DOI:
10.1039/D4PY00061G
(Paper)
Polym. Chem., 2024,
15, 1437-1452
High-performance polyethylene elastomers using a hybrid steric approach in α-diimine nickel precatalysts†
Received
18th January 2024
, Accepted 7th March 2024
First published on 7th March 2024
Abstract
The synthesis of polyethylenes with concurrent high mechanical and elastic properties at elevated reaction temperatures using nickel catalysts proves to be challenging, primarily due to excessive chain walking and transfer reactions. In this study, hybrid steric hindrance, involving flexible cycloalkyl and relatively compact benzhydryl groups, was introduced into the α-diimine structure to prepare a set of nickel complexes, aiming to enhance the catalytic performance and polyethylene properties simultaneously for ethylene polymerization. Upon activation with EASC, these precatalysts displayed exceptionally high activity (up to 4.3 × 107 g mol−1 h−1 at 30 °C) and high thermal stability (activity up to 1.8 × 106 g mol−1 h−1 at 110 °C over a period of 30 min). High to ultra-high molecular weights and a moderate to high number of branches (34–145 per 1000C) along with narrow unimodal dispersity (PDI ≤ 2) across various reaction conditions are the characteristics of the obtained polyethylene. Of significant note is that the polyethylene obtained at high reaction temperature using these precatalysts exhibited significantly distinguished mechanical and elastic properties. Particularly, the PE prepared at 80 °C displayed high tensile strength (σ = 25.9 MPa) concurrently with high elastic recovery (SR = 70%), a combination of mechanical and elastic properties rarely reported in polyethylenes prepared at high temperatures. These results highlight the role of the hybrid steric bulk strategy in effectively controlling the ratio of chain walking and chain growth reactions.
Introduction
Thermoplastic polyolefin elastomers are extensively studied polymers, valued for their broad applications in the automotive, construction, and consumer goods industries. The appeal of these polymers lies in their ability to provide durable, weather-resistant, and cost-effective materials suitable for sealing, gasketing, and impact absorption.1–3 Linear low-density polyethylenes (LLDPE), ethylene/propylene elastomers, polyolefin elastomers, branched polymers, are mainly obtained by transition metal catalyzed copolymerization, primarily using ethylene and α-olefins (such as 1-octene, 1-hexene, 1-butene) as feedstocks.4,5 Incorporation of comonomers introduces branches along the polymer chain, reducing the crystallinity and imparting unique elastic and mechanical properties that are unattainable in the homopolymerization of ethylene alone.6–8 In the mid-1990s, Brookhart introduced late transition metal catalysts using α-diimine as a ligand structure for ethylene polymerization.9,10 These catalysts are notable for their “chain walking” ability, enabling the production of branched polyethylenes using only ethylene as the monomer.11–14 Most importantly, it offers a straightforward, single-step method to produce a thermoplastic elastomer with high elastic and mechanical properties. These catalysts exhibit excellent performance when used at room temperature. However, at industrial operating temperatures (>80 °C), they tend to show high rates of chain transfer and chain walking reactions.7,12,15–18 Excessive occurrences of these reactions (chain transfer and chain walking reactions) during polymerization can lead to highly branched polyethylenes with low molecular weights. This results in polymers that are completely amorphous and have reduced mechanical properties.12,15 Thus, the balance between chain walking and chain growth reactions is significantly important to achieve high performance polyethylenes.16–18 Until now, the development of high-performance catalysts that can maintain high polymerization activity, control the polymer molecular weight and branching density at elevated temperatures, remains a significant challenge in the field.
To date, immense structural modifications have been done in α-diimine nickel catalysts, and changes were mainly made at the N-aryl part, the backbone of the ligand or completely modify the N,N-bidentate ligand framework, for enhancing their resistance against facile decomposition and uncontrolled polymer properties of the resulting polyethylene.19–21 Steric variations at the ortho position of the N-phenyl moiety significantly enhance the catalytic performance. This improvement is attributed to the introduction of steric bulk at the axial position of the metal center, which restricts N-aryl rotation and consequently mitigates associated chain transfer reactions.20 A variety of α-diimine nickel precatalysts have been extensively investigated so far.19,21 Based on the mechanical and elastic properties of polyethylenes, some representative α-diimine nickel precatalysts are shown in Fig. 1.22–33 These catalysts are good for producing high-performance polyethylenes with high mechanical and elastic properties at low temperatures (≤30 °C). However, the mechanical properties of polyethylenes prepared at a higher temperature exhibit low tensile strength but have high associated strain and elastic recovery. This is because of high chain walking and chain transfer reactions as compared to the chain growth reactions, leading to low polymer molecular weights and high branching density. For instance, catalyzed by catalysts A and B at 50 °C, polyethylene exhibits a low tensile strength (2.5 and 3.3 MPa, respectively) but high strain recovery (70% and 87%, respectively).22,23 The tensile strength of the polyethylene prepared at 80 °C using catalyst C decreased to 2.1 MPa, while exhibiting a high strain recovery of up to 71%.24 Similar mechanical and elastic properties were also observed for polyethylene samples prepared using catalysts D, E, and F.25–27 On the other hand, symmetrical α-diimine nickel precatalysts gave better control over the chain walking reactions and promoted chain growth reactions. For instance, the catalyst G-based polyethylene produced at 100 °C exhibited high tensile strength with an elongation of break value of 763% but was not good in elastic recovery (SR = 27%).28 In the case of catalyst H, both the tensile strength and elastic recovery of the polyethylene produced at 70 °C were comparatively good (σ = 9.4 MPa, ε = 680%, SR = 76%).29 More promising results were observed with precatalyst I.30 The polyethylene synthesized at 100 °C demonstrated an excellent combination of mechanical and elastic properties (σ = 10.0 MPa, ε = 1863% and SR = 55%). The polyethylene based on the precatalyst J, containing a cycloalkyl group as the ortho substituent, showed excellent tensile strength; however, it exhibited relatively poor elastic recovery.31 On the other hand, the polyethylene obtained using precatalysts K and L showed notable elastic recovery, however its mechanical properties were relatively inferior.32,33 These polymers do not exhibit optimal performance in applications where there is a simultaneous demand for high mechanical strength and elasticity. Thus, well-balanced steric hindrance in α-diimine nickel catalysts is critical for obtaining high-performance polyethylenes at higher reaction temperatures. Encouraged by the catalytic performance of precatalyst I, we were further interested in incorporating a carbocyclic ring at the ortho position of the N-aryl unit.30 In this study, we have prepared a series of α-diimine nickel complexes. In situ activation of these precatalysts with EASC endowed them with excellent catalytic properties such as high thermal stability (up to 110 °C), high polymerization activity and high to ultra-high molecular weight with a narrow molecular weight distribution across all reaction temperatures. Most importantly, polyethylenes synthesized at both low and high reaction temperatures (80 °C) exhibited high mechanical and elastic properties simultaneously (σ = 25.9 MPa, ε = 1280% and SR = 70%). Polyethylenes with such a combination of high mechanical and elastic properties are rarely achieved.
 |
| | Fig. 1 Structural modifications in α-diimine nickel precatalysts and their impact on the mechanical and elastic properties (data reported of mechanical and elastic properties herein is selected for those polyethylene samples which were prepared at the highest reaction temperature). | |
Results and discussion
Synthesis of ligands and their nickel complexes
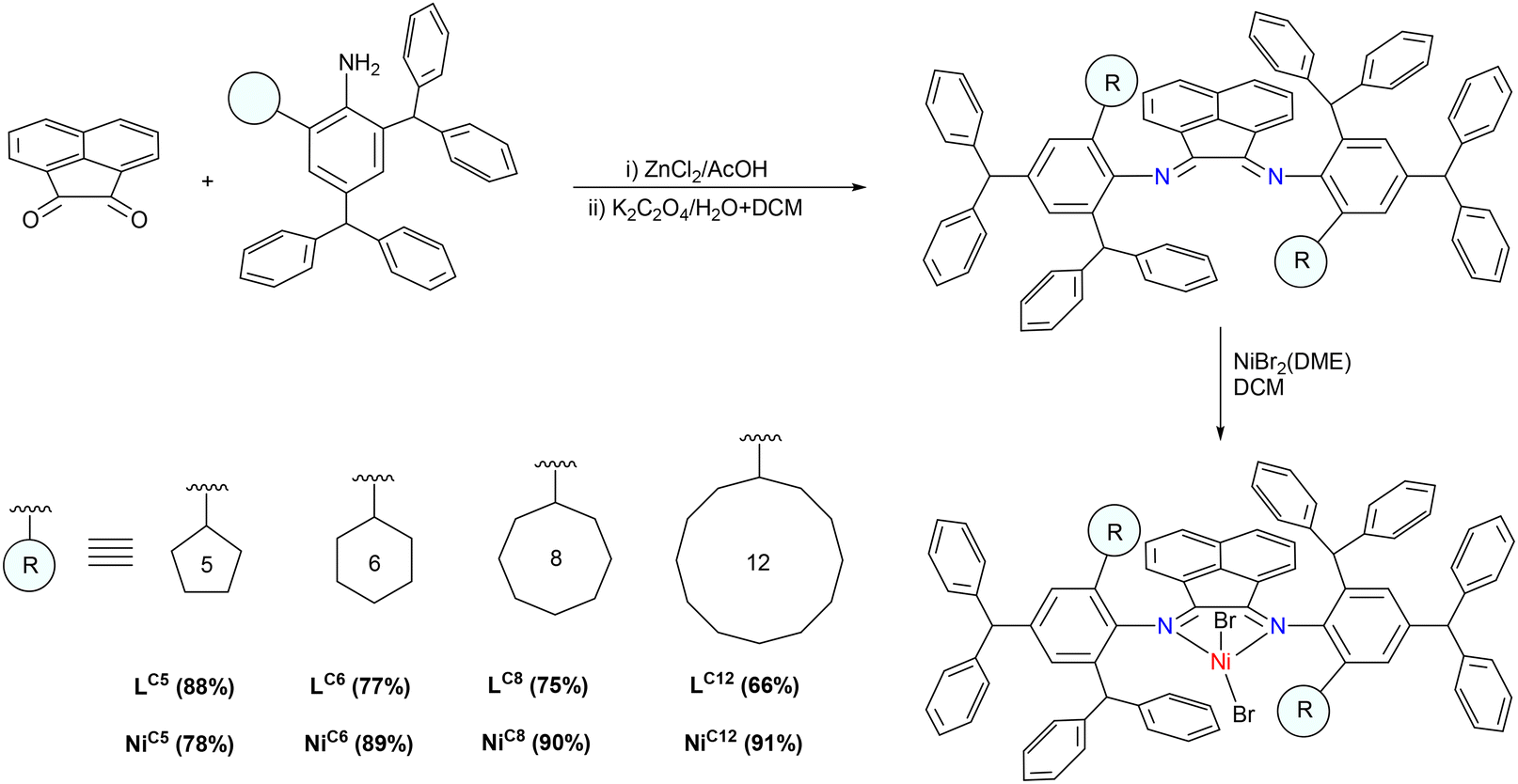
The important parameter in the design of α-diimine nickel precatalysts that demonstrate superior activity and high polymer molecular weights at high temperatures is the steric bulk at the axial sites of the active species.34,35 Introducing steric bulk at the ortho position of the phenyl group in the imine unit generates steric congestion at the axial sites of the active species which in turn hinders the rotation of the N-aryl unit. However, the synthesis of such sterically crowded α-diimines presents a challenge, as the imine unit is prone to facile decomposition. Thus, few examples with high steric hindrance are reported in the literature.20,28,31,33,35 Following a modified procedure, a hybrid steric hindrance composed of a carbocyclic ring and a benzhydryl group was introduced at the axial sites of the metal center. In particular, a series of α-diimine ligands were synthesized using a template method.34 The reaction involved heating of 2,4-dibenzhydryl-6-cycloalkylaniline and 1,2-acenaphthalenedione to reflux in acetic acid with an excess of zinc chloride. The following demetallation reaction led to pure ligands in high to excellent yields, eliminating the need for the labour-intensive purification method of column chromatography (66–88%, Scheme 1). The overnight stirring of these ligands with NiBr2(DME) in dichloromethane (DCM) at room temperature under an inert atmosphere afforded the desired 1,2-bis(2,4-dibenzhydryl-6-cycloalkylphenylimino)acenaphthene-nickel bromide complexes in excellent yields [where cycloalkyl = cyclopentyl for NiC5; cyclohexyl for NiC6; cyclooctyl for NiC8; cyclododecanyl for NiC12]. The structure and purity of these new ligands and their nickel complexes were confirmed through elemental analysis, FTIR, 1H and 13C NMR spectroscopy. Furthermore, the molecular structure of three complexes, NiC5, NiC6, and NiC12, was determined using X-ray diffraction analysis. The 1H NMR spectra of the ligand revealed two distinct sets of peaks, suggesting the rotation of N-aryl groups. This can happen due to the presence of two different ortho substituents on the phenyl group of the imine, leading to the formation of two isomers of ligands: syn and anti. In the 1H NMR spectrum of LC5 (R = cyclopentyl), both syn- and anti-isomers are observed in a ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 at room temperature, respectively (Fig. 2). However, the interconversion between these isomers became notably restricted with the increase of the cycloalkyl ring size in the ligand structure: the syn/anti ratio was 1.1/8.9 for LC6, 0.9/9.1 for LC8, and 0.8/9.2 for LC12. The gradual increase in the size of the cycloalkyl ring restricts the N-aryl rotation due to enhanced steric bulk at the axial sites. Thus, LC12 exhibited the lowest interconversion between the isomers. As nickel complexes are paramagnetic, NMR studies were not conducted to observe isomer interconversions.36 However, we anticipate a similar restriction in the N-aryl rotation in the corresponding nickel complexes at room temperature. The limited N-aryl rotation in α-diimine nickel complexes plays an important role in improving the thermal stability, catalytic activity, and polymer molecular weight at elevated reaction temperatures.34 Moreover, in the FTIR spectra of the ligands, stretching vibrations of the imine bond were observed in the range of 1652–1668 cm−1. These vibrations were approximately 20 cm−1 higher in wavenumber relative to the imine bond vibrations in the nickel complexes.37 The disparity in wave numbers suggests effective coordination between the metal center and the nitrogen atoms of the imine bonds in the ligand.38 Elemental analysis further confirmed the high purity of both the ligands and their respective nickel complexes.
