
Construction of S-scheme heterojunctions of a Ti-doped Ce-MOF and BiOCl for efficient photocatalytic selective oxidation of amines†
Danxia
Zhao
 *ab,
Pengyu
Wu
c,
Huayue
Zhu
ab,
Ru
Jiang
ab,
Jingwei
Chen
a,
Chuhan
Qiu
a,
Shengtao
Jiang
a and
Guoping
Lu
*d
*ab,
Pengyu
Wu
c,
Huayue
Zhu
ab,
Ru
Jiang
ab,
Jingwei
Chen
a,
Chuhan
Qiu
a,
Shengtao
Jiang
a and
Guoping
Lu
*d
aInstitute of Environmental Engineering Technology, Taizhou University, Taizhou 318000, Zhejiang, P.R. China. E-mail: danxia@tzc.edu.cn; danxia@126.com
bInstitute of Environmental Engineering Technology, Taizhou University, Taizhou, 318000, Zhejiang, P. R. China
cc Key Laboratory for Advanced Technology in Environmental Protection of Jiangsu Province, Yancheng Institute of Technology, Jiangsu 224051, P. R. China
dNanjing University of Science and Technology, School of Chemical Engineering, 200 Xiaoling Wei, Nanjing, Jiangsu 210094, P. R. China. E-mail: glu@njust.edu.cn
First published on 23rd January 2024
Abstract
The solar-driven selective oxidation of amines to imines has attracted widespread attention due to its green and non-polluting nature, as well as its high selectivity. In this study, a unique S-scheme BiOCl/UiO-66(Ce/Ti) heterojunction was first constructed as an organic aerobic photocatalyst. Under visible light irradiation with water as the sole solvent, the yield of the imine reached 98%. The S-scheme charge transfer mechanism was verified through in situ X-ray photoelectron spectroscopy and electron paramagnetic resonance measurements, revealing the source of excellent photocatalytic activity. Additionally, the synergistic effect of the interface microenvironment generated by the interaction between OVs and Ce–Ti was found to play a significant role in the photocatalytic oxidation process. This work emphasizes the importance of designing a heterojunction interface microenvironment (IME) for achieving efficient photocatalytic selective oxidation of amines.
Introduction
Solar-driven selective oxidation of organic compounds, such as the oxidation coupling of primary amines, is highly desirable for the synthesis of pharmaceuticals and novel chemical substances.1,2 In particular, aerobic oxidation reactions using molecular oxygen and air as oxidants have attracted significant research interest due to their environmental friendliness and sustainability.3,4 In recent years, considerable efforts have been devoted to the conversion of primary amines to imines using photocatalytic techniques.5,6 To address the issue of severe photogenerated electron–hole recombination and the low oxidation–reduction capacity of individual photocatalysts leading to an overall low photocatalytic reaction efficiency, various heterojunction technologies have been developed. In 2019, Yu et al.7 first proposed the S-scheme heterojunction. Specifically, S-scheme heterojunctions are constructed using staggered oxidation photocatalysts (OPs) and reduction photocatalysts (RPs), effectively preserving active electrons and holes while eliminating inactive ones through recombination, thus achieving effective separation of electron–hole pairs with a strong redox ability. Therefore, S-scheme heterojunctions are suitable for coupling reactions, CO2 reduction, and environmental remediation.8,9BiOCl photocatalysts possess a highly positive VB position, providing photogenerated holes with strong oxidation ability, making them ideal candidates as oxidation semiconductor photocatalyst (OP) materials.10 Additionally, the BiOCl surface exhibits outstanding hydrophilicity, facilitating the adsorption of reactants and thereby exhibiting good photocatalytic activity.11 To construct efficient S-scheme systems based on BiOCl, the selection of a reduction semiconductor photocatalyst (RP) plays a crucial role in the photocatalysis of amines. Metal–organic frameworks (MOFs) are emerging porous crystalline materials composed of metal nodes and organic ligands, which have attracted extensive interest in the field of photocatalysis due to their structural diversity and tunable functionality.12–14 In recent years, UiO-66 has emerged as a standout subfamily within MOFs, demonstrating significant semiconductor properties due to its efficient ligand-to-metal charge transfer (LMCT) induced by light irradiation. For instance, Cavka et al.15 first reported the UiO-66 MOF constructed using terephthalate ligands and Zr(IV)–oxo clusters. However, it exhibited general performance with low efficiency under visible light irradiation. Recently, encapsulating cerium oxide clusters within MOFs has been proven to be a promising approach to achieve highly efficient solar energy conversion photocatalysts, where the HOMO–LUMO gap of the MOFs can be tuned by modifying organic linkers, thereby increasing the absorption range of visible light.16,17 In particular, UiO-66(Ce) is expected to have longer excited states and easier charge–hole separation compared to UiO-66(Zr) due to the low-lying unoccupied 4f orbitals of Ce4+, and the redox cycling between Ce4+ and Ce3+ may also confer more attractive catalytic performance to UiO-66(Ce).18,19
However, due to the inherent defects of UiO-66(Ce), UiO-66(Ce) as a reduction semiconductor photocatalyst is still in its early stages and requires further exploration. Considering that the Ce4+/Ce3+ orbitals could not combine well with the orbitals of organic ligands, this may make LMCT and the generation of reductive photoelectrons more difficult.20 Therefore, additional media are needed to bridge the gap between the Ce4+/Ce3+ cycle and the oxidation of most target molecules. Fortunately, it has been confirmed that the photocatalytic reducibility of UiO-66 can be improved through Ti-mediated electron transfer mechanisms. For example, Zhang et al.21 and Wang et al.22 have demonstrated that the introduction of Ti into UiO-66(Ce) and UiO-66-NH2(Zr) significantly prolongs the excited state lifetime and enhances the catalytic activity. Therefore, it is feasible to consider using UiO-66 as a potential qualified amine selective RP.
