
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluating the electronic structure and stability of epitaxially grown Sr-doped LaFeO3 perovskite alkaline O2 evolution model electrocatalysts†
Chuanmu
Tian
 a,
Clément
Maheu
ab,
Xiaochun
Huang
c,
Freddy E.
Oropeza
d,
Márton
Major
e,
Joachim
Brötz
e,
Marcus
Einert
a,
Wolfgang
Donner
e,
Kelvin Hongliang
Zhang
c and
Jan P.
Hofmann
*a
a,
Clément
Maheu
ab,
Xiaochun
Huang
c,
Freddy E.
Oropeza
d,
Márton
Major
e,
Joachim
Brötz
e,
Marcus
Einert
a,
Wolfgang
Donner
e,
Kelvin Hongliang
Zhang
c and
Jan P.
Hofmann
*a
aSurface Science Laboratory, Department of Materials and Geosciences, Technical University of Darmstadt, Peter-Grünberg-Straße 4, 64287 Darmstadt, Germany. E-mail: hofmann@surface.tu-darmstadt.de
bNantes Université, CNRS, Institut des Matériaux Jean Rouxel, IMN, F-44000 Nantes, France
cState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Siming South Street 422, Xiamen 361005, P. R. China
dPhotoactivated Processes Unit, IMDEA Energy Institute, Parque Tecnológico de Móstoles, Avda. Ramón de la Sagra 3, 28935 Móstoles, Madrid, Spain
eDepartment of Materials and Geosciences, Technical University of Darmstadt, Peter-Grünberg-Straße 2, 64287 Darmstadt, Germany
First published on 4th October 2024
Abstract
In this work, we have investigated the relationships between surface stability, electronic structure and O2 evolution reaction (OER) activity for epitaxial thin film La1−xSrxFeO3 (x = 0, 0.33, 0.8) model electrocatalysts before and after different electrochemical treatments. Cyclic voltammetry (CV) between +1.22 V and +1.92 V vs. RHE results in the continuous enhancement of OER performance of LaFeO3, while for La0.67Sr0.33FeO3 and La0.2Sr0.8FeO3 a gradual decrease of OER performance with increasing number of CV cycles was observed. A combination of atomic force microscopy, X-ray diffraction and X-ray reflectivity reveals that the surfaces of La1−xSrxFeO3 (x = 0, 0.33, 0.8) undergo surface morphology changes during OER treatment. Synchrotron ex situ X-ray photoemission spectroscopy data show a gradual down-shift of the Fermi level (EF) of LaFeO3 with increasing number of CV cycles, while near edge X-ray absorption fine structure spectroscopy (NEXAFS) at the Fe L-edge and O K-edge shows the presence of surface Fe4+ species as well as new hole states near the conduction band minimum upon electrochemical treatment, leading to a further enhancement of the electrochemical activity of LaFeO3. The newly formed hole state in LaFeO3 that appeared after 3 CV cycles remained constant upon progressing OER treatment. On the contrary, the decrease of OER performance of La0.67Sr0.33FeO3 and La0.2Sr0.8FeO3 with increasing CV cycles is attributed to an up-shift of EF along with a decrease of Fe4+ and hole state content after OER treatment. Furthermore, we found that the stability of the OER performance of La1−xSrxFeO3 is closely related to the leaching of Sr during OER, and the stability deteriorates with increasing Sr doping concentration in the pristine samples.
Introduction
Electrochemical water splitting is a promising pathway for large-scale production of green hydrogen and is expected to contribute to solving global energy and environmental issues.1,2 However, the oxygen evolution reaction (OER) limits the efficiency of the overall water electrolysis due to the higher overpotential caused by the kinetically demanding four electron/proton transfer process.3,4 Therefore, the development of highly active and stable OER electrocatalysts is critical to water electrolysis. Currently, IrO2- and RuO2-based catalysts for water electrolysis have been considered as the best OER catalysts in acidic media owing to their excellent OER activity, but their scarcity and high cost hinder their large-scale application.5–7 Hence, cheap, earth-abundant and stable transition metal oxides are becoming more attractive, especially Fe, Co and Ni, when alkaline water electrolysis is considered.8–10 Among them, the perovskite oxides have attracted great attention due to their low cost and tunable electronic structure.11–13 Perovskite oxides have the general formula ABO3, where A is an alkaline earth metal cation (La, Ba, Sr, etc.) and B is a transition metal cation (Fe, Co, Ni, etc.).14 Currently, in order to provide a design basis for optimally performing semiconducting perovskite oxide OER electrocatalysts, significant efforts are devoted to explore the relationship between the electronic structure and electrocatalytic activity of perovskite oxides with activity descriptor concepts such as the eg orbital occupancy,15 p-band centre of the oxygen atom,16 metal–oxygen covalency.17LaFeO3, a typical perovskite oxide, has been widely studied due to its good stability in alkaline electrolytes and adjustable electronic structure.18,19 Over the past decades, a series of strategies has been explored to enhance the electrochemical activity of LaFeO3, including nanostructure engineering to increase the surface area,20 morphology design to expose more active crystal facets,21 and aliovalent doping to increase the electrical conductivity and to modulate electronic structure.22–25 Among them, aliovalent doping is a powerful method to enhance the OER activity of LaFeO3. Recently, we found that doping by Sr can significantly improve the OER performance of LaFeO3.26 We argue that filled electronic states near the Fermi level (EF) improve the orbital overlap with key OER reaction intermediates, favouring their formation. At the same time, the hole states close to the EF increase the electron affinity of the LaFeO3, thereby facilitating charge transfer at the solid–electrolyte interface. Importantly, it has been observed that perovskite electrocatalysts usually undergo surface morphology and compositional changes during OER.27–34 It is then almost impossible to predict the OER performance of electrocatalysts from their pristine (bulk and surface) composition and structure since their active surface phase can strongly differ from the bulk composition and structure.35 Thus, it is critical to investigate the electronic structure and surface properties of the catalytically active surface, ideally under in situ conditions. For example, Wan et al. reported that SrIrO3 undergoes surface reconstruction accompanied by the formation of amorphous layers during electrochemical treatment.36 They attributed the improved OER performance of SrIrO3 to the formation of more active amorphous SryIrOx layer. Another typical perovskite with surface reconstruction is Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF). Fabbri et al. used in situ X-ray absorption spectroscopy (XAS) to reveal that the (Co/Fe)O(OH) layer, which formed by the surface reconstruction of BSCF, can be considered as the OER active surface phase.37
Although we have established the relationship between the electronic structure of pristine Sr-doped LaFeO3 powder-based electrocatalysts and their OER activity,26 the stability of the surface chemical composition, morphology, and electronic structure as the OER progresses still needs to be investigated and verified. Therefore, in order to establish a relationship between the surface properties, i.e., surface chemical composition, atomic and electronic structure including their stabilities, and the OER activity of the Sr-doped LaFeO3, we have employed epitaxial La1−xSrxFeO3 (001) single crystalline films as models to exclude the effect of nanostructure. We prepared a series of well-defined La1−xSrxFeO3 (x = 0, 0.33, 0.8) single crystal films on Nb-doped SrTiO3 (001) substrate via pulsed laser deposition (PLD). Our result show that the OER performance of LaFeO3 increases with increasing CV cycles, while the OER activity of La0.67Sr0.33FeO3 and La0.2Sr0.8FeO3 is gradually decreasing. The surface properties of La1−xSrxFeO3 (x = 0, 0.33, 0.8) were investigated by atomic force microscopy (AFM), X-ray diffraction (XRD) and X-ray reflectivity (XRR) before and after OER treatments. The combination of synchrotron X-ray photoemission spectroscopy (SXPS) and near edge X-ray absorption fine structure spectroscopy (NEXAFS) revealed electronic structure changes of La1−xSrxFeO3 (x = 0, 0.33, 0.8) upon OER treatment in the amount of surface Fe4+ active species, hole state and occupied density of states (DOS) near the Femi level (EF).
Experimental section
Materials synthesis
La1−xSrxFeO3 (x = 0, 0.33, 0.8) targets were synthesized by solid-phase reaction. Stoichiometric amounts of La2O3 (Alfa, >99.999%), Fe2O3 (Alfa, >99.999%) and SrO (Alfa, >99.999%) were mixed in a mortar. After grinding for 2 hours, the powder was pressed into pellets through a hydraulic press at 10 tons for 2 minutes, and finally these pellets were calcined in a muffle furnace (Kejing, KSL-1700) at 1000 °C for 24 h with 5 °C min−1 heating rate, followed by natural cooling to room temperature. La1−xSrxFeO3 (x = 0, 0.33, 0.8, 1.0) single crystal thin films were grown on one-side polished (001)-oriented 0.1% Nb-doped SrTiO3 substrates (HeFei Kejing) by PLD using the respective target. Laser ablation was performed at a repetition rate of 5 Hz and an energy density of 1.0 J cm−2 with a 248 nm KrF excimer laser. To achieve ∼40 nm film thickness, the laser was impinged on the target for 15 min with the substrate temperature 550 °C and oxygen partial pressure of 30 mTorr.Materials characterization
The crystal structure of the pure La1−xSrxFeO3 targets was analysed by X-ray diffraction (XRD) using Cu Kα radiation generated at 40 kV and 44 mA. Thin film growth was examined by high-resolution X-ray diffraction using a rotating Cu anode (45 kV, 190 mA) Rigaku SmartLab diffractometer in θ–2θ geometry, equipped with a Ge (220) double bounce monochromator (selecting the Cu Kα1 wavelength) in parallel beam mode, while the thickness of the films was determined by X-ray reflectivity (XRR) scans on the same diffractometer. The surface morphology of the La1−xSrxFeO3 samples before and after OER treatments were determined by atomic force microscopy (AFM) with a Bruker Icon Dimension AFM in amplitude modulation mode using PPP-Zeihr cantilevers (NanoAndMore GmbH, Wetzlar, Germany). This type of cantilever has a nominal spring constant of 27 N m−1, a nominal resonance frequency of 130 kHz, and a typical tip radius of <7 nm. A free oscillation amplitude A0 ≈ 120 nm of the cantilever tip was adjusted, and a setpoint ratio of Asp/A0 ≈ 0.7 was set to ensure operation in the repulsive tip–sample interaction regime. We scanned the surface with a tip velocity of 2 μm s−1.The SXPS and NEXAFS measurements were performed at the soft X-ray spectroscopy undulator beamline U49-2_PGM-1 at BESSY II synchrotron in Berlin. The beamline provides photons in the energy range of 85–1600 eV. The solid–liquid-interface analysis system (SOLIAS) endstation has been used and spectra were measured at normal emission using a Phoibos 150 (SPECS) energy analyser with a 1D delay line detector.38 An Ar-ion sputter-cleaned Au foil served as a calibration sample by making use of the Au4f7/2 transition located at 84.0 eV. Samples were electrically connected to the ground of the spectrometer. Excitation energies were adjusted for each core level to measure the photoelectrons at identical kinetic energy of 550 eV, which allows investigation of each element at a comparable depth of analysis, i.e., three times the inelastic mean free path at 550 eV kinetic energy (1.2–1.3 nm). The NEXAFS measurements at the Fe L-edge and O K-edge were carried out by recording the Auger electron yield of the respective elements.
Electrochemical measurements were carried out in three-electrode configuration using a PalmSens electrochemical workstation (PalmSens BV, The Netherlands) setup in the double glass cross electrochemistry chamber of the SOLIAS endstation.38 The chamber was continuously purged by argon keeping the sample under an inert, dry, and carbon-free atmosphere and minimizing atmospheric contamination between the electrochemical treatments and the synchrotron-based measurements. A platinum wire was used as a counter electrode and Hg/HgO (1 M KOH) as a reference electrode. Cyclic voltammetry (CV) measurements were performed in 1 M KOH at a scan rate of 10 mV s−1 with the potential from +1.22 to +1.92 V (vs. RHE). Each sample was subjected to four OER treatments, namely total 0 CV (exposure to electrolyte only), total 3 CV (+3 increment), total 10 CV (+7 increment), and total 30 CV (+20 increment) cycles. After each treatment step, the sample was rinsed with MilliQ water and blown dry by Ar, then transferred to the vacuum system of the SOLIAS endstation for spectroscopic investigation.
Results and discussion
Structural characterization
XRD and XRR were employed to characterize the quality of the La1−xSrxFeO3 thin films grown on 0.1% Nb-doped SrTiO3 (001) substrates by PLD. Fig. 1 shows the XRD and XRR results of the La1−xSrxFeO3 in pristine state (black curve) and after 30 CV cycles (i.e., after all four electrochemical treatment steps, red curve). The well-defined oscillations are observed in both XRD and XRR plots for pristine LaFeO3 (LFO) and La0.67Sr0.33FeO3 (LSF-0.33), indicating the high quality of the thin films. However, these oscillations did not appear in the XRD plot (Fig. 1c) of La0.2Sr0.8FeO3 (LSF-0.8), which is attributed to the presence of a large number of oxygen vacancies in the LSF-0.8 sample causing the film not to grow epitaxial, similar as reported for SrCoO3−x and SrFeO3−x.39,40 The peak at 2θ = 46.4° corresponds to the (002) orientation of the 0.1% Nb-doped SrTiO3 single crystal substrate, while the peaks at 2θ = 45.8°, 46.2° and 46.9° correspond to (002) oriented LFO, LSF-0.33 and LSF-0.8, respectively. To determine the thickness of the La1−xSrxFeO3 thin films, we obtained and fitted XRR data as shown in Fig. S1,† and the corresponding fitting parameters are reported in Table S1.† The overall thickness of the pristine LFO, LSF-0.33 and LSF-0.8 single crystal thin films are 37.5 ± 0.5 nm, 38.6 ± 0.2 and 39.2 ± 0.2, respectively. After 30 CV cycles between +1.22 V to +1.92 V (vs. RHE) in 1 M KOH, there is a slight decrease in the thickness of the crystalline layer of all La1−xSrxFeO3 (x = 0, 0.33) thin films and an amorphous layer appears on all La1−xSrxFeO3 (x = 0, 0.33) film surfaces. The corresponding XRR plot of LFO before and after OER treatment is shown in Fig. 1d. We also notice that the distance of the Kiessig fringes in the XRR plot of LFO remain consistent after 30 CV cycles, indicating negligible change of the overall film thickness. At the same time, the XRD (Fig. 1b) and XRR (Fig. 1e) data of LSF-0.33 show that the thickness of the crystalline layer as well as the overall thickness of the film remain unchanged. Fig. 1c shows the XRD results of LSF-0.8 before and after 30 CV cycles; we notice that the peak intensity corresponding to LSF-0.8 increased after 30 CV, which is attributed to a surface reconstruction of LSF-0.8 thin film occurring under OER conditions. Based on the XRD and XRR results, we can state that the OER treatment resulted in an amorphous layer on the surface of La1−xSrxFeO3 (x = 0, 0.33) samples. In addition, Table S1† compiles the roughness of all La1−xSrxFeO3 (x = 0, 0.33, 0.8) samples. After 30 CV cycles, the roughness of LFO and LSF-0.33 increased from 0.61 nm to 0.69 nm and 0.67 to 0.82, respectively, while the roughness decreased from 1.73 nm to 0.41 nm for LSF-0.8.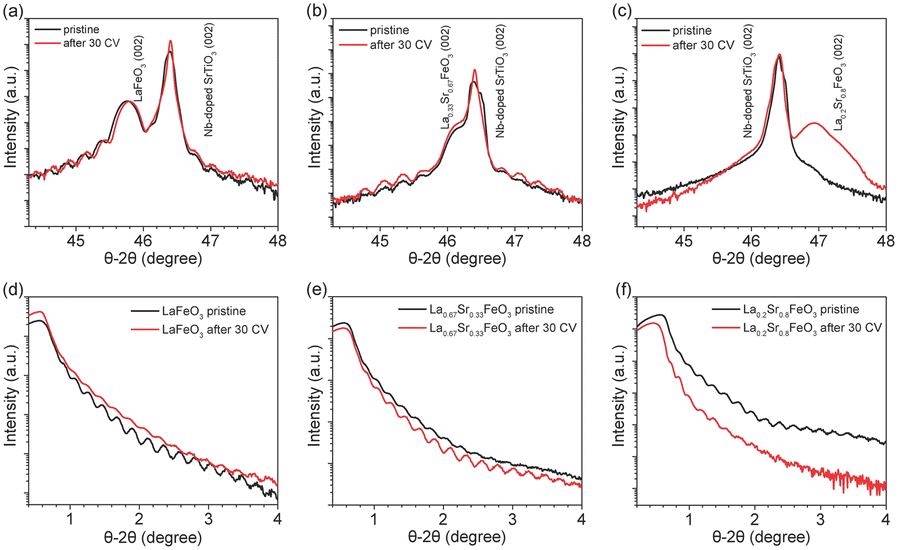 | ||
| Fig. 1 XRD and XRR of La1−xSrxFeO3 thin films in pristine state (black curves) and after 30 CV cycles (red curves). (a) XRD of LaFeO3, (b) XRD of La0.67Sr0.33FeO3, (c) XRD of La0.2Sr0.8FeO3, (d) XRR of LaFeO3, (e) XRR of La0.67Sr0.33FeO3 and (f) XRR of La0.2Sr0.8FeO3. | ||
To further investigate the effect of OER treatment on the surface properties, we carried out AFM measurements on the pristine and OER-treated thin films. As shown in Fig. S2a and c,† the pristine LFO and LSF-0.33 have a flat surface with a root mean square roughness (RMS) of 0.50 nm and 0.30 nm, respectively, which further confirms the high quality of these films grown by PLD. However, the RMS of pristine LSF-0.8 reaches 5.36 nm which is much higher than LFO and LSF-0.33. Such a high RMS value of LSF-0.8 is attributable to the fact that it was not epitaxially grown on the 0.1% Nb–SrTiO3 substrate, which is consistent with the above XRD results. After all four OER treatment steps, the RMS of LFO (Fig. S2b†) remained virtually unchanged. As shown in Fig. S2c,† the RMS of LSF-0.33 increased from the initial 0.30 nm to 1.12 nm after OER treatment. On the contrary, we notice that the RSM of LSF-0.8 (Fig. S2f†) is drastically reduced from 5.36 to 0.54 nm after OER treatment. Among them, the changes in the LSF-0.8 film due to OER treatment were the most pronounced, which is consistent with our XRD and XRR results.
Electronic structure by X-ray photoemission and absorption spectroscopies
The oxidation state of Fe was determined by NEXAFS of the Fe L-edge. Fig. 2a shows the Fe L-edge of LFO after different OER treatment steps. The shape of the spectra of pristine and 0 CV samples are consistent with previously reported results for Fe3+ oxidation state.26 More importantly, the shape and intensity of the Fe L-edge remain consistent after 3 and even 30 CV cycles. To further investigate the oxidation state of Fe, we measured the O K-edge NEXAFS of LFO after different CV cycles, shown in Fig. 2b. The results indicate that a new unoccupied (hole) state emerges at 528.4 eV, which is associated with Fe4+ in agreement with our previous report.26 More interestingly, this hole state exhibits the same behaviour after 3 and even 30 CV cycles. The Fe L-edge of pristine samples of LSF-0.33 (Fig. 2c, black curve) and LSF-0.8 (Fig. S3a,† black curve) also exhibited Fe4+ oxidation state characteristics, with LSF-0.8 being very similar to SrFeO3. However, the linear shape and intensity of the Fe L-edge of LSF-0.33 and LSF-0.8 both show slight changes at different CV cycles, suggesting that the Fe4+ oxidation state still exists after OER treatment. At the same time, the intensity of the hole state features in the O K-edge spectra in LSF-0.33 (Fig. 2d) and LSF-0.8 (Fig. S3b†) samples exhibit a gradually decreasing trend with increasing CV cycles, indicating that the Fe4+ content in LSF-0.33 and LSF-0.8 is gradually decreasing due to the fact that these hole states are closely related to the Fe4+ oxidation state. Additionally, we still observe the presence of the hole state on the O K-edge even after 30 CV cycles, further confirming that Fe4+ is still existent in LSF-0.33 and LSF-0.8 after different OER treatment steps. Taken together, the above results indicate that the OER treatment led to a morphology change of the La1−xSrxFeO3 surface and the appearance of a hole state at LFO, while the hole state content of both LSF-0.33 and LSF-0.8 samples decreased after surface reconstruction. | ||
| Fig. 2 NEXAFS spectra of LaFeO3 and La0.67Sr0.33FeO3 with different CV cycles. (a) Fe L-edge of LaFeO3, (b) O K-edge of LaFeO3, (c) Fe L-edge of La0.67Sr0.33FeO3 and (d) O K-edge of La0.67Sr0.33FeO3. The yellow bars in (a) indicate the changes in the characteristics of Fe3+ and Fe4+ at 708 eV under different CV cycles. | ||
In order to investigate the stability of the surface chemical composition and electronic structure of La1−xSrxFeO3 (x = 0, 0.33, 0.8) with increasing OER treatment steps, we additionally carried out synchrotron XPS (SXPS) measurements. Fig. 3 shows the SXPS core level and VB spectra of LFO after different CV cycles; the XAS and XPS data have been acquired subsequently before each next OER treatment step. The Fe 2p3/2 region of LFO pristine exhibits a characteristic feature located around 710.2 eV along with a satellite peak near 719 eV, in agreement with the Fe3+ oxidation state. After different OER treatments, the characteristics of Fe 2p did not change significantly, but the position of the main peak gradually shifted to lower binding energy with increasing CV cycles. The same shift is also seen in the corresponding La 3d (Fig. 3b) and O 1s (Fig. 3c) core level spectra. In addition, the VB spectrum of LFO in the Fig. 3d show that the distance between the valence band maximum (VBM) and EF decreased with increasing CV number, indicating a downward movement of EF or alternatively an upward movement of the VBM.
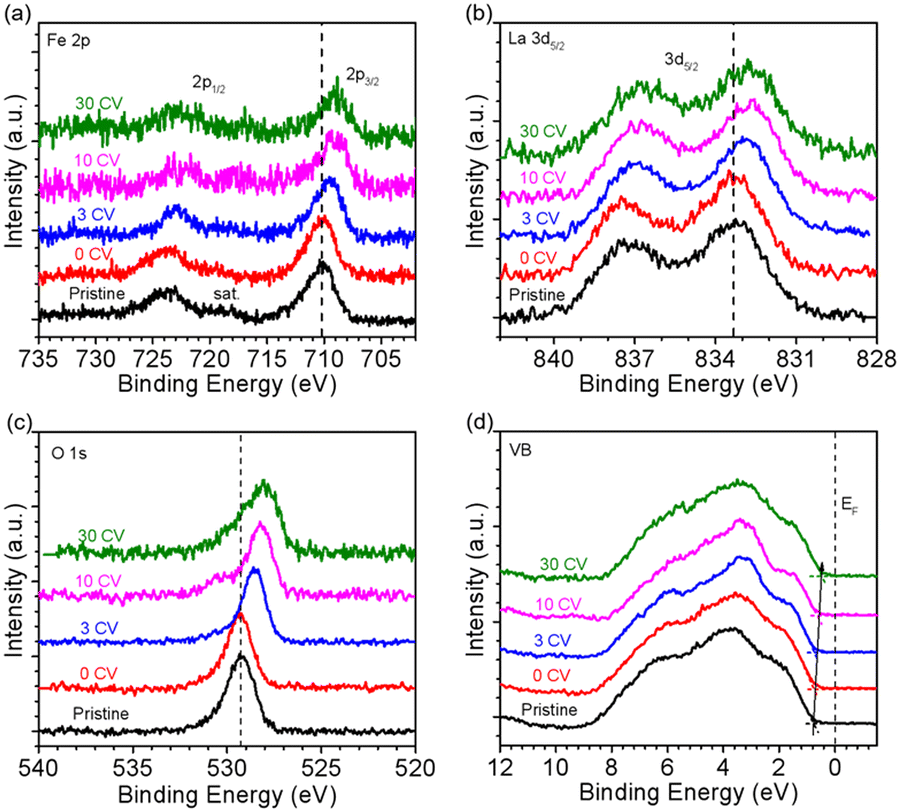 | ||
| Fig. 3 SXPS of LaFeO3 with kinetic energy constant at 550 eV. (a) Fe 2p, (b) La 3d, (c) O 1s and (d) VB spectra after different CV cycles. | ||
Fig. 4 summarizes the shift of the core levels and VB spectra, as well as the corresponding band energy schemes for LFO from the SXPS data. By combining the systematic shift of the binding energies of the Fe 2p, La 3d, and O 1s spectra together with the shift of the VB spectra, we can clarify the downward shift of EF with progressing CV cycling. More importantly, the above XRD and AFM data combined with the NEXAFS results indicate that the shift of EF can be attributed to the formation of Fe4+ species on the LFO surface due to surface reconstruction during the OER process. Such a phenomenon has also been observed in different transition metal oxides, such as, LaNiO3 and SrIrO3.36,41 The SXPS data of LFO-0.33 with different numbers of CV cycles are shown in Fig. S4,† respectively. In contrast to the LFO samples, the core levels of LSF-0.33 (Fe 3d, La 3d, and O 1s) shift to higher binding energies, and the distance between VBM and EF increased with increasing CV cycling, indicating a gradual upward movement of EF in LSF-0.33.
 | ||
| Fig. 4 (a) Shifts of core level binding energy and (b) Fermi level (EF) position of LaFeO3 relative to the VBM BE as a function of CV cycles. | ||
Electrochemical characterization
The OER performance of La1−xSrxFeO3 (x = 0, 0.33, 0.8) thin films were evaluated in a three-electrode setup in the double glass cross electrochemistry chamber of the SOLIAS endstation in 1 M KOH solution at 10 mV s−1 scan rate with a potential range of +1.22 V to +1.92 V vs. RHE. The current density of CVs of all samples was normalized by the geometric area without ohmic compensation. Fig. 5a shows CV plots of LFO under different CV cycles. The results suggest that the electrocatalytic activity of LFO experiences an enhancement with increasing number of CV cycles. On the contrary, the current density of LSF-0.33 (Fig. 5b) as well as LSF-0.8 (Fig. 5c) films show a continuous trend of decreasing OER performance. To compare the OER activity of La1−xSrxFeO3 with different Sr doping concentrations, the current densities of different samples with different CV cycles at +1.8 V (vs. RHE) are shown in Fig. 5d. The initial (3rd CV) OER performance of La1−xSrxFeO3 is enhanced with increasing Sr doping concentration, which is in line with our previously reported results.26 More importantly, we observed a more significant decrease in OER performance for the LSF-0.8 sample compared to LSF-0.33. The current density of LSF-0.33 at +1.8 V (vs. RHE) decreases from 0.085 mA cm−2 (3rd CV) to 0.069 mA cm−2 (30th CV), while LSF-0.8 decreases from 0.303 mA cm−2 (3rd CV) to 0.192 mA cm−2 (30th CV) – a reduction of 18.8% and 41.8%, respectively. | ||
| Fig. 5 OER performance of La1−xSrxFeO3 with different CV cycles. (a) CV polarization curves of LaFeO3, (b) CV polarization curves of La0.67Sr0.33FeO3, (c) CV polarization curves of La0.2Sr0.8FeO3 and (d) current density of La1−xSrxFeO3 (x = 0, 0.33, 0.8) with different CV cycles at +1.8 V (vs. RHE). | ||
It is well known that stability is also an important metric for evaluating OER catalysts. In order to have a more in-depth understanding of OER stability of the La1−xSrxFeO3 (x = 0, 0.33, 0.8) catalysts, we calculated the compositions of La, Sr and Fe (La% + Sr% + Fe% = 100%) from SXPS spectral region integration after different CV cycles, as shown in Table 1. There is a significant increase in La concentration on the surface of the LFO film after OER treatment, which indicated that La gradually aggregated on the surface of LFO during the OER process. Recently, She et al. reported that surface aggregation of A-site cations (La or Sr) in La1−xSrxFeO3 is an important factor contributing to the poor OER stability by blocking catalytically active sites.22 In addition, we observe that the surface Sr content in both LSF-0.33 and LSF-0.8 gradually decreases with increasing CV, while the La content did not change significantly. The reduction of surface Sr concentration in LSF-0.33 and LSF-0.8 is related to the leaching of Sr during OER; a similar phenomenon has been observed in SrIrO3.36 There is a close relationship between current density and Sr concentration as shown in Fig. 6. It can be clearly observed that the leaching of Sr in LSF-0.8 is particularly more pronounced as compared to LSF-0.33, in agreement with the deterioration of the OER performance.
| LFO | LSF-0.33 | LSF-0.8 | ||||||
|---|---|---|---|---|---|---|---|---|
| La (at%) | Fe (at%) | La (at%) | Sr (at%) | Fe (at%) | La (at%) | Sr (at%) | Fe (at%) | |
| a Expected stoichiometry. b By XPS fitting. | ||||||||
| Pristinea | 50.0 ± 0.4 | 50.0 ± 0.2 | 33.5 ± 0.4 | 16.5 ± 0.6 | 50.0 ± 0.5 | 10.1 ± 0.2 | 40.0 ± 0.3 | 49.9 ± 0.3 |
| 0 CVb | 49.1 ± 0.6 | 50.9 ± 0.3 | 33.9 ± 0.6 | 14.8 ± 0.3 | 51.2 ± 0.3 | 12.5 ± 0.3 | 36.4 ± 0.2 | 51.1 ± 0.4 |
| 3 CVb | 62.6 ± 0.5 | 37.3 ± 0.4 | 33.0 ± 0.5 | 10.7 ± 0.4 | 55.7 ± 0.2 | 15.7 ± 0.5 | 22.7 ± 0.4 | 61.5 ± 0.2 |
| 10 CVb | 63.0 ± 0.4 | 37.0 ± 0.5 | 31.5 ± 0.4 | 10.1 ± 0.5 | 58.4 ± 0.4 | 15.8 ± 0.4 | 17.2 ± 0.5 | 67.0 ± 0.4 |
| 30 CVb | 64.5 ± 0.2 | 35.5 ± 0.4 | 31.1 ± 0.2 | 9.4 ± 0.4 | 59.5 ± 0.5 | 14.3 ± 0.2 | 15.1 ± 0.4 | 70.6 ± 0.5 |
 | ||
| Fig. 6 Relationship between current density and Sr concentration of La0.67Sr0.33FeO3 and La0.2Sr0.8FeO3 with different CV cycles. | ||
Such a different OER behaviour of La1−xSrxFeO3 with CV cycling is likely attributed to two factors. The first factor is the emergence of a hole state close to the conduction band minimum. Our recent study shows that the hole state near the conduction band minimum plays a crucial role in the oxidation of water by reducing the energy barrier for electron transfer at the electrode–electrolyte interface.26 The above NEXAFS O K-edge spectra of LSF-0.33 and LSF-0.8 show that the content of the hole state (∼528 eV) near the conduction band gradually decreases with increasing CV cycles, resulting in a decrease of OER performance. The second factor is the occupied density of states (DOS) near the EF. According to the Marcus–Gerischer charge transfer model,42 the increase in the occupied DOS near EF would reduce the energy of donating electrons at the electrode–electrolyte interface, which results in increased performance of the catalyst by a decrease in charge transfer overpotential. It is worth noting that our SXPS results show that the EF of LFO is shifting toward the VBM with increasing number of CV cycles, which indicates that the occupied DOS near the EF is gradually increasing. On the contrary, the EF of the LSF-0.33 shift away from the VBM, resulting in a decrease of occupied DOS near the EF of LSF-0.33. The similar trend of electrochemical activity and occupied DOS near EF indicates that occupied DOS near EF are an important factor for determining the OER performance. Overall, our results highlight that the OER performance of La1−xSrxFeO3 (x = 0, 0.33, 0.8) is closely related to the surface Fe4+ species, the hole state near the CB and the occupied DOS near the EF. Sr leaching during OER is the key factor for the poor stability of LSF-0.33 and LSF-0.8.
Conclusions
We have investigated the surface properties, stability of the electronic structure and OER performance of well-definedLa1−xSrxFeO3 (x = 0, 0.33, 0.8) single crystal films. Our results reveal that operating the electrocatalysts under OER conditions lead to an increase of OER performance of LFO and a decrease of activity of LSF-0.33 and LSF-0.8. A combination of XRD, XRR and AFM measurements shows that the surfaces of La1−xSrxFeO3 (x = 0, 0.33, 0.8) undergo surface morphology and compositional changes upon OER treatment. Additionally, the electronic structure also experiences significant changes especially in the concentration of Fe4+ oxidation state, the hole state and the position of EF with respect to the valence band maximum. The improved activity of LFO is attributed to i) the presence of hole states as well as ii) the increase of the occupied density of states near EF upon OER cycling. Conversely, the reduction of these factors leads to the decrease of OER performance of LSF-0.33 and LSF-0.8. The leaching of Sr is an important factor leading to the observed poor stability of LSF-0.33 and LSF-0.8 under OER conditions, and the OER performance decays faster with increasing Sr doping concentrations at the A-site in the pristine perovskite structure. Our study provides insights into the surface structural and compositional evolution, electronic structure stability and electrochemical properties of transition metal oxides as function of OER treatment and reveals that appropriate doping has substantial impact on the OER stability of transition metal oxides. The study further demonstrates the importance of synchrotron-based investigations for the precise determination and interpretation of electrochemical properties and activities with respect to electronic structure engineering of perovskite electrocatalysts achieving optimized OER performance.Data availability
Data supporting figures and tables in the manuscript and the ESI† is made available in a https://www.zenodo.org repository accessible under DOI (https://doi.org/10.5281/zenodo.13902074).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Helmholtz Zentrum Berlin (HZB) is acknowledged for granting beam time at BESSY II under application 202-10001-ST-1.1-P. R. Golnak (HZB) is acknowledged for technical assistance during beam time at beamline U49-2_PGM-1. We also thank Dr. T. Hellmann and Dr. W. Calvet for setting up SOLIAS. C. M. Tian acknowledges financial support from the China Scholarship Council (No. 202008420222). J. P. Hofmann, M. Einert, and C. Maheu acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation under the Walter Benjamin Program to M. Einert, project no. 469377211 as well as in the priority program SPP2196, no. 423746744 and CRC 1548, FLAIR, project B07, no. 463184206).References
- Z. P. Ifkovits, J. M. Evans, M. C. Meier, K. M. Papadantonakis and N. S. Lewis, Decoupled Electrochemical Water-splitting Systems: A Review and Perspective, Energy Environ. Sci., 2021, 14, 4740 RSC.
- L. Schlapbach and A. Züttel, Hydrogen-Storage Materials for Mobile Applications, Nature, 2001, 414, 353–358 CrossRef CAS.
- Y. Gorlin and T. F. Jaramillo, A Bifunctional Nonprecious Metal Catalyst for Oxygen Reduction and Water Oxidation, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS PubMed.
- E. Fabbri, A. Habereder, K. Waltar, R. Kotz and T. J. Schmidt, Developments and Perspectives of Oxide-Based Catalysts for the Oxygen Evolution Reaction, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- V. Petrykin, K. Macounova, O. A. Shlyakhtin and P. Krtil, Tailoring the Selectivity for Electrocatalytic Oxygen Evolution on Ruthenium Oxides by Zinc Substitution, Angew. Chem., Int. Ed., 2010, 49, 4813–4815 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, Synthesis and Activities of Rutile IrO2 and RuO2 Nanoparticles for Oxygen Evolution in Acid and Alkaline Solutions, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- A. Zagalskaya and V. Alexandrov, Role of Defects in the Interplay between Adsorbate Evolving and Lattice Oxygen Mechanisms of the Oxygen Evolution Reaction in RuO2 and IrO2, ACS Catal., 2020, 10, 3650–3657 CrossRef CAS.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Electrocatalysis for the Oxygen Evolution Reaction: Recent Development and Future Perspectives, Chem. Soc. Rev., 2017, 46, 337 RSC.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Toward the Rational Design of Non-Precious Transition Metal Oxides for Oxygen Electrocatalysis, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- K. X. Zhang and R. Q. Zou, Advanced Transition Metal-Based OER Electrocatalysts: Current Status, Opportunities, and Challenges, Small, 2021, 17, 2100129 CrossRef CAS.
- D. Liu, P. F. Zhou, H. Y. Bai, H. Q. Ai, X. Y. Du, M. P. Chen, D. Liu, W. Fai, K. H. Lo, C. T. Kwok, S. Chen, S. P. Wang, G. C. Xing, X. S. Wang and H. Pan, Development of Perovskite Oxide-Based Electrocatalysts for Oxygen Evolution Reaction, Small, 2021, 17, 2101605 CrossRef CAS.
- C. W. Sun, J. A. Alonso and J. J. Bian, Recent Advances in Perovskite-Type Oxides for Energy Conversion and Storage Applications, Adv. Energy Mater., 2021, 11, 20000549 Search PubMed.
- H. N. Sun, J. Dai, W. Zhou and Z. P. Shao, Emerging Strategies for Developing High-Performance Perovskite-Based Materials for Electrochemical Water Splitting, Energy Fuels, 2020, 34, 10547–10567 CrossRef CAS.
- X. Xu, C. Su, W. Zhou, Y. Zhu, Y. Chen and Z. Shao, Co-doping Strategy for Developing Perovskite Oxides as Highly Efficient Electrocatalysts for Oxygen Evolution Reaction, Adv. Sci., 2016, 3, 1500187 CrossRef.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles, Science, 2011, 334, 1383–1385 CrossRef CAS.
- Y. L. Lee, J. Kleis, J. Rossmeisl, Y. Shao-Horn and D. Morgan, Prediction of Solid Oxide Fuel Cell Cathode Activity With First-Principles Descriptors, Energy Environ. Sci., 2011, 4, 3966–3970 RSC.
- J. J. Song, C. Wei, Z. F. Huang, C. T. Liu, L. Zeng, X. Wang and Z. C. J. Xu, A Review on Fundamentals for Designing Oxygen Evolution Electrocatalysts, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- A. R. Burton, R. Paudel, B. Matthews, M. Sassi, S. R. Spurgeon, B. H. Farnum and R. B. Comes, Thickness Dependent OER Electrocatalysis of Epitaxial LaFeO3 Thin Films, J. Mater. Chem. A, 2022, 10, 1909–1918 RSC.
- Y. W. Dai, J. Yu, Z. B. Zhang, C. Cheng, P. Tan, Z. P. Shao and M. Ni, Interfacial La Diffusion in the CeO2/LaFeO3 Hybrid for Enhanced Oxygen Evolution Activity, ACS Appl. Mater. Interfaces, 2021, 13, 2799–2806 CrossRef CAS.
- Y. X. Li, X. Y. Zhang, Z. J. Wu, H. B. Sheng, C. Li, H. Y. Li, L. X. Cao and B. H. Dong, Coupling Porous Ni Doped LaFeO3 Nanoparticles with Amorphous FeOOH Nanosheets Yields an Interfacial Electrocatalyst for Electrocatalytic Oxygen Evolution, J. Mater. Chem. A, 2021, 9, 23545–23554 RSC.
- J. Dai, Y. L. Zhu, Y. J. Zhong, J. Miao, B. W. Lin, W. Zhou and Z. P. Song, Enabling High and Stable Electrocatalytic Activity of Iron-Based Perovskite Oxides for Water Splitting by Combined Bulk Doping and Morphology Designing, Adv. Mater. Interfaces, 2019, 6, 1801317 CrossRef.
- S. X. She, J. Yu, W. Q. Tang, Y. L. Zhu, Y. B. Chen, J. Sunarso, W. Zhou and Z. P. Shao, Systematic Study of Oxygen Evolution Activity and Stability on La1−xSrxFeO3−δ Perovskite Electrocatalysts in Alkaline Media, ACS Appl. Mater. Interfaces, 2018, 10, 11715–11721 CAS.
- Z. S. Li, L. Lv, J. S. Wang, X. Ao, Y. J. Ruan, D. Z. Zha, G. Hong, Q. H. Wu, Y. C. Lan, C. D. Wang, J. J. Jiang and M. L. Liu, Engineering Phosphorus-doped LaFeO3-δ Perovskite Oxide as Robust Bifunctional Oxygen Electrocatalysts in Alkaline Solutions, Nano Energy, 2018, 47, 199–209 CAS.
- P. Shikha, T. S. Kang and B. S. Randhawa, Effect of Different Synthetic Routes on the Structural, Morphological and Magnetic Properties of Ce Doped LaFeO3 Nanoparticles, J. Alloys Compd., 2015, 625, 336–345 CrossRef CAS.
- R. A. Rincón, E. Ventosa, F. Tietz, J. Masa, S. Seisel, V. Kuznetsov and W. Schuhmann, Evaluation of Perovskites as Electrocatalysts for theOxygen Evolution Reaction, ChemPhysChem, 2014, 15, 2810–2816 Search PubMed.
- Z. C. Shen, Y. B. Zhuang, W. W. Li, X. C. Huang, F. E. Oropeza, E. J. M. Hensen, J. P. Hofmann, M. Y. Cui, A. Tadich, D. C. Qi, J. Cheng, J. Li and K. H. L. Zhang, Increased Activity in the Oxygen Evolution Reaction by Fe4+-induced Hole States in Perovskite La1-xSrxFeO3, J. Mater. Chem. A, 2020, 8, 4407–4415 Search PubMed.
- P. P. Lopes, D. Y. Chung, X. Rui, H. Zheng, H. Y. He, P. F. B. D. Martins, D. Strmcnik, V. R. Stamenkovic, P. Zapol, J. F. Mitchell, R. F. Klie and N. M. Markovic, Dynamically Stable Active Sites from Surface Evolution of Perovskite Materials during the Oxygen Evolution Reaction, J. Am. Chem. Soc., 2021, 143, 2741–2750 CAS.
- J. J. Suo, B. W. Yang, E. Mosconi, H. S. Choi, Y. J. Kim, S. M. Zakeeruddin, F. D. Angelis, M. Grätzel, H. S. Kim and A. Hagfeldt, Surface Reconstruction Engineering with Synergistic Effect of Mixed-Salt Passivation Treatment Toward Efficient and Stable Perovskite Solar Cells, Adv. Funct. Mater., 2021, 31, 2102902 Search PubMed.
- Y. Sun, R. Li, X. X. Chen, J. Wu, Y. Xie, X. Wang, K. K. Ma, L. Wang, Z. Zhang, Q. L. Liao, Z. Kang and Y. Zhang, A-Site Management Prompts the Dynamic Reconstructed Active Phase of Perovskite Oxide OER Catalysts, Adv. Energy Mater., 2021, 11, 2003755 CAS.
- L. K. Gao, X. Cui, C. D. Sewell, J. Li and Z. Q. Lin, Recent Advances in Activating Surface Reconstruction for the High-Efficiency Oxygen Evolution Reaction, Chem. Soc. Rev., 2021, 50, 8428 RSC.
- A. Grimaud, A. Demortiere, M. Saubanere, W. Dachraoui, M. Duchamp, M. L. Doublet and J. M. Tarascon, Activation of Surface Oxygen Sites on an Iridium-Based Model Catalyst for the Oxygen Evolution Reaction, Nat. Energy, 2016, 2, 1–10 Search PubMed.
- Y. Sun, C. R. Wu, T. Y. Ding, J. Gu, J. W. Yang, J. Cheng and K. H. L. Zhang, Direct Observation of the Dynamic Reconstructed Active Phase of Perovskite LaNiO3 for the Oxygen-Evolution Reaction, Chem. Sci., 2023, 14, 5906–5911 RSC.
- M. J. Choi, L. Wang, K. A. Stoerzinger, S. Y. Chung, S. A. Chambers and Y. G. Du, Epitaxial Design of Complex Nickelates as Electrocatalysts for the Oxygen Evolution Reaction, Adv. Energy Mater., 2023, 13, 2300239 CrossRef CAS.
- E. M. Kiens, M. J. Choi, L. H. Wei, Q. Y. Lu, L. Wang and C. Baeumer, Deeper Mechanistic Insights into Epitaxial Nickelate Electrocatalysts for the Oxygen Evolution Reaction, Chem. Commun., 2023, 59, 4562–4577 RSC.
- Y. Y. Li, X. C. Du, J. W. Huang, C. Y. Wu, Y. H. Sun, G. F. Zou, C. T. Yang and J. Xiong, Recent Progress on Surface Reconstruction of Earth-Abundant Electrocatalysts for Water Oxidation, Small, 2019, 15, 1901980 CrossRef PubMed.
- G. Wan, J. W. Freeland, J. Kloppenburg, G. Petretto, J. N. Nelson, D. Y. Kuo, C. J. Sun, J. G. Wen, J. T. Diulus, G. S. Herman, Y. Q. Dong, R. H. Kou, J. Y. Sun, S. Chen, K. M. Shen, D. G. Schlom5, G. M. Rignanese, G. Hautier, D. D. Fong, Z. X. Feng, H. Zhou and J. Suntivich, Amorphization Mechanism of SrIrO3 Electrocatalyst: How Oxygen Redox Initiates Ionic Diffusion and Structural Reorganization, Sci. Adv., 2021, 7, 7323 CrossRef.
- E. Fabbri, M. Nachtegaal, T. Binniger, X. Cheng, B. J. Kim, J. Durst, F. Bozza, T. Graule, R. Schäublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Dynamic Surface Self-Reconstruction is the Key of Highly Active Perovskite Nano-Electrocatalysts for Water Splitting, Nat. Mater., 2017, 16, 925–931 CrossRef CAS.
- T. Mayer, M. Lebedev, R. Hunger and W. Jaegermann, Elementary Processes at Semiconductor/Electrolyte Interfaces: Perspectives and Limits of Electron Spectroscopy, Appl. Surf. Sci., 2005, 252, 31–42 CrossRef CAS.
- S. J. Callori, S. Hu, J. Bertinshaw, Z. J. Yue, S. Danilkin, X. L. Wang, V. Nagarajan, F. Klose, J. Seidel and C. Ulrich, Strain-Induced Magnetic Phase Transition in SrCoO3−δ Thin Films, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 140405 CrossRef.
- S. Roh, S. Lee, M. Lee, Y. S. Seo, A. Khare, T. Yoo, S. Woo, W. S. Choi and J. Hwang, Oxygen Vacancy Induced Structural Evolution of SrFeO3−x Epitaxial Thin Film from Brownmillerite to Perovskite, Phys. Rev. B, 2018, 97, 075104 CrossRef CAS.
- H. Liu, R. R. Xie, Q. X. Wang, J. L. Han, Y. Han, J. Wang, H. Fang, J. Qi, M. Ding, W. X. Ji, B. He and W. M. Lü, Enhanced OER Performance and Dynamic Transition of Surface Reconstruction in LaNiO3 Thin Films with Nanoparticles Decoration, Adv. Sci., 2023, 2207128 CrossRef CAS.
- I. Heller, J. Kong, K. A. Williams, C. Dekker and S. G. Lemay, Electrochemistry at Single-Walled Carbon Nanotubes: The Role of Band Structure and Quantum Capacitance, J. Am. Chem. Soc., 2006, 22, 7353–7359 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00260a |
| This journal is © The Royal Society of Chemistry 2025 |