
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTime-resolved control of nanoparticle integration in titanium-organic frameworks for enhanced catalytic performance†
Carmen
Fernández-Conde
 a,
Yongkun
Zheng
b,
Marta
Mon
b,
Antonio
Ribera
c,
Antonio
Leyva-Pérez
*b and
Carlos
Martí-Gastaldo
*a
a,
Yongkun
Zheng
b,
Marta
Mon
b,
Antonio
Ribera
c,
Antonio
Leyva-Pérez
*b and
Carlos
Martí-Gastaldo
*a
aInstituto de Ciencia Molecular, Universitat de València, c/Catedrático José Beltrán, 2, 46980, Paterna, Spain. E-mail: carlos.martiuv.es
bInstituto de Tecnología Química UPV-CSIC, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Avenida de los Naranjos s/n, 46022, Valencia, Spain. E-mail: anleyva@itq.upv.es
cDepartament de Química Inorgànica, Universitat de València, c/Dr. Moliner, 50, 46100, Burjassot, Spain
First published on 14th December 2023
Abstract
Among the multiple applications of metal–organic frameworks (MOFs), their use as a porous platform for the support of metallic nanoparticles stands out for the possibility of integrating a good anchorage, that improves the stability of the catalyst, with the presence of a porous network that allows the diffusion of substrates and products. Here we introduce an alternative way to control the injection of Au nanoparticles at variable stages of nucleation of a titanium(IV) MOF crystal (MUV-10). This allows the analysis of the different modes of nanoparticle integration into the porous matrix as a function of the crystal formation stage and their correlation with the catalytic performance of the resulting composite. Our results reveal a direct effect of the stage at which the Au nanoparticles are integrated into MUV-10 crystals not only on their catalytic activity for the cyclotrimerization of propargyl esters and the hydrochlorination of alkynes, but also on the selectivity and recyclability of the final solid catalyst, which are far superior than those reported for the same reactions with TiO2 supports.
Introduction
Metal–organic frameworks (MOFs) have gained increasing attention in fields like chemistry, biology, and materials science due to their unparalleled adaptability to different goals enabled by their modular nature.1,2 These hybrid reticular solids are built by the interlinking of inorganic and organic nodes, and offer periodic structures for tailorable porosities, pore chemistry and topological diversity.3 However, MOFs can also suffer from weak chemical, thermal and mechanical stabilities, which can limit their application.4 In this regard, the preparation of composites by combination of MOFs with other materials such as organic polymers, metal nanoparticles, metal oxides, nanocarbons or silica is a versatile route either to overcome some of these limitations or endow them with additional catalytic or optical features.5 The combination, or even synergistic interaction of these components with the uniform pore distribution and periodic distribution of molecular components in the MOF, can lead to new properties not accessible to the simple sum of the individual components.Regarding metal nanoparticles (MNPs), their integration into porous platforms as zeolites, mesoporous silica or activated carbons is often used to prevent their aggregation and catalytic inactivation.6 In the case of molecular frameworks, this route can also benefit from better control of the confinement effects and tailorable interaction with the surface chemistry of the host, both useful in modifying MNP activity for superior catalytic performance.7 Compared to the “ship in a bottle” approach, which involves direct impregnation, the “bottle around ship” or template synthesis method enables the limitations imposed by the porosity metrics of the host on the size of the nanoparticle to be overcome (Fig. 1a). This route relies on the nucleation and growth of MOF crystals by reaction of their molecular components in the presence of surfactant stabilized MNPs, provided that the synthetic conditions used do not prevent the assembly of the framework. This strategy has been used to gain exquisite control over the spatial distribution of the encapsulated nanoparticles and their chemical environment in nanocrystals of MOFs such as ZIF-8,8–10 IRMOF,11,12 HKUST13 or UiO-66,14,15 between 100 and 500 nm in size.
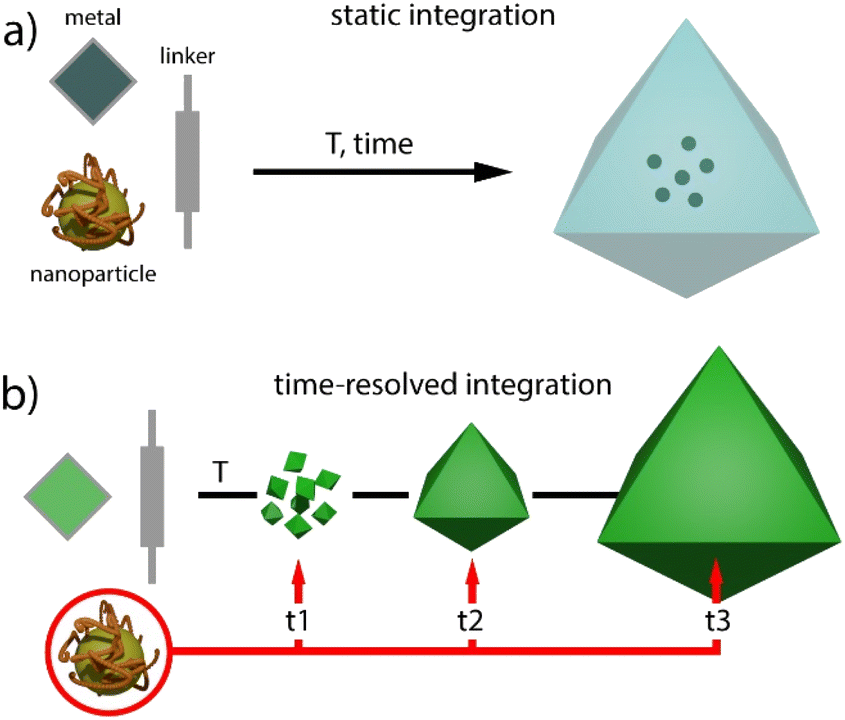 | ||
| Fig. 1 Comparison of (a) static and (b) time-resolved (this work) methods for encapsulating nanoparticles in MOF crystals. | ||
These precedents inspired us to explore alternative routes to control the integration of pre-synthesised MNPs in MOF crystals of micrometric size. To this end, we have adapted the solvothermal synthesis of titanium MOFs under static conditions as an alternative method that enables continuous injection of molecular precursors at a fixed concentration and comparatively shorter reaction times for more homogeneous crystal size distributions. This new method is well suited to gain dynamic control over the injection of MNPs at variable nucleation and crystal growth stages for varying degrees of integration in the resulting MNP@MOF hybrid crystal composites (Fig. 1b). Since one of the main applications of MNPs is catalysis,16,17 we next use the time-resolved encapsulation method to analyse the effect of the injection time on the performance of the resulting catalyst associated with changes in reactant accessibility and/or the chemical nature of the MNP surface.18 Our results reveal changes in conversion, selectivity and catalyst recyclability for the Au-catalyzed cyclotrimerization of propargyl esters19 and the hydrochlorination of alkynes,20 confirming the potential of controlling the injection of nanoparticles at adjustable times for composites with superior catalytic performance.