More importantly, it is encouraging that the formation of abundant oxygen vacancies (OVs) has been discovered for the first time in Ce-MOFs.23 The generation of OVs facilitates the internal transfer of reducible electrons and the formation of hydroxyl groups (˙OH) and superoxide (˙O2−) radicals. Additionally, Ce-MOFs with abundant oxygen vacancies not only provide channels for the transport of charge carriers, facilitating the formation of S-scheme heterojunctions with low interface charge transfer barriers, but also offer more favorable binding sites to achieve tighter and more stable contact interfaces. Inspired by enzymatic reactions, the specific physicochemical environment of the interface microenvironment (IME) may interact with substrates and influence the catalytic process, providing advantages for achieving high activity and selectivity of catalysts. Therefore, by modifying the types and combinations of inorganic nodes and organic linkers, UiO-66 is endowed with different microscopic structures and tunable interface microenvironments, providing a new pathway for efficient primary amine conversion.
In summary, it is hoped that a new S-scheme heteroatom-doped MOF/BiOCl photocatalyst can be constructed through interface engineering and used in aerobic organic reactions. Unfortunately, there have been no reports in this area to date, and the exploration of relevant mechanisms remains blank. In this work, we chose Ce4+ and terephthalic acid as precursors to synthesize UiO-66(Ce), which was then transformed into Ti-doped UiO-66(Ce/Ti) through cation exchange. Its suitable conduction band position and small work function meet the requirements for the formation of an S-scheme system with BiOCl. Furthermore, the abundant surface oxygen vacancies in UiO-66(Ce/Ti) not only promote interfacial charge transfer, but also potentially participate in the internal circulation of Ce4+/Ce3+ and Ti4+/Ti3+, thereby facilitating the generation of additional Lewis acid sites on the surface of the photocatalyst and forming a microenvironment at the BiOCl/MOF interface.
In this work, a novel S-scheme heterojunction of BiOCl/MOF with a strong internal electric field was designed and synthesized for the first time by combining titanium-doped cerium oxo-based UiO-66 MOFs with BiOCl. Compared to using BiOCl alone, the conversion rate of aniline was improved by approximately 3.3 times. The enhanced photocatalytic activity benefits from the favorable S-scheme charge transfer from BiOCl to UiO-66(Ce/Ti) and the electron interactions between Ce and Ti, leading to the conversion of Ce4+ + Ti3+ ⇌ Ce3+ + Ti4+ + OVs. The charge transfer in the S-scheme heterostructure was determined by electron paramagnetic resonance (EPR) spectroscopy and in situ X-ray photoelectron spectroscopy (XPS). The reaction mechanism for the enhanced oxygen photocatalytic performance of the BiOCl/UiO-66(Ce/Ti) catalyst was further revealed through density functional theory (DFT) calculation. It is worth noting that significant charge accumulation occurs at the interface of the heterostructure model, indicating the strong interaction and synergistic effect between UiO-66(Ce/Ti) and BiOCl. The resulting interface microenvironment not only greatly promotes the separation and transport of electrons and holes, but also retains electrons with a high oxidation–reduction ability in the conduction band of UiO-66(Ce/Ti) and holes in the valence band of BiOCl. This study provides insights for the rational design of advanced materials and reaction systems for selective photocatalytic oxidation of amines, highlighting the modulation of IME engineering.
Experimental section
Synthesis of BiOCl/UiO-66(Ce/Ti) heterojunction photocatalysts
The synthetic procedures of S-scheme BiOCl/UiO-66(Ce/Ti) heterojunctions are illustrated in Scheme 1. Firstly, 1,4-benzendicarboxylic acid (H2BDC, 354 mg, 2.13 mmol) and an aqueous of cerium(IV) ammonium nitrate (4 mL, 5.3 M) were used as raw materials to prepare UiO-66(Ce) via the solvothermal method reported in the literature.24 Then, UiO-66(Ce/Ti) rich in oxygen vacancies was prepared using cation exchange.21 On the other hand, BiOCl nanosheets with a two-dimensional morphology were prepared using BiCl3 and KMnO4 as raw materials.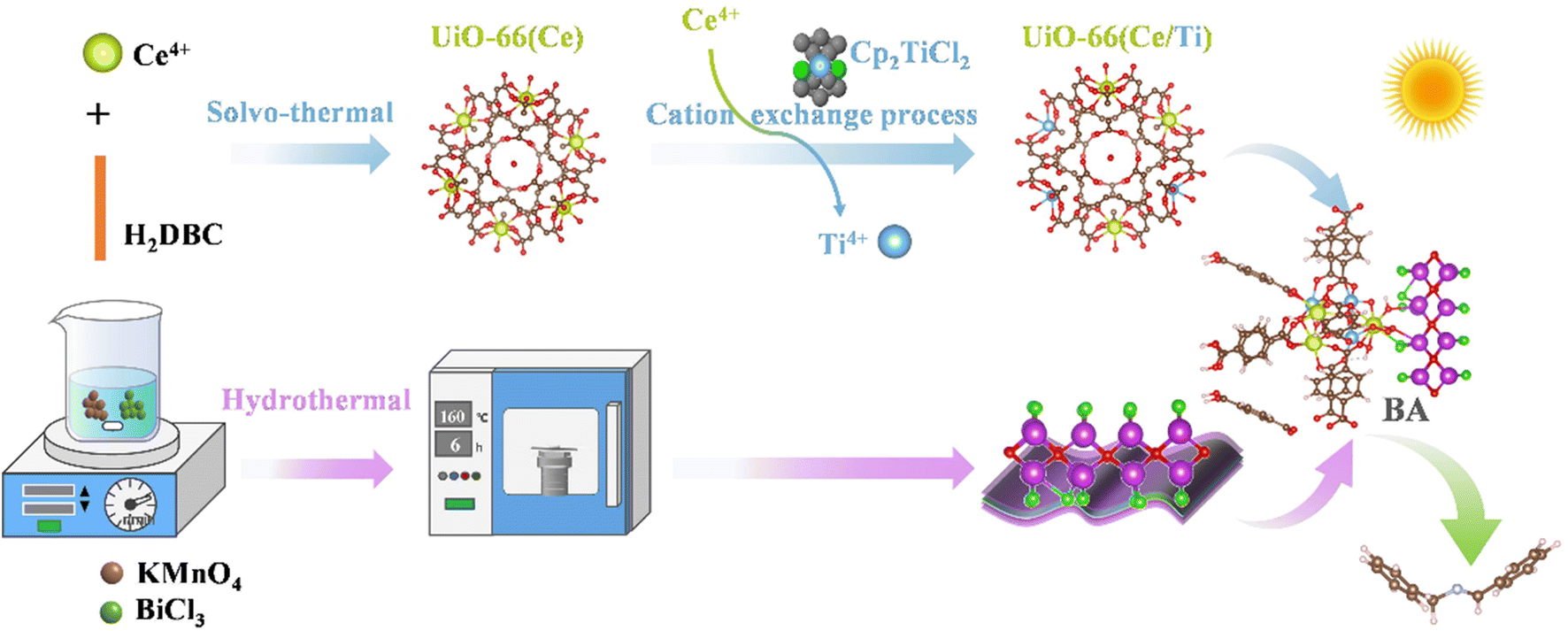 | ||
| Scheme 1 Synthetic illustration of the BCT heterojunction photocatalyst. | ||
The coupling of the MOF with BiOCl nanosheets through the hydrothermal method results in the formation of S-scheme BiOCl/UiO-66(Ce/Ti) (BCT) heterojunction photocatalysts. The detailed preparation methods of BiOCl and UiO-66(Ce/Ti) catalysts are provided in the ESI.†
Characterization of photocatalysts
The as-prepared samples were characterized by X-ray powder diffraction (XRD), Fourier transform infrared (FTIR) spectroscopy, X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray spectroscopy (EDS), high-resolution transmission electron microscopy (HRTEM), UV-vis diffuse reflectance spectroscopy (DRS), photoluminescence (PL) spectroscopy and electrochemical impedance spectroscopy (EIS). Detailed information about experimental instruments and setting parameters is provided in the ESI.†Procedures for the oxidative coupling of amines
In a typical oxidation process, a 10 mL round-bottom flask with two necks was used as the reaction vessel. Firstly, 20 mg of photocatalyst and 0.2 mmol of amines were placed in 3 mL of water with a magnetic stirrer. The system was first furnished with an O2 balloon with stirring for 0.5 h, then the bottom was sealed under O2 protection and irradiated for 6 h with visible light (λ > 400 nm), which was placed on one side of the reaction flask with 8 cm apart. After the reaction, the system was centrifuged and separated from the aqueous phase by extraction with ethyl acetate. The product was quantified using GC-MS analysis. The catalyst after the reaction was obtained by centrifugation and washed four times with ethyl acetate and ethyl alcohol, which could be used for the next cycle after drying.Other testing details and DFT calculation methods related to photocatalytic performance are included in the ESI.†
Results and discussion
Crystal structure and morphology
The phase structure of the samples was investigated using powder X-ray diffraction (XRD) (Fig. S1a†). The XRD pattern of BiOCl was perfectly consistent with the standard pattern (JCPDS NO. 06-0249),25 with the characteristic Bragg reflections attributed to the (001), (002), (101), (110), (102), (003), and (112) planes of BiOCl at 2θ = 12°, 24°, 26°, 32°, 33°, 37°, and 41°, respectively. The XRD spectra of UiO-66(Ce/Ti) were similar to those reported in the literature,21 indicating the successful synthesis of UiO-66. In the XRD pattern of the BCT heterostructure, no noticeable signals from the MOF were detected due to the higher dispersibility and lower MOF content. However, it should be noted that in addition to the peaks of BiOCl, tiny peaks assigned to the MOF were observed, indicating the successful construction of the heterostructure. FTIR spectroscopy (Fig. S1b†) was used to analyze the surface chemical bonds and functional groups of the synthesized samples. Three characteristic peaks were observed in the BiOCl spectrum, with the peak at 524 cm−1 corresponding to the Bi–O bond. The stretching and bending vibrations of hydroxyl groups primarily appeared at around 3414 cm−1, which may be related to crystalline water adsorbed on the BiOCl surface. The peak at 1622 cm−1 was associated with the bending vibration of the OH functional group. Within the spectral range of 3666–3635 cm−1, a broad peak appeared in the MOF, possibly partially masked by the OH groups of adsorbed solvent molecules (3400–2800 cm−1). The asymmetric (1590–1550 cm−1) and symmetric (1390–1370 cm−1) stretching peaks of carboxylic acid groups were present in the spectra of all MOF compounds.26 The peaks for the stretching and flexing vibrations of C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds mainly appeared at around 1152 cm−1–1657 cm−1, 1018 cm−1 and 1108 cm−1, which were related to the stretching vibrations of C–O and CO. The peaks at 510 cm−1 and 671 cm−1 may be associated with the bending and stretching vibrations of Ce–O, Ti–O or M–O–M bonds. It is worth noting that the band at 671 cm−1 is the result of the interaction between Ce3+ and the carbon part of the terephthalic acid ligand (COOH).27 An observation of the FTIR spectrum of BCT suggests that the peak at 1680 cm−1 is likely associated with the stretching vibration of the CO bond, while the peak at 1574 cm−1 is associated with the stretching vibration of the Ce–O bond. The peaks at 1422 cm−1 and 1511 cm−1 are related to the vibrations of functional groups in the UiO-66 structure. The peaks at 779 cm−1 and 733 cm−1 are attributed to the Bi–O–Bi bond and the Bi–Cl bond, respectively, proving the binding effect of the MOF on the BiOCl surface. Furthermore, after the combination of BiOCl and the MOF, the intensity of the MOF band significantly decreases and overlaps with the peaks of BiOCl, confirming the expected formation of a composite material between these two materials.
N bonds mainly appeared at around 1152 cm−1–1657 cm−1, 1018 cm−1 and 1108 cm−1, which were related to the stretching vibrations of C–O and CO. The peaks at 510 cm−1 and 671 cm−1 may be associated with the bending and stretching vibrations of Ce–O, Ti–O or M–O–M bonds. It is worth noting that the band at 671 cm−1 is the result of the interaction between Ce3+ and the carbon part of the terephthalic acid ligand (COOH).27 An observation of the FTIR spectrum of BCT suggests that the peak at 1680 cm−1 is likely associated with the stretching vibration of the CO bond, while the peak at 1574 cm−1 is associated with the stretching vibration of the Ce–O bond. The peaks at 1422 cm−1 and 1511 cm−1 are related to the vibrations of functional groups in the UiO-66 structure. The peaks at 779 cm−1 and 733 cm−1 are attributed to the Bi–O–Bi bond and the Bi–Cl bond, respectively, proving the binding effect of the MOF on the BiOCl surface. Furthermore, after the combination of BiOCl and the MOF, the intensity of the MOF band significantly decreases and overlaps with the peaks of BiOCl, confirming the expected formation of a composite material between these two materials.
The morphological characteristics of the prepared BiOCl, MOF and BCT composite materials were studied using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). In the SEM images, BiOCl shows a two-dimensional nanosheet morphology with a size of approximately 10 nm (Fig. 1a). In the high-resolution TEM (HRTEM) images, the spacing between lattice fringes is 0.275 nm, which is attributed to the (110) plane of the tetragonal phase of BiOCl (Fig. 1f). After the combination of BiOCl and the MOF, the BCT nanosheets aggregate into a more open morphology, like loose flowers (Fig. 1c). The HRTEM image of BCT reveals that the BiOCl nanosheets are closely attached to the porous MOF surface (Fig. 1d), with a clearly visible lattice fringe at the interface. The lattice spacing of the MOF corresponds to its (111) plane and measures 0.24 nm (Fig. 1f). Furthermore, energy-dispersive X-ray (EDX) element mapping (Fig. 2) shows a uniform distribution of the elements Bi, O, Cl, Ti, Ce and N, confirming the successful construction of the heterojunction structure.
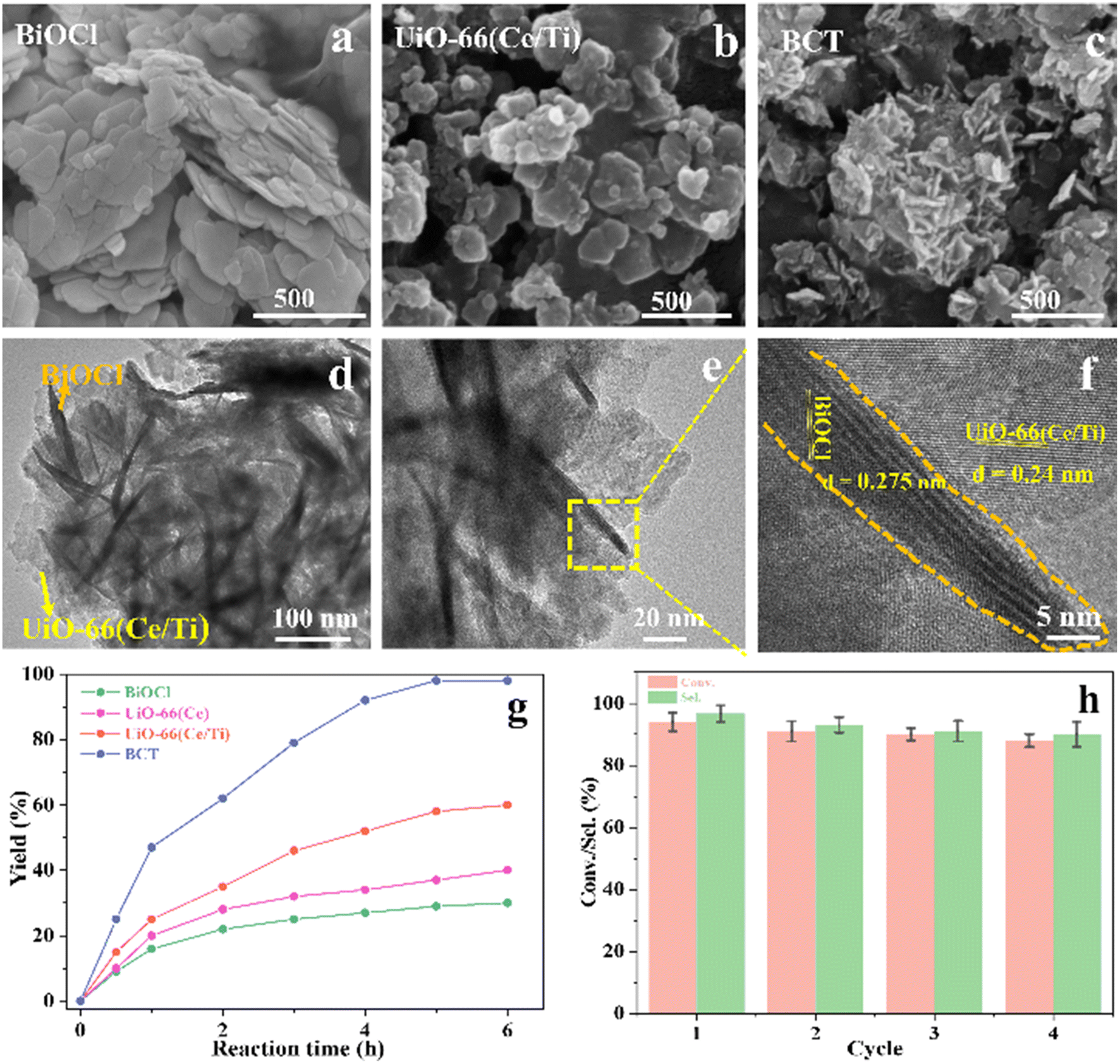 | ||
| Fig. 1 SEM images of the as-prepared samples: (a) BiOCl, (b) UiO-66(Ce/Ti) and (c) BCT. HRTEM images of BCT (d–f). (g) Time course of yields of different catalysts. (h) Photoactivity of recycled BCT. Reaction conditions: 0.2 mmol substrate, 20 mg of catalyst, O2 atmosphere, 3 mL of H2O, λ > 400 nm. Yields were determined by GC-MS. | ||
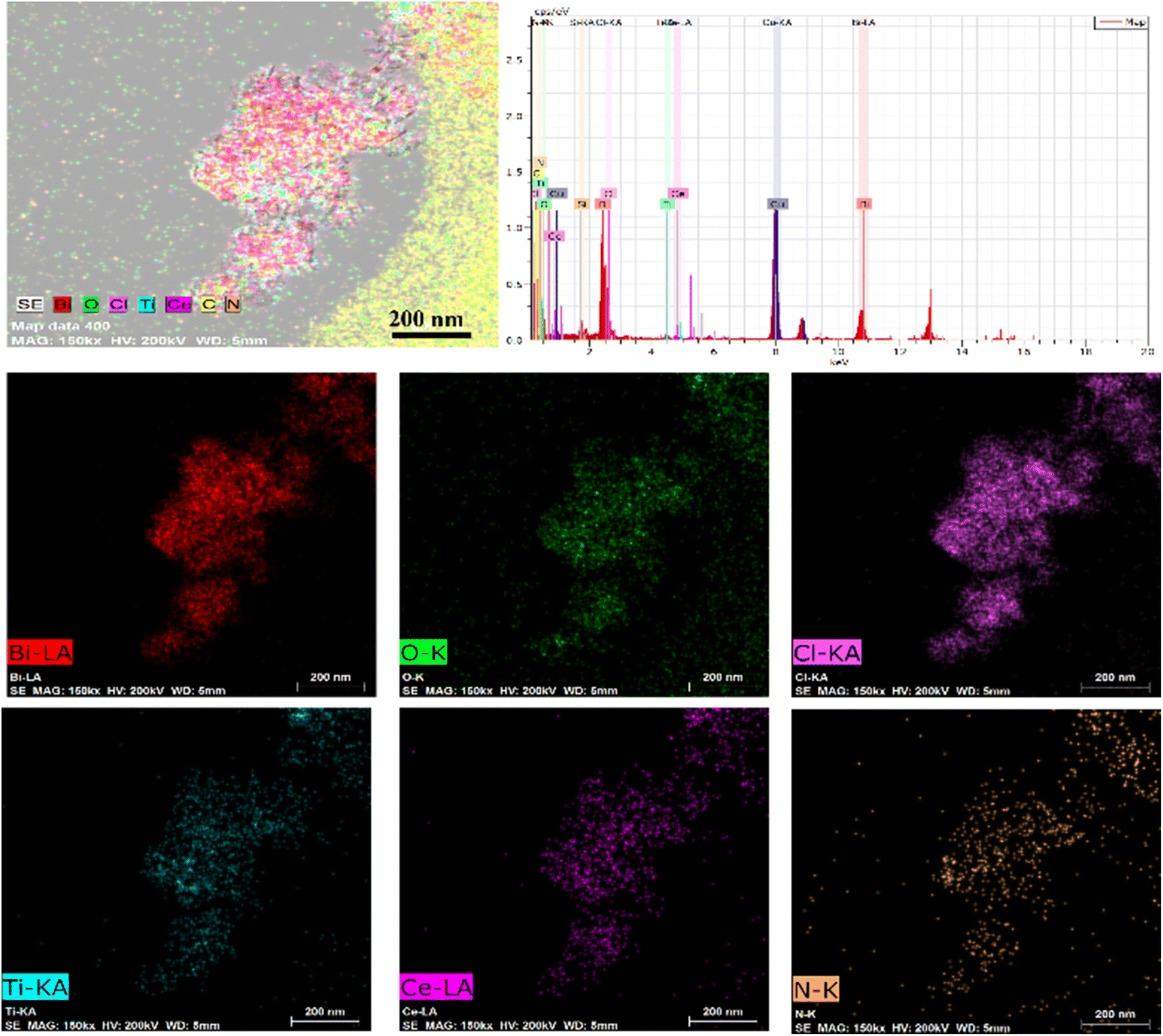 | ||
| Fig. 2 EDS elemental mapping images of Bi, O, Cl, Ti, Ce and N elements. | ||
Photocatalytic oxidation of amines
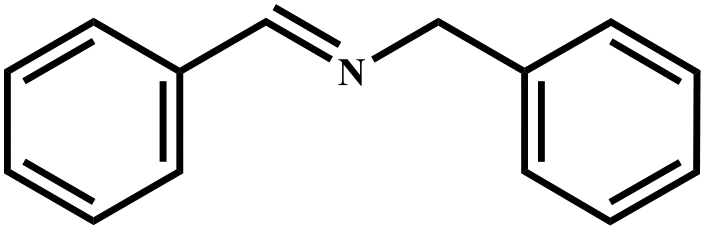
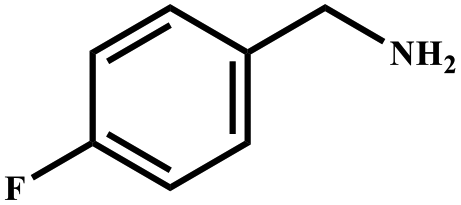
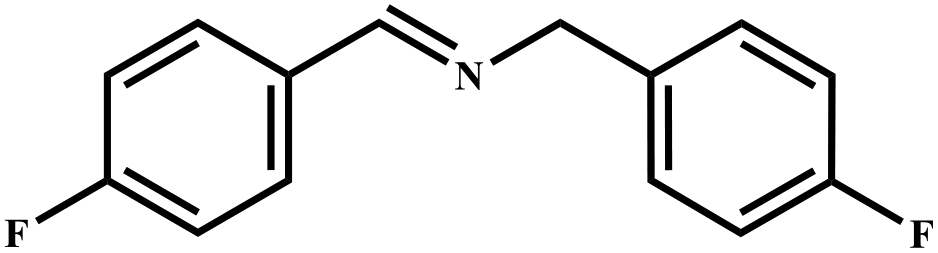
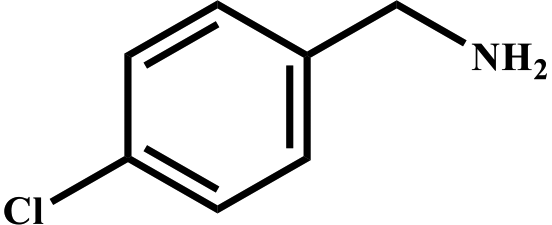
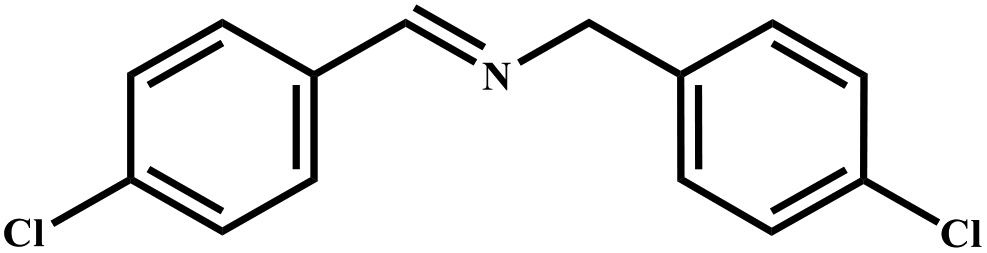
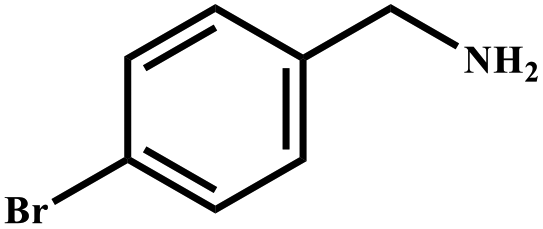
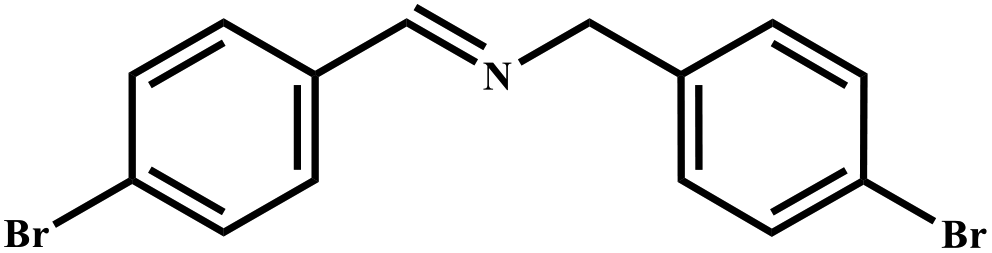
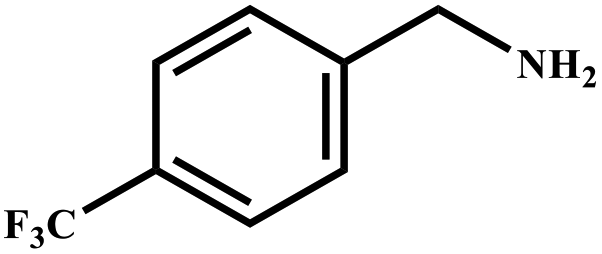
This study for the first time used the photo-selective oxidation of benzylamine (BA) as a model reaction to investigate the effect of interface engineering on the photocatalytic performance of the BCT heterojunction. Initially, control experiments were conducted without the presence of a photocatalyst, light source, or oxygen atmosphere (Table 1, entries 2–4). Obviously, no target product was detected, confirming the essential role of both the photocatalyst and oxygen in the organic transformation. Under visible light (λ > 400 nm) irradiation, when UiO-66(Ce) was used as a photosensitizer, only a 40% product yield was achieved, indicating that cerium alone as the metal node was unable to sustain this high efficiency. However, when Ti was doped into UiO-66(Ce), UiO-66(Ce/Ti) as a photocatalyst achieved a 60% product yield. We speculate that the interaction between benzylamine and UiO-66(Ce/Ti) promotes the formation of surface coordinating species, resulting in a stronger visible light-induced LMCT, and accelerates the oxidation process of benzylamine. Subsequently, BA and BCT reacted under an oxygen atmosphere, resulting in the formation of the corresponding imine after 6 h with a conversion rate as high as 98% and a selectivity of 99% (Table 1, entry 1), highlighting the importance of the heterostructured interface formed by UiO-66(Ce/Ti) and BiOCl. Additionally, the catalytic performance of BCT was compared with several representative photocatalysts in Table S1,† and BCT demonstrated a higher benzylamine conversion rate. Importantly, in contrast to other reported photocatalysts, BCT synthesized in this study achieved a considerable imine yield in a solution with water as the sole medium. The kinetic equation for benzylamine photooxidation can be attributed to a pseudo-first-order kinetic process (Fig. 1g), where BCT exhibited the highest reaction rate. Under the optimal conditions, the scope of substrates was expanded to evaluate the generality of BCT photocatalysts. Amines containing electron-withdrawing para groups (–F, –Cl, –Br, –CF3) and electron-donating para groups (–CH3, –OCH3) were efficiently converted to the desired N-benzylbenzylamine without any other byproducts observed (Table 2, entries 2–5, 8 and 9). Furthermore, amines with electron-donating substituents exhibited higher imine yields through photo-selective oxidation on BCT compared to those with electron-withdrawing substituents. Additionally, due to the spatial effect, the conversion rates of regioisomers consecutively following the order of ortho < meta < para (Table 2, entries 6–11).| Entry | BCT | O2 | hν | Scavengers | Yielda (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 0.2 mmol benzylamine, 20 mg of BCT, O2 atmosphere, and 3 mL of H2O. Yields were determined by GC-MS. | |||||
| 1 | + | + | + | — | 98 |
| 2 | + | + | — | — | — |
| 3 | — | + | + | — | — |
| 4 | + | — | + | — | Trace |
| 5 | + | + | + | BQ (O2˙−) | 8 |
| 6 | + | + | + | β-Carotene (1O2) | 13 |
| 7 | + | + | + | KI (h+) | 40 |
| 8 | + | + | + | IPA (˙OH) | 83 |
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Conv.b (%) | Sel.b (%) |
| a Reaction conditions: benzylamine (0.2 mmol), catalysts (20 mg), H2O (3 mL), O2 atmosphere, temperature (25 °C), visible light (λ > 400 nm), irradiation time (6 h). b Yields were determined by GC-MS. | ||||
| 1 |
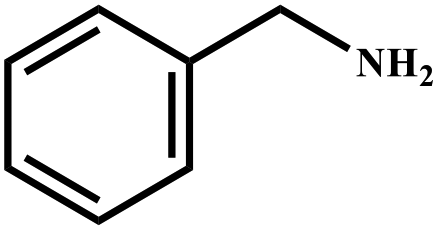
|
|
98 | 99 |
| 2 |
|
|
91 | 99 |
| 3 |
|
|
87 | 99 |
| 4 |
|
|
90 | 99 |
| 5 |
|
|
80 | 99 |
| 6 |
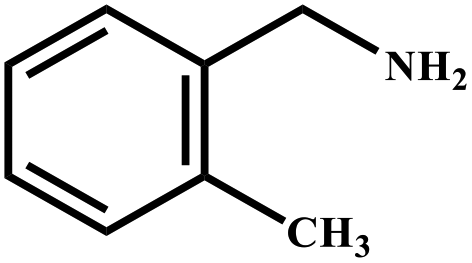
|
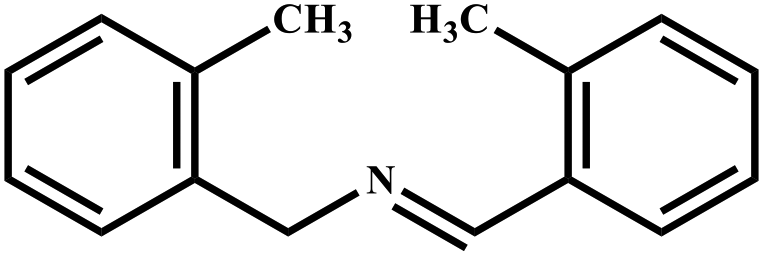
|
78 | 99 |
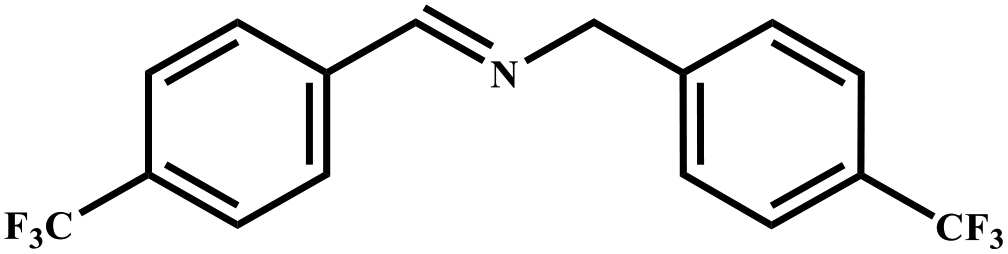
| 7 |
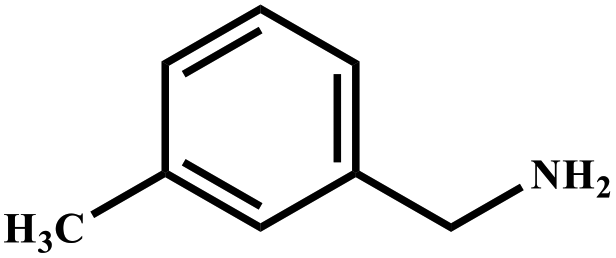
|
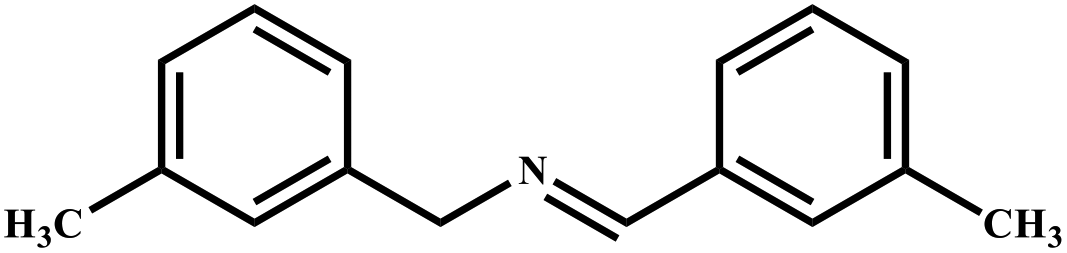
|
85 | 99 |
| 8 |
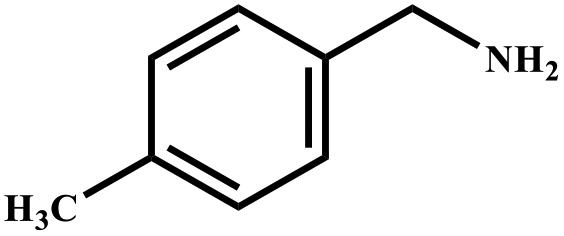
|
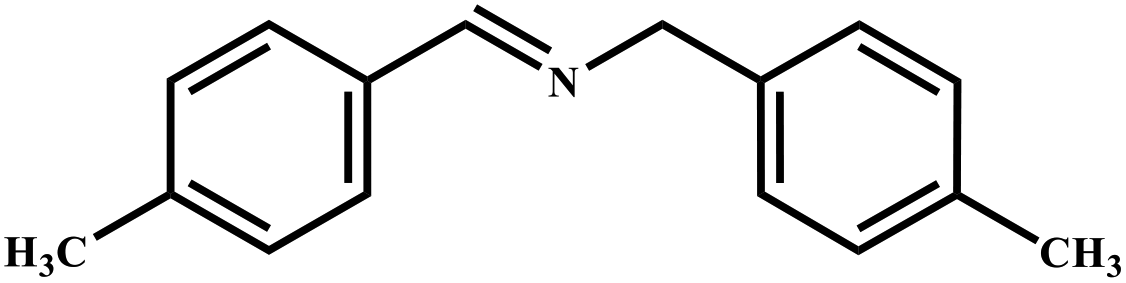
|
92 | 99 |
| 9 |
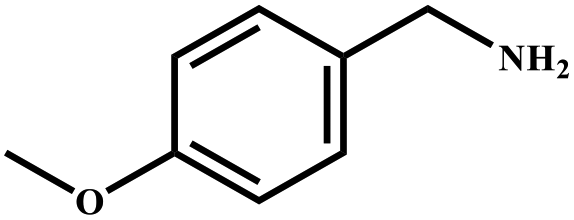
|
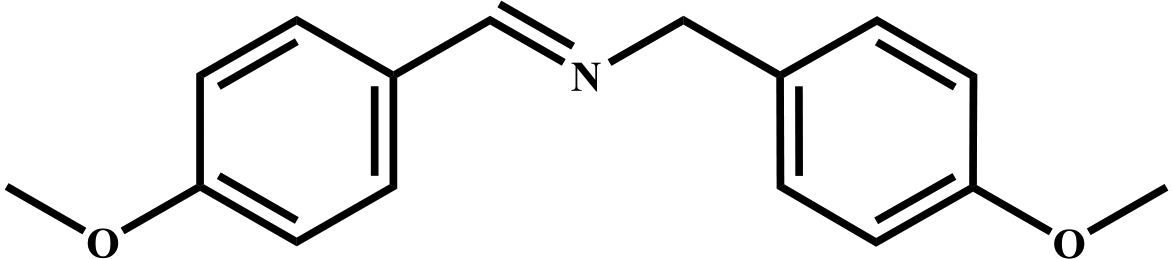
|
98 | 99 |
| 10 |
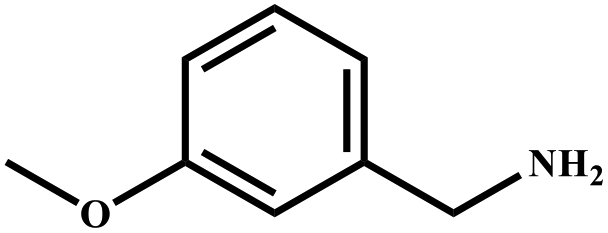
|
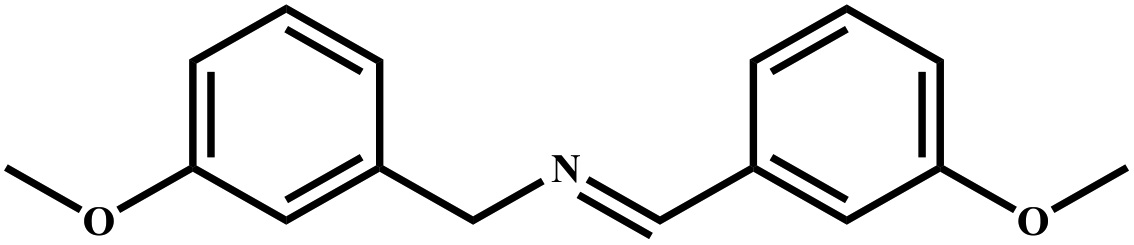
|
96 | 99 |
| 11 |
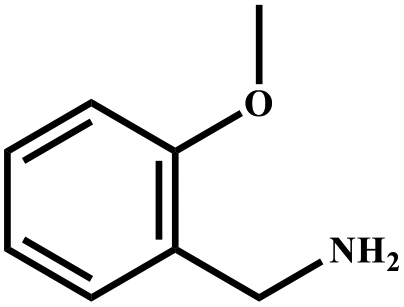
|
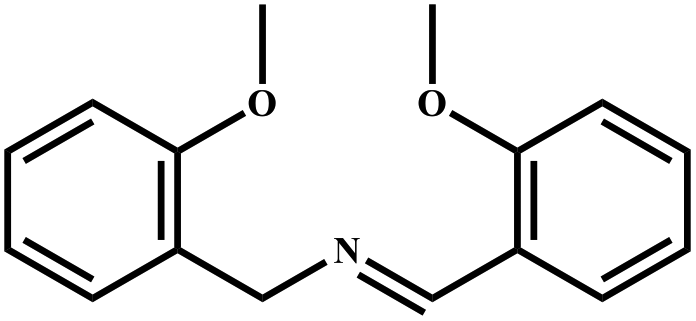
|
92 | 99 |
The durability of BCT photocatalysis was measured through recycling tests, and the data are summarized in Fig. 1h. It is worth mentioning that the conversion rate of BA remained at around 88% over the four cycles, indicating no significant loss of catalytic activity. The XRD patterns and SEM images of BCT, as shown in Fig. S2,† demonstrated no noticeable changes in the morphology compared to before the experiment, confirming the good structural stability of BCT.
Physicochemical properties and electronic structure
Chemical and physical techniques were employed to study the charge separation of typical samples and to elucidate the charge transfer mechanism in the S-scheme heterojunction. The optical absorption characteristics of the samples were investigated by UV-vis diffuse reflectance spectroscopy (UV-vis DRS) and compared with UiO-66(Ce); UiO-66(Ce/Ti) exhibited significantly enhanced optical absorption in the full spectral range. After introducing porous UiO-66(Ce/Ti) into BiOCl, the composite material BCT exhibited strong optical absorption under visible light irradiation (Fig. S3†). The band gap width of the prepared samples can be calculated using the Kubelka–Munk function formula.28 The band gaps of BiOCl, UiO-66(Ce), and UiO-66(Ce/Ti) were 3.02 eV, 2.81 eV, and 2.72 eV, respectively (Fig. 3a). The valence band positions of the prepared samples were evaluated based on the XPS-VB spectrum. The maximum VB positions of BiOCl, UiO-66(Ce), and UiO-66(Ce/Ti) were approximately 2.80 eV, 2.18 eV, and 2.36 eV, respectively (Fig. S4†). The conversion formula for hydrogen electrode potential is ENHE = Φ + VBmax − 4.44,29 where ENHE, Φ, and VBmax represent the potential of the NHE (eV), the electron work function of the instrument (3.88 eV), and the determined maximum valence band value by XPS-VB, respectively. Therefore, the VB potential values of BiOCl, UiO-66(Ce), and UiO-66(Ce/Ti) were 2.24 eV, 1.62 eV, and 1.80 eV (vs. NHE), respectively. The corresponding CB potential values of the above samples were calculated to be −0.78 eV, −1.19 eV, and −0.92 eV using the formula ECB = EVB − Eg.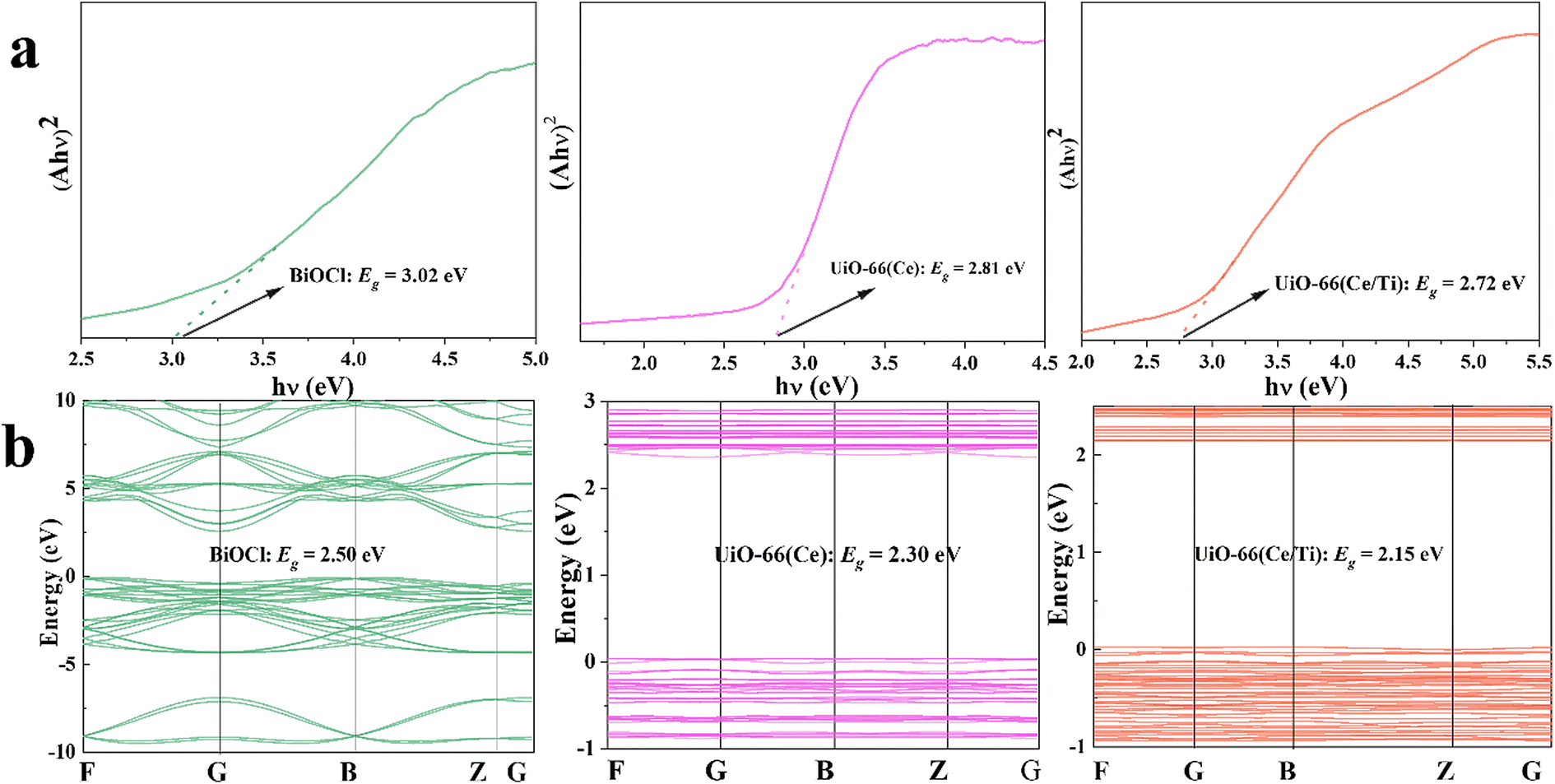 | ||
| Fig. 3 (a) Plots of (αhν)1/2 or (αhν)2vs. (hν) for the band gap energy of the samples calculated using the Kubelka–Munk function formula. (b) Band structures of the samples calculated by DFT simulations. | ||
Mott–Schottky tests were carried out to measure the flat band potential (FB)30 of BiOCl and UiO-66(Ce/Ti), as shown in Fig. S5,† the positive slopes of both BiOCl and the MOF indicate that they are n-type semiconductors.31 The FB potentials are −0.50 V and −0.68 V (relative to NHE) for BiOCl and UiO-66(Ce/Ti), respectively. According to previous reports,32 the CB potentials of BiOCl and UiO-66(Ce/Ti) were calculated to be −0.70 V and −0.88 V, respectively, which is consistent with the VB-XPS spectral results.
The band structures, total density of states (TDOS), and local density of states (LDOS) of BiOCl, UiO-66(Ce), UiO-66(Ce/Ti), and the BiOCl/UiO-66(Ce/Ti) heterojunction were calculated using DFT. From the observation of Fig. 3b, the band gaps of BiOCl, UiO-66(Ce), and UiO-66(Ce/Ti) were 2.50 eV, 2.30 eV, and 2.15 eV, respectively, which are smaller than the experimental values from UV-vis DRS. This phenomenon is due to the limitation of discrete Fourier transformation calculations, which does not consider the discontinuity of the exchange–correlation potential. In Fig. S6,† the valence band maximum (VBM) of BiOCl is mainly determined by the O 2p and Cl 3p states, while the contribution of Bi 6s and Bi 6p states is small. The conduction band minimum (CBM) is mainly controlled by the Bi 6p state. In pure UiO-66(Ce), the VBM is mainly contributed by the O 2p and C 1p states and the CBM is dominated by the Ce 4f state. After Ti atom doping, UiO-66(Ce/Ti) has the same VBM as UiO-66(Ce), while the CBM is mainly contributed by the Ce 4f and Ti 3d states. When BiOCl is coupled with UiO-66(Ce/Ti), the BCT structure exhibits enhanced charge density. Therefore, we speculate that the BCT heterojunction will show enhanced photo-responsiveness.
The work functions (W) of BiOCl and UiO-66(Ce/Ti) were calculated by DFT to be 7.56 and 6.77 eV, respectively (Fig. 4a). The calculated values of the Ef for BiOCl and UiO-66(Ce/Ti) were −7.56 eV and −6.77 eV, respectively (Ef = Vac − W,33 where Vac is the vacuum level, assumed to be 0). It is evident that BiOCl has a larger W (7.56 eV) and a lower Ef (−7.56 eV), while UiO-66(Ce/Ti) has a smaller W (6.77 eV) and a higher Ef (−6.77 eV). Therefore, when they come into contact, electrons can easily transfer from UiO-66(Ce/Ti) to BiOCl, bringing them to the same FE and generating an interface electric field (IEF). When the BCT heterojunction is exposed to light, the electrons of BiOCl and UiO-66(Ce/Ti) are excited from the valence band (VB) to the conduction band (CB), respectively. Under the combined effect of the IEF, interface band bending, and Coulomb interaction, the optically generated electrons in BiOCl flow towards UiO-66(Ce/Ti) and recombine with the optically generated holes in UiO-66(Ce/Ti). This charge transfer behavior follows the S-scheme mechanism (Fig. 4b).
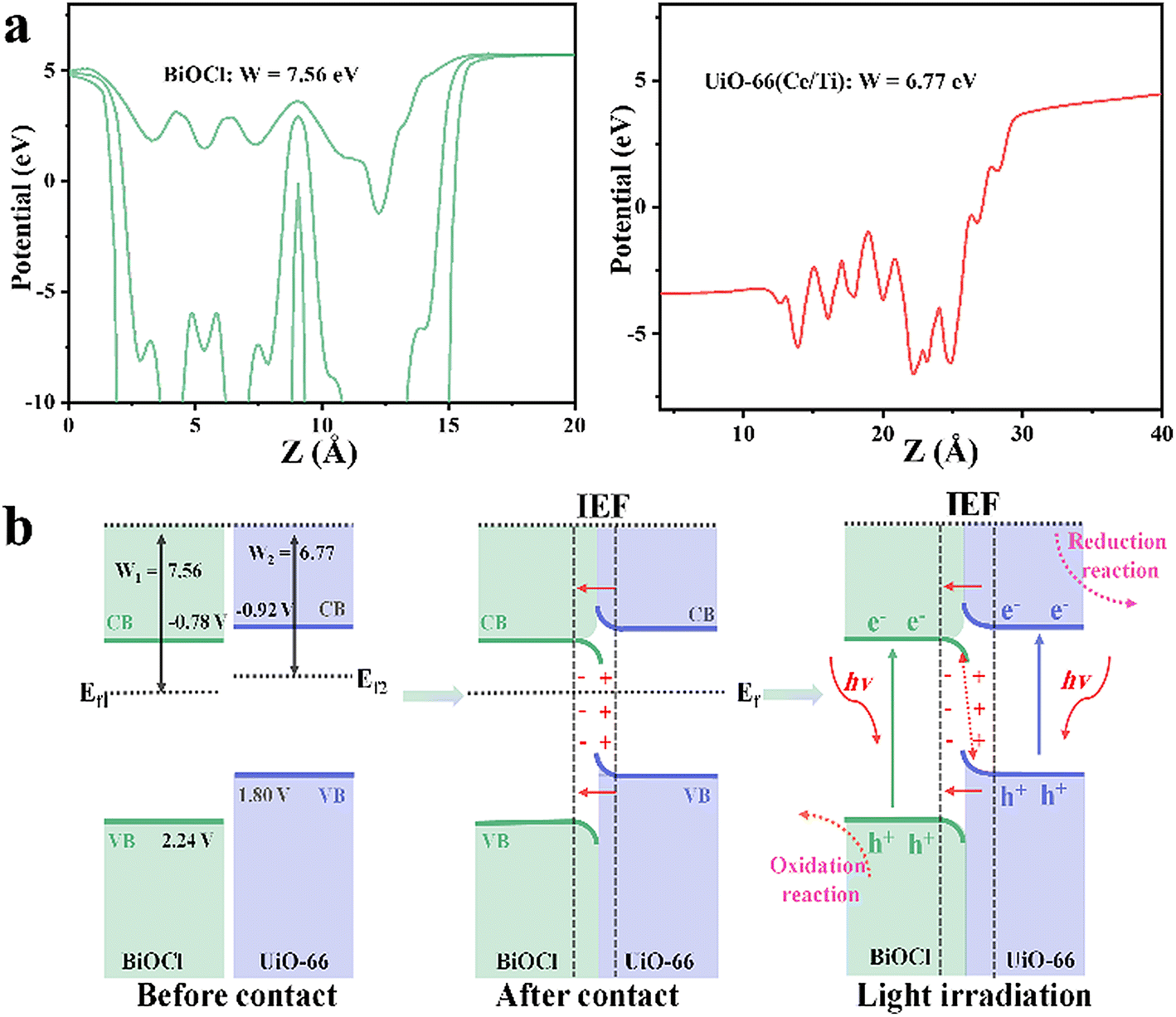 | ||
| Fig. 4 (a) Work functions (W) of BiOCl and UiO-66(Ce/Ti) calculated by DFT. (b) Schematic diagrams of the S-scheme heterojunction. | ||
To identify the surface elemental composition and electronic states of BiOCl, the MOF, and BCT, the X-ray photoelectron spectroscopy (XPS) technique was employed. As shown in Fig. S7,† the XPS survey spectra reveal the presence of Bi, Cl, Ce, Ti, O, C, and N elements in the BCT composite material, which is consistent with the EDS results. The characteristic peaks observed at 164.5 eV and 159.2 eV can be attributed to the binding energies of Bi 4f5/2 and Bi 4f7/2 (Fig. 5a), respectively, in BCT.34 The binding energies of Cl 2p in BCT are located at 199.5 eV and 197.9 eV, corresponding to Cl 2p1/2 and Cl 2p3/2, respectively (Fig. 5b). The high-resolution Ti 2p XPS spectra of BCT, as shown in Fig. 5c, indicate that Ti4+ occupies the majority of proportion. The peak values of 2p3/2 at 459.2 eV and 457.5 eV are attributed to Ti4+ and Ti3+, respectively. The peak values of Ti 2p1/2 at 466.64 eV, 465.04 eV, and 461.5 eV are assigned to Ti4+.26 It is worth noting that the interaction between the MOF and BiOCl may result in a new chemical environment, such as the formation of Ti–O covalent bonds. This leads to the redistribution of titanium electrons, making the Ti 2p1/2 level closer to oxygen atoms compared to the Ti 2p3/2 level. Therefore, the binding energy and peak position of the Ti 2p peak in BCT are increased compared to the pure MOF. Additionally, BCT exhibits two peaks at around 465.0 eV, possibly due to the overlap between the binding energy of Bi 4d3/2 at 466.5 eV and the binding energy of Ti 2p1/2 at 465.0 eV.35 In the Ce 3d XPS spectrum of BCT, the Ce 3d5/2 peaks at BEs of 881.4 eV and 885.9 eV are assigned to Ce3+, while the peaks at 883.6 eV and 887.9 eV are related to Ce4+ (Fig. 5d). The Ce 3d3/2 peaks at 899.0 eV and 904.5 eV belong to Ce3+ species, while the corresponding peaks at 901.2 eV and 908.0 eV are attributed to Ce4+.21,26 The C 1s spectra of BCT are shown in Fig. S7b,† and the peaks at 284.8, 286.3, and 288.9 eV are attributed to C–C/C–O and OC–O, respectively. The O 1s spectrum of BCT (Fig. S7c†) exhibits peaks at 530.2, 531.5, and 533.1 eV, corresponding to lattice oxygen, oxygen vacancies and chemisorbed oxygen, respectively.36 The content of oxygen vacancies in BCT is higher than that in BiOCl, consistent with the variation in the signal intensity of oxygen vacancies in the EPR spectra (Fig. S8†), indicating that the presence of Ti provides more active sites, affecting the adsorption and activation of BA. Besides, the change in the binding energy can be used to investigate the direction of carrier transfer in heterogeneous photocatalysts.37 Compared with BiOCl, the binding energy of Bi 4f and Cl 2p in the BCT composite material significantly shifts to lower energy levels in the dark, indicating an increased electron density in BiOCl. In contrast, the binding energy of Ti 2p and Ce 5d in BCT shifts to higher energy levels in the dark relative to the MOF, indicating an electron transfer from the MOF to BiOCl. When performing in situ irradiation XPS measurements, the binding energies of Bi 4f and Cl 2p in the BCT composite material significantly shift to higher energy levels under light irradiation compared to those in the dark. On the other hand, the binding energies of Ti 2p and Ce 5d in BCT noticeably shift to lower energy levels (Fig. 5c and d). This indicates that when both BiOCl and the MOF are simultaneously excited, photogenerated electrons transfer from BiOCl to the MOF, consistent with the S-scheme charge transfer model.
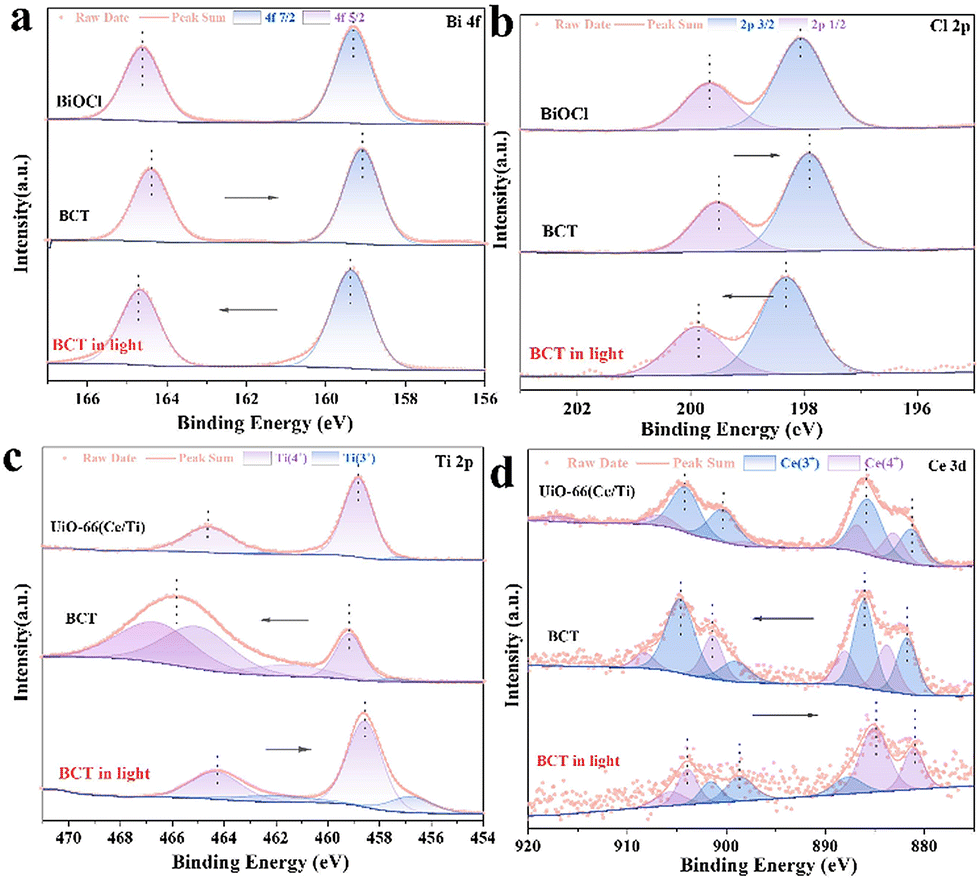 | ||
| Fig. 5 In situ XPS spectra before and after light irradiation: (a) Bi 4f, (b) Cl 2p, (c) Ti 2p, and (d) Ce 3d for the samples. | ||
To further verify the S-scheme mechanism of the BCT heterojunction, the electron paramagnetic resonance (EPR) technique was used to discriminate and semi-quantify the prepared materials, where 5,5-dimethylpyrroline N-oxide (DMPO) captured hydroxyl radicals (˙OH) and superoxide radicals (˙O2−) were used. The signal intensity of DMPO-˙OH increased in the order of MOF < BiOCl < BCT (Fig. 6a), while the signal intensity of DMPO-˙O2− increased in the order of BiOCl < MOF < BCT (Fig. 6b). The signal intensity of BCT was significantly stronger than that of the original BiOCl and MOF, indicating that the photogenerated charge transfer mechanism of the BCT heterojunction conforms to the S-scheme mechanism.37 Photoluminescence (PL) and electrochemical impedance spectroscopy (EIS) also support the above conclusion. The PL intensity of BCT is significantly lower than that of the original BiOCl and MOF, indicating that the construction of an S-scheme heterojunction photocatalyst can effectively suppress the recombination of photo-generated charge carriers (Fig. 6c). The EIS radius of the BCT heterojunction is smaller, indicating a higher photocurrent density compared to the original BiOCl and MOF, reaffirming the efficient separation and transfer efficiency of photo-induced charge carriers on the BCT heterojunction (Fig. 6d).
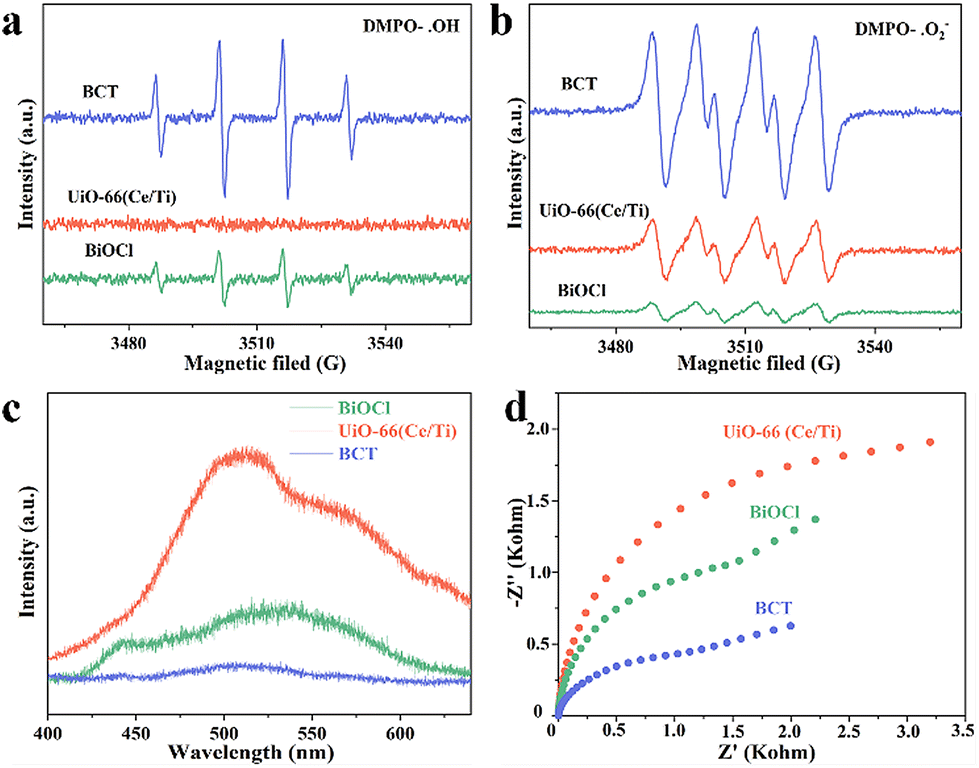 | ||
| Fig. 6 Electron paramagnetic resonance (EPR) spectra of (a) DMPO–OH adducts and (b) DMPO–O2− adducts over BiOCl, UiO-66(Ce/Ti), and BCT under illumination for 2 min. (c) Steady-state photoluminescence (PL) spectra and (d) electrochemical impedance spectroscopy (EIS) spectra of BiOCl, UiO-66(Ce/Ti), and BCT. | ||
Mechanistic investigation
The identification of reactive oxygen species (ROS) is the basis for studying reaction mechanisms. Firstly, we conducted quenching experiments to verify the selective generation of imines by ROS on BCT. Under the optimized conditions, KI, isopropanol (IPA), β-carotene and benzoquinone (BQ) were selected as trapping agents for holes (h+), ˙OH, (singlet oxygen) 1O2, and (superoxide radical anion) ˙O2−, respectively. The conversion of primary amines was almost inhibited when KI, β-carotene, and BQ were added, indicating that h+, 1O2 and ˙O2− played a vital role in the activation of primary amines, respectively (Table 1, entries 5–7). However, IPA hardly inhibited the action (Table 1, entry 8), suggesting that the role of ˙OH can be ignored.Next, EPR measurements were conducted to further detect the existence of 1O2 and ˙O2−. 2,2,6,6-Tetramethyl-piperidine (TEMP) is regarded as a typical trapping agent for 1O2. Under visible light irradiation and an O2 atmosphere, a strong 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet signal was observed after the addition of BCT (Fig. 7b). This signal can be assigned to the 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) radical formed via the reaction of TEMP with 1O2.38 Subsequently, EPR experiments with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the trapping agent for ˙O2− showed a strong specific quartet signal (Fig. 7a), indicating the formation of DMPO/˙OOH derived from the reaction of DMPO with ˙O2−.39,40 As expected, the addition of BA to a mixture of TEMP/BCT and DMPO/BCT attenuated the signal intensity (Fig. 7). The above results demonstrated that BCT is capable of 1O2 and ˙O2− generation, both of which play a key role in the photo-oxidation reaction of amines.
:1:1 triplet signal was observed after the addition of BCT (Fig. 7b). This signal can be assigned to the 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) radical formed via the reaction of TEMP with 1O2.38 Subsequently, EPR experiments with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the trapping agent for ˙O2− showed a strong specific quartet signal (Fig. 7a), indicating the formation of DMPO/˙OOH derived from the reaction of DMPO with ˙O2−.39,40 As expected, the addition of BA to a mixture of TEMP/BCT and DMPO/BCT attenuated the signal intensity (Fig. 7). The above results demonstrated that BCT is capable of 1O2 and ˙O2− generation, both of which play a key role in the photo-oxidation reaction of amines.
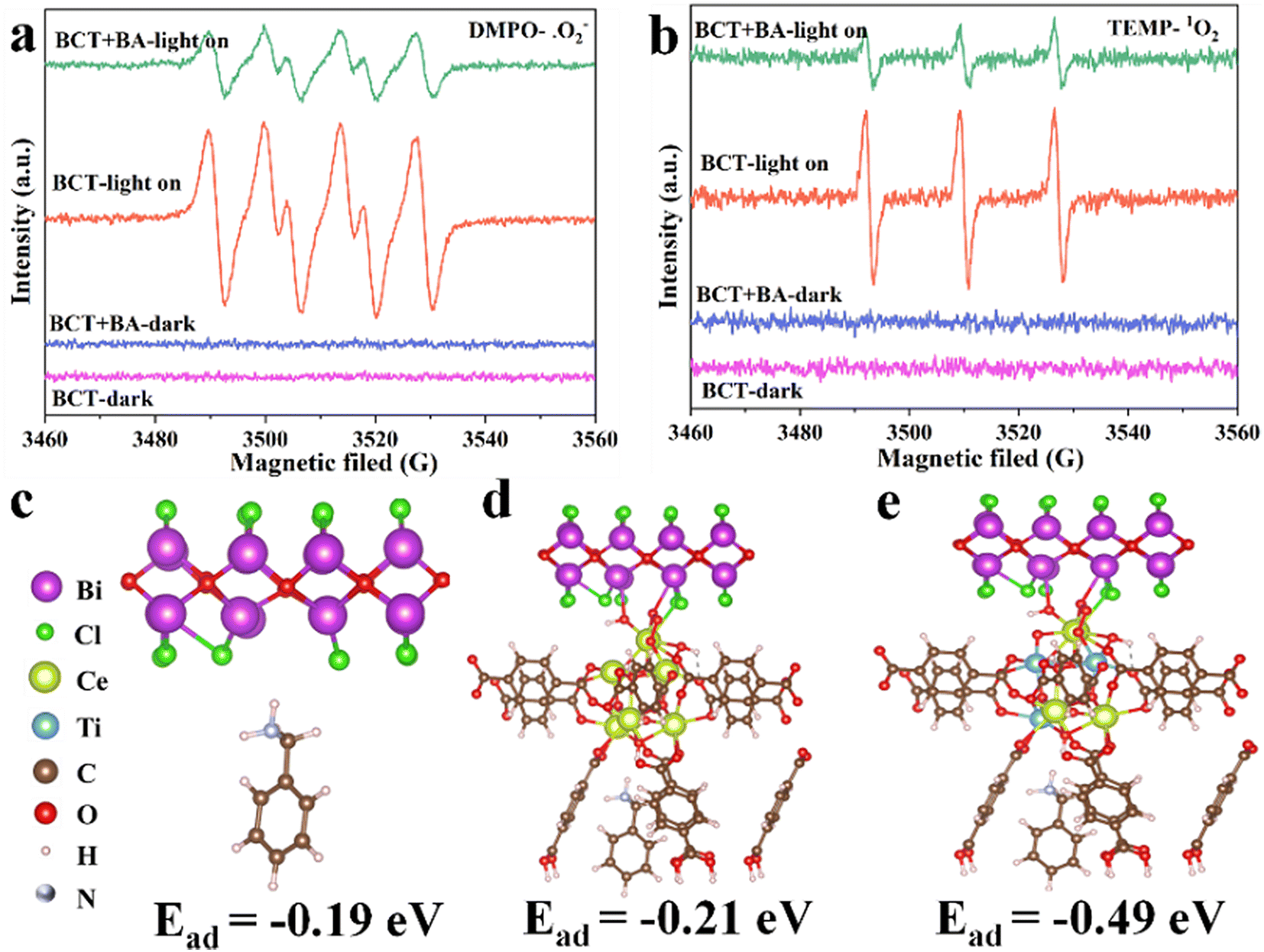 | ||
| Fig. 7 EPR spectra of the samples after mixing (a) TEMP and (b) DMPO solutions with BCT and O2 in the absence or presence of benzylamine under visible light irradiation or in the dark. Optimized adsorption structures: (c) BA on BiOCl, (d) the BiOCl/UiO-66(Ce) heterojunction, and (e) the BiOCl/UiO-66(Ce/Ti) heterojunction. Purple, green, yellow, blue, brown, red, pink, and blue grey atoms are Bi, Cl, Ce, Ti, C, O, H, and N respectively. | ||
Furthermore, the adsorption and activation of BA also play an important role in investigating the selective oxidation mechanism. The EPR spectrum shows that BCT exhibits the highest oxygen vacancy intensity. When BCT adsorbs BA, the oxygen vacancy intensity decreases significantly, indicating that the presence of Ti and oxygen vacancies can affect the adsorption and activation of BA (Fig. S8†), as discussed further in the DFT calculations below.
The calculated adsorption energies of BA on the original BiOCl, BiOCl/UiO-66(Ce), and BiOCl/UiO-66(Ce/Ti) are −0.19 eV, −0.21 eV, and −0.49 eV, respectively, indicating that the adsorption affinity of the heterojunction structure formed by Ti-doped UiO-66(Ce) and BiOCl is significantly enhanced (Fig. 7c–e). This also suggests that the presence of Ti and the formation of OVs will provide more active sites for BA adsorption. Subsequently, a deeper understanding of the adsorption behavior was obtained by calculating the differential charge density (DCD) (Fig. 8a). It was found that a large amount of charge was transferred from BiOCl to BA molecules through the MOF, laying the foundation for the activation of BA and the generation of the corresponding benzylamine free radical cation. In addition, the significant accumulation of charges around the BA molecules and the catalyst surface further confirms the strong affinity of BCT to BA. A large amount of electron enrichment can be observed at the interface of BiOCl/MOF, which may be attributed to the change in the interface electron environment caused by the S-scheme heterojunction, which is very beneficial for the selective oxidation of BA. During the simulation of the activation process of BA, it was found that the activation energy barriers for BA on BCT (0.88 eV, −0.36 eV, −0.42 eV) are significantly lower than those on BiOCl/UIO-66(Ce) (Fig. 8b). The above DFT calculation results indicate that BCT is more conducive to the adsorption and activation of BA, and thermodynamically more favorable for the oxidation of BA.
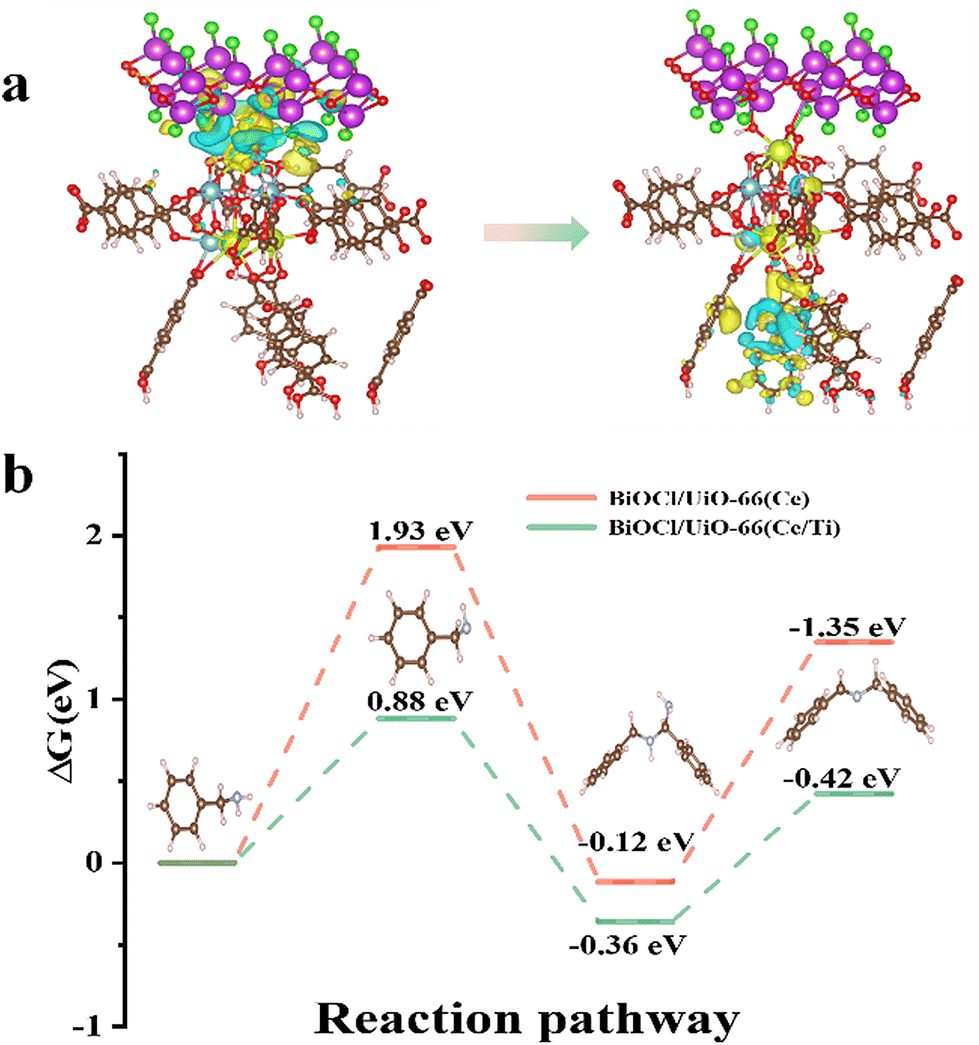 | ||
| Fig. 8 Differential charge densities (DCDs) of (a) BiOCl/UiO-66(Ce/Ti) before and after the adsorption of BA; (b) free energy diagrams for the adsorption and photoconversion of BA on BiOCl/UiO-66(Ce/Ti) and BiOCl/UiO-66(Ce). Purple, green, yellow, blue, brown, red, pink, and blue grey atoms are Bi, Cl, Ce, Ti, C, O, H, and N respectively. | ||
Therefore, combining experiments and DFT calculations, the mechanism of electron transfer and energy transfer may be involved in BA photocatalysis on BCT, as shown in Fig. 9. Under light irradiation conditions, BiOCl and the MOF transition into excited states by excitation of electrons from the valence band (VB) to the conduction band (CB). Subsequently, driven by the Coulomb interaction and the interfacial electric field (IEF), the electrons accumulated in the CB of BiOCl tend to recombine with the holes in the VB of the MOF. This S-scheme band structure is conducive to eliminating the relatively useless electrons in the CB of BiOCl and the holes in the VB of the MOF, thereby promoting spatial separation of photocarrier pairs. Electrons will accumulate in the MOF and generate a singlet excited state of the MOF (MOF*(S1)).
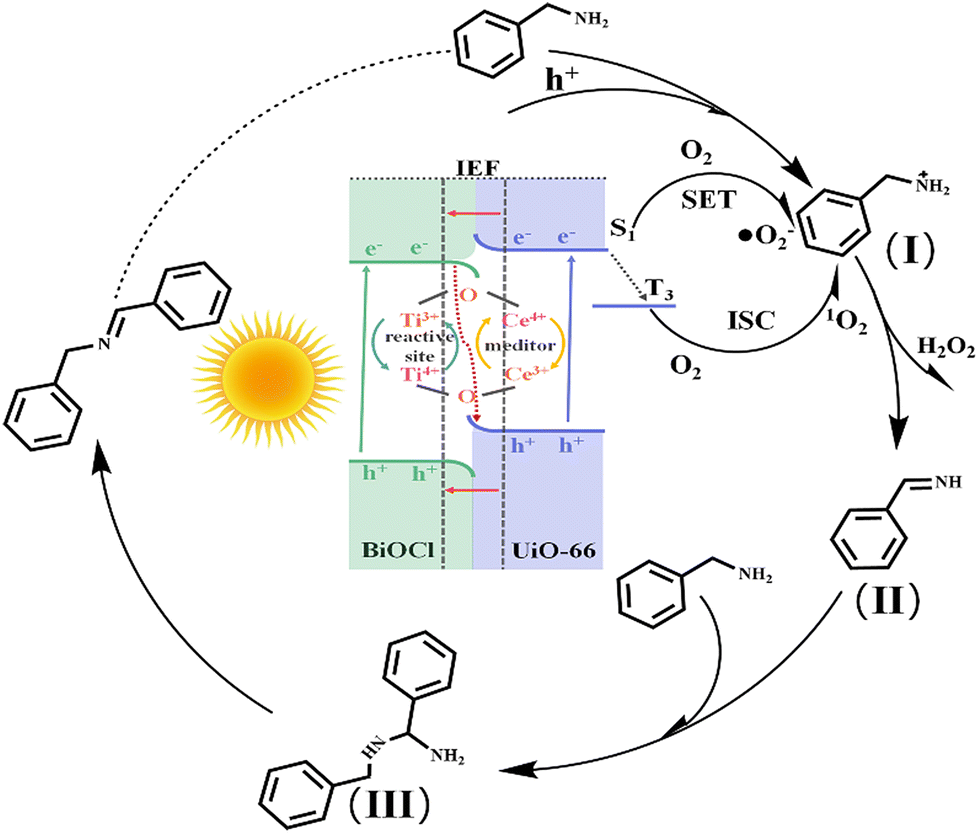 | ||
| Fig. 9 Photocatalytic mechanism for the oxidation of benzylamine using the BiOCl/UiO-66(Ce/Ti) heterojunction under visible light irradiation. | ||
Meanwhile, O2 is directly activated to ˙O2−41 through single electron transfer (SET). In addition, the presence of electron interaction between Ce and Ti leads to the formation of two redox media, Ce4+/Ce3+ and Ti4+/Ti3+, which will promote the generation of more Lewis acid sites on the surface of the photocatalyst. More importantly, the rich oxygen vacancies (OVs) in the MOF are beneficial for the adsorption and activation of BA molecules, thus reducing the overall activation energy barrier for BA oxidation. Therefore, BCT exhibits good activity in the photocatalytic oxidation of BA, which may be attributed to the synergistic effect of the S-scheme band structure and the conversion of Ce4+ + Ti3+ ⇌ Ce3+ + Ti4+ + OVs. On the other hand, MOF(MOF*(S1)) may undergo an intersystem crossing (ISC) to convert to the triple excited state of MOF(MOF*(T3)), and then MOF(MOF*(T3)) activates O2 to form 1O2.42 Meanwhile, the holes aggregated on the VB of BiOCl react with the BA molecules adsorbed on the MOF sites to generate benzylamine radical cation species (I). Active oxygen species (1O2 and ˙O2−) react with the radical cation species (I) to produce NH-imine intermediates (II) and the byproduct H2O2.43 The production of H2O2 in the benzylamine coupling reaction mixture was identified by oxidation using KMnO4 (Fig. S9†).44 It is worth noting that NH-imine exhibits strong reactivity in the photocatalytic process and is therefore easily attacked by amines to form intermediate III. Subsequently, intermediate III removes ammonia with the assistance of a proton to generate the imine product.
Conclusions
In summary, by a simple cation exchange process, Ti doping was introduced into UiO-66(Ce) to obtain UiO-66(Ce/Ti), and its successful coupling with the nanosheets of BiOCl resulted in the fabrication of an S-scheme BiOCl/UiO-66(Ce/Ti) heterojunction. The imine conversion rate of the heterojunction is 2.5 and 3.3 times higher than that of the original UiO-66(Ce) and BiOCl, respectively. The experimental results and DFT calculations indicated that the formation of the BiOCl/UiO-66(Ce/Ti) heterojunction led to the establishment of a strong internal electric field, enhancing the charge separation efficiency, retaining the strong redox ability, and increasing the adsorption and activation ability of amines. Additionally, the electron interaction between Ce and Ti resulted in Ce3+ + Ti4+ + OVs, promoting the generation of more Lewis acid sites on the surface of the photocatalyst and creating a rich oxygen environment at the heterojunction interface, synergistically enhancing the adsorption and activation ability of amines in the S-scheme structure. This study highlights the rational regulation of the microenvironment at the heterojunction interface, providing a new perspective for the design of highly efficient S-scheme heterojunction photocatalysts.Author contributions
Danxia Zhao: investigation, methodology, software and writing – original draft. Pengyu Wu: investigation and writing – original draft. Huayue Zhu: supervision and funding acquisition. Ru Jiang: supervision and writing – review and editing. Jingwei Chen: methodology and data curation. Chuhan Qiu: methodology and data curation. Shengtao Jiang: supervision and funding acquisition. Guoping Lu: writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support by the Zhejiang Provincial Natural Science Foundation of China (Grant No. LTY21B070001), the Scientific and Technological Development Project of Taizhou City (Grant No. 22gya08), the Science and Technology Project of Taizhou (22hba02) and the Science Foundation of Taizhou University for Distinguished Young Scholars (Grant No. 2018JQ001).References
- B. Xu, E. M. Hartigan, G. Feula, Z. Huang, J.-P. Lumb and B. A. Arndtsen, Simple Copper Catalysts for the Aerobic Oxidation of Amines: Selectivity Control by the Counterion, Angew. Chem., Int. Ed., 2016, 55, 15802–15806 CrossRef CAS.
- Y. Sun, H. Shin, F. Wang, B. Tian, C.-W. Chiang, S. Liu, X. Li, Y. Wang, L. Tang, W. A. Goddard III and M. Ding, Highly Selective Electrocatalytic Oxidation of Amines to Nitriles Assisted by Water Oxidation on Metal-Doped α-Ni(OH)2, J. Am. Chem. Soc., 2022, 144, 15185–15192 CrossRef CAS PubMed.
- Y. Liang, J. Wei, X. Qiu and N. Jiao, Homogeneous Oxygenase Catalysis, Chem. Rev., 2018, 118, 4912–4945 CrossRef CAS PubMed.
- N. Zhang, X. Li, H. Ye, S. Chen, H. Ju, D. Liu, Y. Lin, W. Ye, C. Wang, Q. Xu, J. Zhu, L. Song, J. Jiang and Y. Xiong, Oxide Defect Engineering Enables to Couple Solar Energy into Oxygen Activation, J. Am. Chem. Soc., 2016, 138, 8928–8935 CrossRef CAS PubMed.
- S.-D. Wang, C.-L. Lai, Y.-X. Zhang, S.-T. Bao, K.-L. Lv and L.-L. Wen, Effective charge and energy transfers within a metal–organic framework for efficient photocatalytic oxidation of amines and sulfides, J. Mater. Chem. A, 2022, 10, 20975–20983 RSC.
- R. Chen, J.-L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Designed Synthesis of a 2D Porphyrin-Based sp2 Carbon-Conjugated Covalent Organic Framework for Heterogeneous Photocatalysis, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS PubMed.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Ultrathin 2D/2D WO3/g-C3N4 step-scheme H2-production photocatalyst, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- X. Cai, Y. Zang, S. Zang, S. Tang, F. Jing, L. Mo, D. Teng, W. Lin and G. Zhang, Binary pyrene-benzene polymer/Znln2S4 S-scheme photocatalyst for enhanced hydrogen evolution and antibiotics degradation, Appl. Surf. Sci., 2023, 637, 157871 CrossRef CAS.
- G. Chen, Z. Zhou, B. Li, X. Lin, C. Yang, Y. Fang, W. Lin, Y. Hou, G. Zhang and S. Wang, S-scheme heterojunction of crystalline carbon nitride nanosheets and ultrafine WO3 nanoparticles for photocatalytic CO2 reduction, J. Environ. Sci., 2023 DOI:10.1016/j.jes.2023.05.028.
- B. Zhang, J. Zhang, R. Duan, Q. Wan, X. Tan, Z. Su, B. Han, L. Zheng and G. Mo, BiOCl nanosheets with periodic nanochannels for high-efficiency photooxidation, Nano Energy, 2020, 78, 105340 CrossRef CAS.
- Y. Guan, Y. Liu, Q. Lv and J. Wu, Bismuth-based photocatalyst for photocatalytic oxidation of flue gas mercury removal: A review, J. Hazard. Mater., 2021, 418, 126280 CrossRef CAS PubMed.
- A. P. Katsoulidis, D. Antypov, G. F. S. Whitehead, E. J. Carrington, D. J. Adams, N. G. Berry, G. R. Darling, M. S. Dyer and M. J. Rosseinsky, Chemical control of structure and guest uptake by a conformationally mobile porous material, Nature, 2019, 565, 213–217 CrossRef CAS PubMed.
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu, Y. Pan, H.-L. Jiang, D. Mei and C. Zhong, Self-adaptive dual-metal-site pairs in metal-organic frameworks for selective CO2 photoreduction to CH4, Nat. Catal., 2021, 4, 719–729 CrossRef CAS.
- Q. Wang, L. Wang, S. Zheng, M. Tan, L. Yang, Y. Fu, Q. Li, H. Du and G. Yang, The strong interaction and confinement effect of Ag@NH2-MIL-88B for improving the conversion and durability of photocatalytic Cr(VI) reduction in the presence of a hole scavenger, J. Hazard. Mater., 2023, 451, 131149 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- T.-F. Chen, S.-Y. Han, Z.-P. Wang, H. Gao, L.-Y. Wang, Y.-H. Deng, C.-Q. Wan, Y. Tian, Q. Wang, G. Wang and G.-S. Li, Modified UiO-66 frameworks with methylthio, thiol and sulfonic acid function groups: The structure and visible-light-driven photocatalytic property study, Appl. Catal., B, 2019, 259, 118047 CrossRef CAS.
- S. Zheng, H. Du, L. Yang, M. Tan, N. Li, Y. Fu, D. Hao and Q. Wang, PDINH bridged NH2-UiO-66(Zr) Z-scheme heterojunction for promoted photocatalytic Cr(VI) reduction and antibacterial activity, J. Hazard. Mater., 2023, 447, 130849 CrossRef CAS PubMed.
- Y. Xiong, S. Chen, F. Ye, L. Su, C. Zhang, S. Shen and S. Zhao, Synthesis of a mixed valence state Ce-MOF as an oxidase mimetic for the colorimetric detection of biothiols, Chem. Commun., 2015, 51, 4635–4638 RSC.
- R. Dalapati, B. Sakthivel, M. K. Ghosalya, A. Dhakshinamoorthy and S. Biswas, A cerium-based metal–organic framework having inherent oxidase-like activity applicable for colorimetric sensing of biothiols and aerobic oxidation of thiols, CrystEngComm, 2017, 19, 5915–5925 RSC.
- X.-P. Wu, L. Gagliardi and D. G. Truhlar, Metal doping in cerium metal-organic frameworks for visible-response water splitting photocatalysts, J. Chem. Phys., 2018, 150, 041701 CrossRef PubMed.
- Y. Zhang, H. Chen, Y. Pan, X. Zeng, X. Jiang, Z. Long and X. Hou, Cerium-based UiO-66 metal–organic frameworks explored as efficient redox catalysts: titanium incorporation and generation of abundant oxygen vacancies, Chem. Commun., 2019, 55, 13959–13962 RSC.
- A. Wang, Y. Zhou, Z. Wang, M. Chen, L. Sun and X. Liu, Titanium incorporated with UiO-66(Zr)-type Metal–Organic Framework (MOF) for photocatalytic application, RSC Adv., 2016, 6, 3671–3679 RSC.
- X. Zhang, X. Zhang, L. Song, F. Hou, Y. Yang, Y. Wang and N. Liu, Enhanced catalytic performance for CO oxidation and preferential CO oxidation over CuO/CeO2 catalysts synthesized from metal organic framework: Effects of preparation methods, Int. J. Hydrogen Energy, 2018, 43, 18279–18288 CrossRef CAS.
- M. Lammert, M. T. Wharmby, S. Smolders, B. Bueken, A. Lieb, K. A. Lomachenko, D. D. Vos and N. Stock, Cerium-based metal organic frameworks with UiO-66 architecture: synthesis, properties and redox catalytic activity, Chem. Commun., 2015, 51, 12578–12581 RSC.
- D.-X. Zhao, G.-P. Lu and C. Cai, Efficient visible-light-driven Suzuki coupling reaction over Co-doped BiOCl/Ce-doped Bi2O2CO3 composites, Green Chem., 2021, 23, 1823–1833 RSC.
- P. Parnicka, W. Lisowski, T. Klimczuk, A. Mikolajczyk and A. Zaleska-Medynska, A novel (Ti/Ce)UiO-X MOFs@TiO2 heterojunction for enhanced photocatalytic performance: Boosting via Ce4+/Ce3+ and Ti4+/Ti3+ redox mediators, Appl. Catal., B, 2022, 310, 121349 CrossRef CAS.
- K. I. Hadjiivanov, D. A. Panayotov, M. Y. Mihaylov, E. Z. Ivanova, K. K. Chakarova, S. M. Andonova and N. L. Drenchev, Power of Infrared and Raman Spectroscopies to Characterize Metal-Organic Frameworks and Investigate Their Interaction with Guest Molecules, Chem. Rev., 2021, 121, 1286–1424 CrossRef CAS.
- X. Yuan, D. Shen, Q. Zhang, H. Zou, Z. Liu and F. Peng, Z-scheme Bi2WO6/CuBi2O4 heterojunction mediated by interfacial electric field for efficient visible-light photocatalytic degradation of tetracycline, Chem. Eng. J., 2019, 369, 292–301 CrossRef CAS.
- D. Zhao and C. Cai, Preparation of Bi2MoO6/Ti3C2 MXene heterojunction photocatalysts for fast tetracycline degradation and Cr(vi) reduction, Inorg. Chem. Front., 2020, 7, 2799–2808 RSC.
- Z. Jiang, W. Wan, H. Li, S. Yuan, H. Zhao and P. K. Wong, A Hierarchical Z-Scheme α-Fe2O3/g-C3N4 Hybrid for Enhanced Photocatalytic CO2 Reduction, Adv. Mater., 2018, 30, 1706108 CrossRef.
- H.-Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, Visible-Light Photoreduction of CO2 in a Metal-Organic Framework: Boosting Electron-Hole Separation via Electron Trap States, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed.
- C. Cheng, J. Zhang, B. Zhu, G. Liang, L. Zhang and J. Yu, Verifying the Charge-Transfer Mechanism in S-Scheme Heterojunctions Using Femtosecond Transient Absorption Spectroscopy, Angew. Chem., Int. Ed., 2023, 62, e202218688 CrossRef CAS PubMed.
- Y. Xia, B. Cheng, J. Fan, J. Yu and G. Liu, Unraveling Photoexcited Charge Transfer Pathway and Process of CdS/Graphene Nanoribbon Composites toward Visible-Light Photocatalytic Hydrogen Evolution, Small, 2019, 15, 1902459 CrossRef PubMed.
- C. Li, J. Zhang, X. Chen, H. Tao, Y. Zhou and M. Zhu, Upgraded charge transfer by an internal electric field in 2D/2D BiOCl/N-rich C3N5 heterojunctions for efficiently visible-light catalytic NO removal, Chem. Eng. J., 2023, 468, 143753 CrossRef CAS.
- Q. Hu, J. Di, B. Wang, M. Ji, Y. Chen, J. Xia, H. Li and Y. Zhao, In-situ preparation of NH2-MIL-125(Ti)/BiOCl composite with accelerating charge carriers for boosting visible light photocatalytic activity, Appl. Surf. Sci., 2019, 466, 525–534 CrossRef CAS.
- Y. Sun, H. Ji, Y. Sun, G. Zhang, H. Zhou, S. Cao, S. Liu, L. Zhang, W. Li, X. Zhu and H. Pang, Synergistic Effect of Oxygen Vacancy and High Porosity of Nano MIL-125(Ti) for Enhanced Photocatalytic Nitrogen Fixation, Angew. Chem., Int. Ed., 2023, e202316973 Search PubMed.
- L. Wang, B. Cheng, L. Zhang and J. Yu, In situ Irradiated XPS Investigation on S-Scheme TiO2@ZnIn2S4 Photocatalyst for Efficient Photocatalytic CO2 Reduction, Small, 2021, 17, 2103447 CrossRef CAS PubMed.
- X. Liang, Z. Guo, H. Wei, X. Liu, H. Lv and H. Xing, Selective photooxidation of sulfides mediated by singlet oxygen using visible-light-responsive coordination polymers, Chem. Commun., 2018, 54, 13002–13005 RSC.
- Y. Chen, Y. Wang, W. Li, Q. Yang, Q. Hou, L. Wei, L. Liu, F. Huang and M. Ju, Enhancement of photocatalytic performance with the use of noble-metal-decorated TiO2 nanocrystals as highly active catalysts for aerobic oxidation under visible-light irradiation, Appl. Catal., B, 2017, 210, 352–367 CrossRef CAS.
- J. Jiang, Z. Liang, X. Xiong, X. Zhou and H. Ji, A Carbazolyl Porphyrin-Based Conjugated Microporous Polymer for Metal-Free Photocatalytic Aerobic Oxidation Reactions, ChemCatChem, 2020, 12, 3523–3529 CrossRef CAS.
- C. Su, R. Tandiana, B. Tian, A. Sengupta, W. Tang, J. Su and K. P. Loh, Visible-Light Photocatalysis of Aerobic Oxidation Reactions Using Carbazolic Conjugated Microporous Polymers, ACS Catal., 2016, 6, 3594–3599 CrossRef CAS.
- J. Jiang, R. Luo, X. Zhou, Y. Chen and H. Ji, Photocatalytic Properties and Mechanistic Insights into Visible Light-Promoted Aerobic Oxidation of Sulfides to Sulfoxides via Tin Porphyrin-Based Porous Aromatic Frameworks, Adv. Synth. Catal., 2018, 360, 4402–4411 CrossRef CAS.
- V. R. Battula, H. Singh, S. Kumar, I. Bala, S. K. Pal and K. Kailasam, Natural Sunlight Driven Oxidative Homocoupling of Amines by a Truxene-Based Conjugated Microporous Polymer, ACS Catal., 2018, 8, 6751–6759 CrossRef CAS.
- H. Gao, Y. Chen, H. Li, F. Zhang and G. Tian, Hierarchical Cu7S4-Cu9S8 heterostructure hollow cubes for photothermal aerobic oxidation of amines, Chem. Eng. J., 2019, 363, 247–258 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qi02645k |
| This journal is © the Partner Organisations 2024 |