Uniform trehalose nanogels for glucagon stabilization†
Received
1st November 2023
, Accepted 14th February 2024
First published on 13th March 2024
Abstract
Glucagon is a peptide hormone that acts via receptor-mediated signaling predominantly in the liver to raise glucose levels by hepatic glycogen breakdown or conversion of noncarbohydrate, 3 carbon precursors to glucose by gluconeogenesis. Glucagon is administered to reverse severe hypoglycemia, a clinical complication associated with type 1 diabetes. However, due to low stability and solubility at neutral pH, there are limitations in the current formulations of glucagon. Trehalose methacrylate-based nanoparticles were utilized as the stabilizing and solubilizing moiety in the system reported herein. Glucagon was site-selectively modified to contain a cysteine at amino acid number 24 to covalently attach to the methacrylate-based polymer containing pyridyl disulfide side chains. PEG2000 dithiol was employed as the crosslinker to form uniform nanoparticles. Glucagon nanogels were monitored in Dulbecco's phosphate-buffered saline (DPBS) pH 7.4 at various temperatures to determine its long-term stability in solution. Glucagon nanogels were stable up to at least 5 months by size uniformity when stored at −20 °C and 4 °C, up to 5 days at 25 °C, and less than 12 hours at 37 °C. When glucagon stability was studied by either HPLC or thioflavin T assays, the glucagon was intact for at least 5 months at −20 °C and 4 °C within the nanoparticles at −20 °C and 4 °C and up to 2 days at 25 °C. Additionally, the glucagon nanogels were studied for toxicity and efficacy using various assays in vitro. The findings indicate that the nanogels were nontoxic to fibroblast cells and nonhemolytic to red blood cells. The glucagon in the nanogels was as active as glucagon alone. These results demonstrate the utility of trehalose nanogels towards a glucagon formulation with improved stability and solubility in aqueous solutions, particularly useful for storage at cold temperatures.
Introduction
Individuals with type 1 diabetes regularly inject insulin to manage blood glucose levels; however, insulin overdose and irregular eating schedules can lead to hypoglycemia.1 Severe hypoglycemia occurs when blood glucose levels fall below 50 mg dL−1, with complications ranging from weakness, difficulty walking, vision impairment, confusion, and seizures.1–3 Glucagon is currently administered to treat emergency hypoglycemic episodes.4 Glucagon increases blood glucose concentration through binding to hepatic glucagon receptors, which stimulate glycogen breakdown and release of glucose from the liver.5–7 Due to low stability, low solubility at physiological pH, and tendency to form toxic fibrils, glucagon formulations for emergency delivery remain a challenge.8–10
New glucagon formulations have been developed within the past few years, with some receiving FDA approval. In 2019, Eli Lilly released the first nasal rescue glucagon, BAQSIMI™, formulated as a 3 mg dry powder.11–13 Later that year, Xeris Pharmaceuticals released the GVOKE HypoPen® formulated as a solution of glucagon in sulfuric acid and dimethyl sulfoxide (DMSO) in an automatic injector.14,15 In 2021, Zealand Pharma released their automatic injector ZEGALOGUE™, which contains a glucagon analogue, dasiglucagon.16–18 Although these formulations demonstrate great progress and effort towards a stable and efficacious glucagon formulation, their stability in an aqueous pH 7.4 solution at a range of temperatures is limited. For example, BAQSIMI™ is only stable up to 30 °C as a dry powder and cannot be expossed to moisture. GVOKE HypoPen® is only stable up to 25 °C, is formulated in organic solvent, and could cause pain at the injection site from the sulfuric acid. ZEGALOGUE™ is only stable up to 25 °C.11,15,16 Thus, there is still a need for glucagon formulations stable in aqueous solution.
Glucagon has been stabilized by various strategies including chemical modification, covalent attachment of poly(ethylene glycol) (PEG), and addition of excipients including salts, surfactants, and sugars.19–22 However, even with these excipients in solution, glucagon is still susceptible to degradation at neutral or slightly acidic pH (5–7) within ∼26 h in solution.23
During manufacture, storage, and cold chain transport, many biomolecules experience environmental stressors necessitating stabilization.21,24,25 In particular, trehalose has been used as an excipient for RNA, enzymes, insulin, and other proteins, including glucagon in the GVOKE HypoPen®.25–29 Trehalose stabilizes proteins in both solution and lyophilized form;30–32 which is hypothesized to be achieved through vitrification, water entrapment, and/or water replacement mechanisms.33–35 Properties that make trehalose a good stabilizer are its high solubility in water, large hydration number, and a high glass transition temperature (Tg).36 Trehalose polymers, where the disaccharide is a side chain, exhibit superior stabilizing capabilities compared to trehalose.37–41 We have previously shown that proteins retain greater bioactivity against heat and lyophilization in the presence of trehalose polymers as either an excipient, conjugate, hydrogel, or nanogel compared to trehalose.37–39,42 Others have reported that nanoformulations of trehalose polymers are considerably more stable than other nanoparticles in cell culture media and upon storage to deliver RNA and DNA in vivo.27 We previously synthesized a trehalose nanoparticle by utilizing a chemically modified glucagon, thiolated at N-terminus and Lys12. This modification stabilized a glucagon nanoparticle in aqueous solution (pH 7.4) for up to three weeks.29 However, these trehalose nanoparticles were non-uniform in size and morphology when employing glucagon as a crosslinker.29 To translate a formulation to human use, it is desirable that nanoparticles be synthesized with a high level of purity and uniformity.43
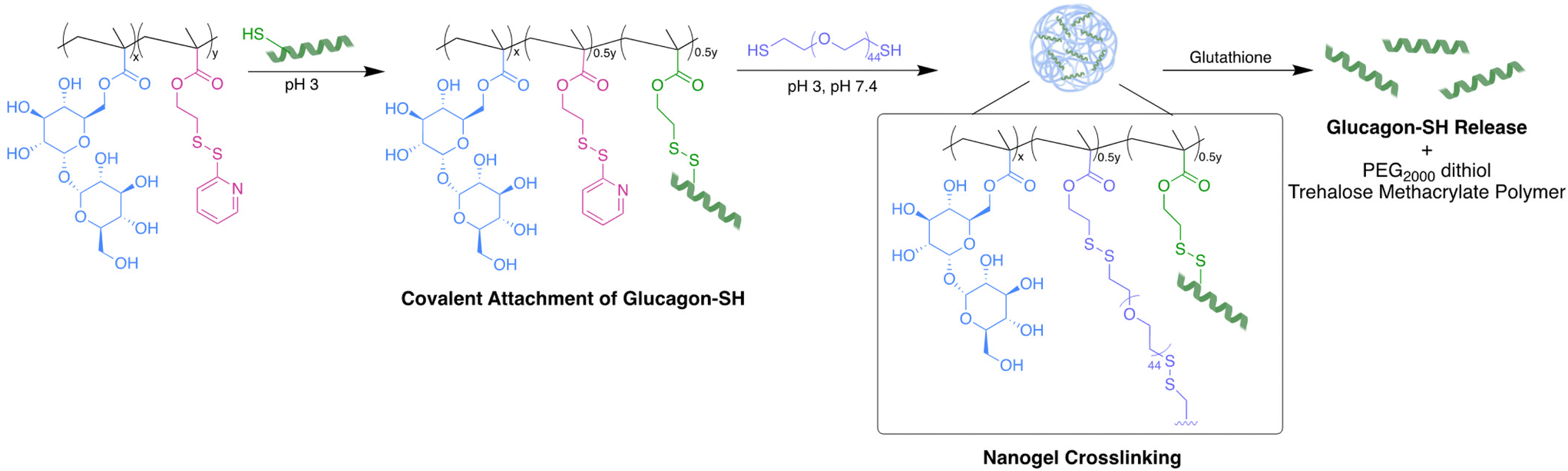
In the study described herein, we aimed to investigate an alternative crosslinker to create uniform trehalose nanoparticles and evaluate the long-term stabilization of glucagon in aqueous solutions over a range of relevant temperatures. Glucagon was site-selectively modified to contain a cysteine at glutamine (Gln) 24 to create a thiol reactive handle for ease of conjugation (glucagon–SH). Previously, Gln24 was replaced with alanine, yielding potency similar to the native peptide.44 Additionally, a crystal structure of a glucagon analogue binding to its receptor including a substitution at Gln24 showed that the modification did not significantly influence receptor binding.45 We copolymerized methacrylate functionalized trehalose with pyridyl disulfide ethyl methacrylate using free radical polymerization conditions forming trehalose polymers with thiol reactive handles. Glucagon–SH was conjugated via a PDS disulfide exchange, followed by crosslinking with PEG2000 dithiol to give pH 7.4 soluble and stable glucagon nanogels uniform in size. The glucagon stability and nanogel uniformity was thoroughly investigated at temperatures mimicking environmental stressors as well as the in vitro biocompatibility and efficacy.
Experimental
Poly(PDSMA-co-TrMA) synthesis
Synthesis of trehalose methacrylate (TrMA) and pyridyl ethyl methacrylate (PDSMA) monomers was conducted as previously published (Fig. S1–S4†).29 The two monomers were copolymerized using free radical polymerization with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PDSMA:TrMA feed ratio resulting in poly(PDSMA-co-TrMA) as previously described.29 Incorporation was determined by 1H NMR spectroscopy (Fig. S5†) and molecular weight was determined by gel permeation chromatography (GPC) (Fig. S6†).
:1 PDSMA:TrMA feed ratio resulting in poly(PDSMA-co-TrMA) as previously described.29 Incorporation was determined by 1H NMR spectroscopy (Fig. S5†) and molecular weight was determined by gel permeation chromatography (GPC) (Fig. S6†).
Empty nanogel synthesis
Poly(PDSMA-co-TrMA) (1.0 mg) was dissolved in 500 μL 10 mM HCl in a 1.5 mL lo bind Eppendorf tube equipped with a micro stir bar. PEG2000 dithiol (0.63 mg, 50% of PDS side chain) was dissolved in 250 μL 10 mM HCl, added to the reaction vial dropwise, and left to stir at 8000 rpm, 4 °C for 3 h. The crude empty nanogel solution was purified against DPBS 50 mM pH 7.4 using a 20 kDa MWCO microdialysis cup at 4 °C for 2 days. After purification, the empty nanogels were filtered using a 0.2 μm PTFE filter until further analysis and stored at 4 °C.
Glucagon nanogel synthesis
Poly(PDSMA-co-TrMA) (1.0 mg) was dissolved in 500 μL 10 mM HCl in a 1.5 mL lo bind Eppendorf tube equipped with a micro stir bar. Glucagon–SH (2.2 mg, 50% of PDS side chain) was dissolved in 250 μL 10 mM HCl and added to the reaction vial. The vial was left to stir at 8000 rpm, 4 °C for 2 h. PEG2000 dithiol (0.63 mg, 50% of PDS side chain) was dissolved in 250 μL 10 mM HCl, added to the reaction vial dropwise, and left to stir at 8000 rpm, 4 °C for 3 h. The crude glucagon nanogel was purified against DPBS 50 mM pH 7.4 using a 20 kDa MWCO microdialysis cup at 4 °C for 2 days. After purification, the glucagon nanogels were filtered using a 0.2 μm PTFE filter and stored at either −20 °C, 4 °C, 25 °C, or 37 °C. All glucagon nanogels were stored at 1 mg mL−1 (polymer) in a solution of DPBS 50 mM pH 7.4 unless otherwise noted for all subsequential experiments. All glucagon nanogels were used immediately unless otherwise noted.
HPLC glucagon quantification
Glucagon incorporation and conjugation was monitored via the disappearance of the glucagon peak during the nanogel formation and reappearance after reducing with tris(2-carboxyethyl)phosphine (TCEP) 100 equiv. using the AUC of the glucagon peak (Fig. S8†) at 220 nm using high performance liquid chromatography (HPLC). Glucagon–SH controls were dissolved in minimal 10 mM HCl then brought to pH 7.4 with 50 mM DPBS.
Thioflavin T (ThT) assay
BSA fibrils prepared at 1 mg mL−1 were used as the positive control by inducing fibrils by heating to 80 °C for 1 h.46 Empty nanogels at 1 mg mL−1 (polymer) in DPBS 50 mM pH 7.4 with 100 equiv. TCEP was used as the negative control. Glucagon nanogels were incubated with 100 equiv. TCEP for 15 min at 4 °C before analysis. 250 μL of ThT solution at 50 μM (in DPBS) was added into a black plate followed by the addition of 50 μL of either glucagon nanogel, empty nanogel, or BSA. The solutions were covered in the dark and incubated at 25 °C for 20 min. Fluorescence intensity was then measured using a plate reader (λex = 450 nm, λem = 482 nm).47
Fetal bovine serum (FBS) glucagon nanogel stability
Glucagon nanogels were incubated with 10% FBS. DLS measurements were taken at 0, 1, 2, and 3 days for samples stored at 4 °C and 25 °C and at 0, 4, 8, and 12 h for samples stored at 37 °C.
Cell lines and maintenance
The mouse embryonic fibroblasts, NIH 3T3 were provided by Professor Andrea M Kasko, Engineering V, University of California Los Angeles and the human liver cancer cells, HepG2, from Professor Yu-Pei, California NanoSystem Institute, UCLA. The mouse embryonic fibroblasts, NIH 3T3, and human liver cancer cells, HepG2, were cultured in Dulbecco's modified Eagle's medium (DMEM) media supplemented with 10% FBS (v/v), 1% Pen Strep, and antibiotics at 37 °C in a standard humidified atmosphere containing 5% carbon dioxide (CO2).
In vitro biocompatibility
The in vitro biocompatibility of empty nanogels and glucagon nanogels were evaluated for 24 h in mouse embryonic fibroblasts, NIH 3T3, by performing the colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) assay. The MTT assay was evaluated for different concentrations: 10, 50, 100, 250, 500, 1000 μg mL−1 for 24 h. 0.7 × 104 cells per well were counted using hemocytometer, seeded in 96-well plates, and incubated at 37 °C overnight. The next day, the cells were treated with different concentrations of the empty nanogel and glucagon nanogel (freshly prepared, 3 month at 4 °C, and 3 months at −20 °C) and incubated at 37 °C for 24 h. Media supplemented with FBS served as a control. After incubation, cell culture media was replaced with serum-free media containing MTT reagent (5 mg per 10 mL) and incubated at 37 °C for 3 h. The MTT-containing media was removed, a solubilizing agent, DMSO, was added to dissolve the formazan crystals, and the absorbance was measured at 570 nm using a plate reader. For normalizing, the absorbance was measured at 630 nm.
Live/dead cell assay
The in vitro biocompatibility of empty nanogels and glucagon nanogels were evaluated for 24 h in mouse embryonic fibroblasts, NIH 3T3, by performing a microscopic live/dead assay. The live/dead assay was evaluated at 4 different concentrations: 10, 100, 500, 1000 μg mL−1. 0.7 × 104 cells per well were counted using a hemocytometer, seeded in 96-well plates, and incubated at 37 °C for 12 h. The next day, the cells were treated with different concentrations of the empty nanogel and glucagon nanogel and incubated for 24 h at 37 °C. After 24 h, the cell culture media was replaced with fresh media and fluorescent probes calcein AM (green fluorescence) and ethidium homodimer-1 (red fluorescence) were added, incubated for 20 min at 37 °C, and imaged using a fluorescent microscope.
In vitro efficacy study
The in vitro efficacy of the glucagon nanogel was evaluated by comparing the level of lactate produced using a Lactate-Glo™ assay kit, Promega. The liver model cell line, HepG2 was used for evaluating the levels of lactate produced. Cells (0.7 × 104 per well) were plated in a 96-well plate and incubated overnight at 37 °C. The next day, the cell culture media was replaced with 1 mg of glucose containing media without FBS and starved for 4 h. After 4 h, the media was replaced with 4.5 mg of glucose containing media with 10% FBS along with the empty nanogel, glucagon–SH (20 μg) and glucagon nanogel (20 μg). The sample of the medium at experimental time points (2, 3, 6, 24, 48, and 72 h) were collected by diluting 4 μl into 96 μl DPBS. The samples were collected and frozen at −20 °C until ready to perform the assay. The samples were thawed and 50 μl was transferred to a white 96-well assay plate. 50 μL of Lactate Detection Reagent (Lactate-Glo™ assay, Promega) was added and incubated for 60 min at 25 °C. Luminescence was recorded using the plate reader.
Metabolic activity
The metabolic activity of the glucagon nanogels were evaluated qualitatively by monitoring the color change of the cell culture media in a liver model cell line, HepG2. Cells (0.3 × 106 per well) were plated into 35 mm cell culture dishes and incubated for 12 h at 37 °C. The next day, the cell culture media was replaced with 1 g L−1 of glucose containing media without FBS and starved for 4 h. The control for the experiment was the cells cultured in the cell culture media supplemented with FBS. After 4 h, the media was replaced with 4.5 g L−1 of glucose containing media with 10% FBS along with native glucagon, glucagon–SH (20 μg mL−1) and glucagon nanogel (20 μg mL−1). The media color change was observed every day and imaged after 72 h.
Hemocompatibility
Hemolytic effects of the glucagon nanogels were determined following a previous report.48 Sheep red blood cells (RBCs) (1 mL, 100%) were resuspended in 5 mL of DPBS (50 mM, pH = 7.4) and centrifuged at 3000 rpm, 4 °C for 10 min. The supernatant was discarded and replenished with more DPBS. This wash was repeated 3–5 more times until the supernatant was visibly clear. The RBCs were resuspended to 8 mL with DPBS. The cells were counted using a hemacytometer and diluted to 2.0 × 107 cells per mL. 250 μL of diluted RBCs were added to clear lo bind Eppendorf tubes followed by the addition of 750 μL of sample. PEG2000 dithiol, poly(PDSMA-co-TrMA), glucagon–SH, and glucagon nanogel at concentrations 5–50 μM were tested (n = 3). Concentrations were based on glucagon–SH. 50 mM DPBS was used as the negative control and 20% Triton X-100 in DPBS was used as the positive control. The samples were incubated at 37 °C for 1 h. Then, the samples were centrifuged at 3000 rpm, 4 °C, for 10 min. 100 μL of supernatant of each sample were aliquoted in a 96-well plate. The absorbance was measured at 540 nm. To determine the hemolysis percentage, we used the following equation: % hemolysis = 100(A − A0)/(ATX − A0) where A is the absorbance reading of the sample, A0 is the negative hemolysis control, and ATX is the positive hemolysis control.
Viscosity measurements
The empty nanogel and glucagon nanogel were prepared as mentioned above and then loaded into a Rheosense syringe. Air bubbles were carefully removed from the syringe. Syringe was loaded into the viscometer and allowed to equilibrate at the measurement temperature for 20 min.
Statistical analysis
All experimental values are reported as the mean ± SD. Graph Pad Prism 8 software was used for statistical analyses. One-way analysis of variance (ANOVA) followed by Turkey's multiple comparison test was employed to compare the means and determine the significance. Statistical significance is denoted by p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****).
Results and discussion
Optimization of glucagon nanogel synthesis
TrMA and PDSMA were copolymerized using free radical polymerization with a feed ratio of 1:1 to yield poly(PDSMA-co-TrMA) with a molecular weight of 9.8 kDa and Đ of 1.9 (1H NMR Fig. S5,† GPC Fig. S6†).29 We first wanted to understand the time course of the disulfide exchange between glucagon–SH and the PDS group of poly(PDSMA-co-TrMA) using the cysteine installed glucagon–SH. We monitored the kinetics via HPLC (Fig. S7†). A solution of poly(PDSMA-co-TrMA) and glucagon–SH in 10 mM HCl were mixed at 4 °C to observe 50% conjugation within 15 minutes and 82% conjugation after 3 h, reaching a plateau. Release of glucagon–SH was confirmed by HPLC, SDS-PAGE, and LC-MS by comparing the purified glucagon nanogel peak to the reduced nanogel peak (Fig. S8–S10†).
A nanoparticle <200 nm was targeted for optimal release of glucagon, clearance, and circulation.49–51 Glucagon nanogel synthesis was first attempted through crosslinking with 10, 20, 30, 40, and 50% PEG2000 dithiol (by PDS group) followed by the covalent attachment of glucagon–SH (to the remaining PDS groups) in 1:1 50 mM DPBS pH 7.4:10 mM HCl and purified by microdialysis using a 20 kDa MWCO filter against 50 mM DPBS pH 7.4 (Fig. S11†). Crosslinking percentages of 10–50% yielded nanogels with sizes >200 nm, with increasing crosslinking percentages yielding smaller nanogel sizes. We hypothesize this was due to the glucagon conjugation to the outside of the nanoparticle; the glucagon could aggregate, causing an increase in nanoparticle size. Although the 50% crosslinking yielded nanogels of 640 nm, we used this percentage in subsequent optimization to synthesize nanogels with 50% glucagon loading (half of the pyridyl disulfide groups). Using a 50% crosslinking, nanogel formation was next attempted with simultaneous glucagon–SH conjugation by mixing both PEG2000 dithiol and glucagon–SH in 1:1 50 mM DPBS pH 7.4:10 mM HCl and purified by microdialysis using a 20 kDa MWCO filter against 50 mM DPBS pH 7.4 (Fig. S12†). This yielded a uniform glucagon nanogel of 360 nm, which led us to believe a smaller size could be achieved by attaching the glucagon–SH first and then crosslinking. To prevent the glucagon from aggregating in solution, the reaction solvent was changed to HCl to ensure any free glucagon in solution would remain soluble throughout purification. Glucagon–SH was then conjugated to poly(PDSMA-co-TrMA) followed by the crosslinking with varying PEG dithiol sizes (600, 1000, and 2000 Da) yielding nanogel sizes of 3190, 310, 150 nm, respectively (Fig. S13–S15†). As the crosslinker length increased, the nanogel size decreased. This could be due to the ability of the larger PEG to better encapsulate the glucagon attached to the trehalose polymer. Thus, PEG2000 dithiol was used as the crosslinker going forward, with adding the glucagon first to minimize aggregation.
With glucagon nanoparticles of 150 nm, the nanogel stability and uniformity to lyophilization was investigated. Lyophilization removes water through sublimation of ice under high vacuum, which has been known to offer a longer shelf-life and ease of transportation of many pharmaceutical products.52–54 DLS and TEM images show the uniformity of the nanogels after lyophilization, reconstituted in 50 mM DPBS pH 7.4, with sizes comparable to the non-lyophilized solution form (Fig. S16 and S17†). Although the lyophilization of the glucagon nanogel could offer additional stabilizing properties, for the scope of this paper we aimed to focus on the solution-based formulation. Formulating the nanogel to be stored in solution removes the reconstitution step for a patient or caregiver during an emergency hypoglycemic event. We see this as clinically advantageous. The optimized nanogel synthesis using a 50% glucagon–SH conjugation followed by crosslinking with 50% PEG2000 in HCl, buffer exchanged to DPBS pH 7.4 was moved forward for the short- and long-term stability experiments (Fig. 1). The glucagon nanogels were stored at −20 °C, 4 °C, 25 °C, and 37 °C to mimic cold chain transport, refrigeration, room temperature, and body temperature. The nanogel uniformity was assessed using DLS and TEM, while the glucagon stability was assessed using HPLC to monitor degradation and ThT to monitor fibrillation.
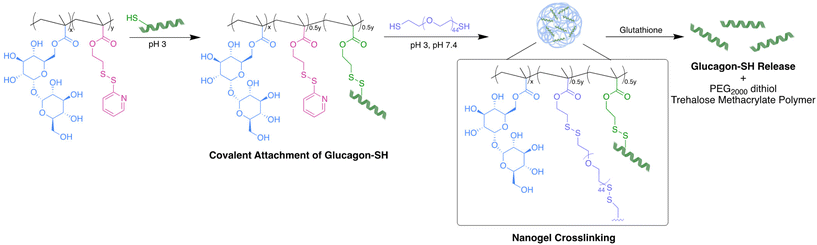 |
| | Fig. 1 Glucagon nanogel formation begins with the covalent attachment of glucagon–SH to poly(PDSMA-co-TrMA), followed by crosslinking with PEG2000 dithiol. In the presence of glutathione, glucagon–SH was released as well as PEG2000 dithiol and trehalose methacrylate polymer. | |
Characterization of glucagon nanogel size
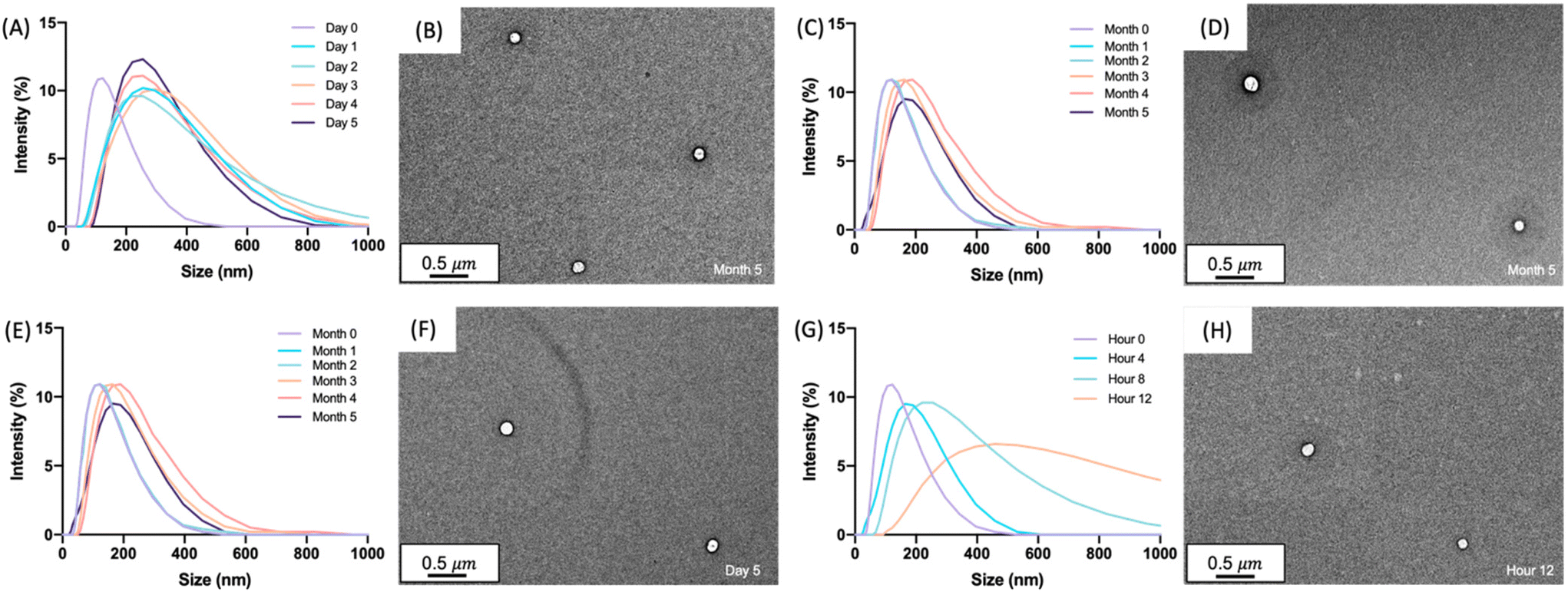
The size and uniformity of the glucagon nanogel was monitored over 5 months at −20 °C and 4 °C, 5 days at 25 °C, and 12 h at 37 °C using DLS and TEM (Fig. 2). At day 0, the glucagon nanogels Z-average (d nm) was 150 by DLS. The glucagon nanogels at −20 °C and 4 °C underwent a small shift in size after day 0 but did not exhibit significant changes in size and uniformity by DLS and TEM (Fig. 2A–D) over time and therefore were deemed stable in size up to 5 months, which was the longest time tested. We attempted to obtain TEM images with more particles in the frame by concentrating the nanogels, however, this caused the nanogels to aggregate. A wider frame of the nanogels at their final time point shows uniformity in size distribution of the nanogels at −20 °C and 4 °C (Fig. S18†). The glucagon nanogel stored at 25 °C exhibited an increased shift of approximately 100 nm after 1 day by DLS, however, after 5 days the glucagon nanogels remained of similar size by both DLS and TEM (Fig. 2E and F). After 12 h at 37 °C, the glucagon nanogels size began shifting dramatically with a large size distribution by DLS (Fig. 2G). However, by TEM, an increase in size was not observed. In a wider frame by TEM, some potential fibrils can be seen after 5 days at 25 °C and after 12 h at 37 °C (Fig. S18†). The discrepancy is likely due to DLS intensity putting an emphasis on larger particle sizes.55 It has been observed that nanoparticles increase in size at increased temperatures due to aggregation.56–58 However, trehalose polymers have been shown to prevent this aggregation.59 Therefore, the glucagon either internally or near the surface could be causing the aggregation observed at 37 °C. The glucagon nanogels at −20 °C and 4 °C can be studied past 5 months to evaluate additional time points; however, the glucagon nanogels formulation at 25 °C and 37 °C will likely need be reformulated before evaluating at longer time points. A larger PEG dithiol crosslinker could potentially be employed to create smaller nanoparticles and improve the stability at higher temperatures, glucagon loading could be lowered, or the trehalose polymer size or trehalose content could be increased. These studies are planned for the future.
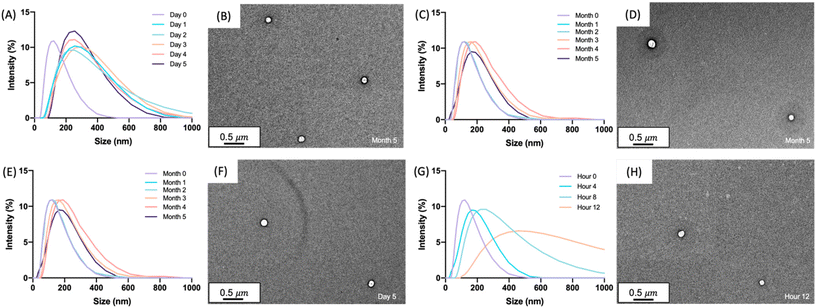 |
| | Fig. 2 Glucagon nanogels (1 mg mL−1) were analyzed by DLS after being stored at (A) −20 °C, (C) 4 °C, (E) 25 °C, and (G) 37 °C for the indicated times. TEM images were taken at the final time point after storage at (B) −20 °C for 5 months, (D) 4 °C for 5 months, (F) 25 °C for 5 days, and (H) 37 °C for 12 hours. DLS intensity was measured at 25 °C. TEM images were taken using a 2% uranyl acetate negative stain. | |
Degradation and fibrillation of glucagon
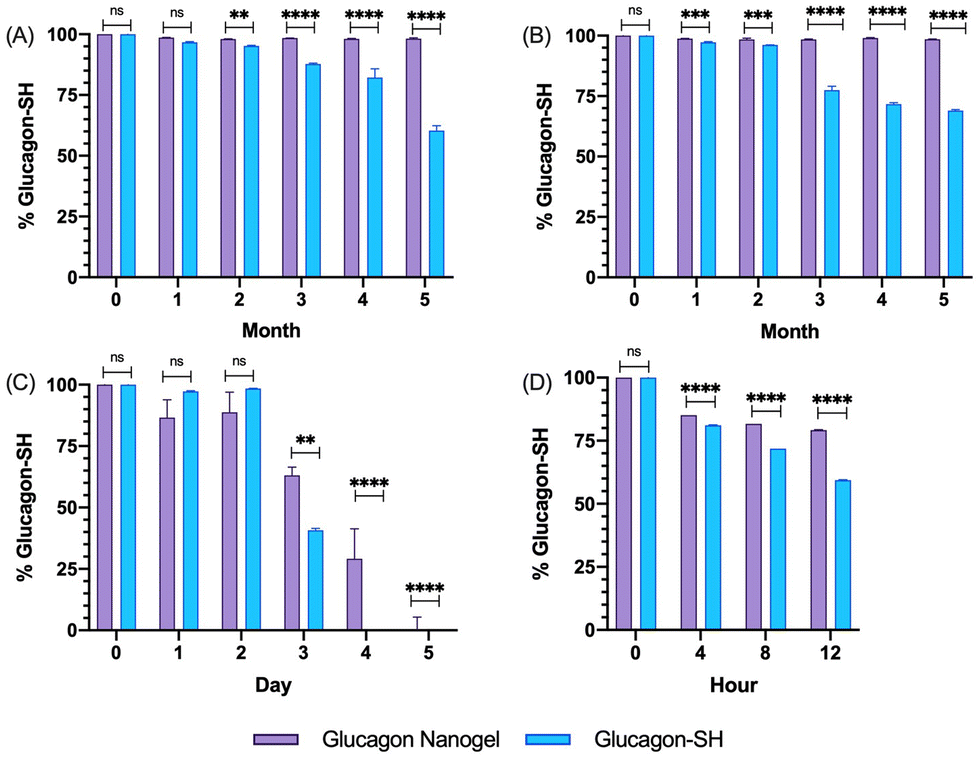
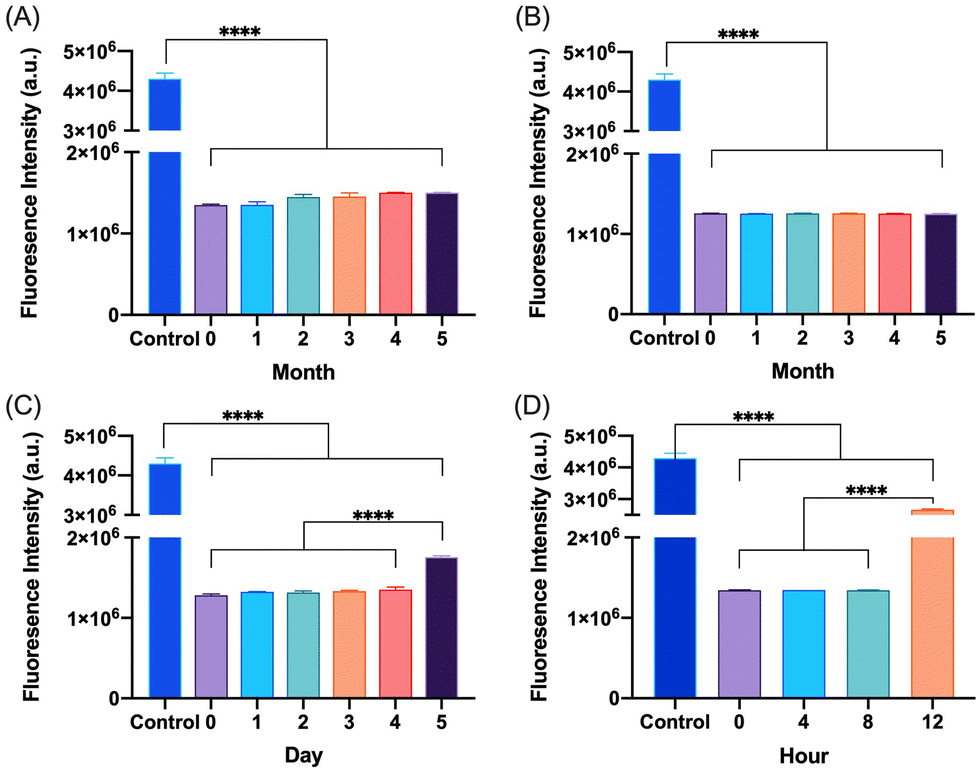
The degradation and fibrillation of the glucagon in the optimized glucagon nanogel formulation was also monitored over 5 months at −20 °C and 4 °C, 5 days at 25 °C, and 12 h at 37 °C using HPLC and ThT assay. A quantitative assessment of glucagon–SH degradation was made by integrating the AUC of the glucagon nanogel after reducing with TCEP, for simplicity, compared to free glucagon–SH at the same temperature and time point. After 2 months at −20 °C, the free glucagon began to degrade significantly more than glucagon in the nanogel (Fig. 3A). After only 1 month at 4 °C, the free glucagon began to degrade significantly more than glucagon nanogel (Fig. 3B). In contrast, the glucagon released from the glucagon nanogel at −20 °C and 4 °C remained above 95% up to 5 months. After 3 days at 25 °C, the free glucagon began to degrade significantly more than glucagon nanogel; however, the glucagon from the nanogel was 63% intact after 3 days (Fig. 3C). After 4 h at 37 °C, the free glucagon began to degrade significantly more than glucagon nanogel; however, the glucagon from the nanogel was also degrading after 4 h (Fig. 3D). Glucagon is known to form fibrils in acidic and alkaline pH solutions within hours at 25 °C.60 Glucagon fibrillation can be assessed qualitatively by measuring the fluorescence with ThT, a fluorescence dye that is known to bind to amyloid fibrils in vitro and can be used to monitor the formation of fibrils in a 96-well plate.8,60 Since the ThT fluorescence is dependent on both acidic and basic pH, inducing a significant decrease in ThT absorbance, a soluble solution of glucagon–SH at either acidic or basic pH was not suitable to be used as a positive control.61 Thus, bovine serum albumin (BSA) fibrils was used as a positive control through induced fibrillation by heating to 80 °C before incubating with ThT, since in this case neutral pH was utilized.46 Both the glucagon released as described above from the nanogels stored at −20 °C and 4 °C exhibited no statistically significant increase in fluorescence over the 5 months of monitoring, indicating no fibril formation (Fig. 4A and B). However, after 5 days at 25 °C, the glucagon nanogels exhibited a significant increase in fluorescence compared to the control and day 0, indicating fibril formation (Fig. 4C). Additionally, after 12 h at 37 °C, the glucagon nanogels exhibited a statistically significant increase in fluorescence compared to the control and day 0, indicating fibril formation (Fig. 4D). We hypothesize that glucagon started forming small molecular weight aggregates that were not detectable by the ThT assays at 3 days at 25 °C, and by day 5 detectable fibrils had begun to form. Likewise, after 4 h at 37 °C less glucagon was observed by HPLC and by 12 h fibrils were observed. It is also possible that some of the glucagon degrades and therefore it not detected at earlier time points. From these studies, it can be concluded that improved glucagon stability against degradation and fibrillation was observed at all temperatures studied. The glucagon nanogels at −20 °C and 4 °C exhibited no significant degradation or fibrillation up to 5 months and are promising for further studied to increase cold chain stability of glucagon.
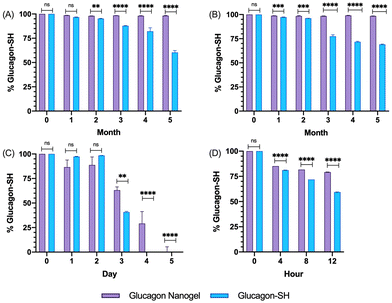 |
| | Fig. 3 Glucagon–SH degradation was assessed by integrating the AUC of the released glucagon–SH using HPLC for glucagon nanogels stored at (A) −20 °C, (B) 4 °C, (C) 25 °C, and (D) 37 °C at various time points. Normalization was done by comparing all samples against t0. Statistical significance was determined by comparing the glucagon–SH released from nanogel to the glucagon–SH control at the respective time and storage temperatures. Statistical significance was determined via a one-way ANOVA with multiple comparisons (p = 0.1 (ns), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****)) (n = 6). | |
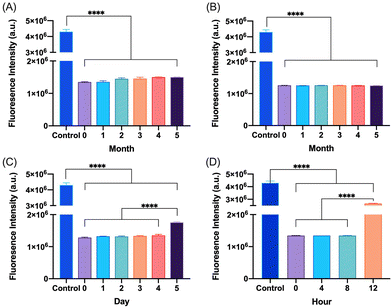 |
| | Fig. 4 Glucagon–SH fibrillation was assessed by measuring the ThT fluorescence of glucagon nanogels stored at (A) −20 °C, (B) 4 °C, (C) 25 °C, and (D) 37 °C at various time points. Statistical significance was determined by comparing each sample to t0 at the respective time and storage temperatures. Unless annotated, there is no statistical significance between t0 and other time points. Statistical significance was determined via a one-way ANOVA with multiple comparisons (****p ≤ 0.0001.) (n = 6). | |
Nanogel stability in cell media conditions
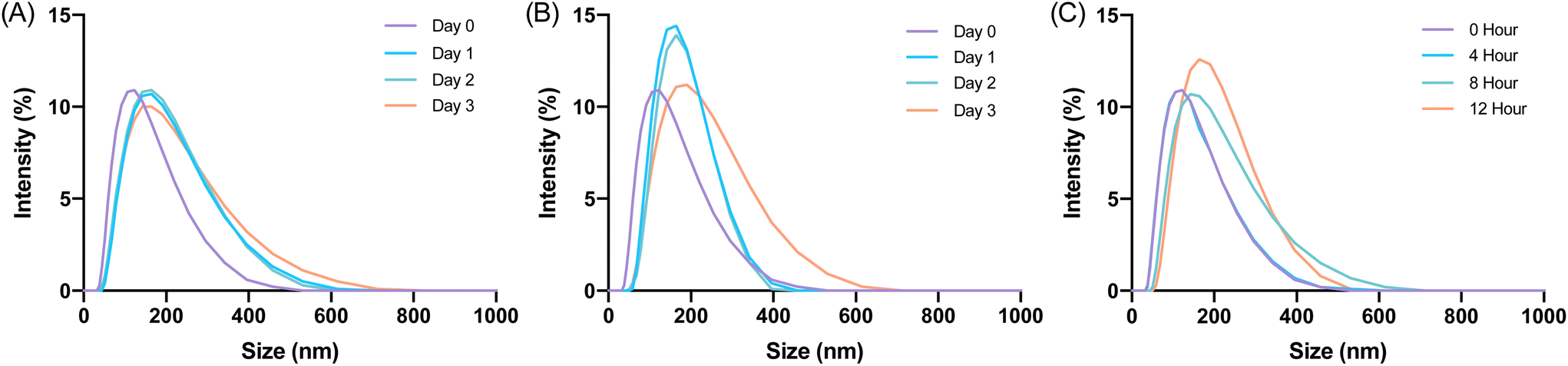
Glucagon nanogel stability was assessed in cell media conditions before beginning in vitro experiments.62 The glucagon nanogels were incubated with 10% FBS and stored at 4 °C, 25 °C, and 37 °C and analyzed by DLS. DLS measurements were taken up to 3 days for the glucagon nanogels stored at 4 °C and 25 °C and 12 h for the glucagon nanogel at 37 °C. The glucagon nanogels did not exhibit any significant change in size at any of the temperatures and time points (Fig. 5). Additionally, the glucagon nanogel remained uniform up until 12 h at 37 °C with 10% FBS, which differs from its DLS size measurement in (Fig. 5Cvs.Fig. 2G). It is possible the proteins and other components in FBS helped to stabilize the aggregation of the nanogels at 37 °C.63
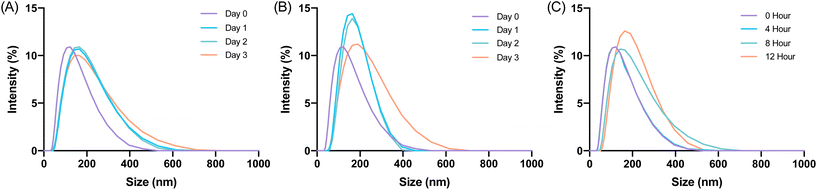 |
| | Fig. 5 Glucagon nanogels were treated with 10% FBS and visualized by DLS for the respective storage temperatures (A) 4 °C, (B) 25 °C, (C) 37 °C at various time points. DLS intensity was measured at 25 °C. | |
In vitro biocompatibility
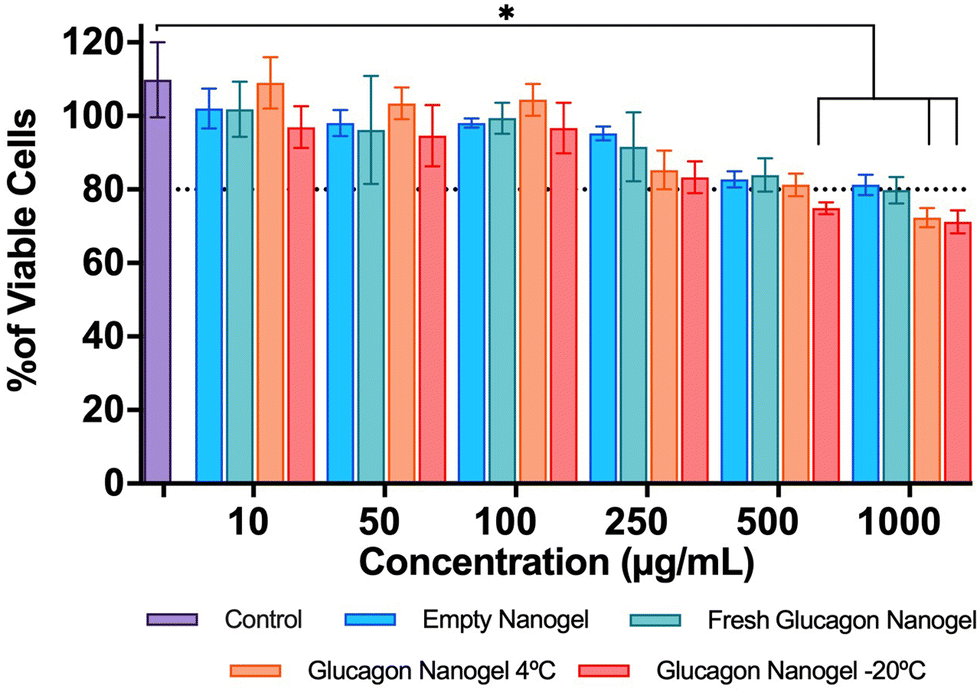
The in vitro biocompatibility of the empty nanogel and glucagon nanogels (freshly prepared, 4 °C for 3 months, and −20 °C for 3 months) were evaluated for 24 h in mouse embryonic fibroblasts, NIH 3T3, by performing the colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) assay. The in vitro biocompatibility was evaluated at 24 h at increasing concentrations of nanogel 10–1000 μg mL−1. The cell viability exposed to empty nanogel and glucagon nanogels were not statistically different from the control except for glucagon nanogels stored for 4 °C for 3 months at 1 mg mL−1, and −20 °C for 3 months at greater than 0.5 mg mL−1 after 24 h, indicating good in vitro biocompatibility of the material (Fig. 6).
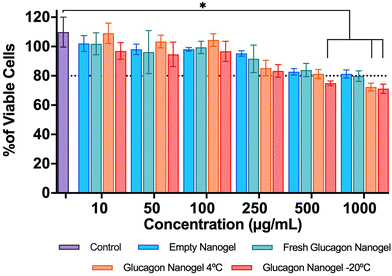 |
| | Fig. 6
In vitro biocompatibility was evaluated after 24 h in mouse embryonic fibroblasts, NIH3T3 by performing the colorimetric MTT assay. Data are represented as mean ± SEM (n = 3). Statistical significance was determined via a one-way ANOVA with multiple comparisons p < 0.05 (*). | |
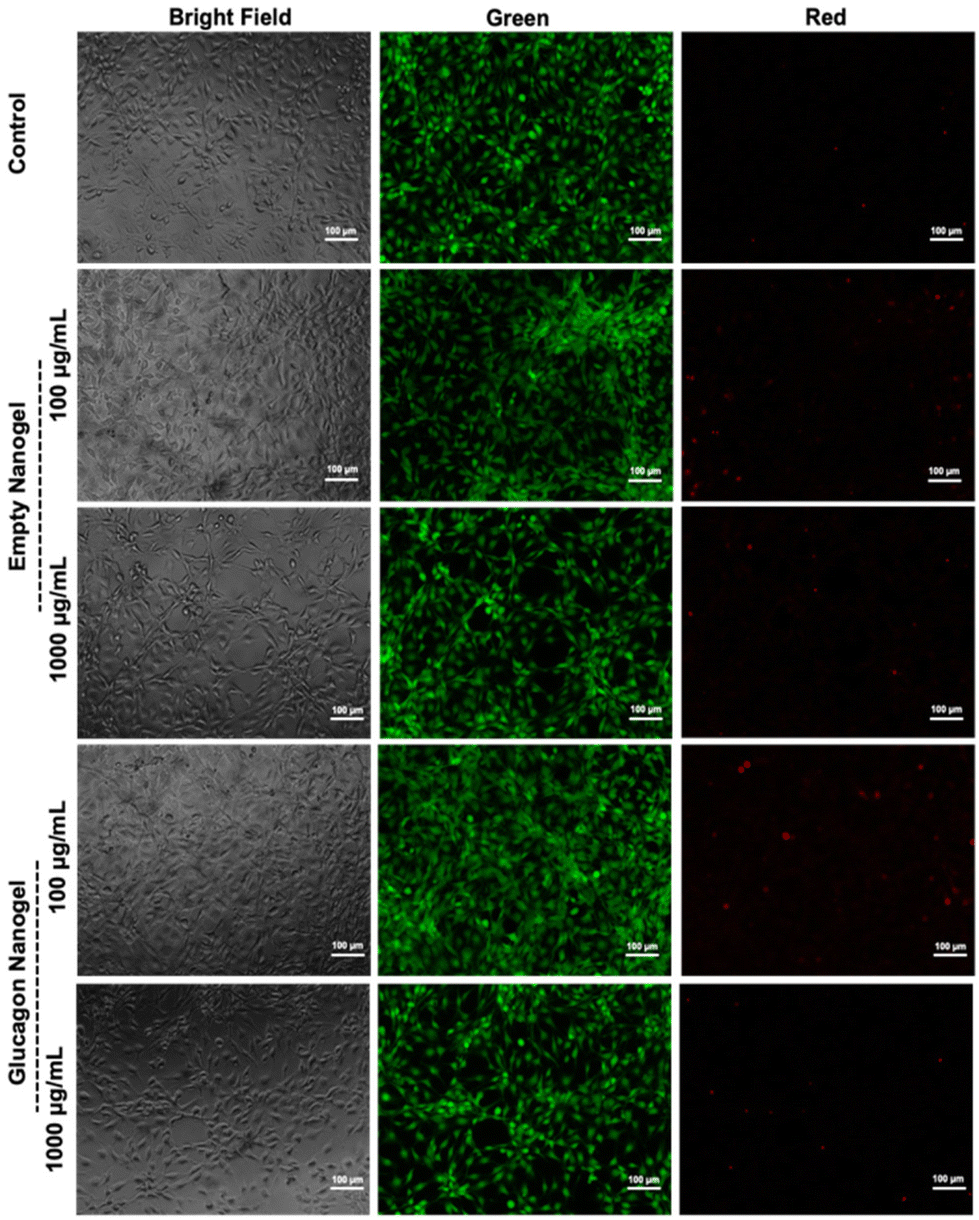
Further, a live/dead assay was used to evaluate the biocompatibility of empty nanogel and freshly prepared glucagon nanogel qualitatively for 24 h (Fig. 7). The calcein AM stains the live cells green and ethidium homodimer-1 stains the dead cells red. The green fluorescence and very minimal red fluorescence indicate the empty nanogel and glucagon nanogel are biocompatible at 10, 100, 500, and 1000 μg mL−1 (Fig. 7 and Fig. S19†).
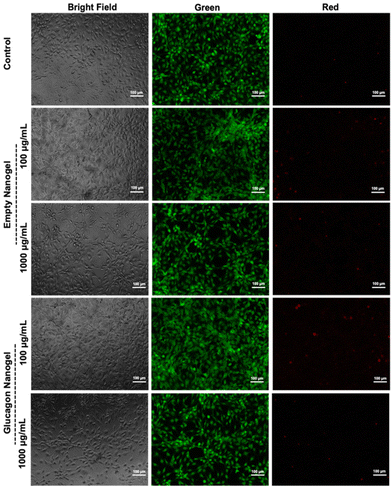 |
| | Fig. 7
In vitro biocompatibility was evaluated after 24 hours in mouse embryonic fibroblasts, NIH 3T3 by performing a live/dead assay using calcein AM and ethidium homodimer. The cells exposed to the empty nanogel and glucagon nanogel were imaged using a bright field and fluorescent microscope (scale bar: 100 μm). | |
In vitro efficacy
The in vitro efficacy of the glucagon nanogel was evaluated by comparing the levels of lactate produced when treated with empty nanogel, glucagon–SH and glucagon nanogel. Glucagon is known to promote the conversion of glycogen to glucose in the liver, producing lactate as a by-product of glycogenolysis (breakdown of the glucose).64 Herein, a liver cell model, HepG2 cells were starved with media containing low glucose, resembling hypoglycemic conditions.3 After starvation, media was replaced containing normal levels of glucose along with empty nanogel, glucagon–SH and glucagon nanogel. The levels of lactate was measured at different time points from 2–72 h using bioluminescence (Fig. 8). The results show that cells treated with glucagon–SH and glucagon nanogel produce significantly higher levels of lactate than the control and empty nanogel. Further, a color change of the cell culture media was observed (phenol red to golden yellow) in samples treated with native glucagon, glucagon–SH, and glucagon nanogel when compared to the control (Fig. S20†). The color change is due to the production of the lactate, an acidic bioproduct of glycogenolysis. These cell studies indicate the activity of the glucagon–SH both free and conjugated to the nanogel, confirming our hypothesis that the modified glucagon is active as native glucagon.
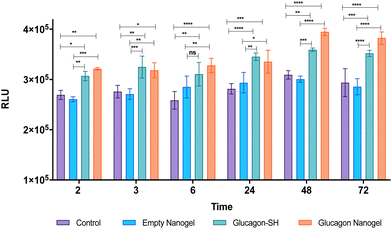 |
| | Fig. 8
In vitro efficacy evaluated in a liver cell line model, HepG2 by comparing the levels of lactate produced from 2–72 h by observing bioluminescence. Statistical significance was determined via a one-way ANOVA with multiple comparisons (p = 0.1 (ns), p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****)) (n = 3). | |
Hemocompatibility
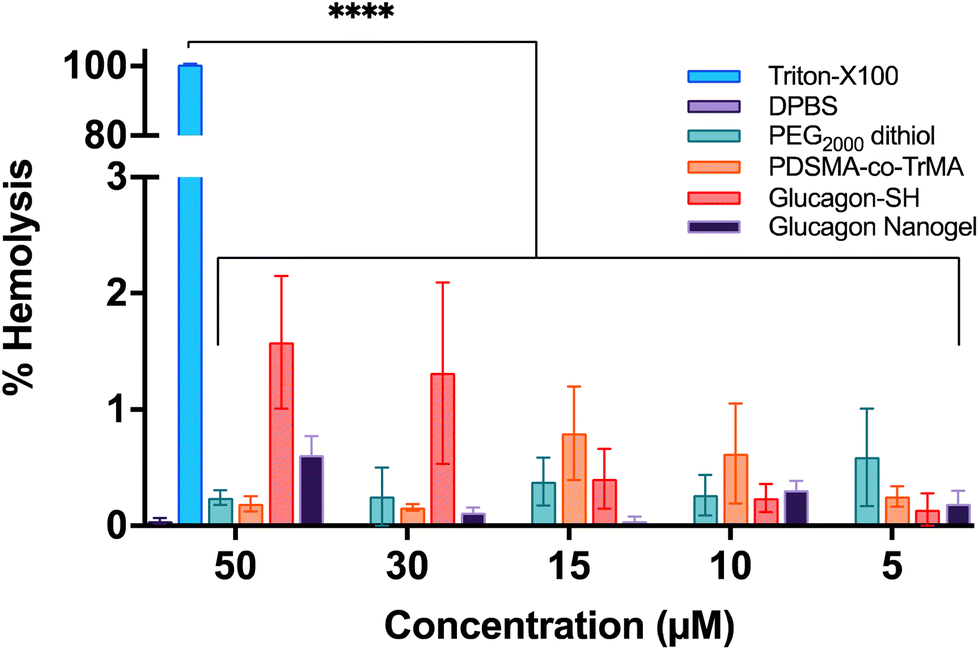
The toxicity of the glucagon nanogels and its components to cells were assessed by measuring the destruction of red blood cells (RBCs). The concentrations of 200–500 μg active ingredient (glucagon) tested were chosen to mimick future and past in vivo experiments.29 Using sheep RBCs, the glucagon nanogels, PEG2000 dithiol, poly(PDSMA-co-TrMA), and glucagon–SH all showed ≤3% hemolysis, indicating the samples are not disrupting the cells (Fig. 9). This is also understood qualitatively by observing a clear supernatant for the samples tested (Fig. S21†). The results show that the nanoparticles, loaded or empty, are non-hemolytic.
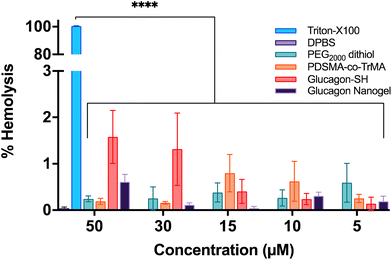 |
| | Fig. 9 Hemocompatibility of the glucagon nanogel and its components were assessed in sheep RBCs. Data are represented as mean ± SEM (n = 3). | |
Viscosity
For the emergency delivery of glucagon, a subcutaneous (SC) route is typically used.15,17 When considering SC injectable therapeutics, one factor to consider is the viscosity of the solution.65–67 The viscosity of formulations that can be SC injected within 10 seconds into humans without pain tolerance concerns is up to 15–20 centipoise (cp).67 At a concentration of 1 mg mL−1 active ingredient (glucagon), the viscosities of the empty and loaded nanogel were measured. At 25 °C with a shear rate of 5000 s−1, viscosity of the empty nanogel was 1.38 ± 0.19 cp and the glucagon nanogel was 1.31 ± 0.06 cp, well below 15–20 cp. These results suggest that viscosities of the nanogels are acceptable for subcutaneous injection of the nanogel formulation into humans.67
Conclusions
In conclusion, we prepared and evaluated a uniform nanogel formulation that solubilized glucagon in aqueous solution at pH 7.4 for long term stabilization at −20 °C and 4 °C. The system was optimized by varying the crosslinker size, crosslinking percentage, and order of addition of the components to yield uniform glucagon loaded nanogels of ∼150 nm in size. The glucagon nanogels were shown to be biocompatible and efficacious in vitro by comprehensive study of hemolysis, colorimetric MTT assay, live/dead assay, and lactate assay. Additionally, this formulation exhibited viscosity suitable for subcutaneous delivery in humans, meaning that the viscosity was much lower than would cause pain tolerance concerns.
Author contributions
E. G. P. conducted the synthesis, conjugation, experimentation, characterization, and hemocompatibility. R. P. S. conducted the in vitro biocompatibility and activity experiments. H. C. L. conducted the viscosity experiments. D. V., J. Y., and A. L. H. provided suggestions and intellectual input for the project. H. D. M. conceived of, over saw the project and suggested intellectual input. E. G. P. wrote and all authors edited the manuscript.
Conflicts of interest
UCLA has filed a US Patent application on trehalose nanogels.
Acknowledgements
This work was funded by National Institutes of Health (NIH R01 DK127908). The authors thank Prof. E Sletten for use of her dynamic light scattering microscope and the UCLA CNSI for use of their transmission electron microscope and dynamic light scattering microscope.
References
- CDC, Low Blood Sugar (Hypoglycemia), https://www.cdc.gov/diabetes/basics/low-blood-sugar.html, (accessed 6 March 2023).
- CDC, How to Treat Low Blood Sugar (Hypoglycemia), https://www.cdc.gov/diabetes/basics/low-blood-sugar-treatment.html, (accessed 6 March 2023).
- J. Morales and D. Schneider, Am. J. Med., 2014, 127, S17–S24 CrossRef CAS PubMed.
- Glucagon & Other Emergency Glucose Products|ADA, https://diabetes.org/healthy-living/medication-treatments/glucagon-other-emergency-glucose-products, (accessed 6 March 2023).
- G. Mattedi, S. Acosta-Gutiérrez, T. Clark and F. L. Gervasio, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 15414–15422 CrossRef CAS PubMed.
- L. Jelinek, S. Lok, G. Rosenberg, R. Smith, F. Grant, S. Biggs, P. Bensch, J. Kuijper, P. Sheppard and C. Sprecher,
et al.
, Science, 1993, 259, 1614–1616 CrossRef CAS PubMed.
- T. D. Müller, B. Finan, C. Clemmensen, R. D. DiMarchi and M. H. Tschöp, Physiol. Rev., 2017, 97, 721–766 CrossRef PubMed.
- S. Onoue, K. Ohshima, K. Debari, K. Koh, S. Shioda, S. Iwasa, K. Kashimoto and T. Yajima, Pharm. Res., 2004, 21, 1274–1283 CrossRef CAS PubMed.
- N. Caputo, J. R. Castle, C. P. Bergstrom, J. M. Carroll, P. A. Bakhtiani, M. A. Jackson, C. T. Roberts, L. L. David and W. K. Ward, Peptides, 2013, 45, 40–47 CrossRef CAS PubMed.
- N. Caputo, M. A. Jackson, J. R. Castle, J. El Youssef, P. A. Bakhtiani, C. P. Bergstrom, J. M. Carroll, M. E. Breen, G. L. Leonard, L. L. David, C. T. Roberts and W. K. Ward, Diabetes Technol. Ther., 2014, 16, 747–758 CrossRef CAS PubMed.
- BAQSIMI® (glucagon) nasal powder | Severe Hypoglycemia Treatment, https://www.baqsimi.com/, (accessed 7 March 2023).
- D. Singh-Franco, C. Moreau, A. D. Levin, D. D. L. Rosa and M. Johnson, Clin. Ther., 2020, 42, e177–e208 CrossRef CAS PubMed.
- J. G. Suico, U. Hövelmann, S. Zhang, T. Shen, B. Bergman, J. Sherr, E. Zijlstra, B. M. Frier and L. Plum-Mörschel, Diabetes Ther., 2020, 11, 1591–1603 CrossRef CAS PubMed.
- M. P. Christiansen, M. Cummins, S. Prestrelski, N. C. Close, A. Nguyen and K. Junaidi, BMJ Open Diabetes Res. Care, 2021, 9, e002137 CrossRef PubMed.
- Gvoke™ (glucagon injection) | Patient Information, https://gvokeglucagon.com, (accessed 7 March 2023).
- Dasiglucagon for BHAP systems, https://www.zealandpharma.com/dasiglucagon-pump, (accessed 7 March 2023).
- N. C. Bailey, J. Dimsits, M. Hammer, D. M. Kendall and T. S. Bailey, Diabetes Technol. Ther., 2022, 24, 231–240 CrossRef CAS PubMed.
- T. Battelino, R. Tehranchi, T. Bailey, K. Dovc, A. Melgaard, J. Y. Stone, S. Woerner, T. von dem Berg, L. A. DiMeglio, D. Kendall and T. Danne, Metab., Clin. Exp., 2021, 116, 154507 CrossRef.
- P. Stigsnaes, S. Frokjaer, S. Bjerregaard, M. van de Weert, P. Kingshott and E. H. Moeller, Int. J. Pharm., 2007, 330, 89–98 CrossRef CAS PubMed.
- L. Matilainen, K. L. Larsen, R. Wimmer, P. Keski-Rahkonen, S. Auriola, T. Järvinen and P. Jarho, J. Pharm. Sci., 2008, 97, 2720–2729 CrossRef CAS PubMed.
- D. J. A. Crommelin, T. J. Anchordoquy, D. B. Volkin, W. Jiskoot and E. Mastrobattista, J. Pharm. Sci., 2021, 110, 997–1001 CrossRef CAS PubMed.
- J. R. Chabenne, M. A. DiMarchi, V. M. Gelfanov and R. D. DiMarchi, J. Diabetes Sci. Technol., 2010, 4, 1322–1331 CrossRef PubMed.
- A. B. Joshi, E. Rus and L. E. Kirsch, Int. J. Pharm., 2000, 203, 115–125 CrossRef CAS PubMed.
