
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly dispersed Pd-based pseudo-single atoms in zeolites for hydrogen generation and pollutant disposal†
Kai
Zhang
a,
Ning
Wang
*d,
Yali
Meng
a,
Tianjun
Zhang
e,
Pu
Zhao
a,
Qiming
Sun
 *ab and
Jihong
Yu
*c
*ab and
Jihong
Yu
*c
aInnovation Center for Chemical Science, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, 215123, P. R. China. E-mail: sunqiming@suda.edu.cn
bJiangsu Key Laboratory of Advanced Negative Carbon Technologies, Soochow University, Suzhou, 215123, Jiangsu, P. R. China
cState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, International Center of Future Science, College of Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: jihong@jlu.edu.cn
dInstitute of Sustainable Energy and Resources, College of Chemistry and Chemical Engineering, Qingdao University, Qingdao 266071, P. R. China. E-mail: wangning2021@qdu.edu.cn
eCollege of Chemistry and Materials Science, Hebei University, Baoding 071002, P. R. China
First published on 30th November 2023
Abstract
Atomically dispersed metal catalysts with excellent activity and stability are highly desired in heterogeneous catalysis. Herein, we synthesized zeolite-encaged Pd-based pseudo-single atoms via a facile and energy-efficient ligand-protected direct H2 reduction method. Cs-corrected scanning transmission electron microscopy, extended X-ray absorption, and pair distribution function measurements reveal that the metal species are close to atomic-level dispersion and completely confined within the intersectional channels of silicalite-1 (S-1) zeolite with the MFI framework. The Pd@S-1-H exhibits excellent activity and stability in methane combustion reactions with a complete combustion temperature of 390 °C, and no deactivation is observed even after 100 h on stream. The optimized bimetallic 0.8Pd0.2Ni(OH)2@S-1-H catalyst exhibits an excellent H2 generation rate from FA decomposition without any additives, affording a superhigh turnover frequency up to 9308 h−1 at 333 K, which represents the top activity among all of the best heterogeneous catalysts under similar conditions. Significantly, zeolite-encaged metal catalysts are first used for Cr(VI) reduction coupled with formic acid (FA) dehydrogenation and show a superhigh turnover number of 2980 mol(Cr2O72−) mol(Pd)−1 at 323 K, surpassing all of the previously reported catalysts. This work demonstrates that zeolite-encaged pseudo-single atom catalysts are promising in efficient hydrogen storage and pollutant disposal applications.
Introduction
Zeolites, a class of crystalline materials with orderly distributed micropores, large specific surface areas, and adjustable acidity/basicity, have been widely used in diverse industrial applications as adsorbents, separation materials, and catalysts.1–4 In recent decades, zeolites have also been regarded as ideal supports to immobilize metal species with small sizes and high dispersity. The rational design and fabrication of zeolite-supported metal catalysts with superior catalytic activity and stability is always a hot topic in heterogeneous catalysis.5–13 Despite the excellent confinement effect of zeolites, zeolite-supported metal catalysts prepared by the conventional impregnation method still often suffer from the formation of large and uneven metal particles as well as unsatisfactory sintering and leaching during the catalytic process. This is because the metal species are inclined to be located on the outer surface of zeolite crystals during the synthetic process. To overcome these shortcomings, several innovative in situ synthetic strategies have been developed for the confined synthesis of ultrasmall metal species within zeolite crystals.14–26 For example, Iglesia and coworkers developed a ligand-stabilized approach to encapsulate a series of small metal nanoparticles with a mean size of 1–2 nm into micropores of various aluminosilicate zeolites.23,24 Corma and coworkers developed a zeolite structure transformation approach to encage subnanometric Pt clusters within MWW zeolites.25 Xiao and coworkers developed a metal-containing-seed-directed synthesis method to fix different kinds of metal nanoparticles with a diameter range of 0.8–3.6 nm within zeolite crystals.22 Our group also reported the encapsulation of subnanometric metal clusters within pure silica zeolites via a ligand-protected method under direct hydrothermal conditions.19,20,26,27 Recently, single- and pseudo-single atom catalysts with extremely high metal dispersity have attracted extensive research interest from researchers due to their nearly 100% atom-utilization efficiency and excellent catalytic properties.28–32 However, such catalysts usually suffer from poor durability and inferior reusability in catalysis. The rational design and fabrication of zeolite-supported metal species with (pseudo)single-atom dispersion while maintaining excellent stability during catalytic reactions and further expanding their catalytic applications remains a nontrivial task but is greatly challenging.Methane, as a typical greenhouse gas, produces a 20-fold higher greenhouse effect than carbon dioxide. The release of unburned methane from exhaust gas has become an environmental issue in recent years, and catalytic methane combustion technology is an effective way to reduce its pollution of the atmosphere.33–36 Supported Pd-based catalysts have been considered the most active catalysts for methane combustion, but they still often suffer from either poor activity or inferior long-term stability toward the complete oxidation of methane due to the severe aggregation of metal species. The development of efficient catalysts with outstanding activity and stability is highly desired. Hydrogen (H2) is considered a green and efficient energy carrier, but efficient and controllable hydrogen storage remains a bottleneck for the future hydrogen economy.37–40 Formic acid (FA, HCOOH) has emerged as a promising liquid-phase hydrogen storage material for portable H2 storage applications.41–44 In addition, FA can also be used as a green and cost-effective reductant for the reduction of toxic pollutants, such as hexavalent chromium (Cr(VI)), which possesses serious mutagenicity and carcinogenicity. The in situ generated H2 from FA dehydrogenation can efficiently reduce Cr(VI) to Cr(III).45,46 Although some heterogeneous catalysts have recently been used for this reaction, few catalysts with both excellent performance and stability have been reported.
In this work, we synthesized zeolite-encaged highly-dispersed pseudo-single Pd and bimetallic Pd–Ni(OH)2 atoms via a facile and energy-saving ligand-protected direct H2 reduction method. The location, pseudo-single atom nature, and precise structure of the metal species were determined by Cs-corrected scanning transmission electron microscopy, extended X-ray absorption, and pair distribution function (PDF) analysis. Due to the extremely high metal dispersion and almost complete exposure of active metal sites, Pd@S-1-H exhibited superior methane combustion activity with a complete combustion temperature of 390 °C, which is lower than that of Pd@S-1-C (416 °C) subjected to calcination and reduction procedures and Pd/S-1-im (513 °C) synthesized by the impregnation method. The catalytic activity of methane combustion over Pd@S-1-H remains unchanged even after 100 h on stream. Taking advantage of the synergistic effect of the bimetallic interface, the optimized bimetallic 0.8Pd0.2Ni(OH)2@S-1-H catalyst exhibits an excellent H2 generation rate from FA decomposition without any additives, affording a superhigh turnover frequency up to 9308 h−1 at 333 K, which is 33 times higher than that over Pd/S-1-im synthesized by the impregnation method, representing the top activity among all of the best heterogeneous catalysts under similar conditions. Significantly, 0.8Pd0.2Ni(OH)2@S-1-H also acts as an efficient catalyst in Cr(VI) reduction coupling with FA dehydrogenation, showing a record turnover number of 2980 mol(Cr2O72−) mol(Pd)−1 at 323 K.
Results and discussion
As shown in Fig. 1A, S-1 zeolite-encaged Pd-based metal samples were prepared by a facile ligand-protection method under in situ hydrothermal conditions at 170 °C for 3 days in reaction gels with molar compositions of 1TEOS/0.4TPAOH/35H2O/a[Pd(en)2]Cl2/b[Ni(en)3](NO3)2, (TEOS = tetraethoxysilane, TPAOH = tetrapropylammonium hydroxide, en = ethylenediamine, a + b = 0.0045, a/b = 10/0, 9/1, 8/2, 7/3, and 0/10). Next, the obtained samples were subjected to two different thermal treatment methods: one was calcinated in air at 550 °C for 6 h and then reduced in H2 flow at 500 °C for 2 h (named xPd(1−x)Ni(OH)2@S-1-C, where x represented the molar ratio of Pd/(Pd + Ni) in the initial synthesis gel); the other was directly reduced in pure H2 flow at 500 °C for 2 h (named xPd(1−x)Ni(OH)2@S-1-H). For comparison, the Pd/S-1-im and Pd–Ni(OH)2/S-1-im samples were also prepared by the incipient wetness impregnation method with the same metal loadings as the Pd@S-1 and Pd–Ni(OH)2@S-1 samples, respectively. Powder X-ray diffraction measurements confirm that the obtained zeolite-encaged Pd-based samples possess an MFI topological structure with high crystallinity (Fig. S1†). Inductively coupled plasma atomic emission spectroscopy (ICP-AES) measurements reveal that all samples possessed a similar molar amount of total metal species, giving total metal loadings of Pd@S-1 and Pd–Ni(OH)2@S-1 samples in the range of 0.64–0.48 wt%. The molar ratios of Pd/Ni in 0.9Pd0.1Ni(OH)2@S-1-H, 0.8Pd0.2Ni(OH)2@S-1-H, and 0.7Pd0.3Ni(OH)2@S-1-H are 0.89/0.11, 0.81/0.19, and 0.72/0.28, respectively, which are similar to the preset ratios in synthetic gels (Table S1†).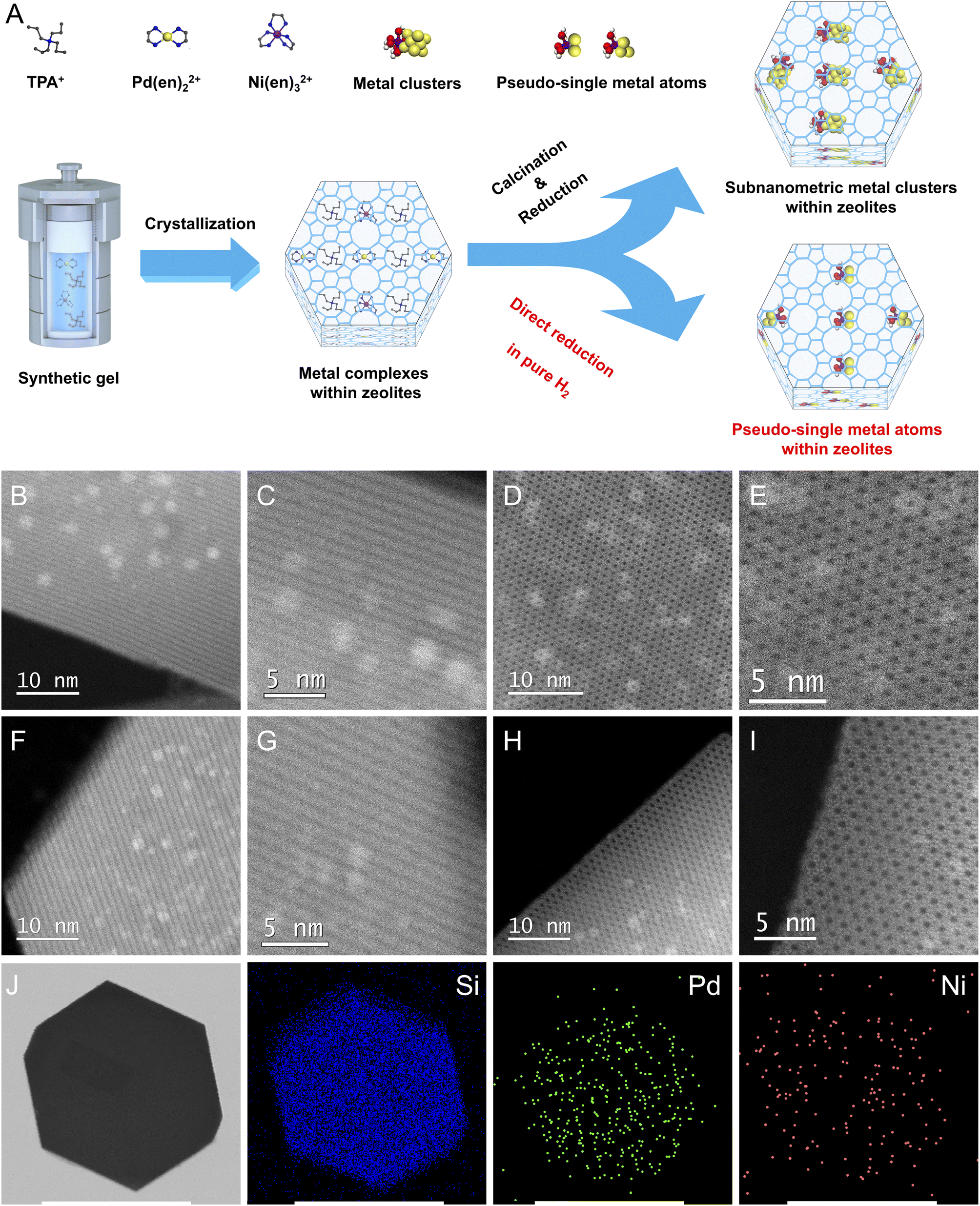 | ||
| Fig. 1 (A) Schematic illustration of the preparation of zeolite-encaged Pd-based catalysts. Cs-corrected STEM images of (B and C) Pd@S-1-C; (D and E) 0.8Pd0.2Ni(OH)2@S-1-C; (F and G) Pd@S-1-H; (H and I) 0.8Pd0.2Ni(OH)2@S-1-H. (J) TEM image of 0.8Pd0.2Ni(OH)2@S-1-H catalyst and corresponding EDX mapping images for Si, Pd, and Ni elements. Scale bar: 250 nm. | ||
Scanning electron microscopy (SEM) measurements demonstrate that all zeolite-supported metal samples possess a uniformly hexagonal prism morphology with average sizes of 200–300 nm. Note that many bright dots corresponding to Pd nanoparticles (NPs) can be clearly observed in Pd/S-1-im, indicating that large amounts of Pd species with large sizes are located outside of S-1 crystals. In contrast, no metal species are detected on the outside of zeolite-encaged metal samples synthesized by the in situ method (Fig. S2†). To obtain precise information about the size and location of the metal species in the samples, Cs-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and high-resolution TEM measurements were performed. As shown in Fig. 1B–I and S3, S4,† homogeneously distributed metal clusters/atoms with subnanometer sizes are completely confined within intersectional void spaces between the straight and sinusoidal channels of S-1 zeolites in the representative Pd@S-1-C, Pd@S-1-H, 0.8Pd0.2Ni(OH)2@S-1-C, and 0.8Pd0.2Ni(OH)2@S-1-H samples, and no metallic species could be observed on the outside of the zeolites. In contrast, the metal NPs are considerably larger in Pd/S-1-im (4–5 nm), and the great majority of metal species are located on the outer surface of zeolite crystals, which is consistent with the SEM observation. The introduction of Ni species does not affect the size of metal clusters in zeolites. Significantly, the Pd@S-1-H and 0.8Pd0.2Ni(OH)2@S-1-H samples subjected to direct H2 reduction possess smaller metal sizes than the corresponding Pd@S-1-C and 0.8Pd0.2Ni(OH)2@S-1-C samples treated by the calcination-reduction process. The metal species in the Pd@S-1-H and 0.8Pd0.2Ni(OH)2@S-1-H samples are in the form of single and pseudo-single atoms. This can be mainly attributed to the fact that during the direct H2 reduction process, the hydrogenolysis of organic templates and ligands and reduction of metal species occur simultaneously; the ammonia gas produced by hydrolysis of organic species (confirmed by H2-temperature-programmed reduction/mass spectrometry analysis, Fig. S5A†) can act as protective agents to restrain the aggregation of metal species, resulting in the formation of single and pseudo-single atoms with decreased size and increased dispersion.
Thermogravimetry analyses reveal that no noteworthy weightlessness can be observed in either Pd@S-1-C or Pd@S-1-H when the temperature is above 300 °C, indicating that almost all organic species were removed by the calcination-reduction and direct H2 reduction processes (Fig. S5B†). N2 adsorption/desorption measurements show that the micropore volume of zeolite-encaged metal samples decreased by approximately 10% compared with that of pure S-1 zeolite (∼0.115 cm3 g−1) because metal species confined within the zeolite matrix caused a slight decrease (∼0.012 cm3 g−1), but large microporous volumes (0.103–0.105 cm3 g−1) and abundant surface areas (399–415 m2 g−1) were still preserved in Pd@S-1-C, Pd@S-1-H, and 0.8Pd0.2Ni(OH)2@S-1-H, respectively (Fig. S6 and Table S1†).
To further identify the electronic and local structure information of metal species encapsulated within S-1 zeolites, the X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra of representative Pd@S-1-C, Pd@S-1-H, and 0.8Pd0.2Ni(OH)2@S-1-H samples are measured. The detailed structural parameters according to EXAFS fittings are summarized in Table S2.† As shown in Fig. 2A and S7,† the Pd K-edge XANES spectra of Pd@S-1-C, Pd@S-1-H, and 0.8Pd0.2Ni(OH)2@S-1-H samples are different from Pd foil and PdO, showing a higher oxidation state than the Pd foil. It can be attributed to the formation of ultrasmall Pd species, which are easy to coordinate with oxygen atoms of zeolite frameworks. A small shift to the higher absorption edge energy and increased intensity of the white line can be observed for Pd@S-1-H in comparison with Pd@S-1-C, suggesting the higher oxidation state of Pd in the Pd@S-1-H sample. Note that a small edge shift to lower energy and slightly decreased intensity of the white line is found in 0.8Pd0.2Ni(OH)2@S-1-H as compared with Pd@S-1, indicating that the Pd species in 0.8Pd0.2Ni(OH)2@S-1-H possess a higher electron density than that in Pd@S-1-H. The Ni K-edge XANES spectrum of 0.8Pd0.2Ni(OH)2@S-1-H is nearly the same as that of the Ni(OH)2 reference spectrum, revealing that the Ni species in the bimetallic clusters exist in the form of a Ni(OH)2 structure (Fig. 2B). In the Pd K-edge Fourier-transformed EXAFS spectra, the significant peaks at ∼2.01 and 2.72 Å were identified as the Pd–O and Pd–Pd metallic bonds, respectively (Fig. 2C). Based on the EXAFS fitting results, the average coordination number (CN) of Pd–O shell for Pd@S-1-H is 2.9, which is higher than that of Pd@S-1-C (1.9) and 0.8Pd0.2Ni(OH)2@S-1-H (2.4), further confirming the higher oxidation state of Pd species in Pd@S-1-H sample. The average CN of the Pd–Pd shell for Pd@S-1-H is just 1.9, which is much smaller than that for Pd@S-1-C (3.5). It indicates that Pd clusters in both Pd@S-1-H and Pd@S-1-C are subnanometer in size, and the average size of Pd clusters in Pd@S-1-H is smaller than that in Pd@S-1-C, in line with the observation of Cs-corrected STEM measurements. Notably, the average CN of the Pd–Pd shell for 0.8Pd0.2Ni(OH)2@S-1-H is as low as 0.9, indicating the Pd species are dimer- or single-atomic dispersion. A small peak at 2.45 Å corresponding to Pd–Ni metallic bond (CN = 0.13) can be also detected, which reveals the partial formation of Pd–Ni alloy structure, and some electrons may transfer from Ni to Pd and form electron-enriched Pd surfaces in 0.8Pd0.2Ni(OH)2@S-1-H. The average valence states of Pd sites in different samples could be disclosed by establishing a linear correlation between the edge energy and the valence state of reference samples. The Pd@S-1-H shows a valence state of Pd (+1.83), which is slightly higher than Pd@S-1-C (+1.67). Notably, the valence state of Pd species in 0.8Pd0.2Ni(OH)2@S-1-H (+1.17) is lower than those of two Pd samples, indicating the higher electron density in 0.8Pd0.2Ni(OH)2@S-1-H (Fig. S8†). In the Ni K-edge EXAFS spectra of 0.8Pd0.2Ni(OH)2@S-1-H is consistent with the Ni(OH)2 reference spectrum, further confirming the formation of the bimetallic Pd–Ni(OH)2 structure (Fig. 2D).
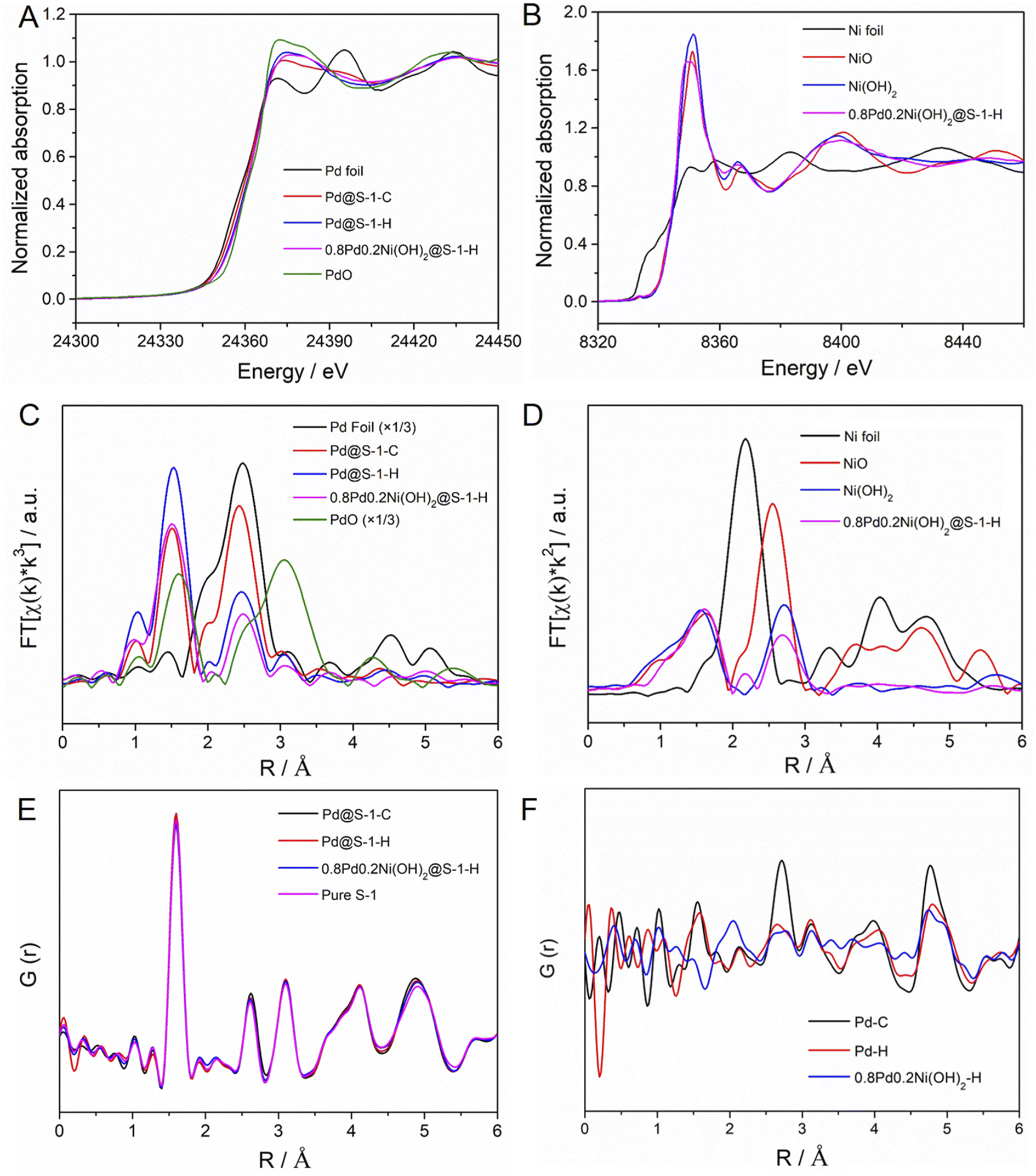 | ||
| Fig. 2 (A) Pd K-edge XANES spectra of Pd@S-1-C, Pd@S-1-H, 0.8Pd0.2Ni(OH)2@S-1-H and Pd foil, PdO standards. (B) Ni K-edge XANES spectra of 0.8Pd0.2Ni(OH)2@S-1-H and Ni foil, NiO, Ni(OH)2 standards. (C) Fourier transform of k3-weighted EXAFS spectra of Pd@S-1-C, Pd@S-1-H, 0.8Pd0.2Ni(OH)2@S-1-H and Pd foil, PdO standards. (D) Fourier transform of k2-weighted EXAFS spectra of 0.8Pd0.2Ni(OH)2@S-1-H and Ni foil, NiO, Ni(OH)2 standards. (E) PDFs of Pd@S-1-C, Pd@S-1-H, 0.8Pd0.2Ni(OH)2@S-1-H, and pure S-1 zeolite samples. (F) d-PDFs of Pd–C, Pd–H, and 0.8Pd0.2Ni(OH)2–H species. | ||
