
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChitosan–saccharide conjugates for eradication of Pseudomonas aeruginosa biofilms†
Priyanka
Sahariah
 *a,
Francesco
Papi
b,
Koi L.
Merz
b,
Olafur E.
Sigurjonsson
c,
Rikke Loiuse
Meyer
de and
Cristina
Nativi
b
*a,
Francesco
Papi
b,
Koi L.
Merz
b,
Olafur E.
Sigurjonsson
c,
Rikke Loiuse
Meyer
de and
Cristina
Nativi
b
aDepartment of Life and Environmental Sciences, Biomedical Centre, University of Iceland, 101 Reykjavik, Iceland. E-mail: prs@chemrsc.org
bDepartment of Chemistry, University of Florence, Sesto F.no, Florence, Italy
cThe REModeL Lab, The Blood Bank, Landspitali University Hospital, Snorrabraut 60, 105 Reykjavik, Iceland
diNANO, Aarhus University, Gustav Weids Vej 14, 8000 Aarhus C, Denmark
eDepartment of Bioscience, Aarhus University, Ny Munkegade 114, 8000 Aarhus C, Denmark
First published on 27th February 2024
Abstract
The problem of antibiotic resistance has raised serious concerns globally and hence the development of new materials which can combat these drug-resistant strains has gained a great deal of attention. Herein, we report the use of a biocompatible material, chitosan, as a scaffold to graft saccharides which can specifically target Pseudomonas aeruginosa. We realized this by synthesizing N-functionalized chitosan conjugates by coupling chitosan to fucose and galactose moieties which intercept Pseudomonas aeruginosa lectins and target the bacterial biofilms. A series of six conjugates containing similar proportions of cationic and sugar moieties were synthesized by direct modification of the chitosan backbone using a method that is highly efficient and reproducible. The conjugates showed a bactericidal effect against both Gram positive and Gram negative bacterial strains. An investigation into the antibiofilm activity of the conjugates revealed the optimum combination of the type and positioning of the functionalities that were highly effective in eradicating Pseudomonas aeruginosa biofilms. 2D and 3D imaging of the conjugate-treated biofilms using confocal laser scanning microscopy (CLSM) allowed us to determine that the conjugates not only acted on the surface but also dispersed into deep layers of the biofilm. Interaction between the conjugates and individual bacterial cells in the biofilm was further confirmed by fluorescence labelling of the conjugates and imaging by CLSM.
Introduction
Antimicrobial resistance has emerged as a global concern with the currently available antibiotics becoming ineffective in controlling infections. Among the WHO priority pathogens, Pseudomonas aeruginosa (P. aeruginosa) has been identified as one of the most life-threating bacteria and has been prioritized for research and development of new antibiotics.1,2P. aeruginosa, a Gram-negative bacterium, causes severe infections in immunocompromised hosts leading to clinically persistent infections such as cystic fibrosis, urinary tract infections, and burn infections.3 The antibiotic resistance in P. aeruginosa is attributed to multiple mechanisms and a variety of virulence factors, of which its ability to form biofilms is an important mechanism by which it can also resist host defenses.4 Biofilms of P. aeruginosa are formed due to the secretion of extracellular polymeric substances which adhere to the bacterial cells forming a matrix. The growing microbial cells embedded in this matrix become less susceptible to antibiotics or host defence systems than the cells in the planktonic form.5 Moreover, biofilm formation is an adaptive mechanism, so the microbial cells can regain antibiotic susceptibility once they lose biofilm protection.6 Hence, developing molecules that can penetrate the biofilm and interact with the microbial cells embedded in the matrix is crutial for displaying activity against P. aeruginosa.Biofilm formation in P. aeruginosa infections is mediated by galactose-specific lectin A (LecA, PA-IL) and fucose-specific lectin B (LecB, PA-IIL). Indeed, the effect of simple derivatives of galactose and fucose on inhibiting the bacterium from producing a biofilm was demonstrated by in vitro and in vivo studies. α-C-Fucosyl and β-O-aryl galactosyl derivatives grafted onto peptide dendrimers showed remarkably increased affinity for LecB and LecA, respectively, up to 100 times and up to 4000 times greater than that of simple carbohydrates, maintaining biofilm inhibition properties.7 Recent studies have also shown the efficiency of other glycopolymers in binding LecA and LecB and inhibiting or disrupting P. aeruginosa biofilm formation.8
Chitosan is a biocompatible linear polymer containing β(1 → 4) linked residues of N-acetyl-2 amino-2-deoxy-D-glucose (glucosamine, GlcN) and 2-amino-2-deoxy-D-glucose (N-acetyl-glucosamine, GlcNAc) residues. Chitosan is endowed with many biological and technological properties such as mucoadhesive, anti-inflammatory, antioxidant, antimicrobial, antifungal, antitumor and wound healing properties.9 In recent years, chitosan has attracted huge attention due to its antimicrobial properties as evidenced by the significant number of publications. An emerging area is the chemical manipulation of the chitosan backbone to modify the free –OH or –NH2 group or to link various biomolecules, all with the intention of enhancing its antimicrobial properties.10 Synthesis of various chitosan conjugates such as chitosan–peptide, chitosan–porphyrin, chitosan–PEG, polyphenol–chitosan, and chitosan–cinnamic acid derivatives has been reported for enhancing the aqueous solubility and antimicrobial activity of the native polymer.11,12 A series of chitosan–antimicrobial peptide (chitosan–AMP) conjugates not only displayed enhanced antimicrobial activity towards Gram-positive and Gram-negative bacteria, but significantly reduced the overall toxicity of the AMPs.13 Chitosan peptide conjugates have also gained interest in the design of antimicrobial coatings, such as the ε-polylysine–chitosan conjugate against Staphylococcus epidermidis, Escherichia coli (E. coli), Staphylococcus aureus (S. aureus), Bacillus subtilis and beer yeast14,15 and peptidoglycan mimetic-chitosan against E. coli.16 Recently, studies have shown the efficiency of chitosan-based conjugates against bacterial biofilms. Conjugation of aminoglycoside antibiotics such as streptomycin to chitosan, damaged established biofilms, and inhibited biofilm formation in Gram positive bacteria Listeria monocytogenes,17 while DNase I supplemented with chitosan (CS) linked with CAZ (CS/CAZ) significantly eradicated Burkholderia pseudomallei biofilm cells.18
Hence, the aim of this study was to rely on the ability of aryl-O-galactoside and C-fucosides to intercept bacterial LecA and LecB and form sugar–chitosan conjugates to treat and disrupt established biofilms of P. aeruginosa. With this aim, we prepared glycosides S1 and S2 which present a five-carbon linker with a terminal carboxylic group (see Scheme 1 and the ESI†). By introducing a specific ratio of the two glycosides on natural chitosan, we aimed to synthesize a series of conjugates and compare their ability to interact with P. aeruginosa biofilms. Moreover, to observe the synergistic effect of antimicrobial cationic functionalities and the two lectins’ ligands, we also introduced S1 and S2 moieties on two cationic chitosan derivatives.
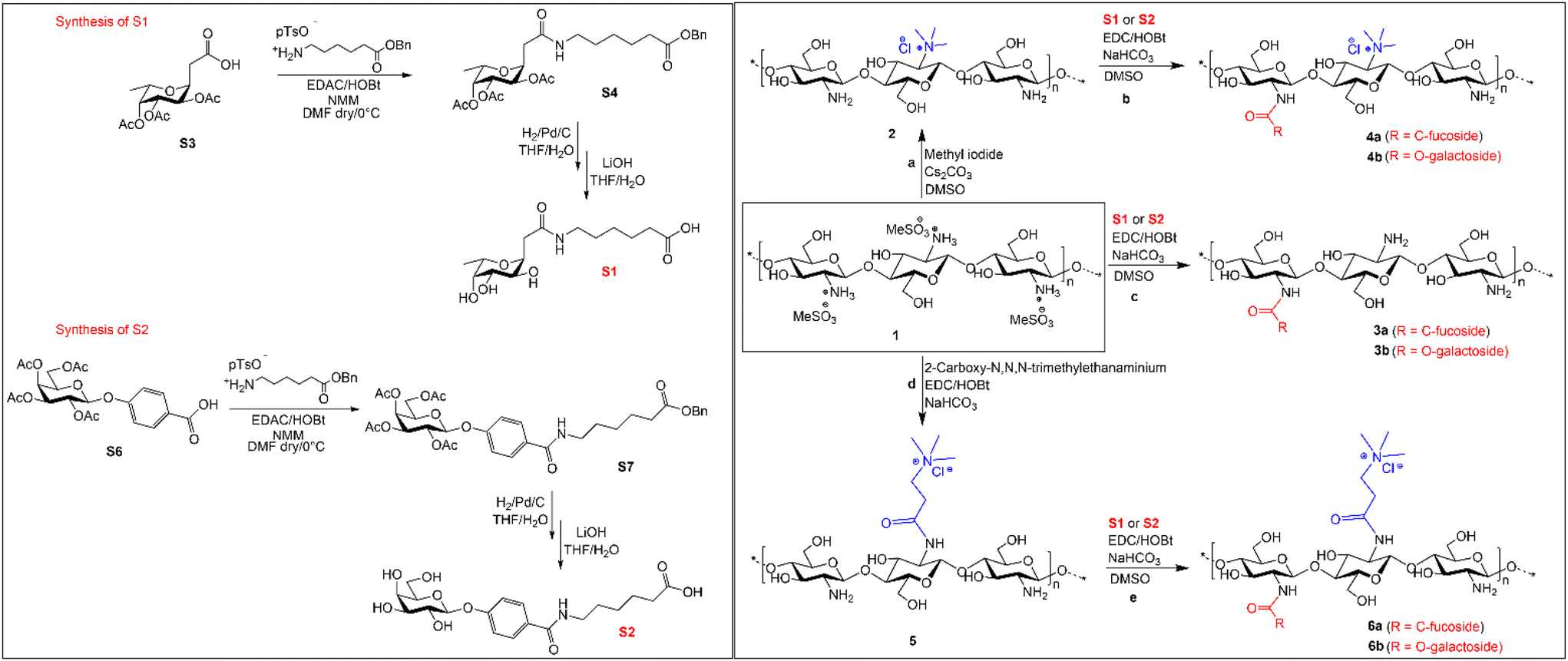 | ||
| Scheme 1 Synthetic scheme for the synthesis of C-fucoside S1, O-galactoside S2 and sugar–chitosan conjugates 6a and 6b from the chitosan mesylate salt. (Details of S1 and S2 synthesis are provided in the ESI.†) | ||
The cationic modifications on chitosan consist in the introduction of the trimethylammoniumyl group either directly at the free amino group or using spacers. We introduced the cationic groups and the sugar moiety each at ∼25% to have at least 50% of the free amino groups on the chitosan backbone. By utilizing the mesylate salt of chitosan, we developed a synthetic method for direct modification of the free amino group in chitosan as well as for linking the glycosyl moieties through amide bond formation (see Scheme 1). Avoiding the use of protective groups, we reduced the complexity of the synthesis and produced a series of six sugar–chitosan conjugates with excellent reproducibility. Antibacterial assessment of the conjugates against both Gram-positive and Gram-negative bacteria allowed us to determine the spectrum of their antibacterial activity. We then utilized the conjugates specifically to interact and eradicate preformed biofilms of P. aeruginosa. Morphological changes in P. aeruginosa biofilms were confirmed visually by confocal laser scanning microscopy (CLSM) imaging of the polymer-treated biofilms. Furthermore, we confirmed that the conjugates interact with P. aeruginosa cells by visualizing the effect of the conjugates on P. aeruginosa cells by fluorescence staining and CLSM.
Experimental
Materials and methods
Chitoceutical Chitosan 95/10 (24701) was purchased from Heppe Medical Chitosan GmbH, Halle, Germany. The degree of acetylation of chitosan was calculated to be 5% by 1H-NMR spectroscopy. The average molecular weight (Mw) and dispersity (D) of the chitosan were determined to be 27 kDa and 2.1, respectively, by gel permeation chromatography. Gram positive bacteria S. aureus (DSM 20231) and Gram-negative bacteria E. coli (ATCC 25922) and P. aeruginosa (PA01 DSM19880) were obtained from DSMZ – German collection of Microorganisms and Cell cultures Gmbh. All other chemicals were purchased from Merck. Silica gel flash column chromatography purifications were performed using Geduran® Si 60 (0.040–0.063 mm). TLC analyses were performed on glass Merck silica gel 60 F254 plates. 1H NMR, 13C NMR and 2D-NMR spectra were recorded on a 500 MHz Bruker AVANCE II at 298 K.Synthetic procedures
Yield: 2.31 g (97.1%). 1H NMR (500 MHz, D2O): δ 2.84 (s, CH3), 3.21 (s, H-2), 3.78–3.96 (m, H-3, H-4, H-5, H-6), 4.90 (H-1, partially overlapped by the water peak) ppm.
Note: the numbering of the protons is according to Fig. 1.
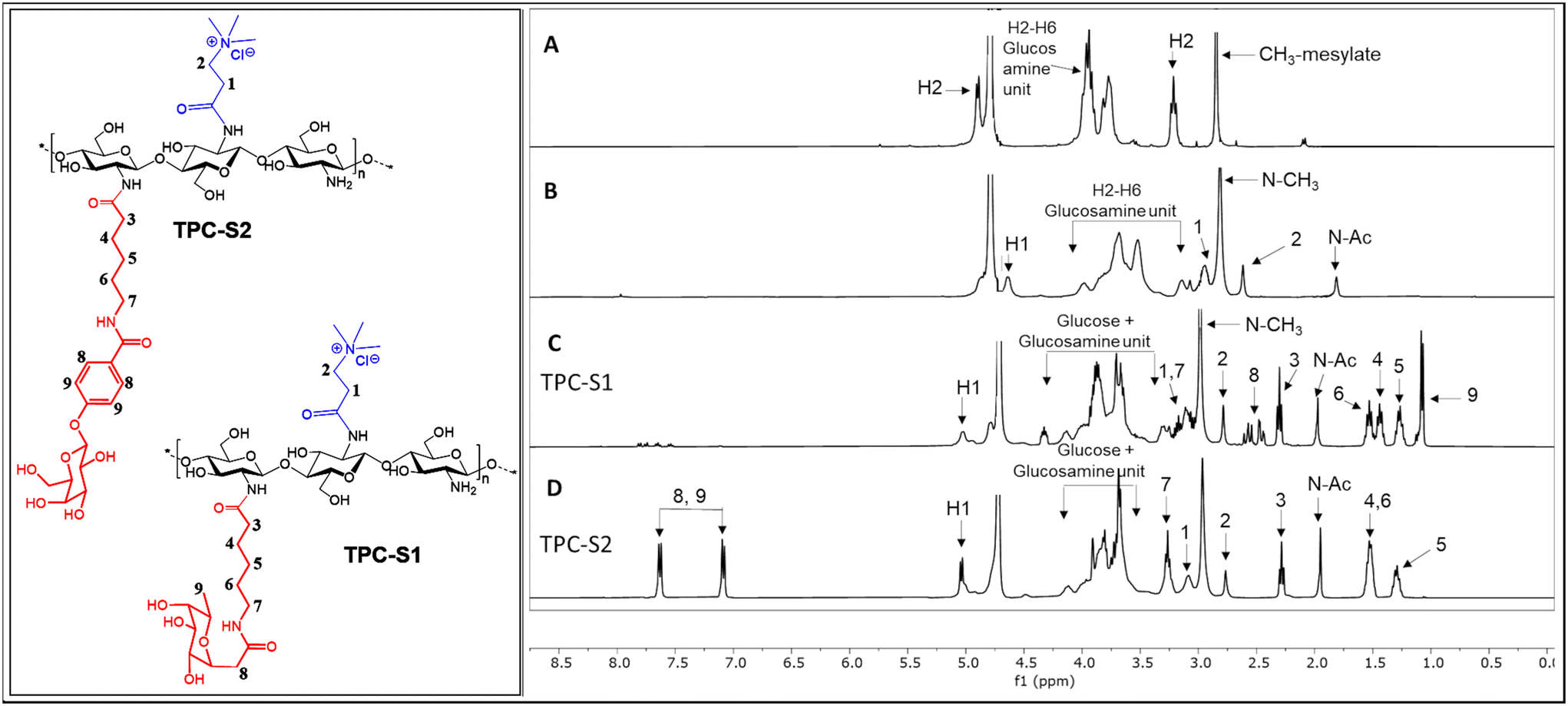 | ||
| Fig. 1 1H-NMR spectra of (A) chitosan mesylate salt; (B) N,N,N-trimethyl N-propanoyl-chitosan (TPC); (C) TPC-S1 conjugate and (D) TPC-S2 conjugate. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O (1:1) at a concentration of 10 mg mL−1. The final spectra were processed using MestreNova software version 14.2.2-28739. The degree of substitution (DS) for the chitosan conjugates and their precursors was evaluated using the integral values in the 1H NMR spectrum and using the equations described below.