- T. Ojha, Q. Hu, C. Colombo, J. Wit, M. van Geijn, M. J. van Steenbergen, M. Bagheri, H. Königs-Werner, E. M. Buhl, R. Bansal, Y. Shi, W. E. Hennink, G. Storm, C. J. F. Rijcken and T. Lammers, Biotechnol. J., 2021, 16, 2000212 CrossRef CAS PubMed.
- M. B. Gelb, K. M. M. Messina, D. Vinciguerra, J. H. Ko, J. Collins, M. Tamboline, S. Xu, F. J. Ibarrondo and H. D. Maynard, ACS Appl. Mater. Interfaces, 2022, 14, 37410–37423 CrossRef CAS PubMed.
- Y. Liu, J. Lee, K. M. Mansfield, J. H. Ko, S. Sallam, C. Wesdemiotis and H. D. Maynard, Bioconjugate Chem., 2017, 28, 836–845 CrossRef CAS PubMed.
- Z. P. Tolstyka, H. Phillips, M. Cortez, Y. Wu, N. Ingle, J. B. Bell, P. B. Hackett and T. M. Reineke, ACS Biomater. Sci. Eng., 2016, 2, 43–55 CrossRef CAS PubMed.
- K. L. Jones, D. Drane and E. J. Gowans, BioTechniques, 2007, 43, 675–681 CrossRef CAS PubMed.
- N. Boehnke, J. K. Kammeyer, R. Damoiseaux and H. D. Maynard, Adv. Funct. Mater., 2018, 28, 1705475 CrossRef.
- C. J. Crilly, J. A. Brom, M. E. Kowalewski, S. Piszkiewicz and G. J. Pielak, Biochemistry, 2021, 60, 152–159 CrossRef CAS PubMed.
- C. Colaço, S. Sen, M. Thangavelu, S. Pinder and B. Roser, Nat. Biotechnol., 1992, 10, 1007–1011 CrossRef PubMed.
- C. Olsson, H. Jansson and J. Swenson, J. Phys. Chem. B, 2016, 120, 4723–4731 CrossRef CAS PubMed.
- L. (Lucy) Chang and M. J. Pikal, J. Pharm. Sci., 2009, 98, 2886–2908 CrossRef PubMed.
- M. A. Mensink, H. W. Frijlink, K. Van Der Voort Maarschalk and W. L. J. Hinrichs, Eur. J. Pharm. Biopharm., 2017, 114, 288–295 CrossRef CAS PubMed.
- A. Lerbret, F. Affouard, A. Hédoux, S. Krenzlin, J. Siepmann, M.-C. Bellissent-Funel and M. Descamps, J. Phys. Chem. B, 2012, 116, 11103–11116 CrossRef CAS PubMed.
- C. Olsson and J. Swenson, J. Phys. Chem. B, 2020, 124, 3074–3082 CrossRef CAS PubMed.
- R. J. Mancini, J. Lee and H. D. Maynard, J. Am. Chem. Soc., 2012, 134, 8474–8479 CrossRef CAS PubMed.
- E. M. Pelegri-O'Day, S. J. Paluck and H. D. Maynard, J. Am. Chem. Soc., 2017, 139, 1145–1154 CrossRef PubMed.
- N. Pradhan, S. Shekhar, N. R. Jana and N. R. Jana, ACS Appl. Mater. Interfaces, 2017, 9, 10554–10566 CrossRef CAS PubMed.
- D. Diaz-Dussan, Y.-Y. Peng, J. Sengupta, R. Zabludowski, M. K. Adam, J. P. Acker, R. N. Ben, P. Kumar and R. Narain, Biomacromolecules, 2020, 21, 1264–1273 CrossRef CAS PubMed.
- K. Debnath, A. K. Sarkar, N. R. Jana and N. R. Jana, Acc. Mater. Res., 2022, 3, 54–66 CrossRef CAS.
- P. H. Panescu, J. H. Ko and H. D. Maynard, Macromol. Mater. Eng., 2019, 304, 1800782 CrossRef.
- Nanoparticle Uniformity, https://www.news-medical.net/life-sciences/Nanoparticle-Uniformity.aspx, (accessed 7 March 2023).
- J. Chabenne, M. D. Chabenne, Y. Zhao, J. Levy, D. Smiley, V. Gelfanov and R. DiMarchi, Mol. Metab., 2014, 3, 293–300 CrossRef CAS PubMed.
- H. Zhang, A. Qiao, L. Yang, N. Van Eps, K. S. Frederiksen, D. Yang, A. Dai, X. Cai, H. Zhang, C. Yi, C. Cao, L. He, H. Yang, J. Lau, O. P. Ernst, M. A. Hanson, R. C. Stevens, M.-W. Wang, S. Reedtz-Runge, H. Jiang, Q. Zhao and B. Wu, Nature, 2018, 553, 106–110 CrossRef CAS PubMed.
- S.-C. How, T.-H. Lin, C.-C. Chang and S. S.-S. Wang, Int. J. Biol. Macromol., 2021, 184, 79–91 CrossRef CAS PubMed.
- H. Levine III, Protein Sci., 1993, 2, 404–410 CrossRef PubMed.
- B. C. Evans, C. E. Nelson, S. S. Yu, K. R. Beavers, A. J. Kim, H. Li, H. M. Nelson, T. D. Giorgio and C. L. Duvall, J. Visualized Exp., 2013, 50166 Search PubMed.
- S. A. Kulkarni and S.-S. Feng, Pharm. Res., 2013, 30, 2512–2522 CrossRef CAS PubMed.
- B. L. Banik, P. Fattahi and J. L. Brown, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 271–299 Search PubMed.
- W. Poon, Y.-N. Zhang, B. Ouyang, B. R. Kingston, J. L. Y. Wu, S. Wilhelm and W. C. W. Chan, ACS Nano, 2019, 13, 5785–5798 CrossRef CAS PubMed.
- T. Ojha, V. Pathak, N. Drude, M. Weiler, D. Rommel, S. Rütten, B. Geinitz, M. J. van Steenbergen, G. Storm, F. Kiessling and T. Lammers, Pharmaceutics, 2019, 11, 433 CrossRef CAS PubMed.
- W. Abdelwahed, G. Degobert, S. Stainmesse and H. Fessi, Adv. Drug Delivery Rev., 2006, 58, 1688–1713 CrossRef CAS PubMed.
- H. Talsma, J. Cherng, H. Lehrmann, M. Kursa, M. Ogris, W. E. Hennink, M. Cotten and E. Wagner, Int. J. Pharm., 1997, 157, 233–238 CrossRef CAS PubMed.
- T. G. F. Souza, V. S. T. Ciminelli and N. D. S. Mohallem, J. Phys.: Conf. Ser., 2016, 733, 012039 CrossRef.
- N. K. Kwon, T. K. Lee, S. K. Kwak and S. Y. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 39688–39698 CrossRef CAS PubMed.
- W. Yu, R. Liu, Y. Zhou and H. Gao, ACS Cent. Sci., 2020, 6, 100–116 CrossRef CAS PubMed.
- L. Pang, Y. Pei, G. Uzunalli, H. Hyun, L. T. Lyle and Y. Yeo, Pharm. Res., 2019, 36, 65 CrossRef PubMed.
- S. Srinivasachari, Y. Liu, G. Zhang, L. Prevette and T. M. Reineke, J. Am. Chem. Soc., 2006, 128, 8176–8184 CrossRef CAS PubMed.
- M. D. Gelenter, K. J. Smith, S.-Y. Liao, V. S. Mandala, A. J. Dregni, M. S. Lamm, Y. Tian, W. Xu, D. J. Pochan, T. J. Tucker, Y. Su and M. Hong, Nat. Struct. Mol. Biol., 2019, 26, 592–598 CrossRef CAS PubMed.
- E. V. Hackl, J. Darkwah, G. Smith and I. Ermolina, Eur. Biophys. J., 2015, 44, 249–261 CrossRef CAS PubMed.
- A. E. Ekkelenkamp, M. M. T. Jansman, K. Roelofs, J. F. J. Engbersen and J. M. J. Paulusse, Acta Biomater., 2016, 30, 126–134 CrossRef CAS PubMed.
- T. L. Moore, L. Rodriguez-Lorenzo, V. Hirsch, S. Balog, D. Urban, C. Jud, B. Rothen-Rutishauser, M. Lattuada and A. Petri-Fink, Chem. Soc. Rev., 2015, 44, 6287–6305 RSC.
- G. Mattedi, S. Acosta-Gutiérrez, T. Clark and F. L. Gervasio, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 15414–15422 CrossRef CAS PubMed.
- S. J. Shire, Curr. Opin. Biotechnol., 2009, 20, 708–714 CrossRef CAS PubMed.
- S. J. Shire, Z. Shahrokh and J. Liu, J. Pharm. Sci., 2004, 93, 1390–1402 CrossRef CAS PubMed.
- C. Berteau, O. Filipe-Santos, T. Wang, H. E. Rojas, C. Granger and F. Schwarzenbach, Med. Devices, 2015, 8, 473–484 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Rajalakshmi P.
Sivasankaran
ab,
Daniele
Vinciguerra
ab,
Rajalakshmi P.
Sivasankaran
ab,
Daniele
Vinciguerra