
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiple stable redox states and tunable ground states via the marriage of viologens and Chichibabin's hydrocarbon†
Yuyang
Dai‡
a,
Zhuofeng
Xie‡
a,
Manling
Bao
a,
Chunmeng
Liu
a and
Yuanting
Su
 *ab
*ab
aCollege of Chemistry, Chemical Engineering and Materials Science, School of Radiation Medicine and Protection, Soochow University, Suzhou 215123, China. E-mail: ytsu@suda.edu.cn
bState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210023, China
First published on 8th March 2023
Abstract
Chichibabin's hydrocarbon and viologens are among the most famous diradicaloids and organic redox systems, respectively. However, each has its own disadvantages: the instability of the former and its charged species, and the closed-shell nature of the neutral species derived from the latter, respectively. Herein, we report that terminal borylation and central distortion of 4,4′-bipyridine allow us to readily isolate the first bis-BN-based analogues (1 and 2) of Chichibabin's hydrocarbon with three stable redox states and tunable ground states. Electrochemically, both compounds exhibit two reversible oxidation processes with wide redox ranges. One- and two-electron chemical oxidations of 1 afford the crystalline radical cation 1˙+ and dication 12+, respectively. Moreover, the ground states of 1 and 2 are tunable with 1 as a closed-shell singlet and the tetramethyl-substituted 2 as an open-shell singlet, the latter of which could be thermally excited to its triplet state because of the small singlet-triplet gap.
Introduction
Diradicaloids have gained considerable attention owing to their synthetic challenges, unique chemical bonding, interesting physical properties, and promising applications in functional materials.1 One of the most noteworthy examples is Chichibabin's hydrocarbon (CH, Fig. 1a), which was first synthesized by Chichibabin in 1907 (ref. 2) and has been of high interest for more than a century. Experimental measurements3 and theoretical studies1a,j,l disclose that CH is best described as a resonance hybrid of open-shell diradical and closed-shell quinoid forms. However, the highly reactive nature of CH and its charged species greatly restricts their development and practical applications. Thus, many efforts have been devoted to preparing its analogues with enhanced stability and stable redox states. Recently, neutral carbon (I),4 dianionic boron (II)5 and dicationic nitrogen (III)6 analogues of CH have been isolated by the groups of Wu, Kubo, Ghadwal, Jana, Marder, Wang and others. Despite these great achievements, these systems typically feature only one or two stable redox states and analogues of CH structurally characterized in all three redox states are limited to II (ref. 5) and III,6c,e while neutral analogues of CH featuring three crystalline redox states remain elusive.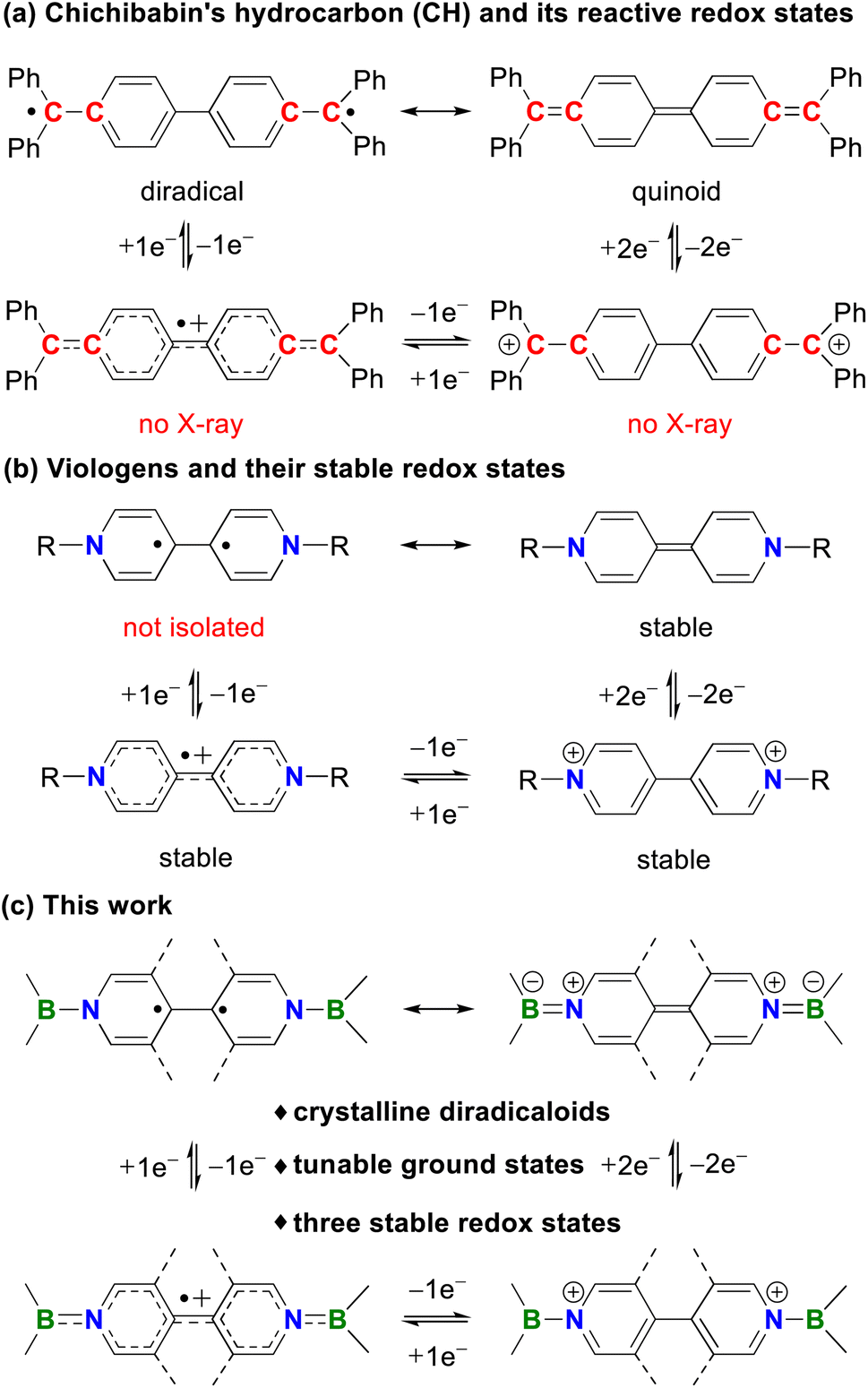 | ||
| Fig. 1 Resonance forms and redox states of (a) Chichibabin's hydrocarbon and (b) viologens. (c) This work. | ||
Organic redox systems featuring multiple redox states are of vital importance in electron transfer processes and offer various applications in photocatalysts, organic electronics, and redox flow batteries.7 Among the most representative examples of such species, one can mention viologens introduced by Michaelis in 1932.8 They can undergo two successive reversible reductions to form cationic radicals and neutral species, respectively (Fig. 1b).9 Although many modifications on bridging backbones and terminal substituents have been developed, neutral species derived from viologens are diamagnetic. This is unfavorable for radical-mediated applications because of the reduced radical utilization efficiency. Thus, it is highly challenging but very important to construct viologens with a diradical neutral state. Recently, the strong sensitivity of the fluorescence and efficient photocatalytic oxidative coupling reaction involved with the diradical neutral state of viologens were reported by Walter10 and He,11 respectively. In spite of these initial advances, such diradicals were only characterized in solution, while their solid-state structures and diradical character are hitherto unknown.
Inspired by these reports, we envision that replacement of both terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in CH with isoelectronic BN bonds could simultaneously lead to bis-BN-based analogues of CH and neutral species of bisboryl-substituted viologens (Fig. 1c), which may combine the diradical character and stable redox states in a single system and allow developing both well-known chemistries. Such a strategy has indeed been randomly employed to construct bis-BN-based analogues of CH, which have been used as key intermediates in 4,4-bipyridine-based organocatalytic addition reactions, but they are closed-shell singlet species and only stable in their neutral states or observed as radical cations in solution.12 Thus, further tuning of the terminal groups and central linkers is highly desirable.
C bonds in CH with isoelectronic BN bonds could simultaneously lead to bis-BN-based analogues of CH and neutral species of bisboryl-substituted viologens (Fig. 1c), which may combine the diradical character and stable redox states in a single system and allow developing both well-known chemistries. Such a strategy has indeed been randomly employed to construct bis-BN-based analogues of CH, which have been used as key intermediates in 4,4-bipyridine-based organocatalytic addition reactions, but they are closed-shell singlet species and only stable in their neutral states or observed as radical cations in solution.12 Thus, further tuning of the terminal groups and central linkers is highly desirable.
By employment of the bulky 1,3,2-diazaborolyl ligand,13 Aldridge14 and our group15 recently isolated BN/carbene- and bis-BN-based analogues of Thiele's hydrocarbon (TH), respectively, both of which are closed-shell singlets in their ground states. Herein, we report the facile isolation, full characterization, and computational studies of the first bis-BN-based analogues of CH, which are also the neutral species of bisboryl-substituted viologens, featuring multiple stable redox states and tunable ground states (Fig. 1c).
Results and discussion
A one-pot reaction of bromoborane (HCNDipp)2BBr (Dipp = 2,6-iPr2C6H3) with potassium graphite in the presence of half the equivalent of 4,4′-bipyridine in toluene at room temperature readily afforded 1 as an orange solid in 64% yield (Scheme 1). Compound 1 is extremely moisture- and oxygen-sensitive but can be stored under an inert atmosphere at room temperature for months. Compound 1 exhibits a well-resolved 1H NMR spectrum at room temperature (Fig. S1†) and resonances for protons on bridging rings appear at a relatively high field (δ = 5.35 and 4.89 ppm). Moreover, even upon heating up to 333 K, no NMR spectral broadening could be observed for 1 (Fig. S2†), which indicates its closed-shell nature.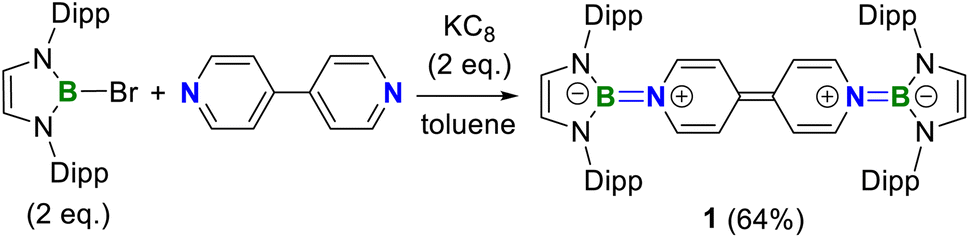 | ||
| Scheme 1 Synthesis of 1. | ||
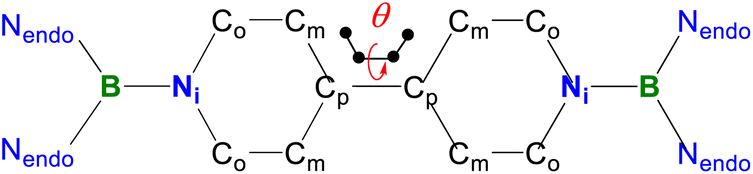
Single crystals of 1 suitable for X-ray diffraction analysis were obtained from its saturated THF/hexane solution mixture. The boron centers in 1 display a trigonal planar geometry (Fig. 2 and S17†). The exocyclic B–Ni bonds (avg. 1.4033(18) Å, Table 1) of 1 are slightly shorter than the endocyclic B–Nendo bonds (avg. 1.4340(18) Å), suggesting somewhat π bonding between B and Ni atoms. Additionally, the central pyridyl rings in 1 are almost coplanar and the Cp–Cp bond (1.374(2) Å) between two pyridyl rings is comparable to those of Bpin (1.375(4) Å)12d and BMes2 (1.391(4) Å)12b substituted analogues, further supporting the closed-shell singlet state for 1.
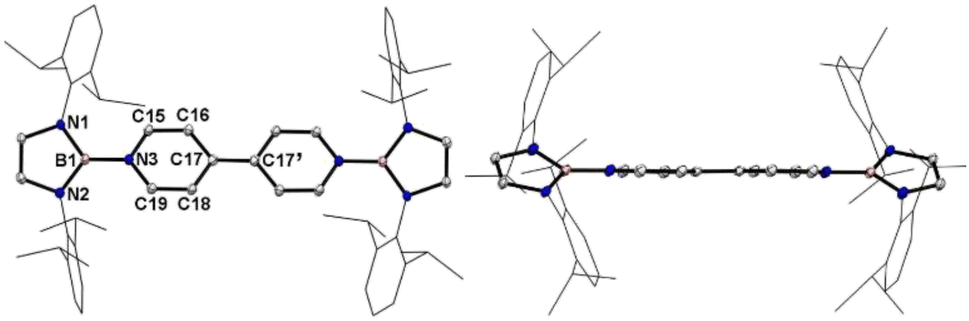 | ||
| Fig. 2 Left: the solid-state structure of 1. Right: side view. Hydrogen atoms are omitted and Dipp groups are simplified as wireframes for clarity. Thermal ellipsoids are set at the 30% probability level. | ||
|
|
||||
|---|---|---|---|---|
| 1 | 1˙+ | 12+ | 2 | |
| B–Nendo | 1.4340(18) | 1.424(5) | 1.407(9) | 1.436(2) |
| B–Ni | 1.4033(18) | 1.476(5) | 1.511(8) | 1.451(2) |
| Ni–Co | 1.4016(18) | 1.378(4) | 1.363(9) | 1.400(2) |
| Co–Cm | 1.3420(19) | 1.354(5) | 1.357(9) | 1.350(2) |
| Cm–Cp | 1.4558(18) | 1.426(5) | 1.389(9) | 1.458(2) |
| Cp–Cp | 1.374(2) | 1.424(7) | 1.479(8) | 1.417(3) |
| BLA | 0.0867(18) | 0.048(5) | 0.019(9) | 0.079(2) |
| θ | 0 | 0 | 27.5 | 53.2 |
To get insight into its electronic structure and ground state, DFT calculations and analyses were carried out using Gaussian 16 (ref. 16) and Multiwfn.17 Geometry optimizations and frequency calculations were performed on the simplified model 1′, in which the Dipp groups were replaced with phenyls. Three electronic states (closed-shell singlet (CS), open-shell singlet (OS), and triplet (T) state) with the (U)B3LYP and (U)PBE0 functionals and Def2SVP and 6-311G(d) basis-sets were considered. However, attempts to obtain the optimized 1′ as an open-shell singlet species (1′-OS) collapsed into a closed-shell singlet (1′-CS) electronic structure. The calculated singlet-triplet energy gaps ΔES–T at different levels are between −25.0 and −28.7 kcal mol−1 for 1′ (Table S2†). The calculated structure of 1′-CS matches well with the X-ray structure, while the triplet structure of 1′-T significantly deviates from the experimental data (Table S3†). Thus, experimental and theoretical studies confirm that the ground state of compound 1 is a closed-shell singlet.
To our delight, the cyclic voltammogram of 1 reveals the first reversible and second quasi-reversible single-electron oxidation processes at E1/2 = −0.99 and −0.29 V (Fig. 3a), respectively, suggesting the targeted three-state redox nature. Such negative potential for the first oxidation wave of 1 is close to those of biscarbene congeners (−1.51 to −1.04 V),4e,f suggesting their similarly strong reducing properties to their all-carbon analogues. However, in contrast to the narrow separations (ΔE < 0.25 V) between two redox events for biscarbene systems, compound 1 exhibits a markedly wide redox range (ΔE = 0.70 V), which indicates the significantly enhanced stability of its radical cation.
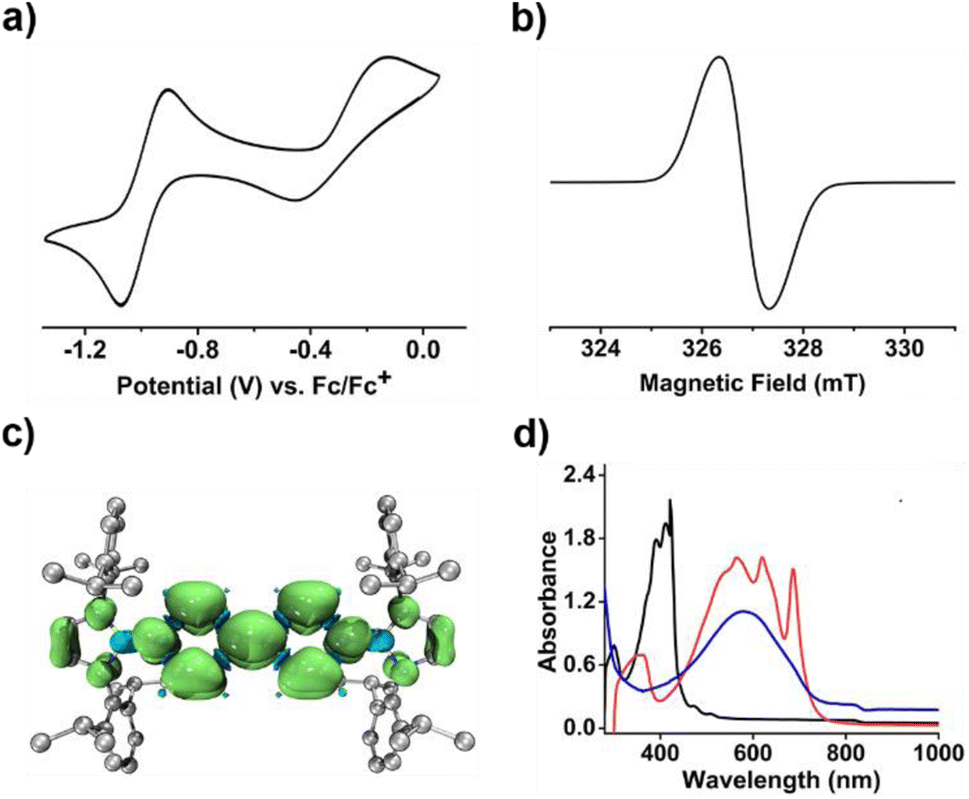 | ||
| Fig. 3 (a) The cyclic voltammogram of 1 in CH2Cl2 (0.1 M [nBu4N][PF6]) measured at a scan rate of 100 mV s−1. (b) The EPR spectrum of 1˙+[SbF6]− in CH2Cl2. (c) The calculated spin density map of 1˙+. (d) The UV-vis spectra of 1 (black), 1˙+ (red) and 12+ (blue). | ||
Next, chemical oxidations of 1 were performed to examine the possibility of generating the corresponding charged species. Treatment of 1 with one equivalent of AgSbF6 in CH2Cl2 at −30 °C immediately afforded a NMR silent deep color solution, from which 1˙+[SbF6]− was isolated as a bluish violet powder in 66% yield (Scheme 2). The EPR spectrum of 1˙+[SbF6]− displays an unresolved broad signal with g = 2.0010 (Fig. 3b), suggesting effective spin delocalization in the entire π-conjugated backbone. As the reaction of 1 with two equivalents of AgSbF6 still gave 1˙+[SbF6]−, stronger oxidant Ag[Al(ORF)4] (RF = C(CF3)3) was employed and compound 1 was successfully doubly oxidized to give 12+·2[Al(ORF)4]− in 52% yield (Scheme 2). The 11B{1H} NMR spectrum of 12+ displays a broad signal at 19.6 ppm (Fig. S9†), which is slightly shifted upfield with respect to that (22.6 ppm) of 1 (Fig. S5†). The 1H NMR spectrum of 12+ shows a greatly downfield shift for protons (δ = 8.23 and 7.82 ppm, Fig. S6†) of central bipyridyl rings relative to those (δ = 5.35 and 4.89 ppm) of 1, which illustrates the increasing aromaticity within the bipyridyl rings.
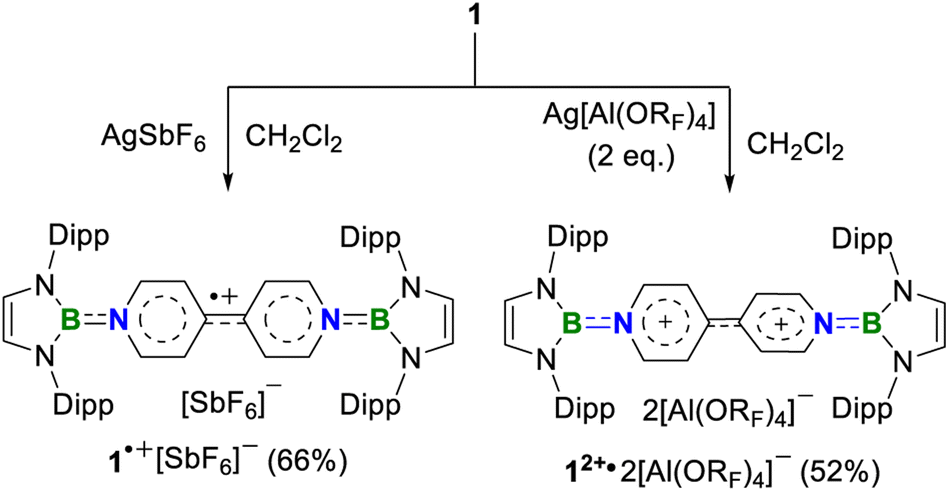 | ||
| Scheme 2 Oxidation reactions of 1. | ||
Crystallographic analysis reveals that the central bipyridyl rings in 1˙+ are also coplanar (Fig. 4 and Table 1), while those in 12+ are distorted to a dihedral angle of 27.5° (Fig. 5 and Table 1). Upon successive one-electron oxidations, the endocyclic B–Nendo bonds are gradually shortened, while the exocyclic B–Ni bonds and central Cp–Cp linkages are stepwise elongated, suggesting the drastically diminishing exocyclic double bond character. Additionally, the bond length alternation (BLA) in central bipyridyl rings is remarkably reduced from 0.087(2) in 1 to 0.048(5) in 1˙+ and further to 0.019(9) in 12+, which supports the progressive aromaticity enhancement.
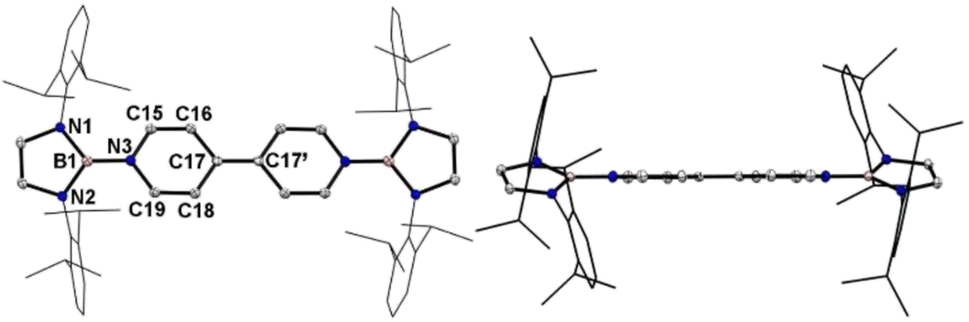 | ||
| Fig. 4 Left: the solid-state structure of 1˙+. Right: side view. Hydrogen atoms are omitted and Dipp groups are simplified as wireframes for clarity. Thermal ellipsoids are set at the 30% probability level. | ||
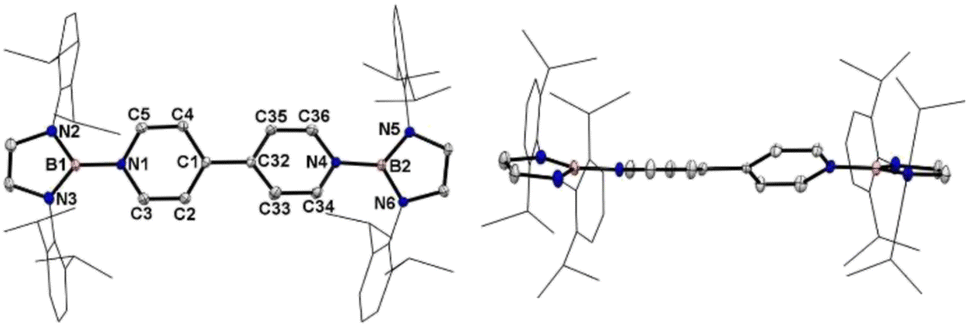 | ||
| Fig. 5 Left: the solid-state structure of 12+. Right: side view. Hydrogen atoms are omitted and Dipp groups are simplified as wireframes for clarity. Thermal ellipsoids are set at the 30% probability level. | ||
Calculations on 1˙+ at the UB3LYP/6-311G(d) level show that the spin density is mainly delocalized across the central bipyridyl moiety with small contributions from two terminal NHB units (Fig. 3c and S21†). The natural population analysis (NPA) on 12+ reveals that two positive charges are highly delocalized with the majority residing on boron atoms (+0.98, Table S5†), which are significantly larger than that of bis(carbodicarbene)[BH]2+ (+0.36) reported by Ong and coworkers,18 and are comparable to those of bis(imino)[BPh]2+ dication (+1.02)19 and borenium-imidazolium (0.94)14 reported by the group of Inoue and Aldridge, respectively. The nucleus independent chemical shifts (NICS) calculations reveal the decreased NICS(1)zz values of central bipyridyl rings from 10.00 in 1′ to −4.24 in 1˙+ and further to −17.88 in 12+ (Table S4†), further hinting at the increasing aromatic character upon progressive one-electron oxidations.
In the UV-vis absorption spectra (Fig. 3d), compound 1 shows a maximum absorption band at 411 nm (HOMO → LUMO) and 1˙+ displays three absorption bands at 564, 620, and 687 nm (SOMO−1(β) → SOMO(β)), while 12+ exhibits an absorbance at 580 nm (HOMO → LUMO). To our knowledge, 1˙+ represents the first example of crystalline radical cations derived from neutral analogues of Chichibabin's hydrocarbon and 12+ is a rare example of three-coordinate diborenium dications.14,18,19 Therefore, the terminal borylation of 4,4′-bipyridine by the bulky 1,3,2-diazaborolyls stabilizes the first neutral analogue of CH featuring three structurally characterized redox states.
As 1 is still a closed-shell singlet, further modification was made to tune the ground state. The distortion of the central linker has been proved as an efficient approach toward the diradical species.1,4,6 Therefore, the replacement of 4,4′-bipyridine by 3,3′,5,5′-tetramethyl-4,4′-bipyridine under the same other conditions was carried out, giving compound 2 as a purple solid in 49% yield (Scheme 3).
 | ||
| Scheme 3 Synthesis of 2. | ||
Like 1, the cyclic voltammogram of 2 also displays two reversible single-electron oxidation processes (E1/2 = −1.54 and −0.99 V, Fig. 6a) with a wide redox range (ΔE = 0.55 V), indicating that 2 also features three stable redox states and stronger reducing ability than 1. However, the UV-vis absorption spectrum of 2 demonstrates a significantly red-shifted maximum absorption band (λmax = 527 nm, Fig. 6b) relative to 1 (λmax = 411 nm), which suggests the increasing diradical character.
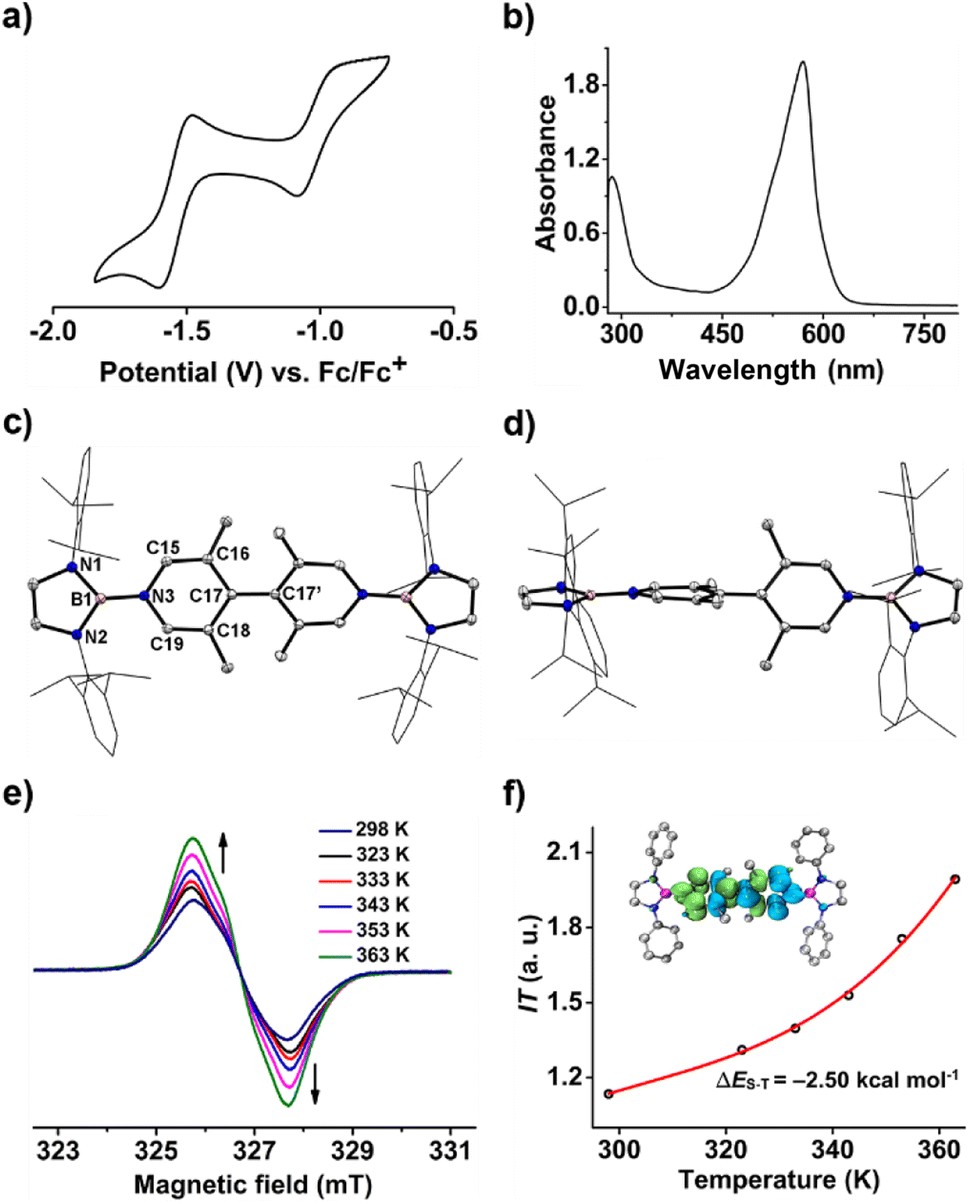 | ||
| Fig. 6 (a) The cyclic voltammogram of 2 in CH2Cl2 (0.1 M [nBu4N][PF6]) measured at a scan rate of 100 mV s−1. (b) The UV-vis spectrum of 2 in toluene. (c) Solid-state structure of 2 with (d) side view. Hydrogen atoms are omitted and Dipp groups are simplified as wireframes for clarity. Thermal ellipsoids are set at the 30% probability level. (e) The variable-temperature EPR spectra of 2 in the solid state. (f) Experimental (black dots) and Bleaney–Bowers fit (red line) of IT–T plots with the calculated spin density distribution map of 2′-OS (inset). | ||
Table 1 shows clear substituent effects on solid-state structures from 1 to 2. For example, in contrast to 1, the exocyclic B–Ni bonds (1.451(2) Å) of 2 are longer than the endocyclic B–Nendo bonds (avg. 1.436(2) Å), illustrating the diminished double bond character in 2 (Fig. 6c). It is noteworthy that bipyridyl rings of 2 are twisted with a dihedral angle of 53.2° (Fig. 6d), which is greatly different from coplanar bipyridyl rings in 1. Concomitantly, the Cp–Cp bond length is elongated from 1.374(2) Å in 1 to 1.417(3) Å in 2, the latter of which is comparable to those (1.398–1.457(7) Å) of analogues of Chichibabin's hydrocarbon with an open-shell singlet ground state,3,4d,4j,6c further supporting the enhanced diradical character.
Similar to 1, compound 2 also displays well-resolved 1H NMR signals at room temperature (Fig. S12†). However, in contrast to 1, obvious NMR spectral broadening was observed for 2 upon heating from 298 K to 333 K (Fig. S13†), which is due to the existence of the thermally populated triplet species (Scheme 3). In addition, the EPR spectrum of 2 at room temperature shows a featureless broad signal centered at a g-value of 2.0018. Variable-temperature EPR measurements of 2 in powder20 demonstrate that the EPR signal intensity increases with increasing temperature (Fig. 6e), which is typical for open-shell singlet diradicaloids.1 Fitting the data by using the Bleaney–Bowers equation gave a singlet-triplet energy gap of −2.50 kcal mol−1 (Fig. 6f), confirming its open-shell singlet ground state.
DFT calculations at the UBH&HLYP/def2-SVP level on 2′, where the Dipp substitutes were replaced with the phenyls, demonstrated that the X-ray data of 2 are between those for 2′-CS and 2′-OS (Table S7†), indicting a singlet ground state with an intermediate diradical character for 2. The calculated ΔES–T (−1.37 kcal mol−1) is consistent with the experiment. The spin density of 2′-OS is distributed through the central bipyridyl rings with the major contribution from two central carbon atoms (Fig. 6f). The diradical character y estimated using the occupancy of the lowest unoccupied natural orbital (LUNO), which represents the degree of the singlet diradical character, is 0.46 calculated at the UBH&HLYP/def2-SVP level. Thus, 2 combines the diradical character of Chichibabin's hydrocarbon and multiple stable redox states of viologens.
Conclusions
In summary, this work reports the bis-BN-based analogues of Chichibabin's hydrocarbon with three stable redox states and tunable ground states. Terminal borylation of 4,4′-bipyridine derivatives by bulky 1,3,2-diazaborolyl ligands allowed both compounds to display two reversible oxidation processes with a wide redox range in electrochemical measurements. One- and two-electron chemical oxidations of 1 gave the crystalline radical cation 1˙+ and dication 12+, respectively. Moreover, modification of the 4,4′-bipyridine with methyl substituents at the meta-carbon positions made the ground-state electronic structures of such neutral species tunable with 1 as a closed-shell singlet and the tetramethyl-substituted 2 as an open-shell singlet with moderate diradical character. EPR and NMR measurements indicated that the excited triplet state of 2 is thermally accessible because of a small singlet-triplet energy gap. This work demonstrates that the strategy of terminal borylation and central distortion of 4,4′-bipyridine may be extended to other bi- or poly-pyridine derivatives to afford novel BN-based radicals with multiple stable redox states and tunable diradical character.Data availability
Detailed experimental procedures and analytical data are available in the ESI.†Author contributions
Xie performed the major synthesis work and properties study. Dai performed some synthetic work and DFT calculations. Bao assisted with the NMR spectra and X-ray single crystallographic diffraction measurements. Liu acquired some funding. Su conceived the concept and prepared the manuscript. All the authors analysed and interpreted the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly acknowledge financial support from the National Natural Science Foundation of China (Grants 22001184, Y. Su and 11904425, C. Liu), Natural Science Foundation of Jiangsu Province (BK20200849), and Entrepreneurship and Innovation Talent Program of Jiangsu Province (Y. Su, and JSSCBS20210664, C. Liu). We thank Dr Liejin Zhou and Dr Yangrong Yao for their help in the VT-NMR characterization and crystallographic refinements, respectively. We also thank the Shanxi Supercomputing Center of China and its Tianhe-2 system for performing calculations.References
- (a) L. Salem and C. Rowland, Angew. Chem., Int. Ed. Engl., 1972, 11, 92–111 CrossRef CAS; (b) A. Rajca, Chem. Rev., 1994, 94, 871–893 CrossRef CAS; (c) F. Breher, Coord. Chem. Rev., 2007, 251, 1007–1043 CrossRef CAS; (d) M. Abe, J. Ye and M. Mishima, Chem. Soc. Rev., 2012, 41, 3808–3820 RSC; (e) J. Casado, R. Ponce Ortiz and J. T. López Navarrete, Chem. Soc. Rev., 2012, 41, 5672–5686 RSC; (f) M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed; (g) Z. Zeng, X. Shi, C. Chi, J. T. López Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC; (h) T. Kubo, Chem. Lett., 2015, 44, 111–122 CrossRef; (i) M. Nakano, Top. Curr. Chem., 2017, 375, 47 CrossRef PubMed; (j) X. Hu, W. Wang, D. Wang and Y. Zheng, J. Mater. Chem. C, 2018, 6, 11232–11242 RSC; (k) T. Stuyver, B. Chen, T. Zeng, P. Geerlings, F. De Proft and R. Hoffmann, Chem. Rev., 2019, 119, 11291–11351 CrossRef CAS PubMed; (l) W. Zeng and J. Wu, Chem, 2021, 7, 358–386 CrossRef CAS; (m) T. Kubo, Bull. Chem. Soc. Jpn., 2021, 94, 2235–2244 CrossRef CAS; (n) Diradicaloids, ed. J. Wu, 1st edn, Jenny Stanford Publishing, 2022 Search PubMed.
- A. E. Tschitschibabin, Ber. Dtsch. Chem. Ges., 1907, 40, 1810–1819 CrossRef CAS.
- L. K. Montgomery, J. C. Huffman, E. A. Jurczak and M. P. Grendze, J. Am. Chem. Soc., 1986, 108, 6004–6011 CrossRef CAS.
- (a) M. Ballester, I. Pascual, C. Carreras and J. Vidal-Gancedo, J. Am. Chem. Soc., 1994, 116, 4205–4210 CrossRef CAS; (b) Z. Zeng, Y. M. Sung, N. Bao, D. Tan, R. Lee, J. L. Zafra, B. S. Lee, M. Ishida, J. Ding, J. T. López Navarrete, Y. Li, W. Zeng, D. Kim, K.-W. Huang, R. D. Webster, J. Casado and J. Wu, J. Am. Chem. Soc., 2012, 134, 14513–14525 CrossRef CAS PubMed; (c) K. Sbargoud, M. Mamada, J. Marrot, S. Tokito, A. Yassar and M. Frigoli, Chem. Sci., 2015, 6, 3402–3409 RSC; (d) J. Wang, X. Xu, H. Phan, T. S. Herng, T. Y. Gopalakrishna, G. Li, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 14154–14158 CrossRef CAS PubMed; (e) D. Rottschäfer, N. K. T. Ho, B. Neumann, H. G. Stammler, M. van Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 5838–5842 CrossRef; (f) D. Rottschäfer, B. Neumann, H.-G. Stammler, D. M. Andrada and R. S. Ghadwal, Chem. Sci., 2018, 9, 4970–4976 RSC; (g) C. Jiang, Y. Bang, X. Wang, X. Lu, Z. Lim, H. Wei, S. El-Hankari, J. Wu and Z. Zeng, Chem. Commun., 2018, 54, 2389–2392 RSC; (h) M. A. Majewski, P. J. Chmielewski, A. Chien, Y. Hong, T. Lis, M. Witwicki, D. Kim, P. M. Zimmerman and M. Stępień, Chem. Sci., 2019, 10, 3413–3420 RSC; (i) Y. Ni, F. Gordillo-Gámez, M. Peña Alvarez, Z. Nan, Z. Li, S. Wu, Y. Han, J. Casado and J. Wu, J. Am. Chem. Soc., 2020, 142, 12730–12742 CrossRef CAS PubMed; (j) A. Maiti, S. Chandra, B. Sarkar and A. Jana, Chem. Sci., 2020, 11, 11827–11833 RSC; (k) A. Maiti, S. Sobottka, S. Chandra, D. Jana, P. Ravat, B. Sarkar and A. Jana, J. Org. Chem., 2021, 86, 16464–16472 CrossRef CAS PubMed; (l) K. Li, Z. Xu, J. Xu, T. Weng, X. Chen, S. Sato, J. Wu and Z. Sun, J. Am. Chem. Soc., 2021, 143, 20419–20430 CrossRef CAS PubMed; (m) T. Nishiuchi, S. Aibara, H. Sato and T. Kubo, J. Am. Chem. Soc., 2022, 144, 7479–7488 CrossRef CAS.
- L. Ji, R. M. Edkins, A. Lorbach, I. Krummenacher, C. Brückner, A. Eichhorn, H. Braunschweig, B. Engles, P. J. Low and T. B. Marder, J. Am. Chem. Soc., 2015, 137, 6750–6753 CrossRef CAS PubMed.
- (a) K. Kamada, S.-i. Fuku-en, S. Minamide, K. Ohta, R. Kishi, M. Nakano, H. Matsuzaki, H. Okamoto, H. Higashikawa, K. Inoue, S. Kojima and Y. Yamamoto, J. Am. Chem. Soc., 2013, 135, 232–241 CrossRef CAS PubMed; (b) X. Zheng, X. Wang, Y. Qiu, Y. Li, C. Zhou, Y. Sui, Y. Li, J. Ma and X. Wang, J. Am. Chem. Soc., 2013, 135, 14912–14915 CrossRef CAS; (c) Y. Su, X. Wang, X. Zheng, Z. Zhang, Y. Song, Y. Sui, Y. Li and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 2857–2861 CrossRef CAS PubMed; (d) X. Wang, Z. Zhang, Y. Song, Y. Su and X. Wang, Chem. Commun., 2015, 51, 11822–11825 RSC; (e) R. Kurata, K. Tanaka and A. Ito, J. Org. Chem., 2016, 81, 137–145 CrossRef CAS PubMed; (f) G. Tan and X. Wang, Acc. Chem. Res., 2017, 50, 1997–2006 CrossRef CAS PubMed; (g) L. Wang, L. Zhang, Y. Fang, Y. Zhao, G. Tan and X. Wang, Chem.–Asian J., 2019, 14, 1708–1711 CrossRef CAS PubMed.
- (a) K. Deuchert and S. Hünig, Angew. Chem., Int. Ed. Engl., 1978, 17, 875–886 CrossRef; (b) T. Nishinaga, Organic Redox Systems: Synthesis, Properties, and Applications, Wiley, 2016 CrossRef; (c) X. Wei, W. Pan, W. Duan, A. Hollas, Z. Yang, B. Li, Z. Nie, J. Liu, D. Reed and W. Wang, ACS Energy Lett., 2017, 2, 2187–2204 CrossRef CAS; (d) P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CrossRef CAS PubMed; (e) S. Lee, J. Hong and K. Kang, Adv. Energy Mater., 2020, 10, 2001445 CrossRef CAS.
- (a) L. Michaelis, Biochem. Z., 1932, 250, 564–567 CAS; (b) L. Michaelis and E. S. Hill, J. Am. Chem. Soc., 1933, 55, 1481–1494 CrossRef CAS.
- Selected recent reviews on viologens: (a) L. Striepe and T. Baumgartner, Chem.–Eur. J., 2017, 23, 16924–16940 CrossRef CAS; (b) K. Madasamy, D. Velayutham, V. Suryanarayanan, M. Kathiresan and K.-C. Ho, J. Mater. Chem. C, 2019, 7, 4622–4637 RSC; (c) K. W. Shah, S.-X. Wang, D. X. Y. Soo and J. Xu, Polymers, 2019, 11, 1839 CrossRef CAS PubMed; (d) M. Kathiresan, B. Ambrose, N. Angulakshmi, D. E. Mathew, D. Sujatha and A. M. Stephan, J. Mater. Chem. A, 2021, 9, 27215–27233 RSC.
- A. N. Woodward, J. M. Kolesar, S. R. Hall, N.-A. Saleh, D. S. Jones and M. G. Walter, J. Am. Chem. Soc., 2017, 139, 8467–8473 CrossRef CAS PubMed.
- B. He, S. Zhang, Y. Zhang, G. Li, B. Zhang, W. Ma, B. Rao, R. Song, L. Zhang, Y. Zhang and G. He, J. Am. Chem. Soc., 2021, 144, 4422–4430 CrossRef PubMed.
- (a) A. Lichtblau, W. Kaim, A. Schulz and T. Stahl, J. Chem. Soc., Perkin Trans. 2, 1992, 1497–1501 RSC; (b) A. Lichtblau, H.-D. Hausen, W. Schwarz and W. Kaim, Inorg. Chem., 1993, 32, 73–78 CrossRef CAS; (c) T. Ohmura, Y. Morimasa and M. Suginome, J. Am. Chem. Soc., 2015, 137, 2852–2855 CrossRef CAS PubMed; (d) D. M. Beagan, V. Carta and K. G. Caulton, Dalton Trans., 2020, 49, 1681–1687 RSC.
- (a) Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS PubMed; (b) Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 CrossRef CAS PubMed; (c) L. Weber, Eur. J. Inorg. Chem., 2017, 3461–3488 CrossRef CAS.
- Y. K. Loh, P. Vasko, C. McManus, A. Heilmann, W. K. Myers and S. Aldridge, Nat. Commun., 2021, 12, 7052 CrossRef CAS PubMed.
- Z. Xie, Y. Dai, M. Bao, Z. Feng, W. Wang, C. Liu, X. Wang and Y. Su, Chem. Commun., 2022, 58, 5391–5394 RSC.
- M. J. Frisch, et al., Gaussion 09, revision D.01, Gaussion, Int., Wallingford, CT, 2013 Search PubMed, see ESI† for geometries, coordinates, and full citation..
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W.-C. Chen, C.-Y. Lee, B.-C. Lin, Y.-C. Hsu, J.-S. Shen, C.-P. Hsu, G. P. A. Yap and T.-G. Ong, J. Am. Chem. Soc., 2014, 136, 914–917 CrossRef CAS PubMed.
- D. Franz, T. Szilvási, A. Pöthig, F. Deiser and S. Inoue, Chem.–Eur. J., 2018, 24, 4283–4288 CrossRef CAS PubMed.
- The VT-EPR spectra of 2 in toluene have been measured at temperature ranging from 293 K to 233 K (Fig. S21†), which also showed higher intensities at higher temperatures, consistent with the conclusion afforded from the solid-state results.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2205846–2205849. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00102d |
| ‡ Y. Dai and Z. Xie contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |