
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDrug catalyzed polymerization yields one pot nanomedicines†
Paul Joshua
Hurst
 a,
Kyle J.
Gassaway
a,
Mohammed Faris
Abouchaleh
a,
Nehal S.
Idris
a,
Chelsea R.
Jones
a,
Chris A.
Dicksion
a,
James S.
Nowick
ab and
Joseph P.
Patterson
*ac
a,
Kyle J.
Gassaway
a,
Mohammed Faris
Abouchaleh
a,
Nehal S.
Idris
a,
Chelsea R.
Jones
a,
Chris A.
Dicksion
a,
James S.
Nowick
ab and
Joseph P.
Patterson
*ac
aDepartment of Chemistry, University of California, Irvine, Irvine, CA 92697, USA. E-mail: patters3@uci.edu
bDepartment of Pharmaceutical Sciences, Irvine, Irvine, CA 92697, USA
cDepartment of Materials Science and Engineering, Irvine, Irvine, CA 92697, USA
First published on 30th January 2024
Abstract
Ring-opening polymerization (ROP) is a powerful method for the synthesis of biocompatible and biodegradable polyester-based amphiphilic block copolymers, which are an excellent nanomaterial class for a wide range of pharmaceutical applications. These block copolymers are synthesized using a catalyst, which is typically purified out. In a separate step, the purified block copolymers are then assembled and drug-loaded for medical use. This multistep process limits the scalability of these nanomaterials restraining their industrial use. Recently, we developed a synchronous polymerization and self-assembly process for polyester-based block copolymer nanomaterials coined Ring-Opening Polymerization-Induced Crystallization-Driven Self Assembly (ROPI-CDSA). In ROPI-CDSA, an organocatalyst facilitates the chain extension of mPEG with L-lactide, yielding semicrystalline self-assemblies. Here, we demonstrate that pharmaceuticals with similar functional groups to ROP organocatalysts can catalyze ROPI-CDSA reactions, resulting in the formation of drug-embedded nanomaterials. The major advantage of this one pot approach is that no additional synthetic steps or purification are required. As a proof-of-principle study, we use two antibiotic drug molecules, chlorhexidine, and trimethoprim, as catalysts. Chlorhexidine acts as a co-initiator and a catalyst leading to drug conjugation whereas trimethoprim acts solely as a catalyst leading to drug encapsulation. The resulting drug-embedded block copolymer nanoparticles retain potent antibacterial activity. We anticipate that this strategy can be extended to other examples of PISA for the scalable production of drug-loaded polymer suspensions.
Introduction
Nanomedicine is a powerful tool for the development of new pharmaceuticals. Embedding drugs within nanomaterials is a strategy used to improve drug efficacy and reduce side-effects. In 1995, Doxil became the first ever FDA approved “nanodrug”.1 Doxil uses lipid vesicles to encapsulate and deliver nanocrystals of the chemotherapy drug doxorubicin. The lipid vesicle delivery system significantly reduces the cardiotoxic side effects of doxorubicin, making it safer than the free drug. More recently, a similar lipid system was used for the mRNA delivery system in the Moderna and Pfizer COVID vaccines.2 The lipid delivery system protects the mRNA from degradation and ensures its uptake into cells. Lipid delivery systems are versatile as they can be used for multiple therapies, however the technology hasn't significantly changed since it was developed in the 1960s.2 Amphiphilic copolymers (e.g. diblock copolymers), which are polymeric analogues of lipids, have been widely studied in academia as next generation drug delivery systems because they offer highly tunable chemical and physical properties.3,4 These properties enable them to be robust, thus allowing them to be used in a wider range of therapeutic applications than lipids. However, block copolymer delivery systems (e.g., micellar self-assemblies) have had limited use in industry as their self-assembly is typically not scalable. Recently, the development of polymerization-induced self-assembly (PISA) has emerged as a one pot solution to the scalability of block copolymer nanoparticles, resulting in suspensions up to 50% polymer wt. compared to around 1% polymer wt. for conventional self-assembly methods (e.g. solvent switch).5–8 Here, one pot refers to multiple transformations that convert starting materials into a final target without the isolation of any intermediates.9 In PISA, a soluble homopolymer is chain-extended by a monomer, that forms the solvophobic block. As a consequence of this polymerization, the growing polymer becomes increasingly less soluble, triggering self-assembly. PISA has been applied to a variety of polymer blocks and polymerization techniques.5,7,10–15 Furthermore, PISA is promising in the application of drug delivery, primarily through encapsulation or post polymerization functionalization.16–20Recently, we developed a one pot scalable synthesis for polyester-based block copolymer nanostructures coined Ring-Opening Polymerization-Induced Crystallization-Driven Self Assembly (ROPI-CDSA),14,21 one of the earliest examples of PISA for ring-opening polymerization (ROPISA).12,15,22–27 To date, ROPISA has applied to core-forming polymers derived from N-caroxyanhydrides,12,22,26L-lactide,14,21 salicylic acid o-carboxyanhydrides,15,23 carbonates,27 and lactones,24 with the processes using L-lactide, carbonates, and lactones containing a semicrystalline core. In our example of ROPI-CDSA, polyethylene glycol is chain extended with L-lactide using organocatalysts in toluene, a selective solvent to form poly(L)-lactide-b-polyethylene glycol (PLLA-b-PEG). The resulting semicrystalline self-assemblies can then be transferred to aqueous solutions via extraction or lyophilization and resuspension. Here, we show that pharmaceuticals with similar functional groups to ring-opening organocatalysts can catalyze ROPI-CDSA reactions, resulting in the formation of drug embedded nanomaterials. The major advantage of this one pot approach is that no additional synthetic steps or purification are required. As a proof-of-principle study, we use two antibiotic drug molecules as catalysts: chlorhexidine and trimethoprim. The resulting drug polymer nanoparticles are then characterized by cryoEM, WAXS, and FTIR. When suspended into water, the drug polymer nanoparticles retain potent activity as demonstrated by minimum inhibitory concentration antibacterial studies.
Results
Synthesis and characterization
ROP can be performed with a wide range of organocatalytic systems including triazabicyclodecene (TBD), diazabicycloundecene (DBU), and a thiourea-based catalyst paired with (−)-sparteine, a tertiary amine.14,21,28–30 The key functional group of TBD, guanidine, is present in a large number of drugs.31 For our study, we selected the bisguanidine, chlorhexidine, and the dihydropyrimidine, trimethoprim. Both drugs contain guanidine-like groups, are affordable, and can be purchased in their free base form (Fig. 1A and B). Furthermore, chlorhexidine and trimethoprim do not contain any amines or alcohols which would be expected to initiate ROP. Drugs containing alcohols have been shown to initiate but not catalyze ROP leading to drug conjugated polymers.32–36 Here, trimethoprim acts strictly as a catalyst as expected leading to the formation of drug-loaded polymer suspensions. However, chlorhexidine acts as both a catalyst and a co-initiator, along with the polyethylene glycol macroinitiator, leading to the formation of drug-conjugated polymer suspensions.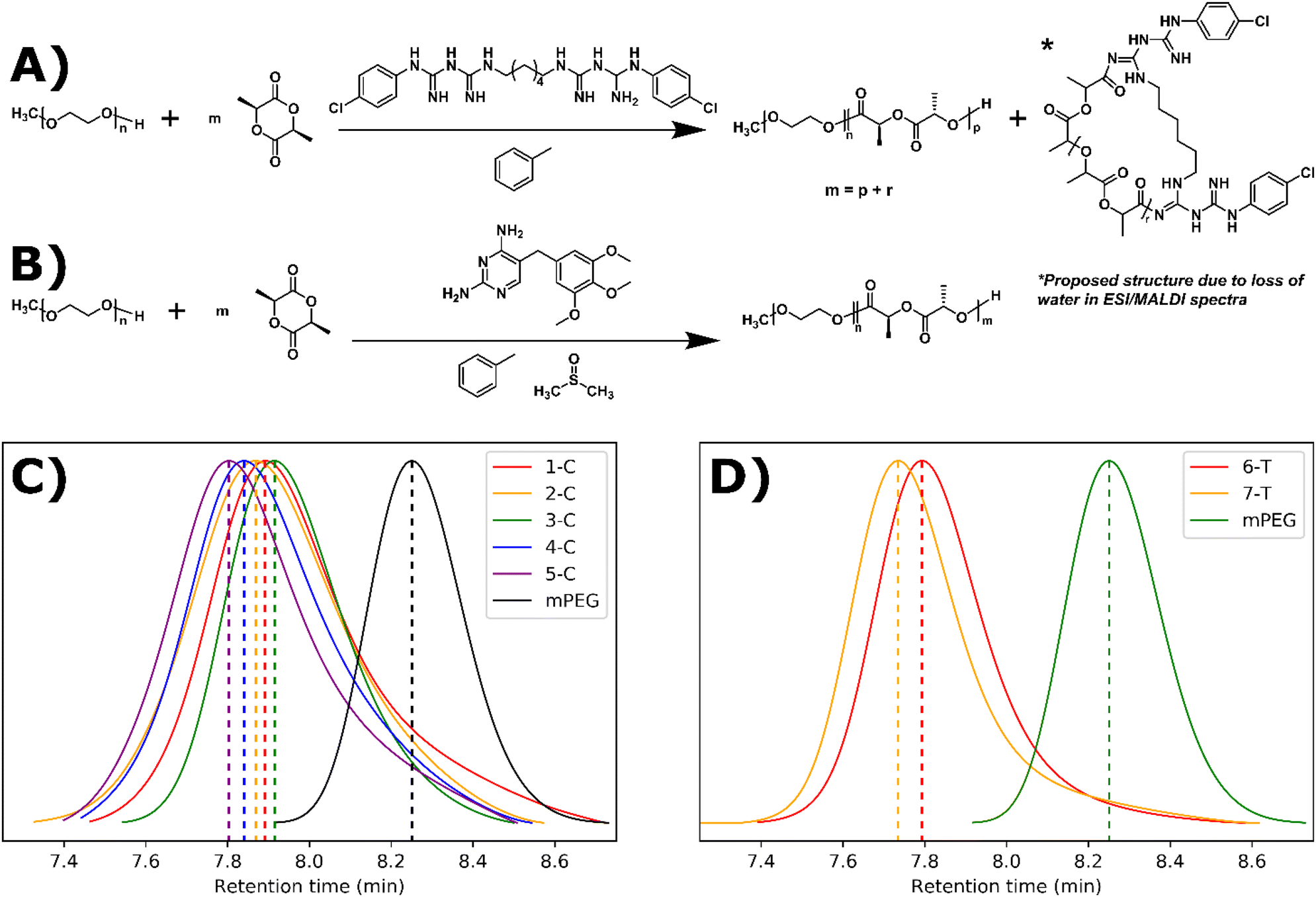 | ||
| Fig. 1 Synthetic scheme and GPC results for guanidine-drug catalyzed ROPI-CDSA: (A) chlorhexidine catalyzed and initiated synthesis of PLLAm-b-PEG45 and PLLAr-chlorhexidine showing a proposed structure for the latter. m, the total equivalents of L-lactide is equal to the equivalents of L-lactide growing off mPEG (p) and the equivalents of L-lactide growing off chlorhexidine (r). (B) trimethoprim catalyzed synthesis of PLLAm-b-PEG45. (C) and (D) GPC data for chlorhexidine and trimethoprim PLLAm-b-PEG45 series respectively. Note that the retention times of the trimethoprim series is lower than the chlorhexidine series, suggesting the trimethoprim-catalyzed polymer series reaches higher molecular weights than the chlorhexidine-catalyzed series. | ||
Chlorhexidine was found to efficiently catalyze the polymerization of L-lactide in toluene with the presence of a mono methylated polyethylene glycol (mPEG) macroinitiator. At 5% molar ratio to the monomer, >95% conversion was achieved in 30 minutes (Fig. 1A and Table 1). At 10% solids wt. (% of solution that is not solvent e.g., polymer/monomer, initiator, and catalyst), self-assembly occurred (as determined by visual inspection of turbidity and later by cryoEM) when using monomer to initiator ratios of 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 68:1 (1-C and 2-C). At 20% solids wt., self-assembly occurred at monomer to initiator ratios of 23:1, 45:1, and 68:1 (3-C, 4-C, and 5-C respectively). Gel permeation chromatography (GPC) was used to track chain extension and dispersity (Đ) (Fig. 1C). The Đ of the resulting polymers were between 1.12 and 1.33, indicating a relatively controlled polymerization. These dispersity values are comparable for those in our previous ROPI-CDSA study where TBD was used as the ROP catalyst.14 However, in a control sample where all conditions are identical to 1-C except chlorhexidine is replaced with DBU, 1-C exhibited a lower molar mass, as indicated by a higher retention time in the GPC data (7.95 min vs. 7.45 min) (Fig. S1†). Samples at 20% solids wt. also show better dispersity and lower retention times by GPC than samples at 10% solids wt., which was different from our previous TBD results.
:1 and 68:1 (1-C and 2-C). At 20% solids wt., self-assembly occurred at monomer to initiator ratios of 23:1, 45:1, and 68:1 (3-C, 4-C, and 5-C respectively). Gel permeation chromatography (GPC) was used to track chain extension and dispersity (Đ) (Fig. 1C). The Đ of the resulting polymers were between 1.12 and 1.33, indicating a relatively controlled polymerization. These dispersity values are comparable for those in our previous ROPI-CDSA study where TBD was used as the ROP catalyst.14 However, in a control sample where all conditions are identical to 1-C except chlorhexidine is replaced with DBU, 1-C exhibited a lower molar mass, as indicated by a higher retention time in the GPC data (7.95 min vs. 7.45 min) (Fig. S1†). Samples at 20% solids wt. also show better dispersity and lower retention times by GPC than samples at 10% solids wt., which was different from our previous TBD results.
| Sample ID | Catalyst (% mol) | Monomer to mPEG Ratio | Solids wt. % | Đ | % Conversionb | Average PLLA DP on mPEGc |
|---|---|---|---|---|---|---|
| a Determined through GPC. b Determined by 1H NMR of crude reaction mixtures. c Determined through 1H NMR following TLC plate chromatography separation in 100% ethyl acetate (bottom spot). | ||||||
| 1-C | Chlorhex. (5) | 45 (90) | 10 | 1.33 | >95 | 18 |
| 2-C | Chlorhex. (5) | 68 (135) | 10 | 1.21 | >95 | 20 |
| 3-C | Chlorhex. (5) | 23 (45) | 20 | 1.12 | >95 | 13 |
| 4-C | Chlorhex. (5) | 45 (90) | 20 | 1.13 | >95 | 24 |
| 5-C | Chlorhex. (5) | 68 (135) | 20 | 1.19 | >95 | 30 |
| 6-T | Trimethop (2.5) | 23 (45) | 20 | 1.12 | 85 | 20 |
| 7-T | Trimethop (2.5) | 45 (90) | 20 | 1.15 | 54 | 25 |
One rationalization for the lower molar mass is that chlorhexidine acts as both a catalyst and a co-initiator with mPEG. This co-initiation would result in a mixture of PLLA-b-PEG and chlorhexidine acylated to PLLA at the bisguanidine. Chlorhexidine has 2 pairs of pKa values of its corresponding conjugate acids (10.3 and 2.2).37 In contrast, the conjugate acid of TBD has pKa ≥ 19.4.38 Due to the twofold symmetry of chlorhexidine, we predicted that chlorhexidine conjugation would either have 2 or 4 active acylation sites. To test this hypothesis, we mixed chlorhexidine with excess vinyl acetate (1:20) following a modified procedure by Hedrick et al.28 Here, the vinyl alkoxide leaving group of the acylation readily rearranges as an aldehyde making the N-acylation irreversible under the experimental conditions. Two equivalents of vinyl acetate reacted with chlorhexidine confirming the presence of two active acylation sites as shown by 1H NMR (Fig. S2, Table S1 and Scheme S1†). Following the acylation with vinyl acetate, 2 equivalents of benzyl alcohol were added. Benzyl alcohol can cleave the amide, undoing the N-acylation. With excess vinyl acetate, this led to a total turnover of 2.75 equivalents of vinyl acetate. This experiment shows that although the alcohol was able to cleave the chlorhexidine amide, it was unable to do so to full conversion. To further test the hypothesis that chlorhexidine acts as a catalyst and an initiator, chlorhexidine was reacted with L-lactide in dichloromethane to produce homopolymers with Đ > 1.25 (Fig. S3 and Scheme S2†). Matrix assisted laser desorption/ionization (MALDI) and electron spray ionization (ESI) mass spectra of these homopolymers revealed the presence of a 2 chlorine isotope pattern consistent with the conjugation of chlorhexidine to the poly(L)-lactide chains (Fig. S4 and 5†). MALDI and ESI data also revealed the presence of chlorhexidine initiated homopolymers in samples 1-C through 5-C (Fig. S6 and 7†). ESI and MALDI mass spectrometry both revealed a loss of 18 for all polymer peaks, indicating the removal of either H2O or NH4. Loss of NH4 is ruled out due to the odd mass values of the peaks. Loss of H2O could be the result of an intramolecular cyclization resulting from a substitution reaction (Scheme S3†). However, we were not able to definitively prove this (see ESI† discussion). 1H NMR and Fourier transform infrared (FTIR) spectra each confirmed that chlorhexidine was chemically modified through the shifting of peaks (e.g. aromatic peak shift), likely through the N-acylation of guanidine groups (Fig. S8†), and the absence of important chlorhexidine stretches, for example 1660 cm−1 in Fig. 2A. These stretches show up in the FTIR spectra of polymer sample (2-C) which has been spiked with chlorhexidine (Fig. S9†). TLC plate chromatography in 100% ethyl acetate was able to separate out the PLLA-b-PEG block copolymer (bottom spot) from the PLLA-chlorhexidine homopolymer (top spot), as verified by NMR (Fig. S10–12†). Following separation, NMR showed the amount of PLLA per PEG unit to give an average degree of polymerization (DP) of PLLA on the resulting PLLA-b-PEG block copolymers (Table 1 and Fig. S10†).
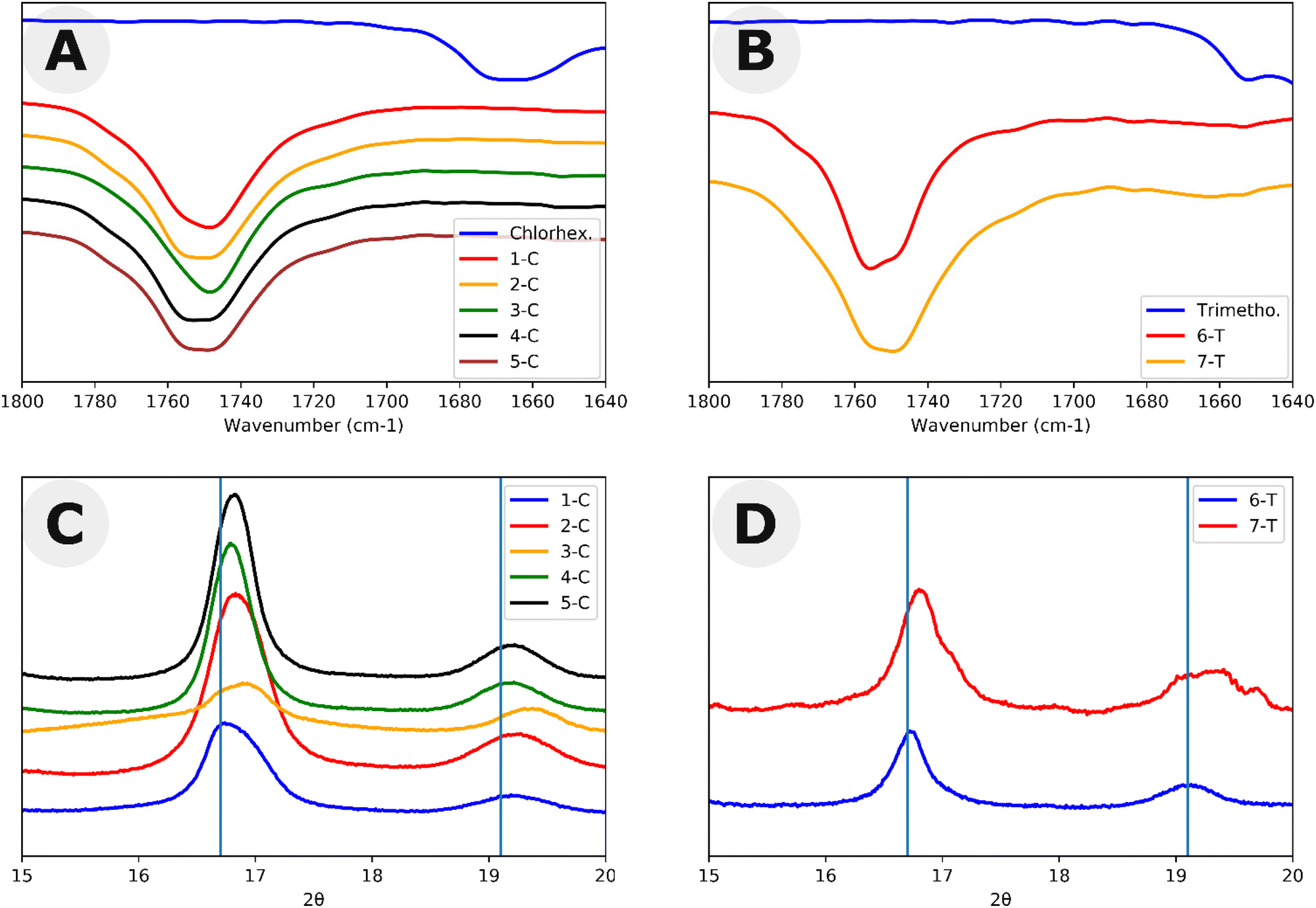 | ||
| Fig. 2 Structural characterization of polymers in this study. FTIR carbonyl spectra for (A) chlorhexidine-catalyzed polymers and (B) trimethoprim-catalyzed polymers. WAXS spectra of the two dominant peaks of (C) chlorhexidine-catalyzed polymers and (D) trimethoprim-catalyzed polymers. Lines represent peaks of 16.7 and 19.1 which are the positions of the two largest WAXS peaks in PLLA. | ||
In addition to the study of chlorhexidine, trimethoprim was found to catalyze the polymerization of L-lactide in a solution of toluene ∼5% DMSO with the presence of a mono methylated polyethylene glycol macroinitiator. Here, a monomer to initiator ratio of 23:1 gave a conversion of about 85%, and a ratio of 45:1 only reached 54% after reaction mixtures were stirred for 2 weeks. 1H NMR peaks of trimethoprim did not shift following the reaction, signifying that there was not any drug conjugation (Fig. S13†). FTIR also indicated the incorporation of many trimethoprim peaks (e.g. around 1700 cm−1), although due to the low relative amounts of trimethoprim, these peaks can be difficult to visualize (Fig. 2B). 6-T and 7-T have similar monomer to mPEG initiator ratios and solids wt% to 3-C and 4-C respectively but have lower retention times and thus higher molar masses despite having lower conversions. These data further suggest that a significant amount of L-lactide polymerizes off the chlorhexidine in the chlorhexidine polymer series.
Structural and morphological studies
Both chlorhexidine and trimethoprim samples produced turbid suspensions in toluene. Unlike previous ROPI-CDSA studies,14,21 none of these samples produced organogels. These mixtures could further be studied through lyophilization and resuspension into water or extraction into water from toluene. Lyophilized powders were studied by FTIR and wide-angle X-ray scattering (WAXS) (Fig. 2 and Fig. S14–17† for full spectra). FTIR shows poly(L)-lactide (PLLA) crystallinity in all samples, as signaled by the dual carbonyl stretch (Fig. 2A and B).39 WAXS of samples showed offsets from 16.7, which is the most stable peak position for the PLLA peak (Fig. 2C and D).39 These offsets, present in all samples, suggest that the semicrystalline structure is slightly different than standard PLLA as well as PLLA-b-PEG in previous ROPI-CDSA studies.14,21,39 The chlorhexidine samples have an estimated crystallinity ranging from 10% (3-C) and 11% (1-C) to 15% (2-C, 4-C) and 16% (5-C), whereas all trimethoprim samples have much lower crystallinity of around 6% for all samples. The lower crystallinity of the trimethoprim catalyzed samples could be a consequence of the lower conversion and presence of DMSO.Cryogenic electron microscopy was also performed on all samples to determine the morphologies after transfer to water (Fig. 3 and Fig. S18†). As shown in our previous work, PLLA based toluene suspensions retain nanomorphology upon transfer to water.14 Samples 1-C through 5-C contained a mixture of morphologies typically seen in PLLA-b-PEG such as lamellae and fibers as well as compound vesicles40 not typically seen (Fig. 3A–D). These compound vesicles could be a consequence of the sample containing a mixture of block copolymer and chlorhexidine-conjugated PLLA polymer. Samples 6-T and 7-T show lamellae and lamellar vesicles containing less morphological variation than 1-C through 5-C (Fig. 3E and F). The differences in morphology are likely a consequence of drug-conjugation present in the chlorhexidine systems, but they could also be caused by differences in polymerization kinetics between chlorhexidine (fast polymerization) and trimethoprim (slow polymerization).21
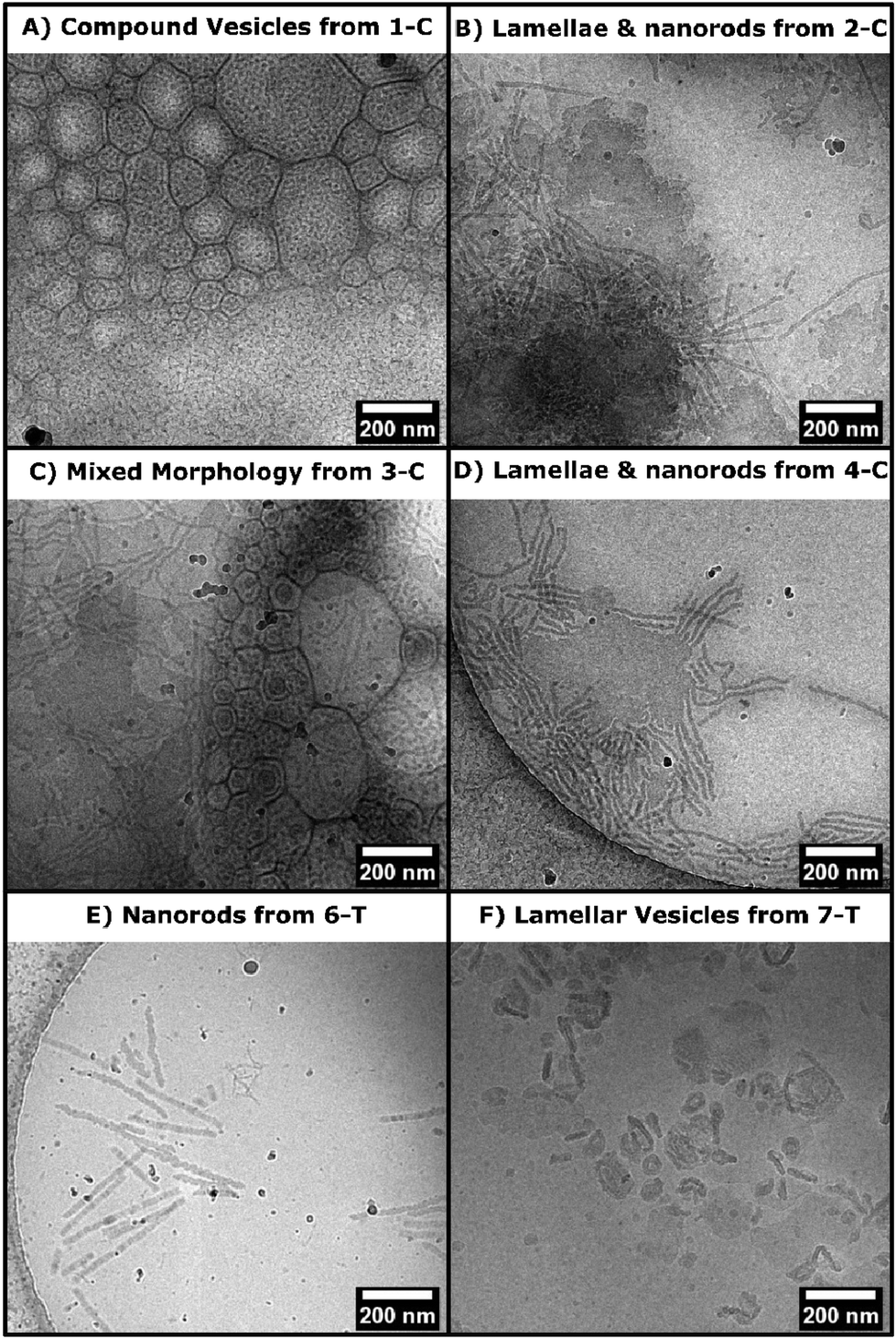 | ||
| Fig. 3 CryoEM micrographs from select drug polymer samples (as labelled on image). A–D) Morphologies seen in samples 1-C to 5-C. E–F) Morphologies seen in samples 6-T and 7-T. | ||
Antibacterial studies
Antibacterial studies were carried out on aqueous resuspensions of the drug/polymer nanoparticles. Three types of bacteria, two Gram positive, B. subtilis and S. epidermidis, and one Gram negative, E. coli, were used in minimum inhibitory concentration (MIC) studies against four samples, two controls: free-base chlorhexidine, and free-base trimethoprim and two experimental samples: 4-C and 6-T. Both experimental samples were prepared from resuspension following lyophilization which ensures that the total amount of drug used in the syntheses are included in the aqueous formulations as no solids are lost during lyophilization (Table 2). Additional studies were performed on extracted samples (Table S4†). MIC values for free chlorhexidine (0.125 and 0.25 μg mL−1) are on the lower end of what is reported in the literature,41–43 but variations in chlorhexidine MIC are common due to variations on testing of the various salt forms as well as the free base form, as well as expected variations in MIC studies.44 The MIC values of free trimethoprim are similar to those reported in the literature.45,46 All polymer samples show antibacterial activity against all three types of bacteria. It should be noted that PLLA-based polymers do not have antibacterial properties.47,48 The polymer samples had higher MIC values than those of the free drugs, which could be from both slower release kinetics, as polymeric formulations prolong drug release (Table 2).49,50 In particular, 4-C has significantly higher MIC values than the free chlorhexidine MIC values, which could be due to the chlorhexidine acylation. The ratio of drug to polymer in all our formulations is commensurate to drug polymer ratios in other antibacterial formulations.49,50 The conjugation efficiency of 4-C and all other chlorhexidine/polymer samples is estimated at 100% as all chlorhexidine is incorporated into polymer, evident by 1H NMR (Fig. S8†) and MALDI/ESI (Fig. S6 and 7†). The encapsulation efficiency of trimethoprim in 6-T was experimentally determined to be approximately 82%. Dialysis of 6-T in aqueous solution also gave a slower release profile when compared to free trimethoprim (Fig. S19†).| Bacteria | Free base chlorhexidine | 4-C | Free base trimethoprim | 6-T |
|---|---|---|---|---|
| B. subtilis (ATCC 6051) | 0.25 | 2 | 0.25 | 0.5 |
| S. epi. (ATCC 14990) | 0.125 | 1 | 0.5 | 1 |
| E. coli (ATCC 10798) | 0.25 | 2 | 0.25 | 1 |
Discussion
In developing the drug-catalyzed ROPI-CDSA, we needed to understand the nature of the ROP catalysis of chlorhexidine and trimethoprim. Generally, as is the case with TBD, ROP organocatalysis proceeds through two mechanisms: dual hydrogen bonding and through an acylation intermediate, with the latter being less energetically favorable.51,52 Previous literature shows that an acyclic analogue of TBD could perform ROP through a dual hydrogen bonding mechanism but at a depressed rate when compared to TBD.51 Our data shows chlorhexidine, which is acyclic, can catalyze ROP of L-lactide; however, some of the resulting PLLA remains tethered to the chlorhexidine (see ESI† discussion). Waymouth et al.,53 showed that TBO, a bicyclic guanidine made of two five membered rings (instead of six membered rings in TBD), could undergo acylation but was unable to deacylate. This observation was rationalized by DFT studies that showed that the acyl group in N-acyl TBO was stabilized due to the adoption of a planar configuration with respect to the guanidine. In contrast, N-acyl TBD adopted a nonplanar configuration, destabilizing the N-acyl bond, allowing for efficient turnover in ROP catalysis. Here, chlorhexidine likely adopts an equilibrium concentration of N-acylated chlorhexidine and free chlorhexidine, enabling for ROP catalysis but with incomplete turnover, resulting in drug polymer conjugation on some of the growing polymer blocks. Even after the chlorhexidine reacts with L-lactide, it remains active, suggesting that the chlorhexidine facilitates ROP catalysis through a dual hydrogen bonding mechanism and facilitates conjugation through an acylation mechanism.For the trimethoprim system, the lack of conjugation with PLLA suggests that trimethoprim either operates through a hydrogen bonding mechanism, through acylation with efficient turnover, like TBD, or a combination of the two. Reacting excess vinyl acetate with trimethoprim did not produce any aldehyde like it did with chlorhexidine, suggesting that acylation is not a mechanistic pathway for trimethoprim catalyzed ROP (Fig. S20†). This lack of acylation may explain why trimethoprim performs ROP at a much slower rate (>10 days) compared to chlorhexidine (30 min) similar to the comparison between the aforementioned acyclic TBD analog and TBD.51
Based on these data we propose that the drug ROPI-CDSA approach produced two different drug delivery systems: a drug-conjugated (prodrug) system with chlorhexidine, with full drug conjugation and an encapsulated system with trimethoprim, with high EE% (>80%) (see ESI† discussion). Typically, a block copolymer-based drug delivery system requires a separate polymerization, purification, and drug conjugation,54,55 or encapsulation step (Fig. 4).56,57 However, we recognize that the exact number of steps necessary and the order of the steps may vary. The drug ROPI-CDSA approach is able to produce block copolymer-based drug delivery system in one pot.
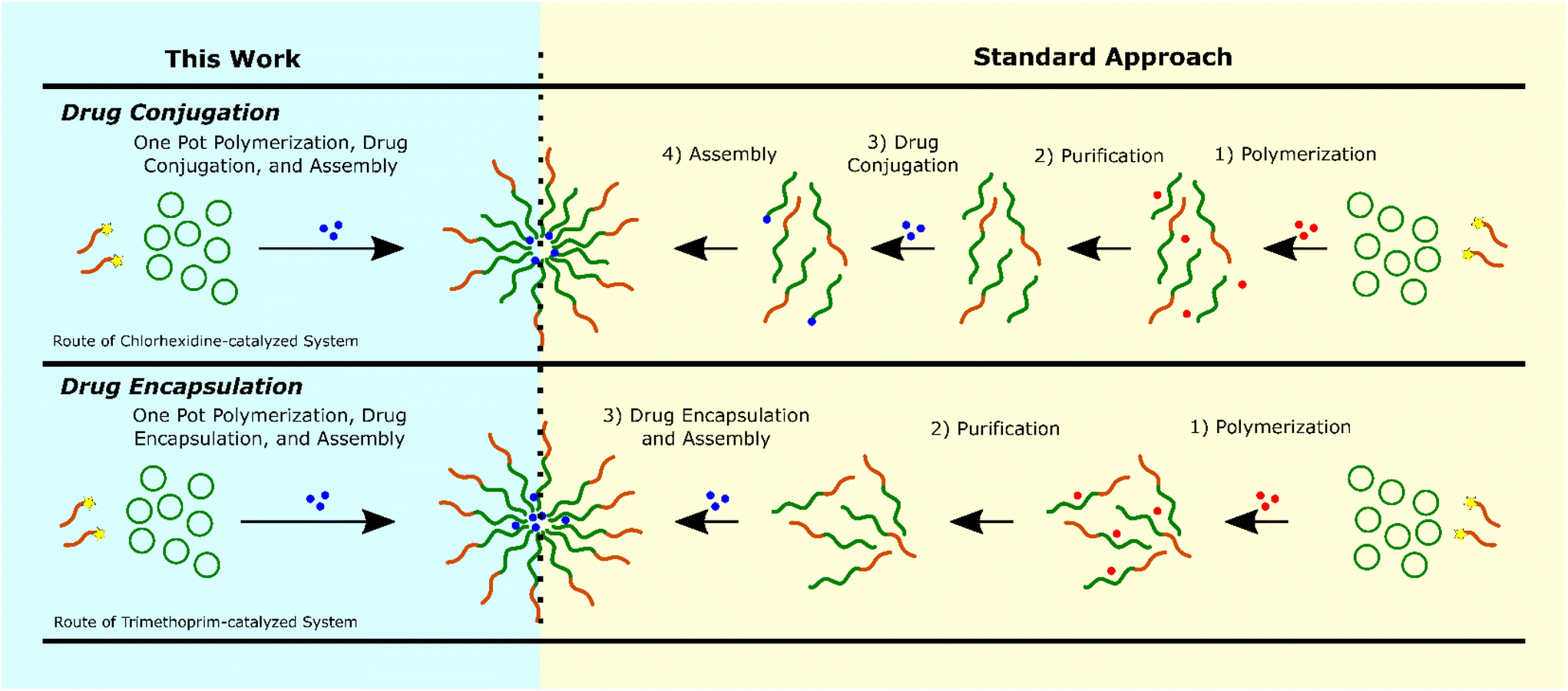 | ||
| Fig. 4 The required synthetic steps for block copolymer-based drug conjugation (top) and drug encapsulation (bottom) with our one pot approach on the left (highlighted in blue) and the current standard approach on the right (highlighted in yellow). Note that this work combines all previous synthetic steps without the need for purification. Additional preparation for both techniques may involve transfer to water or spin-coating which are relatively simple steps listed in this figure. | ||
Conclusion
In summary, we have devised a new, scalable, one pot method for producing nanomedicines coined Drug-catalyzed ROPI-CDSA. We demonstrate that pharmaceuticals can catalyze and create drug carrier systems if they possess the correct functional groups. When paired with a PISA system such as ROPI-CDSA, one pot nanoparticle formulations can be synthesized. With transfer to aqueous suspensions, the resulting polymer drug nanoparticles retained antibacterial activity. Native ROPI-CDSA solutions could also be spin coated to create antibacterial medical devices such as wound sutures and catheters, applications in which chlorhexidine and trimethoprim are currently employed. Future work could involve using other guanidine-based drugs as catalysts such as streptomycin, an antibiotic, which would likely act as a catalyst and an efficient initiator as streptomycin has many alcohol groups or proguanil, an antimalarial drug, which would likely behave similarly to chlorhexidine. We could also branch away from guanidines and explore catalytic behavior among other drugs such as those containing thioureas, ureas, and related compounds as these functional groups are common ROP catalysts.30,58 Among this class are, 2-propylthiouracil, an inexpensive thyroid drug, as well as a slew of more complex urea- and thiourea-containing antitumor and anticancer therapeutics.59 We believe this drug-catalyzed ROPI-CDSA will inspire the development of a wide range of drug-catalyzed reactions to produce one pot nanomedicines, particularly from PISA-based formulations. Most drugs are highly functionalized organic molecules, and through careful selection they can act as organocatalysts. Organocatalysis has played a major role in the development of new small molecule drugs; it is both exciting and timely that organocatalysis can play a role in the development of the next generation of nanodrugs.Experimental
Materials
mPEG45 (MW = 2000) (Sigma-Aldrich) was azeotropically distilled ×2 in toluene and high-vacuumed overnight. L-Lactide (TCI) was recrystallized in toluene ×3. Anhydrous toluene (99.8%), and DBU obtained from Sigma-Aldrich and stored under 4 Å molecular sieves. Benzoic acid (Fisher Chemical), chlorhexidine (Sigma-Aldrich), trimethoprim (MP Biomedicals) were used without further purification with trimethoprim being stored in the dark. DMSO was obtained from a dry solvent still. Vinyl acetate and benzyl alcohol were obtained from Sigma-Aldrich and were degassed and stored under molecular sieves. Chemicals were stored in a dry-N2 atmosphere glovebox. Reactions were performed in a N2 glovebox. 1H NMR spectra were collected on a 500 MHz Bruker Avance spectrometer in CDCl3. 13C and COSY and HMQC spectra were collected on a 600 MHz Bruker Avance spectrometer in CDCl3.Chlorhexidine-catalyzed/co-initiated ROPI-CDSA
Amounts are for 4-C (see Table S2† for all synthetic conditions). mPEG45 (80 mg, 0.04 mmol) was added to a colloidal solution of 5 mol% (relative to L-lactide) of chlorhexidine (45.5 mg, 0.09 mmol) in 1.78 mL of toluene (20% solids w/w). L-Lactide (259 mg, 1.8 mmol) was then added to the resulting clear solution and allowed to stir for 30 minutes at room temperature. The reaction was quenched with 0.05 mL of saturated benzoic acid toluene solution. The reaction mixture was allowed to stir for a day at 400 rpm prior to any structural, morphological, or antibacterial studies. 1H NMR (500 MHz, CDCl3) δ 7.64–7.28 (multiple peaks, chlorhexidine aromatics) 5.16 (q, J = 7.0 Hz, CH, PLLA backbone), 3.72–3.59 (m, CH2 PEG backbone), 3.54 (dd, J = 5.6, 3.6 Hz, CH2, PEG), 3.37 (s, 3H, terminal CH3 PEG), 1.58 (d, J = 6.7 Hz, CH3 PLLA backbone), 1.50 (dd, J = 14.7, 7.0 Hz, terminal CH3 PLLA). Note: other chlorhexidine-based peaks overlap with other polymer peaks.Trimethoprim-catalyzed ROPI-CDSA
Amounts are for 6-T (see Table S3† for all synthetic conditions). 2.5 mol% (relative to L-lactide) of trimethoprim (13.2 mg, 0.045 mmol) was dissolved in 0.1 mL DMSO. mPEG45 (160 mg, 0.08 mmol) was dissolved in toluene. Both solutions were mixed and L-lactide (259 mg, 1.8 mmol) was added. The solution was stirred for 2 weeks at room temperature. Stirring was kept at 400 rpm for reproducibility. 1H NMR (500 MHz, CDCl3) δ 5.16 (q, J = 7.0 Hz, CH, PLLA backbone), 3.72–3.59 (m, CH2 PEG backbone), 3.54 (dd, J = 5.6, 3.6 Hz, CH2, PEG), 3.37 (s, 3H, terminal CH3 PEG), 1.58 (d, J = 6.7 Hz, CH3 PLLA backbone), 1.50 (dd, J = 14.7, 7.0 Hz, terminal CH3 PLLA).Chlorhexidine-catalyzed ROP of PLLA homopolymer
L-lactide (259 mg, 1.8 mmol) was added to a solution of chlorhexidine (75.8 mg, 0.15 mmol) in 2.96 mL of dichloromethane and allowed to stir for 30 minutes. The reaction was quenched with 0.05 mL of saturated benzoic acid in toluene. Different ratios of L-lactide to chlorhexidine were tested keeping all synthetic conditions identical except for altering the amount of L-lactide. 1H NMR (500 MHz, CDCl3) δ 7.64–7.28 (multiple peaks, chlorhexidine aromatics) 5.16 (q, J = 7.0 Hz, CH, PLLA backbone), 1.58 (d, J = 6.7 Hz, CH3 PLLA backbone), 1.50 (dd, J = 14.7, 7.0 Hz, terminal CH3 PLLA). Note: other chlorhexidine-based peaks overlap with other polymer peaks.Acylation control
Following a modified procedure by Hedrick et al.28 20 equivalents of vinyl acetate (344 mg, 4 mmol) was added to a suspension of chlorhexidine (101 mg, 0.2 mmol) in a mixture of dichloromethane (2.0 mL) and DMSO (0.3 mL). Conditions were also varied (see Table S1†). Ratios of the 1H NMR peaks of the aldehyde to the vinyl acetate were compared to measure conversion. Following this acylation, a de-acylation was carried out to the existing solution by adding 2 equivalents of benzyl alcohol (43.3 mg, 0.4 mmol) and then measuring the aldehyde to vinyl acetate ratio again using 1H NMR.TLC Chromatography separation and NMR
Crude polymer solutions were roto-evaporated with gentle heating (30 °C) and dissolved in THF. The resulting solutions were dropped onto a large glass-backed TLC plate repeatedly to guarantee an appreciable amount of material. The TLC plate was then developed in 100% ethyl acetate. Two bands were detected, one with an Rf ≈ 0.8 and another with an Rf ≈ 0.05. Each band was scraped into a beaker and dissolved with THF. The slurries were filtered and the resulting solution was roto-evaporated with gentle heating and high vacuumed to remove residual solvent. 1H NMR, 13C NMR, COSY, and HMQC were collected (CDCl3) from the polymers from the bottom and the top TLC spots as well as a crude reaction mixture.Mass spectrometry (MALDI and ESI)
Matrix assisted laser desorption/ionization (MALDI) mass spectrometry was performed using an AB sciex TOF/TOF 5800 system. A linear low mass positive mode was used to obtain mass spectra. MALDI samples were prepared following a modified procedure by Ji et al.60 Matrix solutions were prepared by dissolving DCIB (3,5-dichloro-2-hydroxy-N-isobutylbenzamide) in THF at 10 mg mL−1. Sample solutions were prepared by dissolving samples in THF at 10 mg mL−1. NaI was dissolved in MeOH at 10 mg mL−1 to form cationization solutions. These three solutions were combined to form a ratio 10:1:1 of matrix, sample, and cationization agent, respectively. A 1-μL volume of each combined solution was pipetted on the target slide.
Electrospray ionization (ESI) mass spectrometry was performed on a Waters LCT Premier operating in ESI+ mode. A stock solution of 1 mg mL−1 was diluted in MS grade MeOH to 5 μg mL−1, and 10 μL were injected through a capillary with a voltage of 3.0 kV, with the desolvation gas at 300 °C and the source at 100 °C.
Structural/crystallinity studies
Wide-angle X-ray scattering (WAXS) patterns were measured on a Rigaku Smart lab X-ray diffractometer in Bragg–Brentano diffraction mode utilizing X-rays generated at 40 kV and 44 mA with Cu Kα irradiation (step size 0.02°, speed 1.0, IS 0.5°, RS1 4.0°, RS2 13 mm). Approximately 20 mg of a lyophilized sample was used in measurements. Crystallinity was estimated using the Smart lab software after peaks were assigned to PLLA-b-PEG. Fourier transform infrared (FTIR) absorbance spectra were collected on a Jasco 4700 FTIR from lyophilized samples. Prior to WAXS and FTIR samples were lyophilized by freezing the toluene solutions (0.5 mL volume) in a round-bottom flask with liquid nitrogen followed by sublimation using a vacuum pump.Resuspension in water
Prior to cryoEM studies and MIC assays, reaction mixtures in organic medium were resuspended in water (chlorhexidine samples) or 95:5 water:DMSO (trimethoprim samples) through either extraction or lyophilization. Extraction was performed by diluting a small volume of the reaction mixture (∼5–10 μL) into uncapped vial of excess water (∼2 mL) and vortexing the solution briefly followed by gentle sonication for 5 minutes. The vials were capped after several hours, allowing the toluene to evaporate. Lyophilization was performed by freezing 0.5 mL of the reaction mixture in liquid nitrogen and putting it under high vacuum on a Schlenk line for 2 hours. The freeze-dried powder was weighed out and resuspended in water through vortexing briefly then gently sonicating for 10 minutes. Both techniques allow for facile control of concentration of antibiotics as no solids material is lost during these procedures.
CryoEM studies
Cryo-TEM samples were prepared from solutions previously prepared onto Quantifoil R2/2 (Electron Microscopy Sciences) grids. Grids were glow discharged for 70 s to increase hydrophilicity prior to sample loading. Vitrification was carried out by an Automatic Plunge Freeze ME GP2 (Leica Microsystems) with 3 μL of sample. Grid preparation was performed at 95–99% humidity and the grids were blotted for 3 s prior to plunging into liquid propane. Samples were then placed on a Gatan CryoEM holder and imaged on a JEOL 2100 TEM using a Schottky type field emission gun operating at 200 keV. Images were recorded using Serial EM software with a Gatan OneView CMOS camera at 4k × 4k resolution. Prior to cryoEM samples prep, reaction mixtures in toluene were extracted and diluted in water to give samples with concentrations ranging from 16–64 μg drug per mL.MIC assays
Bacillus subtilis (ATCC 6051), Staphylococcus epidermidis (ATCC 14990), and Escherichia coli (ATCC 10798) were cultured from glycerol stocks in Mueller-Hinton broth overnight in a shaking incubator at 37 °C. An aliquot of the antibiotic/polymer stock solution (stock solution: 5% DMSO for trimethoprim samples, 100% water for chlorhexidine samples) was diluted with Mueller-Hinton broth to make a 64 μg antibiotic per mL. A 200 μL aliquot of the solution was transferred to a sterile, untreated 96-well plate. Two-fold serial dilutions were made with media across a 96-well plate to achieve a final volume of 100 μL in each well. These solutions had the following concentrations: 64, 32, 16, 8, 4, 2, 1, 0.5, 0.25, 0.125, and 0.0625 μg mL−1. The overnight cultures of each bacterium were diluted with Mueller-Hinton broth to an OD600 of 0.075 as measured for 200 μL in a 96-well plate. The diluted mixture was further diluted to a 1 × 106 CFU mL−1 with Mueller-Hinton media. A 100 μL aliquot of the 1 × 106 CFU mL−1 bacterial solution was added to each well in the 96-well plates, resulting in final bacteria concentrations of 5 × 105 CFU mL−1 in each well. As 100 μL of bacteria were added to each well, the compounds were also diluted to the following concentrations: 32, 16, 8, 4, 2, 1, 0.5, 0.25, 0.125, 0.0625, and 0.03125 μg mL−1. The plate was covered with a lid and incubated at 37 °C for 16 h. The OD600 were measured using a 96-well UV/vis plate reader (MultiSkan GO, Thermo Scientific). The MIC values were taken as the lowest concentration that had no bacteria growth. Each MIC assay was run in triplicate (technical replicates). A single row with just the serial diluted antibiotic and no bacteria was used as a control for opacity. For MIC assays of trimethoprim, the antibiotic stock solution was diluted with Mueller-Hinton broth to make a 16 μg mL−1 solution.Encapsulation efficiency and release study
Following the resuspension procedure described above, 500 μL of a resuspended trimethoprim-catalyzed sample was then transferred into a 500 μL Amicron Ultra 3K centrifugal filter device, which was then inserted in a pre-weighed microcentrifuge tube. Centrifugation at 10000 rpm for 2 minutes separated the supernatant and the concentrated nanoparticle layers. The mass of the supernatant layers was determined after removing the centrifugal filter. The centrifugal filter was inverted upside down into a clean pre-weighted microcentrifuge tube, placed in the centrifuge device and spun for 2 minutes at 10000 rpm to collect the concentrated nanoparticle layer. After removal of the filter, the mass of the concentrated nanoparticle layer was determined. 100 μL of both the supernatant and the concentrated nanoparticle layer layers were separately pipetted into clean 7 mL glass vials and diluted 6-fold with 95% Milli-Q water and 5% DMSO solution. A UV-Vis spectra were collected on the supernatant, concentrated nanoparticle layer as well as some of the pre-centrifuged sample. The concentration of TMP in the concentrated nanoparticle layer was determined using molar extinction coefficients from calibration curves and employing the Beer–Lambert law, followed by a multiplication by a factor of 6 to obtain the original concentration. The average encapsulation efficiency (EE) in two samples was calculated as 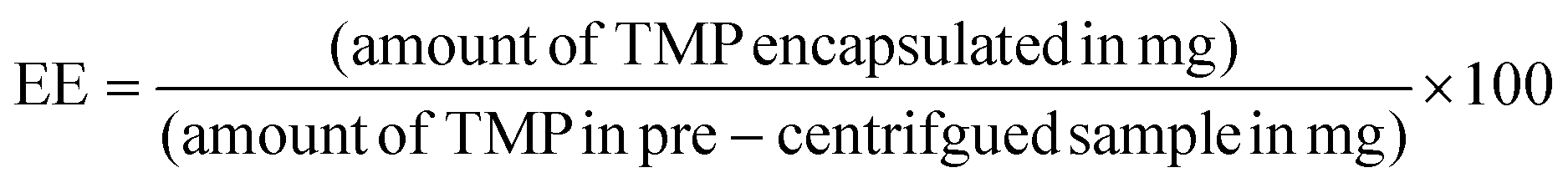 , resulting in an EE of about 82% (averaged from four runs). For the development of a calibration curve, a 0.5 mM stock solution of trimethoprim was prepared by dissolving 7.25 mg in a 50 mL of 95% Milli-Q water and 5% DMSO mixture. Utilizing this stock solution, a standard calibration curves were generated over a range from 0.0078 mM to 0.5 mM.
, resulting in an EE of about 82% (averaged from four runs). For the development of a calibration curve, a 0.5 mM stock solution of trimethoprim was prepared by dissolving 7.25 mg in a 50 mL of 95% Milli-Q water and 5% DMSO mixture. Utilizing this stock solution, a standard calibration curves were generated over a range from 0.0078 mM to 0.5 mM.
For the release study of trimethoprim-loaded samples, following the resuspension procedure described above, trimethoprim catalyzed polymer samples were resuspended to give concentrations of 0.2 mM trimethoprim. These samples along with a free drug control were placed in dialysis membranes and dialyzed in a 90% Milli-Q water and 10% DMSO mixture over a period of 12 hours with aliquots taken every 2 hours and measured by UV/Vis to determine the concentration of remaining trimethoprim inside the dialysis membrane.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. DMR-2238834. The authors acknowledge the use of facilities and instrumentation at the UC Irvine Materials Research Institute (IMRI) supported in part by the National Science Foundation Materials Research Science and Engineering Center program through the UC Irvine Center for Complex and Active Materials (DMR-2011967). Paul Hurst acknowledges the F. Sherwood Rowland Endowment Fund and UCI Department of Chemistry for receiving the UCI Department of Chemistry Rowland fellowship which supported the research in this manuscript. Additionally, we acknowledge Hala Hajjar for help with graphing the MALDI data.References
- Y. C. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- C. Liang, M. Sun, X. Shen, C. Shan, W. Wang, R. Cheng and J. Ye, Org. Process Res. Dev., 2021, 25, 810–816 CrossRef CAS.
- D. B. Wright, M. A. Touve, L. Adamiak and N. C. Gianneschi, ACS Macro Lett., 2017, 6, 925–929 CrossRef CAS PubMed.
- C. E. Boott, J. Gwyther, R. L. Harniman, D. W. Hayward and I. Manners, Nat. Chem., 2017, 9, 785 CrossRef CAS PubMed.
- C. Grazon, P. Salas-Ambrosio, E. Ibarboure, A. Buol, E. Garanger, M. W. Grinstaff, S. Lecommandoux and C. Bonduelle, Angew. Chem., Int. Ed., 2020, 59, 622–626 CrossRef CAS PubMed.
- E. Guégain, C. Zhu, E. Giovanardi and J. Nicolas, Macromolecules, 2019, 52, 3612–3624 CrossRef.
- P. J. Hurst, A. M. Rakowski and J. P. Patterson, Nat. Commun., 2020, 11, 4690 CrossRef CAS PubMed.
- Q. Shi, Y. Chen, J. Yang and J. Yang, Chem. Commun., 2021, 57, 11390–11393 RSC.
- C. Zhu and J. Nicolas, Biomacromolecules, 2022, 23, 3043–3080 CrossRef CAS PubMed.
- H. Phan, M. Cossutta, C. Houppe, C. Le Cœur, S. Prevost, I. Cascone, J. Courty, J. Penelle and B. Couturaud, J. Colloid Interface Sci., 2022, 618, 173–184 CrossRef CAS PubMed.
- J. Cao, Y. Tan, Y. Chen, L. Zhang and J. Tan, Macromol. Rapid Commun., 2021, 42, 2100498 CrossRef CAS PubMed.
- S. Y. Khor, J. F. Quinn, M. R. Whittaker, N. P. Truong and T. P. Davis, Macromol. Rapid Commun., 2019, 40, e1800438 CrossRef PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2019, 40, 1800279 CrossRef PubMed.
- P. J. Hurst, A. A. Graham and J. P. Patterson, ACS Polym. Au, 2022, 2, 501–509 CrossRef CAS PubMed.
- C. Grazon, P. Salas-Ambrosio, S. Antoine, E. Ibarboure, O. Sandre, A. J. Clulow, B. J. Boyd, M. W. Grinstaff, S. Lecommandoux and C. Bonduelle, Polym. Chem., 2021, 12, 6242–6251 RSC.
- S. Yao, J. Yang and J. Yang, Polym. Chem., 2023, 14, 3493–3500 RSC.
- D. Shen, B. Shi, P. Zhou, D. Li and G. Wang, Macromolecules, 2023, 56, 4814–4822 CrossRef CAS.
- S. X. Huang, Z. H. Wang, M. Lin, X. H. Fu and J. Sun, Polym. Chem., 2023, 14, 1801–1808 RSC.
- J. Jiang, X. Zhang, Z. Fan and J. Du, ACS Macro Lett., 2019, 1216–1221 CrossRef CAS PubMed.
- C. E. Ellis, J. D. Garcia-Hernandez and I. Manners, J. Am. Chem. Soc., 2022, 144, 20525–20538 CrossRef CAS PubMed.
- R. C. Pratt, B. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863–7871 CrossRef CAS.
- S.-H. Kim, D. Semenya and D. Castagnolo, Eur. J. Med. Chem., 2021, 216, 113293 CrossRef CAS PubMed.
- R. Tong and J. Cheng, J. Am. Chem. Soc., 2009, 131, 4744–4754 CrossRef CAS PubMed.
- R. Tong and J. Cheng, Angew. Chem., Int. Ed., 2008, 47, 4830–4834 CrossRef CAS.
- R. Tong and J. Cheng, Macromolecules, 2012, 45, 2225–2232 CrossRef CAS.
- Q. Yin, R. Tong, Y. Xu, L. W. Dobrucki, T. M. Fan and J. Cheng, Biomacromolecules, 2013, 14, 920–929 CrossRef CAS PubMed.
- J. Nicolas, Chem. Mater., 2016, 28, 1591–1606 CrossRef CAS PubMed.
- R. R. Gadde, E. F. McNiff and M. M. Peer, J. Pharm. Biomed. Anal., 1991, 9, 1031–1036 CrossRef CAS PubMed.
- I. Kaljurand, T. Rodima, A. Pihl, V. Mäemets, I. Leito, I. A. Koppel and M. Mishima, J. Org. Chem., 2003, 68, 9988–9993 CrossRef CAS PubMed.
- P. Pan and Y. Inoue, Prog. Polym. Sci., 2009, 34, 605–640 CrossRef CAS.
- J. G. E. M. Fraaije and G. J. A. Sevink, Macromolecules, 2003, 36, 7891–7893 CrossRef CAS.
- B. M. a. El-Falaha, A. D. Russell and J. R. Furr, Lett. Appl. Microbiol., 1985, 1, 21–24 CrossRef CAS.
- T. D. Hennessey, J. Periodontal Res., 1973, 8, 61–67 CrossRef PubMed.
- N. Horstmann Risso, C. D. Ottonelli Stopiglia, M. T. Oliveira, S. E. Haas, T. Ramos Maciel, N. Reginatto Lazzari, E. L. Kelmer, J. A. Pinto Vilela and D. V. Beckmann, Int. J. Nanomed., 2020, 15, 6935–6944 CrossRef PubMed.
- J. M. Schuurmans, A. S. N. Hayali, B. B. Koenders and B. H. ter Kuile, J. Microbiol. Methods, 2009, 79, 44–47 CrossRef CAS PubMed.
- S. G. B. Amyes, J. Chemother., 1993, 5, 417–421 CrossRef CAS.
- R. M. E. Richards, R. B. Taylor and Z. Y. Zhu, J. Pharm. Pharmacol., 1996, 48, 981–984 CrossRef CAS PubMed.
- Y. Y. Huang and T. W. Chung, J. Microencapsulation, 2001, 18, 457–465 CrossRef CAS PubMed.
- K. Shameli, M. B. Ahmad, W. M. Z. W. Yunus, N. A. Ibrahim, R. A. Rahman, M. Jokar and M. Darroudi, Int. J. Nanomed., 2010, 5, 573–579 CrossRef CAS PubMed.
- T. C. Moraes Moreira Carraro, C. Altmeyer, N. Maissar Khalil and R. M. Mainardes, J. Mycol. Med., 2017, 27, 519–529 CrossRef CAS PubMed.
- A. F. Radovic-Moreno, T. K. Lu, V. A. Puscasu, C. J. Yoon, R. Langer and O. C. Farokhzad, ACS Nano, 2012, 6, 4279–4287 CrossRef CAS PubMed.
- L. Zhang, R. C. Pratt, F. Nederberg, H. W. Horn, J. E. Rice, R. M. Waymouth, C. G. Wade and J. L. Hedrick, Macromolecules, 2010, 43, 1660–1664 CrossRef CAS.
- L. Simon and J. M. Goodman, J. Org. Chem., 2007, 72, 9656–9662 CrossRef CAS PubMed.
- M. K. Kiesewetter, M. D. Scholten, N. Kirn, R. L. Weber, J. L. Hedrick and R. M. Waymouth, J. Org. Chem., 2009, 74, 9490–9496 CrossRef CAS PubMed.
- V. Taresco, T. F. Abelha, R. J. Cavanagh, C. E. Vasey, A. B. Anane-Adjei, A. K. Pearce, P. F. Monteiro, K. A. Spriggs, P. Clarke, A. Ritchie, S. Martin, R. Rahman, A. M. Grabowska, M. B. Ashford and C. Alexander, Adv. Thermoelectr., 2021, 4, 2000103 CAS.
- M. Salmanpour, G. Yousefi, S. Mohammadi-Samani, M. Abedanzadeh and A. M. Tamaddon, J. Drug Delivery Sci. Technol., 2020, 60, 101933 CrossRef CAS.
- Z. Azhari, P. Smith, S. McMahon, W. Wang and R. E. Cameron, Pharm. Res., 2023, 40(7), 1697–1707 CrossRef CAS PubMed.
- A. Lu, E. Petit, K. Jelonek, A. Orchel, J. Kasperczyk, Y. Wang, F. Su and S. Li, Int. J. Biol. Macromol., 2020, 154, 39–47 CrossRef CAS PubMed.
- B. Lin and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 1645–1652 CrossRef CAS PubMed.
- R. Ronchetti, G. Moroni, A. Carotti, A. Gioiello and E. Camaioni, RSC Med. Chem., 2021, 12, 1046–1064 RSC.
- E. Ji, C. Cummins and G. Fleury, Molecules, 2021, 26, 1412 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Discussion on the nature of the chlorhexidine conjugation to PLLA, drug loading, MIC, GPC, NMR, MALDI, FT-IR, WAXS, ESI-MS and additional CryoEM data. See DOI: https://doi.org/10.1039/d3lp00135k |
| This journal is © The Royal Society of Chemistry 2024 |