
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActive site tuning based on pseudo-binary alloys for low-temperature acetylene semihydrogenation†
Jiamin
Ma‡
,
Feilong
Xing
 ,
Ken-ichi
Shimizu
and
Shinya
Furukawa‡
*
,
Ken-ichi
Shimizu
and
Shinya
Furukawa‡
*
Institute for Catalysis, Hokkaido University, Sapporo 001-0021, Japan. E-mail: furukawa@chem.eng.osaka-u.ac.jp
First published on 26th January 2024
Abstract
The development of an efficient catalytic system for low-temperature acetylene semihydrogenation using nonnoble metals is important for the cost-effective production of polymer-grade pure ethylene. However, it remains challenging owing to the intrinsic low activity. Herein, we report a flexibly tunable catalyst design concept based on a pseudo-binary alloy, which enabled a remarkable enhancement in the catalytic activity, selectivity, and durability of a Ni-based material. A series of (Ni1−xCux)3Ga/TiO2 catalysts exhibiting L12-type pseudo-binary alloy structures with various Cu contents (x = 0.2, 0.25, 0.33, 0.5, 0.6, and 0.75) were prepared for active site tuning. The optimal catalyst, (Ni0.8Cu0.2)3Ga/TiO2, exhibited outstandingly high catalytic activity among reported 3d transition metal-based systems and excellent ethylene selectivity (96%) and long-term stability (100 h) with near full conversion even at 150 °C. A mechanistic study revealed that Ni2Cu hollow sites on the (111) surface weakened the strong adsorption of acetylene and vinyl adsorbate, which significantly accelerated the hydrogenation process and inhibited undesired ethane formation.
Introduction
The selective hydrogenation of a trace amount of acetylene in crude ethylene while inhibiting overhydrogenation to ethane is a major industrial process for producing polymer-grade ethylene toward polyethylene production.1 Pd-based catalysts have been industrially used for the selective hydrogenation of acetylene,2 and the use of noble metals increases the cost of the overall process. Therefore, developing an efficient catalyst using nonnoble metals is urgently required in pure and applied chemistry. In this context, noble-metal-alternative catalysts using 3d transition metals, such as Ni, Co, Cu, and Fe, are promising candidates.Ni-based alloys have been studied as potential candidates as industrial catalysts for acetylene semihydrogenation owing to the relatively high hydrogenation activity of Ni and the enhanced selectivity derived from alloying with less active elements. However, the insufficient low-temperature activity of Ni-based catalysts, compared with that of Pd-based catalysts (e.g., Pd is workable even at <100 °C, whereas Ni typically requires a temperature >200 °C), hampers their industrial application for acetylene semihydrogenation.3 Therefore, a novel catalyst design concept that significantly enhances the hydrogenation activity of Ni without lowering the semihydrogenation selectivity is highly desired. To develop an ideal reaction environment for this purpose, the geometric and electronic states of surface Ni sites should be finely controlled and optimized. Ordered alloys, such as NiM intermetallics (e.g., NiZn,1 NiGa,4 and NiIn5), exhibit specific atomic arrangements and unique electronic states, demonstrating high ethylene selectivity in acetylene semihydrogenation. However, the regulated structures do not allow a flexible modification of the reaction environment. Conversely, solid-solution alloys, such as the Ni–Cu system, can arbitrarily vary the composition ratio, whereas various NixCuy ensembles are generated due to the randomness, which typically results in less selective hydrogenation.
A trimetallic pseudo-binary alloy described as (A1−xA′x)mBn, where a part of active metal A is substituted by a similar third element A′ (close to A in the periodic table), can be a promising candidate to provide high flexibility, tunability, and functionality to the reaction environment owing to the combination of the specific structure derived from intermetallic AmBn and the wide variation in x. Choosing an appropriate structure of parent NimMn, the third element, and its doping amount, x, will facilitate the construction of an optimal and well-tuned active site for highly active and selective acetylene semihydrogenation.
Here, we used Ni3Ga and Cu as the parent intermetallic structure and the third element for active site tuning, respectively. Considering that Ni3Ga exhibits moderately high hydrogenation activity and ethylene selectivity while remaining Ni3 hollow sites on the most stable (111) surface (Fig. 1), there is a large room for active-site modulation to improve catalytic performance. In addition, Cu is known as a diluent for active Ni–Ni ensembles with high solubility. Therefore, doping Cu into the Ni sites of Ni3Ga to form a (Ni1−xCux)3Ga pseudo-binary alloy allows the flexible design and fine-tuning of the reaction environment for efficient semihydrogenation (Fig. 1). Herein, we report that (Ni0.8Cu0.2)3Ga/TiO2 acted as a highly active and selective catalyst for acetylene semihydrogenation, where the optimal Cu doping outstandingly improved the hydrogenation activity while maintaining excellent ethylene selectivity. This resulted in a 48-fold higher specific activity than that of reported 3d transition metal-based catalysts at 150 °C. Moreover, this catalyst is workable even at 100 °C with high performance (100% conversion with 96% selectivity) for the first time. In addition, we showed atomic-level insight into the significant enhancement in the catalytic performance clarified by deep characterization and mechanistic studies.
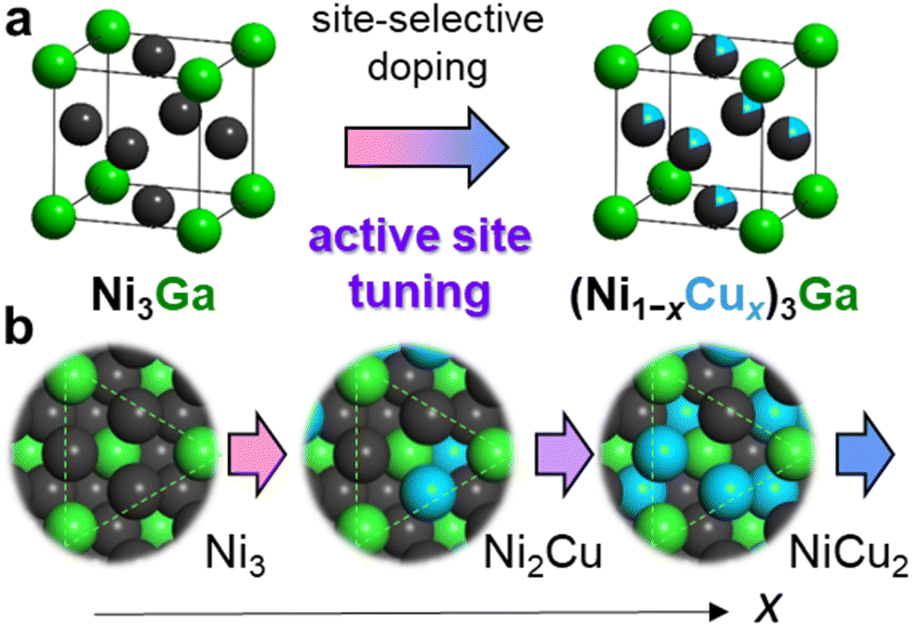 | ||
| Fig. 1 Active site tuning based on pseudo-binary alloy. (a) Site-selective doping of Cu to the Ni sites of intermetallic Ni3Ga to form a (Ni1−xCux)3Ga pseudo-binary alloy. (b) Atomic arrangement of the Ni3Ga (111) surface and the changes in the Ni3 hollow sites upon Cu substitution. | ||
Experimental details
Catalyst preparation
(Ni1−xMx)3Ga/TiO2 (M = Cu, Fe, Co, Zn) and Ni3Ga/TiO2 (Ni: 1 wt%) were prepared through a conventional impregnation method using Ni(NO3)2·6H2O (Wako, 99.9%); Cu (NO3)2·3H2O (Furuya Metal Co. Ltd); Fe(NO3)3·9H2O (Furuya Metal Co. Ltd); Co(NO3)3·6H2O (Wako, 98%); Zn(NO3)2·6H2O (Kanto, 99%); and Ga (NO3)3·nH2O (n = 7–9, Wako, 99.9%) as metal precursors. The TiO2 support (Degussa P25, SBET = 35–65 m2 g−1, anatase + rutile) was added to a vigorously stirred aqueous solution (50 mL H2O per gram of TiO2) containing the precursors of Ni, Ga, and M, followed by stirring for 3 h at 30 °C (Ni: 1 wt%, (Ni + Cu)/Ga = 3). The stoichiometric or small excess Cu was fed for the formation of the desired alloy phase: Cu/(Ni + Cu) = 0, 0.2, 0.33, 0.5, 0.6, 0.67, 0.8 for x = 0, 0.2, 0.25, 0.33, 0.5, 0.6, and 0.75, respectively. The mixture was dried under a reduced pressure at 40 °C using a rotary evaporator. Next, the sample was calcined under flowing air at 400 °C for 1 h and reduced under flowing hydrogen (50 mL min−1) at 600 °C for 1 h. The ramping rate was set at 10 °C min−1 for the calcination and reduction procedures. The corresponding SiO2-supported catalysts were prepared through the pore-filling co-impregnation method. The mixed aqueous solution of the metal precursors was added dropwise to SiO2 (CARiACT G-6, Fuji Silysia, SBET = 673 m2 g−1) to ensure that the solutions only filled the pores of the silica gel (volume of solution: 1.6 mL per gram of silica). The resulting mixture was sealed and aged overnight at room temperature, followed by rapid freezing using liquid nitrogen and subsequent freeze-drying under vacuum at −5 °C overnight. Thereafter, the obtained sample was dried overnight in an oven at 90 °C, followed by calcination and reduction, as mentioned above.Characterization
HAADF-STEM analysis was conducted using a JEOL JEM-ARM200 M microscope equipped with an EDX detector. The XRD patterns of the catalysts were recorded using a MiniFlex 700+D/teX Ultra instrument using a Cu Kα X-ray source. EXAFS measurements of the catalysts were conducted at the BL14B2 and BL01B1 beamlines of SPring-8, Japan Synchrotron Radiation Research Institute (JASRI) using Si (111) double crystals as monochromators. The spectra were recorded at the Ni, Cu, and Ga K-edges in transmission (reference samples and (Ni0.8Cu0.2)3Ga/SiO2 at the Ni and Ga K-edges) and fluorescence (other samples and edges) modes at room temperature. Prior to the measurements, the catalyst was pelletized and pretreated using H2 at 600 °C for 0.5 h in a quartz tube. Afterward, a quartz tube containing the reduced pellet was sealed and transferred to an Argon glove box (O2, <0.01 ppm) to prevent exposure to air. The pellet was sealed in a plastic film bag (Barrier Nylon) with an oxygen absorber (ISO A500-HS: Fe powder). The obtained EXAFS spectra were analyzed using Athena and Artemis software (ver. 0.9.25) implemented in the Demeter package. The k3-weighted EXAFS oscillation was Fourier-transformed in the k range of 3–12 Å−1. The backscattering amplitude and phase shift functions were calculated using FEFF8.Temperature-programmed analyses were conducted using a BELCAT-II (Microtrac BEL) instrument. For C2H2-TPD, the catalyst (100 mg) was pretreated at 600 °C under flowing 5% H2/Ar (20 mL min−1) for 0.5 h and cooled to 50 °C under flowing Ar, followed by cooling to −50 °C using liquid nitrogen with an Ar purge (20 mL min−1). The resulting catalyst was exposed to C2H2 for 1 h at −50 °C. Afterward, Ar was purged for 90 min to remove physisorbed species. Thereafter, the temperature was increased from −50 °C at a ramp rate of 5 °C min−1 under flowing Ar, where the desorbed species were analyzed by quadrupole mass spectrometry placed downstream. C2H4-TPD was performed similarly to C2H2-TPD except that the adsorption and starting temperature was −80 °C. C2H4/H2-TPSR was conducted using the catalyst (80 mg) pretreated with a 5% H2/Ar gas mixture (20 mL min−1) for 0.5 h. After the reduction, the catalyst was cooled to 30 °C using CATCryo-II under a He flow (20 mL min−1), exposed to a mixture of C2H4 and 5% H2/Ar (1 and 20 mL min−1, respectively) at the same temperature for 30 min, and heated from 30 °C to 350 °C at a ramping rate of 10 °C min−1. The formation of ethane (C2H6; m/z = 30) in the outlet gas was analyzed online similarly to TPD.
Catalytic reaction
Acetylene semihydrogenation was conducted in a quartz fixed-bed reactor with an internal diameter of 6 mm at atmospheric pressure. Generally, the catalyst (80 mg) was diluted with quartz sand (2.0 g) loaded into the quartz tube, which was reduced under flowing H2 gas (20 mL min−1) at 600 °C for 0.5 h and cooled to 30 °C under a He flow (20 mL min−1) prior to the catalytic reaction. Subsequently, the catalysts were evaluated by feeding the reactant gas mixture (C2H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2:C2H4:He = 1:10:5/0:39 mL min−1) into the reactor. The temperature dependence of the catalytic performance was evaluated from 100 °C to 250 °C at a ramping rate of 10 °C min−1. The outlet gas was analyzed and quantified using an online gas chromatograph (Shimadzu GC-8A with a column of Unipak S, GL Science) equipped with a thermal conductivity detector. In this study, C2H4 and C2H6 were the only C2 products detected by gas chromatography (GC). No C1 and C4 compounds were detected under the reaction conditions tested in this study. Acetylene conversion and ethylene selectivity were defined by the following equations:
:H2:C2H4:He = 1:10:5/0:39 mL min−1) into the reactor. The temperature dependence of the catalytic performance was evaluated from 100 °C to 250 °C at a ramping rate of 10 °C min−1. The outlet gas was analyzed and quantified using an online gas chromatograph (Shimadzu GC-8A with a column of Unipak S, GL Science) equipped with a thermal conductivity detector. In this study, C2H4 and C2H6 were the only C2 products detected by gas chromatography (GC). No C1 and C4 compounds were detected under the reaction conditions tested in this study. Acetylene conversion and ethylene selectivity were defined by the following equations: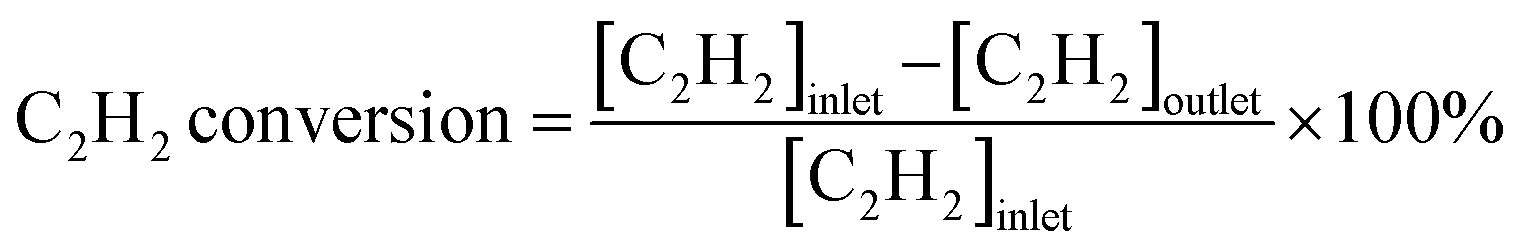 | (1) |
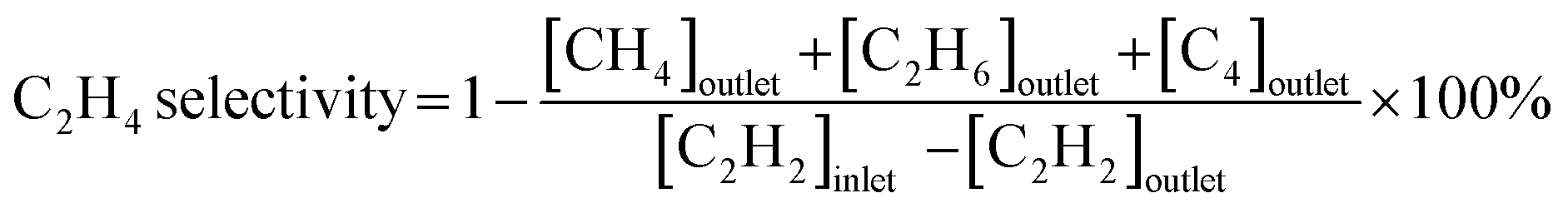 | (2) |
All the catalytic tests for kinetic analysis (Arrhenius-type plots and reaction orders) were performed within a low conversion region (<15%) by adjusting the catalyst amount. All data points were collected four times at 15 min intervals, and their averages were reported.
Hydrogenation of 1-hexyne was performed in a liquid phase batch system using a manner similar to our previous study:6 catalyst, 100 mg (reduced at 400 °C in the reactor); solvent (tetrahydrofuran), 5 mL; alkyne, 0.5 mmol; reaction temperature, 25 °C, H2, 1.0 L (1 atm).
Computational details
Periodic DFT calculations were performed using the CASTEP code7 with Vanderbilt-type ultrasoft pseudopotentials as well as the revised version of the Perdew–Burke–Ernzerhof exchange–correlation functional based on the generalized gradient approximation.8 The plane-wave basis set was truncated at a kinetic energy of 360 eV. A 0.1 eV Fermi smearing was used. The Tkatchenko–Scheffler method was employed to analyze dispersion correlations with a scaling coefficient of sR = 0.94 and a damping parameter of d = 20.9 The reciprocal space was sampled using a k-point mesh with a spacing of 0.04 Å−1, as generated by the Monkhorst–Pack scheme.10 The slab model was constructed using Ni3Ga(111) planes with a thickness of four atomic layers with 13 Å of vacuum spacing. For Cu doping, a part of the Ni atoms in the two surface layers was replaced with Cu so that the Ni/Cu ratio in those layers was four. Geometry optimizations and TS searches were performed on supercell structures using periodic boundary conditions. The convergence criteria for structural optimization and energy calculation were set at (a) a self-consistent field tolerance of 1.0 × 10−6 eV per atom, (b) an energy tolerance of 1.0 × 10−5 eV per atom, (c) a maximum force tolerance of 0.05 eV Å−1, and (d) a maximum displacement tolerance of 1.0 × 10−3 Å. The TS search was performed using the complete linear synchronous transit/quadratic synchronous transit (LST/QST) method.11,12 The convergence criterion for the TS calculations was set at root-mean-square forces on an atom tolerance of 0.1 eV Å−1.Results and discussion
Characterization of the catalysts
First, Ni3Ga and a series of (Ni1−xCux)3Ga catalysts (x = 0.2, 0.25, 0.33, 0.5, 0.6, and 0.75) were prepared through the conventional co-impregnation method using TiO2 as a support (see Methods section for details). Fig. 2a–e shows the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of (Ni0.8Cu0.2)3Ga/TiO2 and the corresponding elemental maps acquired by energy dispersive X-ray (EDX) analysis. Ni, Cu, and Ga were uniformly dispersed on the nanoparticle, suggesting the homogeneous formation of a Ni–Cu–Ga ternary alloy (see Fig. S1† for other nanoparticles). The particle size range was 5–20 nm with a volume-weighted average of 11.4 nm (Fig. 2f). The high-resolution HAADF-STEM image shows lattice fringes with 2.09 Å and 2.55 Å spacings with a dihedral angle of 90° (Fig. 2g), which roughly correlate with the interplanar distances of the (111) and (110) planes of Ni3Ga (2.07 Å and 2.53 Å), respectively. The slightly larger sizes can be attributed to the expansion of the Ni3Ga lattice due to Cu doping to the Ni site (atomic radii: Ni, 1.244 Å; Cu, 1.276 Å). The magnification shows that the atomic arrangement matched with the Ni3Ga-type crystal viewed along the [112] direction (Fig. 2h). Moreover, the superlattice diffraction spots ({110}; indicated by arrows) were observed in the corresponding fast Fourier transform image (FFT; Fig. 2h, lower right inset), which evidences that the ternary alloy exhibited an L12 intermetallic structure but was not a face-centered cubic (fcc) solid-solution alloy. Thereafter, another quantitative analysis for the lattice expansion was conducted by X-ray diffraction (XRD). As shown in Fig. 2i, Ni3Ga/TiO2 exhibited a broad peak at 43.74° (overlapped with TiO2's diffraction), which correlated with the 111 diffraction of intermetallic Ni3Ga. This peak shifted to a lower angle (43.52°) upon Cu doping, again indicating lattice expansion.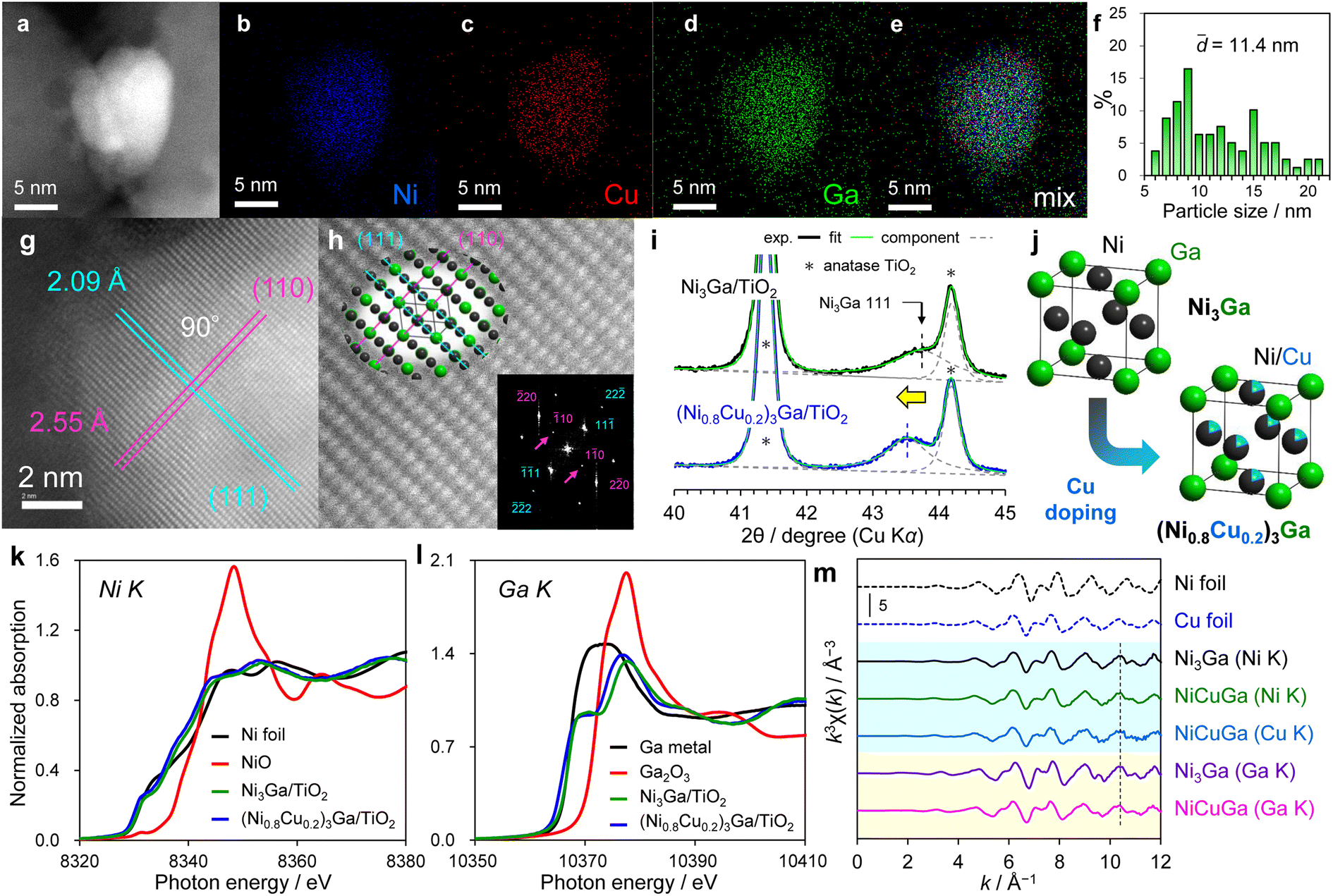 | ||
| Fig. 2 Characterization of (Ni0.8Cu0.2)3Ga/TiO2. (a) HAADF-STEM image and the corresponding elemental maps for (b) Ni, (c) Cu, (d) Ga, and (e) Ni + Cu + Ga. (f) Particle-size distribution. (g) High-resolution image and (h) its magnification overlapped with the Ni3Ga crystal viewed along the [112] direction (upper left) and the corresponding FFT image (lower right). (i) XRD patterns with peak deconvolution. (j) Crystal structure of Ni3Ga and (Ni0.8Cu0.2)3Ga (k) Ni and (l) Ga K-edge XANES spectra. (m) k3-Weighted EXAFS raw oscillations. | ||
Notably, the diffraction angle of the ternary alloy correlated with the theoretical estimation (43.52°; corresponding to 0.5% lattice expansion) using Vegard's law and assuming a solid solution between Ni3Ga and Cu3Ga at a ratio of 0.8:0.2. These results strongly suggested that the (Ni0.8Cu0.2)3Ga pseudo-binary alloy with an L12 structure was formed as nanoparticles on a TiO2 support. The crystallite size of the alloy phase was also estimated using Scherrer's equation (10.6 nm, Table S1†), which correlated with the mean particle size estimated from the HAADF-STEM images. We also performed extended X-ray absorption fine structure (XAFS) analysis for insight into the electronic and geometric states. The X-ray absorption near edge structure (XANES) spectra confirmed that Ni, Cu, and Ga in Ni3Ga/TiO2 and (Ni0.8Cu0.2)3Ga/TiO2 were all in the metallic states (Fig. 2k–l and S2b† for Cu K-edge). In addition, only a minimal difference was observed in the electronic states of Ni and Ga when Cu was doped, indicating that the ligand effect of Cu was extremely minor. The Ni and Cu K-edge EXAFS spectra of (Ni0.8Cu0.2)3Ga/TiO2 exhibited oscillation features similar to those of the Ni K-edge EXAFS of Ni3Ga/TiO2 (Fig. 2m), suggesting that Ni and Cu were present at crystallographically similar sites, i.e., Cu doping to the Ni site. The similarity in the oscillation features was also observed in the Ga K-edge EXAFS spectra, indicating that the original intermetallic structure was retained. Interestingly, the period of the EXAFS oscillation in each edge slightly reduced after Cu doping (Fig. 2m, vertical dotted line).
This correlated with the lattice expansion by Cu doping as indicated by STEM and XRD. We also performed EXAFS curve-fitting analysis to obtain structural information (Table S2 and Fig. S3–S5†). For Ni3Ga/TiO2, Ni–Ni, Ni–Ga, and Ga–Ni scatterings were assigned, in which the coordination number (CN) ratio of Ni–Ga and Ni–Ni was 2.0. This correlated with the L12 intermetallic structure of Ni3Ga. A small contribution of Ga–O (CN = 2.28) was observed, probably due to the interaction with the lattice oxygen of the TiO2 support rather than the presence of Ga oxide because Ga is metallic, as evidenced by XANES. For (Ni0.8Cu0.2)3Ga/TiO2, Ni–Ga, Ni–Ni(Cu), and Ga–Ni(Cu), and Ga–O scatterings were observed similarly to Ni3Ga/TiO2. In addition, Cu–Ga and Cu–Cu(Ni) scatterings were detected in its Cu K-edge spectra with CNs similar to those of Ni–Ga and Ni–Ni(Cu), which correlated with the Cu doping to the Ni site. Furthermore, the interatomic distances of Ni–Ga, Ni–Ni(Cu), and Ga–Ni(Cu) were slightly elongated by Cu doping (+0.01 Å for each, Table S2†). This corresponded to a 0.4% expansion and agreed with that estimated from the peak shift in the XRD pattern (0.5%). Thus, we concluded that the nanoparticulate pseudo-binary alloy of (Ni0.8Cu0.2)3Ga was formed on TiO2.
The (Ni1−xCux)3Ga/TiO2 catalysts with other Cu contents were analyzed by XRD, where peak shifts at lower angles corresponding to the doped Cu content were observed (Fig. S7†). Thus, we successfully prepared a series of (Ni1−xCux)3Ga/TiO2 catalysts with the pseudo-binary alloy structure with a wide range of Cu contents (x = 0, 0.2, 0.25, 0.33, 0.5, 0.6, 0.75).