:D2O (1:1) at a concentration of 10 mg mL−1. The final spectra were processed using MestreNova software version 14.2.2-28739. The degree of substitution (DS) for the chitosan conjugates and their precursors was evaluated using the integral values in the 1H NMR spectrum and using the equations described below.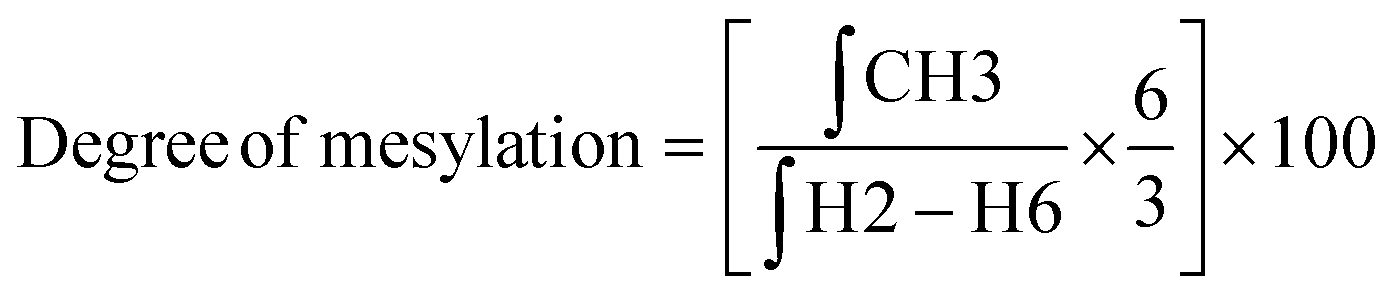 | (1) |
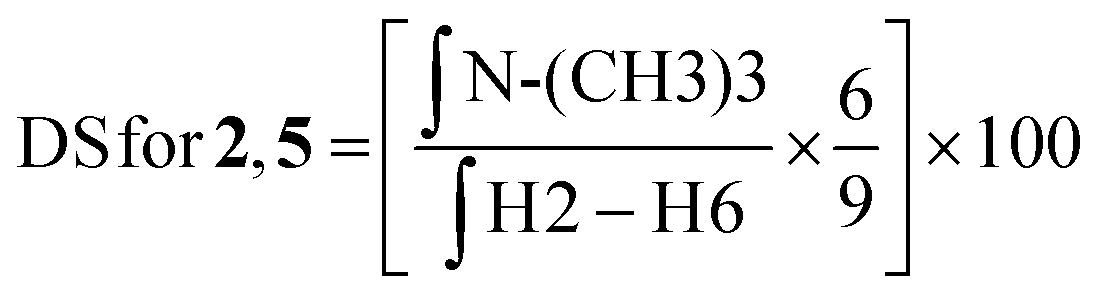 | (2) |
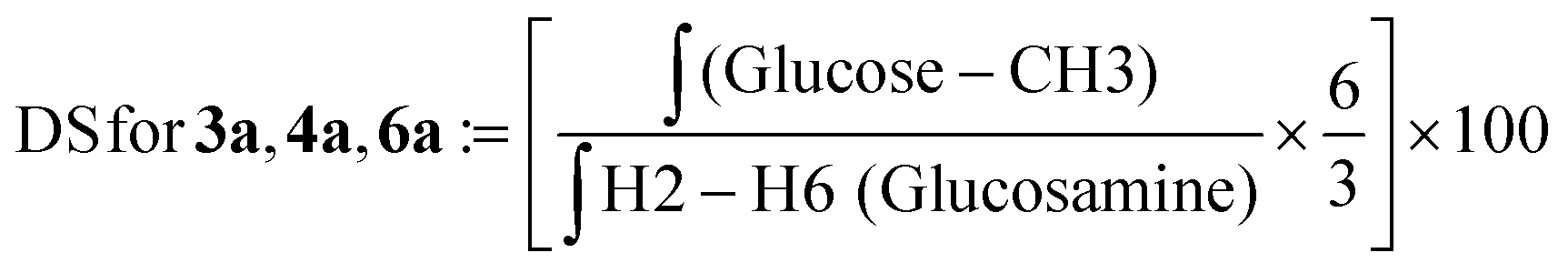 | (3) |
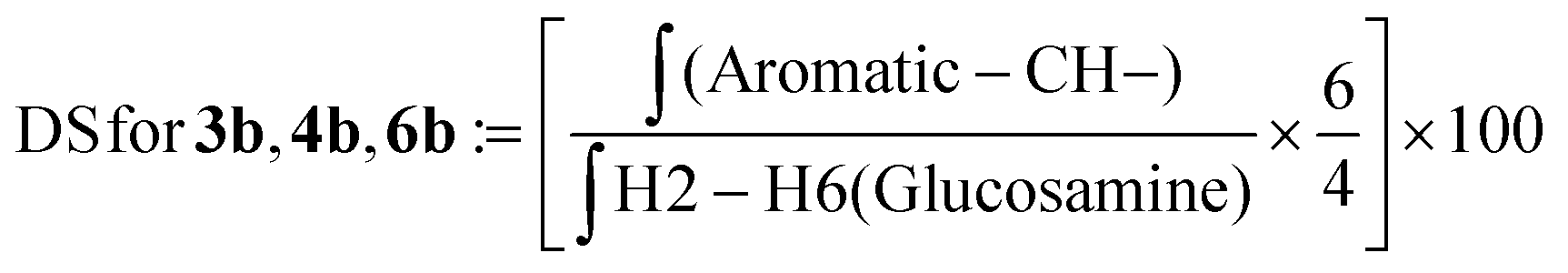 | (4) |
Molecular weight determination
The average molecular weight (Mw) and dispersity (D) for starting chitosan and its derivatives were assessed by gel permeation chromatography using an Agilent 1260 Infinity Multi-Detector GPC/SEC system (Santa Clara, CA, United States) with the triple detection technique. The measurements were performed using the following modules: Agilent 1260 Infinity Standard Degasser (G1322A), Agilent 1260 Infinity Isocratic Pump (G1310B), Agilent 1260 Infinity Standard Autosampler (G1329B), Agilent 1260 Infinity Thermostated Column Compartment (G1316A), Agilent 1260 Infinity GPC/SEC Multi-Detector Suite (G7800A), Agilent 1260 Infinity MDS RID (G7801A), Agilent 1260 Infinity MDS Viscometer (G7802A) and Agilent 1260 Infinity MDS Light Scattering (G7803A). The column set used for the measurement was 2 × PL aquagel-OH Mixed-H 8 μm and 7.5 × 300 mm (PL1149-6800). Agilent polymethylmethacrylate (PMMA) standards were employed to generate the universal calibration. Chitosan conjugates were dissolved in 0.5 M NaNO3/0.01 M NaH2PO4 (pH 2) at a concentration of 1 mg mL−1 before filtering with a syringe filter of 0.45 μm and 25 mm diameter (Corning® syringe filters). The eluent used was 0.5 M NaNO3/0.01 M NaH2PO4 (pH 2) and the samples were run with an injection volume of 100 μL and a flow rate of 1 mL min−1. Triplicate measurements were done for each sample. Data were processed using Agilent GPC/SEC Multi-Detector software (G7852AA). The Mw, Mn and PDI values were determined as calculated by the software.Antibacterial assays
Confocal laser scanning microscopy imaging
The antibacterial effect of chitosan derivatives was visualized by LIVE/DEAD staining of P. aeruginosa biofilms using the membrane-permeable SYTO 60 and membrane-impermeable TOTO-1 stains and confocal laser scanning microscopy (CLSM). The stain combination was used according to a previously published procedure.26 Overnight cultures of P. aeruginosa (3 biological replicates) were prepared as previously described. The cultures were then adjusted to OD = 0.1 using BHI and 160 μl was transferred to the wells of a 96-well microscopy plate (Ibidi catalogue # 89621) for inoculation at 37 °C for 2 h. The bacterial suspension was then carefully removed by aspiration and replaced with 200 μl of BHI and the plate was incubated at 37 °C for 24 h. After 24 h, the supernatant was replaced with 200 μl of fresh BHI and incubated under similar conditions for another 24 h to allow the biofilm to develop further. Solutions of the chitosan conjugates were prepared in BHI at concentrations equal to 0.025 × MBEC and 0.5 × MBEC. After 48 h of incubation, the supernatant was carefully removed from the wells and replaced with 100 μl of the conjugate solutions, whereas the growth controls were treated with fresh BHI. The microscopy plates were then incubated at 37 °C for 24 h. The supernatant in the wells was then replaced with 180 μl of 0.9% NaCl solution and left for 5 min. The removal and addition of fresh NaCl solution was performed three times. Finally, 100 μl of NaCl solution was added to the wells and known concentrations of SYTO 60 and TOTO1 stains were added to the wells to get final concentrations of 10 μM (SYTO60) and 1 μM (TOTO1) in the wells before imaging.To determine the interaction of the conjugates with S. aureus, a bacterial solution (200 μL) with OD = 0.1 was mixed with BHI containing calcofluor (100 μg mL−1) and incubated overnight (37 °C) for growth control. On the other hand, a solution of the conjugate in BHI (150 μL) was incubated with calcofluor (100 μg mL−1) in the dark for 4 hours. After this period, 90 μL of the conjugate solution was added to 10 μL of the bacterial solution (OD = 0.1) and incubated overnight (37 °C). For imaging the cells in CLSM, 10 μL of the growth control cells or conjugate-treated cells was placed on a glass slide, left standing for 20 min and washed with de-ionised water (30 μL × 3). The sample was stained with SYTO60 in 0.9% NaCl solution for 20 min in the dark. After washing the sample with de-ionised water (30 μL × 3), a drop of drop protector was applied for 5 min and covered with a second glass slide and visualized under CLSM. Fluorescence was captured with 405 nm excitation and <560 nm emission for calcofluor. For biofilm visualization, a similar procedure to that described above was followed for preparation.
Haemolytic activity
Human red blood cell (RBC) concentrate (6.1 × 1012 RBCs L−1, total haemoglobin of 208 g L−1, and 0.02 × 109 WBCs L−1) was used for testing the haemolytic activity of the peptide and the conjugates. RBCs (100 μL) were suspended in tris-buffered saline (TBS; 10 mL; pH = 7.2). The samples were prepared in TBS at an initial concentration of 32768 μg mL−1. 50 μL of the samples at the initial concentration were added to the first two wells of a 96-well plate. The samples were then serially diluted two-fold using TBS to have a minimum concentration of 16 μg mL−1. 50 μL of RBC suspension was added to each well and incubated at 37 °C with light shaking for 30 min. Cells treated with TBS and 2% TRITON-X were used as negative and positive controls, respectively. The cell suspensions were centrifuged at 1500 rpm for 10 min, and the supernatant was used for measuring the absorbance of the released haemoglobin at 540 nm with a Thermo Scientific Multiskan Spectrum. The percentage haemolysis was calculated using eqn (5) and the HC50 values were reported.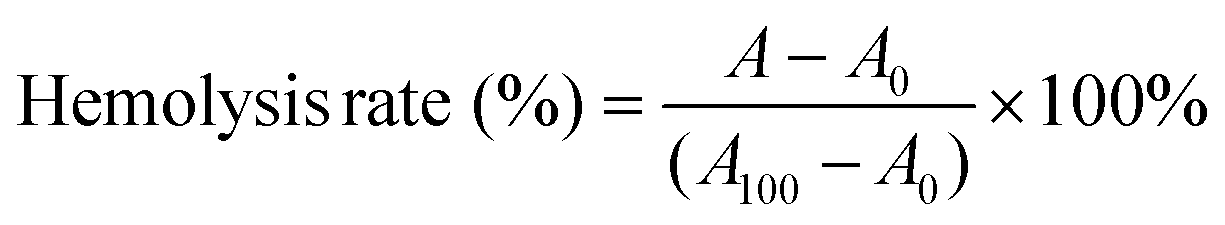 | (5) |
Results and discussion
Multivalent display of carbohydrate epitopes plays a pivotal role in the engagement of the immune machinery. To couple aryl-O-galactoside and C-fucoside epitopes onto chitosan backbone amine groups, prior derivatization with a suitable linker was required.Synthesis of C-fucosyl and aryl-O-galactosyl building blocks
2-(Tri-O-acetyl-α-L-fucopyranosyl) acetic acid (S5) was prepared from commercial L-fucose as described21 (see Scheme 1). EDAC/HOBt-mediated coupling of the 6-(benzyloxy)-6-oxohexan-1-aminium p-toluenesulphonate linker in the presence of NMM and DMF as solvent resulted in the isolation of the S1 precursor, protected as benzyl ester, in 75% yield (S4). Catalytic hydrogenation with Pd/C in THF/H2O and subsequent deacetylation carried out with LiOH in THF/H2O afforded S1 as a white solid in a quantitative yield.Starting from galactose pentaacetate, 4-(tetra-O-acetyl-β-D-galactopyranosyloxy) benzoic acid (S8) was easily isolated as previously reported22 (see Scheme 1). Conversion into galactosyl derivative S2 was achieved under the same conditions described for S1. Amide-coupling with the 6-(benzyloxy)-6-oxohexan-1-aminium p-toluenesulphonate linker resulted in 59% yield, followed by final deprotection steps affording the desired product S2 in 92% yield.
Synthesis of chitosan–sugar conjugates
To link the glycosidic moieties to the chitosan backbone, we performed amide coupling at the amino group of chitosan using the free carboxylic acids of the glycosides S1 and S2. Chemical modification on native chitosan is very difficult owing to its poor aqueous solubility and non-solubility in most organic solvents. Hence, previous studies reported several synthetic strategies such as using protective groups at either one or two of the free –OH groups.23–25 Protected chitosan is then soluble in some organic solvents and hence reactions can be performed in a homogeneous medium for N-modifications with high DS (up to 100%) in the products.25,26 However, introduction and removal of protective groups increases the number of synthetic steps and it results in significant trimming of the polymer chain.27Hence, our study was aimed at introducing low DS glycosides on the chitosan backbone. For this purpose, we optimized a direct synthetic method avoiding the use of protection group. To overcome the solubility issue of chitosan, we prepared the mesylate salt of chitosan, which has excellent aqueous solubility and high solubility in polar organic solvents such as DMF and DMSO. By using the mesylate salt of chitosan, we initially modified the amino group of chitosan to include two cationic groups, namely, the trimethyl amino group and the N,N,N-trimethyl-N′-propanoyl group. For TMC, chitosan was treated with methyl iodide/Cs2CO3 in DMSO, and using calculated equivalents of methyl iodide (MeI) we obtained a DS = 25%. The second chitosan derivative, TPC, was synthesized in two steps: first N-acylation using 3-bromopropanoic acid and then substituting the bromide with the trimethylamino group. The two cationic chitosan derivatives 2 and 5 having a DS of 25% were synthesized as shown in Scheme 1. The purpose of introducing the cationic trimethylamino group on chitosan is for taking advantage of its high aqueous solubility and broad-spectrum antimicrobial properties as observed in previous studies.19,27 We then designed a series of chitosan–sugar conjugates with the aim of having 25–30% of S1 and S2 glycosides grafted on native chitosan as well as on modified chitosan, 2 and 5. We achieved this by reacting the carboxylic residue of S1 and S2 with the free amino group of either native chitosan or the two cationic chitosan derivatives by using EDC/HOBt coupling. Initially, several optimization reactions for the amide coupling reaction were performed as shown in the ESI.† The best results were eventually obtained with EDC/HOBt as coupling reagents in the presence of NaHCO3 and in DMF as solvent. By using 0.5 equivalents of S1 and S2 in the reaction media, we obtained a DS of 25–27% for the desired conjugates (Scheme 1).
Previous studies reported coupling of arginine to chitosan by using EDC/NHS in acetic acid resulting in only 17% DS using an excess of reagent.28 Similar conditions were used for histidine–chitosan linkage resulting in 29% DS using an excess of reagent.29 Our method utilizing the mesylate salt of chitosan provides a more efficient synthetic method. The main advantages of the method are reproducible DS (∼25%) obtained with only half equivalent of the reagent, applicable for reactions with different functional moieties and reactions in homogeneous organic media. As seen in Table 1, we have a higher ratio of free amino groups in CS-S1/S2 (75%) than that in TMC-S1/S2 (48%) and TPC-S1/S2 (48%). Since the trimethyl amino group in chitosan has previously shown high antibacterial activity and improved aqueous solubility,25 the aim of this study was also to observe any synergistic effect of the sugar and the cationic group on the antibacterial activity of the conjugates. Table 1 also shows the weight average molecular weight (Mw) and dispersity of the conjugates as determined by GPC. We observe that despite a reduction in the Mw of the conjugates from the original chitosan due to the synthetic modification, we have a uniform Mw distribution for all the conjugates. The similar Mw values obtained after the synthetic modification also validate the reproducibility of the synthetic procedure using the chitosan mesylate salt. The Mw values for the conjugates are in the range of 7.1–8.1 kDa and the dispersity lies between 1.0 and 1.6 for all the conjugates. A great advantage of introducing the cationic group and S1/S2 onto the chitosan backbone is that they convert an insoluble biopolymer into a modified polymer exhibiting significantly enhanced aqueous solubility, which is a pre-requisite for biological studies.
| Compd no. | Conjugate | Degree of substitution | M w (Da) | M n (Da) | D | ||
|---|---|---|---|---|---|---|---|
| Cationic group (%) | Sugar (%) | Free amine (%) | |||||
| M w = weight average molecular weight; Mn = number average molecular weight; D = dispersity. | |||||||
| 3a | CS-S1 | 0 | S1 (25) | 75 | 8187 | 4947 | 1.65 |
| 3b | CS-S2 | 0 | S2 (25) | 75 | 7680 | 7486 | 1.02 |
| 4a | TMC-S1 | TMC (25) | S1 (27) | 48 | 7880 | 6638 | 1.18 |
| 4b | TMC-S2 | TMC (25) | S2 (27) | 48 | 7148 | 439 | 1.11 |
| 6a | TPC-S1 | TPC (25) | S1 (27) | 48 | 7878 | 5715 | 1.37 |
| 6b | TPC-S2 | TPC (25) | S2 (27) | 48 | 8149 | 6509 | 1.25 |
We characterized the conjugates by 1H-NMR spectroscopy. Fig. 1 shows the comparison of the 1H-NMR spectra of the chitosan mesylate salt, TPC, TPC-S1 and TPC-S2. Compared to spectrum A, B shows the presence of two new peaks for protons 1 and 2. Additionally, the appearance of the N-(CH3)3 peak at 2.81 ppm further confirms the attachment of the N,N,N-trimethylamino propanoyl group to chitosan. Spectra C and D represent TPC-S1 and TPC-S2. In spectrum C for TPC-S1, several new peaks appear in the region of 1.07 to 2.61 ppm, which correspond to protons from the linkers in the sugar (see the ESI†). Moreover, additional peaks from the glucose unit are seen merged with the protons of the glucosamine unit of chitosan. Similarly, for TPC-S2 in spectrum D, we see new peaks from 1.29 to 2.28 ppm and peaks from the glucose unit merge with the glucosamine unit of chitosan. Furthermore, the conjugation of S2 can be confirmed by the appearance of the protons in the aromatic region at 7.08 and 7.62 ppm corresponding to the benzene ring of the aryl moiety. Similarly, the conjugation of S1 and S2 with chitosan and TMC was confirmed from their respective 1H-NMR spectra. All spectra for intermediates and conjugates are provided in the ESI.† The DS for the chitosan derivatives and the chitosan–sugar conjugates was calculated from the integrals in the 1H-NMR spectra using the equations in the Experimental section and the results are given in Table 1.
Antibacterial properties of chitosan–sugar conjugates
We initially screened the series of six conjugates against three bacterial strains – S. aureus, E. coli and P. aeruginosa. The results are reported as the Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC) (Table 2). The conjugates in general displayed higher activity toward Gram positive S. aureus than towards the two Gram negative strains. Of the two Gram negative bacterial strains, P. aeruginosa was more susceptible to the conjugates. As seen in Table 2, TMC-S1 (4a) and TMC-S2 (4b) are the most active of the six conjugates toward P. aeruginosa with an MIC of 256 μg mL−1. Conjugates 4a, 4b, 6a and 6b displayed high activity toward S. aureus (MIC = 64–128 μg mL−1). All the conjugates remained moderately active toward the Gram negative E. coli. Conjugates CS-S1 and CS-S2 remained low in activity and this difference can be justified by the absence of the cationic trimethylamino group unlike the remaining four conjugates. We observed that the presence of the cationic group increases the inhibitory effect of the conjugates by 1–3 fold. These results are consistent with the previously reported studies where chitosan modified with cationic groups such as trimethylammoniumyl and guanidinyl showed enhanced antimicrobial activity toward Gram positive and Gram-negative bacteria compared to native chitosan.10,19,26,30 We further confirmed the accuracy of the inhibitory concentration of the conjugates by determining their minimum bactericidal concentration values. As seen in Table 2, the MBC values are within 1–2 fold dilution of the MIC values of the conjugates. Thus, all the six conjugates showed bactericidal property toward the three tested strains.| Conjugate | S. aureus | E. coli | P. aeruginosa | HC50 (μg ml−1) | HC5 (μg ml−1) | Selectivity HC50/MIC | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC (μg ml−1) | MBC (μg ml−1) | MIC (μg ml−1) | MBC (μg ml−1) | MIC (μg ml−1) | MBC (μg ml−1) | S. aureus | E. coli | P. aeruginosa | |||
| MIC = minimum inhibitory concentration; MBC = minimum bactericidal concentration; HC50 and HC5 = concentrations causing 50% and 5% haemolysis in red blood cells, respectively. | |||||||||||
| CS-S1 (3a) | 512 | 1024 | 2048 | 2048 | 1024 | 1024 | ≥32768 |
1024 | ≥32 | ≥16 | ≥32 |
| CS-S2 (3b) | 256 | 256 | 2048 | 4096 | 512 | 1024 | ≥32768 |
1024 | ≥64 | ≥16 | ≥64 |
| TMC-S1 (4a) | 128 | 256 | 1024 | 2048 | 256 | 256 | 16384 |
512 | 64 | 64 | 64 |
| TMC-S2 (4b) | 64 | 128 | 2048 | 2048 | 256 | 512 | 16384 |
512 | 64 | 8 | 64 |
| TPC-S1 (6a) | 128 | 128 | 1024 | 2048 | 512 | 1024 | 16384 |
512 | 32 | 16 | 32 |
| TPC-S2 (6b) | 128 | 256 | 512 | 1024 | 512 | 1024 | 16384 |
256 | 32 | 32 | 32 |
To assess the preliminary toxicity of the conjugates, we tested them against human red blood cells (RBCs). We determined the lowest concentrations corresponding to haemolysis of the RBCs and calculated the HC50 and HC5 values (Table 2). The conjugates showed non-haemolytic properties. As seen in Table 2, CS-S1 and CS-S2 did not show a haemolytic effect with HC50 values of ≥32768 μg mL−1, while all the remaining four conjugates displayed a very low toxicity of 16384 μg mL−1. The HC5 values for the chitosan–sugar conjugates showed that the toxicity of unmodified chitosan was lower than that of chitosan modified with cationic derivatives. The haemolysis (%) of RBCs caused by the conjugates against the measured concentrations is shown in Fig. 2. In general, a lower toxicity of the conjugates is expected where a non-toxic polymer (chitosan) is combined with a low toxicity sugar molecule. These results are consistent with previous studies where the combination of chitosan even with toxic antimicrobials such as antimicrobial peptides, ciprofloxacin, and colistin resulted in significant overall lowering of toxicity of the conjugates.13,31,32 The lower haemolytic activity gave higher selectivity of the conjugates towards all the bacterial strains in the planktonic state (Table 2).
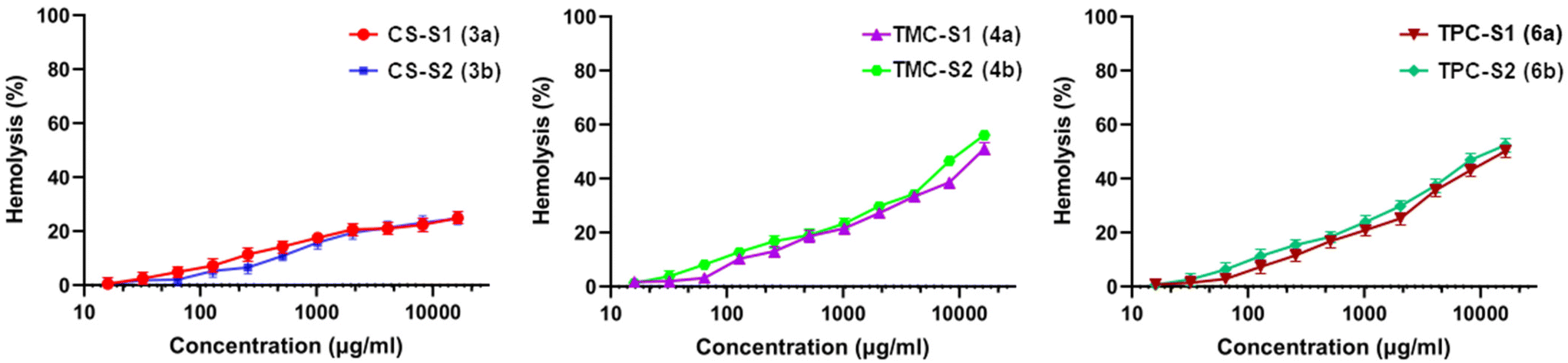 | ||
| Fig. 2 Graphs showing the hemolytic activity (%) against concentration of the chitosan–sugar conjugates. | ||
To determine if the conjugates would be effective in eradicating P. aeruginosa biofilms, we tested all the conjugates within a similar concentration range and assessed the Minimum Biofilm Eradication Concentration (MBEC). As reported in Table 3, TMC-S1, TMC-S2 and TPC-S2 were highly effective in eradicating P. aeruginosa biofilms with MBEC values of 8–16 μg mL−1. Conversely, these three conjugates displayed moderate activity towards P. aeruginosa in the planktonic form. CS-S2 and TPC-S1 conjugates also showed high activity toward biofilms with a MBEC of 64 μg mL−1. The least active of all the conjugates was CS-S1. Since the DS of the sugar moieties is similar in all the conjugates, the MBEC values give us a comparative analysis of the activity of the conjugates against biofilms. Table 3 shows that each series of conjugates (containing S1 and S2 glycosides) displayed similar antibacterial activity toward P. aeruginosa with no difference in their MIC values. However, a clear difference in the antibiofilm activity of the conjugates is observed. All conjugates containing the S2 glycoside (CS-S2, TMC-S2 and TPC-S2) were more effective in eradicating P. aeruginosa biofilms. Significant difference is observed within the CS conjugates where the MBEC of CS-S1 is 4 times that of CS-S2. On the other hand, the MBEC values of TMC-S2 and TPC-S2 conjugates varied only by 2-fold compared to their S1 counterparts. Thus, considering the antibiofilm activity toward P. aeruginosa, we can conclude that the O-galactoside S2 conjugates were more efficient than the C-fucoside S1 conjugates. Few studies reported the capability of chitosan and its conjugates to inhibit biofilms. Conjugates of chitosan with the aminoglycoside antibiotic streptomycin were capable of disrupting the biofilms formed only by Gram positive bacteria but not by Gram negative bacteria such as P. aeruginosa.17 Chitosan derivatives containing different combinations of cationic and hydrophobic groups were also highly effective in eradicating the biofilm population of Staphylococcus aureus.33 Biofilm inhibition up to 50–70% was achieved for P. aeruginosa using chitosan stabilized silver and gold nanoparticles,34 while chitosan itself was capable of inhibiting biofilms of several Gram positive bacteria.35,36 However, no study with chitosan conjugates for complete eradication of P. aeruginosa biofilms has been reported so far. P. aeruginosa biofilms possess high levels of intrinsic antibiotic tolerance and hence cannot be eradicated using the available antibiotics. Our study has shown that by using saccharidic ligands for LecA and LecB to decorate chitosan, we have been able to develop conjugates that effectively disrupt pre-formed P. aeruginosa biofilms.
| Conjugate | Antibacterial activity | Selectivity | ||
|---|---|---|---|---|
| Planktonic MIC (μg ml−1) | Biofilm MBEC (μg ml−1) | Planktonic HC50/MIC | Biofilm HC50/MBEC | |
| MIC = minimum inhibitory concentration; MBEC = minimum biofilm eradication concentration; HC50 = concentration causing 50% haemolysis in red blood cells. | ||||
| CS-S1 (3a) | 1024 | 256 | ≥32 | ≥128 |
| CS-S2 (3b) | 512 | 64 | ≥64 | ≥512 |
| TMC-S1 (4a) | 256 | 16 | 64 | 1024 |
| TMC-S2 (4b) | 256 | 8 | 64 | 2048 |
| TPC-S1 (6a) | 512 | 32 | 32 | 512 |
| TPC-S2 (6b) | 512 | 16 | 32 | 1024 |
The higher antibiofilm activity of TMC-S1/S2 and TPC-S1/S2 than that of CS-S1/S2 conjugates reveals that the presence of the cationic trimethyl amino group contributes positively to the antibiofilm activity. Although, CS-S1/S2 conjugates disrupted the pre-formed P. aeruginosa biofilms, higher antibiofilm activity was observed when the sugar moieties were combined with the trimethylamino groups. This demonstrates the synergistic effect of the two functionalities against P. aeruginosa biofilms. Furthermore, TMC-S1/S2 showed improved activity compared to TPC-S1/S2 conjugates. This indicates that the cationic group that is close to the polymer chain increases the activity, as was also observed in a previous study.27 As seen in Table 3, the selectivity of the conjugates toward P. aeruginosa biofilms is significantly higher than the selectivity toward individual cells.
The MBEC assay identifies the concentration required to kill the last cell in the biofilm. However, antimicrobial compounds might kill most of the bacteria in the biofilm at much lower concentrations. We therefore enumerated the number of viable cells in the biofilms after 24 h treatment with the conjugates at concentrations corresponding to the highest, intermediate and the lowest dilutions used for the MBEC measurement. The graphs in Fig. 3 show the viability of the biofilm population of P. aeruginosa after treatment with different concentrations of the conjugates and for the growth control. Results show that CS-S1, CS-S2, TMC-S1 and TMC-S2 can reduce the biofilm population by 50% (3.5 log difference) at 2–4× dilution of their MBECs, while TPC-S1 and TPC-S2 reduced the bacterial population by 3 log difference at 4–8× dilutions of their MBECs.
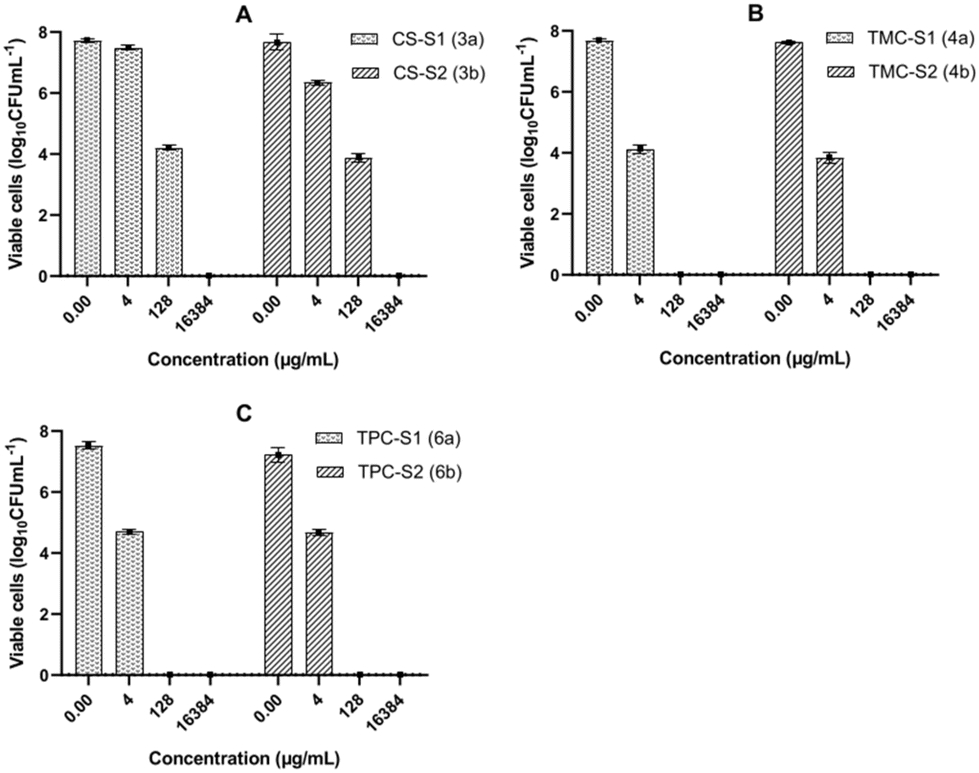 | ||
| Fig. 3 Graphs showing the viability of the biofilm population of P. aeruginosa when treated with different concentrations of (A) CS-S1 and CS-S2; (B) TMC-S1 and TMC-S2; (C) TPC-S1 and TPC-S2 conjugates. Each bar in the graphs represents the mean of three biological replicates and the error bars represent the standard deviation. | ||
The antibacterial effect of conjugates was visualized by live/dead staining of P. aeruginosa biofilms using a confocal laser scanning microscope (CLSM). We used a combination of the membrane-permeable SYTO 60 and membrane-impermeable TOTO-1 stains for staining the cells in the biofilm. The red colour in the images represents live cells while the green colour represents dead cells. Fig. 4 shows the images of P. aeruginosa biofilms treated with the two highly active conjugates TMC-S1 and TMC-S2. The untreated biofilm 1 is uniform in structure containing viable cells (red color). The biofilm treated with a low concentration (0.025 × MBEC) of the conjugate TMC-S1 (0.4 μg mL−1) shows dead cells (green color) scattered over the biofilm (image 2), whereas the biofilm treated with a higher concentration (0.5 × MBEC) of TMC-S1 (8 μg mL−1) contains mostly dead cells (image 3). A similar effect is seen for biofilms (images 4 and 5) treated with low and high concentrations (0.2 μg mL−1 and 4 μg mL−1) of TMC-S2. Hence, the CLSM imaging of treated and untreated biofilms confirmed the strong effect that these conjugates have on biofilm viability.
 | ||
| Fig. 4 2D confocal images of the biofilm population of P. aeruginosa where red = live cells and green = dead cells. (1) Growth control; (2) biofilm treated with 0.025 × MBEC of TMC-S1; (3) biofilm treated with 0.5 × MBEC of TMC-S1; (4) biofilm treated with 0.025 × MBEC of TMC-S2 and (5) biofilm treated with 0.5 × MBEC of TMC-S2. | ||
To confirm the ability of conjugates to penetrate P. aeruginosa biofilms, we performed 3D imaging of TMC-S2-treated biofilms compared to the growth control (Fig. 5). Image 1 shows that the biofilm of the growth control has a thickness of 12.3 μm. When the biofilm was treated with a lower concentration (0.025 × MBEC) of TMC-S2 (image 2), we observed an accumulation of dead cells throughout the biofilm and the thickness of the biofilm was reduced to 6.1 μm. At a higher concentration (0.5 × MBEC), the biofilm (image 3) mostly shows dead cells, with a significantly reduced thickness of 3 μm. Image 3 also confirms that the dead cells are not only present on the surface but also in the deeper layers of the biofilm, suggesting that the conjugates are capable of dispersing into the biofilm matrix.
 | ||
| Fig. 5 3D confocal images of the biofilm population of P. aeruginosa where red = live cells and green = dead cells. (1) Growth control, (2) biofilm treated with 0.025 × MBEC of TMC-S2 and (3) biofilm treated with 0.5 × MBEC of TMC-S2. | ||
To confirm that the conjugates interact with P. aeruginosa, we visualized the effect of the conjugate TMC-S2 on P. aeruginosa cells by fluorescence staining and confocal microscopy. Calcofluor which binds to chitosan37 was used as a fluorescent probe to visualize the conjugate TMC-S2. The bacterial cells were treated either with calcofluor (for growth control) or TMC-S2-calcoflour and stained with SYTO60 to visualize the cells. Fig. 6 shows the 2D CLSM images of individual P. aeruginosa cells untreated and treated with TMC-S2-calcoflour. As seen, the growth control (image 1) contains viable cells (red) while the cells treated with TMC-S2-calcoflour emit pink fluorescence (image 2). Images 3 and 4 show similar observations when biofilms of P. aeruginosa are untreated and treated with TMC-S2-calcoflour. These images confirm that TMC-S2 can interact with P. aeruginosa cells both in the planktonic form and in the biofilm.
 | ||
| Fig. 6 2D confocal images showing P. aeruginosa where red = bacterial cells stained with calcofluor and SYTO60; blue = TMC-S2 stained with calcofluor; pink = overlapping of TMC-S2 with P. aeruginosa suggesting binding. (1) Growth control (planktonic form); (2) cells treated with 0.025 × MBEC of TMC-S2-calcoflour, (3) growth control (biofilm) and (4) biofilm treated with 0.025 × MBEC of TMC-S2-calcoflour. | ||
Conclusions
Development of novel antimicrobials that are based on biocompatible materials and can operate by a different mode of action to target bacterial biofilms, such as those formed by P. aeruginosa, is a promising avenue to address the problem of antibiotic resistance. However, the design and development of bio-based materials endowed with high efficiency against bacteria is often very challenging. In this study, we report the synthesis of a series of novel chitosan–sugar conjugates by direct chemical modification of the chitosan backbone. The synthetic method we developed gave excellent yield and reproducibility. We observed that by attaching either sugar moieties or a combination of sugar and cationic moieties on chitosan the bactericidal effect is enhanced, on both Gram-positive and Gram-negative bacteria. More importantly, we showed that the series of conjugates developed were highly effective in eradicating the biofilm population of P. aeruginosa. Furthermore, these conjugates displayed very low toxicity and hence were highly selective toward P. aeruginosa. Structure–activity relationship analyses revealed that the conjugates containing a combination of sugar (aryl-O-galactoside or C-fucoside) and cationic moieties on chitosan were the most efficient in disrupting the biofilms. We confirmed this by CLSM imaging of the dead cells present in conjugate-treated P. aeruginosa biofilms. 3D imaging showed that the conjugates were capable of dispersing into the biofilm matrix which resulted in a significant reduction in the thickness of the biofilm. By using fluorescence staining and CLSM imaging, we showed that the conjugates do interact with individual bacterial cells in the biofilm.Author contributions
P.S.: conceptualization, investigation, formal analysis, methodology, visualization, writing – original draft; F.P.: synthesis of glycans, writing; K.L.M.: synthesis of sugars; O.E.S.: investigation, formal analysis, writing – review and editing; R.L.M.: conceptualization, writing – review and editing, supervision, project administration, funding acquisition; C.N.: conceptualization, writing – review and editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research work was supported by MUR, Italy (PRIN22 20222BLNZS). We acknowledge the additional support from Erasmus grant, University of Iceland. This manuscript is based upon the work from COST Action Functional Glyconanomaterials for the Development of Diagnostics and Targeted Therapeutic Probes (GLYCONanoPROBES) CA18132 supported by COST (European Cooperation in Science and Technology).References
- W. H. Organization, Prioritization of pathogens to guide discovery, research and development of new antibiotics for drug-resistant bacterial infections, including tuberculosis, Report 9240026436, World Health Organization, 2017 Search PubMed.
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao and E. Wool, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- M. G. Wilson and S. Pandey, Pseudomonas aeruginosa. [Updated 2023 Aug 8]. In StatPearls [Internet], StatPearls Publishing, Treasure Island (FL), 2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557831/ Search PubMed.
- Z. Pang, R. Raudonis, B. R. Glick, T. J. Lin and Z. Cheng, Biotechnol. Adv., 2019, 37, 177–192 CrossRef CAS PubMed.
- P. S. Stewart and J. W. Costerton, Lancet, 2001, 358, 135–138 CrossRef CAS PubMed.
- M. C. Walters 3rd, F. Roe, A. Bugnicourt, M. J. Franklin and P. S. Stewart, Antimicrob. Agents Chemother., 2003, 47, 317–323 CrossRef PubMed.
- G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty, A. Stocker, T. Darbre and J.-L. Reymond, Chem. Sci., 2016, 7, 166–182 RSC.
- D. Boffoli, F. Bellato, G. Avancini, P. Gurnani, G. Yilmaz, M. Romero, S. Robertson, F. Moret, F. Sandrelli, P. Caliceti, S. Salmaso, M. Cámara, G. Mantovani and F. Mastrotto, Drug Delivery Transl. Res., 2022, 12, 1788–1810 CrossRef CAS PubMed.
- I. Aranaz, A. R. Alcántara, M. C. Civera, C. Arias, B. Elorza, A. Heras Caballero and N. Acosta, Polymers, 2021, 13, 3256 CrossRef CAS PubMed.
- P. Sahariah and M. Másson, Biomacromolecules, 2017, 18, 3846–3868 CrossRef CAS PubMed.
- Y. Qin and P. Li, Int. J. Mol. Sci., 2020, 21, 499 CrossRef CAS PubMed.
- M. E. Abd El-Hack, M. T. El-Saadony, M. E. Shafi, N. M. Zabermawi, M. Arif, G. E. Batiha, A. F. Khafaga, Y. M. Abd El-Hakim and A. A. Al-Sagheer, Int. J. Biol. Macromol., 2020, 164, 2726–2744 CrossRef CAS PubMed.
- P. Sahariah, K. K. Sørensen, M. Á. Hjálmarsdóttir, Ó. E. Sigurjónsson, K. J. Jensen, M. Másson and M. B. Thygesen, Chem. Commun., 2015, 51, 11611–11614 RSC.
- C. Liang, F. Yuan, F. Liu, Y. Wang and Y. Gao, Int. J. Biol. Macromol., 2014, 70, 427–434 CrossRef CAS PubMed.
- T. Mohan, K. S. Kleinschek and R. Kargl, Carbohydr. Polym., 2022, 280, 118875 CrossRef CAS PubMed.
- D. Pranantyo, L. Q. Xu, E. T. Kang and M. B. Chan-Park, Biomacromolecules, 2018, 19, 2156–2165 CrossRef CAS PubMed.
- A. Zhang, H. Mu, W. Zhang, G. Cui, J. Zhu and J. Duan, Sci. Rep., 2013, 3, 3364 CrossRef PubMed.
- R. Pakkulnan, N. Thonglao and S. Chareonsudjai, Sci. Rep., 2023, 13, 1059 CrossRef CAS PubMed.
- P. Sahariah, G.-I. Kontogianni, E. Scoulica, O. E. Sigurjonsson and M. Chatzinikolaidou, Carbohydr. Polym., 2023, 312, 120796 CrossRef CAS PubMed.
- F. R. Cockerill, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved Standard, Ninth Edition, M07-A9 Vol. 32 No. 2, 2012 Search PubMed.
- E. M. Johansson, S. A. Crusz, E. Kolomiets, L. Buts, R. U. Kadam, M. Cacciarini, K. M. Bartels, S. P. Diggle, M. Cámara, P. Williams, R. Loris, C. Nativi, F. Rosenau, K. E. Jaeger, T. Darbre and J. L. Reymond, Chem. Biol., 2008, 15, 1249–1257 CrossRef CAS PubMed.
- R. U. Kadam, M. Bergmann, M. Hurley, D. Garg, M. Cacciarini, M. A. Swiderska, C. Nativi, M. Sattler, A. R. Smyth, P. Williams, M. Cámara, A. Stocker, T. Darbre and J.-L. Reymond, Angew. Chem., Int. Ed., 2011, 50, 10631–10635 CrossRef CAS PubMed.
- K. Kurita, H. Ikeda, M. Shimojoh and J. Yang, Polym. J., 2007, 39, 945–952 CrossRef CAS.
- O. V. Rúnarsson, C. Malainer, J. Holappa, S. T. Sigurdsson and M. Másson, Carbohydr. Res., 2008, 343, 2576–2582 CrossRef PubMed.
- B. E. Benediktsdóttir, V. S. Gaware, Ö. V. Rúnarsson, S. Jónsdóttir, K. J. Jensen and M. Másson, Carbohydr. Polym., 2011, 86, 1451–1460 CrossRef.
- P. Sahariah, B. M. Óskarsson, M. Á. Hjálmarsdóttir and M. Másson, Carbohydr. Polym., 2015, 127, 407–417 CrossRef CAS PubMed.
- P. Sahariah, V. S. Gaware, R. Lieder, S. Jónsdóttir, M. Á. Hjálmarsdóttir, O. E. Sigurjonsson and M. Másson, Mar. Drugs, 2014, 12, 4635–4658 CrossRef PubMed.
- B. P. Antunes, A. F. Moreira, V. M. Gaspar and I. J. Correia, Carbohydr. Polym., 2015, 130, 104–112 CrossRef CAS PubMed.
- V. M. Gaspar, J. G. Marques, F. Sousa, R. O. Louro, J. A. Queiroz and I. J. Correia, Nanotechnology, 2013, 24, 275101 CrossRef CAS PubMed.
- D. Yan, Y. Li, Y. Liu, N. Li, X. Zhang and C. Yan, Molecules, 2021, 26, 7136 CrossRef CAS PubMed.
- A. R. Egorov, M. N. Kurasova, O. Khubiev, N. A. Bogdanov, A. G. Tskhovrebov, A. A. Kirichuk, V. N. Khrustalev, V. V. Rubanik, V. V. Rubanik and A. S. Kritchenkov, Mendeleev Commun., 2022, 32, 774–776 CrossRef.
- N. V. Dubashynskaya, A. N. Bokatyi, A. V. Dobrodumov, I. V. Kudryavtsev, A. S. Trulioff, A. A. Rubinstein, A. D. Aquino, Y. A. Dubrovskii, E. S. Knyazeva, E. V. Demyanova, Y. A. Nashchekina and Y. A. Skorik, Int. J. Mol. Sci., 2023, 24, 166 CrossRef CAS PubMed.
- P. Sahariah, M. Másson and R. L. Meyer, Biomacromolecules, 2018, 19, 3649–3658 CrossRef CAS PubMed.
- M. F. Ma, F. Y. Faris Taufeq, S. Suleman Ismail Abdalla and H. Katas, RSC Adv., 2022, 12, 19297–19312 RSC.
- B. Orgaz, M. M. Lobete, C. H. Puga and C. San Jose, Int. J. Mol. Sci., 2011, 12, 817–828 CrossRef CAS PubMed.
- V. Felipe, M. L. Breser, L. P. Bohl, E. Rodrigues da Silva, C. A. Morgante, S. G. Correa and C. Porporatto, Int. J. Biol. Macromol., 2019, 126, 60–67 CrossRef CAS PubMed.
- C. A. Marangon, V. C. A. Martins, M. H. Ling, C. C. Melo, A. M. G. Plepis, R. L. Meyer and M. Nitschke, ACS Appl. Mater. Interfaces, 2020, 12, 5488–5499 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lp00263b |
| This journal is © The Royal Society of Chemistry 2024 |