Results and discussion
Time-resolved synthesis of MUV-10 by continuous injection of molecular precursors
To explore this concept, we chose the titanium framework MUV-10 (MUV = Materials of the Universitat de València) due to its excellent chemical stability in acid and base, and the presence of octahedral and cuboctahedral cavities not big enough to encapsulate nanoparticles with diameters above 1 nm (Fig. 2a). Also, this heterobimetallic TiCa MOF can be isolated as single crystals of micrometric size at the gram scale and displays intrinsic photocatalytic activity.21,22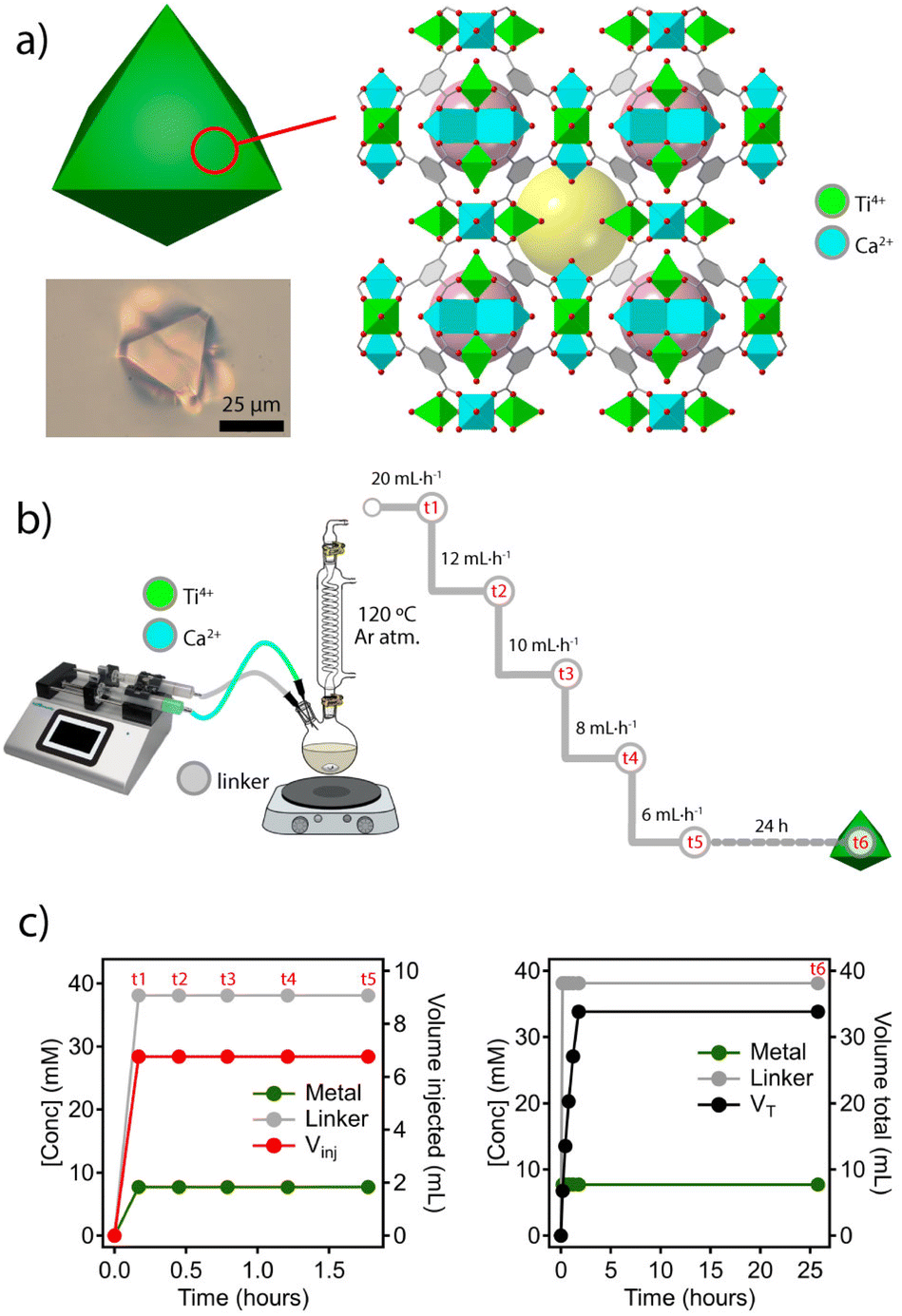 | ||
| Fig. 2 (a) Structure of MUV-10 showing the presence of octahedral (purple) and cuboctahedral (yellow) cavities. A crystal obtained by solvothermal synthesis under static conditions. (b) Experimental design used for the time-resolved synthesis of MUV-10 by continuous injection of molecular precursors. (c) Profile of variation in concentration (left) and volume (right) of the metal and linker injected over time during the first hour of the experiment and (d) variation in concentration and total volumes throughout the experiment. | ||
The solvothermal synthesis reported originally was adapted to a dual syringe infusion pump for the separate injection of stock solutions of CaCl2 and Ti(OiPr4) dissolved in a mixture of DMF and acetic acid, and the linker (H3BTC = benzene-1,3,5-tricarboxylic acid) dissolved in DMF. Both solutions were continuously injected into a round bottom flask preheated at 120 °C under an inert atmosphere and constant stirring. As shown in Fig. 2b, the initial injection feed rate of 20 mL h−1 (t1) was gradually decreased (t2–t4) down to a minimum of 6 mL h−1 (t5). From this point, the reaction was let to proceed for 24 hours without further injection of reagents (t6). This experimental design ensures a constant linker to metal ratio with negligible changes in reaction volume during the nucleation process. A similar approach has been used previously to synthesize nanoparticles of various MOFs with different chemistries for narrow particle size distributions in all cases.23 As shown in Fig. 2c, the concentration of added reagents and their ratio remain constant despite a progressive reduction of the injected volume throughout the process (t1 to t5). Although both the concentration of acetic acid acting as a modulator and the L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M ratio are the same as those used in the solvothermal synthesis reported, the concentration of the linker and metal precursors used was 1000 times higher. We argued that this supersaturation would result in increased nucleation and faster crystal growth rates at a fixed reaction temperature for better control of crystal size and morphology.24 According to our tests, a continuous injection of reagents at a low feed rate seems to facilitate the formation of crystals with much more homogeneous size than faster injections (ESI Section S2.1†). For additional information, all synthetic details are summarized in ESI S2.†
:M ratio are the same as those used in the solvothermal synthesis reported, the concentration of the linker and metal precursors used was 1000 times higher. We argued that this supersaturation would result in increased nucleation and faster crystal growth rates at a fixed reaction temperature for better control of crystal size and morphology.24 According to our tests, a continuous injection of reagents at a low feed rate seems to facilitate the formation of crystals with much more homogeneous size than faster injections (ESI Section S2.1†). For additional information, all synthetic details are summarized in ESI S2.†
After the reaction was completed at t6, the mixture was left to cool down to room temperature and the material was recovered by centrifugation and washed thoroughly with DMF and methanol before analysis. Scanning electron microscopy (SEM) images confirm the expected octahedral morphology for the MUV-10 crystals prepared by time-resolved synthesis, which also present a more homogeneous size dispersion centered at 2 μm, compared to the polydispersity obtained with conventional synthesis (Fig. 3a). On a related note, we also analysed separate aliquots over the 24 hours of reaction to account for the effect of time on the crystal sizes to confirm that this reaction time provides the most homogeneous size dispersion of micrometric crystals (Table S4†).
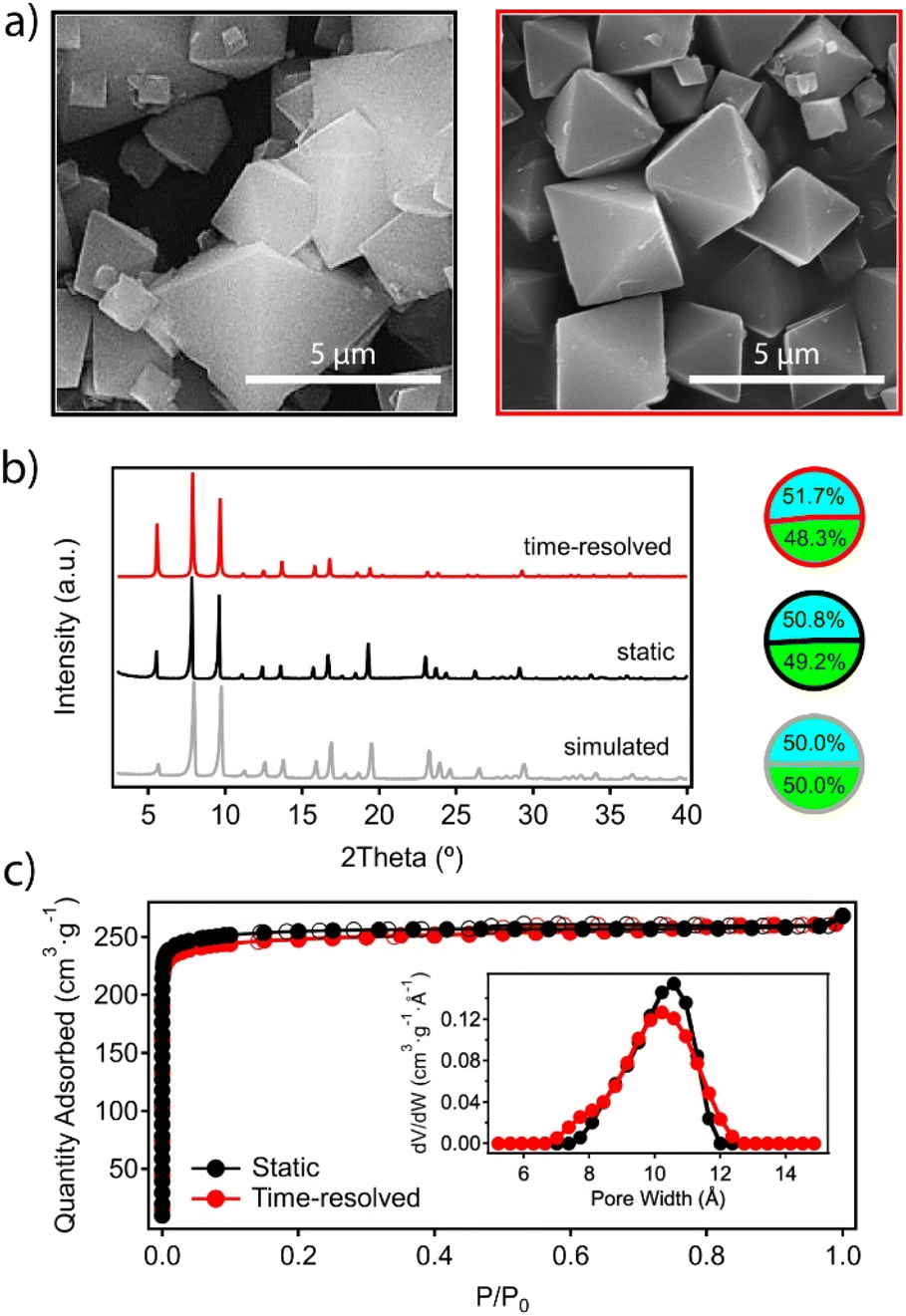 | ||
| Fig. 3 (a) SEM micrographs of MUV-10 crystals prepared by the static (left) and time-resolved (right) methods. (b) Comparison of their PXRD patterns and metal ratios with those expected for an ideal framework. (c) N2 isotherms and PSD plots at 77 K. | ||
One of the main difficulties in controlling the formation of titanium–organic frameworks is the tendency of titanium(IV) in solution to form amorphous oxides by hydrolysis reactions which are difficult to control.25 This reaction competes with the formation of the MOF and could lead to the formation of amorphous species, rich in titanium but undetectable by X-ray diffraction, which would contaminate the bulk material.26 This risk is even higher in an open synthesis system with continuous injection of reagents despite the inert atmosphere. Single-point energy dispersive X-ray (EDX) and ICP-MS analysis of the time-resolved sample rules out this possibility and confirms a Ti:Ca ratio that matches the stoichiometry of the Ti2Ca2 cluster, and does not present significant deviations with respect to the material prepared under static conditions (Fig. 3b and ESI 2.2, Table S5†). The powder X-ray diffraction (PXRD) pattern of the ground crystals also confirms the formation of a crystallographically pure phase corresponding to the simulated structure of the material, which shows no significant differences with respect to that of the material prepared under static conditions (Fig. 3b). The Le Bail refinements of the PXRD patterns of both samples confirm minimal deviations in the cell parameters (ESI Section S2.2, Fig. S2†). Finally, we tested the adsorption capacity of the material by N2 uptake at 77 K after thermal vacuum activation of the washed solid. Fig. 3c confirms that the synthetic method has no effect on the gravimetric surface area, which remains close to 1000 m2 g−1 in both cases. The pore size distribution (PSD) plot shows a broad contribution centered at around 1.1 nm, corresponding to the overlap of the octahedral and cuboctahedral cavities of a very similar size in the MUV-10 framework (Fig. 2a).
These results confirm time-resolved synthesis by continuous injection of molecular precursors to be a method compatible with the synthesis of MUV-10 crystals which, in addition to reducing reaction times from 48 to 24 hours, also results in better control of particle size possibly as a result of maintaining the supersaturation conditions throughout the synthesis.
Time-resolved encapsulation of nanoparticles at varying stages of MUV-10 nucleation/growth
One of the main advantages of this synthetic method is arguably the possibility to inject alternative components at specific growth stages of the system by using an additional injection syringe in the experimental scheme shown in Fig. 2b. Before incorporating the nanoparticles, we chose to separately analyse different aliquots of MUV-10 isolated at three different growth stages at times t1, t3 and t5, without allowing the reaction to run for 24 hours (Fig. 4a). All materials were isolated by centrifugation and washed in the same way for analysis. The SEM images show very clear differences between them. MUV-10-t1 shows very heterogeneous particle sizes with no clear morphological features, suggesting a very early nucleation stage (Fig. 4b). On the other hand, t2 already shows incipient octahedral crystals with less size dispersion, but with faces covered by much smaller and irregular particles. Finally, t5 corresponds to perfectly formed octahedral crystals without the presence of additional materials. These differences are also reflected in the relative percentages of metals in the samples (Fig. 4c). The relative percentage of Ti at t1 is very high, suggesting the dominant presence of titanium oxides at this early stage of nucleation. In contrast, t3 and t5 show Ti:Ca ratios much closer to those expected for the formation of Ti2Ca2 clusters. These relative differences are also transferred to the diffraction and gravimetric adsorption of the isolated aliquots (Fig. 4d and e). While t1 corresponds to the formation of amorphous titanium oxides without significant porosity, t5 corresponds to the formation of phase pure MUV-10 with the expected gravimetric adsorption capacity. On the other hand, although the diffraction of t3 corresponds to that of the MOF, the observed reduction in N2 uptake suggests the presence of an additional amorphous phase associated with the incomplete formation of the framework.
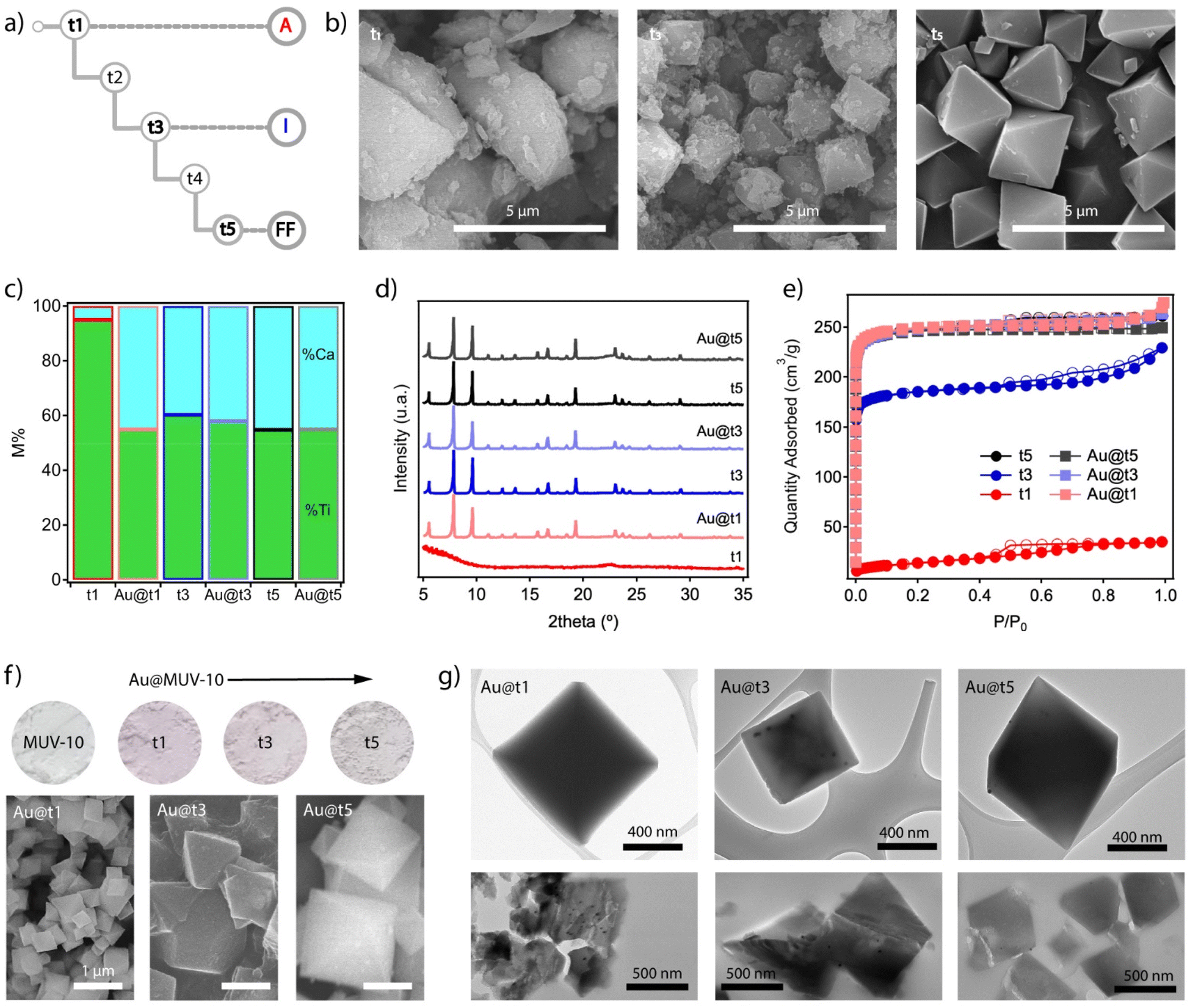 | ||
| Fig. 4 (a) Scheme showing the times for which an aliquot of the material formed during the time-resolved synthesis is extracted according to the procedure described in Fig. 2b. (b) SEM images of the isolated solids at t1, t3 and t5. (c) Variations in the proportions of metals in the solids before (t1, t3, and t5) and after the injection of Au nanoparticles until complete crystal growth (Au@t1, t3, and t5). (d) Diffraction patterns and (e) gas uptake isotherms of all materials confirming the formation of MUV-10 with the expected structure and porosity after Au injection in all cases. (f) Color gradient in the Au@MUV-10 samples compared to that of the material without gold (top) and changes in particle size and morphology as a function of the addition time (bottom). (g) TEM analysis of Au@MUV-10 crystals before (top) and after microtoming them into 0.5 μm thick slices for facilitating the visualization of the nanoparticles (bottom). | ||
This information suggests the presence of three different stages of MUV-10 formation, which we will refer to as amorphous (t1, A), incipient (t3, I) and fully formed (t5, FF). This differentiation between MOF formation states seemed appropriate to study the effect of injecting NPs and analysing their impact on the formation of the final material and possible variations in the distribution of nanoparticles in the resulting crystals. For this purpose, we synthesized Au nanoparticles of sizes between 15 and 20 nm stabilized with polyvinylpyrrolidone (PVP) by following the Turkevich method (ESI Section S3.1†).27 By introducing an additional pump in the scheme shown in Fig. 2b for a third addition channel, we injected 1 mL of a freshly made solution of Au NPs [0.35 mM] in methanol at the points t1, t3 or t5 with a fixed injection feed rate of 20 mL h−1. All reactions were performed according to the reagent injection rates described in the above procedures, followed by a reaction time of 24 hours to ensure a meaningful comparison between the resulting Au@MUV-10 materials.
The first piece of experimental evidence of the incorporation of the nanoparticles is the purple colour of all the materials isolated compared to white MUV-10 (Fig. 4f top). It is worth noting their relative variations in color that seem to indicate either different amounts of Au loaded or changes in its distribution within the solid. To confirm this possibility, the Au@MUV-10 solids were analysed by ICP-MS after digestion in an acid medium. The results confirm the presence of comparable amounts of gold in all samples, with values ranging from 161 to 240 ± 3 ppm. However, the samples show significant differences in particle size and morphology depending on the point at which the nanoparticles were injected. According to the SEM analysis shown in the same figure (Fig. 4f bottom), the most abrupt change is observed for Au@t1, which presents particles much smaller than 1 μm. This suggests a possible effect of the addition of NPs on the early stages of MUV-10 nucleation for the formation of smaller crystals. This effect is much less pronounced in Au@t3 and t5, possibly because the injection has already taken place on crystals with fairly advanced growth, consistent with the characteristics of incipient and fully formed states of formation of the MOF that correspond to these points as described above. We argue that these changes in particle size for comparable Au loadings result in changes in the density of nanoparticles per crystal that translate into the observed colour intensity changes.
To confirm the incorporation of the nanoparticles into the MUV-10 crystals, the samples were also analyzed by transmission electron microscopy (TEM). Compared to similar studies on the encapsulation of NPs between 100 and 500 nm in MOFs,8–15 the high contrast of the crystals and the smaller size of the nanoparticles made the analysis more challenging, especially in the case of Au@t1 for which we did not observe NPs in any of the crystals examined compared to their presence in t3 and t5 (Fig. 4g top). To simplify the analysis and improve the transmittance of the samples, we microtomed them into ≈0.5 μm thick slices which were then deposited on the mesh grid for TEM analysis by following the same protocol reported for the spectroscopic analysis of UiO-68 lamellae at the nanoscale.28 The bottom part of Fig. 4g confirms the presence of NPs in the sections of all crystals, although more clearly in Au@t1 and t3. In the case of t1, where their presence was not evident before the crystal was cut, the high density observed also seems to support the hypothesis that their incorporation takes place in the early stages of MUV-10 formation, relegating the nanoparticles to the inner core of the crystal. This was further confirmed by focused ion beam scanning electron microscope (FIB-SEM) analysis (Fig. S19†). These relative differences between materials are not reflected in their bulk properties. All Au@MUV-10 solids show minimal compositional variations with Ti:Ca ratios close to those expected for MUV-10 (Fig. 4c). Their PXRD patterns are also consistent with the formation of the framework without significant variations in their crystallinity or diffraction lines indicative of the formation of an additional phase (Fig. 4d). The N2 isotherms are also consistent with the behavior expected for MUV-10, with total pore volumes of 0.4 cm3 g−1 and non-hysteretic type I adsorption profiles (Fig. 4e). Au@t1 is the only system that shows a small adsorption tail at P/P0 > 0.9, due to the smaller particle size and corresponding intergrain mesoporosity.
Catalytic activation of electron-deficient alkynes with Au@MUV-10
The results above confirm the formation of Au@MUV-10 crystals with comparable framework properties and Au loading contents. We hypothesized that this scenario was well fitted to study how the relative changes associated with the way in which the nanoparticles are incorporated into the porous support from their injection at the different stages of MUV-10 formation might affect their activity when used as heterogeneous catalysts.We chose to test their ability to activate electron deficient alkynes during the cyclotrimerization reaction of propargyl esters, using 1,2-dichlorobenzene (o-DCB) as a solvent, to forge three new C–C bonds in one pot (Fig. 5a). The experiments were carried out by reacting ethyl propiolate at 120 °C under mechanical stirring in the presence of 0.2 mol% Au NPs encapsulated as Au@t1, t3 or t5 materials. The results after 20 hours (GC-FID analysis, see also Scheme S1 and Table S12†) show that the three materials have very different catalytic activities: while Au@t1 shows a consistent 60% conversion and 10:1 selectivity towards the 1,2,4- (1a) vs. the 1,3,5-trisubstituted isomer product (1b) during 5 reuses, Au@t3 only achieves a ∼45% conversion and ∼5:1 selectivity (products 1a:1b) under the same reaction conditions, and Au@t5 significantly loses catalytic activity and selectivity throughout the reuses, to give ∼30% yield and ∼1:1 selectivity in the fifth use. The chemical structure of the products 1a:1b was confirmed by combined gas chromatography-mass spectrometry (GC-MS) and 1H- and 13C-nuclear magnetic resonance (NMR) analyses.24 These results, obtained with 0.05 mmol of organic substrate, are consistent with those of experiments at other scales (Fig. S25†). It is noteworthy here that the catalytically active surface Au atoms on a 20 nm NP account for just 5% of the total Au atoms,30 and thus the true catalytic loading of Au in the reaction is 0.01 mol% and the turnover number (TON) of the reaction with Au@t1 after 5 cycles is 30000, two orders of magnitude higher than that of any other metal NP or cluster catalyst reported so far for this reaction, either under heterogeneous or homogeneous conditions, as far as we know (Table S13†).31–33 Considering that previous studies with the Au–TiO2 catalyst suggests that the electron-richest the Au NP is, the highest catalytic activity is found,29,34 it is not surprising that the encapsulated Au NPs in the electron-rich MUV-10 MOF show an enhanced catalytic activity.
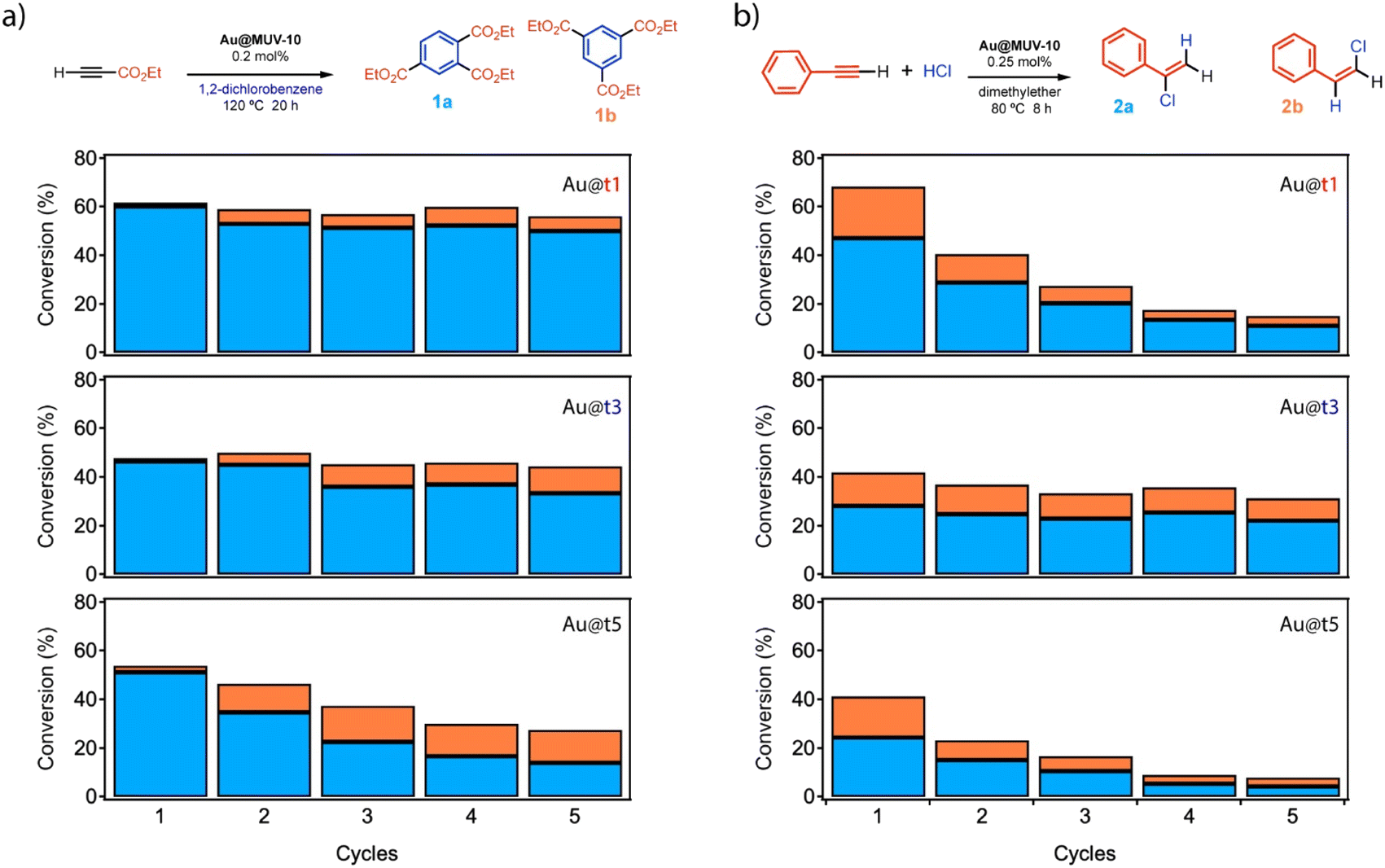 | ||
| Fig. 5 Summary of reaction conditions and product formation by using Au@MUV-10 catalysts prepared by gold injection at different reaction times. (a) Cyclotrimerization reaction of propiolate with 1,2-dichlorobenzene and (b) hydrochlorination of phenylacetylene. | ||
The above results suggest a relationship between the way in which the Au NPs are integrated into the porous support and not only the catalytic activity, but also the selectivity and recyclability of the final solid catalyst.35 The latter displays better endurance when Au NPs are concentrated in the innermost areas of the crystal (Au@t1) compared to the significant loss of activity for Au@t5. In order to check this hypothesis, two new materials prepared in the same way as Au@t1 but with 10× and 5× times higher Au loadings were prepared and tested in catalysis (see ESI Sections S3.3 and S4.1†). The results (Table S12†) confirm that when Au NPs are not completely integrated into the MOF structure, the product 1a:1b selectivity decreases to ∼3:1, values similar to those obtained with Au NPs supported on TiO2 (ref. 29) and also the value observed here for unsupported Au NPs (Table S12†). Indeed, this ∼3:1 selectivity value is, in average, the selectivity observed for the less stable Au@t5 catalyst. In other words, one can conclude that the stability conferred by the MUV-10 MOF framework to the encapsulated Au NPs is essential to achieve a very good catalytic activity (60%), selectivity (10:1) and recyclability (5 uses without catalytic activity depletion). Hot filtration tests (Fig. S26†) showed that there is no catalytic active species in the solution during the reaction for any of the Au@t1–5 MOF catalysts, and thus leaching does not occur and cannot explain the catalytic differences observed between the solid materials.
In order to confirm if the encapsulation of the Au NPs at different stages of MUV-10 MOF formation translates in a high catalytic activity for other organic reactions, we decided to test catalysts Au@t1, Au@t3 and Au@t5 for the hydrochlorination reaction of phenylacetylene (0.25 mol% Au, Fig. 5b).36 This reaction has been reported to be catalyzed by 3 nm diameter size Au NPs supported on TiO2 (ref. 20 and 37) but not by any other solid catalyst or metal NP-based catalyst, as far as we know. The lack of solid metal catalysts for the hydrochlorination reaction is not surprising since the highly aggressive HCl reactant favors leaching of the metal, although the industrially relevant hydrochlorination reaction of acetylene to manufacture vinyl chloride can be performed with Au species supported on activated charcoal supports.37 Here, having in mind the larger size of our Au NPs (20 nm) and their strong encapsulation within the MOF, the leaching could be minimized while the catalytic activity could be tuned by the Au NP/MUV-10 MOF composite structure. Indeed, the results show that, as in the previous reaction, the conversion and selectivity for products 2a:2b follows the order Au@t1 (67%) > Au@t3 (42%) > Au@t5 (41%) (Fig. 5b, for results at five times lower scales see Fig. S27†). GC-MS and NMR analyses confirmed the structure of products 2a:2b.20 However, only Au@t3 was reusable with constant conversion and major selectivity to the Markovnikov product (2a:2b 4:1). If we consider again that only 5% of Au atoms are available on the surface of the 20 nm Au NPs, probably much less because of the tight MOF-Au NP interaction, the catalytic activity must be one order of magnitude higher than that of the supported 3 nm Au NPs, where 38% of Au atoms are available on surface. As for the previous reaction, we performed new experiments to confirm the negligible activity of unsupported Au NPs and the lack of significant leaching as assessed by hot filtration tests, to support the heterogeneous nature of the catalytic reaction (Table S14 and Fig. S28†). At this point, the origin of the decrease in catalytic activity found for the Au@t1 and Au@t5 catalysts throughout the reuses is not clear; however, a tentative explanation would be the competitive polymerization of phenylacetylene on the MOF catalyst under acidic reaction conditions, which might block the access of the reactants to the encapsulated Au NPs.
Conclusions
The possibility of controlling the injection of additional components to MOF crystals at different nucleation stages offers new perspectives for controlling their integration with these molecular supports. This is exemplified here by the time-resolved integration of Au nanoparticles in the titanium framework MUV-10 at variable reaction times. Injection at a very early stage facilitates the incorporation at the inner core of the crystals, which results in catalysts with enhanced activity, selectivity and recyclability for the cyclotrimerization of propargyl esters and the hydrochlorination of alkynes.Our preliminary results suggest that the time-resolved synthesis of MUV-10 can be adapted to other titanium(IV) MOFs and additives in solution for the design of composites of very diverse nature and range of applications.
Data availability
Additional experimental information generated during the current study are available from the corresponding authors on reasonable request.Author contributions
All authors contributed to the discussion, participated in the writing of the original draft, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EU (ERC-2021-COG-101043428), the Generalitat Valenciana (PROMETEU-2021-054 & MFA/2022/026) and the Spanish government (CEX2019-000919-M, PID2020-118117RB-I00, CEX2021-001230-S and PID2020-115100GB-I00). This study forms part of the Advanced Materials programme and was supported by MCIN with funding from European Union NextGenerationEU (PRTR-C17.I1) and Generalitat Valenciana. M. M. thanks MICIIN for a contract under the Juan de la Cierva program (FJC2019-040523-I) and Y. Z. thanks the China Scholarship Council (CSC No. 202009350009) for a PhD fellowship.Notes and references
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Wöll, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60, 23946–23974 CrossRef CAS PubMed.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2008, 38, 481–494 RSC.
- B. Li, J. Ma and P. Cheng, Small, 2019, 15, 1804849 CrossRef PubMed.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- P. Wang, J. Zhao, X. Li, Y. Yang, Q. Yang and C. Li, Chem. Commun., 2013, 49, 3330–3332 RSC.
- X. Liu, L. He, J. Zheng, J. Guo, F. Bi, X. Ma, K. Zhao, Y. Liu, R. Song and Z. Tang, Adv. Mater., 2015, 27, 3273–3277 CrossRef CAS PubMed.
- K. Sugikawa, Y. Furukawa and K. Sada, Chem. Mater., 2011, 23, 3132–3134 CrossRef CAS.
- M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef CAS PubMed.
- T. Tsuruoka, H. Kawasaki, H. Nawafune and K. Akamatsu, ACS Appl. Mater. Interfaces, 2011, 3, 3788–3791 CrossRef CAS PubMed.
- K. M. Choi, K. Na, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 7810–7816 CrossRef CAS PubMed.
- B. Rungtaweevoranit, J. Baek, J. R. Araujo, B. S. Archanjo, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2016, 16, 7645–7649 CrossRef CAS PubMed.
- A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657–1712 CrossRef CAS PubMed.
- P. Escamilla, W. D. Guerra, A. Leyva-Pérez, D. Armentano, J. Ferrando-Soria and E. Pardo, Chem. Commun., 2022, 59, 836–851 RSC.
- F. Garnes-Portolés, R. Greco, J. Oliver-Meseguer, J. Castellanos-Soriano, M. C. Jiménez, M. López-Haro, J. C. Hernández-Garrido, M. Boronat, R. Pérez-Ruiz and A. Leyva-Pérez, Nat. Catal., 2021, 4, 293–303 CrossRef.
- A. Das, C. Dash, M. Yousufuddin, M. A. Celik, G. Frenking and H. V. R. Dias, Angew. Chem., Int. Ed., 2012, 51, 3940–3943 CrossRef CAS PubMed.
- J. Oliver-Meseguer, A. Doménech-Carbó, M. Boronat, A. Leyva-Pérez and A. Corma, Angew. Chem., Int. Ed., 2017, 56, 6435–6439 CrossRef CAS PubMed.
- J. Castells-Gil, N. M. Padial, N. Almora-Barrios, J. Albero, A. R. Ruiz-Salvador, J. González-Platas, H. García and C. Martí-Gastaldo, Angew. Chem., Int. Ed., 2018, 57, 8453–8457 CrossRef CAS PubMed.
- S. Rojas, J. García-González, P. Salcedo-Abraira, I. Rincón, J. Castells-Gil, N. M. Padial, C. Marti-Gastaldo and P. Horcajada, Sci. Rep., 2022, 12, 14513 CrossRef CAS PubMed.
- X. G. Wang, Q. Cheng, Y. Yu and X. Z. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7836–7840 CrossRef CAS PubMed.
- M. J. V. Vleet, T. Weng, X. Li and J. R. Schmidt, Chem. Rev., 2018, 118, 3681–3721 CrossRef.
- H. Assi, G. Mouchaham, N. Steunou, T. Devic and C. Serre, Chem. Soc. Rev., 2017, 46, 3431–3452 RSC.
- J. Castells-Gil, N. M. Padial, N. Almora-Barrios, I. da Silva, D. Mateo, J. Albero, H. García and C. Martí-Gastaldo, Chem. Sci., 2019, 10, 4313–4321 RSC.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- A. Centrone, B. Lerma-Berlanga, A. J. Biacchi, C. Fernández-Conde, G. Pavlidis and C. Marti-Gastaldo, Adv. Funct. Mater., 2023, 33, 2302357–2302365 CrossRef CAS.
- A. Leyva-Pérez, J. Oliver-Meseguer, J. R. Cabrero-Antonino, P. Rubio-Marqués, P. Serna, S. I. Al-Resayes and A. Corma, ACS Catal., 2013, 3, 1865–1873 CrossRef.
- P. Rubio-Marqués, A. Leyva-Pérez and A. Corma, Chem. Commun., 2013, 49, 8160–8162 RSC.
- V. P. Anju, S. K. Barik, B. Mondal, V. Ramkumar and S. Ghosh, ChemPlusChem, 2014, 79, 546–551 CrossRef CAS PubMed.
- H. Miura, Y. Tanaka, K. Nakahara, Y. Hachiya, K. Endo and T. Shishido, Angew. Chem., Int. Ed., 2018, 57, 6136–6140 CrossRef CAS PubMed.
- S. Song, C. Li, T. Liu, P. Zhang and X. Wang, Org. Lett., 2021, 23, 6925–6930 CrossRef CAS PubMed.
- J. Oliver-Meseguer, M. Boronat, A. Vidal-Moya, P. Concepción, M. A. Rivero-Crespo, A. Leyva-Pérez and A. Corma, J. Am. Chem. Soc., 2018, 140, 3215–3218 CrossRef CAS PubMed.
- M. Mon, J. Ferrando-Soria, T. Grancha, F. R. Fortea-Pérez, J. Gascon, A. Leyva-Pérez, D. Armentano and E. Pardo, J. Am. Chem. Soc., 2016, 138, 7864–7867 CrossRef CAS PubMed.
- S. Dérien, H. Klein and C. Bruneau, Angew. Chem., Int. Ed., 2015, 54, 12112–12115 CrossRef PubMed.
- G. Malta, S. J. Freakley, S. A. Kondrat and G. J. Hutchings, Chem. Commun., 2017, 53, 11733–11746 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04678h |
| This journal is © The Royal Society of Chemistry 2024 |