
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanical stability of cis, trans-poly(p-phenylene vinylenes)†
Yurachat
Janpatompong‡
,
Kamil
Suwada‡
,
Michael L.
Turner
 * and
Guillaume
De Bo
*
* and
Guillaume
De Bo
*
Department of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: michael.turner@manchester.ac.uk; guillaume.debo@manchester.ac.uk
First published on 29th March 2023
Abstract
Understanding the stability of conjugated polymers towards mechanical stimuli is critical for optimizing the processing and use of these materials in a range of electronic and optoelectronic devices, such as transistors, light emitting diodes and solar cells. The fast-growing field of mechanochemistry aims to tame the destructive mechanical forces to perform specific chemical changes in mechanophores but it can also be used to probe the stability of conjugated polymers towards destructive mechanical forces. Using ultrasonication it is possible to benchmark the mechanical integrity of phenylenevinylene block copolymers and show, through experimental studies and DFT simulation, that conjugated olefins are highly stable to cis–trans isomerisation upon extensive elongation, a useful property for the development of flexible organic electronics.
Introduction
π-Conjugated polymers have been processed from solution into the active layer of electronic devices such as organic light emitting diodes (OLEDs),1–4 organic photovoltaics (OPVs)5–8 and organic field-effect transistors (OFETs).9–13 The performance of these devices is greatly influenced by the morphology of the active layer, and this is strongly influenced by the kinetics of thin film formation during device fabrication.6,14,15 In addition, there is an increasing focus on printing or roll-to-roll processing of these materials to fabricate flexible light-weight devices over large areas.16 Rod-coil block copolymers are of interest due to the possible long range order of the self-assembled thin film morphologies.17–20 These significantly influence the optoelectronic properties of the conjugated polymer blocks and can dramatically affect the polymer solubility, crystallization, morphology, aggregation and self-assembly.21,22Many modern consumer electronics are designed to be conformable, bent or flexed in use. This means that the individual components (i.e. transistors, diodes, resistors, capacitors) need to be mechanically robust to extension and flexing and the performance of these devices must be stable under repeated mechanical deformation.23 Semiconducting polymers often do not exhibit the same mechanical strength and toughness of traditional engineering polymers as they have rigid planar backbones that are ordered in the solid state but are surrounded by long, flexible side chains to enhance solubility, reducing the cohesive forces within thin films of these materials.24 Although the thickness of the active semiconducting layer of most flexible electronic devices is more than an order of magnitude less than that of other layers, mechanical change or damage within these layers leads to dramatic changes in device performance.25 An understanding of the destructive nature of mechanical energy on the chemical structure of conjugated polymers will help in the design of materials for mechanically robust, functional devices.
Application of elongational force to organic molecules embedded in the polymer backbone can be achieved in solution using high intensity ultrasound (as solvodynamic shear is generated in the surrounding of collapsing cavitation bubbles).26,27 This technique has been used to activate a variety of mechanophores,28 sometimes along unusual reaction pathways,29–31 prepare functional materials,32 carry out in situ catalysis,33,34 or induce the release of small molecules,35 amongst others. Here we investigate the mechanical strength of cis, trans-poly(p-phenylene vinylenes) (PPVs) under tensional force. The mechanical activation was performed in solution using high-intensity ultrasound. We anticipated the possible cis to trans isomerisation of the olefins upon elongation, a phenomenon previously observed by AFM,36,37 but we found that the structural integrity (notably the stereochemistry of the double bonds) and the optical properties are conserved upon mechanical activation. The origin of PPV mechanical strength was investigated computationally by DFT using the Constrained Geometry Simulates External Force (CoGEF) technique.28,38
Results and discussion
Mechanical activation of polymers by ultrasonication requires that the polymer molecular weight is above a limiting value (Mlim) above which mechanochemical coupling is effective. In this work triblock copolymers were prepared with a central cis, trans PPV segment for mechanical activation linked to long PMMA chains to ensure that the molecular weight of the chains exceeded the Mlim to allow for mechanochemical coupling. This is known to be more than 30 kDa for PMMA.39 The preparation of the cis, trans PPV block was achieved via ROMP of substituted paracyclophanediene monomers 1 and 2 (Scheme 1).21 The target length for the PPV block was 10 monomeric units and it was capped at both ends by an α-bromoester functional group. The macroinitiators 5 and 6 were used to grow PMMA blocks from both ends using ATRP. The ROMP process gives alternating cis, trans stereochemistry polymers due to the ring opening of only one vinylene of the cyclophanediene monomer (1, 2) and the choice of cyclophanediene (1 or 2) enables the optoelectronic properties of these triblock copolymers to be modified.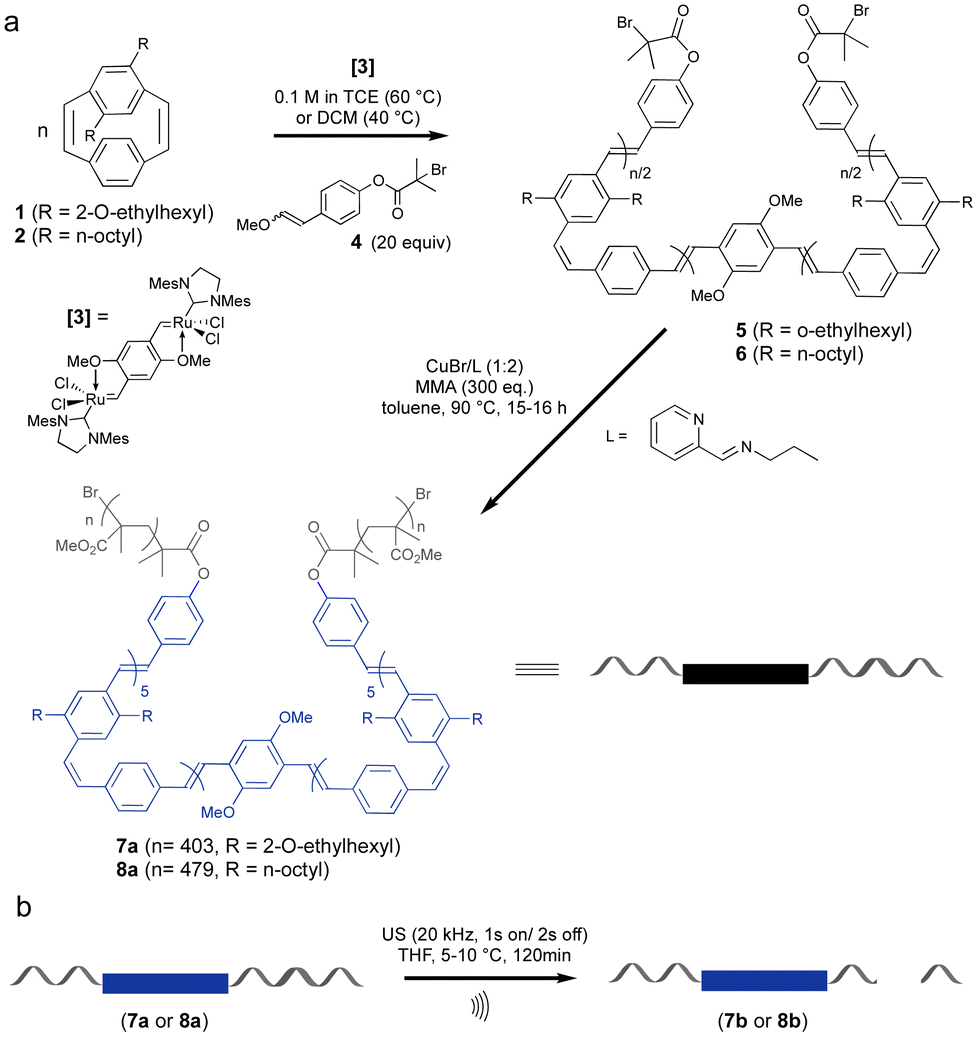 | ||
| Scheme 1 Synthesis (a) and mechanical activation (b) of coil–rod–coil ABA phenylene-vinylene triblock copolymers 7a and 8a. | ||
Incorporation of the α-bromoester end group in 5 and 6 was achieved by quenching the living ROMP of cyclophanediene 1 and 2 with 20 eq. of vinyl ether E/Z-4 (f = 96% and 76%) (Scheme 1). Polymers 5 and 6 were isolated as yellow solids after purification, in yields of 95 and 97%, respectively. The presence of the α-bromoester end groups in polymers 5 and 6 enable these polymers to be used as macroinitiators in ATRP. The PMMA–PPV–PMMA triblock copolymers 7a and 8a were prepared by copper-catalyzed ATRP of MMA at 90 °C.21,40 High molecular weight PMMA segments (Scheme 1) were obtained after heating for 16 and 15 hours, respectively. The polymerization was terminated by exposure of the reaction to air and the polymeric products were isolated by precipitation into methanol, followed by reprecipitation into diethyl ether from chloroform.
The molecular weights of 7a and 8a were determined by size exclusion chromatography (Table 1) in THF, calibrated using polystyrene standards. The number average molar masses (Mn) determined by SEC were 64.3 (Đ = 1.40) and 79.3 kDa (Đ = 1.41) for 7a and 8a, respectively. The 1H NMR spectra of triblock copolymers 7a and 8a are shown in Fig. S5–8.† Resonances relating to PPV and PMMA segments are apparent, with the key signals assigned for each block. The resonances below 2.00 ppm are associated with the 2-(R/S) ethylhexyl and alkyl side chain and the C–CH3 and CH2 of the PMMA segment. An approximate degree of polymerization for the PMMA segment was calculated by integration of the OMe group of MMA (δ = 3.17–3.98 ppm) against the combined phenyl and vinylene protons for 7a (δ = 6.40–7.53 ppm) and methylene protons for 8a (δ = 2.16–2.86 ppm) respectively. The degree of polymerization of the PMMA segment determined by 1H NMR spectroscopy allowed for an absolute Mn value to be calculated (see Table 1). The discrepancy between the apparent Mn from SEC and the absolute Mn calculated from 1H NMR spectroscopy is due to the difference in hydrodynamic volume between the triblock copolymer and the PS standards.41
| x n (PPV)a | x n (PMMA)b | M n (kg mol−1) | Đ | M n (kg mol−1) | λ max (nm) | λ em (nm) | |
|---|---|---|---|---|---|---|---|
| a From [5]/[catalyst 3] or [6]/[catalyst 3]. b Determined by 1H NMR spectroscopy. c Determined by SEC with RI detection (calibrated against narrow Đ PS standards). | |||||||
| 5 | — | — | 5.86 | 1.24 | 5.19 | 451 | 527 |
| 6 | — | — | 6.24 | 1.32 | 4.72 | 382 | 495 |
| 7a | 10 | 806 | 64.31 | 1.40 | 85.85 | 452 | 528 |
| 7b | 10 | 806 | 25.08 | 1.42 | — | 452 | 528 |
| 8a | 10 | 958 | 79.30 | 1.41 | 100.55 | 374 | 493 |
| 8b | 10 | 960 | 25.69 | 1.34 | — | 374 | 493 |
Mechanochemical activation of the polymers 7a and 8a was performed by ultrasonication (20 kHz, 15.6 W cm−2, 1 s on/2 s off) of dilute (∼2 mg mL−1) polymer solutions in the dark, while maintaining the temperature between 5 and 10 °C. The complete cleavage of both polymers was observed after 120 min of sonication (Fig. 1a, Mn = 25.1 and 25.7 kDa for 7b and 8b respectively). This transformation was indicative of the destructive mechanical forces acting on the triblock copolymer backbone, resulting in covalent bond scissions in the PMMA blocks. Despite the broad dispersity of polymers 7a and 8a (Đ ∼1.4), the PPV block should be placed in the central region of the block copolymer in most of the chains. In any case, as force is distributed along the backbone (with its highest intensity in the centre), it is possible to activate a mechanophore that is located off-centre,42,43 especially for a low-force process such as olefin isomerisation (which typically occur ∼1 nN).36,37
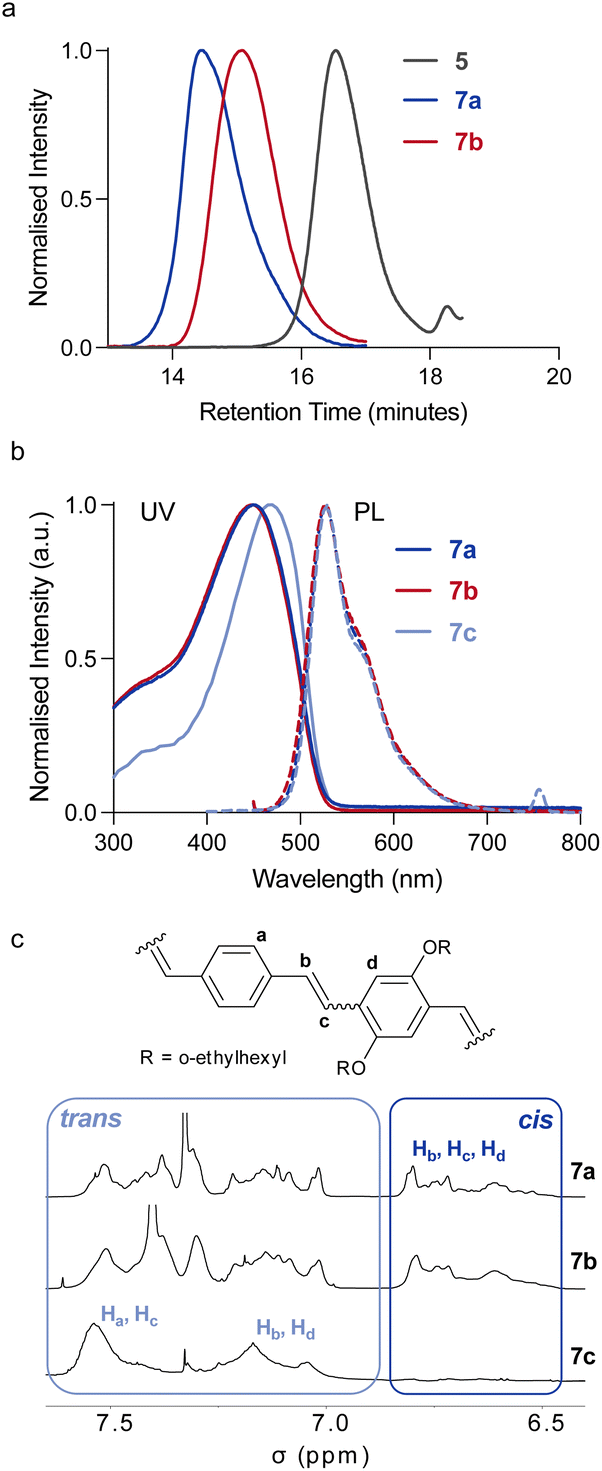 | ||
| Fig. 1 SEC traces (a), UV-PL spectra (b), and partial 1H NMR spectra (500 MHz, CD2Cl2) (c) of pre- and post-sonication polymers 7a and 7b respectively, along with all-trans reference polymer 7c. Spectra aligned on the –OCH3 peak of PMMA. | ||
The 1H NMR spectra of 7a,b and 8a,b pre- and post-sonication showed no substantional changes in the olefin signals (Fig. 1c and Fig. S5–8†), indicating that no cis, trans isomerisation occurred in the PPV block during sonication. This was also confirmed by the absorption and emission spectra of rod–coil triblock copolymer 7 (Fig. 1b), which show no difference between pre- and post-sonication samples (7a and 7b). The UV-vis spectrum exhibits a λmax of 452 nm, with a PLmax at 528 nm while polymer 8 shows a λmax of 374 nm and a PLmax at 493 which is blue shifted from 7 (Table 1).
As the mechanical elongation led to polymer backbone degradation before chemical changes could occur at the PPV block, the scission very likely occurred within the PMMA backbone. To gain better understanding of the PPV moiety under stress, calculations were performed on models of polymers 7a (Fig. 2 and 3) and 8a (Fig. S1†), which were constructed from the initiator and one ROMP monomer unit on each side, capped by pivalate moiety simulating the PMMA attachment point (7′ and 8′, respectively). The elongation of these models was simulated by incrementally increasing the distance between the anchor atoms and optimizing the geometry at each step (CoGEF, DFT B3LYP 6-31G+ (d′, 3p′) level of theory in Gaussian 16). Both models experience a high degree of deformation upon simulated elongation before the scission of a terminal bond is observed (Fig. 2c), which is indicative of a non-selective scission within PMMA block. Both 7′ and 8′ show a similar Fmax value of 5.5 nN (Fig. 2c and S1†), while CoGEF simulation of a PPV segment derived from 7′ (i.e. without pivalate groups, see ESI†) leads to the scission of the C–C bond separating the cis-olefin from the substituent-free ring at a considerably higher force of 6.9 nN, which is beyond the force required to cleave the PMMA backbone (see ESI†).
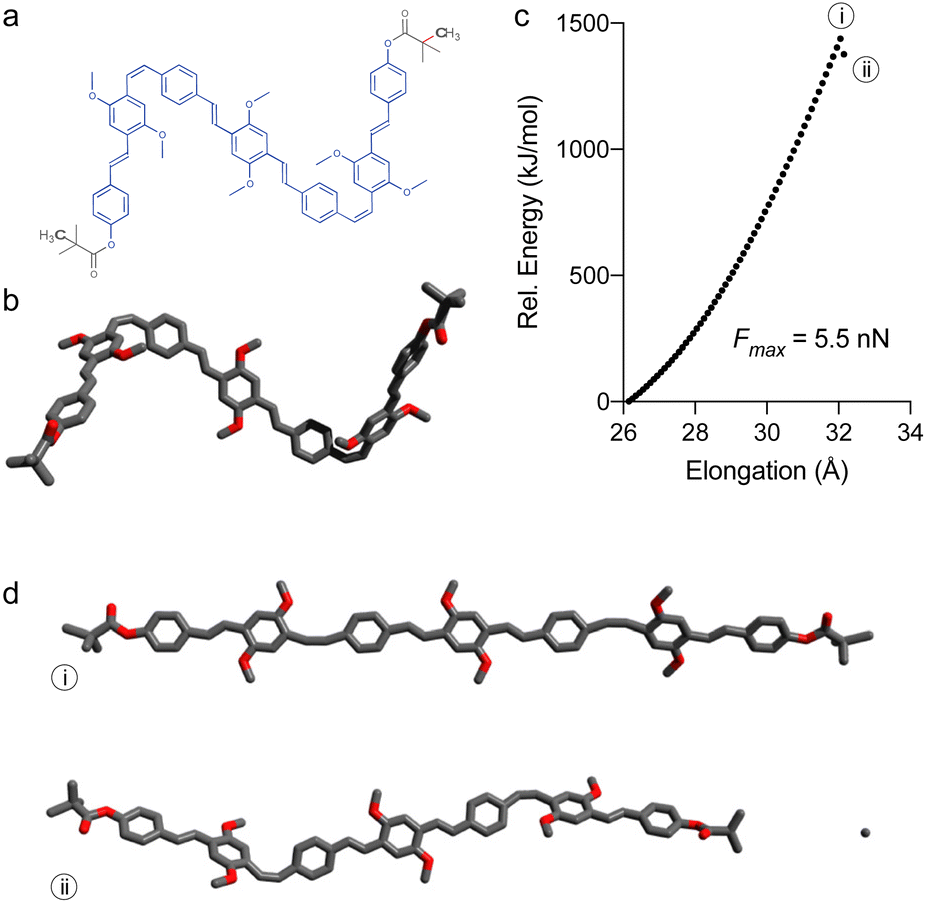 | ||
| Fig. 2 Structure of 7′ used for CoGEF simulations with constrained atoms marked in bold and scissile bond highlighted in red (a), along with its energy minimized structure (b). The evolution of energy upon simulated elongation (CoGEF, DFT B3LYP/6-31G*) of this model (c) reveals the most elongated (Emax) and post-scission structures (d). | ||
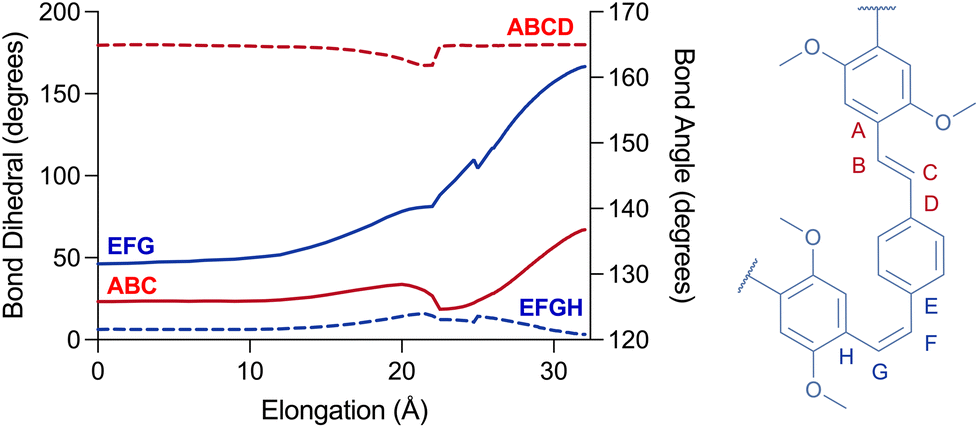 | ||
| Fig. 3 Changes in selected angles (plain) and dihedrals (dashed) upon simulated elongation of 7′. | ||
Remarkably, no isomerisation takes place during the elongation (Fig. 3). Instead, the simulated force led to the distortion of trans-olefins from 129° up to 140° (angle ABC), while the cis-olefins have bent from 131° to nearly 162° (angle EFG) at Emax (i, Fig. 2c and d) for 7′. Moreover, the ring-adjacent bonds (e.g. AB) elongated from ∼1.46 Å to as high as 1.65 Å, while the vinylene bonds (e.g. BC) elongated from ∼1.35 Å up to 1.44 Å. Unsurprisingly, the cis-olefins experienced a higher level of distortion than the trans-isomers. The rings themselves experience a lower deformation as cross-ring distance D–E elongates from 2.86 Å up to 3.09 Å for example. Taken together these observations explain why the PPV polymers do not isomerise during stretching, as the elongational force applied during sonication does not provide the torsional momentum (i.e. the opening of dihedral angle EFGH) required to flip the olefins from Z to E stereochemistry.
Conclusions
The mechanical stability of cis, trans-PPVs was investigated in solution by sonicating PMMA–PPV–PMMA triblock copolymers. The mechanical stress induced by the sonication led to the scission of the PMMA blocks instead of isomerisation or scission of the central conjugated fragment. Despite the effectively higher elongational forces acting on the conjugated segment of the triblock chain, this PPV block maintained its initial photophysical properties as no bond scission or isomerisation occurred. Computational simulation of the elongation process using the CoGEF technique reveals that the elongational force applied during sonication did not provide the torsional momentum required for the isomerisation of olefins. The results bode well for the development of flexible organic electronics utilising PPVs and using mechanochemistry to benchmark stability of conjugated polymers in general.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Development and Promotion of Science and Technology Talents Project (DPST), Thailand for the financial support to Y. J. and the EPSRC for a studentship to K. S. and funding of the NMR spectrometers under grant EP/K039547/1. G. D. B. is a Royal Society University Research Fellow.References
- M. Gross, D. C. Müller, H.-G. Nothofer, U. Scherf, D. Neher, C. Bräuchle and K. Meerholz, Nature, 2000, 405, 661–665 CrossRef CAS PubMed.
- Y. Liu, S. Yan and Z. Ren, Chem. Eng. J., 2021, 417, 128089 CrossRef CAS.
- W. Lu, J. Kuwabara, T. Iijima, H. Higashimura, H. Hayashi and T. Kanbara, Macromolecules, 2012, 45, 4128–4133 CrossRef CAS.
- R. M. Pankow and B. C. Thompson, Polymer, 2020, 207, 122874 CrossRef CAS.
- X. Zhan and D. Zhu, Polym. Chem., 2010, 1, 409–419 RSC.
- B. Zheng, L. Huo and Y. Li, NPG Asia Mater., 2020, 12, 3 CrossRef CAS.
- Y. Xu, Y. Cui, H. Yao, T. Zhang, J. Zhang, L. Ma, J. Wang, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2101090 CrossRef CAS PubMed.
- J.-H. Kim, S. Wood, J. B. Park, J. Wade, M. Song, S. C. Yoon, I. H. Jung, J.-S. Kim and D.-H. Hwang, Adv. Funct. Mater., 2016, 26, 1517–1525 CrossRef CAS.
- J. Yang, Z. Zhao, S. Wang, Y. Guo and Y. Liu, Chem, 2018, 4, 2748–2785 CAS.
- S. Nagasawa, E. Al-Naamani and A. Saeki, J. Phys. Chem. Lett., 2018, 9, 2639–2646 CrossRef CAS PubMed.
- S. Choi, J. W. Jeong, G. Jo, B. C. Ma and M. Chang, Nanoscale, 2019, 11, 10004–10016 RSC.
- M. Pandey, N. Kumari, S. Nagamatsu and S. S. Pandey, J. Mater. Chem. C, 2019, 7, 13323–13351 RSC.
- R. Zhao, Y. Min, C. Dou, B. Lin, W. Ma, J. Liu and L. Wang, ACS Appl. Polym. Mater., 2020, 2, 19–25 CrossRef CAS.
- K. Weng, L. Ye, L. Zhu, J. Xu, J. Zhou, X. Feng, G. Lu, S. Tan, F. Liu and Y. Sun, Nat. Commun., 2020, 11, 2855 CrossRef CAS PubMed.
- M. D. M. Faure and B. H. Lessard, J. Mater. Chem. C, 2021, 9, 14–40 RSC.
- K. Liu, B. Ouyang, X. Guo, Y. Guo and Y. Liu, npj Flexible Electron., 2022, 6, 1–19 CrossRef CAS.
- M. He, F. Qiu and Z. Lin, J. Mater. Chem., 2011, 21, 17039–17048 RSC.
- Y. Tao, B. Ma and R. A. Segalman, Macromolecules, 2008, 41, 7152–7159 CrossRef CAS.
- P. D. Topham, A. J. Parnell and R. C. Hiorns, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1131–1156 CrossRef CAS.
- Y.-C. Chiu, C.-C. Shih and W.-C. Chen, J. Mater. Chem. C, 2015, 3, 551–558 RSC.
- V. Komanduri, D. R. Kumar, D. J. Tate, R. Marcial-Hernandez, B. J. Lidster and M. L. Turner, Polym. Chem., 2019, 10, 3497–3502 RSC.
- D.-H. Jiang, B. J. Ree, T. Isono, X.-C. Xia, L.-C. Hsu, S. Kobayashi, K. H. Ngoi, W.-C. Chen, C.-C. Jao, L. Veeramuthu, T. Satoh, S. H. Tung and C.-C. Kuo, Chem. Eng. J., 2021, 418, 129421 CrossRef CAS.
- J. P. Cachaneski-Lopes and A. Batagin-Neto, Polymers, 2022, 14, 1354 CrossRef CAS PubMed.
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- M. Brinkmann, L. Hartmann, L. Biniek, K. Tremel and N. Kayunkid, Macromol. Rapid Commun., 2014, 35, 9–26 CrossRef CAS PubMed.
- G. De Bo, Macromolecules, 2020, 53, 7615–7617 CrossRef.
- P. A. May and J. S. Moore, Chem. Soc. Rev., 2013, 42, 7497–7506 RSC.
- I. M. Klein, C. C. Husic, D. P. Kovács, N. J. Choquette and M. J. Robb, J. Am. Chem. Soc., 2020, 142, 16364–16381 CrossRef CAS PubMed.
- R. Nixon and G. De Bo, Nat. Chem., 2020, 12, 826–831 CrossRef CAS PubMed.
- Y. Liu, S. Holm, J. Meisner, Y. Jia, Q. Wu, T. J. Woods, T. J. Martinez and J. S. Moore, Science, 2021, 373, 208–212 CrossRef CAS PubMed.
- J. M. Lenhardt, M. T. Ong, R. Choe, C. R. Evenhuis, T. J. Martinez and S. L. Craig, Science, 2010, 329, 1057–1060 CrossRef CAS PubMed.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 2017, 357, 475–479 CrossRef CAS PubMed.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Nat. Chem., 2009, 1, 133–137 CrossRef CAS PubMed.
- P. Michael and W. H. Binder, Angew. Chem., Int. Ed., 2015, 54, 13918–13922 CrossRef CAS PubMed.
- R. Küng, R. Göstl and B. M. Schmidt, Chem. – Eur. J., 2022, 28, e202103860 Search PubMed.
- W. Huang, Z. Zhu, J. Wen, X. Wang, M. Qin, Y. Cao, H. Ma and W. Wang, ACS Nano, 2017, 11, 194–203 CrossRef CAS PubMed.
- M. Radiom, P. Kong, P. Maroni, M. Schäfer, A. F. M. Kilbinger and M. Borkovec, Phys. Chem. Chem. Phys., 2016, 18, 31202–31210 RSC.
- M. K. Beyer, J. Chem. Phys., 2000, 112, 7307–7312 CrossRef CAS.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- B. J. Lidster, J. M. Behrendt and M. L. Turner, Chem. Commun., 2014, 50, 11867–11870 RSC.
- L. Jones, D. L. Pearson, J. S. Schumm and J. M. Tour, Pure Appl. Chem., 1996, 68, 145–148 CrossRef CAS.
- K. L. Berkowski, S. L. Potisek, C. R. Hickenboth and J. S. Moore, Macromolecules, 2005, 38, 8975–8978 CrossRef CAS.
- J. M. Lenhardt, A. L. Black Ramirez, B. Lee, T. B. Kouznetsova and S. L. Craig, Macromolecules, 2015, 48, 6396–6403 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00021d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |