
Potential of catalytic oxidation of kraft black liquor for the production of biosourced compounds†
Léa
Vilcocq
 *a,
Nicolas
Chaussard
ab,
Antonio
Hernández Mañas
ab,
Olivier
Boyron
a,
Manel
Taam
a,
Frédérique
Bertaud
c,
Pascal
Fongarland
a and
Laurent
Djakovitch
*b
*a,
Nicolas
Chaussard
ab,
Antonio
Hernández Mañas
ab,
Olivier
Boyron
a,
Manel
Taam
a,
Frédérique
Bertaud
c,
Pascal
Fongarland
a and
Laurent
Djakovitch
*b
aUniv. Lyon, CNRS, Université Claude Bernard Lyon 1, CPE-Lyon, CP2M – UMR 5128, F-69616, Villeurbanne, France. E-mail: lea.vilcocq@cnrs.fr
bUniv. Lyon, CNRS, Université Claude Bernard Lyon 1, IRCELYON – UMR 5256, F-69626 Villeurbanne Cedex, France. E-mail: laurent.djakovitch@ircelyon.univ-lyon1.fr
cCTP (Centre Technique du Papier), Domaine Universitaire CS90251, 38044 Grenoble Cedex 9, France
First published on 25th May 2023
Abstract
Industrial kraft black liquor from maritime pine was oxidised in aqueous, alkaline medium, under air, at 150 °C, with or without a CuO/TiO2 catalyst. The oxidation products were analysed by HPLC, elemental analysis, SEC, FTIR, NMR. The results showed the depolymerisation of lignin, the formation of phenolic compounds in low yields, with vanillin being the main phenolic compound, and the formation of aliphatic compounds in higher yields, with formic, succinic and tartronic acids being the main identified aliphatic compounds. The presence of the catalyst favoured the formation of phenolic and aliphatic compounds. Replacing kraft black liquor by pure kraft lignin as a starting material did not improve the performances of the catalytic oxidation, indicating that lignin purification may not be necessary under our conditions. Switching from a batch reactor to a fixed bed reactor, operating under similar conditions, did not increase the yields of oxidation products but did increase the productivity. This work demonstrates the potential of kraft black liquor to produce a large panel of compounds, including phenolics, aliphatic acids and oxidised lignin, all of which are valuable in the chemical industry. For the first time, a catalytic process for the chemical valorisation of kraft black liquor is presented.
Introduction
Kraft black liquor (KBL) is an industrial effluent produced during the kraft process in pulp and paper mills. In the kraft process, wood chips are delignified by cooking at 155–165 °C in white liquor, an aqueous solution of soda and sodium sulphide, to produce pulp, a fibrous material rich in cellulose. The liquid residue is called black liquor (BL) and contains the reagents in excess (soda and sodium sulphide), and their by-products (sulphates, sulphites, thiosulphates) as well as degraded hemicellulose and lignin. 180 million tons of paper pulp are produced annually and each ton of pulp produces ca. 7 tons of weak (i.e. diluted) black liquor.1 This bio-waste is currently being burnt to regenerate inorganics in novel white liquor and to recover energy from combustion of organic compounds. The recovery boiler produces ca. 2 tons of biogenic CO2 per ton of pulp.2 Moreover, it is often the bottleneck in the kraft process. Therefore, bypassing some of the black liquor with an alternative process would be beneficial to pulp mills. In the long-term, consideration should be given to converting pulp mills into integrated forest biorefineries producing a wide range of commodities with low emissions of CO2.3During kraft cooking, hemicelluloses are degraded with the peeling reaction to form a broad range of aliphatic acids: formic, acetic, glycolic, lactic and isosaccharinic acids were observed, among others.4 Lignin is also degraded but retains a polymeric structure. However, transformations occur as some ether inter-unit bonds (β-O-4 bonds) are cleaved and new C–C bonds are formed.5 Black liquors also contain high concentrations of inorganics derived from white liquor: sodium sulphate, carbonate or hydroxide are present. The pH is very high (greater than 13).
Separation and valorisation of lignin from black liquor has been attempted in several pathways. Kraft lignin can be recovered by acidification and precipitation6 (LignoBoost process) and commercialised as dispersant, binder, metal adsorbent, etc.7,8 Kraft lignin can be valorised by different processes, such as gasification or pyrolysis, the latter producing bio-oil, biochar and biogas as products.9,10 Under milder reaction conditions, hydrotreatment, solvolysis,11 hydrolysis, or oxidation12 can be applied to depolymerise lignin and produce of monomers in low yields (aromatics, phenolics, aliphatics), oligomers and gases. Among the technical lignins, kraft lignin is the most recalcitrant and usually gives the lowest yields of products of interest, as kraft cooking alters the structure of the lignin.
One of the most interesting compounds produced from lignin is vanillin. This phenolic aldehyde can be obtained by depolymerisation and oxidation of guaiacyl units in lignin.13 It has many applications in fragrances and flavours (F&F) industry and a high market value (20 $ per kg in 2016 according to Fache et al.,14 10–15 $ per kg according to Rinaldi et al. in 2016![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15). The industrial production of vanillin relies for ca. 15% on the oxidation of lignosulfonate with copper sulphate as a catalyst (Borregaard process).14 Besides, the formation of aliphatic acids from lignin oxidation has been reported in a few articles: formic, acetic, succinic, oxalic, and glutaconic acids have been observed.16
15). The industrial production of vanillin relies for ca. 15% on the oxidation of lignosulfonate with copper sulphate as a catalyst (Borregaard process).14 Besides, the formation of aliphatic acids from lignin oxidation has been reported in a few articles: formic, acetic, succinic, oxalic, and glutaconic acids have been observed.16
Many research teams have attempted to increase the productivity of lignin oxidation towards vanillin and phenolics.15,17–25 The oxidant (O2 or H2O2), the solvent (water or other), the pH, the operating conditions (reaction time, temperature, pressure, type of reactor) and the type of lignin, all have influence on the reaction performance. O2 has been identified as the cheapest and greenest oxidant, but also requires high temperatures and is not selective. Mild temperatures (100–190 °C) and moderate partial O2 pressures (20–100 bar) are often applied. High pH is necessary for lignin solubilisation and increased reaction rates. In addition, lower pH values are associated with higher degradation rate of vanillin. Wet oxidation of lignin produces phenolic aldehydes (vanillin, syringaldehyde, p-hydroxybenzaldehyde), phenolic acids (vanillic, syringic, p-hydroxybenzoic acids), phenolic ketones (acetovanillone, acetosyringone, acetophenone), quinones, but also aliphatic monoacids (formic, acetic), diacids (oxalic, succinic, malonic, maleic), hydroxy-acids (lactic). Lignins with a low molecular weight and high β-O-4 content give higher yields in a shorter reaction time. Kraft lignins, with a low β-O-4 content, result in low yields of aromatic products.
A catalyst can also be added to the reaction.26 Metallic salts of Cu, Fe, Mn, Co, Zr have been tested. CuSO4 was identified as an interesting catalyst and has been used in industrial processes such as the Borregaard process. Organometallic complexes such as metallosalens and metalloporphyrins, which mimic the action of lignin peroxidases, are also promising. Solid catalysts have also been developed, based on Cu, Co, Fe, Zn, V, Mo, Re, Pd, Pt oxides or hydroxides, bulk or supported on various materials (Al2O3, C, TiO2, etc.). Copper-based solid catalysts have a high potential for lignin oxidation, as they are active, stable, inexpensive and non-noble materials. In particular, the production of phenolic aldehydes from eucalyptus kraft lignin with CuO catalyst,27 and from softwood lignosulfonate with Cu(OH)2 catalyst28 have been reported.
Another category of interesting compounds in KBL is the mix of aliphatic acids. These ones can be isolated from KBL by various separation techniques (chromatography, membrane filtration, liquid–liquid extraction, distillation) but none of them has been industrialised so far.4 Due to the complexity of the black liquor matrix, several separation operations are necessary to purify the aliphatic compounds. Besides, as an alternative to combustion in boiler, concentration and recovery of inorganics can be achieved by wet oxidation, membrane nanofiltration, or gasification.
Direct conversion of black liquor is challenging because of the complexity of KBL. It is a concentrated, alkaline effluent, combining organic and inorganic components, as well as polymers and monomers with a broad variety of chemical functions. Black liquors can be transformed through different pathways to produce compounds of interest to the fuels and chemical industries: gasification of black liquor has been investigated as an alternative to combustion in recovery boiler.29 Liquefaction of black liquor produces bio-oil as an alternative fuel.30
The chemical valorisation pathway, in which biosourced compounds, either polymers or monomers, are transformed under mild reaction conditions (temperature inferior to 300 °C, pressure inferior to 100 bar), has rarely been applied to crude black liquor. Few examples of catalytic reduction31 or catalytic oxidation32 have demonstrated the (partial) depolymerisation of lignin and sometimes the formation of phenolic compounds from lignin in black liquor, in yields inferior to 10%. In a recent communication, our research teams demonstrated the feasibility of direct oxidation of black liquor to produce low yields of vanillin and phenolics; however with reasonable productivity.33
In this work, the products of the catalytic oxidation of KBL have been studied in detail. The reaction was carried out with air, and a solid CuO/TiO2 catalyst. The formulation of the catalyst was based on elements from the literature and from our experience showing a good stability of TiO234 and CuO35 in aqueous alkaline conditions. The role of different parts of the catalyst (support, metal) was elucidated. Our aim was to demonstrate the potential of KBL for the production of a wide range of biosourced compounds and materials by catalytic oxidation. To this end, reaction products were identified and characterized in detail: phenolic compounds, but also aliphatic compounds and residual lignin. Two types of reactors were compared: a batch stirred reactor with a catalytic slurry and a fixed bed continuous reactor. A study of the productivity of various compounds of interest was also attempted to provide some insights into the techno-economic prospects of KBL oxidation.
Experimental
Materials
Kraft black liquor (KBL) employed in all the experiments was obtained from a kraft pulp mill in France, which uses local maritime pine (Pinus pinaster) as biomass.Kraft Lignin (KL) was extracted from KBL in “Centre Technique du Papier” (CTP). KBL was acidified by bubbling CO2 until pH 9. The lignin suspension obtained was filtrated to collect the lignin. The lignin was purified by acid washing (diluted sulphuric acid in water at 2%) and finally air-dried.
TiO2 Aerolyst 7711 support was kindly provided by Evonik. Cu(NO3)4, NaOH, HCl, acetonitrile HPLC grade, H2SO4, H3PO4, NaH2PO4 were purchased from Alfa Aesar or Sigma Aldrich and used without further purification.
Catalyst preparation and characterisation
The preparation and characterisation of CuO/TiO2 catalyst were described in our previous publication.33 Briefly, TiO2 pellets were impregnated with an aqueous Cu(NO3)4 solution following the incipient wetness impregnation technique. The pellets were dried at 110 °C overnight and then calcined at 550 °C under air flow in a muffle furnace. After calcination, the pellets were crushed and sieved and the 90–200 μm fraction was retained for catalytic tests in batch reactor. For the continuous reactor, the pellets were used without crushing. The catalyst was characterised by XRD, ICP, and N2 physisorption for porosimetry (see ESI† for details).Catalytic oxidation test and post-treatment
The catalytic oxidation of KBL was carried out in a 300 mL stainless steel batch reactor (Vinci Technology). KBL was diluted in NaOH solution (10 g L−1) up to a dry matter content equal to 5 g L−1. The reactor was filled with 150 mL of the diluted KBL solution and 0.75 g of catalyst (equivalent to 5% copper/lignin mass ratio). The reactor was sealed and tightened, purged three times with synthetic air and pressurised with air at 20 bar at room temperature. It was heated up to 150 °C and time zero was set when this working temperature was reached (about 30 min). The reactor was stirred with a mechanical stirrer equipped with a Rushton turbine (1800 rpm). At the end of the reaction time (0 to 90 min), the reactor was cooled in an ice bath, depressurised and opened.The protocol for catalytic oxidation in a continuous reaction was described in a previous communication.33 Briefly, a stainless steel tube was loaded with 20 g of CuO/TiO2 catalyst mixed with silicon carbide to form a bed 12.5 cm high in an isothermal zone between two layers of pure SiC at the inlet and outlet of the reactor. The reactor was sealed, placed in a tubular oven and connected to a feed of diluted KBL (same concentration as for batch reactions), fed with a pump, and air flow (15 NL h−1), fed by a mass flow regulator. After the reactor, a backpressure membrane regulator maintained the reactor pressure at 80 bar. After the backpressure regulator, a sampling valve allowed liquid samples to be taken periodically. The liquid flow was controlled to keep the contact times (defined as the ratio of mass of catalyst to mass liquid flow) as short as possible.
Whatever the reactor used, samples (50–100 mL) of the reaction medium were filtered under vacuum to remove solid residues. In the batch reactor, the catalyst was found in the solid residue; in the continuous reactor, filtration did not produce significant amount of solids. The filtrate was acidified with a 10% HCl solution until pH 1 to precipitate residual lignin. The precipitate was recovered by centrifugation at 4000 rpm for 20 min, washed with HCl solution and water and then dried under vacuum for 9 hours at 60 °C. The recovered solid was called Klason phase (KP), “Klason” designating the acid precipitate in KBL and in product mixtures. The liquid separated in the first centrifugation was called liquid phase (LP).
Products analysis
In the first one, a Phenomenex Kinetex XB C18 column was used and the mobile phase was a gradient of acetonitrile and aqueous H2SO4 solution (10 mM) starting with 5 vol% acetonitrile (0–6 min) and increasing to 30 vol% acetonitrile (ramp from 6 to 16 min and step from 16 to 30 min). Phenolic compounds were identified and quantified by this method using commercial compounds as references. Identification was based on retention time and UV spectrum similarity. Quantification was based on external calibration with four levels of concentration in the range 0.02–0.1 g L−1, at 273 nm.
In the second method, a Phenomenex Synergi RP-Hydro column was used and the mobile phase was a gradient of acetonitrile and aqueous solution buffered at pH 2.6 with a H3PO4/NaH2PO4 buffer, starting from 0 vol% acetonitrile (0–15 min) and increasing to 22 vol% acetonitrile (ramp from 15 to 48 min and step from 48 to 70 min). Aliphatic compounds were identified and quantified by this method using commercial compounds as references. Identification was based on retention time and UV spectrum similarity. Quantification was based on external calibration with four levels of concentration in the range 1–10 mM, at 210 nm. Several types of aliphatic acids can be found in crude KBL: simple carboxylic acids (e.g. formic, acetic), hydroxy-acids (e.g. glycolic, lactic), iso-saccharinic acids. In our case, gluco-isosaccharinic acid (GISA) was detected but not quantified due to the lack of commercial standard.
FT-IR analyses were carried out in an absorbance mode using a Nicolet IS5 equipped with a Thermo Scientific ID7-ATR accessory with diamond crystal. The sample was placed directly on the diamond and the IR signal was recorded with a resolution of 4 cm−1, 32 scans and a spectral range of 4000 cm−1 to 525 cm−1. The spectra obtained with this method were qualitatively analysed to characterise different structures in the lignin.
HSQC NMR spectra were acquired on a Bruker ADVANCE III 400 MHz equipped with a BBFO probe (Z gradient). Samples were prepared by dissolving an aliquot (10–15 mg) in 600 mg of (CD3)2SO. Data were recorded over 48 scans at 50 °C. A standard pulse sequence hsqcedetgpsp.3 was used.
Size Exclusion Chromatography (SEC) analyses were performed on a Malvern Panalytical TDA Viscotek system equipped with three columns (Mixed C, 300 mm × 7 mm I.D. from Agilent Technologies). Klason phase samples were dissolved in THF and filtered at 0.45 μm before analysis. All samples were fully soluble in THF. 100 μL of sample at a concentration of 3 mg mL−1 was eluted in THF at a flow rate of 1 mL min−1 at 35 °C. Polystyrene standards from 500 g mol−1 to 2 million g mol−1 were used for calibration. Online detection was performed using a differential refractive index detector. The OmniSEC 5.12 software from Malvern Panalytical was used for the calculations.
TGA analysis was performed on Mettler Toledo TGA/DSC STAR System apparatus, at 5 °C min−1, from 25 to 1000 °C, air as carrier gas. The moisture degree was determined by the weight loss at 100–115 °C and the ashes content by weight remaining at 1000 °C.
Elementary analysis (CHNS) was performed on a Thermo Scientific FlashSmart apparatus. 2 mg of sample in aluminium crucibles were analysed for each KBL, KL and KP sample. Data were recorded using Eager Smart software.
Calculations
The lignin conversion was calculated considering that KP corresponds to unreacted lignin, according to the formula:where [DM] is the dry matter concentration before starting the reaction (g L−1), %lignin the percentage of pure lignin in the dry matter of KBL (i.e. 31 wt%, see Table 1 for details) and [KP] the Klason phase concentration at the end of the reaction (g L−1).
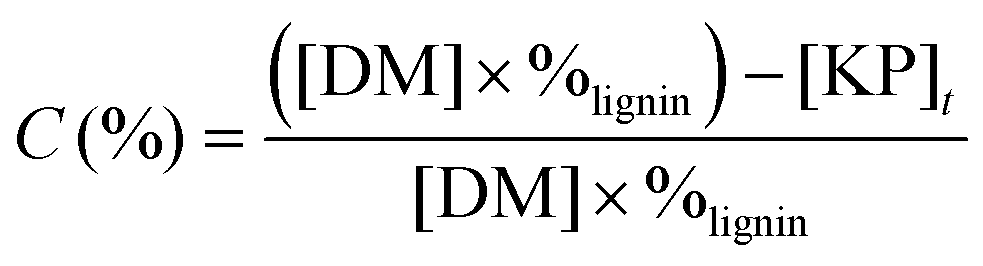
| KBL | KL | |
|---|---|---|
| LHV – lower heating value; AIL – acid insoluble lignin; ASL – acid soluble lignin.a Analysis performed on freeze-dried KBL. | ||
| pH (20 °C) | 13.7 | — |
| Dry matter content (wt%, on crude sample) | 26.0 ± 0.1 | 95.5 |
| Density 20 °C (g cm−3) | 1.11 | — |
| Lignin content (AIL + ASL) (wt%, o.d. sample) | 30.8 ± 0.6 (23.5 + 7.3) | 96.1 ± 0.1 (93.1 + 2.9) |
| Na (wt%, o.d. sample) | 17.6 ± 0.05 | 0.28 ± 0.02 |
| K (wt%, o.d. sample) | 1.35 ± 0.05 | 0.15 ± 0.02 |
| Total sulphur (wt%, o.d. sample) | 0.93 ± 0.02 | 2.2 |
| Ashesa (wt%, o.d. sample) | 39.9 | 2.6 |
| C/H/N/Sa (wt%, o.d. sample) | 31/4/0/1 | 61/6/0.2/2.2 |
| LHVa (MJ kg−1 o.d. sample) | 8.11 ± 0.6 | 23.4 ± 0.2 |
| Molar mass (Mw) (g mol−1) | 4934/498/208 | 2964 |
The yields of phenolic compounds were calculated on lignin mass basis, according to the formula:
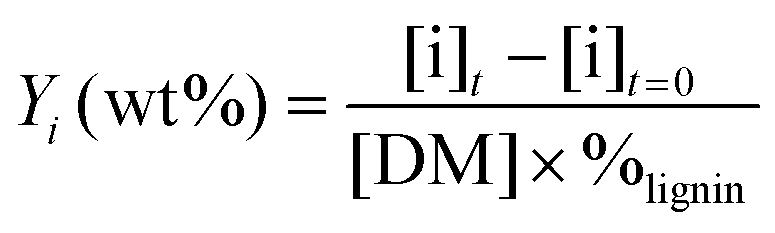
The yields of aliphatic compounds were calculated on carbon basis, according to the formula:
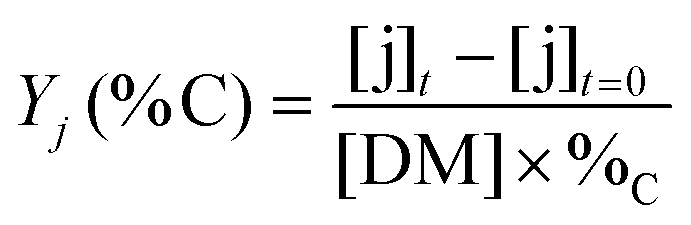
The productivity of the oxidation products was calculated from the mass concentration of the products at the end of the reaction and the reactor features. For the batch reactor, productivity was calculated according to the formula:
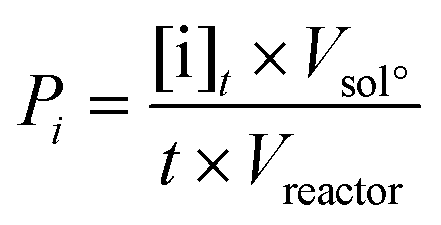
For the continuous reactor, the productivity was calculated following the formula:
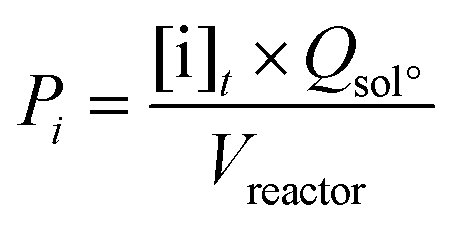
The repeatability of the catalytic test and analytical methods was evaluated on one catalytic test and showed good repeatability of the Experimental methods (see ESI†).
Results and discussion
Characterisation of starting materials
KBL and KL were characterised using a combination of several techniques (Table 1). KBL was characterised either as a solution or as a freeze-dried solid, depending on the technique used. KBL is an alkaline solution (pH = 13.7), concentrated (26 wt% dry matter) and dense (density 1.1). KL has a low moisture content (4.5 wt%). The ashes content varies from 39% in KBL, corresponding to a large amount of inorganic material, to 2.6% in KL, which contains only residual amounts of Na, K, S. The carbon content is 31% of the dry matter of KBL. This corresponds to a highly oxygenated material. The lignin content in KBL was only 31%, which implies that KBL contains ca. 30% non-lignin organic compounds. On the other hand, KL is highly pure with a lignin content of 96%. In KL, the percentage of carbon was 61%, which corresponds to the carbon percentage in a guaiacyl unit (65%), taking into account moisture and inorganic content in KL. The sulphur content is 1% in KBL and 2.2% in KL, showing that sulphur tends to accumulate in lignin, either by physisorption of chemically bound thiol groups, sulphates and sulfones. Neither KBL nor KL contain nitrogen.To determine the carbohydrate content of the starting materials, a post-hydrolysis protocol was performed by adapting protocols from Theander & Westerlund36 and NREL.37 Sugar concentrations before and after post-hydrolysis give the original content of free sugars and polysaccharides in starting materials (Table 2). The total carbohydrate content is below 1.5 wt% for both KBL and KL, indicating that most of the carbohydrates from hemicellulose have been degraded during the kraft cooking.
| Content (wt%, o.d. sample) | KBL | KL |
|---|---|---|
| OS: oligosaccharides. Xylose, galactose and mannose were co-eluted. | ||
| Glucose | 0.18 | 0.04 |
| Xylose/galactose/mannose | 0.04 | 0 |
| Arabinose | 0.08 | 0.11 |
| Gluco-OS | 0.1 | 0 |
| Xylo/galacto/manno-OS | 0.55 | 0.31 |
| Arabino-OS | 0.35 | 0.06 |
| Total carbohydrates | 1.3 wt% | 0.5 wt% |
FTIR analysis was performed on freeze-dried KBL and KL (Fig. S1 in ESI†). FTIR of KBL displays an atypical spectra, badly resolved (bands at 1580 cm−1, 1492 cm−1, 1411 cm−1, 1313 cm−1, 1260 cm−1, 1128 cm−1, 1223 cm−1 and 1003 cm−1), probably due to interaction of lignin moieties with salts.38 However, Klason phase isolated from KBL and re-diluted in deionised water displays characteristic bands: 1423 cm−1, 1509 cm−1 and 1591 cm−1 corresponding to the aromatic rings; 1265 cm−1, 1194 cm−1 and 1027 cm−1 corresponding to guaiacyl moieties (resp. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching; C–C and C–O stretching; aromatic C–H deformation); 1693 cm−1 for unconjugated carbonyl groups as CO in β-position or in COOH; 1452 cm−1 and 1423 cm−1 assigned to C–H stretching in CH2 and CH3 alkyl moieties; 2934 cm−1 assigned to C–H stretching in CH2 and CH3 alkyl moieties and 3410 cm−1 assigned to phenolic O–H stretching. The band at 1761 cm−1 can be attributed to esters.
O stretching; C–C and C–O stretching; aromatic C–H deformation); 1693 cm−1 for unconjugated carbonyl groups as CO in β-position or in COOH; 1452 cm−1 and 1423 cm−1 assigned to C–H stretching in CH2 and CH3 alkyl moieties; 2934 cm−1 assigned to C–H stretching in CH2 and CH3 alkyl moieties and 3410 cm−1 assigned to phenolic O–H stretching. The band at 1761 cm−1 can be attributed to esters.
For KL, bands corresponding to aromatic rings (1426 cm−1, 1510 cm−1 and 1593 cm−1), guaiacyl moieties (1264 cm−1, 1206 cm−1 and 1028 cm−1) unconjugated carbonyl groups (1695 cm−1), C–H stretching in CH2 and CH3 alkyl moieties (2934 cm−1, 1450 cm−1 and 1426 cm−1) and phenolic O–H stretching (3448 cm−1) are observed.
HSQC NMR spectra were recorded for both KBL and KL (see Fig. S5, ESI†). Please note that HSQC spectra of initial KBL, as obtained from freeze-dried black liquor is atypical, like for FTIR spectroscopy, as correlations for inter-unit bonds are not observed. However, proton NMR shows these alkyl moieties clearly (Fig. S6, ESI†).
We previously reported HSQC NMR spectra for KL (see Fig. S7, ESI†).39 Like for KBL, spectra show all characteristic correlations for lignin structure: methoxy groups (δH/δC: 3.76/56.2 ppm); β-O-4 linkages (A) with (Aα) (δH/δC: 4.79/71.7 ppm), (AβG) (δH/δC: 4.74/81.9 ppm), (AβG/H) (δH/δC: 4.29/85.1 ppm) and (Aγ) (δH/δC: 3.48/60.6 ppm), and associated (A′γ) (δH/δC: 4.14/67.6 ppm) and (A′′γ) (δH/δC: 3.67/63.7 ppm); β–β linkages (B) with (Bα) (δH/δC: 4.64/85.6 ppm), (Bβ) (δH/δC: 3.04/54.1 ppm) and (Bγ) (δH/δC: 3.75 and 4.10/71.7 ppm); phenylcoumaran units (β-5 linkages) (C) gave correlations at (Cα) (δH/δC: 5.48/87.4 ppm) and (Cβ) (δH/δC: 3.44/53.9 ppm). Cinnamyl alcohol signal is observed at (Dγ) (δH/δC: 4.08/61.9 ppm). Aromatic region exhibits correlations corresponding to guaiacyl units (G) (G2: δH/δC: 6.91/111.36 ppm, G5: δH/δC: 6.77/116.11 ppm, G6: δH/δC: 6.77/121.74 ppm) and p-hydroxyphenyl units (H) (H2,6: δH/δC: 7.21/128.85 ppm). Correlations corresponding to ferulic derivatives (FA2) (δH/δC: 7.41/109.69 ppm), (p-CA2.6) (δH/δC: 7.42/126.31 ppm) and (CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH) (δH/δC: 5.34/120.94, 5.73/122.88 ppm), to guaiacyl units substituted by carbonyl moieties (G′) (G′2: δH/δC: 7.49/112.24 ppm, G′6: δH/δC: 7.27/121.12 ppm), and in that case residual carbohydrates (δH/δC: 3.02–3.72/68.2–78.5 ppm) are also observed.
CH) (δH/δC: 5.34/120.94, 5.73/122.88 ppm), to guaiacyl units substituted by carbonyl moieties (G′) (G′2: δH/δC: 7.49/112.24 ppm, G′6: δH/δC: 7.27/121.12 ppm), and in that case residual carbohydrates (δH/δC: 3.02–3.72/68.2–78.5 ppm) are also observed.
In summary, raw KBL is a complex material containing lignin, other organic compounds and inorganics in similar proportions. Only traces of polysaccharides and monosaccharides can be found in KBL. KBL is highly oxygenated. Structural analysis revealed the presence of polymers with typical lignin features (aromatic guaiacyl, carbonyl, alkyl and phenolic moieties) and monomers. Furthermore, the characterisation of KL shows that this material is almost purely lignin with structural features similar to those of lignin in KBL.
Catalytic oxidation of kraft black liquor
Oxidation of KBL was carried out in batch mode with or without CuO/TiO2 catalyst. The reaction conditions were chosen on the basis of previous studies by our teams on pure lignin transformation.12,40,41 The temperature was set at 150 °C, the pressure at 20 bar, stirring rate 1800 rpm and the copper/dry matter ratio at 5 wt%.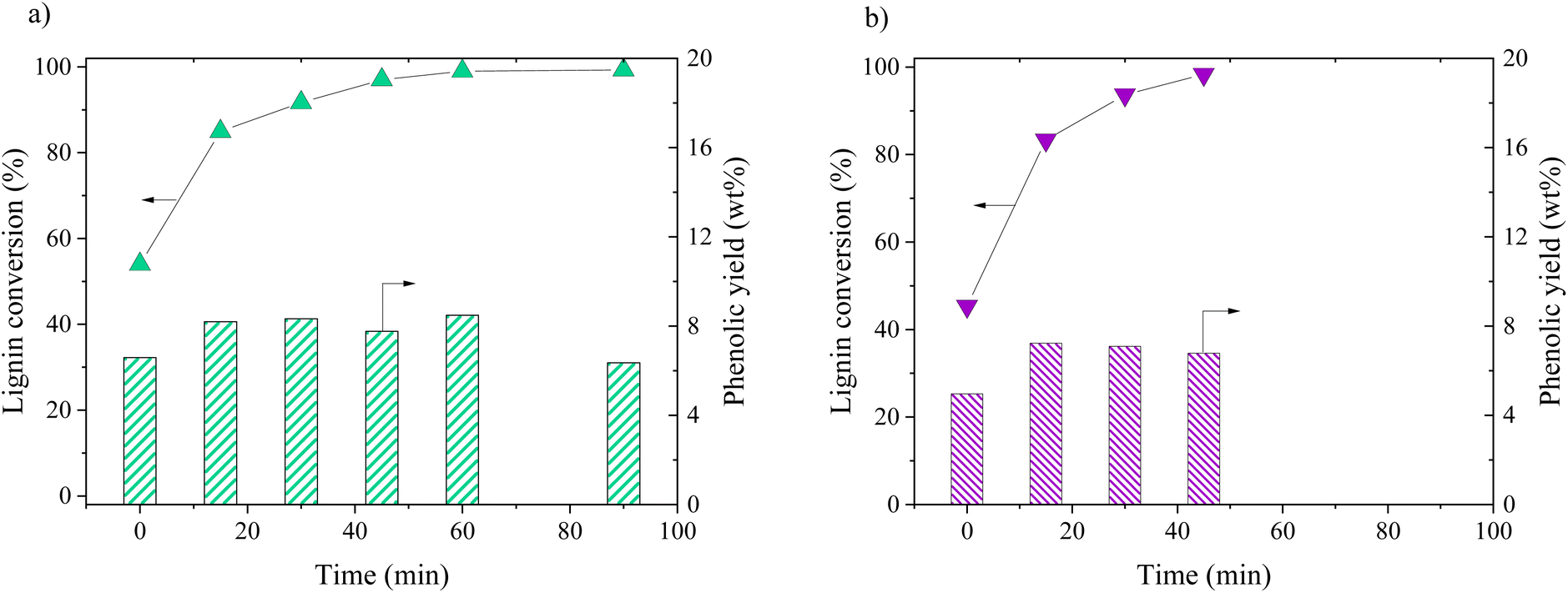 | ||
| Fig. 1 Lignin conversion (symbols) and phenolic yields (bars) as a function of time during KBL oxidation for catalysed (a) and non-catalysed (b) reactions. Operating conditions: 150 °C, 20 bar air, stirring 1800 rpm, dry matter concentration 5 g L−1, copper/dry matter mass ratio 5%. | ||
The composition of the phenolics was detailed for both non-catalysed and catalysed reaction (Fig. 2). The main phenolic compound was vanillin and it was produced with a yield of 2.1 wt% at time zero without catalyst, then increased up to 3.6 wt% in 15 min and was stable between 30 and 45 min. In the presence of CuO/TiO2 catalyst, the vanillin yield started at 3.4 wt%, then increased up to 4.6 wt% at 30 min before slowly decreasing for reaching 2.7 wt% at 90 min. This decrease was attributed to the degradation of vanillin.
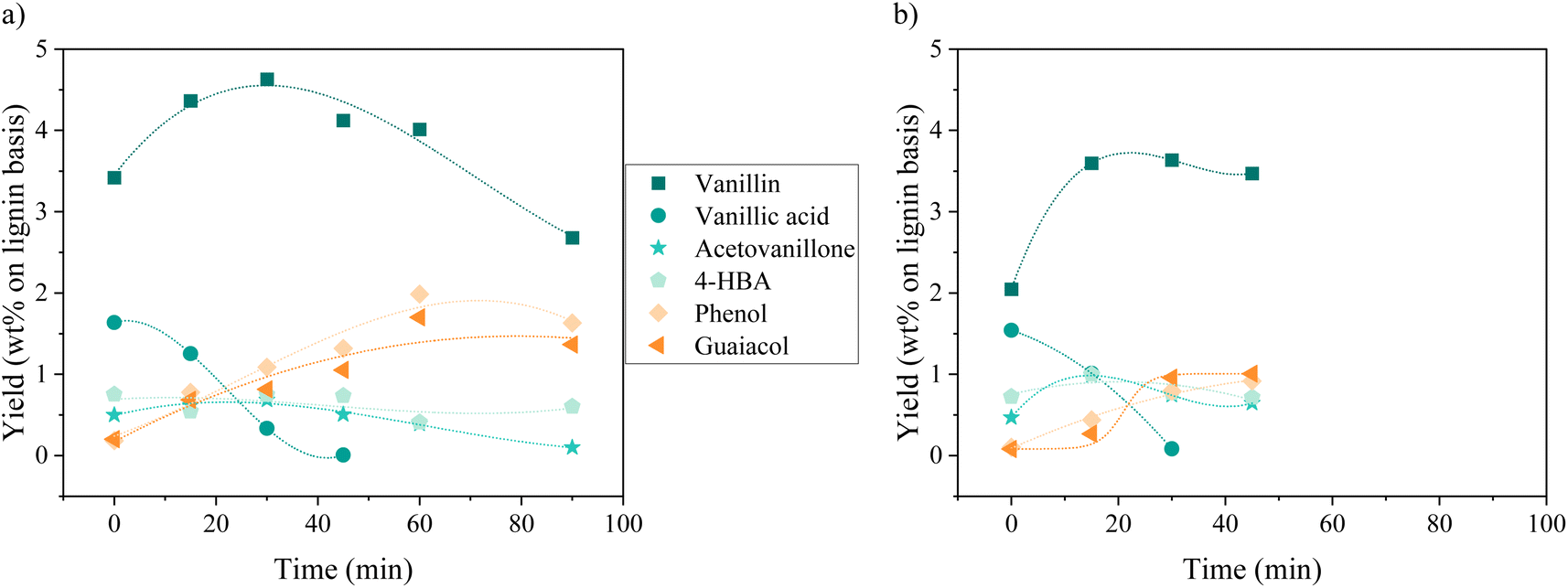 | ||
| Fig. 2 Yields of phenolic compounds as a function of time during KBL oxidation for catalysed (a) and non-catalysed (b) reactions. Operating conditions: 150 °C, 20 bar air, stirring 1800 rpm, dry matter concentration 5 g L−1, copper/dry matter ratio 5%. Lines are only guides for the eyes. 4-HBA: 4-hydroxy-benzoic acid. | ||
Minor phenolic compounds, such as vanillic acid, acetovanillone, 4-hydroxybenzoic acid, phenol and guaiacol were observed and quantified. 4-Hydroxybenzaldehyde was only detected as traces in most of the samples. Other commonly observed compounds26 were not detected: homovanillic acid, anisic acid, benzoic acid and catechol. The yield profiles were different for each phenolic compound. Vanillic acid started with a yield of 1.5 wt% (without catalyst) or 1.6 wt% (with catalyst) and decreased continuously over the reaction time, reaching the lowest level of HPLC quantification (0.1 g L−1) after 30 min for the non-catalytic reaction and after 45 min for the catalytic reaction. For acetovanillone, the initial yield was 0.5 wt% without catalyst, increasing to 1.1 wt% after 30 min and then decreasing, whereas in the presence of catalyst, acetovanillone yield started at 0.5 wt% and then remained stable for 60 min before decreasing. Less oxygenated compounds such as phenol and guaiacol are present as traces at the beginning of the reaction and are then produced up to 0.8 wt% each (without catalyst, after 45 min) and up to 2.0 wt% and 1.7 wt% in the presence of a catalyst in 60 min.
The reaction pathways by which phenolics are produced and consumed during lignin oxidation have been already described in literature and particularly in our previous communication.33 Vanillin, acetovanillone and vanillic acid all come from guaiacyl units in lignin whereas 4-hydroxybenzaldehyde could come from the degradation of other phenolic monomers or from hydroxyphenyl units. Phenol and guaiacol can be formed from the degradation of other phenolics or from a non-oxidative pathway of lignin cleavage. Each phenolic compound has its own degradation rate in oxidative conditions. Particularly, the fast oxidative degradation of vanillic acid when compared to vanillin have already been described under non-catalytic conditions.42 Therefore, it seems possible to tune the composition of the phenolic mix by balancing reaction conditions, and particularly reaction time.
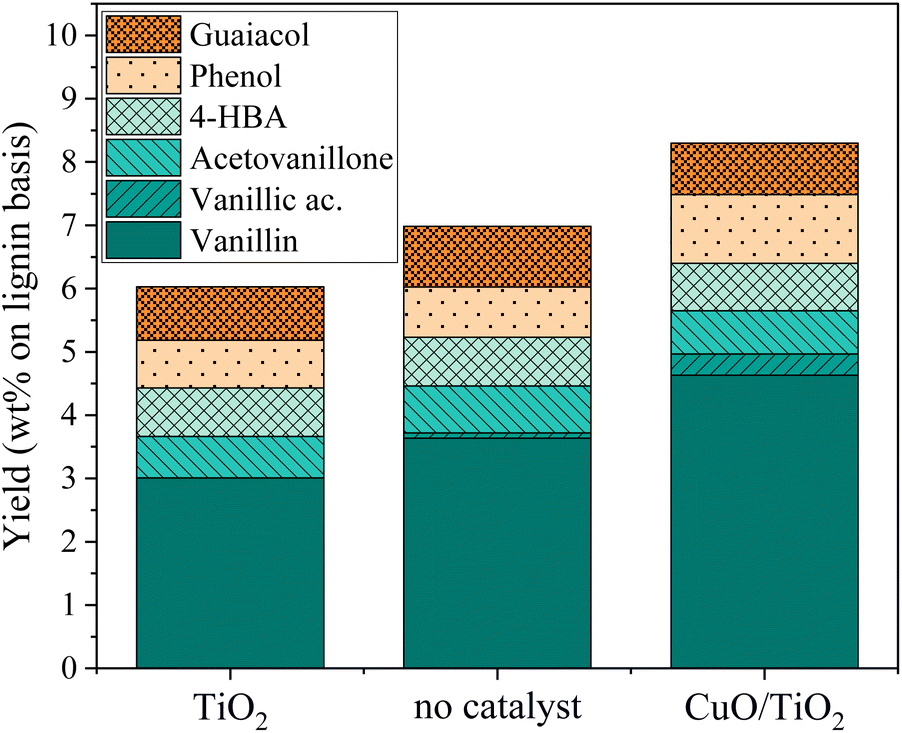
An additional experiment was performed to determine the active species on the CuO/TiO2 catalyst. The TiO2 support was tested alone with the grain size and concentration corresponding to those used for CuO/TiO2 (Fig. 3). The conversion value after 30 min was 89 wt% after 30 min, equivalent to that obtained in the absence of catalyst (94 wt%) or with CuO/TiO2 catalyst (92 wt%). The phenolics yields are also shown on Fig. 3. It can be noted that the values obtained in the presence of TiO2 are similar to those obtained without catalyst, i.e. 6.0 wt% yield with TiO2vs. 7.1 wt% yield without catalyst and 8.3 wt% yield with CuO/TiO2. The detail of the phenolics composition is also similar for both the TiO2 catalyst and the run without catalyst (Fig. 4). This confirms that copper oxide is the only active species of CuO/TiO2 involved in phenolic production during KBL oxidation.
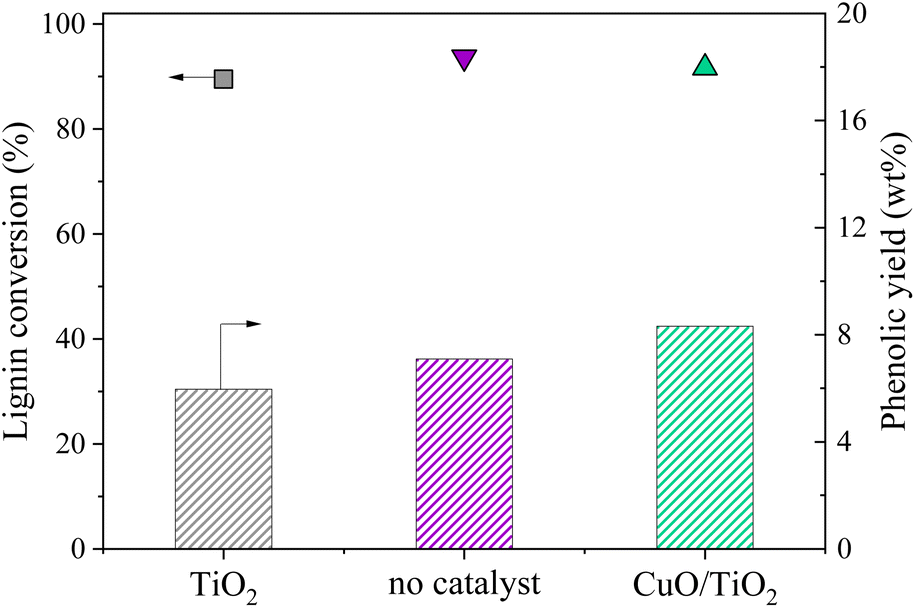 | ||
| Fig. 3 Lignin conversion (symbols) and phenolic yields (bars) during KBL oxidation for non-catalysed and catalysed reactions. Operating conditions: 150 °C, 20 bar air, stirring 1800 rpm, dry matter concentration 5 g L−1, copper/dry matter mass ratio 5%, 30 min. | ||
 | ||
| Fig. 4 Yields of phenolic compounds during KBL oxidation for non-catalysed and catalysed reactions. Operating conditions: 150 °C, 20 bar air, stirring 1800 rpm, dry matter concentration 5 g L−1, copper/dry matter mass ratio 5%, 30 min. | ||
Fig. 5 depicts the production of aliphatic compounds during catalytic and non-catalytic oxidation of KBL. The production of aliphatic compounds starts during the heating ramp and yields simple acids (formic and acetic acids), but also in di-acids such as oxalic, malonic, malic and succinic acids. Hydroxy-acids (glycolic and lactic acids) were not produced at this stage (little degradation even occurred). This production is more important for catalytic oxidation and the distribution of acids is slightly different: more formic acid is produced at the beginning of catalytic oxidation and more tartronic acid in the absence of catalyst. During the course of the reaction, the global yield in aliphatic acids increases drastically during the first 45 min of reaction after which a slower evolution occurs. The main aliphatic products are succinic, tartronic, oxalic, formic and acetic acids. Their production increases continuously during 45 min, but is more important in the presence of a catalyst (48 C% vs. 44 C% total yield). The production of acetic acid is more important in the absence of catalyst whereas the production of glycolic acid is slightly higher in the presence of catalyst. After a long reaction time (90 min), tartronic acid is degraded and more succinic acid is produced.
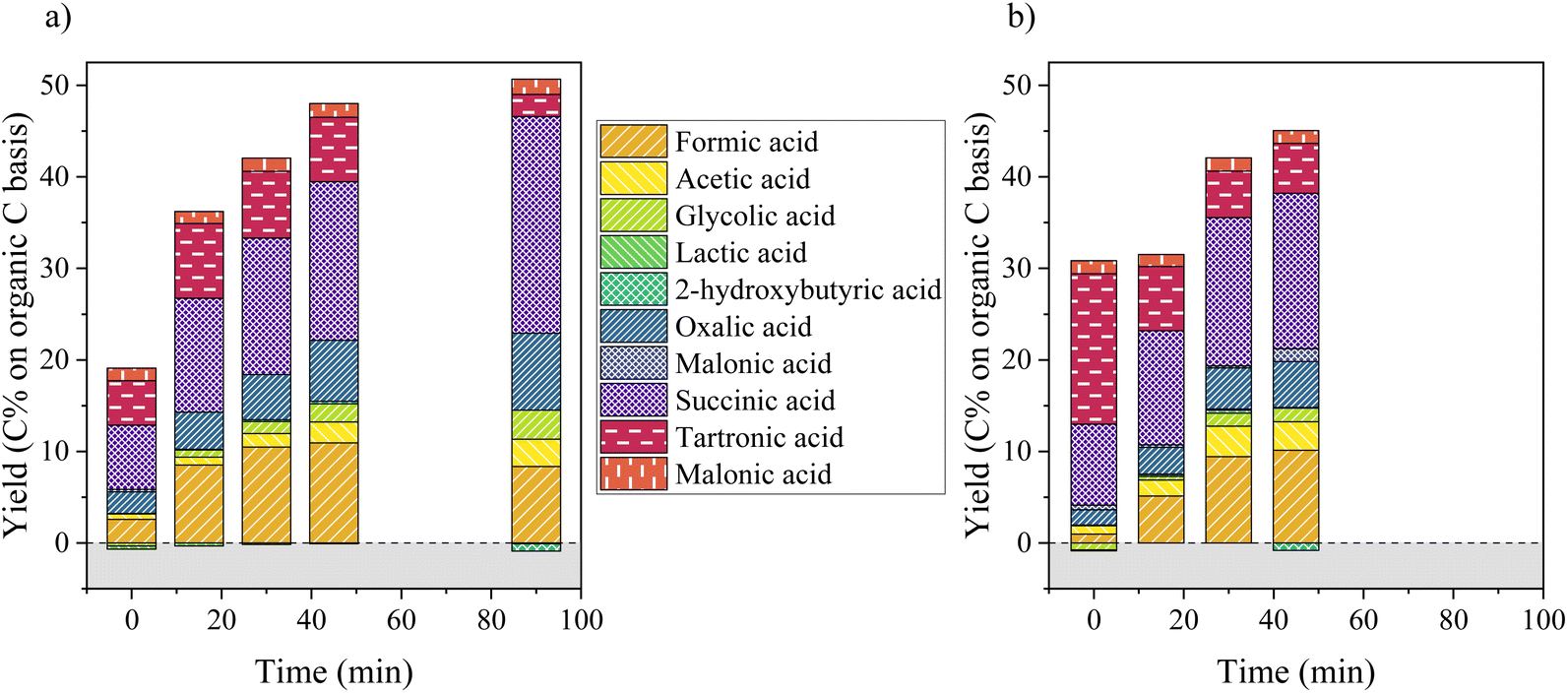 | ||
| Fig. 5 Production of aliphatic compounds during catalytic (a) or non-catalytic (b) oxidation of KBL. Yields are based on carbon content in KBL. Operating conditions: 150 °C, 20 bar air, stirring 1800 rpm, dry matter concentration 5 g L−1, copper/dry matter ratio 5%. | ||
SEC analysis (Table 3 and Fig. 6) shows two peaks corresponding to the presence of two types of compounds in all KP samples: oligomers with an average molar mass (Mw) between 900 and 1300 g mol−1. Monomers, dimers or trimers with an average molar mass inferior to the lowest calibration level (<400 g mol−1). Masses of oligomers decrease with reaction time but are similar for reactions with or without catalyst. The ratio between the oligomer peak and the monomer/dimer/trimer peak also decreases with reaction time, indicating that the KP fractions contain only a minor part of oligomers after 30 min.
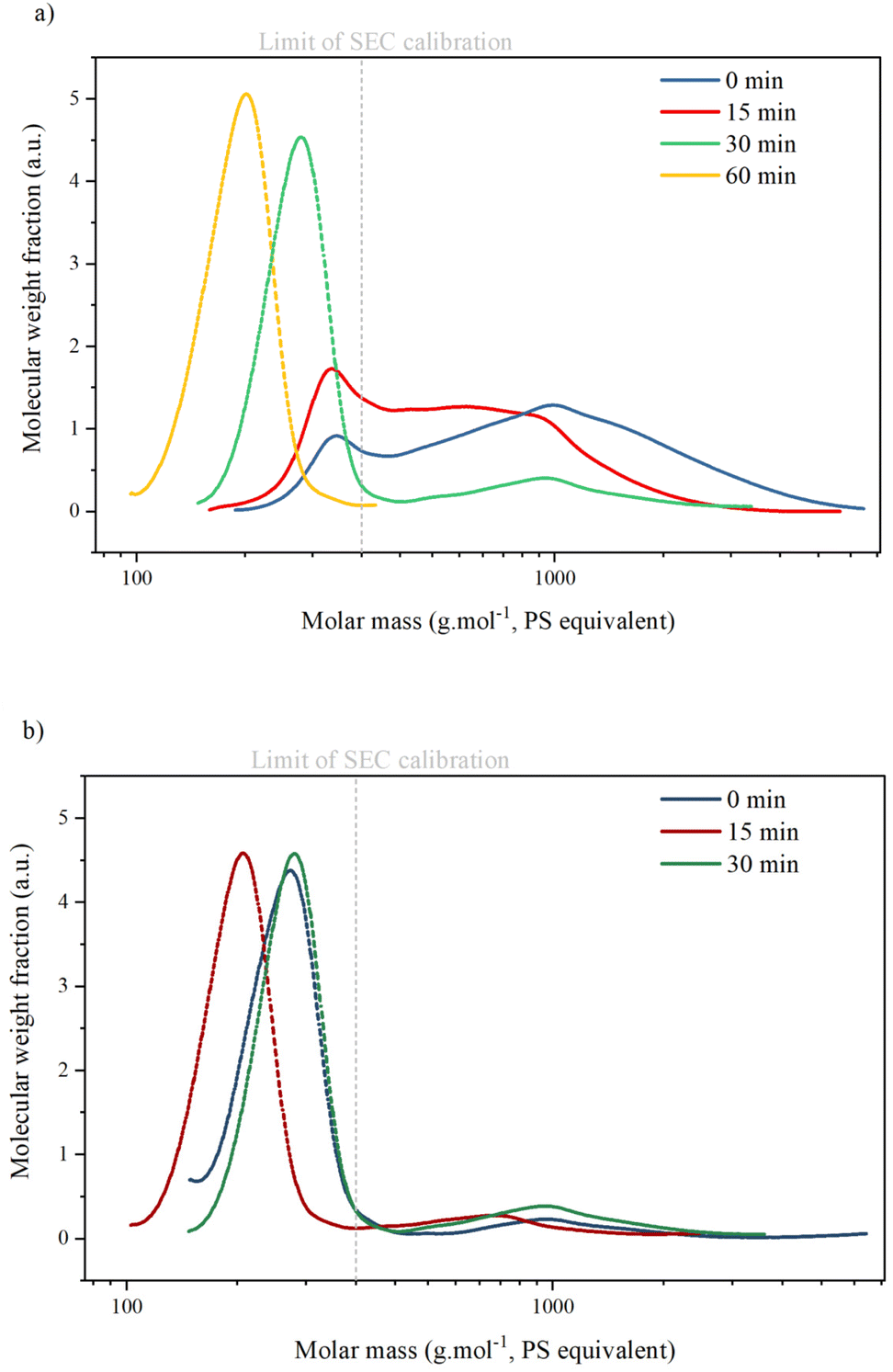 | ||
| Fig. 6 SEC analysis of Klason phases during catalytic (a) or non-catalytic (b) oxidation of KBL. Operating conditions: 150 °C, 20 bar air, stirring 1800 rpm, dry matter concentration 5 g L−1, copper/dry matter ratio 5%. | ||
The values of Mw for monomers are given as an indication only, as calibration with polystyrene is not suitable for lignin analysis: Constant et al. compared several SEC methods for technical lignin analysis and showed that the results obtained were highly dependent on the type of column, solvent and sample preparation.5 Furthermore, SEC is not suitable for the determination of small molar masses. It has already been reported that oligomers of lignin tend to deviate from PSS calibration curves during SEC analysis.43 Therefore, in this paper, SEC results on molar masses were considered only semi-quantitative and used for comparative purpose only.
FTIR spectra of the Klason phases issued from non-catalytic runs (see Fig. S2 in ESI†) still showed characteristic bands corresponding to lignin structure: aromatics at 1423 cm−1, 1509 cm−1 and 1591 cm−1; guaiacyl moieties at 1265 cm−1 and 1027 cm−1; unconjugated carbonyl groups at 1683–1693 cm−1; C–H stretching in CH2 and CH3 alkyl moieties 1452 cm−1 and ca. 1420 cm−1. Surprisingly, for runs at 0 and 15 min spectra showed an increase of the relative intensities of the bands corresponding to C–O stretching (1027 cm−1, 1044 cm−1 and 1076 cm−1). At the same time, the band at 1510 cm−1, characteristic of aromatics, decreased slightly in relative intensity, attesting for a release of aromatic moieties from lignin during the reaction. The band at 1693 cm−1 increased, attesting an oxidation of the material. After 30 min, IR spectrum did not show anymore the increase of the bands associated to C–O stretching, and the band at 1510 cm−1 is almost absent.
FTIR spectra obtained after a catalytic run of 30 min display almost the same spectra as the run carried out in the absence of catalyst. No significant differences were observed; spectra display all bands characteristic from the lignin structure, however with a relative lower intensity of the band at 1510 cm−1.
HSQC NMR were recorded for the Klason phases (see Fig. S8–S10, ESI†) and compared, each other, to that of the Klason phase obtained at a reaction time of 0 min. For reaction carried out in the absence of the catalyst, HSQC spectra of the Klason phase obtained at initial reaction time display all characteristic correlations of lignin structure: In the oxygenated aliphatic regions, correlations are observed for methoxy groups (δH/δC: 3.80/56.3 ppm); β-O-4 linkages (Aα) (δH/δC: 4.79/71.8 ppm), (AβG) (δH/δC: 4.78/82.0 ppm), (AβG/H) (δH/δC: 4.31/84.8 ppm) and (Aγ) (δH/δC: 3.53/60.4 ppm), (A′γ) (δH/δC: 4.12/67.4 ppm) and associated (A′′γ) (δH/δC: 3.63/63.4 ppm); β–β linkages (Bα) (δH/δC: 4.65/85.5 ppm), (Bβ) (δH/δC: 3.08/54.7 ppm) and (Bγ) (δH/δC: 3.72 and 4.11/71.3 ppm); β-5 linkages gave correlations at (Cα) (δH/δC: 5.52/87.6 ppm) and (Cβ) (δH/δC: 3.46/53.8 ppm). Aromatic region exhibits the characteristic correlations corresponding to guaiacyl units (G) (G2: δH/δC: 6.93/111.7 ppm, G5: δH/δC: 6.80/116.2 ppm, G6: δH/δC: 6.78/121.8 ppm); and those of p-hydroxyphenyl units (H2,6: δH/δC: 7.38/126.3 ppm). Correlations corresponding to guaiacyl units substituted by carbonyl moieties are also observed (G′2: δH/δC: 7.64/113.6 ppm, G′6: δH/δC: 7.33/120.3 ppm).
HSQC NMR spectra obtained after 15 min and 30 min reaction times without catalyst show highly degraded structures. Only correlations related to methoxy moieties (δH/δC: 3.85/56.8 ppm) and some of those corresponding to guaiacyl units (G2: δH/δC: 7.03/113.4 ppm, G5: δH/δC: 6.91/116.3 ppm, G′2: δH/δC: 7.49/112.2 ppm, G′6: δH/δC: 7.33/120.3 ppm) and p-hydroxyphenyl units (H2,6: δH/δC: 7.44/125.5 ppm) are observed. No correlations were observed for the inter-unit bonds, but some correlations corresponding to conjugated carboxylic acids were observed (δH/δC: 7.16–6.98/124.2 ppm and 6.85/127.4 ppm).
For the Klason phases issued from reaction conducted in the presence of the catalyst (see Fig. S11, ESI†), HSQC NMR spectra recorded after 30 min reaction showed almost the same situation, displaying only the correlation corresponding the methoxy groups (δH/δC: 3.87/56.8 ppm), and mostly to guaiacyl units substituted by carbonyl moieties (G′2: δH/δC: 7.55/113.1 ppm). Little correlations corresponding to guaiacyl unit were observed (G5: δH/δC: 6.87/115.9 ppm). Like before, correlations attributed to conjugated carboxylic acids (δH/δC: 7.16–6.98/124.2 ppm and 6.85/127.4 ppm) were observed.
Data obtained from HSQC NMR and SEC analyses showed that lignin in KBL is strongly degraded during catalytic oxidation, whatever a catalyst was used or not, with a drastic decrease in oligomer content in accordance with the disappearance of inter-unit moieties. That contrasts in some extent with data obtained from FTIR showing that lignin structure was kept in the Klason phases; however, FTIR and NMR analyses both showed a decrease of the aromatic content that can be related to oxidation of the aromatics in residual lignin, corroborated by elemental analysis and the observation of conjugated carboxylic acids in HSQC NMR experiments. Such a hypothesis is supported by the increase of the band at 1693 cm−1 in FTIR corresponding to carbonyl groups and the relatively intense correlations corresponding mainly to guaiacyl units substituted by carbonyl moieties (G′2: δH/δC: 7.64/113.6 ppm, G′6: δH/δC: 7.33/120.3 ppm).
Catalytic oxidation of kraft lignin (KL) – comparative study
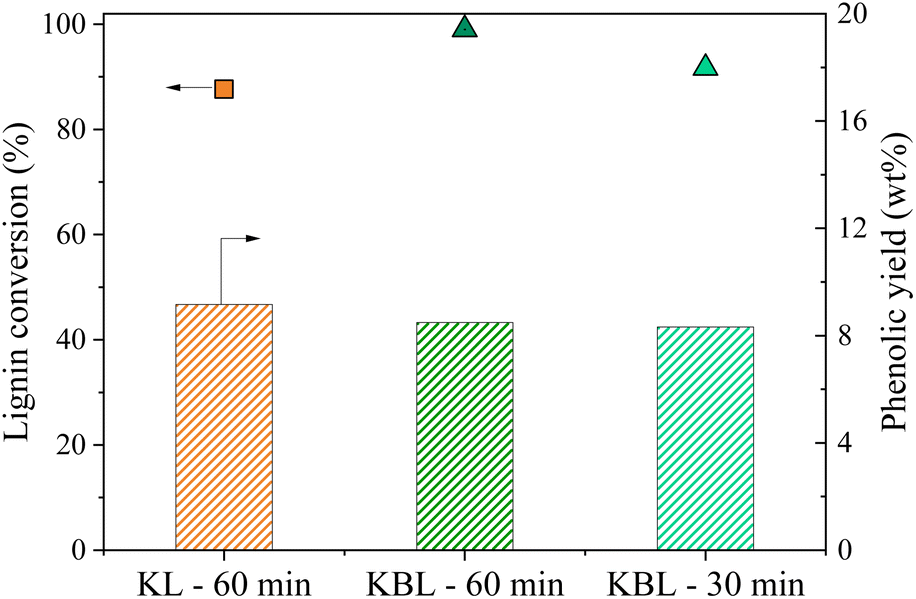 | ||
| Fig. 7 Lignin conversion (symbols) and phenolic yields (bars) during KBL and PL catalytic oxidation. Operating conditions: 150 °C, 20 bar air, stirring 1800 rpm, 30–60 min, dry matter concentration 5 g L−1, copper/dry matter ratio 5%. | ||
The distribution of phenolics was also similar for both starting materials after the same reaction time (Fig. 8). Vanillin was produced with a slightly higher yield from KBL (4.0 wt%) than from KL (3.7 wt%). Other phenolic compounds were detected at the same level of production.
 | ||
| Fig. 8 Yields of phenolic compounds during KBL and KL catalytic oxidation. Operating conditions: 150 °C, 20 bar air, stirring 1800 rpm, 30–60 min, dry matter concentration 5 g L−1, copper/lignin mass ratio 5%. | ||
Our results show that the oxidation of KBL is as efficient as the oxidation of KL for the production of phenolics. Therefore, the lignin purification step may not be necessary for the valorisation of kraft lignin. This finding may have strong implications for future industrial applications.
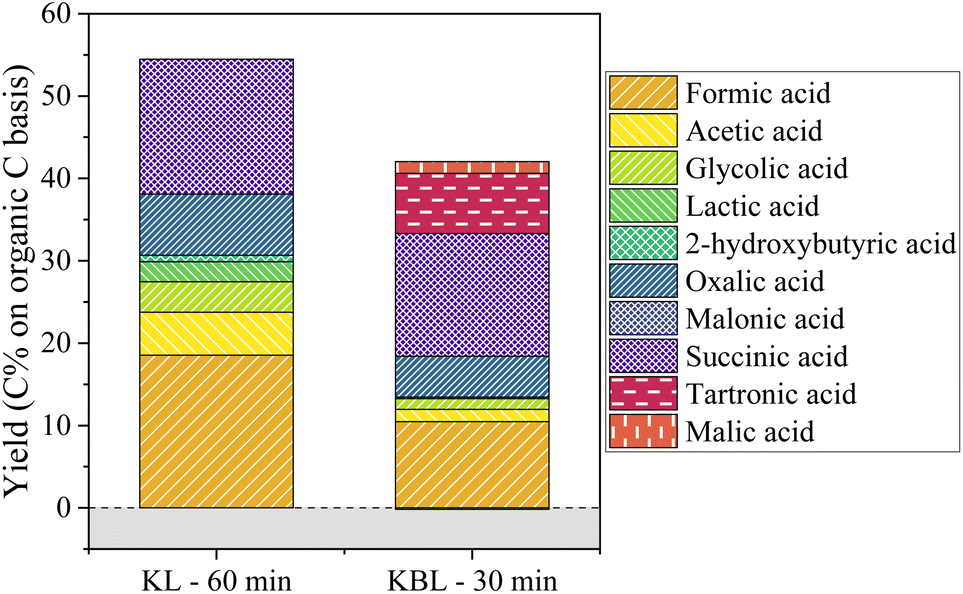 | ||
| Fig. 9 Aliphatic compounds production after PL and KBL oxidation. Operating conditions: 150 °C, 20 bar air, stirring 1800 rpm, 30–60 min, dry matter concentration 5 g L−1, copper/dry matter mass ratio 5%. | ||
The production of aliphatic acids from KL oxidation shows that a large part of aliphatic acids is actually formed from lignin, through ring opening reactions. Although lignin is often seen as a potential source of aromatic compounds, its valorisation into high value aliphatic compounds could also represent an interesting alternative.
Elemental analysis showed that KP from KL oxidation contains 56% of carbon and 0.5% of sulphur, indicating a partial oxidation and a partial desulphurisation of lignin during the reaction, as for KBL.
The SEC chromatogram of KP after KL oxidation shows two peaks corresponding to oligomers (Mw 1000 g mol−1) and monomers/dimers/trimers (Table 4 and Fig. 10). The ratio between the two peaks is higher after KL oxidation than after KBL oxidation, even after 30 min, indicating that lignin degradation is more advanced during KBL oxidation.
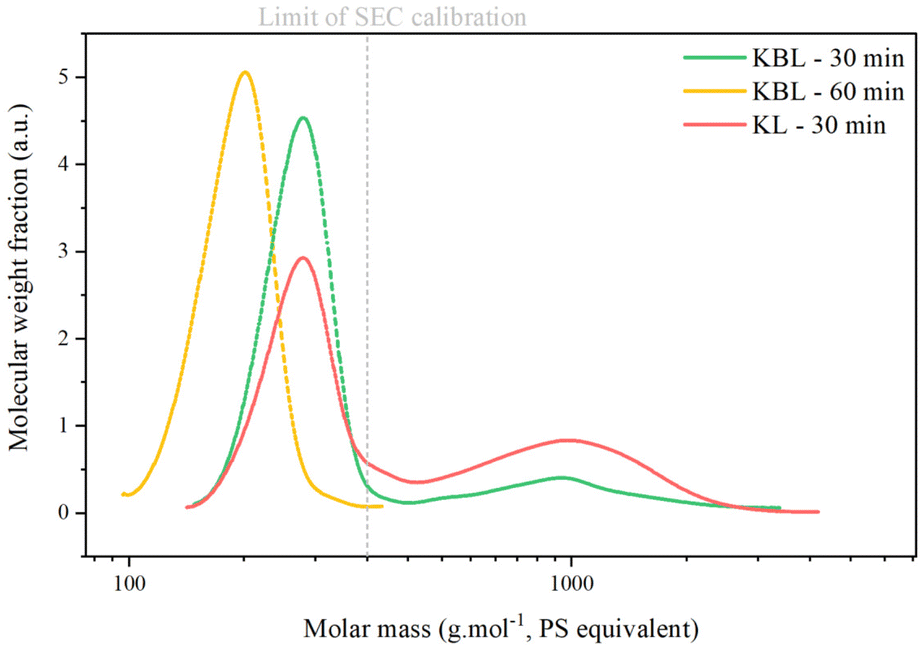 | ||
| Fig. 10 SEC analysis of Klason phases during catalytic oxidation of KL or KBL. Operating conditions: 150 °C, 20 bar air, stirring 1800 rpm, dry matter concentration 5 g L−1, copper/dry matter ratio 5%. | ||
On FTIR spectra (Fig. S4 in ESI†), some degradation of lignin structure is observed in both cases. However, characteristic bands of lignin can still be observed after KL oxidation (1417 cm−1, 1504 cm−1 and 1590 cm−1 corresponding to the aromatic rings; 1267 cm−1, 1184 cm−1 and 1029 cm−1 characteristic of guaiacyl moieties (resp. CO stretching; C–C and C–O stretching; aromatic C–H deformation); 1687 cm−1 for unconjugated carbonyl groups; 1461 cm−1, 1422 cm−1 and 1417 cm−1 assigned to C–H stretching in CH2 and CH3 alkyl moieties) while the relative intensity of the different bands did not fit with that of the original materials.
HSQC NMR spectra of the Klason phases obtained after a 30 min catalytic run, for KBL and after 60 min for KL (Fig. S12, ESI†), showed a strong degradation of lignin during reaction, like observed from FTIR. Regarding KL, like for KBL spectra, only correlation corresponding to methoxy groups (δH/δC: 3.91/56.7 ppm), guaiacyl units substituted by carbonyl moieties (G′2: δH/δC: 7.65/113.1 ppm), and partly to guaiacyl unit (G5: δH/δC: 6.98/116.1 ppm) and p-hydroxyphenyl units (H2,6: δH/δC: 7.58/124.9 ppm) are observed.
Reactor study – batch vs. continuous reactor
In an attempt to intensify the process of catalytic oxidation of KBL, the reaction was transferred to a fixed bed reactor, with the same catalyst, temperature, and KBL concentration. The residence time and contact time were very different from one mode to another because of the intrinsic properties of each reactor: in the batch reactor, reaction times are long and the catalyst is diluted in the reactor whereas in the fixed bed reactor, reaction times can be shortened and the catalyst concentration in the reactor is high.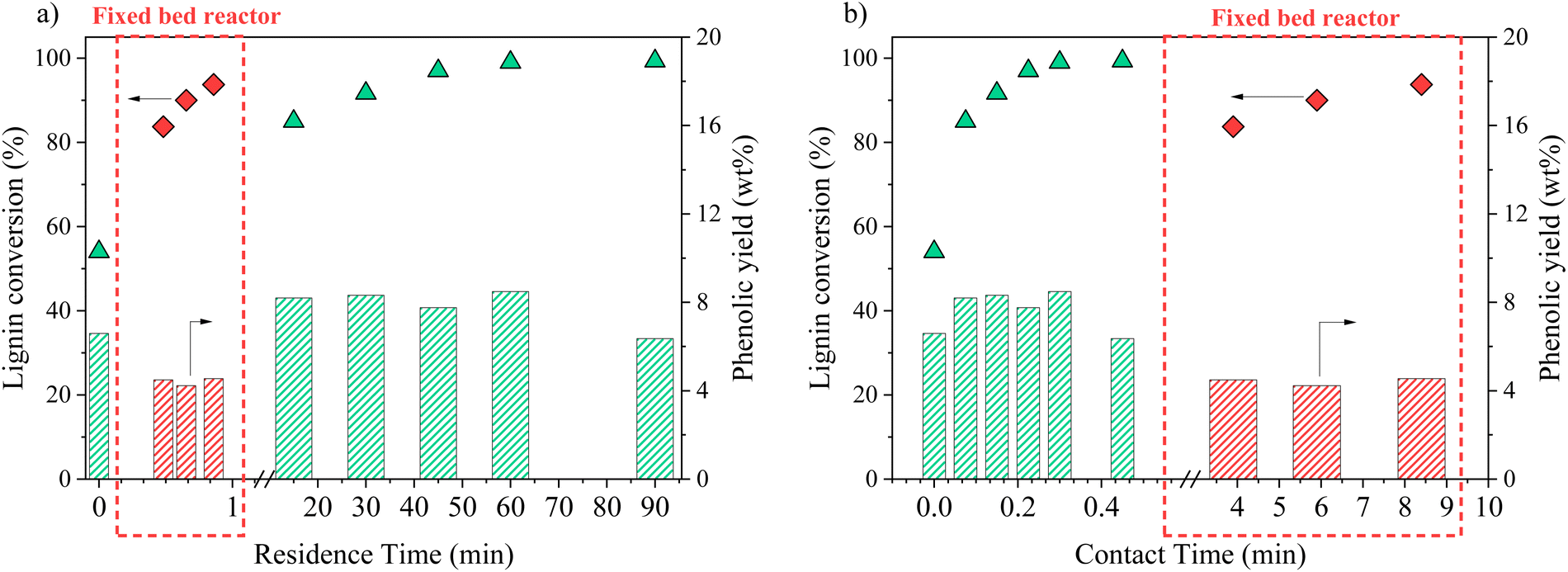 | ||
| Fig. 11 Lignin conversion and phenolics production during KBL catalytic oxidation, represented as a function of residence time (a) or contact time (b). Operating conditions: 150 °C, 20–80 bar air, dry matter concentration 5 g L−1, catalyst CuO/TiO2. | ||
The detailed composition of phenolics yield in both batch and continuous modes is depicted on Fig. 12. In the fixed bed reactor, the yield of vanillin corresponds to the lowest yield observed in the batch reactor at a long contact time; therefore, it seems that vanillin reaches a plateau around 2.5 wt% after a certain contact time. Acetovanillone reaches a very low yield in continuous mode at long contact time. Surprisingly, vanillic acid is still present in continuous mode at long contact times. Phenol and guaiacol are present with very low yields (inferior to 0.5 wt%).
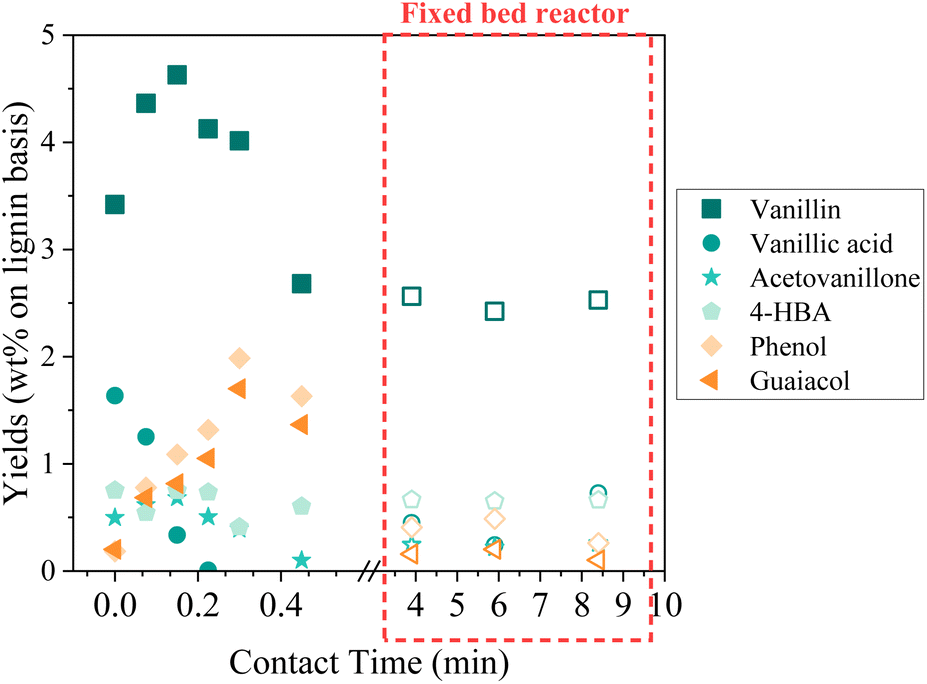 | ||
| Fig. 12 Details of phenolics production during KBL catalytic oxidation. Operating conditions: 150 °C, 20–80 bar air, dry matter concentration 5 g L−1, catalyst CuO/TiO2. | ||
The comparison of batch results and continuous results shows that the production and degradation of phenolics is a complex phenomenon that cannot be fully explained by an effect of contact time, i.e. the CuO/TiO2 catalyst effect. The reactivity of phenolics is most probably influenced by homogeneous reactions in an alkaline, oxidative medium and by mass transfer effects, which differ from one reactor to another.
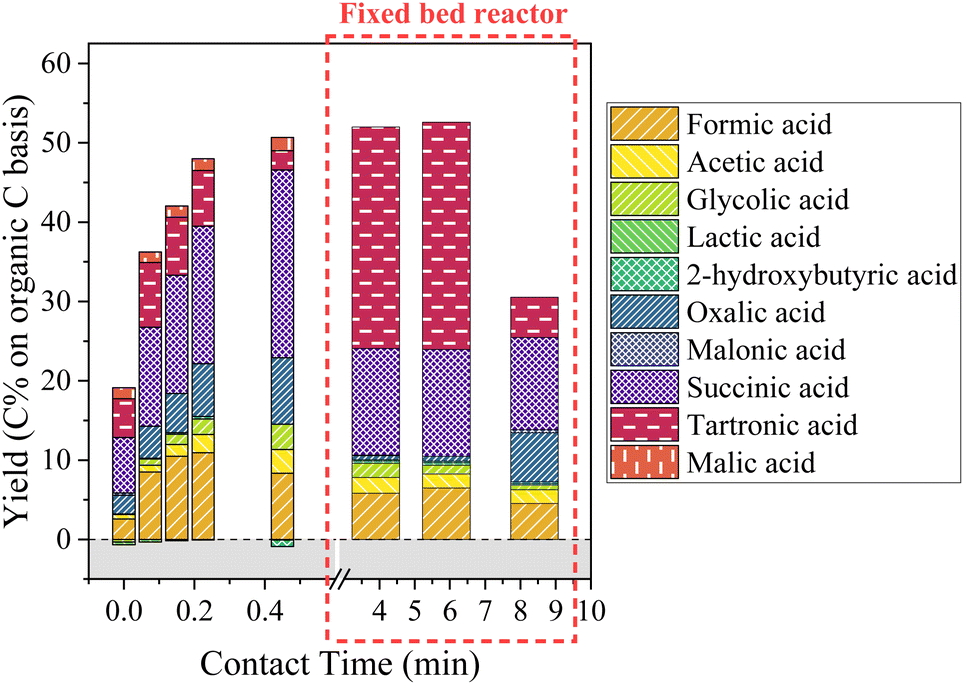 | ||
| Fig. 13 Aliphatic compounds production during KBL catalytic oxidation. Operating conditions: 150 °C, 20–80 bar air, dry matter concentration 5 g L−1, catalyst CuO/TiO2. | ||
Again, the formation and degradation of aliphatic acids seems to be complex and dependent not only on contact time (catalyst activity) but also on homogeneous reactions and/or reactor type.
Productivity for various compounds
Our work describes for the first time the formation of a broad range of products from the catalytic oxidation of black liquor: phenolic compounds, aliphatic compounds and lignin residues have been identified and quantified. This allows a first estimation of the productivity depending on the operating conditions (catalyst, time, starting material, reactor type) can be made (see Table S3 in ESI†). The aim here is not to provide a precise techno-economic analysis but only to draw the reader's attention on the potential of KBL for the production of these compounds.The productivity of vanillin was 88 g h−1 m−3 from isolated kraft lignin (Fig. 14a) but only 46 g h−1 m−3 from KBL under identical operating conditions (see ESI†), due to the lower lignin content in KBL. However, reducing the reaction time is efficient to increase vanillin productivity up to 105 g h−1 m−3 at 30 min (Fig. 14b). The use of a continuous reactor did not lead to an increase in vanillin yield (see below). Nevertheless, it is very efficient to increase the productivity: 730 g h−1 m−3 can be obtained at low contact time (Fig. 14d). These trends can also be observed for phenol productivity, which reaches a maximum of 120 g h−1 m−3 in continuous reactor with KBL. The other phenolic compounds give low individual productivity. However, the mix of all phenolic compounds could also be valorised as a mixture. The global phenolics productivity reaches 220 g h−1 m−3 from lignin, 190 g h−1 m−3 from KBL in batch reactor, 1.27 kg h−1 m−3 in continuous reactor.
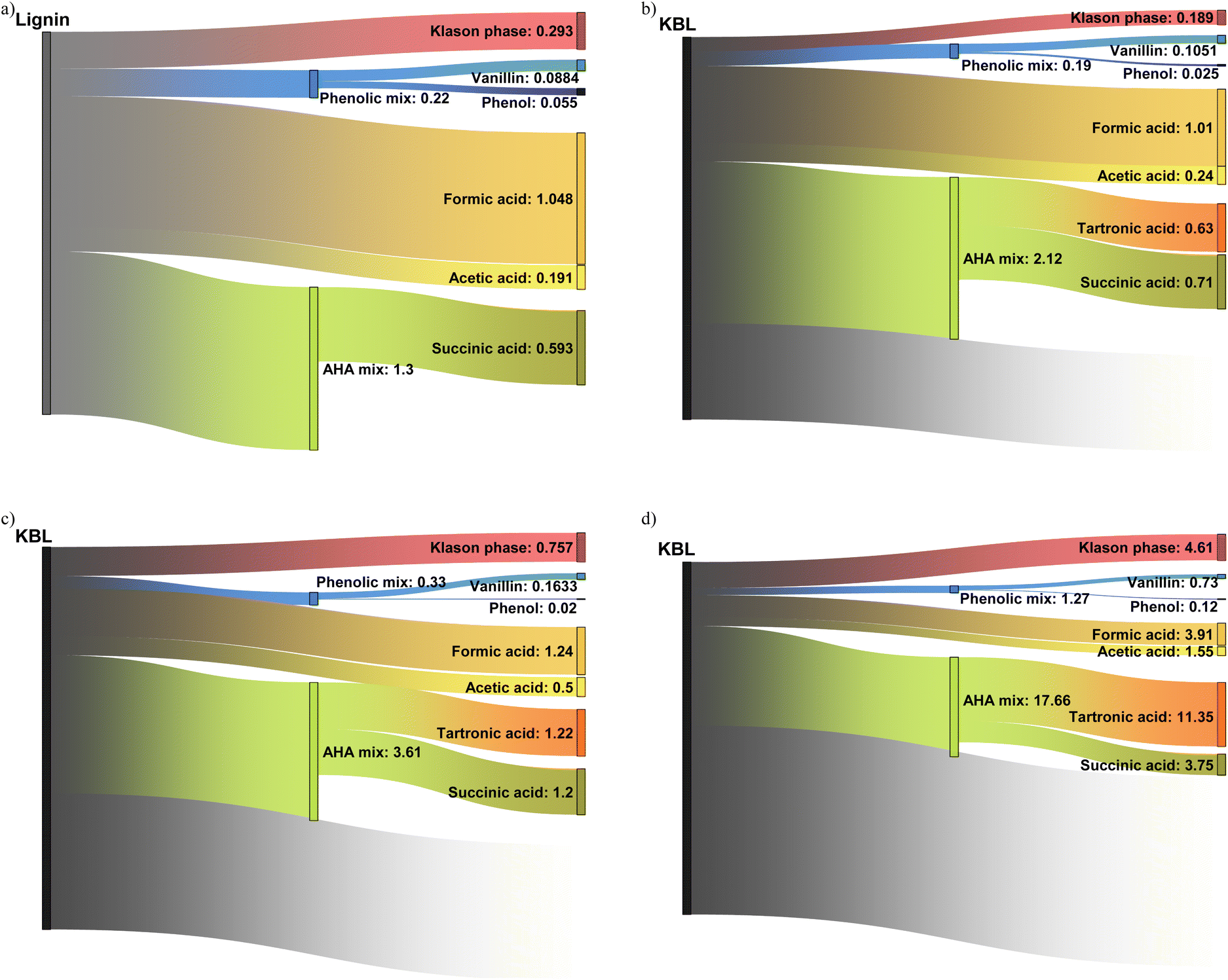 | ||
| Fig. 14 Sankey diagrams representing the productivity in kg m−3 h−1 of commodities from KL and KBL under different operating conditions: starting from KL, reaction time 60 min, catalyst CuO/TiO2 (a); starting from KBL, reaction time 30 min, catalyst CuO/TiO2 (b); starting from KBL, reaction time 15 min, no catalyst (c); starting from KBL, catalyst CuO/TiO2, continuous reactor (d). The grey part represents the non-identified materials and/or gaseous compounds, not analysed in this work. | ||
A phenolics mixture can be isolated from other oxidation products, for example by liquid–liquid extraction.18,44 Separation of one specific phenolic, e.g., vanillin, is more difficult and would require additional purification steps: liquid–liquid extraction, supercritical CO2 extraction, precipitation–crystallisation, membrane separation, solid adsorption have been studied for vanillin recovery after lignin oxidation.45 A combination of several purification steps is usually required. Centrifugal partition chromatography has shown a great potential for separating different phenolic compouds.46 These technologies could be adapted to recover vanillin from the KBL oxidation product mixture, with additional difficulties caused by the high concentration of sulphur and salts.
The yields of aliphatic compounds are much higher than those of phenolics and consequently the productivities are higher. Formic acid, which could be easily recovered by distillation, gives a productivity between 1 kg h−1 m−3 in batch reactor (from lignin or from KBL) and reaches 4.2 kg h−1 m−3 in continuous reactor. Acetic acid productivity reaches 191 g h−1 m−3 from lignin and 240 g h−1 m−3 from KBL in batch reactor, and 1.5 kg h−1 m−3 in continuous reactor. The volatility of acetic acid is also high enough to consider a distillation operation. Tartronic acid is not volatile and would require other techniques of purification (e.g., reactive distillation, electrodialysis, chromatography). Its productivity is null from lignin but reaches 11 kg h−1 m−3 from KBL in continuous reactor, making it a promising target molecule from KBL. Tartronic acid is a valuable compound for fine chemistry, used as a pharmaceutical, anti-corrosion agent, etc.47 Similarly, succinic acid would be demanding in terms of purification but its productivity reaches 593 g h−1 m−3 from lignin, and up to 3.75 kg h−1 m−3 from KBL in a continuous reactor. Succinic acid is a valuable compound that can be used for the synthesis of pharmaceuticals, adhesives, solvents, intermediates for polymer synthesis, and food additives.48 An alternative could be the valorisation of a mixture of hydroxy-acids (AHA mix), which is less complicated to purify.4 The productivity for this mix is the highest in a continuous reactor, reaching 17.86 kg h−1 m−3 from KBL.
The Klason phase could also be valorised as a material. Oxidised lignin can be applied, for example, to heavy metal removal49 or as a plant fertilizer.50 Its productivity reaches 293 g h−1 m−3 from lignin, or alternatively 757 g h−1 m−3 from KBL without catalyst, or 4.61 kg h−1 m−3 from KBL in a continuous reactor. It has the advantage of being easily removed from the product mixture by simple precipitation and filtration or centrifugation.
Finally, the comparison of the productivities from lignin and from KBL at identical reaction times (data in ESI†) evidences that lignin is a more productive starting material, given that KBL only contains ca. 30% of pure lignin (on a dry basis). The costs and energies associated with lignin purification prior to reaction should be taken into account to assess the advantage of using lignin as a starting material for the production of various commodities. The use of a solid CuO/TiO2 catalyst is advantageous for the productivity of phenolics but detrimental for the productivity of Klason phase. The most interesting finding is that the use of a continuous reactor allows the productivity of all products to be drastically increased. This demonstrates the interest of using intensified reactors in future forest biorefineries.
The products obtained from KBL oxidation form a complex mixture, which would require several steps of purification. Some research efforts on separation technologies is necessary to achieve the production of valuable biosourced compounds from KBL.
Conclusions
The oxidation of kraft black liquor was performed with or without a CuO/TiO2 catalyst, in an autoclave or in a fixed bed reactor. The oxidation of pure kraft lignin was also investigated. The oxidation products were systematically analysed.Our results showed that lignin depolymerisation was fast and was mainly due to non-catalytic reactivity. The resulting Klason phases contain degraded, oxidised and desulphurised lignin materials. Increasing the reaction time resulted in increased lignin depolymerisation. Using kraft lignin instead of kraft black liquor did not enhance lignin depolymerisation.
Besides lignin, two families of products were analysed. Phenolic compounds were produced in low yields, with vanillin being the main phenolic compound. Vanillin and its derivatives tend to degrade at long reaction times. Using a CuO/TiO2 catalyst increased the yields of phenolics, with a maximum yield of 8 wt% phenolics reached after 30 min at 150 °C. Aliphatic compounds were produced in higher yields, with a maximum of 50 C% achieved at long reaction times with a copper catalyst. The composition of the aliphatic acids evolves with the reaction time, with tartronic, succinic and formic acids being the main acids analysed. Using kraft lignin instead of kraft black liquor did not lead to higher phenolic yields but slightly increased the yields of aliphatics, with different main acids (no tartronic acid and more oxalic acid).
Switching from an autoclave to a fixed bed reactor resulted in different characteristic times: longer contact times and shorter residence times can be reached in continuous mode. This did not lead to an increase in the yields of phenolic and aliphatic compounds but to a different distribution in each family of products. These results illustrate that kraft liquor oxidation is a complex network of reactions involving catalytic and non-catalytic steps.
Finally, the calculation of productivity showed that a large panel of compounds can be produced from kraft black liquor: small productions of phenolic mix or pure phenolic compounds (phenol, vanillin), together with larger productions of formic acid, acetic acid, mix of hydroxy- and di-acids and/or pure fluxes of succinic and tartronic acids, and oxidised lignin. Additional research effort is needed to purify these compounds, which are of interest in chemical, F&F, pharmaceutical industries. We hope that these results will provide an incentive for the design of new processes for the chemical valorisation of black liquor.
Data availability
The datasets generated during and/or analysed during the current study are not publicly available but are available from the corresponding authors on reasonable request.Author contributions
LV: conceptualisation; methodology; investigation; writing – original draft, review & editing; visualisation; supervision. AHM: conceptualisation; methodology; investigation; writing – review; visualisation. NC: methodology; investigation; writing – review; visualisation. OB: investigation; writing – review; visualisation. MT: investigation; visualisation. FB: methodology; investigation; resources; writing – review. PF: conceptualisation; methodology; writing – review; supervision; funding acquisition. LD: conceptualisation; methodology; investigation; writing – review & editing; visualisation; supervision; project administration; funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Laura Reyes is acknowledged for her help on KBL characterization. IRCELYON scientific and technical services are kindly acknowledged. Chantal Lorentz (IRCELYON) is warmly thanked for her help on NMR experiments. The industrial company Smurfit Kappa, and particularly Jérôme Moindrot, is thanked for providing black liquor. Evonik is thanked for providing TiO2 catalyst support. This work was funded by ANR (project Phenoliq ANR-16-CE06-0007), CNRS and UCBL. The authors thank AXELERA chemical and environmental cluster.Notes and references
- H. Sixta, Handbook of Pulp, Wiley, Lenzing, 2nd edn, 2006, vol. 1–2 Search PubMed.
- K. Kuparinen, E. Vakkilainen and T. Tynjälä, Mitigation Adapt. Strategies Global Change, 2019, 24, 1213–1230 CrossRef.
- M. Moshkelani, M. Marinova, M. Perrier and J. Paris, Appl. Therm. Eng., 2013, 50, 1427–1436 CrossRef CAS.
- L. Reyes, C. Nikitine, L. Vilcocq and P. Fongarland, Green Chem., 2020, 22, 8097–8115 RSC.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- R. C. Andeme Ela, L. Spahn, N. Safaie, R. C. Ferrier and R. G. Ong, ACS Sustainable Chem. Eng., 2020, 8, 13997–14005 CrossRef CAS.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- T. Aro and P. Fatehi, ChemSusChem, 2017, 10, 1861–1877 CrossRef CAS PubMed.
- E. Terrell, L. D. Dellon, A. Dufour, E. Bartolomei, L. J. Broadbelt and M. Garcia-Perez, Ind. Eng. Chem. Res., 2020, 59, 526–555 CrossRef CAS.
- C. Rutten, A. Ramírez and J. Posada Duque, J. Chem. Technol. Biotechnol., 2017, 92, 257–270 CrossRef CAS.
- W. Sebhat, A. El-Roz, A. Crepet, C. Ladavière, D. D. S. Perez, S. Mangematin, C. C. Almada, L. Vilcocq, L. Djakovitch and P. Fongarland, Biomass Convers. Biorefin., 2020, 10, 351–366 CrossRef CAS.
- C. Cabral Almada, A. Kazachenko, P. Fongarland, D. Da Silva Perez, B. N. Kuznetsov and L. Djakovitch, Biomass Convers. Biorefin., 2022, 12, 3795–3808 CrossRef CAS.
- V. E. Tarabanko, D. V. Petukhov and G. E. Selyutin, Kinet. Catal., 2004, 45, 569–577 CrossRef CAS.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- A. G. Demesa, A. Laari, I. Turunen and M. Sillanpää, Chem. Eng. Technol., 2015, 38, 2270–2278 CrossRef CAS.
- L. Das, P. Kolar and R. Sharma-Shivappa, Biofuels, 2012, 3, 155–166 CrossRef CAS.
- S. Kang, X. Li, J. Fan and J. Chang, Renewable Sustainable Energy Rev., 2013, 27, 546–558 CrossRef CAS.
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
- R. Ma, M. Guo and X. Zhang, Catal. Today, 2018, 302, 50–60 CrossRef CAS.
- J.-M. Ha, K.-R. Hwang, Y.-M. Kim, J. Jae, K. H. Kim, H. W. Lee, J.-Y. Kim and Y.-K. Park, Renewable Sustainable Energy Rev., 2019, 111, 422–441 CrossRef CAS.
- M. Otromke, R. J. White and J. Sauer, Carbon Resour. Convers., 2019, 2, 59–71 CrossRef CAS.
- X. Liu, F. P. Bouxin, J. Fan, V. L. Budarin, C. Hu and J. H. Clark, ChemSusChem, 2020, 13, 4296–4317 CrossRef CAS PubMed.
- D. Bourbiaux, J. Pu, F. Rataboul, L. Djakovitch, C. Geantet and D. Laurenti, Catal. Today, 2021, 373, 24–37 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- J. C. Villar, A. Caperos and F. García-Ochoa, Wood Sci. Technol., 2001, 35, 245–255 CrossRef CAS.
- V. E. Tarabanko, N. A. Fomova, B. N. Kuznetsov, N. M. Ivanchenko and A. V. Kudryashev, React. Kinet. Catal. Lett., 1995, 55, 161–170 CrossRef CAS.
- M. Naqvi, J. Yan and E. Dahlquist, Bioresour. Technol., 2010, 101, 8001–8015 CrossRef CAS PubMed.
- M. Huet, A. Roubaud, C. Chirat and D. Lachenal, Biomass Bioenergy, 2016, 89, 105–112 CrossRef CAS.
- A. Garron, P. P. Arquilliere, W. Al Maksoud, C. Larabi, J.-J. Walter and C. C. Santini, Appl. Catal., A, 2015, 502, 230–238 CrossRef CAS.
- H. R. Muddassar, M. H. Sipponen, K. Melin, D. de Kokkonen, O. Pastinen and S. Golam, Ind. Eng. Chem. Res., 2015, 54, 7833–7840 CrossRef CAS.
- A. Hernández Mañas, N. Chaussard, F. Bertaud, L. Vilcocq, P. Fongarland and L. Djakovitch, Ind. Eng. Chem. Res., 2022, 61, 7430–7437 CrossRef.
- J.-P. Lange, Angew. Chem., Int. Ed., 2015, 54, 13186–13197 CrossRef CAS PubMed.
- A. Santos, P. Yustos, A. Quintanilla, G. Ruiz and F. Garcia-Ochoa, Appl. Catal., B, 2005, 61, 323–333 CrossRef CAS.
- O. Theander and E. A. Westerlund, J. Agric. Food Chem., 1986, 34, 330–336 CrossRef CAS.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of Structural Carbohydrates and Lignin in Biomass, 2012 Search PubMed.
- W. Sebhat, A. El Roz, P. Fongarland, L. Vilcocq and L. Djakovitch, Catalysts, 2021, 11, 875 CrossRef CAS.
- C. Cabral Almada, A. Kazachenko, P. Fongarland, D. Da Silva Perez, B. N. Kuznetsov and L. Djakovitch, Catalysts, 2021, 11, 467 CrossRef.
- A. Hernandez Mañas, PhD thesis, Université Claude Bernard Lyon 1, 2020 Search PubMed.
- L. Vilcocq, N. Chaussard, A. Hernández Mañas, F. Bertaud, P. Fongarland and L. Djakovitch, Waste Biomass Valorization, 2023 DOI:10.1007/s12649-023-02082-y.
- F. M. Casimiro, C. A. E. Costa, C. M. Botelho, M. F. Barreiro and A. E. Rodrigues, Ind. Eng. Chem. Res., 2019, 58, 16442–16449 CrossRef CAS.
- G. Zinovyev, I. Sulaeva, S. Podzimek, D. Rössner, I. Kilpeläinen, I. Sumerskii, T. Rosenau and A. Otthast, ChemSusChem, 2018, 11, 3259–3268 CrossRef CAS PubMed.
- P. C. R. Pinto, E. A. B. da Silva and A. E. Rodrigues, Ind. Eng. Chem. Res., 2010, 49, 12311–12318 CrossRef CAS.
- M. I. F. Mota, P. C. Rodrigues Pinto, J. M. Loureiro and A. E. Rodrigues, Sep. Purif. Rev., 2016, 45, 227–259 CrossRef CAS.
- I. L. D. Rocha, A. M. Da Costa Lopes, S. P. M. Ventura and J. A. P. Coutinho, ACS Sustainable Chem. Eng., 2022, 10, 4913–4921 CrossRef CAS PubMed.
- B. Katryniok, H. Kimura, E. Skrzyńska, J.-S. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Green Chem., 2011, 13, 1960 RSC.
- M. Verma, P. Mandyal, D. Singh and N. Gupta, ChemSusChem, 2020, 13, 4026–4034 CrossRef CAS PubMed.
- G. C. Quintana, G. J. M. Rocha, A. R. Gonçalves and J. A. Velásquez, BioResources, 2008, 3, 1092–1102 CAS.
- S. Sutradhar, N. Alam, L. P. Christopher and P. Fatehi, Catal. Today, 2022, 404, 49–62 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst preparation and characterization, FTIR spectra, NMR spectra, HPLC analysis of KBL, productivity values. See DOI: https://doi.org/10.1039/d3gc00388d |
| This journal is © The Royal Society of Chemistry 2023 |