The interatomic structure of the encapsulated metallic clusters in zeolite was further investigated by pair distribution function (PDF) analyses. The experimental PDFs of different S-1 zeolite-encaged metal catalysts and pure S-1 zeolite are shown in Fig. 2E. The peaks at ca. 1.6, 2.6, and 3.1 Å can be attributed to the Si–O, O–O, and Si–Si distances in S-1 zeolites, respectively. The above signals are very similar in Pd@S-1-C, Pd@S-1-H, 0.8Pd0.2Ni(OH)2@S-1-H, and pure S-1 zeolite samples, indicating the introduction of metal species inside the zeolites rarely affect the structure of S-1 zeolites. The differential PDF (d-PDF) analysis is a method based on the difference between two PDFs and can be used for obtaining structural information of guest species.47 Therefore, we can extrapolate the structure of Pd species in S-1 zeolites corresponding to the calculated difference in the experimental PDFs of the Pd@S-1 and pure S-1 zeolite. Fig. 2F shows the calculated d-PDF of encapsulated metal species in zeolite-encage metal catalysts. The peaks at approximately 2.7 and 4.7 Å, assigned to the first and second-shell Pd–Pd bonds can be observed in Pd@S-1-C, indicating the formation of small Pd clusters. In contrast, the first-shell Pd–Pd bond in Pd@S-1-H and 0.8Pd0.2Ni(OH)2@S-1-H are nearly neglected, reflecting the formation of atomically dispersed Pd atoms in the samples subjected to the direct H2 reduction treatment, in accordance with the observation of Cs-corrected STEM and EXAFS results. Notably, a unique peak at about 2.0 Å can be observed in 0.8Pd0.2Ni(OH)2@S-1-H, which is attributed to the formation of the Ni–O bond.
The thermal stabilities at high temperatures under various atmospheres are also investigated over the representative 0.8Pd0.2Ni(OH)2@S-1-H sample. The average particle size of metal species is only 1.6 nm after calcination at 700 °C for 2 h under N2 atmosphere and all metal species remain to be confined within zeolite crystals. In contrast, the mean particle size of metal species in 0.8Pd0.2Ni(OH)2/S-1-im dramatically increases to 6.2 nm and most of the metal NPs are located on the outside of zeolites under the same thermal treatment (Fig. S9†). Moreover, the particle sizes of metal species in 0.8Pd0.2Ni(OH)2@S-1-H are 1.0, 1.8, and 2.2 nm after calcination in H2, O2, and water-vapor treatment at 600 °C, respectively (Fig. S10†). Significantly, all metal species are still encapsulated inside the zeolite crystals after high-temperature thermal treatments. The above results demonstrate that the 0.8Pd0.2Ni(OH)2@S-1-H possesses superior thermal and hydrothermal stability under high temperatures, suggesting its promising applications under practical harsh conditions.
The catalytic performances of Pd@S-1-H, Pd@S-1-C, and Pd/S-1-im catalysts in methane combustion reactions at a GHSV of 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL gcat−1 h−1 are shown in Fig. 3A. The methane conversion improves with the increase of the reaction temperature. Among all of the catalysts, the Pd@S-1-H exhibits the highest methane combustion activity with a complete combustion temperature of 390 °C, which is lower than those of Pd@S-1-C (416 °C) and Pd/S-1-im (513 °C), respectively. The significantly improved activity can be attributed to the Pd@S-1-H possessing decreased sizes of Pd species compared with the Pd@S-1-C and Pd/S-1-im, thus exposing more accessible active sites (especially the corner site) for the reactants. In addition, the unique microenvironment of Pd species and highly dispersed Pdδ+ (0 < δ < 2) species due to the confinement effect of zeolites may also be a key factor affecting the catalytic performance of methane oxidation reactions. The water resistance ability of Pd@S-1-H is further studied through on-stream tests in the presence of 4.5 vol% water vapor. As shown in Fig. S11,† although the catalytic activity of Pd@S-1-H is slightly decreased under the humidity condition as compared to the dry condition, the Pd@S-1-H catalyst still gives an excellent activity with the complete methane conversion temperature at 415 °C under humidity condition. Such a temperature is close to that under dry condition and much lower than that of Pd/S-1-im, indicating the excellent water vapor resistance of the Pd@S-1-H in methane oxidation reactions. Significantly, the Pd@S-1-H exhibits superhigh long-term stability, and 100% of methane conversion remains unchanged even after 100 h on stream at 470 °C. In contrast, the methane conversion of Pd/S-1-im decreases rapidly from 88.0% to 68.7% within 65 h under the same condition (Fig. 3B). When the reaction temperature decreases to 365 °C, the initial methane conversion of Pd@S-1-H is about 81% and the methane conversion increases gradually as the reaction goes on. Note that the methane conversion can be stable at around 89% even after 100 h on stream. The above results indicate that the encapsulation of metal species inside zeolites by using the ligand-protected direct H2 reduction method, can not only enhance the utilization of metal species and catalytic activity, but also remarkably improved the durability during methane combustion reactions.
000 mL gcat−1 h−1 are shown in Fig. 3A. The methane conversion improves with the increase of the reaction temperature. Among all of the catalysts, the Pd@S-1-H exhibits the highest methane combustion activity with a complete combustion temperature of 390 °C, which is lower than those of Pd@S-1-C (416 °C) and Pd/S-1-im (513 °C), respectively. The significantly improved activity can be attributed to the Pd@S-1-H possessing decreased sizes of Pd species compared with the Pd@S-1-C and Pd/S-1-im, thus exposing more accessible active sites (especially the corner site) for the reactants. In addition, the unique microenvironment of Pd species and highly dispersed Pdδ+ (0 < δ < 2) species due to the confinement effect of zeolites may also be a key factor affecting the catalytic performance of methane oxidation reactions. The water resistance ability of Pd@S-1-H is further studied through on-stream tests in the presence of 4.5 vol% water vapor. As shown in Fig. S11,† although the catalytic activity of Pd@S-1-H is slightly decreased under the humidity condition as compared to the dry condition, the Pd@S-1-H catalyst still gives an excellent activity with the complete methane conversion temperature at 415 °C under humidity condition. Such a temperature is close to that under dry condition and much lower than that of Pd/S-1-im, indicating the excellent water vapor resistance of the Pd@S-1-H in methane oxidation reactions. Significantly, the Pd@S-1-H exhibits superhigh long-term stability, and 100% of methane conversion remains unchanged even after 100 h on stream at 470 °C. In contrast, the methane conversion of Pd/S-1-im decreases rapidly from 88.0% to 68.7% within 65 h under the same condition (Fig. 3B). When the reaction temperature decreases to 365 °C, the initial methane conversion of Pd@S-1-H is about 81% and the methane conversion increases gradually as the reaction goes on. Note that the methane conversion can be stable at around 89% even after 100 h on stream. The above results indicate that the encapsulation of metal species inside zeolites by using the ligand-protected direct H2 reduction method, can not only enhance the utilization of metal species and catalytic activity, but also remarkably improved the durability during methane combustion reactions.
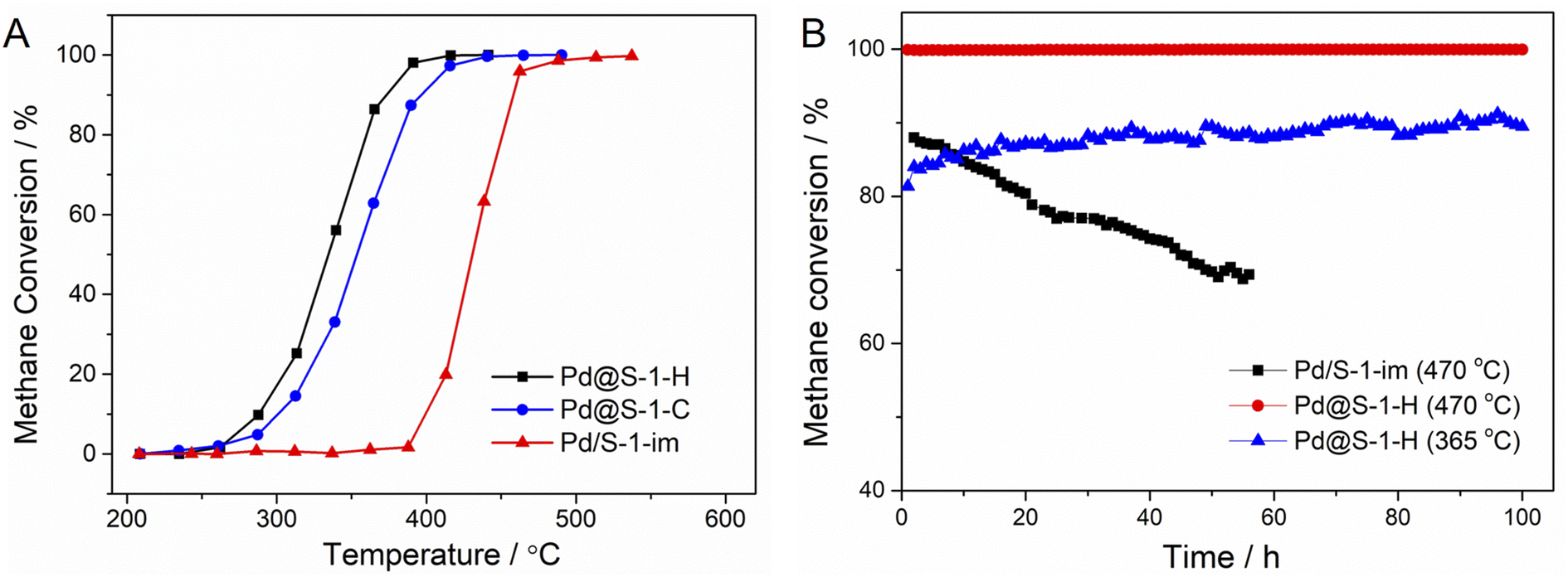 | ||
| Fig. 3 (A) Catalytic performance of methane combustions over Pd@S-1-H, Pd@S-1-C, and Pd/S-1-im catalysts. Reaction condition: 2% CH4–20% O2–78% N2, gas hourly space velocity (GHSV) = 36000 mL gcat−1 h−1. (B) Long-term stability of methane combustions over Pd@S-1-H and Pd/S-1-im catalysts with a GHSV of 36000 mL gcat−1 h−1. | ||
Catalytic activities of H2 generation from FA dehydrogenation over various zeolite-supported metal catalysts were evaluated at 333 K without any additive. As shown in Fig. 4A, the Pd@S-1-H exhibited an excellent H2 generation rate (TOF = 1936 h−1), which is nearly 3 and 7-fold higher than that of Pd@S-1-C (TOF = 651 h−1) and Pd/S-1-im synthesized by impregnation method (TOF = 281 h−1). The activity improvement of Pd@S-1-H can be mainly attributed to the decreased size of Pd species, resulting in more accessible active Pd sites for FA dehydrogenation. The introduction of Ni(OH)2 species into Pd clusters can further enhance the catalytic activities of H2 generation from FA dehydrogenation (Fig. 4B). Among all catalysts, the 0.8Pd0.2Ni(OH)2@S-1-H exhibited the highest H2 generation rate: 147 mL of CO2 and H2 without CO impurity (<10 ppm, Fig. S12†), corresponding to 100% FA conversion (HCOOH = H2 + CO2), could be released within 1.67 min at 333 K without any additive, affording a superhigh initial TOF value of 9308 h−1, which is 4.8 and 33 times higher than those over Pd@S-1-H and Pd/S-1-im, respectively. Given that Ni(OH)2@S-1-H shows no activity for the FA dehydrogenation, the Pd species are responsible for the H2 generation from FA dehydrogenation. As expected, the H2 generation rate of 0.8Pd0.2Ni(OH)2@S-1-H reduction is also about 1.3-fold improvement in comparison with that over 0.8Pd0.2Ni(OH)2@S-1-C. The TOF value of representative 0.8Pd0.2Ni(OH)2@S-1-H represents the top level among all of the previously reported heterogeneous catalysts and is even higher than many results obtained with additives under similar catalytic conditions (Table S3†).27,48,49 In comparison, the H2 generation rate over the physical mixture of Pd@S-1-H and Ni(OH)2@S-1-H are identical to the single Pd@S-1-H, but much lower than that over 0.8Pd0.2Ni(OH)2@S-1-H (Fig. S13†). The above results revealed that the superior catalytic activity of FA dehydrogenation over 0.8Pd0.2Ni(OH)2@S-1-H can be mainly ascribed to the formation of ultrasmall and highly-dispersed metal species coupled with the synergistic effect of the Pd–Ni(OH)2 interface that can remarkably lower the energy barrier of FA dehydrogenation, which is also confirmed by theoretical calculations in our previous work.19 In addition, a more electron-rich Pd surface favors the C–H activation of the Pd-formate intermediate to produce H2 and CO2, accelerating the H2 generation from FA decomposition.
 | ||
| Fig. 4 (A) Volume of the generated gas (CO2 + H2) versus time for FA dehydrogenation over (A) Pd@S-1-C, 0.2Pd0.8Ni(OH)2@S-1-C, Pd@S-1-H, Ni(OH)2@S-1-H, 0.2Pd0.8Ni(OH)2@S-1-H, and Pd/S-1-im; (B) Pd@S-1-H and Pd–Ni(OH)2@S-1-H with various Pd/Ni ratios at 333 K (2 M FA, 3 mmol, nmetal/nFA = 0.012). (C) Volume of the generated gas versus time for FA dehydrogenation at different temperatures over 0.8Pd0.2Ni(OH)2@S-1-H catalyst (2 M FA, 3 mmol, nmetal/nFA = 0.012). Inset of (C): Arrhenius plot (ln TOF vs. 1000/T). (D) Recycling tests for FA dehydrogenation over 0.8Pd0.2Ni(OH)2@S-1-H catalyst at 333 K (nmetal/nFA = 0.012). The TOF values of the first run to five runs are 9308, 9789, 8741, 8945, and 9074 h−1, respectively. | ||
The catalytic activities of catalysts are highly dependent on the reaction temperature, and the H2 generation rates improved along with the increase in catalytic temperatures (Fig. 4C and S14, S15†). The apparent activation energy (Ea) of 0.8Pd0.2Ni(OH)2@S-1-H is calculated as 51.4 kJ mol−1, which is lower than that of Pd@S-1-H catalysts (66.5 kJ mol−1), further indicating the higher activity of H2 generation over the zeolite-encapsulated bimetallic catalytic system than the corresponding monometallic counterpart. The zeolite-encaged Pd-based metal catalysts also possess excellent recycling stability for H2 generation from FA dehydrogenation. No degradation of catalytic activities for FA dehydrogenation can be observed over Pd@S-1-H and 0.8Pd0.2Ni(OH)2@S-1-H after five cycles at 333 K (Fig. 4D and S16, S17†). Significantly, there is no variation in the morphology and crystallinity of zeolites as well as the particle size and distributions of metal species in spent catalysts after recycling reactions, further confirming the excellent stabilities of the zeolite-encapsulated metallic catalyst during FA dehydrogenation (Fig. S18–S20†).
Given the zeolite-encaged Pd-based catalysts exhibit excellent catalytic activity and stability in FA dehydrogenation, such catalyst can be expected to have high activity in the Cr(VI) reduction coupled with FA dehydrogenation. Catalytic activities of Cr(VI) reduction in aqueous solution over the representative Pd@S-1-H and 0.8Pd0.2Ni(OH)2@S-1-H catalysts are evaluated at 323 K by using K2Cr2O7 and FA as a representative Cr(VI) compound and reducing agent, respectively. As shown in Fig. 5A, B and S21,† the peak at ∼350 nm corresponding to Cr(VI) in UV-vis absorption spectroscopy decreases continually as the reaction time is prolonged, and the color of the reaction solution changes from yellow to colorless, indicating the gradual reduction of Cr(VI) in the presence of catalysts (Fig. S22†). 0.02 mmol Cr2O72− can be converted completely to Cr3+ within 10 min over 0.8Pd0.2Ni(OH)2@S-1-H, which is 2.6-fold faster than Pd@S-1-H. In contrast, pure S-1 zeolite was inactive for the Cr(VI) reduction even with the addition of FA, indicating that the Pd species were responsible for the Cr(VI) reduction (Fig. S23†). In addition, neglectable Cr(VI) conversion can be observed over 0.8Pd0.2Ni(OH)2@S-1-H when the FA was replaced by H2 (1 bar), indicating that the in situ generated H2 from FA dehydrogenation is more favorable to the Cr2O72− reduction to Cr3+via an H2 transfer pathway (Cr2O72− + 8H+ + 3H2 → 2Cr3+ + 7H2O) (Fig. S24†). Because the bimetallic Pd–Ni(OH)2 species is more active for the FA dehydrogenation than monometallic Pd species, not unexpectedly, the 0.8Pd0.2Ni(OH)2@S-1-H exhibits the significantly enhanced activity of cascade Cr(VI) reduction coupled with FA dehydrogenation than Pd@S-1-H.
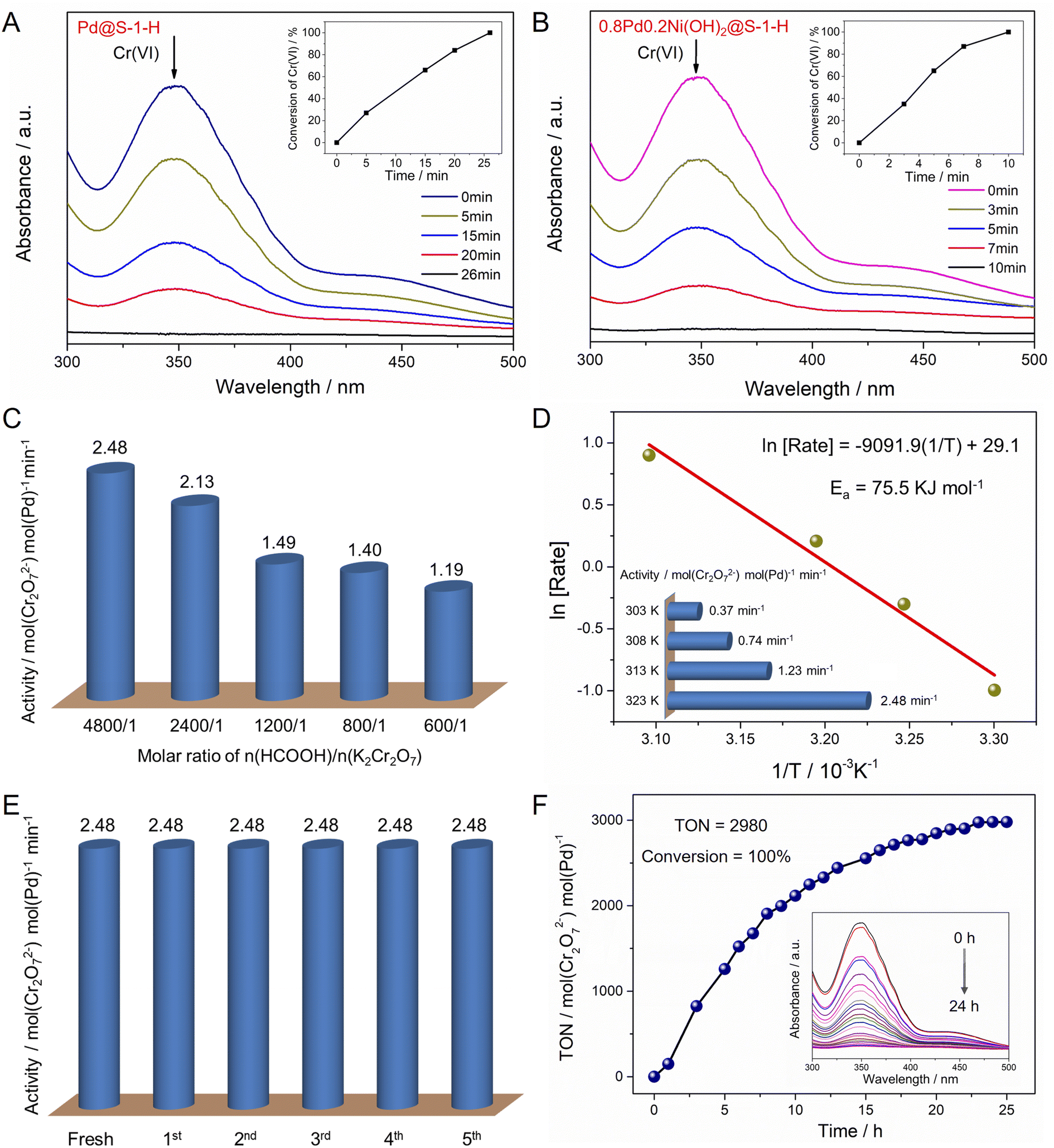 | ||
| Fig. 5 (A and B) UV-vis absorption spectra for the Cr(VI) reduction by FA over Pd@S-1-H and 0.8Pd0.2Ni(OH)2@S-1-H catalysts. Catalytic condition: 28 mg catalyst, 0.2 mL K2Cr2O7 (0.1 M), 4 mL HCOOH (6 M) (n(HCOOH)/n(K2Cr2O7) = 1200/1), 5.8 mL H2O, T = 323 K. (C) Catalytic activity of Cr(VI) reduction over 0.8Pd0.2Ni(OH)2@S-1-H catalyst with different n(HCOOH)/n(K2Cr2O7) ratios. Catalytic condition: 28 mg catalysts, 0.05–0.4 mL K2Cr2O7 (0.1 M), 4 mL HCOOH (6 M) (n(HCOOH)/n(K2Cr2O7) = 4800/1–600/1), 5.95–5.6 mL H2O, T = 323 K. (D) Catalytic activity of Cr(VI) reduction over 0.8Pd0.2Ni(OH)2@S-1-H catalyst at different temperatures. Catalytic condition: 28 mg catalyst, 0.05 mL K2Cr2O7 (0.1 M), 4 mL HCOOH (6 M), 5.95 mL H2O, T = 303–323 K. (E) Recycling stability of Cr(VI) reduction over 0.8Pd0.2Ni(OH)2@S-1-H catalyst. Catalytic condition: 28 mg catalyst, 0.05 mL K2Cr2O7 (0.1 M), 4 mL HCOOH (6 M), 5.95 mL H2O, T = 323 K. (F) Durability test of Cr(VI) reduction over 0.8Pd0.2Ni(OH)2@S-1-H catalyst. Catalytic condition: 28 mg catalyst, 40 mL K2Cr2O7 (0.1 M), 200 mL HCOOH (6 M), 260 mL H2O, T = 323 K. | ||
The molar ratio of n(HCOOH)/n(K2Cr2O7) can affect the catalytic activity of Cr(VI) reduction. The reaction rate of Cr(VI) reduction doubled with the increase of n(HCOOH)/n(K2Cr2O7) ratio from 600/1 to 4800/1 (1.19 vs. 2.48 min−1) (Fig. 5C). The catalytic activity of Cr(VI) reduction over 0.8Pd0.2Ni(OH)2@S-1-H is dependent on the reaction temperature, and the reduction rate of Cr(VI) enhances with the increase of reaction temperatures, affording apparent activation energy of 75.5 kJ mol−1 (Fig. 5D and S25†). Moreover, the catalytic activity of 0.8Pd0.2Ni(OH)2@S-1-H remains unchanged after five consecutive cycles, reflecting its excellent recycling stability during the Cr(VI) reductions (Fig. 5E and S26†). The Pd atoms in the spent 0.8Pd0.2Ni(OH)2@S-1-H catalyst are uniformly dispersed throughout the S-1 crystals, which is much similar to that in a fresh one, indicating the high stability of 0.8Pd0.2Ni(OH)2@S-1-H during Cr(VI) reduction reactions (Fig. S27†). Significantly, the 0.8Pd0.2Ni(OH)2@S-1-H also possesses superior durability for the cascade reduction of Cr(VI) species, 4 mmol K2Cr2O7 can be completely converted to Cr3+ within 24 h over only 28 mg of 0.8Pd0.2Ni(OH)2@S-1-H catalyst, affording a high turnover number of 2980 mol(Cr2O72−) mol(Pd)−1 at 323 K (Fig. 5F), which represents the best stability among all of the previously-reported heterogeneous catalysts (Table S4†).50,51
Conclusions
In summary, we prepared a series of zeolite-encaged Pd-based pseudo-single atoms via a facile and energy-saving ligand-protected direct H2 reduction method. In comparison with the conventional calcination-reduction treatment, the direct H2 reduction process is more energy-saving and able to further reduce the size and improve the dispersity of Pd-based metal species within zeolite micropores. Thanks to the extremely high metal dispersion and confinement effect of zeolites, the Pd@S-1-H exhibited superior activity and durability in methane combustion with a complete combustion temperature of 390 °C that is much lower than Pd/S-1-im (513 °C). By virtue of the synergy between the interface of Pd and Ni(OH)2 species, the 0.8Pd0.2Ni(OH)2@S-1-H catalyst exhibited a superhigh H2 generation rate from FA decomposition, affording a TOF value up to 9308 h−1 at 333 K, which is 33 times higher than that of Pd/S-1-im. Significantly, the zeolite-encaged metal catalysts are for the first time used for the Cr(VI) reduction coupling with FA dehydrogenation reaction, and the 0.8Pd0.2Ni(OH)2@S-1-H gave a record turnover number of 2980 mol(Cr2O72−) mol(Pd)−1 at 323 K. This work provides references for the preparation of high-performance zeolite-encaged atomically dispersed metal catalysts. The superhigh metal utilization, superior catalytic activity and excellent stability of such catalyst suggest important prospects for practical applications in the field of efficient hydrogen storage and pollutant disposal in the future.Author contributions
Q. S., J. Y. and N. W. conceived the idea, designed the experiments, and supervised this work. K. Z., N. W., Y. M., T. Z., P. Y., and Q. S. performed experiments and analysed the data. Q. S. wrote the manuscript, Q. S. and J. Y. revised the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (Grant No. 2022YFA1506000), the National Natural Science Foundation of China (No. 22109110, 22105110, 22288101, 21835002), the Natural Science Foundation of Jiangsu Province (No. BK20210698), the Jiangsu Distinguished Professor Program, and Gusu Talent Innovation and Entrepreneurship Foundation (ZXL2022497).Notes and references
- L.-H. Chen, M.-H. Sun, Z. Wang, W. Yang, Z. Xie and B.-L. Su, Chem. Rev., 2020, 120, 11194–11294 CrossRef CAS PubMed.
- M. Dusselier and M. E. Davis, Chem. Rev., 2018, 118, 5265–5329 CrossRef CAS PubMed.
- Y. Li and J. Yu, Nat. Rev. Mater., 2021, 6, 1156–1174 CrossRef CAS.
- B. M. Weckhuysen and J. Yu, Chem. Soc. Rev., 2015, 44, 7022–7024 RSC.
- Y. Chai, W. Dai, G. Wu, N. Guan and L. Li, Acc. Chem. Res., 2021, 54, 2894–2904 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- L. Liu and A. Corma, Nat. Rev. Mater., 2021, 6, 244–263 CrossRef CAS.
- Z. Qu and Q. Sun, Inorg. Chem. Front., 2022, 9, 3095–3115 RSC.
- Q. Sun, N. Wang and J. Yu, Adv. Mater., 2021, 33, 2104442 CrossRef CAS PubMed.
- H. Wang, L. Wang and F.-S. Xiao, ACS Cent. Sci., 2020, 6, 1685–1697 CrossRef CAS PubMed.
- S.-M. Wu, X.-Y. Yang and C. Janiak, Angew. Chem., Int. Ed., 2019, 58, 12340–12354 CrossRef CAS PubMed.