Catalytic performance in acetylene semihydrogenation
Next, the synthesized (Ni1−xCux)3Ga/TiO2 catalysts were tested in acetylene semihydrogenation in the absence of excess ethylene. Fig. 3a summarizes their catalytic performances. As universal and standard scales for activity, apparent activation energy (Ea) and the temperature in which acetylene conversion reached 50% (T50) were employed, respectively. The catalytic activity exhibited a volcano-type trend with respect to the Cu content with x = 0.25–0.33 as the best. Fig. 3b and c show the temperature dependence of acetylene conversion and ethylene selectivity, respectively, using the representative catalysts, Ni3Ga/TiO2, (Ni0.8Cu0.2)3Ga/TiO2, and (Ni0.67Cu0.33)3Ga/TiO2. Owing to the remarkable enhancement in catalytic activity by Cu doping, the temperature required for the complete conversion drastically decreased from 230 °C to 150 °C and 120 °C, respectively. Moreover, ethylene selectivity was improved by Cu doping, particularly in a low-temperature region (<150 °C; i.e., 90.5% → 96.2% at 150 °C for x = 0.2).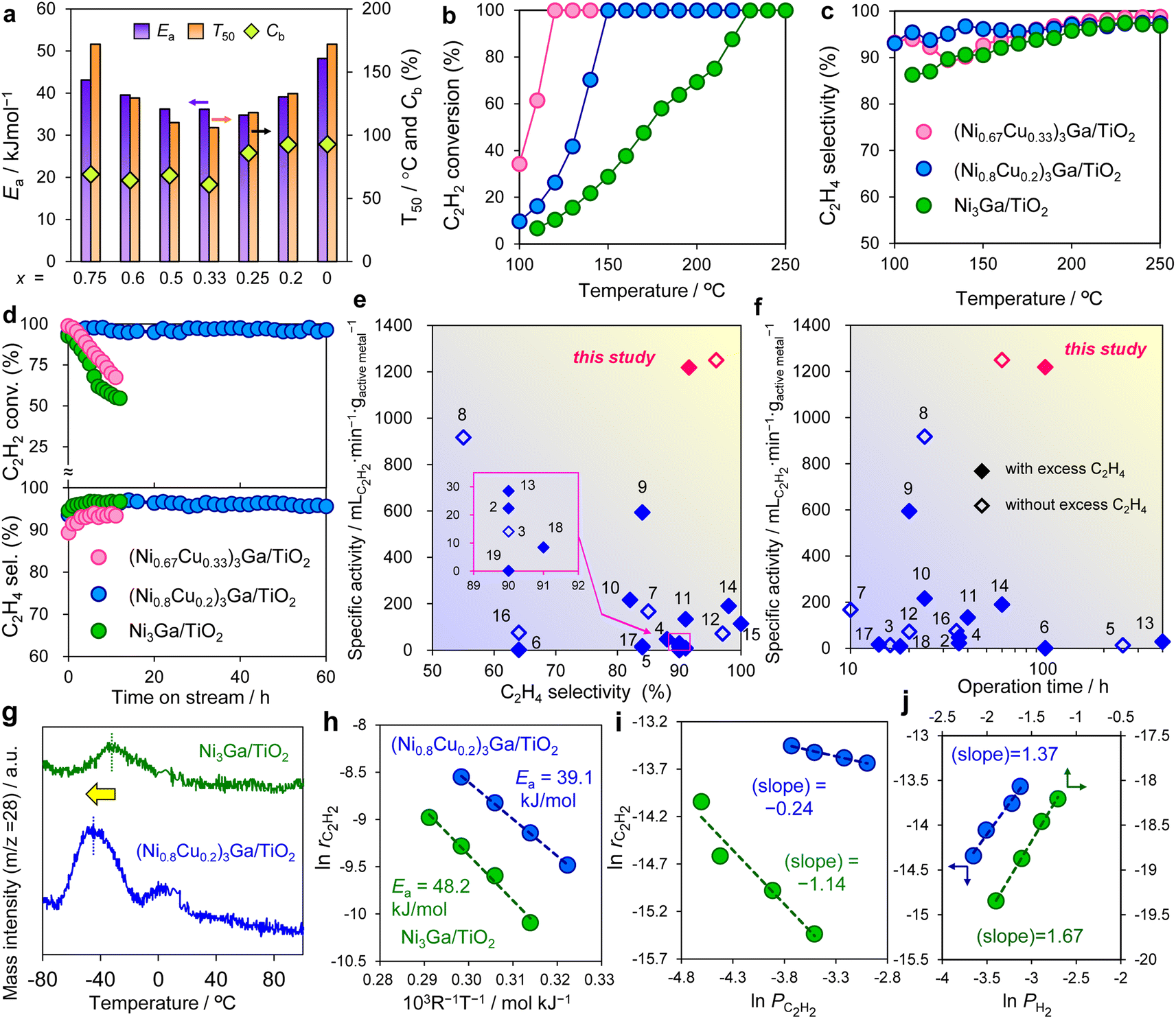 | ||
| Fig. 3 Catalytic performance and mechanistic studies. (a) Ea, T50, and Cb obtained in acetylene semihydrogenation over a series of (Ni1−xCux)3Ga/TiO2 catalysts. (b) C2H2 conversion and (c) C2H4 selectivity as a function of the reaction temperature in the absence of ethylene (C2H2:H2:He = 1:10:39 mL min−1). (d) Long-term stability test (Ni3Ga, 220 °C; (Ni0.8Cu0.2)3Ga, 150 °C; (Ni0.67Cu0.33)3Ga, 145 °C) in the absence of ethylene. (e and f) Comparison with reported systems using 3d transition metal-based catalysts (<200 °C): specific activity at high acetylene conversion (>80%) vs. (e) ethylene selectivity and (f) operation time (numbers correspond to the entries in Table S3, Fig. S11 and S12†). (g) C2H4-TPD profiles. (h) Arrhenius-type plots. (i and j) Reaction orders of (i) acetylene (PC2H2) and (j) hydrogen partial pressures. | ||
We also checked the carbon balance (Cb) in the reaction (the temperature dependences of acetylene conversion, ethylene selectivity, and carbon balance are shown in Fig. S8†). Importantly, a significant decrease in Cb was observed when x ≥0.25 (Fig. 3a), probably because extra Cu caused oligomerization to form green oil. Therefore, we chose x = 0.2 as the optimal Cu content because it afforded the satisfactorily high activity with minimum oligomerization.
A long-term stability test was conducted at the near-complete conversion of acetylene, revealing that rapid deactivation occurred for Ni3Ga and (Ni0.67Cu0.33)3Ga, whereas no deactivation was observed for (Ni0.8Cu0.2)3Ga after at least 60 h (Fig. 3d). Thus, the activity, selectivity, and stability were significantly increased by the catalyst design based on the pseudo-binary alloy and the appropriate amount of Cu doping.
We also examined the catalytic performance in the presence of excess ethylene, a practical condition (C2H2:H2:C2H4:He = 1:10:5:19 mL min−1), in which activity, selectivity, and stability (stable up to 100 h) trends similar to those in Fig. 3b–d were obtained (Fig. S9 and S10†). Fig. 3e and f summarize the comparison of the catalytic performance in acetylene semihydrogenation at low temperatures (≤200 °C) with reported systems using 3d transition metal-based catalysts12–30 (see Table S3, Fig. S11 and S12† for details). The (Ni0.8Cu0.2)3Ga/TiO2 catalyst exhibited outstandingly high specific activity and long-term stability while retaining excellent ethylene selectivity. It is known that catalytic activity and selectivity/stability are typically in tradeoff because excess activity tends to induce undesired side reactions. Therefore, satisfying the upper right region in Fig. 3e and f is a challenging task. In this context, the development of a Ni-based catalyst efficiently working within a low-temperature region is highly innovative.
As a control experiment, we prepared (Ni0.8Cu0.2)3Ga/SiO2 to investigate the suitability of supports for this catalysis. XRD and XAFS confirmed the formation of a pseudo-binary alloy structure in a similar way, as mentioned above (Table S2, Fig. S2, S3, S6, and S13†). The Ni K-edge XANES spectra revealed a slight decrease in the electron density of Ni changing from TiO2 to SiO2 (Fig. S2a†). Using a SiO2 support, the catalytic activity in acetylene semihydrogenation increased (100% conversion at 130 °C), whereas ethylene selectivity significantly decreased (50–75%, Fig. S14†). This trend was more prominent in the presence of excess ethylene, where overhydrogenation to ethane markedly proceeded (the selectivity was −18% at the lowest, Fig. S15†). Thus, SiO2 was unsuitable for selective hydrogenation presumably owing to the reduction in the electron density of Ni. A plausible interpretation is that the low electron density of Ni renders it cationic and strengthens the adsorption of acetylene and ethylene, resulting in high activity but low selectivity. In addition, a series of (Ni0.8M0.2)3Ga/TiO2 catalysts using 3d transition metals other than Cu (M = Fe, Co, Zn) were prepared and similarly tested (Fig. S16†). However, no catalyst showed superior performance to that of (Ni0.8Cu0.2)3Ga/TiO2, confirming the suitability of Cu as a modifier for Ni.
We also tested the catalytic performance of (Ni0.8Cu0.2)3Ga/TiO2 in 1-hexyne hydrogenation in a liquid phase. The reaction proceeded smoothly at room temperature within 6 h and selectively to from 1-hexene without any by-products such as n-hexane and 2-hexenes even after the conversion reached 100% (Fig. S17a†). A similar test was also conducted using 2-hexyne as an internal alkyne, which resulted in selective conversion to cis-2-hexene with high yield (Fig. S17b†). Thus, the developed catalyst is also applicable to selective hydrogenation of longer alkynes.
Mechanistic study
To understand the role of Cu doping in enhanced catalysis, we conducted a mechanistic study. First, the temperature-programmed desorption of ethylene (C2H4-TPD) was performed using Ni3Ga/TiO2 and (Ni0.8Cu0.2)3Ga/TiO2. As shown in Fig. 3g, Ni3Ga showed an ethylene desorption peak centered at −32 °C, whereas that of (Ni0.8Cu0.2)3Ga was observed at −45 °C. This indicated that ethylene adsorption was weakened by Cu doping, which correlated with enhanced ethylene selectivity. Fig. 3h shows the Arrhenius plots obtained during acetylene semihydrogenation and the corresponding Ea (corresponding to those in Fig. 3a), suggesting that the energy barrier of acetylene hydrogenation to ethylene was lowered by doping an appropriate amount of Cu. The reaction orders of acetylene and hydrogen's partial pressures (PC2H2 and PH2, respectively) on the overall conversion rate of acetylene were also estimated (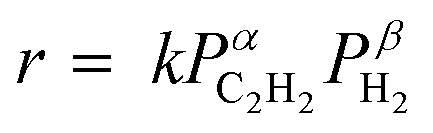 , Fig. 3i and j). For Ni3Ga/TiO2, α and β were largely negative (−1.14) and positive (1.67), respectively. This indicated that the strong adsorption of acetylene on Ni competes with H2 adsorption and/or activation, which is a typical trend in acetylene semihydrogenation. Conversely, for (Ni0.8Cu0.2)3Ga/TiO2, α significantly increased to near zero (−0.24), while β slightly decreased (1.37). Therefore, it was likely that Cu doping to Ni markedly weakened acetylene adsorption, thereby facilitating H2 adsorption/activation. This may have been due to the moderate dilution of the Ni–Ni ensembles (i.e., Ni3 hollow sites on the (111) plane) by Cu. Considering these results, a possible interpretation for the significant enhancement in catalytic activity is the double promotion of the reaction rate by increasing the rate constant (lower Ea) and the hydrogen coverage.
, Fig. 3i and j). For Ni3Ga/TiO2, α and β were largely negative (−1.14) and positive (1.67), respectively. This indicated that the strong adsorption of acetylene on Ni competes with H2 adsorption and/or activation, which is a typical trend in acetylene semihydrogenation. Conversely, for (Ni0.8Cu0.2)3Ga/TiO2, α significantly increased to near zero (−0.24), while β slightly decreased (1.37). Therefore, it was likely that Cu doping to Ni markedly weakened acetylene adsorption, thereby facilitating H2 adsorption/activation. This may have been due to the moderate dilution of the Ni–Ni ensembles (i.e., Ni3 hollow sites on the (111) plane) by Cu. Considering these results, a possible interpretation for the significant enhancement in catalytic activity is the double promotion of the reaction rate by increasing the rate constant (lower Ea) and the hydrogen coverage.
Next, we investigated the origin of the side reaction yielding ethane using the temperature-programmed surface reaction of ethylene hydrogenation (C2H4/H2-TPSR) (Fig. S18†). Unexpectedly, no ethane evolved from 30 °C to 280 °C for Ni3Ga/TiO2 and (Ni0.8Cu0.2)3Ga/TiO2, implying that the overhydrogenation of product ethylene hardly occurred on these catalysts. Therefore, the small amount of byproduct ethane observed in acetylene semihydrogenation appeared to be directly generated from acetylene, as often proposed as a side pathway via an ethylidene intermediate. Thus, the selectivity enhancement shown in Fig. 3c may be attributed to the inhibition of this pathway by Cu doping. The C2H2-TPD experiment revealed that strongly adsorbed acetylene (assignable to di-σ-C2H2) observed on Ni3Ga/TiO2 disappeared when Cu was doped (Fig. S19†). These results demonstrated that the active site tuning by Cu substitution accelerated the desired reaction and minimized the undesired side pathway owing to the change in adsorption behaviour.
DFT calculations
Finally, we performed density functional theory (DFT) calculations to elucidate the roles of Cu substitution in the geometry and energetics of acetylene hydrogenation over the Ni-based catalysts. The Ni3Ga (111) surface was considered as the parent reaction environment for the Cu substitution. As a model of (Ni0.8Cu0.2)3Ga (111), a part of the Ni atoms in the two surface layers was randomly replaced with Cu so that the Ni/Cu ratio in the two layers was 4 (hereinafter referred to as Ni3Ga(111)–Cu). The resulting Ni2Cu ensembles were considered active sites for the stepwise hydrogenation of acetylene to ethylene and ethane (Fig. 4a, S20 and S21† for details). Fig. 4b summarizes the energy landscapes of the stepwise hydrogenation over the Ni-based catalysts. Overall, the adsorbate states were destabilized upon Cu substitution. However, the degree of destabilization differed depending on the conformation of the adsorbates. The higher the CN (atop, 1; bridge, 2; hollow, 3), the higher the destabilization. For example, the vinyl (C2H3) on the hollow and di-σ-C2H2 (C2H4) exhibited increases of 0.39 eV and 0.19 (0.21) eV, respectively, whereas the vinyl and ethyl on the top only exhibited minimal changes.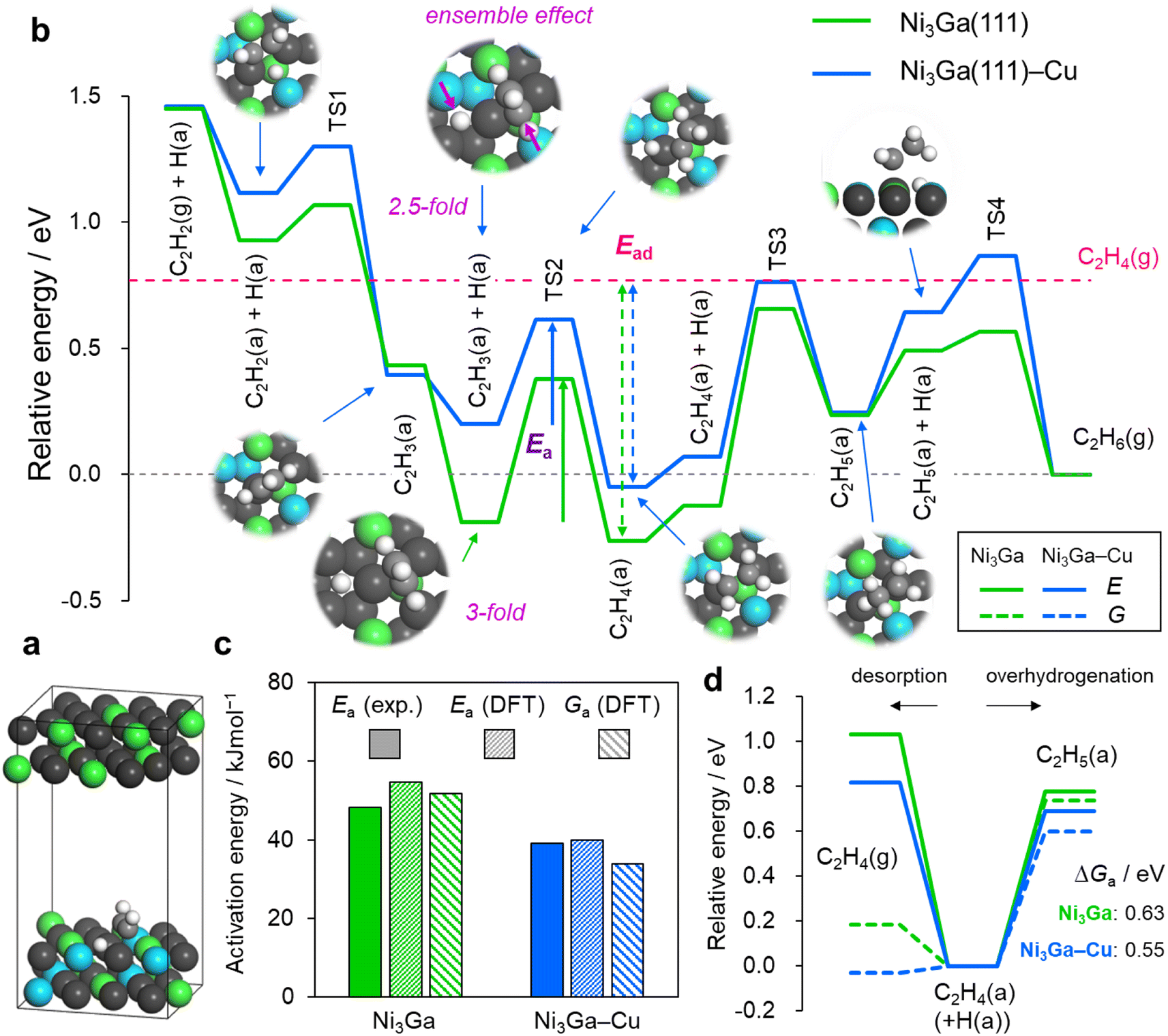 | ||
| Fig. 4 DFT calculations for acetylene hydrogenation. (a) Example of the optimized structure in the slab model: C2H2(a) + H(a) on Ni3Ga(111)–Cu. (b) Energy landscapes of the stepwise hydrogenation from acetylene to ethane. Sum of the bare slab and gas-phase C2H6 was set to zero in energy. (c) Comparison of activation energies. (d) Energy diagrams of ethylene desorption and overhydrogenation. | ||
For the vinyl on the hollow, the coordinating carbon atom was off-centered to be slightly far from the Cu atom, resulting in a “2.5-fold-like” state and significant destabilization. This trend indicated that the ensemble effect by Cu efficiently tuned the adsorptivity.31 Moreover, the minute energy change for the on-top adsorption on Ni suggested that the contribution of the ligand effect of Cu on Ni was extremely minor. For the transition states (TSs), the degree of destabilization was typically intermediate between the initial and final states. Consequently, the energy barrier of the hydrogen attack toward vinyl to form ethylene, i.e., the highest barrier from acetylene to ethylene (the rate-determining step in acetylene semihydrogenation), is decreased. The activation energies calculated for Ni3Ga and Ni3Ga–Cu (electronic and free energies) correlated with those obtained experimentally (Fig. 4c). These results support the validity of the DFT model and reaction pathways/configurations considered for this study. For the overhydrogenation of ethylene, the difference in energy barriers of its desorption and overhydrogenation (ΔE or ΔG) has been typically considered as a scale. Although the changes in the electronic energy of desorption exceeded those of overhydrogenation, they significantly reduced when free energy was considered (Fig. 4d) because desorption can gain a large contribution of translational entropy. Owing to the large ΔG (>0.55 eV), ethylene overhydrogenation appeared to hardly occur over Ni3Ga and Ni3Ga–Cu, correlating with the C2H4/H2-TPSR results and again suggesting the involvement of direct hydrogenation to ethane via the ethylidene intermediate. The DFT calculation also revealed that the adsorbate states of ethylidene on the Ni2Cu site of Ni3Ga–Cu were considerably higher than those on the Ni3 site of Ni3Ga owing to the similar ensemble effect (Fig. S22†). This correlated with the C2H2-TPD results (the absence of strongly adsorbed C2H2) and probably contributed to the enhancement in ethylene selectivity by Cu doping.
Thus, the DFT study adequately explained the roles of Cu doping to Ni in tuning the active site geometry and the resulting catalytic performance, which was consistent with the experimental results. Based on the insights from the DFT calculation, the best x ratio to maximize the fraction of Ni2Cu ensemble sites was 0.33. This also agrees with the highest activity obtained with this ratio. However, to retain the highest Cb for better stability, we chose an x of 0.2 as the optimal value for active site tuning as mentioned above.
Conclusions
In summary, we successfully applied a catalyst design concept based on a pseudo-binary alloy for the fine-tuning of the active property and resulting catalytic performance in acetylene semihydrogenation. A part of the Ni sites in intermetallic Ni3Ga was substituted with Cu to form a series of (Ni1−xCux)3Ga catalysts with various Cu contents (x = 0.2, 0.25, 0.33, 0.5, 0.6, and 0.75). The optimal catalyst, (Ni0.8Cu0.2)3Ga/TiO2, exhibited outstandingly high catalytic activity among reported 3d transition metal-based systems (1250 mLC2H2−1 min−1 g−1), excellent ethylene selectivity (96%), and long-term stability (100 h) with near full conversion even at 150 °C. The appropriate amount of Cu doping formed Ni2Cu hollow sites on the stable Ni3Ga (111) surface, which significantly destabilized the multifold coordination of adsorbates, such as 3-fold vinyl, thereby lowering the energy barrier of acetylene hydrogenation. This destabilization also weakened the strong adsorption of acetylene, which (1) further accelerated the hydrogenation process by increasing hydrogen coverage and (2) improved ethylene selectivity by suppressing the direct hydrogenation pathway via the ethylidene intermediate. Owing to the combination of these enhancements, acetylene semihydrogenation at low temperatures can be performed using Ni-based materials. Thus, the wide tunability and expandability of this catalyst design concept are highly versatile for constructing innovative catalysts. The findings in this study provide a highly efficient catalytic system for the production of polymer-grade ethylene and uncover a new field of catalyst design based on multinary alloys.Data availability
The data supporting this article have been included in ESI.†Author contributions
S. F. and J. M. designed the research and co-wrote the manuscript. J. M. performed all the experiments. F. X. contributed to catalytic performance experiment. S. F. conducted all the computational studies. J. M., F. X., S. F., and K. S. discussed about the data and commented on the manuscript equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by JSPS KAKENHI (Grant Numbers 17H04965 and 20H02517), JST CREST (JPMJCR17J3), JST PRESTO (JPMJPR19T7), and the Collaborative Research Projects of Laboratory for Materials and Structures, Institute of Innovative Research, Tokyo Institute of Technology. The XAFS was performed with the approval of JASRI (No. 2021B1962 and 2022B0302). The computation time was provided by the supercomputer systems in the Institute for Chemical Research, Kyoto University.References
- J. K. N. Felix Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen and C. H. Christensen, Science, 2008, 1320–1323 CrossRef PubMed.
- J. Osswald, R. Giedigkeit, R. E. Jentoft, M. Armbrüster, F. Girgsdies, K. Kovnir, T. Ressler, Y. Grin and R. Schlögl, J. Catal., 2008, 258, 210–218 CrossRef CAS.
- B. Lou, H. Kang, W. Yuan, L. Ma, W. Huang, Y. Wang, Z. Jiang, Y. Du, S. Zou and J. Fan, ACS Catal., 2021, 11, 6073–6080 CrossRef CAS.
- Y. Liu, X. Liu, Q. Feng, D. He, L. Zhang, C. Lian, R. Shen, G. Zhao, Y. Ji, D. Wang, G. Zhou and Y. Li, Adv. Mater., 2016, 28, 4747–4754 CrossRef CAS PubMed.
- Y. Chen and J. Chen, Appl. Surf. Sci., 2016, 387, 16–27 CrossRef CAS.
- M. Miyazaki, S. Furukawa and T. Komatsu, J. Am. Chem. Soc., 2017, 139, 18231–18239 CrossRef CAS PubMed.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 6–9 CrossRef.
- K. Hu, M. Wu, S. Hinokuma, T. Ohto, M. Wakisaka, J. I. Fujita and Y. Ito, J. Mater. Chem. A, 2019, 7, 2156–2164 RSC.
- N. Govind, M. Petersen, G. Fitzgerald, D. King-Smith and J. Andzelm, Comput. Mater. Sci., 2003, 28, 250–258 CrossRef CAS.
- T. A. Halgren and W. N. Lipscomb, Chem. Phys. Lett., 1977, 49, 225–232 CrossRef CAS.
- X. Ge, Z. Ren, Y. Cao, X. Liu, J. Zhang, G. Qian, X. Gong, L. Chen, X. Zhou, W. Yuan and X. Duan, J. Mater. Chem. A, 2022, 19722–19731 RSC.
- B. Fu, A. J. McCue, Y. Liu, S. Weng, Y. Song, Y. He, J. Feng and D. Li, ACS Catal., 2022, 12, 607–615 CrossRef CAS.
- Y. Liu, J. Zhao, J. Feng, Y. He, Y. Du and D. Li, J. Catal., 2018, 359, 251–260 CrossRef CAS.
- C. Lu, A. Zeng, Y. Wang and A. Wang, ACS Omega, 2021, 6, 3363–3371 CrossRef CAS PubMed.
- C. Lu, Y. Wang, R. Zhang, B. Wang and A. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 46027–46036 CrossRef CAS PubMed.
- Y. Wang, B. Liu, X. Lan and T. Wang, ACS Catal., 2021, 11, 10257–10266 CrossRef CAS.
- H. Liu, M. Chai, G. Pei, X. Liu, L. Li, L. Kang, A. Wang and T. Zhang, Chin. J. Catal., 2020, 41, 1099–1108 CrossRef CAS.
- H. Zhou, B. Li, H. Fu, X. Zhao, M. Zhang, X. Wang, Y. Liu, Z. Yang and X. Lou, ACS Sustain. Chem. Eng., 2022, 10, 4849–4861 CrossRef CAS.
- Y. Cao, H. Zhang, S. Ji, Z. Sui, Z. Jiang, D. Wang, F. Zaera, X. Zhou, X. Duan and Y. Li, Angew. Chem., Int. Ed., 2020, 132, 11744–11749 CrossRef.
- X. Shi, Y. Lin, L. Huang, Z. Sun, Y. Yang, X. Zhou, E. Vovk, X. Liu, X. Huang, M. Sun, S. Wei and J. Lu, ACS Catal., 2020, 10, 3495–3504 CrossRef CAS.
- Y. Chai, G. Wu, X. Liu, Y. Ren, W. Dai, C. Wang, Z. Xie, N. Guan and L. Li, J. Am. Chem. Soc., 2019, 141, 9920–9927 CrossRef CAS PubMed.
- J. Gu, M. Jian, L. Huang, Z. Sun, A. Li, Y. Pan, J. Yang, W. Wen, W. Zhou, Y. Lin, H. J. Wang, X. Liu, L. Wang, X. Shi, X. Huang, L. Cao, S. Chen, X. Zheng, H. Pan, J. Zhu, S. Wei, W. X. Li and J. Lu, Nat. Nanotechnol., 2021, 16, 1141–1149 CrossRef CAS PubMed.
- F. Huang, Y. Deng, Y. Chen, X. Cai, M. Peng, Z. Jia, J. Xie, D. Xiao, X. Wen, N. Wang, Z. Jiang, H. Liu and D. Ma, Nat. Commun., 2019, 10, 4431 CrossRef PubMed.
- C. Riley, A. De La Riva, S. Zhou, Q. Wan, E. Peterson, K. Artyushkova, M. D. Farahani, H. B. Friedrich, L. Burkemper, N. V. Atudorei, S. Lin, H. Guo and A. Datye, ChemCatChem, 2019, 11, 1526–1533 CrossRef CAS.
- Y. Chen and J. Chen, Appl. Surf. Sci., 2016, 387, 16–27 CrossRef CAS.
- M. Armbrüster, K. Kovnir, M. Friedrich, D. Teschner, G. Wowsnick, M. Hahne, P. Gille, L. Szentmiklósi, M. Feuerbacher, M. Heggen, F. Girgsdies, D. Rosenthal, R. Schlögl and Y. Grin, Nat. Mater., 2012, 11, 690–693 CrossRef PubMed.
- X. Dai, Z. Chen, T. Yao, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, S. Wei, Y. Wu and Y. Li, Chem. Commun., 2017, 53, 11568–11571 RSC.
- T. Kojima, S. Kameoka, S. Fujii, S. Ueda and A. P. Tsai, Sci. Adv., 2018, 4, eaat6063 CrossRef CAS PubMed.
- Y. Nakaya and S. Furukawa, Chem. Rev., 2023, 123, 5859–5947 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03704e |
| ‡ Present address: Division of Applied Chemistry, Graduate School of Engineering, Osaka University, Osaka 565-0871, Japan. |
| This journal is © The Royal Society of Chemistry 2024 |