
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A library of vinyl phosphonate anions dimerize with cyanostars, form supramolecular polymers and undergo statistical sorting†
Yusheng
Chen
 a,
Anastasia
Kuvayskaya
b,
Maren
Pink
a,
Alan
Sellinger
bc and
Amar H.
Flood
*a
a,
Anastasia
Kuvayskaya
b,
Maren
Pink
a,
Alan
Sellinger
bc and
Amar H.
Flood
*a
aDepartment of Chemistry, Indiana University, 800 E. Kirkwood Avenue, Bloomington, Indiana 47405, USA. E-mail: aflood@indiana.edu
bDepartment of Chemistry, Colorado School of Mines, 1012 14th Street, Golden, Colorado 80401, USA
cNational Renewable Energy Laboratory (NREL), 15013 Denver West Parkway, Golden, Colorado 80401, USA
First published on 29th November 2023
Abstract
Supramolecular dimers are elementary units allowing the build-up of multi-molecule architectures. New among these are cyanostar-stabilized dimers of phosphate and phosphonate anions. While the anion dimerization at the heart of these assemblies is reliable, the covalent synthesis leading to this class of designer anions serves as a bottleneck in the pathway to supramolecular assemblies. Herein, we demonstrate the reliable synthesis of 14 diverse anionic monomers by Heck coupling between vinyl phosphonic acid and aryl bromide compounds. When this synthesis is combined with reliable anion dimerization, we show formation of supramolecular dimers and polymers by co-assembly with cyanostar macrocycles. The removal of the covalent bottleneck opened up a seamless synthetic route to iterate through three monomers affording the solubility needed to characterize the mechanism of supramolecular polymerization. We also test the idea that the small size of these vinyl phosphonates provide identical dimer stabilities across the library by showing how mixtures of anions undergo statistical (social) self-sorting. We exploit this property by preparing soluble copolymers from the mixing of different monomers. This multi-anion assembly shows the utility of a library for programming properties.
Introduction
Supramolecular dimers consist of two molecules linked by non-covalent interactions.1,2 Well-established dimers are characterized by reliable non-covalent linkages that offer high stability and predictable stoichiometries.3–13 Examples are π systems linked by hydrogen bonding arrays,14–16 terpyridine ligands driven by metal-coordination17,18 and π-stacked dimers encapsulated by cucurbit[n]uril hosts.19,20 The discovery of reliable linkage chemistries led to the use of dimers in various applications21–33 and to construct sophisticated assemblies.14,20,34–40 The versatility of these dimers can be amplified when they are covalently modified to create a variety of molecular building blocks.3,5,20,41 For example,35 a variety of architectures were assembled from a series of guest molecules prepared using the same synthetic route. In principle, each building block will have the same dimerization propensity, which allows properties to be defined by the substituent. Despite this promise and the possibility of generating a library of building blocks by covalent modification, there are very few4,42 collections with more than 10 members. The recent discovery of cyanostar-stabilized anion dimers that form with 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometries (Fig. 1)43–47 is a case in point where the synthesis of anion-based building blocks is a bottleneck. To remove this bottleneck, we leverage a vinyl-substituted phosphonic acid that can be readily substituted with aryl groups using Heck coupling for the creation of a 14-member library, with each forming supramolecular links as either dimers or polymers. We use the expedient synthesis and statistical self-sorting of the anions as complementary methods to tune polymer solubility and to showcase the utility of the library.
:2 stoichiometries (Fig. 1)43–47 is a case in point where the synthesis of anion-based building blocks is a bottleneck. To remove this bottleneck, we leverage a vinyl-substituted phosphonic acid that can be readily substituted with aryl groups using Heck coupling for the creation of a 14-member library, with each forming supramolecular links as either dimers or polymers. We use the expedient synthesis and statistical self-sorting of the anions as complementary methods to tune polymer solubility and to showcase the utility of the library.
 | ||
| Fig. 1 Modular synthesis of vinyl phosphonates to form a library of anions provide reliable and interchangeable assembly with cyanostar macrocycles into supramolecular dimers and polymers. | ||
Receptor-stabilized anion–anion dimers are connected by anti-electrostatic hydrogen bonds (AEHB)46–50 that form inside receptors, e.g., cyanostar macrocycles,43,44 Caballero's halogen-bonding receptors51 and others.49,52–56 These dimers have been used to construct supramolecular polymers45,51 leading to tunable, thermally-reversible adhesives.57 A key challenge, however, is preparation of the molecular building blocks. The synthesis and purification of organic hydroxyanions bearing hydrophilic headgroups (R-HPO4−, R-HPO3−) is sometimes unpredictable.57–60 As an example, only two organophosphates57 were synthesized for exploring their dimerization with eight of the other anions43–45,61 coming from commercial sources. These modest numbers suggest that these hydroxyanions fall short of the mark needed for conferring versatile access to subunit variation. Recently, Baran developed a phosphorylation method to access phosphoric acids.60 However, the reagent has limited substrate scope (aliphatic alcohols) and a cost ($500 per g) that inhibits prototyping of self-assembled materials. Ideally, these building blocks should be accessible in one or two efficient synthetic steps from a diversity of commercial building blocks.
Another opportunity that stems from generation of a library in which each member shares the same binding site is statistical self-sorting between members.62–64 This behavior is usually explored by mixing equimolar amounts of different building blocks with the host and evaluating the distribution of homodimers and inter-mixed heterodimers. The monomers can come together exclusively (integrative), segregate exclusively (narcissistic), show purely statistical outcomes (social) or variations thereof. Sorting has been investigated with dimers65–67 and used to generate alternating copolymers68–70 and diblock copolymers.67,71 The extent of sorting with anion–anion dimers, however, has not been reported to our knowledge.