- D. Farrusseng and A. Tuel, New J. Chem., 2016, 40, 3933–3949 RSC.
- N. Wang, Q. Sun and J. Yu, Adv. Mater., 2019, 31, 1803966 CrossRef PubMed.
- Y. Chai, G. Wu, X. Liu, Y. Ren, W. Dai, C. Wang, Z. Xie, N. Guan and L. Li, J. Am. Chem. Soc., 2019, 141, 9920–9927 CrossRef CAS PubMed.
- S. Goel, S. I. Zones and E. Iglesia, J. Am. Chem. Soc., 2014, 136, 15280–15290 CrossRef CAS.
- L. Liu, M. Lopez-Haro, C. W. Lopes, C. Li, P. Concepcion, L. Simonelli, J. J. Calvino and A. Corma, Nat. Mater., 2020, 3, 628–638 CAS.
- Y. Liu, Z. Li, Q. Yu, Y. Chen, Z. Chai, G. Zhao, S. Liu, W.-C. Cheong, Y. Pan, Q. Zhang, L. Gu, L. Zheng, Y. Wang, Y. Lu, D. Wang, C. Chen, Q. Peng, Y. Liu, L. Liu, J. Chen and Y. Li, J. Am. Chem. Soc., 2019, 141, 9305–9311 CrossRef CAS.
- Y. Ma, S. Song, C. Liu, L. Liu, L. Zhang, Y. Zhao, X. Wang, H. Xu, Y. Guan, J. Jiang, W. Song, Y. Han, J. Zhang and P. Wu, Nat. Catal., 2023, 6, 506–518 CrossRef CAS.
- Q. Sun, N. Wang, Q. Bing, R. Si, J. Liu, R. Bai, P. Zhang, M. Jia and J. Yu, Chem, 2017, 3, 477–493 CAS.
- Q. Sun, N. Wang, Q. Fan, L. Zeng, A. Mayoral, S. Miao, R. Yang, Z. Jiang, W. Zhou, J. Zhang, T. Zhang, J. Xu, P. Zhang, J. Cheng, D.-C. Yang, R. Jia, L. Li, Q. Zhang, Y. Wang, O. Terasaki and J. Yu, Angew. Chem., Int. Ed., 2020, 59, 19450–19459 CrossRef CAS PubMed.
- C. Wang, E. Guan, L. Wang, X. Chu, Z. Wu, J. Zhang, Z. Yang, Y. Jiang, L. Zhang, X. Meng, B. C. Gates and F.-S. Xiao, J. Am. Chem. Soc., 2019, 141, 8482–8488 CrossRef CAS.
- J. Zhang, L. Wang, B. Zhang, H. Zhao, U. Kolb, Y. Zhu, L. Liu, Y. Han, G. Wang, C. Wang, D. S. Su, B. C. Gates and F.-S. Xiao, Nat. Catal., 2018, 1, 540–546 CrossRef CAS.
- M. Choi, Z. Wu and E. Iglesia, J. Am. Chem. Soc., 2010, 132, 9129–9137 CrossRef CAS.
- S. Goel, Z. J. Wu, S. I. Zones and E. Iglesia, J. Am. Chem. Soc., 2012, 134, 17688–17695 CrossRef CAS.
- L. Liu, U. Diaz, R. Arenal, G. Agostini, P. Concepcion and A. Corma, Nat. Mater., 2017, 16, 132–138 CrossRef CAS PubMed.
- N. Wang, Q. Sun, R. Bai, X. Li, G. Guo and J. Yu, J. Am. Chem. Soc., 2016, 138, 7484–7487 CrossRef CAS.
- Q. Sun, B. W. J. Chen, N. Wang, Q. He, A. Chang, C.-M. Yang, H. Asakura, T. Tanaka, M. J. Hülsey, C.-H. Wang, J. Yu and N. Yan, Angew. Chem., Int. Ed., 2020, 59, 20183–20191 CrossRef CAS PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- Q. Sun, N. Wang, T. Zhang, R. Bai, A. Mayoral, P. Zhang, Q. Zhang, O. Terasaki and J. Yu, Angew. Chem., Int. Ed., 2019, 58, 18570–18576 CrossRef CAS PubMed.
- X. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- Z. Hou, L. Dai, J. Deng, G. Zhao, L. Jing, Y. Wang, X. Yu, R. Gao, X. Tian, H. Dai, D. Wang and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202201655 CrossRef CAS PubMed.
- T. Li, Y. Yao, Z. Huang, P. Xie, Z. Liu, M. Yang, J. Gao, K. Zeng, A. H. Brozena, G. Pastel, M. Jiao, Q. Dong, J. Dai, S. Li, H. Zong, M. Chi, J. Luo, Y. Mo, G. Wang, C. Wang, R. Shahbazian-Yassar and L. Hu, Nat. Catal., 2021, 4, 62–70 CrossRef CAS.
- A. W. Petrov, D. Ferri, F. Krumeich, M. Nachtegaal, J. A. van Bokhoven and O. Kröcher, Nat. Commun., 2018, 9, 2545 CrossRef.
- W. Wang, W. Zhou, W. Li, X. Xiong, Y. Wang, K. Cheng, J. Kang, Q. Zhang and Y. Wang, Appl. Catal., B, 2020, 276, 119142 CrossRef CAS.
- S. Guan, Y. Liu, H. Zhang, R. Shen, H. Wen, N. Kang, J. Zhou, B. Liu, Y. Fan, J. Jiang and B. Li, Adv. Sci., 2023, 10, 2300726 CrossRef CAS.
- C. Lang, Y. Jia and X. Yao, Energy Storage Mater., 2020, 26, 290–312 CrossRef.
- M. Navlani-García, K. Mori, Y. Kuwahara and H. Yamashita, NPG Asia Mater., 2018, 10, 277–292 CrossRef.
- Q. Sun, N. Wang, Q. Xu and J. Yu, Adv. Mater., 2020, 32, 2001818 CrossRef CAS.
- J. Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- X. Gu, Z.-H. Lu, H.-L. Jiang, T. Akita and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11822–11825 CrossRef CAS.
- Z. Li and Q. Xu, Acc. Chem. Res., 2017, 50, 1449–1458 CrossRef CAS.
- A. Zhang, J. Xia, Q. Yao and Z.-H. Lu, Appl. Catal., B, 2022, 309, 121278 CrossRef CAS.
- Z. H. Farooqi, M. W. Akram, R. Begum, W. Wu and A. Irfan, J. Hazard. Mater., 2021, 402, 123535 CrossRef CAS PubMed.
- P. Veerakumar and K.-C. Lin, Chemosphere, 2020, 253, 126750 CrossRef CAS PubMed.
- T. Iida, D. Zanchet, K. Ohara, T. Wakihara and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2018, 57, 6454–6458 CrossRef CAS PubMed.
- S.-J. Li, Y.-T. Zhou, X. Kang, D.-X. Liu, L. Gu, Q.-H. Zhang, J.-M. Yan and Q. Jiang, Adv. Mater., 2019, 31, 1806781 CrossRef.
- F.-Z. Song, Q.-L. Zhu, N. Tsumori and Q. Xu, ACS Catal., 2015, 5, 5141–5144 CrossRef CAS.
- S. Dai, X. Wu, J. Zhang, Y. Fu and W. Li, Chem. Eng. J., 2018, 351, 959–966 CrossRef CAS.
- L. Tan, T. Ray Jones, J. Poitras, J. Xie, X. Liu and G. Southam, J. Hazard. Mater., 2020, 398, 122945 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05851d |
| This journal is © The Royal Society of Chemistry 2024 |