:6 at room temperature, respectively (Fig. 2). However, the interconversion between these isomers became notably restricted with the increase of the cycloalkyl ring size in the ligand structure: the syn/anti ratio was 1.1/8.9 for LC6, 0.9/9.1 for LC8, and 0.8/9.2 for LC12. The gradual increase in the size of the cycloalkyl ring restricts the N-aryl rotation due to enhanced steric bulk at the axial sites. Thus, LC12 exhibited the lowest interconversion between the isomers. As nickel complexes are paramagnetic, NMR studies were not conducted to observe isomer interconversions.36 However, we anticipate a similar restriction in the N-aryl rotation in the corresponding nickel complexes at room temperature. The limited N-aryl rotation in α-diimine nickel complexes plays an important role in improving the thermal stability, catalytic activity, and polymer molecular weight at elevated reaction temperatures.34 Moreover, in the FTIR spectra of the ligands, stretching vibrations of the imine bond were observed in the range of 1652–1668 cm−1. These vibrations were approximately 20 cm−1 higher in wavenumber relative to the imine bond vibrations in the nickel complexes.37 The disparity in wave numbers suggests effective coordination between the metal center and the nitrogen atoms of the imine bonds in the ligand.38 Elemental analysis further confirmed the high purity of both the ligands and their respective nickel complexes.
 |
| | Scheme 1 Synthesis of ligands and their corresponding nickel complexes bearing cycloalkyl groups. | |
 |
| | Fig. 2
1H NMR spectra of ligands LC5–LC12 at room temperature. | |
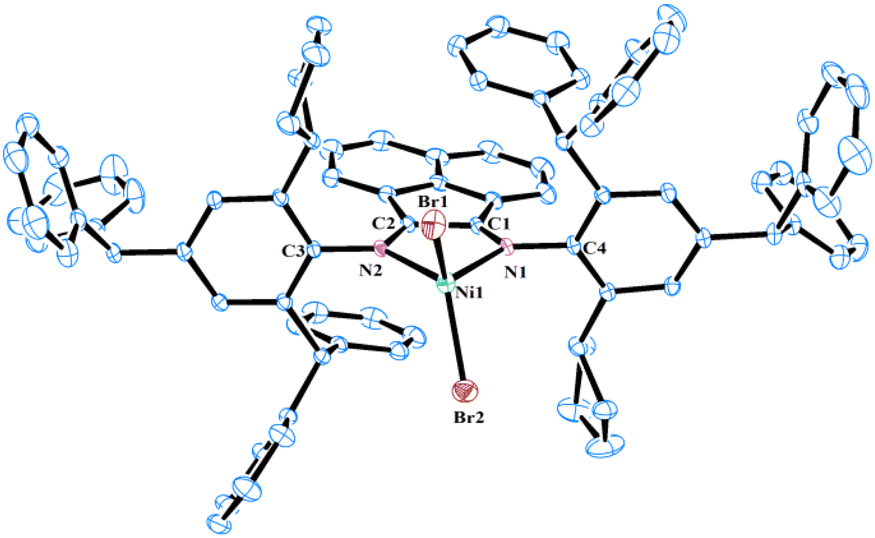
The molecular structures of NiC5, NiC6, and NiC12 were determined using single-crystal X-ray diffraction analysis. Single crystals of these complexes were grown by layering their saturated solutions in dichloromethane with hexane. As depicted in Fig. 3–5, the nickel centers in all three complexes exhibit a distorted tetrahedral geometry. The observed bond angles and lengths are generally consistent with those reported in previous studies of α-diimine nickel precatalysts used in ethylene polymerization.39–41 A comparative analysis of these three structures reveals that the Br1–Ni1–Br2 angle gradually decreases with the increase of the ring size of the cycloalkyl group [124.09(3)° for NiC5, 123.84(4)° for NiC6, and 121.39(5)° for NiC12]. This suggests that the increased steric hindrance from the larger cycloalkyl rings leads to a gradual reduction in the Br1–Ni1–Br2 angle. In contrast, the N1–Ni1–N2 bite angle remains largely unaffected [83.28(9)° for NiC5, 83.74(16)° for NiC6, and 83.2(2)° for NiC12]. It was also observed that the chelate and backbone of acenaphthene are coplanar, and nearly perpendicular to the plane of the N-aryl ring, with average dihedral angles of 85.8° for NiC5, 87.1° for NiC6, and 87.3° for NiC12. Moreover, all these nickel complexes are found in the trans configuration in their solid-state structures, though both cis and trans isomers are detected in solution. The solid-state conformation of cycloalkyl substituents varies with the size of their ring: a half-chair conformation is observed in NiC5, a chair conformation in NiC6, and a mixed chair-boat type conformation in NiC12. However, the cycloalkyl substituents on the N-aryl ring are flexible and can adopt different conformations in solution, which may be advantageous in reducing associative chain transfer reactions by locking the axial positions of the metal center with induced steric hindrance.42,43
 |
| | Fig. 3 Molecular structure of NiC5 with thermal ellipsoids shown at a 30% probability level. All hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Ni1–Br1 2.334(6), Ni1–Br2 2.331(6), Ni1–N1 2.035(2), Ni1–N2 2.042(2), C1–C2 1.513(3), N1–C1 1.282(3), N2–C2 1.281(4), N1–C4 1.441(3) N2–C3 1.450(3); and bond angles (°): Br1–Ni1–Br2 124.09(3), N1–Ni1–N2 83.28(9), N1–Ni1–Br1 115.83(6), N1–Ni1–Br2 103.74(6), N2–Ni1–Br1 106.72(6), N2–Ni1–Br2 116.04(7). | |
 |
| | Fig. 4 Molecular structure of NiC6 with thermal ellipsoids shown at a 30% probability level. All hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Ni1–Br1 2.326(11), Ni1–Br2 2.329(11), Ni1–N1 2.033(4), Ni1–N2 2.021(4), C1–C2 1.509(6), N1–C1 1.291(6), N2–C2 1.295(6), N1–C4 1.424(6), N2-C3 1.440(5); and bond angles (°): Br1–Ni1–Br2 123.84(4), N1–Ni1–N2 83.74(16), N1–Ni1–Br1 116.77(11), N1–Ni1–Br2 102.97(12), N2–Ni1–Br1 106.87(11), N2–Ni1–Br2 115.81(11). | |
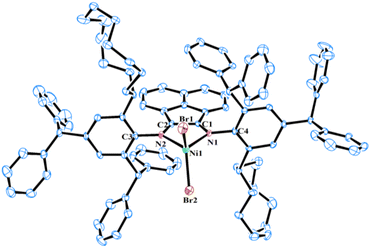 |
| | Fig. 5 Molecular structure of NiC12 with thermal ellipsoids shown at a 30% probability level. All hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Ni1–Br1 2.327(12), Ni1–Br2 2.318(11), Ni1–N1 2.015(5), Ni1–N2 2.040(5), C1–C2 1.509(8), N1–C1 1.281(8), N2–C2 1.270(8), N1–C4 1.453(7), N2–C3 1.456(7); and bond angles (°): Br1–Ni1–Br2 121.39(5), N1–Ni1–N2 83.2(2), N1–Ni1–Br1 110.31(14), N1–Ni1–Br2 113.12(14), N2–Ni1–Br1 113.70(15), N2–Ni1–Br2 108.81(14). | |
Ethylene polymerization
Recently, our group reported synthesis and ethylene polymerization studies using high-performance α-diiminonickel precatalysts with variations in the steric and electronic ortho substituents with the C2 symmetry.30,34 These precatalysts exhibited increased thermal stability, higher polymer molecular weights and a narrower polydispersity index than the conventional α-diimine nickel catalysts, and greatly linked with the systematic variations in the ortho steric and electronic substituents. Most importantly, the effect of reaction conditions was significant on the activity and polymer properties. Considering the influence of reaction conditions, first, the best activator was selected for activation of NiC5 and then the effects of the activator amount, run time and ethylene pressure were systematically examined for ethylene polymerization. Finally, the effect of the reaction temperature and ligand structure was determined. Characterization of the obtained polyethylene was performed through combined GPC, DSC, and high-temperature 1H and 13C NMR spectra. The units for polymerization activity and molecular weights are typically expressed as g mol−1 h−1 and g mol−1 respectively. However, for simple discussion, we omitted these units in the text. Additionally, we systematically examined the impact of temperature and catalyst structure on the mechanical properties.
Selection of the best activator.
The nickel complex NiC5 was initially tested as a precatalyst for ethylene polymerization under typical conditions: 30 °C temperature, 30 min runtime, 1 MPa ethylene pressure, and using 100 mL toluene. Alkylaluminium compounds—ethylaluminum sesquichloride (EASC), diethylaluminum chloride (DEAC), methylaluminoxane (MAO), and modified MAO (MMAO)—used as cocatalysts, have significant influence on the polymerization activity and polymer properties due to their distinct characteristics, such as alkylating ability, Lewis acidity, interaction with active species, and the size of the organoaluminate counter ions.23 The polymerization results in Table 1 clearly demonstrate the dependence of the polymerization activity and polymer properties on the combination of the NiC5 precatalyst with different cocatalysts. Overall, all cocatalysts exhibit high activation efficacy to activate NiC5. MMAO exhibited comparatively low activity, with a minimum value of 7.10 × 106 among all the tested cocatalysts. Replacing it with DEAC led to an increased polymerization activity by a factor of 1.4 (Table 1, entry 3). Significant improvement was evident when EASC and MAO were used as cocatalysts. The EASC afforded the highest activity of 15.4 × 106 (Table 1, entry 4). It is worth noting that despite using much higher amounts of MAO and MMAO compared to the other two cocatalysts (Al/Ni = 1000), their activity could not reach the level achieved with EASC (Al/Ni = 400). Even MMAO, the most expensive cocatalyst, displayed much lower activity, only half that of EASC. Overall, the order of polymerization activity based on the tested alkylaluminium compounds is as follows: EASC > MAO > DEAC > MMAO. These results suggest that Lewis acidity alone may not be the only factor for effective activation of the metal center, as DEAC, despite being more acidic than MAO, resulted in relatively lower activity. On the other hand, the higher acidity of EASC than all other cocatalysts makes it a more effective activator for NiC5. The polymer molecular weight (Mw), molecular weight distribution (Mw/Mn), and melt temperature (Tm) also showed dependency on the nature of the cocatalyst. Although NiC5/EASC displayed the highest activity, it produced polyethylene with lower molecular weights. In general, polymer molecular weights using various cocatalysts were around 1 × 105, with the highest value of 6.1 × 105 achieved for the DEAC-activated system. In all cases, the polymer displayed narrow dispersity, particularly evident with MMAO (Mw/Mn = 1.54) and EASC (Mw/Mn = 1.67), highlighting the single-site chain propagation of active species. The melt temperature of polyethylene obtained with MMAO (Tm = 105 °C) was comparatively higher than those of other cocatalysts (Tm = 91–98 °C). This signifies a reduced degree of chain walking reactions in the NiC5/MMAO system. Given its superior polymerization activity, EASC was selected as the preferred cocatalyst for further investigations.
Table 1 Selection of best activator for the NiC5 precatalyst toward ethylene polymerizationa
| Entry |
Co-cat. |
Al/Ni |
PE (g) |
Act.b (106) |
M
w
(105) |
M
w/Mnc |
T
m
(°C) |
|
General conditions for polymerization: precat (NiC5, 2.0 μmol); time (0.5 h); solvent (toluene, 100 mL); ethylene (1 MPa); temperature (30 °C).
Activity unit (g mol−1 h−1).
GPC results (Mw, Mw/Mn; unit: g mol−1).
DSC results.
|
| 1 |
MAO |
1000 |
15.1 |
15.1 |
6.2 |
1.84 |
98 |
| 2 |
MMAO |
1000 |
7.1 |
7.1 |
3.4 |
1.54 |
105 |
| 3 |
DEAC |
400 |
10.0 |
10.0 |
6.1 |
1.70 |
94 |
| 4 |
EASC |
400 |
15.4 |
15.4 |
2.2 |
1.67 |
91 |
Effect of cocatalyst amount, runtime and ethylene pressure.
In order to improve the polymerization activity of NiC5, the reactions were conducted at various concentrations of EASC (Al/Ni = 200, 300, 400, 450, 500, 550, and 600), while maintaining all other conditions constant. The results in Table 2 demonstrate a strong correlation between the concentration of EASC and both activity and polymer molecular weight. Increasing the cocatalyst from 200 to 300 equiv. resulted in a dramatic improvement in activity by a factor of about 2. Subsequent increments in the cocatalyst concentration led to a linear growth of activity, reaching a maximum value of 24.4 × 106 at 550 (Table 2, entries 1–6). However, using 600 equiv. of EASC, a mere 50 equiv. difference in the cocatalyst concentration, led to a dramatic decrease in activity by a factor of about 1.5 compared to the highest obtained activity (Table 2, entry 7). This is typical behaviour of nickel catalysts used for ethylene polymerization.44 On the other hand, an increase in cocatalyst concentration had a negative impact on polymer molecular weights. The use of 200 equiv. of EASC led to polymers with molecular weights above 1 million g per mol, which linearly decreased with the increase of Al/Ni ratios. Likely, an excess concentration of the aluminum compound in the reaction mixture facilitates chain transfer reactions over chain propagation, resulting in comparatively lower polymer molecular weights. Furthermore, certain reasons remain unknown to us. However, we hypothesize that the overloading of the alkyl aluminum compound (cocatalyst) during its reaction with precatalysts may lead to the decomposition of some active species. Overall, Mw/Mn remained narrow, but became slightly broader at higher cocatalyst concentrations, which also support the occurrence of high chain transfer reactions. Except for the polyethylene obtained at 200 equiv. of cocatalysts (Tm = 101 °C), polymer melt temperatures were less affected with respect to the cocatalyst concentration (Tm = 82–94 °C). The results obtained in this study are consistent with the findings observed in our earlier research and other investigations described in the literature.23,44,45
Table 2 Effect of the cocatalyst amount, runtime and ethylene pressure on ethylene polymerization using NiC5/EASCa
| Entry |
Al/Ni |
t (min) |
PE (g) |
Act.b (106) |
M
w
(105) |
M
w/Mnc |
T
m
(°C) |
|
General conditions for polymerization: precat (NiC5, 1.0 μmol); cocat (EASC); solvent (toluene, 100 mL); ethylene (1 MPa); temperature (30 °C).
Activity unit (g mol−1 h−1).
GPC results (Mw, Mw/Mn; unit: g mol−1).
DSC results.
Ethylene (0.1 MPa).
Ethylene (0.5 MPa).
|
| 1 |
200 |
30 |
4.9 |
9.9 |
11.5 |
1.75 |
101 |
| 2 |
300 |
30 |
9.9 |
19.7 |
9.3 |
1.85 |
87 |
| 3 |
400 |
30 |
10.5 |
21.0 |
8.8 |
1.94 |
87 |
| 4 |
450 |
30 |
11.3 |
22.6 |
7.9 |
1.75 |
82 |
| 5 |
500 |
30 |
11.6 |
23.2 |
7.4 |
1.78 |
91 |
| 6 |
550 |
30 |
12.2 |
24.4 |
6.0 |
1.69 |
94 |
| 7 |
600 |
30 |
7.7 |
15.4 |
4.9 |
1.89 |
90 |
| 8 |
550 |
5 |
3.6 |
42.9 |
2.7 |
1.95 |
101 |
| 9 |
550 |
10 |
4.3 |
26.5 |
3.1 |
1.62 |
96 |
| 10 |
550 |
15 |
6.2 |
24.8 |
3.0 |
1.94 |
103 |
| 11 |
550 |
45 |
14.1 |
18.8 |
6.5 |
1.65 |
84 |
| 12 |
550 |
60 |
16.9 |
16.9 |
6.7 |
1.48 |
97 |
| 13e |
550 |
30 |
2.1 |
4.2 |
6.4 |
1.87 |
76 |
| 14f |
550 |
30 |
6.9 |
13.8 |
6.5 |
2.10 |
98 |
Note that polymerization activity across all cocatalyst concentrations was observed to be above 10 million g per mol per h measured for a 30 min runtime. Thus, to investigate the stability of the active species for longer periods, experiments were conducted over different runtimes (t = 5 min, 10 min, 15 min, 30 min, 45 min, 60 min). The results in Table 2 (entries 6, 8–12) exhibited the effect of reaction time on the polymer yield, activity, and polymer molecular weight. The NiC5/EASC system was extremely active, producing more than 3 g of the polymer in a short duration of 5 min and achieving an exceptionally high activity of 42.9 × 106 (Table 2, entry 8). To the best of our knowledge, this is the highest reported activity for ethylene polymerization to date for 5 min or more reaction time. On the other hand, a consistent decrease in activity was noted with the prolongation of the reaction time (Table 2, entry 12). The activity decreased by a factor of 2.5 from 5 min to 60 min. The decomposition of active species with the prolongation of the reaction time could be a reason for the decrease in polymerization activity. Meanwhile, the increase in reaction viscosity with the progress of polymerization may also somehow contribute to the decrease in catalytic activity over time.46a Although the lowest activity of 16.9 × 106 was obtained over a runtime of 60 min, it still represents exceptionally high activity, highlighting the high stability of the active species at 30 °C.34 Meanwhile, polymer molecular weights gradually increased from 2.7 × 105 to 6.7 × 105 with the prolongation of reaction time (Table 2, entries 6, 8–12). There were fewer variations in the mass distribution of the polymer with changes in the reaction time, indicating a single-site catalytic behavior of the catalysts, consistent with previous similar reports.46b,47
Next, the impact of ethylene pressure was investigated. Table 2 (entries 6, 13 and 14) summarizes ethylene polymerization under 0.1 MPa, 0.5 MPa, and 1 MPa ethylene pressure. There is a noteworthy dependence on pressure for the polymerization activity measured based on 30 min reaction time. Increasing the pressure from 0.1 MPa to 0.5 MPa resulted in a 3.2-fold rise in activity, while doubling the ethylene pressure resulted in an approximately 1.7-fold increase in activity. Previous studies on α-diimine palladium complexes for olefin polymerization have indicated that the catalyst resting state is the alkyl olefin complex and that chain propagation remains unaffected by ethylene pressure.9,48 In contrast, for nickel complexes, the catalyst resting state can be the alkyl olefin complex, the β-agostic complexes, or a combination of both.12,49 In this study, the results suggest that the catalyst resting state primarily involves agostic complexes, thus leading to a first-order dependence on ethylene pressure. Moreover, polymer molecular weights under 0.1 MPa and 0.5 MPa are similar, whereas a slightly lower value was observed at 1 MPa. This disparity may be attributed to increased chain transfer reactions at higher pressure (β-H elimination reactions).50,51
Screening of nickel complexes at various reaction temperatures
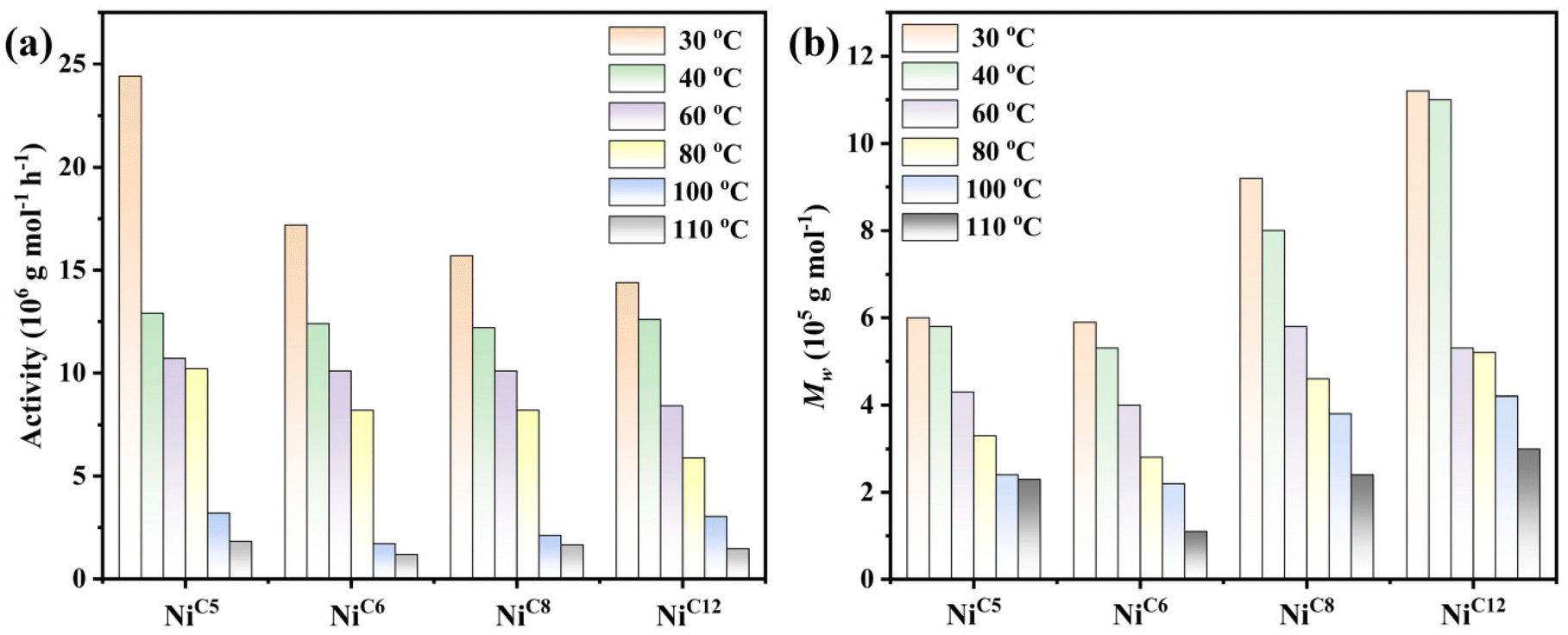
For in situ activation with EASC (550 equiv.), all the prepared complexes were investigated in detail at different reaction temperatures to examine their thermal stability and the most appropriate steric environment at the axial site of the metal center. The tests were carried out in 100 mL of toluene, with a constant ethylene pressure of 1 MPa for a runtime of 30 min. The reaction temperature varied in the range of 30 to 110 °C. The polymerization data are collected in Table 3 and represented graphically in Fig. 6. In general, all precatalysts exhibited exceptionally high activities with a broad range of reaction temperatures from 30 to 110 °C for ethylene polymerization. The complex NiC5 (R = cyclopentyl) was the most active precatalyst. It maintained a high polymerization activity, typically in the level 107 at a wide range of temperature of 30 to 80 °C, with a peak activity of 24.4 × 106 at 30 °C and interestingly, no significant loss in the activity was noted in the temperature range of 40 °C to 80 °C. However, a significant decline was observed when the polymerization was carried out at 100 °C and 110 °C, leading to lower activities as compared to the activity at 80 °C by a factor of 3.2 and 5.7 respectively (Table 3, entries 5 and 6). Overall, the activity dropped with the rise in temperature, consistent with the results seen in the previous reports.30,45 The decline in the activity can be attributed to the deactivation of the active species at higher temperatures28,32,33 and the reduction in the solubility of the ethylene monomer.51b–d It is worth noting that the NiC5 maintained a high level of activity, even at 110 °C, and it provided an exceptionally high activity of 1.8 × 106, indicating the catalyst's strong resistance to thermal degradation (Table 3, entry 6). The activity at a higher temperature is much better than previously reported α-diimine nickel precatalysts studied for 30 min or more reaction time.34,52a This indicates that the incorporation of 2,4-dibenzhydryl-6-cyclopentylaniline in a symmetrical fashion may generate significant steric blockage at the axial sites of the metal center, which in turn may restrict the flexibility of N-aryl rotation, thus preventing somehow the C–H activation of the ortho-substituents.52b,54,55 Moreover, it produced high molecular weight polyethylene at 30 °C (Mw = 6.0 × 105) which gradually decreased with the increase of reaction temperature to the lowest value of Mw = 2.3 × 105 at 110 °C. Although the molecular weight of polyethylene decreased, it still maintained the Mw in the level of 105 across all reaction temperatures. It is generally accepted that a high polymerization reaction increases the chain transfer reactions (via β-H elimination) as compared to the chain growth, leading to relatively lower polymer molecular weights.52c The mass distribution of the polymer remained narrow across the temperature range (Mw/Mn = 1.45–1.81), which strongly agrees with the single site catalytic behavior of polymer chain growth. Likewise, it should be noted that the branching degree of the obtained polyethylene is greatly dependent on the reaction temperature. Higher temperatures increase the branching degree while concurrently reducing the polymer melt temperature. Thus, there is a progressive decrease in the melt temperature, transitioning from 94 °C to 76 °C and the branching degree increased from 47 to 109 per 1000C with the rise in temperature (Table 3, entries 1–6). This increase indicates that the chain walking rate is notably higher at elevated temperatures.46,53
Table 3 Screening of nickel complexes at various reaction temperaturesa
| Entry |
Precat. |
T (°C) |
PE (g) |
Act.b (106) |
M
w
(105) |
M
w/Mnc |
T
m
(°C) |
B
|
|
General conditions for polymerization: precat. (1.0 μmol); cocat. (EASC); solvent (toluene, 100 mL); time (0.5 h); ethylene (1 MPa).
Activity unit (g mol−1 h−1).
GPC results (Mw, Mw/Mn; unit: g mol−1).
DSC results.
Determined by 1H NMR spectra [(2 × IMe/3 × Itotal) × 1000].
Amorphous.
Not determined.
|
| 1 |
NiC5
|
30 |
12.2 |
24.4 |
6.0 |
1.69 |
94 |
47 |
| 2 |
NiC5
|
40 |
6.5 |
12.9 |
5.8 |
1.77 |
88 |
66 |
| 3 |
NiC5
|
60 |
5.4 |
10.7 |
4.3 |
1.75 |
86 |
72 |
| 4 |
NiC5
|
80 |
5.2 |
10.2 |
3.3 |
1.81 |
83 |
89 |
| 5 |
NiC5
|
100 |
1.6 |
3.2 |
2.4 |
1.45 |
82 |
97 |
| 6 |
NiC5
|
110 |
0.9 |
1.8 |
2.3 |
1.75 |
76 |
109 |
| 7 |
NiC6
|
30 |
8.6 |
17.2 |
5.9 |
1.65 |
79 |
72 |
| 8 |
NiC6
|
40 |
6.2 |
12.4 |
5.3 |
1.62 |
73 |
90 |
| 9 |
NiC6
|
60 |
5.1 |
10.1 |
4.0 |
1.63 |
67 |
96 |
| 10 |
NiC6
|
80 |
4.1 |
8.2 |
2.8 |
1.71 |
67 |
112 |
| 11 |
NiC6
|
100 |
0.9 |
1.7 |
2.2 |
1.79 |
65 |
145 |
| 12 |
NiC6
|
110 |
0.6 |
1.2 |
1.1 |
2.53 |
—f |
—g |
| 13 |
NiC8
|
30 |
7.9 |
15.7 |
9.2 |
1.72 |
84 |
34 |
| 14 |
NiC8
|
40 |
6.1 |
12.2 |
8.0 |
1.76 |
84 |
—g |
| 15 |
NiC8
|
60 |
5.1 |
10.1 |
5.8 |
1.66 |
78 |
86 |
| 16 |
NiC8
|
80 |
4.1 |
8.2 |
4.6 |
1.82 |
76 |
88 |
| 17 |
NiC8
|
100 |
1.6 |
2.1 |
3.8 |
1.75 |
73 |
109 |
| 18 |
NiC8
|
110 |
0.8 |
1.7 |
2.4 |
1.75 |
—f |
—g |
| 19 |
NiC12
|
30 |
7.2 |
14.4 |
11.2 |
1.81 |
75 |
60 |
| 20 |
NiC12
|
40 |
6.3 |
12.6 |
11.0 |
1.72 |
75 |
—g |
| 21 |
NiC12
|
60 |
4.2 |
8.4 |
5.3 |
1.81 |
74 |
—g |
| 22 |
NiC12
|
80 |
2.9 |
5.9 |
5.2 |
1.53 |
72 |
77 |
| 23 |
NiC12
|
100 |
1.5 |
3.0 |
4.2 |
1.63 |
68 |
96 |
| 24 |
NiC12
|
110 |
0.7 |
1.5 |
3.0 |
2.01 |
—f |
101 |
 |
| | Fig. 6 Comparison of polymerization activity (a) and polymer molecular weight (b) at different temperatures using different precatalysts. | |
To determine the effect of the ortho-carbocyclic ring of aniline on the catalytic behaviour, NiC5 (R = cyclopentyl), NiC6 (R = cyclohexyl), NiC8 (R = cyclooctyl) and NiC12 (R = cyclododecanyl) were investigated for ethylene polymerization (Table 3, entries 1–24). Under otherwise identical conditions, it was noted that the polymerization activity gradually decreased with the increase of carbocyclic ring size (Fig. 6a). The polymerization activity for NiC5 was 1.4, 1.6 and 1.7 times higher than those of NiC6, NiC8 and NiC12 respectively at 30 °C. The decline in activity indicates that enlarging the ring size was not advantageous in enhancing the polymerization activity. This finding arises due to the excessive steric hindrance resulting from the larger ring size at the axial sites of the active species, which somehow reduces the rate of coordination–insertion of monomers. However, upon comparing the polymerization activity of these catalysts at higher reaction temperatures, specifically equal to or above 40 °C, the differences in the activity were relatively less pronounced. For instance, the polymerization activity obtained at 40 °C was 12.9 × 106 for NiC5, 12.4 × 106 for NiC6, 12.2 × 106 for NiC8, and 12.6 × 106 for NiC12. A similar observation was noted at other reaction temperatures. NiC5 was found to be the most active catalyst across all reaction temperatures. The polymerization activities for all precatalysts gradually decreased with the rise of temperature, consistent with the typical behaviour of α-diimine nickel catalysts at higher temperatures.32,33 Even though the activity dropped, it is noteworthy to mention that all prepared catalysts exhibited exceptional thermal stability compared to the previously reported nickel catalysts in the class of α-diimine.28–33 Based on the activity achieved at 110 °C, the thermal stability of these precatalysts decreased in the order of NiC5 > NiC8 > NiC12 > NiC6. The polymerization activity can be achieved in the range of 1.7–3.2 × 106 at 100 °C and 1.2–1.8 × 106 at 110 °C, indicating exceptional stability and potential of these catalysts to work at higher temperatures. Once again, we assumed that the presence of bulky benzhydryl and carbocyclic rings at the ortho position of the aniline may generate significant steric blockage at the axial sites of the metal center, which in turn may inhibit N-aryl rotation and thereby protect the active species from decomposition.52b,54,55 Consequently, all these complexes exhibit high thermal stability.
Preceding studies have revealed that an increase in steric crowding over the axial sites of the metal center leads to improvement in the molecular weights of polymers in α-diimine nickel precatalysts [Fig. 6b].20 Consistent with this observation, NiC12 bearing the largest steric bulk produced the highest polymer molecular weights, approximately 1.8-fold higher than NiC5 or NiC6, and 1.2-fold higher than NiC8 at 30 °C (Table 3, entries 1, 7, 13 and 19). At 30 °C, the overall trend in the polymer molecular weight is NiC12 > NiC8 > NiC6 ≈ NiC5, corroborating with previous reports.20 Interestingly, polymer molecular weights for NiC12 exceed the 1 million g per mol range at both 30 °C and 40 °C, indicating the positive effect of the increased steric bulk present at the ortho position of aniline. The widely accepted explanation is that the steric bulk facilitates chain growth as well as protects the active species from chain transfer reactions to aluminum compounds, resulting in high molecular weight polyethylene. In line with previous reports,22–27 an increase in reaction temperature had a negative effect on polymer molecular weights, as higher temperatures lowered the energy barrier for chain transfer reactions (β-H elimination as well as transfer to aluminum compounds). Additionally, the increase of temperature can reduce the energy barrier for the N-aryl groups to rotate more freely around the active species. This rotation not only caused the decomposition of active species but also increases the chances of inducing chain transfer reactions. Collectively, these reactions led to relatively lower polymer molecular weights. For example, in the case of NiC12, polymer molecular weights gradually decreased from 11.2 × 105 at 30 °C to 3.0 × 105 at 100 °C, representing a 3.7-fold decline. However, polymer molecular weights decreased with the rise in temperature, and values across all reaction temperatures remained in the range of 105 g mol−1. This is likely due to the incorporation of the steric bulk, which reduces chain transfer processes and maintains a high rate of chain growth reactions. With the exception of polymerization at 110 °C for NiC6 and NiC12, the dispersity of polymers produced from all precatalysts was narrow and unimodal across all reaction temperatures, largely staying below 2. These results suggest the presence of only single-site active species, even when polymerization was performed at higher temperatures, and maintained narrow dispersity. At 110 °C, polymers derived from complex NiC6 showed a bimodal, wider dispersity, suggesting the presence of two active species at higher temperatures. Meanwhile, the dispersity for NiC12-based polyethylene was relatively broad but remained unimodal.28–30
The interest in α-diimine nickel catalysts is primarily driven by their ability to generate branches in polyethylene using only ethylene as the feedstock.28–30 As expected, these nickel precatalysts generate highly branched polyethylene with branching density varying in the range of 34–145 per 1000C. In general, the values of the branching degree are comparable to previously reported “sandwich-type” α-diimine nickel precatalysts,54 but comparatively higher than the symmetrical α-diimine nickel catalysts reported by Long and Guan independently.55–57 The branching density was observed to increase with the rise of reaction temperature, consistent with previous reports.31–33 The polyethylene with a higher branching density can easily melt at low temperatures due to the more amorphous topology of the polymer. Thus, the DSC results exhibited that polymer melt temperatures gradually decreased with the elevation of reaction temperature. In the case of NiC5, the polymer melt temperature decreased from 94 °C to 76 °C, while the branching density increased from 47 to 109 per 1000C with the rise of temperature. Moreover, at lower reaction temperatures, the branching degree of polyethylene was observed to increase with the increasing carbocyclic ring size, suggesting that the increase of steric hindrance at axial sites of the metal center lessened the chain walking phenomenon. Mechanistic studies on α-diiminonickel precatalysts has unveiled various possible resting states, including alkyl olefinic species, β-agostic species, and combinations thereof.33,50,51 Two factors, chain walking and chain growth, mainly determined the branching density of polyethylene.16,17 The steric bulk from the ortho substituent, in general, reduces the rate of both reactions. The steric hindrance induced by the flexible carbocyclic group at the ortho position of aniline moderately facilitates the β-agostic alkyl species and chain walking thereof over the chain growth reaction, therefore, NiC6 and NiC12 generate polyethylene with a comparatively higher branching density as compared to NiC5 at 30 °C. In contrast, the complex NiC8 had a larger carbocyclic ring than that of NiC5 or NiC6 but generated a comparatively less branching density. It is possible that the steric repulsion between the substituents on the phenyl of the imine leads to a conformation of cyclooctyl which can reduce the steric hindrance overall as compared to the cyclopentyl or cyclohexyl bearing nickel complexes. It is also observed that, the difference in the branching degree of polyethylene within the series of prepared nickel precatalysts became smaller at higher temperatures. Likely, rapid conformation changes of the carbocyclic ring reduces the difference in the steric bulk at higher temperatures. Moreover, the branching content of selected polyethylene was determined using high-temperature 13C NMR spectra (Table 3, entries 1 and 5, Fig. 7 and 8 respectively).58 The branching pattern of polyethylene produced at 30 °C was CH3 (57%), 1,4 paired CH3 (12%), 1,6 paired CH3 (6%), CH3CH2 (3%), CH3(CH2)2CH2 (9%), and CH3(CH2)n−1CH2 (13%) branches. The polyethylene obtained at 100 °C exhibited some difference: the amount of the CH3 branch was slightly decreased to 55% and some peaks of CH3(CH2)3CH2 (2%) branches appeared. While the other branching content remained almost the same: 1,4 paired CH3 (12%), 1,6 paired CH3(8%), CH3CH2 (3%), CH3(CH2)2CH2 (8%), and CH3(CH2)n−1CH2 (12%). A similar branching pattern has been reported in previous reports.34,44,46
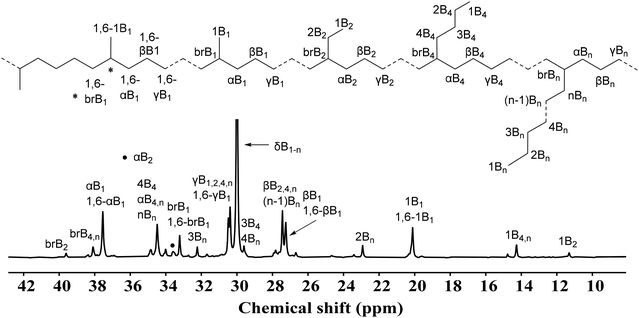 |
| | Fig. 7 High temperature 13C NMR spectrum of polyethylene (Table 3, entry 1). | |
 |
| | Fig. 8 High temperature 13C NMR spectrum of polyethylene (Table 3, entry 5). | |
Life time of NiC5 at higher temperatures
The polymerization results obtained with the NiC5/EASC system in Table 3 (entries 1–6) displayed exceptional thermal stability for 0.5 h reaction time. In order to investigate the stability of the active species at higher temperatures, experiments were additionally conducted over different reaction times and temperatures (Table 4, entries 1–11). To our delight, polymer yields exhibited a strict linear increase at 80 °C (entries 1–3), and approached linearity at 100 °C (entries 4–7) or 110 °C (entries 8–11) with the prolongation of reaction times. However, polymerization activities in the first few mins were high (20 × 106 at 100 °C and 13.3 × 106 at 110 °C for 2 min) and then steadily decreased over a prolongation of time at different temperatures. This decrease can be attributed to the deactivation of active species occurring at higher temperatures and with prolonged reaction times.28,32,33 The reduced solubility of the ethylene monomer in toluene at elevated temperatures also show to somehow decrease the activity at elevated reaction temperatures.51b–d Despite the decrease in activity with the reaction time, polymer yields almost directly increased with the reaction time. This highlights that some catalytic species still remained active at elevated temperatures, showing a high activity of 5.6 × 106 at 80 °C, 1.5 × 106 at 100 °C and 0.7 × 106 at 110 °C over a long period of 2 hours (Table 4, entries 3, 7 and 11). However, polymer molecular weights did not grow linearly with the reaction time. Polymer molecular weights greatly depend on the ratio of chain propagation and chain transfer rates. It is believed that polymerization within a short period of reaction produced extremely high chain growth reactions, while the chain transfer reaction proceeded with prolonged reaction times.23,24 Thus, molecular weights of the resulting polyethylene deviate from the linear relationship of the polymer molecular weight with the reaction time. Moreover, molecular weights of the resulting polyethylene decreased with the rise of reaction temperature; it still maintained the Mw in the level of 105 across all reaction temperatures. Generally, a high polymerization reaction increases the chain transfer reactions (via β-H elimination) as compared to the chain growth, leading to relatively lower polymer molecular weights.28,33 The dispersity of polymer molecular weights remained narrow and unimodal, strongly suggesting single-site active species. The high polymerization activity at elevated temperature, a linear increase of polymer yield and consistently high polymer molecular weights across all reaction temperatures suggest the high thermal stability of the NiC5/EASC system.
Table 4 Ethylene polymerization at longer reaction times using NiC5/EASCa
| Entry |
t (min) |
T (°C) |
PE (g) |
Act.b (106) |
M
w
|
PDIc |
T
m
(°C) |
|
General conditions for polymerization: NiC5 (1.0 μmol); cocat. (EASC); solvent (toluene, 100 mL); ethylene (1 MPa).
Activity unit (g mol−1 h−1).
GPC results (Mw, Mw/Mn; unit: 105 g mol−1).
DSC results.
Not determined.
|
| 1 |
30 |
80 |
5.16 |
10.3 |
3.3 |
1.81 |
82.5 |
| 2 |
60 |
80 |
7.01 |
7.0 |
3.8 |
1.72 |
78.3 |
| 3 |
120 |
80 |
11.1 |
5.6 |
4.0 |
1.70 |
75.4 |
| 4 |
2 |
100 |
0.67 |
20.3 |
1.1 |
2.31 |
—e |
| 5 |
15 |
100 |
1.32 |
5.3 |
1.4 |
2.18 |
—e |
| 6 |
30 |
100 |
1.61 |
3.2 |
2.4 |
1.55 |
82 |
| 7 |
120 |
100 |
3.08 |
1.5 |
3.1 |
2.33 |
—e |
| 8 |
2 |
110 |
0.44 |
13.3 |
0.9 |
2.40 |
—e |
| 9 |
15 |
110 |
0.72 |
2.9 |
1.5 |
2.24 |
—e |
| 10 |
30 |
110 |
0.90 |
1.8 |
2.3 |
1.70 |
76 |
| 11 |
120 |
110 |
1.36 |
0.7 |
2.6 |
2.31 |
—e |
Mechanical properties
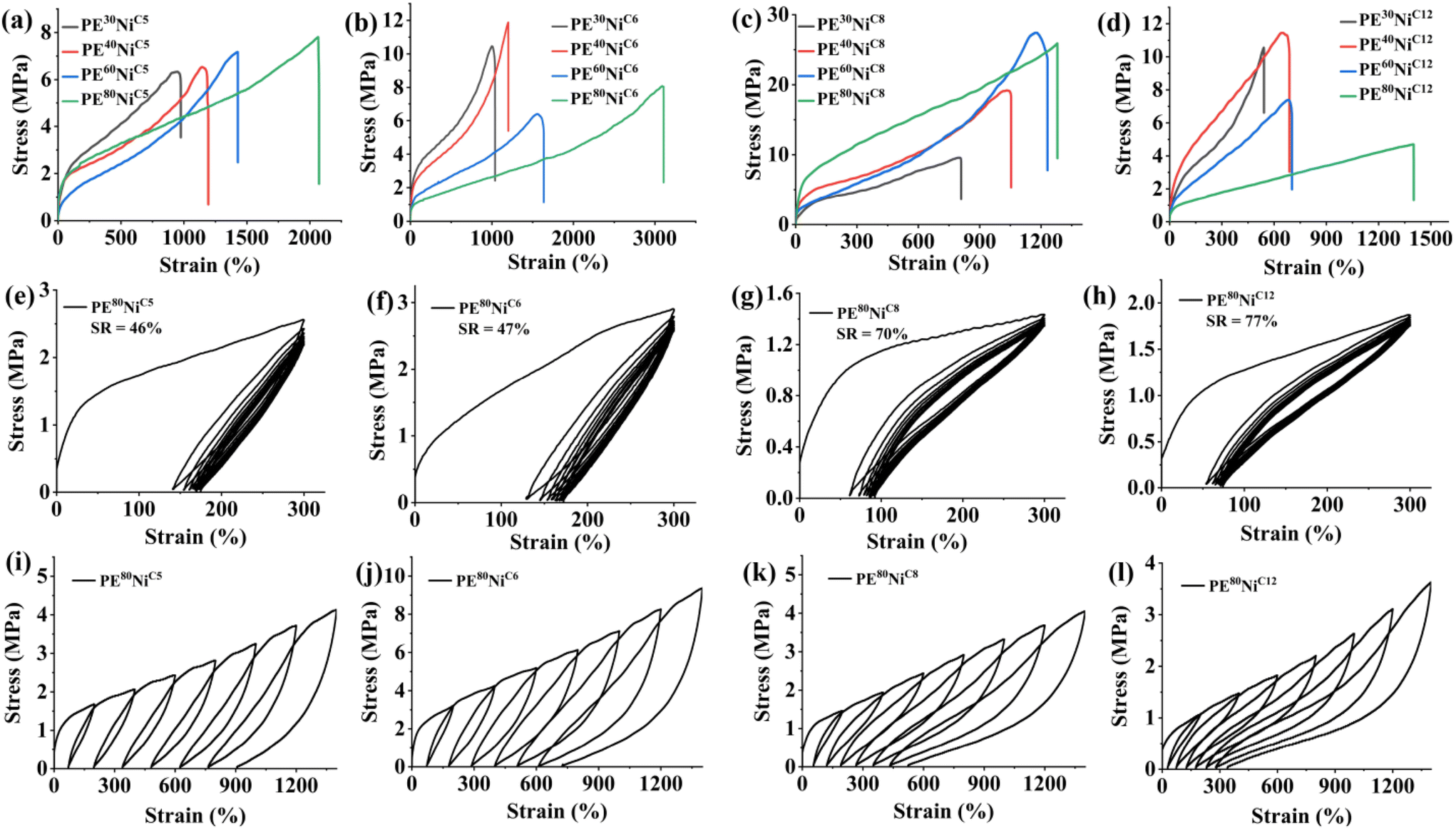
The investigations into the mechanical properties of these polyethylene samples revealed intriguing findings. The molecular weight, branching density and branching content of the resulting polyethylene are critical parameters that greatly influence these properties.59 These parameters can be effectively modulated through adjustments in the catalyst structure and reaction temperature. Thus, we examined the polyethylene samples obtained at various reaction temperatures (30 °C, 40 °C, 60 °C, 80 °C) using different precatalysts (NiC5, NiC6, NiC8, NiC12) for stress–strain response and elastic recovery (Table 5 and Fig. 9). These polyethylenes exhibited a wide range of mechanical properties, contingent upon the precatalyst structure and the specific reaction temperature used for ethylene polymerization. There was a discernible correlation between the ultimate tensile strength (σ) and the maximum elongation at break (ε) with the molecular weight and branching density of the polymer. For polyethylene derived from NiC5 and NiC8, we observed a consistent increase in ultimate tensile strength with the rise of reaction temperature used for polymer synthesis (Fig. 9a and c). Conversely, polyethylene obtained with NiC6 and NiC12 exhibited an inverse relationship (Fig. 9b and d). This suggests that the high branching density, prompted by a higher temperature, is a decisive factor in enhancing the mechanical properties of NiC5 and NiC8 variants. On the other hand, high branching density coupled with relatively low molecular weights appeared to compromise the tensile strength, resulting in a higher strain. Remarkably, the polyethylene sample PE60NiC8 obtained at 60 °C using NiC8, achieved an exceptional ultimate tensile strength of up to 27.4 MPa, accompanied by a maximum elongation at break of 1231% (Table 5). Similarly, the sample PE80NiC8 demonstrated exceptional mechanical properties, with a σ of 25.9 MPa and an ε of 1280%. These values surpass those of commercial polyolefin elastomer (CPOE) and previously reported elastic polyolefins synthesized using late transition metal catalysts. In summary, the ultimate tensile strength and strain at break of these polyethylene samples can be effectively modulated, ranging from 4.7 MPa to 27.4 MPa and 542% to 3105%, respectively, by varying the reaction temperatures and catalytic systems.
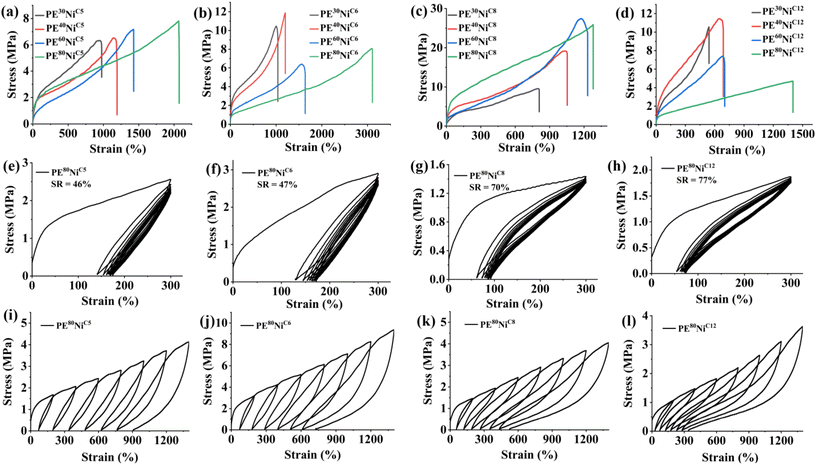 |
| | Fig. 9 Stress–strain curves generated by (a) NiC5, (b) NiC6, (c) NiC8, and (d) NiC12 at various reaction temperatures. Plots of hysteresis experiments of ten cycles at a strain of 300% for PE samples generated by (e) NiC5, (f) NiC6, (g) NiC8, and (h) NiC12 at 80 °C. Stress–strain curves of PE samples generated by (i) NiC5, (j) NiC6, (k) NiC8, and (l) NiC12 at 80 °C during step-cycle tensile deformation at different strains. | |
Table 5 Mechanical properties for polyethylene samples obtained at different temperatures using different nickel precatalysts
| PEa |
M
w
(105) |
T
m (°C)c |
B
|
σ (MPa)e |
ε
(%) |
Max Elong.e (mm) |
SRe (%) |
|
PE samples (Table 3, entries 1–4, 7–10, 13–16, 19–22).
GPC results (Mw unit: g mol−1).
DSC results.
Determined by 1H NMR spectra [(2 × IMe/3 × Itotal) × 1000].
Ultimate tensile strength (σ), elongation at break (ε), strain recovery after ten cycles with fixed strain 300% (SR) were determined using a universal tester.
Not determined.
|
|
PE30NiC5
|
7.92 |
93.7 |
47 |
6.3 |
969 |
91 |
22 |
|
PE40NiC5
|
5.95 |
88.3 |
66 |
6.5 |
1193 |
102 |
29 |
|
PE60NiC5
|
4.33 |
85.7 |
72 |
7.2 |
1428 |
167 |
41 |
|
PE80NiC5
|
3.25 |
82.5 |
89 |
7.8 |
2072 |
205 |
46 |
|
PE30NiC6
|
5.95 |
78.7 |
72 |
10.5 |
1038 |
114 |
34 |
|
PE40NiC6
|
5.25 |
72.9 |
90 |
11.8 |
1201 |
115 |
40 |
|
PE60NiC6
|
4.04 |
67.4 |
96 |
6.4 |
1636 |
152 |
43 |
|
PE80NiC6
|
2.82 |
66.5 |
112 |
8.1 |
3105 |
303 |
47 |
|
PE30NiC8
|
9.18 |
84.2 |
34 |
8.9 |
809 |
78 |
36 |
|
PE40NiC8
|
8.03 |
83.7 |
—f |
19.3 |
1054 |
113 |
41 |
|
PE60NiC8
|
5.77 |
78.4 |
86 |
27.4 |
1231 |
152 |
44 |
|
PE80NiC8
|
4.57 |
76.9 |
88 |
25.9 |
1280 |
161 |
70 |
|
PE30NiC12
|
11.2 |
74.6 |
60 |
10.5 |
542 |
67 |
34 |
|
PE40NiC12
|
11.0 |
75.2 |
—f |
11.4 |
688 |
85 |
47 |
|
PE60NiC12
|
5.27 |
74.1 |
—f |
7.4 |
702 |
88 |
56 |
|
PE80NiC12
|
5.25 |
72.3 |
77 |
4.7 |
1401 |
121 |
77 |
The strain recovery (SR) of these polyethylene samples was quantified through a hysteresis experiment consisting of ten cycles at a fixed strain of 300%. The SR value exhibited a gradual improvement as the reaction temperature increased from 30 °C to 80 °C (Fig. 9e–h, S28, S29,† and Table 5). Polyethylene samples synthesized with NiC5 and NiC6 catalysts displayed moderate strain recovery after the tenth cycle of the hysteresis experiment, with SR values ranging from 22% to 46% and 34% to 47%, respectively. In contrast, the strain recovery of the samples produced with NiC8 and NiC12 catalysts showed significant improvement with the increase of reaction temperature. For example, polyethylene samples PE80NiC8 and PE80NiC12 demonstrated SR values of 70% and 77%, respectively (Fig. 9g and h). Moreover, we assessed the strain recovery of these polyethylene samples at different applied strains, increasing the strain by 200% at each step (Fig. 9i–l). The SR values were plotted against the applied strain for the polyethylene samples synthesized at 80 °C using various precatalysts (Fig. 10). Once again, PE80NiC8 and PE80NiC12 exhibited excellent elastic properties, maintaining SR values above 63% and 76%, respectively, throughout cyclic tensile deformations up to 1400% strain. However, the strain recovery values for the remaining samples exhibited a rapid decline as the applied strain increased. Overall, the elastic properties of PE80NiC8 and PE80NiC12 polyethylene samples exhibited better properties as compared to the polyolefin previously studied by Ricci et al. and are either comparable or slightly inferior to those reported by Coates et al. for multiblock copolymers.60,61 Moreover, the SR values achieved are at par with those of the olefin block copolymers commercialized by Dow.62 It was previously noted in the literature that the polyethylenes prepared at elevated temperatures generally do not exhibit high tensile strength.22–33 This is because higher reaction temperatures tend to result in a greater number of branches, lower molecular weights, and reduced crystallinity. These characteristics collectively make the polymer more amorphous, resulting in a lower tensile strength. In contrast, the polyethylene samples, specifically PE80NiC8, synthesized at higher reaction temperatures (80 °C), displayed exceptional mechanical and elastic properties, including a high tensile strength of 25.9 MPa, excellent elongation at break of 1280%, high strain recovery of 70% after ten cycles with an applied strain of 300%, and the SR values were maintained above 63% throughout cyclic tensile deformations up to 1400% strain. Such a combination of mechanical and elastic properties within a single polymer prepared at high reaction temperature is rarely reported in the literature. Hence, these catalysts offer a unique approach for producing thermoplastic polyethylene elastomers in a single step using only ethylene as the feedstock. It has been established that the polymer molecular weight, branching degree, and branching content, play a significant role in determining the elastic properties of polyethylene. These properties, and subsequently, the elastic properties, can be modulated by altering reaction conditions and catalyst structures. However, the outstanding mechanical properties of these polyethylene samples may also arise from their multiblock microstructure, featuring both hard and soft segments. Prior studies on propylene polymerization have demonstrated that changes in the catalyst geometry can lead to the formation of isotactic (hard segment) and atactic (soft segment) blocks within the polymer.63 Polymers with alternating hard and soft blocks are known to exhibit excellent elastic and mechanical properties. We proposed that the carbocyclic ring at the ortho positions of these α-diimine nickel catalysts can adopt varying conformations, resulting in different steric hindrances during the ethylene polymerization process. If chain transfer reactions occur at a slower rate than the conformational exchange rate of the ortho substituents under the given conditions, it may give rise to the formation of a multiblock polymer with both soft and hard segments. This hypothesis is supported by the 13C NMR spectra, which revealed the presence of short and long chain branches along the polymer chain backbone.
 |
| | Fig. 10 Elastic recovery percentages of PE samples generated at 80 °C by catalysts NiC5, NiC6, NiC8, and NiC12. | |
Experimental section
Synthesis of ligands
LC5
.
Synthesis of ligands is a two-step process. The first step is the one-pot synthesis of zinc complexes. For the LC5, 2,4-dibenzhydryl-6-cyclopentylaniline (0.8 g, 1.62 mmol, 2.27 equiv.) and anhydrous ZnCl2 (0.11 g, 0.81 mmol, 1.14 equiv.) were added into a suspension of acenaphthylen-1,2-dione (0.13 g, 0.71 mmol) in glacial acetic acid (15 mL), and heated to reflux with constant stirring for 4 h. Cooling the reaction mixture to room temperature afforded a red precipitate which was collected by filtration. The resulting red solid was washed with acetic acid (10 ml × 3 times), and then with hexane (10 ml × 3 times) to remove the remaining acetic acid, and finally dried under reduced pressure. The second step was the removal of the coordinated zinc metal to obtain the ligand. For this, the red solid obtained in the first step was dissolved in dichloromethane (10 mL) and then added into a solution of potassium oxalate (0.23 g, 1.26 mmol, 2 equiv.) in water (10 mL). The resulting immiscible solution was stirred vigorously for 1 h at room temperature. The two layers were separated, and the organic layer was washed with water (10 mL × 3 times) and dried with Na2SO4. The removal of all the volatiles under reduced pressure and recrystallization with dichloromethane and hexane afforded an orange crystalline solid (0.71 g, overall yield 88%). FTIR (KBr, cm−1): 3082 (w), 3057 (m), 3023 (m), 2948 (m), 2867 (m), 1664 (ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), m), 1601 (m), 1496 (s), 1447 (m), 1272 (w), 1080 (w), 1032 (w), 930 (w), 833 (w), 778 (w), 742 (w), 697 (vs). 1H NMR (400 MHz, CDCl3, TMS): δ 7.71 (d, J = 8.2 Hz, 1H), 7.57 (d, J = 8.2 Hz, 2H), 7.35–7.18 (m, 23H), 7.18–6.89 (m, 34H), 6.83–6.75 (m, 3H), 6.75–6.56 (m, 9H), 6.31 (dd, J = 9.5, 5.7 Hz, 5H), 6.22 (d, J = 7.1 Hz, 2H), 6.14 (t, J = 7.4 Hz, 2H), 5.68 (s, 1H), 5.62 (s, 2H), 5.53 (s, 3H), 3.22 (t, J = 8.2 Hz, 2H), 2.95 (t, J = 8.1 Hz, 1H), 2.30–2.18 (m, 2H), 2.00 (d, J = 6.9 Hz, 1H), 1.67–1.06 (m, 30H). 13C NMR (100 MHz, CDCl3, TMS): δ 163.2, 147.4, 147.1, 144.6, 144.5, 144.4, 143.8, 141.6, 139.1, 134.2, 132.5, 129.7, 129.6, 129.5, 129.4, 129.4, 129.4, 129.3, 129.2, 128.9, 128.4, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.3, 127.0, 126.6, 126.1, 126.1, 125.8, 125.8, 125.6, 125.5, 125.0, 123.7, 123.3, 56.6, 56.6, 52.4, 51.7, 40.6, 40.0, 35.0, 34.7, 34.0, 31.6, 26.0, 25.8, 25.7, 25.6, 22.7, 14.1. Anal. calcd for C86H72N2 (1133.54): C, 91.13; H, 6.40; N, 2.47. Found: C, 90.64; H, 6.39; N, 2.46.
N), m), 1601 (m), 1496 (s), 1447 (m), 1272 (w), 1080 (w), 1032 (w), 930 (w), 833 (w), 778 (w), 742 (w), 697 (vs). 1H NMR (400 MHz, CDCl3, TMS): δ 7.71 (d, J = 8.2 Hz, 1H), 7.57 (d, J = 8.2 Hz, 2H), 7.35–7.18 (m, 23H), 7.18–6.89 (m, 34H), 6.83–6.75 (m, 3H), 6.75–6.56 (m, 9H), 6.31 (dd, J = 9.5, 5.7 Hz, 5H), 6.22 (d, J = 7.1 Hz, 2H), 6.14 (t, J = 7.4 Hz, 2H), 5.68 (s, 1H), 5.62 (s, 2H), 5.53 (s, 3H), 3.22 (t, J = 8.2 Hz, 2H), 2.95 (t, J = 8.1 Hz, 1H), 2.30–2.18 (m, 2H), 2.00 (d, J = 6.9 Hz, 1H), 1.67–1.06 (m, 30H). 13C NMR (100 MHz, CDCl3, TMS): δ 163.2, 147.4, 147.1, 144.6, 144.5, 144.4, 143.8, 141.6, 139.1, 134.2, 132.5, 129.7, 129.6, 129.5, 129.4, 129.4, 129.4, 129.3, 129.2, 128.9, 128.4, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.3, 127.0, 126.6, 126.1, 126.1, 125.8, 125.8, 125.6, 125.5, 125.0, 123.7, 123.3, 56.6, 56.6, 52.4, 51.7, 40.6, 40.0, 35.0, 34.7, 34.0, 31.6, 26.0, 25.8, 25.7, 25.6, 22.7, 14.1. Anal. calcd for C86H72N2 (1133.54): C, 91.13; H, 6.40; N, 2.47. Found: C, 90.64; H, 6.39; N, 2.46.
LC6
.
Following the same two-step procedure as described for the synthesis of LC5 with only difference in aniline (2,4-dibenzhydryl-6-cyclohexylaniline) afforded the corresponding ligand LC6 as an orange solid (0.64 g, overall yield 77%). FTIR (KBr, cm−1): 3084 (w), 3057 (m), 3023 (m), 2928 (s), 2867 (m), 1668 (ν(CN), m), 1652 (ν(CN), m), 1603 (m), 1496 (s), 1447 (m), 1275 (w), 1244 (w), 1078 (w), 1032 (w), 932 (w), 833 (w), 778 (w), 746 (w), 699 (vs). 1H NMR (400 MHz, CDCl3, TMS): δ 7.54 (d, J = 8.2 Hz, 2H), 7.36–7.26 (m, 8H), 7.25–7.19 (m, 4H), 7.19–7.01 (m, 17H), 7.01–6.87 (m, 6H), 6.74 (dd, J = 5.4, 3.2 Hz, 6H), 6.26 (t, J = 7.5 Hz, 4H), 6.19–6.07 (m, 4H), 5.56 (d, J = 13.7 Hz, 4H), 2.84 (t, J = 10.4 Hz, 2H), 2.09 (s, 2H), 1.68–1.54 (m, 6H), 1.51–0.99 (m, 14H). 13C NMR (100 MHz, CDCl3, TMS): δ 163.70, 146.63, 144.57, 144.47, 143.97, 141.61, 139.06, 135.42, 132.05, 129.95, 129.54, 129.41, 129.36, 129.28, 129.22, 129.08, 128.18, 128.15, 128.02, 127.78, 127.19, 126.66, 126.14, 126.11, 125.89, 125.72, 125.06, 123.49, 56.60, 52.23, 38.55, 34.43, 33.63, 26.68, 25.93. Anal. calcd for C88H76N2 (1161.59): C, 90.99; H, 6.60; N, 2.41. Found: C, 91.14; H, 6.59; N, 2.43.
LC8
.
Following the same two-step procedure as described for the synthesis of LC5 with only difference in aniline (2,4-dibenzhydryl-6-cyclooctylaniline) afforded the corresponding ligand LC8 as an orange solid (0.65 g, overall yield 75%). FTIR (KBr, cm−1): 3086 (w), 3062 (m), 3029 (m), 2920 (s), 2853 (m), 1664 (ν(CN), m), 1599 (m), 1496 (s), 1445 (m), 1277 (w), 1078 (w), 1032 (w), 926 (w), 831 (w), 782 (w), 738 (w), 701 (vs). 1H NMR (400 MHz, CDCl3, TMS): δ 7.55 (d, J = 8.2 Hz, 2H), 7.34–7.17 (m, 18H), 7.17–6.90 (m, 23H), 6.77–6.69 (m, 6H), 6.30 (t, J = 7.5 Hz, 4H), 6.24–6.12 (m, 4H), 5.56 (d, J = 19.0 Hz, 4H), 3.04 (dt, J = 8.9, 4.7 Hz, 2H), 2.05–1.93 (m, 2H), 1.74–1.57 (m, 6H), 1.57–1.30 (m, 16H), 1.25 (d, J = 10.8 Hz, 9H). 13C NMR (100 MHz, CDCl3, TMS): δ 163.18, 146.15, 144.59, 144.54, 143.97, 141.81, 140.12, 138.77, 135.74, 132.29, 129.85, 129.64, 129.42, 129.40, 129.33, 129.11, 128.16, 128.00, 127.75, 127.32, 126.69, 126.11, 125.71, 125.09, 123.32, 56.51, 52.26, 38.99, 31.76, 31.48, 28.42, 27.67, 25.36, 25.25, 25.13. Anal. calcd for C92H84N2 (1217.70): C, 90.75; H, 6.95; N, 2.30. Found: C, 90.99; H, 6.94; N, 2.29.
LC12
.
Following the same two-step procedure as described for the synthesis of LC5 with only difference in aniline (2,4-dibenzhydryl-6-cyclododecylaniline) afforded the corresponding ligand LC12 as an orange solid (0.62 g, overall yield 66%). FTIR (KBr, cm−1): 3088 (w), 3057 (m), 3027 (m), 2928 (s), 2863 (m), 1656 (ν(CN), m), 1603 (m), 1498 (s), 1445 (m), 1034 (w), 926 (w), 835 (w), 778 (w), 738 (w), 699 (vs). 1H NMR (400 MHz, CDCl3, TMS): δ 7.55 (d, J = 8.2 Hz, 2H), 7.32–7.16 (m, 19H), 7.16–7.02 (m, 14H), 7.02–6.91 (m, 8H), 6.73–6.67 (m, 5H), 6.36 (t, J = 7.5 Hz, 4H), 6.28–6.15 (m, 4H), 5.56 (d, J = 20.6 Hz, 4H), 2.95 (d, J = 7.1 Hz, 2H), 1.84 (s, 2H), 1.40 (d, J = 6.8 Hz, 6H), 1.34–1.03 (m, 40H). 13C NMR (100 MHz, CDCl3, TMS): δ 162.59, 146.33, 144.66, 144.56, 143.94, 141.95, 138.16, 133.53, 132.02, 129.70, 129.56, 129.39, 129.36, 129.32, 129.08, 129.02, 128.08, 127.96, 127.75, 127.45, 126.57, 126.05, 125.69, 125.10, 122.96, 56.42, 52.27, 37.00, 31.57, 27.75, 27.23, 26.26, 26.02, 22.63, 22.32, 22.26, 22.14, 20.93, 20.62, 14.08. Anal. calcd for C100H100N2 + EtOH (1375.98): C, 89.04; H, 7.77; N, 2.04. Found: C, 89.39; H, 7.93; N, 2.02.
Synthesis of nickel complexes
NiC5
.
Under a nitrogen atmosphere, (DME)NiBr2 (64 mg, 0.19 mmol) was added into the solution of ligand LC5 (225 mg, 0.19 mmol) in dichloromethane (10 mL) and stirred at room temperature. After around 12 hours of stirring, the reaction mixture was concentrated under reduced pressure, and excess diethyl ether was added to induce precipitates. The resulting deep red solid was further washed with diethyl ether (3 × 5 mL) and filtered. Drying under reduced pressure for at least 24 h afforded NiC5 as a deep red solid. Overall yield: 210 mg, 78%. FTIR (KBr, cm−1): 3088 (w), 3059 (m), 3025 (m), 2958 (m), 2865 (m), 1652 (ν(CN), m), 1601 (m), 1496 (s), 1451 (m), 1291 (w), 1078 (w), 1034 (w), 835 (w), 782 (w), 746 (w), 701 (vs). Anal. calcd for C86H72NiBr2N2 + EtOH (1398.11): C, 75.60; H, 5.62; N, 2.00. Found: C, 75.61; H, 5.49; N, 2.03.
NiC6
.
Adopting the general procedure of NiC5 synthesis, NiC6 was afforded as a deep red solid. Yield: 212 mg, 89%. FTIR (KBr, cm−1): 3082 (w), 3059 (m), 3025 (m), 2926 (m), 2853 (m), 1647 (ν(CN), m), 1619 (m), 1601 (m), 1583 (s), 1498 (s), 1451 (m), 1291 (w), 1248 (w), 1078 (w), 1030 (w), 776 (m), 746 (m), 703 (vs). Anal. calcd for C88H76NiBr2N2 + EtOH (1380.09): C, 76.59; H, 5.55; N, 2.03. Found: C, 76.12; H, 5.77; N, 1.99.
NiC8
.
Adopting the general procedure of NiC5 synthesis, NiC8 was afforded as a deep red solid. Yield: 234 mg, 90%. FTIR (KBr, cm−1): 3084 (w), 3059 (m), 3025 (m), 2920 (s), 2855 (m), 1649 (ν(CN), m), 1621 (m), 1601 (m), 1583 (m), 1493 (s), 1449 (m), 1293 (w), 1076 (w), 1034 (w), 831 (w), 778 (w), 746 (w), 699 (vs). Anal. calcd for C92H84NiBr2N2 + EtOH (1482.27): C, 76.17; H, 6.12; N, 1.89. Found: C, 75.95; H, 5.85; N, 1.93.
NiC12
.
Adopting the general procedure of NiC5 synthesis, NiC12 was afforded as a deep red solid. Yield: 360 mg, 91%. FTIR (KBr, cm−1): 3084 (w), 3059 (m), 3023 (m), 2936 (s), 2863 (m), 1647 (ν(CN), m), 1619 (m), 1601 (m), 1583 (m), 1500 (s), 1471 (m), 1445 (m), 1293 (w), 1080 (w), 1032 (w), 833 (w), 778 (w), 744 (w), 703 (vs). Anal. calcd for C100H100NiBr2N2 + EtOH (1594.48): C, 76.83; H, 6.70; N, 1.76. Found: C, 76.66; H, 6.45; N, 1.80.
Conclusion
In summary, a series of 1,2-diaryliminoacenaphthylene nickel complexes with a hybrid steric hindrance of cycloalkyl and benzhydryl groups were prepared and characterized through FTIR, 1H and 13C NMR spectroscopy, elemental analysis, and single crystal X-ray diffraction. These complexes, upon activation with EASC, exhibited exceptional catalytic performance in ethylene polymerization. In particular, the precatalyst NiC5 demonstrated exceptional polymerization activity, showing a level of 16.9 × 106 to 42.9 × 106 over one hour at 30 °C. More remarkably, it maintained a high activity of 5.6 × 106 at 80 °C, 1.5 × 106 at 100 °C and 0.7 × 106 at 110 °C after two hours, highlighting the high thermal stability of the prepared precatalysts. Variations in the cycloalkyl group ring size were found to influence the catalytic performance and the properties of the resultant polymer. Precatalysts with smaller cycloalkyl rings exhibited superior polymerization activity, whereas larger rings facilitated the chain growth reaction, producing high to ultra-high molecular weight polyethylenes with narrow dispersity. Moreover, the degree of branching in the resultant polyethylene could be modulated in a range of 34–145 per 1000C. These variations significantly impacted the mechanical and elastic properties of the polyethylene. The previously reported polyethylene prepared at a high reaction temperature (≥70 °C) exhibited inferior mechanical properties due to high branching and low molecular weights. In contrast, the herein reported polyethylene produced at higher temperatures not only exhibited excellent mechanical properties (tensile strength σ = 25.9 MPa, elongation at break ε = 1280%) but simultaneously showed remarkable elastic recovery (SR = 70%). These properties align with those of thermoplastic polyolefin elastomers and represent a unique combination of tensile strength and elasticity rarely obtained in polyethylenes synthesized at high temperatures.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work has been financially supported by the Chemistry and Chemical Engineering Guangdong Laboratory (2111018 and 2132012), and Q. M. would like to express gratitude towards the Foreign Youth Talent Program (QN2022030008L) for their support. H. S. would like to express gratitude towards the ANSO Scholarship Program for their support.
References
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93 CrossRef CAS.
- G. Zanchin and G. Leone, Prog. Polym. Sci., 2021, 113, 101342 CrossRef CAS.
- B. Adhikari, D. De and S. Maiti, Prog. Polym. Sci., 2000, 25, 909 CrossRef CAS.
- P. D. Hustad, Science, 2009, 325, 704–707 CrossRef CAS PubMed.
-
A. Gurses, K. Gunes and E. Sahin, in Green Chemistry for Sustainable Textiles, The Textile Institute Book Series, Woodhead Publishing, 2011, vol. 6, pp. 77–91 Search PubMed.
-
C. D. Craver and C. E. Carraher, in Applied Polymer Science: 21st Century, Elsevier, 1st edn, 2000 Search PubMed.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714–719 CrossRef CAS PubMed.
- H. Ohtaki, F. Deplace, G. D. Vo, A. M. LaPointe, F. Shimizu, T. Sugano, E. J. Kramer, G. H. Fredrickson and G. W. Coates, Macromolecules, 2015, 48, 7489–7494 CrossRef CAS.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267–268.2 CrossRef CAS.
- Z. Ye, L. Xu, Z. Dong and P. Xiang, Chem. Commun., 2013, 49, 6235–6255.2 RSC.
- Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059–2062.2 CrossRef CAS PubMed.
- M. C. Baier, M. A. Zuideveld and S. Mecking, Angew. Chem., Int. Ed., 2014, 53, 9722 CrossRef CAS PubMed.
- D. Meinhard, M. Wegner, G. Kipiani, A. Hearley, P. Reuter, S. Fischer, O. Marti and B. Rieger, J. Am. Chem. Soc., 2007, 129, 9182–9191 CrossRef CAS PubMed.
- Z. Hai, Z. Lu, S. Li, Z. Y. Cao and S. Dai, Polym. Chem., 2021, 12, 4643–4653 RSC.
- Y. Zhang, Y. Zhang, X. Hu, C. Wang and Z. Jian, ACS Catal., 2022, 12, 14304–14320 CrossRef CAS.
- J. M. Eagan, O. Padilla-Velez, K. S. O. Connor, S. N. MacMillan, A. M. LaPointe and G. W. Coates, Organometallics, 2022, 41, 3411–3418 CrossRef CAS.
- X. Wang, L. Fan, Y. Ma, C. Guo, G. A. Solan, Y. Sun and W. H. Sun, Polym. Chem., 2017, 8, 2785–2795 RSC.
- M. Qasim, M. S. Bashir, S. Iqbal and Q. Mahmood, Eur. Polym. J., 2021, 160, 110783 CrossRef CAS.
- Q. Mahmood, X. Li, L. Qin, L. Wang and W. H. Sun, Dalton Trans., 2022, 51, 14375–14407 RSC.
- H. Suo, G. A. Solan, Y. Ma and W. H. Sun, Coord. Chem. Rev., 2018, 372, 101–116 CrossRef CAS.
- H. Suo, I. V. Oleynik, C. Huang, I. I. Oleynik, G. A. Solan, Y. Ma, T. Liang and W. H. Sun, Dalton Trans., 2017, 46, 1568 RSC.
- Q. Mahmood, Y. Zeng, E. Yue, G. A. Solan, T. Liang and W. H. Sun, Polym. Chem., 2017, 8, 6416–6430 RSC.
- R. Wu, Y. Wang, R. Zhang, C. Y. Guo, Z. Flisak, Y. Sun and W. H. Sun, Polymer, 2018, 153, 574–586 CrossRef CAS.
- Q. Zhang, R. Zhang, Y. Ma, G. A. Solan, T. Liang and W. H. Sun, Appl. Catal., A, 2019, 573, 73–86 CrossRef CAS.
- R. Zhang, Z. Wang, Y. Ma, G. A. Solan, Y. Sun and W. H. Sun, Dalton Trans., 2019, 48, 1878 RSC.
- Y. Wang, A. Vignesh, M. Qu, Z. Y. Sun and W. H. Sun, Eur. Polym. J., 2019, 117, 254–271 CrossRef CAS.
- J. Fang, X. Sui, Y. Li and C. Chen, Polym. Chem., 2018, 9, 4143–4149 RSC.
- L. Guo, K. Lian, W. Kong, S. Xu, G. Jiang and S. Dai, Organometallics, 2018, 37, 2442–2449 CrossRef CAS.
- L. Qin, X. Wang, Q. Mahmood, Z. Yu, Y. Wang, S. Zou, T. Liang and W. H. Sun, Chin. J. Polym. Sci., 2023 DOI:10.1007/s10118-024-3073-0.
- S. Dai, S. Li, G. Xu, C. Wu, Y. Liao and L. Guo, Polym. Chem., 2020, 11, 1393–1400 RSC.
- K. Lian, Y. Zhu, W. Li, S. Dai and C. Chen, Macromolecules, 2017, 50, 6074–6080 CrossRef CAS.
- L. Guo, W. Sun, S. Li, G. Xu and S. Dai, Polym. Chem., 2019, 10, 4866–4871 RSC.
- L. Wang, M. Liu, Q. Mahmood, S. Yuan, X. Li, L. Qin, S. Zou, T. Liang and W. H. Sun, Eur. Polym. J., 2023, 194, 112112 CrossRef CAS.
- W. Lu, B. Ding, W. Zou and S. Dai, Eur. Polym. J., 2023, 201, 112577 CrossRef CAS.
- F. Zhai and R. F. Jordan, Organometallics, 2017, 36(15), 2784–2799 CrossRef CAS.
- M. Gao, S. Du, Q. Ban, Q. Xing and W. H. Sun, J. Organomet. Chem., 2015, 798, 401–407 CrossRef CAS.
- X. Wang, L. Fan, Y. Ma, C.-Y. Guo, G. A. Solan, Y. Sun and W. H. Sun, Polym. Chem., 2017, 8(18), 2785–2795 RSC.
- L. Guo, W. Kong, Y. Xu, Y. Yang, R. Ma, L. Cong, S. Dai and Z. Liu, J. Organomet. Chem., 2018, 859, 58–67 CrossRef CAS.
- Y. Zhao, S. Li, W. Fan and S. Dai, J. Organomet. Chem., 2021, 932, 121649 CrossRef CAS.
- S. Dai and C. Chen, Angew. Chem., Int. Ed., 2016, 55, 13281–13285 CrossRef CAS PubMed.
- L. Yang, C. Adam, G. Nichol and S. Cockroft, Nat. Chem., 2013, 5, 1006–1010 CrossRef CAS PubMed.
- J. Echeverría, G. Aullon, D. Danovich, S. Shaik and S. Alvarez, Nat. Chem., 2011, 3, 323–330 CrossRef PubMed.
- Q. Mahmood, E. Yue, J. Guo, W. Zhang, Y. Ma, X. Hao and W. H. Sun, Polymer, 2018, 159, 124–137 CrossRef CAS.
- X. Li, L. Qin, Q. Mahmood, Z. Yu, S. Zou, Y. Wang, T. Liang and W. H. Sun, Eur. Polym. J., 2023, 200, 112520 CrossRef CAS.
-
(a) H. U. Moritz, Chem. Eng. Technol., 1989, 12, 71–87 CrossRef CAS;
(b) Q. Mahmood, Y. Zeng, X. Wang, Y. Sun and W. H. Sun, Dalton Trans., 2017, 46, 6934–6947 RSC.
- Y. Zeng, Q. Mahmood, X. Hao and W. H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1910–1919 CrossRef CAS.
- S. A. Svejda, K. L. Johnson and M. Brookhart, J. Am. Chem. Soc., 1999, 121, 10634 CrossRef CAS.
- J. Heinemann, R. Mülhaupt, P. Brinkmann and G. Luinstra, Macromol. Chem. Phys., 1999, 200, 384 CrossRef CAS.
- L. H. Shultz, D. J. Tempel and M. Brookhart, J. Am. Chem. Soc., 2001, 123, 11539–11555 CrossRef CAS PubMed.
-
(a) M. D. Leatherman, S. A. Svejda, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 2003, 125, 3068–3081 CrossRef CAS PubMed;
(b) D. P. Gates, S. A. Svejda, E. Oñate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320–2334 CrossRef CAS;
(c) C. S. Popeney, A. L. Rheingold and Z. Guan, Organometallics, 2009, 28, 4452–4463 CrossRef CAS;
(d) L. S. Lee, H. J. Ou and H. F. Hsu, Fluid Phase Equilib., 2005, 231, 221–230 CrossRef CAS.
-
(a) M. Zada, L. Guo, R. Zhang, W. Zhang, Y. Ma, G. A. Solan, Y. Sun and W. H. Sun, Appl. Organomet. Chem., 2019, 33, 4749 CrossRef;
(b) D. H. Camacho and Z. Guan, Chem. Commun., 2010, 46, 7879–7893 RSC;
(c) D. H. Camacho, E. V. Salo, J. W. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2004, 43, 1821–1825 CrossRef CAS PubMed.
- R. Wu, Y. Wang, R. Zhang, C. Y. Guo, Z. Flisak, Y. Sun and W. H. Sun, Polymer, 2018, 153, 574–586 CrossRef CAS.
- D. Zhang, E. T. Nadres, M. Brookhart and O. Daugulis, Organometallics, 2013, 32, 5136–5143 CrossRef CAS.
- J. L. Rhinehart, L. A. Brown and B. K. Long, J. Am. Chem. Soc., 2013, 135, 16316–16319 CrossRef CAS PubMed.
- D. H. Camacho and Z. Guan, Macromolecules, 2005, 38, 2544–2546 CrossRef CAS.
- C. S. Popeney, A. L. Rheingold and Z. Guan, Organometallics, 2009, 28, 4452–4463 CrossRef CAS.
- S. Jiang, Y. Zheng, I. V. Oleynik, Z. Yu, G. A. Solan, I. I. Oleynik, M. Liu, Y. Ma and W. H. Sun, Molecules, 2023, 28(12), 4852 CrossRef CAS PubMed.
- Z. He, Y. Liang, W. Yang, H. Uchino, J. Yu, W. H. Sun and C. C. Han, Polymer, 2015, 56, 119–122 CrossRef CAS.
- G. Leone, M. Mauri, F. Bertini, M. Canetti, D. Piovani and G. Ricci, Macromolecules, 2015, 48, 1304–1312 CrossRef CAS.
- K. S. O. Connor, A. Watts, T. Vaidya, A. M. LaPointe, M. A. Hillmyer and G. W. Coates, Macromolecules, 2016, 49, 6743–6751 CrossRef.
-
W. Weng, A. H. Dekmezian, E. J. Markel and D. L. Peters, U.S. Patent, 62001184327, 2001 Search PubMed.
- G. W. Coates and R. M. Waymouth, Science, 1995, 267, 217–219 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General consideration and materials, a general procedure for ethylene polymerization, X-ray crystallographic studies, Table S1 crystal data and structural refinements for NiC5, NiC6 and NiC12, 1H and 13C NMR spectra of ligands (Fig. S1–S8), 1H NMR spectra of polyethylene under different conditions (Fig. S9–S27), elastic recovery measurement of different polyethylene samples (Fig. S28 and S29), GPC curves of different polyethylene samples (Fig. S30 and S31), and references. CCDC 2326343 (NiC5), 2326344 (NiC6) and 2326345 (NiC12). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4py00061g |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b,
Rongyan
Yuan
b,
Yizhou
Wang
ac,
Song
Zou
a,
Kainat Fatima
Tahir
abc,
Yanping
Ma
*b,
Rongyan
Yuan
b,
Yizhou
Wang
ac,
Song
Zou
a,
Kainat Fatima
Tahir
abc,
Yanping
Ma