
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ SERS reveals the route regulation mechanism mediated by bimetallic alloy nanocatalysts for the catalytic hydrogenation reaction†
Xiaoxiao
Li‡
,
Jinghua
An‡
 ,
Ze
Gao
,
Chang
Xu
,
Yaoying
Cheng
,
Simin
Li
,
Lu
Li
* and
Bo
Tang
*
,
Ze
Gao
,
Chang
Xu
,
Yaoying
Cheng
,
Simin
Li
,
Lu
Li
* and
Bo
Tang
*
College of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Shandong Normal University, Jinan 250014, P. R. China. E-mail: tangb@sdnu.edu.cn; lilu5252@163.com
First published on 27th February 2023
Abstract
Synthesizing arylamines with high selectivity via hydrogenation of nitroaromatics is a long-standing challenge because of the complex reaction pathways. Revealing the route regulation mechanism is the key to obtain high selectivity of arylamines. However, the underlying reaction mechanism of route regulation is uncertain owing to a lack of direct in situ spectral evidence of the dynamic transformation of intermediate species during the reaction process. In this work, by using in situ surface-enhanced Raman spectroscopy (SERS), we have employed 13 nm Au100−xCux nanoparticles (NPs) deposited on a SERS-active 120 nm Au core to detect and track the dynamic transformation of intermediate species of hydrogenation of para-nitrothiophenol (p-NTP) into para-aminthiophenol (p-ATP). Direct spectroscopic evidence demonstrates that Au100 NPs exhibited a coupling route with the in situ detection of the Raman signal assigned to coupling product p,p′-dimercaptoazobenzene (p,p′-DMAB). However, Au67Cu33 NPs displayed a direct route without the detection of p,p′-DMAB. The combination of X-ray photoelectron spectroscopy (XPS) and density functional theory (DFT) calculations reveals that Cu doping can favor the formation of active Cu–H species owing to the electron transfer from Au to Cu, which can promote the formation of phenylhydroxylamine (PhNHOH*) and favor the occurrence of the direct route on Au67Cu33 NPs. Our study provides direct spectral evidence demonstrating the critical role of Cu in route regulation for the nitroaromatic hydrogenation reaction at a molecular level and clarifies the route regulation mechanism. The results have significant implications for revealing multimetallic alloy nanocatalyst mediated reaction mechanisms and help to guide the rational design of multimetallic alloy catalysts for catalytic hydrogenation reactions.
Introduction
The hydrogenation reactions of nitroaromatic compounds is a typical reaction to prepare arylamines, which are very important industrial intermediates for dyes, medicines agricultural chemicals, and additives.1 However, synthesizing arylamines with high selectivity via hydrogenation reactions is a long-standing challenge because of the complex reaction pathways (Scheme 1).2,3 In the nitroaromatic hydrogenation process, arylamines can be synthesized by the direct route of nitroaromatic → nitrosoaromatic → arylhydroxylamine → arylamines. These metastable reaction intermediates will further undergo the condensation coupling route to form coupling derivatives such as arylazo compounds, which can lower the selectivity of arylamines. Therefore, designing catalysts for highly selective preparation of arylamines and revealing the route regulation mechanism is the key to obtain high selectivity of arylamines. | ||
| Scheme 1 The complex reaction route for the reduction of a nitroaromatic compound to the corresponding aniline. | ||
Gold (Au), which acts as a typical nanocatalyst, has received wide attention and has been regarded as an excellent hydrogenation catalyst owing to its good stability.4 However, the mild catalytic hydrogenation properties of Au nanoparticles (NPs) limit their further applications.5 In this regard, bimetallic alloy NPs have recently been prepared to improve the catalytic hydrogenation performance6–10 by route regulation.11–13 For example, the reaction pathway of hydrogenation reactions of nitroaromatic compounds can be altered by alloying copper into Au NPs under visible-light irradiation.8 However, the underlying reaction mechanism of route regulation is uncertain owing to a lack of direct in situ spectral evidence of the dynamic transformation of intermediate species during the reaction process. Although many techniques are available to study the reaction mechanism, the dynamic transformation of intermediate species during the reaction process is difficult to be tracked in real-time owing to their small adsorption amount and short lifetime.12,14–17 Therefore, a technique with high sensitivity, is needed urgently to in situ study the route regulation mechanism in real-time over bimetallic alloy NPs.
Surface-enhanced Raman spectroscopy (SERS) is a promising tool for in situ detecting surface species owing to its ultrahigh surface single-molecule sensitivity.18–24 During SERS measurement, the Raman signal from surface species can be amplified by using SERS-active Au23,25 or Ag.26,27 By using its high sensitivity, in situ SERS has been used to detect active species in biological systems by research groups including our group.28–35 Recently, it has begun to emerge in the study of the underlying catalytic reaction mechanisms.36–38 For example, by using bifunctional Au@Ni3FeOx structures, the Raman signal from intermediate species (O–O−) during the oxygen evolution reaction on Ni3FeOx NPs can be enhanced and seen because of the SERS enhancement from the Au core.39 Besides, the direct spectral evidence of active intermediate species (*OH and *OOH) in the oxygen reduction reaction was obtained on a metal Pt catalyst by using a Au core as the Raman signal enhancer.40 In addition, with the help of in situ SERS, the hydrogen spillover effect,41 size effect,42,43 as well as crystal effect44 were revealed by using the hydrogenation of para-nitrothiophenol (p-NTP) to para-aminthiophenol (p-ATP) as a model reaction. That said, SERS is a highly surface-sensitive technique and can be employed to in situ track the dynamic transformation of intermediate species on bimetallic alloy NPs.
Herein, with hydrogenation of p-NTP to p-ATP as the model reaction, we used in situ SERS to reveal the route regulation mechanism mediated by Au100−xCux alloy nanocatalysts. Specifically, 13 nm single-component Au100 NPs, as well as bimetallic Au90Cu10 NPs and Au67Cu33 NPs were prepared and deposited on a SERS-active 120 nm Au core covered with a very thin layer of SiO2, forming Au@SiO2@Au100, Au@SiO2@Au90Cu10, and Au@SiO2@Au67Cu33, respectively. Here, the SiO2 shell is essential to prevent the interaction between the 120 nm Au core and analytical targets. Then, the Raman signal of surface intermediate species during hydrogenation of p-NTP to p-ATP on nanocatalysts can be amplified and detected due to the SERS enhancement properties of the 120 nm Au core. Moreover, we studied the changes in the Raman signal of intermediate species by varying the Cu content. As a result, the direct spectral evidence demonstrating that the reaction route of p-NTP to p-ATP regulated from the coupling route on Au100 NPs to the direct route on bimetallic Au67Cu33 NPs, was presented. By combining the in situ spectral evidence, X-ray photoelectron spectroscopy (XPS), and density functional theory (DFT) calculations, the underlying route regulation mechanism mediated by bimetallic Au67Cu33 NPs for the nitroaromatic hydrogenation reaction was further clarified.
Results and discussion
At the beginning, three nanocatalysts, including one single-component Au100 NPs and two bimetallic Au100−xCux alloy NPs with different molar ratios of the Au atom and Cu atom, were prepared according to the literature report.52 Inductively coupled plasma mass spectrometry (ICP-MS) results showed that the molar ratios of Au and Cu for the two bimetallic Au100−xCux NPs are 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 and 67:33, respectively (Table S2†). Thus, Au100 NPs, Au90Cu10 NPs, and Au67Cu33 NPs were successfully obtained. Transmission electron microscopy (TEM) characterization studies further showed that Au100 NPs, Au90Cu10 NPs, and Au67Cu33 NPs possessed almost identical shape and similar size (13 nm) (Fig. 1a and S1a−c†).8 X-ray diffraction (XRD) data of AuCu alloys with different compositions show the typical face-centered cubic crystal phase with a similar diffraction pattern between the standard Au and Cu peaks (Fig. 1b), which is consistent with the reported AuCu alloys in the literature.52 The high-resolution transmission electron microscopy (HRTEM) image of Au67Cu33 and Au90Cu10 shows an interplanar distance of about 0.22 nm, which matches well with the d-spacing value of the metallic AuCu(111) plane (Fig. S2†). Further study of the p-NTP hydrogenation process on Au100 NPs, Au90Cu10 NPs, and Au67Cu33 NPs using in situ SERS is limited due to their less-SERS enhancement properties owing to their small size (Fig. S3†).45 In this regard, SERS-active Au NPs with a diameter of 120 nm were prepared as the SERS enhancer (Fig. S4a and b†).45,46 After that, the Au@SiO2 structure was further prepared by coating 120 nm Au NPs with a thin layer of silica (∼2 nm), which can prevent the interaction between 120 nm Au NPs and analytical targets (Fig. 1c).40,41 Then, Au100 NPs, Au90Cu10 NPs, and Au67Cu33 NPs were deposited on Au@SiO2, forming Au@SiO2@Au100, Au@SiO2@Au90Cu10, and Au@SiO2@Au67Cu33 catalysts, respectively. The UV-vis extinction spectrum results showed that the peak of plasmon resonance was shifted from 604 cm−1 to 625 cm−1 after depositing nanocatalysts on Au@SiO2, indicating the successful decoration of Au@SiO2 with nanocatalysts (Fig. S5†). Fig. 1d shows the TEM characterization and elemental mapping images of a single Au@SiO2@Au67Cu33 catalyst with a 120 nm Au core and many Au67Cu33 NPs as the satellites. Therefore, the Raman signal from surface species on Au100 NPs, Au90Cu10 NPs, or Au67Cu33 NPs can be amplified by using a SERS-active Au core and detected using in situ spectra. These results make sure that the hydrogenation process of p-NTP on Au@SiO2@Au100, Au@SiO2@Au90Cu10, or Au@SiO2@Au67Cu33 catalysts can be investigated by using in situ SERS (Fig. 1e).
:10 and 67:33, respectively (Table S2†). Thus, Au100 NPs, Au90Cu10 NPs, and Au67Cu33 NPs were successfully obtained. Transmission electron microscopy (TEM) characterization studies further showed that Au100 NPs, Au90Cu10 NPs, and Au67Cu33 NPs possessed almost identical shape and similar size (13 nm) (Fig. 1a and S1a−c†).8 X-ray diffraction (XRD) data of AuCu alloys with different compositions show the typical face-centered cubic crystal phase with a similar diffraction pattern between the standard Au and Cu peaks (Fig. 1b), which is consistent with the reported AuCu alloys in the literature.52 The high-resolution transmission electron microscopy (HRTEM) image of Au67Cu33 and Au90Cu10 shows an interplanar distance of about 0.22 nm, which matches well with the d-spacing value of the metallic AuCu(111) plane (Fig. S2†). Further study of the p-NTP hydrogenation process on Au100 NPs, Au90Cu10 NPs, and Au67Cu33 NPs using in situ SERS is limited due to their less-SERS enhancement properties owing to their small size (Fig. S3†).45 In this regard, SERS-active Au NPs with a diameter of 120 nm were prepared as the SERS enhancer (Fig. S4a and b†).45,46 After that, the Au@SiO2 structure was further prepared by coating 120 nm Au NPs with a thin layer of silica (∼2 nm), which can prevent the interaction between 120 nm Au NPs and analytical targets (Fig. 1c).40,41 Then, Au100 NPs, Au90Cu10 NPs, and Au67Cu33 NPs were deposited on Au@SiO2, forming Au@SiO2@Au100, Au@SiO2@Au90Cu10, and Au@SiO2@Au67Cu33 catalysts, respectively. The UV-vis extinction spectrum results showed that the peak of plasmon resonance was shifted from 604 cm−1 to 625 cm−1 after depositing nanocatalysts on Au@SiO2, indicating the successful decoration of Au@SiO2 with nanocatalysts (Fig. S5†). Fig. 1d shows the TEM characterization and elemental mapping images of a single Au@SiO2@Au67Cu33 catalyst with a 120 nm Au core and many Au67Cu33 NPs as the satellites. Therefore, the Raman signal from surface species on Au100 NPs, Au90Cu10 NPs, or Au67Cu33 NPs can be amplified by using a SERS-active Au core and detected using in situ spectra. These results make sure that the hydrogenation process of p-NTP on Au@SiO2@Au100, Au@SiO2@Au90Cu10, or Au@SiO2@Au67Cu33 catalysts can be investigated by using in situ SERS (Fig. 1e).
