
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in radical polymerization of bio-based monomers in aqueous dispersed media†
Elena
Rigo
abc,
Vincent
Ladmiral
 a,
Sylvain
Caillol
a and
Patrick
Lacroix-Desmazes
*a
a,
Sylvain
Caillol
a and
Patrick
Lacroix-Desmazes
*a
aICGM, CNRS, ENSCM, University of Montpellier, 34293 Montpellier, France. E-mail: patrick.lacroix-desmazes@enscm.fr
bOMNOVA Solutions France SAS, 91400 Orsay, France
cSynthomer Ltd., Harlow, CM20 2BH, UK
First published on 9th May 2023
Abstract
This review describes the recent research and development progress in the radical polymerization of bio-based monomers in aqueous dispersed media. The previous review on this subject was published in 2019 by S. Molina-Gutiérrez and coworkers (Green Chem., 2019, 21, 36). This topic is constantly evolving and improving because of the need for greener solutions to replace petroleum-derived monomers and more sustainable procedures to generate aqueous dispersions of polymers. For these reasons, we chose not only to update the previous review, but also to emphasize opportunities and constraints and to present considerations about green chemistry to outline the revolution that is arising.
 Elena Rigo | Elena Rigo obtained her Master's degree in chemistry in 2021 from the University of Toulouse III Paul Sabatier (France). She is currently a PhD student at IGCM (Institute Charles Gerhardt of Montpellier) under the supervision of Dr Patrick Lacroix-Desmazes. Her research is focused on the synthesis of new biobased monomers for radical polymerization in aqueous dispersed media. |
 Vincent Ladmiral | Vincent Ladmiral graduated from the National Graduate School of Chemistry of Montpellier in 1998, and received his PhD from the University of Warwick in 2006. After postdoctoral fellowships at the University of Kyoto, the University of Sydney, the University of New South Wales and the University of Sheffield, he was appointed as CNRS Research Fellow in 2012 in the Institute Charles Gerhardt of Montpellier, and was promoted to Director of Research in 2022. His research interests focus on the structure–property relationship of polymers and materials. |
 Sylvain Caillol | Sylvain Caillol is Research Director at CNRS. He graduated as engineer from the National Graduate School of Chemistry of Montpellier in 1998. He received his PhD degree in 2001 from the University of Bordeaux. Subsequently, he joined the company Rhodia where he headed the Polymer Department in the Research Center of Paris. In 2007, he joined the CNRS at the Institute Charles Gerhardt of the University of Montpellier where he started a new research topic dedicated to green chemistry and biobased polymers. He won the Green Materials Prize in 2018 and 2020 and he entered the list of the world's top scientists (Stanford) in 2021. |
 Patrick Lacroix-Desmazes | Patrick Lacroix-Desmazes graduated in 1992 from the National Graduate School of Chemistry in Montpellier. He obtained his PhD degree in 1996 from the University of Lyon. After working at BP Chemicals in 1997, he joined CNRS in Montpellier and was promoted to CNRS Research Director in 2009. He is the head of the polymer department at the Institute Charles Gerhardt of Montpellier. His research interests cover reversible-deactivation radical polymerizations, the synthesis of polymer colloids and hybrid materials in aqueous media, and the synthesis and use of polymers in supercritical carbon dioxide, as well as green chemistry and circular economy. |
Sustainability spotlightToday, the latex market is mostly dominated by synthetic polymers produced from fossil resources. In the last decade, the problems of climate change, biodiversity loss and pollution associated with the use of fossil resources have raised questions in society. The use of water as a medium of polymerization to produce aqueous polymeric dispersions decreases the use of organic solvents, but still the monomers are petro-sourced. In order to further reduce the environmental impact of polymeric dispersions, a transition towards a bio-based, low carbon footprint chemistry is desirable and chemical producers are looking for greener alternatives. This review describes the recent research and development advancements in the radical polymerization of bio-based monomers in aqueous dispersed media. The monomers for this purpose derive from a vast amount of biomass feedstock that includes vegetable oils and lipids, terpenes, lignin derivatives, carbohydrates and proteins. Beyond the UN Sustainable Development Goals SDG12 (responsible consumption and production), this area of research also promotes industrial innovation (SDG9.4, SDG9.5). |
Introduction
The first latexes were described during the IIIrd century B.C. by Theophrastus, an ancient Greek botanist.1 Latexes exist in their natural form in natural rubber extracted from the rubber tree (Hevea brasiliensis), and consist of an aqueous dispersion of polyisoprene particles stabilized by protein surfactants. During World War II, emulsion polymerization processes were improved to produce synthetic latexes because of the insufficient supply of natural rubber. Today, synthetic latex materials have entered into our everyday life, they surround us and we use them to create many materials such as gloves, swim caps, tires, rubber bands, balloons, sporting equipment, and coatings like paints and adhesives, among others.2Currently, the latex market is mostly dominated by synthetic polymers produced from fossil resources. In the last decade, the problem of fossil fuel depletion and the environmental concerns associated with the use of fossil resources have raised questions in society. In order to contribute to reaching the Paris Agreement long-term temperature goals,3 a transition towards a bio-based, low carbon footprint, and more sustainable chemistry is desirable and chemical producers are looking for greener alternatives. This change in mentality is opening a window of opportunities for bio-based polymers derived from natural raw materials.4 The monomers for this purpose derive from a vast amount of biomass feedstock that includes vegetable oils and lipids, terpenes, lignin derivatives, carbohydrates and proteins.5 The biomass raw materials can be treated by thermal,6 catalytic,7 pyrolysis,8 liquefaction9 and electrochemical10 routes to extract and valorize biosourced building blocks.
For the moment, high bio-based content latexes are rarely employed for the implementation of new industrial applications, because of the limited availability of the bio-based monomers, their cost11 and sometimes their competition with the food industry.12 In their recent review,13 Calvo-Flores and Martin-Martinez described the achievements and challenges to reach a fully bio-based economy starting from bio-based materials. Indeed, the functions found in bio-sourced monomers are often not adapted to radical polymerization. In addition, when additional double bonds are present, they can easily lead to undesirable crosslinking. Thus, functionalization of bio-based platform molecules with groups able to react by radical polymerization is the main approach to prepare bio-based monomers. The final aim is to produce bio-based materials with equal or improved performance in applications in comparison with the current 100% petroleum-based materials. Two different routes have been employed to replace petroleum derivatives: (1) the drop-in approach consisting in the synthesis of strictly identical structures to petro-based monomers from biomass (e.g. bio-based styrene or acrylic acid14,15), and (2) the synthesis of novel monomer structures. In this review, a focus on this latter approach and the current development of new bio-based monomers and their improved properties in comparison with their traditional petroleum equivalents are presented.
This review describes the recent research and development advancements in the radical polymerization of bio-based monomers in aqueous dispersed media. The previous review on this topic was published in 2019 by S. Molina-Gutiérrez and coworkers.16 Another review published in 2020 by Leiza and coworkers summarizes the most relevant monomers, surfactants, crosslinkers, chain transfer agents and nanofillers only for the production of waterborne polymeric dispersions for coating and adhesive applications.17 From 2019 to 2022, several new ideas have emerged and progress has been made in this field. This review outlines the improvements and limitations of this field with a special insight into adherence to green chemistry principles.18 For this purpose and to enrich the discussion, the calculations of some green metrics such as atom economy (% AE), E-factor, C-efficiency, reaction mass efficiency (% RME) and the % of renewable carbon atoms have been added for the most relevant reactions and when deemed necessary (the details of the calculations are given in the ESI†). Indeed, the development of new bio-based monomers is often not synonymous with a more sustainable process because the steps used to extract, purify and synthesize them are more time-consuming and more energy intensive. Therefore, these green metrics can cast some light on the transition from petroleum-based radical polymerizations in aqueous dispersed media to their bio-based counterparts. In particular, atom economy measures the conversion efficiency of a chemical process in terms of the atoms actually incorporated into the desired products, assuming a yield of 100%.19 It is defined as the molecular weight of the desired product divided by the sum of the molecular weights of all the reactants used in the stoichiometric equation, expressed as a percentage. The higher the % AE is, the more sustainable the reaction is, because less reactants end up as waste products. E-factor measures the amount of waste produced in a process. It is defined as the mass ratio of waste to desired product (expressed in kg of waste per kg of product). It takes into account waste byproducts, leftover reactants, solvent losses and spent catalysts (also supported). Hence, a higher E-factor means more waste and consequently, greater negative environmental impact.20 The lower the E-factor is, the more sustainable the process is. Carbon efficiency (C-efficiency) quantifies the number of carbon atoms in the starting materials that ends up in the desired product. It is expressed in % based on the total mass of carbon in the reactants; the higher the C-efficiency, the better.21 Reaction mass efficiency (% RME) measures the efficiency with which the reactant mass ends up in the desired product.22 It is defined as the mass of isolated product divided by the total mass of the reactants and is expressed in %; the higher the % RME, the better. Biobased carbon is the calculation of the amount of renewable carbon (% renewable C) in the final monomer; the higher the % of renewable C, the better. Note that in our metrics calculation, the acrylate and methacrylate groups are not considered as bio-based because their production from biomass is not yet mature/available at the industrial scale.
Radical polymerizations in dispersed aqueous media mainly include emulsion, miniemulsion, microemulsion, dispersion and suspension polymerizations.2,23,24 They can be considered as greener processes because water is used as solvent, thus reducing the volatile organic compounds (VOCs) content.25 In industry, emulsion and suspension polymerizations are the two main employed processes. Emulsion polymerization typically produces sub-micrometer polymer particles called latex,26 which is colloidally stable and directly suitable for applications (e.g. acrylic paints). In contrast, suspension polymerization typically produces beads on the millimeter scale,27,28 mostly used as intermediates for the formation of solid polymers which are then processed (e.g. into polyvinyl chloride pipes). Emulsion polymerization differs from suspension polymerization because water-soluble surfactants are used to emulsify the monomers. Steric stabilizers are used in suspension polymerization to prevent the coagulation of the polymer beads. In emulsion polymerization,26 latex particles are generated through a process called the nucleation stage (interval 1), starting from the decomposition of water-soluble radical initiators in the aqueous phase. These radicals react with the fraction of monomers in water (the monomers used in emulsion polymerization are usually hydrophobic with nonetheless a non-negligible solubility in water; e.g. styrene concentration in water at 50 °C is 4.3 mmol L−1), resulting in oligomeric radicals. These oligomeric radicals become hydrophobic beyond a few units and either enter preformed surfactant micelles swollen with monomer (micellar nucleation) or aggregate to form nuclei that swell with monomer (homogeneous nucleation), that will in turn become particles (nucleation). Droplet nucleation is usually negligible in conventional emulsion polymerization, especially in the presence of micelles, given the much higher total surface area produced by a high concentration of micelles (typically 1018 to 1021 L−1 and around 10 nm in diameter) compared to a lower concentration of large droplets (typically around 1010 to 1012 L−1 and around 10 μm in diameter). When the number of particles increases, so does their capture of oligomers, and at a certain point, all the oligomeric radicals are captured; this is the end of the nucleation period. The next stage is the particle growth in the presence of monomer droplets (interval 2), which act as a reservoir of monomer to supply monomers to the growing latex particles. The monomers diffuse from the monomer droplets to the growing latex particles that are usually the main locus of polymerization. The diffusion rate of the monomer through the aqueous phase is faster than the polymerization rate in the particles (except for monomers exhibiting very low water solubility), i.e. the particles are swollen by monomers and the swelling concentration is determined by the thermodynamic equilibrium (Morton equation). For instance, the saturation concentration of styrene in particles at 50 °C is 5.5 mol L−1 for particles of 94 nm diameter (unswollen particle size). Thus, during interval 2, the monomer concentration in the growing latex particles where polymerization takes place is mostly constant, which in turn leads to a roughly constant rate of polymerization. After complete consumption of the monomer reservoir (monomer droplets), particle growth takes place in the absence of monomer droplets (interval 3). During interval 3, the remaining monomer in the latex particles is consumed, thus the monomer concentration in the latex particles decreases, which in turn decreases the rate of polymerization. A gel effect can however arise during interval 3 (especially for acrylates) due to a higher viscosity within the latex particles. This leads to a lower diffusion and a decrease in the diffusion-controlled rate of termination, thus causing autoacceleration of the polymerization. In seeded emulsion polymerization, as latex particles are already present at the beginning of the polymerization, nucleation (interval 1) does not occur, unless a high concentration of surfactant is used, favoring micellar nucleation (secondary nucleation, leading to a multimodal particle size distribution). Batch emulsion polymerization refers to a process where all reactants are introduced at the beginning of the polymerization. In industry, semi-batch emulsion polymerization is often used, where a fraction of the reactants is introduced at the beginning of the reaction (seed stage during which nucleation is completed) and the remaining fraction is added over the course of the reaction. Starved-feed semi-batch emulsion polymerization refers to a process where the monomers are introduced at a rate lower than the rate of polymerization, i.e. the instantaneous monomer conversion is high (typically higher than 90%), avoiding a composition drift during the course of the copolymerization with monomers of different reactivity ratios (for instance styrene M1 is consumed faster in batch copolymerization with acrylates M2 since reactivity ratios r1 > r2). These conditions also lead to a monomer concentration in the monomer-swollen latex particles that is lower than the saturation concentration (i.e. reaction in interval 3). Semi-batch emulsion polymerization also allows control of the nucleation step, followed by growth of the latex particles, while maintaining good colloidal stability of the latex particles (feed of monomers and surfactants) in order to reach high solids content (typically higher than 40% final solids content for industrial latexes) without major copolymer composition drift. In contrast, polymer beads are formed by polymerization within the monomer droplets in suspension polymerization (oil-soluble radical initiators). In the last four years, the majority of published articles have used emulsion and miniemulsion polymerization processes to form aqueous bio-based polymeric dispersions. Miniemulsion is a variant of emulsion polymerization26,29,30 where small (sub-micrometer, typically less than 300 nm in diameter) monomer droplets are formed through high energy input (e.g. ultrasound, high pressure homogenizer) and the polymerization takes place within the sub-micrometer monomer droplets (usually initiated by oil-soluble radical initiators although water-soluble radical initiators can also be used and droplet nucleation operates thanks to the small size of the monomer droplets), leading to a stable latex. A co-stabilizer (e.g. a hydrophobe such as hexadecane) is used to avoid Ostwald ripening; it retards the monomer diffusion from the smaller droplets to the larger ones to improve the stability of the miniemulsion. Notably, miniemulsion polymerization is well suited for very hydrophobic monomers (such as lauryl methacrylate exhibiting water solubility of 4.71 × 10−5 mmol L−1 at 35 °C), for which the micellar or homogeneous nucleation step in the water phase, typical of conventional emulsion polymerization, is not efficient.
This review is organized into five chapters, one for each category of biomass feedstock (vegetable oils and lipids, terpenes, lignin derivatives, carbohydrates, and proteins) (Fig. 1). Sometimes, categories overlap because they all possess unique characteristics that are useful to achieve the highest components bio-content (e.g. copolymerization of vegetable oil monomers with lignin derivative-based monomers).
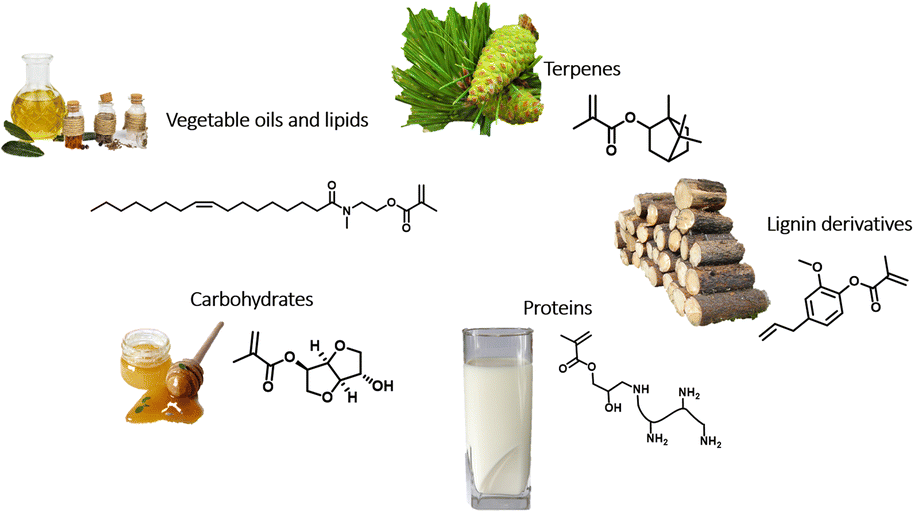 | ||
| Fig. 1 Categories of biomass feedstock considered in this review and illustrative examples of some corresponding bio-based monomers. | ||
Vegetable-oil- and lipid-based polymers
In this section, attention is directed to the recent progress made in the polymerization of fatty acids and their corresponding esters, as well as lipids, and of all the other molecular structures derived from them.Vegetable oils and lipids present several advantages over conventional petro-based raw materials: they are an abundant, low-cost, biodegradable (under certain conditions), hydrophobic, biocompatible and low-toxicity renewable feedstock.31–33 Efforts have been made to transform these natural resources into novel bio-sourced platform molecules and monomers and they mostly derived from the food waste industry.13 The chemical structure of plant-oil-based monomers contains both a polar ester moiety and a hydrophobic chain (C15–C17) often including double bonds, opening the door to many possibilities to perform reactions. Vegetable oils and lipids can find application in the production of polymeric materials with interesting properties and promising uses, including thermosets and latex materials (for coatings and paints), among others. Many characteristics can change the physicochemical properties of these oils such as the stereochemistry of the double bonds, the degree of unsaturation and the length of the fatty acid chains34,35 and can affect the thermochemical, physical and mechanical properties of the final bio-based polymers.36 In addition, the length of the fatty acid chain influences the hydrophobicity of the bio-based monomers, which is an important parameter in radical polymerization in aqueous dispersed media (e.g. ab initio emulsion polymerization of very hydrophobic monomers is usually problematic). Interestingly, the crosslinking density can be controlled through the unsaturation degree of the fatty acid chains (crosslinking can operate during polymerization or after polymerization to increase the gel content and the glass transition temperature, Tg).
Fatty acids and their derivatives
To increase the reactivity of vegetable oils under radical polymerization conditions, modifications and functionalizations are performed on one of the several reactive sites, which include the double bond, the allylic carbons, the ester group, and the carbon alpha to the carbonyl group (Fig. 2). In particular, the internal double bonds present in the lipophilic segment often cause problems upon radical polymerization because of their low reactivity and their tendency to crosslink. Hence, the introduction of more reactive groups is needed in the vegetable oil chain.34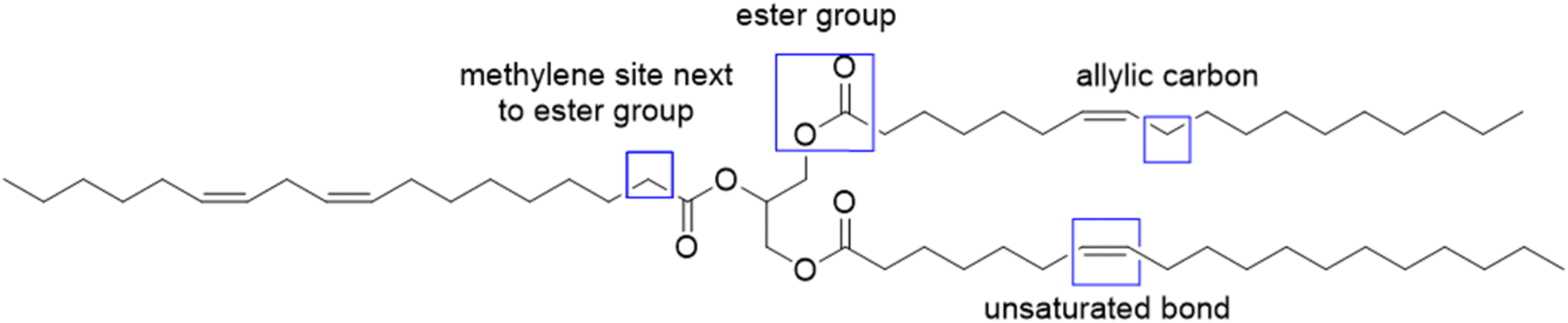 | ||
| Fig. 2 Four main active sites of triglycerides. | ||
Over the last four years, the most used approaches to functionalize oils have been the modification of the ester group and/or the double bonds of the fatty chain.
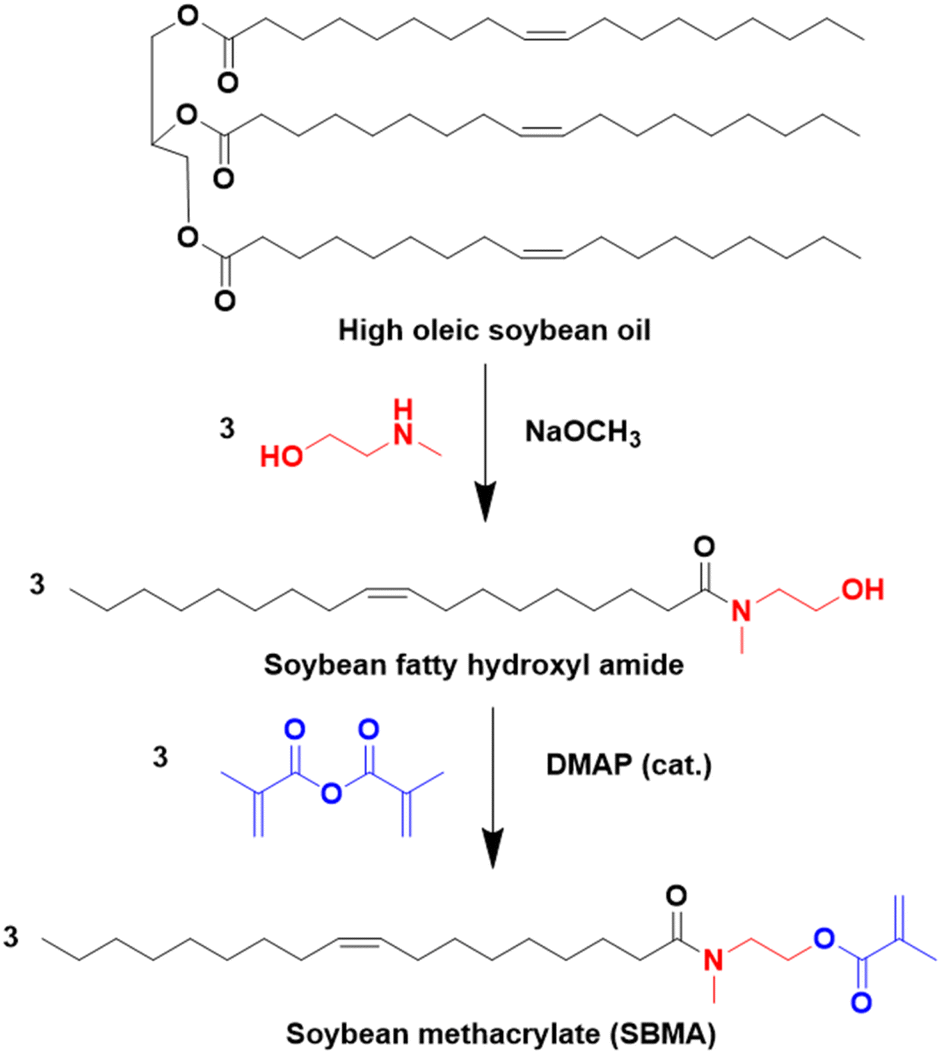 | ||
| Fig. 3 Synthesis of soybean methacrylate (SBMA) from oleic soybean oil. | ||
| Green metrics | |
|---|---|
| % AE | 71 |
| E-factor (kg waste/kg product) | 0.49 |
| % C-efficiency | 77 |
| % RME | 69 |
| % Renewable C | 84 |
Through solution radical polymerization in toluene, a bio-based homopolymer (PSBMA) with soft properties was produced (Tg = −6 °C).38 Thus, SBMA could be a good bio-based resource to replace low Tg units in current acrylic materials. Indeed, the authors reported successful semi-batch emulsion copolymerization of SBMA with various comonomers such as methyl methacrylate, styrene and butyl acrylate (up to 50 wt% SBMA), allowing for good tunability of thermal and mechanical properties in the prepared latexes.37 The authors also emphasized the presence of the alkene functional groups, present in the unsaturated fatty side chain of SBMA (without the need for further functionalization). These double bonds were used for post-polymerization auto-oxidative crosslinking of the dried films at 125 °C to obtain bio-based polymers (thermosets) with promising properties for ultra-strong, ultra-tough, and high Tg (>80 °C) coating applications.
Another approach to substitute the structure of ester groups is transesterification. This approach has been used by Voronov and coworkers,40 in a one-step transesterification process to synthesize a vinyl monomer (HOSBM) from high oleic soybean oil (Fig. 4).41 The calculation of the green metrics (Table 2)40 for this reaction shows that the chemical process suffers mainly from the use of a large amount of solvent (tetrahydrofuran, THF, 150 mL) and a large excess of acrylamide (5.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of hydroxyethyl acrylamide:triglyceride). Although not discussed by the authors, the substitution of THF by Me-THF, a biosourced solvent, could make the process more environmentally sustainable.
:1 ratio of hydroxyethyl acrylamide:triglyceride). Although not discussed by the authors, the substitution of THF by Me-THF, a biosourced solvent, could make the process more environmentally sustainable.
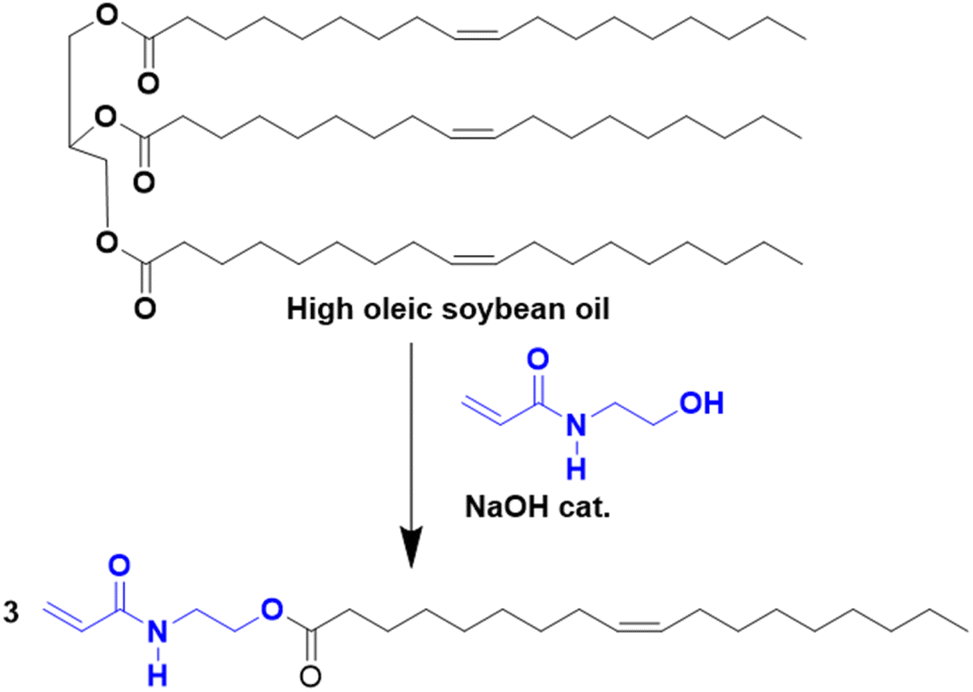 | ||
| Fig. 4 Synthesis of HOSBM by transesterification reaction. | ||
| Green metrics | |
|---|---|
| % AE | 73 |
| E-factor (kg waste/kg product) | 1.2 |
| % C-efficiency | 95 |
| % RME | 68 |
| % Renewable C | 87 |
For the first time, the authors combined the softness and flexibility of HOSBM with the strength of cardanol-based fragments in different monomer ratios to form a fully renewable plant-based cross-linked film from a latex produced by miniemulsion polymerization. Depending on the HOSBM/cardanol monomer ratio, the thermal transition of the latex polymers varied (increasing the HOSBM content, the Tg value increased from −35 to 10 °C) and the mechanical properties of the cross-linked films changed via auto-oxidation at 135 °C (increasing the cardanol monomer content, the amount of unsaturation increased, which resulted in the increase of the cross-linking and of the Tg of the cured films, from 5 to 53 °C and the Young's modulus from 0.9 MPa to 2.3 MPa).
Voronov and his coworkers applied miniemulsion polymerization to copolymerize HOSBM with a wide range of guaiacol and eugenol derivatives to prepare bio-based aromatic latexes using 10 to 75 wt% guaiacol/eugenol derivatives and 25–90 wt% HOSBM. They obtained polymers with molar masses varying from 25000 to 650000 g mol−1 depending on the HOSBM/aromatic derivatives ratios and they demonstrated that the incorporation of aromatic fragments (guaiacol/eugenol) enhanced the rigidity of the film, whereas the vegetable-oil-based units (HOSBM) contributed to softening the polymeric material.42
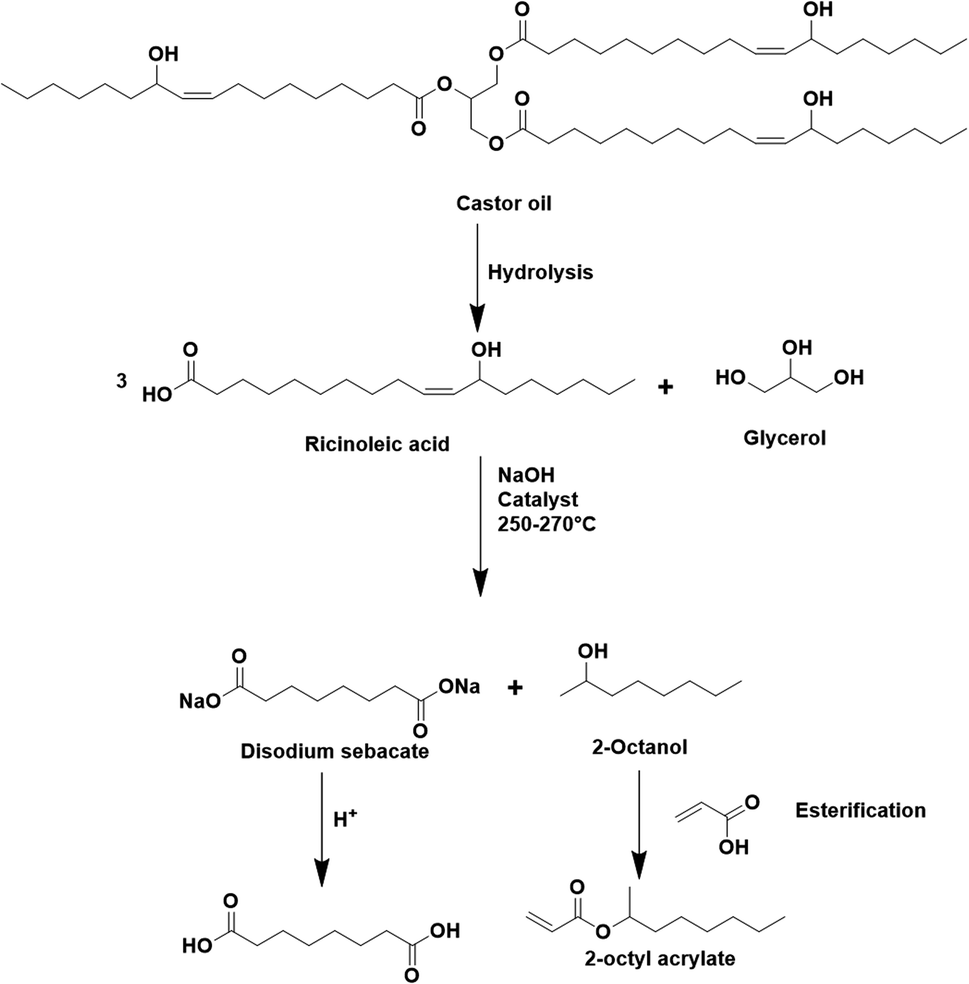 | ||
| Fig. 5 Synthesis of 2-octyl acrylate (OA) from castor oil. | ||
The seeded emulsion homopolymerization of 2-octyl acrylate (OA) was studied by Barquero and coworkers.47 In particular, they investigated the possibility of using OA as a substitute for petrol-based 2-ethylhexyl acrylate (2-EHA). They found that both monomers achieved high instantaneous conversions by seeded emulsion polymerization and that there was no difference in the evolution of latex particle size over time. However, they found that OA formed more gel (final value of 75%) than 2-EHA (up to 58%). The higher gel content in the case of OA was ascribed to intermolecular chain transfer involving the proton on the tertiary carbon next to the oxygen atom (Fig. 6a), followed by termination by combination. Similarly, the seeded emulsion homopolymerization of 2-octyl methacrylate was studied and compared with its petroleum equivalent 2-ethylhexyl methacrylate. In this case, no gel formation was observed due to the termination by disproportionation. However, intermolecular H-abstraction on the tertiary carbon still occurred (Fig. 6b), leading to long chain branching.
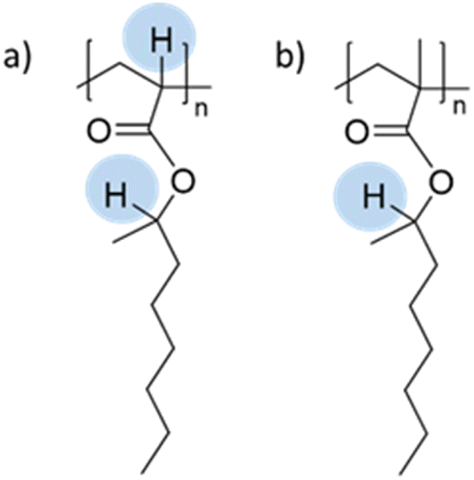 | ||
| Fig. 6 Abstractable hydrogen atoms of (a) 2-octyl acrylate and (b) 2-octyl methacrylate biobased homopolymers. | ||
In another paper,48 Leiza and coworkers described the copolymerization of 2-octyl acrylate (OA) with piperonyl methacrylate (PIPEMA) (Fig. 7a), a bio-based monomer synthesized via methacrylation of piperonyl alcohol (derived from black pepper). The starved-feed seeded semi-batch emulsion copolymerization of OA/PIPEMA/MAA 84/15/1 (weight composition) with final solids content of 50 wt% (copolymer seed latex, 2-octyl acrylate/isobornyl acrylate/acrylic acid in a weight ratio of 88/10/2, with 20 wt% solid content) yields a pressure-sensitive adhesive (PSA) with UV-tunable shear resistance and adhesive properties. This was possible because piperonyl methacrylate has two different functional groups: a methacrylate group able to polymerize via free radical polymerization, and a 1,3-benzodioxole moiety, capable of reacting upon UV light irradiation for post-curing (crosslinking) the final PSA (Fig. 8). High (71 wt%) bio-based monomer content was achieved.
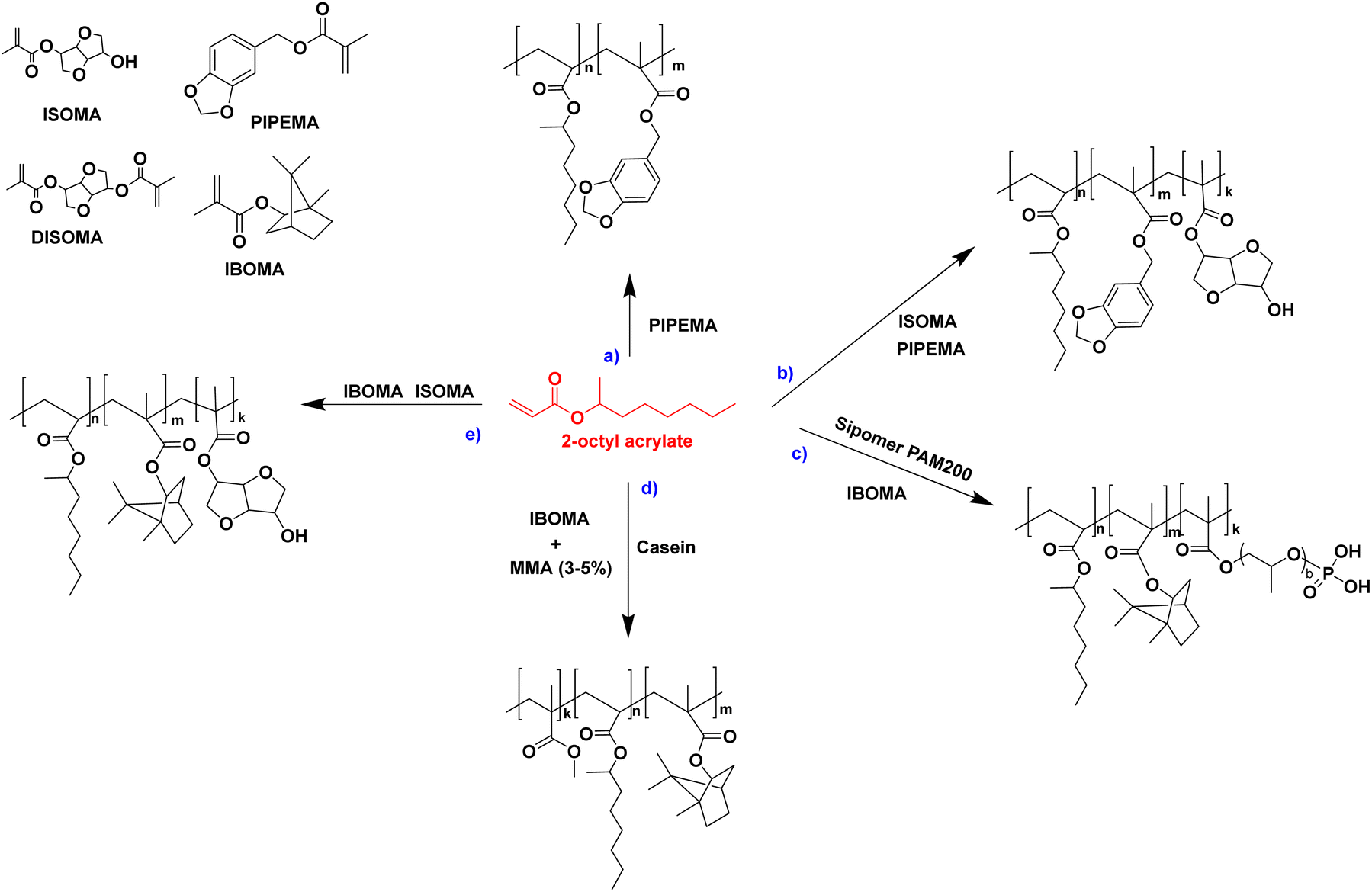 | ||
| Fig. 7 Copolymerization reactions involving 2-octyl acrylate. | ||
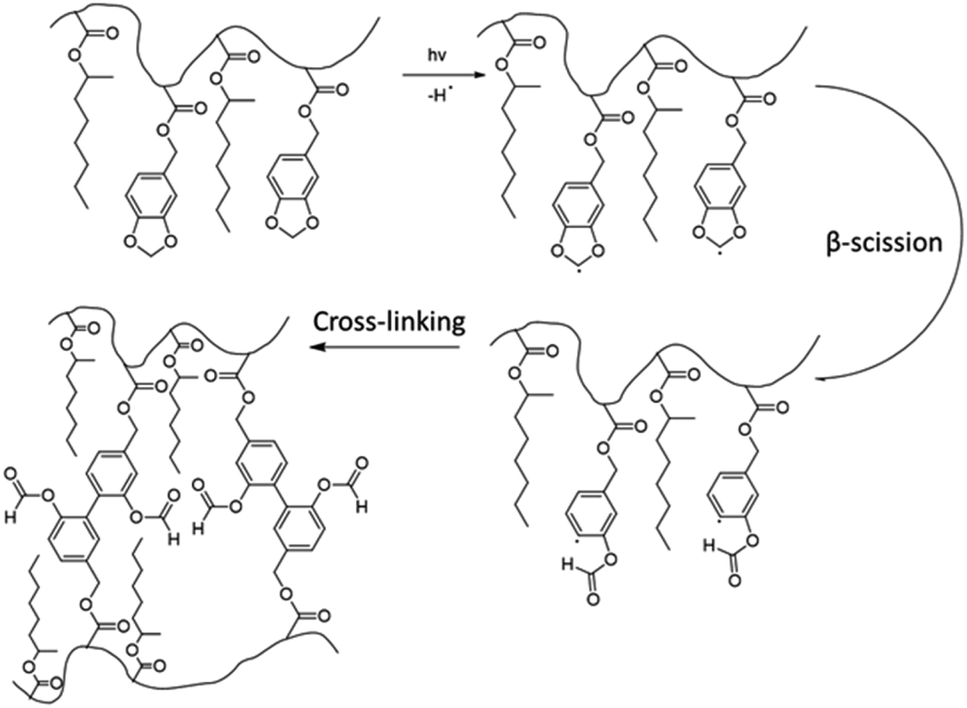 | ||
| Fig. 8 1,3-Benzodioxole photoreaction and formation of a cross-linked PSA. | ||
In order to improve the properties of their bio-based formulation, they decided to add a low content (1 wt%) of isosorbide methacrylate (ISOMA), a bicyclic chemical compound derived from glucose (Fig. 7b).49 They found that the presence of the hydroxyl groups of ISOMA enhanced PSA extensibility and cohesiveness and promoted the removability in water of the PSA tape. This is an interesting feature to take into consideration to avoid harsh conditions (e.g. basic conditions at 85 °C), which increase the economic cost and damage the environment during the washing processes of reusable glass bottles. The tape was completely removed after 40 min of immersion at room temperature and in 10 min at 65 °C.50 An overview of the current strategies and methods developed to create sustainable PSAs was reported by Asua and coworkers in a recent review.51 2-Octyl acrylate was also copolymerized by seeded semi-batch emulsion polymerization with isobornyl methacrylate (IBOMA), another biosourced monomer extracted from pine resin, to produce a bio-based (bio-based contents up to 72%) latex for PSA applications.52 Recently, Leiza and coworkers (Fig. 7c)53 used batch miniemulsion copolymerization of 2-octyl acrylate and isobornyl methacrylate using a phosphate polymerizable surfactant (Sipomer PAM200) to form a novel hybrid CeO2 bio-based (up to 70 wt%) acrylic binder for waterborne coatings. The phosphate groups were able to react with the metal surface, forming a layer at the substrate/coating interface achieving a better corrosion resistance. In addition, CeO2 nanoparticles were homogeneously distributed in the polymer films and acted as corrosion inhibitors. The copolymerization between OA and IBOMA was also performed and involved methacrylated casein (bovine milk protein), an emulsifier capable of being radically polymerized by batch emulsion polymerization (surfmer), and non-bio-based methyl methacrylate (MMA) (Fig. 7d).54 Leiza and coworkers explained that without MMA the polymerization did not occur, because OA and IBOMA are poorly soluble in water. Hence, small amounts (3–4% wbm “weight fraction based on monomers”) of a more water-soluble monomer such as MMA promoted the emulsion polymerization, avoiding the more complicated miniemulsion polymerization process used in their previous work, resulting in latex with high bio-based content (75 wt%). The film formed from this latex showed promising properties in terms of mechanical performance and resistance to solvents as well as biodegradability in organic compost of the casein part of the material. This last property is required to respect the environmental regulations for biodegradable materials. Thus, this film could be a good starting point for the improvement of bio-based latex coatings.
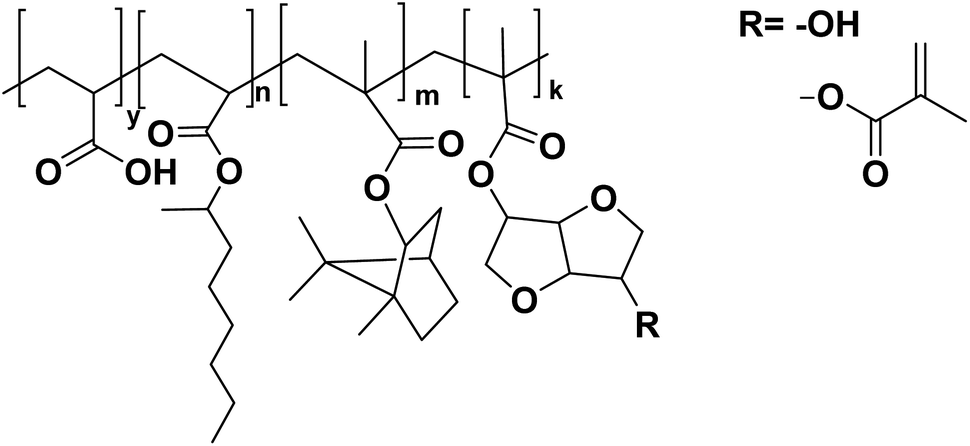
OA and IBOMA, in combination with a mixture (called ISOMAraw) of isosorbide methacrylate (ISOMA) and isosorbide dimethacrylate (DISOMA), were the bio-based monomers employed together with MAA (bringing carboxylic acid functions) to synthesize some ASRs (alkali soluble resins) by emulsion polymerization (Fig. 9). After neutralization with a shot of ammonia, these ASRs (amphiphilic polymers) were used as electrosteric stabilizers for polymer dispersions to create pressure-sensitive adhesives. PSAs were synthesized by semi-batch emulsion copolymerization of OA, IBOMA and ISOMA as monomers, and 2-ethylhexyl thio-glycolate as chain transfer agent to achieve bio-based content up to 71 wt% (Fig. 7e).55 ISOMA and DISOMA are bicyclic monomers obtained from glucose and their synthesis is described in the carbohydrates section (vide infra).50 The DISOMA led to pendant double bonds onto the ASR, thereby allowing the radical grafting of the ASR onto the polymer latex particles to create performant PSA tape. The utilization of ISOMA, with pendant hydroxyl groups, provided hydrogen-bonding interactions in the PSA formulation, which were key to the clean and fast (around 20 min) removal of the adhesive tape from a glass substrate in alkali solutions.
 | ||
| Fig. 9 Alkali soluble resins (ASRs) used as electrosteric stabilizers. | ||
The high hydrophobicity of some bio-based monomers can hinder the straightforward incorporation of such monomers in waterborne dispersions. Llorente and coworkers56 illustrated these difficulties in the case of the copolymerization of OA and IBOMA. They proposed a stability map for OA/IBOMA copolymers with Dowfax 2A1 as surfactant with different monomers ratios and solids content and highlighted the conditions where stable dispersions were achieved. Their scheme showed the formation of stable dispersions when the solids content was lower than 55 wt% and the presence of IBOMA in the formulation was lower than 50% wbm.
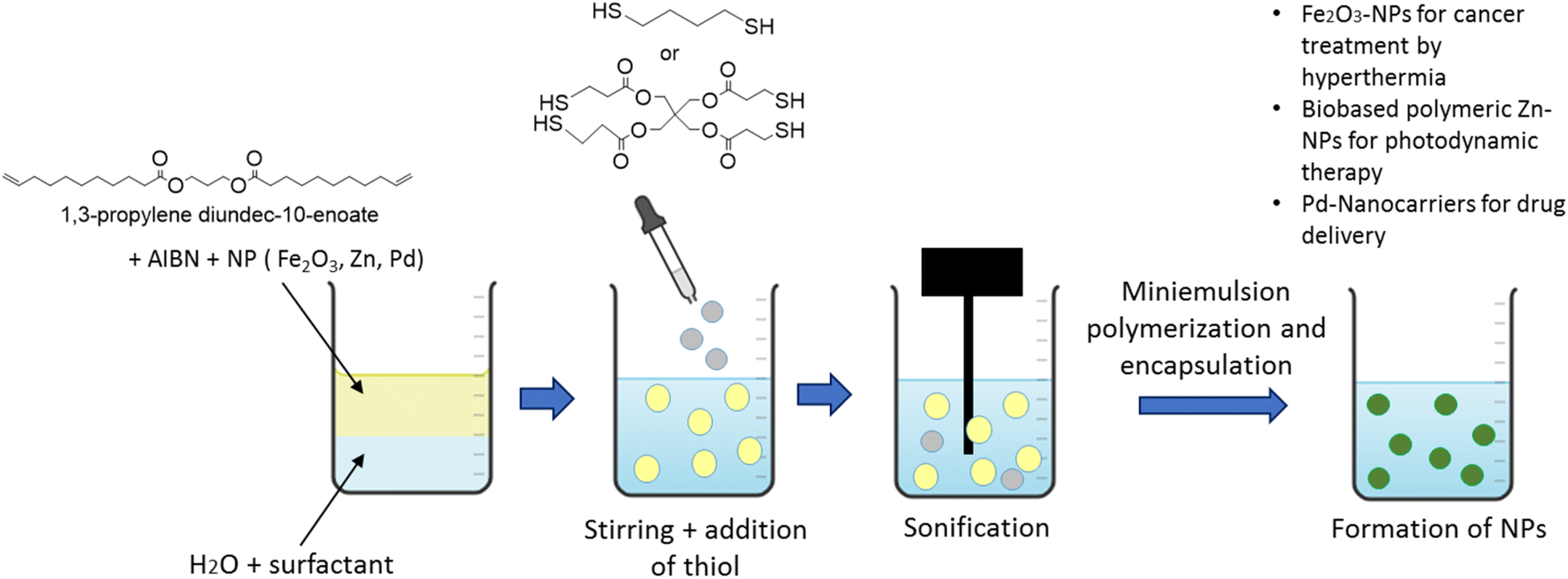 | ||
| Fig. 10 Miniemulsion polymerization by thiol–ene chemistry. | ||
All the described thiol–ene polymerizations were performed under aqueous miniemulsion polymerization conditions, which are not currently amenable to industrial setups due to the cost and the needed specific instrumentation.
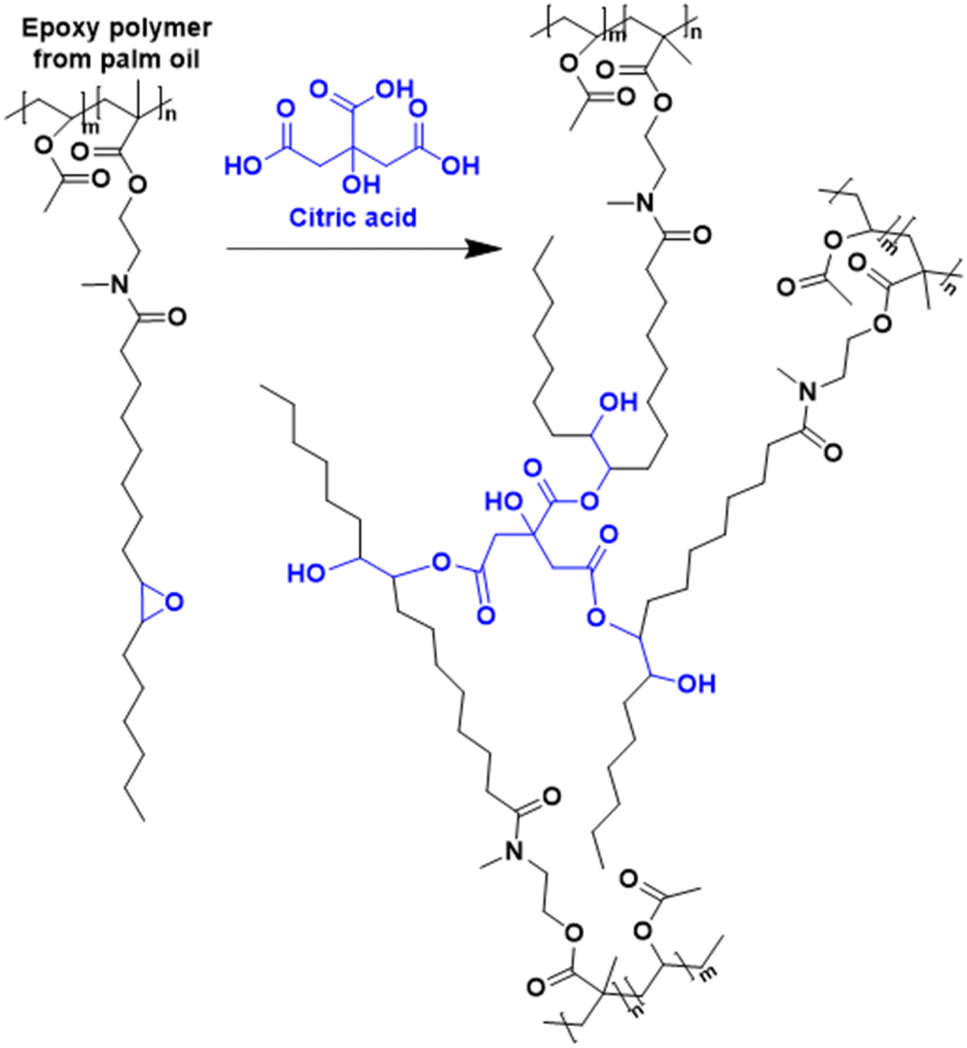 | ||
| Fig. 11 Synthesis of recyclable epoxy resin from palm oil. In blue are the groups capable of curing via transesterification. | ||
Lipid miniemulsion polymerization: the example of cardanol
An interesting bio-based aromatic building block from lipids is cardanol. Cardanol is a by-product derived from cashew nut shell liquid, which is directly extracted from the shell of the cashew nut. It is a phenolic compound bearing an unsaturated hydrophobic alkyl chain (Fig. 12). Its functionalization to prepare cardanol-based monomers was already reported by Molina-Gutiérrez and her coworkers in their previous review,16 and its potential to form bio-based latexes by emulsion copolymerization with MMA was already described.68 Later, hydroxylated cardanol methacrylate was copolymerized with MMA by radical miniemulsion polymerization.69 In the last four years, cardanol methacrylate was functionalized with phosphonic acid on the alkyl side chain by Li and coworkers70 to improve the adhesion properties of the coatings thereof (Fig. 12). This phosphorus-cardanol monomer was copolymerized with MMA (50/50 weight ratio) under aqueous miniemulsion conditions to prepare latex and films possessing 1B adhesion onto steel plates.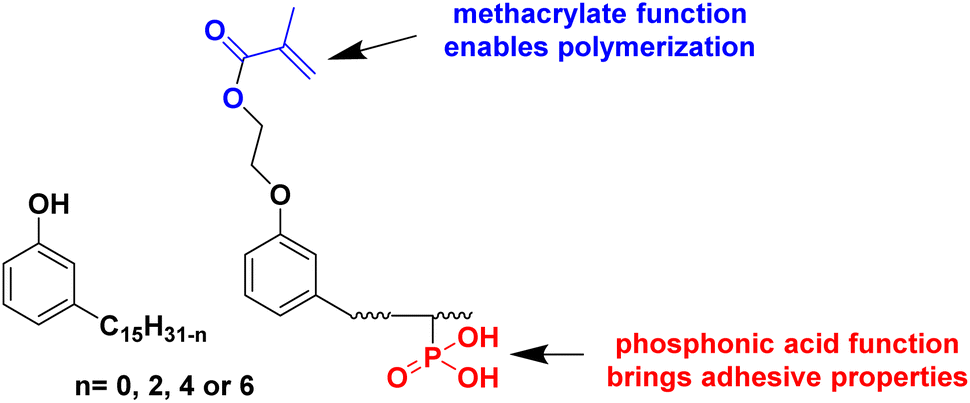 | ||
| Fig. 12 Chemical structure of cardanol and phosphonated cardanol methacrylate. | ||
As outlined, some progress has been made in the field of vegetable-oil and lipid-based monomers and their polymerization. However, as shown by the green metrics, synthesis optimization is key to the industrial scale development of these new bio-based monomers. In addition, because of the high hydrophobicity of the long aliphatic chains, they most often need to be polymerized by miniemulsion polymerization, which is an expensive and non-scalable process. Furthermore, as a consequence of the low reactivity of the internal double bonds, the oil and lipid derivatives should be modified with some petroleum-derived functional groups (e.g. acrylate and methacrylate) that require more expenditure and time. Finally, the internal double bonds can lead to undesirable or ill-controlled cross-linking either during the polymerization or during the processing of the material.
Terpene-based polymers
The wide family of terpenes (consisting of compounds with the formula (C5H8)n) present in leaves, fruits, and flowers of different plants can be another great resource for bio-based monomers.71 In the past few decades, terpenes have been used as flavors and fragrances, and in medical formulations. However, today they are considered the best renewable solution to replace isoprene or other unsaturated hydrocarbon monomers derived from petroleum.72 Terpenes are found in nature in large quantities and their production is simple, consequently their price is comparable to that of their petroleum-based counterparts.Terpenes, like non-biosourced diene monomers (such as butadiene or isoprene), are predominantly polymerized via emulsion processes, rather than homogeneous processes (bulk or solution), to avoid low polymerization rates and low molar masses.
Polyterpenes can be synthesized via radical (thermal or redox initiated) (conventional or controlled), anionic, cationic or coordination polymerization and they can replace fossil-based conjugated diene monomers in elastomer applications.73 The presence of conjugated double bonds in their structures allows them to be polymerized and cross-linked using reactions that are already known and used in industry.
In the last four years, the most studied terpene has been myrcene, the primary component of essential oils derived from bay, cannabis, and hops.72 Like farnesene, another terpene isolated from the oil of perilla, myrcene has been investigated as a biosourced diene and compared to butadiene and isoprene. Myrcene and farnesene are less toxic than butadiene, hence their use in cosmetic and fragrance formulations.74–76 They are also less volatile than butadiene and isoprene. Therefore, they can be polymerized at atmospheric pressure, through a simpler process avoiding the use of expensive pressurized reactors, thus minimizing energy consumption. For all these reasons, the study of terpenes and their utilization has always caught the interest of researchers. The first studies about polymerization of β-myrcene dates back to 1948, and much progress has been made in the study of terpenes since then. Recently, Kalita et al. reported for the first time the preparation of a triblock copolymer poly(styrene)-b-poly(β-myrcene)-b-poly(styrene) (SMS) by reversible addition–fragmentation chain-transfer (RAFT) polymerization with the use of DBTTC (S,S-dibenzyl trithiocarbonate) as chain transfer agent (CTA) with the aim to create a thermoplastic elastomer. They achieved a good rate of polymerization (unlike previous studies77) where poly(β-myrcene) acted as the low Tg block and polystyrene (PS) as the high Tg block (Fig. 13a).78 Styrene was polymerized first by solution RAFT polymerization in toluene. Then, miniemulsion polymerization of myrcene was performed in the presence of 4,4′-azobis(4-cyanovaleric acid) as radical initiator, starting from the pre-formed PS macro-RAFT agent (dioxane and sonication were used to prepare the aqueous miniemulsion in the presence of sodium dodecyl sulfate (SDS) as surfactant). The authors found that the best formulation had a styrene content of 20–30 wt% and they showed that the material behaved as a thermoplastic elastomer with an ultimate tensile strength of ∼3.5 MPa (at 625% strain). The scraps of this triblock copolymer were also successfully recycled and reused, indicating the sustainability of this material. The authors proposed that these triblock copolymers could be modified via different “ene” reactions via its pendant vinyl function to design the material for different specialty applications.
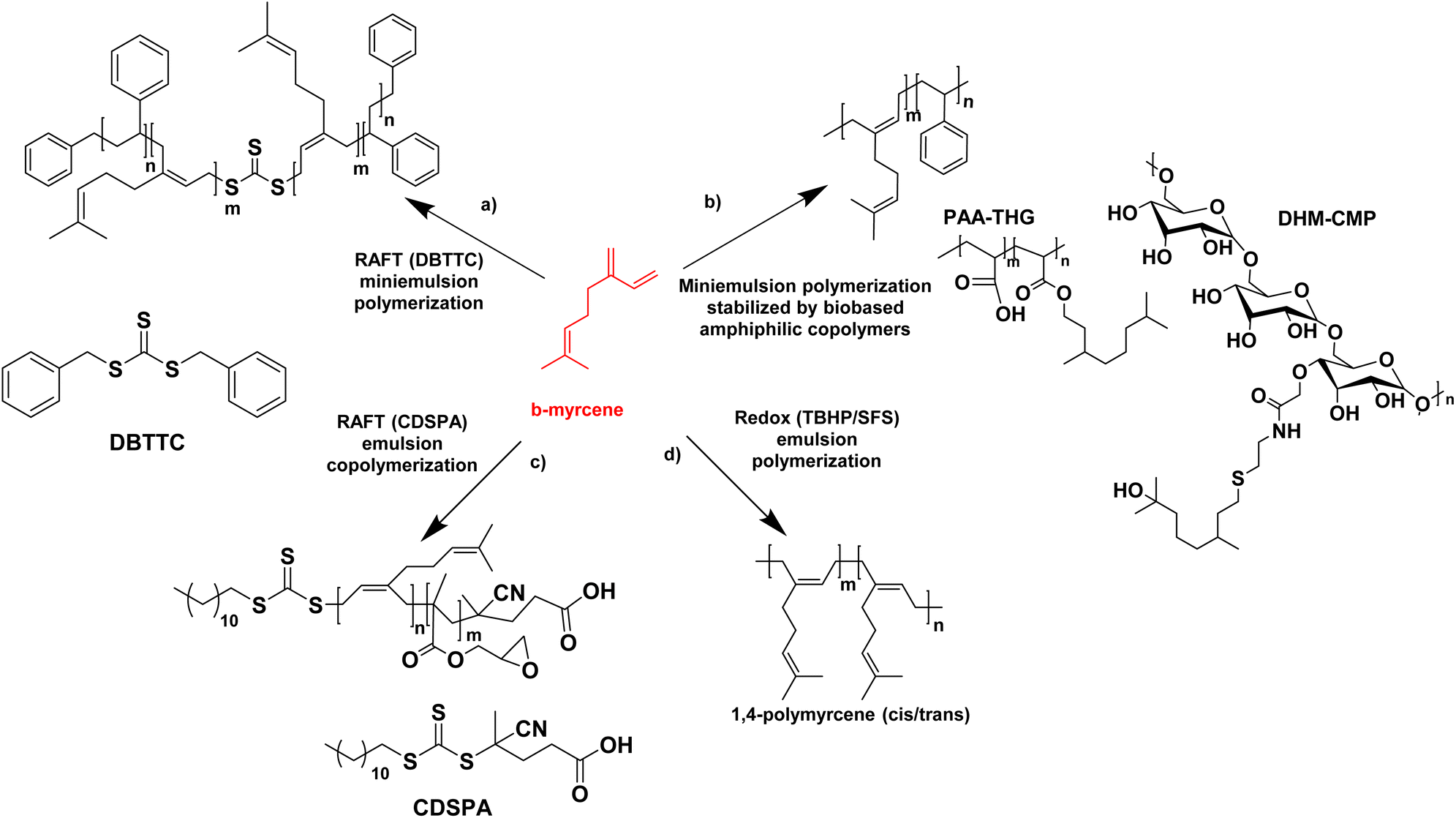 | ||
| Fig. 13 β-Myrcene (mini)emulsion polymerization reactions. | ||
The miniemulsion homopolymerization of myrcene and copolymerization between β-myrcene and styrene was also reported by Save and her coworkers (Fig. 13b).79 Interestingly, the utilization of amphiphilic macromolecular bio-based stabilizers for the synthesis of terpene-based waterborne latex was reported. Two bio-based amphiphilic copolymers were synthesized. The first one was synthesized by the esterification between a hydrophilic backbone constituted by poly(acrylic acid) (PAA) and a hydrophobic part composed of tetrahydrogeraniol (THG). Both are bio-sourced because the production of AA by fermentation from 3-hydroxypropionic acid is widely described on the laboratory scale80 and THG is a natural terpenoid. The second copolymer was made by the amidation reaction between a hydrophilic polysaccharide, carboxymethylpullulan (CMP) produced from pullulan, a natural polysaccharide derived from starch81 and a hydrophobic part composed of a dihydromyrcenol (DHM) terpenoid amino-derivative. The newly synthesized amphiphilic copolymers were employed as stabilizers in the miniemulsion (co)polymerization of β-myrcene and styrene. The monomer conversion was lower in the presence of myrcene with a maximum of 72% conversion for the copolymers with styrene. The synthesized latex particles had a diameter between 171 nm and 306 nm and a Tg of −60 °C for poly(myrcene) and Tg of −14 °C for poly(styrene-co-myrcene) (43 mol% of myrcene in the copolymer). The crosslinked latex particles (e.g. gel content of 77% for poly(styrene-co-myrcene), probably arising from branching reactions due to the presence of unsaturated groups) stabilized with the bio-based amphiphilic copolymers were then tested as Pickering emulsion stabilizers. With all latex particles, stable emulsions were obtained for at least one year. With PS and poly(styrene-co-myrcene) particles stabilized with copolymers with a low degree of substitution in hydrophobic terpene, W/O emulsions were formed whereas O/W emulsions were formed with poly(myrcene) particles (at a weight fraction of dodecane and water of 50/50 w/w).
Saldívar-Guerra and his coworkers (Fig. 13c)82 have developed the RAFT copolymerization of β-myrcene and glycidyl methacrylate (GMA) using CDSPA (4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid) as chain transfer agent and potassium persulfate as radical initiator, under aqueous emulsion polymerization conditions and using semi-continuous addition of the monomers to prepare a bio-based elastomer (monomer conversion in the range of 74–92%). They determined the reactivity ratios of the monomers, and studied the effect of the β-myrcene concentration over the molar mass of the copolymer, as well as the microstructure of the copolymers (predominance of 1,4-cis) (polymerizations in bulk and in toluene solution with a monomer conversion lower than 20%). Gel content studies, on films prepared by casting from the latex, showed that the copolymer films immersed in THF for 48 h did not dissolve, as a result of the cross-linking of the polymer. Gel content of 17–78% increased with increasing GMA content (0–21%) in the feed. The cross-linking of the polymer had already been demonstrated in previous work.83
It is preferable to perform the synthesis of poly(terpenes) at room temperature, because their microstructures, in particular the double bonds, are strongly affected by the temperature of polymerization. Bhowmick and coworkers have shown that the free radical emulsion polymerization of β-myrcene initiated by ammonium persulfate at 70 °C reached up to 96% polymer yield in 20 h but it led to the formation of a mixture of 1,4-cis, 1,4-trans, 1,2-vinyl and 3,4 structures,84 whereas the redox-initiated emulsion polymerization at lower temperature led mainly to 1,4 addition products. With that in mind, the same team investigated not only the redox homopolymerization of β-myrcene but also the homopolymerization of β-ocimene and β-farnesene and the effect of reaction time and redox systems on the monomer conversion and the polymer molar mass.85 Poly(β-myrcene) showed the best results. Thus, they used redox-initiated (tert-butyl hydroperoxide TBHP as oxidant and sodium formaldehyde sulfoxylate SFS as reducing agent, in the presence of Fe-EDTA as catalyst) aqueous emulsion polymerization at room temperature (25 °C) (Fig. 13d). They produced a high molar mass (Mn = 1.68 × 105 g mol−1) poly(β-myrcene) rubber (Tg = −70 °C) containing predominantly (90%) 1,4-cis/trans linkages (and 10% 3,4-structures determined by 2D 1H NMR), with however a maximum polymer yield of only 65%. Importantly, these polymers suffer from chain scission during the polymerization, decreasing the final molecular weight. In contrast, β-ocimene yielded low polymer yield (15–20%) and only oligomers (Mn ∼ 1.5 kg mol−1). Exact information related to the microstructures could not be determined. Finally, β-farnesene also yielded low polymer yield (20–24%) and low molar mass polymers (Mn ∼ 3.2 kg mol−1) (because of the sterically crowded diene group, which is not very suitable for radical propagation) with microstructure dominated mainly by 1,4-cis and 1,4-trans linkages.
Biosourced farnesene was also studied in copolymerization with isobornyl methacrylate (IBOMA). IBOMA is produced by the reaction between camphene and methacrylic acid (camphene is derived from the isomerization of terpenes, such as alpha-pinene, derived from pinesap) (Fig. 14). IBOMA is a very versatile monomer. In the previous section, it was used as comonomer to prepare waterborne latex coating.53,54 Luk and Maríc86 synthesized poly(farnesene) homopolymer by nitroxide-mediated polymerization (NMP) in a miniemulsion using D7 (Dispolreg 007) or NHS-BB (succinimidyl-modified BlocBuilder) as alkoxyamine initiators. However, the final farnesene conversion was limited (30–40%), large particles were obtained (300–400 nm diameter size) in spite of the large amount of used Dowfax 8390 surfactant (15 wt% relative to monomer), the experimental Mn for Mn,target = 30000 g mol−1 was significantly higher than the theoretical Mn and the dispersity Đ was most often larger than 2. Finally, the poly(farnesene) was isolated, dissolved in toluene and used as macroalkoxyamine in the solution copolymerization of an IBOMA/farnesene mixture (10 mol% farnesene relative to IBOMA as NHS-BB requires a small amount of this comonomer to control the polymerization of a methacrylate). Thus, a diblock copolymer poly(farnesene)-b-poly(iBOMA-co-farnesene) was produced by solution block copolymerization, with a modest increase in molar mass from 38900 to 44300 g mol−1 while Đ increased from 1.83 to 2.24 (Fig. 14a). Based on these results, the authors proposed that farnesene and IBOMA might be good biosourced alternatives to butadiene and styrene to make materials similar to SBS thermoplastic elastomer; however further studies have to be carried out in order to reach this goal.
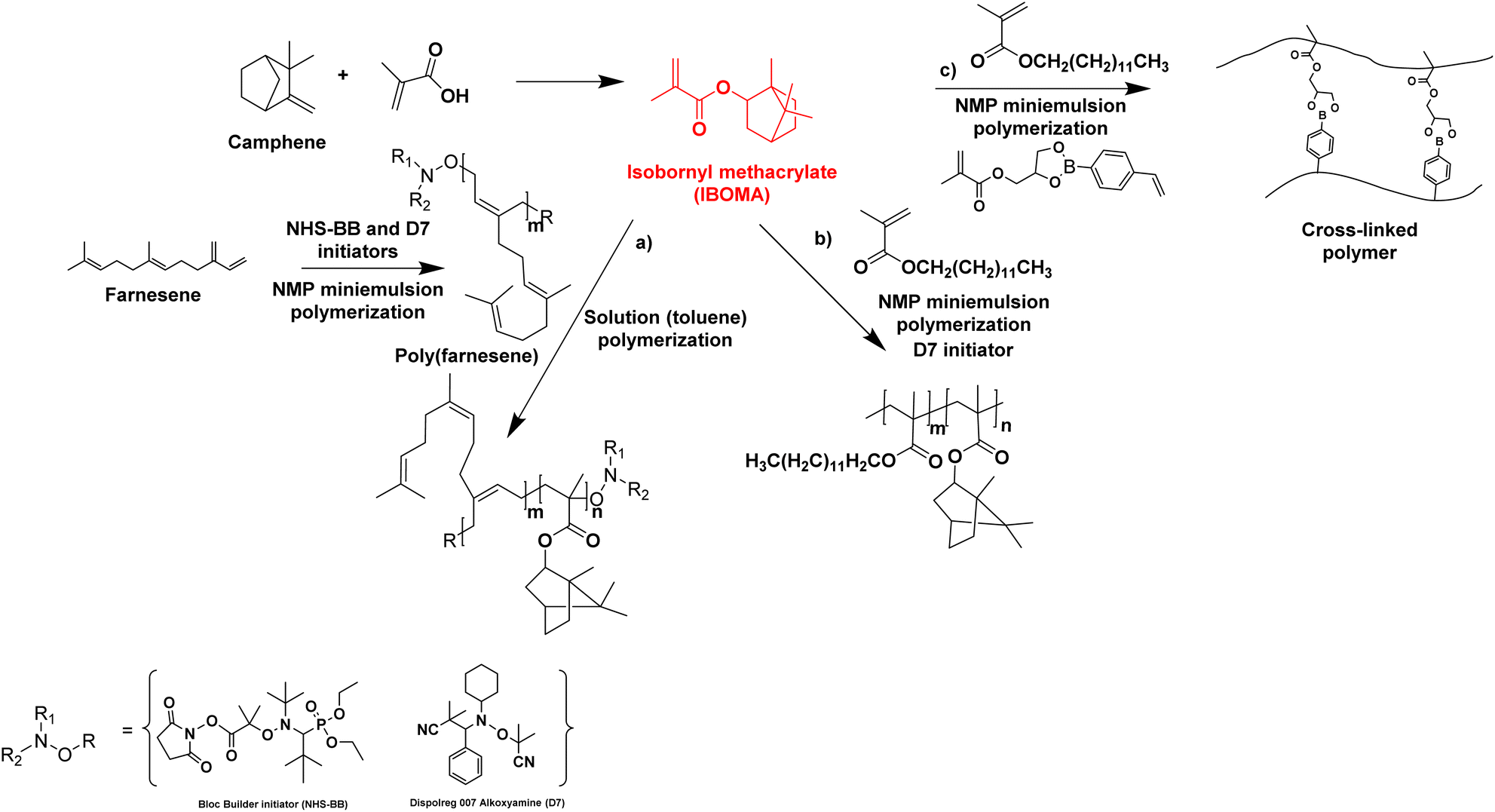 | ||
| Fig. 14 Synthesis of IBOMA from camphene and the use of IBOMA in nitroxide-mediated (miniemulsion) polymerizations. | ||
Maric and his coworkers87 also applied the NMP technique in an aqueous miniemulsion to the copolymerization of IBOMA and methacrylic ester 13.0 (C13MA), a partially biosourced monomer produced from plant-oil (Fig. 14b). Poly(IBOMA) has a high glass transition temperature (Tg = 111–200 °C) and C13MA was incorporated to decrease the Tg (Tg,PC13MA = −46 °C). They succeeded in making statistical and block copolymers with tunable Tg (from −44 to 123 °C) using an oil-soluble alkoxyamine initiator (Dispolreg 007) without any controlling comonomers, reaching conversion up to 92%. The same monomers (IBOMA and C13MA) were copolymerized by miniemulsion with the incorporation of a cross-linker derived from the esterification of a glycerol monomethacrylate (GMMA) and 4-vinylphenylboronic acid (VPBA).88 The new hydrophobic crosslinker was able to react in nitroxide-mediated miniemulsion copolymerization (dimer content from 5 to 15 mol% in the initial feed) to provide cross-linked polymers with a high reprocessing capability in a simple recycling process (80 °C for 45 min) thanks to the boronic ester dynamic covalent bonds (Fig. 14c).
Terpenes and terpenoids may also find application in pressure-sensitive adhesives. Terpenoids are molecules derived from the 5-carbon compound isoprene, sometimes cyclic and with functional groups usually containing oxygen. Du Prez and coworkers89 copolymerized, via a semi-batch aqueous emulsion process, the (meth)acrylate derivatives of four common terpenoids (tetrahydrogeraniol, citronellol, menthol and isoborneol) (Fig. 15) with methyl methacrylate and acrylic acid to prepare a bio-based PSA with excellent properties in terms of tack, peel strength and shear resistance. Tetrahydrogeranyl acrylate was used as an alternative to 2-ethyl hexyl acrylate to prepare a low Tg bio-based polymer (Tg,(poly,tetrahydrogeranyl acrylate) = −73 °C), while isobornyl and menthyl methacrylates were used as alternatives to methyl methacrylate to prepare high Tg bio-sourced polymers (Tg,(poly,isobornyl methacrylate) = 155 °C and Tg,(poly,menthyl methacrylate) = 105 °C). Citronellyl (meth)acrylate, containing allylic hydrogens in the long alkyl chain, was employed as a partially bio-based crosslinker.
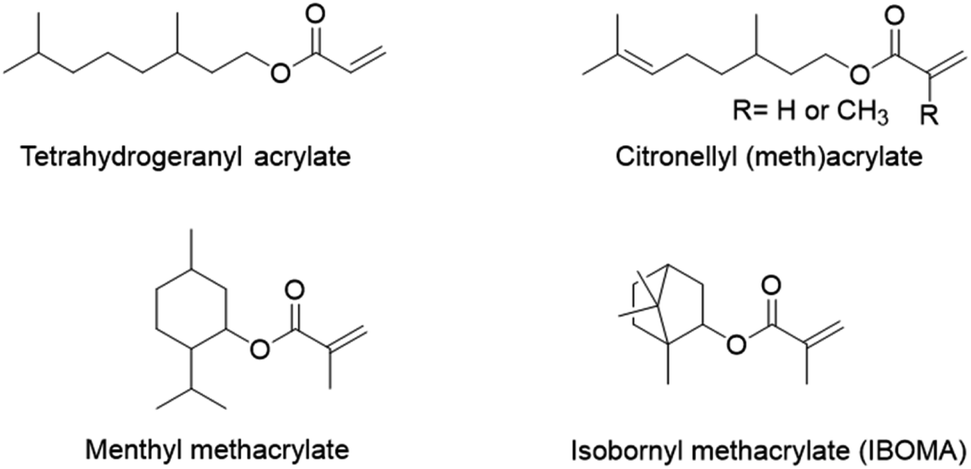 | ||
| Fig. 15 Acrylate and methacrylate derivatives of tetrahydrogeraniol, citronellol, menthol and isoborneol. | ||
Terpenes could become a great resource for future polymer materials, not only because they are abundant and may be seen as cheaper and safer analogs of isoprene and butadiene, but also because they are easier to handle at atmospheric pressure and room temperature. Furthermore, it is possible to control the microstructure of the resulting polymers via tuning the polymerization conditions such as the polymerization temperature. As described above, the combination of terpene derivatives with other monomers (bio-based or not) can lead to polymers featuring a large range of Tg and thus amenable to various applications. Nevertheless, as indicated above, terpenes suffer from low monomer conversion and low molar masses as well as a dependence of the microstructure on the polymerization temperature, which limit their industrial development.
Lignin derivative-based polymers
In contrast to the previously described biomass categories, lignin is an aromatic-rich polymer. It is one of the three main components of lignocellulosic biomass (the other two being cellulose and hemicellulose, which are two carbohydrate polymers). Lignin is one of the most abundant raw materials on Earth.90 Lignocellulose is the non-edible portion of plants; therefore, the use of lignin for the production of platform monomers is not in competition with food supply. For this reason, its depolymerization process has been deeply studied.91 However, to the best of our knowledge, up to now the only industrially available aromatic monomer obtained from lignin is vanillin.13,92In the last four years, research in the field of emulsion polymerization focused on various lignin-derived monomers such as eugenol and 3-allyl-5-vinylveratrole (Fig. 16). These compounds are good starting points for the synthesis of bio-based polymers and latexes.
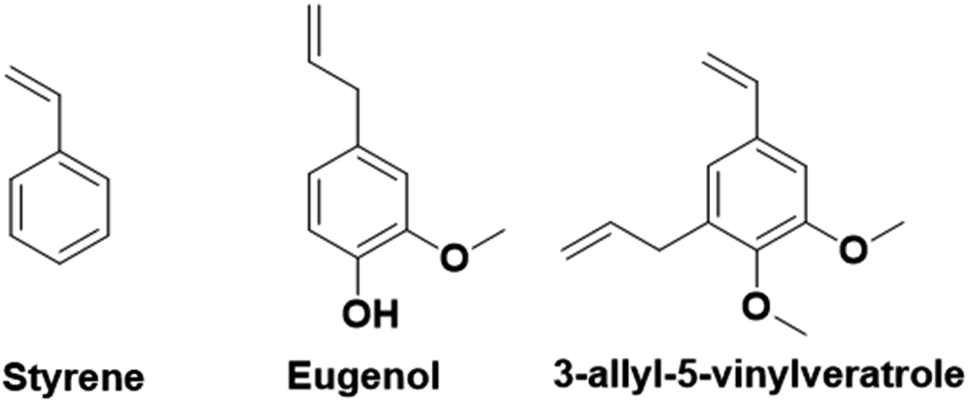 | ||
| Fig. 16 Chemical structures of aromatic bio-based monomers and styrene. | ||
Lignin-derived aromatic monomers, due to their structural similarities to styrene (Fig. 16), which is one of the most widely used fossil-oil derived monomers yet potentially carcinogenic, are becoming attractive alternatives. However, lignin derivatives have greater potential and versatility than the simple styrene molecule. Indeed, they possess an additional reactive site, the phenol function, that can be easily derivatized to obtain a large range of reactive molecules and prepare materials with distinct characteristics such as Tg, film forming properties, or gel content for a large range of applications. Conversely, phenol groups are radical inhibitors and as such are not suitable for radical polymerization. Hence, their modification is necessary prior to performing emulsion radical polymerization. This may limit their industrial applications.
Eugenol derivatives
In laboratories, one of the most studied lignin-derived monomers is eugenol, a molecule that can be obtained from several plants including clove buds, cinnamon bark, tulsi leaves, turmeric, pepper, ginger, oregano, and thyme. In the previous section, we have already described the potential of eugenol derivatives42 in emulsion copolymerization with vinyl monomers from high oleic soybean oil. Molina-Gutiérrez et al.93,94 designed a platform of bio-based monomers derived from eugenol, isoeugenol (an isomer of eugenol), and dihydroeugenol (the hydrogenated form of eugenol). In particular, they functionalized the phenol group with ethoxy(meth)acrylate functions to prepare readily polymerizable monomers.93 According to green metrics (Table 3),93 the reaction performed to functionalize the eugenol was not as green as expected. In fact, the first step was performed at 150–180 °C and the second one used dichloromethane (DCM), a toxic solvent that worsened the final sustainability. However, they proposed another synthetic pathway, where ethyl acetate was used as a “greener” solvent.| Green metrics | |
|---|---|
| % AE | 34 |
| E-factor (kg waste/kg product) | 4.54 |
| % C-efficiency | 49 |
| % RME | 25 |
| % Renewable C | 63 |
For the first time, they succeeded in the aqueous emulsion polymerization of bio-based methacrylate monomers derived from eugenol.95 In particular, they gradually substituted methyl methacrylate with ethoxy dihydroeugenyl methacrylate (because it does not possess any pendent double bonds that could engage in undesirable reactions) in the semi-batch emulsion copolymerization of butyl acrylate, methyl methacrylate and methacrylic acid (BA, MMA, MAA).96 They prepared a latex with a high total solids content of 50 wt% with particle diameter of 173 nm, containing 12% wbm of a lignin-bio-based monomer, with a Tg of −31 °C and a gel content of 69%. The adhesive properties of the polymers were tested and the peel and tack forces were shown to be superior to those measured for a commercial product, demonstrating that these bio-based latexes could be suitable for applications as PSA.
3-Allyl-5-vinylveratrole
In a similar way, Voronov and coworkers conceived a promising platform based on 3-allyl-5-vinylveratrole (AVV), a monomer derived from vanillin.97 First, they performed AVV homopolymerization in toluene solution and confirmed by 1H NMR spectroscopy that the allylic unsaturation was retained during homopolymerization and could thus be utilized post-polymerization to form a cross-linked network. Then, they copolymerized AVV and a vegetable-oil-based acrylic monomer (HOSBM) using a batch miniemulsion polymerization process (solids content about 30%) to prepare fully bio-based latexes (84–86% monomer conversion). They studied the viscoelastic behavior of the resulting polymeric materials, after oxidative curing of the films at 135 °C, to demonstrate that it could be modulated by the monomer ratios of high Tg (AVV) and low Tg (HOSBM) constituents. Thus, they obtained latex films with Tg ranging between 44 and 80 °C (increasing with AVV concentration in the feed), however showing a high gel content (84–88%) in all cases.The synthesis of these lignin-derived platform monomers has opened the door to their homopolymerization and the development of new molecules with a variety of properties and potential applications in materials. However, several constraints remain to be overcome. Firstly, the industrial availability of the bio-based building blocks obtained by the depolymerization of lignin has to be improved. Secondly, many of these building blocks do not possess functional groups adequate for radical polymerization, thus they need to be chemically modified. In addition, molecules like eugenol possess functional groups such as the allyl group, which are prone to undergo secondary reactions (e.g. degradative chain transfer) under radical polymerization conditions. These side reactions need to be studied and controlled to reach the desirable properties for the final materials. Despite these limitations, lignin-based molecules constitute a very promising platform to prepare new bio-based monomers suitable for radical polymerizations in aqueous media. Furthermore, lignin is the most abundant biomass to afford bio-based aromatic monomers.
Carbohydrate-based polymers
Carbohydrates represent one of the most abundant classes of renewable raw materials and they are the starting point for preparing several bio-based platform molecules.98 In particular, from corn, it is industrially possible to extract polysaccharide chains and to easily produce platform bio-based molecules (sugars) by fermentation or other chemical processes. Carbohydrates distinguish themselves from the bio-based building blocks reported in the previous sections by their higher oxygen content, and thus higher hydrophilic character and water-solubility. Vegetable oils, terpenes and lignin derivatives are most often employed in radical polymerizations in aqueous dispersed media as hydrophobic monomers, whereas carbohydrates are mainly hydrophilic species. In this section, we will try to summarize the recent attempts at using carbohydrates as water-soluble or water-insoluble components in radical polymerization, in addition to their use as hydrophilic components. The radical polymerization of carbohydrate derivatives in aqueous dispersed media is actually more varied and complicated to describe than that of hydrophobic bio-based building blocks. Carbohydrates might be used to form the surface of the polymer particles (water-soluble monomers, surfmers and steric stabilizers as well as water-insoluble Pickering stabilizers) or the core of the polymer particles (hydrophobic monomers, seed). In 2015, Smeets et al.99 described the use of carbohydrates as surfactants, initiators, transfer agents and (macro)monomers for the synthesis of carbohydrate-functionalized hybrid latex particles. In this review, we have decided to report some relevant examples, not only when carbohydrates are used as water-soluble (macro)monomers, amphiphilic (surface-active) or hydrophobic monomers, as indicated in the title of the review (“Recent advances in radical polymerization of bio-based monomers in aqueous dispersed media”), but we have also considered off-topic examples which are important to understand the versatility and the complexity of using carbohydrate derivatives in emulsion polymerization. For this reason, the use of carbohydrates as other water-soluble steric stabilizers (macrotransfer agents or macroinitiators, depending on their reactive site), Pickering stabilizers, and as water-insoluble seeds is also mentioned. Two different approaches are employed to use carbohydrates in radical polymerization in aqueous dispersed media as illustrated in Fig. 17. The first one consists in using low molecular weight carbohydrates (sugars) such as monosaccharides, disaccharides or polyols. These sugars can be derivatized to form bio-based monomers with a hydrophilic, surface-active or hydrophobic character. The second approach involves the use of carbohydrates in the form of oligosaccharides or polysaccharides, which can either be water-soluble or water-insoluble. When the polysaccharide is water-soluble, it can be functionalized with reactive groups to convert it into a reactive steric stabilizer, which can participate in radical polymerization as macromonomer, macrotransfer agent or macroinitiator. In the other case, like starch and cellulose, the polysaccharide is water-insoluble; thus, it can be used as a Pickering stabilizer or as a seed in radical polymerization in aqueous dispersed media.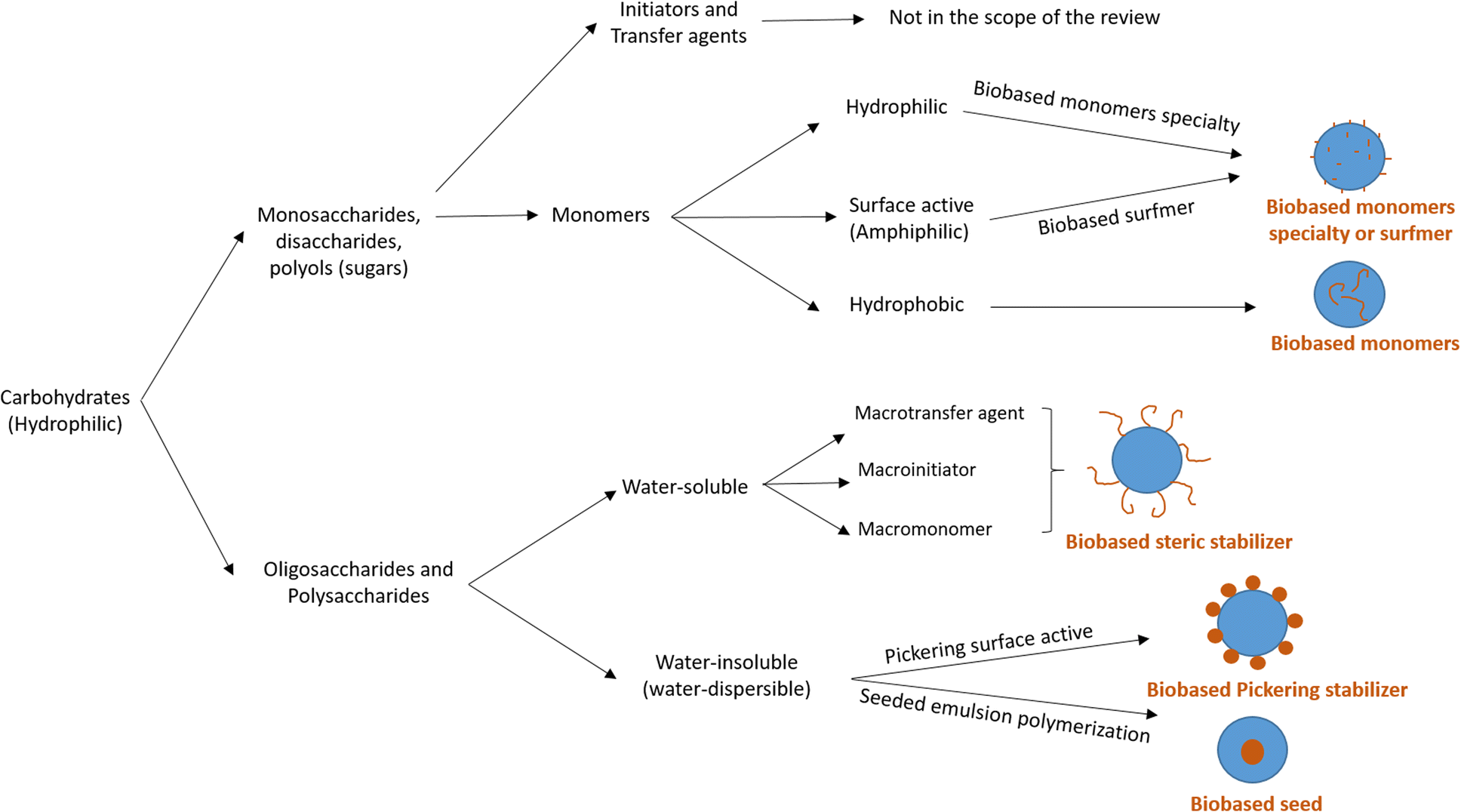 | ||
| Fig. 17 Different strategies to use carbohydrates in radical polymerizations in aqueous dispersed media. | ||
Monomers from sugars (glycomonomer)
| Green metrics | |
|---|---|
| % AE | 94 |
| E-factor (kg waste/kg product) | 14.3 |
| % C-efficiency | 51 |
| % RME | 53 |
| % Renewable C | 55 |
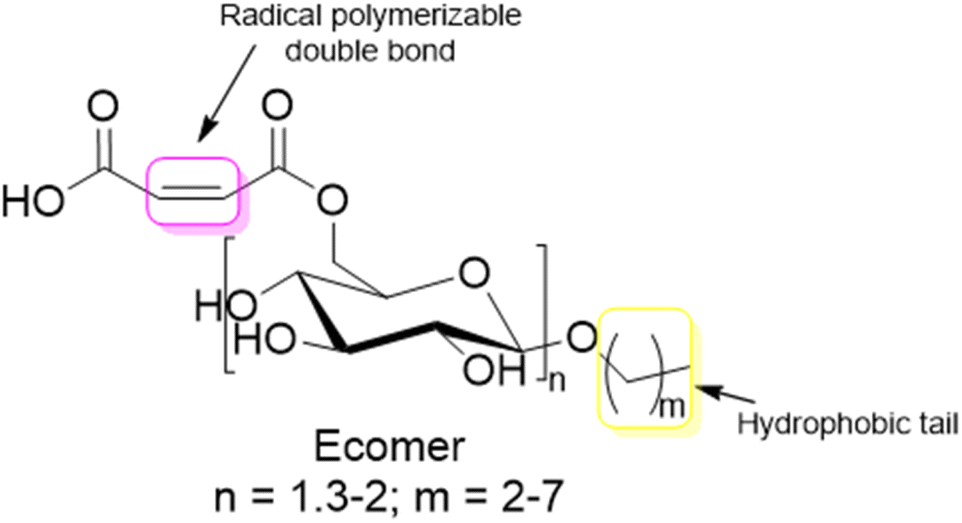 | ||
| Fig. 18 Ecomer® macromonomer structure. | ||
The principal monosaccharide used as platform molecule is glucose. It is one of the most important and most used starting points for the preparation of bio-based monomers such as acrylates (acrylic acid, methacrylic acid, itaconic acid, methylene lactones) as well as derivatives (e.g. esters) of cinnamic acid, fumaric acid, muconic acid and crotonic acid.101 The aqueous radical emulsion polymerization of some hydrophobic itaconic acid derivatives has been studied in the past four years to develop new types of innovative bio-based materials (Fig. 19), for a wide range of applications such as PSA materials and silica-filled bio-based elastomers. Itaconic acid (IA) is a commercially available dicarboxylic acid obtained by the fermentation of glucose using specific filamentous fungi. Its production is currently estimated to be on a 40000 tons per year scale by fungal fermentation,101 hence it is not in competition with the food industry. Vega-Rios and coworkers performed emulsion copolymerization between di-n-butyl itaconate (DBI), a bio-based monomer derived from the esterification of IA and 1-butanol (Fig. 19a), and lauryl methacrylate (LMA), a coconut oil derivative, in order to develop PSA.102 IA was identified as the donor of dicarboxylic acid functional groups (1 wt%) while DBI and LMA were employed as low Tg monomers. For the first time, they synthesized a fully bio-based PSA (25% solids content) and they studied the effect of the monomer weight ratio (DBI:LMA:IA was varied from 99:0:1 to 0:99:1) of the synthesized copolymers on the viscoelastic properties and Tg. The copolymer films with more than 50 wt% DBI showed suitable properties for PSA materials. However, although the authors observed some gel content (68% gel content for 99 wt% LMA content), they did not use any cross-linking agent and therefore the formulation might be improved to create a good PSA material.
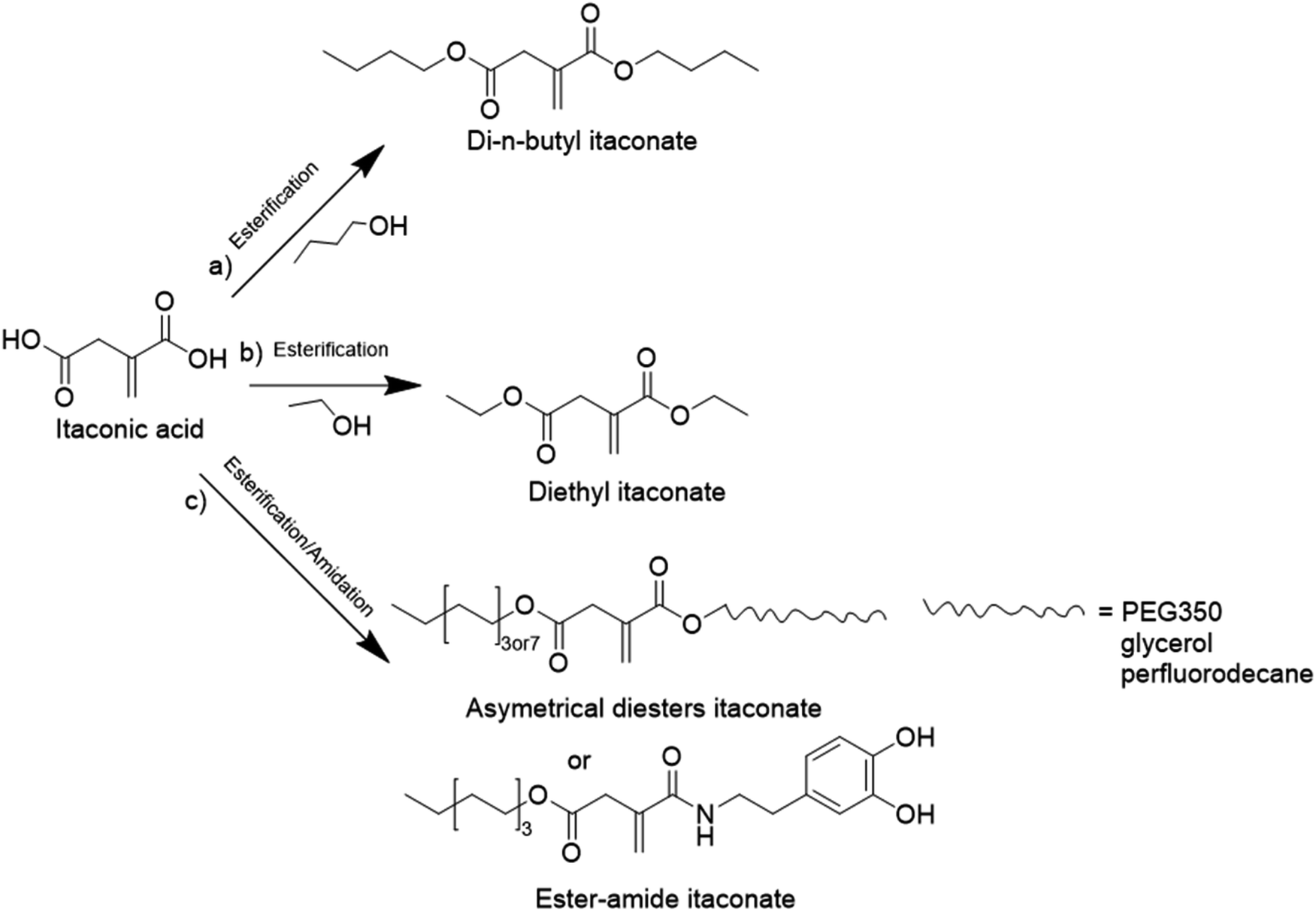 | ||
| Fig. 19 Diesters and ester amide from biosourced itaconic acid. | ||
Another alkyl derivative of IA, diethyl itaconate (Fig. 19b), was studied and polymerized by Lei and coworkers.103 They designed a new sustainable and environmentally friendly route for producing high-performance silica-filled bio-based elastomers from diethyl itaconate, butyl acrylate, and glycidyl methacrylate via emulsion polymerization. Glycidyl methacrylate was introduced in the polymer chains to incorporate epoxy groups for crosslinking with silica nanoparticles. The silica/poly(diethyl itaconate-co-butylacrylate-co-glycidyl methacrylate) bio-based composite presented an outstanding heat oil resistance in comparison with a petroleum-derived silica/poly(acrylate) composite. This article describes an interesting bio-based alternative for the production of elastomer materials using emulsion polymerization as an environmentally friendly process.
Itaconic acid was also esterified to form three asymmetrical diesters and one ester amide.104 The itaconate-diesters carried on one side an octyl or cetyl group and on the other side a poly(ethylene glycol) (PEG350), glycerol or perfluorodecane segment. The ester amide-itaconate was formed by an octyl group on one side and by dopamine on the other side (Fig. 19c). The functionalized itaconates were amphiphilic and they were tested as surfmers for the preparation of polystyrene by emulsion polymerization. It was shown by the authors that all the surfmers introduced in the feed were covalently linked to the polystyrene chains and showed good capacity to sterically stabilize polystyrene latexes of 150 nm diameter. The drawback emphasized by the authors was the large mole fraction (15–20 mol%) of surfmer required to form a stable latex.
Oligosaccharides or polysaccharides
Other studies have been done for the first time on polysaccharides for the development of bio-based latexes. The polysaccharides could be water-soluble or water-insoluble.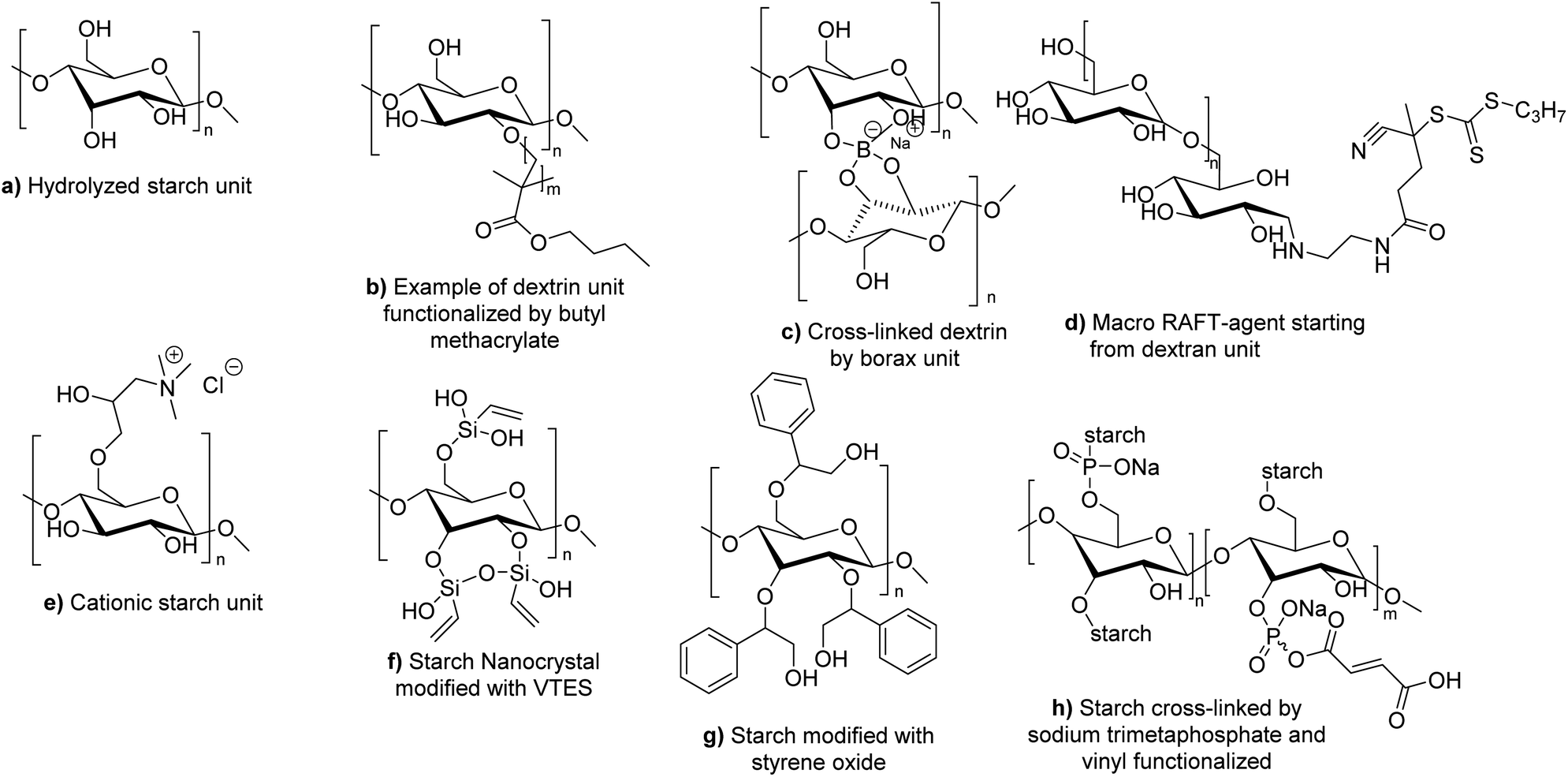 | ||
| Fig. 20 Different forms of modified starch used in radical polymerization in dispersed aqueous media. | ||
To the best of our knowledge, no example of the use of polysaccharides as macromonomers has been reported in the last four years in radical polymerization in dispersed media. However, some examples of macroinitiators have been described. Wu et al. reported the use of hydrolyzed starch (Fig. 20a) as macroinitiator for the synthesis of latex.106 In particular, initiation was triggered by a redox mechanism using CeIV+ ions. The radical formed on the hydrolyzed starch was able to react with BA or MMA units via seeded surfactant-free emulsion polymerization to form block and graft copolymers capable of stabilizing latex particles. The synthesized bio-based latex was tested as binder for paper coating and showed equal properties in comparison with its non-bio-based analogues.
Several examples of polysaccharides used as macrotransfer agents were reported in the last four years. Boufi and coworkers107 reported the use of dextrin, a water-soluble low-molar mass polysaccharide produced by the partial acid hydrolysis of native starch, and composed of D-glucosyl units. Simple adsorption of dextrins is not sufficient to sterically stabilize polymer latexes synthesized by emulsion polymerization of acrylic monomers. In fact, the true steric stabilizer is the amphiphilic copolymer dextrin-graft-poly(acrylates) (Fig. 20b) synthesized in situ during the surfactant-free emulsion polymerization initiated by H2O2/ascorbic acid redox initiator. The chemical grafting of dextrins took place via the H-radical transfer from the produced HO* radicals onto the dextrin units, followed by propagation of the vinylic monomer (butyl methacrylate, BMA, and MMA were successfully tested). Potassium persulfate was also tested as initiator (with various monomers such as butyl methacrylate, styrene, butyl acrylate, and vinyl acetate, as well as for the copolymerization of ethylhexylacrylate and butyl methacrylate) but it resulted in less efficient grafting to dextrin, less efficient steric stabilization and bigger latex particle size. To go further, they introduced borax as cross-linker into the dextrin-stabilized latex (Fig. 20c). Upon water removal (film formation), the reaction between borax and the hydroxyl groups of dextrin generated a cross-linked network. Interestingly, the addition of borax led to an increase of the cohesive strength of the pressure sensitive adhesive (PSA). An enhancement of the peel strength (about 40%) was noted for the PSA latex prepared in the presence of dextrins, compared to a PSA latex produced by emulsion polymerization in the presence of an anionic surfactant. Finally, the peel strength was even substantially enhanced by more than 100% in the presence of a small amount of borax (about 0.5 wt%).
Ferji and coworkers described the synthesis of a core–shell latex using a dextran derivative as polymeric stabilizer.108 Dextran (a polysaccharide synthesized from starch by bacteria) was modified in order to prepare a trithiocarbonate macro-RAFT agent which can act both as initiator under UV-irradiation (365 nm) and reversible chain transfer agent (Fig. 20d). Then, this macro-RAFT agent was involved in the photo RAFT-polymerization-induced self-assembly (photo-PISA) in an aqueous dispersion of 2-hydroxypropyl methacrylate (HPMA), in the absence of external initiator, to form a latex composed of diblock copolymer dextran-b-PHPMA. The final stable nanoparticles presented a spherical morphology. The sphere size increased with the degree of polymerization of PHPMA and varied from 16 to 238 nm.
Oliviera et al. decided to employ cationic starch (CS) derived polymer as hydrophilic macrotransfer agent to stabilize coating binders and as sizing agents (Fig. 20e).109 Cationic starch can be produced by reacting the free hydroxyl groups present in native starch with molecules containing tertiary or quaternary ammonium groups such as 2,3-epoxypropyl trimethyl ammonium chloride.110 They polymerized BA and MMA by surfactant-free emulsion polymerization initiated by a hydrogen peroxide and ferrous sulfate redox system at 85 °C, using cationic starch as grafting agent with different starch contents (from 7 to 20 wt%). Importantly, to obtain a stable latex, it was necessary to enzymatically hydrolyze the starch with α-amylase prior to the start of the polymerization in order to convert starch into oligosaccharides through the cleavage of glycosidic bonds. The authors showed that the increase of cationic starch content in the formulation did not influence the overall monomer conversion (>98%), but it decreased the grafting efficiency (proportion of graft copolymer poly(CS)-graft-poly(BA-co-MMA) recovered after Soxhlet extraction using chloroform as solvent), and also decreased the hydrodynamic diameter of the latex particles (from 201 to 111 nm). Notably, the latexes were colored (brown) in the presence of the modified starch. The authors concluded that the optimal cationic starch concentrations in terms of particle size, water resistance and colloidal stability were in the range between 7 and 10 wt%. The latex films were not cytotoxic after 24 h, but they were cytotoxic after 96 h of incubation. Cytotoxicity was ascribed to the α-amylase enzyme present in the films. In another study, they studied the thermal and mechanical properties of the CS-BA-MMA films.111 They showed that higher grafting percentage (obtained at low CS content in the formulation) improved the thermal stability of the films. The addition of CS to the formulation increased the tensile, elastic modulus and hardness properties of the latex film, but the elongation at break worsened. They concluded that a compromise between the thermal and mechanical properties of the films could be obtained at around 7–10 wt% CS.
Polysaccharides as Pickering stabilizers. The use of natural organic particles as stabilizers for the formation and polymerization of Pickering emulsions has been recently reviewed by Héroguez and coworkers112 and a focus on the use of cellulose, starch and chitin as Pickering stabilizers has been summarized in Cunningham and coworkers' recent review.113 The utilization of biosourced glucose-based stabilizers could bring emulsion polymerization even more in line with the green chemistry principles. It avoids the use of traditional synthetic surfactants that can cause environmental pollution resulting from the leaching out of the surfactants when the latex is used as a binder for architectural coatings or tissue irritation in biomedical applications. For that reason, Boufi and coworkers used 8 wt% (with respect to monomer content) starch nanocrystals (SNC) as Pickering stabilizers for ethylhexylacrylate and butylmethacrylate emulsion copolymerization initiated by H2O2/citric acid redox initiating system at 50 °C.114 In order to increase the stabilization efficiency and solids content (up to 30 wt% in the case of PBMA homopolymer), vinyltriethoxysilane (VTES) functions were grafted onto SNC (Fig. 20f) to obtain reactive nanocrystals in radical polymerization (grafting through). The resulting pressure sensitive adhesive property was increased (improvement in peel strength and energy adhesion of the PSAs) and commercial applications as PSA might be envisaged.
Cunningham and coworkers used a modified form of starch as Pickering stabilizer in the emulsion polymerization of styrene and MMA using ammonium persulfate as water-soluble initiator and di(ethylene glycol) ethyl ether acrylate (DEGEEA) as functional comonomer, and in miniemulsion polymerization of styrene, MMA and BMA using Vazo-52 as oil-soluble radical azo-initiator. Starch modification was performed with styrene oxide to tune its hydrophilic/hydrophobic character (Fig. 20g).115 The results showed that the new Pickering stabilizer was able to perform well only in the case of styrene miniemulsion polymerization (8% wbm of stabilizer) (monomer conversion > 80%), but was unsuitable for the miniemulsion polymerization of methacrylate monomers where coalescence of the latex particles occurred during the polymerization. PMMA stable latexes were obtained by emulsion polymerization (80% monomer conversion) using 8% wbm of Pickering stabilizer, where additional stabilization was likely provided by the water-soluble persulfate initiator.
A more challenging work was described by Schmitt and coworkers.116 They reported a Pickering double oil-in-water-in-oil emulsion using cellulose nanocrystals (CNC) modified with brominated functions (Fig. 21). Through the brominated function, they were able to tune the hydrophobicity of CNC. For example, when CNC-Br20% was used, the CNC reacted as Pickering hydrophilic stabilizers (direct emulsion, i.e. oil-in-water). Conversely, when CNC-Br90% was used, the CNC could be employed as Pickering hydrophobic stabilizer (inverse emulsion). They thus could prepare a double emulsion polymerization stabilized by CNC partially composed of biosourced components. They used water, isopropyl myristate (derived from vegetable-based myristic acid) as oil phase, hydroxyl oligoethylene glycol methacrylate as monomer and tetraethylene glycol diacrylate as cross-linker in the aqueous phase and brominated CNC as Pickering stabilizer. They showed that the final object presented promising encapsulation properties for both hydrophilic and hydrophobic dyes.
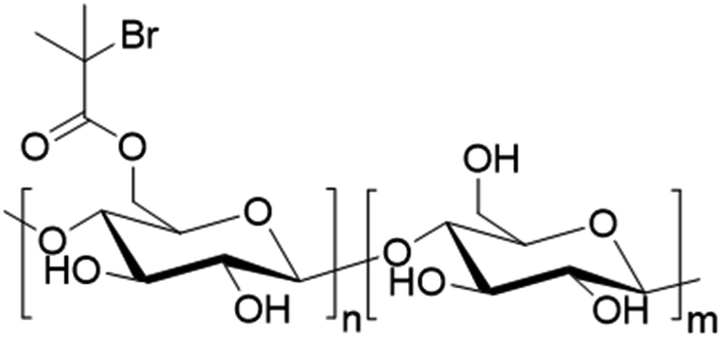 | ||
| Fig. 21 Cellulose nanocrystals (CNC) modified with brominated functions. | ||
Polysaccharides as a seed. Some examples of synthesis of starch core/shell latexes were performed in these last four years using polysaccharides as a seed.
Dubé and coworkers incorporated functionalized starch nanoparticles (SNP) inside an acrylic polymer.117 They first cross-linked starch molecules with sodium trimetaphosphate (a non-toxic cross-linker) (Fig. 20h). Then, they functionalized the cross-linked starch with a water-soluble bio-based macromonomer containing glucose (able to anchor to SNP surfaces via the trimetaphosphate group). The resulting modified SNP were polymerized via a semi-batch emulsion polymerization (17 wt% SNP relative to total polymer weight and 42 wt% solids content) with BA, MMA and AA. Butyl vinyl ether was added as comonomer prone to copolymerize rather than homopolymerize to act as a bridge between SNP and hydrophobic acrylates. The tack, peel strength and shear strength were evaluated to confirm the adhesive properties of the films. The TEM images were used to confirm the core–shell structure of the final latex. In their following article, the authors reported the increase of SNP content in the same semi-batch emulsion formulation (45 wt% SNP and 55 wt% solid content were achieved), although these results were obtained at the expense of adhesive properties and led to an increase of latex viscosity and particle size.118
Dubé and coworkers also worked on cellulose nanocrystals (CNC).119 CNC are highly water-insoluble and their dispersion in water is highly sensitive to the ionic strength of the aqueous phase; hence, their use in emulsion polymerization was limited. In their work, Dubé et al. demonstrated that the use of an atypical emulsion polymerization, which limited the ionic strength, allowed the incorporation of CNC in a colloidally stable nanoparticle. Indeed, they developed a semi-batch emulsion polymerization method where MMA and BA were used as monomers, AIBN as neutral initiator (dissolved in the monomers because it is oil-soluble) and Triton X-405 as neutral surfactant during the first stage of polymerization. The negatively charged sodium dodecyl sulphate (SDS) surfactant and potassium persulfate (KPS) initiator were only used for the second stage of the semi-batch polymerization (feeding). By this method and a precise addition feed of the different components (including the CNC dispersion), they maintained a low ionic strength in the aqueous medium and produced a stable latex with a solids content of 35 wt%. However, droplet nucleation occurred, estimated at 13% of total monomer weight, leading to the accumulation of a polymer gel on the impeller.
As outlined in this section, carbohydrates are an important source of sugar-based components (monomers/surfmers) and polysaccharide-based components (water-soluble steric stabilizers, Pickering stabilizers, water-dispersible seeds) that can be used in radical polymerization in aqueous dispersed media. However, their use for industrial applications presents several drawbacks. First, because of the polar character of carbohydrates, they are more easily introduced at the surface of the polymer particles, and thus the bio-content is often limited to values below 20 wt%, unless special efforts are made to decrease their hydrophilicity in order to incorporate them in the core of the polymer particles. However, these additional efforts might be a barrier for industrial scale-up. Second, due to their polar character and specific structure, carbohydrates may affect important properties such as the latex viscosity, the color of the films and the water sensitivity of the films. In conclusion, the incorporation of carbohydrates at high content in aqueous dispersions is not easy and may not be desirable, as it may badly affect the performance of the materials in some coating applications.
Protein-based polymers
The last option to replace petroleum-based polymers is proteins. Proteins are an attractive option because of their purity, biodegradability, low toxicity, high availability and low price.120 The utilization of proteins in radical polymerization in aqueous dispersed media has been examined in the last four years in emulsion and miniemulsion processes to create wood and paper adhesives, waterborne wood coatings and food packaging. In order to integrate proteins into aqueous dispersions prepared by radical polymerization, various strategies are usually employed, leading to the so-called hybrid-protein latexes. As in the carbohydrates section, the nature of the protein can influence its way of incorporation and the applications of the resulting latex. When the protein is water-soluble, it can react as macrotransfer agent, macroinitiator or macromonomer as well as (macro)surfmer in case of amphiphilic properties (Fig. 22). In contrast, when the protein is water-insoluble, it can be involved as surface Pickering stabilizer or as a seed to form core–shell particles. Because of the diversity and versatility of proteins, as in the carbohydrates section, reporting in this tutorial review the examples where proteins are not only used as (macro)monomer or (macro)surfmer but also as (macro)transfer agent, (macro)initiator, bio-based Pickering stabilizer or bio-based seed, even though the latter are not encompassed in the title of the review, is relevant. The hydrophilic or hydrophobic character of the protein depends on the side groups of its constituent amino acids, such as carboxylic acid, amide, hydroxyl, amine, thiol, alkyl or aryl side groups. Furthermore, the functional groups of some proteins (e.g. amines) can be profitable to introduce some moieties capable of reacting in radical polymerization (e.g. methacrylation with glycidyl methacrylate).121,122 Recent progress in the use of hybrid protein/synthetic polymer nanoparticles for the production of ecofriendly alternatives for industrial applications such as paints, coatings, films and adhesives has been described by Gugliotta and coworkers in a review article.123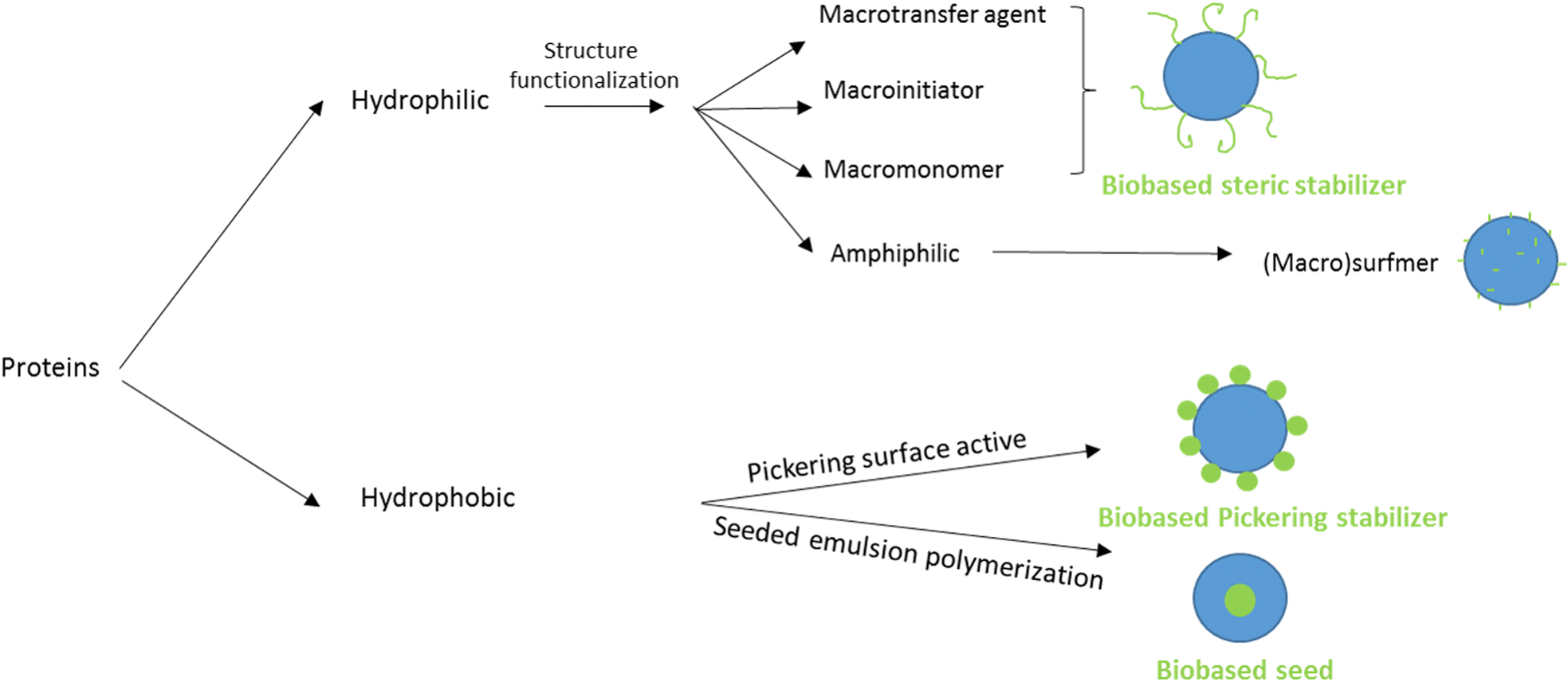 | ||
| Fig. 22 Different strategies to use proteins in radical polymerizations in aqueous dispersed media. | ||
Zein
Zein is a hydrophobic protein obtained as byproduct from corn processing during the production of bioethanol and cornmeal.124,125 It is a positively charged protein126 composed of non-polar amino acids such as leucine, alanine and proline (hydrophobic alkyl lateral groups).127 Its utilization in seeded emulsion polymerization has been described, in particular its power to impart hydrophobicity and compensate for the hydrophilicity of other proteins such as casein used as a seed122 (more details in the Casein section) or polysaccharides such as starch used as a seed (in combination with casein used as bio-macro-emulsifier) (more details in the Casein section below).128Casein
Gugliotta and coworkers focused their research on casein, a protein derived from bovine milk with good biocompatibility, high purity, biodegradability, easy availability and with a high Tg (180 °C). They functionalized native casein with methacrylate functions to prepare a macromonomer. The methacrylated casein was copolymerized with methyl methacrylate (MMA) and ethyl acrylate (EA) by surfactant-free emulsion polymerization to create a hybrid casein-acrylate latex for adhesive applications.121 They did not detail the quantitative composition of the monomer mixture in the emulsion polymerization process. However, the acrylate part (MMA and EA) was in the core and the casein was located around it as steric stabilizer. In the above-mentioned article, Gugliotta and coworkers studied the variation of the content of native casein and methacrylated-casein (from 5 to 30% wbm) and they showed that it influenced the polymerization rate and the final monomer conversion. The increase of native casein concentration favored the polymerization rate and led to higher final monomer conversion. In addition, the latex synthesized with native casein presented a lower grafting degree with the acrylate core whereas the utilization of methacrylated-casein improved the degree of grafting (less free casein) leading to a reduced latex viscosity. The adhesive removal from a glass substrate was also tested in alkaline solution and the latexes synthesized with methacrylated-casein showed better removal capacity compared to those synthesized with native casein.The methacrylated-casein was also employed in surfactant-free emulsion polymerization for the preparation of waterborne latexes composed of a core of acrylate polymer (BA and MMA) and a shell of methacrylated casein. Tannic acid (TA) was added as a cross-linking agent to the resulting latex prior to film formation for 5 days at room temperature, leading to crosslinked films129 (Fig. 23). TA is a natural polyphenolic molecule extracted from plants.130 Cross-linking during the film formation occurred by reaction of tannic acid with the amino groups of casein. The resulting waterborne cross-linked film showed improved water resistance and higher decomposition temperature, tensile stress and elastic modulus compared to the non-crosslinked film. The film showed excellent UV-blocking behavior, which can be interesting for the development of UV-blocking coatings to prevent photo-degradation of some organic materials. Nevertheless, the biodegradation rate of the tannic acid-crosslinked film was shown to be slower compared to that of the non-crosslinked film.
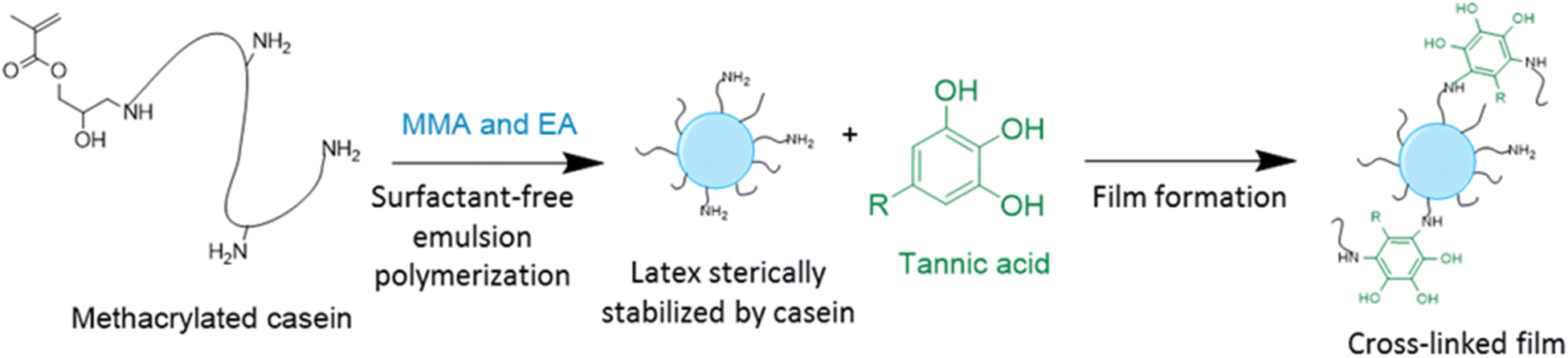 | ||
| Fig. 23 Schematic description of the formation of a cross-linked film from latex particles sterically stabilized by casein in the presence of tannic acid. | ||
The main drawback in the utilization of casein is its high hydrophilicity, imparting low water resistance to the films. For this reason, in another study, Gugliotta and coworkers added hydrophobic zein to mitigate the high hydrophilic character of casein (methacrylated or not) in their latex formulation.122 They prepared waterborne hybrid nanoparticles, by emulsifier-free miniemulsion polymerization of butyl acrylate, methyl methacrylate, or octadecyl acrylate, containing both proteins (at the surface) and acrylate (in the core) that showed enhanced mechanical properties and good soil biodegradation (only the casein part of the composite latex was degraded). The contact angle experiments with water showed that the incorporation of zein in the hybrid particles increased the film surface hydrophobicity compared to the zein-free hybrid casein/synthetic polymer film. In order to demonstrate that the miniemulsion polymerization was an adequate process for zein incorporation, they physically mixed the acrylate latex with both proteins; however, in this case the resulting polymer film presented phase separation due to the incompatibility of the components.
Another example of the use of modified-casein was described by Ronco and coworkers128 where casein was used as a surfmer to stabilize seeded emulsion polymerization. Ronco and coworkers, using the procedure developed by Huang and coworkers,126 modified hydrophilic starch with hydrophobic zein protein by ionic complexation. The new starch/zein bio-particles were hydrophobic and their introduction in the core (seed) of an acrylic polymer (BA/MMA: 80/20 mass ratio) was possible by batch and semi-batch seeded emulsion polymerization. The utilization of modified-casein as surfmer permitted the latex to be stabilized and the final latex bio-content to be increased. The hybrid particles exhibited a multilobular morphology, composed of a core of starch/zein bio-particle aggregates surrounded by many lobules of the acrylic copolymer.
Despite the provided modifications, such as the addition of tannic acid as cross-linker or the use of zein as a water repelling protein, the water resistance of hybrid casein/synthetic polymer films still needs deeper evaluations for applications in coatings.
In the above articles, the bio-content of the hybrid protein/synthetic polymer latex was limited to the protein fraction. To increase the bio-content of the latex particles, Leiza and coworkers combined hydrophilic casein (native or methacrylated) with hydrophobic 2-octyl acrylate and IBOMA as bio-based monomers.54 Thus, they copolymerized OA and IBOMA by emulsion polymerization in batch or semi-batch, in the presence of casein, as described previously (Fig. 7d). Notably, they reported that the addition of about 5% wbm of petro-sourced MMA, with sufficient solubility in water, was necessary to reach high monomer conversion. This is due to the complication of the nucleation step when very hydrophobic monomers such as OA or IBOMA are used in emulsion polymerization.
Soy
Soy protein is a byproduct isolated from the soybean oil industry. Its utilization is gaining increasing importance because it is a renewable, biodegradable and an inexpensive byproduct of the agricultural industry.131 Similar to casein, soy protein presents poor water resistance and low bonding strength. To overcome these disadvantages, it was used as a hydrophilic component in emulsion formulations. For example, Chen and coworkers investigated the potential of soy protein as macromonomer to develop a formaldehyde-free adhesive capable of curing at ambient temperature.132 First, they functionalized soy protein with N-methylolacrylamide and copolymerized the resulting protein monomer with styrene by aqueous emulsion polymerization (Fig. 24). At the end of polymerization and after freeze-drying, 8 wt% (based on soy mass) methylene diisocyanate was added to the polymer as cross-linker to form a soy-based cured-adhesive at room temperature. The resulting material showed high shear strength (1.04 MPa) and high water resistance.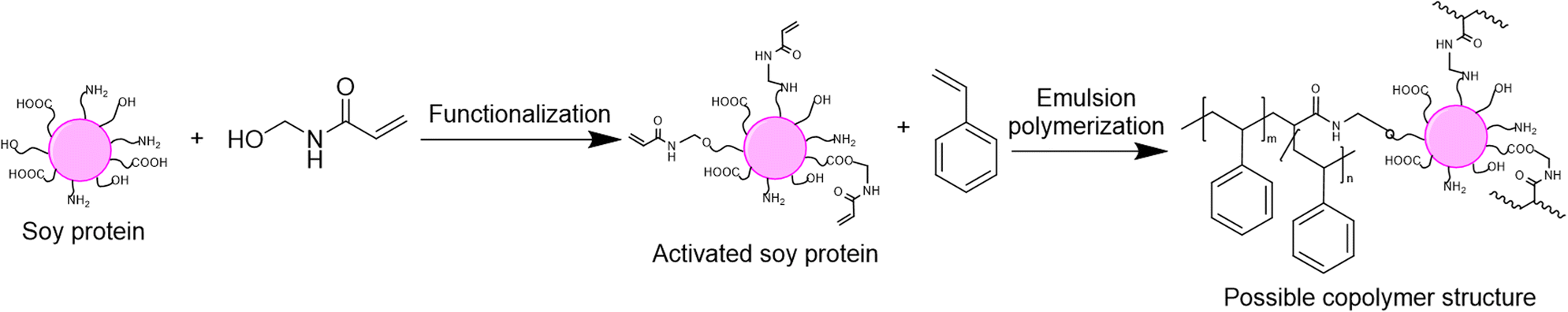 | ||
| Fig. 24 Schematic description of the functionalization of soy protein and its use in emulsion polymerization. | ||
Chuangying and coworkers improved a waterborne wood coating with a modified soy-protein.133 First, they modified the soy protein cutting the disulfide bonds with sodium metabisulfite under alkaline conditions at 85 °C in order to break the secondary and quaternary structure of the protein. In this way, they increased the availability of the amino and hydroxyl groups for transfer reactions. Then, they added the modified soy-protein to a terpolymer latex (methyl methacrylate, butyl acrylate and 2-(methacryloyloxy)ethyl acetoacetate), previously synthesized by emulsion polymerization, composing the base of the waterborne wood coating. According to the authors, the amino and hydroxyl groups of the modified soy-protein reacted with the oxidant and radical initiator (ammonium persulfate) to form radicals which reacted with the residual monomers of the latex, leading to a latex grafted with soy protein. The resulting polyacrylic-soy film exhibited a Tg of 35 °C and desirable elastic modulus (46.9 MPa), tensile strength (4.7 MPa) and elongation at break (187%). In particular, the resulting waterborne coating showed improved pencil hardness (3H) in comparison with the reference coating (HB).
Collagen
Another studied protein is collagen, the most abundant protein found in animals' connective tissues.134 Minari and coworkers investigated the redox surfactant-free emulsion copolymerization of acrylic acid and butyl acrylate (BA/AA: 70/30) in the presence of different concentrations of hydrolyzed collagen (from 15 to 50% wbm).135 The amine groups available on hydrolyzed collagen were able to be activated by the radical initiator (hydrogen peroxide). Thus, the hydrolyzed collagen acted as a macroinitiator for the polymerization of the acrylic monomers. The resulting latex nanoparticles, formed by an acrylic core stabilized by grafted hydrolyzed collagen, presented excellent film formation capability. The authors also tried to neutralize the carboxylic acid groups of the acrylic acid units in order to obtain a more hydrophilic material, and they found that neutralized films exhibited higher deformability and better adhesion properties than the non-neutralized ones.During these last four years, progress has been made in the use of native or modified proteins in polymerization in aqueous dispersed media. The diversity of the hydrophilic character of proteins has permitted the development of their utilization in several ways such as hydrophilic co(macro)monomers, macrotransfer agents, amphiphilic (macro)surfmers or hydrophobic modifiers (zein). However, as in the case of casein, because of the excessive hydrophilicity of proteins, the resulting materials often suffer from a low water resistance and corrosion resistance136,137 as a consequence of the presence of pendant hydrophilic groups on the protein backbone that decrease the product's final quality. Furthermore, the utilization of some proteins such as casein and soy is in competition with the food industry. Interestingly, native proteins are biodegradable; however, their modification often decreases their biodegradation capability. Finally, the bio-content introduced into latexes by proteins is usually low (typically less than 20 wt% of the latex particles), and thus their combination with other categories of hydrophobic bio-based monomers is highly desirable to reach high bio-content.
Conclusions
The progress made in radical polymerization of bio-based monomers in aqueous dispersed media in the last four years has been summarized. The field of bio-based emulsion polymerization is catching the attention of more and more researchers. The variety and the diversity of the five bio-based resource categories offer many opportunities to obtain a large range of reactive molecules and prepare materials with distinct characteristics such as Tg, film forming properties, chemical and mechanical properties or gel content, among others, for a large range of applications. The final aim is to produce sustainable bio-based materials with equal or improved performance in comparison to the current 100% petroleum-based materials. For that reason, when appropriate, the green metrics were calculated in the review. They give the reader an idea of the environmental impact of the process and they will help to find the more sustainable process and greener monomers. In the following table, we have tried to summarize the ratio of renewable carbons (considering that acrylate and methacrylate groups are not bio-based) of the most relevant reported monomers (Table 5).| Family | Monomers | % Renewable C | |
|---|---|---|---|
| Lipids and oils | SBMA |
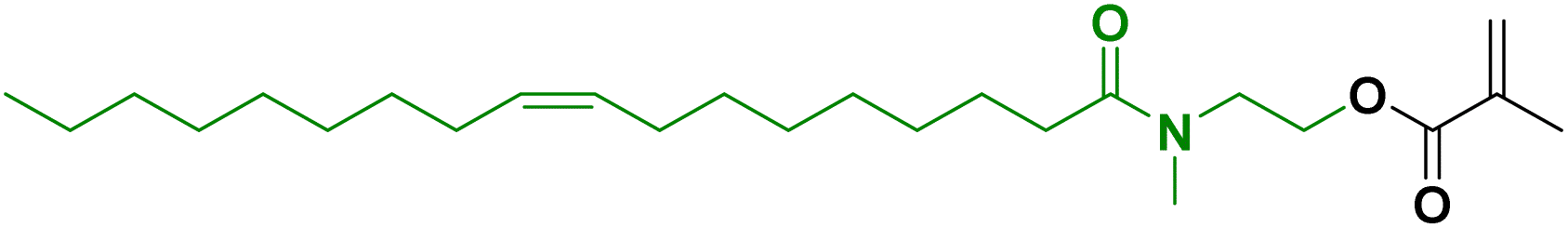
|
84 |
| HOSBM |
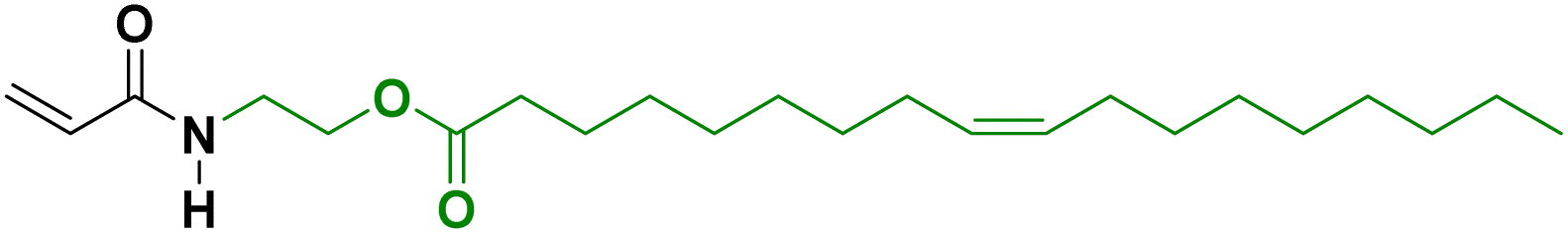
|
87 | |
| 2-Octyl acrylate |
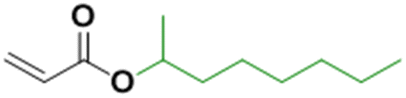
|
73 | |
| Methacrylated cardanol |
|
78 | |
| Terpenes | β-Myrcene |

|
100 |
| IBOMA |
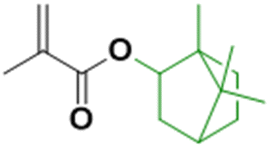
|
71 | |
| Lignin | Methacrylated eugenol |
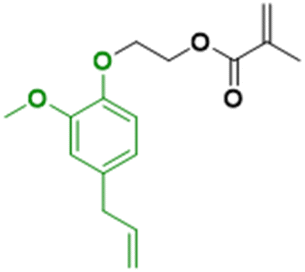
|
63 |
| Carbohydrates | Itaconate derivatives |
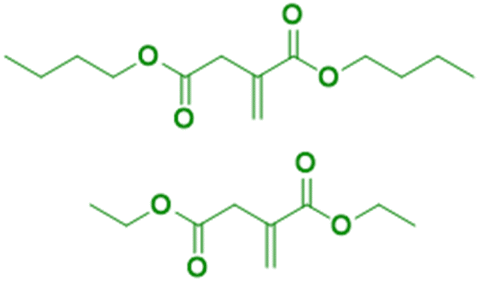
|
100 |
| ISOMA |
|
60 | |
As explained in the review, the addition of radical polymerizable functions such as acrylate and methacrylate groups is necessary because the majority of bio-based platform molecules does not possess suitable radical polymerizable groups.
As shown in Table 5, the % of renewable C is quite high (>55%), reaching 100% for some molecules (such as β-myrcene, which is directly used in emulsion polymerization, or itaconate derivatives, if we consider that ethanol138 and butanol139 can be derived from biomass). The lowest renewable C contents are achieved by eugenol methacrylate and pure ISOMA if we consider that the methacrylate moiety is not bio-based. It is complicated to compare the five categories using only their % of renewable C.
Each category presents advantages and drawbacks. For example, proteins and carbohydrates are highly hydrophilic, they often impart detrimental properties to the resulting films such as low water resistance. On the other hand, vegetable-oil derivatives and lipids are very hydrophobic, and can often only be polymerized using miniemulsion polymerization, which is not easy to scale-up at the industrial level. For those very hydrophobic vegetable oils and lipids, non-aqueous polymerization processes would be probably more adapted (or aqueous suspension polymerization, if well-suited for the targeted application). Terpene derivatives show some drawbacks as well, for example the difficulty to reach high monomer conversion. In addition, their microstructures, in particular the double bonds (e.g. 1,4-cis or 1,4-trans linkages, 1,2- or 3,4-structures), are strongly affected by the temperature of polymerization. From our point of view, the most promising category is the lignin family. The wide variety of phenol derivatives gives rise to interesting new properties and potential applications. Nevertheless, the large-scale commercial availability of lignin-based synthons remains scarce (today the only phenolic monomer produced on a large scale is vanillin) and this might slow down the industrial development of alternative lignin-based aqueous dispersions.
The transition to fully bio-based synthetic latexes is starting. As described above, in recent years, latexes including both petrol-based and bio-based monomers have been prepared, progressively increasing the bio-content of the latex. Ultimately, the goal would be to reach a total substitution of petro-based monomers by sustainable bio-based monomers (with low impact in different environmental categories such as carbon footprint, ecotoxicity, water use and considering an efficient end-of-life management) while ideally reaching better performance or even new properties leading to new applications and markets. The progressive substitution of the petro-sourced monomers by the bio-based ones seems to be a good solution also because of the currently higher cost and low commercial availability of bio-based monomers in comparison to their petroleum counterparts. The challenges ahead will be to find truly sustainable feedstocks and green synthesis paths to form bio-based platform molecules at an economically attractive cost.
Public and industrial interest in these bio-based materials is increasing. Hence, the future of bio-based polymers is undoubtedly promising. The partial replacement of monomers derived from petrochemical sources could increase the sustainability of the polymeric aqueous dispersions, making the derived products more eco-friendly. However, the other components of the formulation such as the initiators, surfactants, and transfer agents have to be taken into account in the pursuit of greener aqueous polymeric dispersions (although they have lower impact due to their lower concentrations in aqueous polymeric formulations). It must be emphasized that, as indicated in a recent review regarding recent advances in polymer colloids including a section on biobased lower carbon footprint latexes, the transition from petro-sourced polymers to biobased alternatives has been slower than predicted from an industrial point of view.140 Therefore, more effort is still required for research on renewable materials available on a large scale. Finally, in some cases depending on the applications (e.g. applications for which recycling is not an option as long as there are no specific collection schemes in place, such as in cosmetics), the objective will be not only to create a fully biobased material but also a material endowed with controlled on-demand degradability, biocompatibility and non-toxicity.141
Author contributions
Data curation: E. Rigo; formal analysis: E. Rigo; funding acquisition: P. Lacroix-Desmazes; project administration: P. Lacroix-Desmazes; supervision: P. Lacroix-Desmazes, V. Ladmiral, S. Caillol; validation: P. Lacroix-Desmazes, V. Ladmiral, S. Caillol; writing – original draft: E. Rigo, P. Lacroix-Desmazes; writing – review & editing: P. Lacroix-Desmazes, V. Ladmiral, S. Caillol.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Synthomer for financial support and for the fruitful discussions (with Dr Sophie Drillieres and Dr Samantha Molina-Gutiérrez). ER thanks Association Nationale de la Recherche et de la Technologie (France) for her PhD grant (CIFRE no. 2021-1361).Notes and references
- P. G. Mahlberg, Bot. Rev., 1993, 59, 1 CrossRef.
- J. C. Daniel and C. Pichot, Les Latex Synthétiques, Tec & Doc-Lavoisier, 2006 Search PubMed.
- H. van Asselt, Earth System Governance, 2021, 9, 100118 CrossRef.
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1 CrossRef CAS.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538 RSC.
- M. Jeguirim, B. Khiari and L. Limousy, Biomass Feedstocks, Elsevier Inc., 2019 Search PubMed.
- M. H. M. Ahmed, N. Batalha, H. M. D. Mahmudul, G. Perkins and M. Konarova, Bioresour. Technol., 2020, 310, 123457 CrossRef CAS PubMed.
- G. Wang, Y. Dai, H. Yang, Q. Xiong, K. Wang, J. Zhou, Y. Li and S. Wang, Energy Fuels, 2020, 34, 15557 CrossRef CAS.
- Q. Li, X. Yuan, X. Hu, E. Meers, H. C. Ong, W. H. Chen, P. Duan, S. Zhang, K. B. Lee and Y. S. Ok, Renewable Sustainable Energy Rev., 2022, 154, 111814 CrossRef CAS.
- F. W. S. Lucas, R. G. Grim, S. A. Tacey, C. A. Downes, J. Hasse, A. M. Roman, C. A. Farberow, J. A. Schaidle and A. Holewinski, ACS Energy Lett., 2021, 6, 1205 CrossRef CAS.
- F. D. Gunstone, Eur. J. Lipid Sci. Technol., 2011, 113, 3 CrossRef CAS.
- A. E. Atabani, A. S. Silitonga, H. C. Ong, T. M. I. Mahlia, H. H. Masjuki, I. A. Badruddin and H. Fayaz, Renewable Sustainable Energy Rev., 2013, 18, 211 CrossRef CAS.
- F. G. Calvo-Flores and F. J. Martin-Martinez, Front. Chem., 2022, 10, 1 Search PubMed.
- J. Spekreijse, J. Le Nôtre, J. van Haveren, E. L. Scott and J. P. M. Sanders, Green Chem., 2012, 14, 2747 RSC.
- G. Hayes, M. Laurel, D. MacKinnon, T. Zhao, H. A. Houck and C. R. Becer, Chem. Rev., 2023, 123, 2609 CrossRef CAS PubMed.
- S. Molina-Gutiérrez, V. Ladmiral, R. Bongiovanni, S. Caillol and P. Lacroix-Desmazes, Green Chem., 2019, 21, 36 RSC.
- M. Aguirre, S. Hamzehlou, E. González and J. R. Leiza, Adv. Chem. Eng., 2020, 56, 139 CAS.
- P. T. Anastas and J. B. Zimmerman, IEEE Eng. Manag. Rev., 2007, 35, 16 Search PubMed.
- S. Fadlallah, P. Sinha Roy, G. Garnier, K. Saito and F. Allais, Green Chem., 2021, 23, 1495 RSC.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437 RSC.
- J. Martínez, J. F. Cortés and R. Miranda, Processes, 2022, 10, 1274 CrossRef.
- J. Andraos, Org. Process Res. Dev., 2005, 9, 149 CrossRef CAS.
- J. Barton and I. Capek, Radical Polymerization in Disperse systems, Ellis Horwood Series in Polymer Chemistry, 1994 Search PubMed.
- P. A. Lovell and M. S. El-Aasser, Emulsion Polymerization and Emulsion Polymers, Wiley, 2006 Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC.
- P. A. Lovell and F. J. Schork, Biomacromolecules, 2020, 21, 4396 CrossRef CAS PubMed.
- B. Brooks, Chem. Eng. Technol., 2010, 33, 1737 CrossRef CAS.
- M. C. C. Pinto, J. G. F. Santos, F. Machado and J. C. Pinto, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, 2013, pp. 600–630 Search PubMed.
- J. M. Asua, Prog. Polym. Sci., 2002, 27, 1283 CrossRef CAS.
- J. M. Asua, Prog. Polym. Sci., 2014, 39, 1797 CrossRef CAS.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893 RSC.
- C. Zhang, T. F. Garrison, S. A. Madbouly and M. R. Kessler, Prog. Polym. Sci., 2017, 71, 91 CrossRef CAS.
- Qurat-ul-Ain, K. M. Zia, F. Zia, M. Ali, S. Rehman and M. Zuber, Int. J. Biol. Macromol., 2016, 93, 1057 CrossRef CAS PubMed.
- A. Gandini and T. M. Lacerda, Proceedings, 2021, 69, 26 Search PubMed.
- A. Kohut, S. Voronov, Z. Demchuk, V. Kirianchuk, K. Kingsley, O. Shevchuk, S. Caillol and A. Voronov, Molecules, 2020, 25, 2990 CrossRef CAS PubMed.
- A. Kohut, S. Voronov, Z. Demchuk, V. Kirianchuk, K. Kingsley, O. Shevchuk and A. Voronov, ACS Symp. Ser., 2020, 1372, 27 CrossRef CAS.
- M. E. Lamm, P. Li, S. Hankinson, T. Zhu and C. Tang, Polymer, 2019, 174, 170 CrossRef CAS.
- L. Yuan, Z. Wang, N. M. Trenor and C. Tang, Macromolecules, 2015, 48, 1320 CrossRef CAS.
- M. Foti, R. Médici and H. J. Ruijssenaars, J. Biotechnol., 2013, 167, 344 CrossRef CAS PubMed.
- Z. Demchuk, W. S. J. Li, H. Eshete, S. Caillol and A. Voronov, ACS Sustainable Chem. Eng., 2019, 7, 9613 CrossRef CAS.
- A. Kohut, Z. Demchuk, K. Kingsley, S. Voronov and A. Voronov, Eur. Polym. J., 2018, 108, 322 CrossRef CAS.
- Z. Demchuk, A.-S. Mora, S. Choudhary, S. Caillol, A. Voronov and A. Voronov Biobased, Ind. Crops Prod., 2021, 162, 113237 CrossRef CAS.
- R. B. N. Prasad and B. V. S. K. Rao, Chemical Derivatization of Castor Oil and Their Industrial Utilization, Elsevier Inc., 2017 Search PubMed.
- E. B. Mubofu, Sustainable Chem. Processes, 2016, 4, 11 CrossRef.
- J. I. García, H. García-Marín and E. Pires, Green Chem., 2014, 16, 1007 RSC.
- M. Checa, S. Nogales-Delgado, V. Montes and J. M. Encinar, Catalysts, 2020, 10, 1 CrossRef.
- M. Barrenetxe, A. Agirre, J. I. Santos, A. Badía, J. R. Leiza and A. Barquero, Macromol. React. Eng., 2022, 16, 2200014 CrossRef CAS.
- A. Badía, J. I. Santos, A. Agirre, M. J. Barandiaran and J. R. Leiza, ACS Sustainable Chem. Eng., 2019, 7, 19122 CrossRef.
- A. Badía, A. Agirre, M. J. Barandiaran and J. R. Leiza, Int. J. Adhes. Adhes., 2021, 108, 102860 CrossRef.
- A. Badía, A. Agirre, M. J. Barandiaran and J. R. Leiza, Biomacromolecules, 2020, 21, 4522 CrossRef PubMed.
- M. A. Droesbeke, R. Aksakal, A. Simula, J. M. Asua and F. E. Du Prez, Prog. Polym. Sci., 2021, 117, 101396 CrossRef CAS.
- A. Badía, J. Movellan, M. J. Barandiaran and J. R. Leiza, Ind. Eng. Chem. Res., 2018, 57, 14509 CrossRef.
- E. González, R. Stuhr, J. M. Vega, E. García-lecina, H. J. Grande, J. R. Leiza and M. Paulis, Polymers, 2021, 13, 6 CrossRef PubMed.
- M. Allasia, M. Aguirre, L. M. Gugliotta, R. J. Minari and J. R. Leiza, Prog. Org. Coat., 2022, 168, 106870 CrossRef CAS.
- A. Badía, M. J. Barandiaran and J. R. Leiza, Eur. Polym. J., 2021, 144, 110244 CrossRef.
- O. Llorente, A. Barquero, M. Paulis and J. R. Leiza, Prog. Org. Coat., 2022, 172, 107137 CrossRef CAS.
- S. P. Bunker and R. P. Wool, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 451 CrossRef CAS.
- J. La Scala and R. P. Wool, J. Am. Oil Chem. Soc., 2002, 79, 59 CrossRef CAS.
- S. Bunker, C. Staller, N. Willenbacher and R. Wool, Int. J. Adhes. Adhes., 2003, 23, 29 CrossRef CAS.
- P. B. Cardoso, T. O. Machado, P. E. Feuser, C. Sayer, M. A. R. Meier and P. H. H. Araújo, Eur. J. Lipid Sci. Technol., 2018, 120, 1700212 CrossRef.
- P. C. M. dos Santos, T. O. Machado, J. V. C. Santin, P. E. Feuser, E. S. Córneo, R. A. Machado-de-Ávila, C. Sayer and P. H. H. de Araújo, J. Appl. Polym. Sci., 2021, 138, 4 Search PubMed.
- P. C. Mattos dos Santos, P. E. Feuser, P. B. Cardoso, B. T. Steiner, E. da S. Córneo, R. Scussel, A. da C. Viegas, R. A. Machado-de-Ávila, C. Sayer and P. H. Hermes de Araújo, J. Biomater. Sci., Polym. Ed., 2018, 29, 1935 CrossRef CAS PubMed.
- N. F. Freire, P. E. Feuser, J. da Silva Abel, R. A. Machado-de-Ávila, R. Lopes Fialho, E. Cabral Albuquerque, C. Sayer and P. H. Hermes de Araújo, Int. J. Polym. Mater. Polym. Biomater., 2022, 71, 349 CrossRef CAS.
- F. Hoelscher, P. B. Cardoso, G. Candiotto, C. Guindani, P. Feuser, P. H. H. Araújo and C. Sayer, J. Polym. Environ., 2021, 29, 3668 CrossRef CAS.
- W. Liu, M. Wu, C. Ma, C. Liu, X. Zhang, Z. Wang and Z. Wang, ACS Sustainable Chem. Eng., 2022, 10, 13301 CrossRef CAS.
- S. Mu, Y. Zhang, J. Zhou, B. Wang and Z. Wang, ACS Sustainable Chem. Eng., 2020, 8, 5296 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965 CrossRef CAS PubMed.
- V. Ladmiral, R. Jeannin, K. Fernandes Lizarazu, J. Lai-Kee-Him, P. Bron, P. Lacroix-Desmazes and S. Caillol, Eur. Polym. J., 2017, 93, 785 CrossRef CAS.
- W. S. J. Li, C. Negrell, V. Ladmiral, J. Lai-Kee-Him, P. Bron, P. Lacroix-Desmazes, C. Joly-Duhamel and S. Caillol, Polym. Chem., 2018, 9, 2468 RSC.
- W. S. J. Li, F. Cuminet, V. Ladmiral, P. Lacroix-Desmazes, S. Caillol and C. Negrell, Prog. Org. Coat., 2021, 153, 106093 CrossRef CAS.
- P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8 CrossRef CAS PubMed.
- A. Behr and L. Johnen, ChemSusChem, 2009, 2, 1072–1095 CrossRef CAS PubMed.
- I. Magaña, R. López, F. J. Enríquez-Medrano, S. Kumar, A. Aguilar-Sanchez, R. Handa, R. Díaz de León and L. Valencia, J. Mater. Chem. A, 2022, 10, 5019 RSC.
- R. L. Melnick and J. Huff, in Reviews of Environmental Contamination and Toxicology, Springer-Verlag, New York, 1992, p. 111 Search PubMed.
- A. M. Api, F. Belmonte, D. Belsito, S. Biserta, D. Botelho, M. Bruze, G. A. Burton, J. Buschmann, M. A. Cancellieri, M. L. Dagli, M. Date, W. Dekant, C. Deodhar, A. D. Fryer, S. Gadhia, L. Jones, K. Joshi, A. Lapczynski, M. Lavelle, D. C. Liebler, M. Na, D. O'Brien, A. Patel, T. M. Penning, G. Ritacco, F. Rodriguez-Ropero, J. Romine, N. Sadekar, D. Salvito, T. W. Schultz, I. G. Sipes, G. Sullivan, Y. Thakkar, Y. Tokura and S. Tsang, Food Chem. Toxicol., 2020, 144, 111631 CrossRef CAS PubMed.
- K. Çelik, B. Togar, H. Türkez and N. Taspinar, Turk. J. Biol., 2014, 38, 253 CrossRef.
- P. Sarkar and A. K. Bhowmick, ACS Sustainable Chem. Eng., 2016, 4, 5462 CrossRef CAS.
- U. Kalita, S. Samanta, S. L. Banerjee, N. C. Das and N. K. Singha, Macromolecules, 2021, 54, 1478 CrossRef CAS.
- M. Save, M. Le Hellaye, V. de Villedon, I. Adoumaz, M. Pillet, L. Atanase, M. Lahcini, E. Deniau, A. Khoukh, V. Pellerin, I. Ly, V. Dulong and V. Schmitt, Biomacromolecules, 2022, 23, 2536 CrossRef CAS PubMed.
- T. W. Abraham, E. Allen, E. C. Bohnert, C. L. Frank, J. J. Hahn and P. Tsobanakis, WO2014144367A1, 2014.
- M. A. J. Finkelman and A. Vardanis, Appl. Environ. Microbiol., 1982, 44, 121 CrossRef CAS PubMed.
- Á. Pablo-Morales, M. E. Treviño and E. Saldívar-Guerra, Macromol. React. Eng., 2022, 54, 2200007 CrossRef.
- P. Sahu, P. Sarkar and A. K. Bhowmick, ACS Sustainable Chem. Eng., 2018, 6, 6599 CrossRef CAS.
- P. Sarkar and A. K. Bhowmick, RSC Adv., 2014, 4, 61343 RSC.
- P. Sahu and A. K. Bhowmick, Ind. Eng. Chem. Res., 2019, 58, 20946 CrossRef CAS.
- S. B. Luk and M. Maríc, ACS Omega, 2021, 6, 4939 CrossRef CAS PubMed.
- S. Tajbakhsh, F. Hajiali and M. Maric, Ind. Eng. Chem. Res., 2020, 59, 8921 CrossRef CAS.
- S. Tajbakhsh, F. Hajiali and M. Marić, ACS Appl. Polym. Mater., 2021, 3, 3402 CrossRef CAS.
- M. A. Droesbeke, A. Simula, J. M. Asua and F. E. Du Prez, Green Chem., 2020, 22, 4561 RSC.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559 CrossRef CAS PubMed.
- S. Sethupathy, G. Murillo Morales, L. Gao, H. Wang, B. Yang, J. Jiang, J. Sun and D. Zhu, Bioresour. Technol., 2022, 347, 126696 CrossRef CAS PubMed.
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevin, Green Chem., 2014, 16, 1987 RSC.
- S. Molina-Gutiérrez, A. Manseri, V. Ladmiral, R. Bongiovanni, S. Caillol and P. Lacroix-Desmazes, Macromol. Chem. Phys., 2019, 220, 1900179 CrossRef.
- R. Morales-Cerrada, S. Molina-Gutierrez, P. Lacroix-Desmazes and S. Caillol, Biomacromolecules, 2021, 22, 3625 CrossRef CAS PubMed.
- S. Molina-Gutiérrez, V. Ladmiral, R. Bongiovanni, S. Caillol and P. Lacroix-Desmazes, Ind. Eng. Chem. Res., 2019, 58, 21155 CrossRef.
- S. Molina-Gutiérrez, W. S. J. Li, R. Perrin, V. Ladmiral, R. Bongiovanni, S. Caillol and P. Lacroix-Desmazes, Biomacromolecules, 2020, 21, 4514 CrossRef PubMed.
- Y. Polunin, T. J. Burns, E. M. Serum, M. P. Sibi and A. Voronov, ACS Sustainable Chem. Eng., 2021, 9, 7003 CrossRef CAS.
- A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1 CrossRef CAS.
- N. M. B. Smeets, S. Imbrogno and S. Bloembergen, Carbohydr. Polym., 2017, 173, 233 CrossRef CAS PubMed.
- A. Badía, M. Kastelijn, J. Scheerder and J. R. Leiza, Prog. Org. Coat., 2020, 147, 105708 CrossRef.
- H. Fouilloux and C. M. Thomas, Macromol. Rapid Commun., 2021, 42, 2000530 CrossRef CAS PubMed.
- C. R. Casas-Soto, A. S. Conejo-Dávila, V. Osuna, D. Chávez-Flores, J. C. Espinoza-Hicks, S. G. Flores-Gallardo and A. Vega-Rios, Polymers, 2022, 14, 632 CrossRef CAS PubMed.
- W. Lei, X. Yang, M. Wang, H. Yang, J. Liu, Z. yang Wei, D. Shi, R. Wang and L. Zhang, Polymer, 2021, 228, 123910 CrossRef CAS.
- S. Chakraborty and S. Ramakrishnan, Langmuir, 2018, 34, 11729 CrossRef CAS PubMed.
- S. Cummings, Y. Zhang, N. Smeets, M. Cunningham and M. Dubé, Processes, 2019, 7, 140 CrossRef CAS.
- S. Cheng, Y. Zhao and Y. Wu, Prog. Org. Coat., 2018, 118, 40 CrossRef CAS.
- M. Aouay, A. Magnin, J.-L. Putaux and S. Boufi, Colloids Surf., A, 2022, 632, 127776 CrossRef CAS.
- V. L. Romero Castro, B. Nomeir, A. A. Arteni, M. Ouldali, J.-L. Six and K. Ferji, Polymers, 2021, 13, 4064 CrossRef CAS PubMed.
- L. D. A. Rodrigues, C. R. Hurtado, E. F. Macedo, D. B. Tada, L. M. Guerrini and M. P. Oliveira, Prog. Org. Coat., 2020, 145, 105693 CrossRef CAS.
- S. C. Alcázar-Alay and M. A. A. Meireles, Food Sci. Technol., 2015, 35, 215 CrossRef.
- L. D. A. Rodrigues, L. M. Guerrini and M. P. Oliveira, J. Therm. Anal. Calorim., 2021, 146, 143 CrossRef CAS.
- H. Dupont, V. Maingret, V. Schmitt and V. Héroguez, Macromolecules, 2021, 54, 4945 CrossRef CAS.
- A. Rigg, P. Champagne and M. F. Cunningham, Macromol. Rapid Commun., 2022, 43, 2100493 CrossRef CAS PubMed.
- E. Ben Ayed, A. Magnin, J.-L. Putaux and S. Boufi, J. Colloid Interface Sci., 2020, 578, 533 CrossRef CAS PubMed.
- J. C. Cazotti, S. E. Smeltzer, N. M. B. Smeets, M. A. Dubé and M. F. Cunningham, Polym. Chem., 2020, 11, 2653 RSC.
- H. Dupont, V. Héroguez and V. Schmitt, Carbohydr. Polym., 2022, 279, 118997 CrossRef CAS PubMed.
- Y. Zhang, M. F. Cunningham, N. M. B. Smeets and M. A. Dubé, Eur. Polym. J., 2018, 106, 128 CrossRef CAS.
- Y. Zhang, M. F. Cunningham, N. M. B. Smeets and M. A. Dubé, Ind. Eng. Chem. Res., 2019, 58, 20987 CrossRef CAS.
- V. A. Gabriel, P. Champagne, M. F. Cunningham and M. A. Dubé, Can. J. Chem. Eng., 2022, 100, 767 CrossRef CAS.
- S. Guilbert and B. Cuq, in Handbook of Biodegradable Polymers, De Gruyter, 2020, p. 299 Search PubMed.
- A. Aguzin, J. I. Jerkovich, J. Trucone, L. I. Ronco, R. J. Minari and L. M. Gugliotta, Lat. Am. Appl. Res., 2020, 50, 115 CAS.
- M. Allasia, M. C. G. Passeggi, L. M. Gugliotta and R. J. Minari, Ind. Eng. Chem. Res., 2019, 58, 21070 CrossRef CAS.
- L. G. Cencha, M. Allasia, L. I. Ronco, G. C. Luque, M. L. Picchio, R. J. Minari and L. M. Gugliotta, Ind. Eng. Chem. Res., 2021, 60, 4745 CrossRef CAS.
- K. Shi, J. L. Kokini and Q. Huang, J. Agric. Food Chem., 2009, 57, 2186 CrossRef CAS PubMed.
- J. W. Lawton, Cereal Chem., 2002, 79, 1 CrossRef CAS.
- S. Li, L. Huang, B. Zhang, C. Chen, X. Fu and Q. Huang, Food Hydrocolloids, 2021, 112, 106341 CrossRef CAS.
- F. A. Momany, D. J. Sessa, J. W. Lawton, G. W. Selling, S. A. H. Hamaker and J. L. Willett, J. Agric. Food Chem., 2006, 54, 543 CrossRef CAS PubMed.
- S. F. Cabrera, A. Pighin, M. L. Chiana, M. C. G. Passeggi, G. D. Ruano, L. M. Gugliotta, L. I. Ronco and R. J. Minari, J. Polym. Sci., 2022, 60, 3420 CrossRef CAS.
- L. G. Cencha, M. Allasia, M. C. G. Passeggi, L. M. Gugliotta and R. J. Minari, Prog. Org. Coat., 2021, 159, 106413 CrossRef CAS.
- J. A. Wilson, J. Chem. Educ., 1934, 11, 670 CrossRef.
- N. Chen, Q. Zeng, Q. Lin and J. Rao, BioResources, 2015, 10, 5071 CAS.
- P. Zheng, Q. Zeng, Q. Lin, M. Fan, J. Zhou, J. Rao and N. Chen, Int. J. Adhes. Adhes., 2019, 95, 102429 CrossRef CAS.
- B. Feng, D. Wang, Y. Li, J. Qian, C. Yu, M. Wang, D. Luo and S. Wei, Polymers, 2020, 12, 1137 CrossRef CAS PubMed.
- G. A. Di Lullo, S. M. Sweeney, J. Körkkö, L. Ala-Kokko and J. D. San Antonio, J. Biol. Chem., 2002, 277, 4223 CrossRef CAS PubMed.
- G. C. Luque, R. Stürtz, M. C. G. Passeggi, L. M. Gugliotta, V. D. G. Gonzalez and R. J. Minari, Int. J. Adhes. Adhes., 2020, 100, 102624 CrossRef CAS.
- C. R. Frihart, T. Coolidge, C. Mock and E. Valle, Polymers, 2016, 8, 394 CrossRef PubMed.
- N. R. N. Masdek, A. A. Rozali, M. C. Murad and Z. Salleh, ISIJ Int., 2018, 58, 1519 CrossRef CAS.
- I. Muñoz, K. Flury, N. Jungbluth, G. Rigarlsford, L. M. Canals and H. King, Int. J. Life Cycle Assess., 2014, 19, 109 CrossRef.
- Y. Guo, Y. Liu, M. Guan, H. Tang, Z. Wang, L. Lin and H. Pang, RSC Adv., 2022, 12, 18848 RSC.
- M. Aguirre, N. Ballard, E. Gonzalez, S. Hamzehlou, H. Sardon, M. Calderon, M. Paulis, R. Tomovska, D. Dupin, R. H. Bean, T. E. Long, J. R. Leiza and J. M. Asua, Macromolecules, 2023, 56, 2579 CrossRef CAS PubMed.
- I. Paul-Pont, J.-F. Ghiglione, E. Gastaldi, A. Ter Halle, A. Huvet, S. Bruzaud, F. Lagarde, F. Galgani, G. Duflos, M. George and P. Fabre, J. Waste Manage., 2023, 157, 242 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00097d |
| This journal is © The Royal Society of Chemistry 2023 |