
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amidoboronates: bringing together the synthesis of BN-heterocycles via a reductive coupling and dynamic covalent chemistry
Anna J.
McConnell
 ab
ab
aOtto Diels Institute of Organic Chemistry, Kiel University, Otto-Hahn-Platz 4, Kiel 24098, Germany
bDepartment of Chemistry and Biology, University of Siegen, Adolf-Reichwein-Strasse 2, 57068 Siegen, Germany. E-mail: anna.mcconnell@uni-siegen.de
First published on 20th June 2023
Abstract
This Perspective describes how amidoboronates open up new chemical space spanning the areas of BN-heterocycles and dynamic covalent chemistry. BN-containing heterocycles offer the potential to access new properties and reactivity compared to their C–C analogues. Amidoboronates are introduced as a new class of B–N heterocycles that can be synthesised in three isomeric forms (meso5, rac5 and rac6) from the reductive coupling of N-aryl iminoboronates. Furthermore, initial investigations on the dynamic covalent chemistry of amidoboronates are discussed, such as the reversibility of C–C bond formation following the reductive coupling and tuning the rac5/rac6 ratio via dynamic covalent B–N and B–O bonds.
 Anna J. McConnell | Anna McConnell obtained a D.Phil. under the supervision of Prof. Paul Beer from the University of Oxford. She completed postdoctoral research stays in the group of Prof. Jacqueline Barton at the California Institute of Technology and at the University of Cambridge with Prof. Jonathan Nitschke. From 2016–2023 she was a Junior Professor at Christian-Albrechts-Universität zu Kiel and in 2023 she joined the University of Siegen as a Junior Professor. Her research focuses on stimuli-responsive metal–organic cages, dynamic covalent chemistry and luminescent complexes. |
Introduction
Given the isoelectronic nature of CC and BN bonds (Fig. 1a), BN-containing heterocycles1–14 have been investigated as CC isosteres. However, the polarity of the B–N bond alters the electronic properties, leading to different reactivity and selectivity compared to their carbon analogues.9,11 Furthermore, B–N bonds can take the form of coordinative or covalent bonds (Fig. 1a), offering additional avenues for accessing new types of chemistry and tuning properties. As a result, BN-heterocycles, such as azaborines, BN-naphthalenes and BN-polyaromatic hydrocarbons (PAHs), have been exploited in applications from materials science as BN-doped nanographenes10 to catalysis.12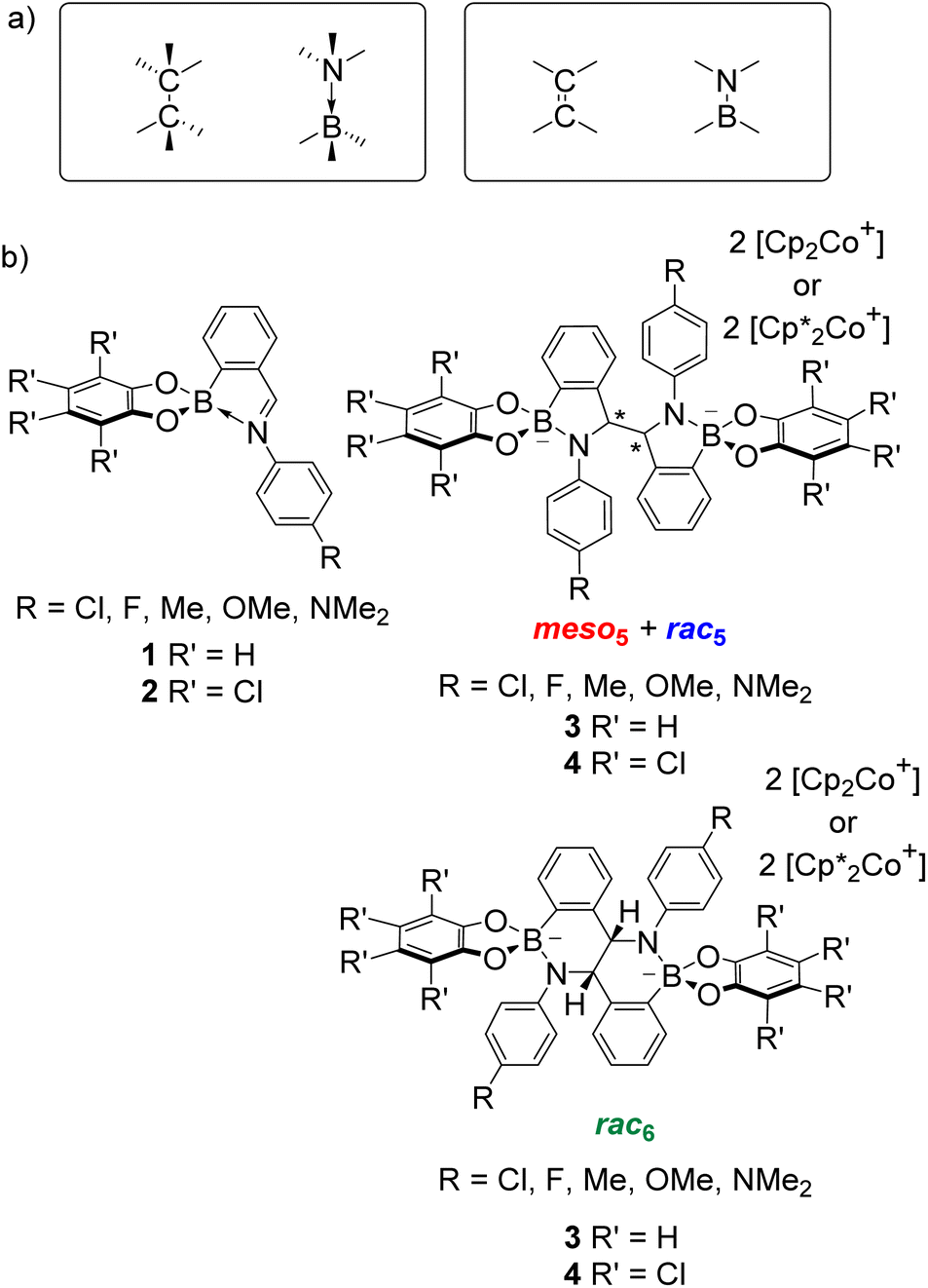 | ||
| Fig. 1 (a) Isoelectronic relationship between CC and BN bonds. (b) Iminoboronates 1–2 for the synthesis of amidoboronates 3–4via a reductive coupling. | ||
Despite the interest in BN-heterocycles, methods for their synthesis (particularly in the quantities required for applications) are still limited compared with the myriad of synthetic methods available in organic chemistry for the synthesis of CC analogues.7,8 Thus, synthetic access to BN-heterocycles is limiting their diversity and the exploration of new chemical space,7 for example in the context of dynamic covalent chemistry.
Dynamic covalent chemistry combines the strength of covalent bonds with the reversibility of bond formation, enabling for example the self-assembly of supramolecular architectures from mechanically interlocked molecules to cages from smaller building blocks under thermodynamic control.15–19 While the dynamic covalent chemistry of a variety of bonds including S–S,16,20–22 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C,23,24 CN,22,24–28 C–O,17,29 Si–O30,31 and B–O19,22,32–34 bonds has been established, the discovery of new types of dynamic covalent bonds, e.g. based on B–N bonds, could open up new avenues for applications.
C,23,24 CN,22,24–28 C–O,17,29 Si–O30,31 and B–O19,22,32–34 bonds has been established, the discovery of new types of dynamic covalent bonds, e.g. based on B–N bonds, could open up new avenues for applications.
This Perspective highlights how these two research fields, B–N heterocycles and dynamic covalent chemistry, have intersected through the synthesis of amidoboronates 3–4 (Fig. 1b), a new class of BN-heterocycles, and established new research directions. A family of amidoboronates has been prepared via the reductive coupling of N-aryl iminoboronates 1 with reduced synthetic effort exploiting the modular synthesis of the iminoboronate substrates and the formation of up to three isomeric amidoboronate products (meso5, rac5 and rac6). Furthermore, the dynamic covalent chemistry of the amidoboronates including the rearrangement between the rac5 and rac6 isomers via dynamic covalent B–N bonds is discussed.
C–C bond formation via reductive couplings
Synthesis of 1,2-diamines and 1,2-diols
Radical-mediated reductive couplings such as the pinacol coupling have been exploited in organic chemistry to form C–C bonds, giving access to 1,2-diamines35–44 and 1,2-diols39,42–52via the reductive coupling of imines and carbonyl compounds, respectively (Scheme 1a). A variety of metal-based reagents including alkali metals,35–37 Mg(I) compounds,53 “GaI”,41 SmI238 and Mn*39 have been used as the stoichiometric reductant. More recently, metal-free reductive couplings45,48,49 have been reported and replacement of the stoichiometric reductant with a photocatalyst has led to the development of photoredox-catalysed reductive couplings.42–44,46,47,50,52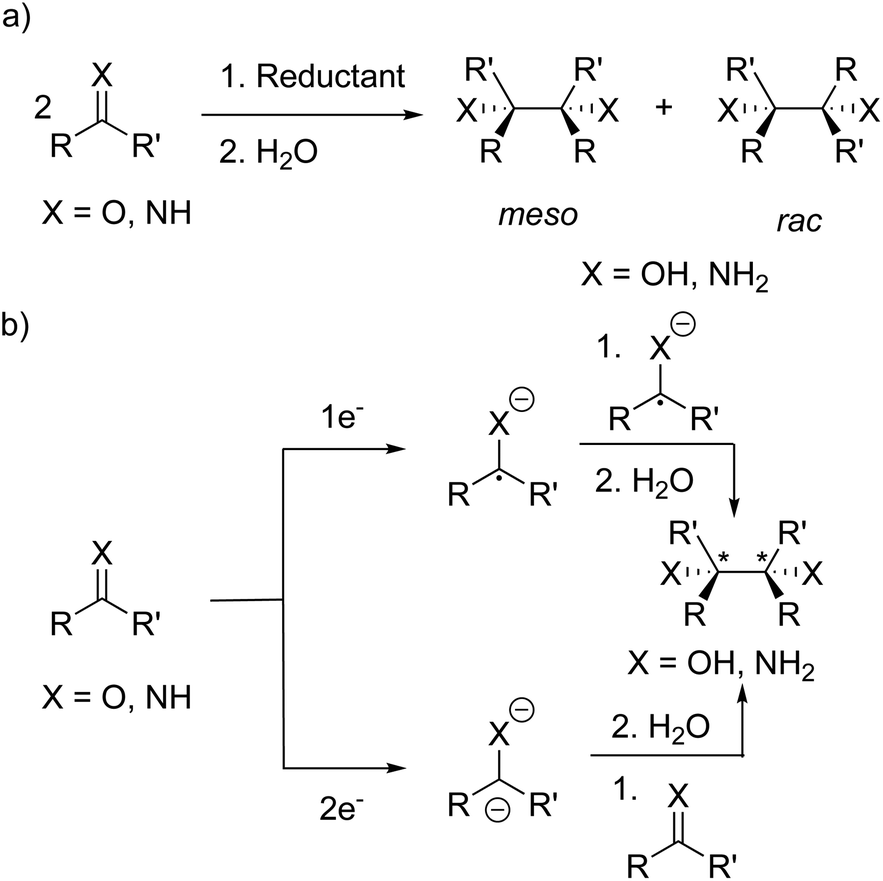 | ||
| Scheme 1 (a) Reductive coupling of aldehydes, ketones and imines giving 1,2-diols and 1,2-diamines, respectively, as a mixture of meso and rac diastereomers. (b) Two proposed mechanisms for the reductive couplings: one electron reduction to the radical anion and recombination of two radical anions; two electron reduction to the dianion and reaction with a second aldehyde/ketone/imine. | ||
Several mechanisms have been proposed for the reductive couplings (Scheme 1b): (a) one electron reduction forming a radical anion and the formation of the dimer via coupling of two radical anions; (b) two electron reduction to the dianion and disproportionation upon reaction with a second CO/CN molecule.35,37 Furthermore, the dimers can form as a mixture of meso and rac diastereomers since the newly formed C–C bond contains two stereogenic centres (when R ≠ R′); in separate studies, Eisch35 and Smith36 investigated the influence of the reaction conditions (e.g. solvent and reductant) on the ratio of the meso and rac diastereomers. In some cases (e.g. with sodium or potassium in THF), only the rac isomer was observed and isomerisation of the initially formed diasteromeric mixture to the rac isomer was hypothesised.35,36 Eisch and co-workers proposed ion-pairing between the radical anion and countercation favours rac isomer formation,35 whereas Smith and co-workers proposed a radical anion/dimeric dianion equilibrium where the rac diastereomer is the thermodynamic product of the reaction.36,37
Synthesis of B–N heterocyclic dimers
Reductive couplings have also been exploited to access BN-heterocyclic dimers following C–C bond formation (Schemes 2 and 3).3,6 Given the electron-deficiency of boron, the reactivity of the boryl as well as carbon-centred radicals needs to be considered;3,5,6 Nozaki and co-workers have reported that anionic ligands and Lewis bases can stabilise boryl radicals, although calculations proposed the carbon-centred radical was the major resonance contributor.5 Subsequently, they reported the reductive coupling of oxazoline-stabilised difluoroborane 5 in the absence of stirring (Scheme 2).3 One electron reduction by KC8 was proposed to form the boryl radical 6a following loss of a fluoride and coupling of the carbon-centred radicals 6b under diffusion control led to dimer 7 formation in 47% yield.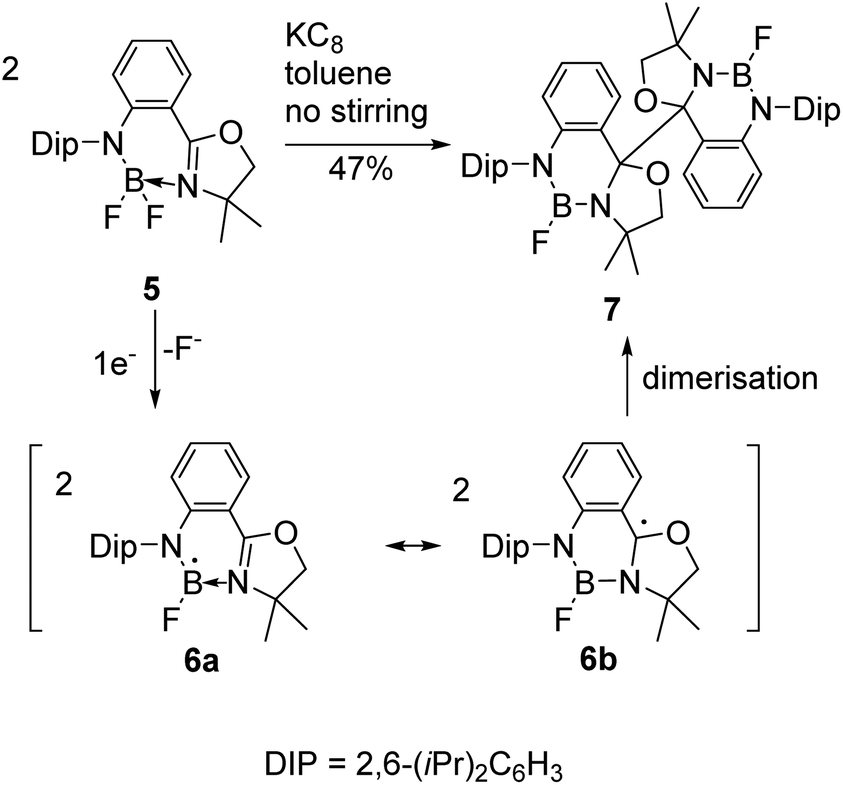 | ||
| Scheme 2 Nozaki's reductive coupling of base-stabilised difluoroborane 5 where one electron reduction by KC8 and loss of fluoride is proposed to form boryl radical 6a. Dimerisation of the carbon-centred radical 6b in the absence of stirring gives 7. | ||
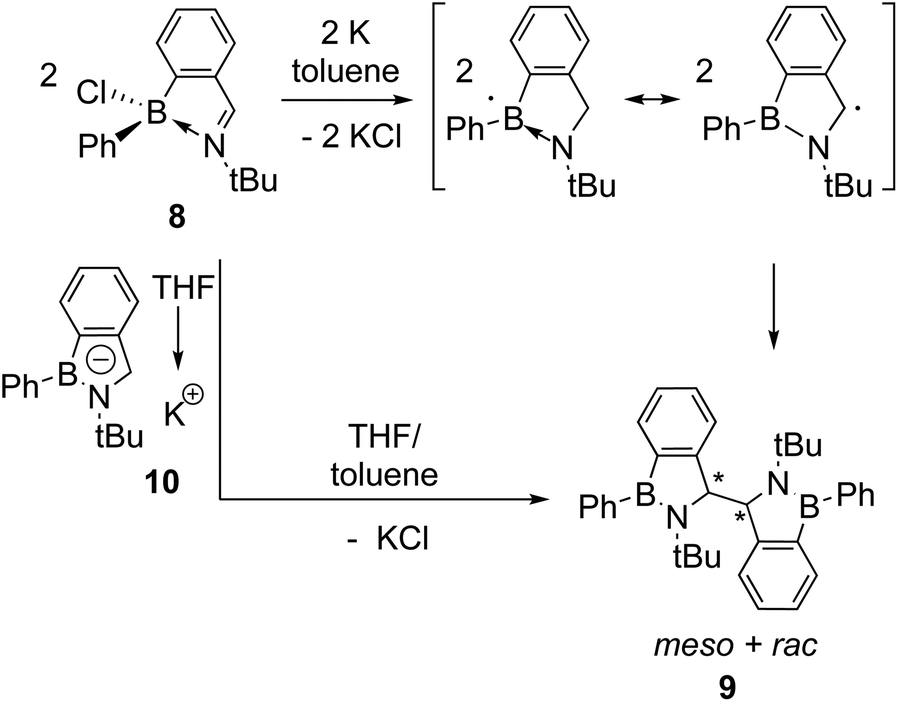 | ||
| Scheme 3 Dostál's reductive coupling of iminochloroborane 8 forming dimer 9 as a mixture of meso and rac diastereomers. The two proposed mechanisms for dimer formation are depicted in analogy to those in Scheme 1. | ||
Similarly, Dostál and co-workers reported the reductive coupling of iminochloroborane 8 with potassium produces a mixture of the meso and rac diastereomeric dimers 9 (Scheme 3).6 The two diastereomers could be separated by fractional crystallisation and the meso isomer was observed to convert to the rac isomer quantitatively upon heating in toluene. Two similar mechanisms to the analogous reductive couplings of imines and carbonyl compounds were proposed, involving the coupling of two carbon-centred radicals following one electron reduction or the reaction of the BN-indenyl anion 10 (formed by 2 electron reduction) with iminochloroborane 8.
Synthesis of amidoboronates
Reductive coupling of N-aryl iminoboronates
N-Aryl iminoboronates can be prepared exploiting dynamic covalent chemistry from three simple building blocks (Scheme 4): an aniline (blue), 2-formylphenyl boronic acid (red) and a diol (black).54–56 Iminoboronates have been used in the self-assembly of macrocycles and cages,55,57 determining the enantiomeric excess of amines and amino acids via fluorescent sensing,58,59 self-healing polymers,60,61 bioconjugation62–66 and drug delivery applications.67–69 These applications typically exploit the dynamic covalent nature of the imine and boronate bonds (see Dynamic Covalent Chemistry), however, the reactivity of the imine has been underexplored.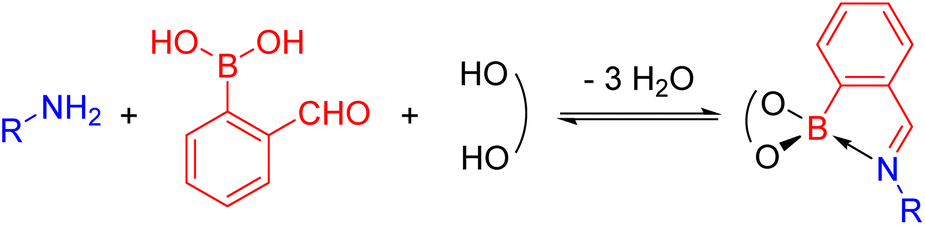 | ||
| Scheme 4 Self-assembly of a N-aryl iminoboronate, the substrate for the reductive couplings, via dynamic covalent chemistry from an amine, 2-formylphenylboronic acid and a diol. | ||
Given the structural similarity of iminoboronates to iminochloroborane 8 (Scheme 3), new BN-heterocycles could be potentially accessed as a mixture of diastereomeric meso5† and rac5† dimers via a reductive coupling of the imine. Furthermore, an advantage of iminoboronates is that a large family of substrates with different steric and electronic properties can be readily prepared by varying the building blocks during the iminoboronate self-assembly. Thus, we investigated the reductive coupling of N-aryl iminoboronates using reductants such as cobaltocene and decamethylcobaltocene, initially focusing on two series of iminoboronates (1 and 2) where the para-substituent of the aniline and the catechol were varied to investigate electronic effects (Scheme 5).70,71
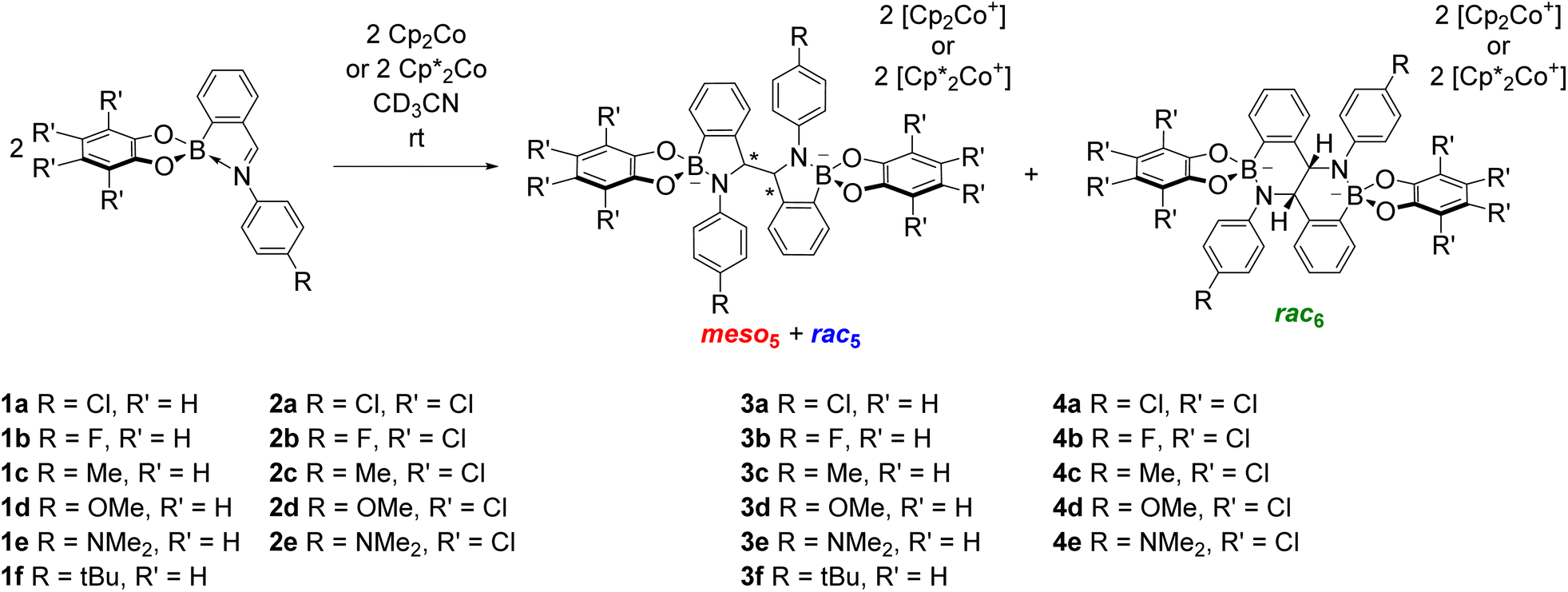 | ||
| Scheme 5 The synthesis of up to three amidoboronate products (meso5, rac5 and rac6) from the reductive coupling of N-aryl iminoboronates (pyrocatechol series: 1a–1e; tetrachlorocatechol series 2a–2e) using cobaltocene or decamethylcobaltocene. | ||
The reductive couplings were initially performed in CD3CN, monitoring the reaction progress by NMR spectroscopy where the loss of the imine signal was observed. In most reductive couplings two new sets of 1H signals appeared including two methine signals between 5–6 ppm, attributed to the formation of a meso5 and rac5 diastereomeric mixture based on subsequent NMR analysis (see below, Solution characterisation). However, it was not possible to quantify the amount of the meso5 and rac5 diastereomers since one of the amidoboronate products typically crystallised from the reaction mixture of the reductive couplings (Table 1). Nevertheless, these crystals enabled the isolation and characterisation of single amidoboronate isomers both in the solid-state and in solution by redissolving the crystals in DMSO-d6.
Solid-state characterisation
X-ray crystal structures of [meso5-3a](Cp2Co)2 and [meso5-3b](Cp2Co)2 were obtained showing the meso isomer in two different conformations, anti and gauche (Fig. 2a and b). X-ray analysis of crystals obtained from the tetrachlorocatechol series (R′ = Cl) revealed the rac5 isomer crystallised as the cobaltocenium and/or decamethylcobaltocenium salts (Fig. 2c).70,71 For 4c, crystal structures of three polymorphs of the cobaltocenium salt were obtained in addition to one crystal structure of the decamethylcobaltocenium salt. The X-ray crystal structures of the meso5 and rac5 isomers confirmed dimerisation via C–C bond formation between the two five-membered rings and Fig. 2 highlights the different stereochemistry around the new C–C bond for the two diastereomers using Newman projections.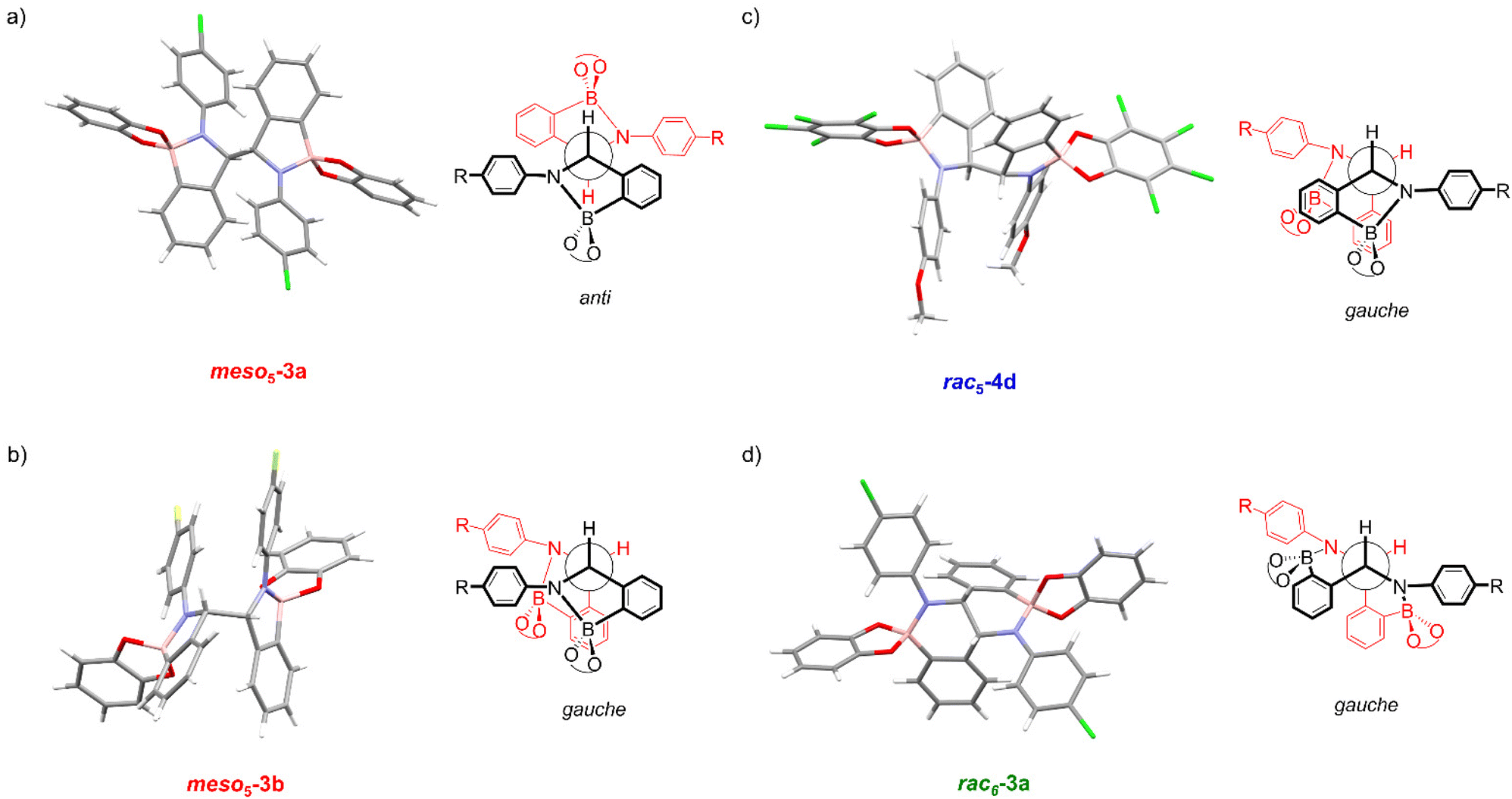 | ||
| Fig. 2 X-ray crystal structures and Newman projections of: (a) [meso5-3a](Cp2Co)2; (b) [meso5-3b](Cp2Co)2; (c) [rac5-4d](Cp2Co)2; (d) [rac6-3a](Cp2Co)2. For clarity, the X-ray structures do not depict the Cp2Co+ countercations and solvent molecules. The Newman projections do not depict the R and catechol substituents and the two halves of the dimer are shown in red and black, particularly to show the change in connectivity in the rac6 isomer. Adapted with permission from ref. 71. | ||
Unexpectedly, X-ray analysis of crystals obtained from analogous reductive couplings of 1a and 1b on separate occasions were not meso5 or rac5 structures but an isomeric and previously unknown B–N heterocyclic scaffold consisting of two fused six-membered rather than five-membered rings (Fig. 2d). Thus, these amidoboronates were named the rac6† product and similar X-ray crystal structures were obtained of [rac6-3c,d,f](Cp2Co)2 in the pyrocatechol series (R′ = H). Since significant quantities of the rac6 product were not observed in the reaction mixtures in CD3CN, further studies in DMSO-d6 (see below, Solution characterisation) probed whether it forms in the solid state only or also in solution.
Unlike the tetrachlorocatechol series where crystals were obtained of the rac5 isomer only, crystals of all three isomers were obtained from the pyrocatechol series (Table 1). Amidoboronates 3a–b containing electron-withdrawing Cl and F aniline substituents crystallised as either the meso5 or rac6 isomer (depending on the reaction conditions) and 3a–d,f crystallised as the rac6 isomer. Although crystals were also obtained from the reductive coupling of 3e, they were not suitable for X-ray analysis. However, subsequent solution studies revealed the rac5 isomer crystallised.
The number of crystal structures of the amidoboronate isomers as well as several iminoboronate starting materials has enabled comparison of different structural parameters (Table 2). Firstly, the formation of covalent B–N bonds in the amidoboronate products is confirmed by the shortening of the B–N bond (1.50–1.55 Å) compared to the dative B–N bond (1.66–1.68 Å) in the iminoboronate. In addition, the two counterions (Cp2Co+ or Cp*2Co+) per dimer indicated the formation of two anionic tetrahedral boron centres. A slight lengthening of the B–O bonds was also observed in the amidoboronates compared to the iminoboronates.
| HC–CH (°) | C–C (Å) | B–N (Å) | B–O (Å) | Ref. | |
|---|---|---|---|---|---|
| a Based on the X-ray crystal structures of three polymorphs of the cobaltocenium salt and one crystal structure of the decamethylcobaltocenium salt. b Based on the X-ray crystal structures of the cobaltocenium and decamethylcobaltocenium salts. | |||||
| 1a | — | — | 1.68 | 1.47–1.46 | 71 |
| 1d | — | — | 1.68 | 1.46–1.47 | 71 |
| 1e | — | — | 1.68 | 1.46–1.48 | 71 |
| 2d | — | — | 1.66 | 1.48–1.49 | 71 |
| meso 5-3a | 179 | 1.56 | 1.55 | 1.51–1.52 | 71 |
| rac 6-3a | 65 | 1.54 | 1.54 | 1.52–1.54 | 71 |
| meso 5-3b | 64 | 1.58 | 1.53 | 1.52–1.53 | 70 |
| rac 6-3b | 65 | 1.54 | 1.53 | 1.52–1.54 | 70 |
| rac 6-3c | 65 | 1.53 | 1.53 | 1.53–1.54 | 70 |
| rac 6-3d | 64 | 1.55 | 1.52 | 1.51–1.54 | 70 |
| rac 6-3f | 64 | 1.54 | 1.54 | 1.51–1.54 | 70 |
| rac 5-4a | 61 | 1.56 | 1.52 | 1.54 | 71 |
| rac 5-4c | 60–70a | 1.54–1.57a | 1.50–1.51a | 1.52–1.56a | 70 |
| rac 5-4d | 68 | 1.55 | 1.51 | 1.53–1.55 | 71 |
| rac 5-4e | 59–66b | 1.55–1.56b | 1.50–1.51b | 1.53–1.56b | 71 |
The X-ray structures also revealed the conformation and bond length of the newly formed C–C bond (Table 2). The thermodynamic stability of the gauche and anti conformations has been reported for dimers formed from carbon-centred radicals.72,73 Indeed, the gauche conformation (59–70°) was observed in all crystal structures except for meso5-3a where the anti conformation (179°) was observed, likely due to different crystallisation conditions. Furthermore, the C–C bond lengths for all isomers (1.53–1.57 Å) were consistent with a sp3-hybridised C–C bond, suggesting bond lengthening due to a contribution from the radical form is minimal at the temperature of the measurements (100 K or 180 K).
Solution characterisation
As revealed by X-ray analysis, up to three isomeric amidoboronates crystallised from the reductive couplings in CD3CN. The solution structure of the meso5, rac5 and rac6 isomers was probed by redissolving the isolated crystals in DMSO-d6. Regardless of the substitution of the amidoboronates, characteristic signals were observed for each product: a methine signal around 5.4 ppm for the meso5 diastereomer; a methine signal around 5.2 ppm and doublet around 7.7 ppm for the rac5 diastereomer; a methine signal around 4.9 ppm and two doublets around 6.9 and 6.8 ppm for the rac6 isomer. Similar shifts were observed for the amidoboronate products in both DMSO-d6 and CD3CN, enabling identification of the isomeric mixtures in the corresponding reductive couplings in CD3CN.The existence of the rac6 product in solution was investigated in time-course NMR experiments (Scheme 6).70 For the reductive coupling of 1f in CD3CN where the rac6 isomer crystallised from the reaction mixture, the rac5 signals were observed to decrease over time and this enabled the characterisation of the remaining isomer in solution, meso5-3f. In analogous reductive couplings with 1b and 1d in DMSO-d6 where crystallisation was prevented, the meso5 and rac5 diastereomers initially formed and the rac5 converted into the rac6 isomer over time, as evidenced by the appearance of a third methine signal consistent with redissolved rac6 crystals. Thus, the rac6 isomer was proposed to form via breakage and rearrangement of the covalent B–N bonds in the rac5 isomer. The following sections introduce dynamic covalent chemistry with a focus on examples relevant to the subsequent discussion of the dynamic covalent chemistry of amidoboronates, including the B–N bonds in more detail.
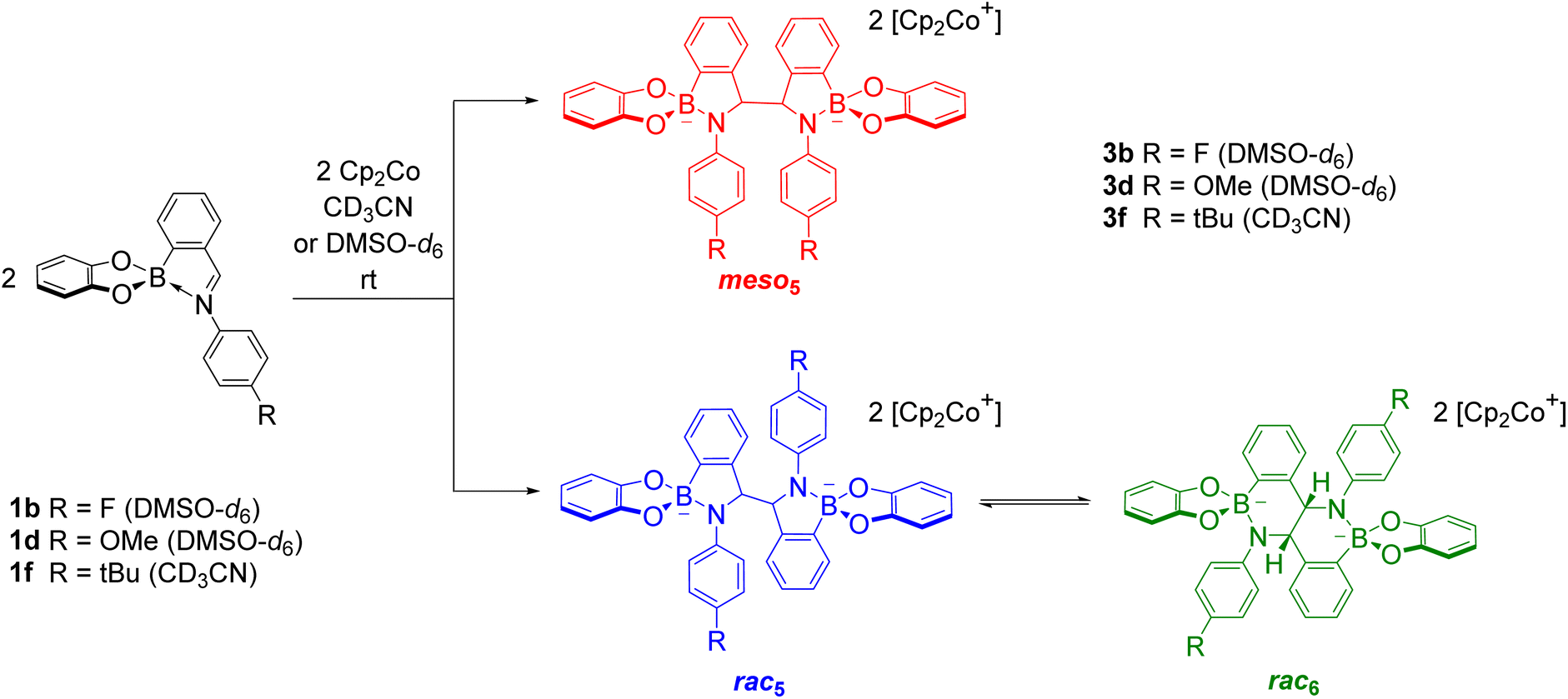 | ||
| Scheme 6 Time-course NMR experiments in CD3CN (for 1f) or DMSO-d6 (for 1b and 1d) suggested the initial formation of the meso5 and rac5 diastereomers followed by interconversion of the rac5 isomer into the rac6via rearrangement of the dynamic covalent B–N bonds. | ||
Dynamic covalent chemistry
Dynamic covalent chemistry15–19 is a diverse research field encompassing a variety of reversible covalent bonds from CN22,24–28 to B–O19,22,32–34 bonds. The combination of several dynamic covalent bonds offers the potential for addressing the different functional groups orthogonally, thus increasing the complexity of the system.
N-Aryl iminoboronates (the substrates for the synthesis of amidoboronates) contain two types of dynamic covalent bonds, an imine and boronate ester, leading to synergistic effects during their self-assembly from an aniline, diol and 2-formylphenyl boronic acid (Scheme 4). Nitschke and co-workers reported the amine and diol subcomponents influence the stability and yield of the resulting iminoboronate;55 synergistic stabilisation of the imine and boronate ester was proposed from the combination of an electron-rich aniline and catecholate due to greater resonance delocalisation over the catecholate, resulting in a B–N dative bond in aprotic solvents. In contrast, incomplete iminoboronate formation was observed with an electron-rich aniline and an electron-rich alkoxide, attributed to increased electron density around the boron centre leading to destabilisation and the absence of a B–N dative bond.
In addition, both the aniline and diol subcomponents can be orthogonally exchanged when a thermodynamically more stable iminoboronate results from exchange.55 More electron-rich amines (e.g. an alkylamine) replaced electron-poor ones (e.g. an aniline) driven by the formation of a more electron-rich imine stabilising the boron centre (Scheme 7a). Furthermore, aliphatic diols were displaced by pyrocatechol due to better delocalisation of the oxygens’ partial negative charge over the aromatic catechol (Scheme 7b).
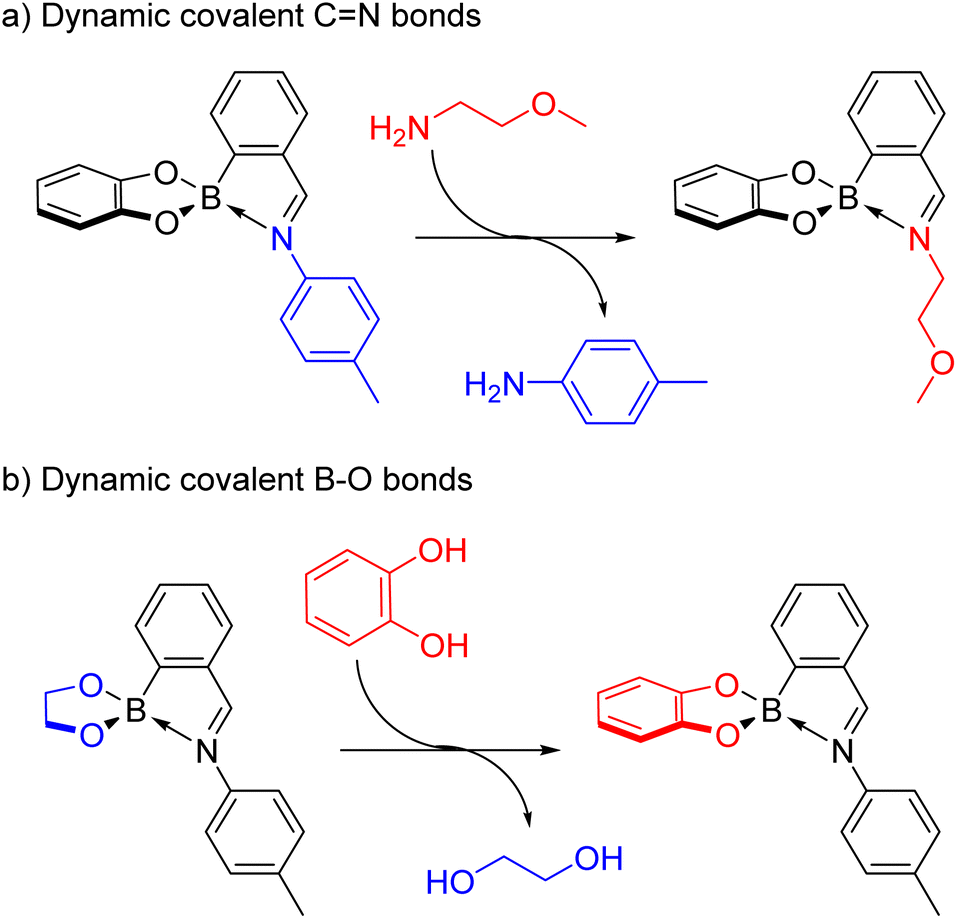 | ||
| Scheme 7 Orthogonal dynamic covalent bonds reported by Nitschke and co-workers in N-aryl iminoboronates involving: (a) imine exchange of an aniline subcomponent for a more electron-rich alkylamine; (b) boronate ester exchange of an aliphatic diol for pyrocatechol. | ||
While combinations of two types of dynamic covalent bonds can be orthogonally exchanged (e.g. imines and boronate esters as in Scheme 7),74 extension to three dynamic covalent bonds introduces additional orthogonality issues. Matile and co-workers investigated the orthogonality of hydrazone (red, Scheme 8a), boronate ester (green) and disulfide (blue) bonds in multicomponent self-assembly 11 as well as model systems like 12 (Scheme 8b) and increasing the stability of the boronate ester was necessary for orthogonality under the acidic hydrazone exchange conditions.22 This was achieved using the boronate esters of benzoboroxoles where the anionic tetrahedral boron centre is proposed to be less electrophilic and intramolecularly stabilised. In model system 12, the boronate ester was stable under the acidic hydrazone exchange conditions and basic disulfide exchange conditions but under the boronate ester exchange conditions (DMSO-d6, 10% D2O, 2% Hünig's base), equilibrium was reached in under 3 min giving a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 mixture of 12 and 13 (Scheme 8b).
:1.1 mixture of 12 and 13 (Scheme 8b).
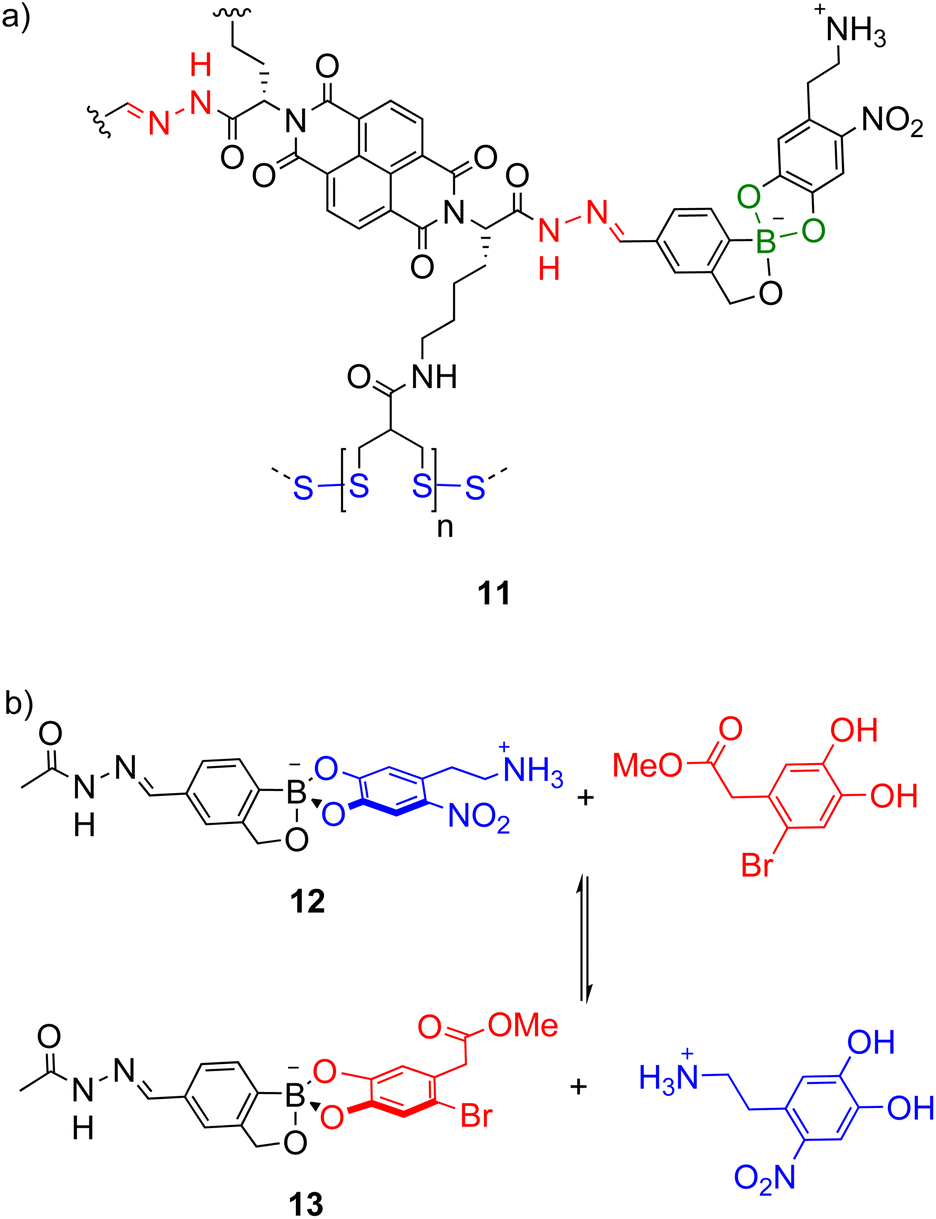 | ||
| Scheme 8 Matile's (a) multicomponent assembly 11 with three types of orthogonal dynamic covalent bonds: hydrazone (red), boronate ester (green) and disulfide (blue); (b) model system 12 for investigating the stability of the boronate ester under the conditions for hydrazone, boronate ester and disulfide exchange. | ||
There could be parallels between the dynamic covalent chemistry of amidoboronates and the examples shown in Schemes 7 and 8. However, there are some differences since reductive coupling of the imine bond removes the possibility of imine exchange in amidoboronates. Nevertheless, there is also the potential to access new types of dynamic covalent bonds in amidoboronates, for example dynamic covalent C–C bonds through the involvement of radicals during the reductive coupling based on literature examples.
While C–C bond formation from the dimerisation and dissociation of organic radicals has been long known (Scheme 9),75–78 its application in dynamic covalent chemistry has only recently emerged.18 To ensure the necessary reversibility, several conditions need to be fulfilled: a relatively low bond dissociation enthalpy to establish the radical/dimer equilibrium under mild conditions; sufficient thermodynamic stabilisation of the radical (e.g. by improving spin delocalisation through expansion of the π system and/or substitution18,79,80) so that the equilibrium shifts towards the radical; suppression of side-reactions given the reactivity of radicals.18 A number of carbon-based radicals including dicyanomethyl,72,73,79,81–83 fluorenyl,80 lactone80,84 and 4-substituted triphenylmethyl85 derivatives have already been shown to undergo reversible C–C bond formation (Scheme 9), enabling the self-assembly of macrocycles72,73,79,82,86,87 and the development of stimuli-responsive materials.81,84
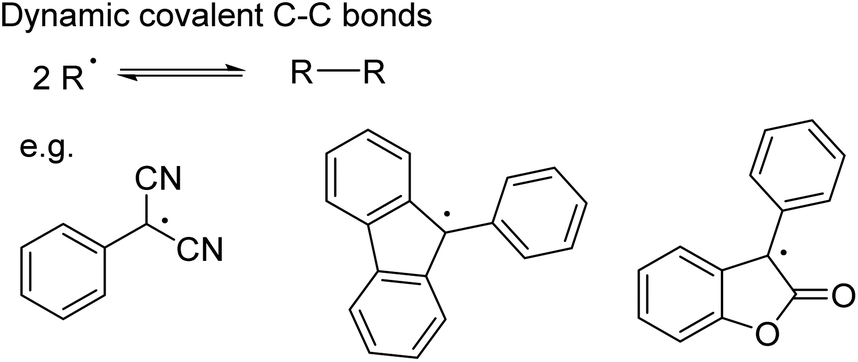 | ||
| Scheme 9 Dynamic covalent C–C bond formation involving radicals, such as dicyanomethyl, fluorenyl and lactone derivatives. | ||
Dynamic covalent chemistry of amidoboronates
Amidoboronates are thus interesting candidates for use in dynamic covalent chemistry applications given the initial implication of dynamic covalent B–N bonds in the rac5/rac6 rearrangement and the potential for dynamic covalent C–C and B–O bonds. The dynamic covalent chemistry of the amidoboronates was explored and of particular interest was the discovery of new chemistry, for example compared to iminoboronates and other reductively coupled dimers.C–C bonds
The reversibility of C–C bond formation was investigated in a series of experiments to gain insight into the reductive coupling mechanism. Based on the analogous reductive coupling of iminochloroborane 8 reported by Dostál and co-workers (see Synthesis of B-N heterocyclic dimers),6 radicals are proposed to be involved. Firstly, isolated crystals of the meso5, rac5 and rac6 isomers were redissolved in DMSO-d6 to probe the existence of a radical/dimer equilibrium. However, there has been thus far no evidence of a conversion between the meso5 and rac isomers at room temperature via reversible C–C bond formation in the redissolved crystals. Furthermore, decomposition was observed upon heating solutions of the redissolved crystals of meso5-3b and rac6-3c at elevated temperatures (90 °C or above).70 This contrasts the finding by Dostál and co-workers that meso-9 can be converted to rac-9 upon heating in toluene (Scheme 3).6However, the addition of the tritylium cation to an isomeric mixture of 3c or 4c led to quantitative regeneration of the corresponding iminoboronates 1c or 2c at room temperature within 10 minutes (Scheme 10). The tritylium cation acts as an electron abstractor leading to oxidative decoupling of the dimers as well as Cp2CoBF4 and the formation of the trityl dimer from subsequent radical coupling. This ability to break the newly formed C–C bond in the dimer has not been reported in the related reductive couplings of imine and carbonyl compounds (see Synthesis of 1,2-diamines and 1,2-diols).
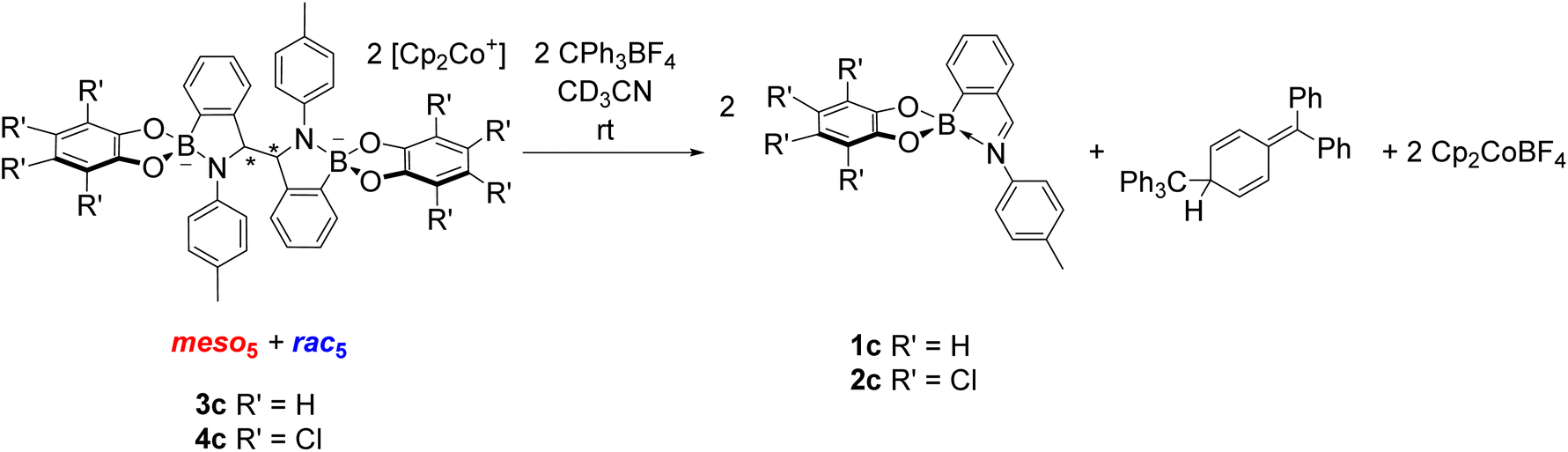 | ||
| Scheme 10 Addition of the tritylium cation to an isomeric mixture of 3c or 4c regenerates the corresponding iminoboronate (1c or 2c) as well as the trityl dimer and Cp2CoBF4. For clarity, only the meso5 and rac5 isomers are depicted as they were the major species observed in the reaction mixture in CD3CN. | ||
In another experiment, the TEMPO radical was added to an isomeric mixture of 4c and upon heating at 70 °C in CD3CN for 3 days, an additional species appeared in the 1H NMR spectrum (Fig. 3a).70 X-ray analysis of crystals obtained from the reaction revealed the formation of 14 where TEMPO rather than the imine is coordinated to the anionic tetrahedral boron centre (Fig. 3b). The formation of an imine was also consistent with the observation of a singlet above 9 ppm for the new species in the NMR spectrum. The formation of 14 was proposed via electron abstraction from the nitrogen lone pair by TEMPO giving a nitrogen-based radical cation, homolytic cleavage of the C–C bond generating an imine and finally, nucleophilic attack of TEMPO− on the boron centre. This contrasts the formation of radicals via homolytic substitution of trigonal catecholborane derivatives by TEMPO.88–90
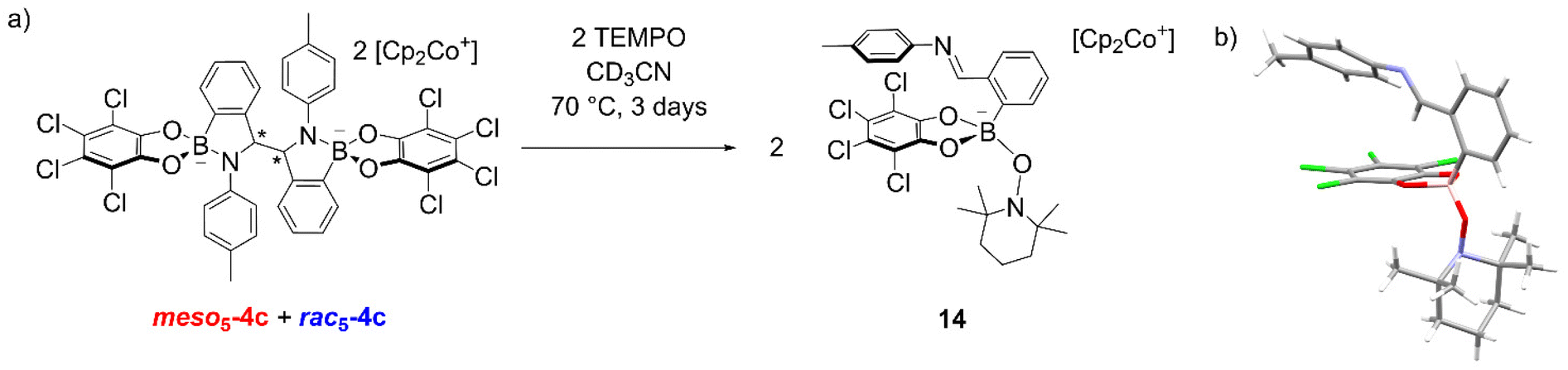 | ||
| Fig. 3 (a) Formation of 14 from the reaction of a 4c isomeric mixture with the TEMPO radical. For clarity, only the major amidoboronate species in solution are depicted; (b) X-ray crystal structure of 14 with Cp2Co+ countercation removed for clarity. | ||
These experiments demonstrated that the new C–C bond can be broken upon addition of an electron abstractor (CPh3+) or a radical (TEMPO). The existence of a radical/dimer equilibrium will be probed in future experiments to investigate whether the reductive coupling can be implemented for the formation of dynamic covalent C–C bonds like in Scheme 9.
B–N bonds
The reductive coupling converts the B–N dative bond observed in iminoboronates 1a–1f and 2a–2e to covalent B–N bonds with anionic tetrahedrally coordinated boron centres. Unexpectedly, these covalent B–N bonds were found to be dynamic as the rearrangement of the rac5 into the rac6 isomer was observed over time in time-course NMR experiments (see Solution characterisation). In contrast, redissolved crystals of meso5-3a–b did not interconvert into either of the rac isomers and there has been no evidence thus far of the formation of a meso6 product.The rac5/rac6 rearrangement was investigated in more detail by redissolving isolated crystals in DMSO-d6 (Scheme 11). Initial investigations suggested increased electron density facilitates cleavage of the B–N bond since the rate of interconversion of 3b,c–f was qualitatively fastest with 3d containing the most electron-donating aniline substituents in the series.70
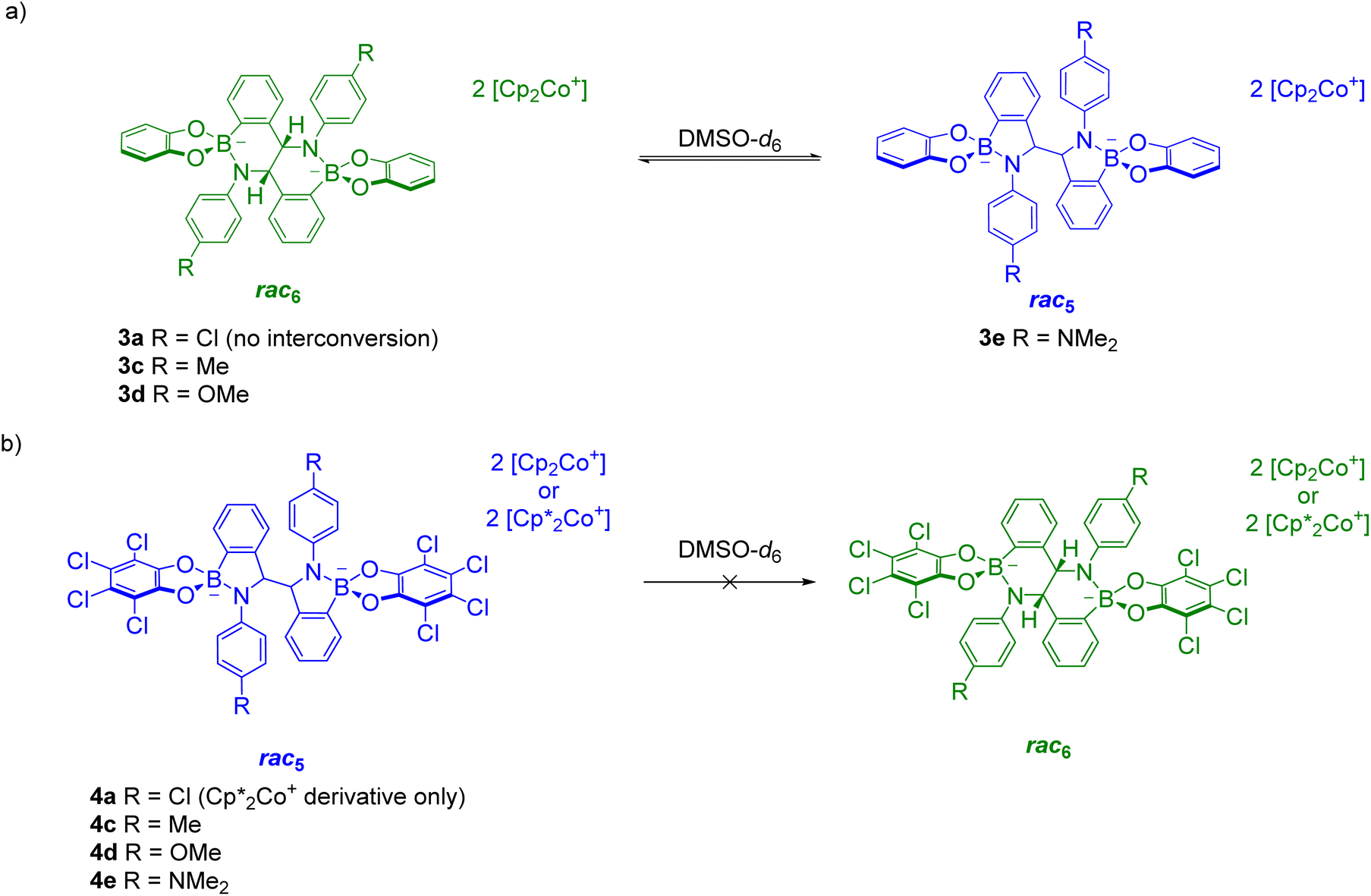 | ||
| Scheme 11 rac 5/rac6 interconversion upon redissolving crystals of (a) rac6-3a,c–d and rac5-3e; (b) rac5-4a,c–e. Adapted with permission from ref. 71. | ||
Further investigations focused on the amount of the rac5 and rac6 isomers following equilibration as a function of the para-substituent on the aniline and the catechol (Fig. 4).71 For the pyrocatechol series, rac6-3a,c–d and rac5-3e crystallised from the reductive couplings. While rac6-3a did not interconvert to the rac5 isomer, the amount of the rac5 isomer for the other derivatives increased with more electron-donating substituents (Scheme 11a, Fig. 4a). A 1:1 rac5/rac6 mixture was obtained for 3e with the most electron-rich NMe2 substituent. In contrast, redissolved rac5 crystals as either the cobaltocenium or decamethylcobaltocenium salt from the tetrachlorocatechol series (4a,c–e) were not observed to interconvert to the rac6 isomer (Scheme 11b, Fig. 4b).
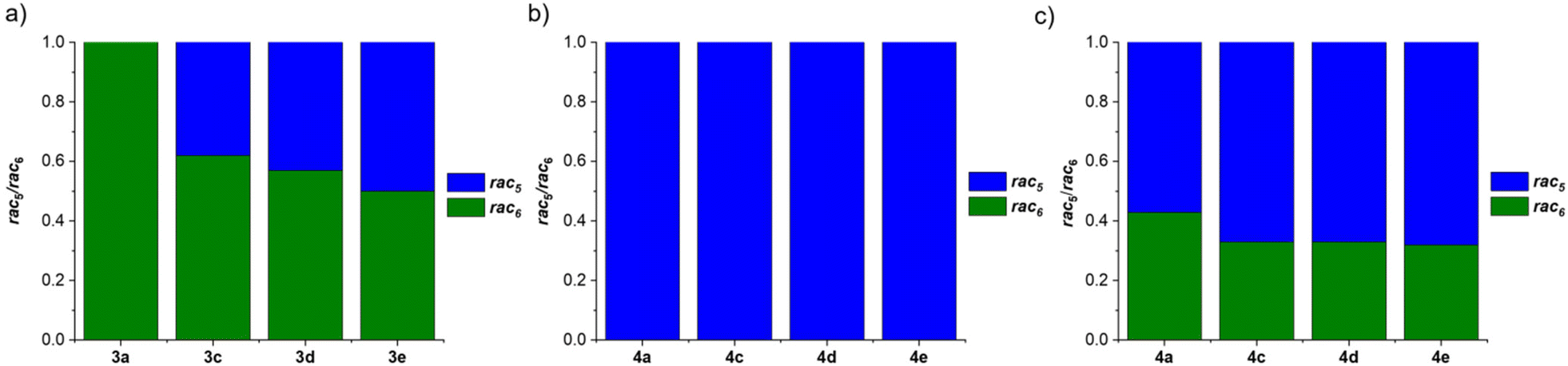 | ||
| Fig. 4 Comparison of the rac5/rac6 isomeric ratios following: (a) equilibration of redissolved crystals of 3a,c–e from the pyrocatechol series; (b) equilibration of redissolved crystals of 4a,c–e from the tetrachlorocatechol series; (c) catechol exchange of pyrocatechol for tetrachlorocatechol in equilibrated rac5/rac6 mixtures from (a). Reprinted with permission from ref. 71. | ||
Conformational analysis of the rac5 and rac6 isomers may provide an explanation for the rac5/rac6 rearrangement (Fig. 5). Of the three most likely conformations (two gauche and one anti), only the gauche conformation (black box) where the anilines are anti to one another has been observed in the X-ray crystal structures (Fig. 2c). A similar gauche conformation (black box, Fig. 5) was also observed in the X-ray crystal structures of the rac6 isomer (Fig. 2d), suggesting that the rac5/rac6 rearrangement occurs via breakage of the B–N bonds and rotation of the phenylboronate ester groups so that the five-membered rings are converted to a fused six-membered ring scaffold. Fig. 5 represents the two halves of the rac5 dimer in red and black to highlight the change of the connectivity in the rac6 isomer where the red nitrogen forms a covalent bond to the boron centre in black and vice versa.
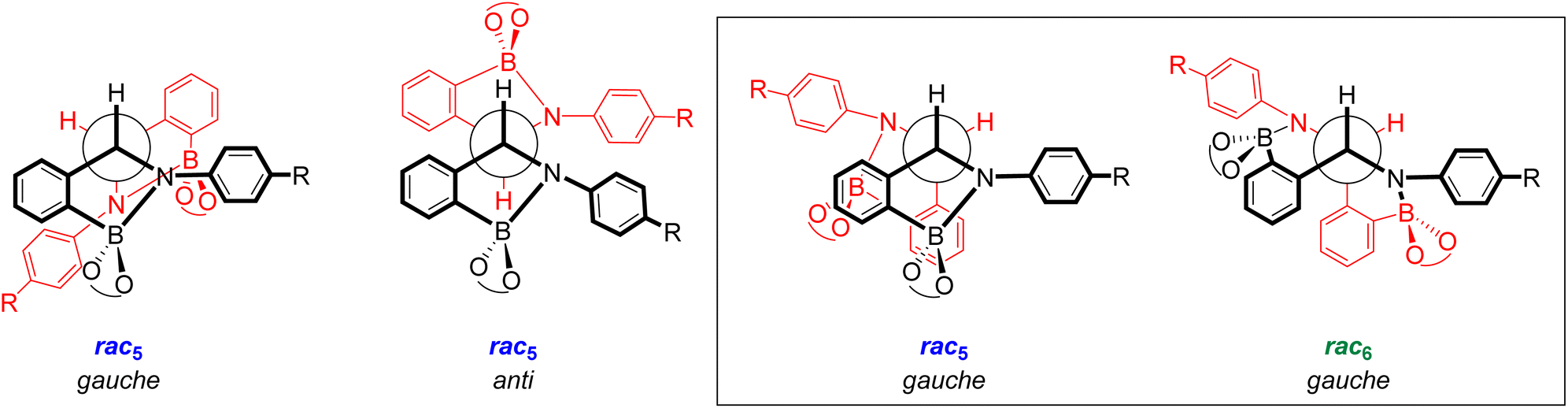 | ||
| Fig. 5 Conformational analysis of the rac5 isomer showing the three possible conformations around the new C–C bond, two gauche and one anti. Black box: the gauche conformations of the rac5 and rac6 isomers observed in X-ray crystal structures. The red and black represent the two halves of the dimer to highlight the change in connectivity upon the rac5/rac6 rearrangement. | ||
Based on the observed interconversions (Scheme 11), electronic effects were proposed to control the rac5/rac6 rearrangement; an electron-withdrawing Cl aniline substituent (3a) or catechol such as tetrachlorocatechol (4a,c–e) resulted in no interconversion of the rac6 and rac5 isomers, respectively, attributed to strengthening of the B–N bond from the reduced electron density. However, increasing the electron density in the B–N bond through more electron-donating aniline substituents is proposed to weaken the B–N bond, resulting in increased rac5/rac6 interconversion.
B–O bonds
The diol subcomponents within boronate ester bonds are known to undergo exchange via dynamic covalent B–O bonds.19,22,55,74 However, in most cases the boron centre has a vacant p orbital (e.g. in iminoboronates in the absence of a BN dative bond, Scheme 7b), whereas in amidoboronates this is filled since the boron is anionic and tetrahedrally coordinated. Nevertheless, Matile and co-workers reported catechol exchange with boronate esters of benzoboroxoles containing anionic tetrahedral boron centres (Scheme 8b).22 Therefore, the dynamic covalent nature of the B–O bonds of amidoboronates was explored in catechol exchange experiments.71Addition of tetrachlorocatechol to equilibrated mixtures of rac-5/rac6-3a,c–e in DMSO-d6 led to complete catechol exchange and the formation of rac5/rac6-4a,c–e (Scheme 12). The rac5/rac6 ratio was changed from a preference for the rac6 isomer prior to catechol exchange (Fig. 4b) to a preference for the rac5 isomer (Fig. 4c). The presence of the rac6 isomer following catechol exchange is attributed to the catechol exchange pathway allowing the direct conversion of rac6-3 to rac6-4 rather than via the rac5/rac6 interconversion since redissolved rac5-4 crystals did not interconvert to the rac6 isomer.
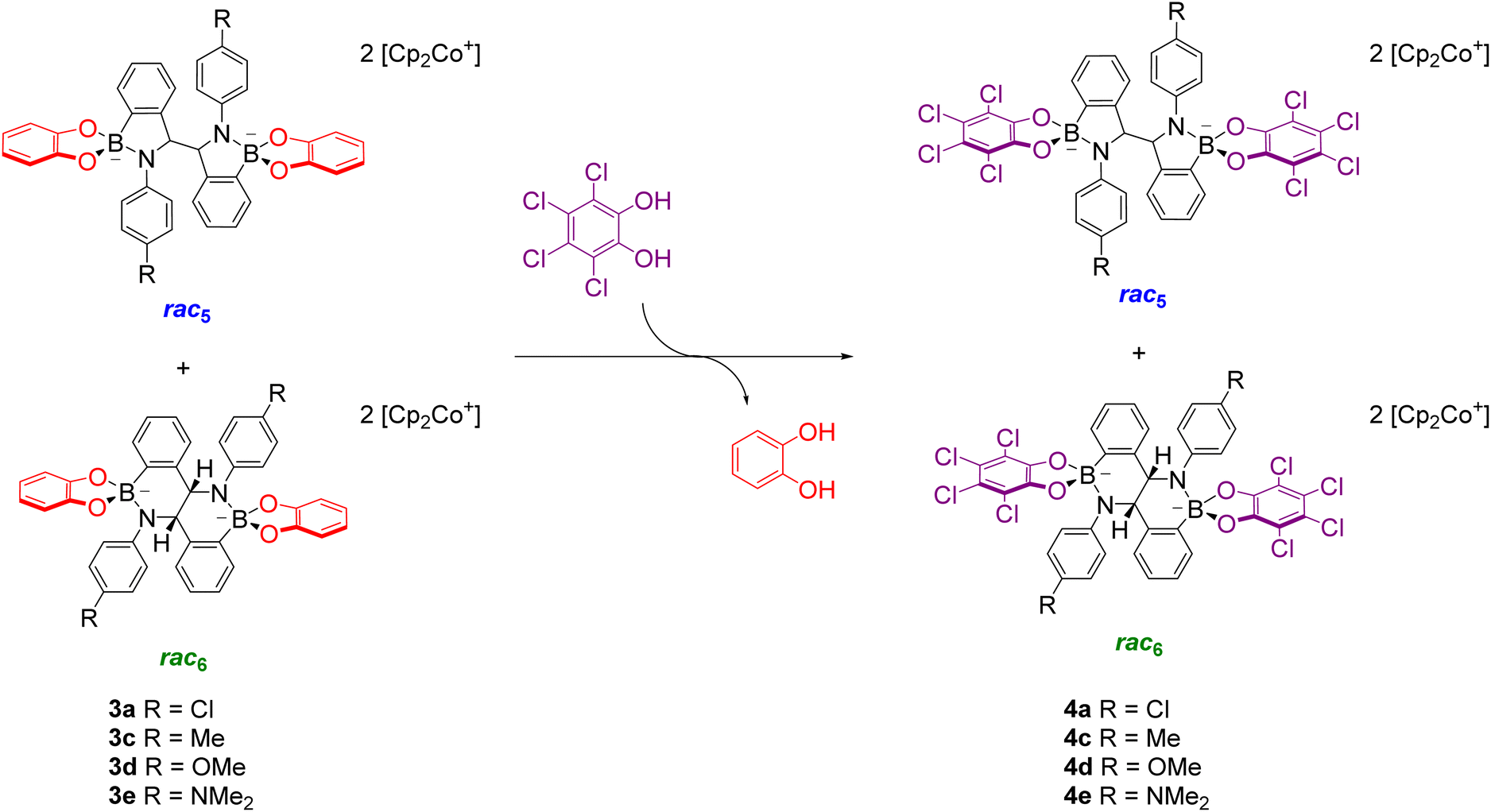 | ||
| Scheme 12 Boronate ester exchange of pyrocatechol for tetrachlorocatechol via dynamic covalent B–O bonds converts an equilibrated mixture of rac5-3 and rac6-3 to rac5-4 and rac6-4. Adapted with permission from ref. 71. | ||
While catechol exchange of pyrocatechol for the more electron-deficient tetrachlorocatechol was observed, addition of excess pyrocatechol to rac5-4d showed no catechol exchange. It was proposed that the tetrachlorocatechol analogues are thermodynamically more stable due to better negative charge delocalisation with tetrachlorocatechol vs. pyrocatechol and this is the driving force for catechol exchange.
Conclusions and outlook
While the number of synthetic methods for BN-heterocycles are limited compared to their carbon analogues, this is an active field with an increasing number of reports particularly given the growing importance of BN-heterocycles in catalysis and materials chemistry applications. On the other hand, dynamic covalent chemistry is an established field but new applications and types of dynamic covalent bonds (e.g. based on radicals18) are emerging.Amidoboronates bring together BN-heterocyclic synthesis and dynamic covalent chemistry and new research directions have been made possible through this intersection of these two seemingly disconnected research fields. Amidoboronates are a new class of BN-containing heterocycles that can be prepared from N-aryl iminoboronate substrates via a reductive coupling (Scheme 13). Their synthesis opens up new chemical space since up to three isomers can be prepared from a single substrate, which itself can be modularly self-assembled from three building blocks using dynamic covalent chemistry. Thus, a large family of new BN-heterocycles was synthesised with reduced effort.
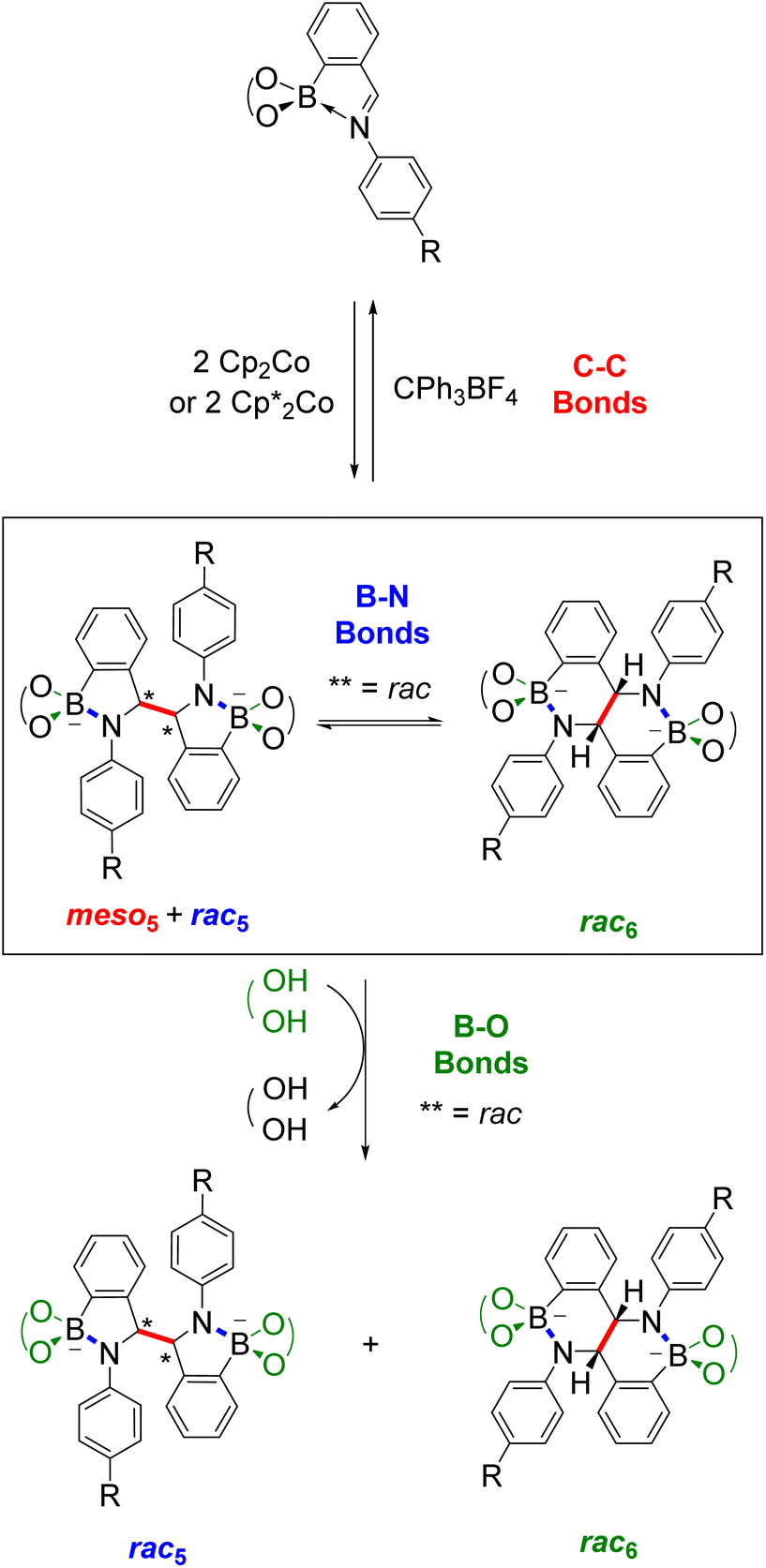 | ||
| Scheme 13 Overview of the synthesis and dynamic covalent chemistry of amidoboronates showing the three types of dynamic covalent bonds (C–C in red, B–N in blue and B–O in green). For clarity, the R groups, catechols and countercations are not depicted. | ||
In addition, initial studies have revealed the interesting and unusual dynamic covalent chemistry of amidoboronates involving C–C, B–N and B–O bonds. Unlike the analogous reductive couplings of carbonyls and imines, C–C bond formation is reversible for amidoboronates since oxidative decoupling of the dimer through the addition of the tritylium cation regenerates the iminoboronate. An unprecedented rearrangement between the rac5 and rac6 isomers via dynamic covalent B–N bonds has also been observed where the aniline para-substituent and the catechol tune the isomeric ratio at equilibrium. Furthermore, boronate ester exchange can be exploited as another tool to control this ratio.
Further exploration of this chemical space will offer new opportunities for applications of this chemistry. Future efforts will focus on exploring the mechanism of the reductive coupling, particularly to control the diastereoselectivity and therefore, selectively synthesise a particular isomer. The three types of dynamic covalent bonds also offer an exciting opportunity for exploring the orthogonality of transformations and the design of more complex stimuli-responsive systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
I would like to thank everyone who has contributed to this project and Patrick Harders for providing feedback on this manuscript. The Deutsche Forschungsgemeinschaft (DFG, project number 447862786) is gratefully acknowledged for financial support.References
- M. J. S. Dewar, V. P. Kubba and R. Pettit, J. Chem. Soc., 1958, 3073–3076 RSC.
- A. J. Ashe and X. Fang, Org. Lett., 2000, 2, 2089–2091 CrossRef CAS PubMed.
- M. Yamashita, Y. Aramaki and K. Nozaki, New J. Chem., 2010, 34, 1774–1782 RSC.
- P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed.
- Y. Aramaki, H. Omiya, M. Yamashita, K. Nakabayashi, S.-i. Ohkoshi and K. Nozaki, J. Am. Chem. Soc., 2012, 134, 19989–19992 CrossRef CAS PubMed.
- M. Hejda, R. Jambor, A. Růžička, A. Lyčka and L. Dostál, Dalton Trans., 2014, 43, 9012–9015 RSC.
- X.-Y. Wang, J.-Y. Wang and J. Pei, Chem. – Eur. J., 2015, 21, 3528–3539 CrossRef CAS PubMed.
- M. M. Morgan and W. E. Piers, Dalton Trans., 2016, 45, 5920–5924 RSC.
- Z. X. Giustra and S.-Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS PubMed.
- K. Matsui, S. Oda, K. Yoshiura, K. Nakajima, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 1195–1198 CrossRef CAS PubMed.
- C. R. McConnell and S.-Y. Liu, Chem. Soc. Rev., 2019, 48, 3436–3453 RSC.
- H. Noda, Y. Asada, M. Shibasaki and N. Kumagai, J. Am. Chem. Soc., 2019, 141, 1546–1554 CrossRef CAS PubMed.
- C.-W. Ju, B. Li, L. Li, W. Yan, C. Cui, X. Ma and D. Zhao, J. Am. Chem. Soc., 2021, 143, 5903–5916 CrossRef CAS PubMed.
- F. Lindl, A. Lamprecht, M. Arrowsmith, E. Khitro, A. Rempel, M. Dietz, T. Wellnitz, G. Bélanger-Chabot, A. Stoy, V. Paprocki, D. Prieschl, C. Lenczyk, J. Ramler, C. Lichtenberg and H. Braunschweig, Chem. – Eur. J., 2023, 29, e202203345 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- R.-C. Brachvogel and M. von Delius, Eur. J. Org. Chem., 2016, 3662–3670 CrossRef CAS.
- D. Sakamaki, S. Ghosh and S. Seki, Mater. Chem. Front., 2019, 3, 2270–2282 RSC.
- S. Chatterjee, E. V. Anslyn and A. Bandyopadhyay, Chem. Sci., 2021, 12, 1585–1599 RSC.
- C. G. Pappas, P. K. Mandal, B. Liu, B. Kauffmann, X. Miao, D. Komáromy, W. Hoffmann, C. Manz, R. Chang, K. Liu, K. Pagel, I. Huc and S. Otto, Nat. Chem., 2020, 12, 1180–1186 CrossRef CAS PubMed.
- N.-M. Phan, E. G. Percástegui and D. W. Johnson, ChemPlusChem, 2020, 85, 1270–1282 CrossRef CAS PubMed.
- S. Lascano, K.-D. Zhang, R. Wehlauch, K. Gademann, N. Sakai and S. Matile, Chem. Sci., 2016, 7, 4720–4724 RSC.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS PubMed.
- C. Liang, S. Kulchat, S. Jiang and J.-M. Lehn, Chem. Sci., 2017, 8, 6822–6828 RSC.
- A.-E. Dascalu, L. Halgreen, A. Torres-Huerta and H. Valkenier, Chem. Commun., 2022, 58, 11103–11106 RSC.
- B. P. Benke, T. Kirschbaum, J. Graf, J. H. Gross and M. Mastalerz, Nat. Chem., 2023, 15, 413–423 CrossRef CAS PubMed.
- Y. Wu, C. Zhang, S. Fang, D. Zhu, Y. Chen, C. Ge, H. Tang and H. Li, Angew. Chem., Int. Ed., 2022, 61, e202209078 CAS.
- K. Caprice, D. Pál, C. Besnard, B. Galmés, A. Frontera and F. B. L. Cougnon, J. Am. Chem. Soc., 2021, 143, 11957–11962 CrossRef CAS PubMed.
- X. Wang, O. Shyshov, M. Hanževački, C. M. Jäger and M. von Delius, J. Am. Chem. Soc., 2019, 141, 8868–8876 CrossRef CAS PubMed.
- D. Hartmann and L. Greb, Angew. Chem., Int. Ed., 2020, 59, 22510–22513 CrossRef CAS PubMed.
- J. Roeser, D. Prill, M. J. Bojdys, P. Fayon, A. Trewin, A. N. Fitch, M. U. Schmidt and A. Thomas, Nat. Chem., 2017, 9, 977–982 CrossRef CAS PubMed.
- S. Ivanova, E. Köster, J. J. Holstein, N. Keller, G. H. Clever, T. Bein and F. Beuerle, Angew. Chem., Int. Ed., 2021, 60, 17455–17463 CrossRef CAS PubMed.
- N. Christinat, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2008, 47, 1848–1852 CrossRef CAS PubMed.
- N. Iwasawa and H. Takahagi, J. Am. Chem. Soc., 2007, 129, 7754–7755 CrossRef CAS PubMed.
- J. J. Eisch, D. D. Kaska and C. J. Peterson, J. Org. Chem., 1966, 31, 453–456 CrossRef CAS.
- J. G. Smith and C. D. Veach, Can. J. Chem., 1966, 44, 2497–2502 CrossRef CAS.
- J. G. Smith and I. Ho, J. Org. Chem., 1972, 37, 653–656 CrossRef CAS.
- E. J. Enholm, D. C. Forbes and D. P. Holub, Synth. Commun., 1990, 20, 981–987 CrossRef CAS.
- R. D. Rieke and S.-H. Kim, J. Org. Chem., 1998, 63, 5235–5239 CrossRef CAS.
- M. Shimizu, I. Suzuki and H. Makino, Synlett, 2003, 1635–1638 CrossRef CAS.
- R. J. Baker, C. Jones, M. Kloth and D. P. Mills, New J. Chem., 2004, 28, 207–213 RSC.
- M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef CAS PubMed.
- C.-M. Wang, P.-J. Xia, J.-A. Xiao, J. Li, H.-Y. Xiang, X.-Q. Chen and H. Yang, J. Org. Chem., 2017, 82, 3895–3900 CrossRef CAS PubMed.
- S. Kundu, L. Roy and M. S. Maji, Org. Lett., 2022, 24, 9001–9006 CrossRef CAS PubMed.
- Z. Qiu, H. D. M. Pham, J. Li, C.-C. Li, D. J. Castillo-Pazos, R. Z. Khaliullin and C.-J. Li, Chem. Sci., 2019, 10, 10937–10943 RSC.
- A. Caron, É. Morin and S. K. Collins, ACS Catal., 2019, 9, 9458–9464 CrossRef CAS.
- M. Liu, L. Tan, R. T. Rashid, Y. Cen, S. Cheng, G. Botton, Z. Mi and C.-J. Li, Chem. Sci., 2020, 11, 7864–7870 RSC.
- M. Yasui, K. Hanaya, T. Sugai and S. Higashibayashi, RSC Adv., 2021, 11, 24652–24655 RSC.
- J. Jo, S. Kim, J.-H. Choi and W.-j. Chung, Chem. Commun., 2021, 57, 1360–1363 RSC.
- Z.-W. Xi, L. Yang, D.-Y. Wang, C.-W. Feng, Y. Qin, Y.-M. Shen, C. Pu and X. Peng, J. Org. Chem., 2021, 86, 2474–2488 CrossRef CAS PubMed.
- Á. Péter, S. Agasti, O. Knowles, E. Pye and D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349–5365 RSC.
- F. Calogero, G. Magagnano, S. Potenti, F. Pasca, A. Fermi, A. Gualandi, P. Ceroni, G. Bergamini and P. G. Cozzi, Chem. Sci., 2022, 13, 5973–5981 RSC.
- C. A. Rosengarten, K. Yuvaraj, L. F. Lim, N. Cox and C. Jones, Chem. – Eur. J., 2023, 29, e202300135 CrossRef CAS PubMed.
- H. E. Dunn, J. C. Catlin and H. R. Snyder, J. Org. Chem., 1968, 33, 4483–4486 CrossRef CAS.
- M. Hutin, G. Bernardinelli and J. R. Nitschke, Chem. – Eur. J., 2008, 14, 4585–4593 CrossRef CAS PubMed.
- R. R. Groleau, T. D. James and S. D. Bull, Coord. Chem. Rev., 2021, 428, 213599 CrossRef CAS.
- A. D. Herrera-España, H. Höpfl and H. Morales-Rojas, Eur. J. Org. Chem., 2022, e202200383 Search PubMed.
- E. G. Shcherbakova, T. Minami, V. Brega, T. D. James and P. Anzenbacher, Angew. Chem., Int. Ed., 2015, 54, 7130–7133 CrossRef CAS PubMed.
- M. Pushina, S. Farshbaf, E. G. Shcherbakova and P. Anzenbacher, Chem. Commun., 2019, 55, 4495–4498 RSC.
- S. Delpierre, B. Willocq, J. De Winter, P. Dubois, P. Gerbaux and J.-M. Raquez, Chem. – Eur. J., 2017, 23, 6730–6735 CrossRef CAS PubMed.
- S. Delpierre, B. Willocq, G. Manini, V. Lemaur, J. Goole, P. Gerbaux, J. Cornil, P. Dubois and J.-M. Raquez, Chem. Mater., 2019, 31, 3736–3744 CrossRef CAS.
- H. Faustino, M. J. S. A. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Chem. Sci., 2016, 7, 5052–5058 RSC.
- M. K. Meadows, E. K. Roesner, V. M. Lynch, T. D. James and E. V. Anslyn, Org. Lett., 2017, 19, 3179–3182 CrossRef CAS PubMed.
- A. J. van der Zouwen, A. Jeucken, R. Steneker, K. F. Hohmann, J. Lohse, D. J. Slotboom and M. D. Witte, Chem. – Eur. J., 2021, 27, 3292–3296 CrossRef CAS PubMed.
- A. M. M. Rangaswamy, M. H. R. Beh, E. Soleimani, S. Sequeira, J. Cormier, K. N. Robertson and D. L. Jakeman, J. Org. Chem., 2022, 87, 13542–13555 CrossRef CAS PubMed.
- M. Zheng, F.-J. Chen, K. Li, R. M. Reja, F. Haeffner and J. Gao, J. Am. Chem. Soc., 2022, 144, 15885–15893 CrossRef CAS PubMed.
- R. Ma, C. Zhang, Y. Liu, C. Li, Y. Xu, B. Li, Y. Zhang, Y. An and L. Shi, RSC Adv., 2017, 7, 21328–21335 RSC.
- A. Biswas, T. Ghosh, P. K. Gavel and A. K. Das, ACS Appl. Bio Mater., 2020, 3, 1052–1060 CrossRef CAS PubMed.
- R. Cheng, G. Li, L. Fan, J. Jiang and Y. Zhao, Chem. Commun., 2020, 56, 12246–12249 RSC.
- E. N. Keyzer, A. Sava, T. K. Ronson, J. R. Nitschke and A. J. McConnell, Chem. – Eur. J., 2018, 24, 12000–12005 CrossRef CAS PubMed.
- P. Harders, T. Griebenow, A. Businski, A. J. Kaus, L. Pietsch, C. Näther and A. J. McConnell, ChemPlusChem, 2022, 87, e202200022 CrossRef CAS PubMed.
- Y. Liu, H. Yi, T. Tao, P. Hoa and C. Chunyan, Angew. Chem., Int. Ed., 2018, 57, 9023–9027 CrossRef PubMed.
- K. Oda, S. Hiroto and H. Shinokubo, J. Mater. Chem. C, 2017, 5, 5310–5315 RSC.
- A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC.
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- H. D. Hartzler, J. Org. Chem., 1966, 31, 2654–2658 CrossRef CAS.
- J. Ohkanda, Y. Mori, K. Maeda and E. Osawa, J. Chem. Soc., Perkin Trans. 2, 1992, 59–63 RSC.
- C. Harnack, W. Krull, M. Lehnig, W. P. Neumann and A. K. Zarkadis, J. Chem. Soc., Perkin Trans. 2, 1994, 1247–1252 RSC.
- T. Kobashi, D. Sakamaki and S. Seki, Angew. Chem., Int. Ed., 2016, 55, 8634–8638 CrossRef CAS PubMed.
- M. Frenette, C. Aliaga, E. Font-Sanchis and J. C. Scaiano, Org. Lett., 2004, 6, 2579–2582 CrossRef CAS PubMed.
- J. P. Peterson, M. R. Geraskina, R. Zhang and A. H. Winter, J. Org. Chem., 2017, 82, 6497–6501 CrossRef CAS PubMed.
- D. Wang, C. Capel Ferrón, J. Li, S. Gámez-Valenzuela, R. Ponce Ortiz, J. T. López Navarrete, V. Hernández Jolín, X. Yang, M. Peña Álvarez, V. García Baonza, F. Hartl, M. C. Ruiz Delgado and H. Li, Chem. – Eur. J., 2017, 23, 13776–13783 CrossRef CAS PubMed.
- B. Adinarayana, D. Shimizu, K. Furukawa and A. Osuka, Chem. Sci., 2019, 10, 6007–6012 RSC.
- K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2015, 54, 6168–6172 CrossRef CAS PubMed.
- W. P. Neumann, A. Penenory, U. Stewen and M. Lehnig, J. Am. Chem. Soc., 1989, 111, 5845–5851 CrossRef CAS.
- G. Wittig, E. Dreher, W. Reuther, H. Weidinger and R. Steinmetz, Justus Liebigs Ann. Chem., 1969, 726, 188–200 CrossRef.
- D. Beaudoin, O. Levasseur-Grenon, T. Maris and J. D. Wuest, Angew. Chem., Int. Ed., 2016, 55, 894–898 CrossRef CAS PubMed.
- S. Fukuzumi and J. K. Kochi, J. Org. Chem., 1980, 45, 2654–2662 CrossRef CAS.
- C. Ollivier, R. Chuard and P. Renaud, Synlett, 1999, 807–809 CrossRef CAS.
- M. Pouliot, P. Renaud, K. Schenk, A. Studer and T. Vogler, Angew. Chem., Int. Ed., 2009, 48, 6037–6040 CrossRef CAS PubMed.
Footnote |
| † In the naming of the isomers, the subscripts 5 and 6 refer to the heterocyclic ring size following dimerisation. |
| This journal is © The Royal Society of Chemistry 2023 |