Herein, we report the synthesis of 14 aromatic-based vinyl phosphonic acids using a mild Heck coupling approach (Fig. 1) and their subsequent quantitative deprotonation and dimerization with cyanostar, which exceeds the handful examined in the prior six publications.43–45,57,61 They were each shown to dimerize as high-fidelity complexes stabilized by a pair of cyanostars with 2:2 linkage stoichiometry at the equivalence point as observed using X-ray structures, 1H NMR spectroscopy and mass spectrometry. As a demonstration of the facile modularity, we iterated on a series of ditopic monomers using covalent synthesis to overcome solubility limits in a supramolecular homopolymer. We also tested the idea that the small size of the vinyl phosphonate would allow anion–anion linkages to have identical intermolecular affinities. We note, however, that small anions often lead to mixtures of macrocycles43,53,61 with a deterioration in the reliability of the 2:2 stoichiometry. Our results show, fortuitously, the ideal outcome of identical affinities. Thus, we leverage this property to convert an insoluble homopolymer into soluble forms of random copolymers using the statistical self-sorting between ditopic members in the library.
Results and discussions
Synthesis of vinyl phosphonate library
All the anions were prepared from the corresponding vinyl phosphonic acids. The acids were synthesized by Heck coupling between vinyl phosphonic acid and various aryl bromides (Scheme 1).72,73 This modular synthetic approach produces target compounds in good to excellent yields and on gram scales from a variety of aromatic halides, including halogenated chromophores and photoswitches (Scheme 1). All the synthesized compounds were isolated predominantly as (Z)-isomers, a trace amount of the (E)-isomer (≈34:1 E/Z ratio) was detectable in the crude mixture of 2. The acids were converted to the phosphonates by addition of stoichiometric amounts of tetrabutylammonium hydroxide (TBAOH) to yield the tetrabutylammonium (TBA+) salts.45,57 The salts were dried under vacuum before complexation with cyanostar macrocycle.
 | ||
| Scheme 1 Summary of vinyl phosphonic acid synthesis using Heck coupling. | ||
Dimerization of fluorene vinyl phosphonate with cyanostar macrocycles
The formation of high-fidelity receptor:anion complexes based on anti-electrostatic hydrogen bonding was examined using dimethylfluorene-vinyl phosphonate 9 (Fig. 2b). Addition of this vinyl phosphonate to a cyanostar solution (1 mM, dichloromethane) confirmed formation of the 2:2 cyanostar:phosphonate complex at the equivalence point (1.0 equiv.). The appearance of the 14.78 ppm signal confirms43,45 formation of self-complementary hydrogen bonds between phosphonate head groups inside the cyanostars (Fig. 2a). We also observe the typical 8-line NMR pattern of the cyanostar dimer in 2:2 complexes.43 The cyanostar macrocycle has a bowl shape and forms a racemic mixture of M and P chiral forms. Dimerization of the macrocycle in anion complexes slows the exchange between M and P chiral forms with both meso (MP) and chiral (MM/PP) combinations observed as diastereomeric proton signals in a 57:43% ratio (e.g., Hc:Hc′)74 while retaining the racemic mixture. We also observe these two diastereomers reflected in the 31P-NMR signals in a 55:45% ratio (Fig. 2c). The similar relative peak intensities support the encapsulation of the two phosphonates inside the pair of cyanostars.
 | ||
| Fig. 2 (a) Titration of dimethyl fluorene vinyl phosphonate 9 into cyanostar (1 mM, CD2Cl2, 298 K, 600 MHz). (b) Structure of tetrabutylammonium salt of dimethyl fluorene vinyl phosphonate. (c) 31P NMR spectrum at equivalence point (1 mM, CD2Cl2, 298 K, 202 MHz). | ||
The 2:2 cyanostar:phosphonate complex was unambiguously confirmed using single-crystal structure determination (Fig. 3). The single crystal was obtained by vapor diffusion of n-hexane in a dichloromethane solution. The anion dimer is located inside the pair of π-stacked cyanostars with two tetrabutylammonium cations situated close to the anionic center. The two α-protons of TBA+ are hydrogen bonded (3.16 and 3.36 Å) with the external oxygen atom (Fig. 3a). The geometry of the phosphonate dimer shows a pair of short OH⋯O hydrogen bonds. The distance between donor and acceptor oxygens (2.46 Å), is consistent with the classification of very strong hydrogen bonding75 and in the range that is typical of hydrogen bonds between anionic dimers (Fig. 3b).48,76 The long and short bond lengths between phosphorous and oxygen atoms can be used to assign single and double bonds (Fig. 3b). Thus, the negatively charged oxygen atom is the hydrogen bond acceptor despite the greater Coulomb repulsion produced from two negatively charged centers so close together. We note that the cyanostar displays the whole-molecular disorder typical of their crystal packing preferences.74 We observed a 60:40 ratio between M and P isomers, which is consistent with the ratio of diastereomers seen in 1H NMR, i.e., meso:chiral = 57:43%. We also observe two positions of the fluorene moiety with a 75:25% occupancy possibility, differing modestly by 11° in their tilt angle.
 | ||
| Fig. 3 (a) Crystal structure of 2:2 cyanostar:phosphonate complex with 9 with the presence of two tetrabutylammonium cations. (b) The pair of self-complementary hydrogen bonds are observed at a O⋯O distance of 2.46 Å. Data collected at 150 K. Non-polar hydrogen atoms and disorder of cyanostar and phosphonate omitted for clarity (CCDC 2263985). | ||
Testing the sterics limits of assembly with cyanostar using vinyl phosphonate
Less bulky substituents on phosphates or phosphonates are known to produce a mixture of 2:2 stoichiometric and other 3:2 cyanostar:anion complexes that lower the reliability of dimerization.43 Therefore, to evaluate the limits of reliability, we tested the smallest analogue in the series, vinyl phosphonate (VP). When 1.0 equivalent of this anion was added to cyanostar, a clear signature for the diastereomeric cyanostar dimer and the 14 ppm hydrogen bonding shows formation of the 2:2 cyanostar:anion complex (Fig. 4a) consistent with the larger fluorene vinyl phosphonate (9). The hydrogen bonding signal at ∼14.5 ppm has low intensity. This phenomenon was also observed in the 2:2 cyanostar:bisulfate dimer in which the anion is small.44
 | ||
| Fig. 4 (a) Titration of vinyl phosphonate (VP) into cyanostar (1 mM, CD2Cl2, 298 K, 600 MHz), the downfield region was subjected to exponential apodization to make hydrogen bond signals easier to observe. (b) 31P NMR spectrum at equivalence point (1 mM, CD2Cl2, 298 K, 202 MHz). (c) High-resolution mass spectrum of cyanostar:vinyl phosphonate complex, ratio of cyanostar and vinyl phosphonate is 1:1. | ||
All evidence supports formation of the target complex with 2:2 stoichiometry. Consistently, the diastereomeric phosphorous signals for the 2:2 complex can be observed by 31P NMR spectroscopy (Fig. 4b) and the parent ion is observed in the high-resolution mass spectrum at 2288.3193 m/z (Fig. 4c). Furthermore, addition of excess anion does not change the 8-line pattern seen in the aromatic region of the 1H-NMR spectra, indicating the 2:2 complex is the exclusive cyanostar-stabilized assembly beyond 1.0 equivalent.
Prior to the equivalence point, we observe a high degree of exchange broadening. The broadened NMR spectra could result from exchange between the 2:2 complex and either 3:2 species43,61 or the uncomplexed macrocycle. The absence of any peaks around 6.9 and 7.2 ppm (ref. 61) as well as the absence of a downfield shifted H-bond signature at around 13 ppm (ref. 43) exclude formation of the higher order 3:2 species (Fig. 4a). Therefore, we attribute the broadening of aromatic signals from 0.2 to 0.6 equivalents to exchange between complexed and uncomplexed macrocycle that occurs at rates close to the NMR time scale. This faster rate of exchange, relative to the larger fluorene analog (Fig. 2), likely results from the low steric profile of the smaller vinyl group.
Solvent dependence of cyanostar–vinyl phosphonate dimers
Solvent polarity is known61 to affect the stoichiometry of complexation between cyanostar and anion. In the more polar acetonitrile solvent, the solvophobic effect favors formation of a triple-stack of macrocycle to form the 3:2 macrocycle:anion complex.61 To test if this behavior also emerges with vinyl phosphonates, the titration was performed in a polar solvent mixture selected to ensure solubility of all components (2:1 v/v dichloromethane: acetonitrile).43,61 Instead of the vinyl phosphonate anion, which displays broad proton signals, phenyl vinyl phosphonate (1) was selected. This anion shows distinct signals and maintains a high-fidelity 2:2 signature in pure dichloromethane. Consistent with previous reports,43,61 the titration shows signatures for both 3:2 and 2:2 complexes that maximize at their respective equivalence points, i.e., 0.67 and >1.0 equivalents (Fig. S85†). These results show solvent polarity plays the same role in directing assembly outcome of vinyl phosphonate systems as with other cyanostar-stabilized anion dimers.
Demonstrating the versatility of vinyl phosphonates as cyanostar-stabilized dimers
The reliable formation of 2:2 cyanostar:anion complexes motivated us to examine if the versatility of the vinyl phosphonates also support their use as a general platform for supramolecular dimerization. A series of monosubstituted aryl vinyl phosphonates were prepared using a variety of π systems, e.g., benzene (1), 4-trifluoromethyl benzene (2), naphthalene (3), anthracene (4), pyrene (5), azobenzene (6), stilbene (7) and carbazole (8).
Assembly of 4-trifluoromethylphenyl vinyl phosphonate (2), with cyanostar is exemplary of dimerization. Upon addition of 1 equivalent of the phosphonate to the cyanostar solution, we observed the characteristic aromatic and hydrogen bonding pattern of the 2:2 dimer (Fig. 5, top). The proton assignments for the complexed dimers were based on their splitting pattern and coupling constants. Interestingly, these vinyl phosphonate signals are also subject to the influence of the meso and chiral cyanostar dimers with intensity ratios for the diastereomers that match cyanostar. Consistent with the complexed architecture, ROESY NMR experiments show correlations between macrocycle protons assigned to the meso dimer (e.g., Hd′ at 8.62 ppm) and the meso form of the aryl-vinyl-phosphonate (e.g., Hf′ at 6.68 ppm) (Fig. S88†). Similar correlations were observed between the macrocycle and phosphonate for the chiral isomers. These correlations confirm that the splitting observed in the aromatic signals for the aryl-vinyl-phosphonate anion stems from meso and chiral forms of the cyanostar dimers.
 | ||
| Fig. 5 Stack of 1H NMR spectra of 2:2 dimers of cyanostar with 1 equivalent of the aryl vinyl phosphonate anions (1 mM, CD2Cl2, 298 K, 600 MHz). Peaks from cyanostar are shown in blue, peaks from phosphonates are shown in black, and the hydrogen bonding signal is colored red. The downfield region (∼14 ppm) was subjected to exponential apodization (3 Hz) to improve the signal-to-noise ratio of the hydrogen-bond region. The remainder of the spectrum apodization was set to 0.3 Hz. All counter-cations are tetrabutylammonium. High-resolution electrospray ionization mass spectra of 2:2 dimers of cyanostar with aryl vinyl phosphonate anions (1 mM, CD2Cl2) at the 1:1 equivalence point. Samples for high resolution ESI mass analysis were directly infused with a constant spray voltage of 2.7 kV. | ||
All other aryl vinyl phosphonates showed identical signatures with high-fidelity formation of 2:2 cyanostar:anion complexes at the equivalence point (Fig. 5). Diastereomeric peaks for the cyanostar dimers of these complexes are located in the same aromatic region (7.3–9.1 ppm). The assembly of anthracene vinyl phosphonate with cyanostar also shows visible splitting of proton Ha into its diastereomers. This enhanced splitting is carried over to splitting in the resonance for the hydrogen bond between each phosphonate dimer. All others show a single broaden peak and are located in a range 14–15.5 ppm.
The high-resolution electrospray ionization (ESI) mass spectra (Fig. 5) are all consistent with 2:2 dimer formation. The parent ions of these complexes were all observed in the range from 2400 to 2900 m/z as a consequence of different substituents. In the case of a few, we also observe multimers that are attributed to aggregation in the electrospray experiment.
We also obtained the crystal structure for the phenyl vinyl phosphonate (1) dimer in both its solvated and fully desolvated state (Fig. S119,† CCDC 2263986 and 2263987), showing a crystal-to-crystal transition of the 2:2 dimer upon desolvation.77
Pure statistical sorting across the modular anion library
We designed the library to introduce various functional groups from the aryl substituents while leaving the linkage chemistry to be determined solely by the vinyl phosphonate without influence from sterics or electronics. The sterics are the same across the library and the calculated ESP values do not differ much suggesting support for this hypothesis (Fig. S134†). To test this idea, we studied the sorting behavior of the anions.62We selected the electron-poor phosphonate (2) with an electron withdrawing group and the electron-rich carbazole phosphonate (8) to test sorting. We expect to see the 2·8 heterodimer (Fig. 6a) together with the homodimers (2·2 and 8·8). When the two vinyl phosphonates are mixed together, we observe a new set of peaks between the parents (Fig. 6b) assigned to the heterodimer 2·8, which was verified by an ESI-MS peak at 2710.5076 m/z (Fig. 6c). The integration ratio of the protons for 2·2, 2·8 and 8·8 is in the 1:2:1 ratio expected of statistical self-sorting (Fig. S125†).62–64 A competition titration supports this interpretation (Fig. S126†) indicating the affinities of anions 2 and 8 are the same. These results confirm our idea that binding of vinyl phosphonates to cyanostar do not depend significantly on their substituents.
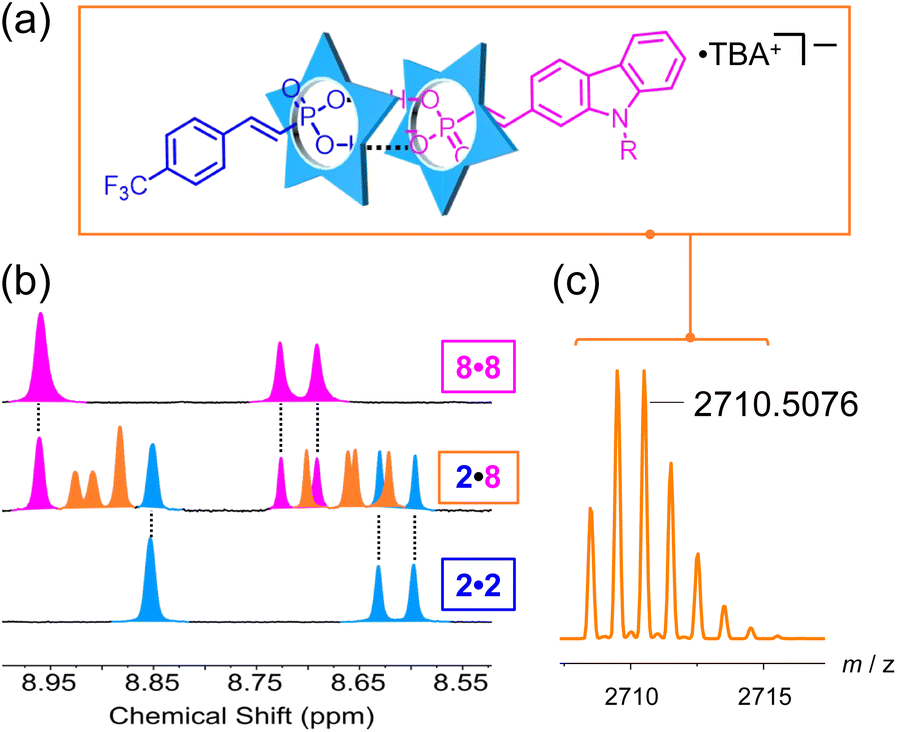 | ||
| Fig. 6 (a) Heterodimer structure containing 2 (blue) and 8 (magenta). (b) 1H NMR spectra of homodimer 2·2 (blue), heterodimer 2·8 (orange) and 8·8 (magenta). (c) High-resolution mass spectrum of the parent heterodimer ion (1 mM, 298 K, 600 MHz, CD2Cl2). | ||
To connect the vinyl phosphonates with different hydroxyanions, we also performed sorting and competition studies between vinyl phosphonate 8 and naphthyl phosphate (P). Interestingly, we also observed statistical self-sorting where the heterodimer was confirmed by 1H NMR and ESI-MS (Fig. S128 and S133†). However, a competition study shows that the 1-naphthyl phosphate binds to cyanostar stronger than the vinyl phosphate (Fig. S129 and S130†).
We leveraged this stronger binding guest to drive depolymerization of a supramolecular polymer formed from ditopic vinyl phosphonate (see below). In this case, the depolymerization was produced by adding 1.0 equiv. of 1-naphthyl phosphate (Fig. S131†).
Polymerization of the ditopic vinyl phosphonate with cyanostar macrocycle
The modular synthesis and reliable dimerization of aryl vinyl phosphonates provide an opportunity to consider formation of supramolecular polymers. For this purpose, dibromofluorene-based building blocks were subjected to Heck coupling to yield the di-vinyl phosphonic acid analogues. The diacid was ionized with TBAOH to obtain the dianion as a divergent ditopic monomer, then mixed with cyanostar macrocycle in a 1:2 ratio to match the ideal stoichiometry of polymers (Fig. 7a). The first ditopic monomer we prepared was dimethyl fluorene bis(vinyl phosphonate) (11). When cyanostar is added to drive anion dimerization, the solubility of the resulting polymer is quite low with irreversible precipitation appearing at about 1 mM, which was also observed in a prior polymer driven by anion dimerization45 and in other supramolecular polymers.37,78 By taking advantage of the modular synthesis, we prepared the fluorene monomer (10) with two hexyl chains in the 9-position but observed precipitation at 2 mM. Subsequently, we prepared a ditopic monomer based on a substituted carbazole bearing one 2-ethylhexyl side chain (13) but again precipitation occurred at 1 mM. Ultimately, use of a fluorene monomer with branched alkyl chains (12), i.e., 2-ethylhexyl, significantly improved polymer solubility. As a consequence, variable concentration studies were performed without precipitation to help determine the likely mechanism of polymerization.
 | ||
| Fig. 7 (a) Scheme of fluorene-based monomer synthesis and supramolecular polymerization. (b) Characterization of polymerization using double logarithmic plots of specific viscosity versus cyanostar concentration with 0.5 equiv. of the diphosphonate 12 (room temperature, CH2Cl2) and the normalized species size in 2 and 10 mM solution based on diffusion NMR. (c) Stack of 1H NMR spectra of variable concentrations complex solution with 2:1 stoichiometry of cyanostar:phosphonate (CD2Cl2, 298 K, 600 MHz). | ||
As observed previously,45,57 a key signature of supramolecular polymerization is the observation of threshold behavior in viscosity as a function of concentration. To determine this threshold concentration, i.e., critical polymerization concentration (CPC), the specific viscosity of the polymer solutions was recorded from 0.1 to 10 mM based on the cyanostar concentration. The double-logarithmic plot of specific viscosity versus concentration (Fig. 7b) shows a linear relationship with an initial slope of 0.29, indicating noninteracting assemblies in solution.79,80 However, at a higher concentration range, the slope changes to 1.31. This stronger concentration dependence indicates the formation of polymers with increased size capable of entaglement under flow.79,80 Consistently, the diffusion coefficient derived from NMR peaks (Hb, Hb′, Hc, Hc′) assigned to the polymers decreases from 9.6 ± 0.3 to 6.2 ± 0.1 × 10−11 m2 s−1 at 2 and 10 mM, respectively. Based on the Stokes–Einstein equation for a spherical particle,45 we calculated that the normalized volume of the average solution species increases 4-fold from 2 to 10 mM (Fig. 7b). Consistent with previous reports,45 variable concentration 1H NMR spectra also provide evidence of the existence of oligomers below CPC (Fig. 7c). At 0.1 mM, multiple peaks appear in the range from 7.3–9.1 ppm, instead of typical 8-line pattern, which indicate the coexistence of oligomers and polymers. As concentration increases, the intensity of peaks assigned to polymer gradually increases concomitant with the disappearance of peaks assigned to oligomers beyond 2 mM, consistent with the CPC.
The intersection of the two trendlines determines the CPC as 1.6 mM based on cyanostar.2 This concentration lies between the previously reported critical polymerization concentration of rigid ditopic phosphate (below 1 mM)57 and flexible ditopic phosphonate (5 mM) system that is driven by the anion dimerization inside the cyanostars.45 According to the relationship between monomer rigidity and critical polymerization concentration predicted by polymer physics, the more rigid monomer gives lower polymerization concentration.81 Therefore, this moderate critical polymerization concentration is consistent with semi-rigid property of the fluorene-based monomer.
Copolymerization between vinyl phosphonate monomers
In addition to using iterative synthesis to yield soluble monomers, the sorting results inspired us to explore copolymerization by doping the high solubility monomers into the insoluble homopolymer (Fig. 8a). The dimethyl-decorated monomer (11) of poorest solubility was selected for forming a copolymer with the fluorene monomer with 2-ethylhexyl side chain (12). When equal amounts of 11 and 12 were mixed with cyanostar, a hydrogen bond signature of the heterodimer 11–12 is observed at 14.5 ppm (Fig. 8a, blue dot), which sits between the two homodimer signals at 14.25 and 14.6 ppm. When the molar percentage of 11 is increased, we observed a change in intensities consistent with statistical sorting. | ||
| Fig. 8 (a) 1H NMR spectra of homopolymers and copolymers involving 11 and 12 (5 mM cyanostar, 298 K, 600 MHz, CD2Cl2). (b) Pictures of solutions and representations of the homopolymer formed with monomer 11 and a copolymer of monomer 11 and 12. | ||
Macroscopically, precipitation of the homopolymer involving 11 occurred after 20 minutes, while no precipitation was observed in the copolymers over the same time frame. Consistent with copolymerization, the diffusion coefficient derived from NMR peaks (Hb, Hb′, Hc, Hc′) of the copolymer formed from a 50–50 mix of 11 and 12 ranges 6.2–6.5 × 10−11 m2 s−1 at 5 mM, indicating the two monomers are converted to one single polymeric assembly instead of different homopolymers. The diffusion coefficient of the homopolymer formed from 12 and the cyanostar (9.3 ± 0.2 × 10−11 m2 s−1, 5 mM) is larger than the copolymer and indicates that the average size is smaller. Based on the observation that polymerization of 11 gives insoluble precipitation, we reason that the copolymer exhibits stronger aggregation than the homopolymer of 12 alone.
Conclusion
Aryl vinyl phosphonates fulfill all the hallmarks of a versatile platform for anion–anion dimerization inside a pair of cyanostar macrocycles including modular access to 14 building blocks that form small molecules, macromolecules, and statistical self-sorting combinations. All the assemblies form 2:2 linkages in high fidelity as confirmed by characteristic 1H NMR signals, mass spectra and X-ray crystal structures. The on-demand synthesis of vinyl phosphonic acids and corresponding vinyl phosphonates allowed access to solubility-tuned ditopic monomers for preparing supramolecular polymers. The concentration-driven polymerization exhibits a critical polymerization concentration of 1.6 mM, which is consistent with prediction from polymer physics for semi-rigid monomers in our anion-driven supramolecular polymerization system. The isosteric vinyl phosphonate headgroups allow statistical self-sorting between building blocks with different aryl substituents. Copolymerization between two monomers provided the basis for tuning macromolecular solubility. The versatility of vinyl phosphonates, their modular synthesis, and their identical intermolecular affinities opens up cyanostar-stabilized anion dimers as a platform for prototyping functional supramolecular materials.
Data availability
The ESI† is available free of charge on the journal website. General methods, synthetic procedure, NMR titrations, 2D NMR spectroscopy, X-ray diffraction analyses, and ESI-MS analyses are included.Author contributions
YC and AK conceived the project under the supervision of AHF and AS; AK synthesized and characterized all phosphonic acids; YC prepared all phosphonates and conducted assembly and crystal growing experiments; MP collected X-ray diffraction data, solved and refined the crystal structures; YC, AK, AS and AHF analyzed the data; YC and AHF wrote and edited the manuscript with input from AK, AS and MP.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Science Foundation (CHE-2105848). This work was authored in part by the National Renewable Energy Laboratory (NREL), operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Synthesis of the organophosphorus acids at Colorado School of Mines was funded by the Separation Science Program, Division of Chemical Sciences, Geosciences, and Biosciences (CSGB) Division, and the Physical Behavior of Materials Program, Materials Sciences and Engineering (MSE) Division, within the Office of Basic Energy Sciences, Office of Science, DOE, through the Materials and Chemical Sciences Research on Critical Materials National Laboratory Program (LAB 20-2304). We thank Xinfeng (Frank) Gao at Indiana University Bloomington for his help with NMR spectroscopy. The 500 MHz NMR spectrometer of the Indiana University NMR facility was supported by NSF grant CHE-1920026 and the Prodigy probe was purchased in part with support from the Indiana Clinical and Translational Sciences Institute funded, in part by NIH Award TL1TR002531. Support for the acquisition of the Bruker Venture D8 diffractometer through the Major Scientific Research Equipment Fund from the President of Indiana University and the Office of the Vice President for Research is gratefully acknowledged. Y. C. acknowledge the support from Slagle Fellowship. We thank the staff of the Indiana University Mass Spectrometry Facility for recording mass spectra. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government.Notes and references
- G. Wu, F. Li, B. Tang and X. Zhang, J. Am. Chem. Soc., 2022, 144, 14962–14975 CrossRef CAS PubMed.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed.
- C. M. Leenders, G. Jansen, M. M. Frissen, R. P. Lafleur, I. K. Voets, A. R. Palmans and E. W. Meijer, Chem.–Eur. J., 2016, 22, 4608–4615 CrossRef CAS PubMed.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- M. Olesinska, G. Wu, S. Gomez-Coca, D. Anton-Garcia, I. Szabo, E. Rosta and O. A. Scherman, Chem. Sci., 2019, 10, 8806–8811 RSC.
- U. Rauwald and O. A. Scherman, Angew. Chem., Int. Ed., 2008, 47, 3950–3953 CrossRef CAS PubMed.
- G. D. Storrier and S. B. Colbran, J. Chem. Soc., Dalton Trans., 1996, 2185–2186 RSC.
- G. D. Storrier, S. B. Colbran and D. C. Craig, J. Chem. Soc., Dalton Trans., 1997, 3011–3028 RSC.
- T. Mes, S. Cantekin, D. W. Balkenende, M. M. Frissen, M. A. Gillissen, B. F. De Waal, I. K. Voets, E. Meijer and A. R. Palmans, Chem.–Eur. J., 2013, 19, 8642–8649 CrossRef CAS PubMed.
- O. Shyshov, S. V. Haridas, L. Pesce, H. Qi, A. Gardin, D. Bochicchio, U. Kaiser, G. M. Pavan and M. Von Delius, Nat. Commun., 2021, 12, 3134 CrossRef CAS PubMed.
- G. Wu, Y. J. Bae, M. Olesińska, D. Antón-García, I. Szabó, E. Rosta, M. R. Wasielewski and O. A. Scherman, Chem. Sci., 2020, 11, 812–825 RSC.
- C. Kulkarni, A. K. Mondal, T. K. Das, G. Grinbom, F. Tassinari, M. F. J. Mabesoone, E. W. Meijer and R. Naaman, Adv. Mater., 2020, 32, e1904965 CrossRef PubMed.
- S. I. S. Hendrikse, L. Su, T. P. Hogervorst, R. P. M. Lafleur, X. Lou, G. A. van der Marel, J. D. C. Codee and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 13877–13886 CrossRef CAS PubMed.
- P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601 CrossRef CAS PubMed.
- M. M. Conn and J. Rebek, Chem. Rev., 1997, 97, 1647–1668 CrossRef CAS PubMed.
- U. S. Schubert and C. Eschbaumer, Angew. Chem., Int. Ed., 2002, 41, 2892–2926 CrossRef CAS PubMed.
- S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- X. Yang, R. Wang, A. Kermagoret and D. Bardelang, Angew. Chem., Int. Ed., 2020, 59, 21280–21292 CrossRef CAS PubMed.
- X. Chen, Z. Huang, R. L. Sala, A. M. McLean, G. Wu, K. Sokolowski, K. King, J. A. McCune and O. A. Scherman, J. Am. Chem. Soc., 2022, 144, 8474–8479 CrossRef CAS PubMed.
- Z. Huang, X. Chen, S. J. K. O'Neill, G. Wu, D. J. Whitaker, J. Li, J. A. McCune and O. A. Scherman, Nat. Mater., 2022, 21, 103–109 CrossRef CAS PubMed.
- Q. Li, H. Zhang, K. Lou, Y. Yang, X. Ji, J. Zhu and J. L. Sessler, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2121746119 CrossRef CAS PubMed.
- Q. Song, Y. Gao, J. F. Xu, B. Qin, M. J. Serpe and X. Zhang, ACS Macro Lett., 2016, 5, 1084–1088 CrossRef CAS PubMed.
- J. R. Kumpfer and S. J. Rowan, J. Am. Chem. Soc., 2011, 133, 12866–12874 CrossRef CAS PubMed.
- B. J. Folmer, R. Sijbesma, R. Versteegen, J. Van der Rijt and E. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- H. Barbero and E. Masson, Chem. Sci., 2021, 12, 9962–9968 RSC.
- M. Nazimuddin, H. Barbero, R. Rabbani and E. Masson, Chem. Sci., 2022, 13, 4388–4396 RSC.
- S. J. K. O'Neill, Z. Huang, M. H. Ahmed, A. J. Boys, S. Velasco-Bosom, J. Li, R. M. Owens, J. A. McCune, G. G. Malliaras and O. A. Scherman, Adv. Mater., 2023, 35, e2207634 CrossRef PubMed.
- K. R. Wang, D. S. Guo, B. P. Jiang and Y. Liu, Chem. Commun., 2012, 48, 3644–3646 RSC.
- D. S. Guo, S. Chen, H. Qian, H. Q. Zhang and Y. Liu, Chem. Commun., 2010, 46, 2620–2622 RSC.
- M. Gaeta, D. Raciti, R. Randazzo, C. M. A. Gangemi, A. Raudino, A. D'Urso, M. E. Fragala and R. Purrello, Angew. Chem., Int. Ed., 2018, 57, 10656–10660 CrossRef CAS PubMed.
- D. S. Guo, K. Chen, H. Q. Zhang and Y. Liu, Chem.–Asian J., 2009, 4, 436–445 CrossRef CAS PubMed.
- F. Su, S. Zhang, Z. Chen, Z. Zhang, Z. Li, S. Lu, M. Zhang, F. Fang, S. Kang, C. Guo, C. Su, X. Yu, H. Wang and X. Li, J. Am. Chem. Soc., 2022, 144, 16559–16571 CrossRef CAS PubMed.
- K. D. Zhang, J. Tian, D. Hanifi, Y. Zhang, A. C. Sue, T. Y. Zhou, L. Zhang, X. Zhao, Y. Liu and Z. T. Li, J. Am. Chem. Soc., 2013, 135, 17913–17918 CrossRef CAS PubMed.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS PubMed.
- H. Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406–409 CrossRef CAS PubMed.
- H.-Q. Peng, W. Zhu, W.-J. Guo, Q. Li, S. Ma, C. Bucher, B. Liu, X. Ji, F. Huang and J. L. Sessler, Prog. Polym. Sci., 2023, 137, 101635 CrossRef CAS.
- M. Gaeta, G. Sortino, R. Randazzo, I. Pisagatti, A. Notti, M. E. Fragala, M. F. Parisi, A. D'Urso and R. Purrello, Chem.–Eur. J., 2020, 26, 3515–3518 CrossRef CAS PubMed.
- C. Capici, Y. Cohen, A. D'Urso, G. Gattuso, A. Notti, A. Pappalardo, S. Pappalardo, M. F. Parisi, R. Purrello, S. Slovak and V. Villari, Angew. Chem., Int. Ed., 2011, 50, 11956–11961 CrossRef CAS PubMed.
- K. Wu, T. K. Ronson, P. Su, Z. Chen, L. Goh, A. W. Heard, X. Li, F. Klautzsch, C. A. Schalley, M. Vinković and J. R. Nitschke, Nat. Synth., 2023, 2, 789–797 CrossRef.
- One paper with 11 members was found among 12 publications on supramolecular dimerization and polymerization. Please see ESI for details.†.
- W. Zhao, B. Qiao, C. H. Chen and A. H. Flood, Angew. Chem., Int. Ed., 2017, 56, 13083–13087 CrossRef CAS PubMed.
- E. M. Fatila, E. B. Twum, A. Sengupta, M. Pink, J. A. Karty, K. Raghavachari and A. H. Flood, Angew. Chem., Int. Ed., 2016, 55, 14057–14062 CrossRef CAS PubMed.
- W. Zhao, B. Qiao, J. Tropp, M. Pink, J. D. Azoulay and A. H. Flood, J. Am. Chem. Soc., 2019, 141, 4980–4989 CrossRef CAS PubMed.
- L. M. Azofra, J. Elguero and I. Alkorta, J. Phys. Chem. A, 2020, 124, 2207–2214 CrossRef CAS PubMed.
- L. M. Azofra, J. Elguero and I. Alkorta, Phys. Chem. Chem. Phys., 2020, 22, 11348–11353 RSC.
- W. Zhao, A. H. Flood and N. G. White, Chem. Soc. Rev., 2020, 49, 7893–7906 RSC.
- Q. He, P. Tu and J. L. Sessler, Chem, 2018, 4, 46–93 CAS.
- L. Chen, Q. Feng, C. Wang, S. Yin and Y. Mo, J. Phys. Chem. A, 2021, 125, 10428–10438 CrossRef CAS PubMed.
- F. Zapata, L. Gonzalez, A. Bastida, D. Bautista and A. Caballero, Chem. Commun., 2020, 56, 7084–7087 RSC.
- T. S. C. MacDonald, B. L. Feringa, W. S. Price, S. J. Wezenberg and J. E. Beves, J. Am. Chem. Soc., 2020, 142, 20014–20020 CrossRef CAS PubMed.
- J. R. Dobscha, S. Debnath, R. E. Fadler, E. M. Fatila, M. Pink, K. Raghavachari and A. H. Flood, Chem.–Eur. J., 2018, 24, 9841–9852 CrossRef CAS PubMed.
- P. S. Lakshminarayanan, I. Ravikumar, E. Suresh and P. Ghosh, Chem. Commun., 2007, 5214–5216 RSC.
- P. Sabater, F. Zapata, A. Bastida and A. Caballero, Org. Biomol. Chem., 2020, 18, 3858–3866 RSC.
- D. A. Cullen, M. G. Gardiner and N. G. White, Chem. Commun., 2019, 55, 12020–12023 RSC.
- W. Zhao, J. Tropp, B. Qiao, M. Pink, J. D. Azoulay and A. H. Flood, J. Am. Chem. Soc., 2020, 142, 2579–2591 CrossRef CAS PubMed.
- C. M. Sevrain, M. Berchel, H. Couthon and P. A. Jaffres, Beilstein J. Org. Chem., 2017, 13, 2186–2213 CrossRef CAS PubMed.
- C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981–8006 CrossRef CAS PubMed.
- M. Ociepa, K. W. Knouse, D. He, J. C. Vantourout, D. T. Flood, N. M. Padial, J. S. Chen, B. B. Sanchez, E. J. Sturgell, B. Zheng, S. Qiu, M. A. Schmidt, M. D. Eastgate and P. S. Baran, Org. Lett., 2021, 23, 9337–9342 CrossRef CAS PubMed.
- E. M. Fatila, M. Pink, E. B. Twum, J. A. Karty and A. H. Flood, Chem. Sci., 2018, 9, 2863–2872 RSC.
- M. M. Safont-Sempere, G. Fernandez and F. Wurthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- P. Mukhopadhyay, A. Wu and L. Isaacs, J. Org. Chem., 2004, 69, 6157–6164 CrossRef CAS PubMed.
- A. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS PubMed.
- H. Barbero, N. A. Thompson and E. Masson, J. Am. Chem. Soc., 2020, 142, 867–873 CrossRef CAS PubMed.
- K. Mahata and M. Schmittel, J. Am. Chem. Soc., 2009, 131, 16544–16554 CrossRef CAS PubMed.
- A. Sarkar, R. Sasmal, C. Empereur-Mot, D. Bochicchio, S. V. K. Kompella, K. Sharma, S. Dhiman, B. Sundaram, S. S. Agasti, G. M. Pavan and S. J. George, J. Am. Chem. Soc., 2020, 142, 7606–7617 CrossRef CAS PubMed.
- F. Wang, B. Zheng, K. Zhu, Q. Zhou, C. Zhai, S. Li, N. Li and F. Huang, Chem. Commun., 2009, 4375–4377 RSC.
- N. Tomimasu, A. Kanaya, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2009, 131, 12339–12343 CrossRef CAS PubMed.
- Z. Huang, L. Yang, Y. Liu, Z. Wang, O. A. Scherman and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 5351–5355 CrossRef CAS PubMed.
- M. Hecht, P. Leowanawat, T. Gerlach, V. Stepanenko, M. Stolte, M. Lehmann and F. Wurthner, Angew. Chem., Int. Ed., 2020, 59, 17084–17090 CrossRef CAS PubMed.
- B. W. McNichols, J. T. Koubek and A. Sellinger, Chem. Commun., 2017, 53, 12454–12456 RSC.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed.
- S. Lee, C. H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704–710 CrossRef CAS PubMed.
- G. A. Jeffrey, An introduction to hydrogen bonding, Oxford University Press, New York, 1997 Search PubMed.
- N. G. White, CrystEngComm, 2019, 21, 4855–4858 RSC.
- The desolvation typically causes bad quality or disruption of order in cyanostar–anion crystals, therefore, this crystal-to-crystal transition of a 2:2 dimer is rare and valuable for further crystal engineering of these assemblies. For more details see ESI.†.
- V. Berl, M. Schmutz, M. J. Krische, R. G. Khoury and J.-M. Lehn, Chem.–Eur. J., 2002, 8, 1227–1244 CrossRef CAS PubMed.
- F. Huang, D. S. Nagvekar, X. Zhou and H. W. Gibson, Macromolecules, 2007, 40, 3561–3567 CrossRef CAS.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- C.-C. Chen and E. E. Dormidontova, Macromolecules, 2004, 37, 3905–3917 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General methods, synthesis characterization. CCDC 2263985, 2263986 and 2263987. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03685e |
| This journal is © The Royal Society of Chemistry 2024 |