
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmphiphilic tetra-PCL-b-PEG star block copolymers using benzoxazinone-based linking groups†
Carolin
Bunk
 ab,
Hartmut
Komber
a,
Michael
Lang
*a,
Nora
Fribiczer
c,
Martin
Geisler
a,
Petr
Formanek
a,
Lothar
Jakisch
a,
Sebastian
Seiffert
c,
Brigitte
Voit
ab and
Frank
Böhme
*a
ab,
Hartmut
Komber
a,
Michael
Lang
*a,
Nora
Fribiczer
c,
Martin
Geisler
a,
Petr
Formanek
a,
Lothar
Jakisch
a,
Sebastian
Seiffert
c,
Brigitte
Voit
ab and
Frank
Böhme
*a
aLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Straße 6, Dresden, Germany. E-mail: boehme@ipfdd.de; lang@ipfdd.de
bOrganische Chemie der Polymere, Technische Universität Dresden, 01062 Dresden, Germany
cDepartment Chemie, Johannes Gutenberg-Universität Mainz, Duesbergweg 10-14, 55128 Mainz, Germany
First published on 29th March 2023
Abstract
In this study, two well-defined amphiphilic tetra-arm star block copolymers with a poly(ε-caprolactone) (ε-CL) core and poly(ethylene glycol) (PEG) arms with different length (800 g mol−1 and 2100 g mol−1) were prepared by a hetero-complementary linking reaction and studied in detail. A pentaerythritol core was used as an initiator for the ring-opening polymerization (ROP) of ε-CL, generating a hydroxy-terminated tetra-arm star polymer (tetra-PCL–OH) with controlled molar mass (Mn ∼ 10 kg mol−1) and low dispersity (Đ < 1.1). After end group esterfication with 2-(4-nitrophenyl)-4-oxo-4H-benzo[d][1,3]oxazine-7-carboxylic acid chloride, linear hydrophilic PEG was attached to the respective tetra-PCL to obtain tetra-PCL-b-PEG star block copolymers. The behavior of the two amphiphilic block copolymers was studied in water by a combination of variable-temperature 1H NMR spectroscopy, DLS, DSC and TEM measurements. In the case of longer PEG arms, spherical micelles form at elevated temperatures and remain stable at room temperature over days. Transmission electron microscopy indicates a possible slow morphology change of spherical micelles into long cylindrical micelles after one week. Suspensions of the star block copolymers with the short PEG arms in water remain cloudy, in contrast to the star block copolymers with long arms.
Introduction
Star block copolymers consist of chemically distinct polymer segments (AB-type arms or A-type and B-type arms) connected by a central branching point called the core.1,2 They represent an attractive class of branched polymers which differ distinctly from their linear analogs in terms of a reduced hydrodynamic volume, lower dispersity (Đ), and high functionality with the same synthesis strategy, molar mass and composition.2–5 There are two main strategies for producing this type of polymer: “arm-first” and “core-first” where either the block-copolymer arms or the central homopolymer star are synthesized first. For the latter strategy, one considers in general that the second block of the arms is polymerized in a subsequent step, while the “grafting-onto” variant of the core-first strategy describes the subsequent attachment of independently functionalized and synthesized polymers, e.g. by “click” chemistry or Steglich esterification.2,6–10 This last approach allows designing star block copolymers with the highest structural control. Star block copolymers with both hydrophilic and hydrophobic components, such as poly(ethylene glycol) (PEG), the most widely used biocompatible polymer for biological applications, and poly(ε-caprolactone) (PCL), a biodegradable and non-toxic industrial polyester, have proven to be useful materials for biomedical applications.11–14 This is due to the amphiphilic nature of PCL-b-PEG copolymers, manifested in their tendency to form self-assembling micelles, vesicles, or gels.3,15–21Micelles prepared from PCL-b-PEG star block copolymers were frequently studied (see ESI Table S3†), since the micelle stability of stars is increased over their linear counterparts.3,16 As shown in Table S3,† all strategies have been used to prepare a broad variety of star polymers with different cores and functionalities providing molar masses from 10 kg mol−1 to beyond 200 kg mol−1. High molecular weight stars with PCL as the outer block (“tail”) first form micelles and then may gel upon increasing polymer volume fraction in water,16 while stars with PEG tails do not gel upon micro-phase separation in water. A crucial point of these materials is that both polymers crystallize at similar temperatures, such that structure formation may depend on solvent quality or additional details of the sample preparation.22 This difficulty is partially visible in the micellization data summarized in ESI Tables S3 and S4† where significant changes in micelle size or critical micelle concentration were observed for copolymers of similar architecture, molar mass and composition. Our interest in tetra-PCL-b-PEG micelles stems from investigations on model amphiphilic co-networks based on tetra-arm PEG and PCL star polymers.23–25 Here, soluble amphiphilic star block copolymers containing the same building blocks are needed to structure the material prior to network formation.
In the present work, such star block copolymers were synthesized in a similar manner as the co-networks by grafting amino-terminated linear PEGs of different molar mass onto benzoxazinone-terminated tetra-arm PCL (see Scheme 1). By using the bi-functional benzoxazinone-based coupling agent 2, which reacts hetero-complementarily with hydroxy and amino groups under mild conditions without any by-products,26–28 uniform star block copolymers can be prepared. Particular attention was paid to the defined structure of the tetra-PCL–OH core 1, its quantitative benzoxazinone functionalization (3), and their subsequent complete reaction with amino-terminated PEG 5 to form uniform tetra-PCL-b-PEG star block copolymers 6. Furthermore, the influence of PEG chain length on the behavior of the star block copolymers in aqueous medium and on the formation and stability of micellar structures was studied in detail.
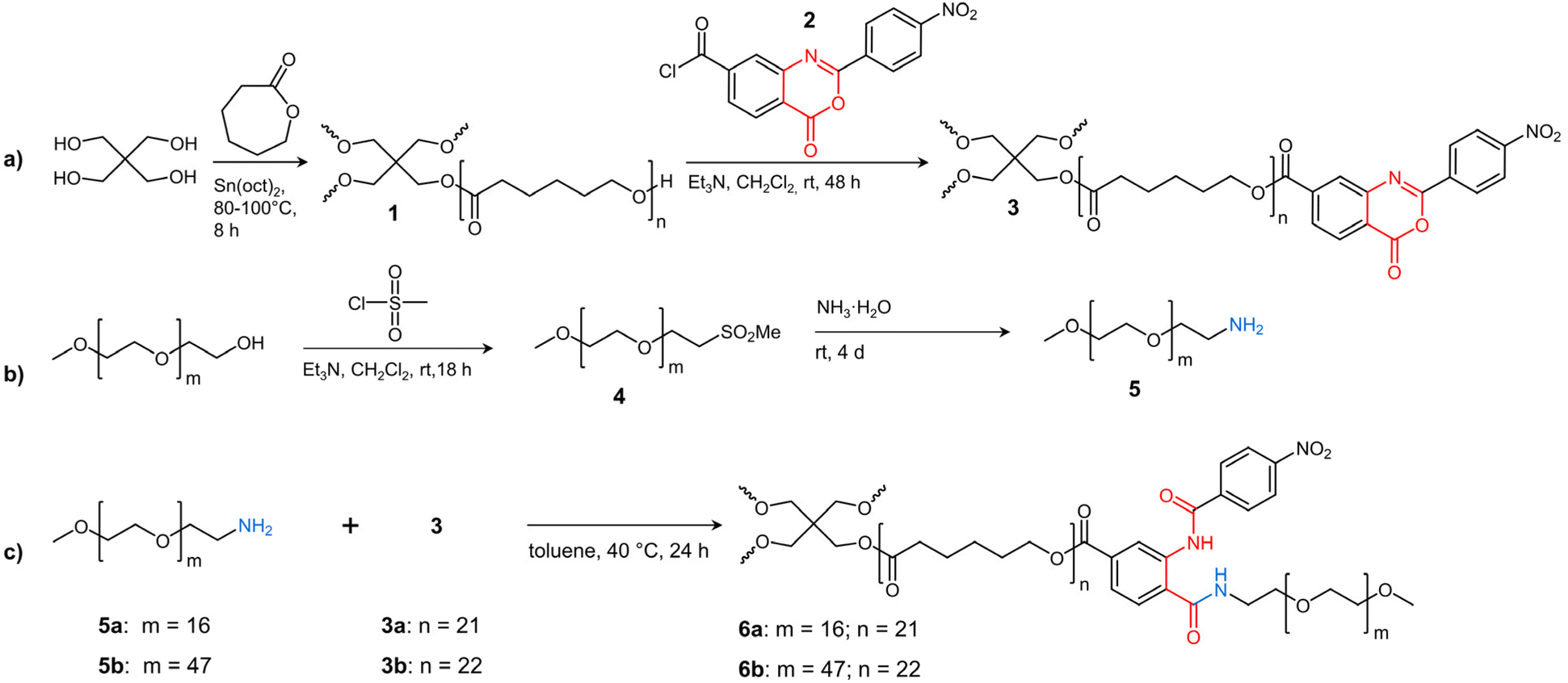 | ||
| Scheme 1 Synthesis of (a) 2-(4-nitrophenyl)-benzoxazinone-terminated tetra-PCL star polymer 3, (b) MeO–PEG–NH25, and (c) amphiphilic tetra-PCL-b-PEG star block copolymer 6. | ||
Experimental section
Materials
All chemicals and solvents (analytical grade) were obtained from Sigma-Aldrich and used as received unless otherwise specified. Methoxy poly(ethylene glycol) (MeO–PEG–OH) (Mn = 2100 g mol−1) was obtained from JenKem Technology (USA) and precipitated twice from THF in cold diethyl ether. Mono-amino-terminated MeO–PEG (MeO–PEG–NH2, 5a, Mn = 822 g mol−1) was obtained from Iris Biotech (Germany). ε-Caprolactone (ε-CL) was dried under reduced pressure over CaH2 for at least 12 hours, then purified by vacuum distillation and stored under nitrogen atmosphere. Tin(II)-2-ethylhexanoate (Sn(oct)2) was purified by vacuum distillation and stored under nitrogen atmosphere. 2-(4-Nitrophenyl)-4-oxo-4H-benzo[d][1,3]oxazine-7-carboxylic acid chloride 2 was synthesized as described earlier.27–29Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn(oct)2:pentaerythritol molar ratio of 87:0.1:1. The synthesis was carried out as follows: first, 136 mg of pentaerythritol and 9.92 g of ε-CL were added to a Schlenk flask at room temperature under nitrogen. The dispersion was stirred at 120 °C for 30 min to obtain a homogeneous mixture. The reaction mixture was cooled to 80 °C and the polymerization was started by injecting the catalyst (Sn(oct)2, 0.2 mL of 0.5 M dry toluene solution) under nitrogen. After 2 h, the reaction temperature was increased to 100 °C for another 6 h. Then, the very viscous reaction mixture was cooled to room temperature to stop the bulk polymerization. The polymer was purified by precipitation from a concentrated CH2Cl2 solution into a 10-fold amount of cold methanol. After filtration and drying in vacuum, a powdery product of 1 was obtained.
:Sn(oct)2:pentaerythritol molar ratio of 87:0.1:1. The synthesis was carried out as follows: first, 136 mg of pentaerythritol and 9.92 g of ε-CL were added to a Schlenk flask at room temperature under nitrogen. The dispersion was stirred at 120 °C for 30 min to obtain a homogeneous mixture. The reaction mixture was cooled to 80 °C and the polymerization was started by injecting the catalyst (Sn(oct)2, 0.2 mL of 0.5 M dry toluene solution) under nitrogen. After 2 h, the reaction temperature was increased to 100 °C for another 6 h. Then, the very viscous reaction mixture was cooled to room temperature to stop the bulk polymerization. The polymer was purified by precipitation from a concentrated CH2Cl2 solution into a 10-fold amount of cold methanol. After filtration and drying in vacuum, a powdery product of 1 was obtained.
1H NMR (CDCl3): δ 4.11 (s; CH2,core), 4.06 (t, 6.7 Hz; CH2OC(O)), 3.65 (t, 6.5 Hz; CH2OH), 2.30 (t, 7.6 Hz; CH2C(O)O), 1.7–1.6 (CH2CH2CH2), 1.38 ppm (m; CH2CH2CH2).
13C NMR (CDCl3): δ 173.3 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 172.7 (CO next to core), 64.0 (CH2OC(O)), 62.4 (CH2OH), 61.9 (CH2,core), 41.9 (Ccore), 34.0 (CH2C(O)O), 32.2 (CH2CH2OH), 28.2 (CH2CH2OC(O)), 25.4 (CH2CH2C(O)O), 24.4 ppm (CH2CH2CH2).
O), 172.7 (CO next to core), 64.0 (CH2OC(O)), 62.4 (CH2OH), 61.9 (CH2,core), 41.9 (Ccore), 34.0 (CH2C(O)O), 32.2 (CH2CH2OH), 28.2 (CH2CH2OC(O)), 25.4 (CH2CH2C(O)O), 24.4 ppm (CH2CH2CH2).
1H NMR (CDCl3): δ 8.52 (d, 8.8 Hz; 19), 8.39 (s; 11), 8.38 (d, 8.8 Hz; 20), 8.34 (d, 8.2 Hz; 14), 8.20 (d, 8.2 Hz; 15), 4.41 (t, 6.7 Hz; 8 next to C9), 4.10 (s; 2), 4.06 (t, 6.7 Hz; 8), 2.35 (t, 7.4 Hz; 4 next to C9), 2.30 (t, 7.4 Hz; 4), 1.85 (m; 7 next to C9), 1.73 (m; 5 next to C9), 1.7–1.6 (5, 7), 1.52 (m; 6 next to C9), 1.38 ppm (m; 6).
13C NMR (CDCl3): δ 173.4 (3), 172.8 (3 next to core), 164.7 (9), 158.0 (16), 155.6 (17), 150.3 (21), 146.3 (12), 138.1 (10), 135.5 (18), 129.4 (15), 129.3 (19), 129.0 (14), 128.9 (11), 123.9 (20), 120.0 (13), 65.8 (8 next to C9), 64.1 (8), 61.9 (2), 42.0 (1), 34.1 (4), 28.3 (7), 25.5 (5), 24.5 ppm (6).
1H NMR (CDCl3): δ 3.62 (s; c), 3.48 (m; b), 3.35 (s; d), 2.84 (t, 5.3 Hz; a), 1.8 ppm (v br; NH2).
13C NMR (CDCl3): δ 73.3 (b), 71.5, 70.5, 70.2 (all c), 58.9 (d), 41.7 ppm (a).
400g. The supernatant was then removed, fresh water added, and centrifuged again. This procedure was carried out 4 times. The purified powder was then freeze-dried to give 6b.
1H NMR (6b, CDCl3). δ 12.62 (s; Ph–NH–CO–Ph), 9.41 (s; 11), 8.36 (d, 8.8 Hz; 20), 8.21 (d, 8.8 Hz; 19), 7.82 (d, 8.2 Hz; 15), 7.75 (d, 8.2 Hz; 14), 7.36 (t, 5.4 Hz; CH2–NH–CO–Ph), 4.41 (t, 6.7 Hz; 8 next to C9), 4.10 (s; 2), 4.05 (t, 6.7 Hz; 8), 3.70–3.50 (a, b, c), 3.37 (s; d), 2.34 (t, 7.4 Hz; 4 next to C9), 2.30 (t, 7.4 Hz; 4), 1.83 (m; 7 next to C9), 1.72 (m; 5 next to C9), 1.7–1.6 (5, 7), 1.50 (m; 6 next to C9), 1.38 ppm (m; 6).
13C NMR (6b, CDCl3): δ 173.2 (3), 172.5 (3 next to core), 168.3 (16), 165.3 (9), 163.0 (17), 149.6 (21), 140.0 (18), 139.5 (12), 133.9 (10), 128.4 (19), 127.3 (14), 124.0 (15), 123.7 (20), 123.2 (13), 121.8 (11), 71.7 (c next to Cd), 70.5–69.0 (b, c), 65.1 (8 next to C9), 63.8 (8), 61.7 (2), 58.7 (d), 41.8 (1), 39.9 (a), 33.9 (4), 28.1 (7), 25.3 (5), 24.3 ppm (6).
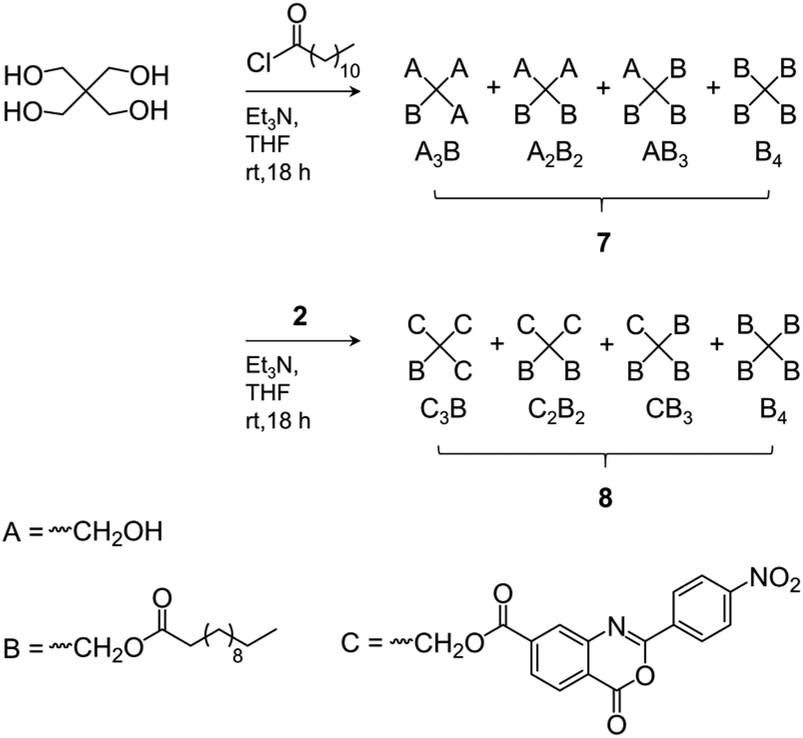 | ||
| Scheme 2 Synthesis of alkyl ester- (7) and benzoxazinone/alkyl ester-substituted (8) pentaerythritol mixtures as model compounds for NMR signal assignments. | ||
Methods
Results and discussion
Synthesis and structural analysis
The synthesis of amphiphilic tetra-PCL-b-PEG star block copolymers 6a and 6b is shown in Scheme 1. First, tetra-PCL–OH 1 was synthesized by ROP of ε-CL using pentaerythritol as initiator and Sn(oct)2 as catalyst, followed by terminal group functionalization with 2-(4-nitrophenyl)-benzoxazinone-based compound 2 by esterification to give 3. Subsequently, the 2-(4-nitrophenyl)-benzoxazinone end groups of 3 were reacted with MeO–PEG–NH2 (5) of different lengths (m = 16 and 47). The synthesis results are summarized in Table 1, and the 1H NMR spectra of 3, 5b, and 6b are shown in Fig. 1. The desired Mn of 1 (∼10 kg mol−1), which forms the hydrophobic core of 6a and 6b, was controlled by the molar ratio of ε-CL to pentaerythritol. Mn,NMR values of 9.3 kg mol−1 and 9.7 kg mol−1 determined by 1H NMR end group analysis (see ESI Fig. S3†) agree very well with the targeted value (Table 1). Functionalization of the terminal hydroxy groups of 1 was carried out with an excess of 2 in solution. The conversion of the terminal CH2OH group, characterized by the methylene signal at 3.64 ppm in the 1H NMR spectrum of 3 (see Fig. 1c), was ≥95%.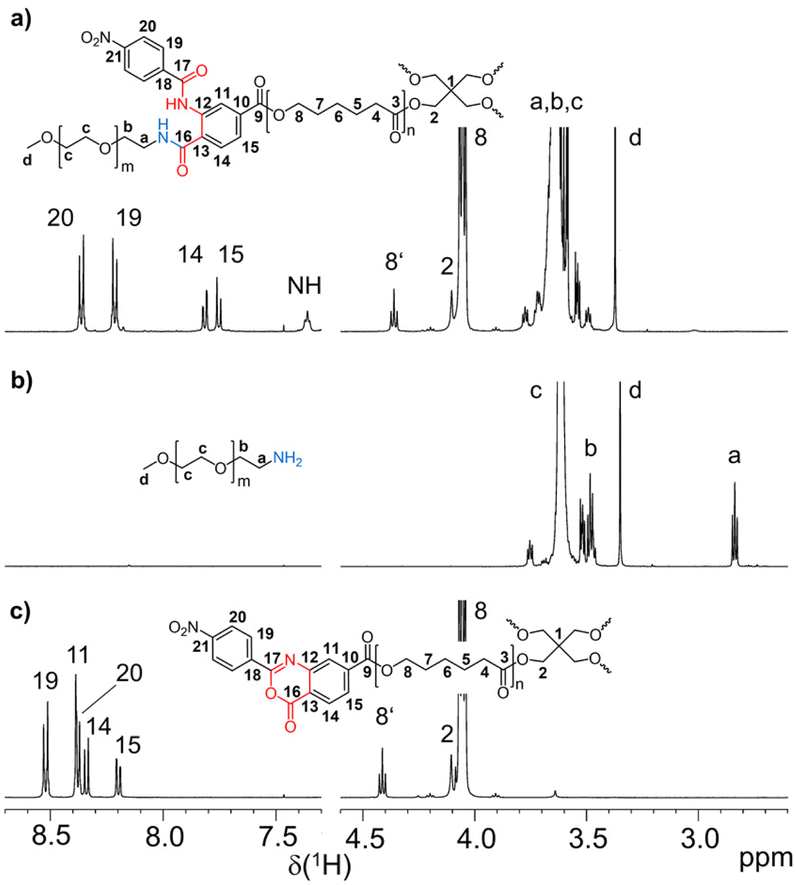 | ||
| Fig. 1 1H NMR spectra (regions) of (a) tetra-PCL-b-PEG star block copolymer 6b, (b) MeO–PEG–NH25b, and (c) benzoxazinone-terminated tetra-PCL star polymer 3 (solvent: CDCl3). | ||
| Polymer |
P
n,NMRa (Ø Pn,arms = ¼ Pn) |
M
n,NMRa (kg mol−1) |
M n,SEC (kg mol−1) | M n,MALDI-TOF (kg mol−1) | M w,SEC (kg mol−1) | M w,MALDI-TOF (kg mol−1) | Đ SEC | Đ MALDI-TOF |
|---|---|---|---|---|---|---|---|---|
| a Based on 1H NMR end group analysis (see ESI Fig. S3, S5, S7 and comments†), Pn is the degree of polymerization (number of polymer units per star). | ||||||||
| 1a | 80 ± 2 | 9.3 ± 0.2 | 8.7 ± 0.6 | 8.6 ± 0.2 | 9.5 ± 0.6 | 8.93 ± 0.16 | 1.09 ± 0.3 | 1.04 |
| 1b | 84 ± 2 | 9.7 ± 0.2 | 10.8 ± 0.6 | 8.9 ± 0.2 | 11.8 ± 0.6 | 9.62 ± 0.11 | 1.09 ± 0.2 | 1.09 |
| 3a | 84 ± 2 | 10.9 ± 0.3 | 9.6 ± 0.7 | 9.7 ± 0.1 | 10.0 ± 0.5 | 10.3 ± 0.2 | 1.04 ± 0.3 | 1.06 |
| 3b | 88 ± 2 | 11.3 ± 0.3 | 10.7 ± 0.5 | 9.5 ± 0.3 | 11.4 ± 0.4 | 9.9 ± 0.3 | 1.07 ± 0.2 | 1.04 |
| 5a | 16 | 0.765 | — | 0.785 | — | 0.825 | — | 1.05 |
| 5b | 47 ± 1 | 2.1 ± 0.1 | 2.1 ± 0.1 | 2.01 ± 0.02 | 2.3 ± 0.1 | 2.04 ± 0.02 | 1.06 | 1.02 |
| 6a | — | 13.5 ± 0.3 | 13.9 ± 0.6 | 11.6 ± 0.7 | 15.5 ± 0.5 | 12.2 ± 0.7 | 1.11 ± 0.04 | 1.05 |
| 6b | — | 19.3 ± 0.4 | 17.6 ± 0.6 | 16.2 ± 0.6 | 17.9 ± 0.6 | 17.1 ± 0.7 | 1.03 ± 0.04 | 1.06 |
The Mn values of 3 shown in Table 1 increased slightly compared to 1. The somewhat reduced dispersity is probably caused by the purification steps performed after functionalization. The results clearly show that no degradation of the tetra-PCL star occurred during the modification. Fig. 1b depicts the 1H NMR spectrum of MeO–PEG–NH2 (5b). Absence of the methyl signal of the mesyl group at 3.08 ppm indicates a high amino functionalization of 5b, which is calculated to be about 90% based on the intensities of the methyl ether (3.35 ppm) and the CH2NH2 (2.84 ppm) signals. A further issue is the perfectness of the tetra-PCL star structure. Mixtures of differently substituted pentaerythritol (7, 8) were obtained by sequential conversion of pentaerythritol with lauryl chloride and compound 2 (Scheme 2). The components of these mixtures serve as model compounds for NMR spectroscopic analysis. Detailed signal assignments are given in the ESI Chapter 6 and 7.† It was found that the 1H NMR spectrum of tetra-PCL–OH 1 is less suitable for quantifying structural defects caused by incomplete conversion of the pentaerythritol core. However, the different substitution patterns of the core can be well distinguished in the 1H NMR spectrum of compound 3. This becomes clear by comparison with the spectrum of mixture 8 (see Fig. 2). The signals of the species with only three PCL arms (CB3) could be identified (∼6 mol%), while lower degrees of substitution (C2B2, C3B) could hardly be detected and are estimated to be <1 mol%. In the 1H NMR spectrum of 6b (see Fig. 1a), the signals of the benzoxazinone terminal groups disappeared completely whereas new signals could be assigned to the newly formed linking benzamide group between the PCL core and the PEG arms. Since all benzoxazinone signals disappear in the spectrum of 6b, it can be assumed that the benzoxazinone groups on the core also participate in the reaction with amino-terminated 5b.
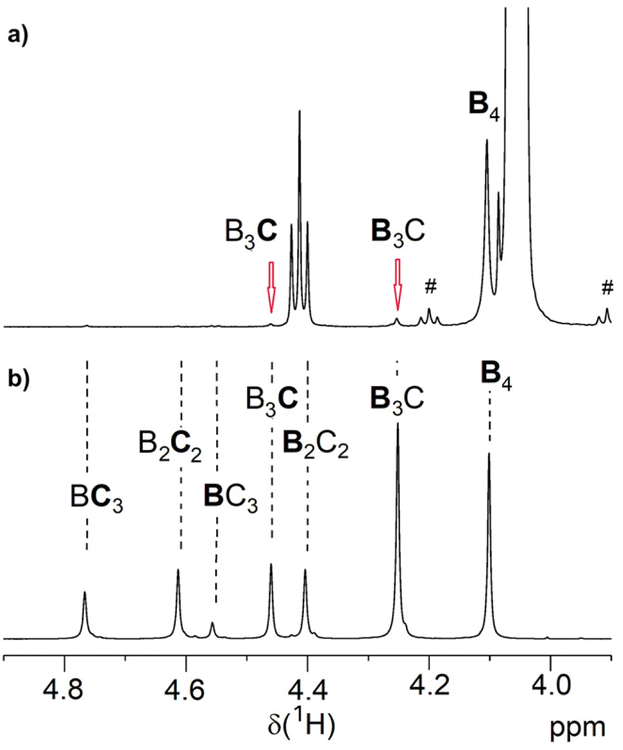 | ||
| Fig. 2 Region of the 1H NMR spectrum of (a) benzoxazinone-terminated star 3a and (b) the corresponding benzoxazinone/alkyl ester-substituted compound mixture 8 (solvent: CDCl3). The abbreviations are explained in Scheme 2 and symbol # marks 13C satellite signals. | ||
The FTIR spectra of 3 and 6b shown in Fig. 3 also confirm the complete conversion of the terminal benzoxazinone groups. In the spectrum of 3, the carbonyl stretching band and the CN stretching band of the benzoxazinone group are clearly visible at 1772 and 1616 cm−1, respectively. After reaction with the amino-terminated PEG 5b, these bands disappear completely in the spectrum of 6b. Instead, amide I (carbonyl stretching band at 1648 cm−1) and amide II (N–H deformation band at 1582 cm−1) bands are visible, which can be attributed to the newly formed benzamide linking group.
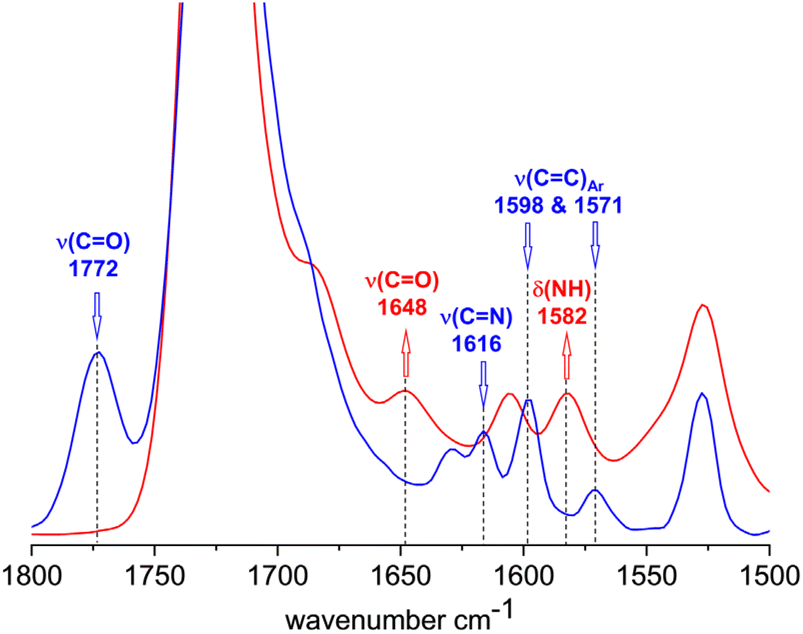 | ||
| Fig. 3 FTIR spectra of 3 (blue) and 6b (red) in the region of unreacted and reacted benzoxazinone group. | ||
MALDI-TOF mass spectra of 1, 3, 6a and 6b are shown in Fig. 4. To exclude mass discrimination effects, the results from the MALDI-TOF analysis were validated by measurements in linear and reflector TOF mode as well as by MD-SEC. Due to an identified slight mass discrimination for 6a, the average molar masses of 6a, 6b were determined from mass spectra measured in linear TOF mode (see ESI Chapter 9†).
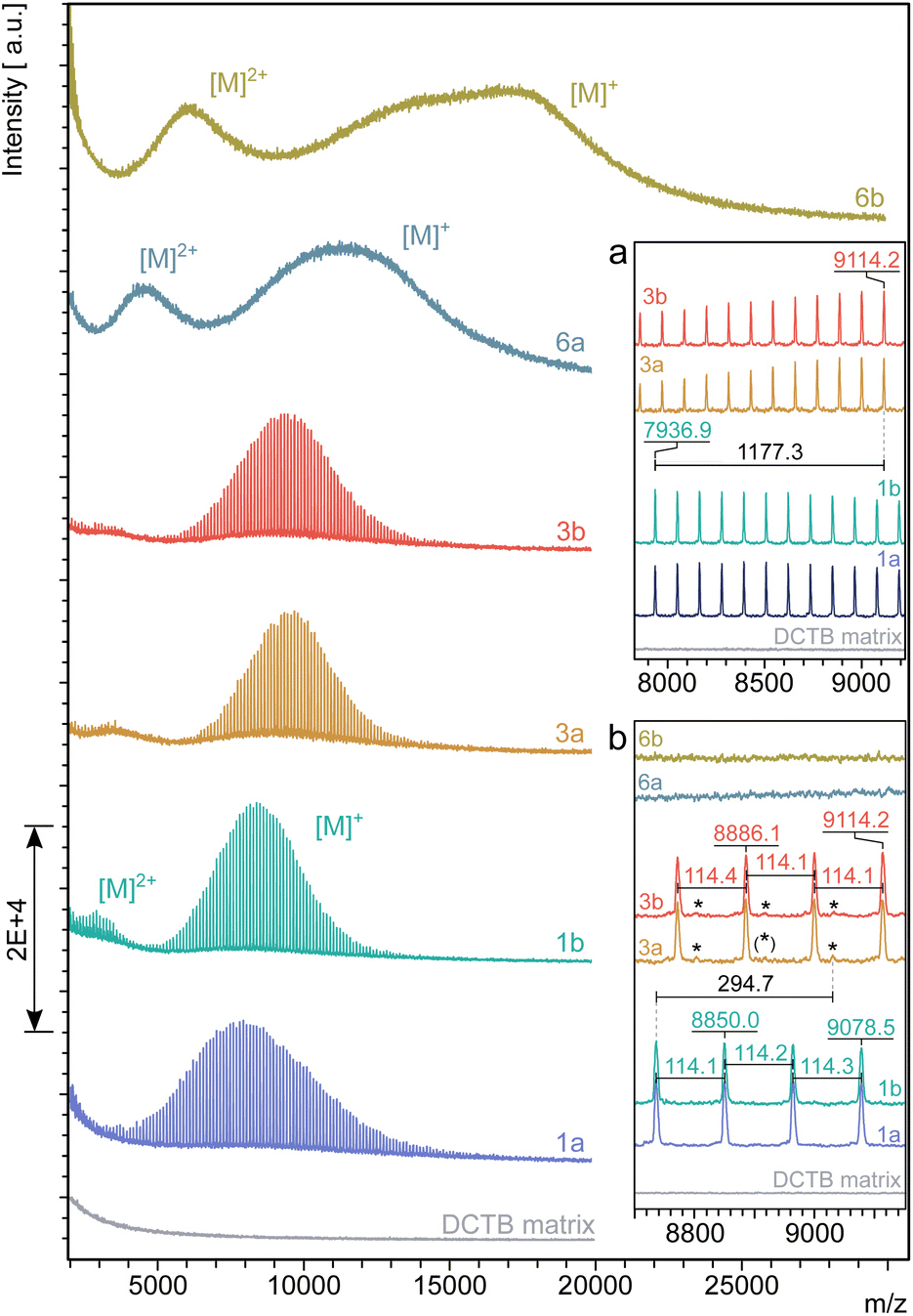 | ||
| Fig. 4 Molar mass distribution determined by MALDI-TOF mass spectrometry of the star polymers 1, 3, 6a and 6b in THF with a shift in the discrete molecule masses between 1 and 3 of their main fractions by Δm/z = 1177.3 (inset a) and the mass differences in between the discrete molecule masses Δm/z = 114 (inset b). The most significant trace impurity found of Δm/z = 294.7 between 1 and 3 is assigned by an asterisk. | ||
The spectra of tetra-PCL-star polymers 1 and 3 are well resolved and allow conclusions about the molecular architecture. This high resolution is not found in the spectra of the star block copolymers 6a and 6b since linking of two polymers with different repeating units usually smears out the discrete signals of the individual polymers.
In the spectra of 1 and 3, the distance between two main peaks corresponds very well to the molar mass of one repeating unit of PCL (Mn = 114.1 g mol−1). The observed peak shift Δm/z = 1177.3 between 1 and 3 (see Fig. 4a) indicates an attachment of four 2-(4-nitrophenyl)-benzoxazinone terminal units per molecule. A small, but significantly resolved trace impurity (see Fig. 4b) of Δm/z = 294.7 may refer to a superimposition of fractions with lower number of modified terminal units (Δm/z = 294.7 [M + K]+, one unit and Δm/z = 867.2 [M + Na]+, three units). For a detailed assignment of trace impurities found by MALDI-TOF MS, see ESI Chapter 9.† The insignificantly low intensity of the mass peaks coming from trace impurities compared to the strongly dominating ones of the main fractions agrees with the findings by 1H NMR spectroscopy and confirms an almost complete termination of the tetra-PCL star polymer 3 with oxazinone groups.
The molar mass measurements confirm the narrow dispersity of the samples even after the third modification step (Table 1). There is a clear shift to higher molar masses from 1 to 3 and especially to 6a and 6b. The MALDI-TOF MS based molar masses tend to be slightly smaller than those determined by NMR spectroscopy and MD-SEC, but in principle show the same trend.
These results, together with the FTIR spectroscopy, MD-SEC and NMR spectroscopy measurements, show that the hetero-complementary conversion between 3 and 5 proceeded quantitatively and that the resulting star block copolymers 6a and 6b are characterized by high molecular uniformity.
Amphiphilic behavior in selective solvent
PCL-b-PEG copolymers are inherently amphiphilic because water is a selective solvent for the hydrophilic PEG block. In water, these polymers tend to form micelles,3,15–20 whereby the micelle geometry is controlled in first instance by the volume fraction of the soluble block33 with only small changes for a low functionality as 4 (number of arms). Since both PEG and PCL have a density close to 1.13 g cm−3, the PEG volume fraction of 6a and 6b are approximately 23% and 44%, respectively. For linear PEG-b-PCL block copolymers, precipitate was found in one work below 12% PEG fraction.34 Vesicles with sizes ranging from 60 nm to 10 μm were found35 for compositions between 9% and 36% PEG.34–37 Cylindrical micelles with a length up to 20 microns may form between 13% and 55% PEG.19,34,35,37–39 Also, a lamellar phase was observed for 11% to 16% PEG19 that could be an intermediate state of vesicle formation. The different particulate structures mentioned above overlap significantly, which complicates the analysis and a discussion of these solutions in addition to possible transitions between different morphologies that can be driven by crystallization40 and possible concentration dependencies of the observed morphologies.41 Similar to the linear block copolymers, we expect that sample 6a may form a broad variety of different morphologies. This complicates a quantitative analysis of particle size by DLS. For block copolymers with a composition comparable to 6b, most of the above mentioned works report only spherical micelles; only sometimes38,39 cylindrical micelles are detected, see also ESI Table S4.†The solubility of the tetra-PCL-b-PEG star block copolymers 6a and 6b in water was investigated at room temperature and at 80 °C. For this purpose, both polymers were suspended in water at a concentration of 10 mg mL−1 at room temperature (see Fig. 5, left). After heating to 80 °C, a clear solution of 6b was obtained within 5 minutes (see Fig. 5, right), which remained stable when cooled back to room temperature. In contrast, 6a remained cloudy over the entire temperature range without the formation of a precipitate (see Fig. 5, right). Both observations agree with the formation of spherical micelles in the low nm range for 6b and the formation of micelles with a size comparable to the wavelength of light or larger for 6a. The equilibrium melting temperature was estimated to be close to 64 °C42,43 for PCL and around 70 °C42–45 for PEG. Crystallization temperatures up to 49 °C46,47 and around 40 °C48,49 were reported for PCL and PEG, respectively. Thus, thermal treatment at 80 °C melts crystalline portions of both polymers, which is a mandatory step for dissolving the star block copolymers. Phase separated PCL domains may crystallize upon cooling back to room temperature. To gain a better insight into the solution and self-assembly behavior of tetra-PCL-b-PEG star block copolymers in water, DSC, (cryo-)TEM, DLS, and 1H NMR measurements were performed on aqueous dispersions of 6a and 6b.
 | ||
| Fig. 5 Suspensions of 6a and 6b in water at room temperature (left) and after 5 min of heating to 80 °C and subsequent cooling to room temperature (right). | ||
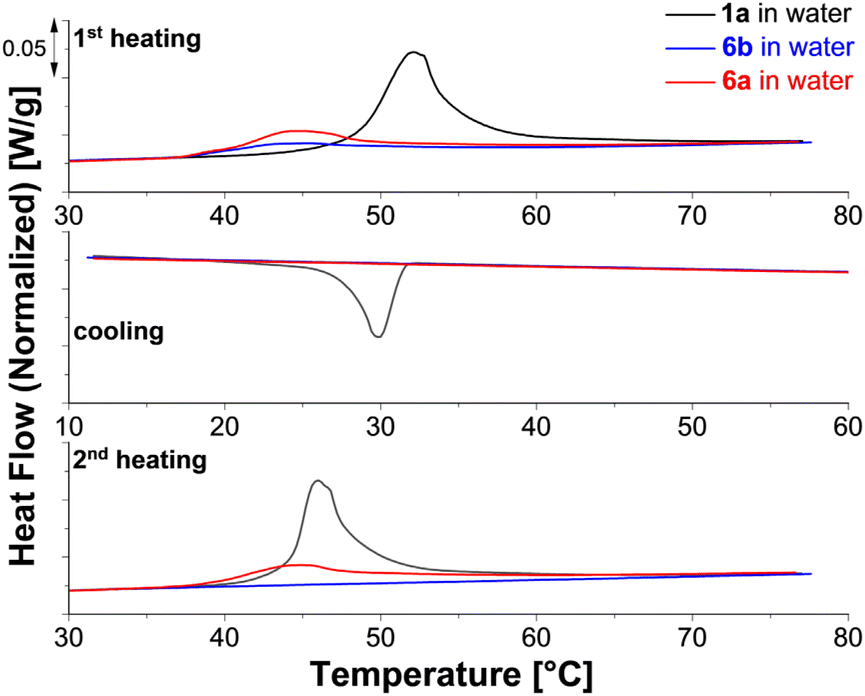 | ||
| Fig. 6 DSC thermograms of 1a (black), 6a (red) and 6b (blue) in water. | ||
Compared to 1, the melting transitions of the star block copolymers 6a and 6b appear in the first heating run at lower temperature (Tm = 45 °C). This difference results in part from the different treatment of the specimens at the end of the synthesis: 6a and 6b are purified in water followed by freeze-drying. Thus, the crystal geometry is here affected by the micelle geometry that is established during this process. During cooling, no crystallization is visible for either copolymer. The second heating scan, recorded after 30 minutes of isothermal annealing at 10 °C, shows a melting peak only for the sample with the shorter PEG block (6a). For explaining this difference, we consider the visible appearance of the samples and the morphologies reported in literature, see ESI Table S4.† Based on this information, we expect mainly small spherical micelles for 6b and larger structures possibly cylindrical micelles or vesicles for 6a. Semi-crystalline vesicles and cylindrical micelles of PCL-b-PEG copolymers with a similar composition as 6a and 6b were analyzed previously.34 Crystallization of vesicles was possible at room temperature, while significant crystallization of cylindrical micelles occurred only significantly below 0 °C. Our observations, therefore, hint towards a significant volume fraction of vesicles in aqueous suspensions of 6a.
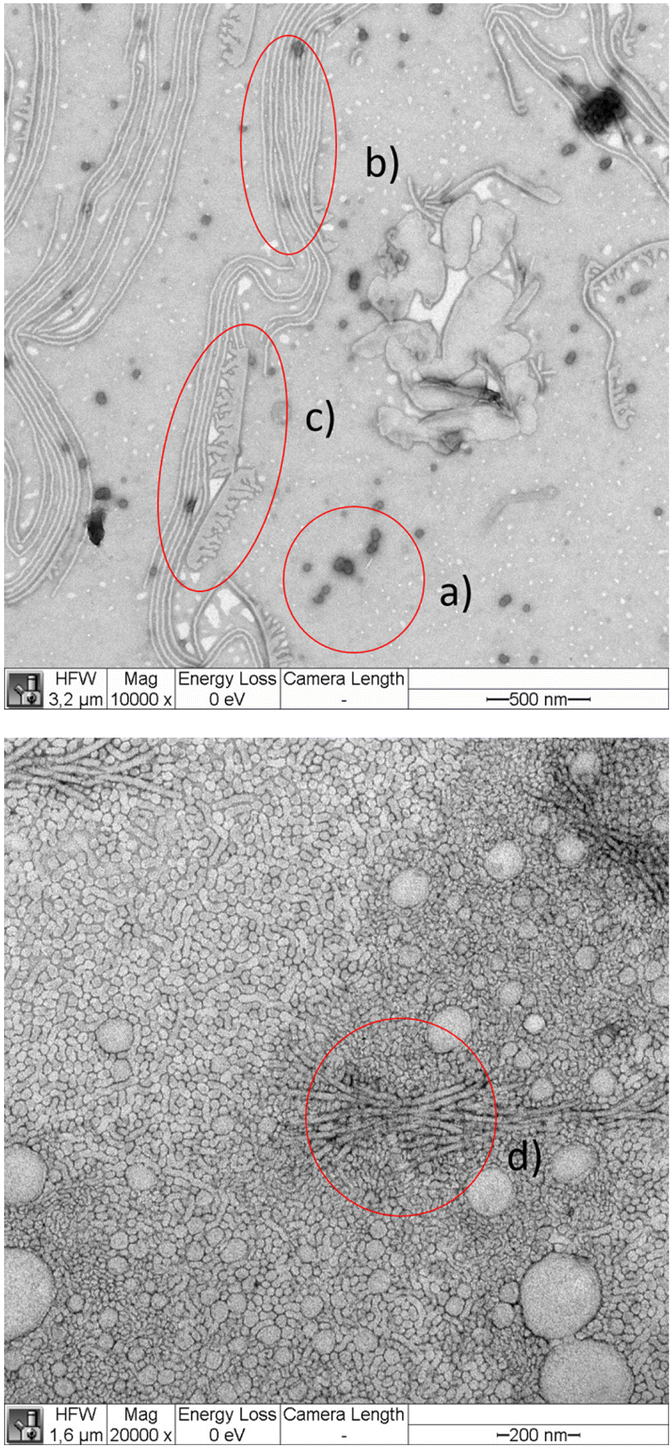 | ||
| Fig. 7 TEM micrograph of 6b adsorbed within 10 seconds from 1 g L−1 solution in water stained with PTA (top) and adsorbed within 10 seconds from 10 g L−1 solution in water stained with UAC (bottom). PTA enters predominantly the PEG corona. Thus, spherical micelles are grey objects with a darker boundary (a), cylindrical micelles as double grey lines (b), while flat surface structures have average grey color with a darker line as boundary (c). UAC stained objects show a similar appearance as PTA stained objects with characteristic super structures (d). | ||
 | ||
| Fig. 8 Cryo-TEM image of a rapidly frozen 10 g L−1 solution of 6b. | ||
We argue that the structures that are visible on the TEM images but do not appear on the cryo-TEM images are most likely surface initiated with additional dependence on the particular stain. The common structure observed in all images are spherical and cylindrical micelles. The diameters of the spherical micelles were determined by manual measurement of typically 30 micelles using the Fiji software. The results are summarized in Table 3. Our data agree with literature data on linear block copolymers that are equivalent to a single arm of our star block copolymers. For instance, the PCL24–PEG44 blocks similar to star arms produce 21.7 ± 5.9 nm sized spherical micelles19 (stained with PTA and prepared using a different protocol) in quantitative agreement with our work (see Table 3).
The diameter of the cylindrical micelles in cryo-TEM was 12 ± 2 nm in 10 g L−1 samples and 13.5 ± 0.7 in 1 g L−1 samples. With UCA, a cylindrical micelle thickness of 21 ± 3 nm was measured in samples adsorbed from 1 g L−1 solution.
We now attempt to correlate the above measurements. Let us assume that the PEG corona of the micelles is not visible in the cryo-TEM images and that the stain does not penetrate the PCL domain. Since PEG is about half of the molar mass of 6b, dry adsorbed undeformed spherical micelles in TEM should have a size approximately 21/3 times larger than the PCL core, i.e. about 20 nm, which is below the size given in Table 3. In the dry adsorbed state, the PEG tails are still swollen by the stain and may develop conformations that are either collapsed due to drying or significantly stretched, when the PEG with the stain inside tends to spread over the substrate. To understand which of these effects is more relevant, we consider Flory's characteristic ratio C∞ of PEG that ranges roughly from 4 to 751 in literature with rotational isomeric state computations around C∞ = 5.52 For a root mean square bond length of 1.46 nm,53 this latter estimate corresponds to a Kuhn segment size of about 8.8 Å with a Kuhn molar mass of 107 g mol−1. The 2100 g mol−1 PEG tails of the star arms consist, therefore, of approximately 20 Kuhn segments. Assuming random walk conformations of the PEG tails as lower bound for the thickness of a PEG corona in non-stretched conformations, we estimate that this layer is at least 4 nm thick and the micelles should appear of at least 23 nm size in TEM. Fully stretched chain conformations on the substrate would lead to a layer of about 17 nm thickness leading to a maximum size of 50 nm in the adsorbed state. Our data lie in between these limits and are in accordance with a weakly stretched PEG corona in the dry adsorbed state or alternatively, a weak penetration of the stain into the PCL domain.
PCL in the crystalline state adopts almost planar zig-zag conformations deviating slightly from the fully extended form54 with a fiber axis of 1.705 nm corresponding to two chemical monomers. Thus, A cylindrical micelle diameter of 12 ± 2 nm refers to about 13 monomers in nearly fully extended conformations. The PCL cores of 6b contain approximately 21 monomers per arm. Thus, the diameter of the cylindrical micelle is presumably a one-folded crystal with about 11 monomers in nearly fully extended conformation. A micelle core of about 15–16 nm is almost midway between a one-folded crystal or a fully extended conformation of PCL arms, and thus, not in accord with an expected crystalline morphology.43 Since also DSC did not show crystallization of 6b, we conclude that the spherical micelles at room temperature consist of an amorphous PCL core.
A crystalline tetra-PCL star with Mn ∼ 10 kg mol−1 and a density around 1.2 g cm−3,54 occupies a volume of about 14 nm3. A sphere with a diameter of 15–16 nm and a volume of order 2000 nm3 provides space for roughly 140 such tetra-PCL star cores and about 1.4 nm2 surface area per PEG tail. In comparison, thinner cylindrical micelles with 12–13 nm diameter provide only about 1.2 nm2 per PEG tail. A similar ratio is obtained when considering amorphous PCL. This reduction refers to an energetic barrier for forming cylindrical micelles as long as the PEG remains dissolved and amorphous, indicating that spherical micelles are at least meta-stable. This barrier can be overcome, for instance, by significant changes of the solvent quality, by adjusting temperature, or when surface adsorption or favorable interactions with some solutes increase the PEG concentration locally, since the solvent quality of water decreases with the PEG volume fraction.55,56 This may explain the lower frequency of cylindrical micelles and short precursors of these in the cryo-TEM images despite of the longer storage time of the solution prior to the measurement (7 days) as compared to the micrographs with UCA and PTA stain (up to 2 days). In consequence, we expect that solutions of 6b immediately after heating beyond the melting temperature develop a dominant portion of spherical micelles but will not exhibit a significant portion of cylindrical micelles.
Samples 6a in comparison to 6b contain shorter PEG tails which stabilize these samples less against morphological changes that might be induced by contact with a surface, staining, or due to temperature changes during sample preparation. Since morphological changes were already a serious issue for 6b, we omitted a similar analysis of 6a.
The slow mode refers to a hydrodynamic radius that increases with concentration between 14–23 nm and 18–34 nm in the case of 6a and 6b, respectively, see ESI Table S1.† Such a behavior is typically attributed to the formation of aggregates or clusters. The nature of these clusters, however, remains unclear: literature data for the solvent quality of acetone regarding both PEG and PCL is scattered around the theta condition, see ESI Table S5.† Furthermore, the relative amplitude of the fast component in the ESI Table S1† does not strongly decay with concentration as one expects for micelle formation or other forms of associations where increasing concentrations favors associations.
For analyzing 6b in water, samples were prepared at 20 g L−1, heated up to 70 °C and cooled back to room temperature. Afterwards, the solution was diluted stepwise to a lowest concentration of 0.025 g L−1. All measurements were conducted within 6 h after preparation starting from the largest concentration. In all samples, monodisperse clusters with a hydrodynamic radius of 20–22 nm are observed, see ESI Table S1.† The size of the dried adsorbed stained micelles in TEM is about 10 to 15 nm less than the particle diameter measured by DLS, which is reasonable when considering the swelling of the densely grafted PEG tails in water, a possible weak penetration of the stain into the PCL, and additional corrections like a weak dispersity of the PEG tails. Up to the end of our measurement time of 6 h, we detect no significant traces of larger species like cylindrical micelles of sample 6b in water.
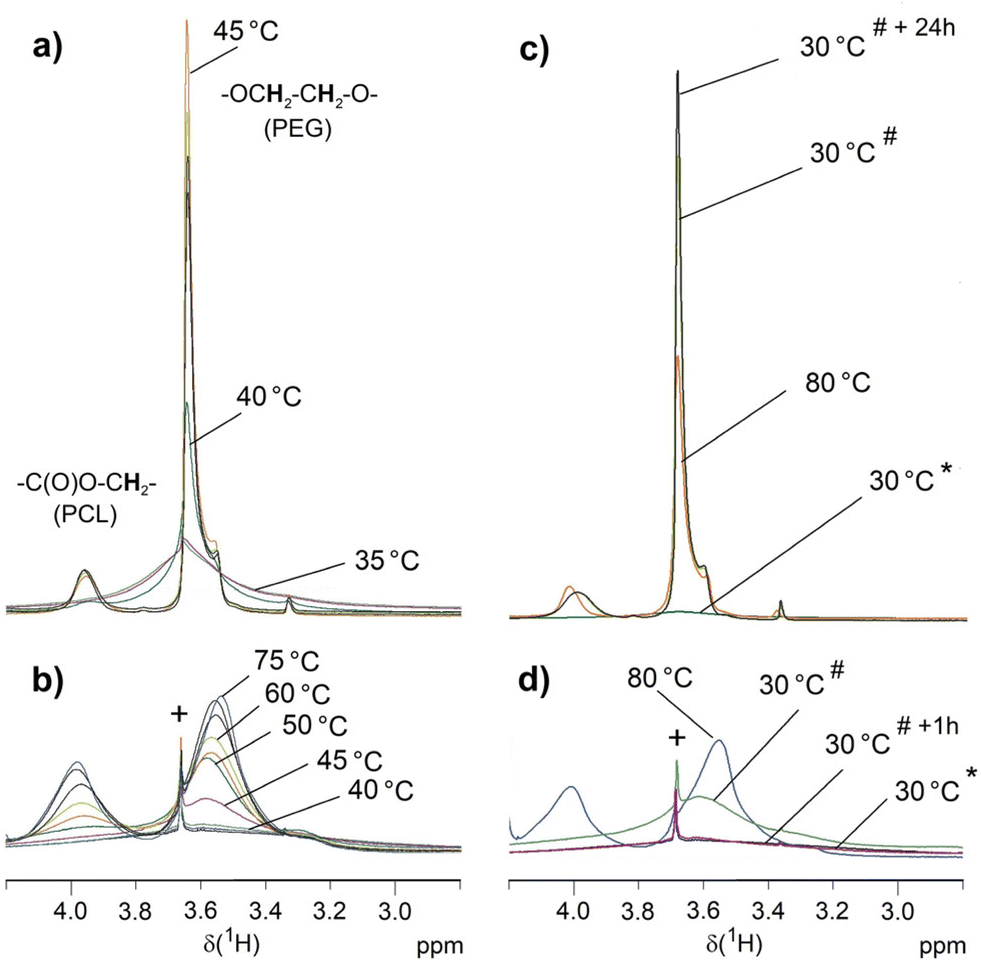 | ||
| Fig. 9 VT 1H NMR measurements on 6b (a and c) and 6a (b and d) in D2O (suspension of 5 g L−1). Only selected temperatures are indicated. The complete series are depicted in the ESI Fig. S19 and S20.† Besides the initial spectra at 30 °C (*), (c) and (d) show additional spectra recorded after cooling down the samples to 30 °C (#) and after keeping the sample for one hour (6a) or 24 hours (6b) at 30 °C (*+1 h, *+24 h). Symbol + marks the signal of a trace of unreacted 5 that is the only compound dissolved at 30 °C. | ||
The findings of these VT 1H NMR experiments correspond to the results of crystallization studies in water by DSC and to the DLS results. The PCL phase of the dispersed samples prior to the first heating is a rigid phase and therefore the spectrum is silent for these signals. The PEG chains are also immobile and only the outermost PEG sphere of 6b seems to show a mobility sufficient to detect a signal. At the melting range of the PCL core (Tm ∼ 45 °C, see Table 3) the corresponding signals appear for both star block copolymers, which is associated with increased mobility. This is confirmed by a reference experiment on the water-insoluble PCL core 1a that also shows resolved signals above Tm. The PEG chains become also more mobile but only for 6b the PEG signal is narrow with a hump probably indicating a still ordered PEG portion. For 6b, micellar structures form in this process, which have high stability. This agrees with the DLS measurements that after heating the sample to 70 °C and subsequent cooling prove stable micellar structures with hydrodynamic radii of about 20 nm at 25 °C for 6b. PCL recrystallization is suppressed in the micellar structures and the 1H NMR signals of PCL remain unchanged after cooling. The two blocks are mobile enough in their entirety in these micellar structures to be quantitatively detected by solution-state NMR techniques as confirmed by the PCL/PEG ratio, which is consistent with that of the CDCl3 solution. The linking groups between both blocks result in well-resolved signals in accordance to the mobility of both blocks. For 6a, the mobility of the short PEG chains seems to be hindered by the larger PCL fraction as indicated by the rather broad signal also at 85 °C. However, the mobility in both blocks is sufficient to largely reflect the PCL/PEG ratio from the CDCl3 solution in the signal integrals and also the signals of the linking groups can be detected. Different to 6b, the increased mobility within the particles during heating does not result in the formation of small micellar structures with a size below the wavelength of visible light in the aqueous environment. Below Tm of PCL, re-formation of a largely immobilized structure occurs, leading to signal collapse for both PCL and PEG in these high-resolution NMR measurements.
In conclusion, these straightforward 1H NMR experiments on suspensions in D2O allow a qualitative evaluation of melting and recrystallization as well as micellar structure formation for these tetra-PCL-b-PEG star block copolymers as demonstrated here for the influence of different PEG chain lengths. The increase in mobility with temperature should be due to both melting PCL structures and solvation of the hydrophilic PEG chains. The formation of small cylindrical micelles or spheres requires a sufficiently high content of the hydrophilic PEG as obviously realized for 6b but not for 6a.
Conclusions
In this work, a combination of “core fist” and “grafting-onto” approach was presented to prepare two uniform tetra-PCL-b-PEG star block copolymers with PCL cores of the same size but with PEG arms of different lengths. The synthesis was carried out by reacting a benzoxazinone-terminated tetra-PCL star with amino-terminated linear PEG. The polymers are characterized by a high degree of uniformity, which can be attributed to the use of structurally uniform starting components and to the quantitative conversion of the reacting end groups. Comprehensive studies of the synthesized star polymers by NMR and ATR-FTIR spectroscopy confirmed the quantitative conversion of the benzoxazinone-terminated tetra-PCL star polymer with the amino-terminated linear PEG chains. The expected molar masses as well as their uniformity were confirmed by MD-SEC measurements and MALDI-TOF MS. In addition, DSC, DLS and 1H NMR measurements in the selective solvent water provided insights into the amphiphilic character. After heating suspensions of the star block copolymers in water above the melting point of PCL (Tm = 45 °C), only the star block copolymer with m = 47 repeating units of PEG and n = 22 repeating units of PCL per arm formed a clear solution with spherical micellar structures in a wide concentration range. The suspension of the star block copolymer with m = 16 and n = 21 in water remained cloudy after heating above Tm. Here large structures form, presumably vesicles that allow recrystallization of the PCL cores upon cooling. DLS measurements of aqueous solutions of the larger star polymer within 6 hours after preparation indicate no formation of cylindrical vesicles. Cryo-TEM images show the formation of some cylindrical micelles after 6 days. This indicates that the spherical micelles are likely a metastable state where the samples lock in during cooling. The studies show that the length of the PEG chains can be used to tune size, stability, and crystallinity of micelles made of star block copolymers in water.With regard to the self-assembly behavior of the star block copolymers, the question arises as to the influence of the relatively large benzoxazinone linker group. In particular, the strong electron-withdrawing effect of the nitro group suggests the formation of strong dipole–dipole interactions at this structural unit. This type of interaction is also thought to occur in the synthesis of related amphiphilic networks and to be responsible for specific deviations in the network connectivities.23 This is the subject of further investigation using various spectroscopic methods directed at the specific interactions of these groups with each other and with the solvents used. In the case of the star polymers studied here, it is expected that in polar solvents such as water the dipole–dipole interactions are suppressed, so that the remaining hydrophobic interactions of the linker groups predominate. The extent to which this influences the association behavior of the star block copolymers in water or other solvents cannot be conclusively clarified from the present studies. Comparative studies with alternative linker groups are planned for this purpose.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed within the collaborative research project “Adaptive polymer gels with model network structure” (FOR2811), funded by the German Research Foundation (DFG), grant numbers 397384169, 423514254, 423478088 and 423373052. We thank Christina Harnisch and Alissa Seifert (IPF Dresden) for SEC measurements. Differential scanning calorimetry experiments were performed by Kerstin Arnold and ATR-FTIR spectroscopy measurements was done by Dr Mikhail Malanin.References
- Y. Zhang, T. Guan, G. Han, T. Guo and W. Zhang, Macromolecules, 2019, 52, 718–728 CrossRef CAS.
- A. S. R. Oliveira, P. V. Mendonça, S. Simões, A. C. Serra and J. F. J. Coelho, J. Polym. Sci., 2021, 59, 211–229 CrossRef CAS.
- P. Dong, X. Wang, Y. Gu, Y. Wang, Y. Wang, C. Gong, F. Luo, G. Guo, X. Zhao, Y. Wei and Z. Qian, Colloids Surf., A, 2010, 358, 128–134 CrossRef CAS.
- K. Ishizu and S. Uchida, Prog. Polym. Sci., 1999, 24, 1439–1480 CrossRef CAS.
- W. Burchard, in Branched polymers II, Springer, 1999, pp. 113–194 Search PubMed.
- A. J. Inglis, P. Pierrat, T. Muller, S. Bräse and C. Barner-Kowollik, Soft Matter, 2010, 6, 82–84 RSC.
- A. M. Bhayo, R. Abdul-Karim, S. G. Musharraf and M. I. Malik, RSC Adv., 2018, 8, 28569–28580 RSC.
- T. Öztürk, A. Kiliçlioğlu, B. Savaş and B. Hazer, J. Macromol. Sci., Part A: Pure Appl.Chem., 2018, 55, 588–594 CrossRef.
- A. Imdad, K. Faheem, R. Sana, P. Samina, A. Shakil, S. Muhammad Raza and M. Muhammad Imran, React. Funct. Polym., 2020, 150, 104553 CrossRef.
- X. Yan, J. Li and T. Ren, RSC Adv., 2018, 8, 29464–29475 RSC.
- Ö. Gökçe Kocabay and O. İsmail, Int. J. Polym. Mater. Polym. Biomater., 2021, 70, 328–337 CrossRef.
- A. Behl, V. S. Parmar, S. Malhotra and A. K. Chhillar, Polymer, 2020, 207, 122901 CrossRef CAS.
- H. Chunyan, C. Zhuo, W. Shengjie, H. Yanfeng, W. Hai, S. Hongfan, K. Deling, L. Xigang, W. Chun, Z. Linhua and Z. Dunwan, Chin. Chem. Lett., 2017, 28, 1905–1909 CrossRef.
- K. S. Shalaby, M. E. Soliman, G. Bonacucina, M. Cespi, G. F. Palmieri, O. A. Sammour, A. A. El Shamy, L. Illum and L. Casettari, Pharm. Res., 2016, 33, 2010–2024 CrossRef CAS PubMed.
- C. Lu, S.-R. Guo, Y. Zhang and M. Yin, Polym. Int., 2006, 55, 694–700 CrossRef CAS.
- C. Lu, L. Liu, S.-R. Guo, Y. Zhang, Z. Li and J. Gu, Eur. Polym. J., 2007, 43, 1857–1865 CrossRef CAS.
- S. Guo and Y. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 1412–1418 CrossRef CAS.
- Z.-X. Du, J.-T. Xu and Z.-Q. Fan, Macromol. Rapid Commun., 2008, 29, 467–471 CrossRef CAS.
- Z.-X. Du, J.-T. Xu and Z.-Q. Fan, Macromolecules, 2007, 40, 7633–7637 CrossRef CAS.
- J. J. Crassous, P. Schurtenberger, M. Ballauff and A. M. Mihut, Polymer, 2015, 62, A1–A13 CrossRef CAS.
- M. Alami-Milani, P. Zakeri-Milani, H. Valizadeh, R. Salehi and M. Jelvehgari, Iran. J. Basic Med. Sci., 2018, 21, 153 Search PubMed.
- N. Brigham, C. Nardi, A. Carandang, K. Allen and R. M. Van Horn, Macromolecules, 2017, 50, 8996–9007 CrossRef CAS.
- C. Bunk, L. Löser, N. Fribiczer, H. Komber, L. Jakisch, R. Scholz, B. Voit, S. Seiffert, K. Saalwächter, M. Lang and F. Böhme, Macromolecules, 2022, 55, 6573–6589 CrossRef CAS.
- M. Lang, R. Scholz, L. Löser, C. Bunk, N. Fribiczer, S. Seiffert, F. Böhme and K. Saalwächter, Macromolecules, 2022, 55, 5997–6014 CrossRef CAS.
- K. Hagmann, C. Bunk, F. Böhme and R. von Klitzing, Polymers, 2022, 14, 2555 CrossRef CAS PubMed.
- L. Jakisch, M. Garaleh, M. Schäfer, A. Mordvinkin, K. Saalwächter and F. Böhme, Macromol. Chem. Phys., 2018, 219, 1700327 CrossRef.
- H. Zhang, L. Jakisch, H. Komber, B. Voit and F. Böhme, Tetrahedron, 2013, 69, 3656–3663 CrossRef CAS.
- L. Jakisch, H. Komber and F. Böhme, Macromol. Mater. Eng., 2007, 292, 557–570 CrossRef CAS.
- L. Jakisch, H. Komber and F. Böhme, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 655–667 CrossRef CAS.
- S. Ishii, H. Kokubo, K. Hashimoto, S. Imaizumi and M. Watanabe, Macromolecules, 2017, 50, 2906–2915 CrossRef CAS.
- K. A. Barrera-Rivera, R. Vera-Graziano, E. López-Sánchez and A. Martinez-Richa, J. Polym. Res., 2015, 22, 1–8 CrossRef CAS.
- Y. Huang, Z. Xu, Y. Huang, D. Ma, J. Yang and J. W. Mays, Int. J. Polym. Anal. Charact., 2003, 8, 383–394 CrossRef CAS.
- S. Jain and F. S. Bates, Science, 2003, 300, 460–464 CrossRef CAS PubMed.
- K. Rajagopal, A. Mahmud, D. A. Christian, J. D. Pajerowski, A. E. Brown, S. M. Loverde and D. E. Discher, Macromolecules, 2010, 43, 9736–9746 CrossRef CAS PubMed.
- J. A. Zupancich, F. S. Bates and M. A. Hillmyer, Macromolecules, 2006, 39, 4286–4288 CrossRef CAS.
- P. P. Ghoroghchian, G. Li, D. H. Levine, K. P. Davis, F. S. Bates, D. A. Hammer and M. J. Therien, Macromolecules, 2006, 39, 1673 CrossRef CAS PubMed.
- J. Zhou, R. Ni and Y. Chau, RSC Adv., 2017, 7, 17997–18000 RSC.
- Y. Geng and D. E. Discher, J. Am. Chem. Soc., 2005, 127, 12780–12781 CrossRef CAS PubMed.
- J.-X. Yang, B. Fan, J.-H. Li, J.-T. Xu, B.-Y. Du and Z.-Q. Fan, Macromolecules, 2016, 49, 367–372 CrossRef CAS.
- A. M. Mihut, J. J. Crassous, H. Schmalz, M. Drechsler and M. Ballauff, Soft Matter, 2012, 8, 3163–3173 RSC.
- N. Fairley, B. Hoang and C. Allen, Biomacromolecules, 2008, 9, 2283–2291 CrossRef CAS PubMed.
- B. Bogdanov, A. Vidts, E. Schacht and H. Berghmans, Macromolecules, 1999, 32, 726–731 CrossRef CAS.
- B. Wunderlich, Macromolecular physics volume 3 Crystal Melting, Academic Press, New York, 2012 Search PubMed.
- C. P. Buckley and A. J. Kovacs, Colloid Polym. Sci., 1976, 254, 695–715 CrossRef CAS.
- C. Luo, W. Chen and Y. Gao, Polym. Sci., Ser. A, 2016, 58, 196–205 CrossRef CAS.
- P. M. Remiro, M. M. Cortazar, M. E. Calahorra and M. M. Calafel, Macromol. Chem. Phys., 2001, 202, 1077–1088 CrossRef CAS.
- R. De Juana and M. Cortazar, Macromolecules, 1993, 26, 1170–1176 CrossRef CAS.
- B. Bogdanov, A. Vidts, A. Van Den Buicke, R. Verbeeck and E. Schacht, Polymer, 1998, 39, 1631–1636 CrossRef CAS.
- P. C. Ashman and C. Booth, Polymer, 1975, 16, 889–896 CrossRef CAS.
- P. K. Kilpatrick, W. G. Miller and Y. Talmon, J. Colloid Interface Sci., 1985, 107, 146–158 CrossRef CAS.
- G. D. Smith, D. Y. Yoon, R. L. Jaffe, R. H. Colby, R. Krishnamoorti and L. J. Fetters, Macromolecules, 1996, 29, 3462–3469 CrossRef CAS.
- G. D. Smith, D. Y. Yoon and R. L. Jaffe, Macromolecules, 1993, 26, 5213–5218 CrossRef CAS.
- H. Lee, R. M. Venable, A. D. MacKerell and R. W. Pastor, Biophys. J., 2008, 95, 1590–1599 CrossRef CAS PubMed.
- Y. Chatani, Y. Okita, H. Tadokoro and Y. Yamashita, Polym. J., 1970, 1, 555–562 CrossRef CAS.
- L. Huang and K. Nishinari, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 496–506 CrossRef CAS.
- W. D. Mkandawire and S. T. Milner, Macromolecules, 2021, 54, 3613–3619 CrossRef CAS.
- M. Rubinstein and R. H. Colby, Polymer physics, Oxford university press, New York, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DLS of 6a and 6b, NMR spectra of 1, 3, 5, 6, 7, and 8, temperature dependent NMR spectra of 6a and 6b, MALDI-TOF and MD-SEC of 1, 3, 5, and 6. See DOI: https://doi.org/10.1039/d3py00078h |
| This journal is © The Royal Society of Chemistry 2023 |