
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic reduction of oxygen to water by non-heme iron complexes: exploring the effect of the secondary coordination sphere proton exchanging site†
Aakash
Santra
,
Avijit
Das
 ,
Simarjeet
Kaur
,
Priya
Jain
,
Pravin P.
Ingole
and
Sayantan
Paria
*
,
Simarjeet
Kaur
,
Priya
Jain
,
Pravin P.
Ingole
and
Sayantan
Paria
*
Department of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi 110016, India. E-mail: sparia@chemistry.iitd.ac.in
First published on 13th February 2024
Abstract
In this study, we prepared non-heme FeIII complexes (1, 2, and 3) of an N4 donor set of ligands (H2L, Me2L, and BPh2L). 1 is supported by a monoanionic bispyridine-dioxime ligand (HL). In 2 and 3, the primary coordination sphere of Fe remained similar to that in 1, except that the oxime protons of the ligand were replaced with two methyl groups and a bridging −BPh2 moiety, respectively. X-ray structures of the FeII complexes (1a and 3a) revealed similar Fe–N distances; however, they were slightly elongated in 2a. The FeIII/FeII potential of 1, 2, and 3 appeared at −0.31 V, −0.25 V, and 0.07 V vs. Fc+/Fc, respectively, implying that HL and Me2L have comparable donor properties. However, BPh2L is more electron deficient than HL or Me2L. 1 showed electrocatalytic oxygen reduction reaction (ORR) activity in acetonitrile in the presence of trifluoroacetic acid (TFAH) as the proton source at Ecat/2 = −0.45 V and revealed selective 4e−/4H+ reduction of O2 to H2O. 1 showed an effective overpotential (ηeff) of 0.98 V and turnover frequency (TOFmax) of 1.02 × 103 s−1. Kinetic studies revealed a kcat of 2.7 × 107 M−2 s−1. Strikingly, 2 and 3 remained inactive for electrocatalytic ORR, which established the essential role of the oxime scaffolds in the electrocatalytic ORR of 1. Furthermore, a chemical ORR of 1 has been investigated using decamethylferrocene as the electron source. For 1, a similar rate equation was noted to that of the electrocatalytic pathway. A kcat of 6.07 × 104 M−2 s−1 was found chemically. Complex 2, however, underwent a very slow chemical ORR. Complex 3 chemically enhances the 4e−/4H+ reduction of O2 and exhibits a TOF of 0.24 s−1 and a kcat value of 2.47 × 102 M−1 s−1. Based on the experimental observations, we demonstrate that the oxime backbone of the ligand in 1 works as a proton exchanging site in the 4e−/4H+ reduction of O2. The study describes how the ORR is affected by the tuning of the ligand scaffold in a family of non-heme Fe complexes.
Introduction
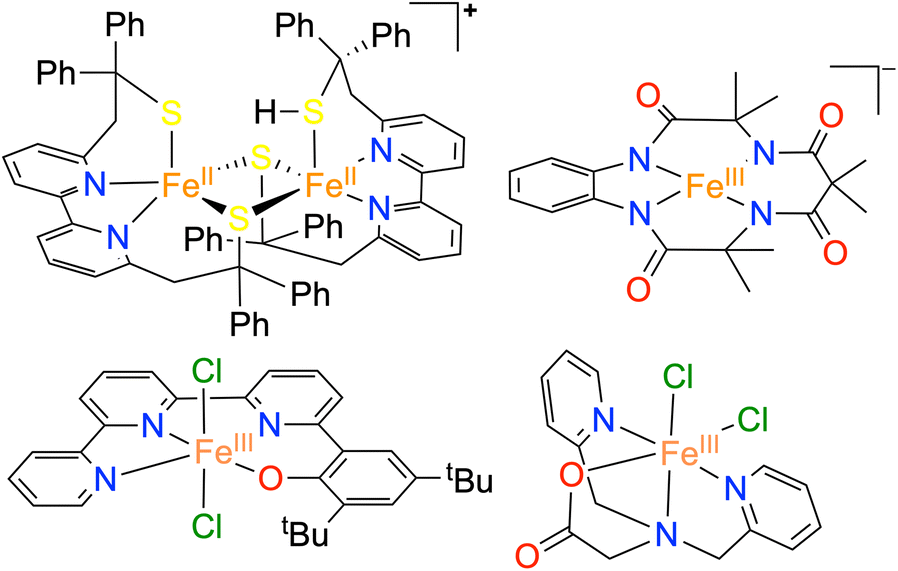
Cytochrome c oxidase (CcO), a terminal oxidase of the respiratory enzymes, is present in the mitochondrial membrane of all eukaryotes and many aerobic bacteria, and is responsible for the four-electron (4e−)/four-proton (4H+) reduction of oxygen (O2) to water (H2O) and protons.1 The exergonic reaction is a reverse process of photosystem II, which is coupled with proton pumping, and the generated chemiosmotic potential in the process is used for ATP synthesis. Multicopper oxidases also perform the oxygen reduction reaction (ORR) to yield H2O.2,3 The development of efficient earth-abundant catalysts for the ORR is crucial, considering its importance in fuel cell technology and metal–air batteries.4 Although Pt and PtM (M = Co, Ni, Cu, etc.) alloys are the most efficient catalysts for the ORR,5 the less natural abundance and high cost of Pt considerably preclude its widespread application.The ORR catalyzed by molecular complexes is particularly interesting because of the clear understanding of the active-site structure, reaction mechanism, and easy tuning of electronic properties around the metal center.6–8 Different coordination complexes of 3d transition metal ions have been explored for electrocatalytic ORR.6–9 Major efforts were made to develop molecular catalysts of different macrocyclic ligands, such as porphyrin, corrole, and phthalocyanine derivatives.6–9 However, a limited number of non-heme complexes are known as ORR catalysts.10–12 Iron is one of the most frequently found metal ions at the active site of versatile nonheme enzymes involved in a variety of small molecule transformation reactions.13,14 The activation of dioxygen at the reduced FeII center and subsequent oxygenation of different substrates are known in versatile non-heme Fe enzymes such as α-keto glutarate dependent oxygenase, catechol dioxygenases, lipoxygenases, pterin-dependent hydroxylases, rieske dioxygenases, etc.13,14 Additionally, a variety of oxygen-bound Fe intermediates have been characterized in the non-heme iron enzymes using small molecule model complexes.15–17 However, examples of molecular nonheme Fe complexes for ORR studies are exceedingly rare.18–23
Nonheme Fe-based molecular homogeneous catalysts reported for ORR studies are depicted in Chart 1. Duboc and coworkers described ORR studies using a dinuclear FeII thiolate-based catalyst. The complex was shown to catalyze the 2e−/2H+ reduction of O2 to H2O2 (∼95%) in the presence of chemical reductants and 4e−/4H+ reduction to H2O (less than ∼10% H2O2) under electrochemical reaction conditions.19 Nam and coworkers reported the selective conversion of O2 to H2O using an FeIII complex of a Tetra-Amido-Macrocyclic-Ligand (TAML) via the formation of a [(TAML)FeV(O)]− reaction intermediate.23 Electrocatalytic ORR using an FeIII complex of an N3O donor set of ligand was described by Machan and coworkers. The catalyst revealed the selective conversion of O2 to H2O via the formation of H2O2 as an intermediate reaction product (2 + 2 reaction pathway).21 Likewise, another nonheme Fe complex of a terpyridine-derived N3O donor set of ligand catalyzed the chemical reduction of O2 to H2O following a 2 + 2 reaction pathway.22 Other than the mentioned complexes, no other nonheme Fe complexes are known as ORR catalysts.
 | ||
| Chart 1 Structural depiction of the reported non-heme iron-based ORR catalysts. | ||
A cross-linked tyrosine residue present around the active site of CcO is thought to play an important role in cleaving the O–O bond of the FeIII–O–O–CuII intermediate.24 Inspired by CcO, the effect of secondary coordination sphere interactions in the biomimetic ORR catalysts, such as the presence of a proton relay site,25–34 positively charged functional groups to exert an electrostatic effect around the redox-active metal ion,35,36 functional groups which can facilitate hydrogen atom transfer (HAT) and proton-coupled electron transfer (PCET) reactions,31,37etc., have been explored to understand the O–O bond cleavage mechanism in different M–porphyrin/corrole complexes. It was established that the presence of such secondary effects could control the key factors of the ORR, such as the reaction rate, overpotential, and product selectivity. However, nonheme complexes describing the effect of secondary coordination sphere interactions on the ORR are scarce38–41 and, to date, are not known with any nonheme Fe complex.
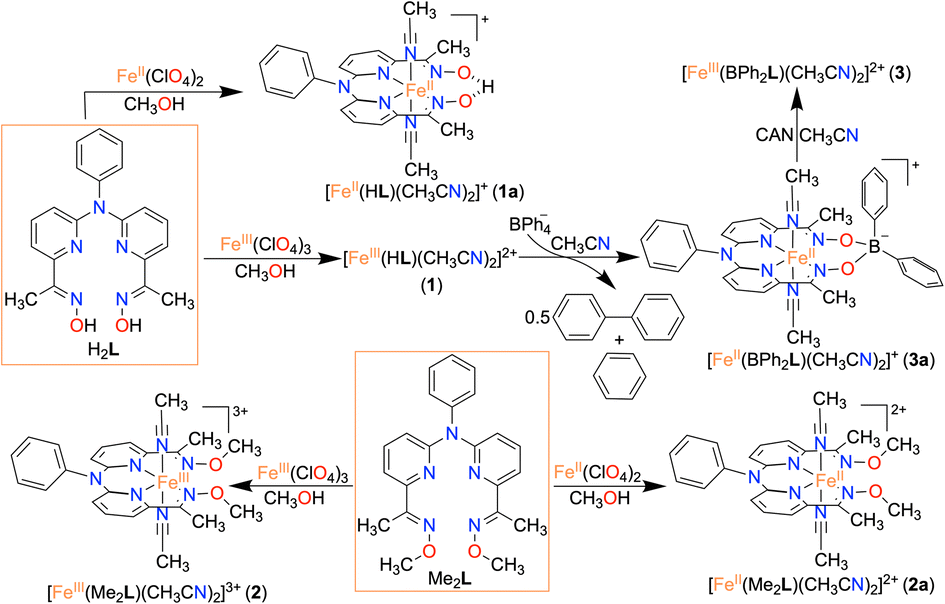
Herein, we report the synthesis and characterization of an FeIII complex (1) of a bis-pyridine-dioxime ligand (H2L). To establish the importance of oxime protons in ORR, another FeIII complex (2) was prepared where the oxime protons of H2L are replaced with methyl groups (Me2L). Additionally, an FeIII complex (3) was prepared where the oxime oxygen atoms of H2L are connected to a −BPh2 group, which creates a macrocyclic ring around Fe (Scheme 1). The electrocatalytic and chemical ORR of 1 has been investigated in oxygen-saturated acetonitrile solution using trifluoroacetic acid (TFAH) as the source of protons. 1 was found to enhance electrocatalytic 4e−/4H+ reduction of O2. Strikingly, no ORR was observed electrocatalytically in the presence of 2 and 3, establishing an important role of oxime protons towards the electrocatalytic ORR of 1. A turnover frequency (TOFmax) of 1.02 × 103 s−1 and an overpotential of 0.98 V were observed for 1. For the chemical ORR, decamethylferrocene (Fc*) was used as the reductant. 1 was found to enhance ORR chemically and revealed 4e−/4H+ selectivity and a kcat value of 6.07 × 104 M−2 s−1. In contrast, the use of 2 resulted in a very slow chemical ORR. Complex 3, however, revealed a chemical ORR and showed 4e−/4H+ selectivity, exhibiting a different rate equation from that of 1. Based on the experimental findings, the catalytic ORR mechanism of the Fe complexes has been discussed. We suggest that the ligand oxime backbone in 1 works as a proton-exchanging site in the catalytic cycle. The study represents the first example of the effect of a secondary coordination sphere proton exchanging site on the ORR study by a molecular non-heme Fe complex.
 | ||
| Scheme 1 Synthesis of the Fe complexes described in this study. | ||
Results and discussion
Synthesis and characterization of iron complexes
Here, we utilized a bispyridine-dioxime ligand system (H2L) for the preparation of Fe complexes. Furthermore, we designed and synthesized a methylated ligand (Me2L), where the oxime protons of H2L are replaced by the –CH3 groups. The preparation and characterization of Me2L are described in the ESI (Fig. S1–S4†).FeIII (1 and 2) and FeII (1a and 2a) complexes were prepared by mixing equimolar amounts of the ligand (H2L or Me2L) and FeIII(ClO4)3·6H2O or FeII(ClO4)2·6H2O in methanol at 25 °C, respectively (Scheme 1). The FeIII complexes are prepared under aerobic conditions. However, FeII complexes were synthesized inside an N2-filled glove box. The ESI-mass spectrum of 1 revealed a molecular ion peak at m/z = 416.08 (Fig. S5†), which corresponds to the deprotonated ligand-coordinated Fe complex. The 1H-NMR spectrum of 1 and 2 in CD3CN revealed broad ligand proton resonances (Fig. S6 and S12†), suggesting the paramagnetic nature of the FeIII complexes. Furthermore, we examined X-band EPR data of 1 in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol/ethanol at 77 K, which showed g values of 2.2, 2.17, and 1.96, confirming the presence of low-spin FeIII in 1 (Fig. 1A). However, a g value of 4.22 was obtained in the EPR spectrum of 2, implying the presence of high-spin FeIII in 2. The EPR data suggest that the methylation of the bispyridine-dioxime ligand causes the change of the FeIII spin-state from S = 1/2 to 5/2. UV-vis spectrum of the FeIII complexes in CH3CN (Fig. 1B) revealed a charge-transfer transition at 468 nm (ε = 1.27 × 103 M−1 cm−1) and 488 nm (ε = 1.27 × 103 M−1 cm−1) for 1 and 2, respectively. Furthermore, both the complexes exhibited ligand-derived π → π* transitions at higher energy (below 360 nm).
:1 methanol/ethanol at 77 K, which showed g values of 2.2, 2.17, and 1.96, confirming the presence of low-spin FeIII in 1 (Fig. 1A). However, a g value of 4.22 was obtained in the EPR spectrum of 2, implying the presence of high-spin FeIII in 2. The EPR data suggest that the methylation of the bispyridine-dioxime ligand causes the change of the FeIII spin-state from S = 1/2 to 5/2. UV-vis spectrum of the FeIII complexes in CH3CN (Fig. 1B) revealed a charge-transfer transition at 468 nm (ε = 1.27 × 103 M−1 cm−1) and 488 nm (ε = 1.27 × 103 M−1 cm−1) for 1 and 2, respectively. Furthermore, both the complexes exhibited ligand-derived π → π* transitions at higher energy (below 360 nm).
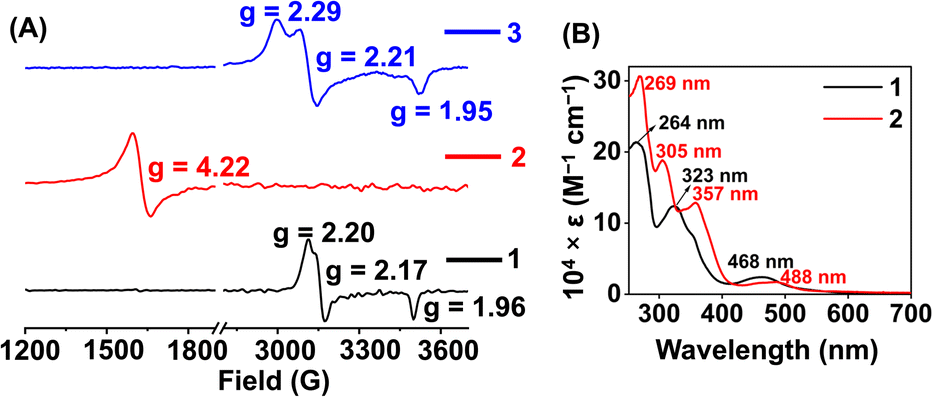 | ||
| Fig. 1 (A) X-band EPR spectra of 1 (5 mM) and 2 (5 mM) in frozen 9:1 methanol/ethanol at 77 K. The EPR data of 3 (3 mM) were recorded in tetrahydrofuran at 77 K. ν = 9.631 GHz, power = 2.71 mW, modulation frequency = 100 kHz, modulation amplitude = 4.91 G. (B) UV-vis spectra of 1 (0.5 mM) and 2 (0.5 mM) in acetonitrile at 25 °C. | ||
The FeII complexes (1a and 2a) were also thoroughly characterized by different spectroscopic techniques. Similar to 1, 1a revealed a molecular ion peak at m/z = 416.08 (Fig. S9†). The solid-state structure of 1a is shown in Fig. 2A, where Fe is coordinated to the two imine and two pyridine nitrogen atoms (dFe−N(imine) = 1.9041(18) and 1.8924(16) Å; dFe−N(pyridine) = 1.9107 (17) and 1.9019(16) Å) of the ligand at the equatorial plane, and axial positions are occupied by the acetonitrile molecules (dFe−N(CH3CN) = 1.9350(17) and 1.9275(16) Å). The X-ray structure of 2a is shown in Fig. 2B. The Fe–Npyridine (1.933(3) and 1.930(2) Å) and Fe–Nimine (1.939(3) and 1.943(3) Å) distances in 2a are slightly longer than the Fe–ligand bond distances observed in 1a. The ligand O–O distance in 2a is found to be 3.042 Å, which is considerably longer than the distance observed in 1a (2.492 Å), implying that the intramolecular hydrogen bonding in 1a makes the ligand cavity smaller.
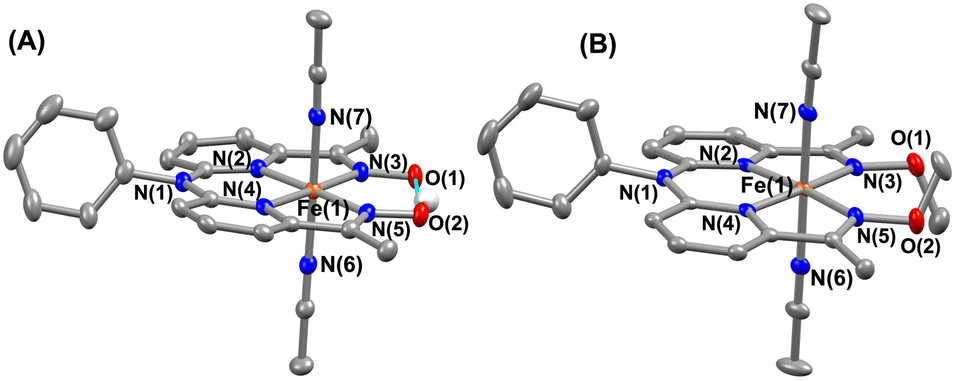 | ||
| Fig. 2 X-ray structure of 1a (A) and 2a (B) with 50% ellipsoid probability. All hydrogen atoms except those attached to the oxime moiety have been omitted for clarity. | ||
The 1H-NMR spectra of the FeII complexes (in CD3CN) are described in Fig. 3C, revealing sharp signals for the ligand proton resonance. The acidic OH proton of the ligand scaffold in 1a appeared at 20 ppm. The methyl protons of the ligand (–(CH3)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–O) were observed at 2.82 ppm and 3.42 ppm in 1a. In 2a, both methyl protons are observed at 3.03 ppm and the –OCH3 protons at 4.00 ppm. Then, we examined the FeII complexes by UV-vis spectroscopy in acetonitrile (Fig. 3A). 1a revealed a strong absorption band at 481 nm, which can be assigned to metal-to-ligand charge-transfer (MLCT).42 In 2a, the MLCT bands are observed at 486 nm. Additionally, ligand-based π → π* transition at around 340 nm was observed in all of the Fe complexes.41,43
N–O) were observed at 2.82 ppm and 3.42 ppm in 1a. In 2a, both methyl protons are observed at 3.03 ppm and the –OCH3 protons at 4.00 ppm. Then, we examined the FeII complexes by UV-vis spectroscopy in acetonitrile (Fig. 3A). 1a revealed a strong absorption band at 481 nm, which can be assigned to metal-to-ligand charge-transfer (MLCT).42 In 2a, the MLCT bands are observed at 486 nm. Additionally, ligand-based π → π* transition at around 340 nm was observed in all of the Fe complexes.41,43
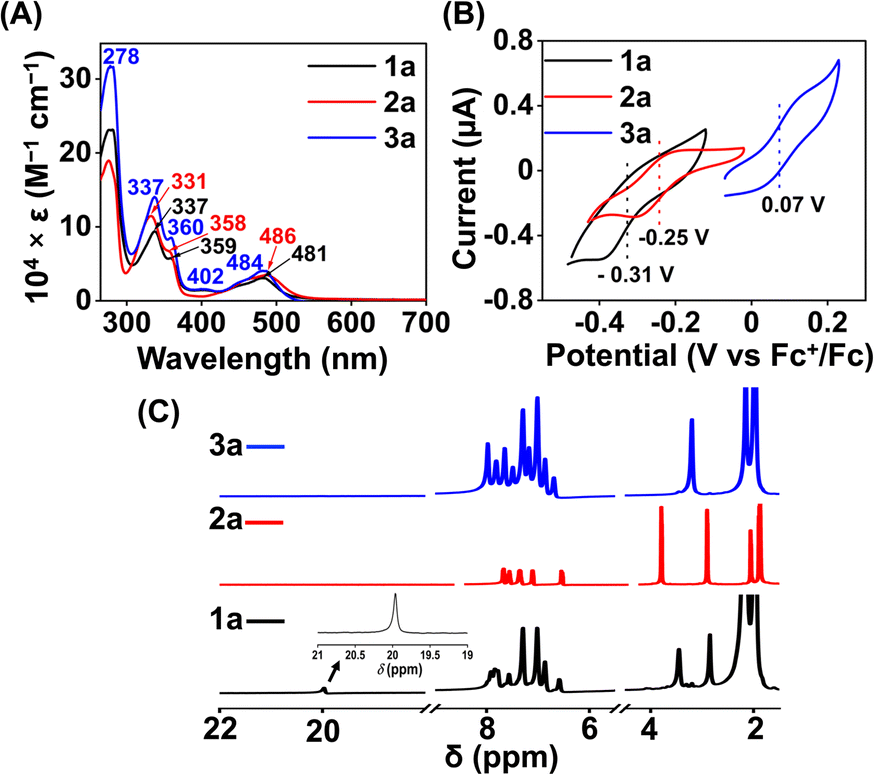 | ||
| Fig. 3 (A) UV-vis spectra of 1a (0.5 mM), 2a (0.5 mM), and 3a (0.5 mM) in acetonitrile at 25 °C. (B) Cyclic voltammogram of the FeII complexes (0.5 mM) in acetonitrile containing 50 mM of nBu4NPF6 as the supporting electrolyte at 25 °C (scan rate 5 mV s−1), using a 3 mm glassy carbon working electrode and Pt wire counter electrode. (C) 1H-NMR spectrum of the FeII complexes in CD3CN at 25 °C. | ||
Interestingly, when we conducted the reaction of 1 with an excess of sodium tetraphenylborate in acetonitrile at 25 °C, the formation of a borylated FeII complex (3a) occurred, where the oxygen atoms of the ligand attached to the −BPh2 moiety and created a six-membered ring around Fe (Scheme 1). Analysis of the reaction solution by GC-mass revealed the formation of biphenyl and benzene as the BPh4− derived product (Fig. S19†). A proposed mechanism for the formation of 3a from 1 has been described in Scheme S2.† The borylated ligand system has been defined as BPh2L throughout the manuscript. The FeII complex (3a) was characterized thoroughly by different spectroscopic techniques. The ESI-mass spectrum of 3a revealed a molecular ion peak at m/z = 580.16 (Fig. S18†), corresponding to a composition of [(BPh2L)Fe]+. The X-ray structure of 3a is described in Fig. 4. Like 1a, a similar coordination geometry around Fe was observed in 3a, and the dFe−N(imine) (1.882(2) and 1.882(2) Å), dFe−N(pyridine) distances (1.906(2) and 1.901(2) Å) were found comparable to that of 1a. The observed Fe–Nimine bond distances of 3a are also comparable to the Fe–Nimine bond lengths reported in the borylated (dioximato)FeII complexes.44 In addition, the Fe–NCH3CN distances of 3a (dFe−N(CH3CN) = 1.950(2) and 1.932(2) Å) are also similar to 1a. The boron atom of the –BPh2 arm in 3a sits 0.497 Å above the plane connecting four donor nitrogen atoms of the ligand. Furthermore, the N(3)–Fe(1)–N(5) angle observed in 3a (97.92(10)°) is also comparable to 1a (97.78(7) °), implying that the structural parameters of 3a are not altered considerably compared to 1a because of the borylated ring formation in 3a. However, one of the axially coordinated acetonitrile molecules in 3a is slightly bent (![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) Fe(1)–N(6)–C(21) = 173.67°) compared to the other axial CH3CN (176.28°), and the axially coordinated CH3CN molecules in 1a, which we suggest is because the phenyl ring of the –BPh2 group remained nearly perpendicular to the plane of the donor nitrogen atoms of the ligand in 3a.
Fe(1)–N(6)–C(21) = 173.67°) compared to the other axial CH3CN (176.28°), and the axially coordinated CH3CN molecules in 1a, which we suggest is because the phenyl ring of the –BPh2 group remained nearly perpendicular to the plane of the donor nitrogen atoms of the ligand in 3a.
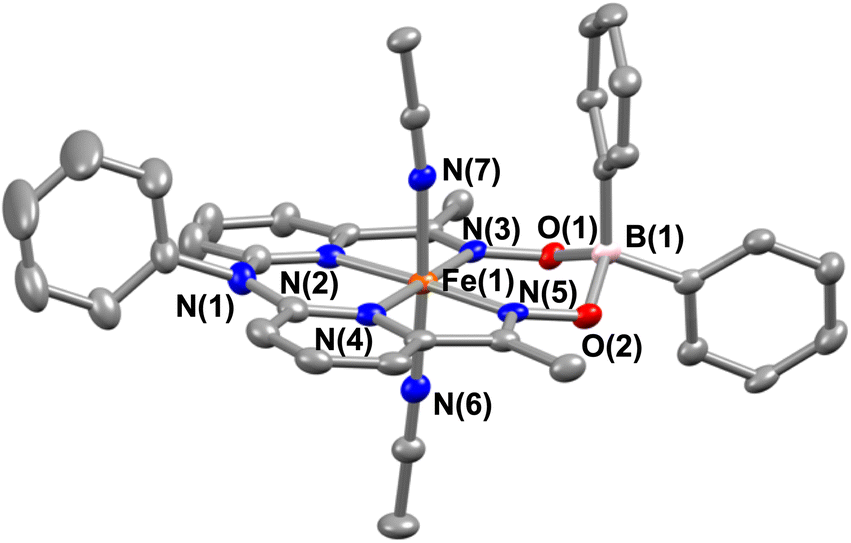 | ||
| Fig. 4 X-ray structure of 3a with 50% ellipsoid probability. All hydrogen atoms have been omitted for clarity. | ||
The electronic spectrum of 3a in acetonitrile is comparable to that of 1a or 2a, as described in Fig. 3A. The 1H NMR spectrum of 3a (Fig. 3C) exhibited features similar to that of 1a or 2a. The methyl protons of the ligand were observed at 3.17 ppm in 3a.
Furthermore, we recorded the X-band EPR spectrum of the FeIII complex (3) of the BPh2L ligand (Fig. 1A), which revealed a rhombic spectrum having g values of 2.29, 2.21, and 1.95. Thus, 3 is also a low-spin complex like 1. The presence of a macrocyclic ring created by the ligand around Fe causes better interactions between Fe and ligand orbitals, likely responsible for the presence of low-spin FeIII in 1 and 3.
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) experiments of the Fe complexes were examined in acetonitrile containing an excess amount of nBu4NPF6 as the supporting electrolyte. 1a revealed FeIII/FeII potential at –0.31 V vs. Fc+/Fc redox couple (Fig. 3B). The value is more than 300 mV anodically shifted than the FeIII/FeII potential of Fe(TPP),34 implying that HL is more electron deficient than the tetraphenyl porphyrin (TPP) system. However, the Eox value of 1a in acetonitrile is comparable to the large family of biomimetic Fe–porphyrin complexes containing different functional groups at the secondary coordination sphere.45 In 2a, the FeIII/FeII peak was observed at −0.25 V vs. Fc+/Fc, which is slightly anodically shifted compred to 1a, suggesting that both Me2L and H2L have similar ligand donor properties. However, in 3a, the FeIII/FeII redox couple shifted anodically at 0.07 V vs. Fc+/Fc (Fig. 3B), implying that the installation of the –BPh2 group caused the ligand system to be more electron deficient. Furthermore, 1 and 2 exhibited redox waves at −0.54 V and −1.0 V vs. Fc+/Fc, respectively, which can be tentatively assigned as the FeII/FeI couple (Fig. S26†).
Electrocatalytic ORR
We examined the electrocatalytic ORR of the Fe complexes in an O2-saturated acetonitrile solution in the presence of TFAH as the proton source. In the presence of an excess of TFAH (20 equiv.), the FeIII/FeII potential of 1 is anodically shifted from −0.31 V to −0.25 V (Fig. 5A). In addition, a similar shift is noticed when CV data of 1a were recorded in the presence of 20 equiv. of TFAH (Fig. S27†). However, in the presence of TFAH, no significant change in the ligand-based π → π* transition was observed at 337 nm in the UV-vis spectrum (Fig. S28†). To further gain insight into the protonation event, we measured the 1H-NMR data of 1a in the presence of an excess amount of TFAH in DMSO-d6 (Fig. S30†). The hydrogen-bonded oxime proton, which was observed at 19.5 ppm, shifted to 11.4 ppm in the presence of an excess of TFAH, demonstrating that both oxime oxygens are protonated in the presence of an excess TFAH. The breakdown of the strong intramolecular hydrogen bonding causes a prominent upfield shift of the oxime protons in the 1H-NMR data. Furthermore, the 1H-NMR spectrum of a ZnIICl complex supported by H2L was recorded in DMSO-d6, which also showed oxime protons at around 11.4 ppm (Fig. S30†). The X-ray structure of the ZnIICl complex showed that both of the oxime oxygen atoms are protonated.41 Thus, we infer that the slight anodic shift of the FeIII/FeII redox potential is because of the protonation of the oxime backbone of 1a, which makes the ligand more electron-deficient. It's important to note here that the protonation of the oxime backbone of Co diimine–dioxime complexes has caused the shift of the electrocatalytic hydrogen evolution peak anodically in the presence of added acid.46 However, the observed anodic shift for 1/1a is not very large compared to the protonation event reported for the iron–porphyrin complexes containing secondary coordination sphere amine groups.34 For example, an FeIII porphyrin complex having tert-amine groups present at the secondary coordination sphere showed a 200 mV anodic shift of FeIII/FeII potential in the presence of p-toluenesulfonic acid.34 Furthermore, it's important to note here that a slight anodic shift of the FeIII/FeII potential of 2/2a was also noted (−0.22 V vs. Fc+/Fc couple) in the presence of an excess TFAH (Fig. 5B and S31†), which we infer as the formation of the hydrogen-bonded adduct, as described in Chart S1.† The addition of TFAH to the acetonitrile solution of 3a resulted in a cathodic shift of FeIII/FeII potential to −0.25 V vs. Fc+/Fc (Fig. S32†), which implies the coordination of TFAH to the Fe center responsible for the cathodic shift. Thus, the FeIII/FeII potential of the Fe complexes of the three ligand systems (H2L, Me2L, and BPh2L) discussed here in the presence of TFAH showed comparable potential values (−0.25 V, −0.22 V, and −0.25 V vs. Fc+/Fc, respectively).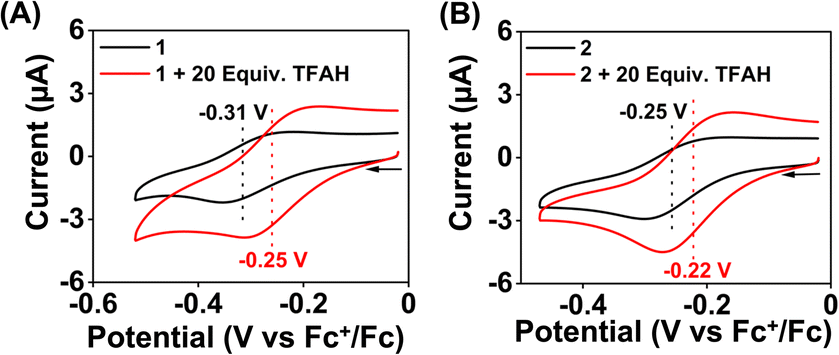 | ||
| Fig. 5 Cyclic voltammogram of 1 (0.5 mM) (A) and 2 (0.5 mM) (B) in the presence and absence of excess TFAH (10 mM) in acetonitrile containing nBu4NPF6 (50 mM) as the supporting electrolyte under a N2 atmosphere (scan rate 0.1 V s−1). A 3 mm glassy carbon working electrode and Pt wire counter electrode were used during the measurements. | ||
Upon cathodic scan, an irreversible current was observed in an O2 saturated reaction solution of 1 (Fig. S33†), suggesting coordination of O2 to the FeII center. In the presence of O2 and TFAH, 1 exhibited a catalytic current at the position of the FeIII/FeII redox couple (Ecat/2 = −0.45 V vs. Fc+/Fc couple, Fig. 6A), and no current enhancement was observed in the absence of O2 (Fig. 6A), confirming the occurrence of catalytic ORR. A rinse test experiment confirms that the electrode-adsorbed material is not responsible for the observed catalytic current (Fig. S34†). The product selectivity for the ORR was evaluated by hydrodynamic voltammetry using an RRDE setup consisting of a glassy carbon disc and Pt ring electrode (Fig. S35†), which showed that 1 shows good selectivity for the 4e−/4H+ reduction of O2. While doing this measurement, it's assumed that transport-limited oxidation of H2O2 would occur in the Pt ring if formed in the ORR. However, a recent study revealed that the H2O2 oxidation at the Pt ring electrode is not transport-limited under the condition mostly employed in the nonaqueous RRDE analysis and could produce lower H2O2 yield if formed during the ORR.47 Nevertheless, in this study, the absence of Pt ring current discards the possibility of the formation of H2O2 as an intermediate reaction product in the ORR of 1.
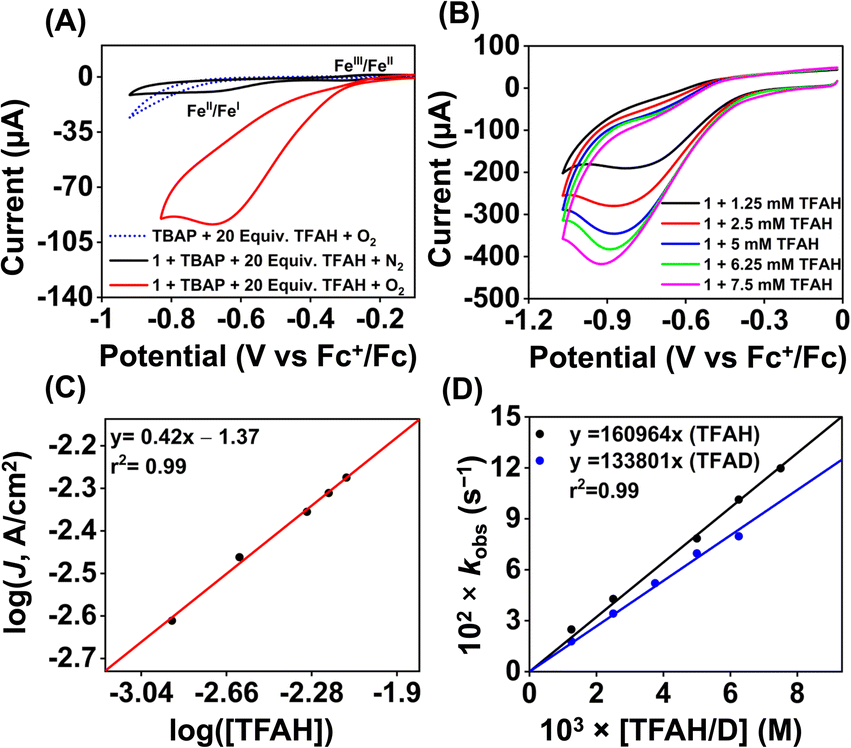 | ||
| Fig. 6 (A) The cyclic voltammogram of 1 (0.5 mM) was measured in an oxygen-saturated acetonitrile solution containing 10 mM of TFAH and 50 mM nBu4NPF6 as the supporting electrolyte at 25 °C (scan rate 0.1 V s−1). The black trace was taken in the absence of oxygen. The dotted blue trace was taken in the absence of 1 under oxygen. (B) Cyclic voltammogram of 1 (0.5 mM) measured at different concentrations of TFAH (1.25–7.5 mM) in oxygen-saturated acetonitrile with a scan rate of 6 V s−1 at 25 °C. (C) A plot of log(J, A cm−2) vs. log([TFAH]) for 1 (0.5 mM) in oxygen saturated acetonitrile containing 50 mM nBu4NPF6 (scan rate 6 V s−1). (D) A plot of kobsvs. [TFAH/D] at a fixed concentration of 1 (0.5 mM) and O2 (6.0 mM) at 25 °C. | ||
In acetonitrile, 1 revealed an effective overpotential (ηeff) of 0.98 V (at Ecat/2 = −0.45 V). The ηeff of 1 is comparable to the Fe–porphyrin catalysts for the ORR.45
Then, we examined the kinetics of electrocatalytic ORR in the presence of varying concentrations of the substrates. The pseudo-first-order rate constants (kobs) were evaluated using eq (1).48,49 Here, ncat (=4) and np (=1) are the numbers of electrons associated with the catalytic process and FeIII/FeII couple. R, T, and F are the Universal gas constant, temperature, and Faraday's constant, respectively. ν is the scan rate.
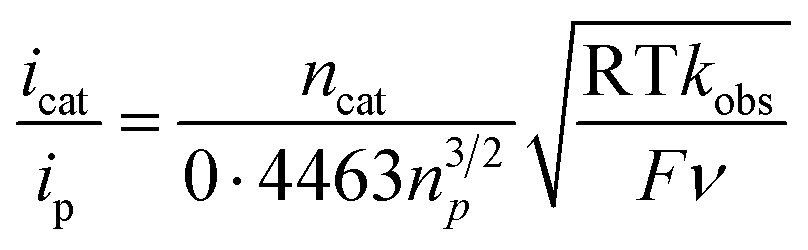 | (1) |
Initially, CV data of 1 (0.5 mM) were recorded in an oxygen-saturated acetonitrile solution containing 10 mM of TFAH at a variable scan rate (Fig. S36†), which revealed no increase of catalytic current above a scan rate of 6 V s−1. Thus, we performed the substrate variation experiments at a scan rate of 6 V s−1, where the pseudo-first-order reaction conditions are maintained in the diffusion layer.50 The catalytic current was found to increase with an increasing concentration of the complex at a constant concentration of TFAH (10 mM) in oxygen-saturated acetonitrile (Fig. S37†). A plot of log(J, A cm−2) vs. log([1]) follows a linear relationship, indicating first-order dependence (Fig. S38†). Likewise, at a constant concentration of 1 (0.5 mM), the catalytic current was found to increase with an increasing concentration of TFAH (ν = 6 V s−1) (Fig. 6B). A plot of log(J, A cm−2) vs. log([TFAH]) also follows a linear relationship (Fig. 6C), inferring first-order dependence. The kobs values at different TFAH concentrations were extracted from the icat (under O2) and ip value (under N2) using eqn (1). A kH+ of 1.60 × 105 M−1 s−1 was obtained from the slope of a plot of kobsvs. [TFAH] (Fig. 6D). Finally, the catalytic current was found to increase with increasing concentrations of O2 at a fixed concentration of 1 (0.5 mM) and TFAH (10 mM), and first-order dependence was established (Fig. S39†). Thus, the rate equation for electrocatalytic ORR follows eqn (2) and (3).
| Rate = kobs[1] | (2) |
| kobs = kcat[O2]1[TFAH]1 | (3) |
A kcat of 2.7 × 107 M−2 s−1 was estimated using eqn (3), and from the slope of Fig. 6D. At a 10 mM TFAH concentration and scan rate of 6 V s−1, a TOF of 1.02 × 103 s−1 has been estimated for 1. Furthermore, we examined electrocatalytic ORR of 1 in the presence of trifluoroacetic acid-d (TFAD), which resulted in a kcat of 2.25 × 107 M−2 s−1 and yielded a kinetic isotope effect (KIE = kHcat/kDcat) of 1.2 (Fig. 6D and S40†). The KIE value is rather small, suggesting the involvement of a proton-coupled electron transfer (PCET) reaction in the electrocatalytic ORR of 1. For the PCET reaction, observation of a small KIE is known in different systems.51
Strikingly, no catalytic current enhancement was observed for 2 at the position of the FeIII/FeII couple when examined in an oxygen-saturated acetonitrile solution in the presence of TFAH (10 mM) (Fig. 7A). As the FeIII/FeII potentials of 2 and 1 are comparable, 2 was also expected to show electrocatalytic activity for the ORR. This unprecedented difference in reactivity implies that the ORR reactivity of these Fe complexes is not solely controlled by the FeIII/FeII potential. Thus, at this point, we presume that the presence of a pseudo-macrocyclic ring in 1 might be a key factor in assisting electrocatalytic ORR. Next, we examined the electrocatalytic behavior of the FeII complex of the BPh2L (3a). However, no catalytic current enhancement was observed at the position of the FeIII/FeII couple of 3a in the presence of O2 and TFAH in acetonitrile at 25 °C (Fig. 7B). Likewise, no electrocatalytic ORR was observed for the FeIII complex (3) in oxygenated acetonitrile in the presence of TFAH. Thus, the result implies that 3a or 3 cannot promote electrocatalytic ORR. In 3a, the introduction of the –BPh2 group resulted in ca. 370 mV anodic shift of the FeIII/FeII potential compared to 1a, which could make the FeII electron-deficient and reduce the O2 affinity of 3a compared to 1a. However, in the presence of TFAH, the position of the FeIII/FeII potential of 3a shifts to −0.25 V vs. Fc+/Fc, similar to that of 1a in the presence of TFAH.
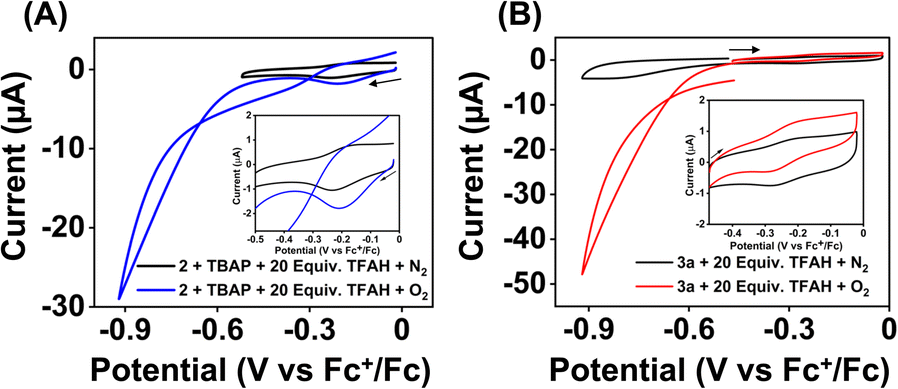 | ||
| Fig. 7 Cyclic voltammograms of 2 (0.5 mM, (A)) and 3a (0.5 mM, (B)) in the presence and absence of oxygen in acetonitrile containing 10 mM of TFAH and 50 mM nBu4NPF6 as the supporting electrolyte at 25 °C. The inset data show a zoomed version of the FeIII/FeII potential region. CV data were recorded using a 3 mm GC electrode at a 100 mV s−1 scan rate. | ||
Next, we correlated the experimental ORR data with the solid-state structures of the FeII complexes, which are summarized in Chart 2. In 2a, the ligand core is considerably opened, and the distance between the two imine nitrogen atoms (3.056 Å) and oxime oxygen atoms (3.042 Å) is substantially elongated compared to the distances observed in 1a (dN(imine)−N(imine) = 2.861 Å, dO(oxime)−O(oxime) = 2.492 Å). This is further indicated by the shortening of the Nimine–Fe–Nimine bond angle in 1a (97.78°) compared to that in 2a (103.87°). In contrast, the ligand cavity in 3a is nearly similar to that of 1a, as indicated by the closely similar Nimine–Nimine and Ooxime–Ooxime distances observed in 1a and 3a. If the presence of a macrocyclic ligand system is the only factor that enhances electrocatalytic ORR, then 3a was also expected to exhibit ORR. Thus, these structural data indicate that the protonated oxime backbone has an important role in the electrocatalytic ORR. Additionally, we also experimentally observed the protonation of the ligand oxime scaffold in the presence of TFAH. These data together suggest that the oxime scaffold works as a proton exchanging site during the electrocatalytic ORR of 1. If the position of the FeIII/FeII potential is the only factor that controls the ORR, then we would have observed electrocatalytic ORR activity of the Fe complexes of three ligand systems presented here.
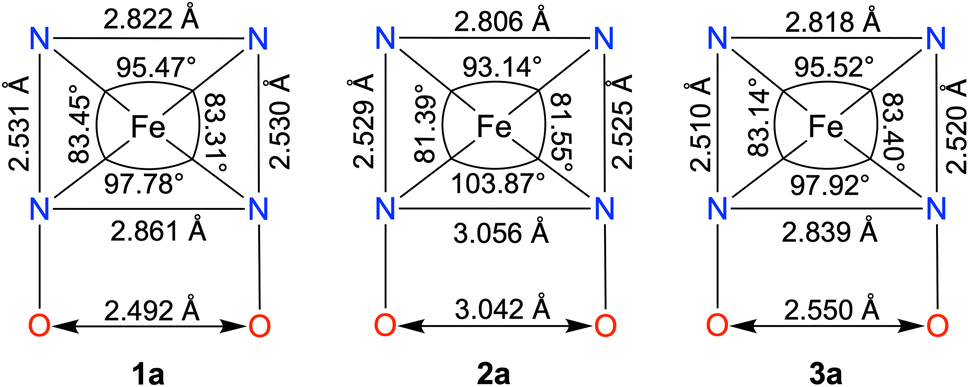 | ||
| Chart 2 A comparison of the structural parameters of the FeII complexes. | ||
Chemical ORR study
We examined the ORR of the FeIII complexes chemically in an O2-saturated acetonitrile solution in the presence of excess amounts of Fc* and TFAH, and the reactions were monitored by UV-vis spectroscopy. Complex 1 is rapidly converted to 1a in the presence of one equiv. of Fc* (Fig. S45†). The addition of catalytic amounts of 1 (0.02 mM) to an oxygen-saturated acetonitrile solution consisting of Fc* (1 mM) and TFAH (20 mM) resulted in an increase of absorbance maxima at 780 nm in the UV-vis spectrum (Fig. 8A), which corresponds to the formation of the decamethylferrocenium cation (Fc*+). The formation of a large excess of Fc*+ suggests the occurrence of catalytic ORR. Investigation of the reaction solution by Ti(O)SO4 assay revealed the absence of detectable H2O2 in the reaction solution (Fig. S46†), which implies that 1 works as a chemical ORR catalyst for the selective 4e−/4H+ reduction of O2 (eqn (4)).| O2 + 4Fc* + 4CF3COOH → 2H2O + 4Fc*+ + 4CF3COO− | (4) |
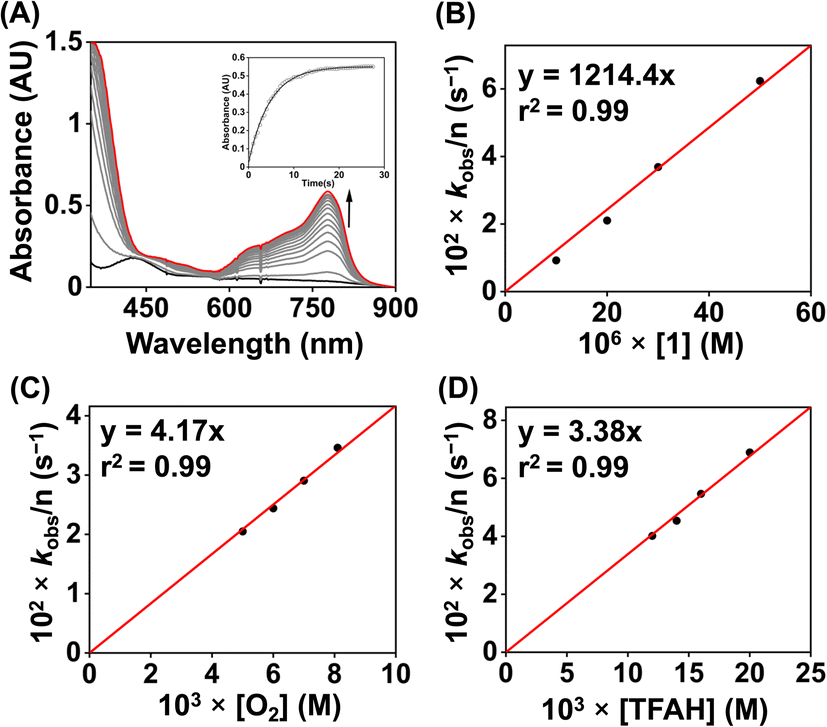 | ||
| Fig. 8 (A) Change of the UV-vis spectrum of an oxygen-saturated acetonitrile solution containing 0.02 mM of 1, 20 mM of TFAH, and 1 mM of Fc* at 25 °C. (B) A plot of kobsvs. [1] at a fixed concentration of O2 (8.1 mM), TFAH (20 mM) and Fc* (1 mM). (C) A plot of kobsvs. [O2] at a fixed concentration of 1 (0.02 mM), TFAH (20 mM), and Fc* (1 mM). (D) A plot of kobsvs. [TFAH] at a fixed concentration of 1 (0.02 mM), Fc* (1 mM), and O2 (8.1 mM). | ||
We further investigated the kinetics of the ORR by UV-vis spectroscopy. The concentration dependence of each reactant was evaluated by varying substrate concentrations under pseudo-first-order reaction conditions and keeping the concentration of the other substrates constant (Fig. S47–S50†). The pseudo-first-order rate constant (kobs) values were extracted from the slope of a plot of ln(ΔA) vs. time (s) at 780 nm (formation of Fc*+). At a fixed concentration of TFAH (20 mM), Fc* (1 mM), and O2 (8.1 mM), it was observed that the kobs values increase with increasing concentration of the complex (1). A plot of kobsvs. [1] follows a linear relationship (Fig. 8B), implying first-order dependence of the complex concentration towards the overall reaction rate. Likewise, we observed that a plot of kobsvs. [O2] and [TFAH] yielded a linear relationship, described in Fig. 8C and D, respectively. However, we noted that the kobs values remained independent upon varying the concentration of Fc*, described in Fig. S51,† suggesting that the reaction rate of the ORR catalyzed by 1 is independent of the Fc* concentration. Thus, based on the kinetic investigations, we propose a rate equation for the chemical ORR catalyzed by 1, described in eqn (5) and (6).
| Rate = ncatd[Fc*+]/dt = kobs[O2]1 | (5) |
| kobs = kcat[complex]1[TFAH]1 | (6) |
Furthermore, the catalytic ORR of 1 was performed in the presence of trifluoroacetic acid-d (TFAD), which exhibited a slowdown of the catalytic reaction and a kinetic isotope effect (KIE) of 1.3 (Fig. S52†). The chemically observed KIE value is close to the value obtained in the electrochemical measurements (Fig. S40†). The obtained KIE value suggests the involvement of a PCET reaction during the chemical ORR.
Additionally, to understand the ORR kinetics, we performed an Eyring analysis for 1 (Fig. 9). For that, we measured the kcat value of the ORR at different temperatures (298–283 K). ΔH‡ and ΔS‡ values of 3.9 kcal mol−1 and −31.4 cal K−1 mol−1, respectively, were obtained for 1. From these values, a ΔG‡ of 13.2 kcal mol−1 at 298 K was estimated. The observed ΔG‡ value for 1 is close to the reported ΔG‡ of the ORR of an Fe(N3Ocarboxylate) (20.5 kcal mol−1 at 298 K),21 Fe(N3Ophenolate) (10.1 kcal mol−1 at 298 K),22 and Co(N2O2) complex (14 kcal mol−1 at 298 K).55
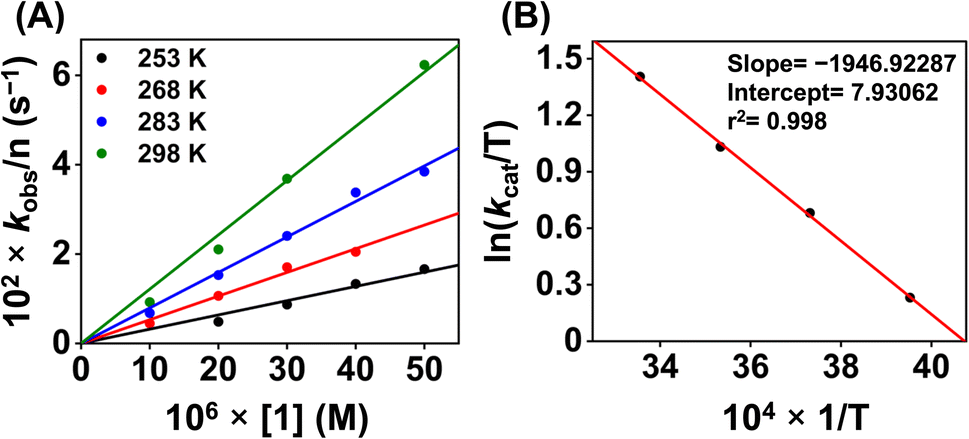 | ||
| Fig. 9 (A) A plot of kobsvs. [1] at different temperatures. (B) A plot of ln(kcat/T) vs. 1/T for the estimation of activation parameters for 1. | ||
Next, we explored the chemical ORR of 2 (0.02 mM) in acetonitrile. In the presence of TFAH (20 mM), Fc* (1 mM), and 2 (0.02 mM) in an oxygen-saturated acetonitrile solution, a very slow formation of Fc*+ was noted compared to the background reaction in the absence of 2 (Fig. S53†). The experiment implies that 2 doesn't enhance the chemical ORR effectively, which is also consistent with the inability of 2 to assist electrocatalytic ORR.
Then, we explored the chemical ORR study of 3 (0.02 mM) in acetonitrile in the presence of Fc* (1 mM) and TFAH (20 mM). Complex 3 was generated in situ by oxidizing 3a using one equiv of ceric ammonium nitrate (CAN). Similar to 1, 3 was also found to be a selective catalyst for 4e−/4H+ reduction of O2 chemically (Fig. S55†). A blank experiment in the absence of 3 showed very little formation of Fc*+ (Fig. S56B†). A comparison of ORR studies revealed the requirement of a larger time for 3 than 1 (Fig. S57†) under identical reaction conditions. Kinetic studies in the presence of varying concentrations of different substrates were conducted, which revealed pseudo-zero-order kinetics behavior in the presence of different substrates (Fig. S58–S61†). The catalytic ORR was found to depend on the concentration of 3 and Fc*, and remained independent of the concentration of TFAH and O2. The initial rate constant (ki) for each of the substrate variation experiments was obtained from the slope of a plot of the [Fc*+] (M) vs. time (s), and the observed ki values were plotted against [substrate] to obtain the substrate dependence plot. The catalytic second-order rate constant (kcat) was obtained using eqn (7) and Fig. 10A.
| Rate = ncatd[Fc*+]/dt = kcat[complex]1[Fc*]1 | (7) |
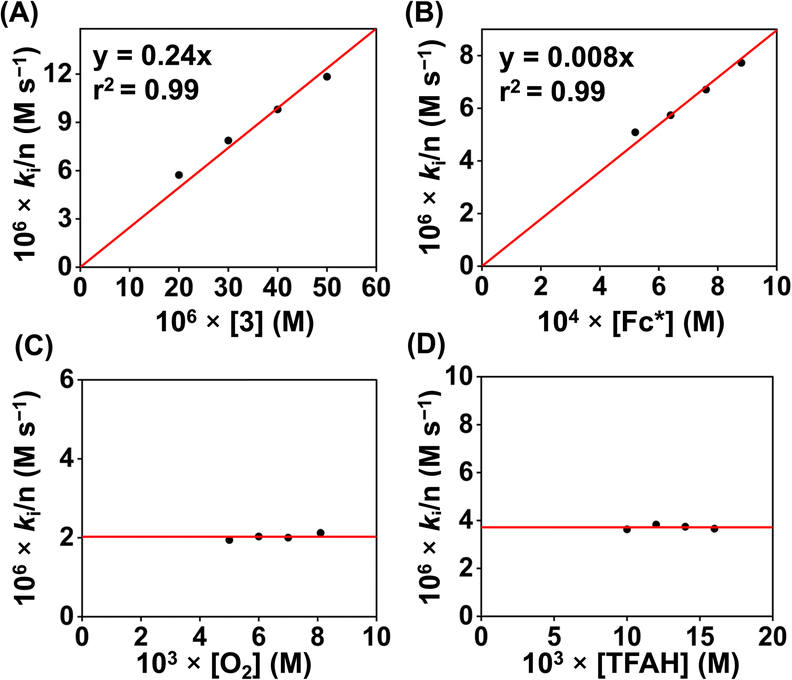 | ||
| Fig. 10 (A) A plot of kivs. [3] at a fixed concentration of O2 (8.1 mM), TFAH (20 mM), and Fc* (1 mM). (B) A plot of kivs. [Fc*] at a fixed concentration of 3 (0.02 mM), TFAH (20 mM), and O2 (8.1 mM). (C) A plot of kivs. [O2] at a fixed concentration of 3 (0.02 mM), TFAH (20 mM), and Fc* (1 mM). (D) A plot of kobsvs. [TFAH] at a fixed concentration of 3 (0.02 mM), Fc* (1 mM), and O2 (8.1 mM). | ||
A kcat value of 2.47 × 102 M−1 s−1 was obtained at 25 °C for 3. A TOF value of 2.4 × 10−1 s−1 has been calculated for 3 (Fig. S58D†). The ORR parameters of 1 and 3 have been described in Table 1.
| Parameters | 1 | 3 |
|---|---|---|
| a Determined by electrochemical measurements. b The value corresponds to TOFmax, measured at a scan rate of 6 V s−1 and TFAH concentration of 10 mM. | ||
| k cat | 6.07 × 104 M−2 s−1, 2.7 × 107 M−2 s−1a | 2.5 × 102 M−1 s−1 |
| k obs (s−1) | 1.02 × 103b | 2.4 × 10−1 |
| η (V) | 0.98 | — |
| ΔH‡ (kcal mol−1) | 3.9 | 8.7 |
| ΔS‡ (cal K−1 mol−1) | −31.4 | −31.5 |
| ΔG‡ (kcal mol−1), 298 K | 13.2 | 18.1 |
Then we explored the Eyring analysis of 3 by measuring the kcat of the ORR at different temperatures (Fig. 11). A ΔH‡ and ΔS‡ value of 8.7 kcal mol−1 and −31.5 cal K−1 mol−1, respectively, were found for the ORR of 3. Using these values, a ΔG‡ of 18.7 kcal mol−1 has been estimated for 3, which is significantly higher than that of 1. Thus, the activation parameters suggest that the kinetic barrier for the ORR of 3 is higher than that for 1.
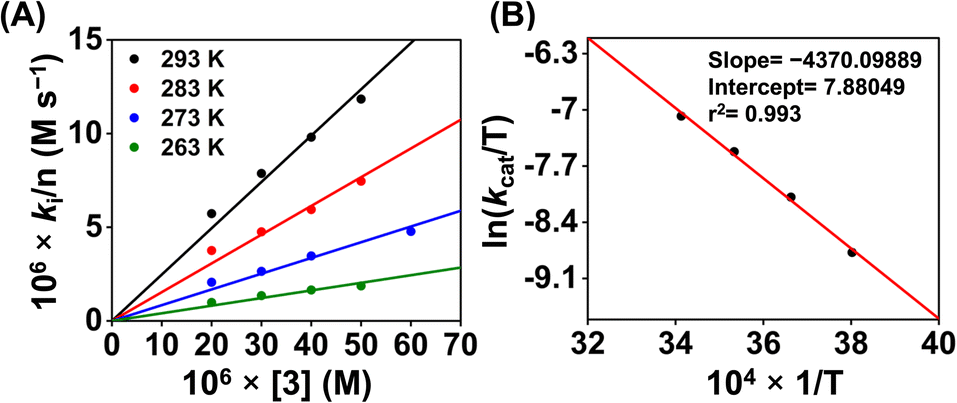 | ||
| Fig. 11 A plot of kivs. [3] (A) at different temperatures. A plot of ln(kcat/T) vs. 1/T for the estimation of activation parameters for 3 (B). | ||
A comparison of the structural parameters of the FeII complexes (1a and 3a) revealed that there are only slight differences in the metrical parameters between 1a and 3a. However, the chemical kinetics of the ORR of the Fe complexes are drastically different. Thus, the apparent reactivity difference can be attributed to the involvement of the oxime protons of the ligand in the PCET reactions for 1. We suggest reversible protonation of the oxime backbone upon reduction of 1 in the presence of TFAH (vide infra). The protonated oxime-backbone is then involved in the intramolecular proton transfer reaction to the Fe–oxygen intermediate(s) formed in the ORR pathway for 1. However, the borylated oxime arm in 2 is more electron deficient and doesn't participate in the reversible protonation reaction; rather, the CV measurement indicates the coordination of TFAH to the Fe center in the presence of an excess TFAH. We propose that this difference in the ligand architecture makes the rate-law of the ORR different for 1 and 3. We recently described the 4e−/4H+ reduction of O2 by a molecular CoIII complex of HL, where the involvement of the protonated oxime arm of the ligand has been described.41 The formation of a peroxo-bridged Co(III) dimer was evident at low temperatures in acetonitrile or acetone. However, no formation of such an intermediate was noted for the complex 1 in acetonitrile at low temperature. However, ORR selectivity and the reaction rate equation for both complexes are the same in acetonitrile. In addition, the kcat values observed in the chemical ORR are also comparable for both Fe and Co complexes in acetonitrile.
Additionally, it has been shown that the catalytic ORR selectivity for 4e−/4H+ reduction can follow a distinct 2 + 2 pathway for molecular Fe complexes.21,22 To explore such a possibility, we initially investigated the reaction of 1 with one equiv. urea·H2O2, which showed a change in a single spectrum of 1 (Fig. S62A†). Furthermore, no significant change was encountered up to the addition of five equiv. of urea·H2O2. However, no decomposition of 1 also occurred, as evident from the UV-vis data. Next, we analyzed the reaction mixture after the addition of an excess of urea·H2O2 (5 equiv.) to 1 (incubated for 5 minutes) without any added proton and electron donor, which revealed unreacted H2O2 in the reaction solution (Fig. S62B†). If 1 simply catalyzes the disproportionation of H2O2 (2H2O2 = 2H2O + O2) via the formation of the Fen+(O) intermediate,56,57 the catalyst structure should be retained after the reaction. However, there is a clear change in the features of 1 in the presence of H2O2. Moreover, if the disproportionation reaction occurs in the mentioned time scale, the reaction is slower than the 4e−/4H+ reduction of O2 catalyzed by 1. Thus, based on this experiment, we suggest that 1 undergoes a structural reorganization in the presence of added H2O2. However, this reactivity may not be relevant to the ORR mechanism of 1 (vide infra).
Next, we examined the reaction of 1 (0.02 mM) with urea·H2O2 (4 mM) in the presence of TFAH (20 mM), and Fc* (1 mM) under a nitrogen atmosphere and the reaction was monitored by UV-vis spectroscopy. The reaction also resulted in the 2e−/2H+ reduction of H2O2 at 25 °C. The formation of Fc*+ is described in Fig. S63.† An induction-like behavior was observed initially in the time trace, which we suggest is because of the structural reorganization that occurred in the presence of H2O2. A pseudo-zero-order kinetics was observed in the presence of varying amounts of the substrates, described in Fig. S64–S71.† An initial rate of the formation of Fc*+ (ki, M s−1) was determined from a plot of [Fc*+] vs. time (s) for different substrates. At a fixed concentration of TFAH and Fc*, the magnitude of ki varies linearly with an increasing concentration of 1 (Fig. S65†). Likewise, ki was found to be proportional to the concentration of urea·H2O2 (Fig. S67†). However, the ki values remained independent of the concentration of TFAH and Fc* (Fig. S69 and S71†). Thus, the H2O2RR catalyzed by 1 follows eqn (8) and (9).
| H2O2 + 2Fc* + 2CF3COOH → 2H2O + 2Fc*+ + 2CF3COO− | (8) |
| Rate = ncatd[Fc*+]/dt = kcat[complex]1[urea·H2O2]1 | (9) |
ORR mechanism
Based on the experimental observations, a possible reaction mechanism of the ORR of 1 is described in Scheme 2. 1e−/1H+ reduction of the FeIII complex (1) results in the formation of protonated FeII complex (1aH) following a PCET pathway. The 1H NMR measurement of 1a in the presence of an excess TFAH also supports the assignment. Then, the binding of O2 to 1aH results in the formation of an FeIII(O2˙) species (1b), which undergoes a PCET reaction to form an FeIII(OOH) complex (1c). The species 1c then undergoes a PCET reaction to form H2O and the FeIII–OH complex (1d).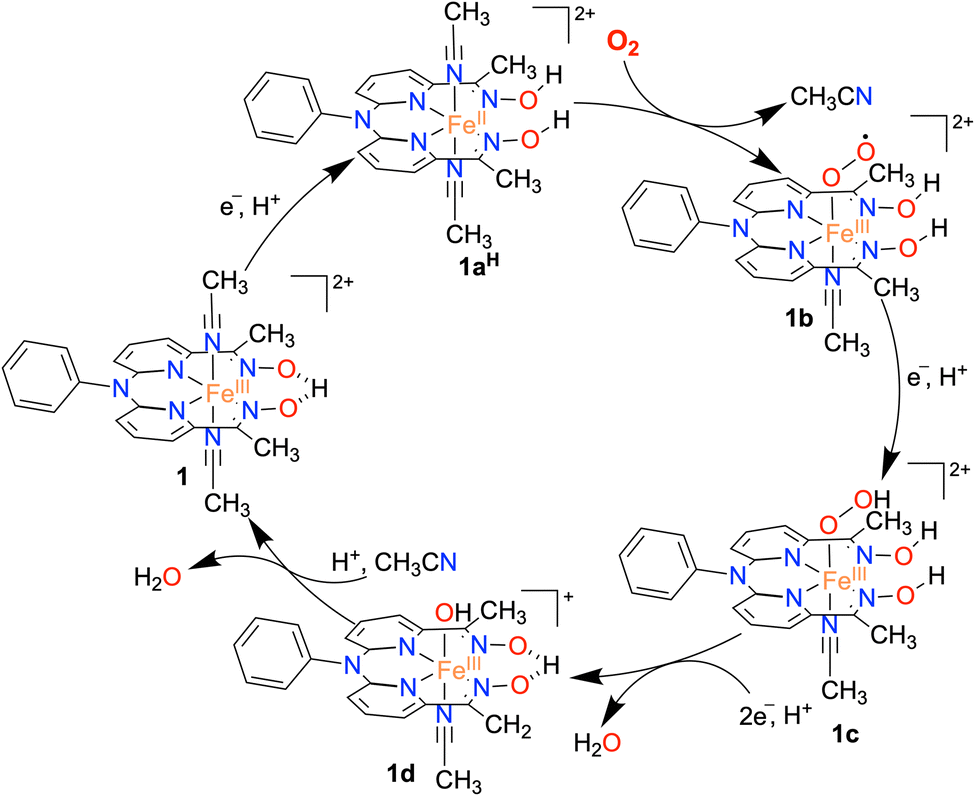 | ||
| Scheme 2 The proposed mechanism of the ORR catalyzed by the Fe complex 1. | ||
Here, we propose that the protonated oxime backbone of the ligand is involved in the intramolecular proton transfer reaction to the distal OH group of the Fe–OOH moiety, possibly in the rate-determining step. 1d then reacts with another equiv. of proton to yield another equiv. of H2O and the starting FeIII complex (1). For 3, we speculate a reaction mechanism similar to that of 1, except for the intramolecular PCET reaction as speculated in the case of 1. Both chemical and electrochemical measurements established that the H2O2 formation via the 2e−/2H+ pathway is not favored in the present study, compared to the reported 2 + 2 pathway of two nonheme Fe ORR catalysts.21,22 We suggest that the protonated oxime backbone assists in the intramolecular proton transfer reaction to the distal oxygen atom of the FeIII–OOH intermediate (1c) and helps in the O–O bond cleavage. Overall, the study highlights the importance of the presence of acidic protons in the vicinity of the redox-active center to assist the ORR. The effect of the secondary coordination sphere on ORR studies has been investigated for different Fe complexes of porphyrin and corrole-based ligand systems.6–9 The presence of tertiary amine group(s) at the secondary coordination sphere increased the rate of the ORR by 2–3 times.34 Likewise, the installation of carboxylic acid,58,59 pendant primary amine and guanidine sites,60 and phenanthroline backbone61 at the secondary coordination sphere of Fe–porphyrin complexes was shown to enhance the ORR. The effect of proton relay sites installed in the Hangman carboxylic acid containing Co/Mn-porphyrin complexes25,28 and Co–corrole complexes consisting of N1,N1-bis(pyridin-2-ylmethyl)ethane-1,2-diamine backbone at the secondary coordination sphere has been investigated.33 Herein, we described the first example of a nonheme iron complex containing a proton relay site at the secondary coordination sphere that can catalyze the 4e−/4H+ reduction of O2.
Conclusions
n summary, we synthesized and thoroughly characterized the Fe complexes of N4 donor ligands (H2L, Me2L, and BPh2L). The X-ray structure of the FeII complexes has been determined, which revealed that similar structural parameters are present in 1a and 3a. However, 2a exhibited different metrical parameters than 1a/3a. The FeIII/FeII reduction potential of 1a and 2a is much more cathodically shifted than 3a, which suggests that FeII is more electron-rich in 1a/2a than in 3a. Electrocatalytic ORR of 1 has been investigated in acetonitrile, which revealed a selective 4e−/4H+ reduction of O2 to H2O with a TOFmax of 1.02 × 103 s−1 and ηeff of 0.98 V. These parameters are often compared to access the catalytic efficiency of molecular complexes in the ORR. The TOF and ηeff of 1 compare well with the reported secondary coordination sphere edited Fe–porphyrin complexes utilized in the ORR study. Strikingly, 2 and 3 were found inactive towards the electrocatalytic reduction of O2. Additionally, we examined the ORR of the FeIII complexes chemically in the presence of a soluble reductant Fc*, which also revealed the occurrence of 4e−/4H+ reduction of O2 for 1 and 3. Detailed kinetic studies of 1 were performed using spectrochemical studies. The catalytic rate equations for the chemical and electrochemical ORR of 1 are identical, demonstrating a similar reaction mechanism. However, complex 2 catalyzed chemical ORR in acetonitrile was very slow. Furthermore, 3 was found to follow different kinetics for the ORR, where the rate of the reaction depends upon the concentrations of the catalyst and reductant, inferring a different rate-limiting step during the chemical ORR of 3 than 1. Furthermore, we noticed that the FeII complexes (1a and 3a) have similar Fe–N bond distances and N–Fe–N bond angles. The only considerable difference between the Fe complexes (1a and 3a) is the absence of oxime groups in 3a. These experimental observations strongly suggest the importance of the oxime groups in assisting the ORR in the case of 1 and is the cause of following a different ORR mechanism from that of 3. In fact, the presence of an intramolecular proton exchanging site in 1 causes a different rate-determining step for 1 than 3 for the chemical ORR. The inactivity of 2 towards the ORR is noteworthy. Despite having a similar E1/2 of 1 and 2, 2 doesn't enhance the ORR. The presence of the protonated oxime scaffolds in 1, therefore, plays a crucial role in accelerating the ORR.The effect of the proton relay source has been examined thoroughly in the case of Fe–porphyrin complexes by incorporating polar protic groups at the secondary coordination sphere of the porphyrin ring. To the best of our knowledge, such a proton relay factor in the ORR reaction of non-heme Fe complexes has not been described before. Thus, this study describes the first example of a nonheme Fe complex for ORR studies that contains a proton binding site at the secondary coordination sphere and accelerates the ORR through the intramolecular proton transfer reaction. The study highlights the ligand design aspects in developing non-heme ORR catalysts and showcases the secondary coordination sphere effect on the selective 4e−/4H+ reduction of O2. Our future efforts will focus on exploring the secondary coordination sphere effect on ORR studies using the nonheme ligands used in this study.
Data availability
All experimental data are available in the manuscript and ESI.†Author contributions
AS and SP designed the project. AS has synthesized and characterized all ligands and Fe complexes described in this study. AS and AD performed the ORR studies. AS, AD, and SK analyzed the data. PJ and PPI assisted in the RRDE experiments. SP supervised the project. AS and SP wrote the original manuscript draft. All authors were involved in writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research work is funded by the Science and Engineering Research Board (SERB, CRG/2022/005842). The authors gratefully acknowledge the Central Research Facility at IIT Delhi for the NMR and EPR measurements. X-ray crystal data of the iron complexes were measured in a DST-FIST-funded Bruker X-ray diffractometer (SR/FST/CSII-027/2014) at the Department of Chemistry, IIT Delhi. AS and AD thank the Indian Institute of Technology Delhi for a doctoral Fellowship.Notes and references
- S. Yoshikawa and A. Shimada, Chem. Rev., 2015, 115, 1936–1989 CrossRef CAS PubMed.
- M. Fernandez-Fernandez, M. A. Sanroman and D. Moldes, Biotechnol. Adv., 2013, 31, 1808–1825 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- H. A. Gasteiger and N. M. Markovic, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- S. Bhunia, A. Ghatak and A. Dey, Chem. Rev., 2022, 122, 12370–12426 CrossRef CAS PubMed.
- M. L. Pegis, C. F. Wise, D. J. Martin and J. M. Mayer, Chem. Rev., 2018, 118, 2340–2391 CrossRef CAS PubMed.
- W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Nat. Rev. Chem, 2017, 1, 0098 CrossRef CAS.
- Y. Zhao, D. P. Adiyeri Saseendran, C. Huang, C. A. Triana, W. R. Marks, H. Chen, H. Zhao and G. R. Patzke, Chem. Rev., 2023, 123, 6257–6358 CrossRef CAS PubMed.
- C. W. Machan, ACS Catal., 2020, 10, 2640–2655 CrossRef CAS.
- A. Das, M. Bera, L. Mallick, B. Chakraborty and S. Paria, in Oxygen Reduction Reaction, ed. K. Sengupta, S. Chatterjee and K. Dutta, Elsevier, 2022, pp. 125–172, DOI:10.1016/B978-0-323-88508-9.00014-8.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed.
- S. Kal and L. Que, JBIC, J. Biol. Inorg. Chem., 2017, 22, 339–365 CrossRef CAS PubMed.
- M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed.
- W. Nam, Acc. Chem. Res., 2015, 48, 2415–2423 CrossRef CAS PubMed.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef CAS PubMed.
- H. S. Soo, A. C. Komor, A. T. Iavarone and C. J. Chang, Inorg. Chem., 2009, 48, 10024–10035 CrossRef CAS PubMed.
- L. Wang, M. Gennari, F. G. Cantu Reinhard, J. Gutierrez, A. Morozan, C. Philouze, S. Demeshko, V. Artero, F. Meyer, S. P. de Visser and C. Duboc, J. Am. Chem. Soc., 2019, 141, 8244–8253 CrossRef CAS PubMed.
- L. Wang, M. Gennari, F. G. Cantu Reinhard, S. K. Padamati, C. Philouze, D. Flot, S. Demeshko, W. R. Browne, F. Meyer, S. P. de Visser and C. Duboc, Inorg. Chem., 2020, 59, 3249–3259 CrossRef CAS PubMed.
- E. N. Cook, D. A. Dickie and C. W. Machan, J. Am. Chem. Soc., 2021, 143, 16411–16418 CrossRef CAS PubMed.
- E. N. Cook, S. L. Hooe, D. A. Dickie and C. W. Machan, Inorg. Chem., 2022, 61, 8387–8392 CrossRef CAS PubMed.
- X. Lu, Y.-M. Lee, M. Sankaralingam, S. Fukuzumi and W. Nam, Inorg. Chem., 2020, 59, 18010–18017 CrossRef CAS PubMed.
- R. B. Gennis, Biochim. Biophys. Acta, Bioenerg., 1998, 1365, 241–248 CrossRef CAS.
- R. McGuire Jr., D. K. Dogutan, T. S. Teets, J. Suntivich, Y. Shao-Horn and D. G. Nocera, Chem. Sci., 2010, 1, 411–414 RSC.
- B. D. Matson, C. T. Carver, A. Von Ruden, J. Y. Yang, S. Raugei and J. M. Mayer, Chem. Commun., 2012, 48, 11100–11102 RSC.
- S. Mukherjee, M. Mukherjee, A. Mukherjee, A. Bhagi-Damodaran, Y. Lu and A. Dey, ACS Catal., 2018, 8, 8915–8924 CrossRef CAS PubMed.
- G. Passard, D. K. Dogutan, M. Qiu, C. Costentin and D. G. Nocera, ACS Catal., 2018, 8, 8671–8679 CrossRef CAS.
- S. Bhunia, A. Rana, P. Roy, D. J. Martin, M. L. Pegis, B. Roy and A. Dey, J. Am. Chem. Soc., 2018, 140, 9444–9457 CrossRef CAS PubMed.
- A. Ghatak, S. Bhakta, S. Bhunia and A. Dey, Chem. Sci., 2019, 10, 9692–9698 RSC.
- A. Singha, A. Mondal, A. Nayek, S. G. Dey and A. Dey, J. Am. Chem. Soc., 2020, 142, 21810–21828 CrossRef CAS PubMed.
- S. Mukherjee, A. Nayek, S. Bhunia, S. G. Dey and A. Dey, Inorg. Chem., 2020, 59, 14564–14576 CrossRef CAS PubMed.
- J. Han, N. Wang, X. Li, W. Zhang and R. Cao, J. Phys. Chem. C, 2021, 125, 24805–24813 CrossRef CAS.
- S. Bhunia, A. Ghatak, A. Rana and A. Dey, J. Am. Chem. Soc., 2023, 145, 3812–3825 CrossRef CAS PubMed.
- R. Zhang and J. J. Warren, J. Am. Chem. Soc., 2020, 142, 13426–13434 CrossRef CAS PubMed.
- D. J. Martin and J. M. Mayer, J. Am. Chem. Soc., 2021, 143, 11423–11434 CrossRef CAS PubMed.
- A. W. Schaefer, M. T. Kieber-Emmons, S. M. Adam, K. D. Karlin and E. I. Solomon, J. Am. Chem. Soc., 2017, 139, 7958–7973 CrossRef CAS PubMed.
- R. L. Shook, S. M. Peterson, J. Greaves, C. Moore, A. L. Rheingold and A. S. Borovik, J. Am. Chem. Soc., 2011, 133, 5810–5817 CrossRef CAS PubMed.
- H. Kotani, T. Yagi, T. Ishizuka and T. Kojima, Chem. Commun., 2015, 51, 13385–13388 RSC.
- A. W. Nichols, E. N. Cook, Y. J. Gan, P. R. Miedaner, J. M. Dressel, D. A. Dickie, H. S. Shafaat and C. W. Machan, J. Am. Chem. Soc., 2021, 143, 13065–13073 CrossRef CAS PubMed.
- A. Das, A. Ali, G. Gupta, A. Santra, P. Jain, P. P. Ingole, S. Paul and S. Paria, ACS Catal., 2023, 13, 5285–5297 CrossRef CAS.
- T. Owen, F. Grandjean, G. J. Long, K. V. Domasevitch and N. Gerasimchuk, Inorg. Chem., 2008, 47, 8704–8713 CrossRef CAS PubMed.
- A. Santra, G. Gupta, B. Biswas, A. Das, D. Ghosh and S. Paria, Inorg. Chem., 2023, 62, 9818–9826 CrossRef CAS PubMed.
- I. Vernik and D. V. Stynes, Inorg. Chem., 1996, 35, 6210–6220 CrossRef CAS.
- M. L. Pegis, B. A. McKeown, N. Kumar, K. Lang, D. J. Wasylenko, X. P. Zhang, S. Raugei and J. M. Mayer, ACS Cent. Sci., 2016, 2, 850–856 CrossRef CAS PubMed.
- P.-A. Jacques, V. Artero, J. Pecaut and M. Fontecave, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20627–20632 CrossRef CAS PubMed.
- D. M. Harraz, S. Weng and Y. Surendranath, ACS Catal., 2023, 13, 1462–1469 CrossRef CAS.
- J. M. Saveant and E. Vianello, Electrochim. Acta, 1963, 8, 905–923 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: fundamentals and applications, John Wiley & Sons, Inc., New York, 2001 Search PubMed.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarstroem, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Rodriguez, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2014, 136, 12544–12547 CrossRef CAS PubMed.
- M. L. Pegis, C. F. Wise, B. Koronkiewicz and J. M. Mayer, J. Am. Chem. Soc., 2017, 139, 11000–11003 CrossRef CAS PubMed.
- A. Rana, Y.-M. Lee, X. Li, R. Cao, S. Fukuzumi and W. Nam, ACS Catal., 2021, 11, 3073–3083 CrossRef CAS.
- Y.-H. Wang, Z. K. Goldsmith, P. E. Schneider, C. W. Anson, J. B. Gerken, S. Ghosh, S. Hammes-Schiffer and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 10890–10899 CrossRef CAS PubMed.
- A. Robert, B. Loock, M. Momenteau and B. Meunier, Inorg. Chem., 1991, 30, 706–711 CrossRef CAS.
- K. Sengupta, S. Chatterjee and A. Dey, ACS Catal., 2016, 6, 1382–1388 CrossRef CAS.
- C. T. Carver, B. D. Matson and J. M. Mayer, J. Am. Chem. Soc., 2012, 134, 5444–5447 CrossRef CAS PubMed.
- J. D. Soper, S. V. Kryatov, E. V. Rybak-Akimova and D. G. Nocera, J. Am. Chem. Soc., 2007, 129, 5069–5075 CrossRef CAS PubMed.
- A. Ghatak, S. Bhunia and A. Dey, ACS Catal., 2020, 10, 13136–13148 CrossRef CAS.
- A. Ghatak, S. Samanta, A. Nayek, S. Mukherjee, S. G. Dey and A. Dey, Inorg. Chem., 2022, 61, 12931–12947 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2290392, 2290393 and 2314421. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06753j |
| This journal is © The Royal Society of Chemistry 2024 |