
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of a gold nanoparticles site in an engineered lipase: an artificial metalloenzyme with enantioselective reductase-like activity†
Carla
Garcia-Sanz
 a,
Blanca
de las Rivas
b and
Jose M.
Palomo
*a
a,
Blanca
de las Rivas
b and
Jose M.
Palomo
*a
aInstituto de Catálisis y Petroleoquímica (ICP), CSIC, c/Marie Curie 2, Campus UAM Cantoblanco, 28049 Madrid, Spain. E-mail: josempalomo@icp.csic.es
bDepartment of Microbial Biotechnology, Institute of Food Science, Technology and Nutrition (ICTAN-CSIC), José Antonio Novais 10, 28040 Madrid, Spain
First published on 11th March 2024
Abstract
The conjugation of gold complexes with proteins has proved to be interesting and effective in obtaining artificial metalloenzymes as catalysts with improved properties such as higher stability, activity and selectivity. However, the design and precise regulation of their structure as protein nanostructured forms level remains a challenge. Here, we have designed and constructed a gold nanoparticles-enzyme bioconjugate, by tailoring the in situ formation of gold nanoparticles (AuNPs) at two specific sites on the structure of an alkalophilic lipase from Geobacillus thermocatenulatus (GTL). For this purpose, two genetically modified variants of GTL were created by inserting a unique cysteine residue into the catalytic active site by replacing the active serine (GTL-114) and into the lid site (GTL-193). The enzyme, after a first protein-gold coordination, induced the in situ formation of AuNPs, generating a homogeneous artificial enzyme. The size and morphology of the nanoparticles in the AuNPs-enzyme conjugate have been controlled by specific pH conditions in synthesis and the specific protein region where they are formed. Reductase activity of all of them was confirmed in the hydrogenation of nitroarenes in aqueous media. The protein area seemed to be key for the AuNPs, with the best TOF values obtained for the bioconjugates with AuNPs in the lid site. Finally, the protein environment and the asymmetric properties of the AuNPs were tested in the reduction of acetophenone to 1-phenylethanol in aqueous medium at room temperature. A high reductive conversion and an enantiomeric excess of up to 39% towards (R)-1-phenylethanol was found using Au-Mt@GTL-114 pH 10 as a catalyst. Moderate enantioselectivity towards the opposite isomer was also observed using the Au-Mt@GTL-193 pH 10 conjugate.
Introduction
Metalloproteins are proteins that contain at least one metal incorporated into their structure, where the metal is necessary for the proper function of the protein.1 They are abundant in natural systems and their metal ions have a wide range of functions, including transport and storage of small molecules (e.g., haemoglobin with an Fe2+ site), stabilisation of protein structure (Zn2+ sites in zinc fingers), signalling (Ca2+ channels in signal transduction), and catalysis.2,3 Metalloproteins involved in catalytic transformations are called metalloenzymes. They can carry out reactions with exceptionally high selectivity and specificity, including thermodynamically difficult reactions, such as the reduction of dinitrogen to ammonium (nitrogenases) or the oxidation of water in photosynthesis.4 Usually, enzymes are designed by nature to catalyse reactions efficiently under physiological conditions, but, when confronted with unnatural substrates, enzymes often exhibit low activity and selectivity.5 Therefore, in recent years the synthesis of new artificial metalloenzymes by combining metal systems and enzymes has been a promising line of research in recent years. This field is expanding not only in terms of its applications, but also in term of design.6 The success of such hybrids is based on the advantages offered by the combination of these two domains. The enzyme provides a three-dimensional structure that is excellent for generating a coordination sphere, which has shown high selectivity in different processes and, the use of metal catalysts, (a coordinated metal or organometallic structure) offers a wide range of catalytic possibilities.7 Therefore, this concept has made it possible to obtain higher activity and selectivity of natural metalloenzymes and metalloenzymes with catalytic activities that do not exist in nature.8,9Several strategies have been described for the design of such artificial metalloenzymes.10 One is the genetic modification of amino acids by altering the function or modifying the environment of metal-coordinated amino acids in natural metalloenzymes. Another strategy is based on the creation of new enzymatic or non-biological activities from non-catalytic protein scaffolds.11,12 Finally, it is also possible to replace the existing metal ion in the metalloenzyme with an alternative one.13–15
Indeed, gold nanoparticles (AuNPs) as catalysts have been found many applications in inorganic reactions (e.g., the water–gas shift reaction, ozone decomposition, selective oxidation of H2 to H2O2) and organic reactions (e.g., selective oxidation/reduction of organic compounds, carbon–carbon coupling).16 In general, reduction of AuNPs particle size increases catalytic activity. However, there is a threshold size below which further reduction inhibits catalysis, and the size and shape of metal nanoparticles have a significant effect on their properties. When AuNPs are used as catalysts, aggregation to form bulk material and catalyst poisoning are the most common problems and there is a need to use stabilisers to avoid nanoparticle agglomeration (such as sodium borohydride or sodium citrate).17
Within this field, the conjugation of Au complexes with proteins and enzymes, generating new types of artificial metalloenzymes, has proven to be interesting and effective in obtaining materials with improved properties such as higher stability, catalytic activity and selectivity, among others.18 Other advantages are based on the applicability of chemical processes in aqueous media, which represent more sustainable conditions for Au chemistry. For this reason, such enzyme-Au bioconjugates are becoming increasingly important due to their wide range of applications in the field of nanomedicine such as diagnostic and sensing methods, photothermal therapy and administration and drug delivery among others.19,20
In fact, only a few works have been reported in the literature on design proteins for the sustainable synthesis of Au nanoclusters.21,22 However, in all cases, very large nanoparticle sizes are obtained, and the presence of an external reducing agent is necessary for their formation. Other strategies are based on the introduction of organometallic Au complexes into the protein structure; however, this method requires the use of solvents and much longer synthesis times.23
Therefore, here we describe a new strategy in the synthesis and design of artificial gold metalloenzymes based on the in situ generation of gold nanoparticles coordinated to the enzyme structure from simple gold salts to create for the first time an enzyme-AuNP conjugate. For this purpose, the genetically modified enzyme Geobacillus thermocatenulatus (GTL), containing a unique cysteine (high affinity to Au) will be used.
GTL is a thermoalkalophilic lipase with a size of 6.5 nm × 6.3 nm × 3.65 nm, which has two different oligopeptide lids (Fig. 1).24 Its enzymatic mechanism is based on the movement of an oligopeptide chain from a closed conformation, in which the active site is inaccessible to the solvent, to an open conformation in which this active site is accessible.25 During this open-closed mechanism, the two oligopeptides move simultaneously. The open conformation of the enzyme has a large active site cavity of 2.78 nm (Fig. 1). Recently, we have shown that the lid region of the enzyme could serve as a “second” active site in the protein, in addition to the native active cavity.26
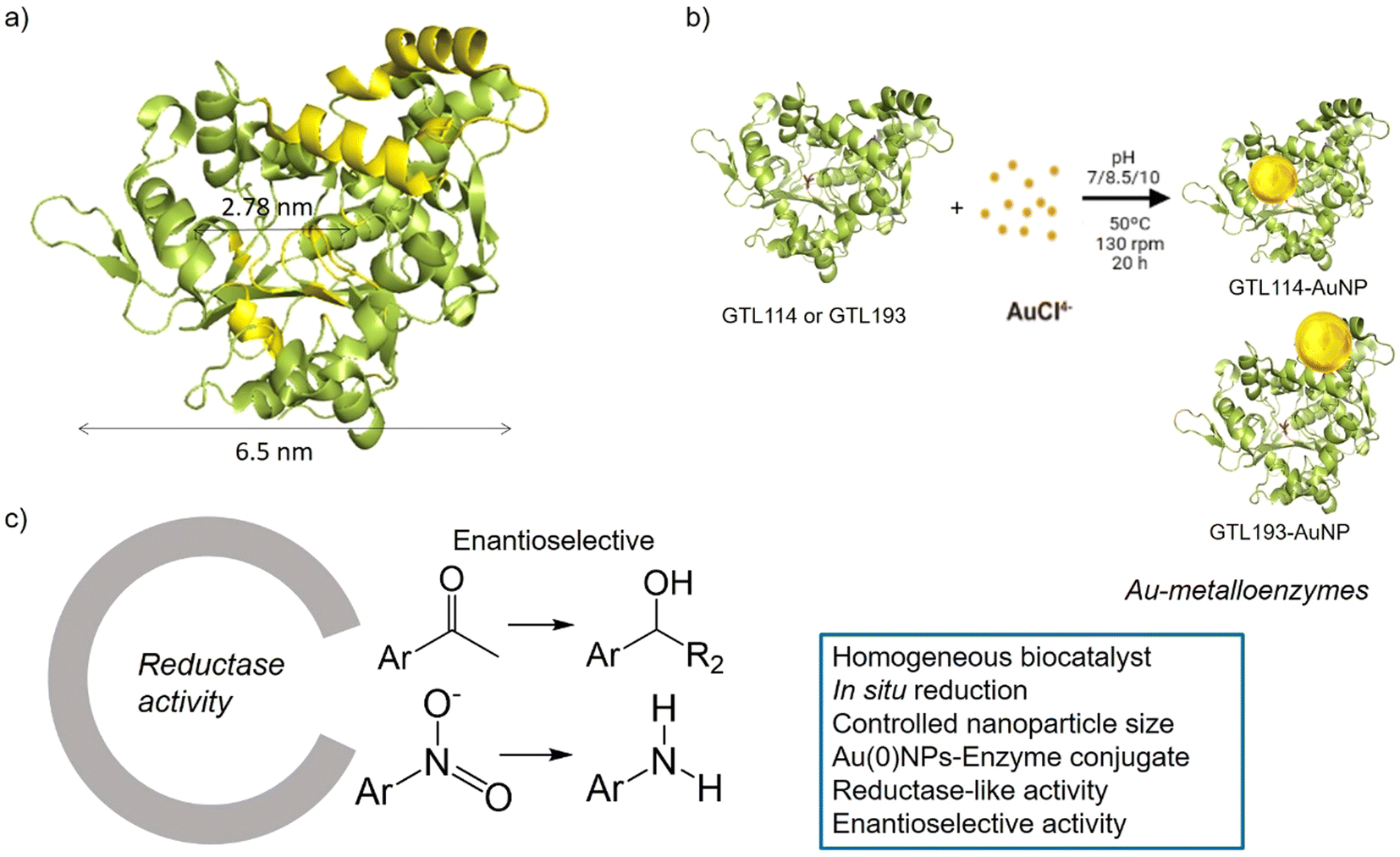 | ||
| Fig. 1 Schematic illustration of the gold-enzyme conjugate synthesis and reductase catalytic activity. (a) GTL structural dimension, (b) Synthetic approach, (c) Novel reductase-like activity and advantages of the artificial metalloenzymes. | ||
The hypothesis is therefore to generate gold nanoparticles in situ, with the enzyme itself acting as a reducing agent (mainly Ser, Thr or hydrophobic residues) (Fig. 1b). In order to focus the formation of these AuNPs, two genetic variants of GTL were used, in which a unique cysteine residue (high affinity Au–S bond) was incorporated in these two areas, the active site (S114C) and the lid site (A193C). Thus, the influence of each enzyme environment on the synthesis, morphology and size of AuNP was studied. Finally, the different enzyme-Au bioconjugates were tested as catalysts in reductive processes, in particular evaluating the second coordination sphere of the enzyme in the chiral properties in asymmetric reduction, transforming an esterase into an enantioselective reductase.
Gold metalloenzymes have been shown to be interesting and effective in arylation and hydroamination reactions.27 Indeed, asymmetric reduction of ketones (to synthesize optically pure alcohols)28 is a crucial step in the synthesis of many pharmaceuticals, agrochemicals, fragrances, and other small organic molecules.29
In this way, innovative systems such as these newly synthesised gold metalloenzymes, exploiting the biocatalytic environment, the robustness of the enzyme and the efficiency of the gold nanoparticles, have been tested in the asymmetric reduction of acetophenone to 1-phenylethanol (Fig. 1).
Results and discussion
Genetic modification of Geobacillus Thermocatenulatus lipase (GTL)
To prepare the two different enzyme areas, genetic variants of Geobacillus Thermocatenulatus lipase (GTL) were first produced by targeted mutagenesis. For this purpose, the targeted introduction of a cysteine (Cys) into their structure was chosen because of the high affinity of this amino acid for bound gold atoms.30 Native GTL contains two cysteines in its complete sequence (C65 and C296). Therefore, prior to the introduction of the cysteine of interest, these native cysteine residues were genetically modified by a conservative change to a serine, overexpressed in E. coli. The new double mutant GTL retained 100% enzymatic activity.25 Next, targeted mutagenesis was performed at positions 114 and 193 of the protein, respectively. In the first case, the catalytic serine 114 was modified by a Cys residue, producing the enzyme variant called GTL-114. In the second case, mutagenesis was performed to introduce a cysteine residue at position 193 (in the lid site),25 producing variant called GTL-193. Structural analysis by fluorescence showed the complete conservation of the three-dimensional structure of the variants compared to the native one and specific activities of the variants were similar to the native GTL (Table S1†).25On the other hand, as mentioned above, GTL is one of the few lipases with two lids, where the area between these two oligopeptide chains was discovered as a new natural active site. For this reason, a targeted mutagenesis was performed at position 193 with cysteine in the oligopeptide, thus designing an enzyme with two active sites. The difference between these two enzyme variants is the possible coordination of Au via the active site (GTL-114) or the lid site (GTL-193).
Synthesis and characterization of AuNP-GTL bioconjugate metalloenzymes
First, both enzyme variants (GTL-114 and GTL 193) were purified. For this purpose, each variant was first adsorbed on a butyl-Sepharose support (a technique that allows the selective reversible immobilisation of lipases in the presence of other proteins).25 Then, once all the lipase variant had been absorbed (tested by enzyme activity), the immobilised derivative was treated in the presence of a buffered solution containing Triton X-100 (pH 7) to selectively desorb the variant and recover the enzyme in solution. This allowed to obtain GTL enzyme fixing the open conformation in all the protein molecules, where they are stabilized by the presence of the detergent molecules (Fig. S1†).24Thus, for the preparation of Au-enzyme conjugates, gold(III) tetrachloride trihydrate (HAuCl4·3H2O) was dissolved in distilled water and the previously obtained enzyme variant solution was added. The synthesis was performed at room temperature and the solution was adjusted to different pHs (3, 7, 8.5 and 10). Gold can absorb light in the visible region of the spectrum due to its surface plasmon resonance (SPR) property.30 Therefore, the formation of metalloenzymes was followed by UV-Vis spectroscopy by monitoring the increase in the band between 520–550 nm, which is characteristic of the formation of AuNPs. No AuNP formation was observed at this temperature in any of the pHs tested (Fig. S2†). In a second approach, the solution was incubated at 50 °C with constant stirring. At this temperature, no Au nanoparticles formation was observed at pH 3 (Fig. S2b†), however, synthesis was obtained in all cases from pH ≥ 7, rapidly indicated by a visible colour change of the solution (Fig. 2 and 3, Fig. S3†). UV analysis showed the formation of a peak between 529 and 541 nm after 2 h of incubation, depending on the GTL mutant used and the pH conditions, although the maximum formation was achieved after 20 h incubation (Fig. 2 and 3, Fig. S3†). In particular, when GTL-114 was used, the highest peak value of 0.23 absorbance (Abs) was obtained in the synthesis at pH 7 (Au-Mt@GTL-114-pH 7) at 533 nm. Higher pH values (8.5 and 10) showed the formation of AuNPs with lower absorbance (0.1) with the maximum wavelength at 544 nm for the other two conjugates (Au-Mt@GTL-114-pH 8.5 and Au-Mt@GTL-114-pH 10) (Fig. 2a). This seems to indicate that the coordination between the enzyme and the AuNPs is slightly different. Indeed, fluorescence analysis showed that the three-dimensional structure of the protein was altered by the gold coordination (Fig. 2b, Table S2†). At pH 7, a clear shift of the maximum by λ = 7 nm (310 nm) from the native enzyme peak (303 nm) was observed in the fluorescence spectrum, suggesting that the presence of Au altered the tertiary structure.
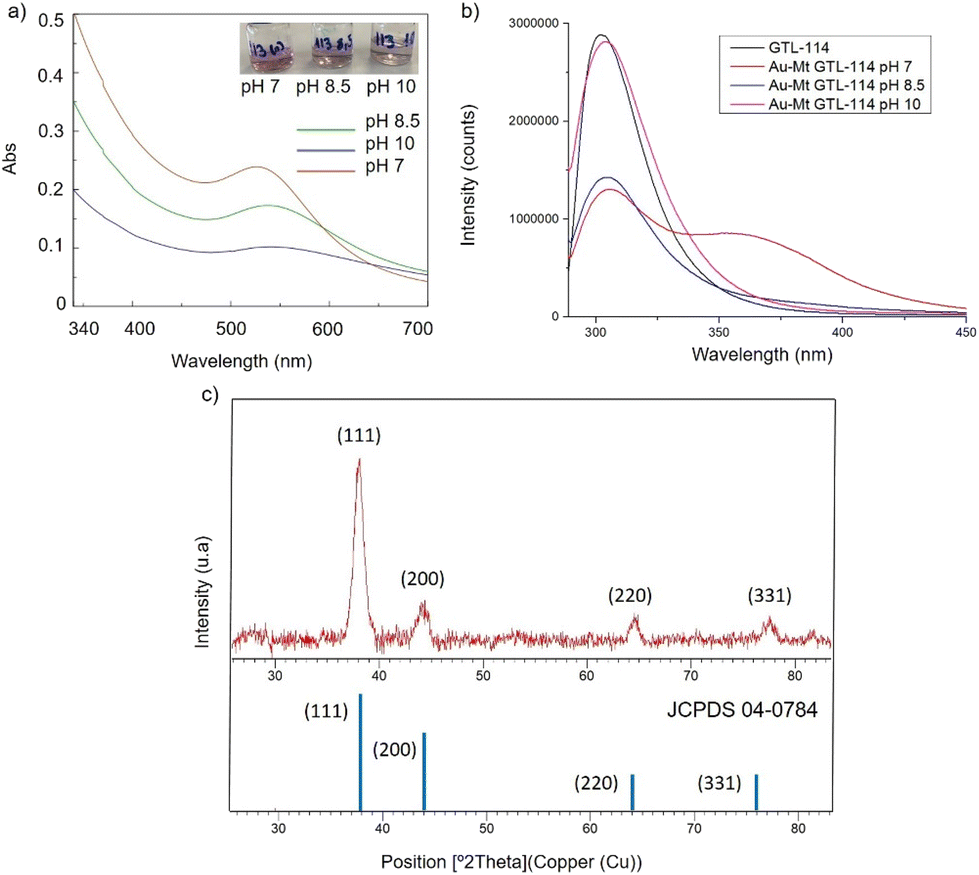 | ||
| Fig. 2 Characterisation of the different GTL-114 gold metalloenzymes. (a) UV-Absorbance spectrophotometer, (b) fluorescence spectra using 280 nm excitation, (c) XRD pattern. | ||
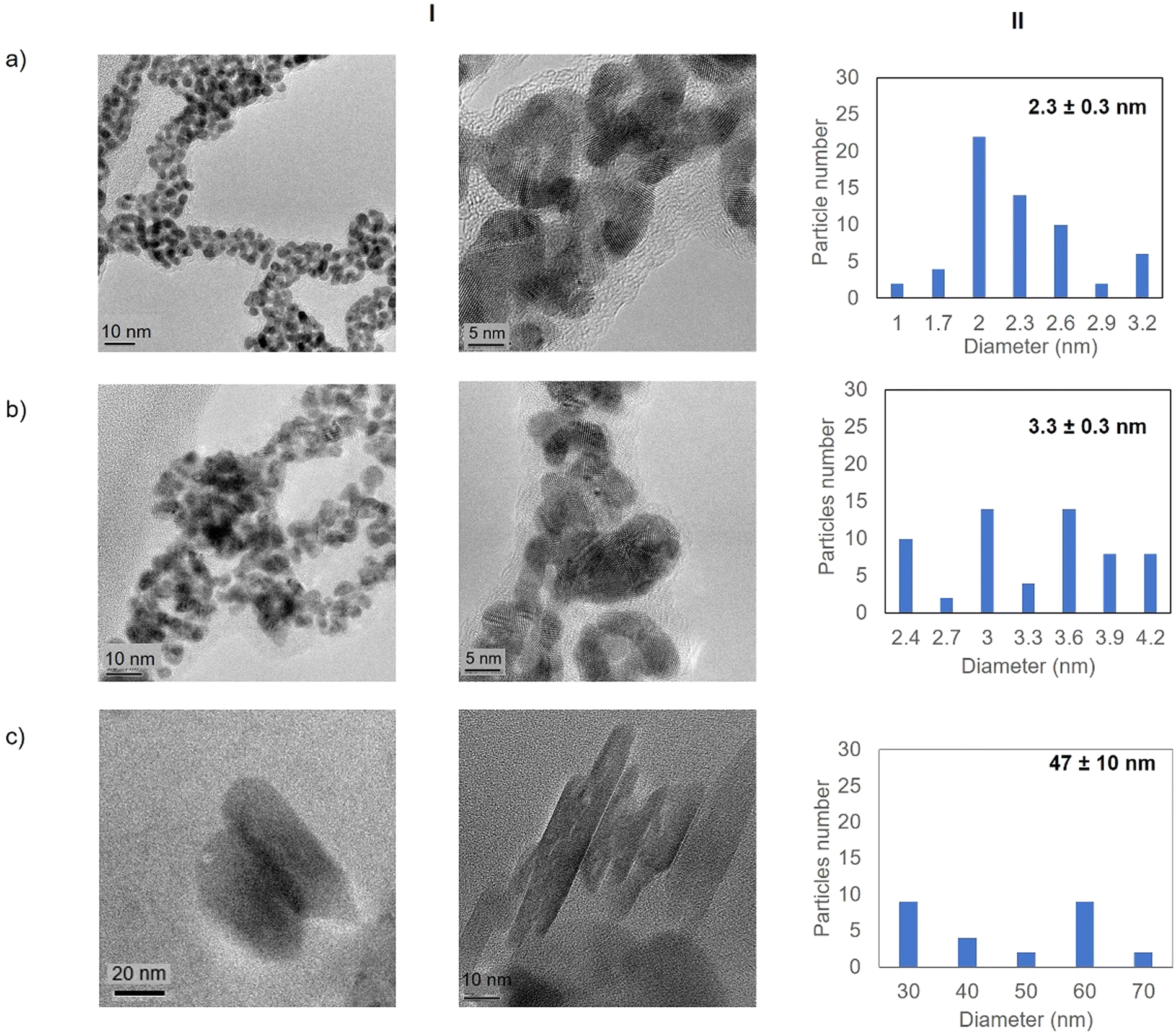 | ||
| Fig. 3 Electronic microscopy characterisation of the different GTL-114 gold metalloenzymes. (a) Au-Mt@GTL-114 pH 7, (b) Au-Mt@GTL-114 pH 8.5, (c) Au-Mt@GTL-114 pH 10. (I) TEM and HR-TEM, (II) nanoparticle size distribution graphic. | ||
At pH 8.5 the protein structure was similar to the obtained at neutral pH. However, at pH 10, the formed gold-variant conserved the same fluorescence intensity as the native enzyme with a slight shift of the spectrum towards the red side (Fig. 2b). In addition, to demonstrate that the gold nanoparticle formation was in all cases uniquely induced by the enzyme producing the enzyme-AuNPs conjugate, synthesis was performed starting from gold salt and amino acid (L-Cys) or cysteine-containing peptide sequences under these conditions in the absence of any reducing agent. Neither AuNPs nor conjugate formation was observed (Fig. S4–S6†). Subsequently, powder X-ray diffraction (XRD) analysis was performed to characterise the gold structures formed in conjugation with the protein (Fig. 2c). The XRD pattern of the sample shows signature for gold (0) species in all cases. The peaks for 2θ at 38.15° (111), 44.37°, 64.5° and 77.5° confirm the presence of gold (0) exclusively in the sample.
High resolution transmission electron microscopy (HR-TEM) revealed the formation of crystalline homogeneously dispersed spherical gold nanoparticles in the GTL-114 metalloenzymes synthesised at pH 7 and pH 8.5 (Fig. 3a and b, Fig. S7†). The average diameter size of the nanoparticles was 2.3 nm and 3.3 nm respectively (Fig. 3a and bII). Indeed, the presence of protein-Au nanoclusters (<2 nm) conjugates is observed in the particle size distribution of the TEM images (Fig. 2aII). NCs exhibit molecular-like properties such as size-depended fluorescence, and this peak around 370 nm in fluorescence spectra (Fig. 2b) seems to indicate a Förster Resonance Energy Transfer (FRET) from Trp of protein to the gold nanocluster.31
However, in the conjugate Au-Mt@GTL-114 pH 10 TEM analysis revealed the formation of Au nanorods structures with a size of about 3 nm diameter and 47 ± 10 nm length (Fig. 3cII).
In the case of GTL-193 (located in the lid site, Fig. 1), UV absorbance showed the highest value for AuNPs formation at 540 nm in Au-Mt@GTL-193-pH 8.5, whereas similar results were obtained in the case of the other two metalloenzymes (Au-Mt@GTL-193-pH 7 and Au-Mt@GTL-193-pH 10) (Fig. 4a), with maximum peak at 533 nm. The effect was also different in terms of enzyme structure, where a higher Trp-quenching was observed in this conjugate (Au-Mt@GTL-193-pH 8.5), whereas a surprising increase (2-fold) in fluorescence intensity was observed for the conjugate synthesised at pH 7 (Fig. 4b). This could indicate that the conformation of the lipase is more open25 due to the conjugation with the gold. In the case of Au-Mt@GTL-193-pH 10, the three-dimensional structure of the lipase was also altered, in this case with a decrease in fluorescence intensity, which means that the enzyme is in a different conformation (probably in a more closed form than the native one). Also, in all cases a similar shift of the maximum of wavelength value of the native enzyme peak was observed as similar than in the previous case. XRD showed as in the previous case the formation exclusively of Au(0) species (Fig. 4c). The chemical and structure of the Au-enzyme bioconjugate was further analyzed by X-ray photoelectron spectroscopy (XPS). Two contributions were found by curve-fitting analysis of the two spin–orbit components of the Au 4f signal (Fig. S8†). The Au 4f 7/2 component of the peak at lower BE values (83.69 eV) is attributed to metallic gold, the latter Au 4f 7/2 component at BE = 85.04 eV can be associated with the Au atoms that are covalently bounded to sulfur groups of the enzyme. In addition, semi-quantitative analysis shows 93% of metallic gold and 7% of metallic gold bounded to sulfur atoms, indicating that thiol groups are grafted onto the gold nanoparticles. This technique allows to demonstrate that AuNPs bind through the cysteine residue in GTL.
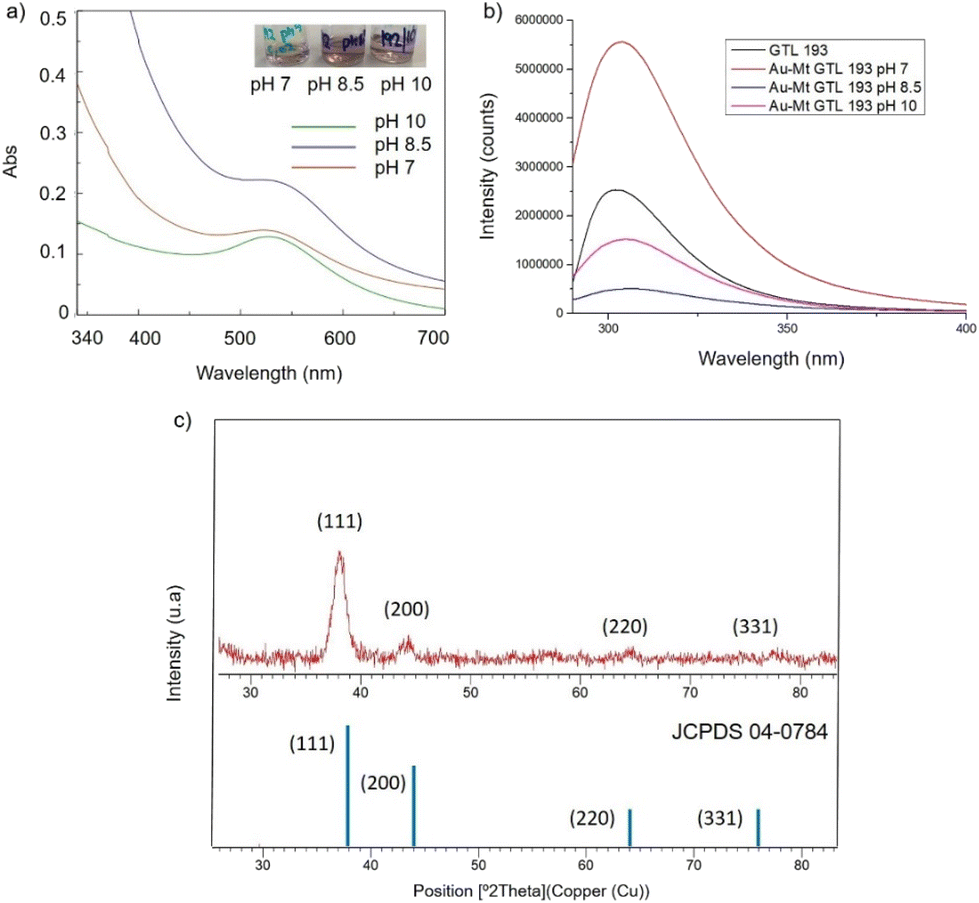 | ||
| Fig. 4 Characterisation of the different GTL-193 gold metalloenzymes. (a) UV-Absorbance spectrophotometer, (b) fluorescence spectra using 280 nm excitation, (c) XRD pattern. | ||
Fig. 5 shows the TEM images for the Au-Mt@GTL-193. In all cases crystalline spherical nanoparticles of Au (0) were formed (Fig. S9†). In Au-Mt@GTL-193 pH 7, nanoparticles with a diameter average size of 10 nm were obtained (Fig. 5a), whereas smaller particles were obtained at the other pHs, with an average diameter size of 3.8 or 3.2 nm for Au-Mt@GTL-193 pH 8.5 (Fig. 5b) and Au-Mt@GTL-193 pH 10 (Fig. 5c), respectively.
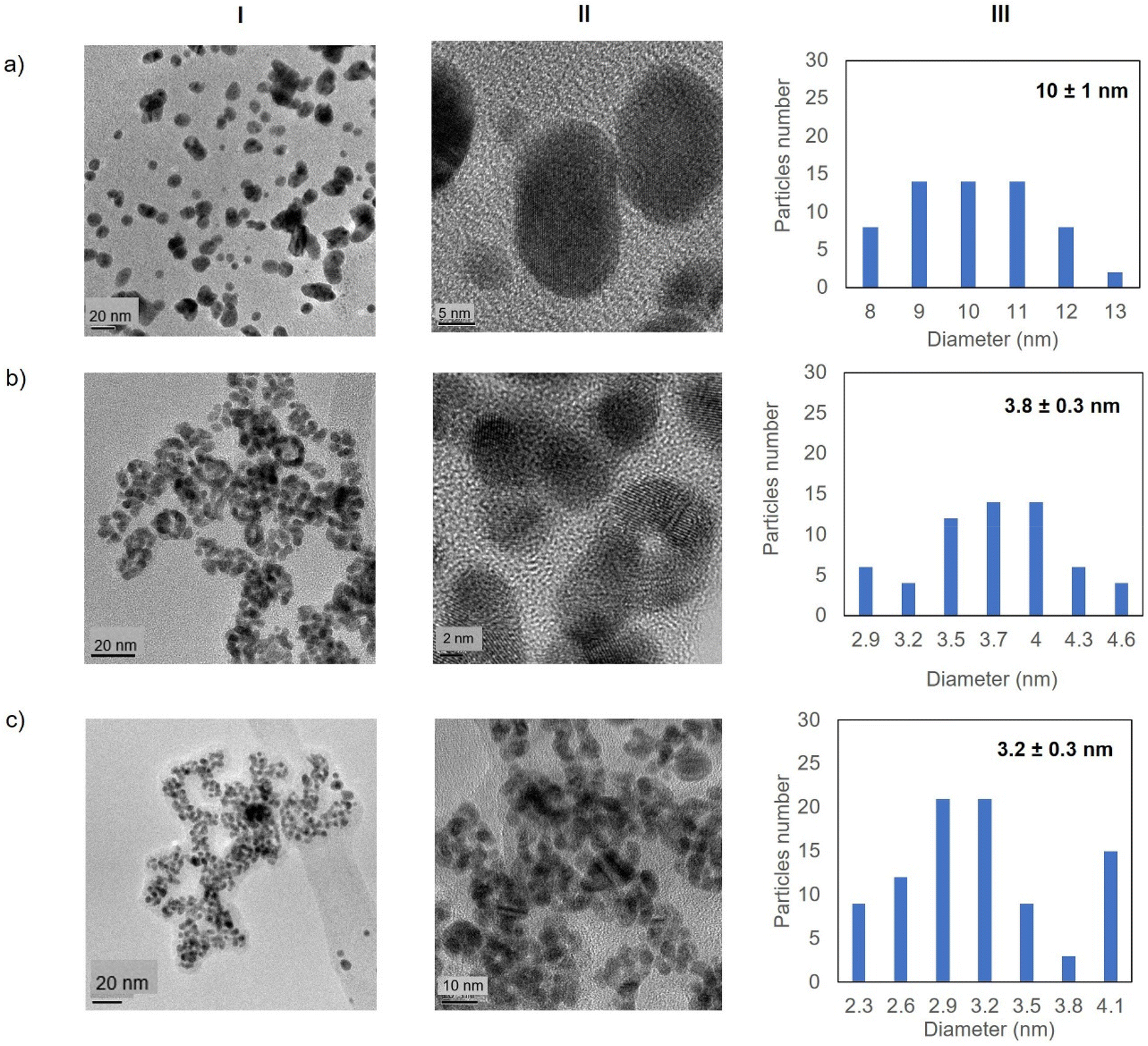 | ||
| Fig. 5 Electronic microscopy characterisation of the different GTL-193 gold metalloenzymes. (a) Au-Mt@GTL-193 pH 7, (b) Au-Mt@GTL-193 pH 8.5, (c) Au-Mt@GTL-193 pH 10. (I) TEM images, (II) HR-TEM images, (III) nanoparticle size distribution graphic. | ||
The Au content in the metalloenzymes was determined by ICP-OES mass analysis. This given Au content of 0.08–0.06 ppm (Table S3†).
Discussion about the Au-metalloenzymes formation by using bioinformatics tools
In order to comprehend the results obtained in the synthesis of Au metalloenzymes, bioinformatics studies of the enzyme structure were developed (Fig. 6, S10†).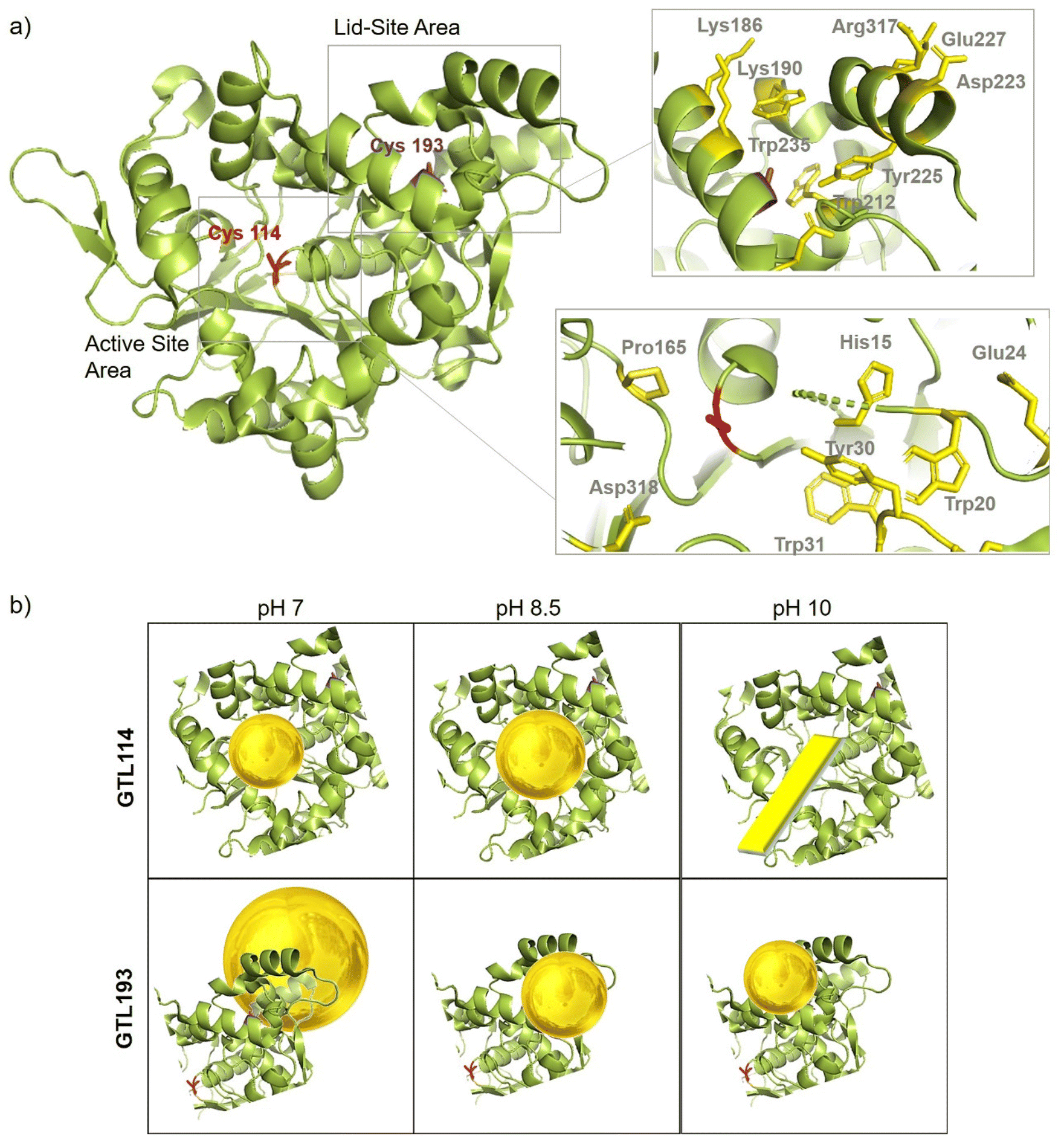 | ||
| Fig. 6 (a) Crystal structure cartoon of the active conformation of GTL, marked in yellow both sites (native active and lid) and two position of engineered variant, Cys114 (red), Cys193 (purple). Cartoon of the peptide sequence and the main protein residues involved in gold coordination (in yellow). (b) Cartoon of the Enzyme-Au conjugates at different pHs and by using the different enzyme variant as scaffold. The protein structure was obtained from the Protein Data Bank (pdb code: 2 W22) and the picture was created using Pymol. | ||
In particular, one of the most interesting results was obtained in the synthesis of the conjugates using the GTL-193 variant. This is located in one of the oligopeptide lid regions representing the “second active site” defined in the enzyme. Two Trp residues (Trp235, Trp212) and two Lys residues (Lys186, Lys190) are located close to the position where Cys was introduced (Fig. 6). However, in the lids peptide more expose to the solvent there are two Asp groups and Arg317. Considering the coordination capacity of the Au with the different groups, the effect of pH seems to be relevant for the position of the Au ions and the subsequent nanoparticle formation. At pH 7 the strongest binding may occur in these three groups more than in cysteine, which could be translated into an effect on the protein opening. This was observed in fluorescence analysis with an increasing in the fluorescence signal. In addition, this change in the protein structure and binding position, where the enzyme structure has less control over the growth of the final nanoparticle is results in the formation of larger nanoparticles. However, this effect changes when increasing pH, as SH has a higher nucleophilic power when deprotonated. This induces the formation and the coordination at this site, which is involved in the coordination of Trp residues and is therefore particularly located between these two oligopeptides. This leads to a decrease in the Trp signal, which can be related to the coordination at this position.
This result is in concordance with our previous studies by incorporating a peptide sequence on this position (Fig. S11†) where the fluorescence intensity also decreased.32
At higher pH, the growth of the nanoparticles could be controlled by the protein structure resulting in smaller nanoparticle sizes. Maybe at pH 10 where the amino residues of lysines are deprotonated, there is more influence in the coordination that accommodate the gold ion, resulting in slightly smaller AuNPs than at 8.5 synthesis (Fig. 5).
In the case of the active site position (GTL114), the oxyanion hole and positions surrounding the cysteine 114 are mainly hydrophobic groups with the presence of Asp 318, Pro165, Tyr30 and Trp20, Trp31 residues. Metal can coordinate on this area at pH 7 or 8.5, with a greater influence on the coordination with Cys at 8.5 but also considering the size of the cavity in the coordination of the gold, the growth of the particle is controlled by the protein structure, resulting in a small size observed in TEM (Fig. 3). Coordination in a more internal position on the hole as at pH 7 was translated into the smallest size of 2.2 nm. At pH 10 the conditions are different, especially for the Trp or Tyr coordination where it is in anionic form. This could be the reason why a different nanostructure is formed, generating a nanorod structure around the oxyanion hole (Fig. 6).
Storage-stability of gold metalloenzymes
These structural changes observed in the enzyme scaffold by the AuNPs formation could also affect to the stability of the final conjugates. To evaluate this, different Au-GTL conjugates were incubated for 14 days at 4 °C and room temperature respectively. After this time, in both experiments the different enzyme solutions did not show any aggregation form (especially relevant in the cold conditions) and the analysis of the AuNPs signal in the UV absorbance or metallic activity assay did not change (Table S4, Fig. S11 and S12†).Metallic activity of the different Au-metalloenzymes in the reduction of nitroarenes
Firstly, the efficiency of the different artificial gold metalloenzymes via gold reductive catalysis was evaluated in the selective hydrogenation of p-nitrophenol (pNP) to p-aminophenol (pAP) (Fig. S13†).The reaction was carried out in an aqueous medium and at room temperature. The free solution of each GTL mutant was used as a control, and no activity was found in any case. It is also important to note that all gold metalloenzymes are homogeneous catalysts, since the catalyst is in the same phase as the reactants, i.e., in solution.
Table 1 shows the TOF (Turnover Frequency) value for the gold metalloenzymes for 1 minute of reaction time. For Au-Mt@GTL-114 (Table 1, entries 1–3), it was found that the catalytic activity increases with increasing pH, with the highest TOF value being obtained for Au-Mt@GTL-114 pH 10 (128 min−1), despite having the largest AuNPs. This is probably due to the presence of nanorods (NRs) in their structure, Gold NRs are known to consist of higher energy side facets and lower energy end facets. Therefore, the relative surface area of the side and end facets, can affect their catalytic efficiency allowing to Au NRs can significantly increase their catalytic activity in comparison with Au nanoparticles.33
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Au content (μg) | Time (min) | TOF value (min−1) | Yield of 2 (%) |
| a Conditions: 1 mM 1 (3 mg), 40 mM (3 mg) NaBH4, 2 mL distilled water, room temperature. | |||||
| 1 | GTL-114 | 0 | 1 | 0 | 0 |
| 2 | GTL-193 | 0 | 1 | 0 | 0 |
| 3 | HAuCl4 | 2 × 10−2 | 1 | 54 | 28 |
| 4 | Au-Mt@GTL-114 pH 7 | 4 × 10−3 | 1 | 43 | 23 |
| 5 | Au-Mt@GTL-114 pH 8.5 | 4 × 10−3 | 1 | 94 | 39 |
| 6 | Au-Mt@GTL-114 pH 10 | 4 × 10−3 | 1 | 128 | 52 |
| 7 | Au-Mt@GTL-193 pH 7 | 7 × 10−5 | 1 | 451 | 33 |
| 8 | Au-Mt@GTL-193 pH 8.5 | 7 × 10−5 | 1 | 297 | 23 |
| 9 | Au-Mt@GTL-193 pH 10 | 7 × 10−5 | 1 | 544 | 40 |
On the other hand, Au-Mt@GTL-193 showed the highest TOF values (Table 1, entries 4–6). This may be due to the fact that in GTL-193 the modification is on the outside (lid site). Therefore, the catalytic centres of the metal are more exposed, leading to a more interaction with the substrate and a higher catalytic activity.
Among them, Au-Mt@GTL-193 pH 10 was found to have the best catalytic activity (544 min−1). This value was almost 2 times higher than those obtained for Au-Mt@GTL-193 pH 7 and Au-Mt@GTL-193 pH 8.5. This can be explained by the size of the nanoparticles, as they have the smaller diameter (3.2 nm) and are therefore they tend to be more effective. In addition, the TOF value for Au-Mt@GTL-193 pH 7 (451 min−1) was also 2 times higher than that obtained for Au-Mt@GTL-193 pH 8.5 (297 min−1).
This high catalytic performance obtained in the Au-metalloenzyme synthesized at pH 10 was attributed to the small nanoparticle size and high dispersion of the Au NPs.
Differences in the degree of crystallinity may go along with the size of the NP and are suggested to be one critical factor determining the overall catalytic activities. Distortion is suggested to induce defects, which for their part influence the association or dissociation of the reagent to the surface of the AuNP, and hence the catalytic activity. Further, it has been reported that amorphous nanostructures with abundant defects exhibited higher catalytic activities compared with crystalline nanostructures.34 In that sense, larger AuNP containing more defects have higher catalytic activities than smaller ones, this could explain the result obtained by the Au-Mt@GTL-193 pH 7.
To the best of our knowledge, Au-Mt@GTL-193 pH 10 shows the highest TOF value (32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 640 h−1) of an Au catalyst in this reaction, reported to date, compared to the highest TOF ∼20000 h−1 found in the literature.35
640 h−1) of an Au catalyst in this reaction, reported to date, compared to the highest TOF ∼20000 h−1 found in the literature.35
Selective reduction of acetophenone to 1-phenylethanol
Finally, the gold metalloenzymes were evaluated as catalysts in the asymmetric reduction of acetophenone (3) to 1-phenylethanol (4) at room temperature (Table 2, Fig. S14†). No product formation was observed when the free native mutant lipases were used as catalysts (Table 2, entries 1–3), nor with the gold salt. On the other hand, all artificial gold metalloenzymes showed reductase-like activity (Table 2, entries 4–9).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Time (h) | C (%) | ee (%)c | Specific activityd (μmoL mg−1 min) |
| a Conditions: 3, 1 mM, 0.05 mgmL−1 (50 μL) NaBH4, 50 μL of catalyst. b Conversion was calculated by HPLC. c Enantiomeric excess (ee) was calculated by HPLC. d Specific activity was defined as μmol mg−1 min−1. | |||||
| 1 | GTL-114 | 2 | 0 | 0 | 0 |
| 2 | GTL-193 | 2 | 0 | 0 | 0 |
| 3 | HAuCl4 | 2 | 0 | 0 | 0 |
| 4 | Au-Mt@GTL-114 pH 7 | 2 | 52 | <5 | 2.5 |
| 5 | Au-Mt@GTL-114 pH 8.5 | 2 | 71 | <5 | 3.4 |
| 6 | Au-Mt@GTL-114 pH 10 | 6 | 39 | 39 (4a, R) | 0.63 |
| 7 | Au-Mt@GTL-193 pH 7 | 2 | 56 | <5 | 2.7 |
| 8 | Au-Mt@GTL-193 pH 8.5 | 2 | 25 | <5 | 1.2 |
| 9 | Au-Mt@GTL-193 pH 10 | 2 | 43 | 16 (4b, S) | 2.1 |
The specific activity varied depending on the mutant and the pH used during the synthesis. The highest specific activity as artificial reductase was found for Au-Mt@GTL-114 pH 8.5 (3.4 μmol mg−1 min). This value was five times higher than that obtained for Au-Mt@GTL-114 pH 10.
On the other hand, there were no remarkable differences in the specific activities obtained between the Au-Mt@GTL-193 metalloenzymes. However, the specific activity for Au-Mt@GTL-193 pH 7 (2.7 μmol mg−1 min) was 2 times higher than that for Au-Mt@GTL-193 pH 8.5. These values represent good specific activities compared to ketoreductases described in the literature, especially considering that no cofactors are required.36,37 Dehydrogenase/reductase (SDR) from Empedobacter brevis, depended of NADH, showed specific activity of 4.2 μmol mg−1 min at pH 7 and 30 °C.37
More interestingly, the gold metalloenzymes synthesised at pH 10 showed enantioselectivity in the reduction process of prochiral ketone (Table 2). Au-Mt@GTL-114 pH 10 showed an enantiomeric excess (ee) of 39% towards (R)-1-phenylethanol whereas Au-Mt@GTL-193 pH 10 showed an opposite enantioselectivity, with an ee of 16% towards (S)-1-phenylethanol.
As far as we known, this is the first example where the simple incorporation of AuNPs (without any ligand) to an enzyme create an enantioselective artificial reductase.
To elucidate the reason of the enantiomer selectivity, molecular docking analysis of the ketone and both enantiomers in the two active sites was performed using mcule software (Fig. 7).38
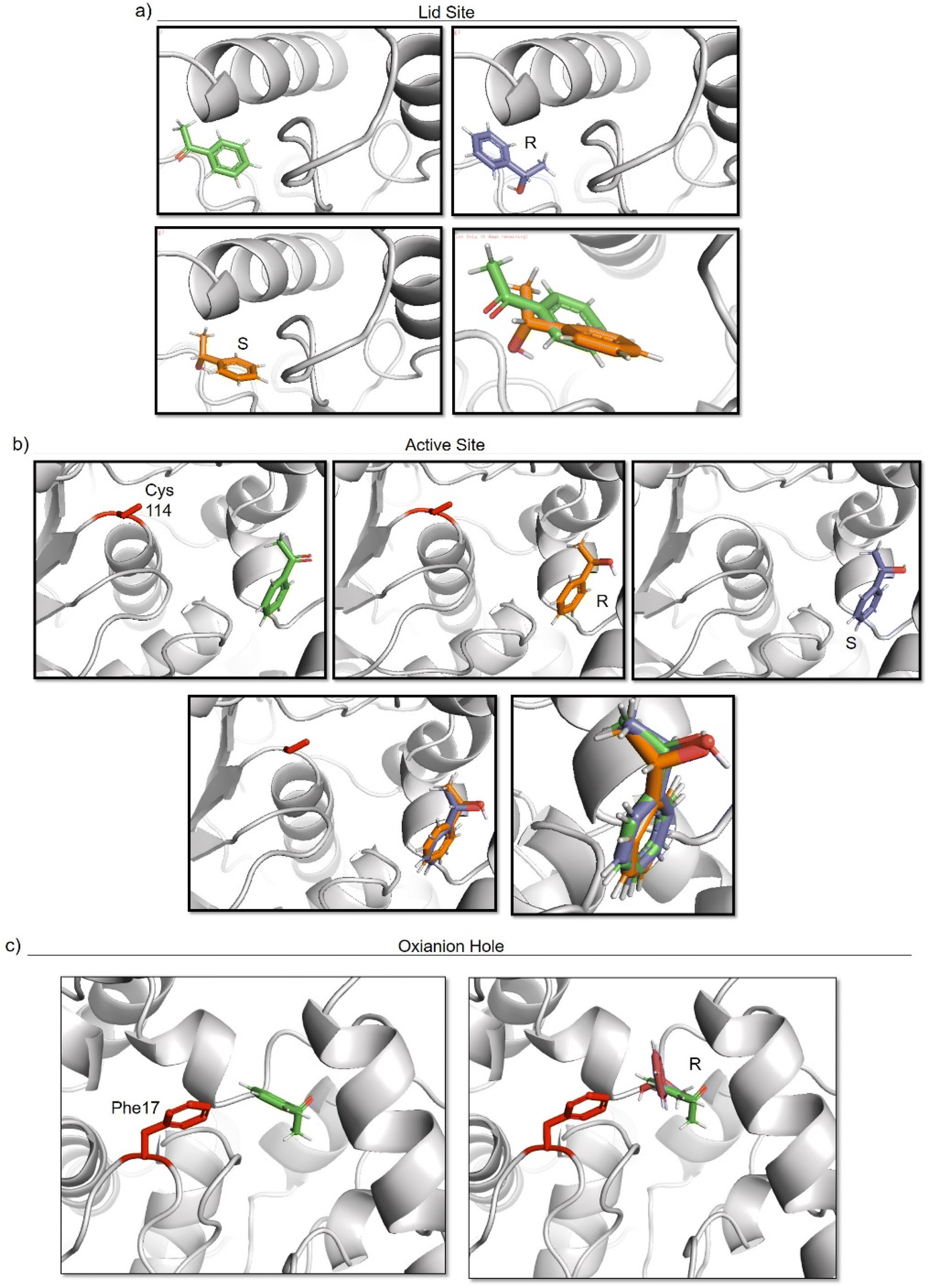 | ||
| Fig. 7 Molecular docking studies of acetophenone and different 1-phenylethanol enantiomers against GTL enzyme selecting two different active site-binding. Docking was performed using Mcule software.37 Images were prepared using pymol. (a) lid site focus on Cys 114, (b) active site focused on cys 114, (c) oxyanion hole focused on Phe 17. | ||
The analysis show that the R-enantiomer is located in a similar position to the acetophenone, and that the formation of this enantiomer by the gold reduction is more preferred than the S enantiomer (Fig. 7a).
Considering the active site S-114 interactions, docking experiments showed that the ketone and both enantiomers showed a very similar binding (Fig. 7b), which will be translated in low selectivity. Considering the formation of AuNPs at pH 7 and 8.5 at this position, this could explain the low selectivity of the environment for this nanoparticle. However, synthesis at pH 10 produce formation of nanorods which are located around the oxyanion hole. This could cause a different coordination of substrate for example by stacking interaction with aromatic ring residues in the oxyanion hole, for example Phe17. Docking experiments considering this amino acid as coordination point was performed, and location of S-enantiomer was more stabilised confirming the experimental selectivity towards this enantiomer (Fig. 7c). In both cases, location of the molecule is in a particular position of the oxyanion hole, stabilized by hydrophobic groups of the protein with respect to the phenyl group on the molecule.
Conclusion
This work demonstrates the successful development of a novel method for the synthesis of a new type of artificial gold metalloenzymes using genetically engineered thermoalkalophilic lipases (GTL-114 and GTL-193) as enzyme scaffolds. In all cases, the formation of nanoparticles in the protein matrix was induced in situ by the enzyme, without the use of an external reducing agent. In addition, it was shown that the mutant and the pH used during the synthesis play an important role in the final formation of the nanoparticles, determining their size and shape. On the other hand, all these Enzyme-Au bioconjugates showed high storage stability and particularly, metal activity in reductive catalysis. For example, in the nitroarene reduction, both Au-Mt@GTL-114 and Au-Mt@GTL-193 synthesized at pH 10 showed the best performance. In the former, because of the nanorods structure formation instead of nanoparticles formed at other pHs, whereas in the latter, based on the smaller size of the crystalline nanoparticles.In the asymmetric reduction of acetophenone to 1-phenylethanol, Au metalloenzymes showed mimetic reductase-like activity and depending on the “active site” of the enzyme where AuNPs are generated, the artificial enzyme showed a different enantiopreference. Au-Mt@GTL-114 pH 10 was able to obtain as main enantiomer (R)-phenylethanol, whereas Au-Mt@GTL-193 pH 10 was able to generate a moderate enantiomeric excess over (S)-phenylethanol, demonstrating that two different enzymatic environments on the same enzyme can induced completely different results.
This technology will make it possible to convert a reductase into an esterase in a very simple way. Furthermore, this method allows the generation of “chirality” in metal nanoparticles induced by the protein environment compared to simple salts or free nanoparticles. Therefore, future research will aim to extend the reaction for its application in the synthesis of complex synthetic structures for pharmaceuticals and fine chemicals.
Experimental
Materials
Butyl-Sepharose® 4 Fast Flow was from GE Healthcare (Uppsala, Sweden). Sodium borohydride, p-nitrophenol (pNP) and p-nitrophenylpropionate (pNPP) were from Alfa Aesar. Triton® X-100, Sodium Phosphate, Tetracloroauric acid(III) 3-hydrate (HAuCl4·3H2O) and Dialysis turbing cellulose (avg. flat 33 mm) were purchased from Sigma Aldrich. Acetophenone, R-phenylethanol and S-phenylethanol were from Fluka. Ac-Gly-Gly-Cys-Gly-Gly-COOH and Ac-Cys-(Asp)4-Asp-COOH were purchased from Isogen (Netherlands).Instrumentation
Synthesis of gold metalloenzymes were carried out using a Opa Q incubator with an OL30 ME orbital. Spectrophotometric analyses were run on a V-730 spectrophotometer (JASCO, Tokyo, Japan). Inductively Coupled Plasma – Optical Emission Spectrometry (ICP-OES) was performed on an OPTIMA 2100 DV instrument (PerkinElmer, Waltham, MA, USA). Au nanoparticles sizes and morphology were determined by transmission electron microscopy (TEM) and high-resolution TEM (HRTEM). Images were obtained on a 2100F microscope (JEOL, Tokyo, Japan) equipped with an energy dispersive X-ray (EDX) detector INCA X-sight (Oxford Instruments, Abingdon, U.K.). Interplanar spacing in the nanostructures was calculated using inversed Fourier transform spectroscopy with the GATAN digital micrograph program (Corporate Headquarters, Pleasanton, CA). X-Ray diffraction (XRD) patterns were obtained using a Texture Analysis D8 Advance Diffractometer (Bruker, Billerica, MA, USA) with Cu Kα radiation Fluorimetric measurements were performed in a Fluoromax Plus fluorimeter (Horiba, Scientific, Tokyo, Japan). Chromatographic analyses were run at 25 °C using an HPLC pump PU-4180 (JASCO, Tokyo, Japan) and an UV-4075 UV-Vis detector (JASCO, Tokyo, Japan).Site-directed mutagenesis, cloning and expression of Geobacillus Thermocatenulatus lipase (GTL)
The gene corresponding to the mature lipase of GTL was cloned into the expression vector pT1GTL. All site-directed mutagenesis experiments were carried out by PCR using mutagenic primers. To introduce the amino acid change, the corresponding primer pair was used as homologous primer pair in a PCR reaction using a specific plasmid as template and Prime Start HS Takara DNA polymerase. The PCR product was digested with DpnI endonuclease which exclusively restricts methylated DNA. Plasmids with mutated GTL were identified by sequencing and then transformed into E. coli BL21 (DE3) cells to express the corresponding proteins. First, C65S was created, and the resulting plasmid was used as a template to create the C65S/C296S-GTL (GTL) double mutant. This plasmid (pT1BGTLmutCys) was used as a template to construct additional mutations, creating the GTL-114 and GTL-193 mutants. The mature GTL lipase gene was cloned into a pT1 expression vector. Cells carrying the recombinant pT1GTL plasmid were grown at 30 °C and overexpression was induced by raising the temperature to 42 °C for 20 hours.Purification of mutant GTL enzymes
In a plastic container, 10 g of the butyl-Sepharose support were incubated with 10 mL of the corresponding enzyme (70 mg) and 190 mL of 25 mM sodium phosphate buffer pH 7 for 24 hours. To check that the enzymes were adsorbing to the support, the decrease in enzyme activity of the supernatants was measured at different times using the pNPP enzyme activity assay. For this, the absorbance (λ = 348 nm) produced by the release of p-nitrophenol (pNP) on hydrolysis of pNPP (50 mM) in sodium phosphate buffer (25 mM, pH 7) was measured. To initiate the reaction, 20 μL of pNPP standard solution (prepared in acetonitrile) was added to 2.5 mL of phosphate buffer. Then, 20 μL of the supernatant were added under magnetic stirring. After 24 hours, i.e. when the enzyme activity is zero as all the enzyme is adsorbed on the support. Finally, the solid was washed with plenty of water, obtaining the desired catalyst. Then the genetically modified GTL enzymes 114 and 193 were desorbed from the support by re-suspending the immobilised enzyme in a 1:10 (w/v) ratio in sodium phosphate buffer (25 mM pH 7) containing 0.5% (v/v) Triton X-100 for 1 hour at room temperature. After the indicated time, the support was removed by centrifugation and the soluble enzymes were obtained in the supernatant.
General procedure for the synthesis, purification and characterisation of gold metalloenzymes
100 μL (70 μg protein) of the mutated GTL enzyme solution was added to 900 μL of a solution containing 0.5 mg mL−1 HAuCl4·3H2O salt. This solution was then adjusted to the desired pH (7/8.5/10 depending on the case) using a 1 mM NaOH solution. Initially, the solution showed a pH value of around 3.3 and a pale greenish-yellow colour. When adjusting the pH (7/8.5/10) the solution became colourless. This solution was kept for 20 hours under constant stirring at 130 rpm at 50 °C.The gold nanoparticles formation was followed by measuring the spectrophotometer the increase of the band around 520–550 nm at different times and observing the colour changes of the solution. After 20 hours, the gold metalloenzyme was obtained in colloidal state. Its colour varies from pink to pale purple depending on the mutant and pH used. Finally, the gold metalloenzymes were purified by dialysis using a membrane with a molecular weight cut-off of 14 kDa was used. The metalloenzymes were kept under agitation for 4 h at room temperature in a beaker containing 1 L of distilled water, changing the water every hour. After this time, purified gold metalloenzymes were obtained. Gold metalloenzymes synthesised were called Au-Mt@GTL-193-pH 7, Au-Mt@GTL-193-pH 8.5, Au-Mt@GTL-193-pH 10, Au-Mt@GTL-114-pH 7, Au-Mt@GTL-114-pH 8.5 and Au-Mt@GTL-114-pH 10, respectively.
Metal activity assay: reduction of 4-nitrophenol to 4-aminophenol
2.7 mg of p-nitrophenol (pNP) were dissolved in 20 mL of distilled water to a concentration of 1 mM. Then, 3.2 mg of solid NaBH4 was added to this solution. After addition, the solution changed from light yellow to strong yellow due to the formation of 4-nitrophenolate ions (the UV peak of the substrate undergoes an immediate shift from 317 to 400 nm). After 30 s, 50 μL of the gold metalloenzyme was added under gentle stirring at room temperature to initiate the reaction. The progress of the reaction was monitored by taking an aliquot of the solution (0.2 mL) at different times by diluting it in 2 mL of distilled water and measuring the UV-Vis absorption spectrum between 500 and 250 nm in a quartz cuvette with 1 cm optical path length.Fluorescence spectroscopy measurements
The measurements of the excitation–emission spectra of the GTL mutants and the synthesised gold metalloenzymes were conducted at room temperature using a quartz cuvette (1 cm path length) with 3 mL of the corresponding sample. An excitation wavelength of 280 nm was used, with excitation and emission bandwidths of 5 nm. Fluorescence emission spectra were recorded between 200 and 500 nm. All spectroscopic measurements were performed using an NDQ-50-50 × 50 M filter.Selective reduction of acetophenone to 1-phenylethanol
0.02 mmol of acetophenone were dissolved in 20 mL of distilled water to a concentration of 1 mM. Then, 50 μL of a 0.5 mg mL−1 NaBH4 solution was added to the previous solution. After 30 s, 50 μL of the gold metalloenzyme was added to initiate the reaction. The reaction was maintained under gentle stirring at room temperature for the indicated reaction time (2 h or 6 h depending on the gold metalloenzyme used in each case).The conversion and the enantiomeric excess (ee) were calculated by chiral HPLC. The reaction was monitored by normal phase HPLC. HPLC analysis was performed using a Thermo HPLC apparatus coupled to a Diode-array detector. The analysis conditions were performed using a Phenomenex Lux 3u Cellulose column (250 × 460 mm) in 90:10 hexane/isopropanol mobile phase at a flow rate of 0.5 mL min−1 and a wavelength of 220 nm. Prior to injection, 600 μL of each sample was extracted with hexane at a 1:1 (v/v) ratio. Hexane phase was directly injected. The extraction coefficient for acetophenone was Kd = 0.52, for (R)-1-phenylethanol Kd = 0.72, and for (S)-1-phenylethanol Kd = 0.79. Under these conditions, the retention times of products were: acetophenone (10.5 min), (R)-1-phenylethanol (13.4 min) and (S)-1-phenylethanol (15.1 min). Pure commercial alcohol enantiomers were used as standards. Enantiomeric excess (ee) was defined as: ee (%) = {(R − S)/(R + S)} × 100.
Data availability
The ESI† includes additional details on enzyme variants characterization (Table S1†), bioinformatics (Fig. S1 and S11†), synthesis of enzyme-AuNPs conjugation (Fig. S2–S10†), stability studies (Fig. S12 and S13, Table S4†), reaction mechanism (Fig. S14 and S15†), fluorescence data (Table S2†) and metal protein content analysis (Table S3†).Author contributions
J. M. P. conceived the project. J. M. P. supervised the project and obtained funding. B. R. performed molecular biology and C. G-S. performed protein purification, synthesis of metalloenzymes. C. G-S. performed gold-enzyme characterization and catalytic reactions. J. M. P. and C. G-S. analysed the data and wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the support from Spanish National Research Council (CSIC) and Ministry of Science (Project AGL2017-84614-C2-2-R) and European Commission for funding the Project Grant No. 101060130, HORIZON-WIDERA-2021-ACCESS-02-01. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- M. Hoppert, in Metalloenzymes, ed. J. Reitner and V. Thiel, Encyclopedia of Earth Sciences Series, Springer, Dordrecht, 2011 Search PubMed.
- J. T. J. Lecomte, D. A. Vuletich and A. M. Lesk, Curr. Opin. Struct. Biol., 2005, 15, 290–301 CrossRef CAS PubMed.
- C. A. Gersbach, T. Gaj and C. F. Barbas, Acc. Chem. Res., 2014, 47, 2309–2318 CrossRef CAS PubMed.
- C. E. Valdez, Q. A. Smith, M. R. Nechay and A. N. Alexandrova, Acc. Chem. Res., 2014, 47, 3110–3117 CrossRef CAS PubMed.
- M. A. Emmanuel, N. R. Greenberg, D. G. Oblinsky and T. K. Hyster, Nature, 2016, 540, 414–417 CrossRef CAS PubMed.
- A. D. Liang, J. Serrano-Plana, R. L. Peterson and T. R. Ward, Acc. Chem. Res., 2019, 52, 585–595 CrossRef CAS PubMed.
- C. Perez-Rizquez, A. Rodriguez-Otero and J. M. Palomo, Org. Biomol. Chem., 2019, 17, 7114–7123 RSC.
- F. Nastri, M. Chino, O. Maglio, A. Bhagi-Damodaran, Y. Lu and A. Lombardi, Chem. Soc. Rev., 2016, 45, 5020–5054 RSC.
- J. C. Lewis, ACS Catal., 2013, 3, 2954–2975 CrossRef CAS.
- J. M. Palomo, Curr. Opin. Green Sustainable Chem., 2021, 29, 100452 CrossRef CAS.
- B. Jiang and M. Liang, Chin. J. Chem., 2021, 39, 174–180 CrossRef CAS.
- Y. Jiang, Z. Ding, M. Gao, C. Chen, P. Ni, C. Zhang, B. Wang, G. Duan and Y. Lu, Chin. J. Chem., 2021, 39, 3369–3374 CrossRef CAS.
- E. N. Mirts, I. D. Petrik, P. Hosseinzadeh, M. J. Nilges and Y. Lu, Science, 2018, 361, 1098–1101 CrossRef CAS PubMed.
- G. Roelfes, Acc. Chem. Res., 2019, 52, 545–556 CrossRef CAS PubMed.
- S. Morra and A. Pordea, Chem. Sci., 2018, 9, 7447–7454 RSC.
- C. Lin, K. Tao, K. Hua, Z. Ma and S. Zhou, Molecules, 2013, 18, 12609–12620 CrossRef CAS PubMed.
- A. Bano, A. Dawood, A. Rida, F. Saira, A. Malik, M. Alkholief, H. Ahmad, M. A. Khan, Z. Ahmad and O. Bazighifan, Sci. Rep., 2023, 13, 12359 CrossRef CAS PubMed.
- J. Naapuri, N. Losada-Garcia, J. Deska and J. M. Palomo, Nanoscale, 2022, 14, 5701–5715 RSC.
- G. Vivo-Llorca, A. Morellá-Aucejo, A. García-Fernández, P. Díez, A. Llopis-Lorente, M. Orzáez and R. Martínez-Máñez, Int. J. Nanomed., 2022, 17, 409–422 CrossRef.
- Y. C. Yeh, Y. B. Creran and V. M. Rotello, Nanoscale, 2012, 4, 1871–1880 RSC.
- A. Aires, I. Llarena, M. Moller, J. Castro-Smirnov, J. Cabanillas-Gonzalez and A. L. Cortajarena, Angew. Chem., Int. Ed., 2019, 58, 6214–6219 CrossRef CAS PubMed.
- C. Lu, B. Maity, X. Peng, N. Ito, S. Abe, X. Sheng, T. Ueno and D. Lu, Commun. Chem., 2022, 5, 39 CrossRef CAS PubMed.
- X. Huang, S. Zhang, Y. Tang, X. Zhang, Y. Bai and H. Pang, Coord. Chem. Rev., 2021, 449, 214216 CrossRef CAS.
- C. Carrasco-López, C. Godoy, B. de Las Rivas, G. Fernández-Lorente, J. M. Palomo, J. M. Guisán, R. Fernández-Lafuente, M. Martínez-Ripolla and J. A. Hermoso, J. Biol. Chem., 2009, 284, 4365–4372 CrossRef PubMed.
- O. Romero, B. de las Rivas, D. Lopez-Tejedor and J. M. Palomo, ChemBioChem, 2018, 19, 369–378 CrossRef CAS PubMed.
- M. Filice, O. Romero, J. Gutiérrez-Fernández, B. De Las Rivas, J. A. Hermoso and J. M. Palomo, Chem. Commun., 2015, 51, 9324–9327 RSC.
- T. C. Chang, K. Tong, T. Yamamoto and K. Tanaka, Angew. Chem., 2021, 60, 12446–12454 CrossRef CAS PubMed.
- A. Prats Luján, M. F. Bhat, T. Saravanan and G. J. Poelarends, ChemCatChem, 2022, 14, 1–5 Search PubMed.
- Y. Gao, S. Yang, Y. Huo and X.-Q. Hu, Adv. Synth. Catal., 2020, 362, 3971–3986 CrossRef CAS.
- D. Fajstavr, A. Karasová, A. Michalcová, P. Ulbrich, N. S. Kasálková, J. Siegel, V. Švorčík and P. Slepička, Nanomaterials, 2021, 11, 1–17 CrossRef PubMed.
- S. Raut, R. Chib, S. Butler, J. Borejdo, Z. Gryczynski and I. Gryczynski, Methods Appl. Fluoresc., 2014, 2, 035004 CrossRef CAS PubMed.
- O. Romero, M. Filice, B. de las Rivas, C. Carrasco-López, J. Klett, A. Morreale, J. A. Hermoso, J. M. Guisan, O. Abian and J. M. Palomo, Chem. Commun., 2012, 48, 9053–9055 RSC.
- L. Khail, S. Sabahat and W. Ahmed, Catal. Lett., 2024, 154, 1018–1025 CrossRef.
- C. Liang, J. Y. Cheong, G. Sitaru, S. Rosenfeldt, A. S. Schenk, S. Gekle, I.-D. Kim and A. Greiner, Adv. Mater. Interfaces, 2022, 9, 2100867 CrossRef CAS.
- K. Shanmugaraj, T. M. Bustamante, C. C. Torres and C. H. Campos, Catal. Today, 2022, 388–389, 383–393 CrossRef CAS.
- H. Zhang, B. Wang, S. Yang, H. Yu and L. Ye, Catalysts, 2022, 12, 1071 CrossRef CAS.
- T. Yamamoto, Y. Nakata, C. Cao, Y. Sugiyama, Y. Asanuma, S. Kanamaru and T. Matsuda, Appl. Microbiol. Biotechnol., 2013, 97, 10413–10421 CrossRef CAS PubMed.
- https://mcule.com/apps/1-clickdocking/results/kZucA9AM6umBxPAdhAcSkH/ .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00573b |
| This journal is © The Royal Society of Chemistry 2024 |