 | ||
| Fig. 1 TEM images (a) and XRD patterns (b) of Au100 NPs, Au90Cu10 NPs and Au67Cu33 NPs. The scale bar in (a) is 20 nm. (c) TEM image of Au@SiO2. (d) TEM image and element mapping images of a single Au@SiO2@Au67Cu33. (e) Illustration of in situ SERS detection of the p-NTP hydrogenation process over Au@SiO2@Au or Au@SiO2@AuCu catalysts. | ||
First, to track the hydrogenation process of p-NTP on single-component Au100 NPs, in situ SERS was used to track the hydrogenation process on a Au@SiO2@Au100 catalyst with sodium borohydride (NaBH4) as a reduction agent. Before the reaction, the catalyst was immersed in the p-NTP aqueous solution overnight, ensuring the adsorption of the p-NTP molecule at the catalyst surfaces with saturation. Then, the reaction started once the catalyst adsorbing p-NTP came into contact with the NaBH4 aqueous solution. During the reaction process, p-NTP can be reduced by the active hydrogen species (H*), which are formed from the dissociation activation of NaBH4 on the catalyst surface.42 And in situ SERS spectra of the hydrogenation reaction of p-NTP on different catalysts were collected at a certain time interval. First, the hydrogenation process of p-NTP on Au@SiO2 was first studied by in situ SERS and is shown in Fig. 2a. At 0 s, the Raman peaks at 1332 cm−1 and 1570 cm−1, assigned to the NO2-stretching and C–C-stretching band in the benzene ring of substrate p-NTP,41 were observed obviously, indicating the successful adsorption of p-NTP on Au@SiO2. However, these peaks did not show any change during the reaction process even on extending the reaction time to 1800 s. And we did not detect any new Raman peaks, indicating that Au@SiO2 is inert in catalyzing the hydrogenation of p-NTP. Thus, Au@SiO2 acted merely as a platform to enhance the Raman signal of surface species. However, for the Au@SiO2@Au100 catalyst, the intensity of Raman peaks associated with substrate p-NTP at 1332 cm−1 and 1570 cm−1, was decreased gradually as the reaction proceeded from 0 s to 1800 s as seen in the in situ SERS spectra (Fig. 2b). Meanwhile, a new peak centered at 1594 cm−1, which was assigned to the stretching coordinate of the benzene ring of product p-ATP,41,47 was detected and the intensity of this new peak was increased gradually with the reaction time. These results indicate that p-NTP can be hydrogenated to p-ATP on Au@Au100. Besides, in addition to the observation of the peaks associated with substrate p-NTP and product p-ATP, three new peaks located at 1142 cm−1, 1388 cm−1 and 1429 cm−1 were detected in the in situ SERS spectra recorded on Au@SiO2@Au100. These three peaks were assigned to the C–N stretching and N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibrational band of coupling intermediate product p,p′-dimercaptoazobenzene (p,p′-DMAB).41 Interestingly, the intensity of peaks assigned to p,p′-DMAB was first increased and then decreased until it disappeared at 1800 s, meaning p,p′-DMAB was the intermediate species during the hydrogenation of p-NTP to p-ATP on the Au@SiO2@Au100 catalyst. The above results confirmed that p,p′-DMAB was directly involved in the p-NTP hydrogenation process on Au100 NPs, indicating the presence of the coupling route synthesizing p-ATP from p-NTP. The control experiment showed that there was no reaction without NaBH4, indicating that NaBH4 acted as the reduction agent in the hydrogenation of p-NTP (Fig. S6†).
N stretching vibrational band of coupling intermediate product p,p′-dimercaptoazobenzene (p,p′-DMAB).41 Interestingly, the intensity of peaks assigned to p,p′-DMAB was first increased and then decreased until it disappeared at 1800 s, meaning p,p′-DMAB was the intermediate species during the hydrogenation of p-NTP to p-ATP on the Au@SiO2@Au100 catalyst. The above results confirmed that p,p′-DMAB was directly involved in the p-NTP hydrogenation process on Au100 NPs, indicating the presence of the coupling route synthesizing p-ATP from p-NTP. The control experiment showed that there was no reaction without NaBH4, indicating that NaBH4 acted as the reduction agent in the hydrogenation of p-NTP (Fig. S6†).
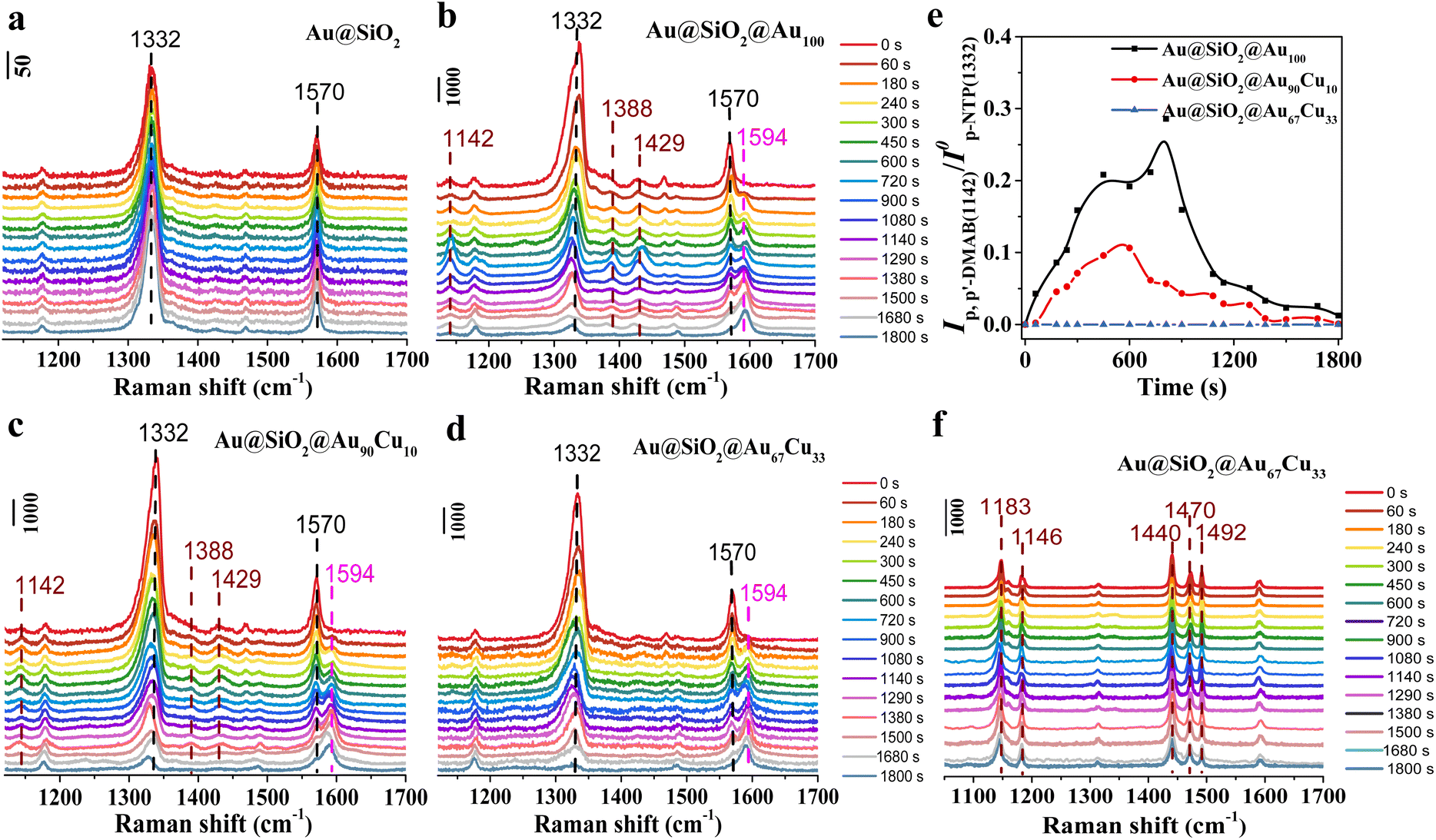 | ||
| Fig. 2 In situ SERS spectra of the hydrogenation reaction of p-NTP in 8.5 × 10−3 M NaBH4 aqueous solution on (a) Au@SiO2, (b) Au@SiO2@Au100, (c) Au@SiO2@Au90Cu10, and (d) Au@SiO2@Au67Cu33. (e) Time-dependent intensity variation curves of the Raman peaks for p,p′-DMAB (at ∼1142 cm−1) on Au@SiO2@Au100, Au@SiO2@Au90Cu10, and Au@SiO2@Au67Cu33. (f) In situ SERS spectra of the hydrogenation reaction of DMAB in 8.5 × 10−3 M NaBH4 aqueous solution on Au@SiO2@Au67Cu33. Note that the SNR of the SERS signal between different catalysts is similar (Table S1†), making sure that the comparison of SERS spectra between different catalysts is valid. | ||
Then, the hydrogenation of p-NTP on Au@SiO2@Au90Cu10 (Fig. 2c) and Au@SiO2@Au67Cu33 (Fig. 2d) catalysts was further studied by in situ SERS with the same measurement conditions as the Au@SiO2@Au100 catalyst. In situ SERS spectra obtained on Au@SiO2@Au90Cu10 were similar to that on Au@SiO2@Au100 except for the observation of the lower intensity of peaks assigned to p,p′-DMAB during the detection time of 1800 s (Fig. 2c). On further increasing the mole percentages of Cu from 10 of Au@SiO2@Au90Cu10 to 33 of Au@SiO2@Au67Cu33, the peaks assigned to p,p′-DMAB were not detected in the in situ SERS spectra recorded on Au@SiO2@Au67Cu33. To compare the discrepancy of chemical behaviors on Au@SiO2@Au100, Au@SiO2@Au90Cu10, and Au@SiO2@Au67Cu33 during the hydrogenation reaction of p-NTP, the initial coverage of p-NTP (Fig. S7†) and time-dependent intensity variation curve of the Raman peaks for p,p′-DMAB (at ∼1142 cm−1) on the three catalysts were plotted (Fig. 2e). Based on the time-dependent coverage variation curves of p-NTP on the three catalysts, we found that the coverage of p-NTP on the three different catalysts decreased when the reaction started. And the coverage of p-NTP on Au@SiO2@Au67Cu33 showed a faster decrease rate, indicating that Cu doping can improve the hydrogenation of p-NTP. The time-dependent intensity variation curve of the Raman peaks for p,p′-DMAB results showed that the peak intensity of p,p′-DMAB decreased in the order of Au@SiO2@Au100 > Au@SiO2@Au90Cu10 > Au@SiO2@Au67Cu33, proposing that the Cu doping can inhibit the formation of coupling product p,p′-DMAB. And p,p′-DMAB might not the reaction intermediate during the hydrogenation of p-NTP to p-ATP over Au@SiO2@Au67Cu33. A control experiment, tracking the hydrogenation of azobenzene (DMAB) on Au@SiO2@Au67Cu33 by in situ SERS, further confirmed this deduction. In the in situ SERS spectra, five peaks located at 1183, 1146, 1440, 1470, and 1492 cm−1, were observed obviously, which are assigned to the C–N and NN stretching modes of DMAB (Fig. 2f). And there was no change for the peaks assigned to DMAB with reaction time,48 confirming that p,p′-DMAB was not the reaction intermediate during the hydrogenation of p-NTP to p-ATP over Au@SiO2@Au67Cu33. These results presented the direct spectral evidence of route regulation from the coupling route on Au100 NPs to the direct route on Au67Cu33 NPs for hydrogenation of p-NTP to p-ATP at a molecular level. That said, the reaction route can be regulated to the direct route owing to a higher Cu concentration in Au67Cu33 NPs.
The regulation of the reaction route from the coupling route to the direct route by alloying Cu in Au may have a great relationship with the change in its electronic structure.49,50 Thus, the XPS characterization studies for Au100 NPs, Au90Cu10 NPs, and Au67Cu33 NPs were conducted and are shown in Fig. 3. Two peaks at binding energies of 84.0 and 87.7 eV were observed for Au100 NPs (Fig. 3a), in accordance with the values for zerovalent Au 4f7/2 and Au 4f5/2 as previously reported.51 However, a slight positive shift of the binding energies of Au 4f7/2 and Au 4f5/2 was observed from the XPS characterization result of Au90Cu10 NPs. With an increase in the Cu concentration, the binding energies of both Au 4f7/2 and Au 4f5/2 for Au67Cu33 NPs were further positively shifted to 84.7 and 88.4 eV, indicating the decrease in the electron density of Au,52 whereas the electron density of Cu increased with the increase in Cu concentration based on the Cu 2p XPS spectra of Au90Cu10 and Au67Cu33 NPs (Fig. 3b). This clearly indicated that electrons were transferred from Au to Cu when these elements form alloy NPs. And a higher Cu concentration in Au100−xCux NPs leads to an increase in the transfer number of electrons from Au to Cu.
 | ||
| Fig. 3 (a) Au 4f XPS spectra of Au100, Au90Cu10 and Au67Cu33 NPs. (b) Cu 2p XPS spectra of Au90Cu10 and Au67Cu33 NPs. | ||
To further understand the critical role of Cu in route regulation from the coupling route on Au100 NPs to the direct route on Au67Cu33 NPs for hydrogenation of p-NTP to p-ATP, density functional theory (DFT) calculations were performed based on Au(111) and AuCu(111). To reduce the complexity of computation, we focused on the hydrogenation process of nitrobenzene (PhNO2) to phenylamine (PhNH2) during DFT calculations. First, the charge distribution of AuCu(111) was analyzed through the charge density difference (Fig. 4a). We found that the charge accumulation is shown as the blue region close to Cu atoms, and the charge depletion is shown as the red region close to Au atoms, meaning that electrons can transfer from Au to Cu. This result is in accordance with the results from XPS characterization studies. The absorption energy of H species, an important active species during hydrogenation of PhNO2, was also calculated on the surface of Au(111) and AuCu(111) here (Fig. 4b). The absorption energy of H species on Au(111) was calculated to be −0.79 eV. However, for AuCu(111), H species is favored to adsorb on the Cu atom with a stronger absorption energy of −1.29 eV. This result proved that it is easier for the active Cu–H species with stronger absorption energy to be formed on AuCu(111). Additionally, the adsorption energies of PhNO2 on AuCu(111) and Au(111) were further calculated, respectively. The results showed that the adsorption energy of PhNO2 on the surface of AuCu(111) (−1.37 eV) was stronger than that of Au(111) (−1.05 eV) (Fig. S8†). The stronger adsorption energy of PhNO2 on AuCu(111) can suppress the formation of coupling products, favoring the formation of PhNH2 with a direct route.8
 | ||
| Fig. 4 (a) Top view of the difference in charge density for AuCu(111), in which blue and red regions indicate electron accumulation and depletion, respectively. (b) Adsorption model of H species on Au(111) and AuCu(111) and their corresponding adsorption energies. Calculated relative energy change for the reaction route on (c) Au(111) and (d) AuCu(111). | ||
To deeply gain insight into the origin of the reaction route regulation by doping Cu in Au for the hydrogenation of nitroaromatics, DFT calculations were further carried out on the reaction steps on Au(111) (Fig. 4c) and AuCu(111) (Fig. 4d), respectively. After being adsorbed on the Au(111) or AuCu(111) surface, *PhNO2 formed PhNOOH* by combining with H*, which then quickly dissociated to form PhNO* and OH*. PhNO* further combined with H* to generate PhNOH*. There are two reaction routes for the further transformation of PhNOH*. (i) PhNOH* can couple with PhNO*, forming PhON = NPh* and facilitating the formation of PhNH2* via the coupling route; (ii) PhNOH* can undergo hydrogenation to form phenylhydroxylamine (PhNHOH*), which favors the occurrence of a direct route to form PhNH2*. However, the Gibbs free energies of the same intermediates on Au(111) and the AuCu(111) surface are different, leading to the existence of different reaction routes between Au(111) and the AuCu(111) surface. For the Au(111) surface, the energy for PhNOH* hydrogenation to PhNHOH* is 0.61 eV, which is higher than that for the coupling reaction between PhNOH* and PhNO* (0.42 eV) (Fig. 4c). This result shows that PhNOH* favors to couple with PhNO*, leading to the appearance of a coupling route at Au(111). For AuCu(111), the formation energy of PhNHOH* (0.10 eV) is much lower than that of coupling intermediate PhON = NPh* (0.30 eV), indicating that AuCu(111) favors the direct route to form PhNH2* by facilitating the formation of PhNHOH* (Fig. 4d). This unambiguously confirms that AuCu(111) can effectively reduce the energy barrier of PhNOH* hydrogenation to PhNHOH*, resulting in the favoring of the occurrence of a direct route on the surface of AuCu(111). In addition, the binding geometries of the reaction intermediates on Au(111) or the AuCu(111) surface, are shown in Fig. S9.†
Based on the results obtained from in situ SERS experiments, XPS characterization studies, and theoretical calculations, the route regulation mechanism mediated by bimetallic Au67Cu33 NPs for the catalytic PhNO2 hydrogenation reaction was proposed. First, NaBH4 is activated on the catalyst surface, forming active hydrogen species. The Cu–H species formed on Au67Cu33 NPs has stronger adsorption energy than Au–H species on Au100 NPs. For Au100 NPs, PhNOH* favors to couple with PhNO*, forming PhON = NPh* and promoting the occurrence of the coupling route. However, the formed active Cu–H species on Au67Cu33 NPs with stronger adsorption energy can promote the direct hydrogenation of PhNOH* to form PhNHOH*, favoring the appearance of a direct route.
Conclusions
In summary, we used in situ SERS to systematically explore the hydrogenation process of p-NTP to p-ATP on Au@SiO2@Au100 as well as bimetallic Au@SiO2@Au90Cu10 and Au@SiO2@Au67Cu33 and obtained the direct spectral evidence of the route regulation mediated by Au67Cu33 NPs. We found that during the hydrogenation process of p-NTP to p-ATP, p,p′-DMAB species was detected and the detected p,p′-DMAB species increased first and then decreased on the Au100 NPs, indicating the presence of the coupling route to from p-ATP. However, p-ATP was the sole product on Au@SiO2@Au67Cu33 NPs, meaning the existence of a direct route during the hydrogenation process of p-NTP to p-ATP. By combining XPS and DFT calculations, we found that Cu doping can favor the formation of active Cu–H species owing to the electron accumulation of Cu, which can promote the formation of PhNHOH* and lead to regulation in the pathway to the direct route on Au67Cu33 NPs. Our study provides direct spectral evidence demonstrating the critical role of Cu in the route regulation for hydrogenation reactions of nitroaromatics on bimetallic Au67Cu33 NPs at a molecular level and clarifies the route regulation mechanism. This result has significant implications for multimetallic alloy nanocatalyst mediated reaction mechanisms and helps to guide the rational design of multimetallic alloy catalysts for catalytic hydrogenation reactions.Data availability
The experimental or computational data associated with this article are placed in the ESI.†Author contributions
The manuscript was written through contributions of all authors. Jinghua An, Lu Li and Bo Tang designed the research, Xiaoxiao Li, Ze Gao and Yaoying Cheng performed the research, Xiaoxiao Li, Ze Gao, Jinghua An, Chang Xu, Simin Li and Lu Li analyzed the data; Xiaoxiao Li, Jinghua An, Lu Li and Bo Tang wrote the paper. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21927811, 22074082, 22002076, and 22106093), the Natural Science Foundation of Shandong Province of China (ZR2019JQ06), the Taishan Scholars Program of Shandong Province (tsqn 201909077), the Local Science and Technology Development Fund Guided by the Central Government Province of China (YDZX20203700002568), and the Key R&D Plan of Shandong Province Grant (2021ZDPT01).Notes and references
- R. S. Downing, P. J. Kunkeler and H. van Bekkum, Catalytic syntheses of aromatic amines, Catal. Today, 1997, 37(2), 121–136 CrossRef CAS.
- P. Serna and A. Corma, Transforming Nano Metal Nonselective Particulates into Chemoselective Catalysts for Hydrogenation of Substituted Nitrobenzenes, ACS Catal., 2015, 5(12), 7114–7121 CrossRef CAS.
- T. Sheng, Y.-J. Qi, X. Lin, P. Hu, S.-G. Sun and W.-F. Lin, Insights into the mechanism of nitrobenzene reduction to aniline over Pt catalyst and the significance of the adsorption of phenyl group on kinetics, Chem. Eng. J., 2016, 293, 337–344 CrossRef CAS.
- X. Liu, H. Q. Li, S. Ye, Y. M. Liu, H. Y. He and Y. Cao, Gold-catalyzed direct hydrogenative coupling of nitroarenes to synthesize aromatic azo compounds, Angew. Chem., Int. Ed., 2014, 53(29), 7624–7628 CrossRef CAS PubMed.
- T. Ishida, T. Murayama, A. Taketoshi and M. Haruta, Importance of Size and Contact Structure of Gold Nanoparticles for the Genesis of Unique Catalytic Processes, Chem. Rev., 2020, 120(2), 464–525 CrossRef CAS PubMed.
- H. L. Jiang, T. Akita, T. Ishida, M. Haruta and Q. Xu, Synergistic catalysis of Au@Ag core-shell nanoparticles stabilized on metal-organic framework, J. Am. Chem. Soc., 2011, 133(5), 1304–1306 CrossRef CAS PubMed.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, Electrochemical CO2 Reduction over Compressively Strained CuAg Surface Alloys with Enhanced Multi-Carbon Oxygenate Selectivity, J. Am. Chem. Soc., 2017, 139(44), 15848–15857 CrossRef CAS PubMed.
- Q. Xiao, S. Sarina, E. R. Waclawik, J. Jia, J. Chang, J. D. Riches, H. Wu, Z. Zheng and H. Zhu, Alloying Gold with Copper Makes for a Highly Selective Visible-Light Photocatalyst for the Reduction of Nitroaromatics to Anilines, ACS Catal., 2016, 6(3), 1744–1753 CrossRef CAS.
- M. Jin, Y. Liu, X. Zhang, J. Wang, S. Zhang, G. Wang, Y. Zhang, H. Yin, H. Zhang and H. Zhao, Selective electrocatalytic hydrogenation of nitrobenzene over copper-platinum alloying catalysts: experimental and theoretical studies, Appl. Catal., B, 2021, 298, 120545 CrossRef CAS.
- F. Tong, X. Liang, F. Ma, X. Bao, Z. Wang, Y. Liu, P. Wang, H. Cheng, Y. Dai, B. Huang and Z. Zheng, Plasmon-Mediated Nitrobenzene Hydrogenation with Formate as the Hydrogen Donor Studied at a Single-Particle Level, ACS Catal., 2021, 11(7), 3801–3809 CrossRef CAS.
- Q. Guan, C. Zhu, Y. Lin, E. I. Vovk, X. Zhou, Y. Yang, H. Yu, L. Cao, H. Wang, X. Zhang, X. Liu, M. Zhang, S. Wei, W.-X. Li and J. Lu, Bimetallic monolayer catalyst breaks the activity–selectivity trade-off on metal particle size for efficient chemoselective hydrogenations, Nat. Catal., 2021, 4(10), 840–849 CrossRef CAS.
- G. Tofighi, X. Yu, H. Lichtenberg, D. E. Doronkin, W. Wang, C. Wöll, Y. Wang and J.-D. Grunwaldt, Chemical Nature of Microfluidically Synthesized AuPd Nanoalloys Supported on TiO2, ACS Catal., 2019, 9(6), 5462–5473 CrossRef CAS.
- S. Dai, T. H. Huang, W. I. Liu, C. W. Hsu, S. W. Lee, T. Y. Chen, Y. C. Wang, J. H. Wang and K. W. Wang, Enhanced CO2 Electrochemical Reduction Performance over Cu@AuCu Catalysts at High Noble Metal Utilization Efficiency, Nano Lett., 2021, 21(21), 9293–9300 CrossRef CAS PubMed.
- D. Combita, P. Concepción and A. Corma, Gold catalysts for the synthesis of aromatic azocompounds from nitroaromatics in one step, J. Catal., 2014, 311, 339–349 CrossRef CAS.
- Y. Z. Chen, Z. U. Wang, H. Wang, J. Lu, S. H. Yu and H. L. Jiang, Singlet Oxygen-Engaged Selective Photo-Oxidation over Pt Nanocrystals/Porphyrinic MOF: The Roles of Photothermal Effect and Pt Electronic State, J. Am. Chem. Soc., 2017, 139(5), 2035–2044 CrossRef CAS PubMed.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, In-situ reconstructed Ru atom array on α-MnO2 with enhanced performance for acidic water oxidation, Nat. Catal., 2021, 4(12), 1012–1023 CrossRef CAS.
- X. Yi, K. Liu, W. Chen, J. Li, S. Xu, C. Li, Y. Xiao, H. Liu, X. Guo, S. B. Liu and A. Zheng, Origin and Structural Characteristics of Tri-coordinated Extra-framework Aluminum Species in Dealuminated Zeolites, J. Am. Chem. Soc., 2018, 140(34), 10764–10774 CrossRef CAS.
- S. Schlucker, Surface-enhanced Raman spectroscopy: concepts and chemical applications, Angew. Chem., Int. Ed., 2014, 53(19), 4756–4795 CrossRef.
- S.-Y. Ding, J. Yi, J.-F. Li, B. Ren, D.-Y. Wu, R. Panneerselvam and Z.-Q. Tian, Nanostructure-based plasmon-enhanced Raman spectroscopy for surface analysis of materials, Nat. Rev. Mater., 2016, 1(6), 16021 CrossRef CAS.
- H. Yin, L.-Q. Zheng, W. Fang, Y.-H. Lai, N. Porenta, G. Goubert, H. Zhang, H.-S. Su, B. Ren, J. O. Richardson, J.-F. Li and R. Zenobi, Nanometre-scale spectroscopic visualization of catalytic sites during a hydrogenation reaction on a Pd/Au bimetallic catalyst, Nat. Catal., 2020, 3(10), 834–842 CrossRef CAS.
- J.-L. Yang, H.-J. Wang, H. Zhang, Z.-Q. Tian and J.-F. Li, Probing Hot Electron Behaviors by Surface-Enhanced Raman Spectroscopy, Cell Rep. Phys. Sci., 2020, 1(9), 100184 CrossRef.
- D. Wang, F. Shi, J. Jose, Y. Hu, C. Zhang, A. Zhu, R. Grzeschik, S. Schlucker and W. Xie, In Situ Monitoring of Palladium-Catalyzed Chemical Reactions by Nanogap-Enhanced Raman Scattering using Single Pd Cube Dimers, J. Am. Chem. Soc., 2022, 144(11), 5003–5009 CrossRef CAS PubMed.
- D. Y. Wei, M. F. Yue, S. N. Qin, S. Zhang, Y. F. Wu, G. Y. Xu, H. Zhang, Z. Q. Tian and J. F. Li, In Situ Raman Observation of Oxygen Activation and Reaction at Platinum-Ceria Interfaces during CO Oxidation, J. Am. Chem. Soc., 2021, 143(38), 15635–15643 CrossRef CAS PubMed.
- H. Ze, X. Chen, X. T. Wang, Y. H. Wang, Q. Q. Chen, J. S. Lin, Y. J. Zhang, X. G. Zhang, Z. Q. Tian and J. F. Li, Molecular Insight of the Critical Role of Ni in Pt-Based Nanocatalysts for Improving the Oxygen Reduction Reaction Probed Using an In Situ SERS Borrowing Strategy, J. Am. Chem. Soc., 2021, 143(3), 1318–1322 CrossRef CAS.
- K. Zhang, L. Yang, Y. Hu, C. Fan, Y. Zhao, L. Bai, Y. Li, F. Shi, J. Liu and W. Xie, Synthesis of a Gold-Metal Oxide Core-Satellite Nanostructure for In Situ SERS Study of CuO-Catalyzed Photooxidation, Angew. Chem., Int. Ed., 2020, 59(41), 18003–18009 CrossRef CAS.
- Y. Li, Y. Hu, F. Shi, H. Li, W. Xie and J. Chen, C-H Arylation on Nickel Nanoparticles Monitored by In Situ Surface-Enhanced Raman Spectroscopy, Angew. Chem., Int. Ed., 2019, 58(27), 9049–9053 CrossRef CAS.
- J. Kelly, R. Patrick, S. Patrick and S. E. J. Bell, Surface-Enhanced Raman Spectroscopy for the Detection of a Metabolic Product in the Headspace Above Live Bacterial Cultures, Angew. Chem., Int. Ed., 2018, 57(48), 15686–15690 CrossRef CAS.
- X. Li, X. Duan, P. Yang, L. Li and B. Tang, Accurate In Situ Monitoring of Mitochondrial H2O2 by Robust SERS Nanoprobes with a Au-Se Interface, Anal. Chem., 2021, 93(8), 4059–4065 CrossRef CAS.
- X. Li, X. Duan, L. Li, S. Ye and B. Tang, An accurate and ultrasensitive SERS sensor with Au–Se interface for bioimaging and in situ quantitation, Chem. Commun., 2020, 56(65), 9320–9323 RSC.
- S. Ye, X. Li, M. Wang and B. Tang, Fluorescence and SERS Imaging for the Simultaneous Absolute Quantification of Multiple miRNAs in Living Cells, Anal. Chem., 2017, 89(9), 5124–5130 CrossRef CAS PubMed.
- S. Lin, H. Ze, X. G. Zhang, Y. J. Zhang, J. Song, H. Zhang, H. L. Zhong, Z. L. Yang, C. Yang, J. F. Li and Z. Zhu, Direct and Simultaneous Identification of Multiple Mitochondrial Reactive Oxygen Species in Living Cells Using a SERS Borrowing Strategy, Angew. Chem., Int. Ed., 2022, 202203511 Search PubMed.
- Q. Li, X. Ge, J. Ye, Z. Li, L. Su, Y. Wu, H. Yang and J. Song, Dual Ratiometric SERS and Photoacoustic Core-Satellite Nanoprobe for Quantitatively Visualizing Hydrogen Peroxide in Inflammation and Cancer, Angew. Chem., Int. Ed., 2021, 60(13), 7323–7332 CrossRef CAS PubMed.
- Z. Liu, S. Li, Z. Yin, Z. Zhu, L. Chen, W. Tan and Z. Chen, Stabilizing Enzymes in Plasmonic Silk Film for Synergistic Therapy of In Situ SERS Identified Bacteria, Adv. Sci., 2022, 9(6), 2104576 CrossRef CAS PubMed.
- J. Liu, Z. Liu, W. Wang and Y. Tian, Real-time Tracking and Sensing of Cu+ and Cu2+ with a Single SERS Probe in the Live Brain: Toward Understanding Why Copper Ions Were Increased upon Ischemia, Angew. Chem., Int. Ed., 2021, 60(39), 21351–21359 CrossRef CAS PubMed.
- C. Zong, M. Xu, L. J. Xu, T. Wei, X. Ma, X. S. Zheng, R. Hu and B. Ren, Surface-Enhanced Raman Spectroscopy for Bioanalysis: Reliability and Challenges, Chem. Rev., 2018, 118(10), 4946–4980 CrossRef CAS PubMed.
- J. Chen, G. Liu, Y. Z. Zhu, M. Su, P. Yin, X. J. Wu, Q. Lu, C. Tan, M. Zhao, Z. Liu, W. Yang, H. Li, G. H. Nam, L. Zhang, Z. Chen, X. Huang, P. M. Radjenovic, W. Huang, Z. Q. Tian, J. F. Li and H. Zhang, Ag@MoS2 Core-Shell Heterostructure as SERS Platform to Reveal the Hydrogen Evolution Active Sites of Single-Layer MoS2, J. Am. Chem. Soc., 2020, 142(15), 7161–7167 CrossRef CAS PubMed.
- H. Zhang, S. Duan, P. M. Radjenovic, Z. Q. Tian and J. F. Li, Core-Shell Nanostructure-Enhanced Raman Spectroscopy for Surface Catalysis, Acc. Chem. Res., 2020, 53(4), 729–739 CrossRef CAS PubMed.
- C. Zhan, X. J. Chen, Y. F. Huang, D. Y. Wu and Z. Q. Tian, Plasmon-Mediated Chemical Reactions on Nanostructures Unveiled by Surface-Enhanced Raman Spectroscopy, Acc. Chem. Res., 2019, 52(10), 2784–2792 CrossRef CAS PubMed.
- C. Hu, Y. Hu, C. Fan, L. Yang, Y. Zhang, H. Li and W. Xie, Surface-Enhanced Raman Spectroscopic Evidence of Key Intermediate Species and Role of NiFe Dual-Catalytic Center in Water Oxidation, Angew. Chem., Int. Ed., 2021, 60(36), 19774–19778 CrossRef CAS PubMed.
- J.-C. Dong, X.-G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z.-L. Yang, D.-Y. Wu, J. M. Feliu, C. T. Williams, Z.-Q. Tian and J.-F. Li, In situ Raman spectroscopic evidence for oxygen reduction reaction intermediates at platinum single-crystal surfaces, Nat. Energy, 2018, 4(1), 60–67 CrossRef.
- H. Zhang, X. G. Zhang, J. Wei, C. Wang, S. Chen, H. L. Sun, Y. H. Wang, B. H. Chen, Z. L. Yang, D. Y. Wu, J. F. Li and Z. Q. Tian, Revealing the Role of Interfacial Properties on Catalytic Behaviors by in Situ Surface-Enhanced Raman Spectroscopy, J. Am. Chem. Soc., 2017, 139(30), 10339–10346 CrossRef CAS PubMed.
- K. Zhang, Y. Liu, Y. Wang, J. Zhao and B. Liu, Direct SERS tracking of a chemical reaction at a single 13 nm gold nanoparticle, Chem. Sci., 2019, 10(6), 1741–1745 RSC.
- J. Wei, S. N. Qin, J. Yang, H. L. Ya, W. H. Huang, H. Zhang, B. J. Hwang, Z. Q. Tian and J. F. Li, Probing Single-Atom Catalysts and Catalytic Reaction Processes by Shell-Isolated Nanoparticle-Enhanced Raman Spectroscopy, Angew. Chem., Int. Ed., 2021, 60(17), 9306–9310 CrossRef CAS PubMed.
- J. Huang, W. Niu, C. Li, C. Tan, P. Yin, H. Cheng, Z. Hu, N. Yang, Q. He, G.-H. Nam and H. Zhang, In-Situ Probing of Crystal-Phase-Dependent Photocatalytic Activities of Au Nanostructures by Surface-Enhanced Raman Spectroscopy, ACS Mater. Lett., 2020, 2(4), 409–414 CrossRef CAS.
- H. Q. Chen, H. Ze, M. F. Yue, D. Y. Wei, Y. L. A, Y. F. Wu, J. C. Dong, Y. J. Zhang, H. Zhang, Z. Q. Tian and J. F. Li, Unmasking the Critical Role of the Ordering Degree of Bimetallic Nanocatalysts on Oxygen Reduction Reaction by In Situ Raman Spectroscopy, Angew. Chem., Int. Ed., 2022, 61(16), e202117834 CAS.
- N. G. Bastus, J. Comenge and V. Puntes, Kinetically controlled seeded growth synthesis of citrate-stabilized gold nanoparticles of up to 200 nm: size focusing versus Ostwald ripening, Langmuir, 2011, 27(17), 11098–11105 CrossRef CAS PubMed.
- W. Xie, B. Walkenfort and S. Schlucker, Label-free SERS monitoring of chemical reactions catalyzed by small gold nanoparticles using 3D plasmonic superstructures, J. Am. Chem. Soc., 2013, 135(5), 1657–1660 CrossRef CAS PubMed.
- L. Zhou, H. Fu, Z. Wang, L. Chen, G. Ren, T. Jiang, C. Gu, L. Liu, W. Zhang, W. Zhang, J. Zhou and J. Han, Isomerization behavior of p-aminoazobenzene directly anchored on MoS2/graphene oxide nanocomposite, Appl. Surf. Sci., 2020, 530, 147216 CrossRef CAS.
- W. Shi, B. Zhang, Y. Lin, Q. Wang, Q. Zhang and D. S. Su, Enhanced Chemoselective Hydrogenation through Tuning the Interaction between Pt Nanoparticles and Carbon Supports: Insights from Identical Location Transmission Electron Microscopy and X-ray Photoelectron Spectroscopy, ACS Catal., 2016, 6(11), 7844–7854 CrossRef CAS.
- G. Chen, C. Xu, X. Huang, J. Ye, L. Gu, G. Li, Z. Tang, B. Wu, H. Yang, Z. Zhao, Z. Zhou, G. Fu and N. Zheng, Interfacial electronic effects control the reaction selectivity of platinum catalysts, Nat. Mater., 2016, 15(5), 564–569 CrossRef CAS PubMed.
- H. Zhu, X. Ke, X. Yang, S. Sarina and H. Liu, Reduction of nitroaromatic compounds on supported gold nanoparticles by visible and ultraviolet light, Angew. Chem., Int. Ed., 2010, 49(50), 9657–9661 CrossRef CAS PubMed.
- X. Zhou, Q. Shen, K. Yuan, W. Yang, Q. Chen, Z. Geng, J. Zhang, X. Shao, W. Chen, G. Xu, X. Yang and K. Wu, Unraveling Charge State of Supported Au Single-Atoms during CO Oxidation, J. Am. Chem. Soc., 2018, 140(2), 554–557 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06808g |
| ‡ Xiaoxiao Li and Jinghua An contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |