
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A mesh reinforced pressure-sensitive adhesive for a linerless label design†
Emily M.
Brogden
 a,
Paul F.
Wilson
b,
Steven
Hindmarsh
c,
Ian
Hands-Portman
d,
Andrew
Unsworth
e,
Evelina
Liarou
a and
Stefan A. F.
Bon
*a
a,
Paul F.
Wilson
b,
Steven
Hindmarsh
c,
Ian
Hands-Portman
d,
Andrew
Unsworth
e,
Evelina
Liarou
a and
Stefan A. F.
Bon
*a
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: s.bon@warwick.ac.uk; Web: https://bonlab.info
bWarwick Manufacturing Group (WMG), University of Warwick, Coventry CV4 7AL, UK
cDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK
dSchool of Life Sciences, University of Warwick, Coventry CV4 7AL, UK
eElectron Microscopy Research Technology Platform (RTP), University of Warwick, Coventry CV4 7AL, UK
First published on 23rd January 2024
Abstract
A concept for an on-demand linerless pressure sensitive adhesive (PSA) label is shown. Containment of a PSA has been achieved by entrapment within a scaffolding 3D hard mesh structure. The label sticks upon instant application of heat and pressure, which softens and deforms the mesh allowing for PSA release. The design eliminates the need for a release liner and release coating in labels offering a more sustainable product. Herein, the mesh-reinforced PSA system was made by film formation of a binary polymer latex mixture consisting of ‘hard’ (high glass transition temperature, Tg,hard) polystyrene particles and a ‘soft’ (low glass transition temperature Tg,soft) poly(n-butyl acrylate)-based PSA latex of similar particle diameter, onto a model polyethylene terephthalate (PET) facestock. The system was annealed above Tg,hard to fuse the polystyrene colloids, creating a 3D interconnected open cellular network. The porous scaffold was shown by scanning electron microscopy, X-ray computed tomography, and confocal microscopy. The linerless PSA label is in a dormant, ‘non-stick’ state at room temperature, showing excellent blocking resistance under storage conditions. Adhesion is activated on demand with heat (T > Tg,hard) and light pressure. The adhesive behavior of the linerless PSA labels was probed using peel, shear strength and tack, its performance being promising.
1. Introduction
A pressure-sensitive adhesive (PSA) is designed to adhere to a surface upon contact when light pressure is applied, with tack being used as a common measure of adhesion.1 One relevant class of PSAs are polymer films cast from water-based dispersions of polymer colloids, also known as polymer latexes. Besides characteristic low values for the glass transition temperature (Tg) of the polymer, that is around −50 °C, the chemical composition and molecular weight distributions of the macromolecules are important in optimizing tack performance.2 Waterborne latex-based PSAs are often made from n-butyl acrylate or 2-ethylhexyl acrylate and have small amounts of comonomers which can provide high Tg segments (routinely styrene or methyl methacrylate) as well as comonomers which can promote wetting and adhesion, or secondary reversible interactions to improve cohesion (for example (meth)acrylic acid).3–6Commercially, these PSAs are used for labels. A typical label has several layers: a topcoat, a facestock with a printed image or text, the PSA, and a liner which contains a release coating, often made using high molecular weight silicones, to facilitate its removal before application.7 The liner stops the PSA from sticking during storage. In the context of environmental sustainability a big drawback is this release liner, which is effectively waste after it has been peeled off. These silicone-coated liners are energy intensive to produce, single use, and difficult to recycle. One obvious way around using a liner is to apply the release coating to the facestock, away from the adhesive layer. This strategy has been used since the 1950s and can be of use for labels in a roll format.8–10 A considerable downside is that printing on such labels post roll-up is complicated because of the presence of the release coating.
A key desirable for the label-making industry, therefore, is to develop linerless labels, that is no liner and no release coating. The obvious question is how to accomplish this? What we are looking for is an adhesive layer which can be activated and stick on demand. One early example one can think of is that of moisture activatable postage stamps, originating back to the mid-19th century. A clear downside here is that the ability to adhere to a substrate heavily depends on the moisture level. Stamps can snap off when conditions are too dry, or they can readily be detached under wet conditions. Think for example of using steam to remove stamps undamaged from old mail items by philatelists.
A solution to achieve a more consistent level of adhesion after activation is to hide and protect a higher performance PSA with a sacrificial ‘hard’ hydrophilic protective polymeric top layer which can be activated, softened and de-wetted by moisture.11,12 Suitable hydrophilic materials include gelatin, poly(vinyl alcohol) and poly(ethylene glycol) amongst others. A conceptually different and interesting approach was reported by Empereur and coworkers who made silicone liner-free PSAs by microencapsulating the PSA.13 Applying pressure ruptures the capsules and releases the adhesive. Einsla and coworkers also reported an elegant strategy.14 They blended the concepts of a waterborne PSA with hot melt adhesives and dispersed coarse agglomerates of poly(styrene-b-isoprene-b-styrene) into an adhesive film providing a temporary scaffold, which upon heat activation, restructured.
Herein, we would like to report the concept of providing a temporary mesh-like scaffold for the adhesive which can be softened upon activation (see it as a hardened sponge that contains the PSA, see Fig. 1b). Such a construct will prevent the embedded adhesive from sticking when you do not want it to stick. In other words, provide it with interim block resistance. We decided to build our 3D scaffold from fused hard (high Tg) polymer latex particles and took our inspiration from studies reported throughout the 1990s where block resistance was enhanced in waterborne coatings upon using a mixture of hard (high Tg) and soft (low Tg) polymer dispersions. We hypothesized that we could build an annealed interconnected mesh originating from hard latex particles which would contain the PSA (soft polymer) in situ through film formation of dispersed blends of hard and soft polymer colloids, see Fig. 1a and b. This scaffold should be mechanically robust at room temperature and resist compression upon applied pressure, alleviating the need for a release liner and coating. The 3D support needs to become easily deformable when heated above its Tg, hereby making it compressible like a sponge and releasing the PSA matrix (Fig. 1c). In other words, a linerless heat-triggered label that sticks on demand.
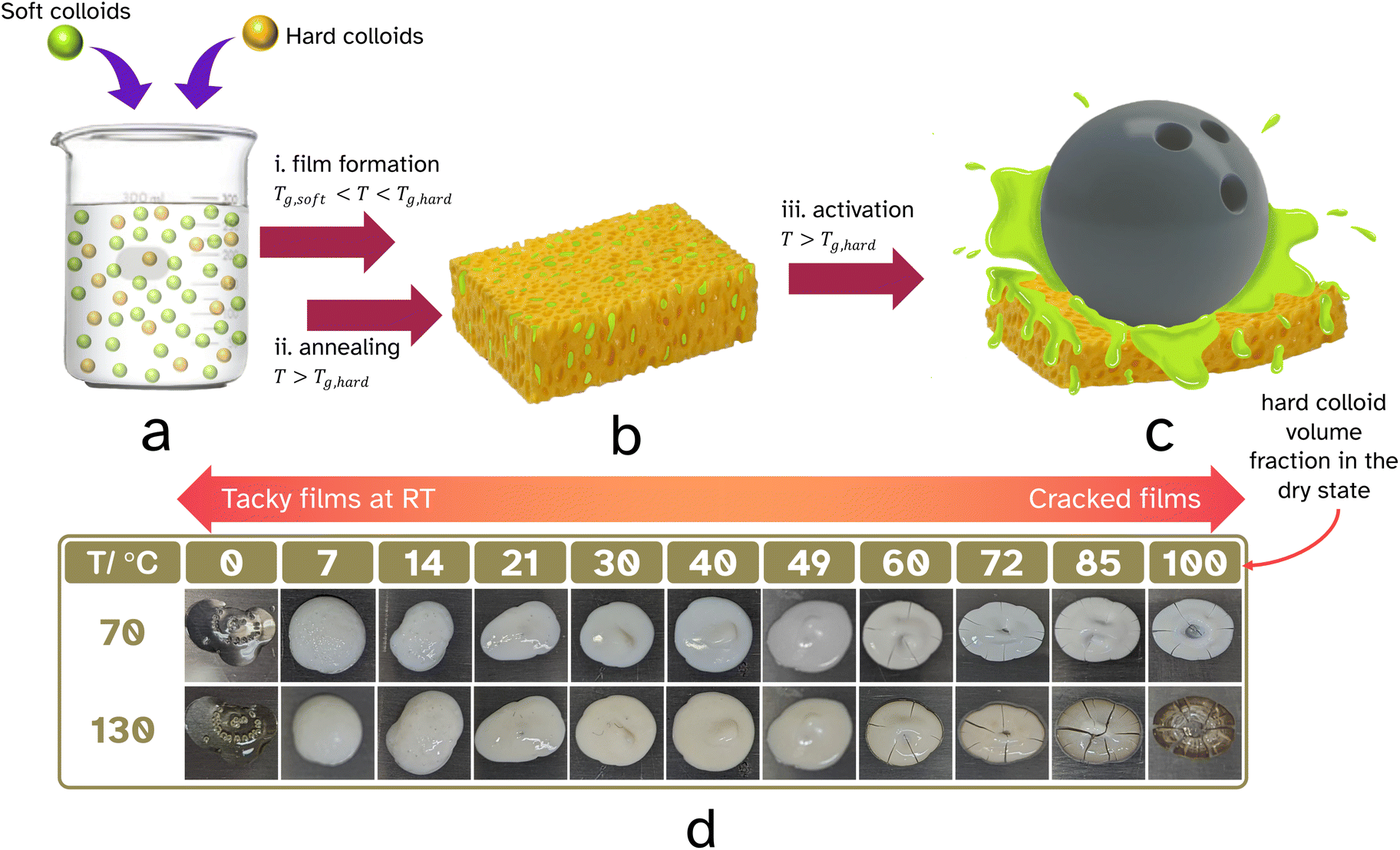 | ||
| Fig. 1 A graphical depiction of the formation of the linerless PSA. A binary mixture of hard, high Tg,hard and soft, low Tg,soft colloids, a, is film formed, Tg,soft < T < Tg,hard, and annealed, T > Tg,hard, to create a hard percolating network containing the soft PSA, b. Only upon activation with high temperature, T > Tg,hard, and pressure is the release of the soft PSA triggered enabling adhesion to the desired substrate, c. The hard–soft colloid ratio was determined by film forming droplets of various volume fractions of PS-latex and PSA-latex at 70 °C and annealing at 130 °C. Visual inspection of cracks determined the upper suitable ratio and tackiness at room temperature determined the lower suitable ratio. | ||
Cavaillé and coworkers reported in 1991 the preparation of polymer films using a binary mixture of poly(n-butyl acrylate) and poly(styrene) latexes.15 Dynamic mechanical analysis showed that the elastic modulus is reinforced by introducing higher volume fractions of the hard poly(styrene) particles. Data showed the appearance of a clear plateau at 50% indicating a 3D percolated hard phase. The extent of reinforcement was further enhanced upon annealing at temperatures above the Tg of the hard latex particles. At around the same time Friel filed a European patent on the concept of reinforcing the block resistance of a waterborne coating with hard latex particles.16 Later studies for example by Heuts and coworkers,17 Eckersley et al.,18 Feng and coworkers,19 and Ottewill et al.20 expanded on this concept. What was interesting to us is that in some cases the hard polymer phase was reported not to be randomly dispersed throughout the soft matrix and seemed to have formed its percolating network through means of aggregation. The origin of this ‘phase separation’ remains elusive.
The idea of creating a mesh-type structure in PSAs has been previously explored. Deplace and coworkers made ‘soft–soft nanocomposite’ PSA mesh-type films from polymer colloids with a core–shell morphology in which the shell was cross-linked to a greater extent.21 This led to a percolating network structure on the length scale of the diameter of the latex particles which greatly enhanced the adhesion energy. Such cellular PSA film morphologies were shown to effectively reduce creep, while in peel tests, the viscoelastic dissipation remained unchanged. Alternatively, addition of ‘hard’ poly(methyl methacrylate) latex particles of considerably smaller diameter, either as a blend or grafted onto the surface of the PSA colloids, improved shear resistance.22 Similarly, a marked increase in tack adhesion energy upon addition of ‘hard’ nano-sized components was also seen when small amounts of ‘soft’ latex particles armored with LAPONITE® clay discs were blended with a conventional PSA water-based dispersion.23 An interesting concept where PSA films could be switched off was shown by Gurney and coworkers.24 They added small ‘hard’ latex particles to a water-based PSA which upon film formation formed a surface-layer percolating structure on the length scale of the diameter of the ‘soft’ latex. Annealing of this network through exposure to heat removed the tackiness permanently.
To build a structural ‘hard’ sponge-like scaffold that can withstand light compression and thus have block resistance is essential for our strategy (Fig. 1a–c). Two important questions that need answering are: what should the volume fraction of ‘hard’ colloids be so that upon film formation a percolated structure is formed? To what extend should this continuous open cellular structure be annealed so that mechanical strength is achieved? Percolation theory25 establishes the minimum threshold beyond which a macroscopic structure is built, and goes back to the gellation theory of Flory (1941)26 and Stockmayer (1943).27 Interesting experiments by Fitzpatrick and coworkers in 1974 looked at randomly closed packed mixtures of monodisperse conducting and non-conducting spheres.28 They found that conductivity was lost below roughly 30 vol% of conducting spheres. Powell confirmed an in silico site-percolation threshold of 0.31 for randomly packed hard spheres of equal diameter, a value later confirmed by Ziff and Torquato.29,30 The scenario gets complicated when the mixtures of spheres are not of uniform diameter, or when the spheres are not randomly placed. He and Ekere showed that the percolation threshold would drop if mixtures of isolating large and conducting small spheres were used.31 One can invisage that non-randomness in packing of binary mixtures of spheres also results in a lowering of the percolation threshold. An important feature to keep in mind is the structural rigidity of the resulting hard mesh. This can be reinforced by annealing and coalescing the hard spheres. Importantly a non-random porous structure at a specific volume fraction of hard component potentially could provide a more robust scaffold.
In this work we will show that indeed it is possible to fabricate a linerless label using the concept of a 3D interconnected mesh that holds a PSA and can become compressible to allow for adhesion on demand when heated for a short period.
2. Experimental
2.1. Materials
Methacrylic acid (contains 250 ppm MEHQ as inhibitor, 99%), Brij L23 solution (30% (w/v) in H2O), ammonium persulfate (reagent grade 98%), 4-styrenesulfonic acid sodium salt, aluminum oxide activated (neutral, Brockmann I), acetone (puriss., ≥99%), sodium dodecyl sulfate (BioReagent, suitable for electrophoresis, for molecular biology, ≥98.5%), 2-acrylamido-2-methyl-1-propanesulfonic acid sodium salt solution (50 wt% in water), sodium persulfate (purum p.a., ≥99.0%), dibutyl disulfide (97%), vinyl acetate (contains 3–20 ppm hydroquinone as inhibitor, ≥99%), and toluene (puriss ≥99.7%) were purchased from Sigma Aldrich. Aluminum oxide activated (basic, Brockmann I) was purchased from Honeywell Fluka. Sodium bicarbonate anhydrous (≥99.5%, lab reagent grade) was purchased from Fisher Scientific. Styrene (contains 10–15 ppm 4-tert-butyl-catechol, 99%) was purchased from Alfa Aesar Chemicals. Lakeland PAE 136 was a gift from Lakeland Laboratories. Hostasol methacrylate, that is 2-(6-methacryloyloxyhexyl)-thioxantheno[2,1,9-dej]isoquinoline-1,3-dione, was synthesized using a procedure reported by Winnick.32Methacrylic acid was filtered through activated aluminium oxide (neutral) prior to use. Styrene and vinyl acetate were filtered through activated aluminium oxide (basic) before use. All other chemicals were used as purchased with no further purification. Deionized water was used in all reactions and analysis. The substrate used to cast films on and make tapes was Mylar® A polyethylene terephthalate (PET), 50 μm thick. The ‘soft’ PSA polymer latex (64 wt% solids content) was kindly provided by UPM Raflatac. It is a typical poly(n-butyl acrylate)-based latex used as a pressure-sensitive adhesive (PSA-latex).
2.2. Methods
The ‘hard’ colloids are a poly(styrene-co-methacrylic acid) latex (PS-latex) stabilized by a mixture of anionic and nonionic surfactants, which was synthesized as follows. Lakeland PAE 136 (0.32 g) in water (105 g) was added to the reactor with 4-styrenesulfonic acid sodium salt (0.08 g). This was degassed, via nitrogen bubbling, for 30 min, together with each of the following in separate round bottom flasks: monomer mixture (styrene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methacrylic acid 97:3, 95 g), brij L23 (30% (w/v) in H2O, 9.2 g in water 5.6 g) and ammonium persulfate (0.2 g) in water (8 g). Once degassed, the monomer charge (8 g) was added to the reaction vessel. The initiator charge (8 ml) was added at t = 0. Feed 1 (monomer, 25.962 ml h−1) and feed 2 (aqueous brij solution, 4.10 ml h−1) began at t = 20 min and fed for 3 h. The reaction mixture was stirred throughout at 220 rpm and heated via inbuilt water jacket at 70 °C. Samples were taken throughout the reaction to monitor the conversion and particle size. The total reaction time was 4 h and 20 min. The latex had a final conversion of 95% calculated by gravimetry, an average hydrodynamic diameter of 204 nm with a polydispersity index of 4.3%. The final solids content was 40 wt% and the glass transition temperature measured by dynamic scanning calorimetry was 102.7 °C using the half height analysis method. A PS-latex tagged with hostasol methacrylate was made using a similar procedure whereby 0.0252 g of hostasol methacrylate was added to the monomer stock prior to the polymerization.
:methacrylic acid 97:3, 95 g), brij L23 (30% (w/v) in H2O, 9.2 g in water 5.6 g) and ammonium persulfate (0.2 g) in water (8 g). Once degassed, the monomer charge (8 g) was added to the reaction vessel. The initiator charge (8 ml) was added at t = 0. Feed 1 (monomer, 25.962 ml h−1) and feed 2 (aqueous brij solution, 4.10 ml h−1) began at t = 20 min and fed for 3 h. The reaction mixture was stirred throughout at 220 rpm and heated via inbuilt water jacket at 70 °C. Samples were taken throughout the reaction to monitor the conversion and particle size. The total reaction time was 4 h and 20 min. The latex had a final conversion of 95% calculated by gravimetry, an average hydrodynamic diameter of 204 nm with a polydispersity index of 4.3%. The final solids content was 40 wt% and the glass transition temperature measured by dynamic scanning calorimetry was 102.7 °C using the half height analysis method. A PS-latex tagged with hostasol methacrylate was made using a similar procedure whereby 0.0252 g of hostasol methacrylate was added to the monomer stock prior to the polymerization.
The Poly(vinyl acetate) colloids (PVAc-latex) were synthesised as follows. Mixtures of sodium bicarbonate (0.1122 g) in water (157.04 g), vinyl acetate (99.75 g) and dibutyl disulfide (0.2389 g), 2-acrylamido-2-methyl-1-propanesulfonic acid sodium salt solution (50 wt% in water, 1.5184 g) in water (11.6216 g) and sodium persulfate (0.3547 g) in water (3.6333 g) were prepared. The sodium bicarbonate in water was added to the reactor and all mixtures and the reactor were purged under nitrogen for 30 minutes. The reactor was heated to 60 °C and stirred at 250 rpm. The initiator solution (4 ml) was injected into the reactor as feed 1 (vinyl acetate and dibutyl disulfide, 21.41 ml h−1) and feed 2 (2-acrylamido-2-methyl-1-propanesulfonic acid sodium salt in water, 2.5 ml h−1) began. The feeds were stopped after 5 h and the reaction continued for another 2 h to ensure high monomer conversion. The latex had a final conversion of >99% calculated by gravimetry, an average hydrodynamic diameter of 194 nm with a polydispersity index of 14.2%. The final solids content was 37 wt% and the glass transition temperature measured by dynamic scanning calorimetry was 27.4 °C using the equal areas analysis method due to the appearance of enthalpic recovery.
Dynamic light scattering (DLS). The average hydrodynamic particle diameter, dz, and dispersity were recorded using the Anton Paar Litesizer 500 (0.3–2000 nm). A disposable cuvette was washed twice with deionized water passed through a hydrophilic PTFE syringe filter with a 200 nm pore size. Each sample was diluted with water containing SDS (8 mM) until the mixture only had a slight blue haze. Measurements were ran at 25 °C, with an equilibration time of 4 min, repeated a minimum of 3 times, each with an average of 6 runs. Each run had a measurement time of 10 s and the measurement angle was 175 °C. The average hydrodynamic diameter and average polydispersity index was calculated by averaging the values of the repeats.
Gravimetric analysis. Half of each sample removed from the reactor (roughly 0.5 g from 1.0 mL), using a degassed syringe, was used for gravimetric analysis. The weight of the aluminium gravimetry pan, P, was measured. The sample was then syringed into the pan immediately after being removed from the reactor and the maximum mass of the pan and the sample, WP, were recorded. The sample was dried at room temperature for 12 hours and then dried at 105 °C in a vacuum oven for 12 hours. The dry mass of the pan, DP, was then recorded. This was used to calculate the solids content, SC, at each time point using eqn (1).
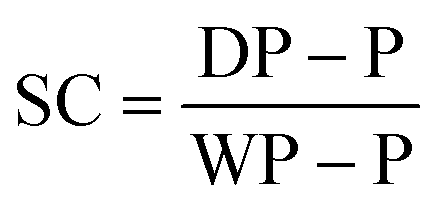 | (1) |
The SC was then used to calculate the instantaneous conversion, Xm,inst, at each time point using eqn (2), where Mt,sol is the mass of all solid components, not including polymer, Mt,tot is the mass of all components and Mt,mon is the cumulative mass of monomer at the time of sampling, t.
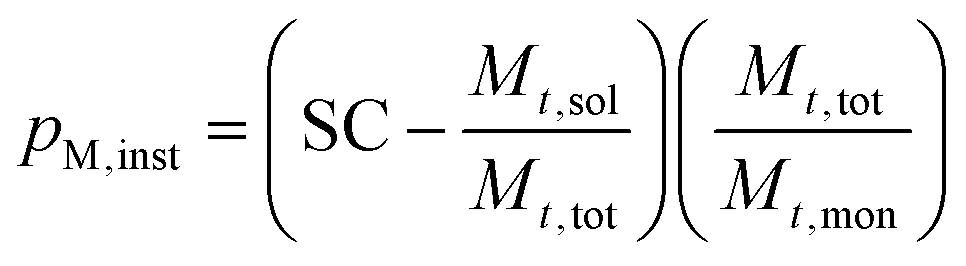 | (2) |
Using pM,inst, the cumulative conversion, pM,cum, was then calculated using eqn (3), where Mtot,M is the total mass of the monomer used for the reaction.
 | (3) |
Dynamic scanning calorimetry (DSC). DSC measurements were carried out on a TA Instruments DSC2500. Samples of PS-latex, PSA-latex and PVAc-latex were freeze dried overnight using a Frozen in Time Lablyo −85 freeze drier. Approximately 10 mg of a freeze-dried sample was weighed in a Tzero Hermetic Aluminium pan (temperature range −180 °C to 600 °C, 40 μL capacity) and sealed with a Tzero Hermetic lid. For the 34.6 vol% PS-latex and 65.4 vol% PSA-latex blend in the dried film, the corresponding wet latex mixture was loaded into the DSC pan and dried in air at room temperature. Three heating and cooling cycles were performed at a rate of 10 K min−1. The glass transition temperature of the material was taken from the third heating cycle using the midpoint for the PS-latex, PSA-latex and the PS-PSA blend. Equal areas analysis was used instead for the PVAc-latex, because of enthalpic recovery. See Fig. S5–S7 and S10.†
Polymer etching. A 34.6 vol% PS and 65.4 vol% PVAc film, Film 1, was cast with a wet height of 100 μm at a speed of 70 mm s−1 at 70 °C onto a sheet of Mylar® A PET. The film was dried at 70 °C for 10 min after casting, and then removed from the heat. Once cooled, it was then annealed at 130 °C for 60 min. The film was then placed in a shallow layer of acetone for 60 min to etch out the PVAc. The acetone was removed by pipetting, and the remaining film was allowed to dry before imaging.
To analyze the quantity of PVAc etched out, four samples were considered. The first sample was a 32.1 vol% PS and 67.9 vol% PVAc mixture. The second and third samples were the two colloidal mixtures of the PS-latex and the PVAc-latex individually. The fourth sample was the remains of Film 1 after acetone etching. The pans were left to dry in a fume hood for 48 h. Thermal gravimetric analysis (TGA) was performed to determine the percentage of PVAc etched from the film. Each sample was ran under nitrogen (50 ml min−1) and heated to 180 °C (10 K min−1) for 30 min to ensure samples were completely dry prior to analysis. The samples were then heated to 600 °C at 10 K min−1. See Fig. S13 and S14 and Table S1† for full analysis.
Scanning electron microscopy (SEM). SEM was performed on a Zeiss Gemini high-resolution scanning electron microscope. The image in Fig. 3a was recorded using the InLens detector at an accelerating voltage of 3 kV, 20 μm aperture, and a working distance between 3 and 7 mm. A small square of etched Film 1 was stuck to an SEM stub using a carbon adhesive dot. Carbon sputtering (one evaporation for 2000 ms) was performed prior to loading the sample into the microscope to minimize charging.
 | ||
| Fig. 2 Images of the adhesive film in use. Prior to activation the film is non-tacky and flexible (a and b). Using a hot glass jar with the label at room temperature (c) the label can be applied with a small amount of pressure (d) causing good adhesion (e). The label can be peeled off after use, leaving minimal residue behind which is easy to peel away (f). | ||
 | ||
| Fig. 3 Visualization of the mesh structure in an acetone etched PVAc-PS model film (Film 1) a–c. (a) SEM image of the top surface, (b) a tilt adjusted image of the cross section after cutting away a section using FIB SEM with a film height of approximately 20 μm, and (c) micro-CT 3D reconstruction of the etched film. Visualization of a similar mesh structure in a PSA-PS film (Film 2) d–f. (d) SEM image of the top surface of Film 2, annealed for 60 min (e) SEM image of the top surface (prepared similarly to Film 2 but with only a 19 min annealing time) with a clean crack after freezing in liquid nitrogen and (f) a cross-section of Film 2, where the PS is tagged with hostasol methacrylate (green), taken using confocal microscopy (the original top surface of the cast film is at the top of the image). | ||
A film of 34.3 vol% PS and 65.7 vol% PSA film, Film 2, was cast with a wet height of 200 μm at a speed of 50 mm s−1 at 70 °C onto a silicone wafer. The film was dried at 70 °C for 10 min after casting, and then removed from the heat. Once cooled, it was then annealed at 130 °C for either 0, 19, 60 or 120 min. After cooling, the samples were frozen in liquid nitrogen and the film cracked apart. Small sections of the wafers were stuck to an SEM stub using copper tape. Carbon sputtering (one evaporation for 2000 ms) was performed prior to loading the sample into the microscope to minimize charging. The images in Fig. 3e and S14† were recorded using the InLens detector at an accelerating voltage of 2 kV, 20 μm aperture, and a working distance between 5 and 8 mm.
Focused ion beam scanning electron microscopy (FIB-SEM). A Tescan Amber was used for FIB SEM. It has two columns; a 30 kV column with a Field Emission Gun (FEG) used in taking scanning electron microscopy images (SEM), and a 30 kV ion column with a Ga+ ion source to allow focused ion beam (FIB) milling.
A trench was dug in the etched Film 1 using 30 kV at 2 nA, followed by cleaning the face to image using 30 kV at 250 pA. Then it was imaged by SEM, Fig. 3b, using 2 kV at 100 pA with a working distance of 5.93 mm. The same sample of Film 2 (annealed for 60 min) after cracking with liquid nitrogen was imaged before, Fig. 3d, and after a trench was dug, Fig. S13,† using 30 kV at 750 pA, followed by cleaning the face to image using 30 kV at 250 pA. Images were taken with SEM using 2 kV at 100 pA with a working distance of 3.55 mm.
X-ray mictrotomography (X-ray CT). A corner cut sample of the etched Film 1 was imaged using a Zeiss 620 Versa. Zeiss Scout and Scan, Zeiss Reconstructor, and Avizo 3D 2021.2 software were used. The exposure voltage was 80 kV, the exposure power was 10 W and the exposure time was 30 s. The voxel (3D pixel) size was 0.149 μm and 3201 projections were taken. The source detector distance was 100.14 mm. Zeiss Scout and Scann, Zeiss Reconstructor and Avizo 3D 2021.2 software were used to compile and analyse the data.
Confocal microscopy. A hostasol methacrylate tagged PS-latex was prepared with a PSA-latex under the same conditions as Film 2. The sample was then frozen in liquid nitrogen to create clean cracks to image a cross section of the film. The film was set upright in dental wax on a glass slide and images were collected on a Zeiss lsm980 running Zen Blue with 20× air lens, 488 nm excitation, emission collected from 490–580 nm.
90° Peel test. A Shimazdu EZ-LX universal testing machine was used with a 500 N tensile jig in the upper position and a peel rolling jig in the lower position. A picture of this set up is provided in Fig. S1.†
Films with varying volume fractions of PS and PSA were cast onto Mylar® A PET at 100 μm wet height with a speed of 70 mm s−1 at 70 °C. The films were heated for 10 min at 70 °C then cooled to room temperature. The films, regardless of PS content, were annealed at 130 °C for 60 min. A second sheet of Mylar® A PET was then applied to the top of the film with a simple cylindrical hand roller at 130 °C and the system was left to equilibrate at 130 °C for 10 min before cooling. The system was then cut orthogonally to the casting direction to obtain strips of tape with dimensions 2 cm in width and 10 cm long. These tapes were attached via double-sided sticky tape to a polypropylene sheet, with dimensions slightly wider than the tape, for support. The tape was then threaded into the peel testing attachment and the top side of the PET substrate was held in place with the upper jig. The test was begun and the jig was pulled upwards at a 90° angle to the horizontal tape at 50 mm min−1. The rolling jig enabled the substrate to move horizontally as the tape was pulled upwards, maintaining the 90° peel angle. The force required to raise the jig was recorded as a function of time, t, and stroke, mm. For each film, 5 identical tapes were tested. The average peel force was calculated using the force data between 20 and 80 mm, then averaging the result from each of the 5 tapes.
Shear strength test. A Shimazdu EZ-LX Universal Testing Machine was used with two 500 N tensile jigs for shear strength tests. A picture of this set up is provided in Fig. S2.†
The strips of film prepared for peel testing were cut further into 2 × 2 cm lap joints, see Fig. 4b. The adherand strip was attached to the upper jig, and the substrate was attached to the lower jig. The test was begun by pulling the upper jig at a rate of 5 mm min−1. Stress, average force divided by area of adhesive joint, was then plotted as a function of strain, stroke divided by length of adhesive joint in the direction parallel to movement. The data was plotted only when the stress exceeded 0.1 N cm−1 to excluded any regions where the tape was not pulled taut enough prior to testing. The data was fit with a third order polynomial when the stress exceeded 0.1 N cm−1 before the stress–strain curve began to plateau. The maximum gradient of this fit in the region specified was taken as a value for the modulus. This was done for each of the five repeats and an average was calculated for each system. The shear strength was determined by using the trapezium rule to calculate the area under the stress–strain curves. The average shear strength was calculated from the 5 repeats to ensure repeatability.
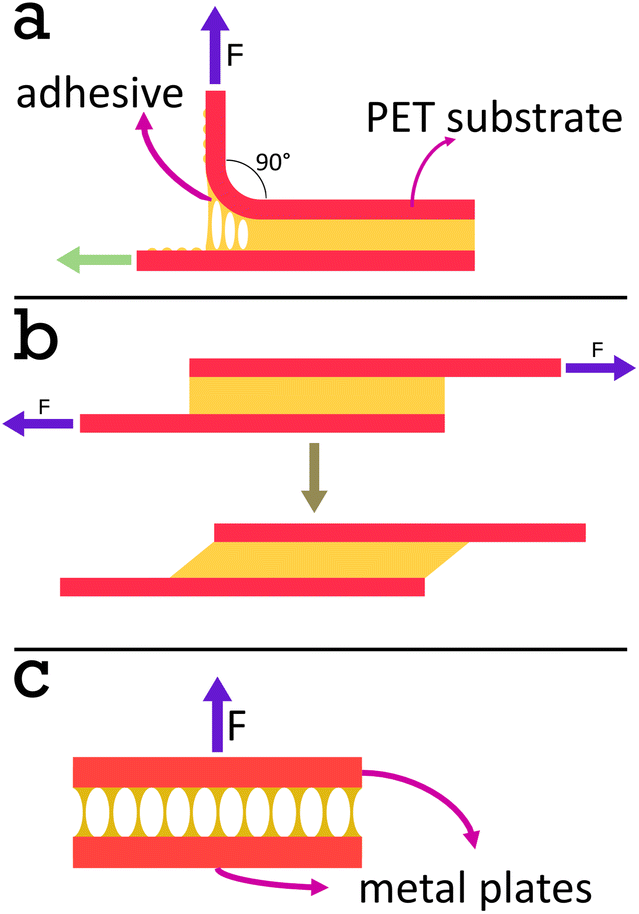 | ||
| Fig. 4 Diagrams of the adhesive tests used. In a peel test (a) the substrate on the top side is pulled up at a 90° angle and the rolling jig allows the rest of the tape to move horizontally (green arrow), maintaining the same angle. A shear test (b) is conducted when two substrates are joined using the adhesive to make a lap joint and pulled apart in the direction parallel to the substrates. A tack test (c) involves pulling apart two substrates joined using the adhesive, with the pulling direction perpendicular to the substrates. | ||
Blocking resistance test. A film composed of 34 vol% PS and 66 vol% PSA, Film 3, was cast onto Mylar® A PET at 200 μm wet height with a speed of 50 mm s−1 at 70 °C. The film was heated for 10 min at 70 °C then cooled to room temperature. The film was annealed at 130 °C for 60 min. It was then cut into 2 cm width strips as for peel testing and two strips were placed on top of each other, so that an adhesive–adhesive interface was created. Various weights were applied equally over the films (see Fig. 7 and S3† for set up) and left for 7 days at 25 °C. After this time a peel test was conducted on the two strips with the same testing conditions as reported in section 2.2.6.
Tack test. An Elcometer 4340 Automatic Film Applicator with a Casting knife film applicator was used for film formation of the colloid mixtures using a custom metal template (Fig. S4†) and 8 mm diameter disposable stainless steel plates. Films with various volume fractions of PS and PSA were cast directly from the wet latex mixtures. PS-latex was freeze-dried (as described in section 2.2.2) and dissolved in toluene (15 wt% solids content) for 3 days before casting to avoid cracks and bubbles in the PS-only film.
The results reported in Fig. 9a are films cast at 70 °C with a wet height of 500 μm and heated at this temperature for 22 min, then cured at 130 °C for 60 min. The results reported in Fig. 9b, were prepared similarly but not annealed.
Tack testing was performed on a Discovery Hybrid Rheometer HR-3 equipped with a Peltier plate to heat the 8 mm lower plate and an Upper Peltier Plate (UPP) to heat the 8 mm upper plate.
Tests conducted varying the PS volume fraction were conducted as follows. First, the sample was loaded onto the rheometer at 65 °C. The sample was then conditioned at 130 °C for 3 minutes. The upper plate was lowered at 5 μm s−1 with a terminating axial force of 0.1 N, where the gap at this stage is taken as the sample height. The top plate then pressed into the sample for 5 s with a maximum force of 10 N to mimic the activation step. The plates were held to equilibrate for 3 min then the temperature was decreased to 65 °C at a rate of 10 °C min−1. The upper plate was raised at 0.1 μm s−1 until complete adhesive failure was achieved (axial force of 0 N).
Tests conducted varying the testing temperature of the reference PSA-latex films were conducted as follows. The sample was conditioned at the testing temperature for 3 min. The upper plate was lowered at 5 μm s−1 with a terminating axial force of 0.1 N. The top plate then pressed into the sample for 5 s with a maximum force of 10 N. The plates were held to equilibrate for 10 minutes then the upper plate was raised at 10 μm s−1 until complete adhesive failure was achieved (axial force of 0 N).
In both testing regimes the load when the top plate was lifted up was divided by the contact area to obtain stress and the displacement of the probe was divided by the initial sample height to obtain stress. In the case of the soft UPM-only films the work of adhesion, Wadh, was obtained using eqn (4).33
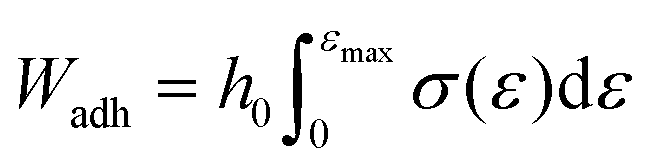 | (4) |
For rheological measurements at 25 °C, the sample was loaded onto the rheometer at 25 °C. If the sample had been annealed the temperature was then raised to 130 °C and the upper plate was lowered at 5 μm s−1 with a terminating axial force of 0.1 N, where the gap at this stage is taken as the sample height. If the sample was not annealled this step was done at 25 °C. Prior to a frequency sweep an amplitude sweep was conducted to determine the linear viscoelastic regime (LVER), this was done at 1 Hz. The frequency sweeps were measured between 0.01 Hz and 100 Hz with the displacement amplitude lying within the LVER and reported in Table S2.†
For rheological measurements at 130 °C, the sample was loaded onto the rheometer at 25 °C. The temperature was then raised to 130° and the upper plate was lowered at 5 μm s−1 with a terminating axial force of 0.1 N, where the gap at this stage is taken as the sample height. Prior to a frequency sweep an amplitude sweep was conducted to determine the linear viscoelastic regime (LVER), this was done at 1 Hz. The frequency sweeps were measured between 0.01 Hz and 100 Hz with the displacement amplitude lying within the LVER and reported in Table S3.†
3. Results and discussion
3.1. Polymer colloid synthesis and properties
The concept of our linerless adhesive is based upon having a 3D mesh structure in place as a temporary scaffold. In this paper we aim to build such structure during the drying of films of binary mixtures of water-based dispersions of polymer colloids, one being the pressure sensitive adhesive (PSA) (soft, low Tg), the other the percolating mesh forming component (hard, high Tg). The soft polymer latex was a commercial n-butyl acrylate based waterborne PSA, PSA-latex. Table 1, gives details of the solids content, particle size and distribution, and the glass transition temperature for all colloids used in this work. The PS-latex, was made by semi-batch emulsion polymerization of styrene using 3 wt% methacrylic acid as a functional comonomer. A PS-latex tagged with hostasol methacrylate was also made for confocal studies (see section 3.2, Fig. 3f). A model ‘soft’ poly(vinyl acetate) based latex, PVAc-latex, was synthesized to allow for selective etching and detailed morphological analysis of the mesh. The instantaneous and cumulative overall monomer conversions of both the PS-latex and the PVAc-latex were monitored by gravimetry during synthesis (Fig. S8 and S9†). The final latexes had monomer conversions exceeding 99%. The particle diameters were also monitored as a function of reaction time (Fig. S8 and S9†). In the case of the PS-latex there is a reasonably linear correlation between the average particle diameter and the cube root of monomer conversion (Fig. S8†), suggesting a short particle nucleation period, and absence of coagulation and secondary nucleation events, to produce a monodisperse, optically opalescent latex. In the case of the PVAc-latex the average particle diameter deviates from linearity upwards, Fig. S9,† suggesting some later stage microcoagulation which increases the particle size dispersity. No macroscopic coagulation was observed and the PVAc-latex remained stable for months.| Colloid | SC/% | d z /nm | Dispersity/% | T g/°C |
|---|---|---|---|---|
| PSA-latex | 64 | 235 | 22.8 | −39.8 |
| PS-latex | 40 | 204 | 4.3 | 102.7 |
| PVAc-latex | 37 | 194 | 14.2 | 27.4 |
3.2. Film and label formation and microstructure analysis
The film formation of the polymer latexes and mixtures of the PS-latex and the PSA-latex were screened at 70 °C. The dried systems were subsequently annealed at 130 °C for 60 min (see Fig. 1d). It is important to mention that in our experiments the average particle diameters of the latexes were comparible, 194–235 nm. Film formation of the PSA-latex alone resulted as expected in a tacky film at room temperature, both before and after annealing. Film formation of the PS-latex alone resulted as expected in a cracked, brittle film due to the particles’ resistance to deformation below Tg,hard. Annealing did not really repair the cracks. Ideally, gradual introduction of the ‘hard’ PS particles into the soft PSA will at one point introduce a percolating network of ‘hard’ particles, the idea being that the annealing step would fuse these together hereby mechanically reinforcing the targeted and desired 3D mesh structure. This introduces then the important property of blocking resistance of the final binary film at room temperature, that is the ability to prevent sticking to a substrate upon application of pressure. This leads to an important question: what volume fraction of PS colloids is required and still would allow for a good-quality film to be produced? Films with ≥60 vol% of PS particles resulted in cracked films, Fig. 1d, where the volume fractions quoted, here and throughout the paper, refer to the composition of the films after drying and not in the wet state. The PSA was able to act as a binder for the PS colloids and thus reduce the need for the PS particles to deform, lowering stress and resulting in preventing crack propagation. After heating at 70 °C, blends with less than or equal to 14 vol% PS-latex were tacky to the touch, with the others showing some tackiness upon applying light pressure. After the annealing step, however, the samples of 21 vol% PS particles and above were not tacky. This suggests the ideal range to achieve blocking resistance at room temperature is between 21–60 vol% of PS in the dried state.As was discussed in the introduction, in the field of waterborne coatings it has previously been observed that when drying a blend of ‘hard’ and ‘soft’ latexes, the blocking resistance of the resulting film was greatly improved.18 In our system, the annealing step above Tg,hard enables the PS particles to deform and thus fuse together. This would create a hard percolating network which after cooling, is incompressible at room temperature unless very high force is applied. Upon further heating, above Tg,hard, this mesh softens and is easily broken and deformed, allowing the soft, tacky polymer to be squeezed out and wet the substrate leading to good adhesion.
We casted a 200 μm thick wet film of a mixture of 34.3 vol% PS-latex and 65.7 vol% PSA-latex (total solids content of the wet mixture was 53.2 wt%) onto logo containing PET as facestock at 70 °C. The label was annealed at 130 °C. Fig. 2 shows a selection of images from Video S2.† In Fig. 2a we show that the label does not stick to itself and therefore shows excellent blocking resistance at room temperature. In Fig. 2b we show that the label has good flexibility and that the mesh reinforced adhesive does not delaminate from the facestock. Fig. 2c and d show the labelling process of a preheated glass jar. Fig. 2e shows that the label is well adhered to the jar, with Fig. 2f showing that the label can be peeled off if needed. Our system operates differently from a traditional PSA. The moment our stick-on-demand label is applied at elevated temperature the ‘soft’ PSA component by itself would have limited adhesion energy (see later Fig. 9b). Its ability to adhere is supported by the softened PS phase which is above its Tg. One could say at this moment its features are that of a hot melt adhesive. Upon cooling the adhesion energy of the PSA phase increases to a level which allows the label to remain adhered.
DSC analysis of a dried film composed of 34.6 vol% PS and 65.6 vol% of PSA shows two clear Tg's (Fig. S10†), that is −40.6 °C and 102.0 °C indicating complete imiscibility and thus a possibility for a hard network structure. A similar conclusion can be drawn from the optical appearance of the dried films of particle blends, as these appear opaque (Fig. 1d). We wanted to get more details on this potential mesh structure by carrying out microstructural analysis. The idea was to etch the soft component out selectively from the labels. For this we made a model system in which we replaced the ‘soft’ PSA-latex with the PVAc-latex. Poly(vinyl acetate) can easily and selectively be dissolved in acetone. The resulting brittle film, Film 1, was imaged after acetone etching via scanning electron microscopy (SEM), focused ion beam SEM (FIB-SEM) and X-ray CT, Fig. 3a–c. Imaging the top surface of the film by SEM, Fig. 3a, indeed showed a continuous fused mesh structure. Additionally, when a section of the mesh is cut away with FIB-SEM the network also looks to percolate the entire height of the film, Fig. 3b. We also confirmed this fused network exists in the 3D rendering of the micro-CT scan, Fig. 3c, Fig. S11 and Video S1† for a full cross section fly through, which details the percolation throughout a large area, 0.9 mm2, of Film 1. Using 3D sections (0.057 mm × 0.015 mm × 0.07 mm) of the mesh structure throughout the sample an average volume fraction can be calculated as 49.9 vol% with a range from 49.1 to 52.1 vol%. Additionally, calculating the volume fraction of 2D slices as a function of height shows no clear trend, Fig. S12,† suggesting there is limited or no vertical stratification of either PS or PVAc particles throughout the film. The volume fraction calculated is higher than the real volume fraction of PS colloids, 34.6 vol%. This is explained partly by the reduction in height of the film during the etching process, as the mesh is no longer supported by the continuous PVAc phase and therefore compresses (seen in the reduction of expected height from 40 μm to 19 μm in Fig. 3b). This would take the volume fraction of percolated network roughly at 25 vol% at original non-compressed height. It should also be mentioned that according to thermogravimetrical analysis, 18 wt% of PVAc is remaining and therefore was only partially etched out (see TGA analysis in Fig. S15 and S16 and Table S1†). This means that some of the PS phase must have been lost upon etching, which makes sense as not all would have been an integral (isolated non-connected fragments or sole particles) or robust part of the 3D mesh structure.
An interesting observation from looking at Fig. 3a–c is that a rough estimate for the diameter of the rope-like mesh structure is around 1–3 μm, whereas the original PS-latex had a particle diameter of around 200 nm. This leads to the question, at what point during the film formation process do the ‘hard’ PS colloids cluster into aggregates or at what point does the PS phase coarsen? There are three possible explanations. Firstly, there could be already some order in the wet binary mixture of latexes as a result of differences between interaction potentials between PS-PS particles, PS-PSA or PS-PVAc particles, and PSA-PSA or PVAc-PVac particles. This would mean that the latex blend in the wet state is not random. Secondly, there is aggregation upon film formation, potentially due to an increased electrolyte concentration as water evaporates and selective loss of colloidal stability. Thirdly, the hard PS domains coarsen when the film is cured above both Tg's, a common phenomenon and for example seen previously in work by Limousin et al. when a styrene–acrylamide hard core and methyl methacrylate–butyl acrylate–styrene soft shell latex is film formed and then annealed.34
To ensure this mesh structure is present in our actual adhesive label, made from a blend of PS-latex and PSA-latex, SEM microstructure analysis of Film 2 was also performed. Fig. 3d shows the top surface of Film 2 as casted and annealed for 60 min. No solvent etching has taken place. There is a clear surface texture on the same length scale as observed in our PVAc model system, particularly when compared to a non-annealed equivalent film, Fig. S14.† To investigate if this structure was only a surface effect or present throughout the bulk, samples similar to Film 2, but with 19 min (Fig. 3e) and 120 min (Fig. S14†) annealing times, were frozen in liquid nitrogen to create clean cracks prior to SEM analysis. Since the PSA has not been etched out it is difficult to determine if a mesh structure persists in the films. FIB-SEM was conducted on samples of Film 2 which were annealed for 0 and 60 min, respectively. Those which had been annealed were relatively easy to mill and trenches could be dug to show internal structure (Fig. S13†). Within the bulk of the film there seems to be air pockets or voids with unusual shapes which hints at the existence of an internal structure throughout the film. The samples which were not annealled proved difficult to mill and imaging suggests the lack of a rigid bulk structure. To resolve any ambiguity, a hostasol methacrylate tagged PS-latex was used and incorporated into a film prepared under the same conditions as Film 2 with a 60 min annealing time. A cross section of a crack (created by freezing with liquid nitrogen) was imaged using confocal microscopy, Fig. 3f. The film height is roughly 100 μm (corresponding well with the expected height based on solids content of the binary mixture) and demostrates clearly that there is a coarsened interpenetrating mesh relatively homogeneously distributed throughout the film, of a microstructure similar to the one observed in the model PVAc system.
3.3. Adhesive performance of mesh reinforced labels
Various adhesive performance tests, peel, shear strength, and tack, were performed on the films made from our mixtures of PS-latex and PSA-latex, see Fig. 4. We analysed films of our PSA-latex, its blend with PS-latex (both annealed and non-annealed), and the PS-latex by itself using oscillatory rheometry measurements to determine the storage and loss shear moduli, G′ and G′′, and tanδ at room temperature and 130 °C, in the linear viscoelastic regimes. G′ (1 Hz) for the PSA at room temperature is 0.0539 MPa, comfortably below the Dahlquist criterion. The same holds for the PS at 130 °C, where G′ (1 Hz) has a value of 0.0377 MPa. See also Fig. S21 and S22 and Tables S2 and S3.†
 | ||
Fig. 5 90° peel testing of PS-PSA films, 2 cm in width, where the PS volume fraction ( 0.00 vol%, 0.00 vol%,  10.15 vol%, 17.31 vol%, 10.15 vol%, 17.31 vol%,  25.00 vol%, 25.00 vol%,  35.64 vol%, 35.64 vol%,  50.10 vol%) in the dry state is varied. (a) The average force measured as a function of peel distance, stroke, for each repeat and (b) the average peel force calculated as a function of hard volume fraction in the PS-PSA film. 50.10 vol%) in the dry state is varied. (a) The average force measured as a function of peel distance, stroke, for each repeat and (b) the average peel force calculated as a function of hard volume fraction in the PS-PSA film. | ||
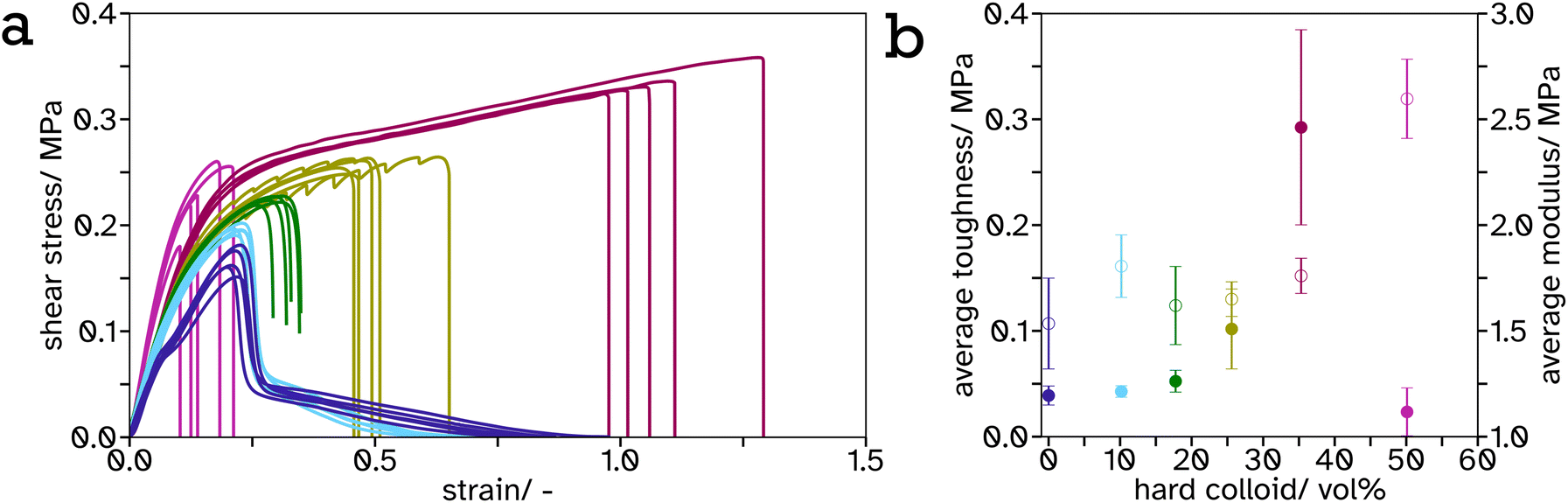 | ||
Fig. 6 Shear strength testing of PS-PSA films joining two PET strips together with a joint dimension of 2 × 1.8 cm and varying the PS fraction ( 0.00 vol%, 0.00 vol%,  10.15 vol%, 10.15 vol%,  17.31 vol%, 17.31 vol%,  25.00 vol%, 25.00 vol%,  35.64 vol%, 35.64 vol%,  50.10 vol%). (a) The average stress, σ, as a function of strain, ε, (b) the average toughness, closed circle, calculated by integrating the stress–strain curves using the trapezium rule, and the average moduli, open circle, calculated by finding the maximum of the derivative of the initial slope fitted using a 3rd order polynomial, as a function of hard content in the films. 50.10 vol%). (a) The average stress, σ, as a function of strain, ε, (b) the average toughness, closed circle, calculated by integrating the stress–strain curves using the trapezium rule, and the average moduli, open circle, calculated by finding the maximum of the derivative of the initial slope fitted using a 3rd order polynomial, as a function of hard content in the films. | ||
As the PS-latex content increases, particularly above the percolation threshold (estimated 31 vol% for spheres of equal size so slightly lower for this system with a size ratio of 1.15), this effect is most visible as the material begins to viscoelastically deform below this threshold but above this threshold there is a drastic drop in stress at a particular strain. This can be attributed to the PS mesh percolating structure across the entire material and since the test was performed below Tg,hard, it exhibits more solid-like properties. The shear strength or toughness, determined as the area under a stress–strain curve, is a good indicator of the energy required to be absorbed by the material to cause complete failure. The toughness was plotted against PS volume fraction, Fig. 6b, and shows an exponential increase from 0 to 35.64 vol%. This is likely again due to the formation of the PS mesh across the entire sample creating a structure which is more difficult to break. There is a distinct drop in toughness after 35.64 vol%. This is likely due to the lack of adhesion between the adhesive and the substrate as a result of decreased soft, tacky polymer, and the structure being predominately hard so more difficult to compress to release the tacky polymer when activated. This creates weaker interfaces at the lap joint which are the cause of the lap joint failure, as opposed to cohesive failure in the lower hard content systems.
It is a common observation with PSAs that peel force and shear strength are inversely proportional. This is also echoed in our linerless label system, creating structure to increase blocking resistance at ambient temperature reduces the systems ability to flow thus decreasing peel force. This can be seen from the frequency sweeps at room temperature, Fig. S21 and Table S2,† where the storage modulus at 1 Hz increases greatly, from 0.23 MPa to 36.78 MPa, after a film of PS (34.3 vol%) in PSA (65.7 vol%) is annealed. Similar results have been reported by Gurney and coworkers when using small hard particles and large soft particles with a size ratio of 5.4.24 A balance has been reached between these properties in the 35.64 vol% PS in PSA system such that it has a peel force between masking tape (2.64 ± 0.15 N) and cellotape (8.60 ± 0.57 N) for easy removal but a high shear strength so the label will not fall off or be easily rubbed off in transport or storage.
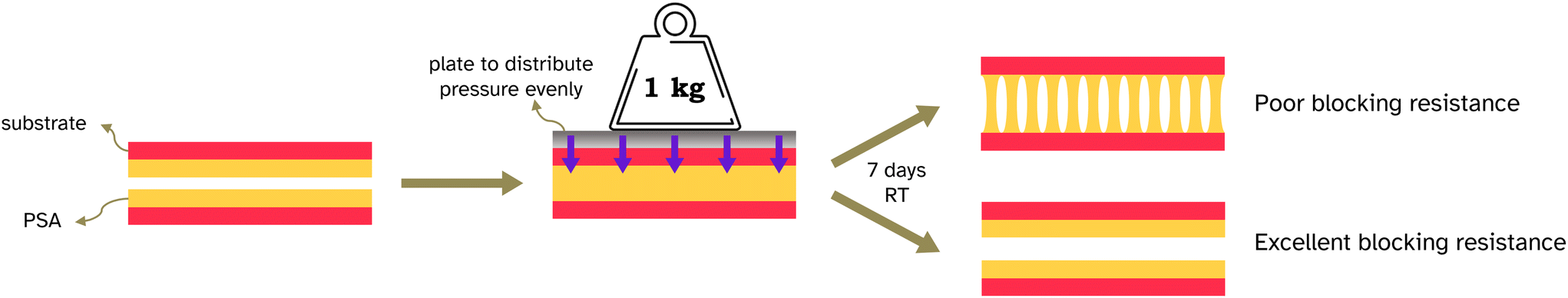 | ||
| Fig. 7 Diagram of the blocking test conducted. | ||
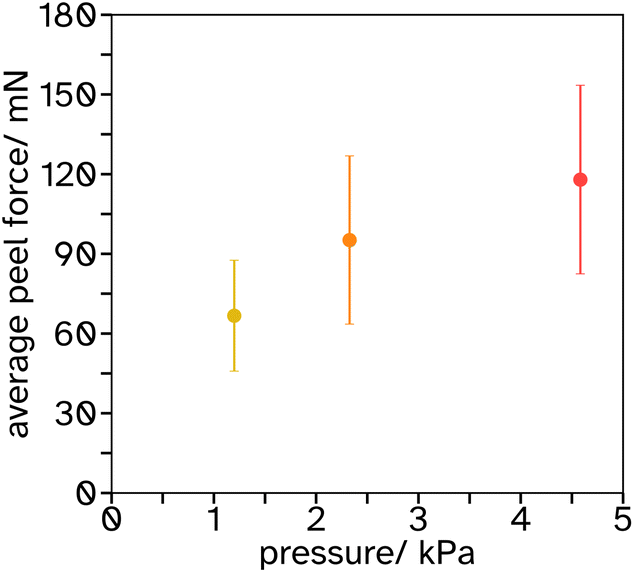 | ||
| Fig. 8 Results of peel tests when no activation of a 34.24 vol% PS in a PSA-latex film has occured, the average peel force is plotted as a function of pressure applied over a period of 7 days. | ||
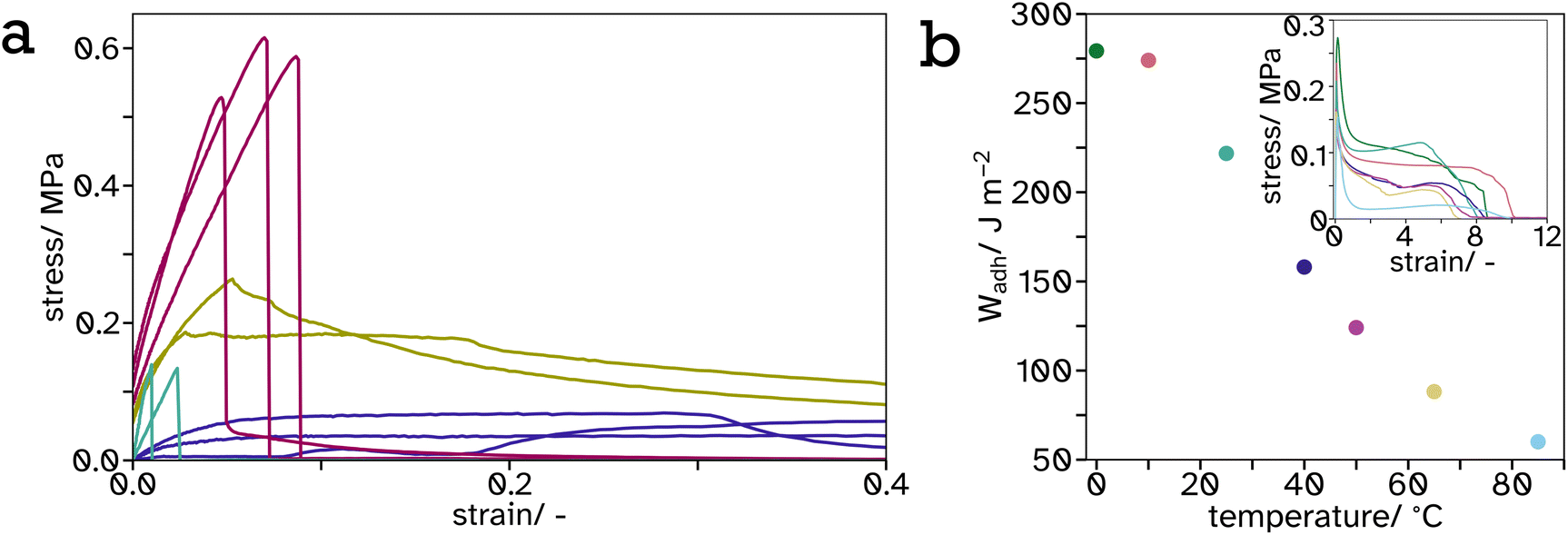 | ||
Fig. 9 Tack curves, measured at 65 °C, represented as stress–strain curves (a) of  0, 0,  25, 25,  34 and 34 and  100 vol% hard PS-latex in soft PSA-latex films. They are only shown here to 0.4 strain to highlight the behaviour of the harder samples, see Fig. S17† for full graph. The work of adhesion, Wadh, calculated from the area under the stress–strain curves (inset), plotted as a function of temperature (b) for tack tests of pure soft PSA-latex films. 100 vol% hard PS-latex in soft PSA-latex films. They are only shown here to 0.4 strain to highlight the behaviour of the harder samples, see Fig. S17† for full graph. The work of adhesion, Wadh, calculated from the area under the stress–strain curves (inset), plotted as a function of temperature (b) for tack tests of pure soft PSA-latex films. | ||
4. Conclusions
We demonstrated that it is possible to fabricate a linerless adhesive by having a PSA embedded into a hard 3D interconnected open cellular mesh as a support structure. Here we achieved this by film formation of mixtures of hard and soft latex particles. Analysis of the mesh showed a coarsened structure of the hard phase, the exact origin of which remains unexplained. We believe that our concept is applicable to a wide range of PSA systems, especially if the structural mesh support can also be manufactured by different means than in situ phase separation and coarsening. We hope that the idea will enthuse people to design similar systems, so that release liners and release coatings can be phased out. Of course, one can also try to find a greener alternative for the chemical composition of the hard and soft components.Author contributions
Conceptualization (SAFB), Data curation (EMB, PFW, SAFB, SH, EL, AU, IHP), Formal analysis (EMB, PFW, SAFB, IHP), Funding acquisition (SAFB), Investigation (EMB), Methodology (EMB, PFW, SAFB, SH, EL, AU, IHP), Project administration (EMB, SAFB), Supervision (SAFB), Validation (EMB, SAFB), Visualization (EMB), Writing – original draft (EMB, SAFB) Writing – review & Editing (EMB, PFW, SAFB, SH, EL, AU, IHP).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The X-Ray Computed Tomography (XCT) data used in this article was acquired using the Free-at-Point-of-Access scheme at the National Facility for X-Ray Computed Tomography (NXCT) and carried out at the Centre for Imaging, Metrology, and Additive Technologies (CiMAT) at the University of Warwick under the EPRSC Project Number (EP/T02593X/1). We thank the Polymer Characterization Research Technology Platform (RTP) at the University of Warwick for providing use of the following equipment: Anton Paar Litesizer 500 (0.3-2000 nm), TA Instruments DSC2500 and Shimazdu EZ-LX universal testing machine. We thank the Electron Microscopy Research Technology Platform (RTP) at the University of Warwick for providing use of the following equipment: Zeiss Gemini high-resolution scanning electron microscope and Tescan Amber scanning electron microscope. We thank Caterina Dasmi for her illustrations used in Fig. 1a–c. We thank UPM Raflatac for the donation of PSA-latex and contributing financially in parts to the work.References
- C. Creton, MRS Bull., 2003, 28, 434–439 CrossRef CAS.
- M. Sasaki, Y. Kashihara, Y. Urahama, T. Hirai, S. Fujii and Y. Nakamura, J. Appl. Polym. Sci., 2021, 138, 50767 CrossRef CAS.
- P. K. Dhal, A. Deshpande and G. Babu, Polymer, 1982, 23, 937–939 CrossRef CAS.
- M. D. Gower and R. A. Shanks, Macromol. Chem. Phys., 2005, 206, 1015–1027 CrossRef CAS.
- R. S. Gurney, A. Morse, E. Siband, D. Dupin, S. P. Armes and J. L. Keddie, J. Colloid Interface Sci., 2015, 448, 8–16 CrossRef CAS PubMed.
- M. Heydari, F. Sharif and M. Ebrahimi, RSC Adv., 2021, 11, 20557–20569 RSC.
- H. C. Tung, US Pat., US5455092A, The Dow Chemical Company, 1995 Search PubMed.
- E. W. Huber, US Pat., US2845728A, Topflight Corporation, 1958 Search PubMed.
- P. Lavanchy, US Pat., US3051588A, Johnson and Johnshon, 1962 Search PubMed.
- T. N. Gaunt and J. Waddington, UK Pat., EP0387916A2, John Waddington PLC, 1985 Search PubMed.
- E. N. De Nagybacczon, UK Pat., WO1990001764A1, Enagy Ltd, 1990 Search PubMed.
- K. O. Henderson, WO Pat., WO2016109199A1, Avery Dennison Corporation, 2016 Search PubMed.
- J. Empereur, M. N. Belgacem and D. Chaussy, Macromol. Mater. Eng., 2008, 293, 167–172 CrossRef CAS.
- M. Einsla, W. Griffith, D. Himmelberger, S. Zolynski, S. Zhang, T. Powell and D. Malotky, J. Appl. Polym. Sci., 2019, 136, 47048 CrossRef.
- J. Y. Cavaillé, R. Vassoille, G. Thollet, L. Rios and C. Pichot, Colloid Polym. Sci., 1991, 269, 248–258 CrossRef.
- J. M. Friel, UK Pat., EP0466409A1, Rohm and Haas Co., 1992 Search PubMed.
- M. P. J. Heuts, R. A. le Fêbre, J. L. M. van Hilst and G. C. Overbeek, in Film Formation in Waterborne Coatings, ed. T. Provder, M. A. Winnik and M. W. Urban, ACS Symposium Series, American Chemical Society, Washington, DC, 1996, vol. 648, pp. 271–285 Search PubMed.
- S. T. Eckersley and B. J. Helmer, J. Coat. Technol., 1997, 69, 97–107 CrossRef CAS.
- J. Feng, M. A. Winnik, R. R. Shivers and B. Clubb, Macromolecules, 1995, 28, 7671–7682 CrossRef CAS.
- R. H. Ottewill, H. J. M. Hanley, A. R. Rennie and G. C. Straty, Langmuir, 1995, 11, 3757–3765 CrossRef CAS.
- F. Deplace, M. A. Rabjohns, T. Yamaguchi, A. B. Foster, C. Carelli, C.-H. Lei, K. Ouzineb, J. L. Keddie, P. A. Lovell and C. Creton, Soft Matter, 2009, 5, 1440 RSC.
- A. Bellamine, E. Degrandi, M. Gerst, R. Stark, C. Beyers and C. Creton, Macromol. Mater. Eng., 2011, 296, 31–41 CrossRef CAS.
- T. Wang, P. J. Colver, S. A. F. Bon and J. L. Keddie, Soft Matter, 2009, 5, 3842 RSC.
- R. S. Gurney, D. Dupin, J. S. Nunes, K. Ouzineb, E. Siband, J. M. Asua, S. P. Armes and J. L. Keddie, ACS Appl. Mater. Interfaces, 2012, 4, 5442–5452 CrossRef CAS PubMed.
- S. Kirkpatrick, Rev. Mod. Phys., 1973, 45, 574–588 CrossRef.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3083–3090 CrossRef CAS.
- W. H. Stockmayer, J. Chem. Phys., 1944, 12, 125–131 CrossRef CAS.
- J. P. Fitzpatrick, R. B. Malt and F. Spaepen, Phys. Lett. A, 1974, 47, 207–208 CrossRef.
- M. J. Powell, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20, 4194–4198 CrossRef.
- R. M. Ziff and S. Torquato, J. Phys. A: Math. Theor., 2017, 50, 085001 CrossRef.
- D. He and N. N. Ekere, J. Phys. D: Appl. Phys., 2004, 37, 1848–1852 CrossRef CAS.
- F. Tronc, M. Li, J. Lu, M. A. Winnik, B. L. Kaul and J. Graciet, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 766–778 CrossRef CAS.
- F. Deplace, C. Carelli, S. Mariot, H. Retsos, A. Chateauminois, K. Ouzineb and C. Creton, J. Adhes., 2009, 85, 18–54 CrossRef CAS.
- E. Limousin, N. Ballard and J. M. Asua, Prog. Org. Coat., 2019, 129, 69–76 CrossRef CAS.
- Z. Zhu, Z. Yang, Y. Xia and H. Jiang, Mechanics of Soft Materials, 2022, vol. 4, p. 7 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the set up of peel, shear strength, and blocking tests (Fig. S1–S3), custom metal template for casting onto 8 mm parallel rheometry plates (Fig. S4), dynamic scanning calorimetric data of polymer latexes and mixtures (Fig. S5–S7 and S10), monomer conversion and hydrodynamic particle diameter data of synthesized polymer latexes (Fig. S8 and S9), 2D projection and full 3D render from micro-CT scan of model mesh (Fig. S11), micro-CT porosity analysis of model mesh (Fig. S12), FIB-SEM of the adhesive film (Fig. S13), SEM of the adhesive film varying annealing time (Fig. S14), thermal gravimetric analysis of poly(vinyl acetate)-based model mesh systems (Fig. S15 and S16, Table S1), optical images of fibrils pulled during peel testing (Fig. S17), raw data from peel tests after blocking analysis (Fig. S18), tack test data (Fig. S19 and S20), rheometer frequency sweeps (Fig. S21 and S22, Tables S2 and S3); two videos videoS1.mp4 showing a 3D render fly through for the micro CT data of the mesh structure, and videoS2.mp4 practical demo of prototype label use. See DOI: https://doi.org/10.1039/d3lp00224a |
| This journal is © The Royal Society of Chemistry 2024 |