
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis without solvent: consequences for mechanochemical reactivity
Lauren E.
Wenger
 and
Timothy P.
Hanusa
*
and
Timothy P.
Hanusa
*
Department of Chemistry, Vanderbilt University, Nashville, Tennessee 37235, USA. E-mail: t.hanusa@vanderbilt.edu
First published on 3rd November 2023
Abstract
Solvents are so nearly omnipresent in synthetic chemistry that a classic question for their use has been: “What is the best solvent for this reaction?” The increasing use of mechanochemical approaches to synthesis—by grinding, milling, extrusion, or other means—and usually with no, or only limited, amounts of solvent, has raised an alternative question for the synthetic chemist: “What happens if there is no solvent?” This review focuses on a three-part answer to that question: when there is little change (“solvent-optional” reactions); when solvent needs to be present in some form, even if only in the amounts provided by liquid-assisted (LAG) or solvate-assisted grinding; and those cases in which mechanochemistry allows access to compounds that cannot be obtained from solution-based routes. The emphasis here is on inorganic and organometallic systems, including selected examples of mechanosynthesis and mechanocatalysis. Issues of mechanochemical depictions and the adequacy of LAG descriptions are also reviewed.
 Lauren E. Wenger | Lauren E. Wenger completed her Bachelor studies in Chemistry and Molecular Biology at Goshen College (Goshen, Indiana) in 2018. There, her research focused on organic synthesis in the lab of Prof. Douglas Schirch and on medicinal chemistry for neglected disease during an internship at AbbVie Pharmaceuticals. She joined the lab of Prof. Timothy Hanusa at Vanderbilt University (Nashville, Tennessee) in 2019. Her graduate studies focus on the mechanochemical synthesis of main group organometallic complexes. Her research interests include the development of sustainable methodologies for organometallic and organic synthesis. |
 Timothy P. Hanusa | Timothy P. Hanusa received his bachelor's degree in chemistry from Cornell College (Mount Vernon, Iowa). He received his PhD in 1983 from Indiana University, Bloomington, working on heteroboranes under the direction of Lee J. Todd. After postdoctoral research on organolanthanides with William J. Evans at the University of California, Irvine, he joined the chemistry faculty at Vanderbilt University in Nashville, Tennessee in 1985, where he is now Professor of Chemistry. His research has focused on organometallic complexes of the main group elements, and on mechanochemical approaches to organometallic synthesis across the periodic table. |
Introduction
The “Fourth Way,” an approach to self-development proposed by the philosopher G. I. Gurdjieff in the early 20th century, was intended as a harmonization of the “ways”, or principles, of the body, emotions, and mind.1 The “Fourth Way” was not meant to be a sophisticated combination of the other three, but a state of consciousness that existed apart from them. In a somewhat parallel, although less esoteric manner, synthetic chemistry has three basic ways of conducting reactions—solvothermally, electrochemically, and photochemically—along with a “fourth way,” i.e., mechanochemically. Like Gurdjieff's Way, a mechanochemical reaction, defined by IUPAC as one “that is induced by the direct absorption of mechanical energy”,2 is a distinct approach to conducting reactions. Mechanochemical processes have been known for far longer than the philosophical “Way”, however, with examples dating from antiquity and even pre-history (e.g., the rubbing of wooden sticks to create fire), and their development has been detailed elsewhere in various reviews.3 Even with such an extended timeline, only in the last quarter century has mechanochemistry come into its own as a systematically explored discipline of synthetic methodology.4Apart from the application of mechanical force, synthetic mechanochemical reactions are typically characterized by the complete, or nearly complete, absence of solvent. This fact alone has significant consequences, as solvents play many well-known roles in synthesis, including creating homogeneous reaction mixtures, dispersing heat,5 stabilizing charged intermediates,6 and controlling reaction rates.7 In addition, the outcome of a reaction can be modified by solvents’ shifting of equilibrium,8 altering of product selectivities,9 and modifying ligand binding.10 Given these critical functions, it should not be surprising that one of the classic concerns in chemical synthesis has long been: “What is the best solvent for this reaction?”
If this question is difficult to answer, the problem becomes even more complex if an alternative is asked, i.e., “What happens if there is no solvent?” At a macroscopic level, there are positive changes, such as avoiding the waste and toxicity associated with solvent use.6,11 In addition, many reactions receive their activation energy through thermal transfer, and much of the added energy goes to heating the bulk of the solvent, with only a fraction of the total energy being transferred to the reagents. Consequently, grinding and ball milling reactions generally require less total energy input than solution reactions that involve external heating,12 and mechanochemically initiated syntheses in general are considered to be “greener” than solution-based counterparts.13
When one moves beyond the broad characteristics of mechanochemistry and considers specific reactions, however, the consequences of removing solvents are not as readily categorized; there may be little to no change, or the outcome of a reaction can be altered completely in either positive or negative directions, as detailed below. This review examines selected recent developments in mechanochemically driven reactions, with a particular focus on the consequences of solvent removal in inorganic and organometallic systems. Examples of mechanosynthesis and mechanocatalysis are discussed as well.
1. Mechanochemical depictions
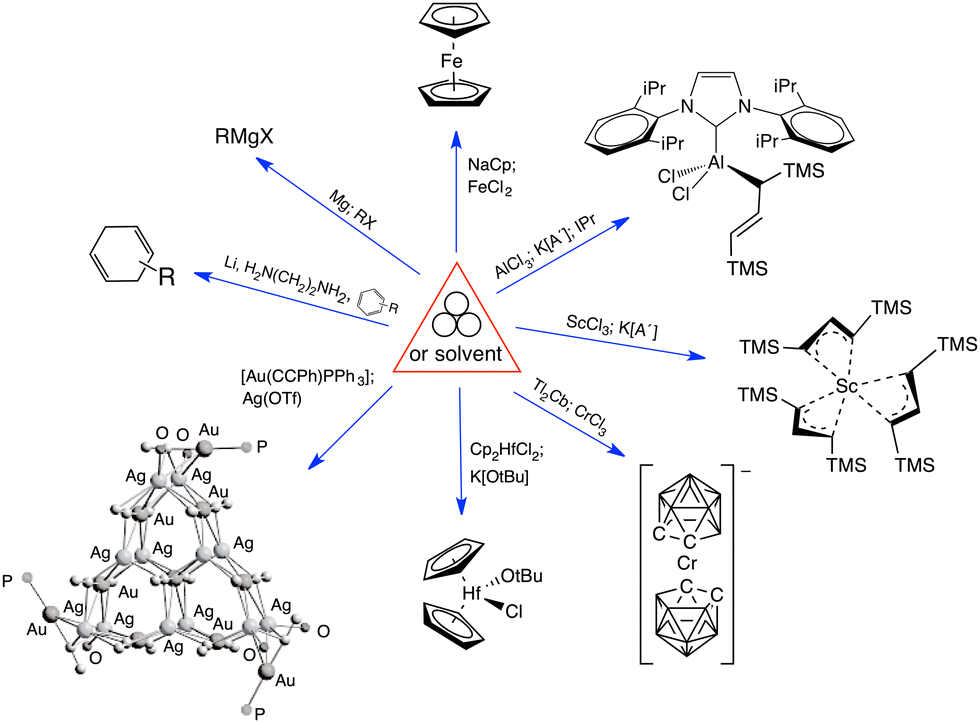
Mechanochemical processes encompass a range of variables, which can be broadly classified as external factors, specific to the reaction equipment, and internal factors, which are particular to the chemical environment and reagents used. External factors include the type of grinder or mill used (e.g., mortar and pestle, mixer mill, planetary mill, twin extrusion, RAM (resonant acoustic mixing));14 the composition and size of the milling jar (e.g., polymethylmethacrylate, stainless steel, zirconia, silicon carbide, and reactive metals like nickel or copper); the size, number, and composition of grinding balls (with similar variants as the milling jar); and the time and speed of milling. Internal factors include the use of additives (e.g., liquid-assisted grinding (LAG), discussed below, and solid grinding agents) and the solid-state structure of the reagents. The temperature inside the mill could be either an external or internal factor, depending on whether it reflects the operation of an external heat source or an exothermic reaction.Attempts have been made to demarcate mechanochemical reactions and their variables through the use of visual imagery, following long-standing approaches in other areas of chemistry. The delta character (Δ), for example, has its origins in alchemical representations of fire,15 but it is universally recognized today as indicating the addition of heat. The use of three circles grouped in a triangular arrangement (Fig. 1(a)), often positioned over an arrow, has been proposed as a graphical symbol for the addition of mechanical energy.16 To retain simplicity, the three-circle symbolism is not meant to indicate the external means of mechanochemical initiation—whether by impact, stretching, reactive extrusion, or any other—nor the amount of energy applied, and this is not fundamentally different from the use of Δ to represent the addition of thermal energy, where neither the source of the heat (a heating mantle, heat exchangers, microwave radiation, etc.) nor its quantity is indicated. The 3-circle symbol has gained some currency among workers in the mechanochemical field, including appearances in journal art.17
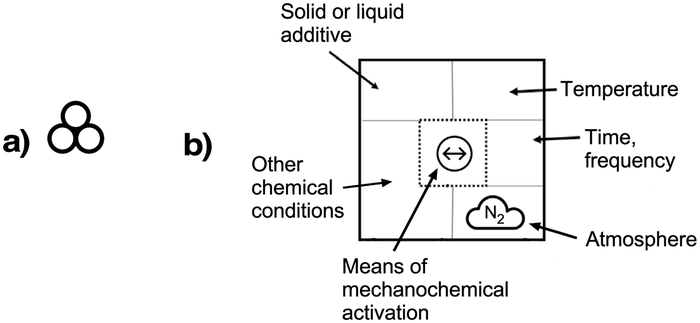 | ||
| Fig. 1 Proposed graphics for mechanochemical reactions: (a) the three-circle symbol, designed to represent the general application of mechanical energy;16 (b) a detailed representation that specifies experimental conditions; as examples, the symbol in the middle represents a vibratory ball mill, and the cloud shape with the enclosed letters “N2” indicates a nitrogen atmosphere.18 | ||
Another, more detailed representation for mechanochemical reactions has been suggested that explicitly denotes the experimental conditions employed (Fig. 1(b)).18 The graphic comprises a box with designated areas for the type of equipment used, any additives present, the temperature, and atmospheric conditions. Specific icons have been suggested for use, and the interested reader is advised to consult the original paper for a list of proposed symbols.
2. Mechanochemistry and additives
The earliest recorded mechanochemical reaction (the grinding of cinnabar (HgS) in a copper vessel to afford quicksilver (Hg), described by the Greek philosopher Theophrastus, ca. 300 BC)19 was assisted by the use of vinegar (acetic acid). The addition of small amounts of liquids (formalized under the term “liquid-assisted grinding”, LAG) or grinding agents such as salts, silica, or polymers has become a widespread practice in synthetic mechanochemistry. Such additives can aid in mixing, prevent aggregation of particles,20 and stabilize intermediates, which can direct the formation of polymorphs21 and modify product distribution.22 The introduction of solvents through solvated (or in the case of water, hydrated) species (i.e., “solvate-assisted grinding” (SAG)), can yield results different from reactions in which liquid is added separately, as in classic LAG.23 Polymer-assisted grinding (POLAG) has grown in popularity, as the technique gives benefits comparable to LAG, while avoiding the potential to form unwanted solvated products, and it also helps control particle size.24 Using room temperature ionic liquids as LAG additives (i.e., in “ILAG”) has been explored as a variation of LAG.25Salt additives (e.g., alkali metal halides) have been successfully used in the preparation of otherwise inaccessible products, although seemingly small variations in their composition can affect reaction outcomes in ways still poorly understood. For example, the mechanochemically induced transformation of the macrocycle [{P(μ-NtBu)}2(μ-NtBu)]2 into its adamantoid isomer P4(NtBu)6 is strongly dependent on the amount of LiCl present in the reaction mixture: no reaction is observed at a 10% weight loading after 90 min of milling, but the yield is quantitative at 20% loading.26 Raising the LiCl amount to 33% drops the yield to 30%, a result ascribed to the dilution effect of the excess salt. Interestingly, the same 20% loading that is effective with LiCl produces only 6% yield if either the halide (LiBr) or the alkali metal (NaCl) is changed. Although essential to the reaction, the exact role that the LiCl plays is unclear, and calculations of the reaction transformation in the presence of LiCl suggest that it does not lower the activation barrier.
Cocrystallisation studies provided some of the first detailed information about the connection between reagent solvation, LAG, and mechanochemical reactivity. The attempted cocrystallisation of citric acid and caffeine found that the dry grinding of anhydrous caffeine and anhydrous citric acid did not lead to a cocrystal.27 However, when water was added in small amounts to the anhydrous reagents, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cocrystal forms.28 Interestingly, the cocrystal could also be prepared by first preparing a caffeine hydrate (caffeine·(H2O)0.8) and grinding it with anhydrous citric acid (Fig. 2). In effect, the hydrate functions as a reservoir of LAG quantities of water.
:1 cocrystal forms.28 Interestingly, the cocrystal could also be prepared by first preparing a caffeine hydrate (caffeine·(H2O)0.8) and grinding it with anhydrous citric acid (Fig. 2). In effect, the hydrate functions as a reservoir of LAG quantities of water.
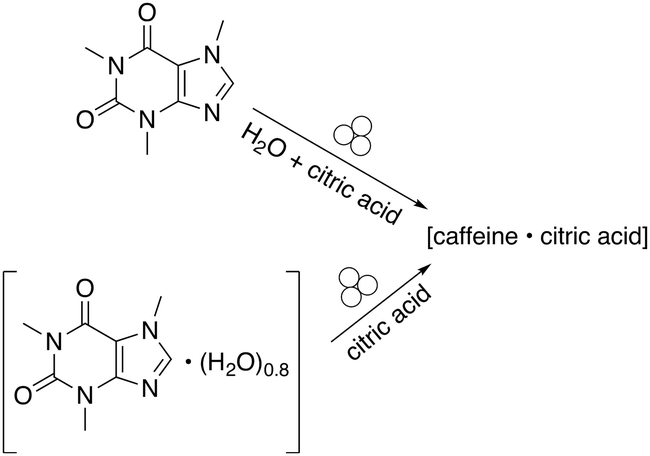 | ||
| Fig. 2 Cocrystal formation using either liquid water or a hydrate.28 | ||
As the LiCl example above suggests, the quantity of additive used in a mechanochemical system matters, and this is true for LAG-enabled reactions in general. Exactly how much solvent should be added depends first of all on the scale of the reaction—too much, and the reaction environment becomes a paste or slurry, if not an outright solution. The first widely accepted attempt to quantify the solvent amount in LAG reactions, i.e., the “η” scale, took this into account.28 With “η” set equal to the μL of liquid divided by the mg of total solid reagents in the milling vessel, the LAG region is generally considered to be 0 < η ≲ 2, as shown in Fig. 3.†
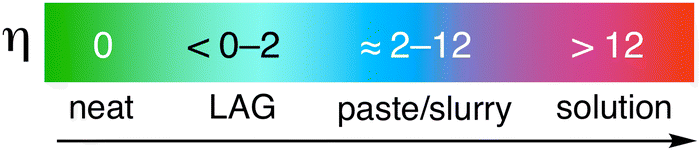 | ||
| Fig. 3 Approximate ranges of solvent addition for mechanochemical reactions on the η scale (μL liquid added/mg of total reagents) for LAG reactions. Distinctive chemistry occurs in the region before paste-like mixtures are evident (η ≲ 2). | ||
The η parameter was originally applied to a system involving cocrystal formation of organic solids, including theophylline and caffeine as pharmaceutical ingredients and L-malic or L-tartaric acid as pharmaceutical cocrystal formers.28 Although a cocrystal does not form upon dry grinding, LAG reactions employing a wide range of solvents produced cocrystals at η = 0.25 for nearly all LAG solvents used. The authors found no clear correlation between solubility of the precursors in the LAG solvent and the ability to form a cocrystal,28 and in fact within the LAG region, reactant solubility did not affect the reaction outcome.21
In the LAG regime, especially as η approaches 0, the solvent may function primarily as a lubricant, promoting molecular diffusion.29 Such amounts of solvent have also been described as “catalytic”,30 and the solvents’ role in this range can be complex and even inhibitory.31 It should not be surprising that the η scale, which is agnostic about the relative molar masses of the LAG solvent and other reagents, or the polarity, basicity, or hydrogen-bonding capability of the solvent, is not able (nor was it intended) to represent fine details of LAG-assisted reactions.
For some types of reactions, particularly organometallic systems, the ratio of the number of molar equivalents of solvent to the reagents can be more informative of the environment at the metal center than the solvent volume to mass ratio (η value). Relatively low values of η may mask the multiple equivalents of solvent that can be present per metal center in a reaction. For instance, in the preparation of ferrocene from iron(II) chloride and sodium cyclopentadiene (2NaCp + FeCl2 → Cp2Fe + 2NaCl), an η value of 1 when using THF as the LAG solvent corresponds to an average of 3.7 molecules of THF per iron center. At this point, the iron is effectively coordinatively saturated, and the reaction can begin without any external activation (either mechanochemically or with heat; see illustrations of this situation in the ESI of ref. 23).
In the synthesis of the bulky Group 14 amides M{N(SiMe3)2}2 (M = Ge, Sn, Pb) from Li{N(SiMe3)2} and GeCl2·(1,4-dioxane), SnCl2, or PbCl2, the groups of Garcia and García-Álvarez explicitly tracked the number of equivalents of solvate molecules per metal center (to which they gave the name “ηsolv”).32 For the germanium reaction, ηsolv = 1 by virtue of the coordinated dioxane molecule, and it was found that the yields of the Sn and Pb amides were also improved at a ratio of one equivalent of dioxane per mole of metal halide. Other than indicating that the amount of solvent was in the LAG region, the corresponding η values for these reactions (0.21, 0.19, and 0.16 for Ge, Sn, and Pb, respectively) were less informative.
3. Classification of mechanochemical reactions
We have found it useful to partition the role that solvents play in the outcome of mechanochemical reactions into three general categories:Type 1: the use of solvent is effectively optional; i.e., the outcomes of solution and mechanochemical reactions are similar.
Type 2: only a solvent-containing synthesis gives the desired product.
Type 3: only the solid-state synthesis gives the desired product, or provides a new product not observed from solution synthesis.
These are of course broad classifications, but are meant to indicate that mechanochemical activation is not a panacea for synthetic difficulties, but is an alternative approach—a “fourth way”— that in many cases can provide products with minimal solvent use and/or generate products that are not isolable from solution-based reactions. The categories provide a framework in which to think about mechanochemical reactions and the not always-obvious ways they can differ from their counterparts in solution (and for that matter, from reactions in the melt).33 It should be noted that these divisions are based on empirical reaction outcomes—whether the reaction mechanisms are similar or appreciably change when the solvent is removed is a different question.
The “Types” are capable of additional refinement; for example, type 1 reactions could be further subdivided into two subtypes, type 1a, which has no significant difference in product selectivity between solution and solid-state, and type 1b, where solution and solid-state reactions may give the same product(s), but the selectivity differs significantly. Examples of each type will be given in the following sections, and it should be noted that precise distinctions are not always possible. As a point of clarification, however, reactions are considered type 1 if the only major difference between the mechanochemical and solution reactions is the use of solvent in the latter. This would not include compounds that can be prepared either in the solid state or in solution, but which require different reagents in each case. For example, the tris(pentamethylcyclopentadienyl) complex (C5Me5)3Y can be prepared mechanochemically by grinding (C5Me5)2Y(μ-Ph)2BPh2 and K[C5Me5], but in benzene solution the same reagents produce the bis(pentamethylcyclopentadienyl) complex (C5Me5)3Y(C6H5).34 (C5Me5)3Y can be synthesized in methylcyclohexane solution, but from [(C5Me5)2Y(μ-H)]2 and 1,2,3,4-tetramethylfulvene instead. In addition, there are cases in which the use of solvents in LAG quantities can appreciably increase the yield, but their use is not always absolutely required; such reactions are also considered to be type 1.
Type 1a reactions: solvent is effectively optional; product is the same
Halide metathesis, which benefits from the thermodynamic driving force of the formation of an alkali metal salt as a by-product (MR + M′X → M′R + MX; M = Li, Na, K), is often found in “solvent-optional” reactions. An early organometallic example is the synthesis of ferrocene from iron(II) chloride and cyclopentadienyl salts of potassium or thallium. Ferrocene is produced in good yield (84%) from a 15 minutes mechanochemical reaction.35 This is largely the same as the classic solution method employing NaCp and FeCl2 in THF (73% yield, 75 min).36 Preparation of complexes with indenyl or allyl ligands (e.g., [Ni(Ind)2]37 and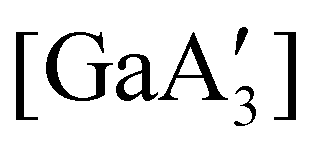 ;38 A′ = [1,3-(SiMe3)2C3H3]−) are included in this group, as are the bis(dicarbollyl) complexes of cobalt(III), iron(III) and chromium(III),39 and heteroleptic Cp/OR complexes of group 4 metals, [Cp2−xM(OtBu)2+x] (M = Zr, Hf).40 Transformation of the trimeric [{tBuZnOtBu}3] into the tetrameric cubane [{tBuZnOtBu}4] occurs both in solution and the solid state,41 and even more complicated structures such as the mixed metal aggregate [Ag12Au10(CCPh)17(OTf)5(PPh3)3], can be formed by mixing separately prepared acetone solutions of [Au(CCPh)PPh3] and Ag(OTf), or by simply grinding the two solid metal reagents together.42
;38 A′ = [1,3-(SiMe3)2C3H3]−) are included in this group, as are the bis(dicarbollyl) complexes of cobalt(III), iron(III) and chromium(III),39 and heteroleptic Cp/OR complexes of group 4 metals, [Cp2−xM(OtBu)2+x] (M = Zr, Hf).40 Transformation of the trimeric [{tBuZnOtBu}3] into the tetrameric cubane [{tBuZnOtBu}4] occurs both in solution and the solid state,41 and even more complicated structures such as the mixed metal aggregate [Ag12Au10(CCPh)17(OTf)5(PPh3)3], can be formed by mixing separately prepared acetone solutions of [Au(CCPh)PPh3] and Ag(OTf), or by simply grinding the two solid metal reagents together.42
The long-known Grignard reagents are the most widely used reagents for preparing C–C bonds.43 Historically, these highly active species have required an environment strictly free of water, an inert atmosphere, rigorously dried solvents, and pre-activated magnesium. Several groups have used ball milling to improve upon the preparation and use of Grignard reagents through direct insertion of Mg into an R–X bond by mechanochemical activation of Mg metal. Harrowfield and coworkers first used ball milling under solvent-free conditions to activate Mg, resulting in Grignard and McMurry reactions.44 Birke and coworkers used ball milling to dechlorinate 1,3,5-chlorobenzene to benzene through in situ Grignard formation, followed by reduction with n-butyl amine as the H donor.45 Our group used mechanochemistry to study the activation of C–F bonds with Mg.46 Ito and coworkers were the first to prepare and use Grignard reagents under air; the Grignard reagent could be isolated and combined with a variety of electrophiles to produce coupled products without the need for inert atmosphere.47 The yield is low for a strictly dry reaction (e.g., 6% yield for the nucleophilic addition product derived from the reaction of bromobenzene and Mg with benzaldehyde), and LAG with THF improves the yield to >90%. The outcome with LAG using a non-basic solvent such as hexanes, however, is even worse than when the reagents are used dry (1% yield).
Another unexpected transformation of a reaction that has traditionally been conducted in solution is the Birch reduction. Used to dearomatize arenes into 1,4-cyclohexadiene derivatives, the standard method requires liquid ammonia, inert atmospheres, and low temperatures to protect the alkali metal from unwanted oxidation. A mechanochemical equivalent has been developed that can be conducted in air and at room temperature, and features reduced reaction times (as short as 1 min). The substrate scope is broad and the reaction is scalable to gram amounts.48
Cyclodehydrogenation, used to create polycyclic aromatic hydrocarbons, has been known for over a century,49 but depending on the substrate, suffers from limited yield and undesirable byproducts. When optimized, the cyclodehydrogenation of 1,1′-binaphthyl can provide perylene quantitatively, for example, but at the cost of harsh conditions; i.e., requiring potassium metal in a pressure vessel with degassed and dehydrated THF at 85 °C under an inert gas atmosphere, with a reaction time of 12 h.50 In contrast, milling lithium wire with the substrate can be done in air at room temperature, and reaction times are reduced to 5–30 min. If THF is used in supra-LAG quantities (e.g., η = 3–10), yields can be >90%. Without THF, yields are cut in half, but reactions are still complete in minutes.51
In all these cases, the mechanochemical benefits of reduced or eliminated solvent use, shorter reaction times, and the like are on full display (Fig. 4).
 | ||
| Fig. 4 Examples of type 1a, “solvent optional” mechanochemical reactions. Clockwise from the top: [Cp2Fe]; [(NHC)AlCl2A′]; [[{1,3-(SiMe3)2C3H3}]3Sc]; [Me4N][Cr(C2B9H11)2]; [Cp2HfCl(OtBu)]; [Ag12Au10(CCPh)17(OTf)5(PPh3)3]; Birch reductions; Grignard reagents (references in the text). | ||
Type 1b reactions: solvent is effectively optional; shifts in product selectivity occur
Mechanochemistry can be used to alter product selectivity compared to similar reactions in solution. There are enough examples of this phenomenon that it has already been the subject of review.9 Here we will discuss several cases to illustrate the sometimes-subtle shifts in reaction outcome with solvent removal.The heteroleptic Cp′/OR complexes of group 4 metals, 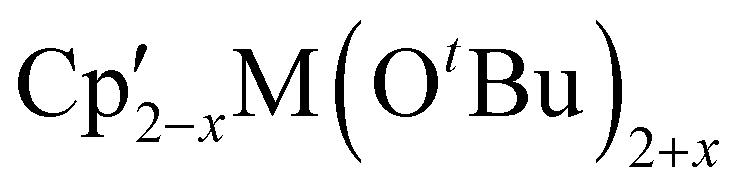 (M = Zr, Hf) are CVD precursors for thin films of zirconia52 and hafnia53 and have been used as polymerization initiators for α-olefins,54 lactide,55 and ε-caprolactone.55c A halide metathetical route for their preparation from Cp2MCl2 (M = Zr, Hf) and K[OtBu] works both in the solid state and in solution.40 For the hafnium system, use of a 1:1 ratio of reagents cleanly produces the monoalkoxide product Cp2HfCl(OtBu) either in hexanes or by ball milling (K[OtBu] + Cp2HfCl2 → Cp2HfCl(OtBu) + KCl). In this case, the mechanochemical reaction is complete in considerably less time than the solution equivalent (15 min vs. 18 h), a partial result of the higher concentration of reagents in the solid-state environment.
(M = Zr, Hf) are CVD precursors for thin films of zirconia52 and hafnia53 and have been used as polymerization initiators for α-olefins,54 lactide,55 and ε-caprolactone.55c A halide metathetical route for their preparation from Cp2MCl2 (M = Zr, Hf) and K[OtBu] works both in the solid state and in solution.40 For the hafnium system, use of a 1:1 ratio of reagents cleanly produces the monoalkoxide product Cp2HfCl(OtBu) either in hexanes or by ball milling (K[OtBu] + Cp2HfCl2 → Cp2HfCl(OtBu) + KCl). In this case, the mechanochemical reaction is complete in considerably less time than the solution equivalent (15 min vs. 18 h), a partial result of the higher concentration of reagents in the solid-state environment.
Considering only the formation of Cp2HfCl(OtBu), the reaction of Cp2HfCl2 with K[OtBu] would be classified as type 1a, and it is so depicted in Fig. 4. However, if an attempt is made to form Cp2Hf(OtBu)2 by increasing the K[OtBu]:Cp2HfCl2 ratio to 2:1, the reaction becomes an example of type 1b. A mixture of the stoichiometrically expected bis(alkoxide) and the monoalkoxide chloride is formed with either solution or mechanochemical methods, but in different amounts. In hexanes solution, the Cp2Hf(OtBu)2:Cp2HfCl(OtBu) ratio is 6.1:1, but with ball milling, the ratio inverts, with the monoalkoxide in excess (0.7:1). Increasing the equivalents of K[OtBu] relative to Cp2HfCl2 to 3:1, however, rapidly (15 min) produces Cp2Hf(OtBu)2 as the sole hafnium-containing product under ball milling conditions.
The situation is even more complex with the titanium analogue of the Cp′/OR systems. Heteroleptic Cp′/OR complexes of Ti have been used as polymerization initiators for styrene56 and lactide,55a and a representative example of these compounds is the titanocene alkoxide Cp2Ti(OtBu)2.57 Owing to weaker metal-ring binding than in the Zr and Hf systems, the reaction of Cp2TiCl2 and two equivalents of K[OtBu] yields four products: the initially expected Cp2Ti(OtBu)2, along with CpTi(OtBu)3, Cp3Ti(OtBu), and Ti(OtBu)4 (Fig. 5).40,58 In THF solution, the major product (70%) is the anticipated product based on the stoichiometry of the reagents, i.e., Cp2Ti(OtBu)2. However, the use of hexanes solution shifts the product distribution towards products with greater loss of the Cp ligands. Curiously, grinding the dry reagents together gives a product distribution similar to that of the hexanes reaction, at least for the two major species (Cp2Ti(OtBu)2 and CpTi(OtBu)3), suggesting that the mechanochemistry environment mimics a less polar environment than that provided by ethers. Using an excess (3 or 4 eq.) of K[OtBu] gives CpTi(OtBu)3 as the major product for all conditions, with Ti(OtBu)4 as the minor product. Once again, the product distribution from the mechanochemistry reaction (88:12) more closely tracks the hexanes outcome (93:7) than that from THF (60:40).40
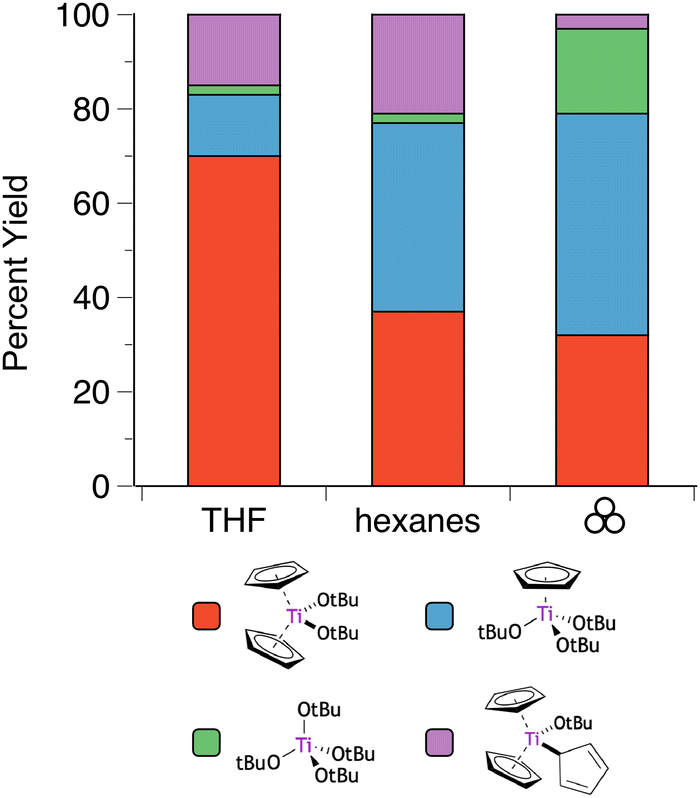 | ||
| Fig. 5 Product selectivity for reactions of K[OtBu] and Cp2TiCl2 (2:1 molar ratio) under solution and mechanochemical conditions.40 | ||
A different sort of product distribution change between solution and mechanochemical preparations is found in the case of bulky allyl complexes of arsenic and antimony. 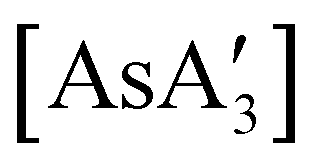 and
and 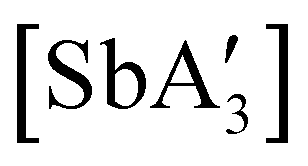 are generated in hexanes solution or mechanochemically from AsI3 and SbCl3, respectively, and 3 equiv. K[A′].59 The
are generated in hexanes solution or mechanochemically from AsI3 and SbCl3, respectively, and 3 equiv. K[A′].59 The 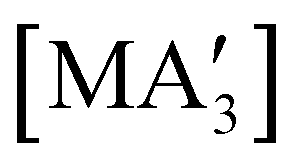 complexes are formed as two diastereomers, one of C1 (R,S,S) symmetry and one of C3 (R,R,R) symmetry, and the C1:C3 ratio varies with the preparation method (Fig. 6). The asymmetric C1 form is the major product from both solution and dry grinding methods for As and Sb, but compared to preparation in hexanes solution, the mechanochemical route increases the relative amount of C1 by a factor of 3.3 for As and 1.5 for Sb. The difference in selectivity has been attributed to the asymmetric environment around the As or Sb centers that is provided by the layered crystal lattice of each metal precursor, an asymmetry that disappears when the reagents are dissolved.
complexes are formed as two diastereomers, one of C1 (R,S,S) symmetry and one of C3 (R,R,R) symmetry, and the C1:C3 ratio varies with the preparation method (Fig. 6). The asymmetric C1 form is the major product from both solution and dry grinding methods for As and Sb, but compared to preparation in hexanes solution, the mechanochemical route increases the relative amount of C1 by a factor of 3.3 for As and 1.5 for Sb. The difference in selectivity has been attributed to the asymmetric environment around the As or Sb centers that is provided by the layered crystal lattice of each metal precursor, an asymmetry that disappears when the reagents are dissolved.
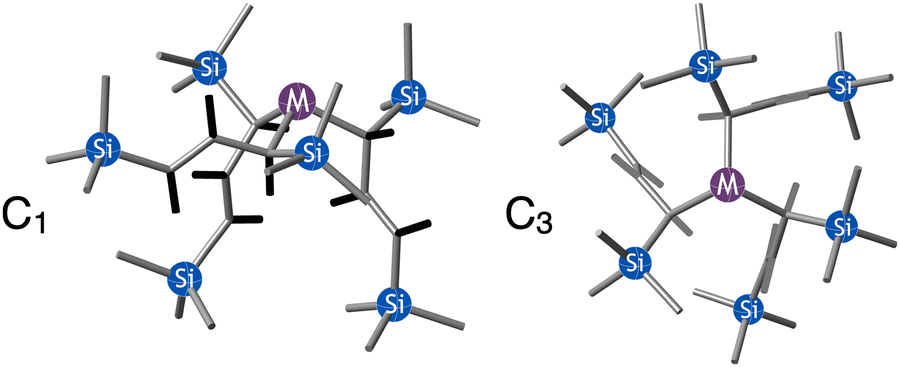 | ||
Fig. 6 Diastereomeric forms of 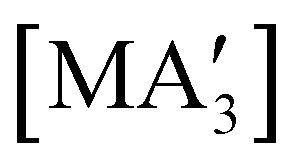 . The C1 forms were crystallographically characterized for M = As, Sb; the C3 forms were calculated.59 . The C1 forms were crystallographically characterized for M = As, Sb; the C3 forms were calculated.59 | ||
Sometimes reagent ratios can be adjusted to bring the results from mechanochemical reactions in line with expectations from stoichiometry. For example, depending on the R group, calcium amides [Ca(NR2)2] can serve as hydrocarbon-soluble sources of Ca2+ ions for organometallic synthesis,60 as non-nucleophilic bases for enolizations,61 and they have roles in catalysis.62 The bis(trimethylsilyl)amido derivative, [Ca(N(TMS)2)2], is an exemplar of this class,63 and multiple synthetic routes have been developed for it,64 including metathetical preparations starting from various calcium salts.60a,61a,65 These solvent-based syntheses are marked by relatively long reaction times (ranging from 3 hours to 5 days),64d the frequent formation of solvated species (with THF, DME, or Et2O), and most seriously with the salt metathesis methods, the simultaneous generation of calciate species, [MCa(NR)3] (M = Li, K; R = N(TMS)2). A mechanochemical version of this system, which involves the milling of a 1:2 ratio of CaI2 and K[NR2], also produces a mixture of [Ca(NR2)2] and [KCa(NR2)3], but when the starting ratio of CaI2 and K[NR2] is lowered to 1:1, a calciate-free product mixture forms during a 10 min grind. The use of the larger amounts of CaI2 evidently serve to suppress the formation of the calciate.
Type 2 reactions: only a solvent-containing synthesis gives the desired product
Serious issues can arise when solvent is removed from a reaction environment. Some of these are matters of scale, as reactions that might be conducted successfully on a sub-gram laboratory scale can fail when they are attempted on multigram or larger amounts.66 Some of these issues could potentially be addressed through different equipment choices, for example, specially designed mills or screw extrusion devices, or resonant acoustic mixers, which can be built to handle kilograms of reagents.14,67 Apart from the basic issues of mixing, there are fundamental chemical issues that must be addressed, such as the dispersion of heat in highly exothermic reactions, which solvents can do efficiently. Critical reaction intermediates may be stabilized in the presence of solvents, and solvent removal may lead to deleterious changes in reaction mechanisms and outcomes.Unless there are special considerations (e.g., the final product must be a solvate or hydrate), the number of reagents that cannot form the desired product under mechanochemical activation and strictly require a solution environment may be relatively small. For example, the palladium β-diketonate Pd(hfac)2 (hfac = hexafluoroacetylacetonate) is readily formed between Na2[PdCl4] and Na[hfac] in solution.68 Grinding the solids together at room temperature for 2 h, however, leaves only an intimate mixture of the reagents.69 The reaction can be made to occur by heating the ground reagents and ultimately subliming the product, although such heating is not necessary in solution.
Liquid-assisted grinding and the related technique of solvate-assisted grinding (SAG)23 have been used to extend the reach of mechanochemical reactions. The organic cocrystal formation experiments with hydrates that were used to establish the η scale have their counterparts in coordination chemistry. For example, experiments to form calcium urea sulfate ([Ca(urea)4]SO4) from anhydrous calcium sulfate and urea have found that essentially no reaction occurs between the reagents when they are ground at either room temperature or at 70 °C. Addition of LAG quantities of water to the reagents makes little difference. With the use of a hydrated sulfate CaSO4·xH2O (x = 0.5, 2), however, the reaction is quantitative after an hour of milling in a mixer mill at both room temperature and 70 °C, demonstrating the importance of coordinated water to the reaction progress.31b A related result was observed in the formation of Ca[urea]4(H2PO4)2 from the milling of urea, urea phosphate, and a calcium source (CaCO3 or Ca(OH)2). Although the reaction will proceed with CaCO3, and thus is not technically a type 2 reaction as defined here, the rate is much faster with Ca(OH)2. A neutralization reaction between Ca(OH)2 and urea phosphate generates water, which then autocatalyzes the reaction even beyond the rate of externally added water.17c
The use of LAG or SAG to rescue reactions from undesirable outcomes is also known for organometallic systems. Previous work by our group and others has demonstrated that the preparation of substituted transition metal allyl complexes 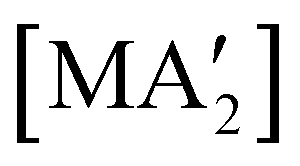 of chromium,70 iron,70a and cobalt71 from the appropriate dichloride or acetylacetonate and K[A′] proceeds in high yield (≥65% with the dichlorides) in THF solution. When attempts are made to prepare these complexes in the solid state with the dichlorides, dry grinding gives very low yields of the desired metal complexes (≤12%), and instead promotes an undesired redox reaction to afford the substituted hexadiene {A′}2 and reduced metal (Fig. 7).23 However, when small amounts of solvent are introduced to the mechanochemical reactions through LAG, the yield increases with the amount of solvent added, as shown in Fig. 8. This illustrates the need for at least small quantities of solvent in these reactions to obtain the allyl complexes in practical yields.
of chromium,70 iron,70a and cobalt71 from the appropriate dichloride or acetylacetonate and K[A′] proceeds in high yield (≥65% with the dichlorides) in THF solution. When attempts are made to prepare these complexes in the solid state with the dichlorides, dry grinding gives very low yields of the desired metal complexes (≤12%), and instead promotes an undesired redox reaction to afford the substituted hexadiene {A′}2 and reduced metal (Fig. 7).23 However, when small amounts of solvent are introduced to the mechanochemical reactions through LAG, the yield increases with the amount of solvent added, as shown in Fig. 8. This illustrates the need for at least small quantities of solvent in these reactions to obtain the allyl complexes in practical yields.
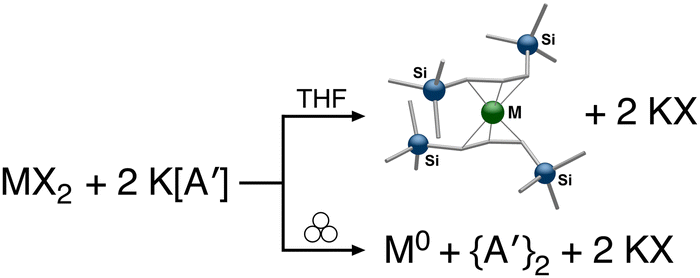 | ||
Fig. 7 Reaction of MX2 (M = Cr, Fe, Co) with K[A′] in THF produces the intended 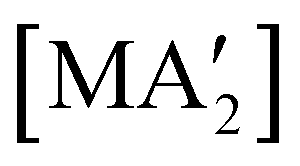 complexes via halide metathesis. Grinding the reagents together without solvent leads to a redox process with metal reduction and ligand coupling. complexes via halide metathesis. Grinding the reagents together without solvent leads to a redox process with metal reduction and ligand coupling. | ||
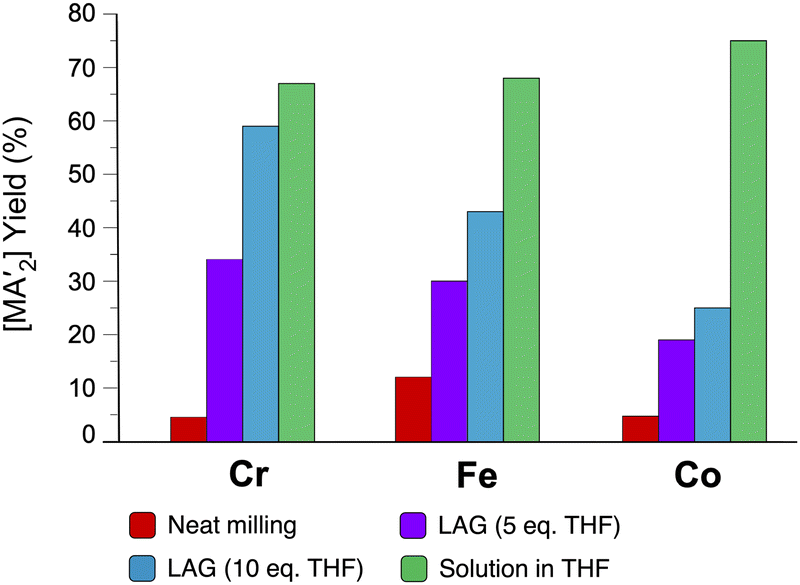 | ||
Fig. 8 Yields of 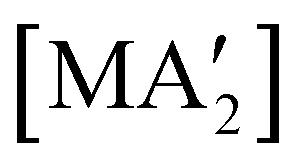 complexes (M = Cr, Fe, Co) under conditions of neat milling, two regions of LAG, and in THF solution.23 complexes (M = Cr, Fe, Co) under conditions of neat milling, two regions of LAG, and in THF solution.23 | ||
The effectiveness of LAG as a synthetic enhancement is clearly not uniform. An illustration of this is provided by the synthesis of  , a bis(allyl) complex closely related to the just-described
, a bis(allyl) complex closely related to the just-described 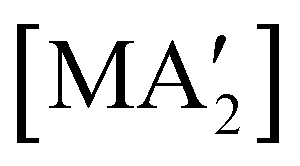 compounds. In a detailed investigation of its synthesis, a wide variety of nickel halide precursors, including solvates of THF, pyridine (py), DME, and water, were used under neat milling, milling with LAG, and solution conditions. The identity of the nickel salt remains a key variable in the outcome of the reaction, regardless of whether or not LAG is used.
compounds. In a detailed investigation of its synthesis, a wide variety of nickel halide precursors, including solvates of THF, pyridine (py), DME, and water, were used under neat milling, milling with LAG, and solution conditions. The identity of the nickel salt remains a key variable in the outcome of the reaction, regardless of whether or not LAG is used. 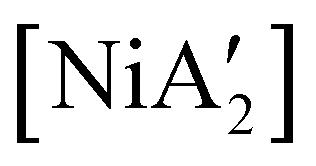 can be prepared in modest to good yields by combining soluble Ni(acac)271b or the modestly soluble [Ni(dme)Br2]72 with K[A′] in THF solution. In contrast, grinding [Ni(dme)Br2] and K[A′] in the absence of solvent provides only a trace amount (3%) of
can be prepared in modest to good yields by combining soluble Ni(acac)271b or the modestly soluble [Ni(dme)Br2]72 with K[A′] in THF solution. In contrast, grinding [Ni(dme)Br2] and K[A′] in the absence of solvent provides only a trace amount (3%) of 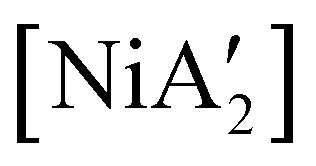 ,23 although adding THF in LAG quantities (η = 0.6) boosts the yield to >50%, approaching solution-based outcomes. In contrast, the reaction of NiCl2 and K[A′] under ball-milling conditions produces only a trace of the
,23 although adding THF in LAG quantities (η = 0.6) boosts the yield to >50%, approaching solution-based outcomes. In contrast, the reaction of NiCl2 and K[A′] under ball-milling conditions produces only a trace of the 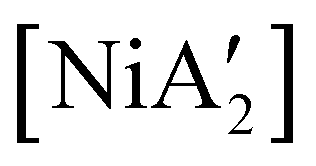 complex (the coupled allyl ligand {A′}2 and nickel black are the major products), and attempting LAG with 5 (η = 0.70) or 10 (η = 1.40) equiv. of THF yields only 1.5% and 2%, respectively, of
complex (the coupled allyl ligand {A′}2 and nickel black are the major products), and attempting LAG with 5 (η = 0.70) or 10 (η = 1.40) equiv. of THF yields only 1.5% and 2%, respectively, of 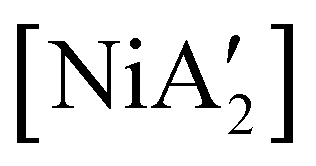 .
.
More complex is the behavior of pyridine as a regular solvent, as a LAG additive, and as a solvated nickel precursor, i.e., [Ni(py)4Cl2]. In contrast to the behavior in THF, the reaction of anhydrous NiCl2 and K[A′] in pyridine solution does gives 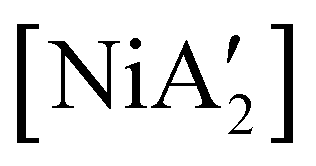 in low yield, but {A′}2 is formed as well, indicating that both metathetical and redox reaction pathways are involved. Interestingly, with the use of pyridine in LAG quantities (5 equiv. per metal center, η = 0.70), the yield of
in low yield, but {A′}2 is formed as well, indicating that both metathetical and redox reaction pathways are involved. Interestingly, with the use of pyridine in LAG quantities (5 equiv. per metal center, η = 0.70), the yield of 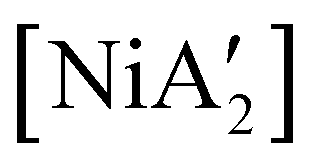 from [Ni(OH2)2Cl2] increases to 46%, with no concomitant formation of {A′}2. Finally, use of the pyridine solvate [Ni(py)4Cl2] in a dry grinding reaction (with an effective η = 0.55) proves the most successful of all, giving the nickel complex in 69% yield with no {A′}2 observed.
from [Ni(OH2)2Cl2] increases to 46%, with no concomitant formation of {A′}2. Finally, use of the pyridine solvate [Ni(py)4Cl2] in a dry grinding reaction (with an effective η = 0.55) proves the most successful of all, giving the nickel complex in 69% yield with no {A′}2 observed.
LAG is beneficial in the formation of Grignard reagents; although some investigators have worked under solvent-free conditions, as noted above, the addition of THF, 2-MeTHF or THP can greatly increase yields. For example, the Bolm group generated Grignard reagents in air (use of LiOH as an additive was beneficial), then combined them with gaseous CO2 or sodium methyl carbonate in the presence of LAG quantities of 2-MeTHF to produce carboxylic acids in a one-pot, three-step mechanochemical process.73 Nickel-catalyzed Kumada–Tamao–Corriu coupling reactions between mechanochemically synthesized organomagnesium nucleophiles and aryl tosylates under ball-milling conditions will proceed with LAG amounts of THF.47 Even more recently, the Ito group has been able to translate this chemistry to calcium-based heavy Grignard reagents; commercially-available calcium can be activated in situ by ball milling in air with an aryl halide, without the need for toxic and/or strong reducing agents (e.g. liquid NH3, lithium biphenylide) or inert atmospheres,74 conditions that are rigorously required for the solution-based counterparts.75
Type 3 reactions: only the solid-state synthesis gives the desired product, or provides a new product not observed from solution synthesis
A compelling feature of mechanochemistry is the ability to prepare novel compounds that are not accessible from solution routes.76 Apart from “green” benefits like reduced use of solvent and energy, access to previously unobtainable compounds has become one of the most exciting frontiers of synthetic mechanochemistry.16,77A straightforward example of this is the preparation of di(indenyl)beryllium, (C9H7)2Be.78 Unlike the presumably similar beryllocene, Cp2Be, that readily forms in Et2O solution,79 attempts to form the indenyl analogue in solution from BeBr2 and K[Ind] were unsuccessful, yielding only various solvated beryllium bromides. In contrast, dry grinding of a 2:1 mixture of K[Ind] and BeBr2 produced the desired (C9H7)2Be in high yield. It could be that the solvating power, driven by high Be⋯O interaction energy, that allows ethers to dissolve BeBr2 also prevents further reaction with the potassium indenide. X-ray crystallography of (C9H7)2Be revealed a monomeric, mixed-hapticity (η5/η1) metallocene, very similar to the related beryllocene. Analogous mechanochemical means were used to prepare a bulky mono(indenyl) species [Be(1,3-TMS2Ind)Br], where the indenyl group binds in an η5 manner. The same reaction again fails in solution.
An initially unanticipated development in mechanochemical synthesis is that it is possible to form solvate-free products from compounds originally made in solution, and these need not be the same as those produced mechanochemically from the same reagents. Examples are found in the chemistry of main-group allyl complexes. For instance, although π-bound, or η3, allyls are commonly found with the alkali metals and the heavier metals of group 2 (calcium, strontium, and barium), beryllium and magnesium complexes commonly display σ-bound, or η1, conformations, and the latter was thought at one point to be the preferred binding mode for magnesium.80 In fact, halide metathesis of MgBr2 with K[A′] in ethers produces the corresponding monomeric adducts 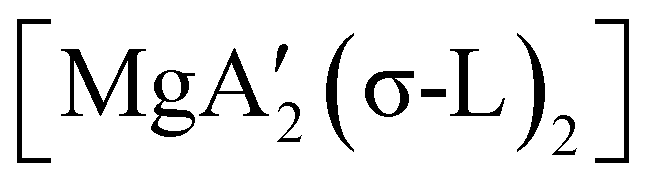 (L = THF, Et2O).10 With prolonged drying under vacuum, the Et2O adduct can be desolvated to yield the dimeric
(L = THF, Et2O).10 With prolonged drying under vacuum, the Et2O adduct can be desolvated to yield the dimeric 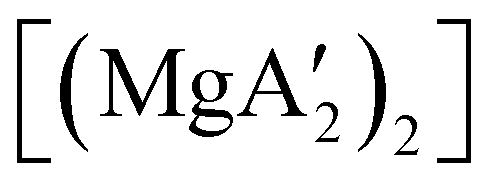 (Fig. 9(a)).10
(Fig. 9(a)).10
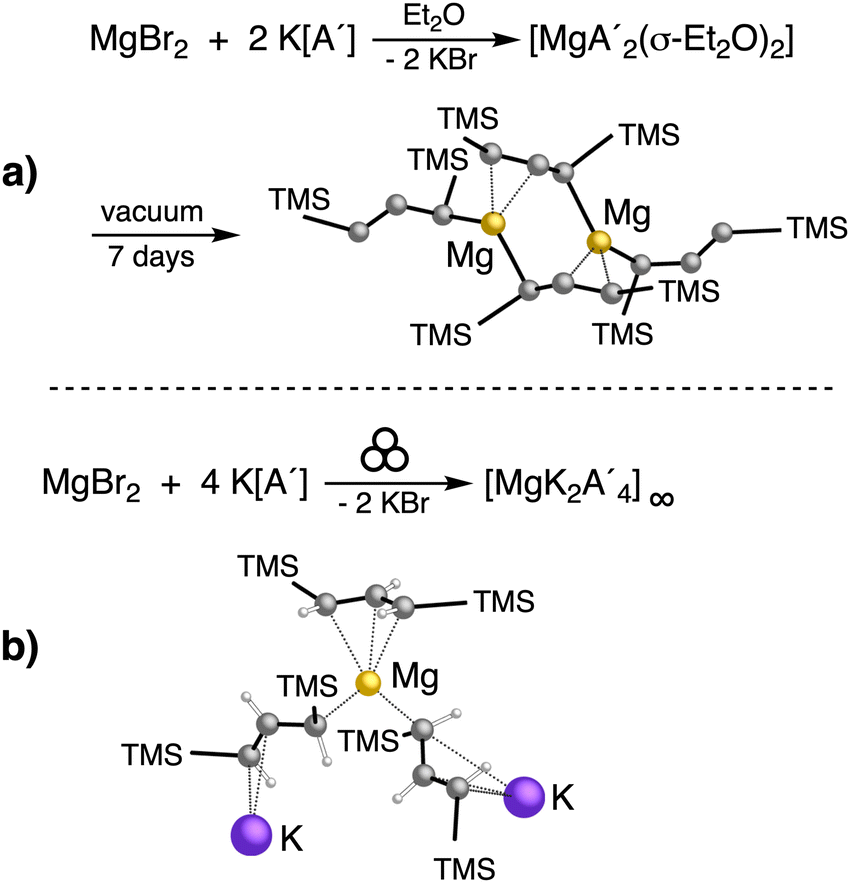 | ||
| Fig. 9 Reaction between MgBr2 and the potassium salt of the bulky ligand [A′]: (a) in ethers, a monomer with η1-bonded allyl ligands is formed. The ethers can be removed from the diethyl etherate adduct to form a dimeric complex with bridging and terminal allyl ligands; (b) the mechanochemical outcome, leading to a coordination polymer that displays an η3-bound allyl ligand. | ||
Owing to the activity displayed by the dimer as a polymerization initiator (see Section 4), a solvent-free mechanochemical route was attempted in order to avoid the lengthy desolvation process. A 10 min milling of a 1:2 ratio of MgBr2 and K[A′], followed by extraction of the ground mixture with hexanes did not lead to the formation of the neutral 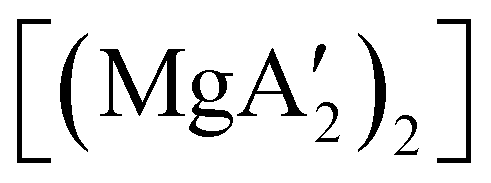 , but rather to the magnesiate product
, but rather to the magnesiate product 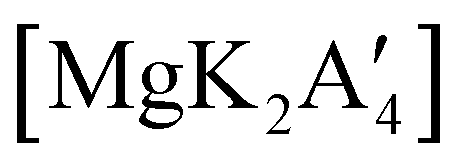 , which forms a coordination polymer in the solid state (Fig. 9(b)).81 Intriguingly, one of the ligands is η3-bound to the magnesium, which is possible only because Mg is formally 4-coordinate (the allyl occupying 2 coordination sites) which is compatible with an sp3 hybridized metal center in a way that higher-coordinate, usually solvated Mg centers are not. Two features of the synthetic outcomes are notable: (1) solvent-free versions of molecules obtained from solution-based reactions may not be the same as those generated mechanochemically; (2) although a 1:2 ratio of MgBr2 and K[A′] was ground together, a balanced equation can be written if a 1:4 ratio were operational (Fig. 9(b)). Such nonstoichiometric outcomes are noted elsewhere in this review, and are perhaps a reflection of the high-energy, far from equilibrium conditions that are often present in mechanochemical reactions.
, which forms a coordination polymer in the solid state (Fig. 9(b)).81 Intriguingly, one of the ligands is η3-bound to the magnesium, which is possible only because Mg is formally 4-coordinate (the allyl occupying 2 coordination sites) which is compatible with an sp3 hybridized metal center in a way that higher-coordinate, usually solvated Mg centers are not. Two features of the synthetic outcomes are notable: (1) solvent-free versions of molecules obtained from solution-based reactions may not be the same as those generated mechanochemically; (2) although a 1:2 ratio of MgBr2 and K[A′] was ground together, a balanced equation can be written if a 1:4 ratio were operational (Fig. 9(b)). Such nonstoichiometric outcomes are noted elsewhere in this review, and are perhaps a reflection of the high-energy, far from equilibrium conditions that are often present in mechanochemical reactions.
An even more complex relationship between solution-based and mechanochemical reactions is found in a set of tin(II) allyl complexes. First prepared in THF solution from tin(II) chloride and 3 equiv. K[A′], the anionic tris(A′)tin(II) species was isolated as a monomeric THF adduct, 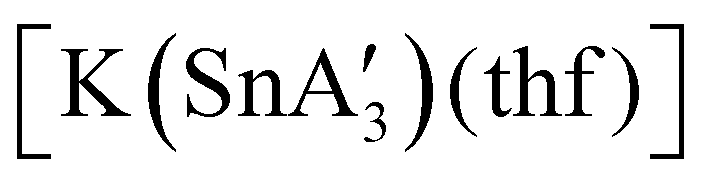 .82 Curiously, from milling a 1:2 ratio of SnCl2 to K[A′], the same stannate can be obtained, but now in the form of a coordination polymer
.82 Curiously, from milling a 1:2 ratio of SnCl2 to K[A′], the same stannate can be obtained, but now in the form of a coordination polymer 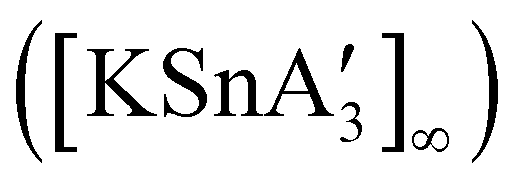 , as the THF is missing that is evidently critical for the formation of the monomeric species (Fig. 10). The solvated and unsolvated forms contain three σ-bound allyls at tin, each of which are π-bound to the potassium cation; this μ:η2:η1-bonding arrangement of each allyl to the metals is isostructural with the related beryllium83 and zinc84 tris(A′) complexes.
, as the THF is missing that is evidently critical for the formation of the monomeric species (Fig. 10). The solvated and unsolvated forms contain three σ-bound allyls at tin, each of which are π-bound to the potassium cation; this μ:η2:η1-bonding arrangement of each allyl to the metals is isostructural with the related beryllium83 and zinc84 tris(A′) complexes.
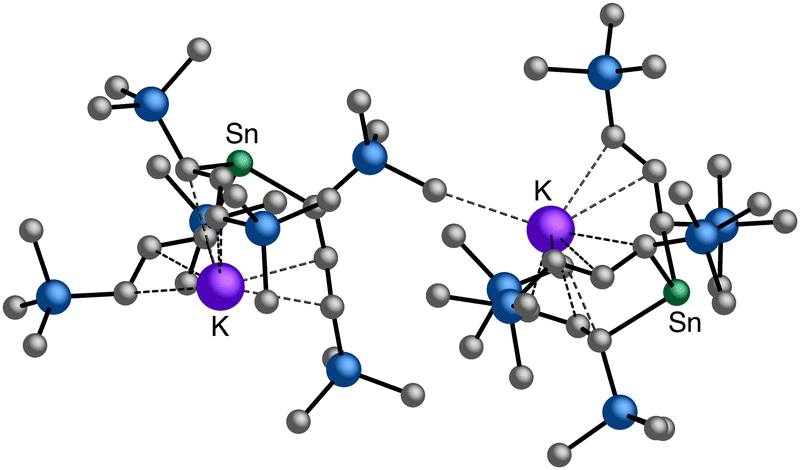 | ||
Fig. 10 Section of the coordination polymer of 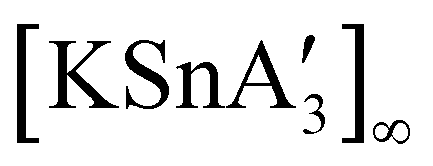 . The K⋯C(H3) contact (3.20 Å) does not exist in the solvated, solution-derived complex, although the coordination environment around the Sn(II) center is the same. . The K⋯C(H3) contact (3.20 Å) does not exist in the solvated, solution-derived complex, although the coordination environment around the Sn(II) center is the same. | ||
If the SnCl2:K[A′] ratio is adjusted to 1:3, to match that used in solution, a redox reaction occurs on 5 min of grinding, yielding a chiral Sn(IV) tetraallyl complex 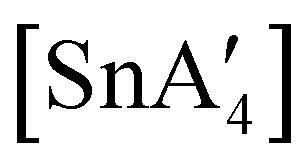 , in addition to
, in addition to 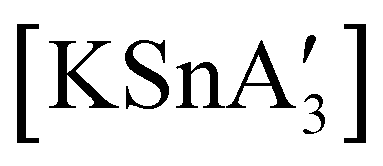 . Extending the grinding to 15 min produces a stereochemical transition to a meso (R,S,R,S) version of the same tetraallyl complex, along with the chiral-
. Extending the grinding to 15 min produces a stereochemical transition to a meso (R,S,R,S) version of the same tetraallyl complex, along with the chiral-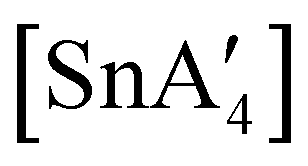 and
and 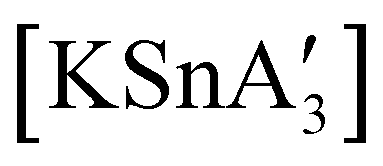 .85 The disproportionation of Sn(II) into Sn(IV) and Sn(0) appears to be vital to the success of the synthesis, as attempts to prepare
.85 The disproportionation of Sn(II) into Sn(IV) and Sn(0) appears to be vital to the success of the synthesis, as attempts to prepare 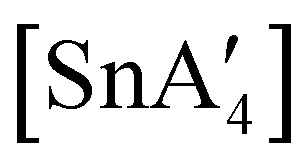 directly from tin(IV) halides are unsuccessful. In such cases, reduction–oxidation occurs, resulting in {A′}2 and tin metal as the major products.38b
directly from tin(IV) halides are unsuccessful. In such cases, reduction–oxidation occurs, resulting in {A′}2 and tin metal as the major products.38b
Metal organic frameworks (MOFs) have emerged as versatile candidates in heterogeneous catalysis, and can be used to generate invaluable organic compounds.86 In addition, MOFs can be designed with specific structural motifs that deliver improved electrochemical and physical properties, and are tunable both in terms of porosity and chemical functionality.87 Although mechanochemistry is often used to prepare MOFs that are already known in solution,67c this is not always the case, and there are topologically novel MOFs that are not available from solution-based reactions.88 As an example, mercury(II) imidazolate MOF [Hg(Im)2] was first isolated as a 3D structure with an interpenetrated diamondoid structure by Masciocchi and coworkers via a solution synthesis (Fig. 11(a)).89 Later, the MOF was prepared with mechanochemical means, which afforded a novel, more stable polymorph, based on square-grid layers.90 Attempts to reproduce the solution method afforded the same square-grid structure, with no evidence of the diamondoid structure, except as a short-lived intermediate in the mechanochemical synthesis (Fig. 11(b)). While the “disappearing polymorphs” phenomenon is a known problem for organic syntheses and pharmaceuticals, this was its first documented occurrence in metal-containing coordination compounds.90
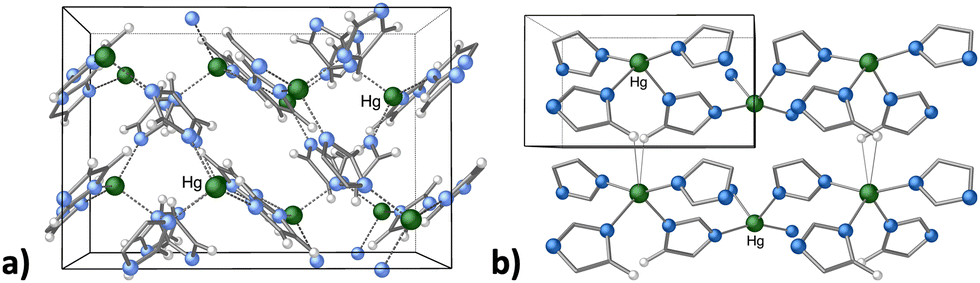 | ||
| Fig. 11 (a) Portion of the crystal structure of dia-Hg(Im)2, prepared in aqueous solution; (b) portion of the crystal structure of sql-Hg(Im)2, prepared with mechanochemical means. | ||
4. Mechanochemistry and catalysis
Mechanochemistry is a promising means to prepare new organometallic complexes to be used as catalytic initiators. In principle, the absence of coordinated solvent leaves open coordination sites for substrates to bind more easily, without needing to displace a ligand first. This hypothesis was explored with various magnesium allyls, whose preparation is discussed above, and their initiation of polymerization of methyl methacrylate (MMA). Room temperature polymerizations of MMA were performed using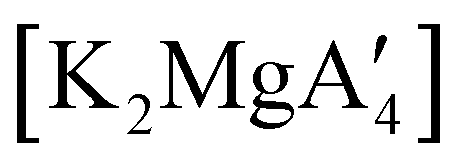 ,
, 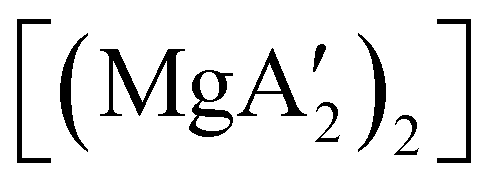 and
and 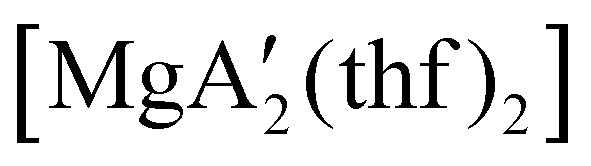 . At a monomer:initiator ratio of 100:1, the yield of PMMA for each initiator after 24 h was 72%, 19%, and 0%, respectively.81 Interestingly,
. At a monomer:initiator ratio of 100:1, the yield of PMMA for each initiator after 24 h was 72%, 19%, and 0%, respectively.81 Interestingly, 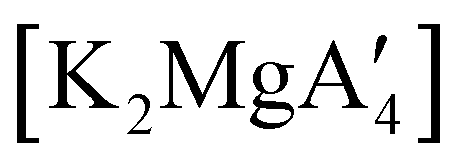 was more active at lower temperature (0 °C) and at lower concentration (1000:1), giving PMMA in 89% yield after 1 h; in contrast,
was more active at lower temperature (0 °C) and at lower concentration (1000:1), giving PMMA in 89% yield after 1 h; in contrast, 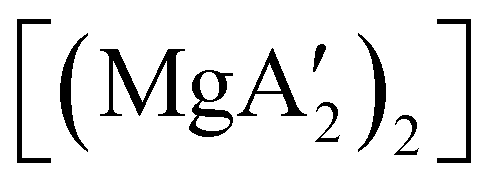 was less active, and did not initiate polymerization under these conditions. This system clearly demonstrates the enhanced ability of unsolvated magnesium allyls to polymerize MMA.
was less active, and did not initiate polymerization under these conditions. This system clearly demonstrates the enhanced ability of unsolvated magnesium allyls to polymerize MMA.
Even though 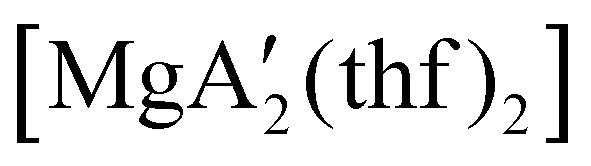 is inactive for MMA polymerization, the calcium analogue
is inactive for MMA polymerization, the calcium analogue 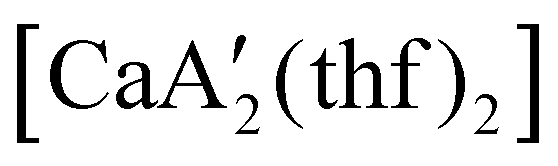 is quite active, producing isotactic PMMA in 77% yield after 30 seconds at 0 °C in toluene.64,91 Activity is reduced in THF solution to 18% conversion under the same conditions, probably due to ion solvation effects of polar solvents.92 As net negative charges correlate with anionic polymerization activity,93 an anionic calciate was sought by milling a mixture of CaI2 and K[A′]. The crystal structure of
is quite active, producing isotactic PMMA in 77% yield after 30 seconds at 0 °C in toluene.64,91 Activity is reduced in THF solution to 18% conversion under the same conditions, probably due to ion solvation effects of polar solvents.92 As net negative charges correlate with anionic polymerization activity,93 an anionic calciate was sought by milling a mixture of CaI2 and K[A′]. The crystal structure of 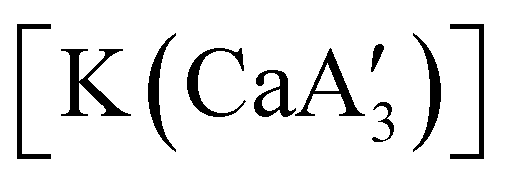 reveals it to be a coordination polymer, with η3-bound allyl ligands around calcium; one allyl is terminally bound on calcium, and two bridge calcium and potassium in a μ2-η3:η3 mode (Fig. 12).
reveals it to be a coordination polymer, with η3-bound allyl ligands around calcium; one allyl is terminally bound on calcium, and two bridge calcium and potassium in a μ2-η3:η3 mode (Fig. 12). 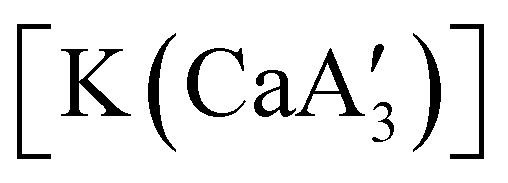 was identified as a polymerization initiator for both MMA and isoprene and compared to
was identified as a polymerization initiator for both MMA and isoprene and compared to 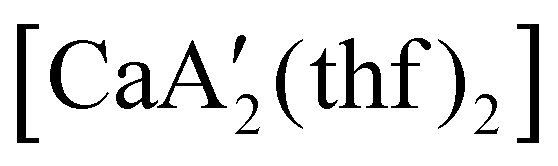 .
. 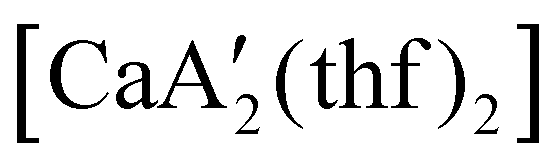 was more active for MMA polymerization, with a turnover frequency of 317 min−1 at 0 °C, while
was more active for MMA polymerization, with a turnover frequency of 317 min−1 at 0 °C, while 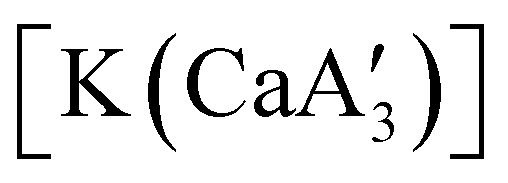 was slower at 61 min−1 at 0 °C. Both produced isotactic-enriched PMMA. The results were the inverse for polymerization of isoprene. While
was slower at 61 min−1 at 0 °C. Both produced isotactic-enriched PMMA. The results were the inverse for polymerization of isoprene. While 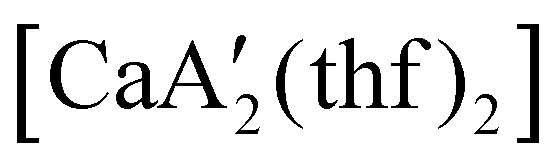 was inactive,
was inactive, 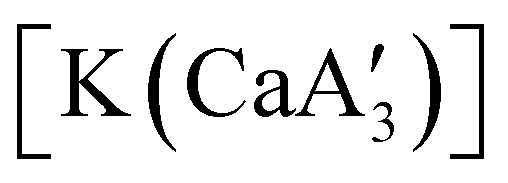 produced high molecular weight, isotactic-enriched poly(isoprene) in 98% yield after 12 h at room temperature. The role of coordinating solvents is the likely cause of the inactivity of
produced high molecular weight, isotactic-enriched poly(isoprene) in 98% yield after 12 h at room temperature. The role of coordinating solvents is the likely cause of the inactivity of 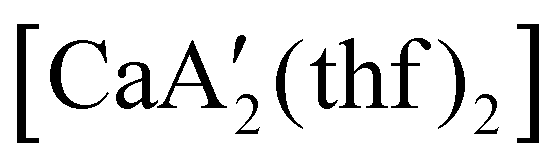 , as the addition of one equivalent of THF to
, as the addition of one equivalent of THF to 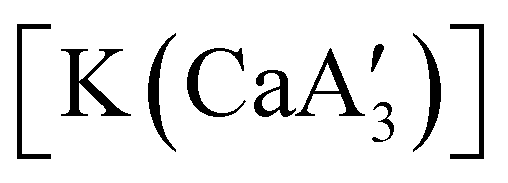 completely inhibits activity. The room temperature and atmospheric pressure used in these studies represent the mildest conditions yet reported for any calcium-based initiator for isoprene polymerization.93
completely inhibits activity. The room temperature and atmospheric pressure used in these studies represent the mildest conditions yet reported for any calcium-based initiator for isoprene polymerization.93
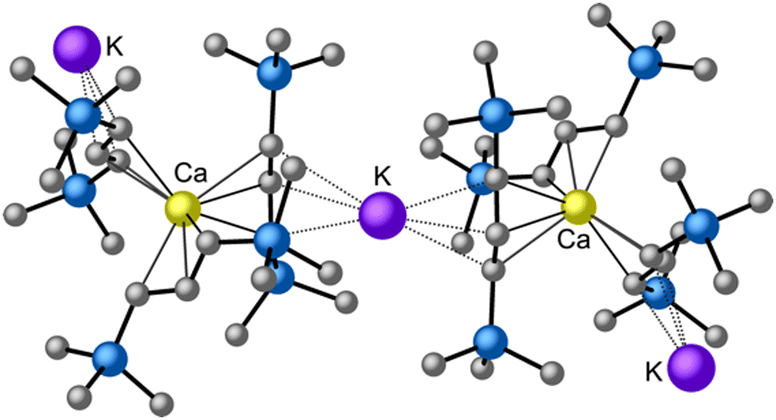 | ||
Fig. 12 Section of the coordination polymer of 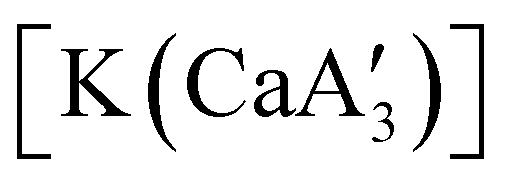 . Mechanochemically generated, it is active as an isoprene polymerization initiator; addition of THF inhibits its reactivity. . Mechanochemically generated, it is active as an isoprene polymerization initiator; addition of THF inhibits its reactivity. | ||
Owing to the low cost and toxicity, and high natural abundance of aluminium,94 the use of aluminium-based polymerization initiators is increasing in popularity. The desire to produce an unsolvated complex that could potentially serve as an initiator was a target in the preparation of a substituted tris(allyl)aluminium complex. The parent tri(allyl)aluminium is known only as a base adduct, [Al(C3H5)3·S] (S = THF, pyridine, OPPh3).95 Base-free 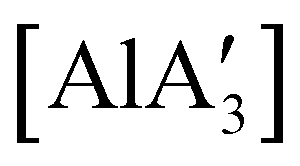 can only be accessed through mechanochemical methods, and various attempts to prepare it in solution have been unsuccessful.96 In stoichiometric reactions, e.g., an insertion reaction with benzophenone,
can only be accessed through mechanochemical methods, and various attempts to prepare it in solution have been unsuccessful.96 In stoichiometric reactions, e.g., an insertion reaction with benzophenone, 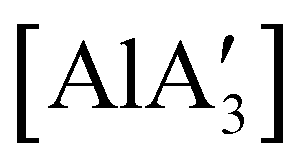 was found to be more reactive than [Al(C3H5)3·THF], a difference ascribed to the lower coordination number of the Al center. On the possibility that the improved reactivity would be maintained in a catalytic context, both
was found to be more reactive than [Al(C3H5)3·THF], a difference ascribed to the lower coordination number of the Al center. On the possibility that the improved reactivity would be maintained in a catalytic context, both 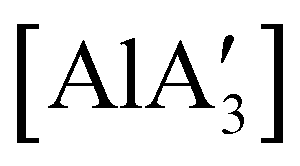 and related compounds of the form
and related compounds of the form 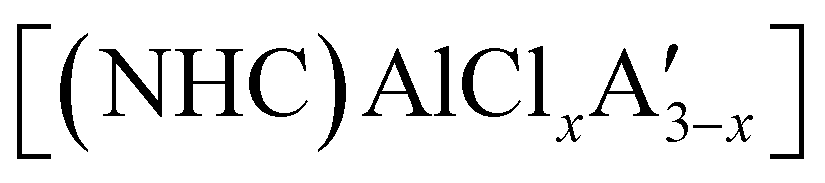 (NHC = IMes or IDipp and x = 1–3) were studied as initiators of L-lactide polymerization.97 Six NHC-containing complexes,
(NHC = IMes or IDipp and x = 1–3) were studied as initiators of L-lactide polymerization.97 Six NHC-containing complexes, 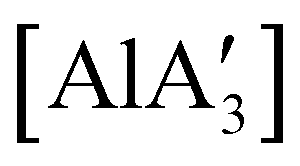 , and [Al(OiPr)3] (as a reference), were tested, and all of them initiated polymerization to some degree. Activity of the NHC complexes increased with increased allyl incorporation, but
, and [Al(OiPr)3] (as a reference), were tested, and all of them initiated polymerization to some degree. Activity of the NHC complexes increased with increased allyl incorporation, but 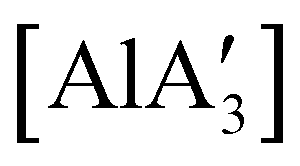 was the most effective, displaying 81% conversion after 120 h. While initiation was slow compared to [Al(OiPr)3] (comparable conversion was seen in 27 h), the polylactide generated from
was the most effective, displaying 81% conversion after 120 h. While initiation was slow compared to [Al(OiPr)3] (comparable conversion was seen in 27 h), the polylactide generated from 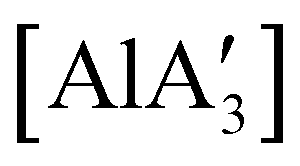 had higher molecular weight (Mn) and lower polydispersity than that produced from [Al(OiPr)3].97
had higher molecular weight (Mn) and lower polydispersity than that produced from [Al(OiPr)3].97
Direct mechanosynthesis and mechanocatalysis
Numerous mechanochemical reactions involve direct mechanosynthesis or direct mechanocatalysis, in which elemental metal additives (in the form of turnings, powder, or foil) or components of the milling assembly (vials and balls) are incorporated into products or catalyse organic transformations.77a,98 Direct mechanocatalysis is simpler than using catalyst initiators used in homogenous catalysis because many initiators have a limited shelf life, are available with erratic quality,99 often require inert atmospheres, and are difficult to recover. In contrast, certain reactions that are air- and moisture-sensitive in homogenous catalysis can be conducted in ambient atmospheres with direct mechanocatalysis.47,100 Thus, direct mechanosynthesis/catalysis is often a safer, cheaper, and easier alternative to solvothermal methods. Various transition metals including Pd and Ag have been used in these reactions.98b,101 There is substantial interest in catalytic reactions based on earth-abundant metals,102 although only a few of them have been studied via direct mechanochemistry.103Catalysis in the ball mill
Mechanochemistry invites creativity in reaction set-up. The Mack group has explored a variety of alternative methods of reaction environments to increase recyclability, decrease waste, and reduce the need for designer catalysts, by using simple metal foils as heterogeneous catalysts. This method was first introduced in the cyclopropanation of various alkenes with diazoacetate derivatives. Using silver foil and stainless-steel milling media, they were able to prepare cyclopropanation products with high diastereomeric selectivity (up to 98:2 d.r.) and high yield (up to 96%).104 The yield and selectivity were in good agreement with the results of silver(I) homogeneous catalysis, while avoiding the need for toxic solvents, refluxing conditions, and inert atmospheres.105 Silver foil out-performed CuI and Pd(OAc)2 catalysts, and stainless steel milling jars/balls were found to be important to maximize yield over copper, Teflon, or nickel jars. Silver foil was easily recycled, allowing for reproducible yields and diastereoselectivity over five catalytic runs. A broad substrate scope was achieved by modifying both the diazoacetate derivative and alkene.104
Cycloadditions of alkynes were also demonstrated using nickel pellets that serve as both the milling media and heterogeneous catalyst, without the need for additional ligands or inert atmospheres.103e Although nickel-catalysed cycloadditions of alkynes typically lead to substituted benzene rings via [2+2+2] cycloaddition, the mechanochemical route allows the inclusion of an additional equivalent into the ring to produce substituted cyclooctatetraene compounds via [2+2+2+2] addition. Nickel incorporation is vital to the reaction, whether in the form of the jar material, ball bearings, foil, powder, or pellets. The combination of nickel pellets and a stainless-steel jar was most successful, providing 94% yield after 16 hours. The use of many smaller (3 mm) nickel pellets was found to be more effective than a single large nickel ball. Using many balls simulated a planetary mill environment in the mixer mill by increasing the amount of shear friction from the balls.103e
This method was further extended to silver- and copper foil-catalysed cyclopropenation of internal and terminal alkynes with diazoacetate compounds, by lining stainless steel vials with silver or copper foil, respectively. This [2+1] cycloaddition was tested on a wide substrate scope. Building on this work and previous research on mechanochemical Sonogashira coupling,103d the Mack group attempted a one-pot, three-component coupling of a terminal alkyne, aryl halide, and a diazoacetate, catalysed by silver foil and palladium(II). Yields were excellent (80–95%) across a variety of substrates. This was expanded by using a one-pot domino Sonogashira coupling, followed by cyclopropenation to prepare a library of fully substituted cyclopropane compounds. Much better results were found by adding the diazoacetate after the initial Sonogashira reaction, but without isolation or purification of intermediates, resulting in good yields (60–75%) over two steps for the fully substituted cyclopropane products.106
Recent work from the Ito group involves the thorough exploration of mechanochemical Pd-catalyzed cross couplings, including Suzuki–Miyaura, Sonogashira, and Buchwald–Hartwig reactions.20,107 In many cases, their mechanochemical routes provide access to the same coupled products accessible via solution routes, but the mechanochemical routes provide distinct advantages, such as ease of reaction set-up, shorter reaction times, increased substrate scope, and tolerance towards air. The first step in the catalytic cycle of cross-couplings is the oxidative addition of the aryl halide to a Pd(0) species formed in situ. Pd(0) species are very sensitive to atmospheric oxygen, so reactions typically require an inert atmosphere. Kubota and Ito demonstrated that oxidative addition to Pd(0) can be done in air in the solid state, as gaseous oxygen diffuses inefficiently in solid-state reaction mixtures.100a Following this study, Pd-catalysed cross couplings were performed between aryl halides and arylboronic acids, carbazoles, aryl amines, amides, and thiols.20,107 Another exciting benefit of mechanochemical cross-couplings is an expanded substrate scope. Since solubility is no longer a factor to consider, solid-state cross-couplings are possible with insoluble polycyclic aryl halides, which are not amenable to solution methods.107e The use of LAG proved important in these systems through either a liquid reagent or a liquid additive for solid-state reactions. An olefin additive was used to act as a dispersant for the palladium catalyst, suppressing the aggregation of Pd nanoparticles and stabilizing the active Pd(0) species as a monomer, which promoted difficult solid-state C–C cross-couplings.20
Conclusions
Although the term is often overused, a genuine “paradigm shift” in synthetic chemistry was marked by the change from using water as the sole solvent to employing organic liquids (ethers, arenes) in the mid-19th century.108 The removal of all or most of the solvent from reactions in mechanochemically induced reactions has the potential to unleash a similar consequential shift, with far-reaching consequences for energy consumption, scaling, and most especially, new chemical species that have been inaccessible from traditional solvent-based approaches. The use of liquid-assisted and solvate-assisted grinding to modify the outcome of a reaction is certain to become even more widespread, and the changes in mechanism(s) of reaction will have to become an area of intensive study if the potential of mechanochemical synthesis is to be fully exploited. And although mechanochemistry may represent a “fourth way” of conducting reactions, it is also one that can be combined with others, as recent results blending mechanical approaches with photochemistry and electrochemistry have shown.109 It is clear that tremendous advances can be anticipated for mechanochemically enhanced, (nearly) solvent-free organic, inorganic, and organometallic synthetic chemistry.Author contributions
L. E. W. and T. P. H. wrote the manuscript jointly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the National Science Foundation (CHE-2155144) is gratefully acknowledged.Notes and references
- P. D. Ouspensky, in The Fourth Way, A. A. Knopf, New York, 1957 Search PubMed.
- IUPAC Compendium of Chemical Technology (the Gold Book), ed. A. A. McNaught and A. Wilkinson, Blackwell Scientific, Oxford, 1997 Search PubMed.
- (a) L. Takacs, JOM, 2000, 52, 12–13 CrossRef CAS; (b) L. Takacs, Chem. Soc. Rev., 2013, 42, 7649–7659 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- D. Tan and F. García, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC.
- J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC.
- A. A. Gečiauskaitė and F. García, Beilstein J. Org. Chem., 2017, 13, 2068–2077 CrossRef PubMed.
- A. Tuulmets and D. Panov, J. Organomet. Chem., 1999, 575, 182–186 CrossRef CAS.
- J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed.
- S. C. Chmely, C. N. Carlson, T. P. Hanusa and A. L. Rheingold, J. Am. Chem. Soc., 2009, 131, 6344–6345 CrossRef CAS PubMed.
- J. M. Andersen and H. F. Starbuck, J. Org. Chem., 2021, 86, 13983–13989 CrossRef CAS PubMed.
- D. Tan, L. Loots and T. Friščić, Chem. Commun., 2016, 52, 7760–7781 RSC.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
- F. Effaty, L. Gonnet, S. G. Koenig, K. Nagapudi, X. Ottenwaelder and T. Friščić, Chem. Commun., 2023, 59, 1010–1013 RSC.
- E. J. Holmyard, Alchemy, Penguin Books, Harmondsworth, England, 1957 Search PubMed.
- N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352–2362 RSC.
- (a) S. Lukin, L. S. Germann, T. Friščić and I. Halasz, Acc. Chem. Res., 2022, 55, 1262–1277 CrossRef CAS PubMed; (b) M. M. Mokhtar, J. M. Andersen, E. A. Kister, J. X. Hopkins, T. Estier, F. Hamilton, H. Guan, J. Mack and R. A. Haley, Eur. J. Org. Chem., 2023, e202300492 CrossRef; (c) P. A. Julien, L. S. Germann, H. M. Titi, M. Etter, R. E. Dinnebier, L. Sharma, J. Baltrusaitis and T. Friščić, Chem. Sci., 2020, 11, 2350–2355 RSC; (d) B. G. Fiss, A. J. Richard, G. Douglas, M. Kojic, T. Friščić and A. Moores, Chem. Soc. Rev., 2021, 50, 8279–8318 RSC; (e) B. Karadeniz, D. Žilić, I. Huskić, L. S. Germann, A. M. Fidelli, S. Muratović, I. Lončarić, M. Etter, R. E. Dinnebier, D. Barišić, N. Cindro, T. Islamoglu, O. K. Farha, T. Friščić and K. Užarević, J. Am. Chem. Soc., 2019, 141, 19214–19220 CrossRef CAS PubMed.
- A. A. L. Michalchuk, E. V. Boldyreva, A. M. Belenguer, F. Emmerling and V. V. Boldyrev, Front. Chem, 2021, 9 DOI:10.3389/fchem.2021.685789.
- E. R. Caley and J. F. C. Richards, Theophrastus on Stones, Ohio State University Press, Columbus, OH, 1956, pp. 204–205 Search PubMed.
- T. Seo, T. Ishiyama, K. Kubota and H. Ito, Chem. Sci., 2019, 10, 8202–8210 RSC.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed.
- L. Chen, M. Regan and J. Mack, ACS Catal., 2016, 6, 868–872 CrossRef CAS.
- H. P. DeGroot and T. P. Hanusa, Organometallics, 2021, 40, 3516–3525 CrossRef CAS.
- D. Hasa, G. Schneider Rauber, D. Voinovich and W. Jones, Angew. Chem., Int. Ed., 2015, 54, 7371–7375 CrossRef CAS PubMed.
- (a) T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66–73 CrossRef PubMed; (b) P. J. Beldon, L. Fábián, R. S. Stein, A. Thirumurugan, A. K. Cheetham and T. Friščić, Angew. Chem., Int. Ed., 2010, 49, 9640–9643 CrossRef CAS PubMed.
- Y. X. Shi, K. Xu, J. K. Clegg, R. Ganguly, H. Hirao, T. Friščić and F. García, Angew. Chem., Int. Ed., 2016, 55, 12736–12740 CrossRef CAS PubMed.
- S. Karki, T. Friščić, W. Jones and W. D. S. Motherwell, Mol. Pharmaceutics, 2007, 4, 347–354 CrossRef CAS PubMed.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC.
- M. Rodrigues, B. Baptista, J. A. Lopes and M. C. Sarraguça, Int. J. Pharm., 2018, 547, 404–420 CrossRef CAS PubMed.
- (a) T. Friščić, J. Mater. Chem., 2010, 20, 7599–7605 RSC; (b) M. Tireli, M. Juribašić Kulcsár, N. Cindro, D. Gracin, N. Biliškov, M. Borovina, M. Ćurić, I. Halasz and K. Užarević, Chem. Commun., 2015, 51, 8058–8061 RSC.
- (a) M. Arhangelskis, D.-K. Bučar, S. Bordignon, M. R. Chierotti, S. A. Stratford, D. Voinovich, W. Jones and D. Hasa, Chem. Sci., 2021, 12, 3264–3269 RSC; (b) I. Brekalo, V. Martinez, B. Karadeniz, P. Orešković, D. Drapanauskaite, H. Vriesema, R. Stenekes, M. Etter, I. Dejanović, J. Baltrusaitis and K. Užarević, ACS Sustainable Chem. Eng., 2022, 10, 6743–6754 CrossRef CAS.
- J. A. Cabeza, J. Fernandez Reynes, F. García, P. García-Álvarez and R. García-Soriano, Chem. Sci., 2023 10.1039/D3SC02709K.
- K. Budny-Godlewski, M. K. Leszczyński, A. Tulewicz, I. Justyniak, D. Pinkowicz, B. Sieklucka, K. Kruczała, Z. Sojka and J. Lewiński, ChemSusChem, 2021, 14(18), 3887–3894 CrossRef CAS PubMed.
- D. H. Woen, C. M. Kotyk, T. J. Mueller, J. W. Ziller and W. J. Evans, Organometallics, 2017, 36, 4558–4563 CrossRef CAS.
- V. D. Makhaev, A. P. Borisov and L. A. Petrova, J. Organomet. Chem., 1999, 590, 222–226 CrossRef CAS.
- G. Wilkinson, Org. Synth., 1956, 36, 31–32 CrossRef CAS.
- (a) C. S. B. Gomes, P. T. Gomes and M. T. Duarte, J. Organomet. Chem., 2014, 760, 101–107 CrossRef CAS; (b) H. P. Fritz, F. H. Köhler and K. E. Schwarzhans, J. Organomet. Chem., 1969, 19, 449–452 CrossRef CAS.
- (a) C. K. Gren, T. P. Hanusa and W. W. Brennessel, Polyhedron, 2006, 25, 286–292 CrossRef CAS; (b) N. R. Rightmire, PhD Thesis., Vanderbilt University, 2015.
- A. P. Borisov, V. D. Makhaev, A. Y. Usyatinskii and V. I. Bregadze, Izv. Akad. Nauk, Ser. Khim., 1993, 1715–1717 CAS.
- N. C. Boyde, N. R. Rightmire, E. J. Bierschenk, G. W. Steelman, T. P. Hanusa and W. W. Brennessel, Dalton Trans., 2016, 45, 18635–18642 RSC.
- J. Lewiński, M. Dutkiewicz, M. Lesiuk, W. Śliwiński, K. Zelga, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2010, 49(44), 8266–8269 CrossRef PubMed.
- M. C. Blanco, J. Cámara, M. C. Gimeno, A. Laguna, S. L. James, M. C. Lagunas and M. D. Villacampa, Angew. Chem., Int. Ed., 2012, 51, 9777–9779 CrossRef CAS PubMed.
- (a) V. Grignard, C. R. Acad. Sci., 1900, 130, 1322–1324 CAS; (b) T. Banno, Y. Hayakawa and M. Umeno, J. Organomet. Chem., 2002, 653, 288–291 CrossRef CAS; (c) D. Seyferth, Organometallics, 2009, 28, 1598–1605 CrossRef CAS.
- J. M. Harrowfield, R. J. Hart and C. R. Whitaker, Aust. J. Chem., 2001, 54, 423–425 CrossRef CAS.
- V. Birke, C. Schutt, H. Burmeier and W. Ruck, Fresenius Environ. Bull., 2011, 20, 2794–2805 CAS.
- I. R. Speight and T. P. Hanusa, Molecules, 2020, 25, 570 CrossRef CAS.
- R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, J. Jiang, S. Maeda, H. Takaya, K. Kubota and H. Ito, Nat. Commun., 2021, 12, 6691 CrossRef CAS PubMed.
- Y. Gao, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2023, 62, e202217723 CrossRef CAS PubMed.
- R. Scholl and J. Mansfeld, Ber. Dtsch. Chem. Ges., 1910, 43, 1734–1746 CrossRef CAS.
- M. Rickhaus, A. P. Belanger, H. A. Wegner and L. T. Scott, J. Org. Chem., 2010, 75, 7358–7364 CrossRef CAS PubMed.
- K. Fujishiro, Y. Morinaka, Y. Ono, T. Tanaka, L. T. Scott, H. Ito and K. Itami, J. Am. Chem. Soc., 2023, 145, 8163–8175 CrossRef CAS.
- A. Sartori, N. El Habra, C. De Zorzi, S. Sitran, M. Casarin, G. Cavinato, C. Sada, R. Gerbasi and G. Rossetto, Chem. Vap. Deposition, 2012, 18, 151–158 CrossRef CAS.
- J. Niinistö, M. Putkonen, L. Niinistö, F. Song, P. Williams, P. N. Heys and R. Odedra, Chem. Mater., 2007, 19, 3319–3324 CrossRef.
- (a) T.-J. Hsiao and J.-C. Tsai, J. Appl. Polym. Sci., 2010, 116, 2040–2049 CrossRef CAS; (b) A. V. Grafov, C. L. Firme, I. A. Grafova, F. Benetollo, M. L. Dias and M. J. M. Abadie, Polymer, 2005, 46, 9626–9631 CrossRef CAS.
- (a) Z. R. Turner, J.-C. Buffet and D. O’Hare, Organometallics, 2014, 33, 3891–3903 CrossRef CAS; (b) J.-C. Buffet, G. R. Harris, J. J. Coward, T. A. Q. Arnold, Z. R. Turner and D. O’Hare, J. Organomet. Chem., 2016, 801, 87–95 CrossRef CAS; (c) Y. Ning, Y. Zhang, A. Rodriguez-Delgado and E. Y. X. Chen, Organometallics, 2008, 27, 5632–5640 CrossRef CAS.
- J.-C. Tsai, J. C. Kuo and Y.-C. Chen, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2304–2315 CrossRef.
- M. W. Carson, e-EROS Encyclopedia of Reagents for Organic Synthesis, 2003, pp.1–2 Search PubMed.
- K. C. Ott, E. J. M. De Boer and R. H. Grubbs, Organometallics, 1984, 3, 223–230 CrossRef CAS.
- N. R. Rightmire, D. L. Bruns, T. P. Hanusa and W. W. Brennessel, Organometallics, 2016, 35, 1698–1706 CrossRef CAS.
- (a) P. S. Tanner, D. J. Burkey and T. P. Hanusa, Polyhedron, 1995, 14, 331–333 CrossRef CAS; (b) E. D. Brady, S. C. Chmely, K. C. Jayaratne, T. P. Hanusa and V. G. Young, Jr., Organometallics, 2008, 27, 1612–1616 CrossRef CAS; (c) A. G. M. Barrett, M. R. Crimmin, M. S. Hill, G. Kociok-Kohn, D. J. MacDougall, M. F. Mahon and P. A. Procopiou, Organometallics, 2008, 27, 3939–3946 CrossRef CAS.
- (a) X. He, B. C. Noll, A. Beatty, R. E. Mulvey and K. W. Henderson, J. Am. Chem. Soc., 2004, 126, 7444–7445 CrossRef CAS PubMed; (b) X. He, E. Hurley, B. C. Noll and K. W. Henderson, Organometallics, 2008, 27, 3094–3102 CrossRef CAS.
- (a) H. Bauer, M. Alonso, C. Färber, H. Elsen, J. Pahl, A. Causero, G. Ballmann, F. De Proft and S. Harder, Nat. Catal., 2018, 1, 40–47 CrossRef CAS; (b) H. Bauer, M. Alonso, C. Fischer, B. Roesch, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2018, 57, 15177–15182 CrossRef CAS PubMed; (c) J. P. Davin, J.-C. Buffet, T. P. Spaniol and J. Okuda, Dalton Trans., 2012, 41, 12612–12618 RSC.
- (a) M. P. Coles, Coord. Chem. Rev., 2015, 297–298, 2–23 CrossRef CAS; (b) M. Westerhausen and W. Schwarz, Z. Anorg. Allg. Chem., 1991, 604, 127–140 CrossRef CAS.
- (a) P. B. Hitchcock, M. F. Lappert, G. A. Lawless and B. Royo, J. Chem. Soc., Chem. Commun., 1990, 1141–1142 RSC; (b) S. R. Drake, D. J. Otway and S. P. Perlepes, Main Group Met. Chem., 1991, 14, 243–256 CAS; (c) D. C. Bradley, M. B. Hursthouse, A. A. Ibrahim, K. M. Abdul Malik, M. Motevalli, R. Möseler, H. Powell, J. D. Runnacles and A. C. Sullivan, Polyhedron, 1990, 9, 2959–2964 CrossRef CAS; (d) M. M. Gillett-Kunnath, J. G. MacLellan, C. M. Forsyth, P. C. Andrews, G. B. Deacon and K. Ruhlandt-Senge, Chem. Commun., 2008, 4490–4492 RSC; (e) M. Westerhausen, Inorg. Chem., 1991, 30, 96–101 CrossRef CAS.
- (a) S. Krieck, P. Schueler, H. Goerls and M. Westerhausen, Inorg. Chem., 2018, 57, 13937–13943 CrossRef CAS PubMed; (b) E. D. Brady, T. P. Hanusa, M. Pink and V. G. Young, Jr., Inorg. Chem., 2000, 39, 6028–6037 CrossRef CAS; (c) A. D. Frankland, P. B. Hitchcock, M. F. Lappert and G. A. Lawless, J. Chem. Soc., Chem. Commun., 1994, 2435–2436 RSC; (d) A. D. Frankland and M. F. Lappert, J. Chem. Soc., Dalton Trans., 1996, 4151–4152 RSC; (e) P. B. Hitchcock, M. F. Lappert, G. A. Lawless and B. Royo, J. Chem. Soc., Chem. Commun., 1990, 1141–1142 RSC.
- J. F. Reynes, V. Isoni and F. García, Angew. Chem., Int. Ed., 2023, 62, e202300819 CrossRef CAS PubMed.
- (a) D. J. am Ende, S. R. Anderson and J. S. Salan, Org. Process Res. Dev., 2014, 18, 331–341 CrossRef CAS; (b) A. A. L. Michalchuk, K. S. Hope, S. R. Kennedy, M. V. Blanco, E. V. Boldyreva and C. R. Pulham, Chem. Commun., 2018, 54, 4033–4036 RSC; (c) H. M. Titi, J.-L. Do, A. J. Howarth, K. Nagapudi and T. Friščić, Chem. Sci., 2020, 11, 7578–7584 RSC.
- S. Okeya, S. i Ooi, K. Matsumoto, Y. Nakamura and S. Kawaguchi, Bull. Chem. Soc. Jpn., 1981, 54, 1085–1095 CrossRef CAS.
- V. Makhaev and L. Petrova, Inorg. Chim. Acta, 2021, 518, 120231 CrossRef CAS.
- (a) J. D. Smith, T. P. Hanusa and V. G. Young, Jr., J. Am. Chem. Soc., 2001, 123, 6455–6456 CrossRef CAS PubMed; (b) C. N. Carlson, J. D. Smith, T. P. Hanusa, W. W. Brennessel and V. G. Young, Jr., J. Organomet. Chem., 2003, 683, 191–199 CrossRef CAS.
- (a) J. D. Smith, K. T. Quisenberry, T. P. Hanusa and W. W. Brennessel, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, m507–m508 CrossRef; (b) M. Schormann, S. Garratt and M. Bochmann, Organometallics, 2005, 24, 1718–1724 CrossRef CAS.
- K. T. Quisenberry, J. D. Smith, M. Voehler, D. F. Stec, T. P. Hanusa and W. W. Brennessel, J. Am. Chem. Soc., 2005, 127, 4376–4387 CrossRef CAS PubMed.
- V. S. Pfennig, R. C. Villella, J. Nikodemus and C. Bolm, Angew. Chem., Int. Ed., 2022, 61, e202116514 CrossRef CAS PubMed.
- P. Gao, J. Jiang, S. Maeda, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2022, 61, e202207118 CrossRef CAS PubMed.
- A. Koch, Q. Dufrois, M. Wirgenings, H. Görls, S. Krieck, M. Etienne, G. Pohnert and M. Westerhausen, Chem. – Eur. J., 2018, 24, 16840–16850 CrossRef CAS PubMed.
- F. Cuccu, L. De Luca, F. Delogu, E. Colacino, N. Solin, R. Mocci and A. Porcheddu, ChemSusChem, 2022, 15, e202200362 CrossRef CAS PubMed.
- (a) J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed; (b) G.-W. Wang, Chin. J. Chem., 2021, 39, 1797–1803 CrossRef CAS.
- R. F. Koby, N. D. Schley and T. P. Hanusa, Angew. Chem., Int. Ed., 2021, 60, 21174–21178 CrossRef CAS PubMed.
- E. O. Fischer and H. P. Hofmann, Chem. Ber., 1959, 92, 482–486 CrossRef CAS.
- S. A. Solomon, C. A. Muryn and R. A. Layfield, Chem. Commun., 2008, 3142–3144 RSC.
- R. F. Koby, A. M. Doerr, N. R. Rightmire, N. D. Schley, B. K. Long and T. P. Hanusa, Angew. Chem., Int. Ed., 2020, 59, 9542–9548 CrossRef CAS PubMed.
- R. A. Layfield, F. García, J. Hannauer and S. M. Humphrey, Chem. Commun., 2007, 5081–5083 RSC.
- N. Boyde, N. Rightmire, T. Hanusa and W. Brennessel, Inorganics, 2017, 5, 36 CrossRef.
- C. K. Gren, T. P. Hanusa and A. L. Rheingold, Organometallics, 2007, 26, 1643–1649 CrossRef CAS.
- R. F. Koby, T. P. Hanusa and N. D. Schley, J. Am. Chem. Soc., 2018, 140, 15934–15942 CrossRef CAS PubMed.
- K. K. Gangu and S. B. Jonnalagadda, Front. Chem., 2021, 9, 747615 CrossRef CAS PubMed.
- I. Thomas-Hillman, A. Laybourn, C. Dodds and S. W. Kingman, J. Mater. Chem. A, 2018, 6, 11564–11581 RSC.
- (a) A. D. Katsenis, A. Puškarić, V. Štrukil, C. Mottillo, P. A. Julien, K. Užarević, M.-H. Pham, T.-O. Do, S. A. J. Kimber, P. Lazić, O. Magdysyuk, R. E. Dinnebier, I. Halasz and T. Friščić, Nat. Commun., 2015, 6, 6662 CrossRef CAS PubMed; (b) T. Stolar, L. Batzdorf, S. Lukin, D. Žilić, C. Motillo, T. Friščić, F. Emmerling, I. Halasz and K. Užarević, Inorg. Chem., 2017, 56, 6599–6608 CrossRef CAS PubMed; (c) M. Arhangelskis, A. D. Katsenis, N. Novendra, Z. Akimbekov, D. Gandrath, J. M. Marrett, G. Ayoub, A. J. Morris, O. K. Farha, T. Friščić and A. Navrotsky, Chem. Mater., 2019, 31, 3777–3783 CrossRef CAS.
- N. Masciocchi, G. Attilio Ardizzoia, S. Brenna, F. Castelli, S. Galli, A. Maspero and A. Sironi, Chem. Commun., 2003, 2018–2019 RSC.
- I. R. Speight, I. Huskić, M. Arhangelskis, H. M. Titi, R. S. Stein, T. P. Hanusa and T. Friščić, Chem. – Eur. J., 2020, 26, 1811–1816 CrossRef CAS PubMed.
- K. T. Quisenberry, R. E. White, T. P. Hanusa and W. W. Brennessel, New J. Chem., 2010, 34, 1579–1584 RSC.
- G. Odian, Principles of Polymerization, John Wiley, New York, 3rd edn, 1991 Search PubMed.
- R. F. Koby, A. M. Doerr, N. R. Rightmire, N. D. Schley, W. W. Brennessel, B. K. Long and T. P. Hanusa, Chem. – Eur. J., 2021, 27, 8195–8202 CrossRef CAS PubMed.
- J. Gao, D. Zhu, W. Zhang, G. A. Solan, Y. Ma and W.-H. Sun, Inorg. Chem. Front., 2019, 6, 2619–2652 RSC.
- C. Lichtenberg, D. Robert, T. P. Spaniol and J. Okuda, Organometallics, 2010, 29, 5714–5721 CrossRef CAS.
- N. R. Rightmire, T. P. Hanusa and A. L. Rheingold, Organometallics, 2014, 33, 5952–5955 CrossRef CAS.
- (a) L. E. Wenger, N. M. Shawver, R. F. Koby, W. W. Brennessel, B. K. Long and T. P. Hanusa, Organometallics, 2023, 42, 283 CrossRef CAS; (b) L. E. Wenger, N. M. Shawver, W. W. Brennessel, B. K. Long and T. P. Hanusa, Organometallics, 2022, 41, 3718–3723 CrossRef CAS.
- (a) A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS; (b) S. Hwang, S. Grätz and L. Borchardt, Chem. Commun., 2022, 58, 1661–1671 RSC; (c) W. Pickhardt, S. Grätz and L. Borchardt, Chem. – Eur. J., 2020, 26, 12903–12911 CrossRef CAS PubMed.
- W. A. Carole and T. J. Colacot, Chem. – Eur. J., 2016, 22, 7686–7695 CrossRef CAS PubMed.
- (a) K. Kubota, R. Takahashi and H. Ito, Chem. Sci., 2019, 10, 5837–5842 RSC; (b) Y. Pang, T. Ishiyama, K. Kubota and H. Ito, Chem. – Eur. J., 2019, 25, 4654–4659 CrossRef CAS PubMed; (c) D. C. Waddell, T. D. Clark and J. Mack, Tetrahedron Lett., 2012, 53, 4510–4513 CrossRef CAS.
- A. C. Jones, J. A. Leitch, S. E. Raby-Buck and D. L. Browne, Nat. Synth., 2022, 1, 763–775 CrossRef.
- R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef CAS PubMed.
- (a) Y. Sawama, M. Niikawa and H. Sajiki, J. Synth. Org. Chem., Jpn., 2019, 77, 1070–1077 CrossRef CAS; (b) Y. Sawama, M. Niikawa, Y. Yabe, R. Goto, T. Kawajiri, T. Marumoto, T. Takahashi, M. Itoh, Y. Kimura, Y. Sasai, Y. Yamauchi, S.-I. Kondo, M. Kuzuya, Y. Monguchi and H. Sajiki, ACS Sustainable Chem. Eng., 2015, 3, 683–689 CrossRef CAS; (c) T. L. Cook, J. A. Walker and J. Mack, Green Chem., 2013, 15, 617–619 RSC; (d) D. A. Fulmer, W. C. Shearouse, S. T. Medonza and J. Mack, Green Chem., 2009, 11, 1821–1825 RSC; (e) R. A. Haley, A. R. Zellner, J. A. Krause, H. Guan and J. Mack, ACS Sustainable Chem. Eng., 2016, 4, 2464–2469 CrossRef CAS.
- L. Chen, M. O. Bovee, B. E. Lemma, K. S. M. Keithley, S. L. Pilson, M. G. Coleman and J. Mack, Angew. Chem., Int. Ed., 2015, 54, 11084–11087 CrossRef CAS PubMed.
- J. L. Thompson and H. M. L. Davies, J. Am. Chem. Soc., 2007, 129, 6090–6091 CrossRef CAS PubMed.
- L. Chen, D. Leslie, M. G. Coleman and J. Mack, Chem. Sci., 2018, 9, 4650–4661 RSC.
- (a) T. Seo, K. Kubota and H. Ito, J. Am. Chem. Soc., 2023, 145, 6823–6837 CrossRef CAS PubMed; (b) K. Kubota, T. Seo and H. Ito, Faraday Discuss., 2023, 241, 104–113 RSC; (c) K. Kubota, K. Kondo, T. Seo and H. Ito, Synlett, 2022, 898–902 CrossRef CAS; (d) K. Kubota, T. Endo, M. Uesugi, Y. Hayashi and H. Ito, ChemSusChem, 2022, 15, e202102132 CrossRef CAS PubMed; (e) T. Seo, N. Toyoshima, K. Kubota and H. Ito, J. Am. Chem. Soc., 2021, 143, 6165–6175 CrossRef CAS PubMed; (f) K. Kubota, R. Takahashi, M. Uesugi and H. Ito, ACS Sustainable Chem. Eng., 2020, 8, 16577–16582 CrossRef CAS; (g) T. Seo, K. Kubota and H. Ito, J. Am. Chem. Soc., 2020, 142, 9884–9889 CrossRef CAS PubMed; (h) R. Takahashi, K. Kubota and H. Ito, Chem. Commun., 2020, 56, 407–410 RSC; (i) K. Kubota, T. Seo, K. Koide, Y. Hasegawa and H. Ito, Nat. Commun., 2019, 10, 111 CrossRef PubMed.
- T. S. Kuhn, The Structure of Scientific Revolutions, University of Chicago Press, Chicago, IL, 2nd edn, 1970 Search PubMed.
- V. Martinez, T. Stolar, B. Karadeniz, I. Brekalo and K. Užarević, Nat. Rev. Chem., 2023, 7, 51–65 CrossRef CAS PubMed.
Footnote |
| † What value of η should represent the LAG region has varied over time, and given the indifference of the metric to the identity of the solvent added, precise boundaries could hardly be expected. Nevertheless, the original paper that defined LAG (ref. 28) concluded: “In our experience, LAG would correspond to η levels … below 1 μL mg−1.” That value has increased over the years, and a region from η = 0–2 is now commonly cited as the region where distinctive LAG reactivity is to be expected (ref. 21). |
| This journal is © The Royal Society of Chemistry 2023 |