
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural organic battery cathodes comprised of organic redox active polymers, reduced graphene oxide, and aramid nanofibers†
Suyash S.
Oka
 a,
Ratul Mitra
Thakur
a,
Alexandra D.
Easley
b,
Micah J.
Green
ab and
Jodie L.
Lutkenhaus
*ab
a,
Ratul Mitra
Thakur
a,
Alexandra D.
Easley
b,
Micah J.
Green
ab and
Jodie L.
Lutkenhaus
*ab
aArtie McFerrin Department of Chemical Engineering, Texas A&M University, College Station, TX 77843, USA. E-mail: jodie.lutkenhaus@tamu.edu; Tel: +1(979)-845-2682
bDepartment of Materials Science & Engineering, Texas A&M University, College Station, TX 77843, USA
First published on 9th September 2023
Abstract
Structural batteries are increasingly being studied because of their multifunctionality, combining good energy storage and mechanical properties. By storing energy within the object's structural elements, these may lead to substantial mass and volume savings in electrified transportation, cube satellites, and aerospace applications. However, most studies on structural batteries focus on conventional cathode materials such as lithium iron phosphate (LFP), lithium cobalt oxide (LCO), or nickel manganese cobalt oxide (NMC). These metal oxide active materials are challenged by their poor rate capabilities at higher C-rates, poor adhesion to current collectors, and, in some cases, materials supply chain issues. Organic radical polymers are a promising alternative because they exhibit rapid charge transfer at high C-rates, have improved adhesion, and are sourced from earth-abundant elements. In this work, structural battery cathodes using the redox-active polymer PTMA (poly(2,2,6,6-tetramethyl-piperidenyloxyl-4-yl methacrylate)) as the electrochemically active material are examined. PTMA-based slurries are thermally crosslinked to inhibit PTMA's dissolution on a structural current collector comprised of mechanically strong reduced graphene oxide (rGO) nanosheets and branched aramid nanofibers (BANFs). High rate capabilities at 25C-rate are observed owing to the fast redox reaction kinetics of PTMA, which led to an excellent specific power of 4310 W kg−1, which is higher than other commercial and structural electrodes. The structural electrode's specific modulus (4.33 GPa cm3 g−1) was superior to that of other commercial systems. Importantly, the PTMA-based active material did not delaminate from the structural electrode during cycling. This investigation focuses on developing structural organic battery electrodes that possess superior mechanical strength while not compromising on electrochemical performance. This work provides a pathway for utilizing earth-available, redox-active polymers as active materials in fast-charging, structural batteries.
1. Introduction
Structural batteries offer the possibility of storing energy within the structural bodies of transportation vehicles and aerospace technologies such as cube satellites and spaceships.1–4 These structural batteries can achieve significant mass and volume savings by reducing the need for separate energy storage and structural elements.1,2 As a requirement, the structural battery should comprehensively possess excellent energy storage performance, mechanical strength, modulus, and toughness to resist mechanical failure and deformation, i.e., a high degree multifunctional performance.1,2,5–8The majority of structural batteries use lithium-ion battery (LIB) chemistries with carbon fiber (CF) supports.9–13 LIBs themselves have high specific energies but cannot be utilized for structural energy storage without a mechanically supportive framework.6,9,14 Asp et al. investigated CF-based structural LIBs in which CF was used as the anode and commercial lithium iron phosphate (LFP) was used as the cathode along with an epoxy-based solid battery electrolyte (SBE)10,11,15,16 Pint et al. reported an LFP-coated CF fabric and fabricated a structural cathode lamina that showed superior tensile strength and modulus.13 Yao et al. reported a glass-ceramic Na3PS4-based high-modulus, solid-state electrolyte for sodium–sulfur (Na–S) batteries with a pyrene-tetraone (PTO) with Na3PS4-based cathode and Na15Sn4-based anode.17–19
Current structural LIBs use active materials such as LFP, nickel manganese cobalt oxide (NMC), and graphite with limited rate capabilities (<5C), preventing them from being used in fast-charging applications.5,8,9,12,13 Fast-charging batteries are important for electrified transportation and its widespread adoption,20 but finding an active material that can reversibly exchange charge at high C-rates remains a challenge. For example, non-structural electrodes have shown higher C-rates (>10C) by utilizing V2O5 with a conducting polymer21 or lithium nickel manganese oxide (LiNi0.5Mn1.5O4) on a free-standing carbon nanofiber (CNF) support,22 but the mechanical properties were not examined.
Organic batteries comprised of redox-active polymers are growing in interest because they do not rely upon potentially toxic and strategic elements such as cobalt or nickel that may cause supply chain limitations moving ahead.23–25 Because of their rapid redox reaction kinetics, redox-active polymers possess excellent fast-charging capabilities that could cater to growing electric vehicle and consumer electronics demand. Redox-active polymer-based organic batteries exhibit good rate capabilities as high as ∼650C,26 which are much higher than those of many inorganic LIBs.25,27 One such redox-active polymer is poly(2,2,6,6-tetramethylpiperidinyloxy-4-yl methacrylate) (PTMA), which consists of a 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) nitroxide radical pendant group on a methacrylate backbone.23,24 The redox chemistry of PTMA has been well-known for nearly 20 years.23,28 The nitroxide radical undergoes a redox reaction by converting to an oxoammonium cation during the charge/discharge process, exchanging an anion in the process, thus storing electrochemical energy.23,24 The theoretical capacity of PTMA (Ctheo) depends on the nitroxide radical content in the synthesized polymer and is thus 111 mA h g−1PTMA with a 100% radical functionalization.24,25 Nakahara et al., for the first time, reported energy storage using linear PTMA with a discharge capacity of 77 mA h g−1PTMA.23 Vlad et al. investigated the melt polymerization of TEMPO-methacrylate in the presence of nano-carbon and reported a capacity of 40 mA h g−1PTMA at a C-rate of 20C.29 Hatakeyama Sato et al. prepared a flexible battery device based on poly(2,2,6,6-tetramethyl-piperidinyloxy-4-yl acrylamide) (PTAm) and single-walled carbon nanotubes (SWCNTs) containing an aqueous electrolyte showing a capacity of 80 mA h g−1PTMA at 10C.26 Zhang et al. reported layered electrodes with covalently grafted pyrene on PTMA and rGO to extend π–π interactions between moieties that delivered 3600 W kg−1PTMA specific power at 20C.30 Suguro et al. prepared a different polymer poly(4-vinyloxy-2,2,6,6-tetramethylpiperidine-N-oxyl) (PTVE) bearing a TEMPO group, but with a vinyl ether backbone that demonstrated a high capacity of 114 mA h g−1PTVE at a low C-rate of 0.6C.31 All the organic battery electrode studies have focused on improving their electrochemical performance; however, none have examined their use in structural energy storage as electrodes that should possess superior mechanical strength and load-bearing capabilities.
Our group has also extensively studied PTMA's redox reaction for energy storage through multiple characterization techniques.32–35 We have previously reported a co-polymer of PTMA and glycidyl methacrylate (GMA) (PTMA–GMA) to improve PTMA's stability in battery electrolytes and characterized the effects of GMA content.34 PTMA–GMA's nitroxide radical FTIR spectral peak around 1355 cm−1 remained intact before and after curing, confirming the preservation of the active radical species during curing.34 Despite demonstrating a high discharge capacity of 104 mA h g−1 at 0.1C and 88 mA h g−1 at 1C, the electrochemical performance significantly diminished above 5C. The PTMA–GMA/Al foil electrodes demonstrated a discharge capacity of only 16 mA h g−1 at 10C, which could be due to the delamination occurring at the electrode-aluminum foil interface. However, the morphology of electrodes after galvanostatic cycling and their applicability as structural energy storage electrodes was not investigated. Wang et al. investigated the mechanism of energy storage in PTMA using electrochemical quartz crystal microbalance with dissipation monitoring (EQCM-D) by measuring PTMA's mass changes during charge and discharge.35 In that study, the conversion of PTMA to an oxoammonium cation (PTMA+) during charging was observed through the accompanying mass change by doping of by anion from the electrolyte.35 During discharging, the PTMA+ was converted back to PTMA with de-doping of the anion.35 However, this past work utilized metallic current collectors, which exhibited poor adhesion with PTMA, resulting in internal stresses and delamination during high-rate cycling.25,36,37 Also, there have been no reports of utilizing PTMA as a cathode active material in a structural battery platform.
Inspired by our past work on reduced graphene oxide (rGO)/branched aramid nanofiber (BANF) structural electrodes for supercapacitors and batteries,12,38–44 we hypothesized that the rGO/BANF platform could be utilized as a current collector. rGO is a two-dimensional (2D), carbon-based nanosheet that possesses good electrical conductivity and high chemical and thermal stability. BANFs are comprised of Kevlar® [poly(p-phenylene terephthalamide)] nanofibers, an ultra-strong, synthetic, para-aramid-based fiber, that exhibit an excellent tensile strength (∼3.8 GPa) and modulus (∼90 GPa). The rGO/BANF composite consists of structured rGO layers with randomly oriented BANFs laying flatly between them, with numerous hydrogen bonding and π–π interactions between the two materials that lead to good mechanical and electrical properties.38–45 We have previously proven the evidence of non-covalent interactions between rGO nanosheets and aramid nanofibers (ANFs) using XPS, where a small peak at 291 eV was attributed to non-covalent interactions.40,44 In a separate study, we predicted that PTMA, rGO, super P, and PMMA are mutually compatible due to having similar Hansen solubility parameters.32 Elsewhere, non-covalent interactions between PTMA and carbonaceous materials have been shown.26,32
Thus far, structural batteries have utilized inorganic mixed-metal oxides such as LFP, LCO, and NCM as battery cathode active materials, while graphite and silicon have been their anode counterparts.46–52 However, poor rate capabilities (i.e., low capacities at C-rates >4C) and significant capacity deterioration during long-term cycling remain important challenges to overcome.48,50,52 One reason for this detrimental performance fade is the poor adhesion of the active material to the CF, which leads to delamination because of stresses generated during the expansion and shrinkage of the active material during the lithium-ion intercalation process.20,52 For example, CF possesses an sp2-hybridized, highly graphitic structure that contributes to the poor adhesion with active materials due to the CF's hydrophobic surface. In contrast, the rGO/BANF composite contains residual hydrophilic groups that may provide superior adhesion.10 Further, a structural battery electrode using an organic redox-active polymer-based electrochemically active material has not been examined previously.
Here, we report the fabrication of structural cathodes using PTMA–GMA as the battery active material and rGO/BANF composite as a carbon-based conductive and mechanically strong current collector. More specifically, the free-standing rGO/BANF composite film was first prepared by vacuum filtration followed by thermal reduction. PTMA–GMA, conductive super P carbon black, and PMMA binder were slurry-cast onto the surface of rGO/BANF composites and thermally crosslinked to prepare the structural cathode. The electrochemical performance of these composite structural cathodes was studied as a function of composition in a lithium metal half-cell configuration. Adhesion between the PTMA–GMA electrode and the structural rGO-BANF current collector was examined using cross-sectional microscopy, which complimented long-term cycling results. The mechanical properties were examined using tensile testing, allowing for comparisons of energy, power, and modulus to the existing literature to understand the multifunctional behavior. Tradeoffs in the structural electrode using an organic redox-active polymer as the active material with mechanical properties and electrochemical performance are identified. Taken together, this study reveals how a structural current collector integrated with an organic redox-active polymer can benefit both fast-charging response and mechanical performance.
2. Materials and methods
2.1. Materials
2,2,6,6-Tetramethylpiperidin-4-yl-methacrylate (TMPM) was purchased from Tokyo Chemical Industry. Graphite was purchased from Bay Carbon, and Kevlar® 69 from Thread Exchange. 2,2′-Azobis(2-methylpropionitrile) (AIBN), glycidyl methacrylate (GMA), 3-chloroperoxybenzoic acid (mCPBA), potassium hydroxide (KOH), dimethyl sulfoxide (DMSO), poly(methyl methacrylate) (PMMA), 1 M lithium hexafluorophosphate (LiPF6) in ethylene carbonate (EC): diethyl carbonate (DEC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), and 2-n-butoxyethylacetate (BCA) were purchased from Sigma-Aldrich. AIBN was recrystallized using methanol by heating to 50 °C and cooling it to room temperature and dried under vacuum overnight at room temperature. Aluminum foil (0.75 mm × 19 mm) was purchased from Alfa Aesar. Whatman glassfiber membrane separator (∼0.21 mm) and Super P carbon were purchased from VWR. Lithium foil was procured from MTI.
:1, v/v), and 2-n-butoxyethylacetate (BCA) were purchased from Sigma-Aldrich. AIBN was recrystallized using methanol by heating to 50 °C and cooling it to room temperature and dried under vacuum overnight at room temperature. Aluminum foil (0.75 mm × 19 mm) was purchased from Alfa Aesar. Whatman glassfiber membrane separator (∼0.21 mm) and Super P carbon were purchased from VWR. Lithium foil was procured from MTI.
2.2. Materials synthesis
PTMA–GMA was synthesized as reported previously.23,34 Briefly, TMPM monomer was polymerized using a free-radical process using AIBN at 60 °C in the presence of 1% GMA as a crosslinker in the reaction mixture. The PTMPM-GMA copolymer was oxidized using mCPBA to the nitroxide radical functionality to obtain PTMA–GMA, which was then vacuum dried. PTMA–GMA's dispersity was 2.62 and number average molecular weight (Mn) was 39235 g mol−1, measured using gel permeation chromatography (GPC) using 1 mg mL−1 solution of PTMA–GMA in tetrahydrofuran (THF) as a solvent. EPR spectroscopy revealed a radical content of about 70% (Fig. S1, ESI†). Graphene oxide (GO) was synthesized from graphite using a modified Hummers’ method, as reported before,44 and was dispersed in DMSO at a concentration of 1 mg mL−1. This process yielded multi-layered GO sheets of about 10–15 μm lateral size.44 BANFs were fabricated by the dissolution of chopped Kevlar® threads (0.5 g) in DMSO (50 mL) and KOH (0.5 g), as previously reported. The mixture was stirred for 14 days until a dark red dispersion (10 mg mL−1) was obtained. BANFs prepared in this manner are 30–40 nm in diameter and 5–10 μm in length.12,44 This highly viscous dispersion was diluted to 0.2 mg mL−1 dispersion. Then, 50 mL of BANF/DMSO (0.2 mg mL−1) and 30 ml of GO/DMSO (1 mg mL−1) dispersions were mixed and stirred for 1 h at room temperature and then heated at 80 °C for 2 h, followed by vacuum filtration to obtain free-standing GO/BANF (75/25 wt/wt%) films. They were dried overnight at ambient conditions, then under vacuum at 80 °C for 3 days, and last thermally reduced at 200 °C for 2 h under vacuum to obtain reduced graphene oxide/BANF (rGO/BANF) films.
2.3. Structural cathode fabrication
To make structural cathodes, PTMA–GMA, PMMA, and Super P carbon with a mass ratio of PTMA–GMA:Super P:PMMA = 50:40:10 were homogenized using a hand-held mortar and pestle and were wet-mixed to prepare a slurry using BCA as the solvent. The resulting slurry was doctor-bladed onto an rGO/BANF film using an automated film applicator (Elcometer 4340 automatic applicator) with a blade thickness of 120 μm at room temperature (∼23 °C). Electrodes of two other compositions (PTMA–GMA/Super P/PMMA = 30/60/10 wt/wt% and PTMA–GMA/Super P/PMMA = 70/20/10 wt/wt%) were also fabricated using the slurry casting method for composition tests. The electrodes were air-dried at ambient conditions for 1 h and then vacuum-dried at room temperature overnight. The PTMA–GMA coated on rGO/BANF film electrodes were cross-linked at 175 °C for 3 h to inhibit the dissolution of PTMA in battery electrolytes and enhance its electrochemical stability.
2.4. Physical and chemical characterization
The electrode thickness after drying was measured using a height gauge (TESA μ-HITE) instrument. Cross-sectional scanning electron microscopy (SEM) was performed using a JEOL SM-7500 SEM. X-ray photoelectron spectroscopy (XPS) was performed using an Omicron ESCA probe with Mg Kα radiation (hν = 1253.6 eV). Survey scans were performed with 1.0 eV steps and dwell time = 50 ms within a range of 10–1100 eV. High-resolution scans were performed with 0.05 eV steps and dwell time = 250 ms. A Shirley-type background correction was applied, and curve fitting was performed using a Gaussian–Lorentzian peak shape. C 1s peak for sp2-hybridized carbon atoms (284.5 eV) was used to calibrate all XPS spectra.2.5. Mechanical characterization
The electrodes were cut into 2 mm × 20 mm rectangular strips for tensile testing. The thickness of all PTMA–GMA/rGO/BANF samples was about 21–27 μm, PTMA–GMA/Al foil samples was about 29–33 μm, while the uncoated rGO/BANF film was 13–15 μm, and the uncoated Al foil was about 20–21 μm. Static uni-axial tensile tests were performed using a DMA Q800 (TA Instruments) at constant rate strain mode with a strain rate of 0.1% min−1, at a preload force of 0.01 N at room temperature with 35–45% relative humidity. The elastic region in the stress vs. strain curves was used to calculate the tensile modulus of samples.2.6. Electrochemical characterization
The PTMA–GMA active material loading in all electrodes was 0.9–1.0 mg cm−2. The PTMA–GMA on rGO/BANF electrodes were cut into 16 mm diameter circular discs using a die cutter (MTI) and tested in a half-cell configuration using a lithium metal foil as the reference and counter electrode (diameter 14 mm, thickness = 0.75 mm, mass = 0.07 g) in a CR-2032-coin cell at room temperature (∼23 °C). A Whatman glassfiber (GF) membrane was used as a separator (diameter = 16 mm, thickness = 0.21 mm). The coin cell comprised of a stainless-steel disk (diameter = 16 mm, thickness = 1 mm, and mass = 2.5 g) spacer along with a stainless-steel spring (diameter ≈ 16 mm, thickness ≈ 1 mm, and mass = 0.7 g), stainless steel top and bottom shells (diameter = 7.6 cm, thickness ≈ 16 mm, and mass ≈ 437 g) and a polypropylene (PP) gasket. 160 μL of 1 M LiPF6 in EC:DEC (1:1 v/v) was used as the electrolyte. All coin cells were assembled in an MBraun glovebox with an inert environment (99.998% Ar) with O2 and moisture at ≤0.1 ppm each. The lithium foil anode, separator, PTMA–GMA structural cathode, and electrolyte were stacked and crimped at 1000 psi. PTMA–GMA structural cathodes were tested in a potential CV window of 3–3.9 V vs. Li/Li+. Before testing, all cells were conditioned using cyclic voltammetry (CV) at 5 mV s−1 for 10 cycles, followed by 3 cycles of constant current–constant voltage (CC–CV) at 0.1C and 3.8 V during charging and −0.1C and 3.4 V during discharging. CV was performed at a scan rate of 1 mV s−1 to identify the redox behavior of PTMA from the oxidation and reduction reaction potentials and the peak separation. Charge–discharge currents for each C-rate were calculated from the theoretical capacity of PTMA (111 mA h g−1). Galvanostatic charge/discharge (GCD) cycling was carried out at varying C-rates (5 cycles each at 1–25C, and then repeated at 1C) for rate capability testing, and 500 cycles at a C-rate of 5C were performed for long-term performance testing using a potentiostat (Solartron Interface 1287). CVs were also taken after rate capability and long-term performance GCD testing to verify the stability of PTMA to extreme fast charging currents and extended cycling. Electrochemical impedance spectroscopy (EIS) was conducted on fresh and galvanostatically tested electrodes using a Gamry interface 1000. A 10 mV AC amplitude with a frequency range from 1000 kHz to 10 mHz at 3.66 V vs. Li/Li+ was used.
3. Results and discussion
PTMA–GMA, super P conductive additive, and PMMA binder were uniformly coated on the surface of rGO/BANF films using a doctor blade and thermally crosslinked at 175 °C for 3 h to obtain PTMA–GMA structural cathodes, Fig. 1A. The PMMA binder was demonstrated to possess Hansen solubility parameters similar to PTMA, PTMA+, and super P carbon and hence was chosen instead of the conventional poly vinylidenefluoride (PVdF) or poly tetrafluoroethylene (PTFE).32 The thickness of the rGO/BANF film was 13–15 μm, and the thickness of the PTMA–GMA coating was 8–12 μm. The cross-sectional image in Fig. 1A shows a typical morphology, with a uniform PTMA–GMA composite electrode atop a layered rGO/BANF film. We first sought to identify electrodes that bore the highest active material loading, while balancing the electrochemical performance. If the PTMA–GMA loading is too low, the overall specific energy of the electrode would be diminished. However, if the PTMA–GMA loading is too high, the conductivity of the electrodes would suffer, and the resulting energy storage performance would be poor. Therefore, electrode compositions of 30, 50, and 70 wt% PTMA–GMA (with the balance being super P and PMMA) on rGO/BANF films were examined. The presence of PTMA–GMA for each electrode composition was verified using X-ray photoelectron spectroscopy (XPS), Fig. 1B and Table S1 (ESI†). The carbon atomic weight % decreased, and the nitrogen and oxygen content increased in samples containing 30 wt% PTMA–GMA to 70 wt% PTMA–GMA, confirming the increased presence of active material. High-resolution XPS spectra of 50 wt% PTMA–GMA on rGO/BANF films for N 1s, C 1s, and O 1s peaks are shown in Fig. 1C–E, respectively, Fig. S2 (ESI†) shows spectra for other electrode compositions. The high-resolution N 1s peak was deconvoluted to show the presence of the nitroxide radical (˙N–O, 401 eV) and the oxoammonium cation group (+N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 405.6 eV), further confirming the presence of PTMA–GMA.
O, 405.6 eV), further confirming the presence of PTMA–GMA.
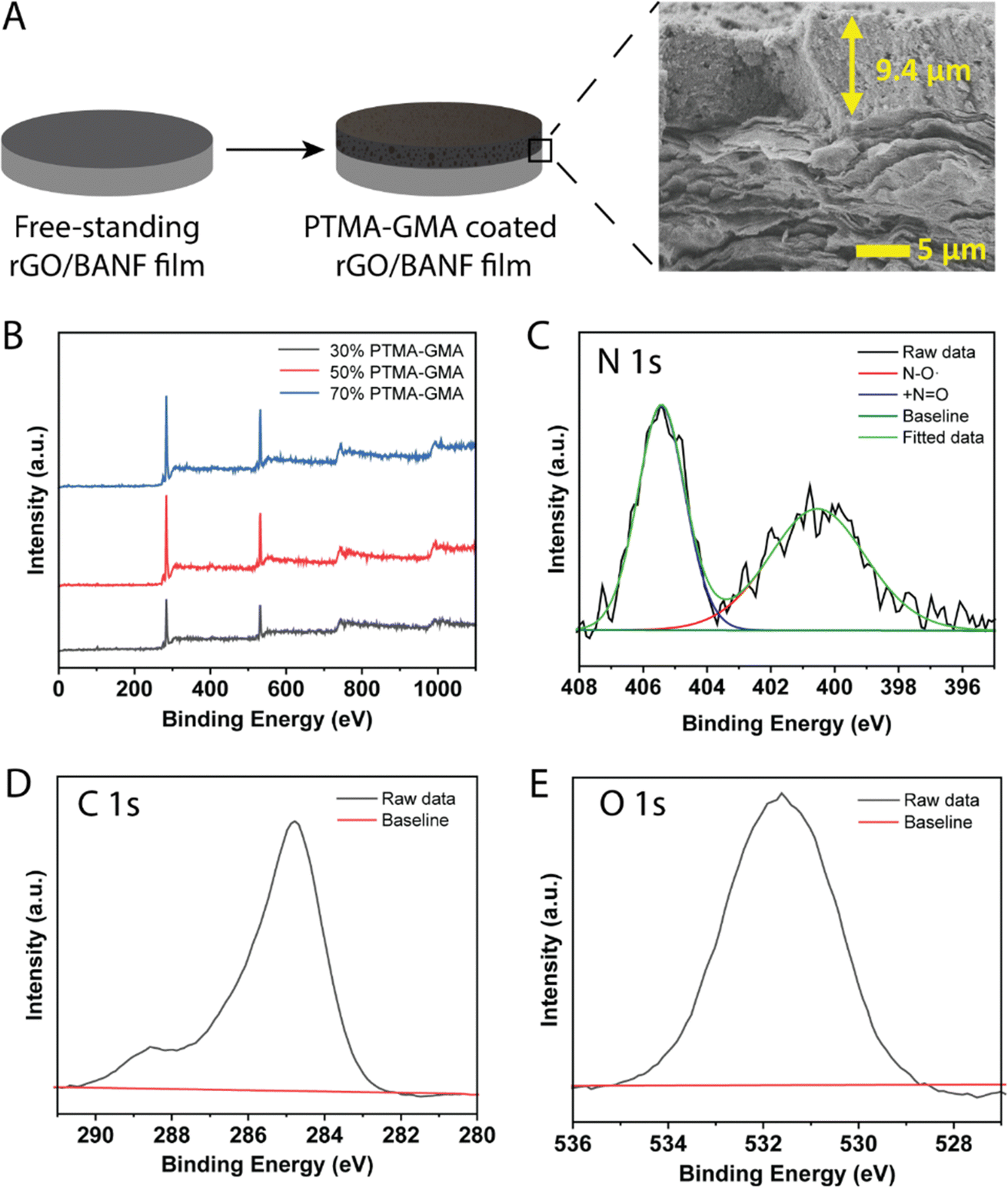 | ||
| Fig. 1 (A) Schematic representation of structural cathode preparation. (B) XPS Survey scans for 30 wt%, 50 wt%, 70 wt% PTMA–GMA on rGO/BANF. High-resolution XPS spectra for 50 wt% PTMA–GMA on rGO/BANF (C) N 1s peak with deconvolution, (D) C 1s peak, and (E) O 1s peak. | ||
The electrochemical performance of PTMA–GMA-based structural cathodes was investigated in a two-electrode lithium metal half-cell using 1 M LiPF6 in EC/DEC (1:1 v/v) as the electrolyte. All data are reported based on the PTMA–GMA mass loading and the mass of PF6− counter-anion is not considered in the calculations. Based on a nitroxide radical content of 70% from EPR spectroscopy (Fig. S1, ESI†), the maximum achievable discharge capacity was about 77 mA h g−1PTMA.34 The rate capability performance was studied for varying compositions 30 wt% PTMA–GMA, 50 wt% PTMA–GMA, and 70 wt% PTMA–GMA on rGO/BANF films (Fig. 2). The PMMA binder content was constant throughout these compositions at 10 wt%, with the balance being super P carbon. Fig. 2A demonstrates the discharge capacities at different C-rates from 1C to 25C. Both 30 wt% and 50 wt% PTMA–GMA electrodes showed good rate capabilities up to 25C where they retained 79% and 77% of their capacities relative to that of 1C; these electrode compositions also recovered 100% of their initial capacities upon returning to 1C. However, the capacity of the 70 wt% PTMA–GMA electrodes faded significantly above 2C, where they retained only 48% capacity of their initial 1C capacity at 5C and failed to demonstrate any capacity above 5C. This could be due to an insufficient carbon content, which leads to poor conductivity.
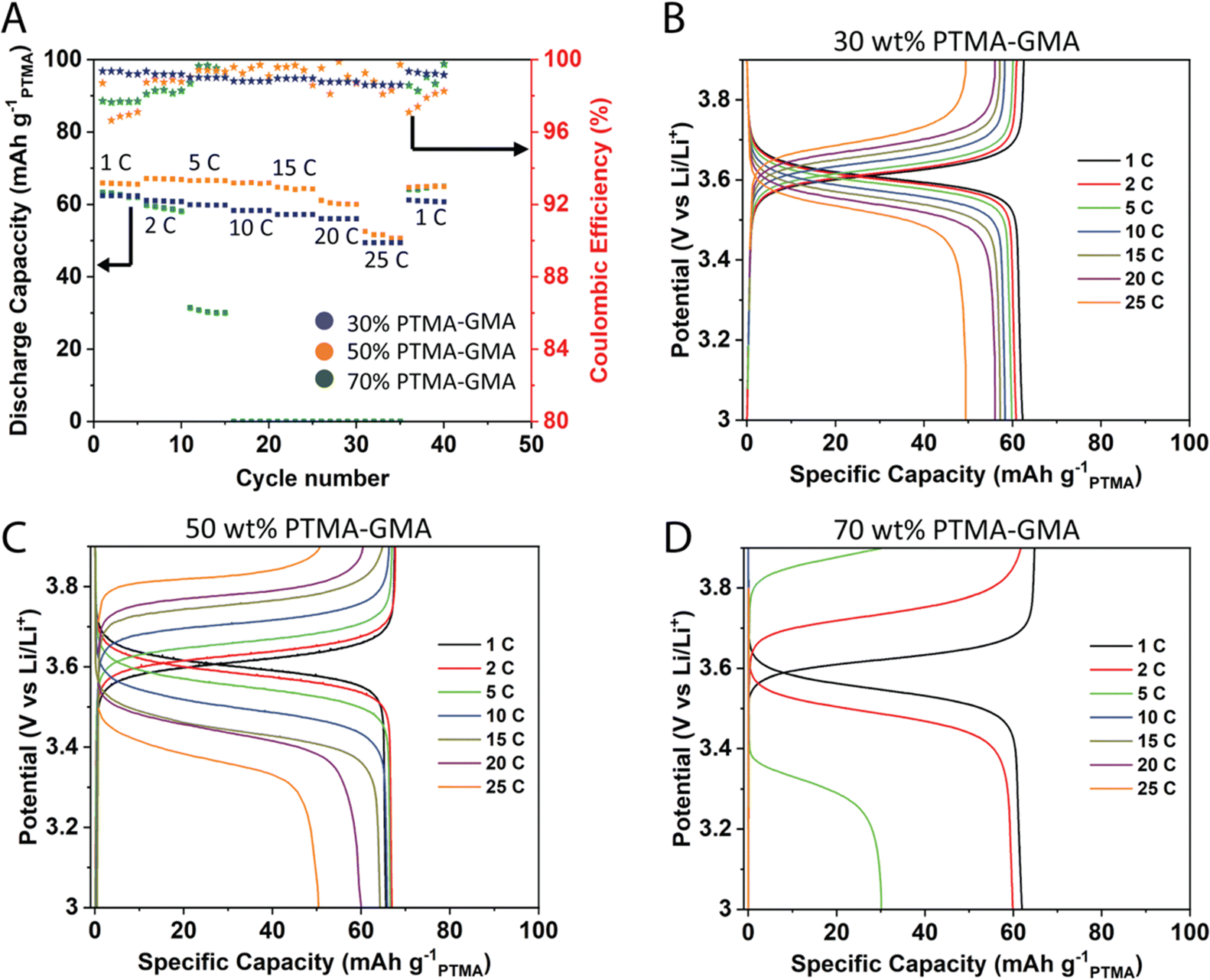 | ||
| Fig. 2 (A) GCD testing showing different C-rates for 30 wt% PTMA–GMA, 50 wt% PTMA–GMA, and 70 wt% PTMA–GMA electrodes on rGO/BANF. GCD curves at different C-rates for (B) 30 wt% PTMA–GMA, (C) 50 wt% PTMA–GMA, (D) 70 wt% PTMA–GMA electrodes on rGO/BANF. The cell configuration was a lithium metal half-cell, and the electrolyte was 1 M LiPF6 in EC/DEC (1:1 v/v) as the electrolyte. Specific capacities are reported based on the PTMA–GMA mass loading. | ||
Fig. 2B–D shows the GCD curves for the three electrode compositions. The difference between the charging and discharging plateau voltages is a qualitative measure of the reversibility in the cell. The difference in plateau voltages was the least for 30 wt% PTMA–GMA electrodes (17.5 mV at 2C) and largest for 70 wt% PTMA–GMA electrodes (255 mV). This observed trend is attributed to the relative super P carbon content in the electrode, in which the 30 wt% PTMA–GMA electrode contained the most super P carbon and was, therefore, the most conductive. Taken together, the 50 wt% PTMA–GMA electrode possessed the best combination of rate capability and active material loading and was, therefore, selected for further investigation. This may be attributed to the good balance of conductivity (due to having sufficient carbon additive) and crosslinking of the network (due to having sufficient PTMA–GMA).
Fig. 3A demonstrates the rate capability performance for the two electrodes in the same cell configuration described above. To compare the rGO/BANF current collector to a more traditional current collector, GCD was aslo performed for 50 wt% PTMA–GMA coated on rGO/BANF and aluminum (Al) foil. The PTMA–GMA structural cathode exhibited a discharge capacity of 66 mA h g−1 at a C-rate of 1C. It also showed an 81% capacity retention at a high C-rate of 25C compared to 1C, Fig. S3a (ESI†). On Al foil, the 50 wt% PTMA–GMA showed a discharge capacity of about 60 mA h g−1 at 1C (10% lower than rGO/BANF film substrate) and capacity retention of only 72%. Fig. 3B and Fig. S3b (ESI†) shows the long-term GCD cycling performance at 5C to examine the longevity of the two electrodes. After 500 cycles, the PTMA–GMA structural cathode retained 93% of its initial capacity, whereas the electrode with PTMA–GMA on Al foil retained only 80% of its initial capacity. The final specific capacities for PTMA–GMA on rGO/BANF and on Al foil were 56 mA h g−1 and 42 mA h g−1. Fig. 3C and D show CV profiles for the 50 wt% PTMA–GMA cathode on rGO/BANF and on Al foil, respectively, before and after rate capability testing. Before testing, in an unconditioned state, both electrodes exhibited larger ΔEp values. The inadequate utilization of PTMA–GMA active material plays a role in the larger ΔEp values in the initial CV cycles. However, after conditioning and galvanostatic testing, as the electrolyte can fully penetrate the electrode and can more easily access the electrode structure, both electrodes exhibited redox peaks of smaller ΔEp values. The retention of the general peak shape and half-wave potential suggests that the active material did not decompose during the process. The ΔEp values changed from 0.284 V before testing to 0.178 V after testing for the 50 wt% PTMA–GMA on rGO/BANF; for Al foil as the current collector, the ΔEp values changed from 0.37 V to 0.27 V. A lower ΔEp value signifies superior reversibility of the redox process, implying that the rGO/BANF, as a substrate, improved the reversibility of the PTMA–GMA electrode.
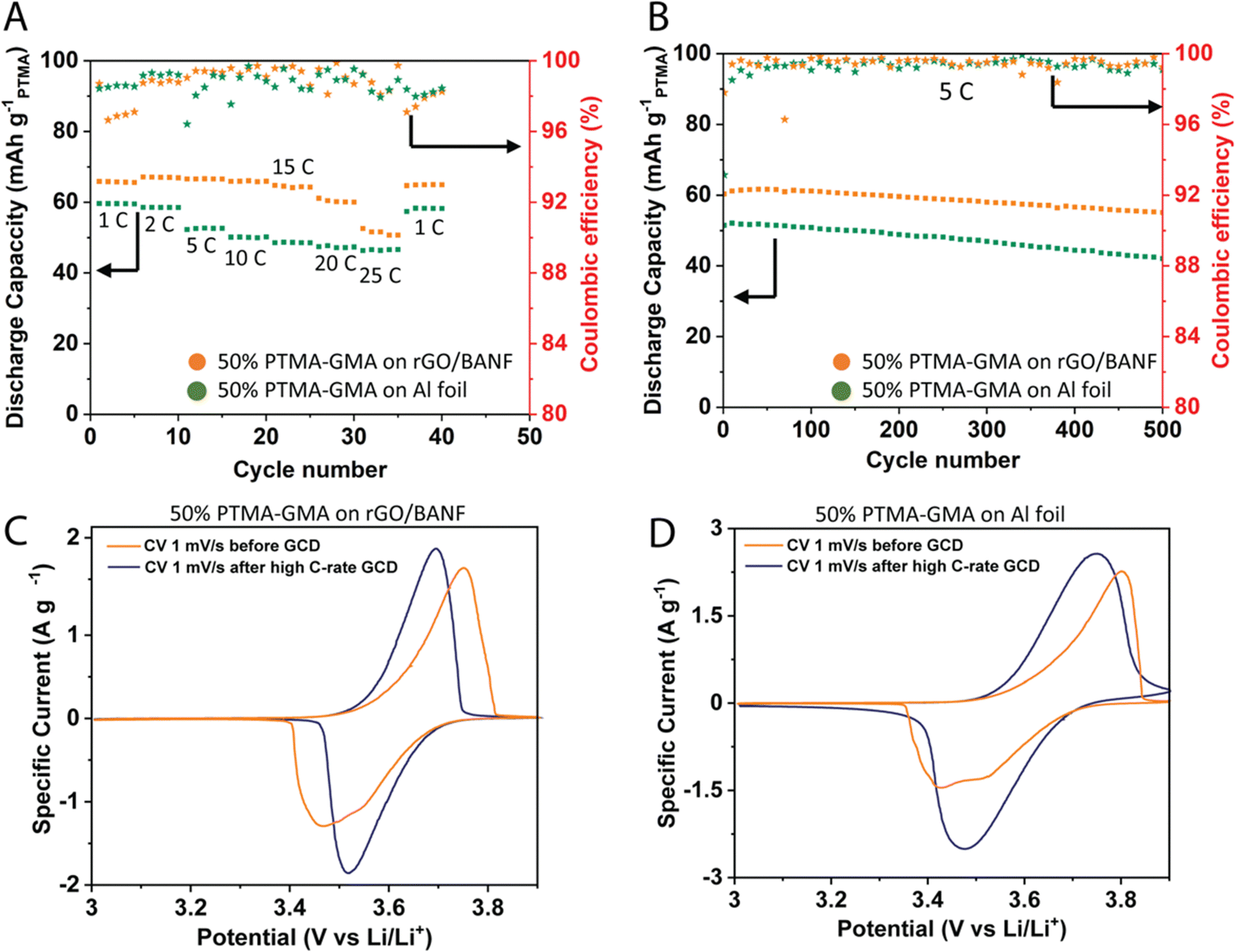 | ||
| Fig. 3 (A) GCD testing at different C-rates for 50 wt% PTMA–GMA on rGO/BANF film, and aluminum foil. (B) Long-term GCD testing at 5C rate for 50 wt% PTMA–GMA on rGO/BANF film, and aluminum foil. CV before GCD testing, and after different C-rate GCD testing for 50 wt% PTMA–GMA on (C) rGO/BANF and (D) aluminum foil. | ||
This excellent rate capability and long-term cycling capacity retention could be due to superior adhesion between PTMA–GMA slurry and rGO/BANF film substrate through π–π interactions that prevent the delamination of active material from the current collector. The BANF backbone in the current collector may be able to bear and transfer loads from internal stresses that result during the charge–discharge cycling of electrodes. The rGO/BANF film outperforms aluminum foil (Fig. 3A and B) as a structural current collector because of its superior cohesive properties with polymeric active materials like PTMA–GMA. Fig. S4a and b (ESI†) demonstrates the GCD rate capability performance at varying C-rates (1–25C), and the CV profiles before and after high C-rate GCD cycling respectively of the 30 wt% PTMA–GMA on rGO/BANF film. It showed a 21% capacity fade at 25C as compared to 1C, while retaining 93% of its initial capacity during repeated cycling at 1C. The CV comparison showed the stability of PTMA–GMA before and after high C-rate GCD for this electrode composition. Fig. S4c and d (ESI†) demonstrates the GCD rate capability performance at varying C-rates, and the CV profiles before and after high C-rate GCD cycling respectively of the 70 wt% PTMA–GMA on rGO/BANF film. Even though it retained 100% of its initial capacity during repeated cycling at 1C, the rate capability at higher C-rates was poor with this 70% active material electrode configuration wherein it only retained 50% capacity at 5C as compared to 1C and lost all its capacity from 10C and above. This could be due to poor conductivity and insufficient quantity of carbon in the electrode to provide electron conductive pathways.
In a comparable previous study, the electrochemical performance of Al foil-based electrodes diminished largely above 5C-rate and those electrodes were not investigated for morphology changes, or delamination.34 On the contrary, in the current study, the structural integrity of the structural and Al foil-based cathodes was examined using cross-sectional SEM (Fig. 4A–D) after rate capability testing. For rGO/BANF as the substrate, some swelling of the PTMA–GMA coating due to wetting by the electrolyte was observed, wherein, after cycling, the thickness of the coating increased from 9.4 μm to 13.6 μm. However, any expansion or swelling of the rGO/BANF current collector was not observed. In comparison, the thickness of PTMA–GMA on Al foil increased much more from 9.2 μm to 17 μm. Notably, Fig. 4C and D shows evidence of delamination of PTMA–GMA from the Al foil current collector, pointing to poor adhesion between the two. Complete delamination was not observed, but partial delamination sites were evident in the cross-sections. Partial delamination may result from localized stresses generated during cycling that propagate further each cycle. This may be the reason why delamination was observed on the aluminum foil, but only a minor increase in charge transfer resistance was observed in the EIS spectra. Even a localized region becoming electrochemically inaccessible by losing intimate contact with the current collector could result in a diminished GCD performance and this may be the case with Al foil electrodes. Similarly, cross-sectional SEM images after long-term cycling for 500 cycles at 5C also show delamination for Al current collectors, Fig. S5 (ESI†). Taken together, the superior adhesion between PTMA–GMA and the rGO/BANF substrate promotes improved cycling behavior, as evidenced in Fig. 3A and B.
 | ||
| Fig. 4 Cross-sectional scanning electron microscopy images of 50 wt% PTMA–GMA slurry coated on rGO/BANF film (A) before and (B) after rate capability cycling. Similarly, for 50 wt% PTMA–GMA slurry coated on Al foil, (C) before and (D) after rate capability cycling. | ||
Fig. 5A and B shows the EIS spectra at the half-wave potential (E1/2) for 50 wt% PTMA–GMA on rGO-BANF before and after rate capability testing, Fig. 5B shows similar EIS spectra for the Al foil control sample. The fitted equivalent circuit is demonstrated in Fig. 5C. It is challenging to interpret the origin of the second, low-frequency semi-circle, which could be due to inhomogeneities. After rate capability testing, RCT1 decreased from 97 Ω to 50 Ω because of electrode conditioning and high utilization of active material during the GCD process, and RCT2 decreased from 54 Ω to 18 Ω because of the conditioning process in which the electrolyte was able to further penetrate the PTMA–GMA coating. On the contrary, for PTMA–GMA coated on Al foil, the resistance increased from 70 Ω to 82.8 Ω. The lower charge transfer resistance for PTMA–GMA on rGO/BANF is a result of the better adhesion between the two, which facilitates electron transfer across the PTMA–GMA|rGO/BANF interface.
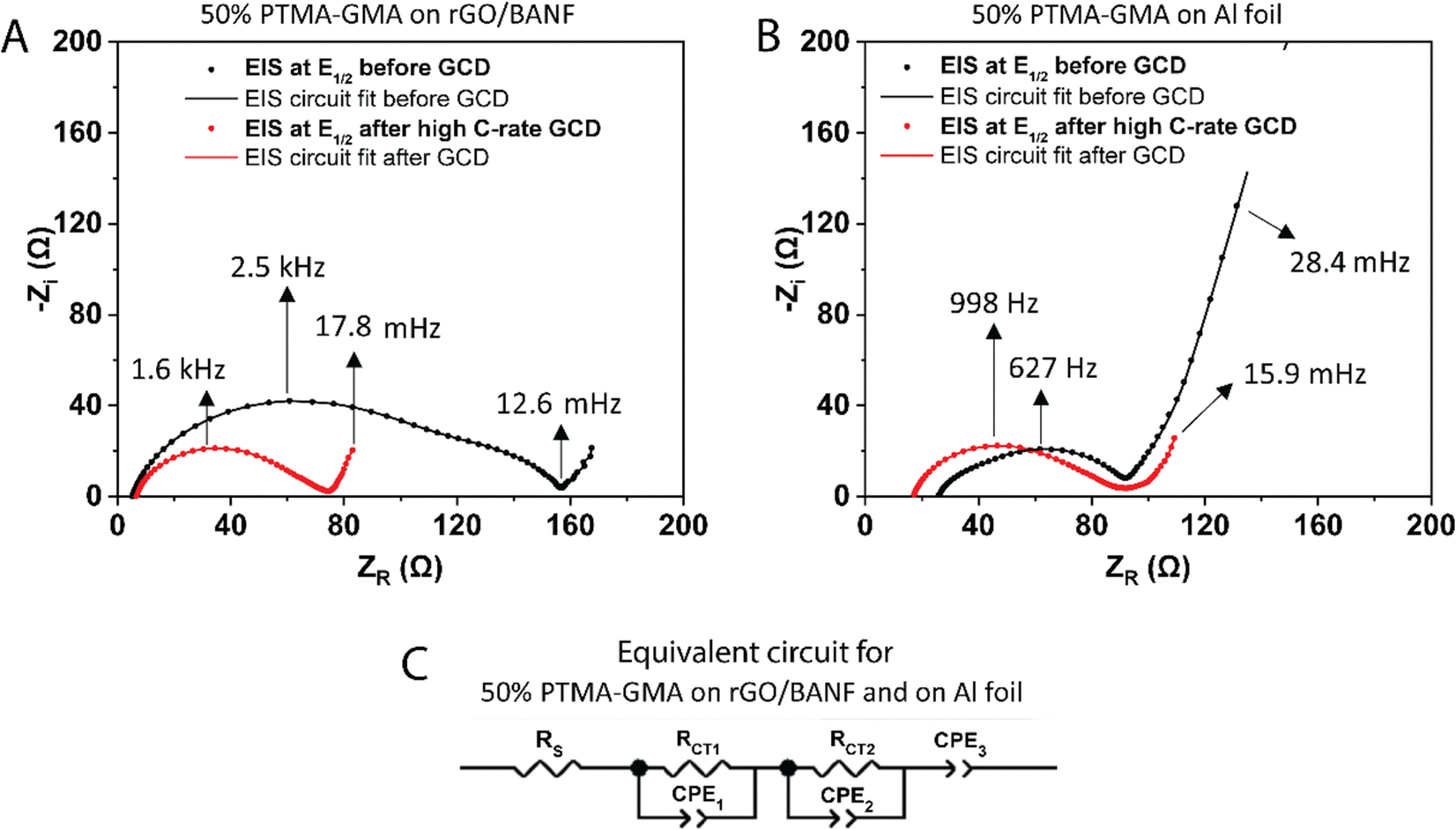 | ||
| Fig. 5 EIS at the half-wave potential (E1/2 = 3.66 V) for 50 wt% PTMA–GMA on (A) rGO/BANF and (B) Al foil before and after rate capability testing. (C) Fitted equivalent circuit for 50 wt% PTMA–GMA on rGO/BANF and Al foil. | ||
The mechanical properties of PTMA–GMA-based structural cathodes were evaluated using tensile testing, and representative stress vs. strain curves are shown in Fig. 6A. Fig. 6B and C shows box plots of the ultimate tensile strength (UTS) and Young's modulus, respectively, comparing 50 wt% PTMA–GMA on rGO/BANF, 50 wt% PTMA–GMA on Al foil, rGO/BANF alone, and Al foil alone. The 50 wt% PTMA–GMA on rGO/BANF showed a 10% higher ultimate tensile strength as compared to 50 wt% PTMA–GMA on Al foil. This may be a result of the BANFs in the structural current collector, which possess superior mechanical properties owing to hydrogen bonding and π–π interactions with rGO12,38–45 that render the composite film mechanically stronger. Further, good adhesion between PTMA–GMA and the structural current collector likely contributed to the improved UTS. As for the modulus, rGO/BANF current collectors generally exhibited a lower Young's modulus compared to Al foil, but delamination of PTMA–GMA from Al foil remained an issue.
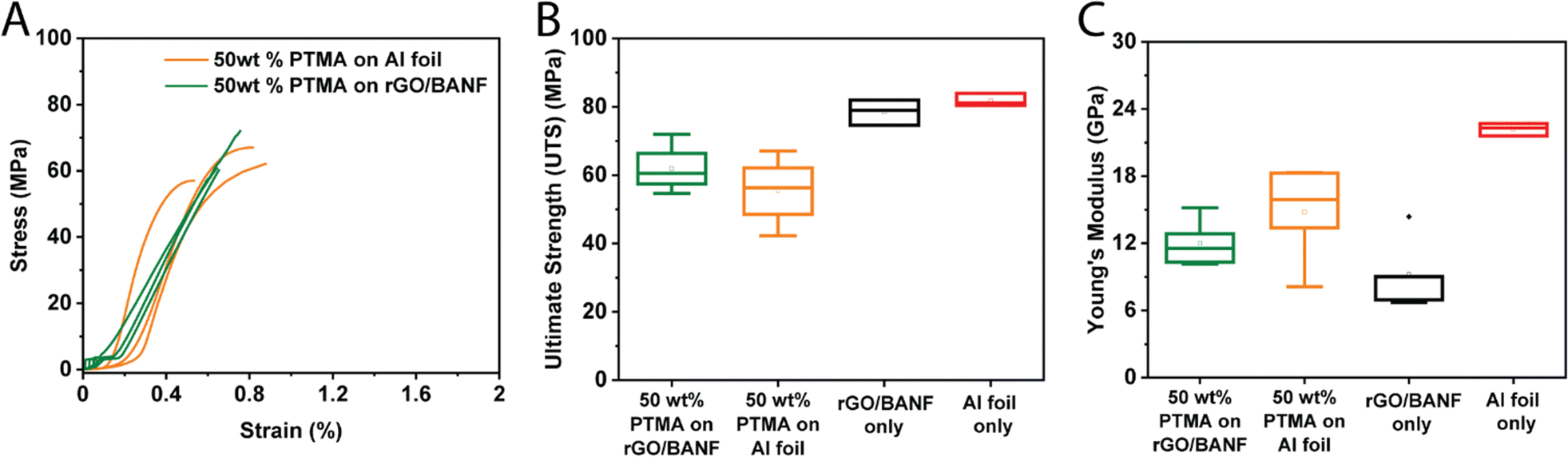 | ||
| Fig. 6 (A) Representative stress–strain curves for 50 wt% PTMA–GMA on rGO/BANF and Al foil. Box plots for (B) ultimate tensile strength (UTS) and (C) Young's modulus, comparing 50 wt% PTMA–GMA on rGO/BANF or aluminum foil, rGO/BANF alone, and Al foil alone. The rectangular shapes extend from the first to the third quartile. The open squares represent the mean, whereas the line inside the rectangular shapes represents the median. The whiskers indicate the maximum and minimum values. Each box corresponds to 7–9 data points. | ||
To illustrate the combined multifunctional features of this structural organic battery cathode, we compared the specific modulus, specific energy, and specific power of 50 wt% PTMA–GMA on rGO/BANF with other structural electrode-level investigation reports10,12,14,53–58 that compute these values in a similar way in an Ashby plot and a Ragone plot (Fig. 7A and B, respectively). More specifically, PTMA–GMA on rGO/BANF structural cathodes were compared against CF-impregnated LiS2,56 rGO/BANF/LFP,12 commercial LFP,55 commercial NCM,53 commercial graphite,54 CF/LFP/structural battery electrolyte (SBE) pouch cell,10 CF/LCO,14 extruded carbon fiber reinforced polymer (CFRP) composite anode,57 and CF/LFP,58 Table S2 (ESI†). Our PTMA–GMA on rGO/BANF structural cathodes demonstrated a specific modulus of 4.33 GPa cm3 g−1 (Fig. 7A), which was far higher than that of commercial NMC, LFP, LCO, and graphite electrodes (highest reported was 1 GPa cm3 g−1).53 Only CF-based structural electrodes showed a higher specific modulus of ∼10 GPa cm3 g−1.10,56 The 50 wt% PTMA–GMA-based structural organic battery cathode demonstrated its highest specific energy of 231 W h kg−1 at 1C, which was slightly lower than that of commercial lithium-ion battery cathodes (>450 W h kg−1).53 The lower specific energy for PTMA–GMA is not surprising because its theoretical capacity is lower than that of commercial active materials. However, PTMA possesses extremely fast redox kinetics, allowing it to charge and discharge at higher C-rates.23,34,35 Shown in Fig. 7B, our PTMA–GMA on rGO/BANF structural cathodes demonstrated its highest specific power of 4310 W kg−1 at a C-rate of 25C, which was superior to other commercial (∼600 W kg−1) or CF-based structural electrodes (50 W kg−1).53,55,57,58 We also compared the electrochemical performance to other non-structural TEMPO-based organic battery cathode reports, Table S2 (ESI†).23,26,29–31
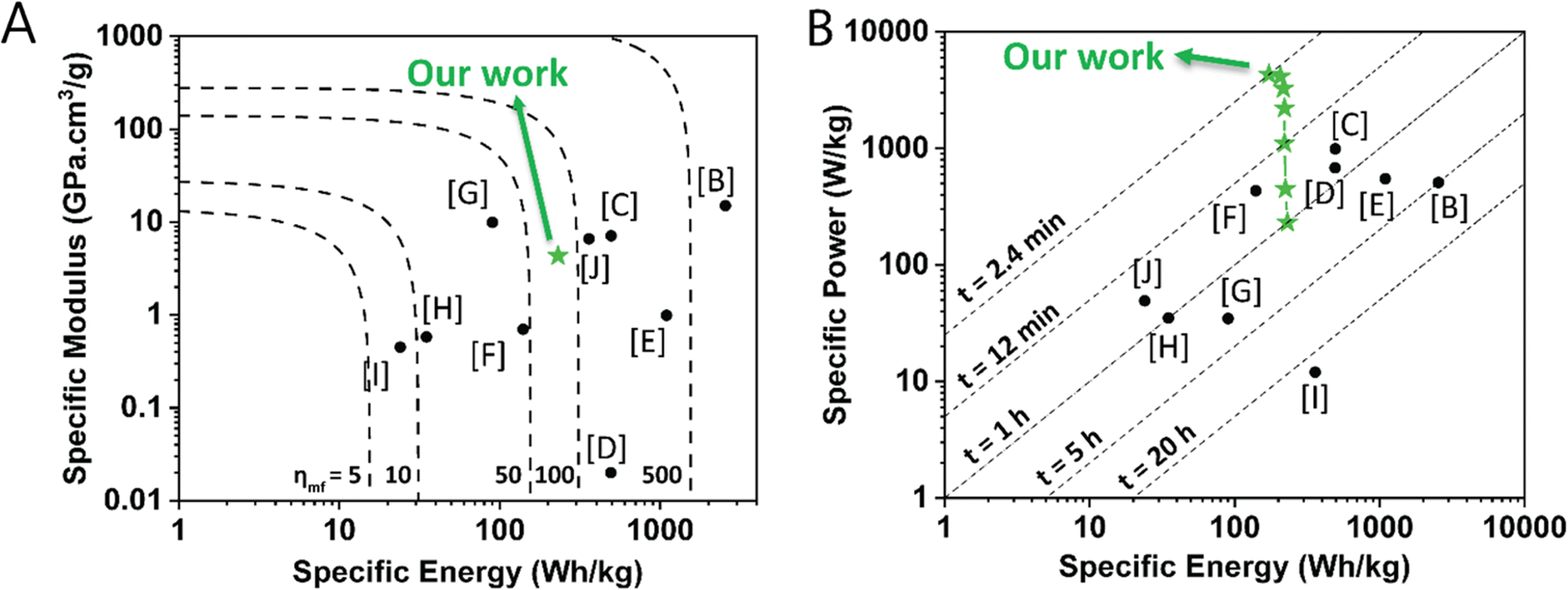 | ||
| Fig. 7 (A) Ashby plot comparing specific modulus vs. specific energy (dashed lines indicate multifunctional efficiency) and (B) Ragone plot comparing specific power vs. specific energy (dashed lines indicate charging times) of the 50 wt% PTMA–GMA on rGO/BANF structural cathode (green stars) with other structural electrodes from the literature. All values were calculated and normalized based on active material mass (e.g., PTMA–GMA in our case). The letters refer to the reference; further details are provided in Table S2 (ESI†). | ||
This work highlights several areas for further improvement. Specifically, it is desirable to maximize the active material content. We explored up to 70 wt% PTMA–GMA, but this value is still low compared to LIBs. Because redox-active polymers are not very conductive and possess a higher internal resistance, the redox-active polymer needs a conductive additive. Therefore, improving electronic conductivity without sacrificing active material content is an important future direction. This may be also achieved through the novel synthesis of redox-active polymers with lower internal resistance and higher theoretical capacity.
4. Conclusions
A structural energy storage electrode based on the organic redox-active polymer PTMA was demonstrated. By using a structural current collector comprised of rGO nanosheets and BANFs, a high-modulus electrode with good interfacial adhesion between the collector and the redox-active polymer was obtained. The numerous non-covalent interactions between rGO and BANF and with the active PTMA material were responsible for the improved interfacial adhesion when compared to an Al current collector. The 50 wt% PTMA–GMA on rGO/BANF structural electrode retained about 77% of its capacity at 25C-rate as compared to 1C, as well as a 93% capacity after 500 galvanostatic cycles at 5C. The 50 wt% PTMA–GMA on rGO/BANF also exhibited the best combination of specific power (∼4310 W kg−1 at 25C-rate) and mechanical properties (specific modulus and UTS of 4.33 GPa cm3 g−1 and 64 ± 8 MPa, respectively) as compared to other compositions tested. This specific power was noteworthy because it was far higher than that of other commercial and structural electrodes. The benefit of the rGO/BANF current collector was demonstrated, in which the polymer adhered to the surface even after high-rate and long-term cycling; in comparison, the polymer readily delaminated from an Al current collector. This investigation has demonstrated a structural organic battery electrode with a superior mechanical strength that does not compromise on the electrochemical performance. Our future work will focus on improving the electrochemical performance of structural organic battery electrodes through a higher active-mass loading (lowering the mass of additives), which may require alternative substrates or organic active materials. As for the mechanical properties, these will be further improved by using CFs, which have higher modulus and UTS as compared to the rGO/BANF platform. We will also plan to translate this concept to structural organic battery pouch cells to gain further insights into device-level multifunctional performance.Author contributions
S. S. Oka prepared the original draft; J. L. Lutkenhaus and M. J. Green reviewed and edited the article; S. S. Oka prepared the rGO/BANF composite and conducted the PTMA–GMA coating on rGO/BANF platform experiments, XPS runs and analysis, SEM imaging, all the electrochemical and mechanical testing. R. M. Thakur and A. E. Easley conducted the synthesis of PTMA–GMA and EPR analysis. All authors approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Air Force Office of Scientific Research Grant No. FA9550-22-1-0388. The authors would like to thank Texas A&M University Materials Characterization Core Facility (RRID:SCR_022202), as well as Dr Yordanos Bisrat (Materials Characterization Facility, TAMU) for assistance with cross-sectional SEM imaging, and Dr Jing Wu (Materials Characterization Facility, TAMU) for assistance with XPS. We acknowledge Texas A&M University Materials Characterization Core Facility (RRID:SCR_022202).Notes and references
- D. Pandey, K. Sambath Kumar, L. N. Henderson, G. Suarez, P. Vega, H. R. Salvador, L. Roberson and J. Thomas, Small, 2022, 18, 2107053 CrossRef CAS PubMed.
- W. Chen, J. Liang, Z. Yang and G. Li, Energy Procedia, 2019, 158, 4363–4368 CrossRef.
- R. A. Marsh, S. Vukson, S. Surampudi, B. V. Ratnakumar, M. C. Smart, M. Manzo and P. J. Dalton, J. Power Sources, 2001, 97–98, 25–27 CrossRef CAS.
- V. Knap, L. K. Vestergaard and D.-I. Stroe, Energies, 2020, 13, 4097 CrossRef CAS.
- H. Zhou, H. Li, L. Li, T. Liu, G. Chen, Y. Zhu, L. Zhou and H. Huang, Mater. Today Energy, 2022, 24, 100924 CrossRef CAS.
- L. E. Asp, M. Johansson, G. Lindbergh, J. Xu and D. Zenkert, Funct. Compos. Struct., 2019, 1, 042001 CrossRef CAS.
- L. E. Asp and E. S. Greenhalgh, Compos. Sci. Technol., 2014, 101, 41–61 CrossRef CAS.
- J. Snyder, R. Carter, E. Wong, P. Nguyen, K. Xu, E. Ngo and E. Wetzel, International SAMPE Technical Conference, 2007, p. 18.
- B. J. Hopkins, J. W. Long, D. R. Rolison and J. F. Parker, Joule, 2020, 4, 2240–2243 CrossRef.
- L. E. Asp, K. Bouton, D. Carlstedt, S. Duan, R. Harnden, W. Johannisson, M. Johansen, M. K. G. Johansson, G. Lindbergh, F. Liu, K. Peuvot, L. M. Schneider, J. Xu and D. Zenkert, Adv. Energy Sustainability Res., 2021, 2, 2000093 CrossRef.
- S. Ekstedt, M. Wysocki and L. E. Asp, Plast., Rubber Compos., 2010, 39, 148–150 CrossRef CAS.
- P. Flouda, S. Oka, D. Loufakis, D. C. Lagoudas and J. L. Lutkenhaus, ACS Appl. Mater. Interfaces, 2021, 13, 34807–34817 CrossRef CAS PubMed.
- K. Moyer, N. A. Boucherbil, M. Zohair, J. Eaves-Rathert and C. L. Pint, Sustainable Energy Fuels, 2020, 4, 2661–2668 RSC.
- P. Liu, E. Sherman and A. Jacobsen, J. Power Sources, 2009, 189, 646–650 CrossRef CAS.
- W. Johannisson, N. Ihrner, D. Zenkert, M. Johansson, D. Carlstedt, L. E. Asp and F. Sieland, Compos. Sci. Technol., 2018, 168, 81–87 CrossRef CAS.
- S. Leijonmarck, T. Carlson, G. Lindbergh, L. E. Asp, H. Maples and A. Bismarck, Compos. Sci. Technol., 2013, 89, 149–157 CrossRef CAS.
- F. Hao, X. Chi, Y. Liang, Y. Zhang, R. Xu, H. Guo, T. Terlier, H. Dong, K. Zhao, J. Lou and Y. Yao, Joule, 2019, 3, 1349–1359 CrossRef CAS.
- X. Chi, Y. Zhang, F. Hao, S. Kmiec, H. Dong, R. Xu, K. Zhao, Q. Ai, T. Terlier, L. Wang, L. Zhao, L. Guo, J. Lou, H. L. Xin, S. W. Martin and Y. Yao, Nat. Commun., 2022, 13, 2854 CrossRef CAS PubMed.
- J. Zhang, Z. Chen, Q. Ai, T. Terlier, F. Hao, Y. Liang, H. Guo, J. Lou and Y. Yao, Joule, 2021, 5, 1845–1859 CrossRef CAS.
- M. Li, M. Feng, D. Luo and Z. Chen, Cell Rep. Phys. Sci., 2020, 1, 100212 CrossRef CAS.
- D. Chao, X. Xia, J. Liu, Z. Fan, C. F. Ng, J. Lin, H. Zhang, Z. X. Shen and H. J. Fan, Adv. Mater., 2014, 26, 5794–5800 CrossRef CAS PubMed.
- X. Fang, M. Ge, J. Rong and C. Zhou, ACS Nano, 2014, 8, 4876–4882 CrossRef CAS PubMed.
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS.
- J. Kim, J. H. Kim and K. Ariga, Joule, 2017, 1, 739–768 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- K. Hatakeyama-Sato, H. Wakamatsu, R. Katagiri, K. Oyaizu and H. Nishide, Adv. Mater., 2018, 30, 1800900 CrossRef PubMed.
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed.
- J.-K. Kim, G. Cheruvally, J.-H. Ahn, Y.-G. Seo, D. S. Choi, S.-H. Lee and C. E. Song, J. Ind. Eng. Chem., 2008, 14, 371–376 CrossRef CAS.
- A. Vlad, J. Rolland, G. Hauffman, B. Ernould and J.-F. Gohy, ChemSusChem, 2015, 8, 1692–1696 CrossRef CAS PubMed.
- K. Zhang, Y. Hu, L. Wang, M. J. Monteiro and Z. Jia, ACS Appl. Mater. Interfaces, 2017, 9, 34900–34908 CrossRef CAS PubMed.
- M. Suguro, S. Iwasa, Y. Kusachi, Y. Morioka and K. Nakahara, Macromol. Rapid Commun., 2007, 28, 1929–1933 CrossRef CAS.
- A. D. Easley, L. M. Vukin, P. Flouda, D. L. Howard, J. L. Pena and J. L. Lutkenhaus, Macromolecules, 2020, 53, 7997–8008 CrossRef CAS.
- S. Wang, A. D. Easley and J. L. Lutkenhaus, ACS Macro Lett., 2020, 9, 358–370 CrossRef CAS PubMed.
- S. Wang, A. M. G. Park, P. Flouda, A. D. Easley, F. Li, T. Ma, G. D. Fuchs and J. L. Lutkenhaus, ChemSusChem, 2020, 13, 2371–2378 CrossRef CAS PubMed.
- S. Wang, F. Li, A. D. Easley and J. L. Lutkenhaus, Nat. Mater., 2019, 18, 69–75 CrossRef CAS PubMed.
- K.-G. Kim, G. H. Park, D. O. Agumba and S. Y. Kim, Surf. Interfaces, 2022, 34, 102297 CrossRef CAS.
- A. Wang, R. Tan, C. Breakwell, X. Wei, Z. Fan, C. Ye, R. Malpass-Evans, T. Liu, M. A. Zwijnenburg, K. E. Jelfs, N. B. McKeown, J. Chen and Q. Song, J. Am. Chem. Soc., 2022, 144, 17198–17208 CrossRef CAS PubMed.
- A. Patel, D. Loufakis, P. Flouda, I. George, C. Shelton, J. Harris, S. Oka and J. L. Lutkenhaus, ACS Appl. Energy Mater., 2020, 3, 11763–11771 CrossRef CAS.
- S. Aderyani, P. Flouda, J. L. Lutkenhaus and H. Ardebili, J. Appl. Phys., 2019, 125, 185106 CrossRef.
- P. Flouda, X. Feng, J. G. Boyd, E. L. Thomas, D. C. Lagoudas and J. L. Lutkenhaus, Batteries Supercaps, 2019, 2, 464–472 CrossRef CAS.
- P. Flouda, A. H. Quinn, A. G. Patel, D. Loufakis, D. C. Lagoudas and J. L. Lutkenhaus, Nanoscale, 2020, 12, 16840–16850 RSC.
- P. Flouda, S. A. Shah, D. C. Lagoudas, M. J. Green and J. L. Lutkenhaus, Matter, 2019, 1, 1532–1546 CrossRef.
- P. Flouda, J. Yun, D. Loufakis, S. A. Shah, M. J. Green, D. C. Lagoudas and J. L. Lutkenhaus, Sustainable Energy Fuels, 2020, 4, 2301–2308 RSC.
- S. R. Kwon, J. Harris, T. Zhou, D. Loufakis, J. G. Boyd and J. L. Lutkenhaus, ACS Nano, 2017, 11, 6682–6690 CrossRef CAS PubMed.
- D. Loufakis, P. Flouda, S. Oka, J. Pombo, D. C. Lagoudas and J. L. Lutkenhaus, J. Phys. Chem. C, 2022, 126, 20293–20301 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- P. Barai, T. Rojas, B. Narayanan, A. T. Ngo, L. A. Curtiss and V. Srinivasan, Chem. Mater., 2021, 33, 5527–5541 CrossRef CAS.
- C. Busà, M. Belekoukia and M. J. Loveridge, Electrochim. Acta, 2021, 366, 137358 CrossRef.
- M. Marinaro, M. Weinberger and M. Wohlfahrt-Mehrens, Electrochim. Acta, 2016, 206, 99–107 CrossRef CAS.
- N. Vicente, M. Haro, D. Cíntora-Juárez, C. Pérez-Vicente, J. L. Tirado, S. Ahmad and G. Garcia-Belmonte, Electrochim. Acta, 2015, 163, 323–329 CrossRef CAS.
- B. Wang, J. B. Bates, F. X. Hart, B. C. Sales, R. A. Zuhr and J. D. Robertson, J. Electrochem. Soc., 1996, 143, 3203–3213 CrossRef CAS.
- J. Zhang, F. Wu, X. Dai, Y. Mai and Y. Gu, ACS Sustainable Chem. Eng., 2021, 9, 1741–1753 CrossRef CAS.
- H. Zhong, M. Sun, Y. Li, J. He, J. Yang and L. Zhang, J. Solid State Electrochem., 2016, 20, 1–8 CrossRef CAS.
- H. Zheng, L. Zhang, G. Liu, X. Song and V. S. Battaglia, J. Power Sources, 2012, 217, 530–537 CrossRef CAS.
- J. Lu, W. Li, C. Shen, D. Tang, L. Dai, G. Diao and M. Chen, Ionics, 2019, 25, 4075–4082 CrossRef CAS.
- W. Huang, P. Wang, X. Liao, Y. Chen, J. Borovilas, T. Jin, A. Li, Q. Cheng, Y. Zhang, H. Zhai, A. Chitu, Z. Shan and Y. Yang, Energy Storage Mater., 2020, 33, 416–422 CrossRef CAS.
- A. Thakur and X. Dong, Manuf. Lett., 2020, 24, 1–5 CrossRef.
- K. Moyer, C. Meng, B. Marshall, O. Assal, J. Eaves, D. Perez, R. Karkkainen, L. Roberson and C. L. Pint, Energy Storage Mater., 2020, 24, 676–681 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00519d |
| This journal is © The Royal Society of Chemistry 2023 |