
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFerrocene-triazole conjugates: do we know why they are biologically active?
Mariola
Koszytkowska-Stawińska
* and
Włodzimierz
Buchowicz
 *
*
Faculty of Chemistry, Chair of Organic Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: wlodzimierz.buchowicz@pw.edu.pl
First published on 21st December 2022
Abstract
The bioorganometallic chemistry of ferrocene has been gaining significance in recent years. This review presents ferrocene-triazole conjugates displaying significant biological properties. The conjugates have been synthesized via azide–alkyne cycloaddition reactions. The data are summarized according to the type of activity (anticancer, antibacterial and/or antifungal, antiprotozoal, and other effects). The results of studies concerning the understanding of the role of the ferrocene core in their biological activity are highlighted. While generally the mode of action of these organometallic species remains unclear, the importance of redox properties of ferrocene has been postulated in several cases.
1 Introduction
Ferrocene was serendipitously discovered in a laboratory 71 years ago.1,2 This air-stable, sparingly water-soluble3,4 organometallic compound is usually considered as exhibiting low toxicity.5,6 While ferrocene derivatives have not been found in nature, their bioorganometallic chemistry commenced with the discovery of anticancer properties of ferrocene polyamides7 and cationic ferrocenium species.8,9 Ferroquine10 and ferrocifens11–14 are the most widely known ferrocene derivatives displaying significant biological activity.Ferrocene-triazole hybrids have attracted considerable scientific attention since the ferrocene-benzotriazole conjugate was found to possess antitumor activity in vivo.5,6,15 Several reasons for interest in 1H-1,2,3-triazoles might be emphasized.
From the preparative point of view, the 1H-1,2,3-triazole platform can be easily prepared via the azide–alkyne cycloaddition of two readily accessible substrates.16 Owing to the specific electron density, the 1H-1,2,3-triazole ring might serve as an isostere of an amide or ester bond, or of a heterocyclic moiety.17 1H-1,2,3-Triazoles can also be considered as isosteres of a carboxylic acid moiety or rigid analogues of olefins. Moreover, by H-bonding or π–π stacking interactions, the 1H-1,2,3-triazole moiety might serve as a centre of interactions with a biological target.17,18 There are also numerous examples of 1H-1,2,3-triazoles with biological properties,19–21 including commercial or investigational drugs.22 The 1H-1,2,3-triazoles are not metabolized.23 Thus, they can be considered as risk-free building blocks in target drug candidates.
The ferrocene-1H-1,2,3-triazole conjugates discussed in this review have been synthesized using copper- or ruthenium-catalysed azide–alkyne cycloaddition (click-chemistry), as shown in Scheme 1. Two general approaches have been used: with a ferrocene-bearing azide (method 1), or alternatively with a ferrocene-bearing alkyne (method 2). In both cases, the azide/alkyne group has been linked directly to ferrocene (methods 1a, 2a), or separated from ferrocene with a linker of variable length and structure. These synthetic routes resulted in a considerable variety of conjugates with diverse structures and properties.
 | ||
| Scheme 1 General synthetic approaches for ferrocene-1H-1,2,3-triazole conjugates of biological interest. CuAAC – copper catalysed azide–alkyne cycloaddition. RuAAC – ruthenium catalysed azide–alkyne cycloaddition. | ||
In this review the literature since 2012 has been covered. Only sparse examples of biologically active ferrocene-1H-1,2,3-triazole conjugates were collected in the previous review on ferrocene-derived triazoles.24 Recently, more general accounts of the biological properties of ferrocene-organic hybrids have also been found.25–30 Nonetheless, the concept of ferrocene-1H-1,2,3-triazole conjugates was not discussed in detail in these publications.
The biologically active conjugates are reviewed in Tables 1–4 according to the type of their activity (anticancer, antibacterial and/or antifungal, antiprotozoal, and other effects). The synthetic route is also shown in the first column of each table. The results of studies dealing with the mechanism of action of the examined compounds (including the biological role of the ferrocene moiety) are presented in detail.
| Entry/synthesisb structure and number of compounds examined | IC50 or EC50 [μM] (cell line) number of cancer cell lines studied | Ref. | ||
|---|---|---|---|---|
| a IC50 or EC50 [μM] for the most active compound for the selected cell line is given. b See Scheme 1 for general click-chemistry synthetic approaches. c See text for details on the mechanism of action. d Synthesized by the post-click C–H arylation. e These compounds displayed also antimicrobial activity (R = C; MIC = 4.68 mg mL−1, Micrococuc luteus). f The corresponding ruthenocenes were also obtained and found to display comparable activity. g Synthesized using post-click amide coupling. Fc = ferrocenyl. | ||||
| 1.1/1a |
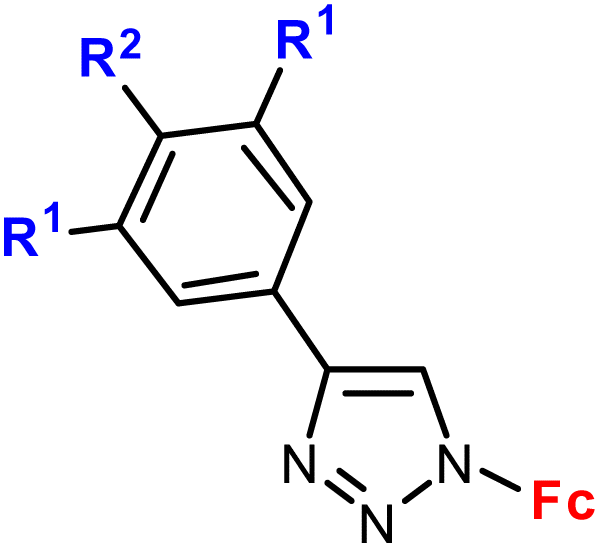
|
R1 = H or OH | 48.9 (HCC38) | 31 |
| R2 = H or OH (2 compounds) | R1 = H, R2 = OH | |||
| 2 cell lines | ||||
1.2/1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
|
R = H, Me, Cl, Ph (4 compounds) | 7.9 ± 0.6 (Raji) | 32 |
| R = Cl | ||||
| 4 cell lines | ||||
| 1.3/1a |
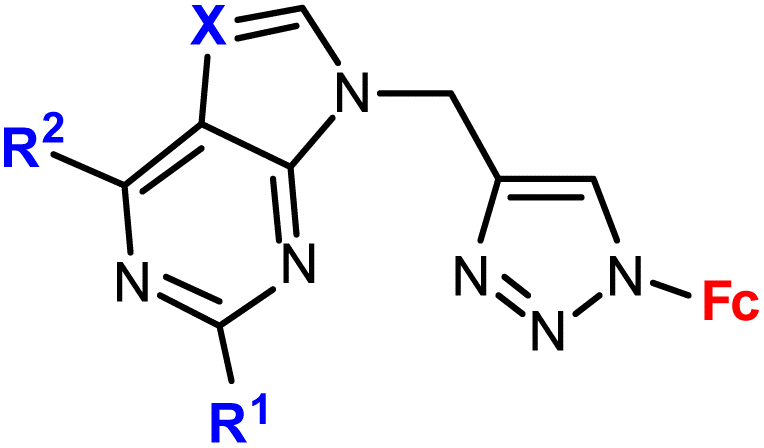
|
R1 = H, NH2 | 9.07 ± 1.21 (SW620) | 33 |
| R2 = Cl, NH2 | R1 = H, R2 = Cl, X = CH | |||
| X = CH or N (4 compounds) | 4 cell lines | |||
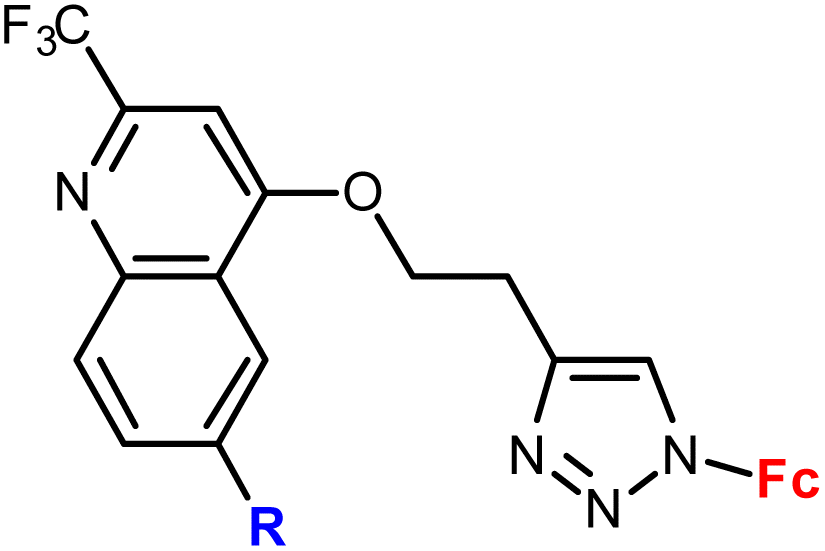
| 1.4/1a |
|
45.47 ± 4.52 (SW620) | 33 | |
| 4 cell lines | ||||
| 1.5/1a |
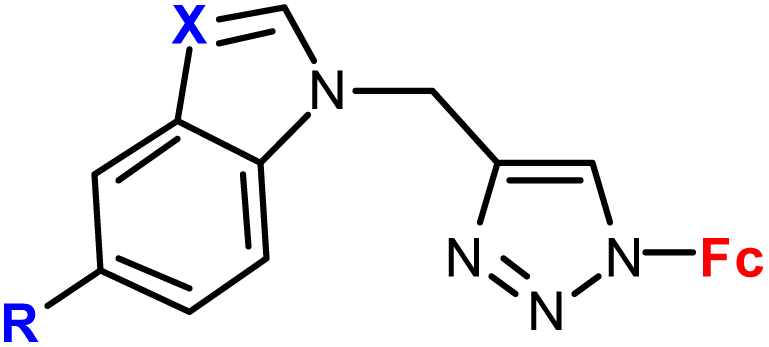
|
R = H or I | 71.79 ± 3.44 (HepG2) | 33 |
| X = CH or N (3 compounds) | R = I, X = CH | |||
| 4 cell lines | ||||
| 1.6/1bc |
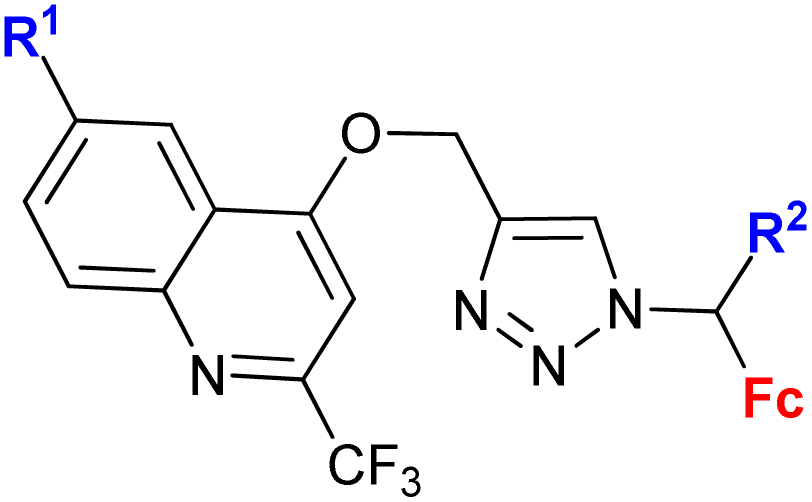
|
R1 = H, Me, Cl, Ph | 95.89 ± 2.4 (K562) | 32 |
| R2 = H or Me (8 compounds) | R1, R2 = Me | |||
| 4 cell lines | ||||
| 1.7/1bc |
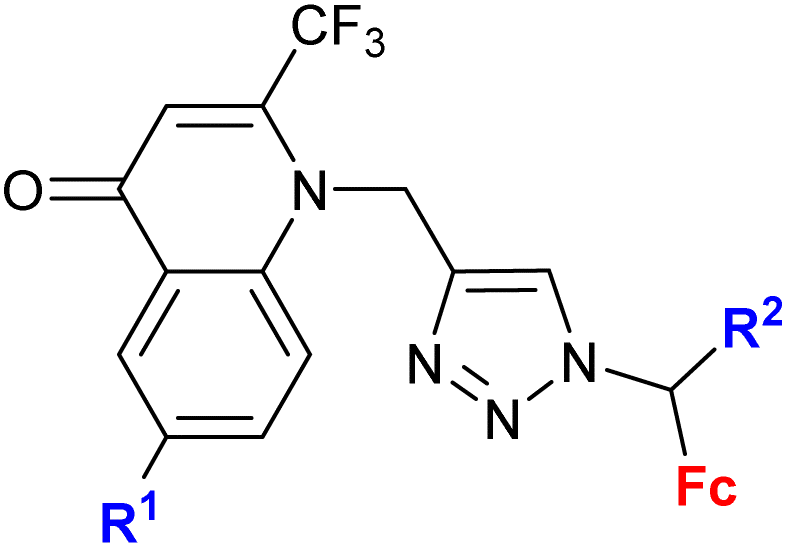
|
R1 = H, Me, Cl, Ph | 7.73 ± 2.4 (K562) | 32 |
| R2 = H or Me (4 compounds) | R1, R2 = H | |||
| 4 cell lines | ||||
| 1.8/1b |
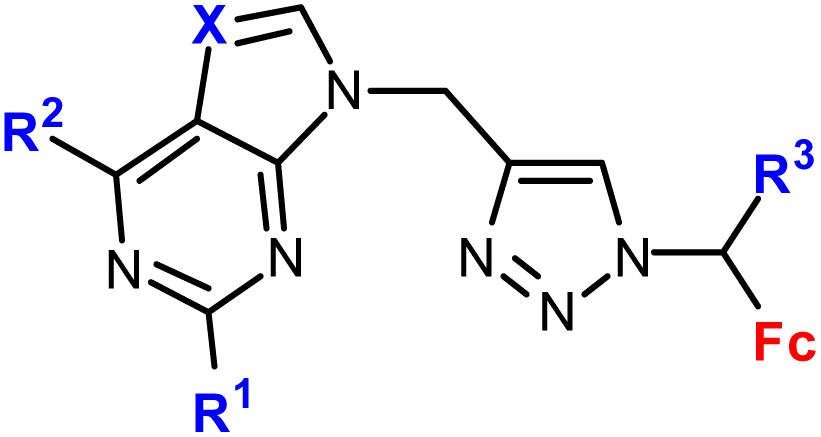
|
R1 = H, Cl, NH2 | 14.38 ± 3.02 (SW620) | 33 |
| R2 = H, Cl, NH2 | R1, R3 = H, R2 = Cl, X = CH | |||
| X = CH, N | 4 cell lines | |||
| R3 = H or Me (8 compounds) | ||||
| 1.9/1b |
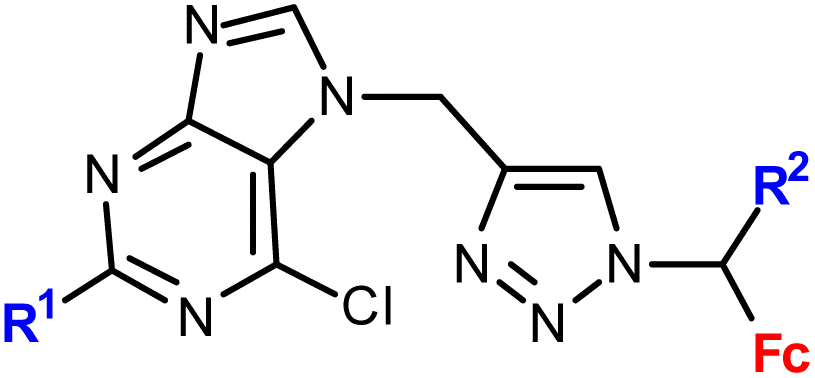
|
R1 = H or NH2 | 15.50 ± 3.24 (SW620) | 33 |
| R2 = H or Me (3 compounds) | R1 = H, R2 = Me | |||
| 4 cell lines | ||||
| 1.10/1b |
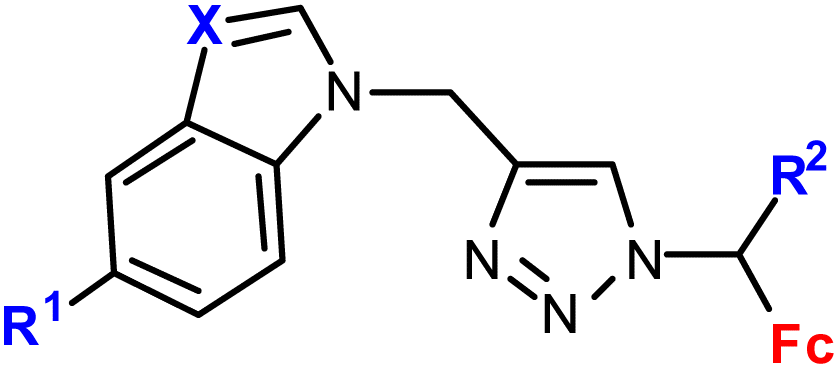
|
R1 = H or I | 53.55 ± 5.05 (HepG2) | 33 |
| R2 = H or Me | R1, R2 = H, X = CH | |||
| X = CH or N (6 compounds) | 4 cell lines | |||
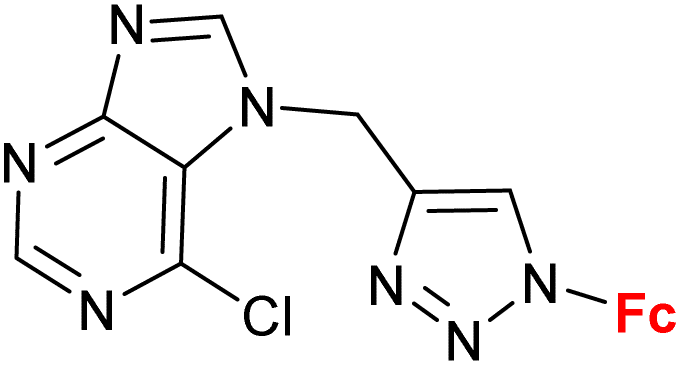
| 1.11/1b |
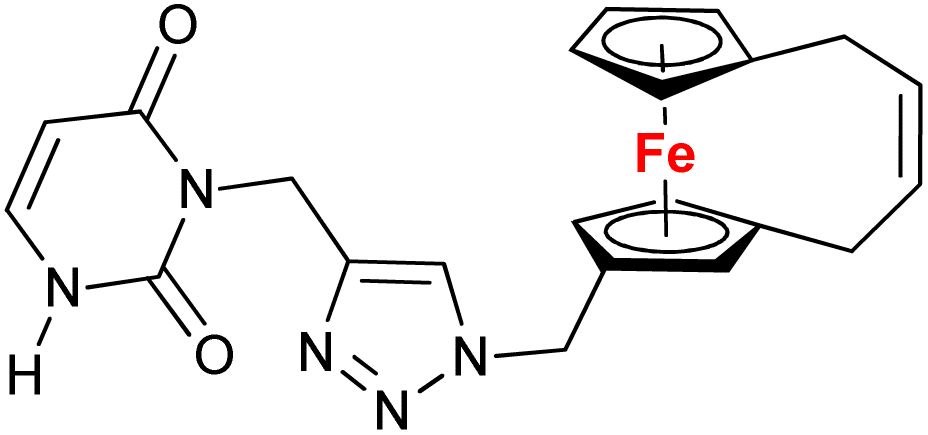
|
10.9 ± 1.7 (A549) | 34 | |
| 3 cell lines | ||||
| 1.12/1b |
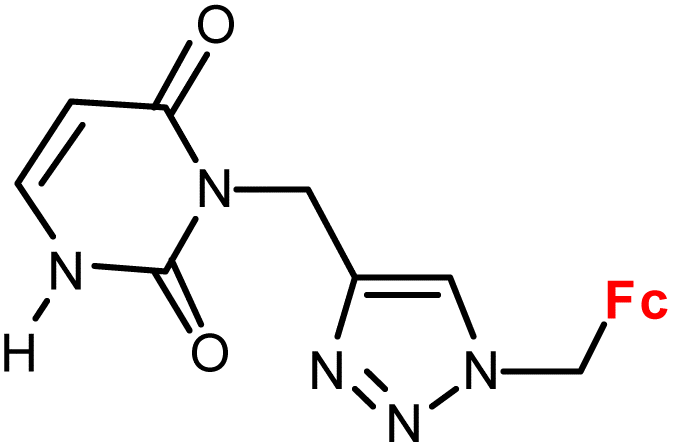
|
28.5 ± 2.2 (A549) | 34 | |
| 3 cell lines | ||||
| 1.13/1bc |
|
R1 = H, Me, Cl, Br, I | 11.9 ± 6.4 (K562) | 35 |
| R2 = H or Me (10 compounds) | R1 = I, R2 = H | |||
| 4 cell lines | ||||
| 1.14/1bc |
|
R1 = H, Me, Cl, Br, I | 2.0 ± 0.8 (Raji) | 35 |
| R2 = H or Me (10 compounds) | R1 = H, R2 = Me | |||
| 4 cell lines | ||||
| 1.15/1b |
|
R = H or Me (2 compounds) | 46.17 ± 9.65 (RD) | 36 |
| 3 cell lines | ||||
| 1.16/1b |
|
M = PdCl2 or ∅ (2 compounds) | 5.73 ± 0.03 (A549) | 37 |
| M = ∅ | ||||
| 4 cell lines | ||||
| 1.17/1a or 1bc |
|
R = Fc, FcCH2 or FcCH(CH3) | 14.0 ± 1.4 (HeLa) | 38 |
| or Cu2+/Zn2+ complexes | (3 ligands, 6 complexes) | R = FcCH(CH3), Cu2+ complex | ||
| 5 cell lines | ||||
| 1.18/1b or 1cc |
|
16 compounds for details see Scheme 2 | 0.7 ± 0.1 (HepG2) | 39 and 40 |
| 5 cell lines | ||||
| 1.19/1b |
|
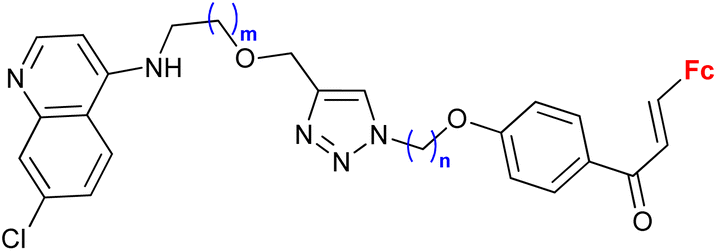
0.37 (HeLa) m = 2, n = 5 | 41 | |
| m = 1 or 2; n = 2–6, 8 (5 compounds) | 1 cell line | |||
| 1.20/2a |
|
R1 = H or OH | 15.3 (HCC38) | 31 |
| R2 = H or OH | R1 = H, R2 = OH, R3 = H | |||
| R3 = H or C6H4–OH-4d (5 compounds) | 2 cell lines | |||
| 1.21/2a |
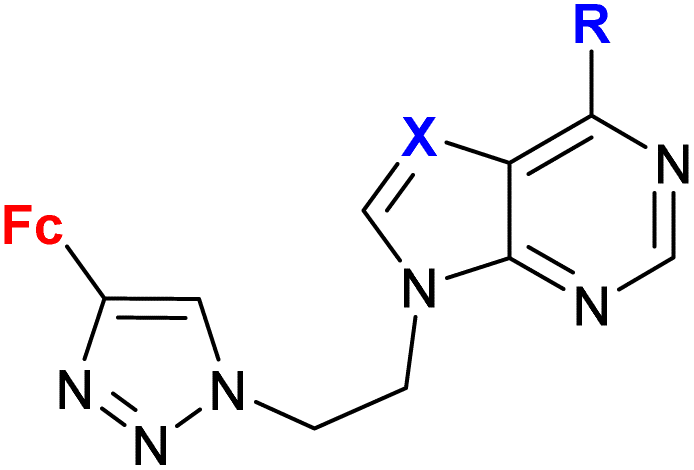
|
R = Cl, pyrrolidin-1-yl, morpholin-1-yl | 45.78 ± 9.11 (CFPAC-1) | 33 |
| X = CH or N (6 compounds) | R = morpholin-1-yl, X = CH | |||
| 4 cell lines | ||||
| 1.22/2a |
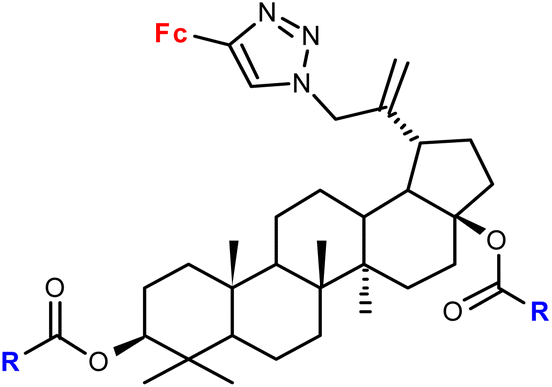
|
R = Me, Et, Ph (3 compounds) | 48.71 ± 3.55 (MS) | 42 |
| R = Me | ||||
| 3 cell lines | ||||
| 1.23/2ae |
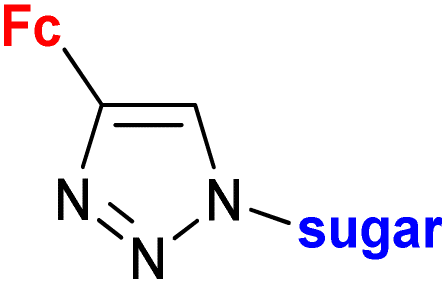
|
Sugar = protected: (A) 5-deoxy-β-D-ribofuranos-5-yl, (B) 5-deoxy-α-D-xylofuranos-5-yl, (C) 6-deoxy-α-D-glucofuranos-6-yl, (D) 6-deoxy-α-D-galactopyranos-6-yl | 5.3 (MDA-MBA-23) | 43 |
| R = A | ||||
| 5 cell lines | ||||
| 1.24/2a or 2b |
|
R1 = Fc or FcCH2f |
38 ± 0.5 (MCF-7) | 44 |
| R2 = CH2SCF3 or C(CF3)2OH (3 compounds) | R1 = FcCH2, R2 = C(CF3)2OH | |||
| 4 cell lines | ||||
| 1.25/2a or 2b |
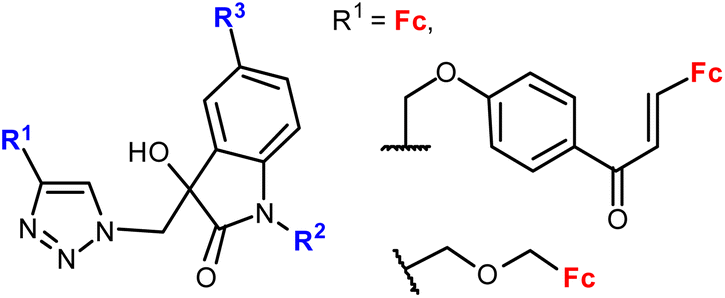
|
20.26 (MDA-MB-231) | 45 | |
| R2 = Me or Bn; R3 = H, Cl, Br, Me (24 compounds) | R1 = FcCH2OCH2, R2 = Me, R2 = Br | |||
| 2 cell lines | ||||
| 1.26/2b |
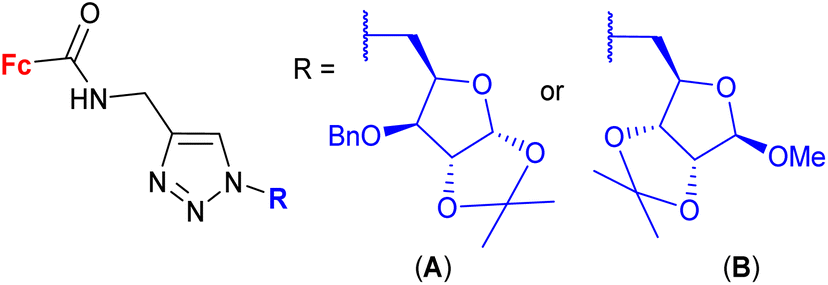
|
2.82 ± 0.14 (MDA-MB-231) | 46 | |
| R = A | ||||
| 4 cell lines | ||||
| 1.27/2b |
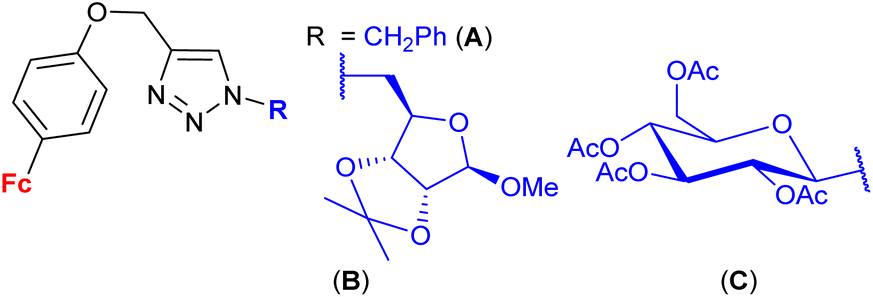
|
9.0 ± 0.03 (MCF-7) | 47 | |
| R = B | ||||
| 3 cell lines | ||||
| 1.28/2b |
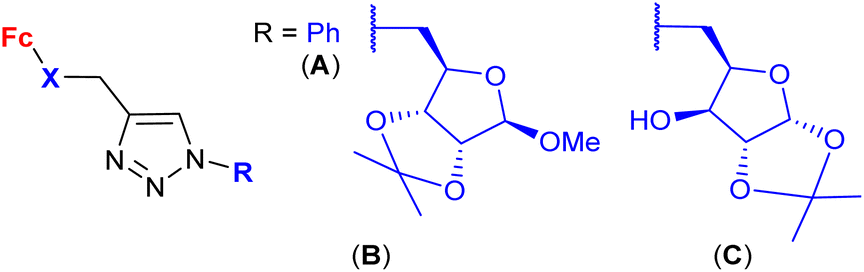
|
2.9 ± 0.25 (A549) | 48 | |
| X = S or Se (6 compounds) | R = B, X = Se | |||
| 4 cell lines | ||||
| 1.29/2bc |
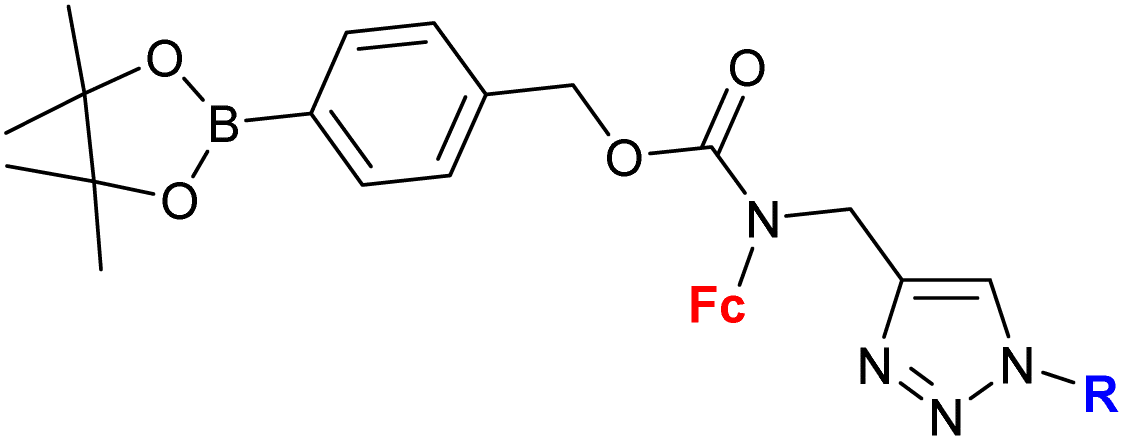
|
(7 compounds, for details see Scheme 3) | 5 ± 1 (A2780, DU-145) | 49–51 |
| 3 cell lines | ||||
| 1.30/2c |
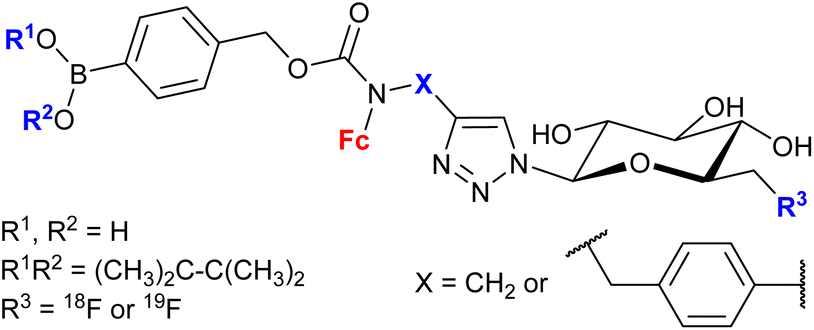
|
20 ± 7 (MCF-7) | 52 and 53 | |
| (6 compounds) | R1, R2 = Me2C-CMe2, R3 = 19F, X = 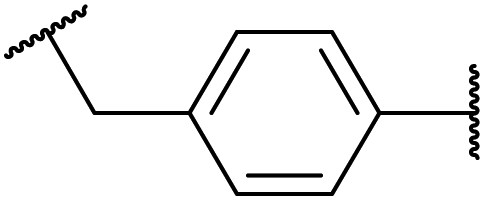 |
|||
| 5 cell lines | ||||
| 1.31g |
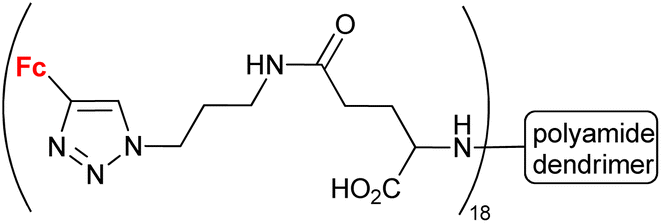
|
5.4 (HeLa) | 54 | |
| 4 cell lines | ||||
| Entry/synthesisb structure and number of compounds examined | Activity (bacterial or fungal strain) | Ref. | ||
|---|---|---|---|---|
| a Representative values of MIC for each class of compounds if given by the authors. b See Scheme 1 for general click-chemistry synthetic approaches. Fc = ferrocenyl. | ||||
| 2.1/2a |
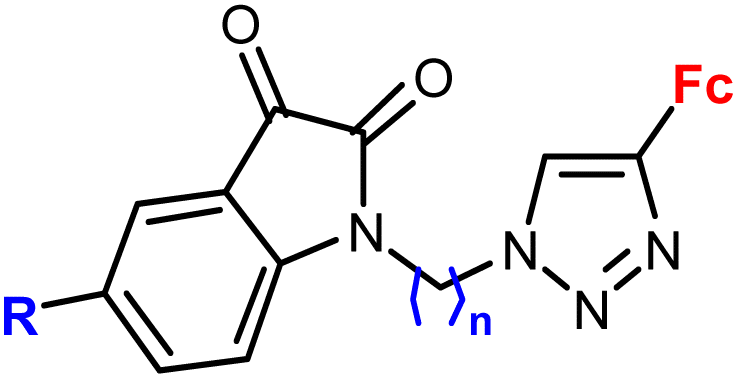
|
R = H, F, Me, Cl | MIC = 105–211 μM (M. tuberculosis mc27000) | 55 |
| n = 2 or 3 (8 compounds) | R = Cl, n = 3 | |||
| 2.2/2a |
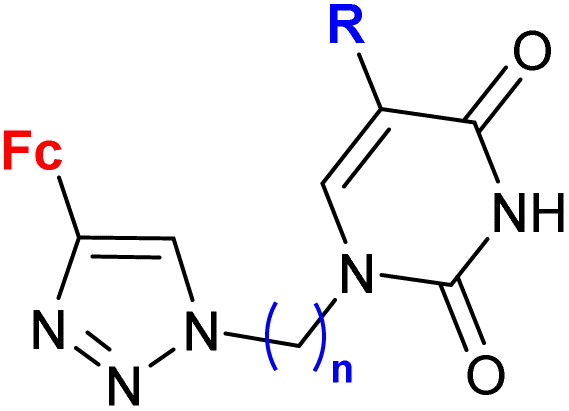
|
R = H, Cl, Br | MIC = 26.67 ± 1.67 μM (M. tuberculosis mc26230) | 56 |
| n = 2–6, 8 (18 compounds) | R = Br, n = 3 or 6 | |||
| 2.3/2b |
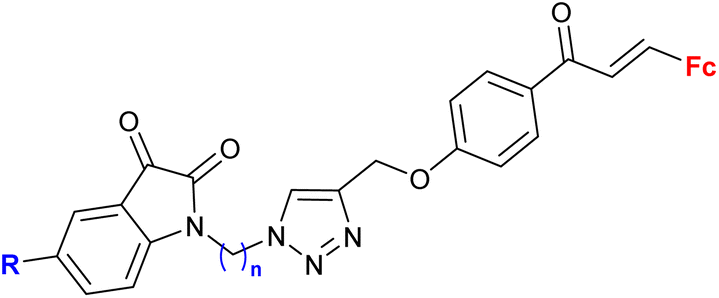
|
MIC > 155 μM (M. tuberculosis mc27000) | 55 | |
| R = H, F, Me, Cl, n = 2 or 3 (8 compounds) | R = Cl, n = 3 | |||
| 2.4/2b |
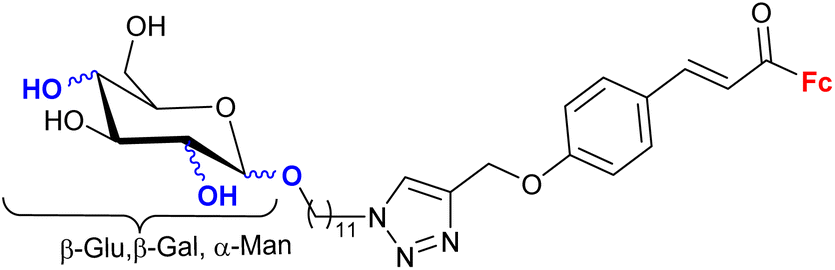
|
Active in vitro as ampicillin against E. coli or S. aureus | 57 | |
| 2 bacterial strains | ||||
| 2 fungal strains | ||||
| 2.5/2b |
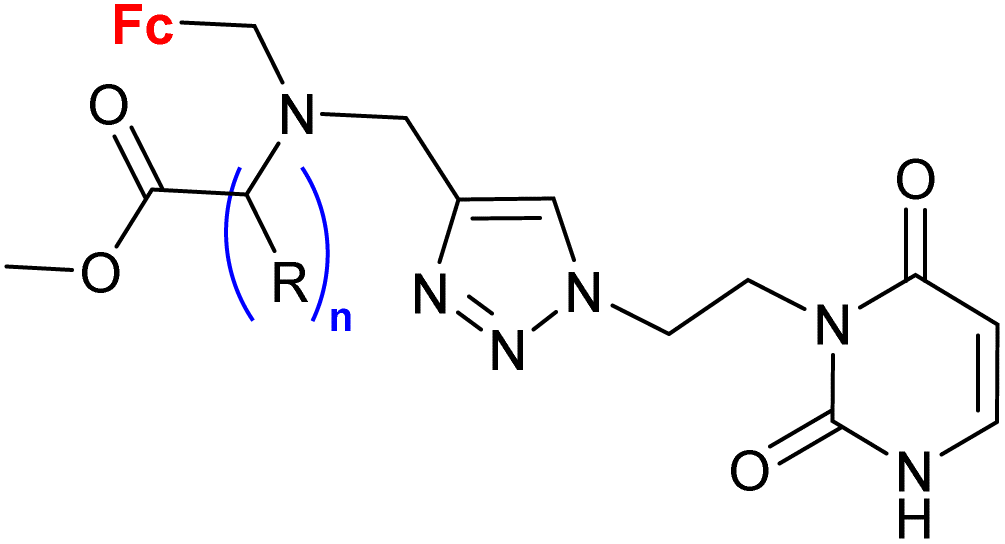
|
15 mm growth inhibition zone (C. guilliermondii IBA 155) | 58 | |
| R = H or (S)-Me; n = 1–3 (4 compounds) | R = (S)-Me, n = 1 | |||
| 15 bacterial strains | ||||
| 6 yeast strains | ||||
| 2.6/2b |
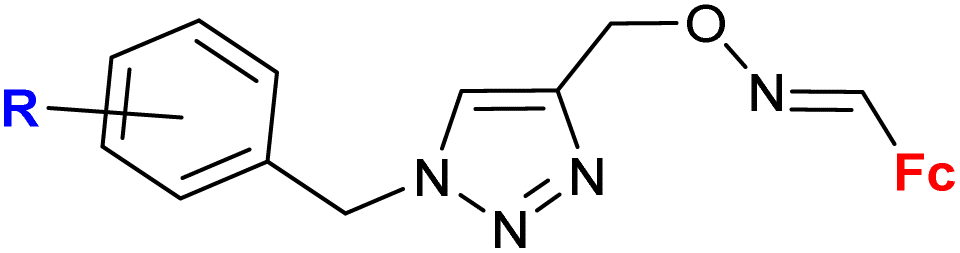
|
MIC = 7.81 μg mL−1 (S. aureus) | 59 | |
| R = H, 4-F, 4-Cl, 4-Br, 4-CF3, 4-OMe, 3-CF3, 3-Cl (8 compounds) | R = 4-F | |||
| 4 bacterial strains | ||||
| 1 fungal strain | ||||
| 2.7/2b |
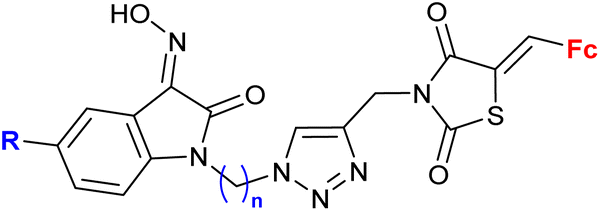
|
MIC = 4 μg mL−1 (e.g. B. subtilis, M. smegmatis, E. coli, or C. albicans) | 60 | |
| R = Me, OMe, Cl, F, Br; n = 3 or 4 (10 compounds) | n = 3: R = Me or OMe | |||
| n = 4: R = Me or Cl, 8 bacterial strains | ||||
| 2 fungal strains | ||||
| 2.8/2b |
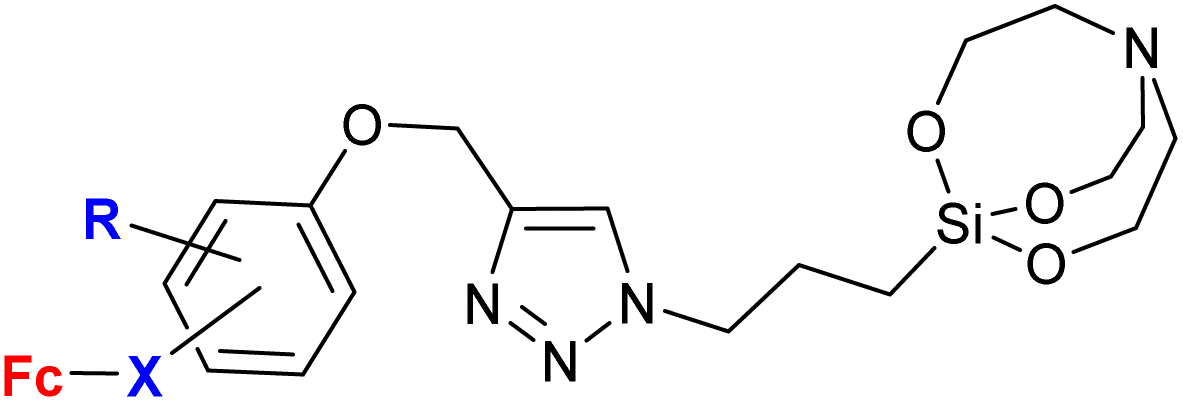
|
31.25 μM (C. albicans) | 61 | |
R = H or OMe; X = –C(O)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– or –CHC(O)– (6 compounds) CH– or –CHC(O)– (6 compounds) |
R = OMe-4, X = (–CHC(O)−)-3 |
|||
| 7 fungal strains | ||||
| 6 bacterial strains | ||||
| Entry/synthesisb structure and number of compounds examined | IC50 (μM) (protozoa strain) | Ref. | ||
|---|---|---|---|---|
| a Representative values of IC50 (μM) for each class of compounds. b See Scheme 1 for general click-chemistry synthetic approaches. Fc = ferrocenyl. | ||||
| 3.1/1b |
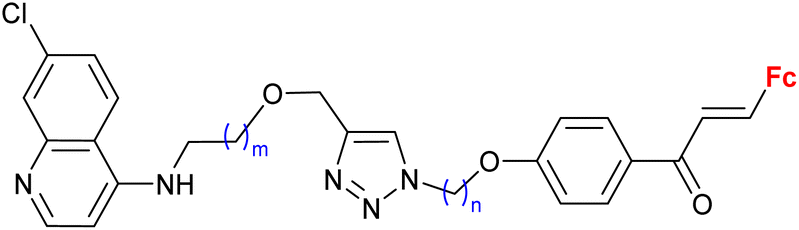
|
0.37 ± 0.03 μM (P. falciparum W2) m = 2, n = 5 | 41 | |
| m = 1 or 2; n = 2–6, 8 (12 compounds) | ||||
| 3.2/1b |
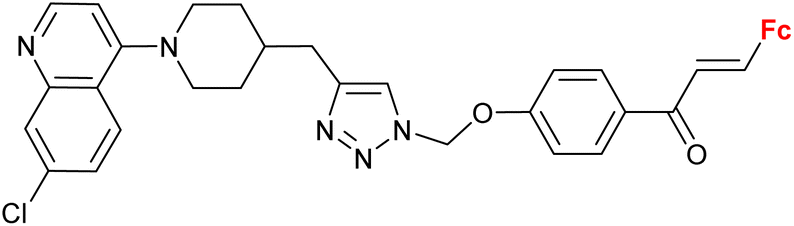
|
2.55 ± 0.25 (P. falciparum W2) n = 2 | 41 | |
| n = 2–6, 8 (6 compounds) | ||||
| 3.3/1b |
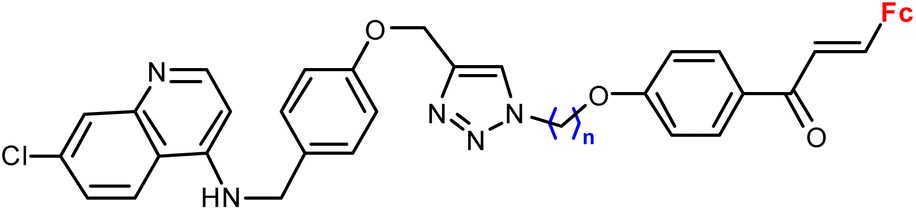
|
1.16 ± 0.04 (P. falciparum W2) n = 8 | 41 | |
| n = 2–6, 8 (6 compounds) | ||||
| 3.4/2a |
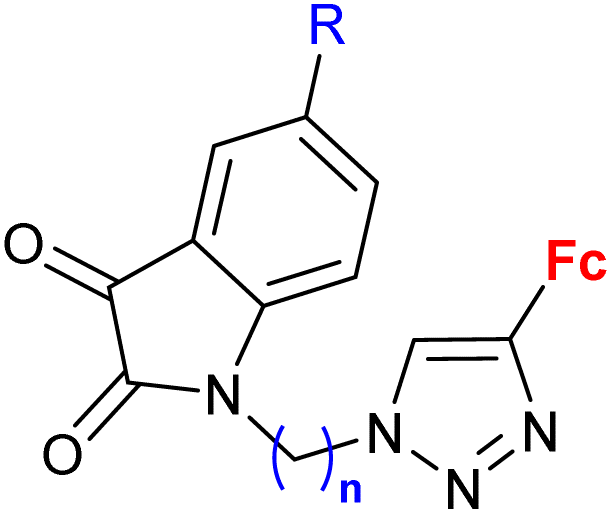
|
R = H, F, Cl, Me n = 2, 3 (7 compounds) | 3.76 (P. falciparum 3D7) R = F, n = 3 | 66 |
| 2 protozoa strains | ||||
| 3.5/2b |
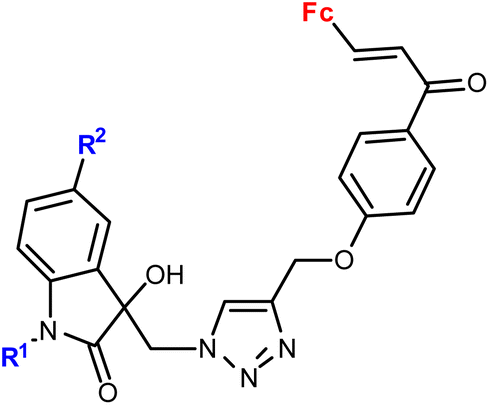
|
R1 = Me, Bn | 28.9 (T. vaginalis) R1 = Bn, R2 = Me | 67 |
| R2 = H, Cl, Br, Me (8 compounds) | ||||
| 3.6/2b |
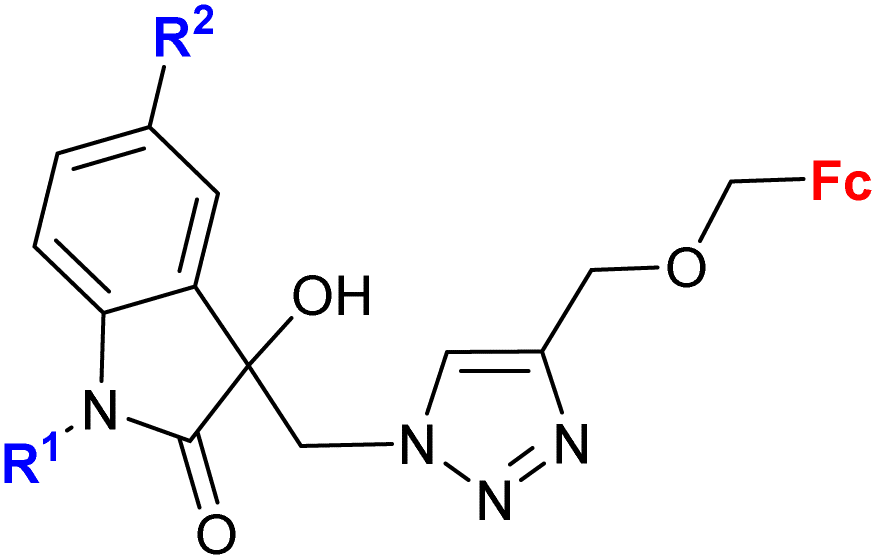
|
R1 = Me, Bn | 27.0 (T. vaginalis) R1 = Bn, R2 = Cl | 67 |
| R2 = H, Cl, Br, Me (8 compounds) | ||||
| 3.7/2b |
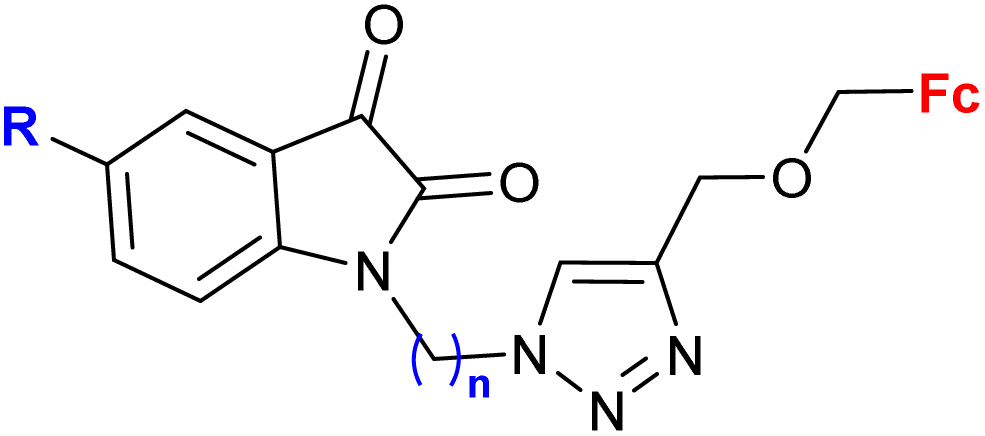
|
R = H, F, Cl, Me n = 2–6, 8 (24 compounds) | 2.26 (T. vaginalis) R = Me, n = 5 | 68 |
| 3.8/2b |
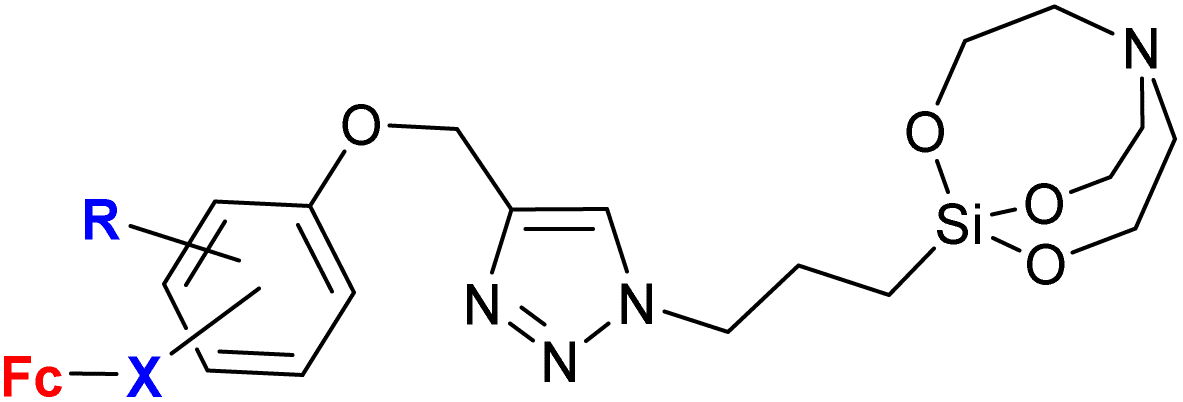
|
0.57 (G. lamblia) R = H, X = –CHC(O)– |
61 | |
| R = H or OMe; X = –C(O)CH– or –CHC(O)– (6 compounds) |
ortho isomer | |||
| 2 protozoa strains | ||||
| Entry/synthesisb structure and number of compounds examined | Effect/object of study | Ref. | ||
|---|---|---|---|---|
| a Representative examples of activity. b See Scheme 1 for general click-chemistry synthetic approaches. c See the text for details on the mechanism of action. Fc = ferrocenyl. | ||||
| 4.1/1a |
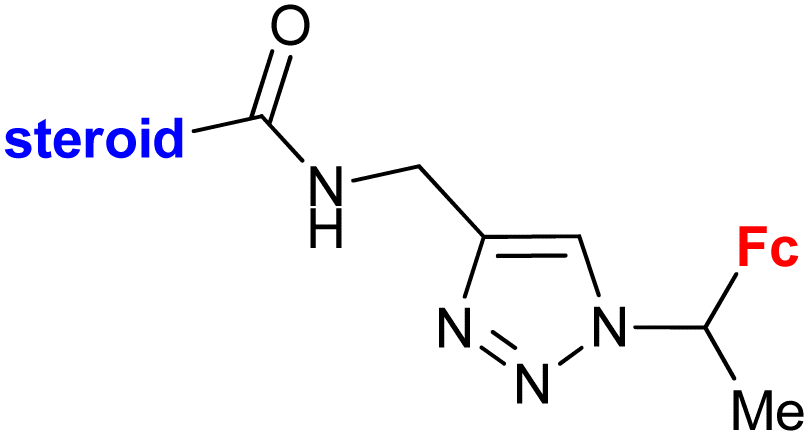
|
Inhibitory activity towards enzymes of the estrogen biosynthesis: the cytochrome P450-dependent aromatase, steroid sulfatase (STS), 17β-hydroxysteroid dehydrogenases type 1 (17β-HSD1) relative conversion = 65 ± 13% (17β-HSD1) steroid = 4-aza-5α-androst-16-en-3-one-17-yl | 69 | |
| Steroid = 4-aza-5α-androst-16-en-3-one-17-yl or 3-methoxy-estra-1,3,5(10),16-tetraene-17-yl | ||||
| 4.2/1b |
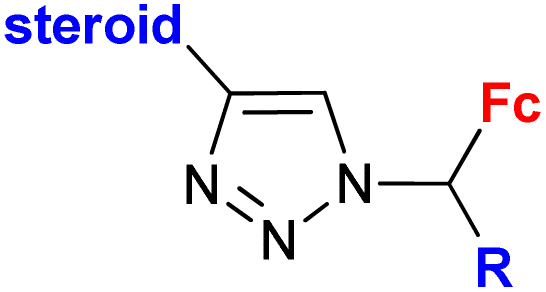
|
Inhibitory activity towards enzymes of the estrogen biosynthesis: the cytochrome P450-dependent aromatase, steroid sulfatase (STS), 17β-hydroxysteroid dehydrogenases type 1 (17β-HSD1) IC50 0.0350 ± 0.0058 μM (STS) steroid = 3,17β-dihydroxy-estra-1,3,5(10)-triene-17α-yl R = Me | 69 | |
| Steroid = estradiol-17α-yl, 3,17β-dihydroxy-estra-1,3,5(10)-triene-17α-yl, 17β-hydroxy-18a-homo-19-nor-androst-4-ene-3-one-17α-yl R = H or Me (3 compounds) | ||||
| 4.3/2a |
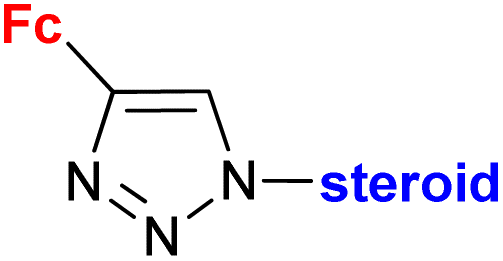
|
Inhibitory activity towards enzymes of the estrogen biosynthesis: the cytochrome P450-dependent aromatase, steroid sulfatase (STS), 17β-hydroxysteroid dehydrogenases type 1 (17β-HSD1) IC50 4.63 ± 1.51 μM (STS) steroid = 3α-hydroxy-5α-androstane-17-one-2β-yl | 69 | |
| Steroid = 3α-hydroxy-5α-androstane-17-one-2β-yl or 17α-hydroxy5α-androstane-16β-yl | ||||
| 4.4/2ac |
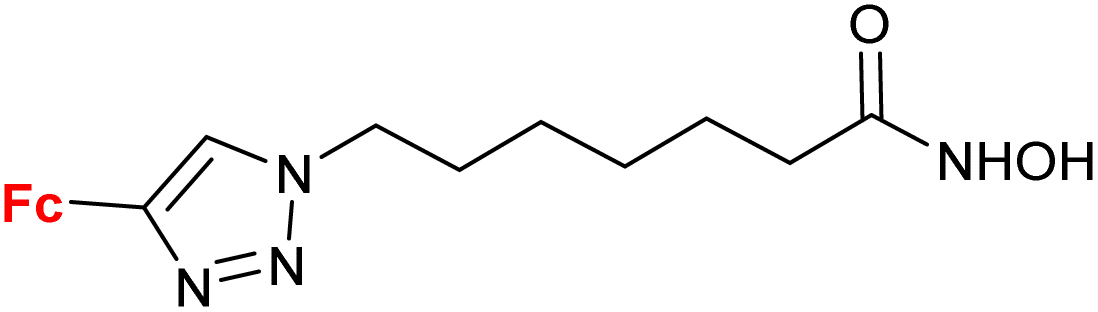
|
Inhibition of histone deacetylase (HDAC1, HDAC2, HDAC3 or HDAC6) IC50 0.0026 ± (0.0002) μM (HDAC6) | 70 | |
| 4.5/2ac |
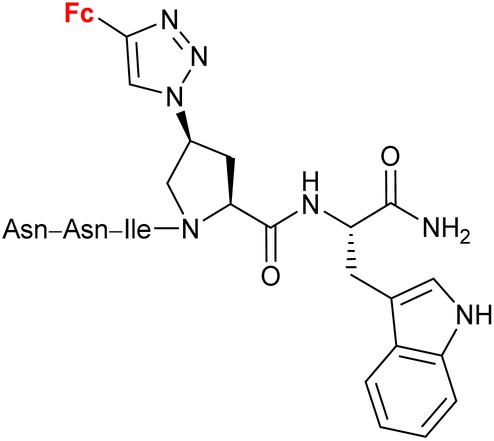
|
Inhibitory activity towards protein gp120 | 71–74 | |
| (11 compounds, lead structure shown, for details see Scheme 5) | ||||
| 4.6/2a |
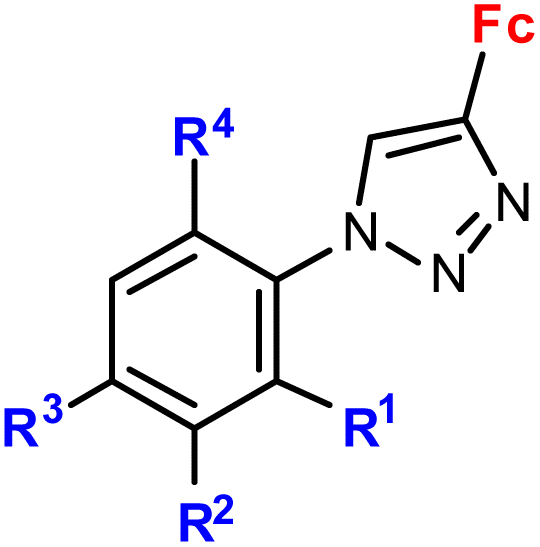
|
R1 = H, NO2 | Low toxicity, non-inflammatory effect (BV-2 cells, RMC cells) and antioxidant activity (RMC cells) | 75 and 76 |
| R2 = H, Cl | ||||
| R3 = H, F, Br | ||||
| R4 = H or –C6H4–NO2-4 (5 compounds) | ||||
| 4.7/2a or 2bc |
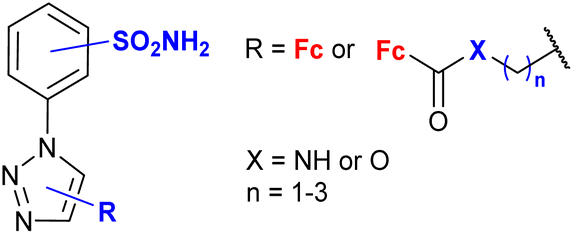
|
inhibition of carbonic anhydrase Ki 3.2 nM ± 5–10% (CA II) R = 5-Fc, 3-SO2NH2 | 77 | |
| 3-SO2NH2 or 4-SO2NH2 (13 compounds) | ||||
| 4.8/2c |
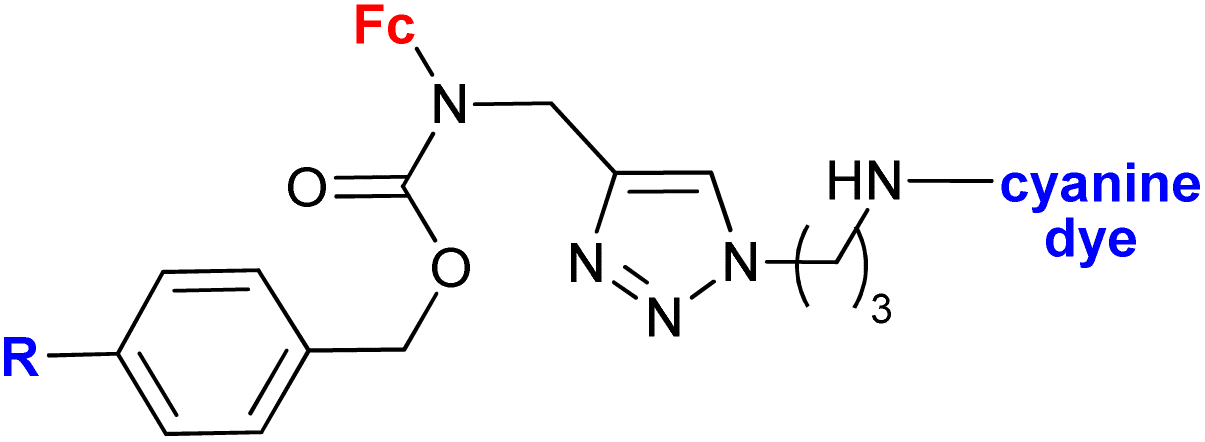
|
In vitro accumulation in A2780 cells, in vivo accumulation in tumours (AR42J or PC-3 tumour-bearing mice) | 78 | |
| 4.9/2c |
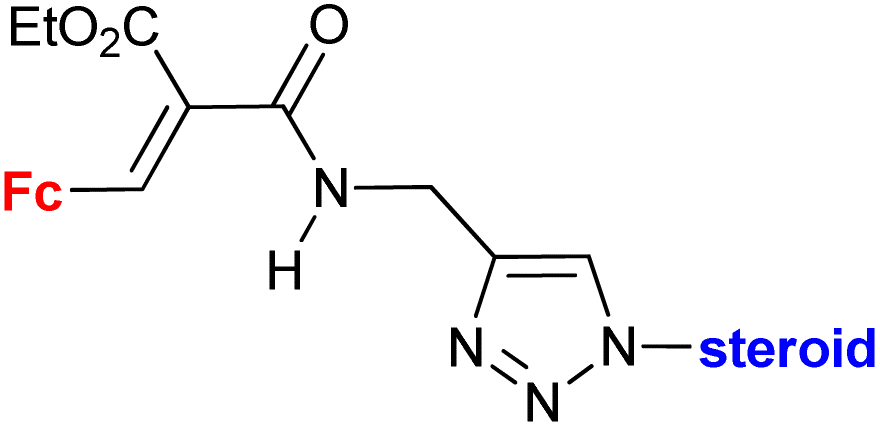
|
Inhibitory activity towards enzymes of the estrogen biosynthesis: the cytochrome P450-dependent aromatase, steroid sulfatase (STS), 17β-hydroxysteroid dehydrogenases type 1 (17β-HSD1) IC50 2.39 ± 1.10 μM (STS) steroid = 17α-hydroxy-5α-androstane-16β-yl | 69 | |
| Steroid = 3α-hydroxy-5α-androstan-17-one-2β-yl, 3α,5α-dihydroxyandrostan-17-one-6β-yl, 17α-hydroxy-5α-androstane-16β-yl | ||||
2 Conjugates with anticancer activity
By far the largest number of compounds was tested for anticancer activity (Table 1). In a majority of reports covered in this section, the following structural features were noted: (a) a non-hydrolysing connection (i.e. other than ester or amide) between ferrocene and the organic part of the molecule (for exceptions, see entries 1.15, 1.16, 1.26 and 1.29), (b) mono-substitution of ferrocene (for exceptions, see entries 1.11 and 1.18), or (c) the 1,4-disubstitution pattern of the 1H-1,2,3-triazole ring (for a few conjugates with the 1,5-disubstitution pattern, see entry 1.18; for an unusual conjugate with a 1,4,5-trisubstituted-1H-1,2,3-triazole ring, see entry 1.20). Post click-synthesized conjugates were surprisingly rare (entries 1.20 and 1.30). Interestingly, studies concerning conjugates bearing structural motifs of natural origin were very ubiquitous (entries 1.1 and 1.20 – polyhydroxy phenols; entries 1.11, 1.12, 1.13 and 1.14 – nucleobases; entry 1.18 – alkaloids; or entries 1.16, 1.26, 1.27 and 1.28 – carbohydrates). It is worth pointing out entry 1.11 with an ansa[4]-ferrocene (ferrocenophane) conjugate that appeared more active than its unbridged counterpart (entry 1.12).The reported values of IC50 were in the micromolar range with the highest anticancer activity determined for the aminoquinoline conjugate (IC50 0.37 μM, entry 1.19). We would like to emphasize that the activities of several conjugates collected in Table 1 are of the same order of magnitude as those of ferrocifen type derivatives.11–14
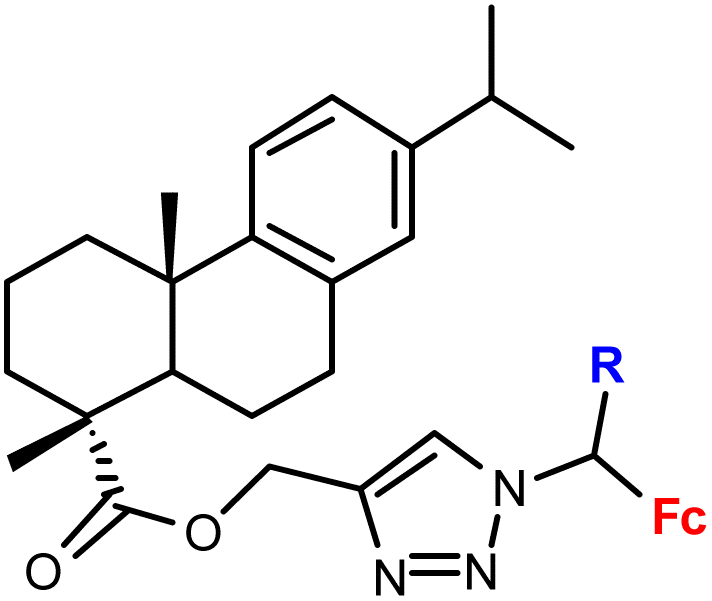
In a majority of studies, the mechanism of the observed biological effect was not investigated, that is understanding of the role of the ferrocene moiety is generally lacking. The notable exceptions include the report by Maračić et al.32 on quinoline–ferrocene conjugates (entries 1.2, 1.6 and 1.7) where low antioxidant activity of the most active compounds was determined (entries 1.2 and 1.7). Furthermore, the cell cycle and intracellular generation of ROS (reactive oxygen species) were studied revealing accumulation in the G0/G1 phase for the two compounds. However, only one compound (entry 1.2) was found to significantly increase the ROS generation in Raji cells.32 The physicochemical properties, such as solubility and metabolic stability in liver microsomes, of purine- and purine isostere-tagged ferrocenes were thoroughly investigated (entries 1.3, 1.4, 1.5, 1.8, 1.9, 1.21).33 However, conclusions on the mode of action of these cytotoxic conjugates were not given.
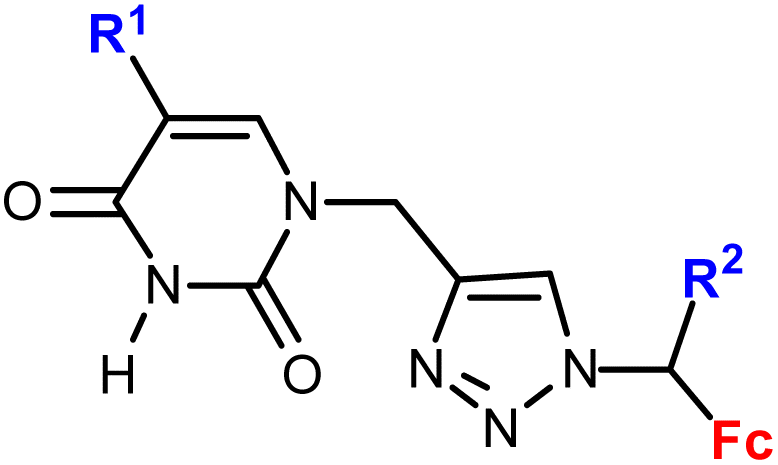
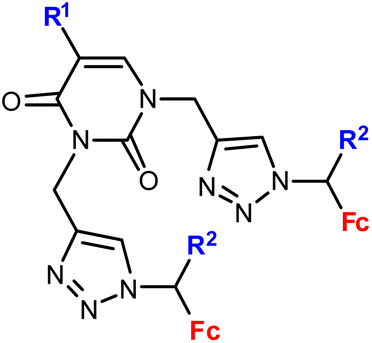
Djaković et al. studied mono- and bis-ferrocene uracil conjugates (entries 1.13 and 1.14, respectively).35 Selected bis-ferrocene derivatives (entry 1.14) were further evaluated for intracellular ROS accumulation, mitochondrial membrane dysfunction activity, and apoptosis induction. The results suggested that the activity might be due to the disruption of the mitochondrial membrane potential and the ability to induce apoptosis in cancer cells.
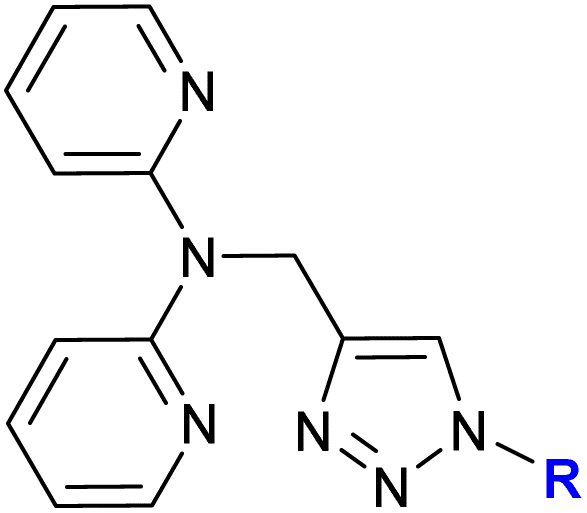
Detailed studies on the mechanism of cytotoxicity of ferrocenyl dipyridylamines, Zn(II) or Cu(II) complexes (entry 1.17), revealed an increase in the number of cells in the S and G2/M phase of the cell cycle. The authors concluded that the Cu(II) complex did not interfere with DNA synthesis but induced replication stress in the cancer cells, probably as a result of complex–protein interactions.38
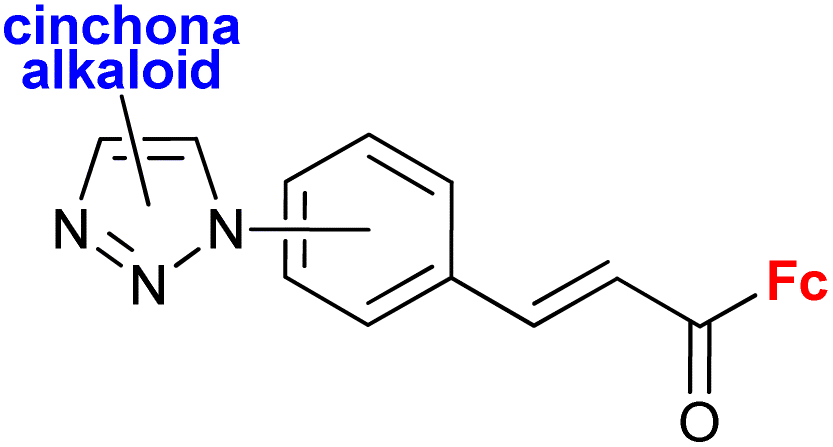
A series of 1H-1,2,3-triazole-tethered hybrids 1–7 bearing a cinchona alkaloid-ferrocene couple was designed and prepared (entry 1.18; Scheme 2).39,40 Interestingly, besides the 1,4-disubstitution pattern of the 1H-1,2,3-triazole scaffold, 1,5-isomers (compounds 1) were also employed. It is worth noting that the 1,5-isomer (compound 1d, IC50 = 9.9 ± 0.5 μM) was less active than the corresponding 1,4-isomer (compound 2d, IC50 = 0.7 ± 0.1 μM). Taking into account the connectivity between ferrocene and 1H-1,2,3-triazole, three groups of derivatives could be classified: (a) compounds 1–3 with an ortho- or para-substituted chalcone linker, (b) derivatives 4–6 with a methylene linker, and (c) derivative 7 without any linker. Examination of all the derivatives in terms of their in vitro cytostatic effect toward hepatocellular carcinoma (HepG2) cells or colon cancer (HT-29) cells revealed high inhibitory activity of the chalcone-containing compounds 1–3 with IC50 values ranging from 0.7 ± 0.1 μM (compound 1d) to 14.3 ± 14.5 μM (compound 2b) in the HepG2 cell line, or 1.5 ± 0.2 μM (compound 1d) to 6.6 ± 7.7 μM (compound 3d) in the HT-29 cell line.39 It is worth mentioning that the observed inhibitory activity of compounds 1–3 was much higher than that of their parent ferrocene-chalcone azides. This observation might suggest a considerable contribution of the cinchona alkaloid moiety to the inhibitory effect of hybrids 1–3. Next, further examination of compounds 2a–d addressed their activity against multidrug resistant (MDR) cancer cell lines of non-small cell lung carcinoma NCI-H460/R, colorectal carcinoma DLD1-TxR or glioblastoma U87-TxR.40 Compounds 2c and 2d were the most interesting among the tested derivatives because they showed significantly higher cytotoxicity towards MDR glioblastoma U87-TxR cells (2c, IC50 3.13 ± 0.06 μM; 2d, IC50 2.30 ± 0.02 μM) or colorectal carcinoma DLD1-TxR cells (2d, IC50 4.43 ± 0.06 μM) than towards the corresponding sensitive counterparts of the evaluated cancer cells. The anticancer activity of compound 2d was postulated to be a consequence of its pro-oxidative effect, reflected by the extreme increase in the hydrogen peroxide and peroxynitrite anion concentrations in colorectal carcinoma DLD1-TxR cells or glioblastoma U87-TxR cells.
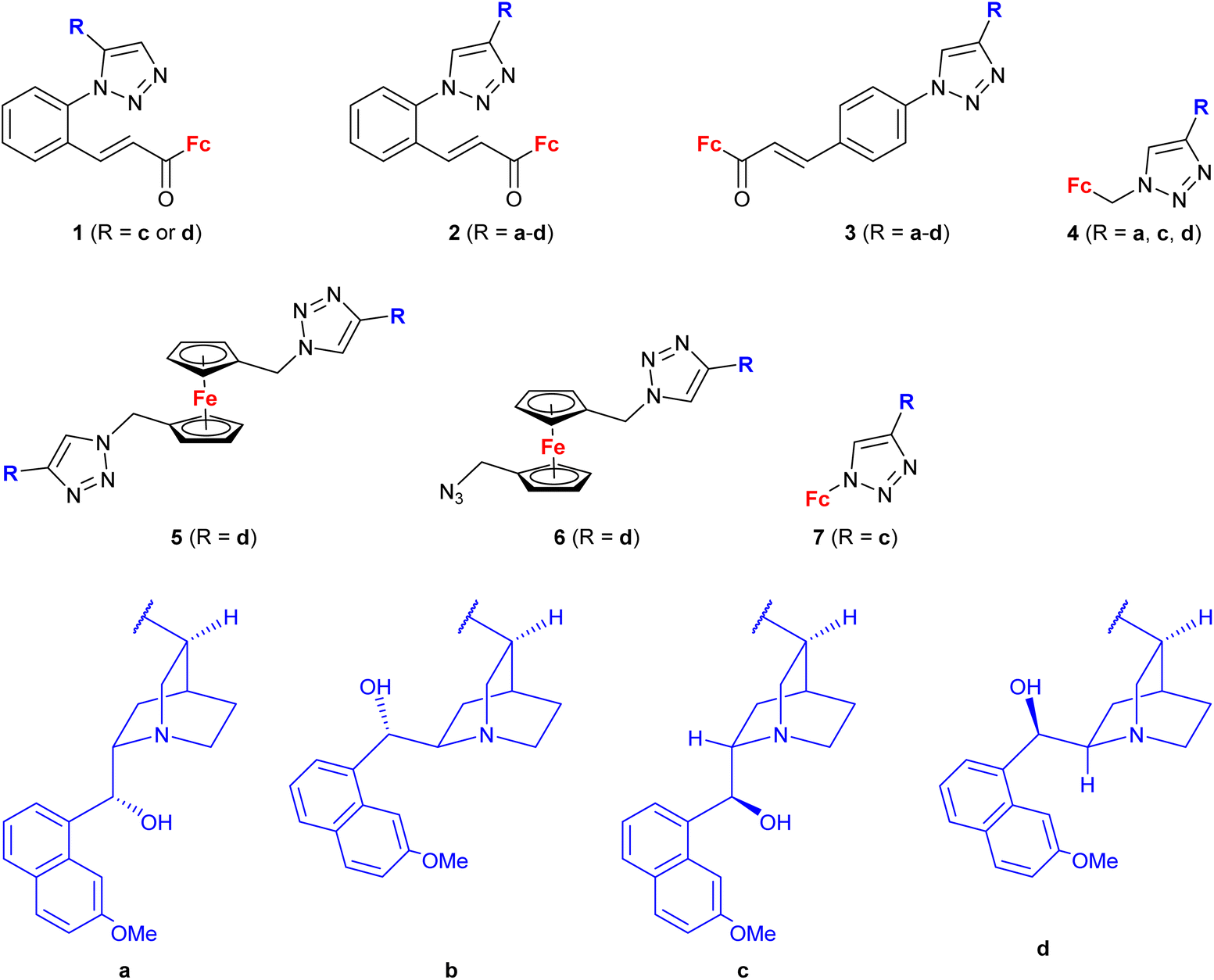 | ||
| Scheme 2 1H-1,2,3-Triazole-tethered cinchona alkaloid-ferrocene hybrids (Table 1, entry 1.18).39,40 Fc = ferrocenyl. | ||
Mokhir and co-workers developed a group of N-alkylaminoferrocene-based prodrugs 8 (entry 1.29; Scheme 3) targeting the lysosomes of cancer cells (compound 8a49) or mitochondria of cancer cells (compounds 8b–g50,51). Compound 8a was toxic towards BL-2 cells (IC50 27 ± 3 μM). Coumarin-induced ROS quenching was postulated to play a role in the moderate cytotoxicity of this compound. Accumulation of compound 8a in cancer cell-specific lysosomes and its activation in these organelles was detected.49 Among compounds 8b–e, bearing a clinically approved drug carboplatin, conjugate 8b was found to accumulate in the mitochondria of A2780 cells more efficiently than the parent carboplatin.50 This was reflected by the substantially higher anticancer effect of conjugate 8b towards A2780 cells than that of carboplatin at all incubation times (e.g. IC50 at 96 h: 20 ± 1 vs >82 ± 7 μM, respectively). The synergistic effect of the platinum and the ferrocene components was postulated for compound 8b owing to the fact that its activity was significantly higher than that of an equimolar mixture of reference compound 8c and carboplatin. Compound 8f exhibited higher efficiency of cellular uptake (BL-2 cell line) than reference compound 8c. Moreover, it exerted overall stronger effects on the cell viability in all studied cancer cell lines (BL-2, A2780, DU-145) than the reference compound 8c, compound 8g, or ferrocene, but it was less potent than an unspecific positive control (i.e. a mixture of FeCl3 and 8-hydroxyquinoline).51 Analogous to the reference compound 8c and butyltriphenylphosphonium iodide, compound 8f was found to kill the cancer cells mostly via necrosis and apoptosis with the former cell death phenotype being more prominent. All tested compounds were more active than ferrocene used as a control.
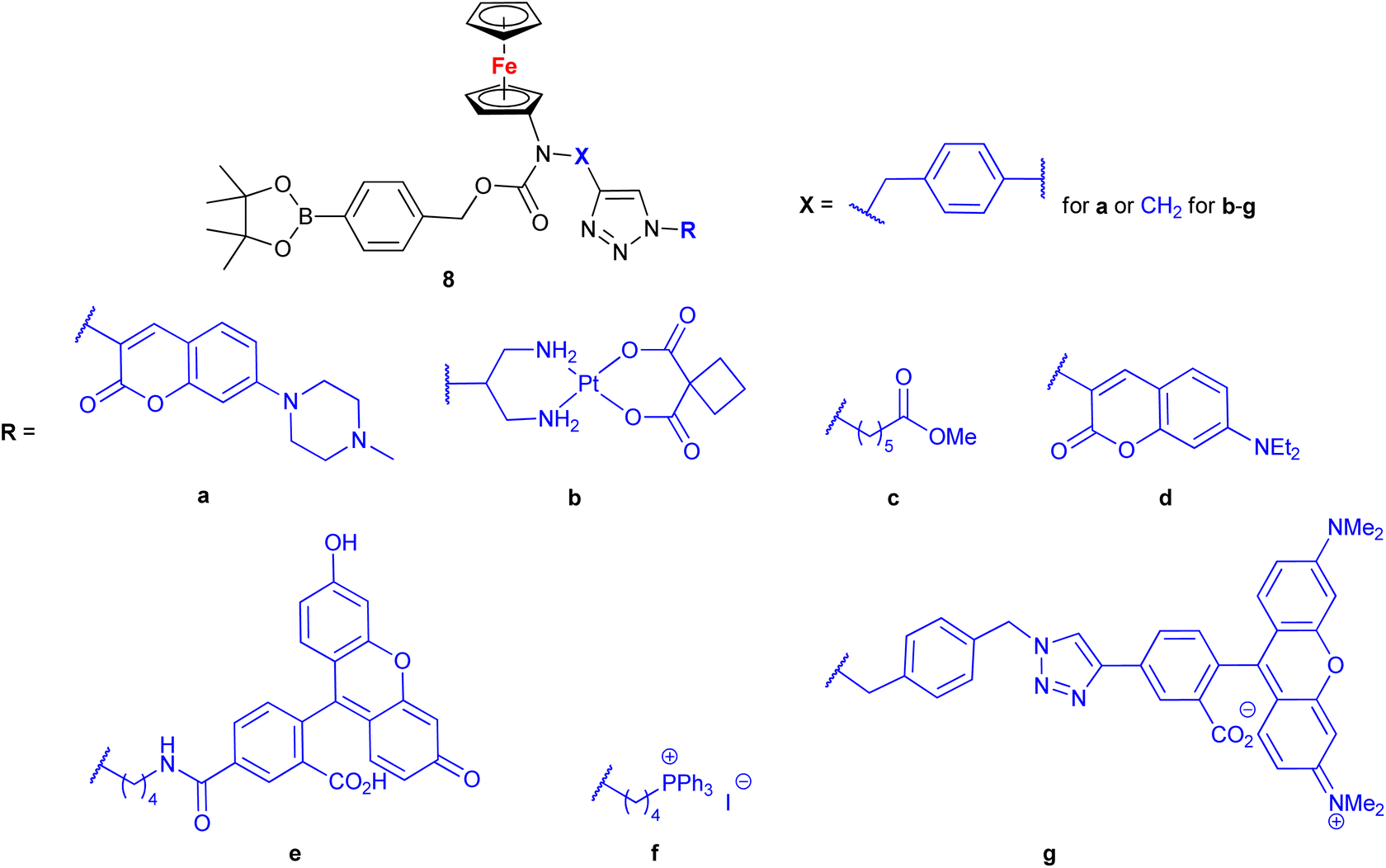 | ||
| Scheme 3
N-Alkylaminoferrocene-based prodrugs targeting the lysosomes of cancer cells (compound 8a49) or mitochondria of cancer cells (compounds 8b–g50,51). Table 1, entry 1.29. | ||
3 Conjugates with antibacterial and/or antifungal activity
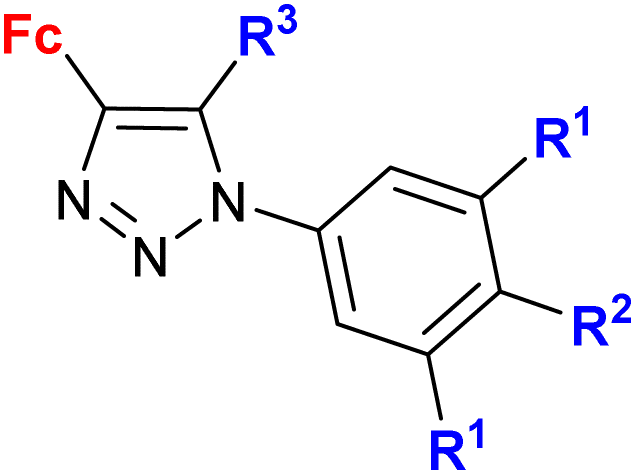
Ferrocene-1H-1,2,3-triazole conjugates have been tested for antimicrobial activity in a limited number of studies (Table 2). In view of their structural features, molecules combining ferrocene and a scaffold with well-established antitubercular activity, such as isatin (entries 2.1 and 2.3) or isatin oxime (entry 2.7), are worth noting.The mechanism of action of compounds summarized in Table 2 was not studied. Some conjugates were reported to show higher (entry 2.6) or comparable activity (entries 2.6 and 2.7) as the reference drugs (streptomycin sulphate for antibacterial activity or fluconazole for antifungal activity). The contribution of the ferrocene core to the antimicrobial activity of the conjugates was revealed by comparative assays employing a structurally resembling non-ferrocene counterpart (entry 2.7).60 Moreover, the authors concluded that suitable substitution of the isatin oxime ring plays an important role in the observed activity. The same research group reported also several 3-ferrocenylidene-2-oxoindole conjugates with moderate antibacterial activity.62 Weak, ultrasound-assisted antibacterial activity (E. coli) was also reported for a ferrocene-triazole-linked porphyrin.63
With the exception of a dendrimer (Table 1, entry 1.31), all examples presented here thus far belonged to the category of small molecules. A different approach was adopted by Lakouraj et al. who obtained a ferrocene-embedded polymer 11 containing azine and xanthone scaffolds as potential pharmacophores (Scheme 4).64 A click-chemistry approach was used to assemble the target polymer 11 from monomers 9 and 10. Polymer 11 showed antibacterial activity towards E. coli, P. aeruginosa, K. pneumoniae, B. subtilis, and S. aureus with a MIC concentration of 125 μg mL−1 and MBC concentrations ranging from 250 to 500 μg mL−1. Additionally, at 100 μg mL−1 polymer 11 showed a 20–40% decrease in the viability of HeLa cells, MCF-7 cells, or Saos cells. Based on its unique antibacterial characteristics, potential industrial applications of polymer 11 were suggested, for instance, as a component of chemical coatings in medical devices used in hospitals.
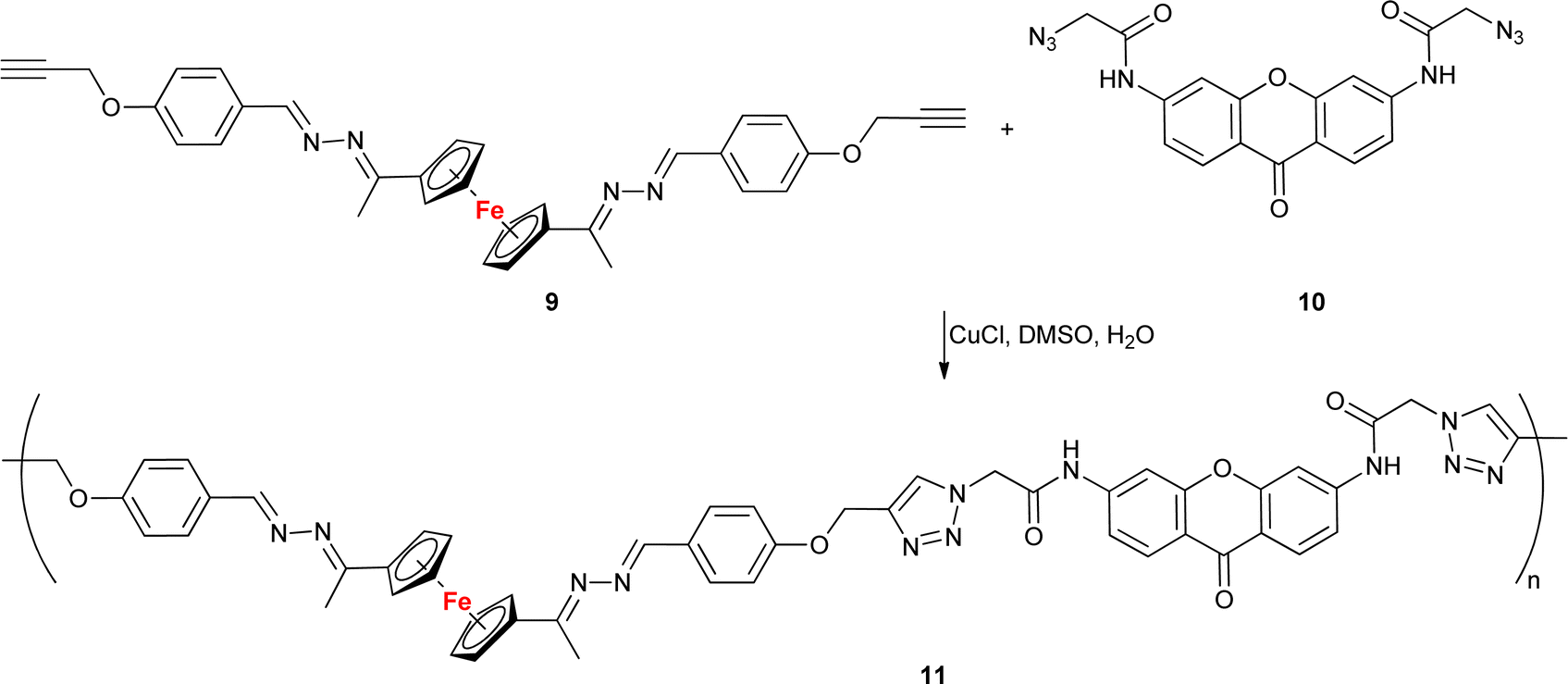 | ||
| Scheme 4 Click-assembled polymer 11 composed of ferrocene, azine and xanthone scaffolds as potential pharmacophores.64. | ||
4 Conjugates with antiprotozoa activity
Ferroquine completed the second phase of clinical trials with patients suffering from uncomplicated P. falciparum malaria.65 Therefore, constant interest in potential ferrocene-antimalarial drugs bearing the 7-chloro-4-quinolinyl moiety has been observed (Table 3, entries 3.1, 3.2 and 3.3). Studies devoted to antiprotozoa activity (including antimalarial activity) of the isatin- or isatin derivative-bearing conjugates could be considered as a promising field (entries 3.4, 3.5, 3.6 and 3.7).The study of a library of conjugates combining the 7-chloro-4-quinolinyl moiety and a ferrocene chalcone (in its ferrocene carboxaldehyde-originated variant, entries 3.1, 3.2, and 3.3) allowed the identification of the carbon chain as the most advantageous linker between these two pharmacophores (entry 3.1 vs. entries 3.2 and 3.3).41 Although the most promising conjugates did not reach antiprotozoa activity of chloroquine or ferroquine, some of them showed IC50 < 1 μM.
Evaluating several isatin-bearing conjugates as potential agents against T. vaginalis (entries 3.5, 3.6 and 3.7), Singh et al. found the isatin substitution pattern as the key factor determining the activity of the assayed conjugates (entry 3.7 vs. entries 3.5 and 3.6).67 The conjugates were not active against normal human flora consisting of non-pathogenic strains: Lactobacillus reuteri (ATCC 23272), Lactobacillus acidophilus (ATCC 43560), and Lactobacillus rhamnosus (ATCC 53103). However, none of them was as active as the reference metronidazole.
A significantly different approach by Singh et al. resulted in ferrocene-chalcones linked through 1H-1,2,3-triazole to organosilatranes. These structurally unique compounds possessed strong activity against G. lamblia (entry 3.8). The same compounds were also tested against some bacterial and fungal strains, but showed only weak activity (see Table 2, entry 2.8).
5 Conjugates with other types of activity
This section includes conjugates evaluated as inhibitors of cancer-associated enzymes (Table 4 entries 4.3 and 4.4) or HIV-1-associated enzymes (entry 4.5), and neuroprotective or anti-inflammatory agents (entry 4.6). Among these structurally diverse conjugates, a broad panel of benzenesulfonamides (entry 4.7) or oligopeptides (entry 4.5) are worth noting.Salmon et al. showed the significant contribution of the ferrocenyl group to the activity of the benzenesulfonamide-bearing conjugates (Table 4, entry 4.7) towards selected carbonic anhydrases when compared to the corresponding ferrocene lacking counterparts, that is phenyl-containing compounds.77 Moreover, several ruthenocene analogues were also studied to reveal higher or comparable activity of ruthenocene vs. ferrocene systems. The 1,5-disubstitution pattern of the 1H-1,2,3-triazole ring was identified as one of the factors determining high enzyme inhibitory activity. The benzene substitution pattern of the conjugates affected their enzyme inhibitory activity. However, water solubility and in vitro metabolic stability did not have an effect on their lipophilicity or in vitro permeability.
Peptide mimetic 12 (entry 4.5, Scheme 5) with nanomolar affinity for the HIV-1 surface protein gp120 and strong potency for inhibiting host cell infection was designed by the research group of Chaiken.79 This compound was then used to identify the minimum-length active dual-antagonist sequence 1380 and a family of inhibitors of HIV-1 entry into host cells, including short-peptides 14–16 and cyclopeptides 17 and 18.71–74 The docking examination of the identified inhibitors suggested the crucial role of the ferrocene moiety in their binding to protein gp120.72 The examined inhibitor–protein complexes were stable when their ferrocene moiety was buried in the protein site 2 hydrophobic cavity by several hydrophobic interactions with the protein residues. Interestingly, the 1,5-disubstituted 1H-1,2,3-triazole analogue of peptide p1 was inactive. In the light of in silico recognized instability of the corresponding peptide–protein complex, these experimental findings confirmed the importance of the 1,4-disubstituted 1,2,3-triazole pattern to retain the ferrocene moiety inside the protein site 2 hydrophobic cavity. Cyclopeptides 17 and 18 greatly resisted in vitro proteolysis as demonstrated by their stability against trypsin and chymotrypsin.74
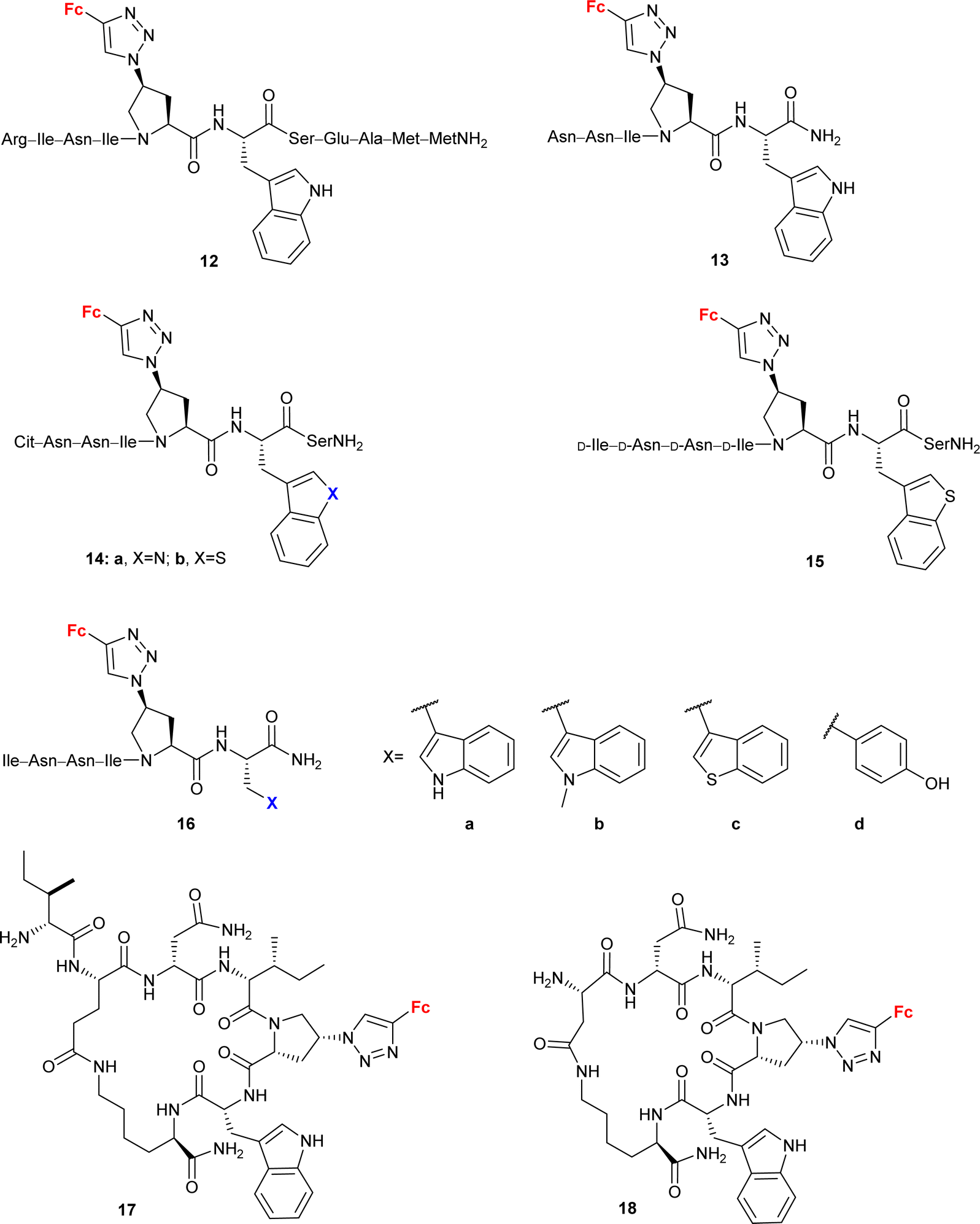 | ||
| Scheme 5 Peptide mimetics with nanomolar affinity for the HIV-1 surface protein gp120 and strong potency for inhibiting host cell infection (Table 4, entry 4.5).71–74 Fc = ferrocenyl. | ||
6 Challenges and outlook
The preparative versatility of the regioselective 1H-1,2,3-triazole-forming click-reaction can be considered as one of the factors stimulating the recent advances in the field of ferrocene-originating drug development. The undoubtedly broad synthetic scope of this approach has been employed to obtain a variety of new conjugates resulting from the attachment of ferrocene to a medically important molecule (e.g. isatin, a nucleobase, a carbohydrate, or an alkaloid framework), or by combining ferrocene (or a ferrocene-based privileged structure, such as ferrocene chalcones) with a structural unit(s) with well-documented pharmacophore activity (e.g. sulfonamide group or polyhydroxybenzene moiety). The latter strategy afforded a variety of structurally diverse de novo conjugates. The majority of new molecular entities discussed in this review have been evaluated for their anticancer potency. In general, the data summarized in this review suggest the advisability of comparative studies on structure–activity optimizations in terms of (a) the ferrocene-1H-1,2,3-triazole distance, (b) the construction of a ferrocene-1H-1,2,3-triazole linker (when present), and (c) the 1H-1,2,3-triazole ring orientation/substitution pattern.The 1,5-disubstitution of the 1H-1,2,3-triazole ring could be considered as a worthwhile option since certain conjugates with this structural motif were reported to show greater biological potency than their 1,4-disubstituted counterparts. Interestingly, conjugates with more complex substitution patterns of the ferrocene scaffold than mono-substitution (including 1,1′-disubstitution or ansa-ferrocene, for instance) were infrequent. The activity of the ansa-ferrocene conjugate34 suggested that structural modifications of the ferrocene core are an attractive route to new biologically active entities.
The effect of individual structural components of the conjugates on their biological activity was studied rather occasionally. When the ferrocene effect was examined, comparative in vitro SAR studies involved ferrocene-lacking counterparts of the target conjugates or their analogues with an aromatic substitute of ferrocene.60,69,77 On the other hand, SAR studies evaluating a potential contribution of the 1H-1,2,3-triazole moiety employed the 1H-1,2,3-triazole-lacking counterpart43 of the target conjugates or their amide or carbamide analogs.46,47,77 In some cases, a comparison of the results seems rather difficult owing to the significant structural differences between the assayed conjugates (for instance, 1H-1,2,3-triazole-containing conjugates vs. chalcone-containing conjugates72).
Assays evaluating ferrocene-containing intermediates (azide34,39,58 or alkyne,59 or their precursor47,59) were also reported. Interestingly, Kocsis et al. gained a considerable increase in the cytostatic effect of the moderately active ferrocene-containing azide by linking it with cinchona alkaloid (Scheme 2, compound 1d, IC50 = 0.7 ± 0.1 μM vs ferrocene-containing azide, IC50 = 19.1 ± 1.3 μM).39 Studies on conjugates with promising activity resulting from combining inactive ferrocene-originating 1H-1,2,3-triazole precursors with natural compounds (carbohydrates or nucleobases) are worth highlighting.34,47,58,59 However, the ineffectiveness of ferrocene linking with an active conjugate precursor was also reported.37,55
Unfortunately, studies on the mode of action of these conjugates were not routine, that is, in many cases the biological results were limited to simple screening of the selected cell lines. Mechanistic investigations were pursued only for some classes of compounds displaying anticancer activity (Table 1). In most cases, the results suggested that the redox properties of the ferrocene core were important for the activity. In particular, Mokhir and co-workers identified N-alkylaminoferrocenium cations as active anticancer species.49–51 This finding recalls the results of the first successful biological studies on ferrocenes that revealed the anticancer activity of some cationic derivatives.8,9
The redox properties of the ferrocene-triazole conjugates have been examined by cyclic voltammetry in several studies.31,33,37,38,43,47,54,59,60,75,76 This technique has been employed mainly as a tool to characterize the new compounds rather than to find the relationship between the oxidation potential and biological activity. In two reports antioxidant activities were correlated with oxidation potentials.33,76 However, in other studies such correlation was explicitly not observed31 or not discussed.
Although infrequent, in silico studies of the mechanism of action of active conjugates brought added value to bioassay-based screening. Molecular docking analyses revealed a considerable contribution of ferrocene and 1H-1,2,3-triazole cores to the biological activity of the conjugates. Chaiken et. al suggested hydrophobic interactions of both ferrocene and 1H-1,2,3-triazole with Env gp120 protein residues.72 On the other hand, Khan et al. detected H-bonding interactions of 1H-1,2,3-triazole with cyclooxygenase-2 or cPLA2 protein residues.76 Furthermore, H-bonding interactions between ferrocene and cPLA2 protein residues via the ferrocene iron atom were also suggested.
Having in mind an approximate character of such analysis at the current stage of these studies, we inspected the collected conjugates in terms of their potential to be selected as a hit compound in the early drug discovery process. The assayed series of compounds discussed in our review was rather small. For this reason, our selection was based on one-digit micromolar potency as a cut-off, in contrast to the 1 μM potency commonly employed in a real hit identification process.81 In the light of the above criterion, within compounds reported to display anticancer activity, undoubtedly promising potency was exhibited by conjugates derived from quinoline (entries 1.2 and 1.19), deazapurine (entry 1.3), 4-quinolone (entry 1.7), uracil (entry 1.14), pyridine (entry 1.16), cinchona alkaloids (entry 1.18), carbohydrates (entries 1.23, 1.26, 1.27 and 1.28), phenylboronic acid (entry 1.29) and the polyamide dendrimer (entry 1.31). Interestingly, linking uracil with ansa-ferrocene (entry 1.11) or 5-iodouracil with ferrocene (entry 1.13) afforded conjugates endowed with promising potential exemplified by their ca. 10 μM potency. Fluorophenyl- (entry 2.6) and isatin oxime-bearing conjugates (entry 2.7) were noted as worth considering in the field of antibacterial drug development. On the other hand, ferrocene-conjugated derivatives of quinoline (entries 3.1–3.3), isatin (entries 3.4 and 3.7) and silatrane (entry 3.8) were found to possess antiprotozoa potency. Indisputably, our observation that quinoline- and isatin-bearing conjugates were reported to display a broad spectrum of biological activity (anticancer, antibacterial and antiprotozoa) might be an incentive to intensify research on ferrocene derivatives as potential drugs.
Taking into account that some contradictory data on the cytotoxicity of ferrocene have been reported,5,6,9,43,46,51,53 it seems reasonable that ferrocene or its simple derivatives should be used as reference compounds in biological studies to facilitate the discovery of promising structural modifications.
Abbreviations
| Asn | Asparagine |
| D-Asn | D-Asparagine |
| AR42J | Rat pancreatic cancer cells |
| Arg | Arginine |
| A2780 | Human ovarian adenocarcinoma cells |
| A549 | Lung carcinoma cell line |
| BL-2 | Burkitt lymphoma cells |
| BV-2 | Murine microglial cell line |
| Cit | Citrulline |
| CFPAC-1 | Pancreatic carcinoma cell line |
| DLD1-TxR | Multidrug resistant colorectal carcinoma cell line |
| DU-145 | Human prostate carcinoma cells |
| Glu | Glutamic acid |
| HCC38 | Breast carcinoma cell line |
| HeLa | Cervical carcinoma cell line |
| HepG2 | Human hepatocyte carcinoma cell line |
| HL-60 | Human promyelocytic leukemia cell line |
| HT-29 | Colon cancer |
| Ile | Isoleucine |
| D-Ile | D-Isoleucine |
| K562 | Chronic myelogenous leukemia cell line |
| Met | Methionine |
| MCF-7 | Hormone-dependent breast cancer cell line |
| MDA-MB-231 | Hormone-independent breast cancer cell line |
| MS | Melanoma cell line |
| NCI-H460/R | Multidrug resistant non-small cell lung carcinoma cell line |
| PC-3 | Human prostate cancer cell line |
| Raji | Burkitt's lymphoma cell line |
| RD | Rhabdomyosarcoma cell line |
| RMC | Rat mesangial cells |
| ROS | Reactive oxygen species |
| SAR | Structure–activity relationship |
| Ser | Serine |
| Saos | Human osteosarcoma cell line |
| SW620 | Colorectal carcinoma cell line |
| U87-TxR | Multidrug resistant glioblastoma cell line |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Warsaw University of Technology.References
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- P. Štěpnička, Dalton Trans., 2022, 51, 8085–8102 RSC.
- I. M. Kolthoff and F. G. Thomas, J. Phys. Chem., 1965, 69, 3049–3058 CrossRef CAS.
- D. Osella, M. Ferrali, P. Zanello, F. Laschi, M. Fontani, C. Nervi and G. Cavigiolio, Inorg. Chim. Acta, 2000, 306, 42–48 CrossRef CAS.
- I. H. Hall, A. E. Warren, C. C. Lee, M. D. Wasczcak and L. G. Sneddon, Anticancer Res., 1998, 18, 951–962 CAS.
- L. V. Snegur, Y. S. Nekrasov, N. S. Sergeeva, Z. V. Zhilina, V. V. Gumenyuk, Z. A. Starikova, A. A. Simenel, N. B. Morozova, I. K. Sviridova and V. N. Babin, Appl. Organomet. Chem., 2008, 22, 139–147 CrossRef CAS.
- V. J. Fiorina, R. J. Dubois and S. Brynes, J. Med. Chem., 1978, 21, 393–395 CrossRef CAS PubMed.
- P. Köpf-Maier, H. Köpf and E. W. Neuse, Angew. Chem., Int. Ed. Engl., 1984, 23, 456–457 CrossRef.
- P. Köpf-Maier, H. Köpf-Maier and E. W. Neuse, J. Cancer Res. Clin. Oncol., 1984, 108, 336–340 CrossRef PubMed.
- C. Biot, G. Glorian, L. A. Maciejewski, J. S. Brocard, O. Domarle, G. Blampain, P. Millet, A. J. Georges, H. Abessolo, D. Dive and J. Lebibi, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS PubMed.
- S. Top, J. Tang, A. Vessières, D. Carrez, C. Provot and G. Jaouen, Chem. Commun., 1996, 955–956 RSC.
- S. Top, A. Vessières, G. Leclercq, J. Quivy, J. Tang, J. Vaissermann, M. Huché and G. Jaouen, Chem. – Eur. J., 2003, 9, 5223–5236 CrossRef CAS PubMed.
- A. Vessières, S. Top, P. Pigeon, E. Hillard, L. Boubeker, D. Spera and G. Jaouen, J. Med. Chem., 2005, 48, 3937–3940 CrossRef PubMed.
- D. Plażuk, A. Vessières, E. A. Hillard, O. Buriez, E. Labbé, P. Pigeon, M.-A. Plamont, C. Amatore, J. Zakrzewski and G. Jaouen, J. Med. Chem., 2009, 52, 4964–4967 CrossRef PubMed.
- L. V. Popova, V. N. Babin, Y. A. Belousov, Y. S. Nekrasov, A. E. Snegireva, N. P. Borodina, G. M. Shaposhnikova, O. B. Bychenko, P. M. Raevskii, N. B. Morozova, A. I. Iiyina and K. G. Shitkov, Appl. Organomet. Chem., 1993, 7, 85–94 CrossRef CAS.
- The Nobel Prize in Chemistry 2022 was awarded to Carolyn R. Bertozzi, Morten Meldal and K. Barry Sharpless “for the development of click chemistry and bioorthogonal chemistry” during the reviewing of our contribution (5th October 2022).
- E. Bonandi, M. S. Christodoulou, G. Fumagalli, D. Perdicchia, G. Rastelli and D. Passarella, Drug Discovery Today, 2017, 22, 1572–1581 CrossRef CAS PubMed.
- K. Bozorov, J. Zhao and H. A. Aisa, Bioorg. Med. Chem., 2019, 27, 3511–3531 CrossRef CAS PubMed.
- D. Dheer, V. Singh and R. Shankar, Bioorg. Chem., 2017, 71, 30–54 CrossRef CAS PubMed.
- N. K. Verma, D. Mondal and S. Bera, Curr. Org. Chem., 2020, 23, 2305–2572 CrossRef.
- A. Rani, G. Singh, A. Singh, U. Maqbool, G. Kaur and J. Singh, RSC Adv., 2020, 10, 5610–5635 RSC.
- Carboxyamidotriazole (FDA UNII identifier, 6ST3ZF52WB; anticancer), cefatrizine (FDA UNII identifier, 8P4W949T8K; antibacterial), cefmatilen (FDA UNII identifier, T750UM24H8; antibacterial), rufinamide (FDA UNII identifier, WFW942PR79; anticonvulsant), tazobactam (FDA UNII identifier, SE10G96M8W; antibacterial), radezolid (FDA UNII identifier, 53PC6LO35W; antibacterial), mubritinib (FDA UNII identifier, V734AZP9BR; anticancer), solithromycin (FDA UNII identifier, 9U1ETH79CK; antibacterial), suvorexant (FDA UNII identifier, 081L192FO9; anti-insomnia), tradipitant (FDA UNII identifier, NY0COC51FI; anti-itchiness), flortanidazole F-18 (FDA UNII identifier, U6954O3II9; radioactive tumor hypoxia tracer). NCATS Inxight Drugs, https://drugs.ncats.io/, (accessed September 7, 2022).
- D. K. Dalvie, A. S. Kalgutkar, S. C. Khojasteh-Bakht, R. S. Obach and J. P. O'Donnell, Chem. Res. Toxicol., 2002, 15, 269–299 Search PubMed.
- V. Ganesh, V. S. Sudhir, T. Kundu and S. Chandrasekaran, Chem. – Asian J., 2011, 6, 2670–2694 CrossRef CAS PubMed.
- G. Jaouen, A. Vessières and S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817 RSC.
- K. Kowalski, Coord. Chem. Rev., 2016, 317, 132–156 CrossRef CAS.
- M. Patra and G. Gasser, Nat. Rev. Chem., 2017, 1, e0066 Search PubMed.
- R. Wang, H. Chen, W. Yan, M. Zheng, T. Zhang and Y. Zhang, Eur. J. Med. Chem., 2020, 190, e112109 CrossRef PubMed.
- V. Raičević, N. Radulović and M. Sakač, Eur. J. Inorg. Chem., 2022, e202100951 Search PubMed.
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931–5986 CrossRef CAS PubMed.
- D. Plażuk, B. Rychlik, A. Błauż and S. Domagała, J. Organomet. Chem., 2012, 715, 102–112 CrossRef.
- S. Maračić, J. Lapić, S. Djaković, T. Opačak-Bernardi, L. Glavaš-Obrovac, V. Vrček and S. Raić-Malić, Appl. Organomet. Chem., 2019, 33, e4628 CrossRef.
- V. Rep, M. Piškor, H. Šimek, P. Mišetić, P. Grbčić, J. Padovan, V. Gabelica Marković, D. Jadreško, K. Pavelić, S. Kraljević Pavelić and S. Raić-Malić, Molecules, 2020, 25, e1570 CrossRef PubMed.
- M. Mazur, M. Mrozowicz, W. Buchowicz, M. Koszytkowska-Stawińska, R. Kamiński, Z. Ochal, P. Wińska and M. Bretner, Dalton Trans., 2020, 49, 11504–11511 RSC.
- S. Djaković, L. Glavaš-Obrovac, J. Lapić, S. Maračić, J. Kirchofer, M. Knežević, M. Jukić and S. Raić-Malić, Appl. Organomet. Chem., 2021, 35, e6052 CrossRef.
- L. V. Anikina, D. A. Shemyakina, L. V. Pavlogradskaya, A. N. Nedugov and V. A. Glushkov, Russ. J. Org. Chem., 2014, 50, 1180–1183 CrossRef CAS.
- S. B. Deepthi, R. Trivedi, L. Giribabu, P. Sujitha and C. G. Kumar, Inorg. Chim. Acta, 2014, 416, 164–170 CrossRef CAS.
- S. Jakopec, N. Pantalon Juraj, A. Brozovic, D. Jadreško, B. Perić, S. I. Kirin and S. Raić-Malić, Appl. Organomet. Chem., 2022, 36, e6575 CrossRef CAS.
- L. Kocsis, I. Szabó, S. Bősze, T. Jernei, F. Hudecz and A. Csámpai, Bioorg. Med. Chem. Lett., 2016, 26, 946–949 CrossRef CAS PubMed.
- A. Podolski-Renić, S. Bősze, J. Dinić, L. Kocsis, F. Hudecz, A. Csámpai and M. Pešić, Metallomics, 2017, 9, 1132–1141 CrossRef PubMed.
- A. Singh, J. Gut, P. J. Rosenthal and V. Kumar, Eur. J. Med. Chem., 2017, 125, 269–277 CrossRef CAS PubMed.
- V. A. Glushkov, D. A. Shemyakina, N. K. Zhukova, L. V. Pavlogradskaya, M. V. Dmitriev, D. V. Eroshenko, A. R. Galeev and I. G. Mokrushin, Russ. J. Org. Chem., 2019, 55, 1690–1697 CrossRef CAS.
- R. Trivedi, S. B. Deepthi, L. Giribabu, B. Sridhar, P. Sujitha, C. G. Kumar and K. V. S. Ramakrishna, Eur. J. Inorg. Chem., 2012, 2267–2277 CrossRef CAS.
- M. Maschke, M. Lieb and N. Metzler-Nolte, Eur. J. Inorg. Chem., 2012, 5953–5959 CrossRef CAS.
- A. Singh, S. T. Saha, S. Perumal, M. Kaur and V. Kumar, ACS Omega, 2018, 3, 1263–1268 CrossRef CAS PubMed.
- S. B. Deepthi, R. Trivedi, L. Giribabu, P. Sujitha and C. G. Kumar, Dalton Trans., 2013, 42, 1180–1190 RSC.
- S. B. Deepthi, R. Trivedi, L. Giribabu, P. Sujitha, C. G. Kumar and B. Sridhar, New J. Chem., 2014, 38, 227–236 RSC.
- S. Panaka, R. Trivedi, K. Jaipal, L. Giribabu, P. Sujitha, C. G. Kumar and B. Sridhar, J. Organomet. Chem., 2016, 813, 125–130 CrossRef CAS.
- S. Daum, M. S. V. Reshetnikov, M. Sisa, T. Dumych, M. D. Lootsik, R. Bilyy, E. Bila, C. Janko, C. Alexiou, M. Herrmann, L. Sellner and A. Mokhir, Angew. Chem., Int. Ed., 2017, 56, 15545–15549 CrossRef CAS PubMed.
- V. Reshetnikov, S. Daum, C. Janko, W. Karawacka, R. Tietze, C. Alexiou, S. Paryzhak, T. Dumych, R. Bilyy, P. Tripal, B. Schmid, R. Palmisano and A. Mokhir, Angew. Chem., Int. Ed., 2018, 57, 11943–11946 CrossRef CAS PubMed.
- V. Reshetnikov, H. G. Özkan, S. Daum, C. Janko, C. Alexiou, C. Sauer, M. R. Heinrich and A. Mokhir, Molecules, 2020, 25, e2545 CrossRef PubMed.
- J. Toms, V. Reshetnikov, S. Maschauer, A. Mokhir and O. Prante, J. Labelled Compd. Radiopharm., 2018, 61, 1081–1088 CrossRef CAS PubMed.
- S. Daum, J. Toms, V. Reshetnikov, H. G. Özkan, F. Hampel, S. Maschauer, A. Hakimioun, F. Beierlein, L. Sellner, M. Schmitt, O. Prante and A. Mokhir, Bioconjugate Chem., 2019, 30, 1077–1086 CrossRef CAS PubMed.
- D. L. Bertuzzi, C. B. Braga, G. Perli and C. Ornelas, Eur. J. Inorg. Chem., 2022, e202101084 CAS.
- K. Kumar, S. Carrère-Kremer, L. Kremer, Y. Guérardel, C. Biot and V. Kumar, Organometallics, 2013, 32, 5713–5719 CrossRef CAS.
- A. Singh, C. Biot, A. Viljoen, C. Dupont, L. Kremer, K. Kumar and V. Kumar, Chem. Biol. Drug Des., 2017, 89, 856–861 CrossRef CAS PubMed.
- M. R. E. Aly, I. H. El Azab and A. A. Gobouri, Monatsh. Chem. Verw. Teile Anderer Wiss. Chem. Mon., 2018, 149, 505–517 CrossRef CAS.
- M. Daniluk, W. Buchowicz, M. Koszytkowska-Stawińska, K. Jarząbek, K. N. Jarzembska, R. Kamiński, M. Piszcz, A. E. Laudy and S. Tyski, ChemistrySelect, 2019, 4, 11130–11135 CrossRef CAS.
- Y. Swetha, E. R. Reddy, J. R. Kumar, R. Trivedi, L. Giribabu, B. Sridhar, B. Rathod and R. S. Prakasham, New J. Chem., 2019, 43, 8341–8351 RSC.
- S. Yagnam, R. Trivedi, S. Krishna, L. Giribabu, G. Praveena and R. S. Prakasham, J. Organomet. Chem., 2021, 937, e121716 CrossRef.
- G. Singh, A. Arora, P. Kalra, I. K. Maurya, C. E. Ruizc, M. A. Estebanc, S. Sinha, K. Goyal and R. Sehgal, Bioorg. Med. Chem., 2019, 27, 188–195 CrossRef CAS PubMed.
- S. Yagnam, E. Rami Reddy, R. Trivedi, N. V. Krishna, L. Giribabu, B. Rathod, R. S. Prakasham and B. Sridhar, Appl. Organomet. Chem., 2019, 33, e4817 CrossRef.
- E. Yu. Rogatkina, A. N. Rodionov, S. E. Mazina and A. A. Simenel, J. Porphyrins Phthalocyanines, 2021, 25, 31–36 CrossRef CAS.
- M. M. Lakouraj, V. Hasantabar, H. Tashakkorian and M. Golpour, Polym. Adv. Technol., 2018, 29, 2784–2796 CrossRef CAS.
- Sanofi, A Randomized, Open Label, Parallel-group, Single Dose Regimen, Phase 2a Study, to Investigate the Clinical and Parasiticidal Activity and the Pharmacokinetics of 3 Dose Levels of Artefenomel (OZ439) Given in Combination With Ferroquine (FQ) and FQ Alone, in African Patients With Uncomplicated Plasmodium Falciparum Malaria, clinicaltrials.gov, 2022.
- K. Kumar, B. Pradines, M. Madamet, R. Amalvict, N. Benoit and V. Kumar, Eur. J. Med. Chem., 2014, 87, 801–804 CrossRef CAS PubMed.
- A. Singh, D. Zhang, C. C. Tam, L. W. Cheng, K. M. Land and V. Kumar, J. Organomet. Chem., 2019, 896, 1–4 CrossRef CAS.
- A. Singh, G. Fong, J. Liu, Y.-H. Wu, K. Chang, W. Park, J. Kim, C. Tam, L. W. Cheng, K. M. Land and V. Kumar, ACS Omega, 2018, 3, 5808–5813 CrossRef CAS PubMed.
- B. E. Herman, J. Gardi, J. Julesz, C. Tömböly, E. Szánti-Pintér, K. Fehér, R. Skoda-Földes and M. Szécsi, Biol. Futura, 2020, 71, 249–264 CrossRef CAS PubMed.
- J. Spencer, J. Amin, R. Boddiboyena, G. Packham, B. E. Cavell, S. S. Syed Alwi, R. M. Paranal, T. D. Heightman, M. Wang, B. Marsden, P. Coxhead, M. Guille, G. J. Tizzard, S. J. Coles and J. E. Bradner, MedChemComm, 2012, 3, 61–64 RSC.
- K. Kamanna, R. Aneja, C. Duffy, P. Kubinski, D. Rodrigo Moreira, L. D. Bailey, K. McFadden, A. Schön, A. Holmes, F. Tuzer, M. Contarino, E. Freire and I. M. Chaiken, ChemMedChem, 2013, 8, 322–328 CrossRef CAS PubMed.
- R. Aneja, A. A. Rashad, H. Li, R. V. Kalyana Sundaram, C. Duffy, L. D. Bailey and I. Chaiken, J. Med. Chem., 2015, 58, 3843–3858 CrossRef CAS PubMed.
- A. A. Rashad, K. Acharya, A. Haftl, R. Aneja, A. Dick, A. P. Holmes and I. Chaiken, Org. Biomol. Chem., 2017, 15, 7770–7782 RSC.
- A. A. Rashad, R. V. Kalyana Sundaram, R. Aneja, C. Duffy and I. Chaiken, J. Med. Chem., 2015, 58, 7603–7608 CrossRef CAS PubMed.
- A. Haque, M.-F. Hsieh, S. I. Hassan, Md. S. Haque Faizi, A. Saha, N. Dege, J. A. Rather and M. S. Khan, J. Mol. Struct., 2017, 1146, 536–545 CrossRef CAS.
- C.-Y. Cheng, A. Haque, M.-F. Hsieh, S. Imran Hassan, M. S. H. Faizi, N. Dege and M. S. Khan, Int. J. Mol. Sci., 2020, 21, e3823 CrossRef PubMed.
- A. J. Salmon, M. L. Williams, Q. K. Wu, J. Morizzi, D. Gregg, S. A. Charman, D. Vullo, C. T. Supuran and S.-A. Poulsen, J. Med. Chem., 2012, 55, 5506–5517 CrossRef CAS PubMed.
- H. Gizem Özkan, J. Toms, S. Maschauer, O. Prante and A. Mokhir, Eur. J. Inorg. Chem., 2021, 5096–5102 CrossRef.
- H. Gopi, S. Cocklin, V. Pirrone, K. McFadden, F. Tuzer, I. Zentner, S. Ajith, S. Baxter, N. Jawanda, F. C. Krebs and I. M. Chaiken, J. Mol. Recognit., 2009, 22, 169–174 CrossRef CAS PubMed.
- M. Umashankara, K. McFadden, I. Zentner, A. Schön, S. Rajagopal, F. Tuzer, S. A. Kuriakose, M. Contarino, J. LaLonde, E. Freire and I. Chaiken, ChemMedChem, 2010, 5, 1871–1879 CrossRef CAS PubMed.
- M. P. Gleeson, A. Hersey, D. Montanari and J. Overington, Nat. Rev. Drug Discovery, 2011, 10, 197–208 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |