
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Physical removal of PAXHs from highly contaminated soil by density differentiation: studying the effectiveness on the molecular level†
Ruoji
Luo
and
Wolfgang
Schrader
 *
*
Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany. E-mail: wschrader@kofo.mpg.de
First published on 8th November 2023
Abstract
Contaminated soils from industrial sites, such as for coal mining or manufactured gas production, can contain polycyclic aromatic hydrocarbons (PAHs) with a concentration higher than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg kg−1, which require an integrated approach for remediation. A physical treatment by separating organic contaminants from soil materials using the density difference could lower the cost for the upcoming chemical and/or biological treatment. In our study, a highly PAH contaminated soil was separated in a 39% (w/w) calcium chloride solution (ρ = 1.4 g cm−3) via stirring, aeration or ultrasonication. Both first and second methods could separate soil materials from organic particles efficiently. The light fraction comprised around 10% of the total soil weight but 80% of solvent extractable organics (SEO). Optical and transmission electron microscopic analysis showed the light fraction, which consisted of mainly black solid aggregates (BSA), differed strongly from soil materials. Additionally, the original contaminated soil, its light and heavy fractions and the corresponding water phase together with the manually separated BSA were analyzed on the molecular level using ultrahigh resolution mass spectrometry (HRMS) with different atmospheric pressure ionization (API) methods, such as electrospray (ESI) and atmospheric pressure photo ionization (APPI). Results showed that SEO, which were primarily associated with BSA and successfully separated through physical method, contained mainly condensed aromatic ring structures of pure hydrocarbons and nitrogen heterocycles with low oxygen content.
000 mg kg−1, which require an integrated approach for remediation. A physical treatment by separating organic contaminants from soil materials using the density difference could lower the cost for the upcoming chemical and/or biological treatment. In our study, a highly PAH contaminated soil was separated in a 39% (w/w) calcium chloride solution (ρ = 1.4 g cm−3) via stirring, aeration or ultrasonication. Both first and second methods could separate soil materials from organic particles efficiently. The light fraction comprised around 10% of the total soil weight but 80% of solvent extractable organics (SEO). Optical and transmission electron microscopic analysis showed the light fraction, which consisted of mainly black solid aggregates (BSA), differed strongly from soil materials. Additionally, the original contaminated soil, its light and heavy fractions and the corresponding water phase together with the manually separated BSA were analyzed on the molecular level using ultrahigh resolution mass spectrometry (HRMS) with different atmospheric pressure ionization (API) methods, such as electrospray (ESI) and atmospheric pressure photo ionization (APPI). Results showed that SEO, which were primarily associated with BSA and successfully separated through physical method, contained mainly condensed aromatic ring structures of pure hydrocarbons and nitrogen heterocycles with low oxygen content.
Environmental significancePolyaromatic hydrocarbons are one of the most dangerous contamination to the environment and human beings. After more than one and a half centuries of industrialization those type of compounds are almost omnipresent in the environment. The standard analytical tool is based on a list of only 16 model compounds that can be easily analyzed but do only tell a small part of the contamination story. Here, a physical method of density differentiation is investigated that allows the simple removal of PAH contaminations from soil. Ultrahigh resolution mass spectrometry shows much more details about the real contamination and give a detailed perspective on the effectiveness of this method. |
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are widespread contaminants in soils and sediments worldwide.1 Their concentration varies widely from below 5 μg kg−1 in the rural area to over 1000 mg kg−1 in the industrial sites, such as for the coal mining or manufactured gas production.2–7 Considering their mutagenic and carcinogenic effects, 16 selected parent PAHs, which are involved in the priority contaminant list suggested by the United States Environmental Protection Agency (U.S. EPA), have been monitored over more than four decades.8,9Until 1990s, excavation and landfilling were recommended as a method to treat heavily PAH contaminated soils from manufactured gas plant sides.10 However, the PAHs were not really eliminated. During the years a wide range of remediation techniques such as thermal, physical, chemical, and biological treatments have been applied for PAH contaminated soil.10,11 Among them, the bioremediation is the most commonly implemented method due to its safe, eco-friendly and cost-effective character. The integrated approach, meaning a combination of more than one remediation techniques, presents the second most frequently used method for especially some highly contaminated soils.12 For instance, soils in the “hot-spot” zones of industrial areas, with PAH concentrations higher than 10000 mg kg−1, are not amenable to most bioremediation.13 Accordingly, a thermal, physical or chemical pretreatment step was advised.
Physical treatments such as soil washing or using solvent extraction are feasible cleanup techniques for highly contaminated soils with PAHs.5 Besides, the usage of density difference as a physical parameter found quiet applications in soil analysis and treatment. For example, heavy metals with a density between 5 and 9 g mL−1 could be precipitated from soil minerals (ρ ≈ 2.5–3.0 g mL−1) using a heavy liquid.14,15 Quite the other way around, by applying a liquid with a proper density lighter organic materials could float and be separated from the soil minerals.16 Ghosh et al. separated PAHs contaminated sediment samples into light and heavy fractions using cesium chloride with a specific gravity of 1.8. Results showed that the light fraction consisting of mainly organic particles, which contributed 5–20% of the total mass but 60–95% of the PAHs.17–19 Using the same solution Richardson and Aitken could recover more than 50% of the PAHs in the light fraction separated from a manufactured gas plant soil, which constituted less than 2% of the total mass.6 Even by suspending contaminated soils into two-fold amount of water could lead to a separation of maximum 76% of the PAHs for tar oil contaminated soils.7 The higher removal rate of PAHs by the simple density separation is very promising for highly PAH contaminated soils. However, in the previous studies conclusions were made based on the quantitative analysis of 16 EPA PAHs. Other alkylated and high molecular weight PAHs as well as polycyclic aromatic heterocycles containing N, S or O (PAXHs, X = N, S, O), which could also be present in such a contamination are not observed when targeting only the standard 16 PAHs,20 thus limiting the method and the information gain.
A versatile tool for the non-targeted soil analysis is ultrahigh resolution Fourier transform mass spectrometry (FTMS)21 in combination with different atmospheric pressure ionization (API) methods, such as electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI), atmospheric pressure photo ionization (APPI)22,23 and atmospheric pressure laser ionization (APLI), in negative and positive mode. This delivers multidimensional information about naphthenic type compounds24,25 or different polyaromatic compounds in ultracomplex samples such as crude oil26,27 and soil.28,29 Negative mode ESI FTMS analysis of dissolved organic matter (DOM)30–34 or soil organic matter (SOM)35–37 helped to understand the soil biogeochemistry and global carbon cycling. Even molecular characterization of natural organic matter (NOM) in groundwater can be accomplished.38 However, a non-targeted analysis of PAH contaminated soil using FTMS was not reported.39,40
In this work we compared different physical separation methods for a highly PAXH contaminated soil. PAXH tend to agglomerate and form sticky balls, which are non-water soluble and can be separated from soil compounds by density differentiation. Here, the method is presented and detailed analyses of the original soil, its fractions and corresponding water phase on the molecular level using ultrahigh resolution MS with different API methods reveal the effectiveness of the method.
Materials and methods
Sample
One highly PAXH contaminated soil sample, obtained from German Ruhrgebiet was provided as a gift, was air dried, ground in a mortar, sieved through 2 mm sieve, and stored in the fridge at 4 °C.Physical separation and following treatment
The contaminated soil was density separated using a calcium chloride (93%, Sigma-Aldrich, Germany) solution with a specific gravity of 1.4 by means of stirring, aeration or ultrasonication (Fig. 1). 80 mL 39% (w/w) calcium chloride solution were poured into a 100 mL baker or filter funnel (pore size: 16–40 μm) containing 5 g of contaminated soil. Then the mixture was either stirred or aerated with gentle nitrogen gas from the bottom for 5 min, or ultrasonicated for 30 min. Afterwards the mixture was equilibrated overnight. Subsequently the light fraction floated on the top were decanted on a filter paper (0.5 μm particle retention, Macherey-Nagel, Germany) and washed with pure water five times to remove calcium chloride. The calcium chloride solution was liquid–liquid extracted with 3 × 40 mL dichloromethane (DCM, 99%, Sigma-Aldrich, Germany). The heavy fraction was washed same way as the light fraction.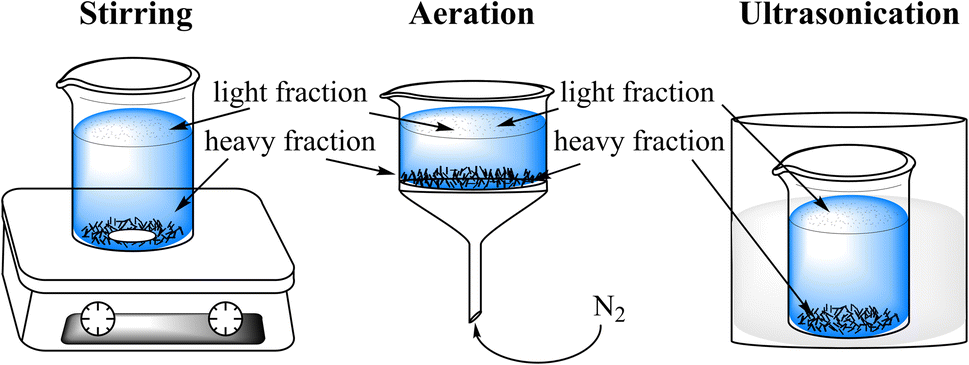 | ||
| Fig. 1 Physical separation of contaminated soil in calcium chloride solution using stirring, aeration or ultrasonication. The calcium chloride solution was liquid–liquid extracted with 3 × 40 mL dichloromethane (DCM, 99%, Sigma-Aldrich, Germany). The heavy fraction was washed as the light fraction. | ||
The original contaminated soil and the separated fractions were Soxhlet extracted using DCM for over 300 cycles.29 In addition the manually picked black solid aggregates (BSA) were extracted as described above. Furthermore, because of the low amount of organic matter found after the liquid–liquid extraction in the calcium chloride solution, a Soxhlet extraction of the original contaminated soil using pure water was performed. Eventually, solvents used for the liquid–liquid and Soxhlet extractions were rotary-evaporated and samples were stored in the fridge at 4 °C prior to the analysis.
Spectrometric analysis
The light and heavy fractions were characterized using Hitachi HF-2000 transmission electron microscopy (TEM) equipped with a cold-field emission gun and a Noran energy dispersive X-ray (EDX) detector.Mass spectrometric and data analysis
Mass spectrometric analysis was performed on a research type FT Orbitrap MS (Thermo Fisher Scientific, Bremen, Germany) with a LTQ Tune Plus 2.7.0 data processing system (Thermo Fisher Scientific, Bremen, Germany). Samples were diluted to 250 μg mL−1 in toluene (99.8%, Acros Organics, Belgium):methanol (99.8%, J.T. Baker, VWR, Germany) (1:1, v/v) or chlorobenzene (99%, Acros Organics, Belgium) for ESI or APPI measurements, respectively. The sprayer voltage was set to +4/−4 kV for the positive and negative mode ESI. The diluted samples were infused with a flow rate of 5 μL min−1 at 5, 2 and 1 arb (arbitrary unit) sheath, auxiliary and sweep gas flow for both polarities. For the positive mode APPI samples were infused with a flow rate of 20 μL min−1 under the irradiation from a Krypton VUV lamp (Syagen, Tustin, CA, U.S.A.) with a photon emission at 10.0 and 10.6 eV. The vaporizer temperature, sheath, auxiliary and sweep gases were set to 350 °C, 20, 5 and 2 arb, correspondingly. Mass spectra were recorded at a mass resolution of 480000 at m/z 400 (FWHM) using spectra stitching method41,42 in a mass range from 125–1200, using a 30 Da mass windows with 5 Da overlap.
The data were recorded by Xcalibur 2.2 (Thermo Fisher Scientific, Bremen, Germany) and further processed using Composer v1.5.0 (Sierra Analytics, Modesto, CA, U.S.A.) with the following chemical constraints: 0 < C < 200, 0 < H.< 1000, 0 < N < 3, 0 < S < 3, 0 < O < 11, 0 < double bond equivalent (DBE) < 100, maximum mass error < 1.5 ppm. It has to be noted that due to the use of a salt solution the formation of Ca-adducts are possible. This was considered during data interpretation but no addition of Ca was found. Therefore, here Ca was not used for the overall determination of the class distributions.
Results and discussion
Physical separation
The contaminated soil was separated via density difference into light (ρ < 1.4 g cm−3) and heavy (ρ > 1.4 g cm−3) fractions. It is shown in Fig. 2 that the light fractions from stirring, aeration and ultrasonication consist of only 10%, 11% and 4% of the total soil weight, whereas the majority (over 85%) of the soil constituents that comprise sand, silt, and clays remained in the heavy fraction. However, considering about the solvent extractable organics (SEO) the result was just reversed. Both the light fractions after stirring and aeration contained over 80% of the SEO, and the corresponding heavy fractions contributed the minor rest portion. Only after ultrasonication, the amount of SEO found in the heavy fraction was higher than in the light fraction. This was probably due to a compact layer of heavier soil formed initially, which entrained the lighter material, thus preventing them from floating.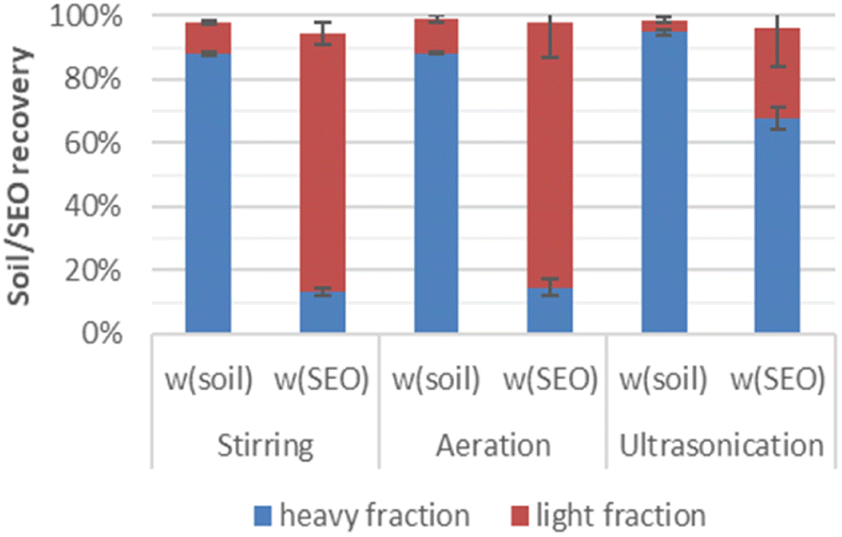 | ||
| Fig. 2 Mass and SEO distributions in the light and heavy fractions after physical separation through stirring, aeration or ultrasonication in 39% CaCl2 solution (ρ = 1.40 g cm−3). | ||
Separation techniques applied for the density differentiation include shaking in combination of centrifugation,6 agitation,7 centrifugation,14,17–19 ultrasonication.15,16 The amount of PAH associated with the light fraction ranges from 42% to 90% of the total PAH. In our study, both stirring, and aeration provided sufficient density separation.
The amount of SEO transferred during the separation into the calcium chloride solution was negligible, so that it did not provide enough sample volume to complete the mass spectrometric measurement. Nonetheless, it might be interesting to see what and which part from the contamination can be transferred into the water phase during the separation. Therefore, the original contaminated soil was Soxhlet extracted using only water. The resulting SEO was less than 0.1% of the total soil weight in comparison to the total soil (Table 1). This indicates a strong hydrophobic character of the SEO originated from the contaminated soil.
| Sample | Percentage of SEO in the sample |
|---|---|
| Orig. soil | 7.3% ± 0.5% |
| String_light | 60.3% ± 3.5% |
| String_heavy | 1.1% ± 1.0% |
| Aeration_light | 54.0% ± 3.2% |
| Aeration_heavy | 1.2% ± 0.2% |
| Ultrasonication_light | 53.6% ± 9.1% |
| Ultrasonication_heavy | 5.2% ± 0.3% |
| Soxhlet extraction of orig. soil using water | 0.1% |
| Soxhlet extraction of BSA | 77.6 ± 1.2% |
The original soil was highly contaminated with organic matter. Around 7% of the total soil weight were extractable using DCM (Table 1). The percentages were increased to 60% and 56% for the light fractions from stirring and aeration, respectively. The weight ratios of the extract to the corresponding heavy fractions were decreased from 7% (compared to the original contaminated soil) to 1.1% and 1.2%. Although the light fraction after ultrasonication weighed less than after stirring or aeration, it consisted of comparable amount of SEO (56%). Additionally, the result of the Soxhlet extraction of manually selected pure BSA from the original contaminated soil showed, that up to 80% of BSA were DCM extractable. This implies that the main SEO was derived from BSA.
Results showed that a simple physical separation through stirring or aeration could lead to an efficient removal of organic matter, which aggregates and has a different density than soil materials. These two physical methods can easily be scaled up for the industrial use and compared to ultrasonication, which anyway showed lower separation efficiency, require less energy.
Soil particle characterization
As shown in Fig. 3a and d, the heavy fraction differed strongly from the light fraction observable already from the optic view. The heavy fraction manifested a clay brown color. In contrast, the light fraction was almost black. Furthermore, the TEM-EDX results revealed, that the heavy fraction contained primarily irregular Si-dominated, Si–Al or Ca natural mineral particles (Fig. 3c).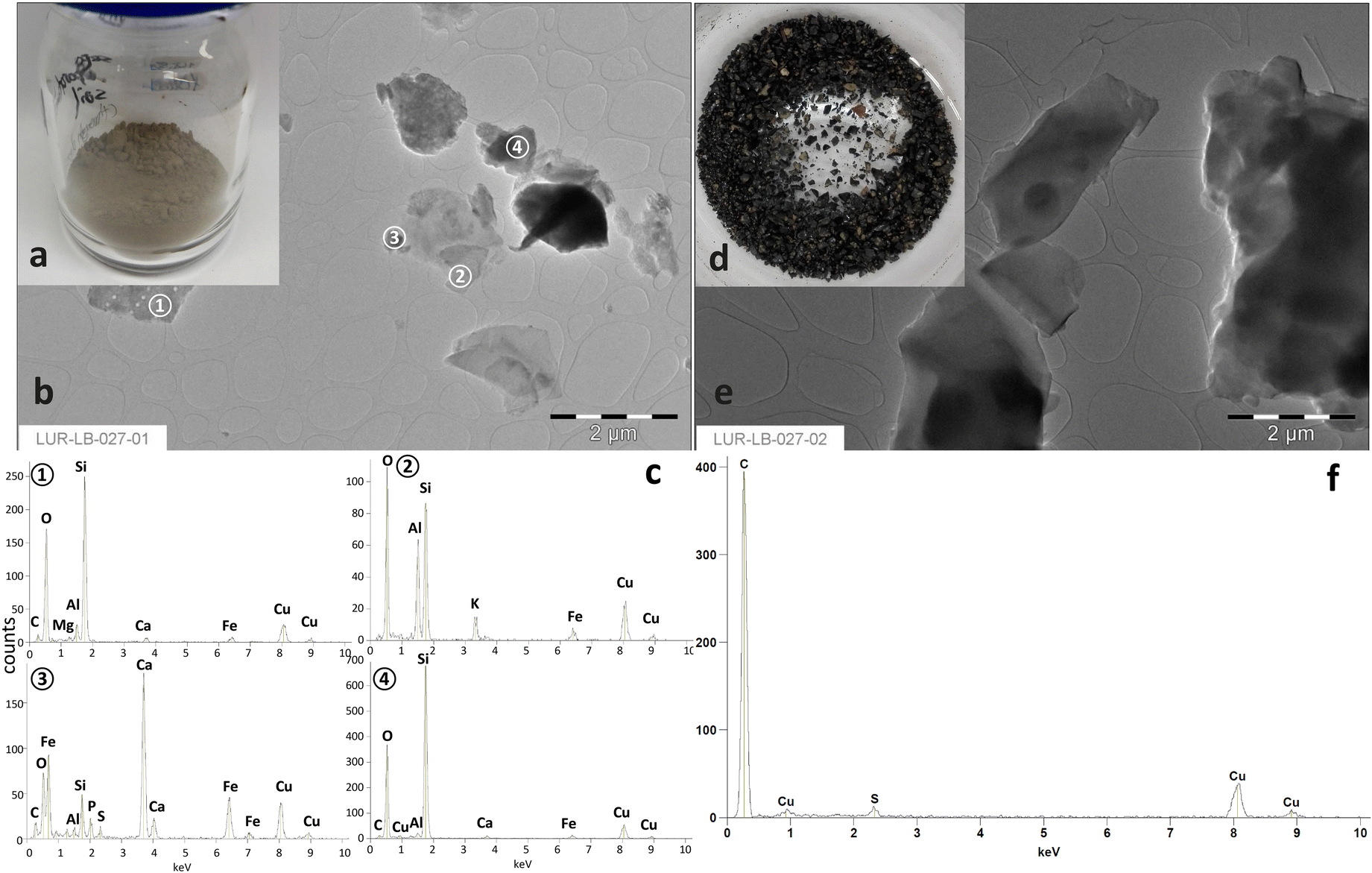 | ||
| Fig. 3 The separated heavy (a) and light (d) fractions from the original soil with their TEM images (b and e) and corresponding EDX results (c and f). | ||
Instead, organic particles dominated in the light fraction (Fig. 3f). The signal for Cu was derived from the copper grid used for fixation of soil particles. The result further confirmed that the particles from the light fraction contained high amount of organic matters, which were successfully removed from soil minerals through simple physical separation. This is in good accordance with the results obtained from Trellu et al.7 However, it needs deeper examinations on the molecular level to judge, whether the rest SEOs adsorbed on soil particles differ from the separated BSA.
FT Orbitrap mass spectra
Soil is a complex mixture consisting of inorganic, organic materials and macro- and microorganisms. A non-targeted analysis of the organic extract from soil is a sophisticated task. Depending highly on the location where the soil is coming from, its content of soil organic matter can vary widely. Their existence can influence the analysis of organic contaminants such as PAXHs in the non-targeted approach. The complexity can be deciphered using ultrahigh-resolution MS. In earlier investigations, negative mode ESI was frequently utilized for the study of organic matter in or derived from soil, which contain mostly polar constituents and high number of oxygen per molecule.30–36However, it is difficult to completely analyze all compounds in soil by using a single ionization method, especially when mostly nonpolar PAHs are of interest. Hence, it is essential to compare different ionization methods for encompassing all detectable classes.
The positive mode APPI mass spectrum of BSA separated from the contaminated soil is shown in the top row of Fig. 4. The results allow a differentiation between natural organic matter from soil and PAH contamination caused by a coking plant,43 but are comparable to data for organic-carbon rich particles analyzed by Ghosh et al.17 Here, radical hydrocarbon class compounds were predominant in the spectrum throughout the mass range between 125 and 1200 Da with most of them have a high aromaticity including the 16 EPA PAHs in lower mass range. The distribution with small aliphatic side chains are a pattern known for being pyrogenic rather than petrogenic or biogenic.
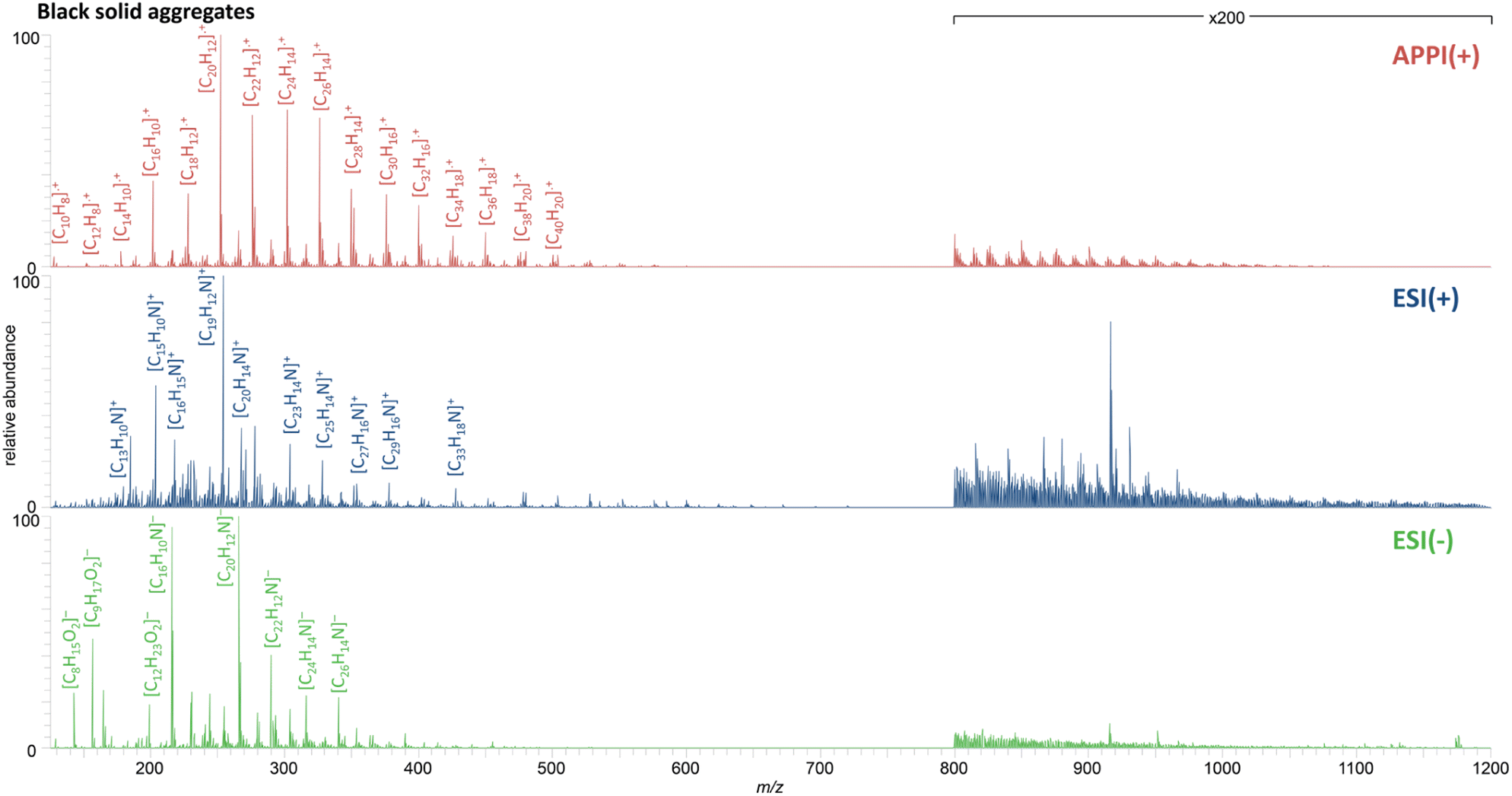 | ||
| Fig. 4 Recombined mass spectra for the Soxhlet extract from BSA, analyzed by FT Orbitrap MS using positive mode APPI (top), ESI (middle) and negative mode ESI (bottom). | ||
Analogous distributions were acquired for the original contaminated soil and its fractions (Fig. S-1†), which illustrate that the soil was heavily contaminated with PAHs that were mainly associated with BSA.
Not only pure PAHs were identified as contaminants, results of positive and negative mode ESI (Fig. 4, middle and bottom) demonstrate that also nitrogen containing PAHs are present. Further, with both polarities applied, the PANHs found in the contaminated soil can be categorized as basic pyridine-containing or neutral pyrrole-containing PAXHs.44,45
Although the proportion of water soluble organics to the total SEO using DCM was small (<1%, Fig. 2), it delivered valuable information about which contaminants can be transferred directly into the water phase, such as groundwater or surface water close to the contamination side, before any remediation techniques have been taken place.
The water extract of contaminated soil contained more polar PANHs in comparison with the DCM extracts of BSA, the contaminated soil as well as its fractions (Fig. S-2†). Additionally, the signals were shifted towards lower mass ranges (<300 Da). Same trend was observed in the positive and negative mode ESI spectra, where low molecular weight basic pyridine-like PANHs and oxygenated PA(N)Hs were detected, respectively. The results revealed that by monitoring only the 16 EPA PAHs might not be sufficient to examine the total risk of PAH contaminated sides, since higher molecular weight PAHs are also present in addition to other more polar heteroatom containing PAHs or transformation products.
Class distribution
The elemental compositions detected using API FTMS were categorized into classes according to the number of heteroatoms (N, S and O) per molecule and summarized in a class distribution plot for each sample. Pure hydrocarbons without any heteroatoms were grouped in the HC class. A total of 20 compound classes were detected in the positive mode APPI for Soxhlet extracts of BSA, contaminated soil and its fractions after stirring using DCM (Fig. 5). The number of classes for the water extract (Fig. 5, orig. soil*) exceeded 20 classes. However, in order to compare the water extract with other samples, only classes were selected, which were found for the DCM extracts. APPI can ionize pure and heteroatom containing PAH efficiently and generate two types of ions: radical cations and protonated molecules.26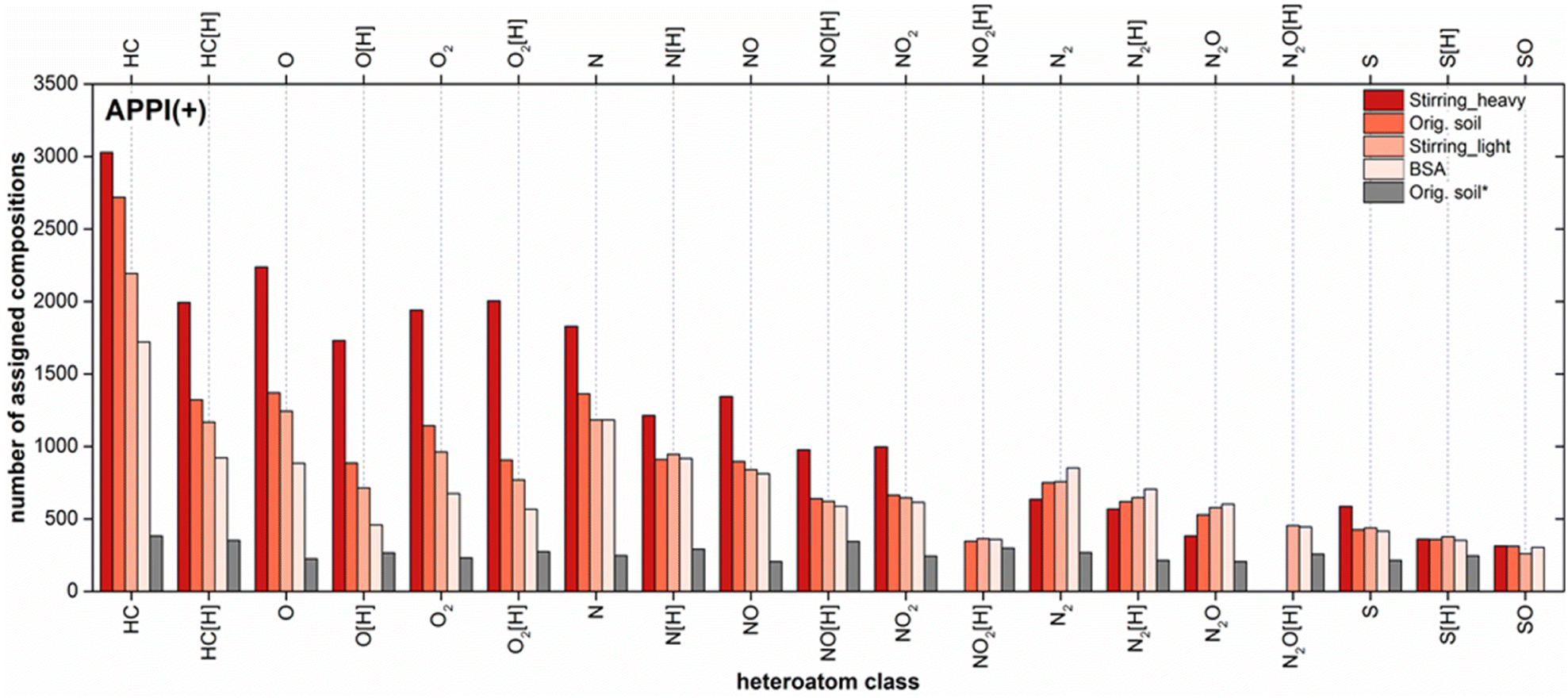 | ||
| Fig. 5 Class distributions for the heavy fraction after stirring, original soil, light fraction after stirring and BSA (from dark to light red) as well as the water extract (selected classes only) denoted as orig. soil*, analyzed using positive mode APPI FT Orbitrap MS. | ||
The highest number of total assigned compositions was found for the heavy fraction after stirring, which was reduced from 22149, 16168 (orig. soil), 15161 (stirring_light) to 13377 (BSA). Even in the water extract a total number of 9093 chemical compositions were assigned. When comparing classes within one sample of the Soxhlet extracts using DCM, highest assignments were detected in the radical hydrocarbon (HC) class, followed by radical and protonated O, O2, N, NO, NO2, N2 and S classes. When comparing same classes between DCM extracts, the number of assigned compositions in each class reduced gradually from the heavy fraction to original soil to light fraction and finally to BSA.
Similar results were gained for the soil fractions after aeration (Fig. S-3†). However, because of an insufficient separation through ultrasonication, the difference between the original soil and its fractions was not as prominent as from other two methods (Fig. S-4†). Nevertheless, among the DCM extracts BSA always displayed lowest assignments in each class. This indicates that, with regard to the chemical composition BSA are simpler than the original soil and its fractions.
In the ESI(+) measurements the total number of assigned compositions was lower than in the APPI(+) measurements, even though more polar classes were detected (Fig. S-4–S-7†).
Similar results were obtained compared to the APPI(+) measurements: the highest assignments were found for the heavy fractions, followed by the original soil, light fractions and lastly BSA. Here, basic pyridine-like PANHs (N, N2) and their oxygenated classes (NO, NO2) with NSOx and SOx classes were the most abundant classes. Besides, protonated molecules radical ions were also detected for such as HC, N and N2 classes, which implies on the other hand the presence of highly condensed aromatic structures in the sample.46
In the ESI(−) measurements the total assignments was lowered to around 10000 for the heavy fractions (Fig. S-8–S-10†). In spite of this, negative mode data allowed a better examination of 5-membered nitrogen containing heterocycles or acidic species such as carboxylic acids, which trace the influence of natural weathering or chemical/biological remediation processes.47 Here, the difference between the heavy fraction and the original soil was minor, but the number of assigned compositions in each class was still higher than the corresponding light fraction and BSA. In the DCM extracts the most abundant classes were O2–4, NO2–3 and N2O2, but a clear shifting towards highly oxidized classes such as O6–8 or NO6 were observed in the water extract. Results indicate that because of better solubility highly weathered oxygen containing and polar PAXHs could be easily transferred into the water phase.
DBE vs. carbon number plot
In Fig. 6, the major compound classes, namely radical HC and protonated N classes, from the positive mode APPI and ESI measurements for the different samples were compared in Kendrick plots. Here, each composition from one class can be visualized by its DBE and number of carbon atoms per molecule. A maximum DBE of 70 with 95 carbon atoms per molecule was detected as radical ions from the HC class. In the N class high molecular weight azaarenes with a maximum DBE of 72 and 94 carbon atoms per molecule were discovered, which exceeded the largest azaarenes reported by Tian et al. for PAH contaminated soils.48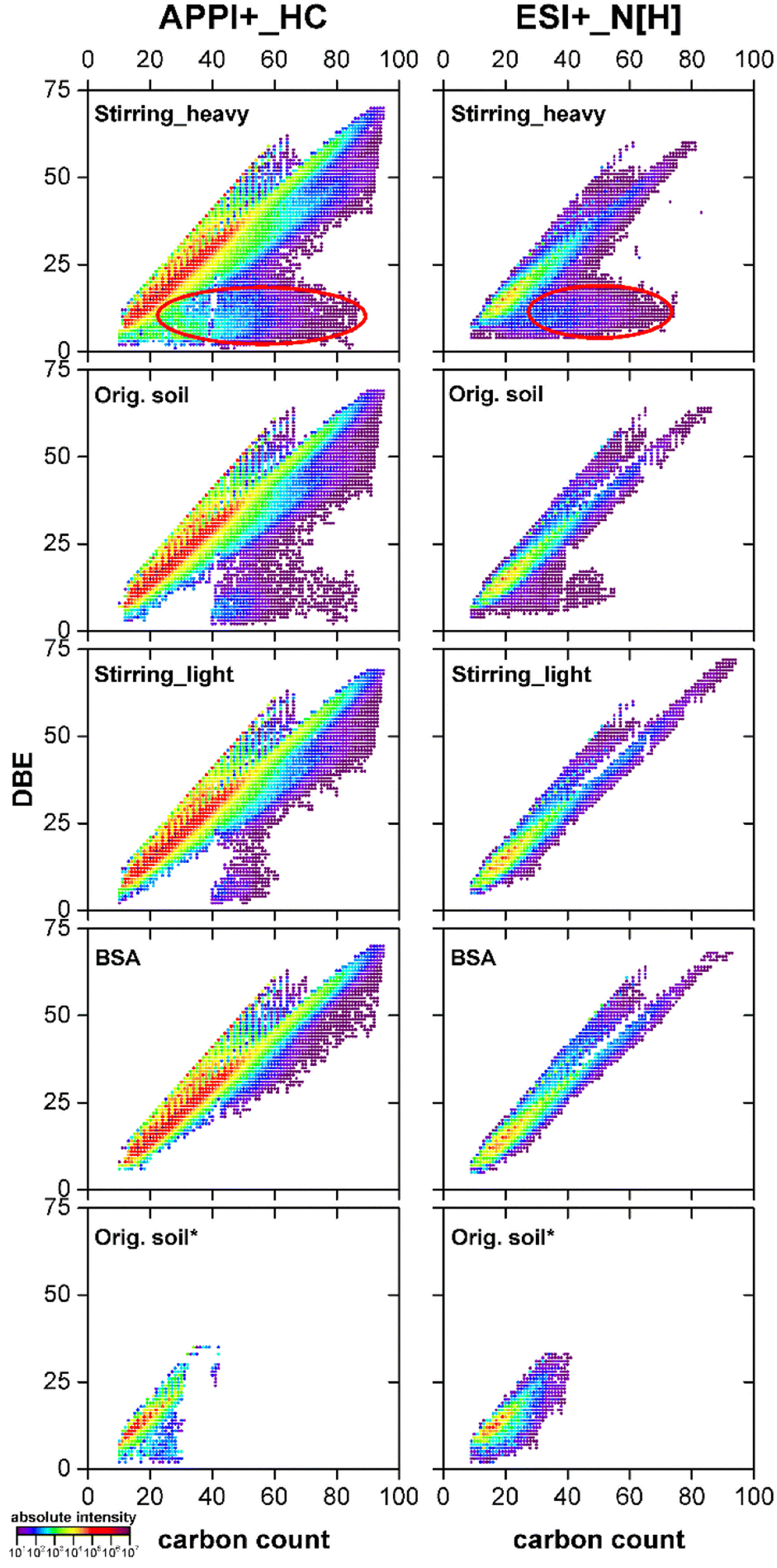 | ||
| Fig. 6 DBE vs. carbon number plots for radical hydrocarbon (left column) and protonated nitrogen (right column) classes from the heavy fraction after stirring, original soil, light fraction after stirring, BSA and original soil after Soxhlet extraction with water (from top to bottom), analyzed using positive mode APPI and ESI Orbitrap MS. For details of this analysis see previous results shown before.28 | ||
In the HC and N classes for BSA only highly condensed aromatic hydrocarbons and N-heterocycles were detected, which represent the identity of the contaminants on a molecular level.28 From the light fraction to original soil and to the heavy fraction, increasing amount of additional compositions with relative lower DBE (<25) showed up. Compared with e.g. the 16 PAHs, these compositions (highlighted in red cycles, Fig. 6) contained more saturated side chains or naphthenic ring systems. Likewise, in the heavy fractions after aeration and ultrasonication, where the main soil particles stayed, more compositions with high carbon number and low DBE were found in comparison to the corresponding light fractions (Fig. S-11†). They were distinct from the chemical point of view from the real contaminants, suggesting their natural origin.
The occurrence of natural hydrocarbons of biogenic origin in soil was investigated by different researchers.49,50 Here, because of the analytical technique used, results were mainly restricted to sum parameters such as TPH or the 16 EPA PAH related compounds with low molecular weight. Using ultrahigh resolution FTMS with different API methods, more compositional information especially polar compounds can be gained.
Moreover, Kendrick plots for the water extract (orig. soil*, Fig. 6) demonstrated that also low molecular weight parent PAXHs could be transferred into water phase easily.
Van Krevelen plot
Another graphical method frequently used for the study of natural organic matter in the environment is the van Krevelen plot.51,52 Recently a new concept called aromaticity index (AI) is incorporated into the van Krevelen plot, which represents the density of C–C double bond in a molecule indicating its aromaticity.53In Fig. 7 the van Krevelen plots for BSA and the unique compositions in the heavy fraction as well as the water extract after subtraction of common compositions from SBA were compared. The compositions in BSA existed largely in ranges of lower H/C and O/C ratio. 2648 out of 3970 compositions of HC and Ox classes for BSA were below the gray line with AI greater than 0.5 (Table 2), which reflects the presence of aromatic cores in the compositions. On the top of this, around 60% of them (1530) possessed condensed aromatic ring structures (CARS), which exhibit below the black line with AI greater than 0.67. CARS were also detected in volcanic ash soil,30 DOM from boreal lakes31 and paddy soils.34 There, CARS pervaded throughout the O/C range of 0 to 0.8 and mainly located between 0.2 and 0.6. Instead, results from our study showed that nearly 90% of the CARS (1370) detected in BSA had an O/C lower than 0.1 and they mainly belonged to pure HC as well as low oxygenated classes (O1–3). The absolute number of compositions with AI > 0.5 or >0.67 was slightly increased for the heavy fraction after stirring. However, due to the large increase in compositions with lower AI, the percentages of compositions with AI > 0.5 and >0.67 reduced from 67% to 41% and from 38% to 21%, respectively. The percentages of aromatic and condensed aromatic structures in the water extract were further reduced to 33% and 14%. Similar result was obtained for the NOx classes (Fig. S12 and Table S-1†).
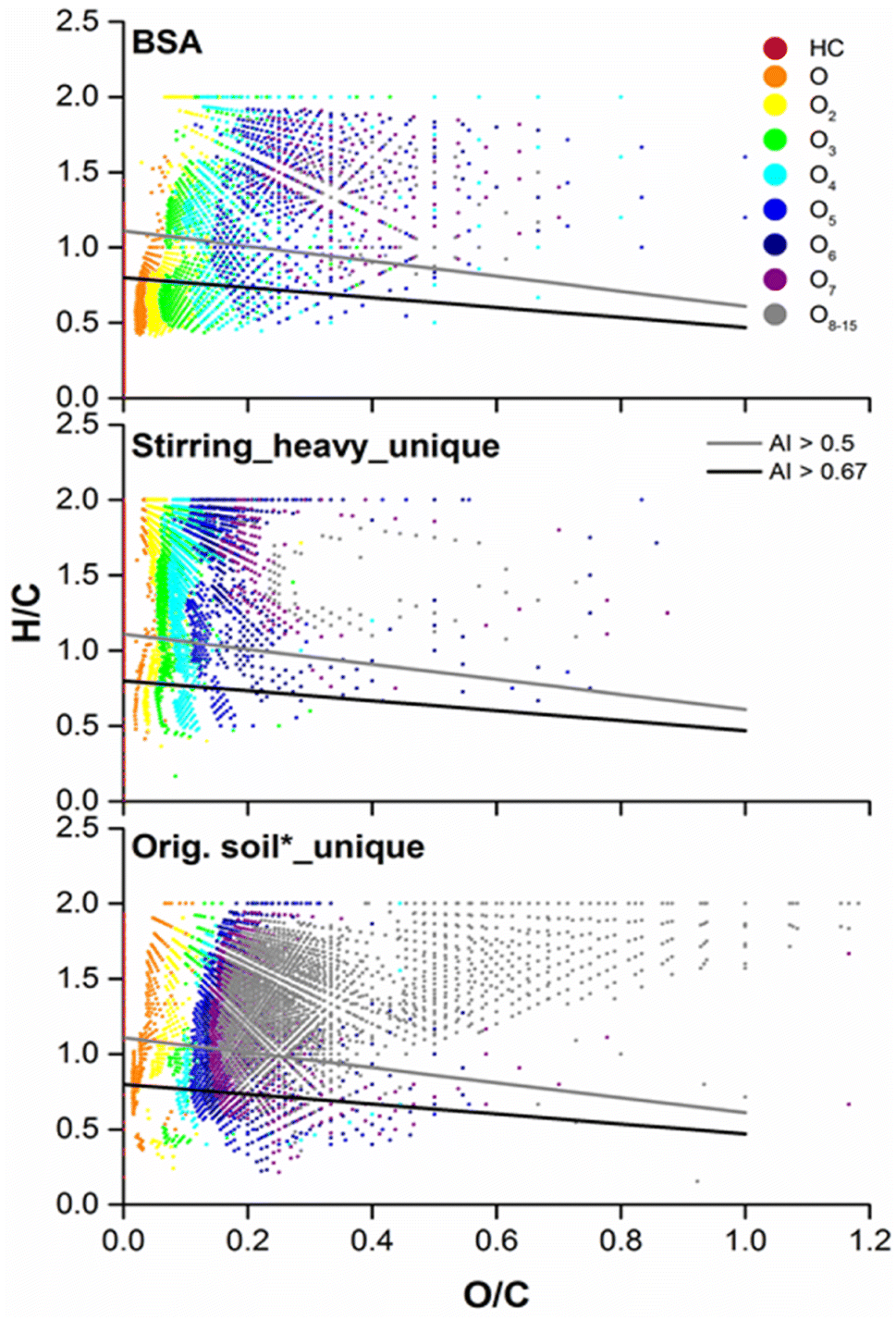 | ||
| Fig. 7 Van Krevelen plots for Soxhlet extract from BSA (top), the unique compositions from the heavy fraction after stirring (middle), and the original soil Soxhlet extracted using water (bottom, denoted as orig. soil*), compared with BSA, analyzed by both positive mode APPI (HC) and negative mode ESI (Ox) FT Orbitrap MS. Compositions below the gray or black line have a aromaticity index (AI) higher than 0.5 or 0.67, respectively. | ||
| HC + Ox classes | Number of compositions in | ||
|---|---|---|---|
| Total | AI > 0.5 | AI > 0.67 | |
| BSA | 3970 | 2648 (67%) | 1530 (38%) |
| Stirring heavy | 7643 | 3125 (41%) | 1633 (21%) |
| Orig. soil* | 6523 | 2150 (33%) | 928 (14%) |
Combined the results obtained from Fig. 6 and 7, we propose that BSA, which were characterized as the main contamination source containing a high amount of pure and nitrogen containing CARS with/without short side chains, could be separated from natural soil material quiet efficiently through physical methods. This simple and cheap separation step via density difference is recommended to be implemented prior to other chemical or biological remediation techniques for soils highly contaminated with PAXHs, which present as BSA.
Considering the 16 EPA priority PAH, the results also show that the contamination of industrial sites are much higher and the complexity is more extreme than what can be covered by these standard compounds. It should be strongly considered to expand the range of compounds that are routinely covered because nobody really knows about the toxic effects of such a diverse and complex mixture.
Conclusions
In this study we investigated different physical separation techniques for the removal of PAXHs from the heavily contaminated soil. Over 80% of SEO could be removed from the contaminated soil using stirring and aeration in a 39% (w/w) calcium chloride solution. Most of the SEO in the lighter fraction were associated with BSA. The BSA were then analyzed and compared to the original contaminated soil as well as soil fractions using FTMS with different API in a non-targeted approach. By combining the results obtained from positive mode APPI and positive/negative mode ESI the BSA were characterized as containing highly aromatized structures, which include pure PAHs and PAXHs (X = N, O and S). Around 67% of the assigned elemental compositions from HC and Ox classes and over 40% of N and NOx class compounds comprise condensed aromatic structures. The short length of side chain for these PAXHs detected in the BSA indicates that the contamination has a pyrogenic origin.Results show that by applying comprehensive non-targeted analysis using API-FTMS a deeper understanding of the soil contamination, including the source and determination of contaminants as well as the corresponding remediation efficiency, can be achieved.
Author contributions
Investigation, data interpretation, formal analysis, writing – original draft – R. L.; conceptualization, funding acquisition, project administration, writing – review & editing, supervision – W. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This generous funding by the Bundesministerium für Wissenschaft und Energie through the ZIM Project KF 3121303SK4 is gratefully acknowledged. The authors thank Dr Klaus Moraw (Ökoplan GmbH, Dinslaken) for providing the real contaminated soil sample and Dr David Stranz (Sierra Analytics, Modesto, CA, USA) for access to new data handling software. The authors thank the Electron Microscopy group of the Max-Planck Institut für Kohlenforschung for their work. Open Access funding provided by the Max Planck Society.Notes and references
- S. A. Stout, S. D. Emsbo-Mattingly, G. S. Douglas, A. D. Uhler and K. J. McCarthy, Beyond 16 priority pollutant PAHs: a review of PACs used in environmental forensic chemistry, Polycyclic Aromat. Compd., 2015, 35, 285–315, DOI:10.1080/10406638.2014.891144.
- W. Wilcke, Global patterns of polycyclic aromatic hydrocarbons (PAHs) in soil, Geoderma, 2007, 141, 157–166, DOI:10.1016/j.geoderma.2007.07.007.
- K. C. Jones, J. A. Stratford, K. S. Waterhouse and N. B. Vogt, Organic contaminants in Welsh soils: polynuclear aromatic hydrocarbons, Environ. Sci. Technol., 1989, 23, 540–550, DOI:10.1021/es00063a005.
- E. Morillo, A. S. Romero, C. Maqueda, L. Madrid, F. Ajmone-Marsan, H. Grcman, C. M. Davidson, A. S. Hursthouse and J. Villaverde, Soil pollution by PAHs in urban soils: a comparison of three European cities, J. Environ. Monit., 2007, 9, 1001–1008, 10.1039/b705955h.
- Z. Gong, X. Wang, Y. Tu, J. Wu, Y. Sun and P. Li, Polycyclic aromatic hydrocarbon removal from contaminated soils using fatty acid methyl esters, Chemosphere, 2010, 79, 138–143, DOI:10.1016/j.chemosphere.2010.01.037.
- S. D. Richardson and M. D. Aitken, Desorption and bioavailability of polycyclic aromatic hydrocarbons in contaminated soil subjected to long-term in situ biostimulation, Environ. Toxicol. Chem., 2011, 30, 2674–2681, DOI:10.1002/etc.682.
- C. Trellu, A. Miltner, R. Gallo, D. Huguenot, E. D. van Hullebusch, G. Esposito, M. A. Oturan and M. Kästner, Characteristics of PAH tar oil contaminated soils—black particles, resins and implications for treatment strategies, J. Hazard. Mater., 2017, 327, 206–215, DOI:10.1016/j.jhazmat.2016.12.062.
- L. Keith and W. Telliard, ES&T special report: priority pollutants: I-a perspective view, Environ. Sci. Technol., 1979, 13, 416–423, DOI:10.1021/es60152a601.
- L. H. Keith, The source of U.S. EPA's sixteen PAH priority pollutants, Polycyclic Aromat. Compd., 2015, 35, 147–160, DOI:10.1080/10406638.2014.892886.
- S. Kuppusamy, P. Thavamani, K. Venkateswarlu, Y. B. Lee, R. Naidu and M. Megharaj, Remediation approaches for polycyclic aromatic hydrocarbons (PAHs) contaminated soils: technological constraints, emerging trends and future directions, Chemosphere, 2017, 168, 944–968, DOI:10.1016/j.chemosphere.2016.10.115.
- S. Gan, E. V. Lau and H. K. Ng, Remediation of soils contaminated with polycyclic aromatic hydrocarbons (PAHs), J. Hazard. Mater., 2009, 172, 532–549, DOI:10.1016/j.jhazmat.2009.07.118.
- S. V. Mohan, T. Kisa, T. Ohkuma, R. A. Kanaly and Y. Shimizu, Bioremediation technologies for treatment of PAH-contaminated soil and strategies to enhance process efficiency, Rev. Environ. Sci. Biotechnol., 2006, 5, 347–374, DOI:10.1007/s11157-006-0004-1.
- A. P. Khodadoust, R. Bagchi, M. T. Suidan, R. C. Brenner and N. G. Sellers, Removal of PAHs from highly contaminated soils found at prior manufactured gas operations, J. Hazard. Mater., 2000, 80, 159–174, DOI:10.1016/S0304-3894(00)00286-7.
- K. J. Henley, Improved heavy-liquid separation at fine particle sizes, Am. Mineral., 1977, 62, 377–381 Search PubMed.
- J. Cotter-Howells, Separation of high density minerals from soil, Sci. Total Environ., 1993, 132, 93–98, DOI:10.1016/0048-9697(93)90263-6.
- C. Cerli, L. Celi, K. Kalbitz, G. Guggenberger and K. Kaiser, Separation of light and heavy organic matter fractions in soil — testing for proper density cut-off and dispersion level, Geoderma, 2012, 170, 403–416, DOI:10.1016/j.geoderma.2011.10.009.
- U. Ghosh, J. S. Gillette, R. G. Luthy and R. N. Zare, Microscale location, characterization, and association of polycyclic aromatic hydrocarbons on harbor sediment particles, Environ. Sci. Technol., 2000, 34, 1729–1736, DOI:10.1021/es991032t.
- U. Ghosh, J. R. Zimmerman and R. G. Luthy, PCB and PAH speciation among particle types in contaminated harbor sediments and effects on PAH bioavailability, Environ. Sci. Technol., 2003, 37, 2209–2217, DOI:10.1021/es020833k.
- M. F. Khalil, U. Ghosh and J. P. Kreitinger, Role of weathered coal tar pitch in the partitioning of polycyclic aromatic hydrocarbons in manufactured gas plant site sediments, Environ. Sci. Technol., 2006, 40, 5681–5687, DOI:10.1021/es0607032.
- J. T. Andersson and C. Achten, Time to say goodbye to the 16 EPA PAHs? Toward an up-to-date use of PACs for environmental purposes, Polycyclic Aromat. Compd., 2015, 35, 330–354, DOI:10.1080/10406638.2014.991042.
- S. K. Panda, J. T. Andersson and W. Schrader, Characterization of supercomplex crude oil mixtures: what is really in there?, Angew. Chem., Int. Ed., 2009, 48, 1788–1791, DOI:10.1002/anie.200803403.
- T. J. Kauppila, H. Kersten and T. Benter, The Ionization Mechanisms in Direct and Dopant-Assisted Atmospheric Pressure Photoionization and Atmospheric Pressure Laser Ionization, J. Am. Soc. Mass Spectrom., 2014, 25, 1870–1881, DOI:10.1007/s13361-014-0988-7.
- T. J. Kauppila, J. A. Syage and T. Benter, Recent Developments in Atmospheric Pressure Photoionization-Mass Spectrometry, Mass Spectrom. Rev., 2017, 36, 423–449, DOI:10.1002/mas.21477.
- M. P. Barrow, M. Witt, J. V. Headley and K. M. Peru, Athabasca Oil Sands Process Water: Characterization by Atmospheric Pressure Photoionization and Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry, Anal. Chem., 2010, 82, 3727–3735, DOI:10.1021/ac100103y.
- J. V. Headley, M. P. Barrow, K. M. Peru, B. Fahlman, R. A. Frank, G. Bickerton, M. E. McMaster, J. Parrott and L. M. Hewitt, Preliminary fingerprinting of Athabasca oil sands polar organics in environmental samples using electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry, Rapid Commun. Mass Spectrom., 2011, 25, 1899–1909, DOI:10.1002/rcm.5062.
- A. Gaspar, E. Zellermann, S. Lababidi, J. Reece and W. Schrader, Impact of different ionization methods on the molecular assignments of asphaltenes by FT-ICR mass spectrometry, Anal. Chem., 2012, 84, 5257–5267, DOI:10.1021/ac300133p.
- Y. Cho, H. N. Abed and S. Kim, Molecular Level Investigation of Oil Sludge at the Bottom of Oil Tank in Ratawi Oil Field by Atmospheric Pressure Photo Ionization Ultrahigh-resolution Mass Spectrometry, Bull. Korean Chem. Soc., 2020, 41, 450–453, DOI:10.1002/bkcs.11991.
- R. Luo and W. Schrader, Getting a better overview of a highly PAH contaminated soil: a non-targeted approach assessing the real environmental contamination, J. Hazard. Mater., 2021, 42, 643–658, DOI:10.1016/j.jhazmat.2021.126352.
- R. Luo and W. Schrader, Development of a Non-Targeted Method to Study Petroleum Polyaromatic Hydrocarbons in Soil by Ultrahigh Resolution Mass Spectrometry Using Multiple Ionization Methods, Polycyclic Aromat. Compd., 2022, 1–16, DOI:10.1080/10406638.2020.1748665.
- R. W. Kramer, E. B. Kujawinski and P. G. Hatcher, Identification of black carbon derived structures in a volcanic ash soil humic acid by Fourier transform ion cyclotron resonance mass spectrometry, Environ. Sci. Technol., 2004, 38, 3387–3395, DOI:10.1021/es030124m.
- A. M. Kellerman, T. Dittmar, D. N. Kothawala and L. J. Tranvik, Chemodiversity of dissolved organic matter in lakes driven by climate and hydrology, Nat. Commun., 2014, 5, 3804, DOI:10.1038/ncomms4804 , https://www.nature.com/articles/ncomms4804#supplementary-information.
- J. Guigue, M. Harir, O. Mathieu, M. Lucio, L. Ranjard, J. Lévêque and P. Schmitt-Kopplin, Ultrahigh-resolution FT-ICR mass spectrometry for molecular characterisation of pressurised hot water-extractable organic matter in soils, Biogeochemistry, 2016, 128, 307–326, DOI:10.1007/s10533-016-0209-5.
- G. Fleury, M. Del Nero and R. Barillon, Effect of mineral surface properties (alumina, kaolinite) on the sorptive fractionation mechanisms of soil fulvic acids: Molecular-scale ESI-MS studies, Geochim. Cosmochim. Acta, 2017, 196, 1–17, DOI:10.1016/j.gca.2016.09.029.
- X.-M. Li, G.-X. Sun, S.-C. Chen, Z. Fang, H.-Y. Yuan, Q. Shi and Y.-G. Zhu, Molecular chemodiversity of dissolved organic matter in paddy soils, Environ. Sci. Technol., 2018, 52, 963–971, DOI:10.1021/acs.est.7b00377.
- M. M. Tfaily, R. K. Chu, N. Tolić, K. M. Roscioli, C. R. Anderton, L. Paša-Tolić, E. W. Robinson and N. J. Hess, Advanced solvent based methods for molecular characterization of soil organic matter by high-resolution mass spectrometry, Anal. Chem., 2015, 87, 5206–5215, DOI:10.1021/acs.analchem.5b00116.
- M. M. Tfaily, R. K. Chu, J. Toyoda, N. Tolic, E. W. Robinson, L. Pasa-Tolic and N. J. Hess, Sequential extraction protocol for organic matter from soils and sediments using high resolution mass spectrometry, Anal. Chim. Acta, 2017, 972, 54–61, DOI:10.1016/j.aca.2017.03.031.
- N. N. Solihat, T. Acter, D. Kim, A. F. Plante and S. Kim, Analyzing Solid-Phase Natural Organic Matter Using Laser Desorption Ionization Ultrahigh Resolution Mass Spectrometry, Anal. Chem., 2019, 91, 951–957, DOI:10.1021/acs.analchem.8b04032.
- M. Jylha-Ollila, H. Laine-Kaulio, J. Schilder, P. Niinikoski-Fusswinkel, T. Kekalainen, J. Janis and H. Koivusalo, Carbon Budget and Molecular Structure of Natural Organic Matter in Bank Infiltrated Groundwater, Groundwater, 2021, 59, 644–657, DOI:10.1111/gwat.13087.
- M. Lu, Z. Zhang, W. Qiao, Y. Guan, M. Xiao and C. Peng, Removal of residual contaminants in petroleum-contaminated soil by Fenton-like oxidation, J. Hazard. Mater., 2010, 179, 604–611, DOI:10.1016/j.jhazmat.2010.03.046.
- M. Lu, Z. Zhang, W. Qiao, X. Wei, Y. Guan, Q. Ma and Y. Guan, Remediation of petroleum-contaminated soil after composting by sequential treatment with Fenton-like oxidation and biodegradation, Bioresour. Technol., 2010, 101, 2106–2113, DOI:10.1016/j.biortech.2009.11.002.
- A. D. Southam, T. G. Payne, H. J. Cooper, T. N. Arvanitis and M. R. Viant, Dynamic range and mass accuracy of wide-scan direct infusion nanoelectrospray Fourier transform ion cyclotron resonance mass spectrometry-based metabolomics increased by the spectral stitching method, Anal. Chem., 2007, 79, 4595–4602, DOI:10.1021/ac062446p.
- A. Vetere and W. Schrader, Mass spectrometric coverage of complex mixtures: exploring the carbon space of crude oil, ChemistrySelect, 2017, 2, 849–853, DOI:10.1002/slct.201601083.
- N. Hertkorn, M. Frommberger, M. Witt, B. P. Koch, P. Schmitt-Kopplin and E. M. Perdue, Natural organic matter and the event horizon of mass spectrometry, Anal. Chem., 2008, 80, 8908–8919, DOI:10.1021/ac800464g.
- X. Chen, B. Shen, J. Sun, C. Wang, H. Shan, C. Yang and C. Li, Characterization and comparison of nitrogen compounds in hydrotreated and untreated shale oil by electrospray ionization (ESI) Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS), Energy Fuels, 2012, 26, 1707–1714, DOI:10.1021/ef201500r.
- J. Tong, J. Liu, X. Han, S. Wang and X. Jiang, Characterization of nitrogen-containing species in Huadian shale oil by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry, Fuel, 2013, 104, 365–371, DOI:10.1016/j.fuel.2012.09.042.
- L. M. Guricza and W. Schrader, Electrospray ionization for determination of non-polar polyaromatic hydrocarbons and polyaromatic heterocycles in heavy crude oil asphaltenes, J. Mass Spectrom., 2015, 50, 549–557, DOI:10.1002/jms.3561.
- H. Chen, A. Hou, Y. E. Corilo, Q. Lin, J. Lu, I. A. Mendelssohn, R. Zhang, R. P. Rodgers and A. M. McKenna, 4 years after the Deepwater Horizon spill: molecular transformation of Macondo well oil in Louisiana salt marsh sediments revealed by FT-ICR mass spectrometry, Environ. Sci. Technol., 2016, 50, 9061–9069, DOI:10.1021/acs.est.6b01156.
- Z. Tian, J. Vila, H. Wang, W. Bodnar and M. D. Aitken, Diversity and abundance of high-molecular-weight azaarenes in PAH-contaminated environmental samples, Environ. Sci. Technol., 2017, 51, 14047–14054, DOI:10.1021/acs.est.7b03319.
- Z. D. Wang, C. Yang, Z. Yang, B. Hollebone, C. E. Brown, M. Landriault, J. Sun, S. M. Mudge, F. Kelly-Hooper and D. G. Dixon, Fingerprinting of petroleum hydrocarbons (PHC) and other biogenic organic compounds (BOC) in oil-contaminated and background soil samples, J. Environ. Monit., 2012, 14, 2367–2381, 10.1039/c2em30339f.
- M. Vecchiato, T. Bonato, A. Bertin, E. Argiriadis, C. Barbante and R. Piazza, Plant residues as direct and indirect sources of hydrocarbons in soils: current issues and legal implications, Environ. Sci. Technol. Lett., 2017, 4, 512–517, DOI:10.1021/acs.estlett.7b00464.
- S. A. Visser, Application of Van Krevelen's graphical-statistical method for the study of aquatic humic material, Environ. Sci. Technol., 1983, 17, 412–417, DOI:10.1021/es00113a010.
- W. C. Hockaday, A. M. Grannas, S. Kim and P. G. Hatcher, The transformation and mobility of charcoal in a fire-impacted watershed, Geochim. Cosmochim. Acta, 2007, 71, 3432–3445, DOI:10.1016/j.gca.2007.02.023.
- B. P. Koch and T. Dittmar, From mass to structure: an aromaticity index for high-resolution mass data of natural organic matter, Rapid Commun. Mass Spectrom., 2006, 20, 926–932, DOI:10.1002/rcm.2386.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3em00379e |
| This journal is © The Royal Society of Chemistry 2024 |