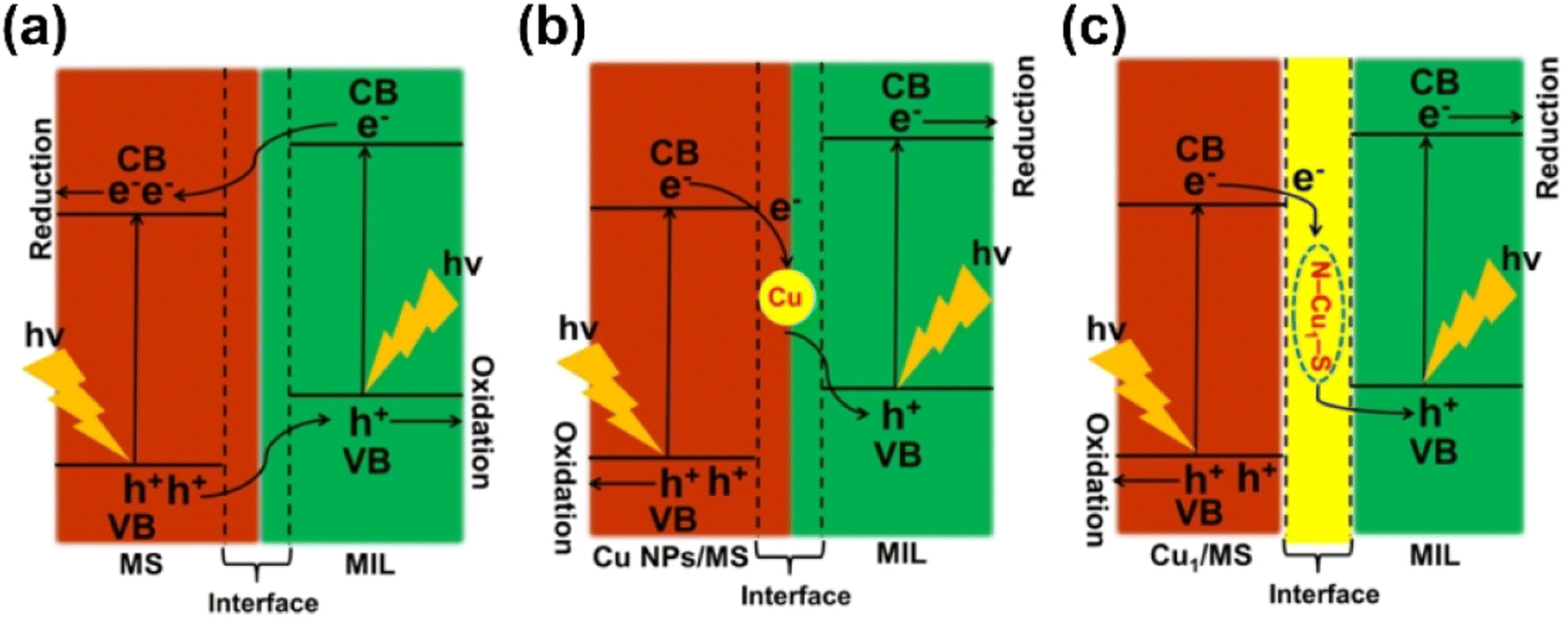 | ||
| Fig. 45 Electron-transfer process models of (a) MS (MoS2)/MIL (MIL-125-NH2), (b) Cu NPs as the electron bridge, and (c) Cu SA as the electron bridge. Reproduced from ref. 376 with the permission of Wiley, copyright 2023. | ||
Nowadays, the effect of the first coordination shell, which comprises non-metal atoms directly bonded to the central atoms, has been substantially learned. In fact, chemical interaction, which is a specific electrostatic force by nature, could also have a long-range influence. This implies that the atoms or groups in the second coordination shell, which are not directly bonded to central metal atoms, might also play an important role in the modulation of the performance of the central atoms, as already reported in some SAC systems for CO2 transformation and other catalytic reactions.377–382 We should emphasize that the non-metal species beyond the first coordination shell might also have an effect by acting as isolated active centers or adsorption sites,383 which would not be discussed in this section but introduced in Section 4.4 when reviewing the synergy between single-atom sites and other active centers.
As mentioned in Section 3.1, simultaneously regulating the near- and long-range coordination environment has been reported to effectively modulate the electronic structure of single-atom Zn sites. ZnN4 sites are decorated with an axial thiophene-S ligand in the first coordination shell, as the surrounding phosphorus atoms are constructed in the carbon matrix of a hollow carbon support (denoted as ZnN4S1/P-HC).174 Such a dual manipulation of the microenvironment at different levels is able to synergistically contribute to the increase in electron localization around the Zn sites. Thus, the adsorption of the *COOH intermediate is strengthened, while the energy barrier for the formation of H2 is still kept high enough to eliminate the competitive HER during an electrochemical CO2 reduction reaction in H2O solvent, resulting in an enhanced performance (Fig. 46). This successful example should be attributed to the remote electron induction effect, which might be an important and efficient strategy for supplementary modulation of the electronic structures of central atoms. In the cases of sulfur-doping into the outer coordination shell of Zn–N4 (ref. 173) and Fe–N4,384 a proton-feeding effect has been proposed as a more important contribution to the reinforced electroreduction of CO2 compared to that of only the electronic effect on the central atoms, although the latter is also making a difference. As for S-doped Zn–N4 sites, the dissociation of bicarbonate (HCO3− → H+ + CO32−) is believed to be enhanced with the introduction of S atoms, leading to the accelerating formation of protons, which then participate in the activation of CO2 toward *COOH. In contrast, the S-doped effect on Fe–N4 is considered to promote the activation of H2O, which offers protons for the continuous formation of the *COOH intermediate.
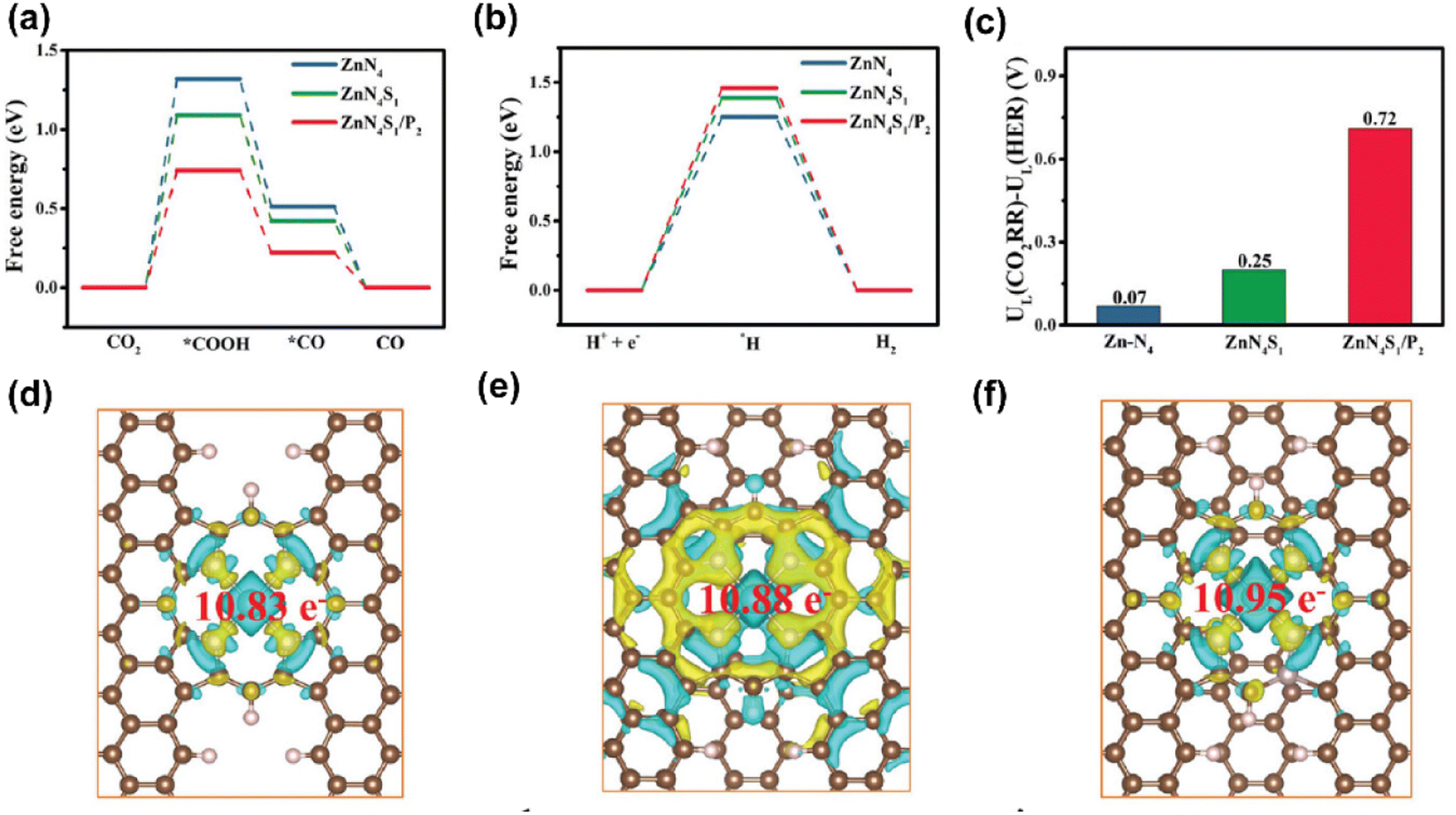 | ||
| Fig. 46 Calculated free energy diagrams for (a) CO2 reduction and (b) the HER. (c) Limiting potential differences for different active moieties at U = 0 V versus RHE. The charge density difference of (d) ZnN4, (e) ZnN4S1, and (f) ZnN4S1/P2 models. The iso-surfaces in yellow and cyan represent electron accumulation and repulsion, respectively. The iso-surfaces were set to be 0.04 e Å−3. Reproduced from ref. 174 with the permission of Wiley, copyright 2023. | ||
In this section, we provide a critical discussion about the effect of the coordination environment and its regulation for tuning the catalytic performance of SACs in CO2 transformation processes. Different coordination endows central single atoms with distinctly varied electronic and geometric structures and accordingly reinforces the charge transfer and modulates specific reaction kinetics. By engineering the coordination microenvironment at different levels, including changing the coordination number, constructing axial coordination, and doping heteroatoms other than carbon and nitrogen, the intrinsic catalytic properties of central atoms could be well regulated with optimized energy bands, electron orbitals and correspondingly modulated intermediate adsorption and transformation. Furthermore, the non-metal atoms beyond the first shell might also have an effect on the central atoms through either remote electronic induction or other kinetic reinforcements to specific reaction procedures. As a result, all these efforts to regulate the coordination environment contribute to the highly controllable properties and performances of SACs, offering excellent opportunities for their widespread use in various reactions under different conditions.
4.3 Support effect
The most significant difference between homogeneous metal-based molecular catalysts and heterogeneous SACs is the application of supports. Supports, basically speaking, are designed and prepared for fixing the central atoms onto specific locations of solid surfaces, thus preventing the loss of active sites during heterogeneous reactions under harsh conditions. Beyond the original intention of support construction, the chemical properties and catalytic performances could be also modulated by controlling the features of supports and the synergistic effects between supports and active centers. In particular, the supports used in electrochemical and photochemical processes are always responsible for charge transfer385 and light-induced charge separation,51,54 respectively. In some cases, the supports could also play a vital role in the adsorption, activation and transfer of certain reactive moieties, such as the most common hydrogen and oxygen species.77,120,226 We should emphasis that the supports discussed in this section would be limited to their intrinsic topological and electronic properties, aiming at their effect on the single-atom sites but not involving active centers other than single atom sites grafted onto them. The synergy between single atom sites and other active centers would be elaborated in the next section.Generally, the effect of supports might be able to be interpreted based on three aspects:17 (i) the dispersion and location of central atoms for the optimal stability and activity of active centers; (ii) the chemical interaction between the substrate and central atoms leading to the structure change of active interfaces; (iii) porous structures and channels for the transportation of reactants and products.132 These aspects could either exert influence independently or co-exist and convolute in a given catalyst. Thus, it might not be an easy task to design a suitable support for SACs without comprehensive understanding of the complex physicochemical factors.
First, the selection and engineering of supports should take the loading and stabilization of single atoms into consideration. As mentioned in Section 3.1, a porous organic polymer affords an excellent support for the anchoring of single atom iridium and provides a suitable chemical environment for quasi-homogeneous reduction of CO2 into formate.101 The much higher performance of this support in comparison with that of traditional activated carbon and popular C3N4 might be attributed to the abundant functional groups, which could be readily introduced during the preparation processes. Relatively high loading of Ir single atoms thus could be chelated onto the support with a satisfying durability. On the other hand, vacancy or defect sites have been extensively reported to be used to seize the single atoms and tune the intrinsic activity of SACs.235,386,387 Single-atom Ir sites could be anchored on the In2O3 substrate, as In2O3 is partially reduced with H2 with the formation of oxygen vacancies prior to being subjected to wet-chemical impregnation.59 Beyond the effect on the fixing of single atoms, the oxygen vacancies even present a synergy with single-atom sites during the activation of CO2 and C–C coupling. In this way, such an Ir SAC realizes effective ethanol production from liquid-phase CO2 hydrogenation, as mentioned in Section 3.1. To tailor the local nitrogen coordination environment of the central single atom Ni for the construction of an efficient Ni–N moiety, Chen et al. adopted a preparation strategy of treating precursors by using low-pressure plasma induced in a microwave oven.388 In this way, the coordination environment of Ni-SACs could be facilely tuned by varying the treatment duration. A longer treatment prompts the reconstruction of the coordinative nitrogen species around Ni atoms, forming a Ni–N2 configuration with neighboring nitrogen vacancies. The resulting active centers with a highly defective local pyridinic N environment afford abundant spare sites for bonding CO2 and thus reduce the energy barrier for the formation of intermediate COOH* and CO*, delivering an enhanced performance in CO2 electroreduction. The formation of defect sites could be also realized by the chemical interaction between metal species and substrates rather than the preparation processes. For instance, on certain crystal facets of CeO2, the presence of Pd could highly promote the formation of oxygen vacancies by providing dissociated H species under a H2 atmosphere, which then facilitate the removal of surface oxygen atoms.121 The oxygen vacancies could serve as adsorption sites for CO2, where CO2 is further activated and goes through stepwise hydrogenation into methanol. A similar effect of chemical interaction between metals and substrates has been also reported in a Cu–TiO2 system for photochemical reduction of CO2.231 Besides inducing the spontaneous formation of oxygen vacancies, such an interaction even affects the spatial distribution of Cu single atoms, which form a configuration featuring neighboring sites engaging in the catalytic transformation of intermediate species.
Recently, with the assistance of the well-defined metal-coordinated phthalocyanines (Pc) as platform molecules supported on carbon-based substrates, the regulation of isolated metal sites based on the effect of chemical interaction between metal species and substrates has been extensively studied in the electrochemical reduction of CO2.330,389–399 As reported in many cases, the metal-Pc sites are loaded on carbon-based substrates, which feature high specific area and provide remarkable electrical conductivity and stability due to the inherent π-conjugated systems in them. However, a promoted strong d–π conjugation between the d orbitals of the metal centers and the π orbitals of the substrate always causes electron transfer from the metal center toward the substrate, i.e., the strong delocalization of electrons introduced by the support. This turns out to be detrimental for the activation of CO2 molecules on metal sites of SACs and vastly hampers the catalytic transformation. To solve this problem, Wang et al. introduced cyano groups (−CN) onto the C3N4 substrate for attenuating the d–π conjugation of single-atom Ni sites.394 As a consequence of the rupture of an aromatic heterocycle near the Ni atoms, the CN groups break the integrity of the conjugated plane between Ni and substrates, therefore confining the electrons to the vicinity of the central metal. The increased localization of electrons in Ni not only favors the adsorption of CO2, but also lowers the free energy barrier for the formation of the intermediate *COOH in the electroreduction of CO2, as predicted by the theoretical calculations and proved by the experimental results (Fig. 47). Interestingly, in an earlier report, the decoration on the Pc ligands by molecular engineering of the pendant groups into –OCH3 and –CN instead of the substrates gives a different result, in which the methoxy-decorated single-atom NiPc sites exhibit much higher performance than that of –CN due to the lower energy barrier for the formation of *COOH.395
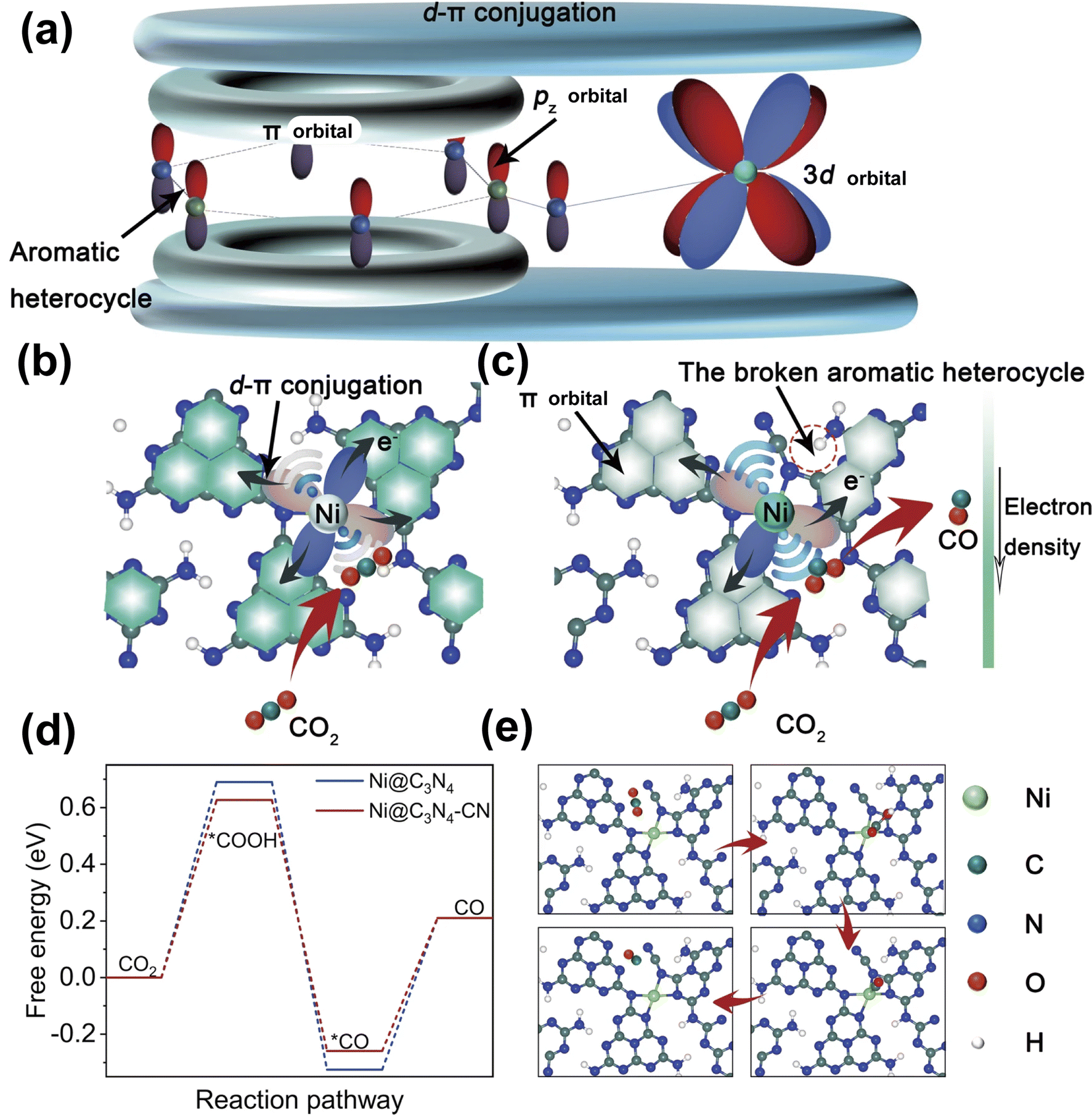 | ||
| Fig. 47 (a) Schematic of d–π conjugation. The effect of d-π conjugation on CO2 activation over (b) Ni–C3N4 and (c) Ni–C3N4–CN. (d) Free energy diagram of CO2 conversion to CO. (e) Structure and adsorption configurations of key intermediates on Ni–C3N4–CN. Reproduced from ref. 394 with the permission of Nature Springer, copyright 2022. | ||
The geometric structure of supports, such as the morphology of crystals121,283 and the curvature of supports,170,171,400 has been also considered as critical factors affecting the electronic structure of single-atom sites via changing the metal-support interaction. The former has been substantially studied in various catalytic systems, and could be rationalized with the distinct properties of different facets, e.g., interatomic spacing, symmetry, lattice-matching, internal strain, defect sites, etc.37,73,385 And the latter has been also more and more recognized in catalytic reactions recently, especially those with carbon-based substrates in electrochemical systems.401,402 Based on the regulation of curvature, the optimization of performance in CO2 reduction has been realized over Zn single-atom sites supported on carbon nanofibers and nano-onions.170,171 By using carbon nanotubes (CNTs), an ideal substrate with an adjustable surface curvature, for supporting molecular centers of single atoms, like CoPc, Su et al. studied the enhancement effect of the strain change on CO2 reduction396 (Fig. 48). The strain change of molecular sites, which is induced by the variation of local curvature, could be implemented by selecting CNTs with different diameters, i.e., the longer the diameter, the smaller the curvature. The largest curvature occurs in a single-wall CNT (SWCNT), where the best performance of methanol production is also shown, as the CoPc has been supported beforehand. The better performance could be attributed to the significantly stronger adsorption of CO and promoted activation of the CO bond in adsorption sites, which both facilitate the deep protonation toward methanol. More intrinsically, the strain induced by the curvature of substrates brings about the distortion of geometry of the adduct CoPcH4(CO), leading to a lower free energy than that of the flat analogue. Inspiringly, such an effect on the basis of strain modulation could even be extended to FePc and NiPc for the oxygen reduction reaction and CO2 reduction reaction, respectively.
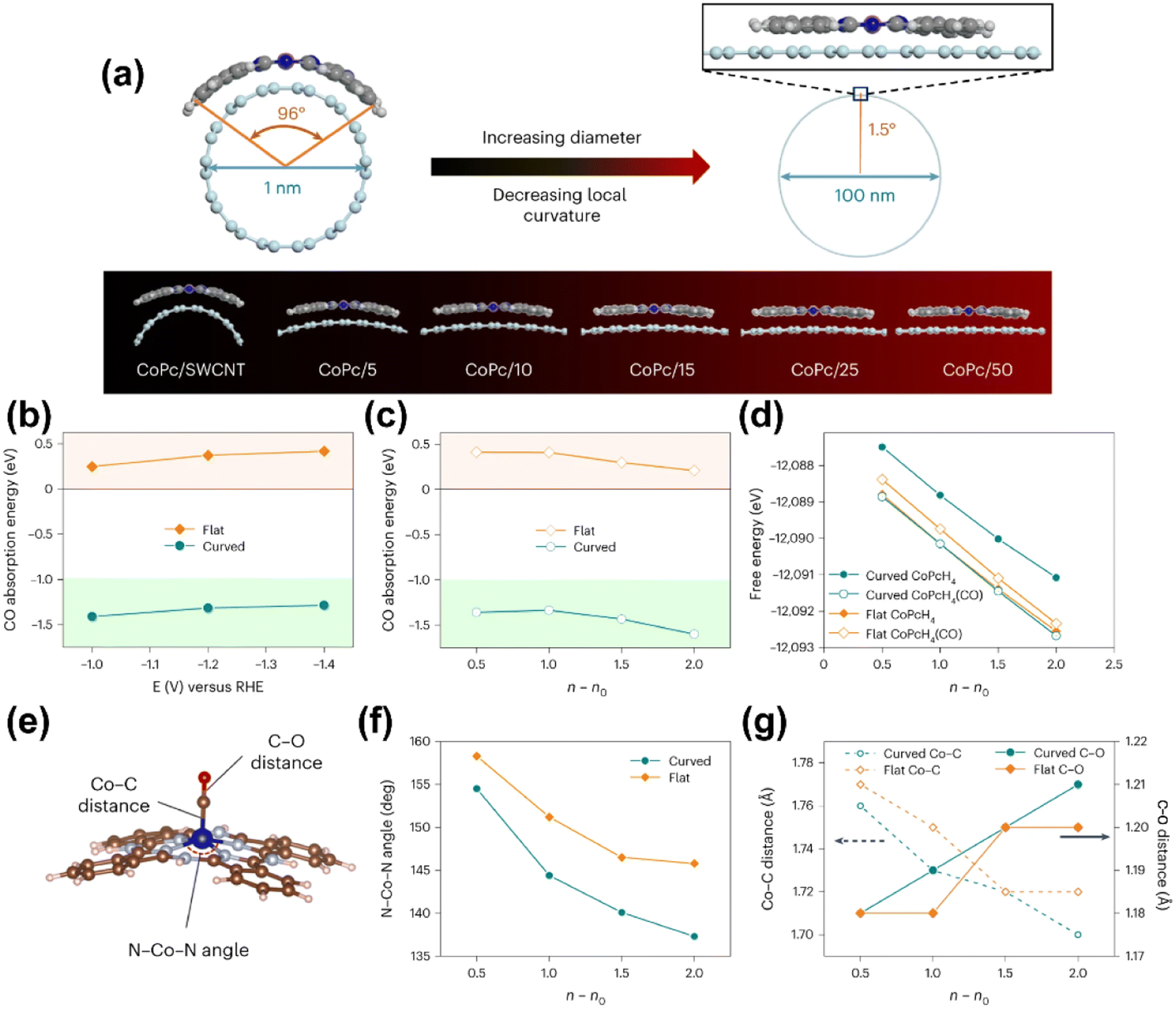 | ||
| Fig. 48 (a) Illustration of the structural distortion of CoPc on different-diameter CNTs. Absorption energy versus potential (URHE, V) (b) and n – n0 (c), where n is the number of electrons added (n – n0 = 1 indicates that 1 electron has been added to the neutral system). (d) Free energies of curved and flat CoPcH4 with and without CO adsorption versus n – n0. (e) Representative N–Co–N angle and Co–C and C–O distances of CoPcH4. (f) N–Co–N angle (degrees) versus n – n0 for the curved and flat structures. (g) Co–C and C–O distances (Å) versus n – n0. Reproduced from ref. 396 with the permission of Nature Springer, copyright 2023. | ||
Apart from the effect on fixing single-atom species and modulating the properties and behaviors of active sites via metal-support interaction, the texture features of supports, like pores and channels, have also shown significant influence on the performance of SACs, such as controlling the diffusion for enhanced mass transfer. Such regulation of diffusion kinetics could not only accelerate reactions by enriching the reactants to a higher local concentration and increasing the probability of contact with active centers, but also shift the reaction equilibrium to a positive orientation and prevent secondary side reactions by evacuating the products in time. Reticular materials, MOFs and COFs, and other porous carbon-based materials have been extensively used for this purpose,403 either as precursors that go through subsequent pyrolysis and other post-treatment,145,404 or directly as substrates for encapsulating or anchoring single-atom metal species.103,107,266–270 Rather than their role in the photochemical catalytic reactions for light harvesting and charge dynamics as mentioned in Section 3.1,226,242 the exploitation of their rigid, robust and multifunctional porous framework is more universal and widely realized in non-photochemical CO2 transformation processes.405 By using polystyrene spheres (PS) as a hard template during the synthesis of iron-doped ZIF-8, highly ordered hierarchical porous single-atom Fe catalysts are fabricated as an excellent platform to investigate the pore size effect on electrochemical CO2 reduction in detail (Fig. 49).404 The pore size could be facilely modulated by changing the size of PS, and a delicately designed porous structure with larger mesopores and macropores significantly creates an advantageous environment with a high local CO2 concentration around the Fe single-atom active centers, which might be attributed to the intrinsically tuned mass transfer for enrichment of CO2 inside the catalyst. Such a porous structure together with the single-atom sites helps to achieve higher performance, especially in the limited mass transfer region. In fact, whether in the thermochemical or the electrochemical or the photochemical processes, increasing the contact between reactants and active centers by enhancing mass transfer yet without affecting the intrinsic features of single-atom sites is always an efficient strategy for improving the apparent performance. The effect of mass transfer could be altered by using the operation conditions, and thus it might be not so significant for those cases that aim at studying the reaction kinetics of SACs rather than diffusion kinetics. However, it would still be an important subject to optimize the diffusion properties of SACs for their practical application in a realistic scenario. Besides the influence on the regulation of the concentration of guest molecules via controlling diffusion, the porous structures might also make a difference in modulating the density of active sites, as reported in a Cu SAC with single Cu atoms embedded onto two-dimensional carbon nitride406 (Fig. 49). The nitrogen-appended nanometric pores afford high loading of single-atom Cu species and confine multiple Cu sites in adjacent locations through a metal ion exchange process between Li+ or Na+ and Cu2+. These Cu sites are kept insulated from each other with a spatial distance beyond that of conventional Cu–Cu, and homogeneously coordinated with two nitrogen atoms within the nanopores of supports. It is manifested that these single-atom Cu sites cooperatively alter the reaction energy profiles into having a much lower barrier for the stepwise formation of CO* and CH* intermediates, resulting in a higher yield of CH4 in the electrochemical CO2 reduction reaction. In the design and fabrication of such a SAC, the porous structure plays a vital role in the confinement of active sites for cooperative catalysis.
 | ||
| Fig. 49 (a) Illustration of the synthesis route of FeNC and hierarchical porous (HP)-FeNC; (b–e) SEM images of HP-FeNC. Reproduced from ref. 404 with the permission of Wiley, copyright 2023. (f) Synthetic scheme of various Cu-CN SACs (poly (heptazine imide) and poly (triazine imide) are denoted as PHI and PTI, respectively) samples through metal ion exchange with Li+ and Na+, respectively. (g) Reaction process of the CO2 reduction reaction on PHI-nCu (n = 1, 2, 3). (h) Local density of states of Cu-d orbitals on PTI-nCu (n = 1, 2, 3). (i) Reaction process of the CO2 reduction reaction on PTI-nCu (n = 1, 2, 3). (j) Charge density difference of CH* absorbed on PTI-2Cu. The yellow and azure areas indicate electron accumulation and loss, respectively. The white, gray, silver, and blue balls represent H, C, N, and Cu atoms, respectively. Reproduced from ref. 406 with the permission of Wiley, copyright 2023. | ||
In summary, besides the fundamental use in surface adsorption, as well as the specific use for charge generation and transfer in electrochemical and photochemical cases, the delicate regulation of supports provides extra opportunities for the optimization of SACs based on their effect on the central single-atom sites. The functional groups, defective sites and other chemical moieties help the dispersion and stabilization of single-atom active centers for durable application in various CO2 transformation reactions under varied operating conditions and redox atmospheres. The chemical interaction between single-atom sites and substrates, which could be tuned by certain modifications in terms of both geometry and electrons, plays a critical role in regulating the local structure of central atoms, thereby leading to varied energy features for the adsorption and transformation of reactant and intermediate species. Furthermore, thanks to the porosity engineering of substrates, the local concentration of reactants could be well-regulated near the single-atom active centers, creating a specific microenvironment for the positive reactions. The confinement effect on the active sites derived from the porous structure, on the other hand, might contribute to synergistic and cooperative catalysis by controlling the density and location of active centers.
4.4 Synergy with other types of active centers
Despite their unique performance in some specific reactions, SACs are not panacea in all cases, especially those which need the cooperation between multiple active sites. Although the arrangement of peer single-atom sites, the regulation of the coordination environment and the modification of supports could make a difference, it still seems that the “Lone Ranger” should find its companies, as the old saying goes, “Many hands make light work”. To date, for more extensive use of SACs in CO2 transformation reactions, many other types of active centers have been introduced in the SAC systems to not only reinforce the reactions the SACs have already realized, but also break through the limitation of the current SACs into more complicated reactions. In these cases, the synergy between single-atom sites and other types of active centers should be the focus of study and design. We should emphasis that some components function as both active centers and substrates anchoring single-atom metal species, which might have an obscure boundary sometimes. In this section, we will try to focus more on their role as active components, while we could not deny the support effect in some cases as well.The combinations of single-atom sites and other metal-containing species grafted on the single-atom catalysts, such as metal nanoparticles, nanoclusters, and metal-based compounds (oxides, carbides, nitrides, sulfides, phosphides, etc.) in three/two/one-dimensional morphology, have been reported for the application in various reactions407,408 including CO2 transformation reactions. As already mentioned in Section 3.1, single-atom Sb with Cu NPs,205 isolated Ni–N with Cu NPs,164 single-atom Pb with Cu NPs,211 single-atom Cu doped SnO2,212 single-atom M doped gold and silver NPs (M = Cu, Ni, Pd, Pt, Co, Rh, and Ir),409 single-atom Co with Ti3C2Tx-MXene,272 single-atom Cu with a Au–Cu alloy,234 single-atom Ag chains with MnO2,236etc., have already manifested their advantages in the electrochemical and photochemical reduction of CO2 with respect to the experimental and theoretical results. There is a well-known term to define the cases where single atoms form an interface with metal NPs, a single-atom alloy (SAA), in which the NPs function as both primary supports and active components.410 In fact, metal alloying is a broadly adopted strategy to improve the performance of a single metal, but is always limited by the side reactions induced by the introduced second metals, such as the HER promoted by platinum-group metals in CO2 electroreduction. Thus, Chhetri et al. developed a series of dual-site catalysts featuring Pd and Pt single-atoms alloyed with Cu crystals, by which they promote the hydrocarbon production without prompting the HER411 (Fig. 50). Owing to the suitable binding energy of both *CO and H*, metallic Cu species are responsible for the primary formation of CH intermediates and C–C coupling for the production of C2+ hydrocarbons, as Pd single-atom sites, instead of promoting the HER, theoretically afford a more stable adsorption of CO* and even suppressed the HER when CO* pre-exists on the top site of Pd. The experimental results further confirm the advantages of such a dual-site catalyst with an optimized configuration of single atoms and metal nanoparticles, where CH4 and C2H4 are preferentially produced over varied morphologies of Cu crystals. Regarding the different interfaces between two metal species in the alloys, there has also been thorough investigation of the mechanisms. Depending on the loading amount of Sn, the Cu–Sn alloy could form distinct interfaces, i.e., a single-atom surface alloy in Cu99Sn1 and Cu97Sn3 and core–shell bulk alloy in Cu70Sn30 (ref. 412) (Fig. 50). Such a configuration of interfaces has a great effect on the electronic structures of Cu atoms. As for the surface alloy with a left shift of the d-band center away from the Fermi energy level, the binding energy of intermediate species is thus weakened for more favored CO production, as the desorption of *CO is enhanced with a lower energy barrier. In contrast, the bulk alloy offers a much higher energy barrier for the formation of *COOH, thus making CO2 through the dominant OCHO* intermediated route for HCOOH production. In a word, delicate design and fabrication of SAA catalysts have been believed to be an efficient method to break the linear scaling relationships shown in single metals, enhance specific procedures and adjust the reaction kinetics, in which the single-atom sites might function as not only the active centers but also the modifiers of metal NPs.413
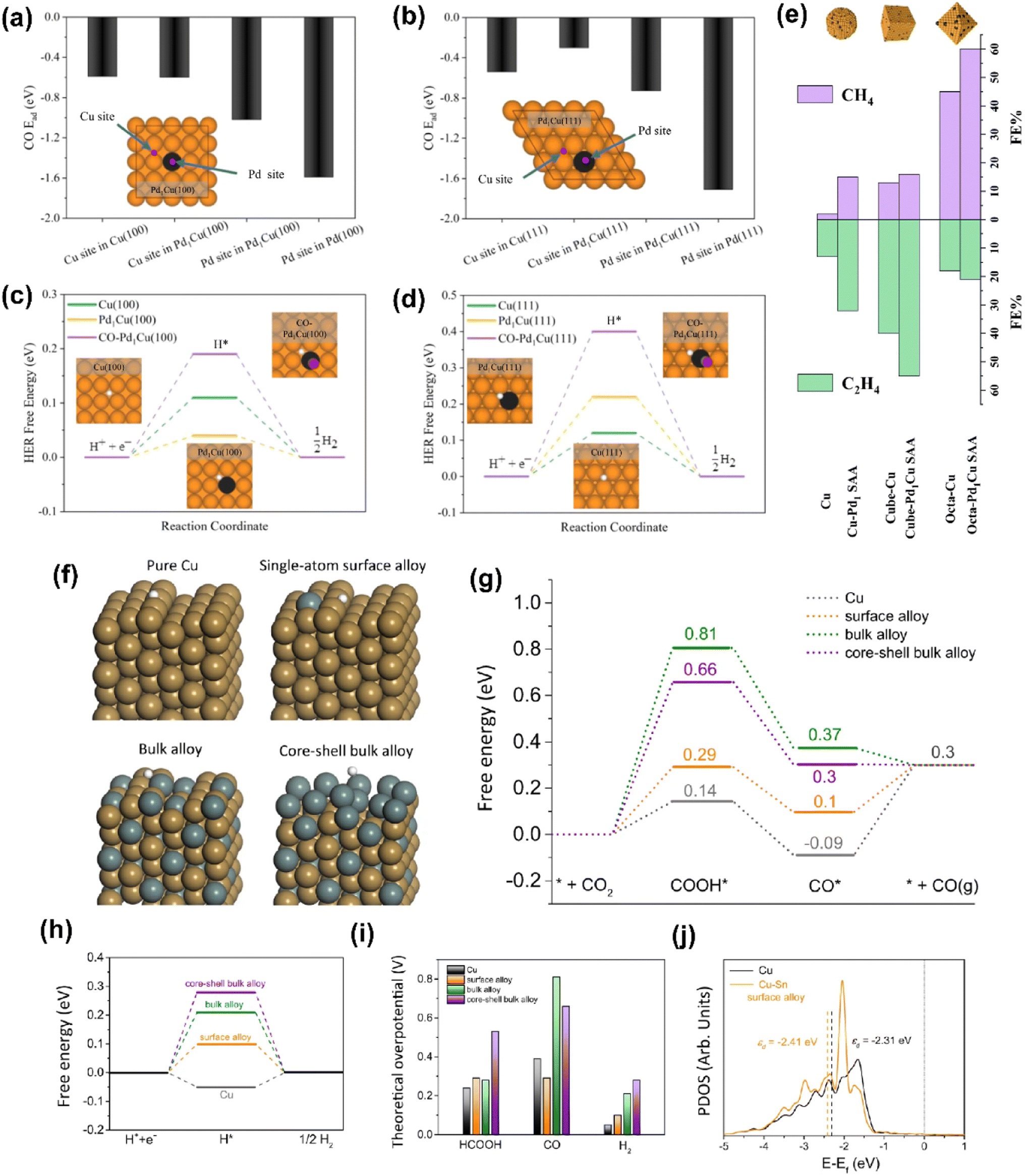 | ||
| Fig. 50 The adsorption energy of *CO over the (a) (100) and (b) (111) facets of bare Cu, Pd1Cu, and bare Pd, respectively. The free energy diagram of the HER over the (c) (100) and (d) (111) facets of bare Cu, Pd1Cu, and Pd1Cu in the presence of CO molecule adsorption over the Pd site (CO–Pd1Cu). Color code: Cu (orange), Pd (black), C (gray), O (red) and H (white). (e) The FE distribution of CH4 and C2H4 obtained using Cu and Pd1Cu SAAs (at −1.1 V vs. RHE). Reproduced from ref. 411 with the permission of Nature Springer, copyright 2023. (f) The theoretical model structures on stepped facets of pure Cu, the Cu–Sn surface alloy, the bulk alloy and the core–shell bulk alloy with adsorbed H*. The dark goldenrod and slate gray balls represent Cu and Sn atoms, respectively. (g) The calculated free energy diagrams of CO2-to-CO conversion. (h) The calculated free energy diagrams of the HER. (i) The theoretical overpotentials of HCOOH, CO, and H2 production. (j) The projected density of states of d orbitals of active Cu atoms at the step edges on pure Cu and the Cu–Sn surface alloy. The dotted gray line indicates the Fermi level. The d-band centers (εd) are denoted by dashed lines. Reproduced from ref. 412 with the permission of Nature Springer, copyright 2023. | ||
In some cases, on the other hand, the single-atom sites do not directly come into contact with the other metal active centers, but afford a tandem process by solely catalyzing certain reactions.414 For instance, Cu NPs and isolated Cu–N4 sites co-exist on a carbon-based substrate for the CO2 electroreduction to C2+ products415 (Fig. 51). The single-atom sites seem to not be responsible for the formation of *CO from CO2 due to the much higher energy barrier than that of Cu NPs according to the theoretical calculations. In contrast, the atomic Cu site could efficiently promote H2O dissociation to provide *H, as verified by the high HER performance of the intact Cu–N–C catalysts. Since the reaction energy for protonation of *CO to *CHO is closely correlated with the *H coverage on the Cu NP surface and properly increased coverage leads to a lower energy barrier for the formation of *CHO, the key to the optimized catalytic performance might be the facilitated generation of *H from atomic Cu sites, which is then transferred to Cu NPs through the carbon matrix. The *H coverage on Cu NPs is accordingly increased, resulting in accelerated protonation and higher activity for C2+ production. Coincidentally, by regulating the preparation conditions, Fe NPs and pyrrole-type Fe–N4 sites could be co-anchored on the less-oxygenated carbon supports for electrochemical CO2 reduction416 (Fig. 51). But the difference is that the isolated Fe–N4 sites are in charge of the adsorption and initial activation of the CO2 molecule, while Fe NPs reinforce the proton transfer from dissociation of water to the active centers for the formation of *COOH, as proved by the kinetic isotope effect in H2O and D2O. The theoretical simulation based on an Fe13-cluster-containing model also implies that the introduction of Fe NPs possibly further favors the stabilization of the *COOH intermediate, thereby boosting the formation of CO. Inspired from these two examples, we should find out if the synergy between such insular active centers might not have on effect by changing their own properties like in the cases of the above mentioned SAA catalysts, but playing to their strengths for the best reaction coupling.
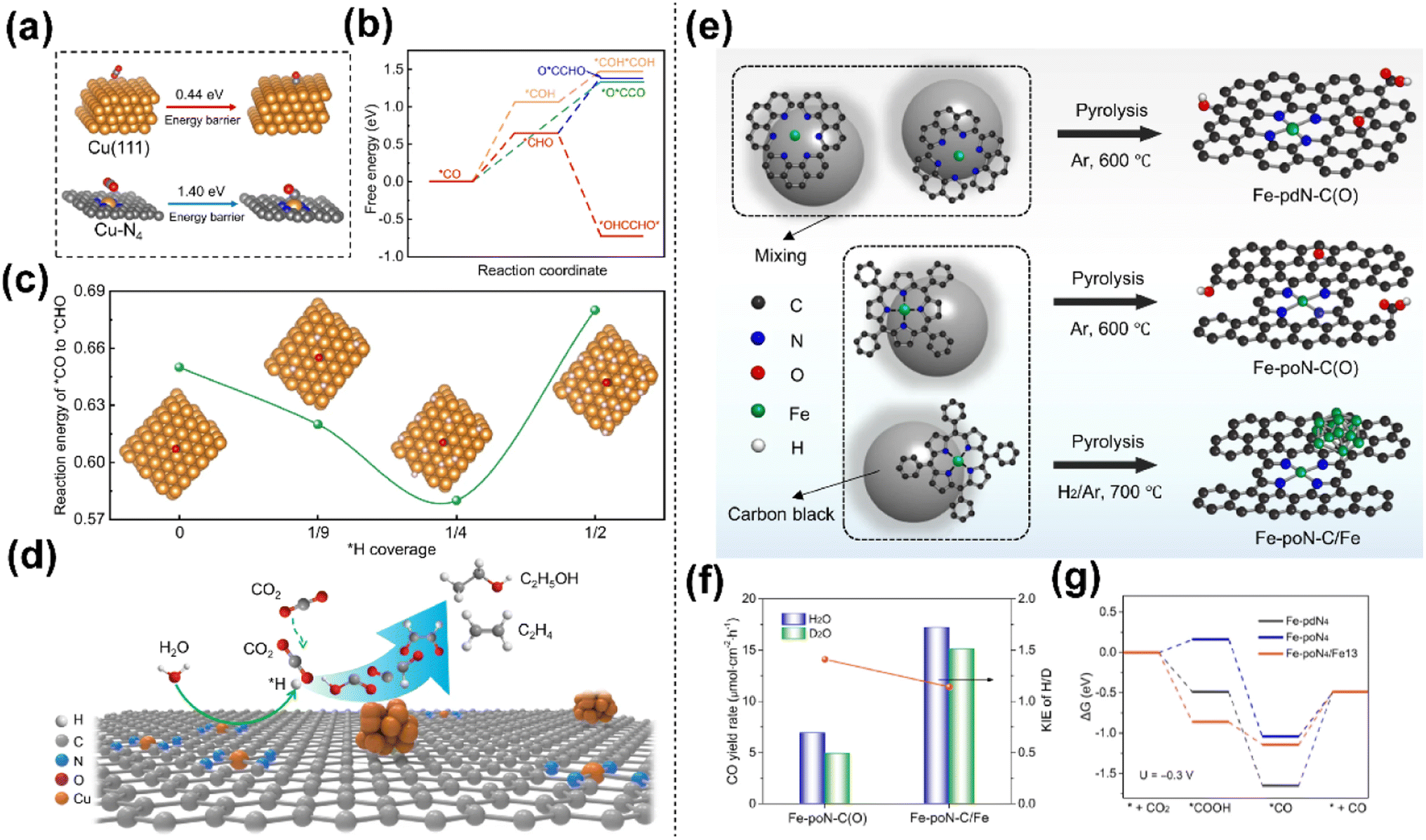 | ||
| Fig. 51 (a) The energy barrier of CO2 conversion to *CO over Cu(111) and Cu–N4. (b) The free energy diagram for the CO2RR to describe the possible C–C coupling step from *CO over Cu(111). (c) Reaction energy of *CO hydrogenation to *CHO on Cu(111) as a function of *H coverage. (d) Proposed reaction mechanism for CO2 reduction to C2 products on Cu1/CuNP. Reproduced from ref. 415 with the permission of Nature Springer, copyright 2023. (e) Schematic illustration for the synthesis of FeSA-pdN-C(O), FeSA-poN-C(O) and FeSA-poN-C/FeNP. (f) Kinetic isotope effect (KIE) values and CO yield rates of FeSA-poN-C(O), and FeSA-poN-C/FeNP. (g) Free energy diagrams at −0.3 V. Reproduced from ref. 416 with the permission of Nature Springer, copyright 2023. | ||
Besides cooperating with metal species, single-atom active sites could be also combined with non-metal active centers. We have demonstrated the synergy between the single atoms and vacancy sites of the substrates in the above sections.59,121,152,215,231,235,295 Indeed, the formation of vacancies could not only change the coordination environment of central atoms but also afford the adsorption and activation of reactants, thus promoting specific reaction kinetics. Non-metal species, in addition to their role in coordination as ligands, could function as active sites as well. For instance, a photocatalyst comprising P and Cu dual sites anchored on graphitic carbon nitride (P/Cu SAs@CN) has been employed in CO2 reduction with a high C2H6 evolution rate of up to 616.6 μmol g−1 h−1.417 The single-atom Cu sites are in a charge-enrichment stage for the adsorption and activation of CO2 molecules, while the isolated P atoms serve as hole capture sites for the accelerated light-driven formation and transfer of charge. In this way, such a catalyst realizes kinetically enhanced C–C coupling between *CO and *COH intermediates and thus achieve a higher C2H6 production. We should notice that, in this process, P atoms do not coordinate with or modify Cu sites, but function as independent active centers. In fact, non-metal elements applied in photocatalytic CO2 transformation are not rare at all, as non-metal semiconductors have exhibited significant light-to-electron activity intrinsically.54,242 Future investigations into the activity of non-metal species in more reactions might require greater efforts for the analysis of local structures, as it would be difficult to distinguish the activity from the coordination and support effects and reach an unambiguous conclusion.
In this section, we demonstrate the cooperation between the single-atom sites and other types of active centers and show how the synergy has an effect. By closely contacting and forming an alloy with well-regulated interfaces, the cooperative sites could break the scaling relationship in a single metal, retrofitting the reaction kinetics. In a separate position, the cooperative sites could afford their own activity for coupling into a tandem process with enhanced procedures. In particular, the non-metal sites have been proved to be able to serve as independent active centers in some light-driven reactions. The introduction of other active centers remarkably breaks through the limitation of SACs in more complicated reactions and is expected to fully exploit the unique advantages of SACs in terms of both structures and performances.
5. Research methods
In the above sections, we have reviewed significant progress on the SACs for chemical conversion of CO2 from the perspective of reaction types, focusing on the distinctive effect that SACs have on the catalytic performance. In fact, the synthetic methods, the characterization techniques and the computational approaches for modelling for structure analysis and mechanism investigation, which are just as important as the catalytic performance, should be also paid great attention. There should be a positive feedback loop in an ideal research process, that is, reasonable design and ingenious synthesis offering preliminary SACs, then accurate evaluation and advanced characterization studies giving a fundamental structure–performance relationship, followed by in-depth investigations into mechanisms providing explanations regarding internal logic and modeling theory, and eventually the obtained insights in turn propelling reasonable design and synthesis (Fig. 52). As the loops are executed over and over again, the catalysts are correspondingly getting closer to the optimum. In comparison with other types of heterogeneous catalysts, SACs possess more potential to realize such desired loops due to their commonly well-defined active sites. Furthermore, as a simple linear triatomic molecule, the conversion of CO2 embraces mazy pathways involving multiple key intermediate species and rate-determining steps. Hence, the incorporation of SACs into CO2 conversion systems, despite bringing greater challenges to penetrating into the inherent understanding, provides more opportunities for accessing the rewarding experience of catalysis studies. Herein, we will concentrate on the crucial issues in terms of material synthesis, characterization, and mechanisms in the systems of CO2 conversion with SACs as catalysts, and very general research studies that are not related to CO2 conversion will be circumvented as possible. | ||
| Fig. 52 Crucial issues of SAC research for CO2 conversion and a positive loop encompassing them. | ||
5.1 Material synthesis
From the perspective of methodology, the synthetic routes of SACs can be divided into two categories: one is the bottom-up strategy, i.e., establishing a substrate initially followed by loading SAs or the corresponding metal precursors onto it, such as precipitation, impregnation, deposition, and adsorption; another is the top-down strategy, i.e., fabricating a metal-substrate mosaic preliminarily followed by transformation into SAC structures, such as pyrolysis, etching, and redispersion. Generally, the choice of synthetic method is highly dependent on not only the features of metal elements and supports, but also the reaction conditions and desired performance. For instance, for the synthesis of a SAC operated at a relatively higher temperature and in a complex redox atmosphere in a thermochemical catalytic system, oxides always more suitably serve as a support rather than carbonitride, since the stronger electronegativity of oxygen atoms favors the enhanced bonding between SAs and supports. Commonly, reducible oxides with the potential of creating vacancies by removing partial oxygen atoms have been applied in these cases. This is rather important for the application of SACs in thermal catalytic CO2 hydrogenation involving C–C coupling that a second type of active sites other than metal SAs, such as Ov, is highly required. In this case, defect engineering is supposed to take into account the synthesis of SACs, so that the synergy effect derived from the collaboration between SAs and Ov could be feasibly cast on both the activation of CO2 and the formation of C–C bonds.59,284 In the meantime, the SA sites always present distinct valence states and electronic structures from that of their NP and NC counterparts via coordinating with neighboring atoms, in which the inherence of supports (e.g., crystal faces and acidity/basicity) contribute to a great extent through the metal-support interactions.49,353,418 For accurately controlling such interactions, meticulous selection of synthetic temperature, solvent, precursors and other relevant parameters should be the focus during the whole preparation procedure.As for typical electrochemical reduction reactions, moderate reaction conditions as well as flexible carbon supports featuring high specific surface area and electricity/heat conductivity endows the synthesis of SACs with more chances for modulating the electronic structure of metal sites. A number of strategies such as defect engineering,152,215 heteroatom doping,173 structural modification,170,171 and functional group attachment210 have be applied in the synthesis process for the regulation of microenvironments. Such variations of chemical microenvironments not only change the electronic states and hybridized orbitals of active sites, but also impose appreciable effects on the mass transfer of reactants, intermediates and products, leading to the corresponding tuned performances thermodynamically and kinetically.132,378,379,385,419 To realize these goals, superior carbon supports such as graphene and CNTs with abundant functional groups have been applied in the bottom-up routes, in which metal SAs could be immobilized via either electrostatic interaction with metal ions as precursors, or π–π interaction with certain metal–organic complex precursors. Furthermore, one-step or multi-step pyrolysis processes with MOFs or certain other metal–organic assemblages as precursors offer opportunities for the construction of more types of SACs, such as various M–N–C (M = metal) materials.
Light-driven reduction reactions involve the generation and transfer of photoexcited electrons, in which the introduced SACs can function as not only active sites for the adsorption and activation of CO2, but also promoters for the modulation of the energy bands of semiconductors. Given that both oxides (e.g., TiO2) and carbon-based240,241 (e.g., g-C3N4) materials can serve as substrates for embedding metal SAs, plus that the MOFs and COFs can be also used when extra photosensitizers and sacrificed hole scavengers exist,226 the synthesis of SACs for photochemical reduction is just like the combination of electro- and thermo-catalysis, although the targets are not completely identical. For instance, silver SAs featuring a chain-like arrangement on the surface of manganese oxides, form a unique structure just like a Z-scheme heterojunction with the combination of neighboring oxide species, resulting in an enhanced performance due to the well-matched energy bands for electron transfer.236
Indeed, no matter which system the SACs are applied in, there are some common goals for propelling the practical application of SACs in CO2 conversion towards an industrial scale: to extend the range of loadings and types,332,420,421 to lower the cost of preparation,43 and to expand the scale of production.43,421 It was reported that a versatile strategy combining impregnation and secondary step annealing could be used for synthesizing ultra-high-density SACs with metal contents up to 23 wt% for 15 metals on chemically distinct carriers.421 This method could be even translated into a standardized, automated protocol executed by a robotic platform, enabling the reproducible preparation (Fig. 53), which further indicated the feasibility and scalability of this strategy. In addition, a straightforward and cost-effective three-dimensional (3D) printing approach has been reported for fabricating a library of SACs in a simple and economic way.43 It is also expected that this 3D-printing technique to make a difference in large-scale commercial production of SACs, thereby steering the use of these materials to a broader spectrum of industrial applications. Notably, aside from the retrofitted conventional synthetic methods, burgeoning machine learning (ML) and artificial intelligence (AI) techniques have been also providing new paradigms for the highly efficient approaches for massive production of SACs.422 Once a large and high-quality database is established, the explosion of efficiency would be brought about by high throughput screening and automated optimization based on reliable prediction of the structure–performance relationship.
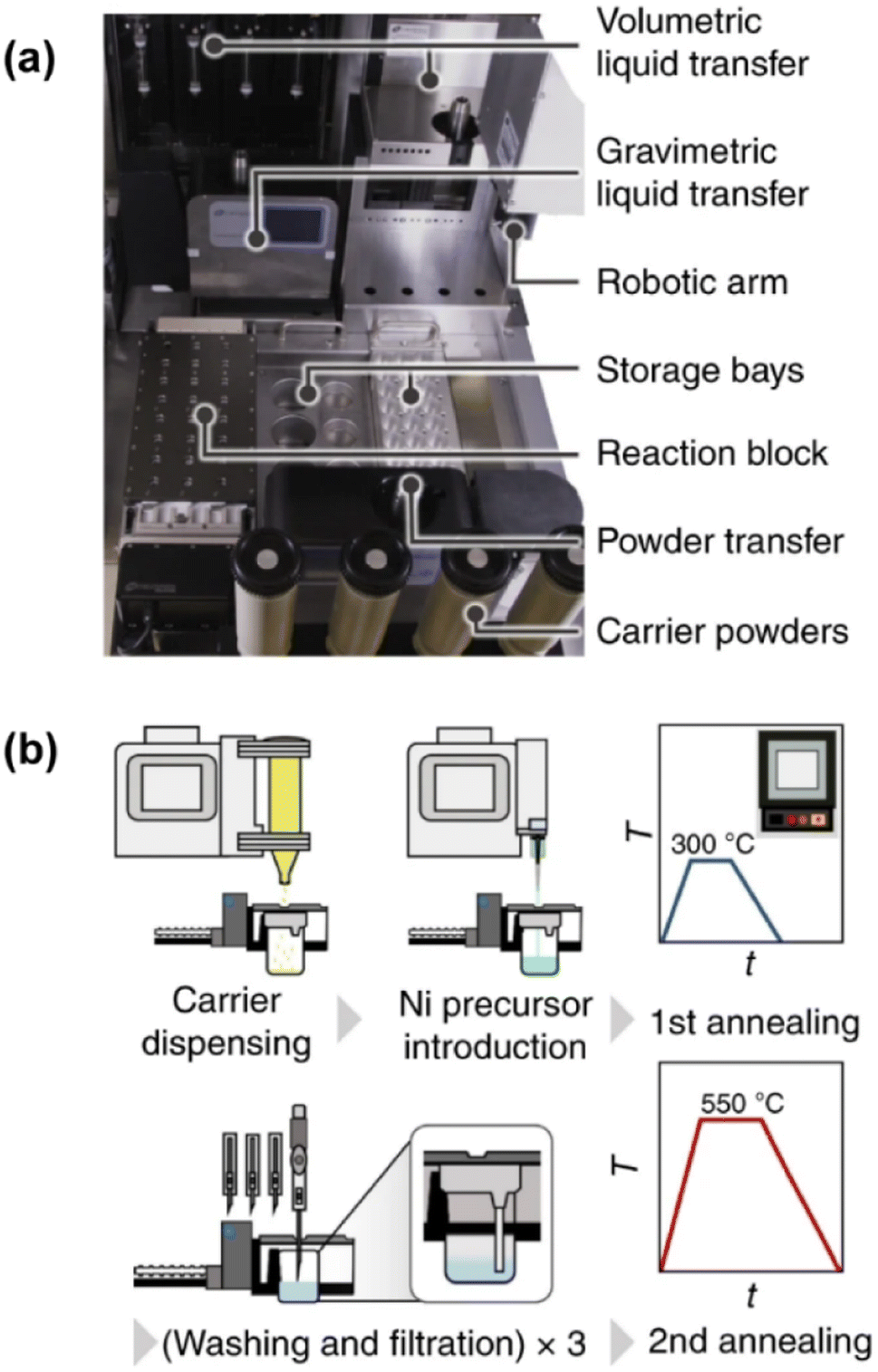 | ||
| Fig. 53 (a) Photograph of the robotic synthesis platform and assignment of tools to unit operations. (b) Flowsheet of the synthesis protocol.421 Reproduced from ref. 421 with permission from Springer Nature, copyright 2022. | ||
5.2 Characterization
The rapid development of atom-scale characterization has propelled the identification of SA species and deepened the proposition of the SAC concept, which was simultaneously inspired by a number of experimental observations. Aberration-corrected scanning transmission electron microscopy (STEM)21 and scanning tunneling microscopy (STM)423 offer opportunities for directly observing the dispersion and arrangement as well as certain surficial properties regarding electronic behaviors. The XAS technique exhibits advantages in obtaining the information about the refined structure including the spatial distance and coordination state between the central atom and neighboring atoms, the electronic states of specific orbitals, the disorder degree of materials, and the geometric configuration with regard to symmetry, thereby being widely applied in most of the recent studies on SACs and could be well combined with theoretical modeling and calculations. The solid state nuclear magnetic resonance (ssNMR) technique specializes in the investigation of SACs based on isotropic chemical shift, chemical shift anisotropy, dipole–dipole coupling, J-coupling, and quadrupole interaction of certain metals and supports, exhibiting unparalleled advantages for the identification of specific metal SAs and understanding of certain interactions between active centers and intermediate species. The infrared (IR) technique, on the other hand, can be used in the studies on SACs via a more indirect approach, e.g., CO-adsorption spectra are commonly acquired to reflect the form of metal species, since metal sites with different geometric and electronic properties possess a characteristic CO-adsorption configuration (on-top, bridged, geminal, etc.) thereby displaying varied absorbance band positions.37 In particular, Mössbauer spectroscopy has been developed to track the dynamic changes in the electronic and coordination structures of iron and tin SACs recently,157,210,424 which are both commonly used as active components for CO2 reduction. This could be attributed to its high energy resolution and feasible acquirement of quantitative information and chemical states, spin state, and coordination symmetry of “selected” elements (Fe, Sn, Ni, Au, etc.).Nowadays, the advancement of various characterization studies is no longer limited to discrimination of SA species from their nano-counterparts (NCs or NPs), but steps forward to in-depth explorations in terms of dynamic evolution, electronic state and host–guest interactions, all of which are of great concerns about the practical application of SACs in CO2 conversion. In order to achieve these goals, higher temporal and spatial resolution under harsh conditions is in great demand, which might be afforded by the incorporation of state-of-the-art devices and upgraded research strategies.425 On the other hand, the innovative exploitation of computational science and big data has promisingly ignited the revolutionary development of characterization techniques by not only predicting the most probable structure of SACs referring to the existing experimental data426,427 but also assisting the collection and analysis of data and accordingly increasing the efficiency and accuracy.428,429 Martini et al. provided an example of tracking the evolution of SACs for the CO2 electrocatalytic reduction using operando XAS and machine learning, while addressing the nature, stability, and evolution of the Ni active sites during the reaction.430 The combination of unsupervised and supervised machine learning (SML) approaches is shown to be able to decipher the X-ray absorption near edge structure (XANES) of the transition-metal based single-atom sites, decoupling the roles of diversified metal sites coexisting in the working catalyst with quantitative structural information about the local environment of active species and the interplay between metal species and adsorbates, such as CO (Fig. 54). Overall, it is plausible to suppose that the intersection between advanced characterization techniques and modern computational science could shed light on the precise analysis of sophisticated SACs.
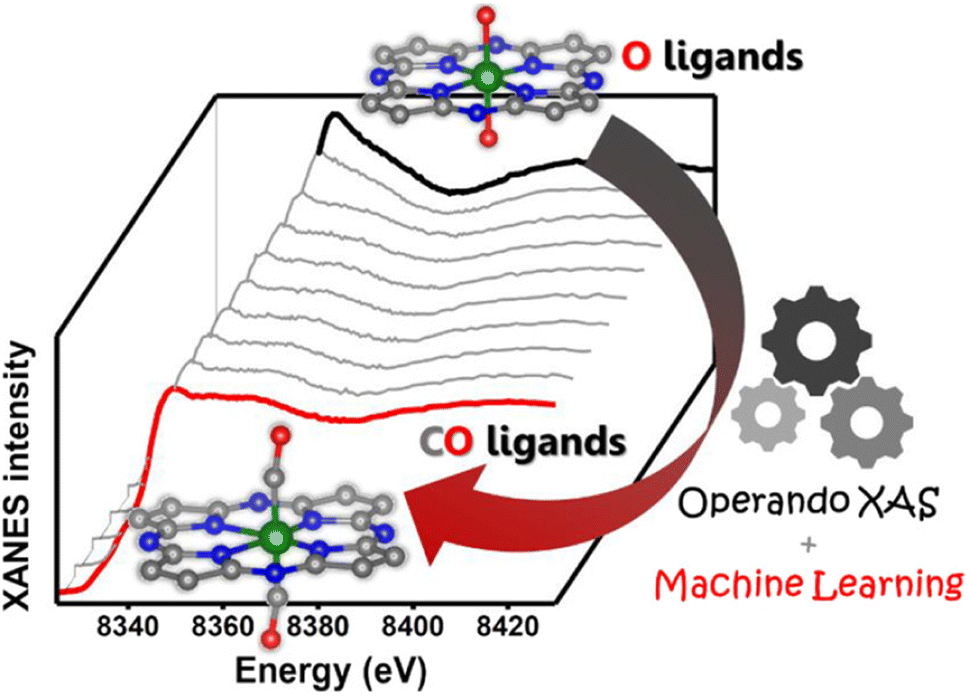 | ||
| Fig. 54 Schematic of tracking the evolution of SACs for CO2 electrocatalytic reduction with a combination of operando XAS and machine learning. Reproduced from ref. 430 with permission from American Chemical Society, copyright 2023. | ||
5.3 Computation and modeling
SACs integrating the merits of homogeneous and heterogeneous catalysis endow the catalytic research studies with more chances to dive into inherent and microscopic perspectives. As for CO2 conversion, just CO2 activation and protonation have involved a complex reaction network, especially in the cases where the formation of main products encompasses multi-step electron and proton transfer. During the reaction process, the active sites can even evolve with the change in redox atmospheres, leading to perceptible variation of reaction pathways. To this end, a complementary approach aside from the experimental observation has been expected to bring a potential solution, which is theoretical simulation based on computational technology.In general, most recent studies show that the initial adsorption and activation of CO2 molecules that break the CO bonds are the potential limited steps of CO2 reduction to 2e− products (e.g., CO and HCOOH), no matter what type of energy input it is. The ambiguities are whether the intermediate binds via a carbon atom (e.g., *COOH) or oxygen atoms (HCOO*) or both carbon and oxygen atoms in a bridged form, and whether the intermediate is formed through coupled proton-electron transfer or a charged state.158 Key descriptors such as the band structure (d-band center for transition metals amd s- and p-bands for main group metals), electronic density and state of (s-, p-, d-, f-) orbitals and charge transfer, are commonly used for predicting and explaining the structure–performance relationships due to their decisive role in intramolecular bond breaking and regeneration, and adsorption and desorption at active interfaces.186,188 The situations are even more complicated for CO2 reduction towards more than 2e− products, particularly those involving C–C coupling. CO or *CO is always considered as the key intermediate, while either the consecutive protonation step via intermediate species such as *CHO and *COH or the dimerization step between CO is probably the limited step.158 Besides, the CO insertion mechanism is also proposed in the thermo-catalytic C–C coupling process in the presence of multifunctional components,59,284 as C–C couplings between other types of intermediates (e.g., CHO) have been also reported for some electro- and photo-catalytic processes.163,295 A common circumstance is that isolate metal sites in SACs cannot be in charge of all these reactions alone, but have an effect with the incorporation of neighboring active sites (e.g., second type of metal and vacancy).59,295 Indeed, multi-electron processes are more likely to be influenced by factors other than only kinetics and thermodynamics, such as coverage of the intermediate, mass transfer, and pH,158 which could be attributed to the inherent complexity. All these mechanism studies have been subjected to a long-term restriction in traditional heterogeneous catalysts because of the difficulty in clarifying the realistic structures and discriminating the role of uneven active centers. Although the computational chemistry has ignited buzz among the scholars hammering at reaction kinetics with the development of computer science, it was not until the concept of SACs that computational chemistry really breaks through the shackles of complex systems and shows great strength in the study of carbon dioxide conversion. On the basis of the principles of quantum chemistry, the Schrodinger equation and various computational methods with specific approximations, researchers might be able to provide valuable information about electron orbitals, density of states, energy profiles of transition states and other intrinsic parameters on a molecular or even atomic scale. Such parameters further push forward the understanding of the interplay between the active centers and guest molecules, contributing to the insight of inherent kinetics, which are always hazed due to the limited experimental approaches. More importantly, in comparison to heterogeneous catalysts with uneven structures, the theoretical studies on SACs could be more easily generalized, providing opportunities for modelling and accordingly developing new catalysts. For instance, Qi et al. combined the first-principles calculations and an artificial intelligence approach to high-throughput screen the stability and activity of 3d, 4d, and 5d transition metal SAs on eight defective metal oxide surfaces during the CO2 reduction reaction (Fig. 55).431 By evaluating the energies of 232 catalytic systems, only 28 of them remain stable with the adsorption of intermediates (*COOH, *OCHO, *CO, *CHO, and *H), although 100 kinds of SACs could be stably anchored in a vacuum. The stability during adsorption could further be reasonably attributed to the electronegativity and number of outer electrons of the SA, the d-band center of metal oxides, and the relative coordination number of the adsorbed species together. As the free energy diagram could be given for various CO2 reduction reactions, the catalytic performance should be also predicted in such modelling systems. Similarly, SACs based on different metals embedded on different substrates can also be modelled and mapped based on the theoretical parameters from computation.432 Such modelling strategies, in turn, have been extensively used for screening efficient SACs, potentially paving the way for wide-spread application of SACs in CO2 transformation and even other specific reactions.
 | ||
| Fig. 55 Schematic diagram of structures for 29 kinds of transition-metal SACs with eight different types of oxide supports. Reproduced from ref. 431 with the permission of Nature Springer, copyright 2022. | ||
6. Conclusions and future outlook
Given that we all look forward to an earth suitable for human habitation for a long time, the solution to excessive carbon emission is in an urgent demand for assuaging climate change and environment deterioration. Chemical CO2 conversion not only offers a potential approach to partially replace the fossil routes by transforming CO2 into valuable products, but also renders enhanced carbon recycling as combined with efficient carbon capture in the future. Burgeoning SACs featuring extremely high atom utilization efficiency as well as unique electronic and geometric properties integrate the merits of homogeneous and heterogeneous catalysts, providing a powerful tool to tackle the critical problems regarding efficiency and economy of CO2 conversion. In the above sections, we have reviewed some critical progress and provided a comprehensive summary of SAC application in the widely adopted CO2 reduction reaction via thermochemical, electrochemical, and photochemical routes, as well as some miscellaneous synthetic reactions involving CO2 conversion. We further offer a multi-level understanding of SACs from a molecular point of view in terms of the central atom, coordination environment, support effect and synergy with other active centers. The ingenious design of materials with regard to the reasonable regulation of the chemical microenvironment and rational explanations toward reaction mechanisms in charge of the specific structure–performance relationships of SACs were highlighted. We also addressed some general issues of research methods for SACs from the perspective of material synthesis, characterization techniques and computation-assisted theoretical modelling in chemical conversion of CO2 based on the summaries of current research results, expecting to inspire further advances in this field.To date, the ‘young and vigorous’ SAC research studies are still in the prime of their lives, and their rapid growth has made great difference in the studies on quite a number of catalysis systems, especially chemical CO2 conversion. Despite holding significant promise, there is still much work to be accomplished before the SACs stepping into a more in-depth stage for both scientific explorations and industrial targets. Herein, we will briefly present our outlook on several important orientations for future research studies of SACs in CO2 conversion.
6.1 From an empirical paradigm to rational design
Over a long time, studies on catalysis have been more likely to follow an empirical paradigm, given that achieving thorough understanding of energy and mass transfer processes is limited due to the complicated nature of chemical reactions and the scarcity of advanced technical approaches. A lot of depletion of temporal and financial resources therefore emerged inevitably. Thanks to the development of characterization studies, the assistance of computational science, and the relatively well-defined active sites of SACs, more general theories are expected to be proposed to guide the rational design of new catalytic materials and reaction processes. Such a ‘rationale’ indicates accurate control and constructing SACs on the basis of abstract descriptors derived from the inherence of reactions rather than only concrete performances. In order to achieve this, we not only urge the further advancement of in situ and operando techniques combined with theoretical calculations but also of a large open-access database including structural information, and kinetic and thermodynamic factors of CO2 conversion according to existing research studies. A milestone is supposed to be providing multiscale modeling of SACs with certain uniformity and quantifiable parameters. Once the model was established, the design of SACs would become simple screening via machine learning or other reasonable analysis methods based on big data. The underlying challenges are data mining in the catalytic science of SACs and a way to guarantee the consistency of predictions and actuality.6.2 From scattered studies to integrated systems
Currently, most studies on SACs for CO2 conversion are still restrained in several specific fields, i.e., thermo-, photo-, and electro-catalysis, and the corresponding up- and down-stream processes (e.g., up: capture and down: separation) are separated and conducted alone. That favors the research studies to some extent due to their specialty concentrating on one target, whereas quite a few potentials of integrated systems are neglected despite also making sense in the meantime. Fortunately, there have been plenty of studies dedicated to combine catalytic systems via various routes featuring different forms of energy inputs,50,433,434 although most of them are still in the preliminary or developmental stages. Furthermore, miscellaneous disciplines such as engineering issues, materials science, and automation technologies could be also involved in highly efficient integrated systems, e.g., electrolytes might play a vital role in an electro-catalytic process; enhanced mass transfer could be realized either by engineering or with the innovation of the material; the extensive exploitation of embodied artificial intelligence like robots and disembodied artificial intelligence like ChatGPT might greatly liberate the manpower from the burdensome and endangered tasks in labs and render a balance between efficiency and safety. In general, harmony is the core of such an integrated system, which can be depicted with a ‘barrel effect’, i.e., highly effective SACs might function as a longer slab, but the volume of a ‘barrel’ is more likely to be determined by the possibly inconspicuous short slabs. Thus, it would be an explicit goal to construct ‘a barrel with slabs of nearly equal length’ from a global perspective. In this regard, the necessary interdisciplinarity should make a great difference.6.3 From laboratory research to commercial application
An ultimate goal of the studies on SACs for CO2 conversion is stepping out of labs one day, toward massive production and sustainable processes by making profits. However, as more is different,435 there is still a long way to go for the practical use of SACs for CO2 conversion. Economic analysis should be the first step. We must know what the market really needs, how much the cost is, and the potential income over a period. Then, we should consider how to cutdown the cost and increase the production by both scientific and engineering methods, e.g., via upgrading the device, improving energy efficiency, and optimizing operation conditions. Another common concern is how to realize the scalability of production, which not only needs massive preparation of SACs but is also associated with a complex scaling effect. Such a scalable process might create new demands for the SAC design and application in turn. Moreover, it could be with more essential factors that the durability and reliability of SACs become successful in some practical cases, especially when operated under harsh conditions. We should pay more attention to the long-term running status of SACs, particularly the dynamic evolution of both electronic and geometric structures, so that we could ensure the stability of SACs and pave a promising way to their feasible commercial application.Author contributions
X. Shang performed the literature search, analyzed the published results, and wrote the manuscript. X. Yang, G. Liu, T. Zhang and X. Su provided key advice and supervised the preparation of the text.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We gratefully acknowledge the financial support from the National Key R&D Program of China (2022YFA1506200), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB36030200 and No. XDA29040600), the CAS Project for Young Scientists in Basic Research (YSBR-022), the National Natural Science Foundation of China (21978286, 22208021, and U19A2015), the Youth Innovation Promotion Association CAS, and the Young Top-notch Talents of Liaoning Province (XLYC2007082).References
- M. R. Allen, D. J. Frame, C. Huntingford, C. D. Jones, J. A. Lowe, M. Meinshausen and N. Meinshausen, Nature, 2009, 458, 1163–1166 CrossRef CAS.
- S. I. Seneviratne, M. G. Donat, A. J. Pitman, R. Knutti and R. L. Wilby, Nature, 2016, 529, 477–483 CrossRef CAS.
- S. Fuss, J. G. Canadell, G. P. Peters, M. Tavoni, R. M. Andrew, P. Ciais, R. B. Jackson, C. D. Jones, F. Kraxner, N. Nakicenovic, C. Le Quéré, M. R. Raupach, A. Sharifi, P. Smith and Y. Yamagata, Nat. Clim. Change, 2014, 4, 850–853 CrossRef CAS.
- S. Ding, M. J. Hülsey, J. Pérez-Ramírez and N. Yan, Joule, 2019, 3, 2897–2929 CrossRef CAS.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Müller, Energy Environ. Sci., 2012, 5, 7281–7305 RSC.
- Y. Li, J. Wang, S. Fan, F. Wang, Z. Shen, H. Duan, J. Xu and Y. Huang, J. Energy Chem., 2021, 53, 168–174 CrossRef CAS.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- C. Ampelli, S. Perathoner and G. Centi, Philos. Trans. R. Soc., A, 2015, 373, 20140177 CrossRef.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS.
- Y. Li, X. Cui, K. Dong, K. Junge and M. Beller, ACS Catal., 2017, 7, 1077–1086 CrossRef CAS.
- N. Mac Dowell, P. S. Fennell, N. Shah and G. C. Maitland, Nat. Clim. Change, 2017, 7, 243–249 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed.
- A. S. Lindsey and H. Jeskey, Chem. Rev., 1957, 57, 583–620 CrossRef CAS.
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283–3297 RSC.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem, 2018, 2, 65–81 CrossRef CAS.
- X. Li, X. Yang, Y. Huang, T. Zhang and B. Liu, Adv. Mater., 2019, 31, 1902031 CrossRef CAS PubMed.
- X. Li, Y. Huang and B. Liu, Chem, 2019, 5, 2733–2735 CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- J. J. Rooney and G. Webb, J. Catal., 1964, 3, 488–501 CrossRef CAS.
- H. S. Taylor and E. F. Armstrong, Proc. R. Soc. London, Ser. A, 1925, 108, 105–111 CAS.
- W. R. Patterson and J. J. Rooney, Catal. Today, 1992, 12, 113–129 CrossRef CAS.
- K. Asakura, H. Nagahiro, N. Ichikuni and Y. Iwasawa, Appl. Catal., A, 1999, 188, 313–324 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS.
- S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS.
- T. Maschmeyer, F. Rey, G. Sankar and J. M. Thomas, Nature, 1995, 378, 159–162 CrossRef CAS.
- J. T. Yates, S. D. Worley, T. M. Duncan and R. W. Vaughan, J. Chem. Phys., 1979, 70, 1225–1230 CrossRef CAS.
- W. Jing, H. Shen, R. Qin, Q. Wu, K. Liu and N. Zheng, Chem. Rev., 2022, 123, 5948–6002 CrossRef.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS.
- J. Kim, H.-E. Kim and H. Lee, ChemSusChem, 2018, 11, 104–113 CrossRef CAS.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS.
- X. He, Q. He, Y. Deng, M. Peng, H. Chen, Y. Zhang, S. Yao, M. Zhang, D. Xiao, D. Ma, B. Ge and H. Ji, Nat. Commun., 2019, 10, 3663 CrossRef PubMed.
- Y. Chen, J. Lin, B. Jia, X. Wang, S. Jiang and T. Ma, Adv. Mater., 2022, 34, 2201796 CrossRef CAS.
- T. Zhang, Nano Lett., 2021, 21, 9835–9837 CrossRef CAS PubMed.
- R. Lang, X. Du, Y. Huang, X. Jiang, Q. Zhang, Y. Guo, K. Liu, B. Qiao, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 11986–12043 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- L. Wang, D. Wang and Y. Li, Carbon Energy, 2022, 4, 1021–1079 CrossRef CAS.
- H. B. Zhang, G. G. Liu, L. Shi and J. H. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- J. Zhang, C. Liu and B. Zhang, Small Methods, 2019, 3, 1800481 CrossRef.
- L. Zhang, Y. Wang, Z. Niu and J. Chen, Small Methods, 2019, 3, 1800443 CrossRef.
- F. Xie, X. Cui, X. Zhi, D. Yao, B. Johannessen, T. Lin, J. Tang, T. B. F. Woodfield, L. Gu and S.-Z. Qiao, Nat. Synth., 2023, 2, 129–139 CrossRef.
- Z. Li, B. Li and C. Yu, Adv. Mater., 2023, 2211221 CrossRef CAS.
- C. B. Hiragond, N. S. Powar, J. Lee and S.-I. In, Small, 2022, 18, 2201428 CrossRef CAS PubMed.
- Y. Wang, L. R. Winter, J. G. Chen and B. Yan, Green Chem., 2021, 23, 249–267 RSC.
- C. Xu, A. Vasileff, Y. Zheng and S.-Z. Qiao, Adv. Mater. Interfaces, 2021, 8, 2001904 CrossRef CAS.
- X. Su, X.-F. Yang, Y. Huang, B. Liu and T. Zhang, Acc. Chem. Res., 2019, 52, 656–664 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS.
- Y. Song, X. Zhang, K. Xie, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1902033 CrossRef CAS.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim, H. Kim and S.-I. In, Energy Environ. Sci., 2022, 15, 880–937 RSC.
- Ž. Kovačič, B. Likozar and M. Huš, ACS Catal., 2020, 10, 14984–15007 CrossRef.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS.
- C. Wang, E. Guan, L. Wang, X. Chu, Z. Wu, J. Zhang, Z. Yang, Y. Jiang, L. Zhang, X. Meng, B. C. Gates and F.-S. Xiao, J. Am. Chem. Soc., 2019, 141, 8482–8488 CrossRef CAS.
- X. Yang, X. Su, X. Chen, H. Duan, B. Liang, Q. Liu, X. Liu, Y. Ren, Y. Huang and T. Zhang, Appl. Catal., B, 2017, 216, 95–105 CrossRef CAS.
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- L. P. Ding, T. T. Shi, J. Gu, Y. Cui, Z. Y. Zhang, C. J. Yang, T. Chen, M. Lin, P. Wang, N. H. Xue, L. M. Peng, X. F. Guo, Y. Zhu, Z. X. Chen and W. P. Ding, Chem, 2020, 6, 2673–2689 CAS.
- X. Ye, C. Yang, X. Pan, J. Ma, Y. Zhang, Y. Ren, X. Liu, L. Li and Y. Huang, J. Am. Chem. Soc., 2020, 142, 19001–19005 CrossRef CAS PubMed.
- M. Yang, J. Yu, A. Zimina, B. B. Sarma, L. Pandit, J. D. Grunwaldt, L. Zhang, H. Xu and J. Sun, Angew. Chem., Int. Ed., 2023, 62, e202216803 CrossRef CAS.
- X. Zhang, G. Zhang, W. Liu, F. Yuan, J. Wang, J. Zhu, X. Jiang, A. Zhang, F. Ding, C. Song and X. Guo, Appl. Catal., B, 2021, 284, 119700 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef.
- B. Liang, J. Ma, X. Su, C. Yang, H. Duan, H. Zhou, S. Deng, L. Li and Y. Huang, Ind. Eng. Chem. Res., 2019, 58, 9030–9037 CrossRef CAS.
- X. L. Liu, M. H. Wang, H. R. Yin, J. T. Hu, K. Cheng, J. C. Kang, Q. H. Zhang and Y. Wang, ACS Catal., 2020, 10, 8303–8314 CrossRef CAS.
- C. Zhou, J. Shi, W. Zhou, K. Cheng, Q. Zhang, J. Kang and Y. Wang, ACS Catal., 2019, 10, 302–310 CrossRef.
- Z. Li, Y. Qu, J. Wang, H. Liu, M. Li, S. Miao and C. Li, Joule, 2019, 3, 570–583 CrossRef CAS.
- Y. Ni, Z. Chen, Y. Fu, Y. Liu, W. Zhu and Z. Liu, Nat. Commun., 2018, 9, 3457 CrossRef PubMed.
- Y. Zhang, Z. Zhang, X. Yang, R. Wang, H. Duan, Z. Shen, L. Li, Y. Su, R. Yang, Y. Zhang, X. Su, Y. Huang and T. Zhang, Green Chem., 2020, 22, 6855–6861 RSC.
- M. T. Arslan, G. Tian, B. Ali, C. Zhang, H. Xiong, Z. Li, L. Luo, X. Chen and F. Wei, ACS Catal., 2022, 12, 2023–2033 CrossRef CAS.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019 CrossRef CAS.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- V. Dieterich, A. Buttler, A. Hanel, H. Spliethoff and S. Fendt, Energy Environ. Sci., 2020, 13, 3207–3252 RSC.
- S. Overa, T. G. Feric, A.-H. A. Park and F. Jiao, Joule, 2021, 5, 8–13 CrossRef.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2094–2100 CrossRef CAS.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2449–2455 Search PubMed.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS PubMed.
- Y. Tang, C. Asokan, M. Xu, G. W. Graham, X. Pan, P. Christopher, J. Li and P. Sautet, Nat. Commun., 2019, 10, 4488 CrossRef PubMed.
- L. DeRita, J. Resasco, S. Dai, A. Boubnov, H. V. Thang, A. S. Hoffman, I. Ro, G. W. Graham, S. R. Bare, G. Pacchioni, X. Pan and P. Christopher, Nat. Mater., 2019, 18, 746–751 CrossRef CAS PubMed.
- M. Moliner, J. E. Gabay, C. E. Kliewer, R. T. Carr, J. Guzman, G. L. Casty, P. Serna and A. Corma, J. Am. Chem. Soc., 2016, 138, 15743–15750 CrossRef CAS PubMed.
- G. S. Parkinson, Z. Novotny, G. Argentero, M. Schmid, J. Pavelec, R. Kosak, P. Blaha and U. Diebold, Nat. Mater., 2013, 12, 724–728 CrossRef CAS PubMed.
- S. Wei, A. Li, J.-C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W.-C. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed.
- S. Li, Y. Xu, H. Wang, B. Teng, Q. Liu, Q. Li, L. Xu, X. Liu and J. Lu, Angew. Chem., Int. Ed., 2023, 62, e202218167 CrossRef CAS.
- P. Du, R. Qi, Y. Zhang, Q. Gu, X. Xu, Y. Tan, X. Liu, A. Wang, B. Zhu, B. Yang and T. Zhang, Chem, 2022, 8, 3252–3262 CAS.
- M.-M. Millet, G. Algara-Siller, S. Wrabetz, A. Mazheika, F. Girgsdies, D. Teschner, F. Seitz, A. Tarasov, S. V. Levchenko, R. Schlögl and E. Frei, J. Am. Chem. Soc., 2019, 141, 2451–2461 CrossRef CAS PubMed.
- H. Liang, B. Zhang, P. Gao, X. Yu, X. Liu, X. Yang, H. Wu, L. Zhai, S. Zhao, G. Wang, A. P. van Bavel and Y. Qin, Chem Catal., 2022, 2, 610–621 CrossRef CAS.
- Y. Li, Z. Zhao, W. Lu, H. Zhu, F. Sun, B. Mei, Z. Jiang, Y. Lyu, X. Chen, L. Guo, T. Wu, X. Ma, Y. Meng and Y. Ding, Appl. Catal., B, 2023, 324, 122298 CrossRef CAS.
- N. H. M. D. Dostagir, R. Rattanawan, M. Gao, J. Ota, J.-y. Hasegawa, K. Asakura, A. Fukouka and A. Shrotri, ACS Catal., 2021, 11, 9450–9461 CrossRef CAS.
- Y. Jiang, Y. Sung, C. Choi, G. Joo Bang, S. Hong, X. Tan, T.-S. Wu, Y.-L. Soo, P. Xiong, M. Meng-Jung Li, L. Hao, Y. Jung and Z. Sun, Angew. Chem., Int. Ed., 2022, 61, e202203836 CrossRef CAS.
- H. Xin, L. Lin, R. Li, D. Li, T. Song, R. Mu, Q. Fu and X. Bao, J. Am. Chem. Soc., 2022, 144, 4874–4882 CrossRef CAS PubMed.
- X. Chen, X. Su, H.-Y. Su, X. Liu, S. Miao, Y. Zhao, K. Sun, Y. Huang and T. Zhang, ACS Catal., 2017, 7, 4613–4620 CrossRef CAS.
- M. Fan, J. D. Jimenez, S. N. Shirodkar, J. Wu, S. Chen, L. Song, M. M. Royko, J. Zhang, H. Guo, J. Cui, K. Zuo, W. Wang, C. Zhang, F. Yuan, R. Vajtai, J. Qian, J. Yang, B. I. Yakobson, J. M. Tour, J. Lauterbach, D. Sun and P. M. Ajayan, ACS Catal., 2019, 9, 10077–10086 CrossRef CAS.
- D. Kim and J. Han, Appl. Energy, 2020, 264, 114711 CrossRef CAS.
- M. Liu, Y. Xu, Y. Meng, L. Wang, H. Wang, Y. Huang, N. Onishi, L. Wang, Z. Fan and Y. Himeda, Adv. Energy Mater., 2022, 12, 2200817 CrossRef CAS.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- S. Enthaler, J. von Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207–1217 RSC.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- R. Sun, Y. Liao, S.-T. Bai, M. Zheng, C. Zhou, T. Zhang and B. F. Sels, Energy Environ. Sci., 2021, 14, 1247–1285 RSC.
- X. Shao, X. Yang, J. Xu, S. Liu, S. Miao, X. Liu, X. Su, H. Duan, Y. Huang and T. Zhang, Chem, 2019, 5, 693–705 CAS.
- G. H. Gunasekar, J. Shin, K.-D. Jung, K. Park and S. Yoon, ACS Catal., 2018, 8, 4346–4353 CrossRef CAS.
- Z. Li, T. M. Rayder, L. Luo, J. A. Byers and C.-K. Tsung, J. Am. Chem. Soc., 2018, 140, 8082–8085 CrossRef CAS.
- K. Park, G. H. Gunasekar, S.-H. Kim, H. Park, S. Kim, K. Park, K.-D. Jung and S. Yoon, Green Chem., 2020, 22, 1639–1649 RSC.
- G. M. Eder, D. A. Pyles, E. R. Wolfson and P. L. McGrier, Chem. Commun., 2019, 55, 7195–7198 RSC.
- Z.-Z. Yang, H. Zhang, B. Yu, Y. Zhao, G. Ji and Z. Liu, Chem. Commun., 2015, 51, 1271–1274 RSC.
- B. An, L. Zeng, M. Jia, Z. Li, Z. Lin, Y. Song, Y. Zhou, J. Cheng, C. Wang and W. Lin, J. Am. Chem. Soc., 2017, 139, 17747–17750 CrossRef CAS.
- K. Mori, T. Taga and H. Yamashita, ACS Catal., 2017, 7, 3147–3151 CrossRef CAS.
- Q. Wang, S. Santos, C. A. Urbina-Blanco, W. Y. Hernández, M. Impéror-Clerc, E. I. Vovk, M. Marinova, O. Ersen, W. Baaziz, O. V. Safonova, A. Y. Khodakov, M. Saeys and V. V. Ordomsky, Appl. Catal., B, 2021, 290, 120036 CrossRef CAS.
- G. Ren, J. Sun, S. Zhai, L. Yang, T. Yu, L. Sun and W. Deng, Cell Rep. Phys. Sci., 2022, 3, 100705 CrossRef CAS.
- S. Zhai, L. Zhang, J. Sun, L. Sun, S. Jiang, T. Yu, D. Zhai, C. Liu, Z. Li and G. Ren, Front. Chem., 2022, 10, 957412 CrossRef CAS PubMed.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411–417 CrossRef CAS PubMed.
- T. J. Deka, A. I. Osman, D. C. Baruah and D. W. Rooney, Environ. Chem. Lett., 2022, 20, 3525–3554 CrossRef CAS.
- J. Nyári, M. Magdeldin, M. Larmi, M. Järvinen and A. Santasalo-Aarnio, J. CO2 Util., 2020, 39, 101166 CrossRef.
- S. Sollai, A. Porcu, V. Tola, F. Ferrara and A. Pettinau, J. CO2 Util., 2023, 68, 102345 CrossRef CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- S. T. Bai, G. De Smet, Y. Liao, R. Sun, C. Zhou, M. Beller, B. U. W. Maes and B. F. Sels, Chem. Soc. Rev., 2021, 50, 4259–4298 RSC.
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS.
- N. H. M. D. Dostagir, C. Thompson, H. Kobayashi, A. M. Karim, A. Fukuoka and A. Shrotri, Catal. Sci. Technol., 2020, 10, 8196–8202 RSC.
- F. Jiang, S. Wang, B. Liu, J. Liu, L. Wang, Y. Xiao, Y. Xu and X. Liu, ACS Catal., 2020, 10, 11493–11509 CrossRef CAS.
- R. Sen, A. Goeppert and G. K. Surya Prakash, Angew. Chem., Int. Ed., 2022, 61, e202207278 CrossRef CAS PubMed.
- H. Zhao, R. Yu, S. Ma, K. Xu, Y. Chen, K. Jiang, Y. Fang, C. Zhu, X. Liu, Y. Tang, L. Wu, Y. Wu, Q. Jiang, P. He, Z. Liu and L. Tan, Nat. Catal., 2022, 5, 818–831 CrossRef CAS.
- W. Wu, Y. Wang, L. Luo, M. Wang, Z. Li, Y. Chen, Z. Wang, J. Chai, Z. Cen, Y. Shi, J. Zhao, J. Zeng and H. Li, Angew. Chem., Int. Ed., 2022, 61, e202213024 CrossRef CAS.
- X.-K. Wu, H.-M. Yan, W. Zhang, J. Zhang, G.-J. Xia and Y.-G. Wang, J. Energy Chem., 2021, 61, 582–593 CrossRef CAS.
- Y. Chen, H. Li, W. Zhao, W. Zhang, J. Li, W. Li, X. Zheng, W. Yan, W. Zhang, J. Zhu, R. Si and J. Zeng, Nat. Commun., 2019, 10, 1885 CrossRef PubMed.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y.-M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B.-M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C.-J. Yang and K. Caldeira, Science, 2018, 360, eaas9793 CrossRef PubMed.
- X. Tan, X. Sun and B. Han, Natl. Sci. Rev., 2022, 9, nwab022 CrossRef PubMed.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- S. Liang, L. Huang, Y. Gao, Q. Wang and B. Liu, Adv. Sci., 2021, 8, 2102886 CrossRef CAS PubMed.
- Y. Cheng, S. Yang, S. P. Jiang and S. Wang, Small Methods, 2019, 3, 1800440 CrossRef.
- C. Jia, Z. Shi and C. Zhao, Curr. Opin. Green Sustainable Chem., 2022, 37, 100651 CrossRef CAS.
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- L. Wang, L. Wang, L. Zhang, H. Liu and J. Yang, Trends Chem., 2022, 4, 1135–1148 CrossRef CAS.
- J. Zhang, W. Cai, F. X. Hu, H. Yang and B. Liu, Chem. Sci., 2021, 12, 6800–6819 RSC.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- Z. Zhao, Z. Chen and G. Lu, J. Phys. Chem. C, 2017, 121, 20865–20870 CrossRef CAS.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315–1316, 10.1039/C39840001315.
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461–7467 CrossRef CAS PubMed.
- J. Schneider, H. Jia, K. Kobiro, D. E. Cabelli, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2012, 5, 9502–9510 RSC.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- P. Su, K. Iwase, S. Nakanishi, K. Hashimoto and K. Kamiya, Small, 2016, 12, 6083–6089 CrossRef CAS PubMed.
- S. Liu, H. B. Yang, S.-F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed.
- D. Xi, J. Li, J. Low, K. Mao, R. Long, J. Li, Z. Dai, T. Shao, Y. Zhong, Y. Li, Z. Li, X. J. Loh, L. Song, E. Ye and Y. Xiong, Adv. Mater., 2022, 34, 2104090 CrossRef CAS PubMed.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang, J. Liu and C. He, Nat. Commun., 2020, 11, 593 CrossRef CAS PubMed.
- R. Boppella, M. Austeria P, Y. Kim, E. Kim, I. Song, Y. Eom, D. P. Kumar, M. Balamurugan, E. Sim, D. H. Kim and T. K. Kim, Adv. Funct. Mater., 2022, 32, 2202351 CrossRef CAS.
- M. D. Hossain, Y. Huang, T. H. Yu, W. A. Goddard Iii and Z. Luo, Nat. Commun., 2020, 11, 2256 CrossRef CAS PubMed.
- H. Li, H. Li, P. Wei, Y. Wang, Y. Zang, D. Gao, G. Wang and X. Bao, Energy Environ. Sci., 2023, 16, 1502–1510 RSC.
- X. Sun, Y. Tuo, C. Ye, C. Chen, Q. Lu, G. Li, P. Jiang, S. Chen, P. Zhu, M. Ma, J. Zhang, J. H. Bitter, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 23614–23618 CrossRef CAS PubMed.
- Z. Zhang, C. Ma, Y. Tu, R. Si, J. Wei, S. Zhang, Z. Wang, J.-F. Li, Y. Wang and D. Deng, Nano Res., 2019, 12, 2313–2317 CrossRef CAS.
- X. Li, Y. Zeng, C.-W. Tung, Y.-R. Lu, S. Baskaran, S.-F. Hung, S. Wang, C.-Q. Xu, J. Wang, T.-S. Chan, H. M. Chen, J. Jiang, Q. Yu, Y. Huang, J. Li, T. Zhang and B. Liu, ACS Catal., 2021, 11, 7292–7301 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- Y. Liang, J. Zhao, Y. Yang, S.-F. Hung, J. Li, S. Zhang, Y. Zhao, A. Zhang, C. Wang, D. Appadoo, L. Zhang, Z. Geng, F. Li and J. Zeng, Nat. Commun., 2023, 14, 474 CrossRef CAS PubMed.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- R. Qiu, J. Jia, L. Peng, R. Li, S. Yan, J. Li, J. Zhang, D. T. Sun, Z. Lan, T. Xue, G. Xu, L. Cui, Z. Lv, C. Li, Y. Hong, Y. Guo, B. Ren, S. Yang, J. Li and B. Han, Green Chem., 2023, 25, 684–691 RSC.
- L. Ding, N. Zhu, Y. Hu, Z. Chen, P. Song, T. Sheng, Z. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202209268 CrossRef CAS PubMed.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- D.-L. Meng, M.-D. Zhang, D.-H. Si, M.-J. Mao, Y. Hou, Y.-B. Huang and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 25485–25492 CrossRef CAS PubMed.
- L. Yan, X.-D. Liang, Y. Sun, L.-P. Xiao, B.-A. Lu, G. Li, Y.-Y. Li, Y.-H. Hong, L.-Y. Wan, C. Chen, J. Yang, Z.-Y. Zhou, N. Tian and S.-G. Sun, Chem. Commun., 2022, 58, 2488–2491 RSC.
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S.-F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed.
- D. Chen, L.-H. Zhang, J. Du, H. Wang, J. Guo, J. Zhan, F. Li and F. Yu, Angew. Chem., Int. Ed., 2021, 60, 24022–24027 CrossRef CAS PubMed.
- J. Rosen, G. S. Hutchings, Q. Lu, R. V. Forest, A. Moore and F. Jiao, ACS Catal., 2015, 5, 4586–4591 CrossRef CAS.
- S. Li, S. Zhao, X. Lu, M. Ceccato, X.-M. Hu, A. Roldan, J. Catalano, M. Liu, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2021, 60, 22826–22832 CrossRef CAS PubMed.
- Z. Hao, J. Chen, D. Zhang, L. Zheng, Y. Li, Z. Yin, G. He, L. Jiao, Z. Wen and X.-J. Lv, Sci. Bull., 2021, 66, 1649–1658 CrossRef CAS PubMed.
- M. Fang, X. Wang, X. Li, Y. Zhu, G. Xiao, J. Feng, X. Jiang, K. Lv, Y. Zhu and W.-F. Lin, ChemCatChem, 2021, 13, 603–609 CrossRef CAS.
- N. Wang, Z. Liu, J. Ma, J. Liu, P. Zhou, Y. Chao, C. Ma, X. Bo, J. Liu, Y. Hei, Y. Bi, M. Sun, M. Cao, H. Zhang, F. Chang, H.-L. Wang, P. Xu, Z. Hu, J. Bai, H. Sun, G. Hu and M. Zhou, ACS Sustainable Chem. Eng., 2020, 8, 13813–13822 CrossRef CAS.
- W. Zheng, D. Wang, W. Cui, X. Sang, X. Qin, Z. Zhao, Z. Li, B. Yang, M. Zhong, L. Lei, Q. Zheng, S. Yao, G. Wu and Y. Hou, Energy Environ. Sci., 2023, 16, 1007–1015 RSC.
- C. Hu, Y. Zhang, A. Hu, Y. Wang, X. Wei, K. Shen, L. Chen and Y. Li, Adv. Mater., 2023, 35, e2209298 CrossRef PubMed.
- M. Ma and Q. Tang, J. Mater. Chem. C, 2022, 10, 15948–15956 RSC.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- J. Han, P. An, S. Liu, X. Zhang, D. Wang, Y. Yuan, J. Guo, X. Qiu, K. Hou, L. Shi, Y. Zhang, S. Zhao, C. Long and Z. Tang, Angew. Chem., Int. Ed., 2019, 58, 12711–12716 CrossRef CAS PubMed.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 6, 8177 CrossRef PubMed.
- S. Wang, P. Zhou, L. Zhou, F. Lv, Y. Sun, Q. Zhang, L. Gu, H. Yang and S. Guo, Nano Lett., 2021, 21, 4262–4269 CrossRef CAS PubMed.
- M. Wang, Y. Yao, Y. Tian, Y. Yuan, L. Wang, F. Yang, J. Ren, X. Hu, F. Wu, S. Zhang, J. Wu and J. Lu, Adv. Mater., 2023, 35, e2210658 CrossRef PubMed.
- B. Zhang, J. Zhang, J. Shi, D. Tan, L. Liu, F. Zhang, C. Lu, Z. Su, X. Tan, X. Cheng, B. Han, L. Zheng and J. Zhang, Nat. Commun., 2019, 10, 2980 CrossRef PubMed.
- N. Zhang, X. Zhang, L. Tao, P. Jiang, C. Ye, R. Lin, Z. Huang, A. Li, D. Pang, H. Yan, Y. Wang, P. Xu, S. An, Q. Zhang, L. Liu, S. Du, X. Han, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 6170–6176 CrossRef CAS PubMed.
- J. Liu, X. Kong, L. Zheng, X. Guo, X. Liu and J. Shui, ACS Nano, 2020, 14, 1093–1101 CrossRef CAS PubMed.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71–129 CAS.
- Y. Wang, W. Qiu, E. Song, F. Gu, Z. Zheng, X. Zhao, Y. Zhao, J. Liu and W. Zhang, Natl. Sci. Rev., 2018, 5, 327–341 CrossRef CAS.
- B. I. Lundqvist, O. Gunnarsson, H. Hjelmberg and J. K. Nørskov, Surf. Sci., 1979, 89, 196–225 CrossRef CAS.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- S. Liu, Z. Li, C. Wang, W. Tao, M. Huang, M. Zuo, Y. Yang, K. Yang, L. Zhang, S. Chen, P. Xu and Q. Chen, Nat. Commun., 2020, 11, 938 CrossRef CAS PubMed.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2020, 10, 1902338 CrossRef CAS.
- Q. Wang, K. Liu, J. Fu, C. Cai, H. Li, Y. Long, S. Chen, B. Liu, H. Li, W. Li, X. Qiu, N. Zhang, J. Hu, H. Pan and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 25241–25245 CrossRef CAS PubMed.
- Q. Wang, M. Dai, H. Li, Y. R. Lu, T. S. Chan, C. Ma, K. Liu, J. Fu, W. Liao, S. Chen, E. Pensa, Y. Wang, S. Zhang, Y. Sun, E. Cortes and M. Liu, Adv. Mater., 2023, e2300695 CrossRef PubMed.
- Z. Zhang, J. Zhu, S. Chen, W. Sun and D. Wang, Angew. Chem., Int. Ed., 2022, e202215136 Search PubMed.
- W. Guo, X. Tan, J. Bi, L. Xu, D. Yang, C. Chen, Q. Zhu, J. Ma, A. Tayal, J. Ma, Y. Huang, X. Sun, S. Liu and B. Han, J. Am. Chem. Soc., 2021, 143, 6877–6885 CrossRef CAS PubMed.
- M. Jia, S. Hong, T.-S. Wu, X. Li, Y.-L. Soo and Z. Sun, Chem. Commun., 2019, 55, 12024–12027 RSC.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji, X. Zheng, Y. Wang, L. Zheng, C. Chen, D. Wang, J. Zhang and Y. Li, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS PubMed.
- Z. Wang, C. Wang, Y. Hu, S. Yang, J. Yang, W. Chen, H. Zhou, F. Zhou, L. Wang, J. Du, Y. Li and Y. Wu, Nano Res., 2021, 14, 2790–2796 CrossRef CAS.
- J. Guo, W. Zhang, L.-H. Zhang, D. Chen, J. Zhan, X. Wang, N. R. Shiju and F. Yu, Adv. Sci., 2021, 8, 2102884 CrossRef CAS PubMed.
- Q. Hao, H.-X. Zhong, J. Z. Wang, K.-H. Liu, J.-M. Yan, Z.-H. Ren, N. Zhou, X. Zhao, H. Zhang, D.-X. Liu, X. Liu, L.-W. Chen, J. Luo and X.-B. Zhang, Nat. Synth., 2022, 1, 719–728 CrossRef.
- N. Zhang, X. Zhang, Y. Kang, C. Ye, R. Jin, H. Yan, R. Lin, J. Yang, Q. Xu, Y. Wang, Q. Zhang, L. Gu, L. Liu, W. Song, J. Liu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 13388–13393 CrossRef CAS PubMed.
- Y. Li, B. Wei, M. Zhu, J. Chen, Q. Jiang, B. Yang, Y. Hou, L. Lei, Z. Li, R. Zhang and Y. Lu, Adv. Mater., 2021, 33, 2102212 CrossRef CAS PubMed.
- L. Zhang, J. Feng, S. Liu, X. Tan, L. Wu, S. Jia, L. Xu, X. Ma, X. Song, J. Ma, X. Sun and B. Han, Adv. Mater., 2023, 35, e2209590 CrossRef PubMed.
- Z. Zeng, L. Y. Gan, H. Bin Yang, X. Su, J. Gao, W. Liu, H. Matsumoto, J. Gong, J. Zhang, W. Cai, Z. Zhang, Y. Yan, B. Liu and P. Chen, Nat. Commun., 2021, 12, 4088 CrossRef CAS PubMed.
- X. Wang, N. Fu, J. C. Liu, K. Yu, Z. Li, Z. Xu, X. Liang, P. Zhu, C. Ye, A. Zhou, A. Li, L. Zheng, L. M. Liu, C. Chen, D. Wang, Q. Peng and Y. Li, J. Am. Chem. Soc., 2022, 144, 23223–23229 CrossRef CAS PubMed.
- J. Li, H. Zeng, X. Dong, Y. Ding, S. Hu, R. Zhang, Y. Dai, P. Cui, Z. Xiao, D. Zhao, L. Zhou, T. Zheng, J. Xiao, J. Zeng and C. Xia, Nat. Commun., 2023, 14, 340 CrossRef CAS PubMed.
- H. Shang, T. Wang, J. Pei, Z. Jiang, D. Zhou, Y. Wang, H. Li, J. Dong, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 22465–22469 CrossRef CAS PubMed.
- Z. Jiang, T. Wang, J. Pei, H. Shang, D. Zhou, H. Li, J. Dong, Y. Wang, R. Cao, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Energy Environ. Sci., 2020, 13, 2856–2863 RSC.
- P. Huang, M. Cheng, H. Zhang, M. Zuo, C. Xiao and Y. Xie, Nano Energy, 2019, 61, 428–434 CrossRef CAS.
- X. Zu, X. Li, W. Liu, Y. Sun, J. Xu, T. Yao, W. Yan, S. Gao, C. Wang, S. Wei and Y. Xie, Adv. Mater., 2019, 31, 1808135 CrossRef PubMed.
- Y. Deng, J. Zhao, S. Wang, R. Chen, J. Ding, H.-J. Tsai, W.-J. Zeng, S.-F. Hung, W. Xu, J. Wang, F. Jaouen, X. Li, Y. Huang and B. Liu, J. Am. Chem. Soc., 2023, 145, 7242–7251 CrossRef CAS PubMed.
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li, C.-W. Pao, J. Xiao, C. Xia and J. Zeng, Nat. Nanotechnol., 2021, 16, 1386–1393 CrossRef CAS PubMed.
- Y. Jiang, J. Shan, P. Wang, L. Huang, Y. Zheng and S.-Z. Qiao, ACS Catal., 2023, 13, 3101–3108 CrossRef CAS.
- G. Shi, Y. Xie, L. Du, X. Fu, X. Chen, W. Xie, T.-B. Lu, M. Yuan and M. Wang, Angew. Chem., Int. Ed., 2022, 61, e202203569 CrossRef CAS PubMed.
- H. Zou, G. Zhao, H. Dai, H. Dong, W. Luo, L. Wang, Z. Lu, Y. Luo, G. Zhang and L. Duan, Angew. Chem., Int. Ed., 2022, 62, e202217220 CrossRef PubMed.
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS.
- S. Chen, B. Wang, J. Zhu, L. Wang, H. Ou, Z. Zhang, X. Liang, L. Zheng, L. Zhou, Y.-Q. Su, D. Wang and Y. Li, Nano Lett., 2021, 21, 7325–7331 CrossRef CAS PubMed.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed.
- M. Ren, X. Guo and S. Huang, Chem. Eng. J., 2022, 433, 134270 CrossRef CAS.
- S. Back and Y. Jung, ACS Energy Lett., 2017, 2, 969–975 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- S. Kong, X. Lv, X. Wang, Z. Liu, Z. Li, B. Jia, D. Sun, C. Yang, L. Liu, A. Guan, J. Wang, G. Zheng and F. Huang, Nat. Catal., 2022, 6, 6–15 CrossRef.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- L. Ju, X. Tan, X. Mao, Y. Gu, S. Smith, A. Du, Z. Chen, C. Chen and L. Kou, Nat. Commun., 2021, 12, 5128 CrossRef CAS PubMed.
- H. Lin, S. Luo, H. Zhang and J. Ye, Joule, 2022, 6, 294–314 CrossRef CAS.
- V. N. Gopalakrishnan, J. Becerra, E. F. Pena, M. Sakar, F. Béland and T.-O. Do, Green Chem., 2021, 23, 8332–8360 RSC.
- H. Zhang, W. Tian, X. Duan, H. Sun, Y. Huang, Y. Fang and S. Wang, Chin. J. Catal., 2022, 43, 2301–2315 CrossRef CAS.
- A. Khandelwal, D. Maarisetty and S. S. Baral, Renewable Sustainable Energy Rev., 2022, 167, 112693 CrossRef CAS.
- Y. Wang, E. Chen and J. Tang, ACS Catal., 2022, 12, 7300–7316 CrossRef CAS PubMed.
- X. Li, W. Bi, Z. Wang, W. Zhu, W. Chu, C. Wu and Y. Xie, Nano Res., 2018, 11, 3362–3370 CrossRef CAS.
- B.-H. Lee, E. Gong, M. Kim, S. Park, H. R. Kim, J. Lee, E. Jung, C. W. Lee, J. Bok, Y. Jung, Y. S. Kim, K.-S. Lee, S.-P. Cho, J.-W. Jung, C.-H. Cho, S. Lebègue, K. T. Nam, H. Kim, S.-I. In and T. Hyeon, Energy Environ. Sci., 2022, 15, 601–609 RSC.
- Z. Jiang, W. Sun, W. Miao, Z. Yuan, G. Yang, F. Kong, T. Yan, J. Chen, B. Huang, C. An and G. A. Ozin, Adv. Sci., 2019, 6, 1900289 CrossRef PubMed.
- L. Yuan, S.-F. Hung, Z.-R. Tang, H. M. Chen, Y. Xiong and Y.-J. Xu, ACS Catal., 2019, 9, 4824–4833 CrossRef CAS.
- Y. Yu, X. a. Dong, P. Chen, Q. Geng, H. Wang, J. Li, Y. Zhou and F. Dong, ACS Nano, 2021, 15, 14453–14464 CrossRef CAS PubMed.
- X. Xiong, C. Mao, Z. Yang, Q. Zhang, G. I. N. Waterhouse, L. Gu and T. Zhang, Adv. Energy Mater., 2020, 10, 2002928 CrossRef CAS.
- J. Ding, X. Liu, M. Shi, T. Li, M. Xia, X. Du, R. Shang, H. Gu and Q. Zhong, Sol. Energy Mater. Sol. Cells, 2019, 195, 34–42 CrossRef CAS.
- Y. J. Wang, G. L. Zhuang, J. W. Zhang, F. Luo, X. Cheng, F. L. Sun, S. S. Fu, T. B. Lu and Z. M. Zhang, Angew. Chem., Int. Ed., 2022, 62, e202216592 CrossRef PubMed.
- Y. Cao, L. Guo, M. Dan, D. E. Doronkin, C. Han, Z. Rao, Y. Liu, J. Meng, Z. Huang, K. Zheng, P. Chen, F. Dong and Y. Zhou, Nat. Commun., 2021, 12, 1675 CrossRef CAS PubMed.
- H. Chen, Y. Xiong, J. Li, J. Abed, D. Wang, A. Pedrazo-Tardajos, Y. Cao, Y. Zhang, Y. Wang, M. Shakouri, Q. Xiao, Y. Hu, S. Bals, E. H. Sargent, C.-Y. Su and Z. Yang, Nat. Commun., 2023, 14, 1719 CrossRef CAS PubMed.
- S. Cao and J. Yu, J. Photochem. Photobiol., C, 2016, 27, 72–99 CrossRef CAS.
- P. Kumar, R. Boukherroub and K. Shankar, J. Mater. Chem. A, 2018, 6, 12876–12931 RSC.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- J. Luo, H. Han, X. Wang, X. Qiu, B. Liu, Y. Lai, X. Chen, R. Zhong, L. Wang and C. Wang, Appl. Catal., B, 2023, 328, 122495 CrossRef CAS.
- Y. Zhao, Z. Han, G. Gao, W. Zhang, Y. Qu, H. Zhu, P. Zhu and G. Wang, Adv. Funct. Mater., 2021, 31, 2104976 CrossRef CAS.
- P. Huang, J. Huang, S. A. Pantovich, A. D. Carl, T. G. Fenton, C. A. Caputo, R. L. Grimm, A. I. Frenkel and G. Li, J. Am. Chem. Soc., 2018, 140, 16042–16047 CrossRef CAS PubMed.
- Y. Yang, F. Li, J. Chen, J. Fan and Q. Xiang, ChemSusChem, 2020, 13, 1979–1985 CrossRef CAS PubMed.
- Z. Han, Y. Zhao, G. Gao, W. Zhang, Y. Qu, H. Zhu, P. Zhu and G. Wang, Small, 2021, 17, 2102089 CrossRef CAS PubMed.
- P. Chen, B. Lei, X. a. Dong, H. Wang, J. Sheng, W. Cui, J. Li, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 15841–15852 CrossRef CAS PubMed.
- H. Cao, J. Wang, J.-H. Kim, Z. Guo, J. Xiao, J. Yang, J. Chang, Y. Shi and Y. Xie, Appl. Catal., B, 2021, 296, 120362 CrossRef CAS.
- P. Sharma, S. Kumar, O. Tomanec, M. Petr, J. Zhu Chen, J. T. Miller, R. S. Varma, M. B. Gawande and R. Zbořil, Small, 2021, 17, 2006478 CrossRef CAS PubMed.
- T. Tong, B. Zhu, C. Jiang, B. Cheng and J. Yu, Appl. Surf. Sci., 2018, 433, 1175–1183 CrossRef CAS.
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS PubMed.
- L. Cheng, H. Yin, C. Cai, J. Fan and Q. Xiang, Small, 2020, 16, 2002411 CrossRef CAS PubMed.
- Y. Li, B. Li, D. Zhang, L. Cheng and Q. Xiang, ACS Nano, 2020, 14, 10552–10561 CrossRef CAS PubMed.
- M. Ma, Z. Huang, D. E. Doronkin, W. Fa, Z. Rao, Y. Zou, R. Wang, Y. Zhong, Y. Cao, R. Zhang and Y. Zhou, Appl. Catal., B, 2022, 300, 120695 CrossRef CAS.
- S. Hu, P. Qiao, X. Yi, Y. Lei, H. Hu, J. Ye and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202304585 CrossRef CAS PubMed.
- Q. Chen, G. Gao, Y. Zhang, Y. li, H. Zhu, P. Zhu, Y. Qu, G. Wang and W. Qin, J. Mater. Chem. A, 2021, 9, 15820–15826 RSC.
- A. J. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS.
- B.-C. He, C. Zhang, P.-P. Luo, Y. Li and T.-B. Lu, Green Chem., 2020, 22, 7552–7559 RSC.
- S. Ji, Y. Qu, T. Wang, Y. Chen, G. Wang, X. Li, J. Dong, Q. Chen, W. Zhang, Z. Zhang, S. Liang, R. Yu, Y. Wang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 10651–10657 CrossRef CAS PubMed.
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Adv. Mater., 2018, 30, 1704624 CrossRef PubMed.
- Z. Wang, J. Yang, J. Cao, W. Chen, G. Wang, F. Liao, X. Zhou, F. Zhou, R. Li, Z.-Q. Yu, G. Zhang, X. Duan and Y. Wu, ACS Nano, 2020, 14, 6164–6172 CrossRef CAS PubMed.
- J. Yang, Z. Wang, J. Jiang, W. Chen, F. Liao, X. Ge, X. Zhou, M. Chen, R. Li, Z. Xue, G. Wang, X. Duan, G. Zhang, Y.-G. Wang and Y. Wu, Nano Energy, 2020, 76, 105059 CrossRef CAS.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed., 2016, 55, 14310–14314 CrossRef CAS PubMed.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu, Y. Pan, H.-L. Jiang, D. Mei and C. Zhong, Nat. Catal., 2021, 4, 719–729 CrossRef CAS.
- M. Kou, W. Liu, Y. Wang, J. Huang, Y. Chen, Y. Zhou, Y. Chen, M. Ma, K. Lei, H. Xie, P. K. Wong and L. Ye, Appl. Catal., B, 2021, 291, 120146 CrossRef CAS.
- Q. Zhang, S. Gao, Y. Guo, H. Wang, J. Wei, X. Su, H. Zhang, Z. Liu and J. Wang, Nat. Commun., 2023, 14, 1147 CrossRef CAS PubMed.
- G. Wang, C.-T. He, R. Huang, J. Mao, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 19339–19345 CrossRef CAS PubMed.
- Y.-H. Chen, M.-Y. Qi, Y.-H. Li, Z.-R. Tang, T. Wang, J. Gong and Y.-J. Xu, Cell Rep. Phys. Sci., 2021, 2, 100371 CrossRef CAS.
- Y. Tang, Y. Wei, Z. Wang, S. Zhang, Y. Li, L. Nguyen, Y. Li, Y. Zhou, W. Shen, F. F. Tao and P. Hu, J. Am. Chem. Soc., 2019, 141, 7283–7293 CrossRef CAS PubMed.
- A. V. Tavasoli, M. Preston and G. Ozin, Energy Environ. Sci., 2021, 14, 3098–3109 RSC.
- W. L. Luyben, Ind. Eng. Chem. Res., 2014, 53, 14423–14439 CrossRef CAS.
- M. Akri, S. Zhao, X. Li, K. Zang, A. F. Lee, M. A. Isaacs, W. Xi, Y. Gangarajula, J. Luo, Y. Ren, Y.-T. Cui, L. Li, Y. Su, X. Pan, W. Wen, Y. Pan, K. Wilson, L. Li, B. Qiao, H. Ishii, Y.-F. Liao, A. Wang, X. Wang and T. Zhang, Nat. Commun., 2019, 10, 5181 CrossRef PubMed.
- J. Wu, J. Gao, S. Lian, J. Li, K. Sun, S. Zhao, Y. D. Kim, Y. Ren, M. Zhang, Q. Liu, Z. Liu and Z. Peng, Appl. Catal., B, 2022, 314, 121516 CrossRef CAS.
- D. Shen, Z. Li, J. Shan, G. Yu, X. Wang, Y. Zhang, C. Liu, S. Lyu, J. Li and L. Li, Appl. Catal., B, 2022, 318, 121809 CrossRef CAS.
- S. Kim, J. Lauterbach and E. Sasmaz, ACS Catal., 2021, 11, 8247–8260 CrossRef CAS.
- L. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. Lou, L. Dong, L. Henderson, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Nat. Energy, 2020, 5, 61–70 CrossRef CAS.
- J. Goldemberg, Science, 2007, 315, 808–810 CrossRef CAS PubMed.
- F. J. Caparrós, L. Soler, M. D. Rossell, I. Angurell, L. Piccolo, O. Rossell and J. Llorca, ChemCatChem, 2018, 10, 2365–2369 CrossRef.
- X. Ye, J. Ma, W. Yu, X. Pan, C. Yang, C. Wang, Q. Liu and Y. Huang, J. Energy Chem., 2022, 67, 184–192 CrossRef CAS.
- K. Zheng, Y. Li, B. Liu, F. Jiang, Y. Xu and X. Liu, Angew. Chem., Int. Ed., 2022, 61, e202210991 CrossRef CAS PubMed.
- X. Shang, G. Liu, X. Su, Y. Huang and T. Zhang, EES Catal., 2023, 1, 353–368 RSC.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- G. Sun, Y. Cao, D. Li, M. Hu, X. Liang, Z. Wang, Z. Cai, F. Shen, B. Chen and K. Zhou, Appl. Catal., A, 2023, 651, 119025 CrossRef CAS.
- J. Wang, D. Deng, Q. Wu, M. Liu, Y. Wang, J. Jiang, X. Zheng, H. Zheng, Y. Bai, Y. Chen, X. Xiong and Y. Lei, ACS Nano, 2023, 17, 18688–18705 CrossRef CAS PubMed.
- J. Ding, H. Bin Yang, X.-L. Ma, S. Liu, W. Liu, Q. Mao, Y. Huang, J. Li, T. Zhang and B. Liu, Nat. Energy, 2023, 8, 1386–1394 CrossRef CAS.
- X. Li, S.-G. Han, W. Wu, K. Zhang, B. Chen, S.-H. Zhou, D.-D. Ma, W. Wei, X.-T. Wu, R. Zou and Q.-L. Zhu, Energy Environ. Sci., 2023, 16, 502–512 RSC.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- N. Li, B. Wang, Y. Si, F. Xue, J. Zhou, Y. Lu and M. Liu, ACS Catal., 2019, 9, 5590–5602 CrossRef CAS.
- S. Yu and P. K. Jain, Nat. Commun., 2019, 10, 2022 CrossRef PubMed.
- Y. Shen, C. Ren, L. Zheng, X. Xu, R. Long, W. Zhang, Y. Yang, Y. Zhang, Y. Yao, H. Chi, J. Wang, Q. Shen, Y. Xiong, Z. Zou and Y. Zhou, Nat. Commun., 2023, 14, 1117 CrossRef CAS PubMed.
- J. Hong, M. Li, J. Zhang, B. Sun and F. Mo, ChemSusChem, 2019, 12, 6–39 CrossRef CAS PubMed.
- Y. Quan, R. Yu, J. Zhu, A. Guan, X. Lv, C. Yang, S. Li, J. Wu and G. Zheng, J. Colloid Interface Sci., 2021, 601, 378–384 CrossRef CAS PubMed.
- X. Shang, G. Liu, X. Su, Y. Huang and T. Zhang, ACS Catal., 2022, 12, 13741–13754 CrossRef CAS.
- J. Zuo, W. Chen, J. Liu, X. Duan, L. Ye and Y. Yuan, Sci. Adv., 2020, 6, eaba5433 CrossRef CAS PubMed.
- X. Peng, L. Zeng, D. Wang, Z. Liu, Y. Li, Z. Li, B. Yang, L. Lei, L. Dai and Y. Hou, Chem. Soc. Rev., 2023, 52, 2193–2237 RSC.
- S. Saini, P. K. Prajapati and S. L. Jain, Catal. Rev., 2022, 64, 631–677 CrossRef CAS.
- X. Shang, H. Zhuo, Q. Han, X. Yang, G. Hou, G. Liu, X. Su, Y. Huang and T. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202309377 CrossRef CAS PubMed.
- Q.-W. Song, R. Ma, P. Liu, K. Zhang and L.-N. He, Green Chem., 2023, 25, 6538–6560 RSC.
- Y. Yang and J.-W. Lee, Chem. Sci., 2019, 10, 3905–3926 RSC.
- T. Li, F. Chen, R. Lang, H. Wang, Y. Su, B. Qiao, A. Wang and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 7430–7434 CrossRef CAS PubMed.
- I. Ro, J. Qi, S. Lee, M. Xu, X. Yan, Z. Xie, G. Zakem, A. Morales, J. G. Chen, X. Pan, D. G. Vlachos, S. Caratzoulas and P. Christopher, Nature, 2022, 609, 287–292 CrossRef CAS PubMed.
- S. Feng, X. Lin, X. Song, B. Mei, J. Mu, J. Li, Y. Liu, Z. Jiang and Y. Ding, ACS Catal., 2021, 11, 682–690 CrossRef CAS.
- S. Fu, S. Yao, S. Guo, G.-C. Guo, W. Yuan, T.-B. Lu and Z.-M. Zhang, J. Am. Chem. Soc., 2021, 143, 20792–20801 CrossRef CAS PubMed.
- M. T. Jensen, M. H. Rønne, A. K. Ravn, R. W. Juhl, D. U. Nielsen, X.-M. Hu, S. U. Pedersen, K. Daasbjerg and T. Skrydstrup, Nat. Commun., 2017, 8, 489 CrossRef PubMed.
- R. Sang, Y. Hu, R. Razzaq, G. Mollaert, H. Atia, U. Bentrup, M. Sharif, H. Neumann, H. Junge, R. Jackstell, B. U. W. Maes and M. Beller, Nat. Commun., 2022, 13, 4432 CrossRef CAS PubMed.
- C. Wang, Q. Song, K. Zhang, P. Liu, J. Wang, J. Wang, H. Zhang and J. Wang, Chem. Commun., 2019, 55, 1299–1302 RSC.
- Q. Yang, C.-C. Yang, C.-H. Lin and H.-L. Jiang, Angew. Chem., Int. Ed., 2019, 58, 3511–3515 CrossRef CAS PubMed.
- J. Xu, H. Xu, A. Q. Dong, H. Zhang, Y. T. Zhou, H. Dong, B. Tang, Y. F. Liu, L. X. Zhang, X. J. Liu, J. Luo, L. J. Bie, S. Dai, Y. H. Wang, X. H. Sun and Y. G. Li, Adv. Mater., 2022, 34, 2206991 CrossRef CAS PubMed.
- J. Sittiwong, O. Opasmongkolchai, P. Srifa, B. Boekfa, P. Treesukol, W. Sangthong, T. Maihom and J. Limtrakul, Mol. Catal., 2023, 535, 112855 CrossRef CAS.
- X. Zhang, X. Zhu, S. Bo, C. Chen, M. Qiu, X. Wei, N. He, C. Xie, W. Chen, J. Zheng, P. Chen, S. P. Jiang, Y. Li, Q. Liu and S. Wang, Nat. Commun., 2022, 13, 5337 CrossRef CAS PubMed.
- X. Wei, Y. Liu, X. Zhu, S. Bo, L. Xiao, C. Chen, T. T. T. Nga, Y. He, M. Qiu, C. Xie, D. Wang, Q. Liu, F. Dong, C. L. Dong, X. Z. Fu and S. Wang, Adv. Mater., 2023, 35, e2300020 CrossRef PubMed.
- D. Zhao, Z. Chen, W. Yang, S. Liu, X. Zhang, Y. Yu, W.-C. Cheong, L. Zheng, F. Ren, G. Ying, X. Cao, D. Wang, Q. Peng, G. Wang and C. Chen, J. Am. Chem. Soc., 2019, 141, 4086–4093 CrossRef CAS PubMed.
- H. Shi, H. Wang, Y. Zhou, J. Li, P. Zhai, X. Li, G. G. Gurzadyan, J. Hou, H. Yang and X. Guo, Angew. Chem., Int. Ed., 2022, 61, e202208904 CrossRef CAS PubMed.
- B. Peng, H. Liu, Z. Liu, X. Duan and Y. Huang, J. Phys. Chem. Lett., 2021, 12, 2837–2847 CrossRef CAS PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- G. A. Somorjai and J. Y. Park, Angew. Chem., Int. Ed., 2008, 47, 9212–9228 CrossRef CAS PubMed.
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- X. Chang, Z.-J. Zhao, Z. Lu, S. Chen, R. Luo, S. Zha, L. Li, G. Sun, C. Pei and J. Gong, Nat. Nanotechnol., 2023, 18, 611–616 CrossRef CAS PubMed.
- Z.-J. Zhao, S. Liu, S. Zha, D. Cheng, F. Studt, G. Henkelman and J. Gong, Nat. Rev. Mater., 2019, 4, 792–804 CrossRef.
- S. Jiao, X. Fu and H. Huang, Adv. Funct. Mater., 2022, 32, 2107651 CrossRef CAS.
- A. S. Varela, W. Ju, A. Bagger, P. Franco, J. Rossmeisl and P. Strasser, ACS Catal., 2019, 9, 7270–7284 CrossRef CAS.
- T. Dai, Z. Wang, X. Lang and Q. Jiang, J. Mater. Chem. A, 2022, 10, 16900–16907 RSC.
- H. Jin, R. Zhao, P. Cui, X. Liu, J. Yan, X. Yu, D. Ma, W. Song and C. Cao, J. Am. Chem. Soc., 2023, 145, 12023–12032 CrossRef CAS PubMed.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- Q. Chang, Y. Liu, J.-H. Lee, D. Ologunagba, S. Hwang, Z. Xie, S. Kattel, J. H. Lee and J. G. Chen, J. Am. Chem. Soc., 2022, 144, 16131–16138 CrossRef CAS PubMed.
- P. Brimley, H. Almajed, Y. Alsunni, A. W. Alherz, Z. J. L. Bare, W. A. Smith and C. B. Musgrave, ACS Catal., 2022, 12, 10161–10171 CrossRef CAS.
- Y.-X. Zhang, S. Zhang, H. Huang, X. Liu, B. Li, Y. Lee, X. Wang, Y. Bai, M. Sun, Y. Wu, S. Gong, X. Liu, Z. Zhuang, T. Tan and Z. Niu, J. Am. Chem. Soc., 2023, 145, 4819–4827 CrossRef CAS PubMed.
- L. Li, K. Yuan and Y. Chen, Acc. Mater. Res., 2022, 3, 584–596 CrossRef CAS.
- H. Li, R. Li, G. Liu, M. Zhai and J. Yu, Adv. Mater., 2023, 2301307 CrossRef PubMed.
- T. Pu, J. Ding, F. Zhang, K. Wang, N. Cao, E. J. M. Hensen and P. Xie, Angew. Chem., Int. Ed., 2023, e202305964 CAS.
- J. Ding, F. Li, J. Zhang, Q. Zhang, Y. Liu, W. Wang, W. Liu, B. Wang, J. Cai, X. Su, H. B. Yang, X. Yang, Y. Huang, Y. Zhai and B. Liu, J. Am. Chem. Soc., 2023, 145, 11829–11836 CrossRef CAS PubMed.
- T. Ding, X. Liu, Z. Tao, T. Liu, T. Chen, W. Zhang, X. Shen, D. Liu, S. Wang, B. Pang, D. Wu, L. Cao, L. Wang, T. Liu, Y. Li, H. Sheng, M. Zhu and T. Yao, J. Am. Chem. Soc., 2021, 143, 11317–11324 CrossRef CAS PubMed.
- J. Pei, T. Wang, R. Sui, X. Zhang, D. Zhou, F. Qin, X. Zhao, Q. Liu, W. Yan, J. Dong, L. Zheng, A. Li, J. Mao, W. Zhu, W. Chen and Z. Zhuang, Energy Environ. Sci., 2021, 14, 3019–3028 Search PubMed.
- J. Wang, E. Kim, D. P. Kumar, A. P. Rangappa, Y. Kim, Y. Zhang and T. K. Kim, Angew. Chem., Int. Ed., 2022, 61, e202113044 CrossRef CAS PubMed.
- J. Zhu, M. Xiao, D. Ren, R. Gao, X. Liu, Z. Zhang, D. Luo, W. Xing, D. Su, A. Yu and Z. Chen, J. Am. Chem. Soc., 2022, 144, 9661–9671 CrossRef CAS PubMed.
- H. Ou, S. Ning, P. Zhu, S. Chen, A. Han, Q. Kang, Z. Hu, J. Ye, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202206579 CrossRef CAS PubMed.
- Q. Zhang, D. Liu, Y. Zhang, Z. Guo, M. Chen, Y. Chen, B. Jin, Y. Song and H. Pan, J. Energy Chem., 2023, 87, 509–517 CrossRef CAS.
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., Int. Ed., 2021, 60, 7382–7388 CrossRef CAS PubMed.
- J. Chen, Y. Zha, B. Liu, Y. Li, Y. Xu and X. Liu, ACS Catal., 2023, 7110–7121, DOI:10.1021/acscatal.3c00586.
- H. Wang, X. Bi, Y. Yan, Y. Zhao, Z. Yang, H. Ning and M. Wu, Adv. Funct. Mater., 2023, 33, 2214946 CrossRef CAS.
- W. Yang, Z. Jia, B. Zhou, L. Chen, X. Ding, L. Jiao, H. Zheng, Z. Gao, Q. Wang and H. Li, ACS Catal., 2023, 13, 9695–9705 CrossRef CAS.
- Y. Lou, F. jiang, W. Zhu, L. Wang, T. Yao, S. Wang, B. Yang, B. Yang, Y. Zhu and X. Liu, Appl. Catal., B, 2021, 291, 120122 CrossRef CAS.
- W. Xia, Y. Xie, S. Jia, S. Han, R. Qi, T. Chen, X. Xing, T. Yao, D. Zhou, X. Dong, J. Zhai, J. Li, J. He, D. Jiang, Y. Yamauchi, M. He, H. Wu and B. Han, J. Am. Chem. Soc., 2023, 145, 17253–17264 CrossRef CAS PubMed.
- J. M. Wang, Q. Y. Zhu, J. H. Lee, T. G. Woo, Y. X. Zhang, W.-D. Jang and T. K. Kim, Nat. Commun., 2023, 14, 3808 CrossRef CAS PubMed.
- P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680–699 CrossRef CAS PubMed.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 1998, 37, 3387–3388 Search PubMed.
- M. Huser, M.-T. Youinou and J. A. Osborn, Angew. Chem., Int. Ed., 1989, 28, 1386–1388 Search PubMed.
- Y. Ren, Y. Tang, L. Zhang, X. Liu, L. Li, S. Miao, D. Sheng Su, A. Wang, J. Li and T. Zhang, Nat. Commun., 2019, 10, 4500 CrossRef PubMed.
- Z. Chen, Z. Liu and X. Xu, Nat. Commun., 2023, 14, 2512 CrossRef CAS PubMed.
- L. Chen, R. R. Unocic, A. S. Hoffman, J. Hong, A. H. Braga, Z. Bao, S. R. Bare and J. Szanyi, JACS Au, 2021, 1, 977–986 CrossRef CAS PubMed.
- C.-S. Hsu, J. Wang, Y.-C. Chu, J.-H. Chen, C.-Y. Chien, K.-H. Lin, L. D. Tsai, H.-C. Chen, Y.-F. Liao, N. Hiraoka, Y.-C. Cheng and H. M. Chen, Nat. Commun., 2023, 14, 5245 CrossRef CAS PubMed.
- Y. Zeng, J. Zhao, S. Wang, X. Ren, Y. Tan, Y.-R. Lu, S. Xi, J. Wang, F. Jaouen, X. Li, Y. Huang, T. Zhang and B. Liu, J. Am. Chem. Soc., 2023, 145, 15600–15610 CrossRef CAS PubMed.
- X. Ren, J. Zhao, X. Li, J. Shao, B. Pan, A. Salamé, E. Boutin, T. Groizard, S. Wang, J. Ding, X. Zhang, W.-Y. Huang, W.-J. Zeng, C. Liu, Y. Li, S.-F. Hung, Y. Huang, M. Robert and B. Liu, Nat. Commun., 2023, 14, 3401 CrossRef CAS PubMed.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- L. Sévery, J. Szczerbiński, M. Taskin, I. Tuncay, F. Brandalise Nunes, C. Cignarella, G. Tocci, O. Blacque, J. Osterwalder, R. Zenobi, M. Iannuzzi and S. D. Tilley, Nat. Chem., 2021, 13, 523–529 Search PubMed.
- X. Cao, L. Zhao, B. Wulan, D. Tan, Q. Chen, J. Ma and J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202113918 CrossRef CAS PubMed.
- X. Ren, S. Liu, H. Li, J. Ding, L. Liu, Z. Kuang, L. Li, H. Yang, F. Bai, Y. Huang, T. Zhang and B. Liu, Sci. China: Chem., 2020, 63, 1727–1733 Search PubMed.
- Q. Zhang, H. J. Tsai, F. Li, Z. Wei, Q. He, J. Ding, Y. Liu, Z.-Y. Lin, X. Yang, Z. Chen, F. Hu, X. Yang, Q. Tang, H. B. Yang, S.-F. Hung and Y. Zhai, Angew. Chem., Int. Ed., 2023, n/a, e202311550 Search PubMed.
- P. Song, B. Hu, D. Zhao, J. Fu, X. Su, W. Feng, K. Yu, S. Liu, J. Zhang and C. Chen, ACS Nano, 2023, 17, 4619–4628 Search PubMed.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H.-L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 CrossRef CAS PubMed.
- X. Zhao, S. Huang, Z. Chen, C. Lu, S. Han, C. Ke, J. Zhu, J. Zhang, D. Tranca and X. Zhuang, Carbon, 2021, 178, 488–496 CrossRef CAS.
- Z. Li, R. Wu, S. Xiao, Y. Yang, L. Lai, J. S. Chen and Y. Chen, Chem. Eng. J., 2022, 430, 132882 CrossRef CAS.
- X. Wang, Y. Wang, X. Sang, W. Zheng, S. Zhang, L. Shuai, B. Yang, Z. Li, J. Chen, L. Lei, N. M. Adli, M. K. H. Leung, M. Qiu, G. Wu and Y. Hou, Angew. Chem., Int. Ed., 2021, 60, 4192–4198 CrossRef CAS PubMed.
- Z. Chen, C. Wang, X. Zhong, H. Lei, J. Li, Y. Ji, C. Liu, M. Ding, Y. Dai, X. Li, T. Zheng, Q. Jiang, H.-J. Peng and C. Xia, Nano Lett., 2023, 23, 7046–7053 Search PubMed.
- W. Zhang, D. Liu, T. Liu, C. Ding, T. Chen, Y. Li, X. Liu, L. Wang, C. Li, J. He, T. Ding and T. Yao, Nano Res., 2023, 16, 10873–10880 CrossRef CAS.
- W. Zheng, J. Yang, H. Chen, Y. Hou, Q. Wang, M. Gu, F. He, Y. Xia, Z. Xia, Z. Li, B. Yang, L. Lei, C. Yuan, Q. He, M. Qiu and X. Feng, Adv. Funct. Mater., 2020, 30, 1907658 CrossRef CAS.
- Y. Zhou, Q. Zhou, H. Liu, W. Xu, Z. Wang, S. Qiao, H. Ding, D. Chen, J. Zhu, Z. Qi, X. Wu, Q. He and L. Song, Nat. Commun., 2023, 14, 3776 CrossRef PubMed.
- Y.-N. Gong, L. Jiao, Y. Qian, C.-Y. Pan, L. Zheng, X. Cai, B. Liu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 Search PubMed.
- Z. Feng, C. Tang, P. Zhang, K. Li, G. Li, J. Wang, Z. Feng and C. Li, J. Am. Chem. Soc., 2023, 145, 12663–12672 CrossRef CAS PubMed.
- J. Ding, Z. Teng, X. Su, K. Kato, Y. Liu, T. Xiao, W. Liu, L. Liu, Q. Zhang, X. Ren, J. Zhang, Z. Chen, O. Teruhisa, A. Yamakata, H. Yang, Y. Huang, B. Liu and Y. Zhai, Chem, 2023, 9, 1017–1035 Search PubMed.
- G. Wang, Y. Wu, Z. Li, Z. Lou, Q. Chen, Y. Li, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2023, 62, e202218460 CrossRef CAS PubMed.
- Y. Zhou, F. Wei, H. Qi, Y. Chai, L. Cao, J. Lin, Q. Wan, X. Liu, Y. Xing, S. Lin, A. Wang, X. Wang and T. Zhang, Nat. Catal., 2022, 5, 1145–1156 CrossRef CAS.
- Y. Gao, B. Liu and D. Wang, Adv. Mater., 2023, 35, e2209654 CrossRef PubMed.
- P. Song, P. Zhu, X. Su, M. Hou, D. Zhao and J. Zhang, Chem. - Asian J., 2022, 17, e202200716 CrossRef CAS PubMed.
- J. Zhao, S. Ji, C. Guo, H. Li, J. Dong, P. Guo, D. Wang, Y. Li and F. D. Toste, Nat. Catal., 2021, 4, 523–531 CrossRef CAS.
- Y. Chen, R. Gao, S. Ji, H. Li, K. Tang, P. Jiang, H. Hu, Z. Zhang, H. Hao, Q. Qu, X. Liang, W. Chen, J. Dong, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3212–3221 CrossRef CAS PubMed.
- H. Liu, Q. Lei, R. Miao, M. Sun, C. Qin, L. Zhang, G. Ye, Y. Yao, B. Huang and Z. Ma, Adv. Funct. Mater., 2022, 32, 2207408 CrossRef CAS.
- W. Ni, H. Chen, J. Zeng, Y. Zhang, H. A. Younus, Z. Zeng, M. Dai, W. Zhang and S. Zhang, Energy Environ. Sci., 2023, 16, 3679–3710 RSC.
- S. Chen, X. Li, C.-W. Kao, T. Luo, K. Chen, J. Fu, C. Ma, H. Li, M. Li, T.-S. Chan and M. Liu, Angew. Chem., Int. Ed., 2022, 61, e202206233 CrossRef CAS PubMed.
- D. Liu, Q. He, S. Ding and L. Song, Adv. Energy Mater., 2020, 10, 2001482 CrossRef CAS.
- N. Zhang, C. Gao and Y. Xiong, J. Energy Chem., 2019, 37, 43–57 CrossRef.
- Y. Li, Z. He, F. Wu, S. Wang, Y. Cheng and S. Jiang, Mater. Rep.: Energy, 2023, 3, 100197 CAS.
- C. Jia, S. Li, Y. Zhao, R. K. Hocking, W. Ren, X. Chen, Z. Su, W. Yang, Y. Wang, S. Zheng, F. Pan and C. Zhao, Adv. Funct. Mater., 2021, 31, 2107072 CrossRef CAS.
- E. Boutin, M. Wang, J. C. Lin, M. Mesnage, D. Mendoza, B. Lassalle-Kaiser, C. Hahn, T. F. Jaramillo and M. Robert, Angew. Chem., Int. Ed., 2019, 58, 16172–16176 CrossRef CAS PubMed.
- Y. Song, J.-J. Zhang, Z. Zhu, X. Chen, L. Huang, J. Su, Z. Xu, T. H. Ly, C.-S. Lee, B. I. Yakobson, B. Z. Tang and R. Ye, Appl. Catal., B, 2021, 284, 119750 CrossRef CAS.
- M. Zhu, R. Ye, K. Jin, N. Lazouski and K. Manthiram, ACS Energy Lett., 2018, 3, 1381–1386 CrossRef CAS.
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D. J. Miller, J. Lu, S.-T. Lee and Y. Li, Chem, 2017, 3, 652–664 CAS.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- Q. Wang, K. Liu, K. Hu, C. Cai, H. Li, H. Li, M. Herran, Y.-R. Lu, T.-S. Chan, C. Ma, J. Fu, S. Zhang, Y. Liang, E. Cortés and M. Liu, Nat. Commun., 2022, 13, 6082 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- J. Su, C. B. Musgrave, Y. Song, L. Huang, Y. Liu, G. Li, Y. Xin, P. Xiong, M. M.-J. Li, H. Wu, M. Zhu, H. M. Chen, J. Zhang, H. Shen, B. Z. Tang, M. Robert, W. A. Goddard and R. Ye, Nat. Catal., 2023, 6, 818–828 CrossRef CAS.
- K. Chen, M. Cao, G. Ni, S. Chen, H. Liao, L. Zhu, H. Li, J. Fu, J. Hu, E. Cortés and M. Liu, Appl. Catal., B, 2022, 306, 121093 CrossRef CAS.
- K. Chen, M. Cao, Y. Lin, J. Fu, H. Liao, Y. Zhou, H. Li, X. Qiu, J. Hu, X. Zheng, M. Shakouri, Q. Xiao, Y. Hu, J. Li, J. Liu, E. Cortés and M. Liu, Adv. Funct. Mater., 2022, 32, 2111322 CrossRef CAS.
- W. Zhu, S. Liu, K. Zhao, G. Ye, K. Huang and Z. He, Small, 2023, 2306144 Search PubMed.
- T. Wang, J. Wang, C. Lu, K. Jiang, S. Yang, Z. Ren, J. Zhang, X. Liu, L. Chen, X. Zhuang and J. Fu, Adv. Mater., 2023, 35, 2205553 CrossRef CAS PubMed.
- D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu, B. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS.
- Y. Wang, Z. Bao, M. Shi, Z. Liang, R. Cao and H. Zheng, Chem. - Eur. J., 2022, 28, e202102915 CrossRef CAS PubMed.
- H. Li, F. Pan, C. Qin, T. Wang and K.-J. Chen, Adv. Energy Mater., 2023, 13, 2301378 CrossRef CAS.
- C. Jia, Y. Zhao, S. Song, Q. Sun, Q. Meyer, S. Liu, Y. Shen and C. Zhao, Adv. Energy Mater., 2023, 2302007 CrossRef CAS.
- C. Wang, Y. Chen, D. Su, W.-L. Man, K.-C. Lau, L. Han, L. Zhao, D. Zhan and X. Zhu, Adv. Mater., 2023, 2303179 CrossRef CAS PubMed.
- S. Roy, Z. Li, Z. Chen, A. C. Mata, P. Kumar, S. C. Sarma, I. F. Teixeira, I. F. Silva, G. Gao, N. V. Tarakina, M. G. Kibria, C. V. Singh, J. Wu and P. M. Ajayan, Adv. Mater., 2023, 2300713 Search PubMed.
- X. Zhang, M. Zhang, Y. Deng, M. Xu, L. Artiglia, W. Wen, R. Gao, B. Chen, S. Yao, X. Zhang, M. Peng, J. Yan, A. Li, Z. Jiang, X. Gao, S. Cao, C. Yang, A. J. Kropf, J. Shi, J. Xie, M. Bi, J. A. van Bokhoven, Y.-W. Li, X. Wen, M. Flytzani-Stephanopoulos, C. Shi, W. Zhou and D. Ma, Nature, 2021, 589, 396–401 CrossRef CAS PubMed.
- Y. Guo, M. Wang, Q. Zhu, D. Xiao and D. Ma, Nat. Catal., 2022, 5, 766–776 CrossRef CAS.
- M.-J. Cheng, E. L. Clark, H. H. Pham, A. T. Bell and M. Head-Gordon, ACS Catal., 2016, 6, 7769–7777 CrossRef CAS.
- B. Kim, Y. C. Tan, Y. Ryu, K. Jang, H. G. Abbas, T. Kang, H. Choi, K.-S. Lee, S. Park, W. Kim, P.-P. Choi, S. Ringe and J. Oh, ACS Energy Lett., 2023, 8, 3356–3364 CrossRef CAS.
- M. Chhetri, M. Wan, Z. Jin, J. Yeager, C. Sandor, C. Rapp, H. Wang, S. Lee, C. J. Bodenschatz, M. J. Zachman, F. Che and M. Yang, Nat. Commun., 2023, 14, 3075 CrossRef CAS PubMed.
- W. Ren, X. Tan, J. Qu, S. Li, J. Li, X. Liu, S. P. Ringer, J. M. Cairney, K. Wang, S. C. Smith and C. Zhao, Nat. Commun., 2021, 12, 1449 CrossRef CAS PubMed.
- Y. Cao, S. Chen, S. Bo, W. Fan, J. Li, C. Jia, Z. Zhou, Q. Liu, L. Zheng and F. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202303048 CrossRef CAS PubMed.
- R. Xia, J. Cheng, Z. Chen, Z. Zhang, X. Zhou, J. Zhou and M. Zhang, Adv. Mater., 2023, 2304920 CrossRef CAS PubMed.
- J. Feng, L. Zhang, S. Liu, L. Xu, X. Ma, X. Tan, L. Wu, Q. Qian, T. Wu, J. Zhang, X. Sun and B. Han, Nat. Commun., 2023, 14, 4615 CrossRef CAS PubMed.
- C. Wang, X. Wang, H. Ren, Y. Zhang, X. Zhou, J. Wang, Q. Guan, Y. Liu and W. Li, Nat. Commun., 2023, 14, 5108 CrossRef CAS PubMed.
- G. Wang, Z. Chen, T. Wang, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2022, 61, e202210789 CrossRef CAS PubMed.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef CAS.
- X. Li, L. Liu, X. Ren, J. Gao, Y. Huang and B. Liu, Sci. Adv., 2020, 6, eabb6833 CrossRef CAS PubMed.
- J. Zheng, K. Lebedev, S. Wu, C. Huang, T. Ayvalı, T.-S. Wu, Y. Li, P.-L. Ho, Y.-L. Soo, A. Kirkland and S. C. E. Tsang, J. Am. Chem. Soc., 2021, 143, 7979–7990 CrossRef CAS PubMed.
- X. Hai, S. Xi, S. Mitchell, K. Harrath, H. Xu, D. F. Akl, D. Kong, J. Li, Z. Li, T. Sun, H. Yang, Y. Cui, C. Su, X. Zhao, J. Li, J. Pérez-Ramírez and J. Lu, Nat. Nanotechnol., 2022, 17, 174–181 CrossRef CAS PubMed.
- M. Tamtaji, H. Gao, M. D. Hossain, P. R. Galligan, H. Wong, Z. Liu, H. Liu, Y. Cai, W. A. Goddard and Z. Luo, J. Mater. Chem. A, 2022, 10, 15309–15331 RSC.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- R. Chen, J. Zhao, Y. Li, Y. Cui, Y.-R. Lu, S.-F. Hung, S. Wang, W. Wang, G. Huo, Y. Zhao, W. Liu, J. Wang, H. Xiao, X. Li, Y. Huang and B. Liu, J. Am. Chem. Soc., 2023, 145, 20683–20691 CrossRef CAS PubMed.
- Y. Hu, F. Zhan, Q. Wang, Y. Sun, C. Yu, X. Zhao, H. Wang, R. Long, G. Zhang, C. Gao, W. Zhang, J. Jiang, Y. Tao and Y. Xiong, J. Am. Chem. Soc., 2020, 142, 5618–5626 CrossRef CAS PubMed.
- Y. Chai, X. Han, W. Li, S. Liu, S. Yao, C. Wang, W. Shi, I. da-Silva, P. Manuel, Y. Cheng, L. D. Daemen, A. J. Ramirez-Cuesta, C. C. Tang, L. Jiang, S. Yang, N. Guan and L. Li, Science, 2020, 368, 1002–1006 CrossRef CAS PubMed.
- Y. Liu, Z. Li, Q. Yu, Y. Chen, Z. Chai, G. Zhao, S. Liu, W.-C. Cheong, Y. Pan, Q. Zhang, L. Gu, L. Zheng, Y. Wang, Y. Lu, D. Wang, C. Chen, Q. Peng, Y. Liu, L. Liu, J. Chen and Y. Li, J. Am. Chem. Soc., 2019, 141, 9305–9311 CrossRef CAS PubMed.
- Z. Ji, M. Hu and H. L. Xin, Sci. Rep., 2023, 13, 14132 CrossRef CAS PubMed.
- A. Annys, D. Jannis and J. Verbeeck, Sci. Rep., 2023, 13, 13724 CrossRef CAS PubMed.
- A. Martini, D. Hursán, J. Timoshenko, M. Rüscher, F. Haase, C. Rettenmaier, E. Ortega, A. Etxebarria and B. Roldan Cuenya, J. Am. Chem. Soc., 2023, 145, 17351–17366 CrossRef CAS PubMed.
- R. Qi, B. Zhu, Z. Han and Y. Gao, ACS Catal., 2022, 12, 8269–8278 CrossRef CAS.
- M. Sun, T. Wu, Y. Xue, A. W. Dougherty, B. Huang, Y. Li and C.-H. Yan, Nano Energy, 2019, 62, 754–763 CrossRef CAS.
- C. Song, Z. Wang, Z. Yin, D. Xiao and D. Ma, Chem Catal., 2022, 2, 52–83 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- P. W. Anderson, Science, 1972, 177, 393–396 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |