
Highly selective cross ketonization of renewable acids over magnesium oxide†
Tejas
Goculdas
ab,
Siddharth
Deshpande
a,
Weiqing
Zheng
 a,
Raymond J.
Gorte
ac,
Sunitha
Sadula
*a and
Dionisios G.
Vlachos
*ab
a,
Raymond J.
Gorte
ac,
Sunitha
Sadula
*a and
Dionisios G.
Vlachos
*ab
aCatalysis Center for Energy Innovation, 221 Academy St., Newark, DE 19716, USA. E-mail: vlachos@udel.edu; sunithak@udel.edu
bDepartment of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy Street, Newark, DE 19716, USA
cDepartment of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, PA 19104, USA
First published on 14th December 2022
Abstract
The rising demand for linear alkylbenzene surfactants (LAS) poses an environmental threat as LAS are industrially produced from petroleum using corrosive acid catalysts. Cross ketonization is a promising route of converting furans and fatty acids to oleo-furansulfonates, precursors for renewable, performance-enhanced LAS replacements. Here, we explore various alkaline earth metals and mixed metal oxides for the cross-ketonization reaction and find that MgO achieves ∼90% yield by minimizing the decarboxylation side reaction. We illustrate the role of molecular structure on the cross-ketonization reaction and the role of complex formation over alkaline-earth metal oxides in minimizing side reactions. We overcome catalyst deactivation by regeneration over multiple runs. This research demonstrates the effectiveness of heterogeneous catalysts for cross ketonization toward developing renewable surfactants.
1. Introduction
Biomass is a renewable resource and relatively abundant. The United States can produce between 991 million and 1147 million dry tons of biomass annually.1 There has been intense research on converting inedible biomass to furans,2–5 platform molecules that can be upgraded to commodity chemicals, such as adipic acid, p-xylene, butadiene, lubricant base oils, and surfactants, among others. Of these, higher molecular weight products are produced by C–C coupling reactions, such as aldol condensation and acylation. These include the synthesis of C28 and C33 lubricants of furfural6 and biomass-derived oleo-furansulfonates (OFS) surfactants.7OFS are synthesized by acylating biobased furan with fatty acids, followed by hydrodeoxygenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and sulfonation. Park et al. tested the performance of OFS in key detergency properties; they found that OFS outperform LAS in essential metrics such as critical micelle concentration, Kraft point, and stability in hard water conditions.7 Therefore, biomass-derived OFS were identified as a performance-enhanced replacement for petroleum-derived LAS. The C–C coupling reaction producing 2-alkoylfuran via Friedel–Crafts acylation is challenging (Fig. 1).7 One could use a fatty acid anhydride over a porous Brønsted acid zeolite, such as Al-SPP, to achieve a 90% yield to 2-alkoylfuran. However, this pathway produces an equimolar amount of fatty acid. Furthermore, the high boiling point of fatty acid makes separating the desired ketone product energy intensive and expensive. In an alternative route, trifluoroacetic anhydride (TFAA) transforms lauric acid to lauric–trifluoroacetic anhydride, which then acylates the furan. While the yield is high, this chemistry generates a waste stream of corrosive trifluoroacetic acid. Attempts with zeolites and other acid catalysts resulted in a lower 2-alkanoyl product yield. Additionally, thermodynamic calculations show that the acylation with longer chains is equilibrium limited, resulting in low yields.8 Therefore, a heterogeneously catalyzed, irreversible pathway is desired.
O group and sulfonation. Park et al. tested the performance of OFS in key detergency properties; they found that OFS outperform LAS in essential metrics such as critical micelle concentration, Kraft point, and stability in hard water conditions.7 Therefore, biomass-derived OFS were identified as a performance-enhanced replacement for petroleum-derived LAS. The C–C coupling reaction producing 2-alkoylfuran via Friedel–Crafts acylation is challenging (Fig. 1).7 One could use a fatty acid anhydride over a porous Brønsted acid zeolite, such as Al-SPP, to achieve a 90% yield to 2-alkoylfuran. However, this pathway produces an equimolar amount of fatty acid. Furthermore, the high boiling point of fatty acid makes separating the desired ketone product energy intensive and expensive. In an alternative route, trifluoroacetic anhydride (TFAA) transforms lauric acid to lauric–trifluoroacetic anhydride, which then acylates the furan. While the yield is high, this chemistry generates a waste stream of corrosive trifluoroacetic acid. Attempts with zeolites and other acid catalysts resulted in a lower 2-alkanoyl product yield. Additionally, thermodynamic calculations show that the acylation with longer chains is equilibrium limited, resulting in low yields.8 Therefore, a heterogeneously catalyzed, irreversible pathway is desired.
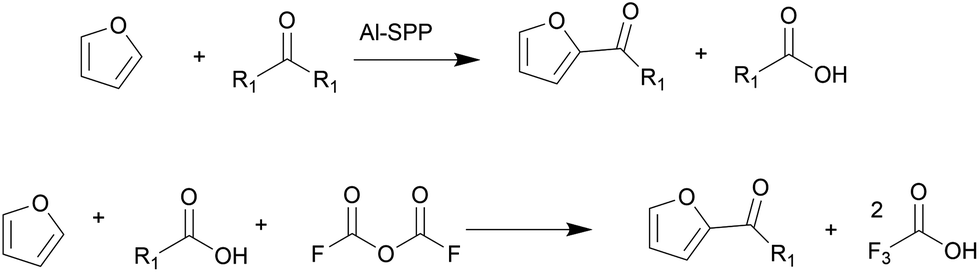 | ||
| Fig. 1 Pathways to oleo-furansulfonate bio-surfactant. R represents the n-C11H23 chain. | ||
Ketonization is an established irreversible C–C coupling reaction due to the abundance of oxygenates derived from coconut husks and palm kernels.9 It has been demonstrated for various substrates using heterogeneous, environmentally friendly catalysts10–12 and for making different products, including jet fuels, precursors for branched-chain lubricants,6,9,13 and ketone bio-waxes.14 Chen et al. utilized cross-ketonization between 2-furoic and C4–C6 acids to synthesize short-chain (C4–6) 2-alkoylfuran in 80% yield using a nanoparticle MgO catalyst. However, this short chain does not possess detergent properties.15 Recently Nguyen et al. produced 2-dodecanoyl furan via cross-ketonization of 2-furoic acid and lauric acid (C12) over earth-abundant, commercially-available iron oxides.16 2-Furoic acid can be produced from commercial biomass-derived furfural, and lauric acid is made commercially from vegetable oils.17–19 High lattice energy commercial metal oxides were screened. They found that in situ pre-reduction of the mixed iron oxide created the active Fe2+ sites for ketonization.16 The irreversible cross-ketonization strategy also offers ease of separation and recyclability of solid catalysts. However, the 2-alkanoyl furan yield was low (43%) due to the 2-furoic acid decarboxylation (a side reaction), as depicted in Fig. 2. A large molar excess of 2-furoic acid (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was needed, and a ∼30% carbon loss was seen. While promising, improvements are necessary.
:1) was needed, and a ∼30% carbon loss was seen. While promising, improvements are necessary.
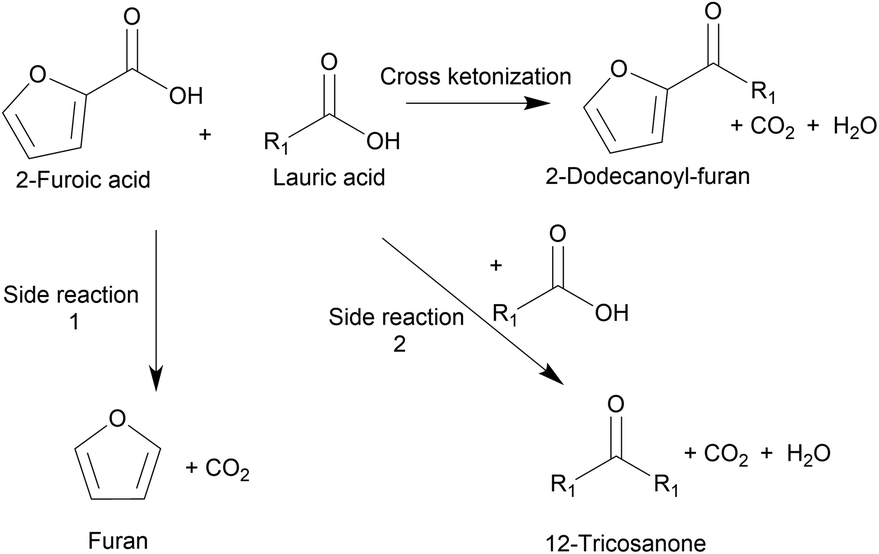 | ||
| Fig. 2 Scheme for the overall ketonization reaction between 2-furoic acid and lauric acid (R1 = C11H23). The side reactions 1 and 2 are the decarboxylation of 2-furoic acid to furan and the lauric acid self-ketonization to 12-tricosanone. | ||
Here we seek to address three critical gaps for the cross-ketonization reaction of 2-furoic acid and fatty acid: (1) explore additional basic and doped metal oxide catalysts to improve 2-product yield, (2) understand the reaction process, specifically the decarboxylation side reaction and (3) provide mechanistic insights into basic oxides. We identify magnesium oxide as a highly selective catalyst, obtaining close to 90% cross-ketone yield under optimized conditions. This catalytic performance is retained over four regeneration cycles. We then extend the study to other fatty and aromatic acids to gain insights into the ketonization chemistry. XRD and FT-IR ex situ studies on the recovered catalyst elucidate metal-acid complexation. Complexation reduces decarboxylation and is consequently identified as the key to MgO's superior performance over other catalysts. Computational studies provide atomistic insights into this complexation and the rate-limiting step over alkaline earth oxides.
2. Experimental methods
2.1 Materials
Furoic acid (FA), lauric acid (LA), n-dodecane, methylene chloride, and chloroform were purchased from Sigma Aldrich. Dimethyl sulfoxide (DMSO) was acquired from Fisher Scientifics. The metal oxides, iron(II, III) oxide (Fe3O4), magnesium oxide (MgO), calcium oxide (CaO), barium oxide (BaO), and hydrotalcite (Mg-Al2O3) were obtained from Sigma Aldrich. The hydrated metal salts used for the synthesis of doped catalysts were also obtained from Sigma Aldrich. The chemicals were all used as received. Detailed characterization for the commercial MgO is given in Fig. S1–S4.†2.2 Catalytic reaction and product analysis
The reactions were carried out in a 100 ml Parr reactor. The reactants, 2-furoic acid, lauric acid, n-dodecane (solvent), and the catalyst, were placed in a glass liner with a magnetic stir bar. After loading the glass liner in the Parr reactor, the reactor was closed, purged three times with nitrogen, and then pressurized to the desired pressure. Experimental details are given in our previous publication.16 After a designated reaction time, the reactor was cooled in an ice bath, depressurized, and opened. Due to the low solubility of 2-furoic acid in n-dodecane, DMSO was added to the post-reaction solution to extract the unreacted 2-furoic acid for quantification. The products and remaining reactants were identified and quantified via a gas chromatogram mass spectrometer (GCMS) equipped with a DB-5 column and a gas chromatogram with a flame ionization detector (GC-FID) equipped with an HP-1 column, respectively. Calibration curves were developed whenever the pure compounds were commercially available. The calibration data is given in Fig. S5–S9.† Because 2-dodecanoyl furan is not commercially available, quantification was performed based on the effective carbon number method (ECN)20 using hexane and 2-acetyl furan standards. The method was verified for 2-hexanoyl furan in our previous work16 and dodecaphenone in this work. Lauric acid was the limiting reagent, so its initial and final concentrations calculated the ketones’ yields.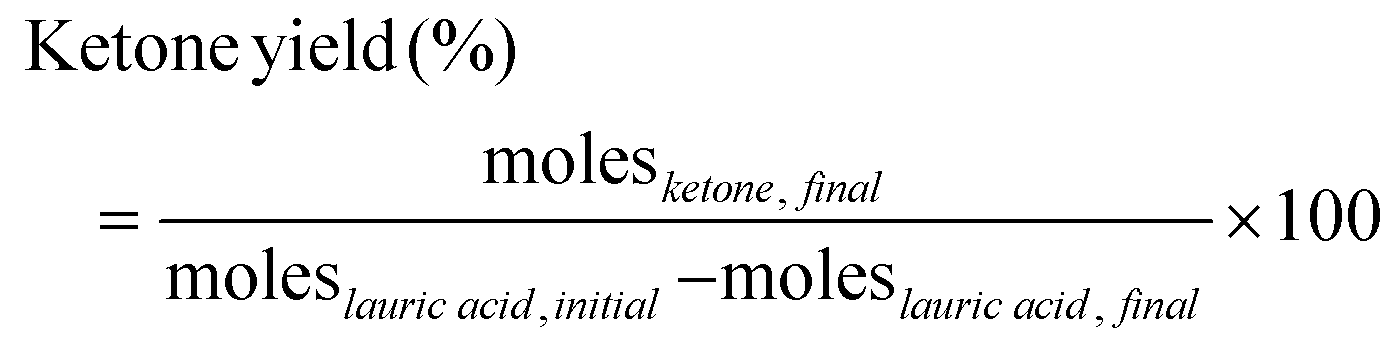 | (1) |
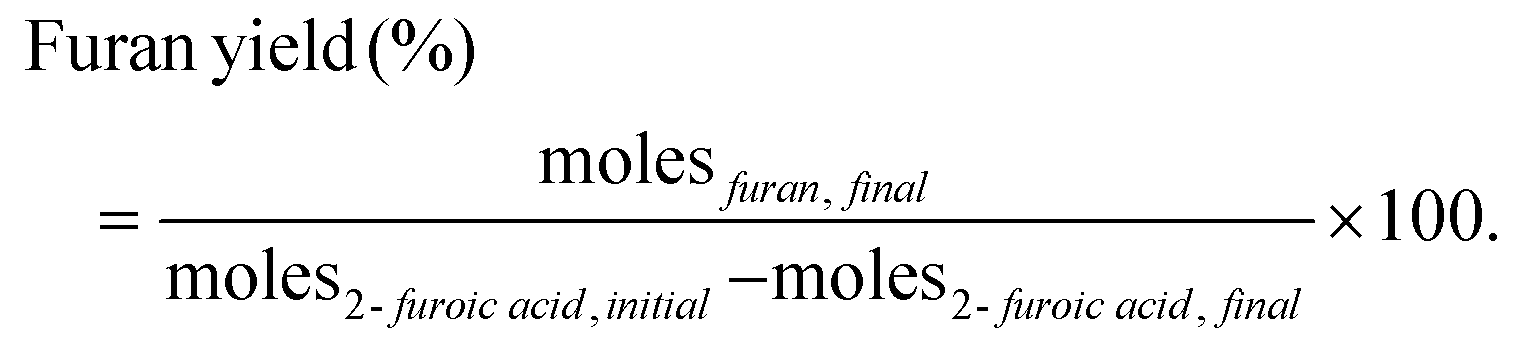 | (2) |
2.3 Catalyst synthesis
The doped metal oxide catalysts Ce0.8Fe0.2O3, Ce0.7Zr0.3O3, Mg3Al1O4.5, Ce0.1Mg3Al0.9O4.5 were prepared using co-precipitation.21 Appropriate amounts of the hydrated salts for each mixed metal oxide were first dissolved in DI water.13,15,22,23 Then the solution was added to another container with DI water. The pH of the solution was raised to 8–9 through the simultaneous addition of a strong base, either ammonium or sodium hydroxide. The metal oxides started precipitating out due to the elevated pH. The colloidal solution was then placed in an oil bath and heated at temperatures ranging from 80–90 °C overnight. The solution was then filtered out, and the precipitate was dried overnight in an oven. The resulting powder was then calcinated to burn off any impurities.2.4 Catalyst characterization
The crystalline phase of the catalysts was identified using an X-ray diffractometer (Bruker D8) with Cu Kα radiation (λ = 1.54056 Å) at 40 kV and 40 mA. Each catalyst's surface area and pore volume were measured using N2 physisorption at 77 K using a Micromeritics ASAP 2020 Brunauer–Emmett–Teller (BET) Analyzer. Thermogravimetric analysis (TGA) was performed with a TA Q600 TGA/DSC instrument with a ramp rate of 20 °C min−1 in air within a temperature range of 20–800 °C. Fourier transform infrared (FTIR) data was collected with a Thermo Fisher FTIR/ATR by scanning the sample from 400 to 4000 nm.2.5 Catalyst recycling
For the recycling experiments, the spent catalyst was filtered from the post-reaction solution and then washed multiple times with chloroform due to its high volatility and ability to solubilize organic material. The recovered catalyst was then dried in an oven at 100 °C overnight. The filtered solution was then analyzed using a Rigaku Supermini 200 WDXRF machine to detect the presence of Mg2+ ions. Finally, the spent material was regenerated by calcination in air at 600 °C. Due to material losses during filtration and collection, two reaction runs were conducted to recover sufficient catalyst for the next reusability experiment. Around 20% loss of recovered catalyst is attributed to the solid residue left on the filter paper that is difficult to scrape and the 2-furoic acid-MgO complex.2.6 ICP-MS analysis
Magnesium species concentration in the post-reaction filtrates was measured using ICP-MS. One milliliter of the filtrate was heated in a vacuum oven at 200 °C to evaporate the n-dodecane. The remaining solids were dissolved in a 20 mL mixture containing 50% (v/v) ethanol, 2.5% (v/v) HNO3, and 47.5% (v/v) water and sonicated in a water bath at 50 °C for 30 min. The mixture was diluted 50 times using a 5% (v/v) HNO3 solution for ICP-MS. Six standards containing 1 ppb, 10 ppb, 50 ppb, 100 ppb, 500 ppb, and 1000 ppb of Mg (SPEX CertiPrep) were used for calibration. The detailed ICP-MS sample preparation procedure was reported by Nguyen et al.162.7 Metal-carboxylate complex synthesis
Complexes of the metal oxides with lauric and 2-furoic acid were prepared, following a literature procedure.24 1.5:1 molar ratio of acid to metal oxide was added to a 100 ml Parr reactor with 30 ml of the n-dodecane under 20 bar of N2 at a stirring rate of 800 rpm. The temperature was raised to 250 °C for 2 h to ensure complete complex formation. The reaction was quenched in an ice bath, and the solids were collected by vacuum filtration. The solids were washed multiple times with chloroform to remove traces of unreacted acids. Then the solids were dried in an oven overnight. The solids were then analyzed using XRD and FTIR to confirm metal oxide conversion to the metal-acid complex form.
2.8 Computational methods
Atomistic simulations were performed using periodic Density Functional Theory (DFT) with the Vienna Ab Initio Simulation Package (VASP 5.4.1). The MgO (001) oxide surface is considered to understand the complexation reaction by acetic acid. The energies and geometries of the most stable configurations on MgO (001) were obtained through minimization of the total energy with respect to geometry using the generalized gradient approximation calculations (GGA-PBE). The projected augmented wave (PAW) method accounts for the core electrons on the valence electron density. A vacuum equivalent to at least 15 Å is included between any two successive slabs, and surface relaxation is allowed in the top two layers. A plane-wave energy cutoff of 400 eV is used for the calculations. A minimum k-point grid sampling of 3 × 3 × 1 is employed. The electronic occupancies are determined according to a Methfessel–Paxton scheme with an energy smearing of 0.1 eV. The cutoff for electronic convergence is 10−4, and structures are fully relaxed when the Hellmann–Feynman forces are smaller than 0.05.3. Results and discussion
3.1 Understanding the decarboxylation side reaction
Initial experiments investigated the effect of temperature, time, and catalyst on the decarboxylation reaction. A set of time-dependent blank reactions were performed with 2-furoic acid under different reaction temperatures (Fig. 3a), and only traces of furan were observed at 200 and 250 °C (<4% yield). However, at 316 °C, fast decarboxylation occurred, resulting in nearly 46% furan yield in 90 min. Further, the time-dependent data indicates that the furan yield increases with increasing time. Interestingly, the furan yield over the iron oxide catalyst was nearly double (80%) (Fig. 3b). Therefore, to further optimize product selectivity, a new catalyst is desired that either suppresses decarboxylation (hence decreasing furan yield) or efficiently catalyzes cross-ketonization at a lower reaction temperature. Corma and coworkers and others13,15,22,23 showed that MgO is an effective catalyst for the self-ketonization reaction for short and long-chain carboxylic acids. Taking motivation from these studies, MgO was screened for the cross ketonization of 2-furoic and lauric acid. Results show a higher 2-dodecanoyl furan yield (57% vs. 43%) and lower furan yield (20% vs. 80%) (Fig. 3b) compared with iron oxide, indicating that MgO effectively promotes cross ketonization but importantly, it minimizes the decarboxylation of 2-furoic acid. To gather more data on the decarboxylation reaction over MgO, the reaction temperature was varied from 280 °C to 400 °C (Fig. 3c). At 280 °C, negligible ketone product or furan formed even at complete conversion. However, with increasing the temperature from 320 °C to 400 °C, the furan yield reached a maximum of 70%. Interestingly, the desired ketone product, 2-dodecanoyl furan, increased from 320 °C to 350 °C (67%), and then the yield dropped sharply to 30% at 400 °C due to the formation of cracking products (Fig. S10†). An optimum reaction temperature of 350 °C was selected for further studies. Under all conditions, self ketonization is negligible.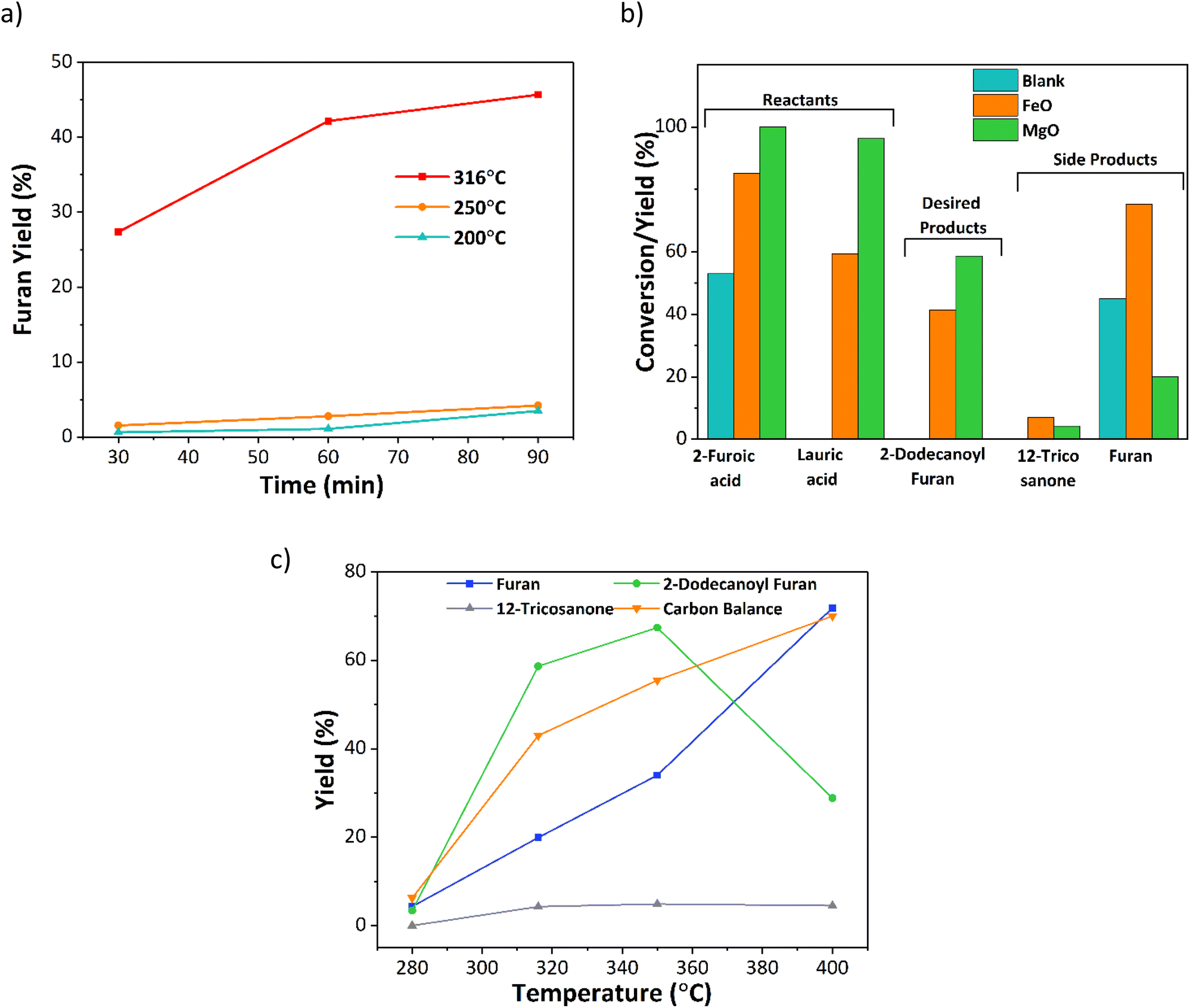 | ||
| Fig. 3 2-Furoic acid decarboxylation to furan. (a) Time and temperature-dependent blank experiments with 2-furoic acid without catalyst. Reaction conditions: 0.25 M 2-furoic acid, 30 ml dodecane, 20 bar N2, 800 rpm. (b) Decarboxylation and cross ketone yield over mixed iron oxide, MgO, and without any catalyst. Reaction conditions: 0.25 M 2-furoic acid, 0.05 M lauric acid, 90 min, 316 °C, 30 ml dodecane, 20 bar N2, 800 rpm. (c) Effect of temperature on cross ketonization of 2-furoic acid and lauric acid over MgO. Reaction conditions: 0.25 M 2-furoic acid, 0.05 M lauric acid, 90 min, 280–400 °C, 30 ml dodecane, 20 bar N2, 800 rpm. | ||
3.2 Catalyst screening and optimization
Based on prior knowledge,14,21,25,26 different metal oxides for the ketonization of 2-furoic acid and lauric acid were screened at 350 °C for 90 min. CaO, BaO, La2O3, and Mg–Al hydrotalcite were tested due to their basicity and activity in self-ketonization.27,28 Ce/Fe, Ce/Zr, and Mg–Al doped with ceria were tested to investigate the impact of doped mixed metal oxides.21,25,29 Ga2O3 was selected due to its similarity to MgO in atomic size. The results in Fig. 4a indicate that only MgO and Mg containing mixed metal oxides were active for cross ketonization except for the strongly basic La2O3(∼40% yield of cross ketone). The poor selectivity of other mixed metal oxides in Fig. 4b is expected as the amphoteric oxides and transition state oxides like CeO2, Fe3O4, and ZrO2 also performed poorly for cross ketonization in a recent publication.16 Interestingly, alkaline earth metals CaO and BaO, while inactive for ketonization (10% and 0%), retarded decarboxylation (45% and 33% furan yield). Ga2O3 resulted in a higher furan yield and did not show any ketonization activity.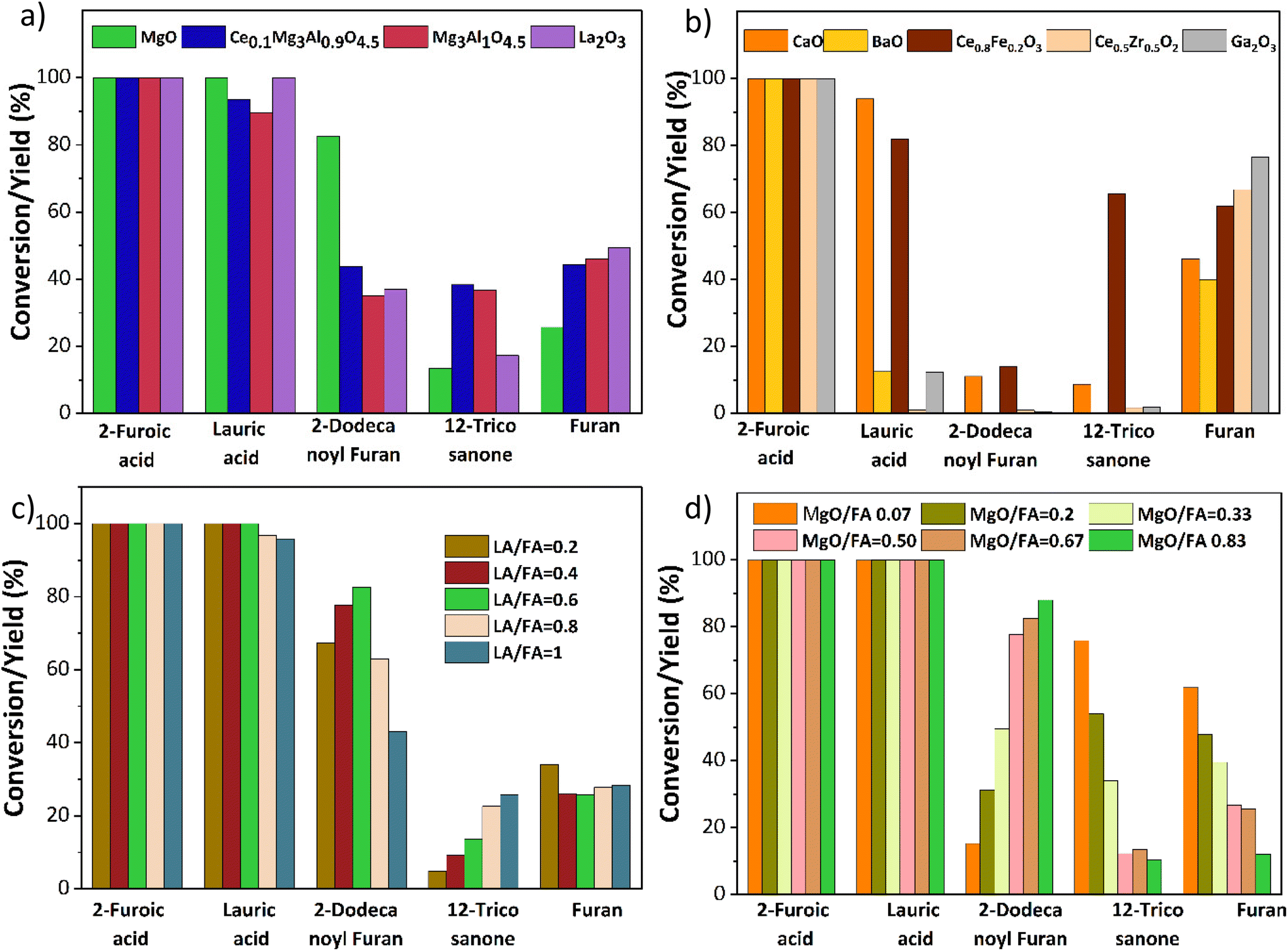 | ||
| Fig. 4 Catalyst screening and optimization of reaction parameters for the cross ketonization of 2-furoic acid and lauric acid. (a) Mg-containing oxides are selective for cross ketonization. (b) Mixed metal and basic oxides show negligible cross ketone yields. Reaction conditions: 0.25 M 2-furoic acid, 0.15 M lauric acid, 30 ml dodecane, 350 °C, 90 min, 20 bar N2, 800 rpm, 0.2 g catalyst. (c) Varying molar ratio of 2-furoic acid and lauric acid. Reaction conditions: 0.25 M 2-furoic acid, 0.05–0.25 M lauric acid, 30 ml dodecane, 350 °C, 90 min, 20 bar N2, 800 rpm, 0.2 g MgO catalyst. (d) Effect of catalyst loading Reaction conditions: 0.25 M 2-furoic acid, 0.15 M lauric acid, 30 ml dodecane, 350 °C, 90 min, 20 bar N2, 800 rpm, 0.02–0.25 g MgO catalyst. | ||
The MgO-catalyzed cross-ketonization conditions were then optimized to improve 2-dodecanoyl furan yield. Fig. 4c shows that a cross ketone yield of 82% was obtained at a molar ratio of 2-furoic acid to lauric acid of 0.6. A recent study by Ignatchenko et al. on cross ketonization of acetic acid and iso-butyric acid found that an equimolar amount achieves maximum product yield.30 Due to the decarboxylation side reaction, our system under study requires more (1.66 times) 2-furoic acid. However, we achieved a higher 2-dodecanoyl yield over MgO with much lower 2-furoic acid than iron oxide (1.6 times vs. 5 times).16 Increasing the lauric acid to 2-furoic acid molar ratio above 0.6 increased the yield of the 12-tricosanone side product, indicating that self-ketonization (Fig. 2, side reaction 2) is promoted.
Finally, the catalyst loading was varied from 0.02 g to 0.25 g (Fig. 4d). The lower catalyst loading of 0.02 g resulted in a lower yield of the cross-ketone product and a higher furan yield of 60%. At the highest loading of 0.25 g, a 2-dodecanoyl furan yield of 86% was achieved. The yield of furan was inversely correlated with the catalyst loading, minimized to 10% at the highest loading. This performance would be expected if carboxylate formation was the limiting factor for decarboxylation. In summary, upon parameter optimization, we achieved 86% of 2-dodecanoyl furan yield at 350 °C, 0.6 lauric acid to 2-furoic acid molar ratio 90 min, and 0.25 g catalyst. This yield is much higher than published literature (43%), with three times lower 2-furoic acid.16 After establishing MgO as the most selective catalyst for ketonization, the cross-ketonization chemistry was expanded to other scientifically relevant molecules.
3.3 Leveraging the chemistry to other fatty acids and aromatic acids
Trans-esterification of triglycerides found in coconut oil is a common production method for lauric acid. However, this process co-produces fatty acids of different chain lengths, such as myristic (C14) and palmitic (C16) acids.31 To reduce separation costs, rather than pure lauric acid, the use of longer chain fatty acids may also open the possibility for mixed fatty acid streams. As shown in Fig. 5a, with increasing alkyl chain length of fatty acid from C12 to C18, the cross-ketone product yield decreased. As expected, the yield of furan was nearly the same for all the different chain lengths. Other authors also reported similar results for the self-ketonization product with increasing fatty chain length.32–34 Complete conversion of reactant acids indicates complexation with the catalyst, and higher temperatures or longer reaction times may be needed to produce a higher yield of 2-alkanoyl furan.35 | ||
| Fig. 5 Effects of fatty acid chain length and aromatic acid structure (a) 0.25 M 2-furoic acid, 0.15 M fatty acid, 30 ml dodecane, 350 °C, 90 min, 20 bar N2, 800 rpm, 0.2 g MgO catalyst. Impact of different ring chemistry (benzoic acid) and aromatic acids with α-hydrogens (2-furyl propionic acid, 3-phenyl propionic acid). (b) 0.25 M aromatic acid, 0.15 M lauric acid, 30 ml dodecane, 350 °C, 90 min, 20 bar N2, 800 rpm, 0.2 g MgO catalyst. (c) A decarboxylation mechanism for 2-furoic acid is reported in the literature.35 (d) Structures of the different aromatic acids tested. | ||
Since decarboxylation is an important side reaction, to investigate the impact of the electronic properties of the aromatic ring on this reaction, aromatic substrates, such as benzoic acid, 3-phenyl propionic acid, and 2-furyl-3-propionic were also investigated. Interestingly, Fig. 5b shows that only 2-furoic acid undergoes decarboxylation. According to the 2-furoic acid decarboxylation mechanism reviewed by Ramirez et al. via experiments and calculations, the reaction proceeds in a concerted asynchronous mechanism, and the critical factors are the presence of the heteroatom on the aromatic ring and the location of the –COOH group.35 Mechanism 1 (Fig. 5c) involves a four-membered ring transition state as the favored one. The acid hydrogen transfer to the carbon bearing the carboxylic moiety in the furan ring dominates the process, and polarization of the O–H bond is the determining factor in the decomposition process. The heteroatom should be located at the α- or C2 position with respect to the COOH group. This is illustrated in Fig. 5b; even though the –COOH group is connected to the aromatic ring, benzoic acid did not show any decarboxylation due to the lack of the heteroatom. Among the other aromatic acids tested (Fig. 5b), 2-furyl 3-propionic acid and 3-phenyl propionic acid did not show any decarboxylation reaction due to the location of the COOH group away from the aromatic ring (Fig. 5d).
Interestingly, 2-furyl 3-propionic acid and 3-phenyl propionic acid also undergo self-ketonization. Both aromatic acids have α-hydrogens, and as a result, they form the enolic component required for ketonization. The role of α-hydrogens is discussed below.
3.4 Catalyst leaching and deactivation
An important aspect of any catalyst is its reusability. Therefore, the potential leeching of Mg2+ and the activity of the spent MgO were investigated. ICP-MS detected magnesium at around 60 ppm in all three replicates (Table S1†). Fresh 2-furoic acid and lauric acid were added to the filtered product solution and brought to reaction conditions to determine the activity of the Mg2+. No cross or self-ketonization product was seen, but a high yield of furan (68%) was observed (Fig. S11a and b†). The high furan yield is expected in the absence of MgO (Fig. 3a). It is evident that the Mg2+ ions are stable complexes in solution that do not catalyze the cross ketonization reaction. The spent MgO catalyst was recovered by filtration and washed with chloroform to dissolve any residual organic species. The washed catalyst was air dried overnight and used without further treatment for a second run. Fig. 6a shows that the 2-dodecanoyl furan yield significantly dropped from 83% to 18%, indicating a loss of catalyst activity. This can be attributed to the formation of MgCO3, as seen in Fig. 6b. Carbonate formation is well known for deactivating ketonization catalysts. The CO2 produced during the ketonization reaction blocks the active site required for ketonization.36–38 This is consistent with literature results where basic oxides convert to the carbonate form instead of releasing CO2 after ketonization.23,39 Specifically, MgO becomes inactive during the time on stream for the self-ketonization of pentanoic acid. However, it is worth noting that the spent catalyst was active for self-ketonization (60%). This could be explained by the partial decomposition of MgCO3 (TGA profiles Fig. 6c) and the spent catalyst undergoing decomposition around the reaction temperature (350 °C). The weight loss below 200 °C can be attributed to the desorption of water, another byproduct of ketonization. Catalyst deactivation could also be attributed to the unreacted magnesium 2-furoic acid complexes that were detected on the recovered catalyst using FTIR (Fig. 6d).24,40 This deactivation was observed by Huang et al. where 2-furoic acid produced in situ through the Cannizzaro reaction blocked the active sites of the calcium catalyst by forming metal carboxylates.41 Despite deactivation through the 2-furoic acid complex formation and carbonate deposition, the spent catalyst was successfully recycled for four runs via regeneration by calcination in air, as shown in Fig. 6a. The regenerated material displayed no organic impurities (Fig. 6c).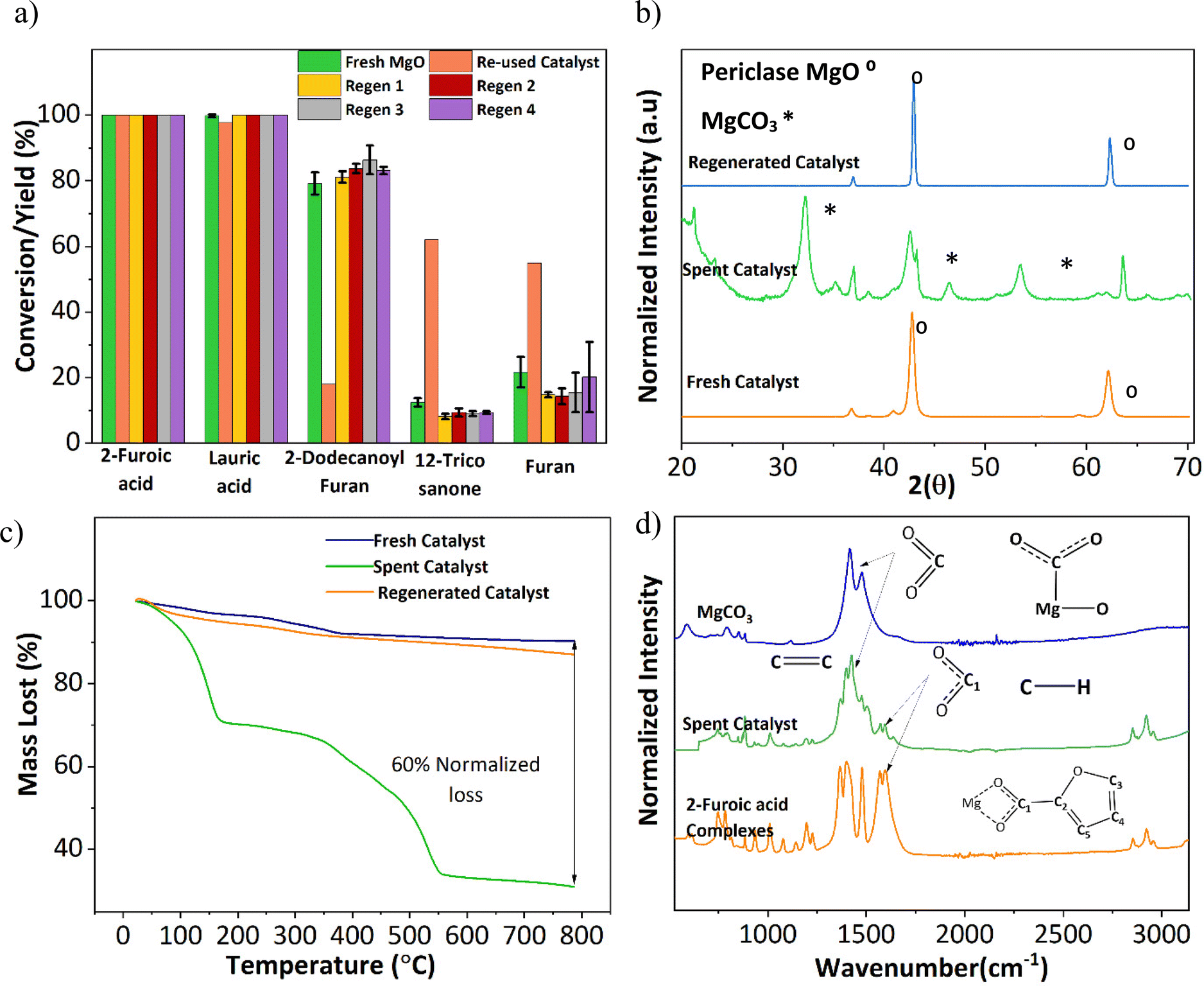 | ||
| Fig. 6 Recyclability of the MgO catalyst and sources of catalyst deactivation. (a) MgO catalyst recyclability with and without regeneration. Reaction Conditions:0.25 M 2-furoic acid, 0.15 M lauric acid, 30 ml dodecane, 350 °C, 90 min, 20 bar N2, 800 rpm, 0.2 g catalyst. Catalyst regeneration was performed by calcination at 600 °C. (b) XRD of the spent, fresh, and regenerated MgO catalyst. The regenerated material shows no carbonate impurities. (c) TGA profiles of the fresh, spent, and regenerated catalyst TGA run from 20–800 °C at a ramp rate of 20 °C min−1 under N2 gas. (d) FTIR spectra of the spent catalyst, pure MgCO3, and pure complexes of 2-furoic acid as references for their identification of the spent material. | ||
The formation of the 2-furoic acid complex also resulted in a low carbon balance of 75% for 2-furoic acid. The complex was identified in the solids of the spent catalyst by FTIR (Fig. 6d) and TGA (Fig. S12†). However, the lost carbon was recovered by a semi-batch approach increasing the total carbon efficiency. The solid material recovered by centrifugation and vacuum filtration after the reaction was transferred to lauric acid with the solvent and brought to reaction conditions. The 2-furoic acid in the complex was converted into the desired cross ketone product (Fig. S13a†). The carbon balance for 2-furoic acid and the overall system was raised to 89% and 93%, respectively, as illustrated in Fig. S13b.† This method results in a lower selectivity to the desired cross ketone product as we use an excess of lauric acid. However, 12-tricosanone, the side product from lauric acid self-ketonization, can be a valuable precursor to lubricants, base oil, and jet fuel alkanes.6,13
In summary, the catalyst undergoes slight leaching but homogenous cross ketonization does not occur. An enhancement in decarboxylation is seen. The MgO gets partially deactivated towards cross ketonization via carbonate formation and complexation with 2-furoic acid. Reaction temperature is sufficiently high to enable in situ partial removal of CO2, rationalizing the high performance of the catalyst. We consider these points further below.
3.5 Reaction mechanism
Depending on the metal oxide's basic strength and lattice energy, ketonization can go through either a surface catalyzed or bulk reaction mechanism. The metal oxides with low lattice energies or high basicity, such as rare and alkaline earth metal oxides, including MgO, CaO, BaO, CdO, and La2O3, favor the bulk mechanism.27,42 These metal oxides strongly interact with the acids forming bulk metal–carboxylate complexes, which then decompose to yield the ketone product and bulk metal carbonate. On the other hand, high lattice energy oxides, such as ZrO2, TiO2, and Fe2O3, facilitate ketonization14 catalyzed by solid surfaces. It is also possible in some cases that both mechanisms are simultaneously operative, and depending on the reaction conditions, one of them dominates. For example, Snell et al. reported that CeO2, typically considered a surface ketonization catalyst, promoted the ketonization reaction through either the bulk or the surface mode depending on reaction temperature.43 However, mechanistic studies for the MgO catalyzed ketonization reaction are limited to short chain carboxylic acids, specifically acetic acid. Moreover, the ketonization reaction mechanism over basic metal oxides has not been explored for furan-based acids, where the side reactions, such as decarboxylation, play a key role in determining the desired cross ketonization product selectivity. For example, the cross ketonization conducted by Zhao et al. between 2-furoic acid and hexanoic acid over nanoparticle MgO was performed at 260 °C, too low a reaction temperature for the decarboxylation reaction to occur. The authors also proposed the surface ketonization mechanism, and the bulk metal–carboxylate complex was not observed. To our knowledge, the reaction mechanism for the MgO catalyzed cross ketonization reaction with α-hydrogen lacking reactants, such as the 2-furoic acid, and explicit complex formation for MgO has not been studied so far. Most notably, the role of the bulk metal–carboxylate complexation in minimizing decarboxylation of 2-furoic acid side reaction has not been elucidated.43,44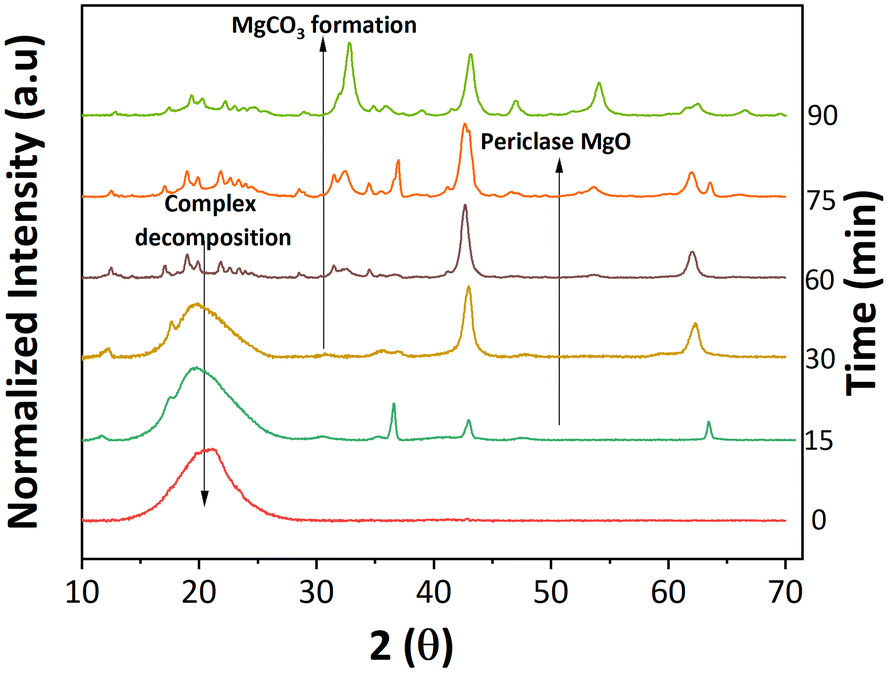 | ||
| Fig. 7 Depicting the morphological changes of the MgO catalyst at different reaction times. XRD data of the recovered catalyst at the corresponding reaction time. | ||
Interestingly, the crystalline phase of MgO was recovered at 75 min. This reappearance can be explained by the subsequent ketonization of the complexes, which is consistent with the higher cross ketone yield at longer reaction times (Fig. S17†). The concurrent changes to the catalyst surface were studied with SEM. The catalyst transformed from cubic periclase MgO to a spherical structure, taking on a spongy appearance as the reaction proceeds (Fig. S18†).
MgO suppressed the decarboxylation through complexation even without lauric acid, as shown in Fig. S19.† To further confirm the complexation effect, the lowest reaction temperature required for metal–carboxylate complexation was investigated and found to be 150 °C (Table S2 and Fig. S20†). From the data shown in Fig. 3c, the furan yield is much lower (<5%) at 200 °C, and one would not expect any decarboxylation at 150 °C. The significance of complexation was further illustrated in our catalyst screening and in Nguyen et al.'s publication, where metal oxides that do not show complexation, such as Ga2O3, CeO2, Ce/Zr, and Ce/Fe, resulted in higher furan yield (60–80%), compared to the alkaline earth metals (30–40%).16
Interestingly, a turnover number <1 was obtained at optimal reaction conditions as the moles of catalyst used are greater than those of the limiting reactant lauric acid (Table S2†). The complexation between MgO and 2-furoic acid minimizes the decarboxylation (Fig. S19 and S20†) side reaction. At lower catalyst loadings, the turnover number is 4.8 but the decarboxylation dominates due to an insufficient amount of MgO to complex with the acid. In summary, the formation of the metal–carboxylate complex plays a key role in controlling the 2-furoic acid decarboxylation side reaction by out-competing the decarboxylation, resulting in efficient utilization of 2-furoic acid and high cross-ketone product yield. These experiments provided a macroscopic view of the bulk mechanism. Due to experimental limitations, computations were used to obtain further insights.
Fig. 8a (top inset) shows the model considered for 11/12 ML of acetic acid on the CaO (001) surface. The average adsorption energy of acetic acid as a function of coverage is reported in Fig. 8b (top inset). On both, the adsorption energy increases till a certain coverage, then decreases, due to the trade-off between the strong dissociative binding nature of the acetic acid and the adsorbate–adsorbate repulsion.
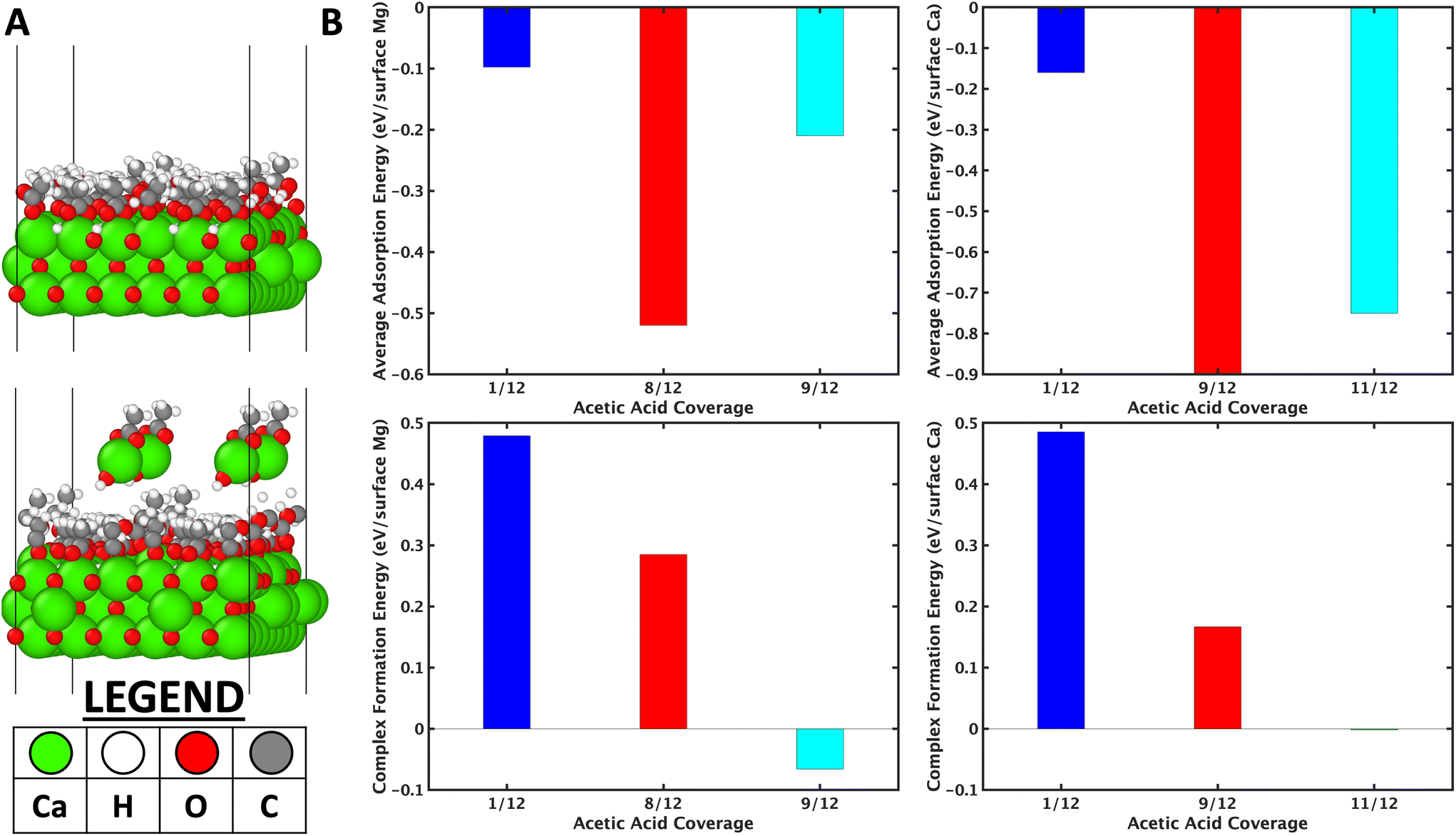 | ||
| Fig. 8 (a) Image showing the simulation for the 11/12 ML model of acetic acid adsorbed on CaO (001) surface and the corresponding complex formation (top and bottom images, respectively). (b) The top row shows the average adsorption energy as a function of coverage of acetic acid, and the bottom row shows the corresponding complex formation energy for a given coverage. | ||
Further, the average coverage is higher on CaO because of its larger lattice constant of (Ca–Ca distance of 3.42 Å vs. Mg–Mg distance of 3.01 Å). Complex formation was then considered by removing one unit of CH3COO–M(OH) (M = Mg/Ca) from the surface and comparing the thermodynamic complex formation energy. The complex formation energy (Fig. 8b lower insets) decreases as a function of acetic acid coverage. On analysis of the high coverage structures, the dissociative adsorption of acetic acid-forming surface hydroxyls (see Fig. 8a top inset, for example) is hypothesized to stabilize the vacancy formed due to etching the complex. The higher the coverage of the acetic acid, the higher the number of hydroxyls on the surface; however, very high coverages are prevented by repulsive adsorbate–adsorbate interactions (Fig. 8b top inset). The importance of surface hydroxyls, observed in DFT simulations, was also confirmed by utilizing MgSO4, MgCl2, and Mg-acetate (MgAc) salts for cross ketonization reaction. It is observed in Fig. S21† that all these Mg precursors show high furan and low cross ketone yields compared to MgO. CaO and BaO were observed to form complexes at room temperature (Table S3†) compared to 150 °C on MgO. This observation also explains the lower furan yields over CaO (Fig. S19†) than MgO.
O group, and OH stretches compared to the pure reactants. The separated solids and filtrate were exposed to the higher reaction temperature of 350 °C. The metal–carboxylate solids decomposed into the ketone products at 350 °C, as shown in Fig. 9a, indicating that metal–carboxylate complex decomposition is the rate-limiting step. The ketonization performance of similar alkaline earth oxides, such as CaO and BaO, was correlated with their metal–carboxylate complexes’ stability to further confirm this hypothesis. Complex formation for three metal oxides showed that CaO and BaO form complexes readily at room temperature vs. MgO at 150 °C. Interestingly, CaO and BaO showed low ketone yields (10% and 0%, Fig. 4a). TGA was performed for the three metal-lauric acid complexes to understand the difference in activity, and the results are shown in Fig. 9b. The relative decomposition order followed exactly the inverse order of their ketonization activity. The barium complex was the most stable, followed by calcium and then magnesium. The MgO complex was the most unstable or reactive, resulting in a higher ketone yield (Fig. S23†). The metal-complexes with 2-furoic acid followed the same thermal trend (Fig. S24†). This set of experiments confirmed that complex decomposition is the rate-limiting step and explains the low ketonization activity of alkaline earth metals CaO and BaO. Nguyen et al. using isotopic labeling of the carbonyl group taking part in ketonization, demonstrated the β-keto acid mechanism for ketonization in our system.16 Snell et al. showed that a single mechanism could be used to describe surface or bulk ketonization regardless of the metal oxide used. In this vein, the β-keto acid mechanism observed by Nguyen et al. can be generalized to the bulk ketonization over MgO.16,43 Therefore, it can be asserted that β-keto acid formation and the following C–C coupling step take place during the rate-limiting complex decomposition.
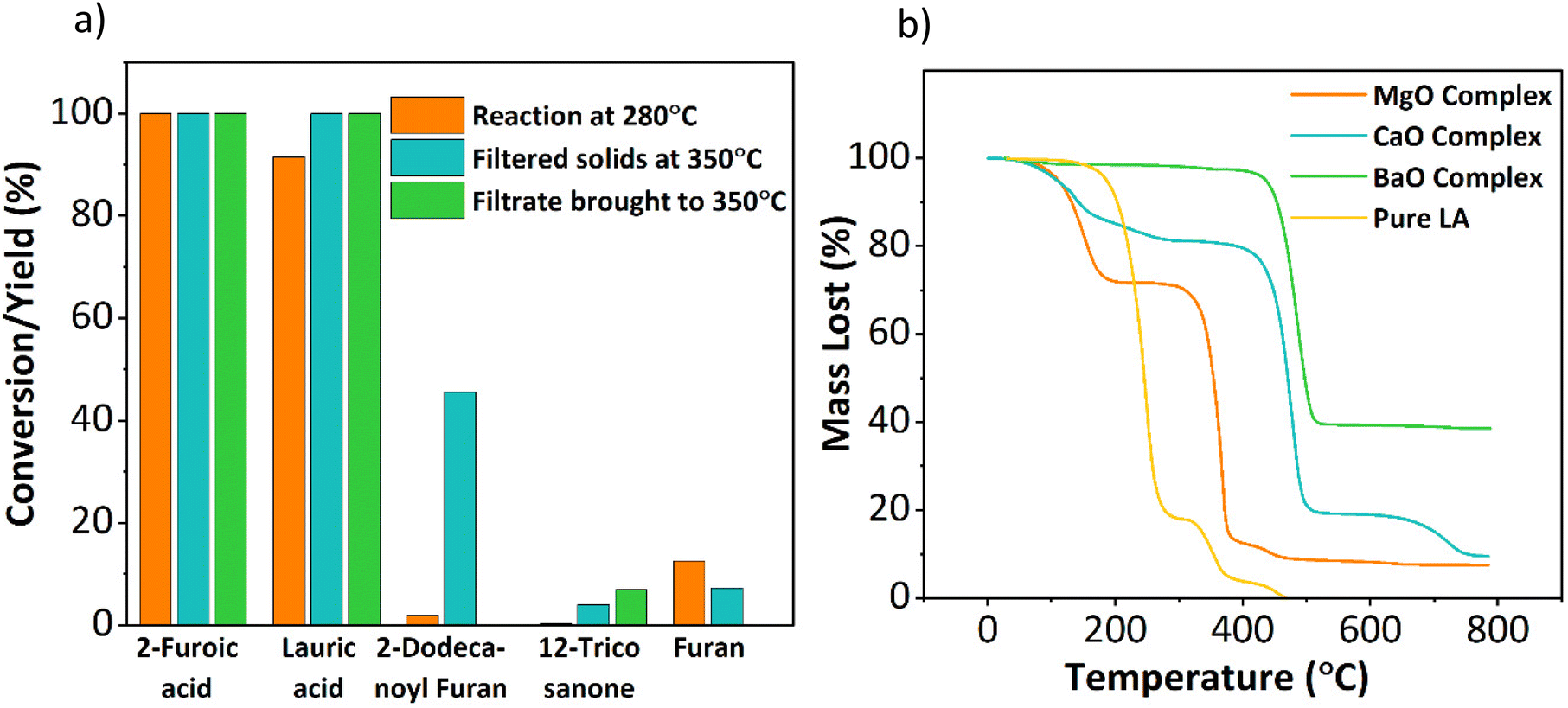 | ||
| Fig. 9 (a) Effect of temperature on metal–carboxylate decomposition. A reaction was first conducted at 280 °C, and the reactivity of the solids recovered, and the filtrate was compared at 350 °C. Initial reaction conditions: 0.25 M 2-furoic acid, 0.15 M lauric acid, 30 ml dodecane, 350 °C, 90 min, 20 bar N2, 800 rpm, 0.2 g MgO catalyst. 1.9 g solids recovered, 50% added to 30 ml dodecane, 20 ml of filtrate recovered and brought to 350 °C, 20 bar N2, 800 rpm. (b) TGA data of the metal complexes with pure lauric acid as a reference. TGA runs at a ramp rate of 20 °C min−1 under N2 gas from 20–800 °C. | ||
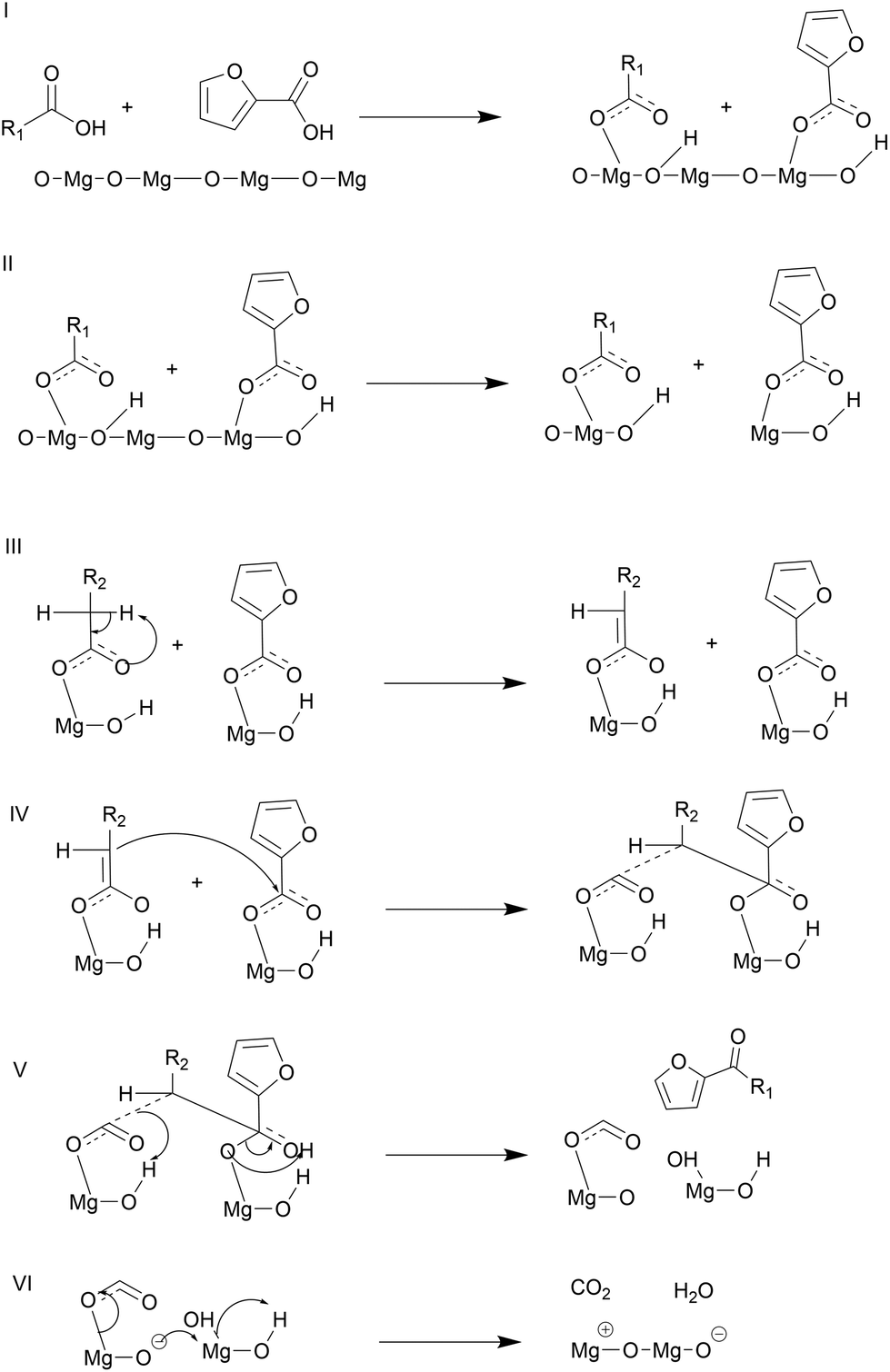 | ||
| Fig. 10 Proposed reaction mechanism for cross ketonization of lauric and 2-furoic acid based on computational and experimental evidence presented in this work. | ||
4. Conclusions
In this work, we significantly improved the yield of the oleo-furansulfonate precursor, 2-dodecanoylfuran. We also provided mechanistic insights into the cross-ketonization of biomass-derived furoic acid and vegetable-oil-derived lauric acid. Among the tested metal oxide, mixed, and doped metal oxide catalysts, earth-abundant, commercially available magnesium oxide is the most active and highly selective toward the cross-ketonization resulting in 86% 2-dodecanoyl yield at 350 °C, 0.6 lauric acid to 2-furoic acid molar ratio, 0.25 g catalyst, and 90 min. This yield is much higher than published literature (43%). XRD and FTIR indicate that catalyst deactivation is due to the formation of MgCO3 and the unreacted magnesium-2-furoic acid complex. However, the catalyst was regenerated through calcination and successfully recycled for four cycles. The chemistry was extended to different chain length fatty acids and aromatic acids to investigate the effect of molecular structure on ketonization product yield and decarboxylation side reaction. Increased fatty acid chain length results in lower ketone product yield. A longer reaction time and higher temperature are required to achieve maximum product yield. The critical factors for decarboxylation are the stability of the aromatic ring and the distance of the carboxylic group from the furan ring. By examining the decarboxylation mechanism and metal–carboxylate complex formation mechanism using different characterization techniques, it was concluded that complex formation outcompetes the 2-furoic acid decarboxylation reaction. XRD studies on the catalyst structure at different reaction times revealed that the cross ketonization mechanism proceeds through a bulk mechanism involving the formation of intermediate complexes with the metal oxide. Complexation is key in minimizing the prominent decarboxylation side reaction. Computational studies revealed the importance of the lattice constant of the metal oxide as well as the presence of surface hydroxyl groups in the complexation mechanism to proceed. This was confirmed experimentally by testing the activity of magnesium salts and the complexation over CaO and BaO. The thermal decomposition was identified as the rate-limiting step, explaining the low activity of CaO and BaO. We finally proposed a reaction mechanism for cross ketonization over MgO. The insights obtained here can guide future work on process and catalyst optimization for this path to renewable surfactants.Data and materials availability
All data needed to evaluate the conclusions in the paper are in the paper and/or the ESI.† Additional data regarding this paper may be requested from the authors.Conflicts of interest
The authors declare no conflicts.Acknowledgements
This work was supported as part of the Catalysis Center for Energy Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, and Office of Basic Energy Sciences under award number DE-SC0001004.References
- M. Langholtz, B. Stokes and L. Eaton, 2016 billion-ton report: Advancing domestic resources for a thriving bioeconomy (Executive Summary), 2016, vol. 12 Search PubMed.
- C. B. B. Farias, F. C. G. Almeida, I. A. Silva, T. C. Souza, H. M. Meira, R. D. C. F. Soares da Silva, J. M. Luna, V. A. Santos, A. Converti, I. M. Banat and L. A. Sarubbo, Electron. J. Biotechnol., 2021, 51, 28–39 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- P. Foley, A. Kermanshahi-pour, E. S. Beach and J. B. Zimmerman, Chem. Soc. Rev., 2012, 41, 1499–1518 RSC.
- F. Deng and A. S. Amarasekara, Ind. Crops Prod., 2021, 159, 113055 CrossRef CAS.
- A. M. Norton, S. Liu, B. Saha and D. G. Vlachos, ChemSusChem, 2019, 12, 4780–4785 CrossRef CAS.
- D. S. Park, K. E. Joseph, M. Koehle, C. Krumm, L. Ren, J. N. Damen, M. H. Shete, H. S. Lee, X. Zuo, B. Lee, W. Fan, D. G. Vlachos, R. F. Lobo, M. Tsapatsis and P. J. Dauenhauer, ACS Cent. Sci., 2016, 2, 820–824 CrossRef CAS.
- M. Koehle, Z. Zhang, K. A. Goulas, S. Caratzoulas, D. G. Vlachos and R. F. Lobo, Appl. Catal., A, 2018, 564, 90–101 CrossRef CAS.
- D. Li, F. Yao and Q. X. Guo, Energy Fuels, 2009, 23, 564–568 CrossRef.
- E. V. Fufachev, B. M. Weckhuysen and P. C. A. Bruijnincx, ACS Sustainable Chem. Eng., 2020, 8, 11292–11298 CrossRef CAS.
- R. Kumar, N. Enjamuri, S. Shah, A. S. Al-Fatesh, J. J. Bravo-Suárez and B. Chowdhury, Catal. Today, 2018, 302, 16–49 CrossRef CAS.
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. E. Resasco, ACS Catal., 2013, 3, 2456–2473 CrossRef CAS.
- A. Corma, M. Renz and C. Schaverien, ChemSusChem, 2008, 1, 739–741 CrossRef CAS.
- B. Boekaerts and B. F. Sels, Appl. Catal., B, 2021, 283, 119607 CrossRef CAS.
- S. Chen and C. Zhao, ACS Sustainable Chem. Eng., 2021, 9, 10818–10826 CrossRef CAS.
- H. Nguyen, Y. Wang, D. Moglia, J. Fu, W. Zheng, M. Orazov and D. G. Vlachos, Catal. Sci. Technol., 2021, 11, 2762–2769 RSC.
- K. J. Zeitsch, in The chemistry and technology of furfural and its many by-products, ed. K. J. Zeitsch, Elsevier, Amsterdam, 1st edn, 2000, ch. 19, vol. 13, pp. 159–163 Search PubMed.
- Y. J. Ng, P. E. Tham, K. S. Khoo, C. K. Cheng, K. W. Chew and P. L. Show, Bioprocess Biosyst. Eng., 2021, 44, 1807–1818 CrossRef CAS.
- A. J. Dijkstra, Lauric Oils, Elsevier Ltd., 1st edn, 2015 Search PubMed.
- J. T. Scanlon and D. E. Willis, J. Chromatogr. Sci., 1985, 23, 333–340 CAS.
- F. Lu, B. B. Jiang, J. Wang, Z. Huang, Z. Liao and Y. Yang, Mol. Catal., 2018, 444, 22–33 CrossRef CAS.
- G. A. H. Mekhemer, S. A. Halawy, M. A. Mohamed and M. I. Zaki, J. Catal., 2005, 230, 109–122 CrossRef CAS.
- S. A. Khromova, A. A. Smirnov, S. A. Selishcheva, R. G. Kukushkin, V. O. Dundich, L. I. Trusov and V. A. Yakovlev, Catal. Ind., 2013, 5, 260–268 CrossRef.
- O. Back and P. Marion, US Pat., US10906857B2, 2021, 2, 1–29.
- B. Jiang, Z. Xi, F. Lu, Z. Huang, Y. Yang, J. Sun, Z. Liao, J. Wang and Y. Yang, Catal. Sci. Technol., 2019, 9, 6335–6344 RSC.
- R. W. Snell and B. H. Shanks, ACS Catal., 2013, 4, 512–518 CrossRef.
- Y. Yamada, M. Segawa, F. Sato, T. Kojima and S. Sato, J. Mol. Catal. A: Chem., 2011, 346, 79–86 CrossRef CAS.
- H. Ling, Z. Wang, L. Wang, C. Stampfl, D. Wang, J. Chen and J. Huang, Catal. Today, 2020, 351, 58–67 CrossRef CAS.
- S. Ding, H. Wang, J. Han, X. Zhu and Q. Ge, Ind. Eng. Chem. Res., 2018, 57, 17086–17096 CrossRef CAS.
- A. V. Ignatchenko, J. S. Deraddo, V. J. Marino and A. Mercado, Appl. Catal., A, 2015, 498, 10–24 CrossRef CAS.
- V. O. Ezigbo and A. Emmanuella, Afr. J. Educ. Sci. Technol., 2018, 3, 59–62 Search PubMed.
- B. Boekaerts, M. Vandeputte, K. Navaré, J. Van Aelst, K. Van Acker, J. Cocquyt, C. Van Caneyt, P. Van Puyvelde and B. F. Sels, Green Chem., 2021, 23, 7137–7161 RSC.
- T. N. Pham, D. Shi and D. E. Resasco, J. Catal., 2014, 314, 149–158 CrossRef CAS.
- K. Lee, Y. Kim and M. Choi, ACS Sustainable Chem. Eng., 2018, 6, 13035–13044 CrossRef CAS.
- B. C. Ramírez, R. Ramírez, R. M. Domínguez, D. Domínguez, A. Herize, M. Maŕ, M. Tosta, T. Cordova and G. Chuchani, Int. J. Chem. Kinet., 2007, 39, 298–306 CrossRef.
- J. H. Miller, G. R. Hafenstine, H. H. Nguyen and D. R. Vardon, Ind. Eng. Chem. Res., 2022, 61, 2997–3010 CrossRef CAS.
- M. Renz, Eur. J. Org. Chem., 2005, 979–988 CrossRef CAS.
- M. Renz and A. Corma, Eur. J. Org. Chem., 2004, 2036–2039 CrossRef CAS.
- C. Liu, A. M. Karim, V. M. Lebarbier, D. Mei and Y. Wang, Top. Catal., 2013, 56, 1782–1789 CrossRef CAS.
- I. A. Lutsenko, M. E. Nikiforova, K. A. Koshenskova, M. A. Kiskin, Y. V. Nelyubina, P. V. Primakov, M. V. Fedin, O. B. Becker, V. O. Shender, I. K. Malyants and I. L. Eremenko, Russ. J. Coord. Chem., 2021, 47, 881–890 CrossRef CAS.
- R. Huang, J. Chang, H. Choi, J. M. Vohs and R. J. Gorte, Catal. Lett., 2022, 1, 1–10 Search PubMed.
- R. Szabo, M. Crozet and P. Vanelle, Synthesis, 2008, 127–135 CAS.
- R. W. Snell and B. H. Shanks, ACS Catal., 2013, 3, 783–789 CrossRef CAS.
- J. C. Kuriacose and S. S. Jewur, J. Catal., 1977, 50, 330–341 CrossRef CAS.
- L. J. Bellamy, The Infrared Spectra of Complex Modules, 2019 Search PubMed.
- R. Pestman, R. M. Koster, A. Van Duijne, J. A. Z. Pieterse and V. Ponec, J. Catal., 1997, 168, 265–272 CrossRef CAS.
- O. Nagashima, S. Sato, R. Takahashi and T. Sodesawa, J. Mol. Catal. A: Chem., 2005, 227, 231–239 CrossRef CAS.
- A. Pulido, B. Oliver-Tomas, M. Renz, M. Boronat and A. Corma, ChemSusChem, 2013, 6, 141–151 CrossRef CAS PubMed.
- S. Wang and E. Iglesia, J. Catal., 2017, 345, 183–206 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04400e |
| This journal is © The Royal Society of Chemistry 2023 |