
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular design for sub-micromolar enzyme-instructed self-assembly (EISA)†
Qihui
Liu
a,
Thomas
Ntim
a,
Zhiyuan
Wu
a,
Hailey A.
Houson
b,
Suzanne E.
Lapi
b and
Jonathan S.
Lindsey
 *a
*a
aDepartment of Chemistry, North Carolina State University, Raleigh, NC 27695-8204, USA. E-mail: jlindsey@ncsu.edu
bDepartment of Radiology, Heersink School of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294, USA
First published on 7th June 2024
Abstract
Enzyme-instructed self-assembly (EISA) has been explored for many applications in the life sciences including imaging and therapeutics. To date, use of the strategy is limited by the relatively high concentration – often in the mM regime – required for assembly. Here, a porphyrin–peptide conjugate (1) was designed as a water-soluble, EISA substrate. Compound 1 is built around a trans-AB porphyrin chassis that is equipped with a monodisperse 1 kDa polyethylene glycol (PEG) linker (a surrogate for a potential targeting agent) and an enzymatically triggerable self-assembly motif. The latter is comprised of two identical self-assembly peptides (also known as hydrogelators) attached to the 2,6-positions of an aryl group. The peptides have sequence GffY and in protected form contain tyrosine as an O-phosphoester GffY(p). Treatment of 1 with alkaline phosphatase (ALP) gave rise to the self-assembly (or aggregation) process as characterized by absorption spectroscopy. A control compound without the phosphoester trigger was synthesized and examined for the self-assembly process using fluorescence and absorption spectroscopy. A concentration-dependent study showed that at 10 nM approximately 50% of porphyrin–peptide conjugate was in the aggregated form. The nanomolar assembly suggests possible applications of EISA substrates in chemotherapy, although a further decrease in concentration to the picomolar regime may be required for use in targeted molecular radiotherapy, where very low mass dosing is typical.
Introduction
Enzyme-instructed self-assembly (EISA) is a process that integrates enzymatic reaction and self-assembly (Fig. 1). Recently, peptides have been investigated as the substrate of EISA given that peptides have good bioactivity, biodegradability, and biocompatibility.1 Since the first use of a small peptide as a substrate of EISA,2 various peptides and their analogues have been explored, along with various enzyme reactions for triggering the process.3–7 Peptide-based EISA can potentially increase the therapeutic efficacy with prolonged life-time, site-selective formation and lower possibility of drug resistance due to the polymorphism of peptide assemblies.8 The application of EISA has expanded to many fields, including therapeutics,5,9–14 imaging,15,16 and drug encapsulation.17,18 However, the typical concentration for assembly of the EISA substrates is in the 10 μM – 10 millimolar regime, which limits the use of the strategy in many fields.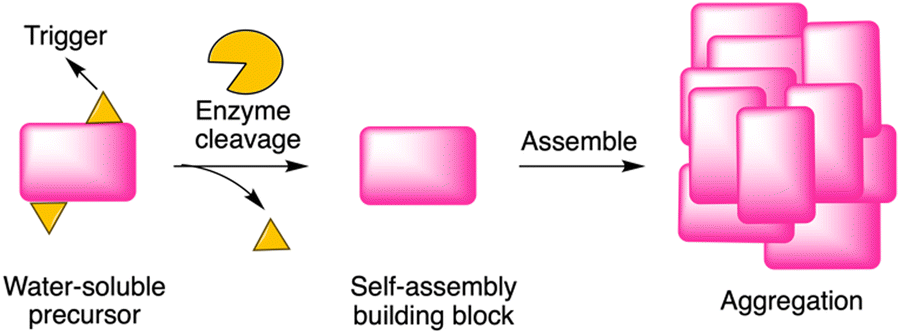 | ||
| Fig. 1 Illustration of EISA, where a water-soluble monomer is converted to a macromolecular assembly. | ||
Porphyrin compounds have many attractive features for use in the design of EISA substrates. The features include stable structures, facility for molecular tailoring, strong absorption of visible light, and photoactive properties. The molecular tailoring includes substitution of functional groups at the perimeter of the porphyrin macrocycle (meso- or β-positions) as well as chelation of diverse metals in the central cavity. Nearly 20 years ago, we designed and synthesized free base porphyrin I (Chart 1) for studies of what is now termed EISA.19 The phosphorylated free base porphyrin I exhibited satisfactory solubility in water (>1 mM) yet gave rise to precipitates upon treatment with the enzyme shrimp ALP. We envisaged that the porphyrins or analogues could be used in molecular brachytherapy, which would entail incorporation of a radionuclide; the enzymatic treatment by phosphatases upregulated in the tumor extracellular space would engender immobilization of the radionuclide. Still, there were perceived limitations to the molecular design and/or studies of I: (1) the concentration examined of I was 68 μM, which is likely ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fold higher than relevant for radiotherapy; and (2) the enzyme treatment was carried out in Tris–HCl buffer at pH 9.0, which is not physiologically relevant. Several other substrates for EISA strategies were examined,20–24 yet each proved to have idiosyncratic limitations.
000 fold higher than relevant for radiotherapy; and (2) the enzyme treatment was carried out in Tris–HCl buffer at pH 9.0, which is not physiologically relevant. Several other substrates for EISA strategies were examined,20–24 yet each proved to have idiosyncratic limitations.
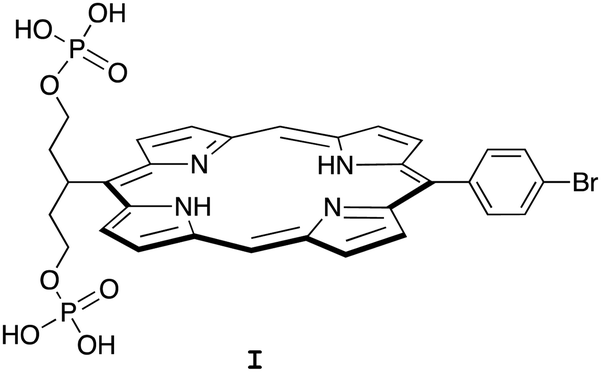 | ||
| Chart 1 Structure of an early porphyrin for EISA studies. | ||
In the ensuing years, porphyrins have drawn increased attention as building blocks in supramolecular assembly processes.25 Porphyrin–peptide conjugates also have been prepared for studies in photodynamic therapy.26 Given such advances, here we designed a porphyrin–peptide structure shown in conceptual form in Fig. 2 Panel A. The molecule in concept bears a porphyrin hub (potentially carrying a radionuclide), multiple self-assembly peptide precursors (for assembly upon enzymatic action), and a cancer targeting agent (CTA). Upon exposure to an enzyme overexpressed in a tumor region, the unveiling of the self-assembly peptides is expected to elicit the self-assembly process. (Equivalent terms in use for the self-assembly peptides and the subsequent assembly are hydrogelators and hydrogelation, respectively.)
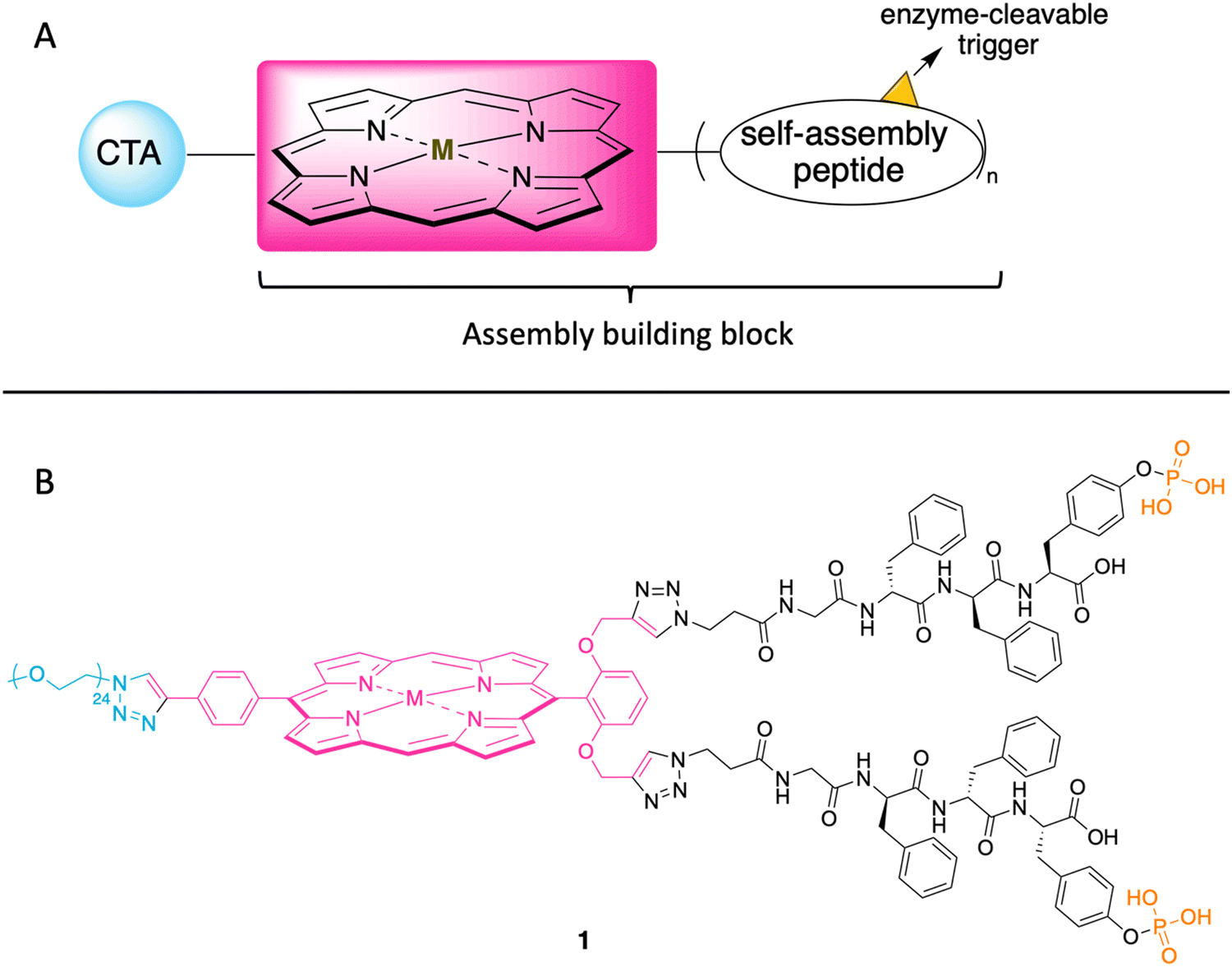 | ||
| Fig. 2 (A) Illustration of the cancer targeting porphyrin–peptide as EISA substrate. (B) Structure of the designed porphyrin–peptide molecule 1 (M = Zn) with a PEG group in lieu of a CTA. | ||
The structure of a model analogue is shown Fig. 2 Panel B. The peptide precursor GffY(p) is a phosphorylated tetrapeptide, where the resulting dephosphorylated peptide is GffY. The N-terminus capped tetrapeptide has been found to be an effective hydrogelator.27,28D-Amino acids in peptides can improve the biostability of short peptide structures.29 Upon the treatment of a suitable phosphatase (e.g., ALP), the peptide will be dephosphorylated and transform into the self-assembly peptide GffY. The peptide precursors are attached to a porphyrin, which also bears a PEG linker (attached via a clickable handle) as a surrogate of a CTA to provide water solubility. The target molecule is expected to exhibit the following features: (1) targeting tumor tissue; (2) delivering a radionuclide; and (3) aggregating and immobilizing upon exposure to a corresponding enzyme that is overexpressed at the tumor.
In this paper, we report the synthesis of 1 and analogues that lack the phosphoester or the PEG moiety. The compounds are tested for EISA via absorption and fluorescence spectroscopy. We note that the term EISA used herein refers to enzyme-instructed self-assembly and not evaporation-induced self-assembly.30 Studies of assembly as a function of concentration have revealed assembly at sub-micromolar concentrations.
Results
Synthesis of pegylated porphyrin–peptide conjugates
The precursor of the self-assembly peptide bearing an azido group on the N-terminus, 2, was synthesized following standard solid-phase peptide synthesis protocols using fluorenylmethoxycarbonyl-protected amino acids (Fmoc-AAs) and coupling in the presence of N,N-diisopropylethylamine (DIPEA) followed by the Fmoc cleavage protocol with piperidine in dimethylformamide (DMF) (Scheme 1). The tetrapeptide bears two D-phenylalanine residues, one phosphoester-derivatized L-tyrosine residue, and one glycine as a spacer. The D-amino acids provide resistance toward protease digestion. Upon treatment with a phosphatase, the phosphoester group will be cleaved, enabling peptide–peptide intermolecular assembly. The phosphoester peptide was synthesized using a chlorotrityl-terminated (2-CTC) resin, with hexafluorophosphate benzotriazole tetramethyl uronium and N-hydroxybenzotriazole (HBTU/HOBt) reagents for coupling and DIPEA as a base. Fmoc-protected amino acids and 3-azidopropanoic acid were loaded in order, with use of phosphoester-derivatized Fmoc-tryosine(PO3H2) in the first step. Cleavage from the resin was achieved upon acidolytic treatment with trifluoroacetic acid, triisopropylsilane, and water (TFA/TIS/H2O: v/v/v, 95:2.5:2.5). The crude product was purified by use of preparative reversed-phase high-performance liquid chromatography (RP-HPLC) to give phosphoester peptide 2. An analogous peptide lacking the phosphoester, 2′, was prepared using Fmoc-tryosine(tert-Bu), in the first step. The tert-butyl protecting group was removed during the resin cleavage step.
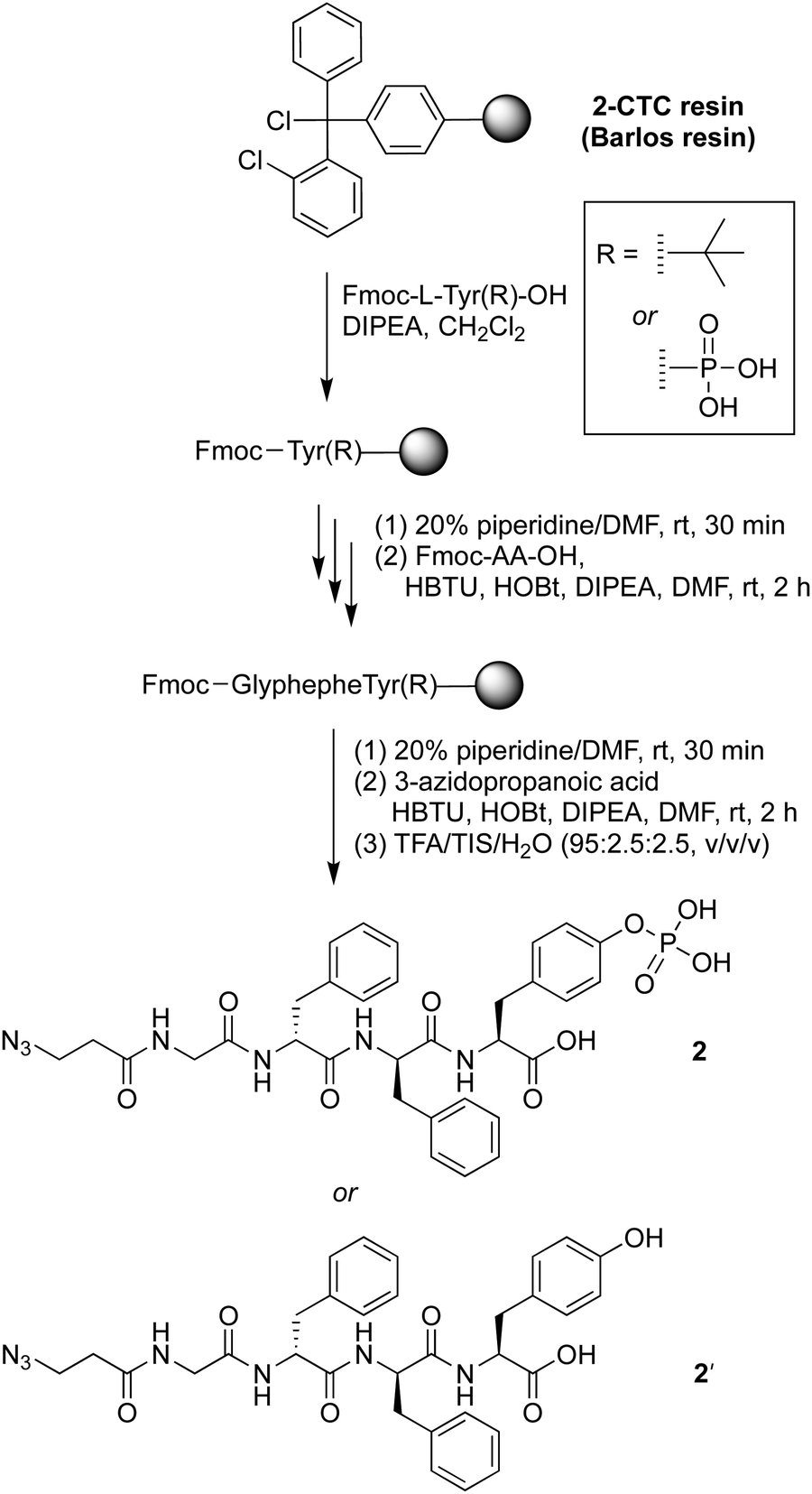 | ||
| Scheme 1 Synthesis of the precursor of the self-assembly peptide 2 and control 2′. | ||
The attachment of phosphoester-peptide 2 to porphyrin 3 was achieved via copper-catalyzed click chemistry31 (Scheme 2). Compound 3 is a zinc porphyrin that bears two terminal ethynes and one triisopropylsilyl (TIPS)-protected ethyne. Porphyrin 3 has been used as a molecular chassis to attach PEG groups (for aqueous solubilization) and folic acid (for cancer targeting).32 The presence of the zinc chelate was attractive in blocking chelation of the copper reagent, which would be expected by the free base porphyrin.31,32 The click reaction was carried out using CuBr and the water-soluble ligand tris(3-hydroxypropyltriazolylmethyl)amine (THPTA)33 in aqueous dimethyl sulfoxide (DMSO) at room temperature. The reaction mixture following the click reaction was washed to remove salts, and the resulting crude product 4 was used in the next step without further purification. The TIPS group was removed with tetra-n-butylammonium fluoride (TBAF) in tetrahydrofuran (THF) and DMSO to give compound 5. As a surrogate for a cancer-targeting agent, a monodisperse PEG24-azide reactant (1114 Da) was tethered to 5via copper-catalyzed click chemistry to yield 1. An analogous series of reactions was carried out using peptide 2′, which lacks the tyrosinyl phosphoester, affording porphyrin–peptide 1′.
 | ||
| Scheme 2 Synthesis of pegylated porphyrin–peptide conjugate 1 and control 1′. | ||
Enzymatic study with ALP
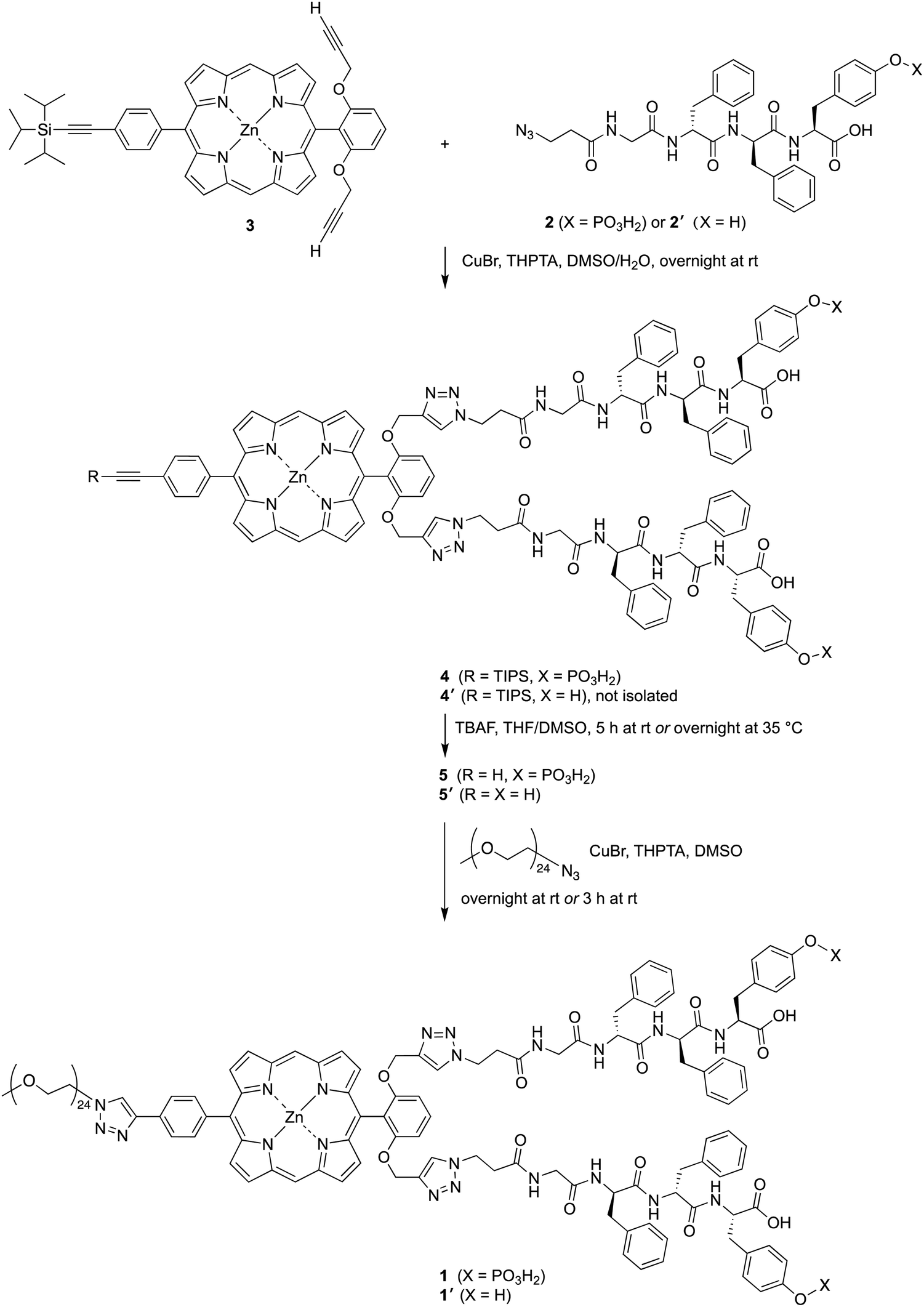
Samples of 1 were treated with ALP and examined in three ways: (1) absorption spectroscopy, (2) filtration through membranes of defined pore size, and (3) RP-HPLC analysis. The results from each method are described below.(1) A stock solution of 1 in DMSO was diluted with phosphate-buffered saline (PBS) to prepare sample solutions with different concentrations (0.10, 0.20, 0.50, 1.0, 2.0, and 5.0 μM). Each sample was treated with 2 U mL−1 of ALP and incubated overnight at 37 °C. The absorption spectra across the range 300–700 nm, where the porphyrin absorbs strongly, were measured before and after the enzyme treatment for each sample (Fig. 3). For samples with concentration <0.50 μM (Fig. 3 Panels A–B), the intensity of the Soret band of 1 at 420 nm decreased after enzyme treatment, yet precipitation or other signs of aggregation were not observed. When the concentration of the sample was ≥0.5 μM (Fig. 3 Panels C–F), the intensity of the Soret peak decreased and a new peak appeared at 455 nm after the enzyme treatment. The new peak at 455 nm indicates aggregation. The Q band (at ∼550 nm) also underwent a bathochromic shift and broadening, albeit of lesser magnitude than that of the higher-energy Soret band. Compound 1′, which lacks the two phosphoester moieties, is prone to aggregation in PBS, consistent with the observed results for ALP treatment of 1.
 | ||
| Fig. 3 Absorption spectra of 1 in PBS at different concentrations. Panel A: 0.1 μM; Panel B: 0.2 μM; Panel C: 0.5 μM; Panel D: 1.0 μM; Panel E: 2.0 μM; Panel F: 5.0 μM. Blue curves represent spectra before ALP treatment, red curves represent spectra after ALP treatment. | ||
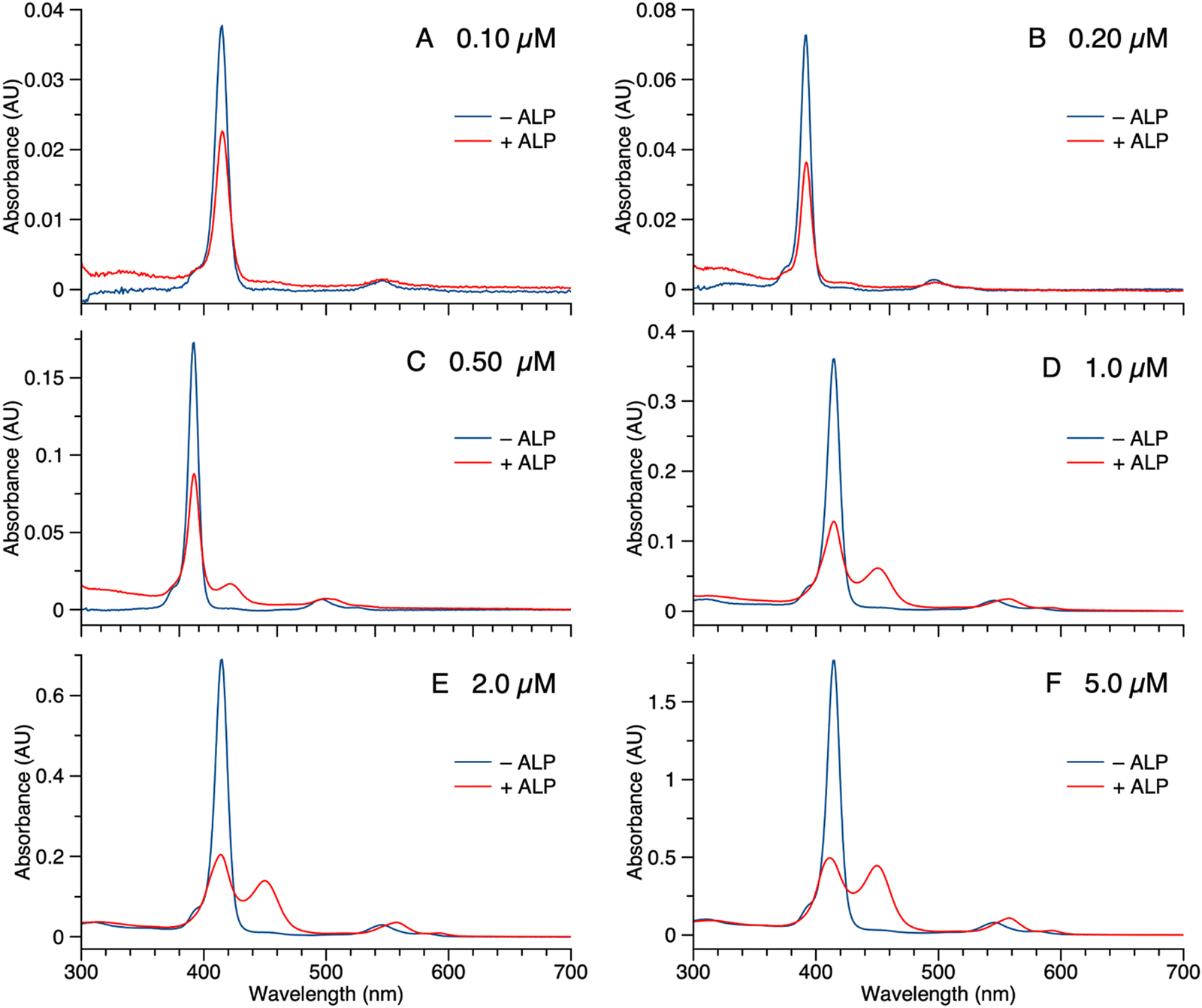
The potent solubilizing agent DMSO was added to the sample of 1 (5.0 μM in PBS) after enzyme treatment to assess reversibility of the aggregation process. After adding DMSO to dilute the concentration by half, the peak at 455 nm almost disappeared and the intensity of the peak at 420 nm increased (Fig. 4 panel A). The color change of the solution during the treatment is shown in Fig. 4 panel B. The color changes (monomeric, pink; aggregated, yellow; disaggregated, lighter pink due to dilution) accompanying sequential treatment with enzyme and DMSO provide visible cues consistent with molecular aggregation. Note that the cuvette photos shown in Fig. 4 were collected at 5 μM solely to show the visibly evident color changes. Absorption spectral changes can be observed at 50-fold lower concentrations as shown in Fig. 3.
 | ||
| Fig. 4 (A) Absorption spectra of 1 (5 μM) in PBS following treatment of 2 U mL−1 ALP overnight at 37 °C (red curve) and sample after ALP treatment upon addition of DMSO (black curve). (B) Color change of the solution of 1 (5.0 μM) before and after ALP treatment, and then dilution with DMSO. | ||
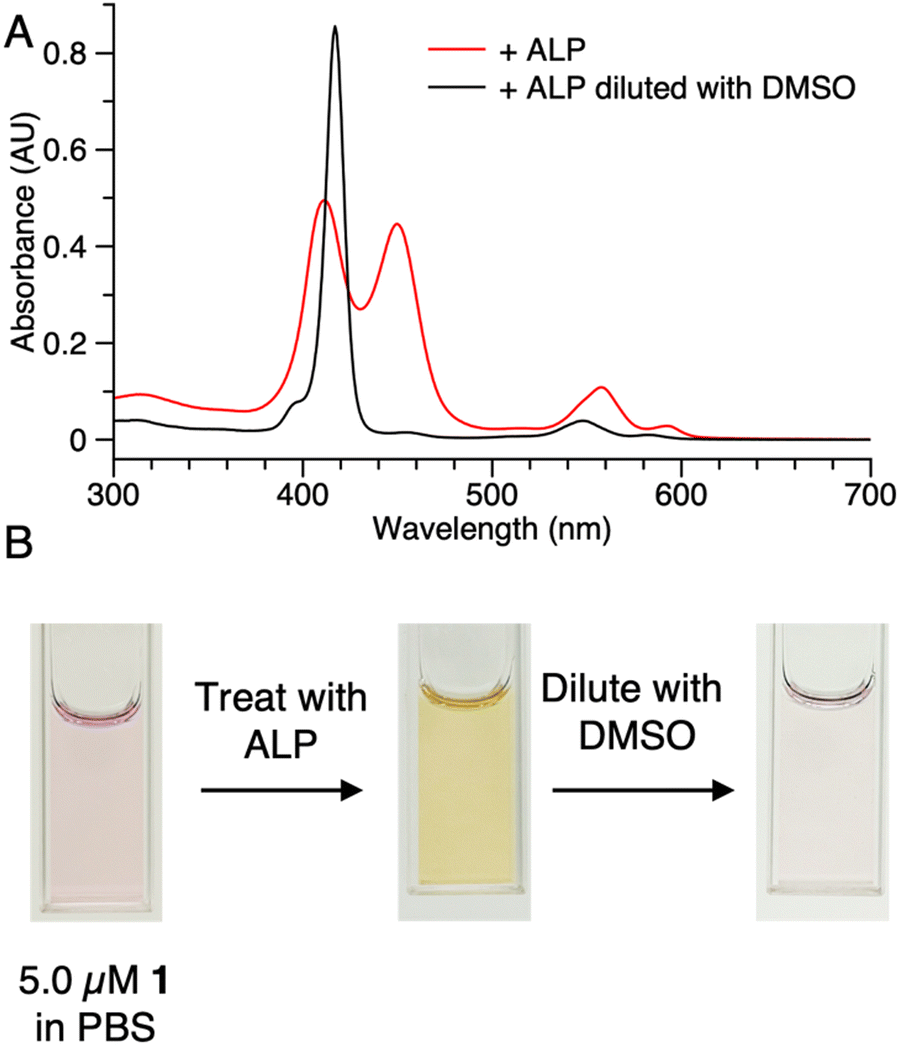
(2) Samples of 1 (5 μM in PBS) with or without ALP treatment were centrifuged in the presence of membrane filters (50 kDa cutoff). In the sample without ALP treatment (NE in Fig. 5 Panel A), the colored constituents passed through the 50 kDa membrane and were collected as the filtrate (bottom tube), while the colored constituents in the sample after enzyme treatment (E in Fig. 5 Panel A) did not pass through the membrane (top tube). No color was observed in the filtrate of sample E. Absorption spectral analysis following centrifugation of sample E showed no absorption in the bottom tube while that from the top (diluted by 20-fold with PBS) showed absorption commensurate with the aggregated porphyrin–peptide conjugate (Fig. 5 Panel B).
 | ||
| Fig. 5 Samples of 1 (5 μM) with or without ALP treatment followed by centrifugation in tubes containing a membrane filter (molecular weight cutoff 50 kDa). (A) NE = without ALP treatment; E = with ALP treatment; Samples were centrifuged for 10 min at 10000 rpm. (B) Absorption spectra of the bottom collection (red curve) and top collection (blue curve) of E after the centrifugation. Sample from the top tube was diluted by 20-fold with PBS before the measurement. | ||
(3) The enzymatic cleavage was monitored with RP-HPLC. A solution of 1 (100 μM in PBS) was treated with 2 U mL−1 ALP and incubated at 37 °C. Periodic aliquots were analyzed by RP-HPLC (5–95% MeCN/10 mM NH4HCO3 in H2O) as shown in Fig. 6. The chromatograph after 30 min of ALP treatment shows peaks from 1 (peak I), partially cleaved product (peak II), and fully cleaved product (peak III). The reaction is essentially complete after incubation for 6 hours (>95% by integration). The chromatogram of the unphosphorylated sample 1′ is shown for comparison. Note that the RP-HPLC traces shown in Fig. 6 were collected from samples at 100 μM as required given the dilution protocols to obtain sufficient material for detection.
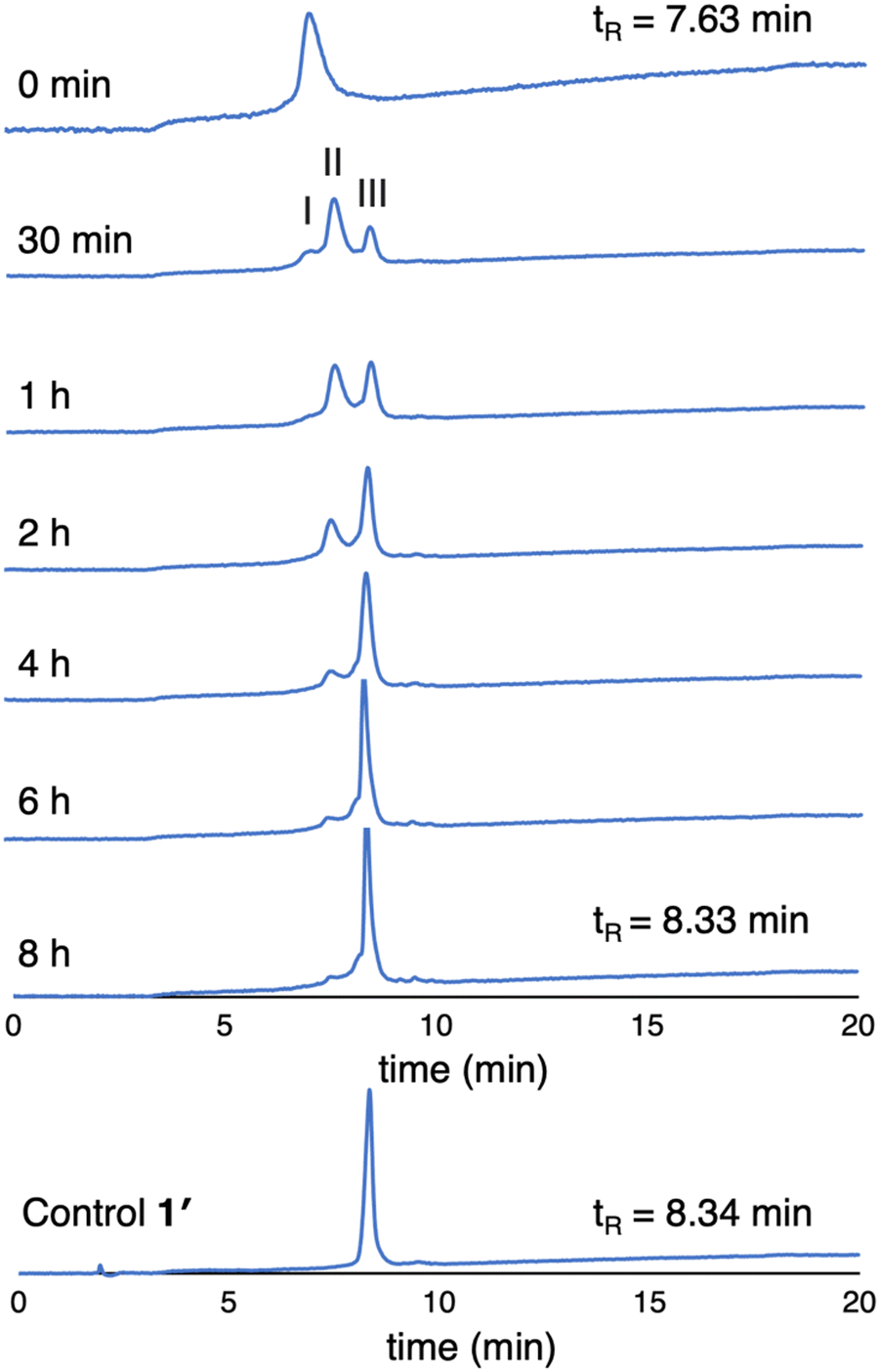 | ||
| Fig. 6 RP-HPLC traces (λabs 420 nm) monitoring dephosphorylation of 1 upon treatment with ALP over 8 hours (100 μM, 2 U mL−1 ALP). The trace at 0 min is prior to treatment with ALP. | ||
Studies also were carried out with the unphosphorylated porphyrin–peptide conjugates. Samples of 1′ (0.5 μM in DMSO or PBS) were analyzed by absorption spectroscopy (Fig. 7 Panel A). The sample in DMSO displayed a Soret band at 419 nm with no sign of aggregation. In PBS, broad peaks at 417 nm and 455 nm were observed, indicating aggregation of the sample. Samples of 5′ (0.5 μM in DMSO or PBS) also were analyzed for comparison (Fig. 7 Panel B). The spectrum of the sample in DMSO was identical with 1′, while in PBS the Soret band split into 3 broad peaks at 409 nm, 419 nm and 455 nm. The spectral differences between 5′ and 1′ might result from different stacking patterns, as porphyrin 5′ does not contain a PEG chain.
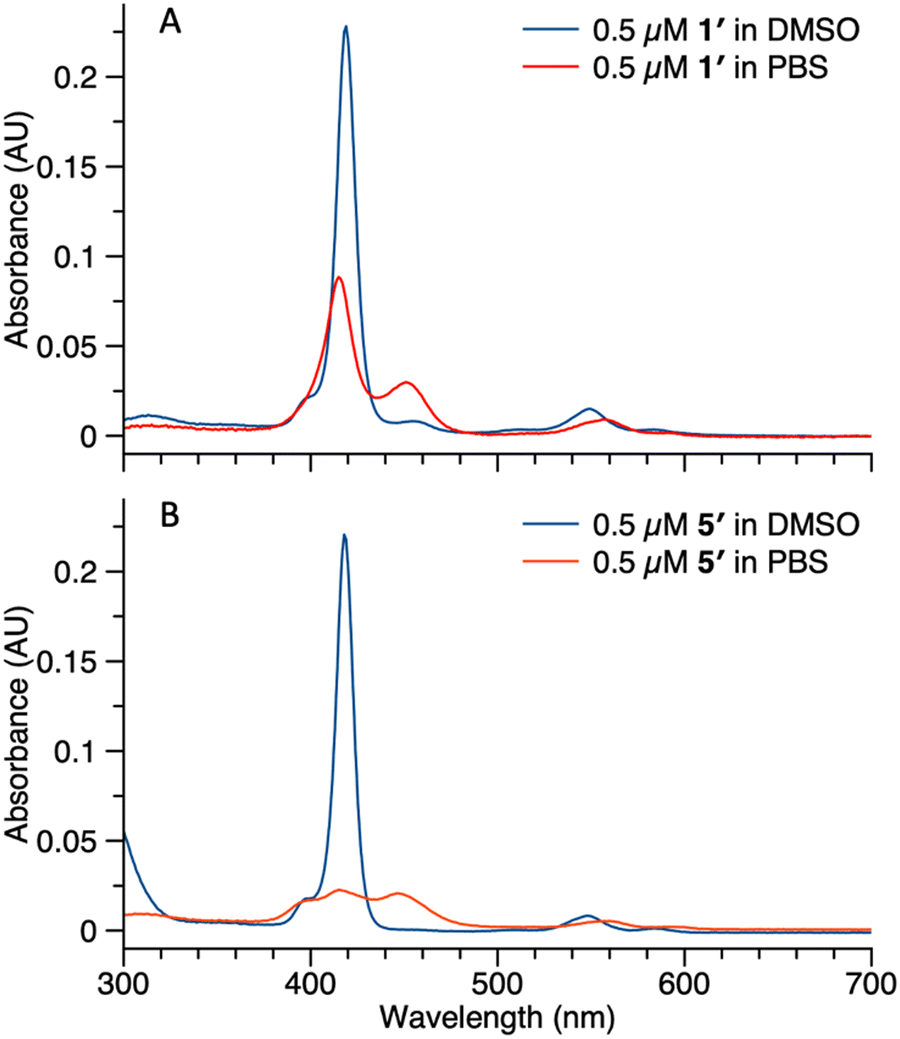 | ||
| Fig. 7 Absorption spectra of compound 1′ (panel A) and 5′ (panel B) in DMSO (blue curve) or PBS (red curve). Concentrations for all samples were 0.5 μM. | ||
Fluorescence study in the sub-micromolar regime
Examination of aggregation of small samples is difficult at concentrations below 1 μM. The presence of the porphyrin may afford an almost ideal case that enables extension into the sub-micromolar regime, given the strong absorption and readily detectable fluorescence. A zinc porphyrin typically exhibits molar absorption coefficient εSoret = 500000 M−1 cm−1, which affords absorption A = 0.5 in a 1-cm pathlength cuvette.34 The values determined here for 1 are consistent with expectation, where εSoret = 460000 M−1 cm−1 in DMSO and εSoret = 350000 M−1 cm−1 in PBS. With large quantities of sample, longer pathlength cuvettes could in principle be employed.35 To probe aggregation at sub-micromolar concentrations, fluorescence spectroscopy was employed given the known fluorescence properties of meso-substituted zinc porphyrins (e.g., emission wavelength λem ∼ 590 and 640 nm, fluorescence quantum yield Φf ∼ 0.030)34 and the typical quenching of fluorescence that arises upon intermolecular aggregation. While the Φf value for a zinc porphyrin may be regarded as quite low, the fluorescence of a zinc porphyrin consists of two distinct bands in the red region (e.g., 590 and 640 nm) where little else emits.
Thus, porphyrin 1′ at sub-micromolar concentrations was examined by fluorescence spectroscopy. The ratio of maximum fluorescence intensity of samples in PBS to that in DMSO was plotted against concentration (Fig. 8). The ratio decreased as the concentration increased in the range of 1.0 nM to 316 nM, which implies that 1′ aggregates more in PBS as the concentration increases. Even at 316 nM, the sample did not aggregate completely in PBS (retained ∼30% of the maximum fluorescence). Fluorescence spectra corresponding to Fig. 8 are provided in the ESI.† The long PEG chain in 1′ imparts hydrophilicity and may contribute to incomplete aggregation. We note that the PEG chain typically occupies a distribution of conformations that include kinking and helical coiling due to the preferred gauche geometry of the –OCH2CH2O– unit and hydrogen bonding with water.36,37 By contrast, the porphyrin–peptide conjugate without a PEG chain, 5′, at concentrations of 31.6 nM, 100 nM, and 316 nM, gave <2% of the maximum fluorescence intensity in PBS versus DMSO. The fluorescence intensity at concentrations >1 μM or <1 nM was not measured due to instrumental limitations.
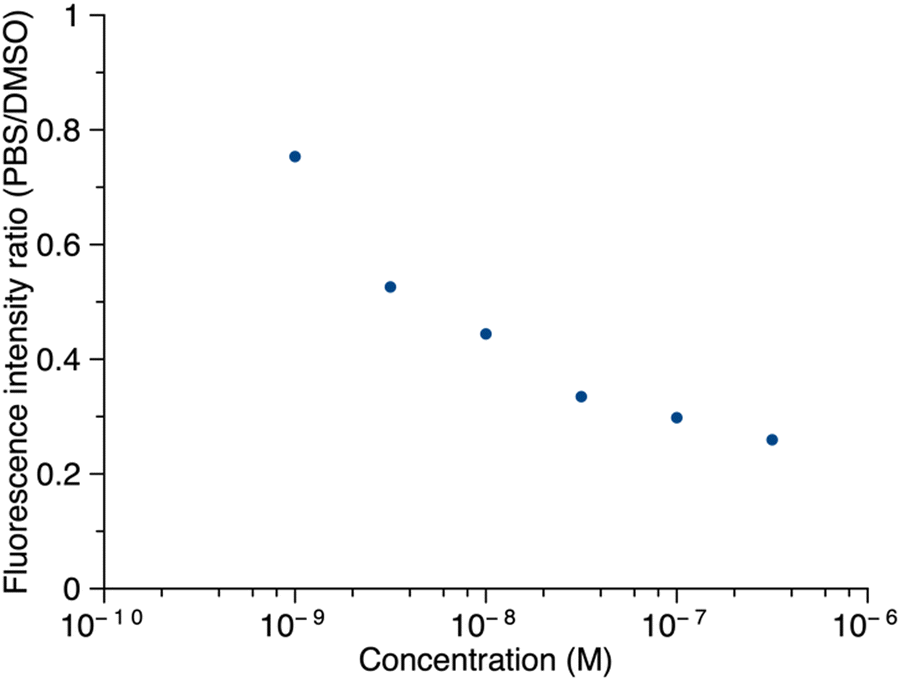 | ||
| Fig. 8 The ratio of the fluorescence intensity of 1′ in PBS versus DMSO as a function of concentration. In DMSO, λabs = 419 nm, λem = 641 nm; in PBS, λabs = 415 nm, λem = 645 nm. | ||
In summary, the data taken together are consistent with aggregation of the porphyrin–peptide conjugates upon dephosphorylation (or the absence of phosphorylation) in aqueous PBS at neutral conditions. The aggregation is reversible as shown by addition of the potent solubilizing agent DMSO. The presence of the zinc(II) porphyrin enables analysis both by absorption spectroscopy and fluorescence spectroscopy. Absorption spectroscopy could be applied in the concentration regime of 5–0.5 μM, whereas fluorescence spectroscopy was applied in the concentration regime extending to 1 nM. Data in support of the aggregation include profound alteration of the porphyrin absorption spectrum, quenching of the fluorescence, and absence of filtration through 50 kDa-cutoff membranes. The fluorescence studies indicated at least 50% of the sample was aggregated at a concentration of 10 nM. Evidence for the phosphatase-induced cleavage of the tyrosine-O-phosphoester was obtained by RP-HPLC analysis. Studies using dynamic light scattering (DLS) spectroscopy were attempted in an effort to gain information concerning the size distribution of the aggregates, but such studies were uninformative for these dilute samples.
Outlook
The development of molecules that undergo EISA offers great promise in the life sciences particularly as genomic and other studies provide information concerning the availability of enzymes in distinct tissues. Realization of such promises likely will require assembly of minute quantities of EISA substrates in dilute physiological milieu. Here, aggregation at concentrations as low as 10 nM has been detected by fluorescence spectroscopy of the porphyrin. The present molecular design combines a trans-AB-porphyrin bearing two self-assembly tetrapeptides (GffY) wherein the latter are in the O-phosphoester state prior to enzymatic treatment. The peptides employed here have been used previously and also have been termed hydrogelators. Many studies of hydrogelation or assembly have been carried out at concentrations substantially greater than those herein, which in some cases may reflect limits of detection given the absence of a readily detected entity such as a porphyrin. Regardless, important applications such as molecular radiotherapy administered intravenously likely require concentrations at least 100-times lower, such as 100 pM or less. More potent self-assembling motifs may be required to enter that regime.One interesting aspect of EISA is that the activated compound may not require the interaction with another hydrogelator to promote retention in the target tissue. Here we demonstrated that at the very low concentration of 0.1 uM, the activated form of 1 came out of solution apparently without forming a gel superstructure. This could potentially allow a compound to concentrate in a tissue by its mere insolubility in interstitial fluid. This concept would hold significant value for treatment schemes which rely on administration of a drug to a body cavity. This includes intra-peritoneal drug delivery for ovarian cancer which has metastasized to the omentum and/or intercranial administration following tumor resection.
Another intriguing prospect of EISA for future studies is the potential to eliminate cellular efflux of a therapeutic after internalization into a cell. A current weakness of several therapeutics including some targeted radiotherapies is that much of the therapeutic agent which is bound or even internalized into the cell is subsequently re-released either due to degradation or recycling of the receptor. This limits the amount of dose which can be sustained long-term in the target tissue. If a compound could become trapped upon internalization, there is potential to achieve higher tumor concentrations. The molecular design challenges may be quite subtle. Porphyrin–peptides 5 and 5′ lack the PEG linker present in 1 and 1′ and appear to be far less soluble. Achieving a balance between solubility prior to enzymatic cleavage and aggregation following enzymatic cleavage is essential, has been little investigated, and is expected to require substantial empirical study, particularly to engineer EISA substrates for function in the sub-nanomolar regime. Consideration of the rich composition of molecules in the biological milieu also is important. Finally, while our focus here is possible therapeutics applications, EISA can support other scientific and technological objectives such as nanostructure construction.38–40
Experimental section
Materials
All Fmoc-protected amino acids and 2-CTC resin were purchased from Chemimpex. TBAF (1 M in THF) was purchased from Oakwood Chemicals. mPEG24-N3 was purchased from Broadpharm. Alkaline phosphatase from bovine intestinal mucosa was purchased from Sigma-Aldrich. Centrifugation of samples was carried out using Amicon centrifugation devices (molecular weight cutoff 50 kDa). The synthesis of TIPS-porphyrin-alkyne (3)32 is reported elsewhere. The pH of PBS was 7.4.Synthesis
:1 v/v) with DIPEA (2.0 equiv.) as the base. After loading the first amino acid on the resin, methanol was used to ensure that all the active sites of the resin were protected. The resin was treated with 20% piperidine in DMF for 30 min to remove the Fmoc group. The following amino acid couplings were performed in DMF using the Fmoc-protected amino acid (3.0 equiv.), HOBt (3.0 equiv.) and HBTU (3.0 equiv.) as the coupling reagents, and DIPEA (6.0 equiv.) as the base. After the installation of the last amino acid, 3-azidopropanoic acid was installed following the same coupling protocol. The peptide derivative was cleaved using 95% TFA, 2.5% TIS, and 2.5% H2O for 1 h. Then, ice-cold ethyl ether was added to precipitate the crude peptide. The latter was purified by preparative RP-HPLC [Buchi® PrepPure C850, Buchi® column (10 μm C18 100 Å, LC column 150 × 10 mm), linear gradient with 30–80% MeCN/H2O, 0.1% TFA] to yield a white solid: 1H NMR (500 MHz, DMSO-d6) δ 2.40 (t, J = 6.5 Hz, 2H), 2.61–2.71 (m, 2H), 2.75–2.86 (m, 2H), 2.89–2.98 (m, 1H), 2.98–3.07 (m, 1H), 3.48 (t, J = 6.5 Hz, 2H), 3.53–3.65 (m, 1H), 3.65–3.77 (m, 1H), 4.41 (td, J = 4.8, 8.6 Hz, 1H), 4.48 (td, J = 4.0, 8.8 Hz, 1H), 4.57 (td, J = 4.6, 8.8 Hz, 1H), 7.00–7.28 (m, 14H), 7.98 (d, J = 8.2 Hz, 1H), 8.11 (d, J = 8.5 Hz, 1H), 8.15 (t, J = 5.7 Hz, 1H), 8.38 (d, J = 8.1 Hz, 1H). 13C{1H} NMR (176 MHz, DMSO-d6) δ 34.7, 36.7, 38.0, 38.2, 40.9, 42.3, 47.3, 54.0, 54.1, 54.1, 120.1, 120.1, 126.6, 126.7, 128.5, 128.5, 129.7, 129.7, 130.7, 133.4, 138.0, 138.1, 150.7, 150.7, 168.7, 170.3, 171.0, 171.2, 173.2. 31P NMR (202 MHz, DMSO-d6) δ −6.25. ESI-MS obsd 710.2324, calcd 710.2334 [(M + H)+, M = C32H36N7O10P].
:5)]. The sample was homogeneous by RP-HPLC. MALDI-MS obsd 3188.34, calcd 3188.23 (M+, M = C153H195N21O46P2Zn). ESI-MS obsd 1593.1171, calcd 1593.1093 [(M − 2H)2−, M = C153H195N21O46P2Zn].
:4)]. The sample was homogeneous by RP-HPLC. MALDI-MS obsd 3029.05, calcd 3028.30 [(M + H)+, M = C153H193N21O40Zn]. ESI-MS obsd 1513.1503, calcd 1513.1430 [(M − 2H)2−, M = C153H193N21O40Zn].
000 rpm using Amicon membrane filters (50 kDa cutoff). The filtrate and the unfiltered material were examined by absorption spectroscopy.
Conflicts of interest
The authors declare competing financial interests.Acknowledgements
This work was supported by NC State University. NMR and mass spectrometry measurements were carried out in the Molecular Education, Technology, and Research Innovation Center (METRIC) at NC State University.References
- T.-T. Wang, Y.-Y. Xia, J.-Q. Gao, D.-H. Xu and M. Han, Pharmaceutics, 2021, 13, 753 CrossRef CAS PubMed.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS PubMed.
- X. Qin, W. Xie, S. Tian, J. Cai, H. Yuan, Z. Yu, G. L. Butterfoss, A. C. Khuong and R. A. Gross, Chem. Commun., 2013, 49, 4839–4841 RSC.
- A. Tanaka, Y. Fukuoka, Y. Morimoto, T. Honjo, D. Koda, M. Goto and T. Maruyama, J. Am. Chem. Soc., 2015, 137, 770–775 CrossRef CAS PubMed.
- W. Du, X. Hu, W. Wei and G. Liang, Bioconjugate Chem., 2018, 29, 826–837 CrossRef CAS PubMed.
- J. Gao, J. Zhan and Z. Yang, Adv. Mater., 2020, 32, 1805798 CrossRef CAS PubMed.
- B. J. Kim and B. Xu, Bioconjugate Chem., 2020, 31, 492–500 CrossRef CAS PubMed.
- P. Huang, Y. Gao, J. Lin, H. Hu, H.-S. Liao, X. Yan, Y. Tang, A. Jin, J. Song, G. Niu, G. Zhang, F. Horkay and X. Chen, ACS Nano, 2015, 9, 9517–9527 CrossRef CAS PubMed.
- X. Du, J. Zhou, H. Wang, J. Shi, Y. Kuang, W. Zeng, Z. Yang and B. Xu, Cell Death Dis., 2017, 8, e2614 CrossRef CAS PubMed.
- Z. Feng, H. Wang, X. Chen and B. Xu, J. Am. Chem. Soc., 2017, 139, 15377–15384 CrossRef CAS PubMed.
- Z. Feng, H. Wang, R. Zhou, J. Li and B. Xu, J. Am. Chem. Soc., 2017, 139, 3950–3953 CrossRef CAS PubMed.
- Y. Wang, X. Li, D. Zheng, Y. Chen, Z. Zhang and Z. Yang, Adv. Funct. Mater., 2021, 31, 2102505 CrossRef CAS.
- B. Jana, S. Jin, E. M. Go, Y. Cho, D. Kim, S. Kim, S. K. Kwak and J.-H. Ryu, J. Am. Chem. Soc., 2023, 145, 18414–18431 CrossRef CAS PubMed.
- Y. Gao, J. Shi, D. Yuan and B. Xu, Nat. Commun., 2012, 3, 1033 CrossRef PubMed.
- J. Zhou, X. Du, C. Berciu, H. He, J. Shi, D. Nicastro and B. Xu, Chem., 2016, 1, 246–263 CAS.
- Y. Wang, Y. Zhang, X. Li, C. Li, Z. Yang and L. Wang, Chem. – Asian J., 2018, 13, 3460–3463 CrossRef CAS PubMed.
- C. Liang, X. Yan, R. Zhang, T. Xu, D. Zheng, Z. Tan, Y. Chen, Z. Gao, L. Wang, X. Li and Z. Yang, J. Controlled Release, 2020, 317, 109–117 CrossRef CAS PubMed.
- Z. Yao, K. E. Borbas and J. S. Lindsey, New J. Chem., 2008, 32, 436–451 RSC.
- D. L. Lee, H.-L. M. Chin, C. G. Knudsen, G. L. Mayers, D. S. Rose, R. K. Skogstrom, T. Palzkill, H. Fujita, Y. Zhang, Z. Wu and J. S. Lindsey, Proc. SPIE, 2020, 11477, 1147708 Search PubMed.
- Y. Zhang, Z. Wu, I. Takashima, K.-U. Nguyen, N. Matsumoto and J. S. Lindsey, New J. Chem., 2020, 44, 14266–14277 RSC.
- Z. Wu, J. Dou, K.-U. Nguyen, J. C. Eppley, K. Siwawannapong, Y. Zhang and J. S. Lindsey, Molecules, 2022, 27, 8682 CrossRef CAS PubMed.
- D. Sato, Z. Wu, J. Dou, J. Son and J. S. Lindsey, New J. Chem., 2023, 47, 8223–8242 RSC.
- J. Son, Z. Wu, J. Dou, H. Fujita, P.-L. D. Cao, Q. Liu and J. S. Lindsey, Molecules, 2023, 28, 4143 CrossRef CAS PubMed.
- H. Wei, J. Min, Y. Wang, Y. Shen, Y. Du, R. Su and W. Qi, J. Mater. Chem. B, 2022, 10, 9334–9348 RSC.
- Q. Zou, M. Abbas, L. Zhao, S. Li, G. Shen and X. Yan, J. Am. Chem. Soc., 2017, 139, 1921–1927 CrossRef CAS PubMed.
- H. Wang, C. Ren, Z. Song, L. Wang, X. Chen and Z. Yang, Nanotechnology, 2010, 21, 225606 CrossRef PubMed.
- C. Ou, J. Zhang, X. Zhang, Z. Yang and M. Chen, Chem. Commun., 2013, 49, 1853–1855 RSC.
- J. Shi, X. Du, D. Yuan, J. Zhou, N. Zhou, Y. Huang and B. Xu, Biomacromolecules, 2014, 15, 3559–3568 CrossRef CAS PubMed.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
- F. Dumoulin and V. Ahsen, J. Porphyrins phthalocyanines, 2011, 15, 481–504 CrossRef CAS.
- H. A. Houson, Z. Wu, P.-L. D. Cao, J. S. Lindsey and S. E. Lapi, Mol. Pharmaceutics, 2024, 21, 2441–2455 CrossRef CAS PubMed.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- M. Taniguchi, J. S. Lindsey, D. F. Bocian and D. Holten, J. Photochem. Photobiol., C, 2021, 46, 100401 CrossRef CAS.
- Q. Liu, M. Taniguchi, S. Goel and J. S. Lindsey, Dyes Pigm., 2024, 223, 111914 CrossRef CAS.
- R. Begum and H. Matsuura, J. Chem. Soc., Faraday Trans., 1997, 93, 3839–3848 RSC.
- D. Sato, Z. Wu, H. Fujita and J. S. Lindsey, Organics, 2021, 2, 161–273 CrossRef CAS.
- R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19–24 CrossRef CAS PubMed.
- L. Gao, J. Wu and D. Gao, ACS Nano, 2011, 5, 6736–6742 CrossRef CAS PubMed.
- J. Tan, D. Liu, Y. Bai, C. Huang, X. Li, J. He, Q. Xu and L. Zhang, Macromol., 2017, 50, 5798–5806 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fluorescence detection data. See DOI: https://doi.org/10.1039/d4nj01798f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |