
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom sandwich complexes to dendrimers: journey toward applications to sensing, molecular electronics, materials science, and biomedicine†
Didier
Astruc

Univ. Bordeaux, ISM, UMR CNRS No. 5255, 351 Cours de la Libération, 33405 Talence Cedex, France. E-mail: didier.astruc@u-bordeaux.fr
First published on 3rd May 2023
Abstract
This review links various areas of inorganic chemistry around the themes developed by our research group during the last four decades. It is firstly based on the electronic structure of iron sandwich complexes, showing how the metal electron count dictates their reactivities, with various applications (via C–H activation, C–C bond formation) as reducing and oxidizing agents, redox and electrocatalysts and precursors of dendrimers and catalyst templates through bursting reactions. Various electron-transfer processes and consequences are explored, including the influence of the redox state on the acidity of robust ligands and the possibility to iterate in situ C–H activation and C–C bond formation to build arene-cored dendrimers. Examples of how these dendrimers are functionalized are illustrated using the cross olefin metathesis reactions, with application to the synthesis of soft nanomaterials and biomaterials. Mixed and average valence complexes give rise to remarkable subsequent organometallic reactions, including the salt influence on these reactions. The stereo-electronic aspect of these mixed valencies is pointed out in star-shaped multi-ferrocenes with a frustration effect and other multi-organoiron systems, with the perspective of understanding electron-transfer processes among dendrimer redox sites involving electrostatic effects and application to redox sensing and polymer metallocene batteries. Dendritic redox sensing is summarized for biologically relevant anions such as ATP2− with supramolecular exoreceptor interactions at the dendrimer periphery in parallel with the seminal work on metallocene-derived endoreceptors by Beer's group. This aspect includes the design of the first metallodendrimers that have applications in both redox sensing and micellar catalysis with nanoparticles. These properties provide the opportunity to summarize the biomedical (mostly anticancer) applications of ferrocenes, dendrimers and dendritic ferrocenes in biomedicine (in particular the contribution from our group, but not only). Finally, the use of dendrimers as templates for catalysis is illustrated with numerous reactions including C–C bond formation, click reactions and H2 production reactions.
 Didier Astruc | Didier Astruc born in Versailles, studied in Rennes, where he obtained two doctorate degrees with prof. René Dabard, then was a NATO post-doctoral fellow with prof. Richard R. Schrock at M. I. T., Cambridge, USA, and later spent a sabbatical year with prof. K. Peter C. Vollhardt at the University of California, Berkeley. He is professor of chemistry at the University of Bordeaux, member of the Institut Universitaire de France and of the French, German (Leopoldina) and several European Academies, and Fellow of the Royal Society of Chemistry and Chemistry Europe. His presents interests are in nanomaterials and hydrogen energy. |
I. Introduction
Shortly following Pauson's report 70 years ago of ferrocene with an erroneous 10-electron electronic structure1 violating the century-old Langmuir's 18-electron rule,2 the aromatic ground for the extraordinary robustness of ferrocene was established, justifying Woodward's proposal of the ferrocene name.3 The subsequent flourishing chemistry was more that of a tridimensional super-aromatic organic compound than that representative of organo-transition-metal chemistry.4 The outstanding ferrocene stability, connected to its 18-electron structure,4–6 allowed formation of a rather stable cation, 17-electron ferricenium in a variety of salts, in spite of the loss of one electron from an essentially non-bonding orbital (and Fe–Cp distance only slightly increasing compared to ferrocene), whereas most organometallic compounds do not withstand such a monoelectronic oxidation, which provokes their breakdown.7 The stability of late transition-metal sandwich complexes in at least two oxidation states was used at the beginning of our research to provide a variety of electron and proton transfer processes. The latter reaction type was at the origin of the discovery of starburst reactions leading to stars and dendrimers. Then, some analogies between metallodendrimers and metal nanoparticles led us to examine and exploit their supramolecular interplay with applications to sensing, molecular electronics, materials science, catalysis and biomedicine. Herein, a review of this interdisciplinary research path and its applications is proposed.II. Applications of the 17 and 19-electron late transition-metal sandwich complexes as reservoirs of electron holes and electrons
There is a rare truly organometallic reaction of ferrocene that deserved interest, namely its Lewis-acid induced cyclopentadienyl substitution by arenes upon mild heating leading to cationic arene complexes [FeCp(η6-arene)]+X− with Cp = η5-C5H5, X− = PF6−, in which the robust 18-electron sandwich Fe(II) structure is preserved.8 The cationic nature of these arene Fe(II) complexes allowed an “Umpolung” of arene reactivity including, for instance, reversible deprotonation of the 18-electron form, which turned out to be synthetically productive (Scheme 1, bottom). The stability of their neutral 19-electron Fe(I) form such as 1a facilitated by the presence of six methyl substituents on the arene allowed its full characterization including inter alia X-ray crystal structure,91H NMR (paramagnetism),9 redox potentials,10 EPR (rhombic distortion)11 and He(I) photoelectron spectroscopy (extremely low value of the ionization potential).12 The exceptional electron richness of this new family of transition-metal sandwich complexes that were the first characterized stable 19-electron complexes with the new stable Fe(I) oxidation state in monometallic complexes yielded a variety of electron-transfer reactions starting from that with O2 upon short contact with air (Scheme 1).12,13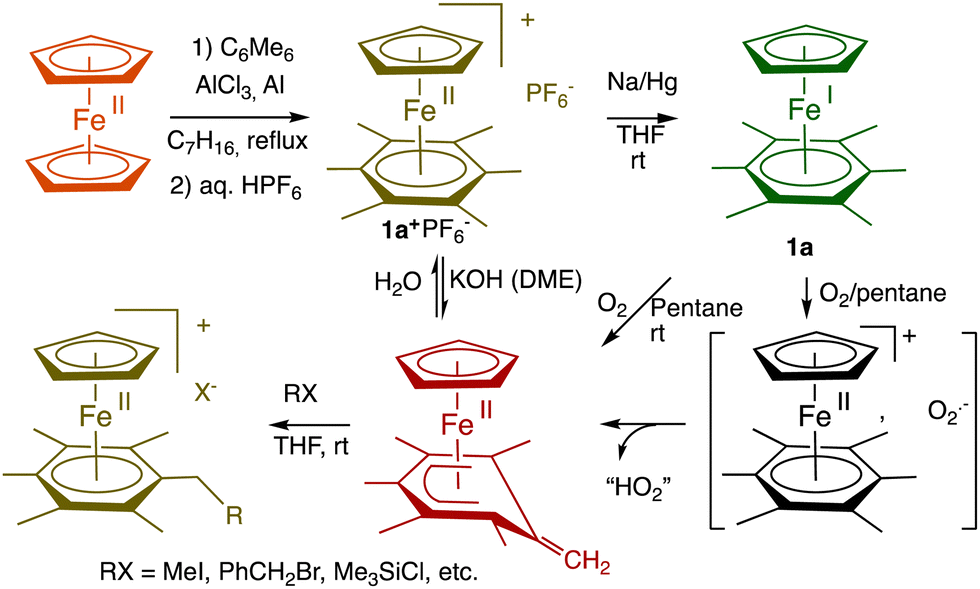 | ||
| Scheme 1 Synthesis and reaction with O2 of the 19-electron Fe(I) complex 1a. Due to the large difference (about 1 V) between the two redox systems Fe(I)/Fe(II) and O2/O2˙−, electron transfer from Fe(I) to O2 is very exergonic and fast, followed by deprotonation by O2˙− within the intermediate ion cage (from O2˙− EPR).14,15 In THF, this cage superoxide reaction is totally inhibited by NaPF6 due to ion exchange between the two ion pairs [Fe(II)+, O2˙−] and [Na+,PF6−],16 (for a review of salt effects, see ref. 17) which is reminiscent of the function of superoxide dismutase enzyme.18 | ||
Among other remarkable electron-transfer reactions of 1a and of the analogue [FeCp*(η6-C6Me6)], 1b, in which Cp* = η5-C5Me5, were the single, double and triple reduction of C60 depending on the reaction stoichiometry,19 the generation of N-heterocyclic carbenes of well-known catalytic importance20 and the stabilization of a new nanocomposite family by reduction of gold nanoclusters.21
Catalytic electron-transfer reactions of these Fe(I) complexes of interest were the redox catalysis of nitrates and nitrites to ammonia22,23 and the electron-transfer-chain (ETC) catalyzed (electrocatalytic) arene substitution in the series [FeCp(arene)]PF6 by three 2-electron phosphorus donors24 and CO exchange by phosphines in Vollhardt's fulvalene heterobimetallic carbonyl complexes (Scheme 2).25,26
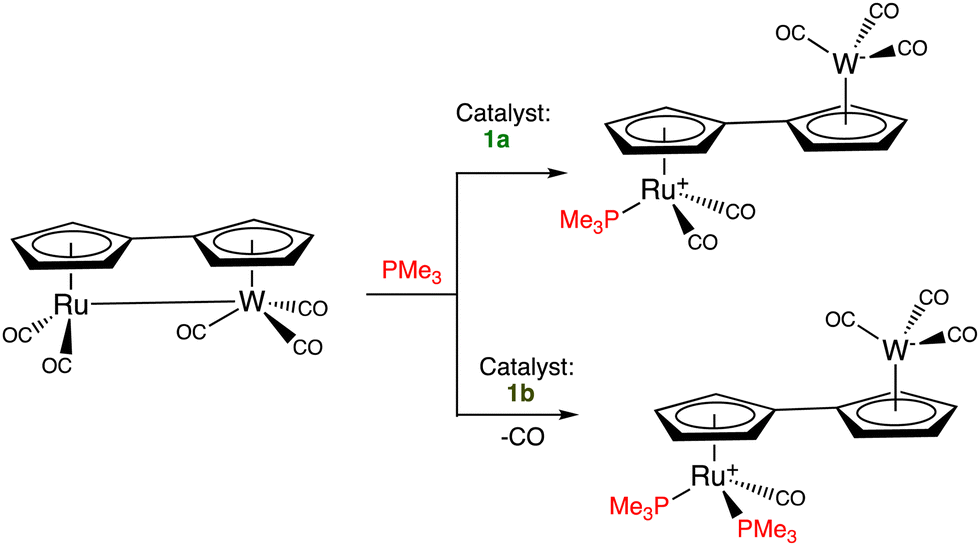 | ||
| Scheme 2 ETC-catalyzed PMe3 ligand addition in the fulvalene heterobimetallic complex [RuW(η10,μ2-C10H8)(CO)5]. The reaction depends on the Fe(II/I) redox potential of the electron-reservoir ETC catalyst (1avs. [Fe(η5-C5Me5)(η6-C6Me6)], 1b). With the more electron-rich electron reservoir 1b as ETC catalyst, two PMe3 ligands are introduced whereas, with 1a, only one PMe3 ligand is added. ETC catalysis was pioneered by Henry Taube.27 | ||
In both types of reactions, the availability of a library of Fe(I) complexes with different values of their Fe(II)/Fe(I) redox potential was very useful to determine the very fate of reactions. More generally, for applications in redox chemistry, a scale gathering useful transition metal sandwich complexes is represented in Fig. 1.
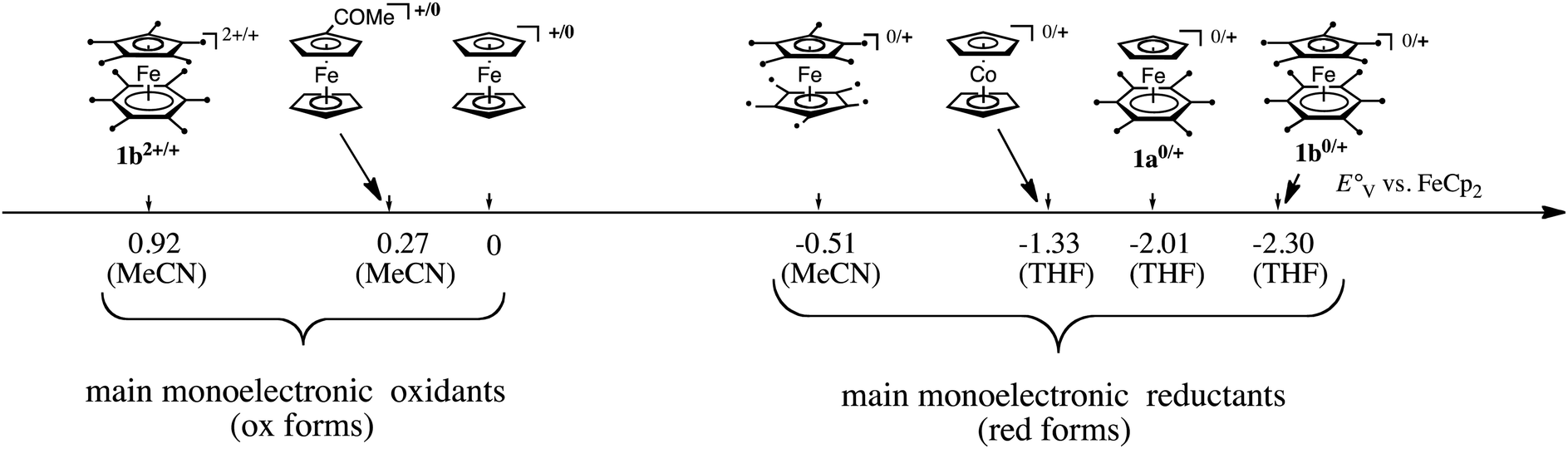 | ||
| Fig. 1 Redox potential scale of useful late transition-metal sandwich complexes as redox reagents. | ||
Finally, another useful property of the transition-metal sandwich complexes that were fully methylated on both ligands such as 1b and decamethylmetallocenes (Co, Fe) was their ability to serve as medium-independent references in cyclic voltammetry for the determination of redox potentials.28,29 Indeed, the permethylated sandwich systems are more reliable than the traditional ferricenium/ferrocene redox system that is subjected to redox potential variations due to the presence of nucleophilic solvent that interact with the cationic Fe center, whereas the permethylation of the sandwich rings sterically prevents such interactions.
In ferrocene chemistry, access to the 19-electron species occurs at a cathodic potential that is too negative for applications, even if a salt of the 19-electron hexa(t-butyl) ferrocene anion was recently characterized at −30 °C.30 On the other hand, in the sandwich series [Fe(η5-C5R5)(η6-C6Me6)]n+ (n = 0–2), the 3 oxidation states are accessible (Scheme 3)31 and could be isolated with suitable counter anions for 1b2+. Consequently, the 17-electron Fe(III) and 19-electron Fe(I) forms have been used as reservoirs of electron hole and electrons, respectively, for redox catalysis and ETC catalysis,32 (electrocatalysis) (Scheme 2).27
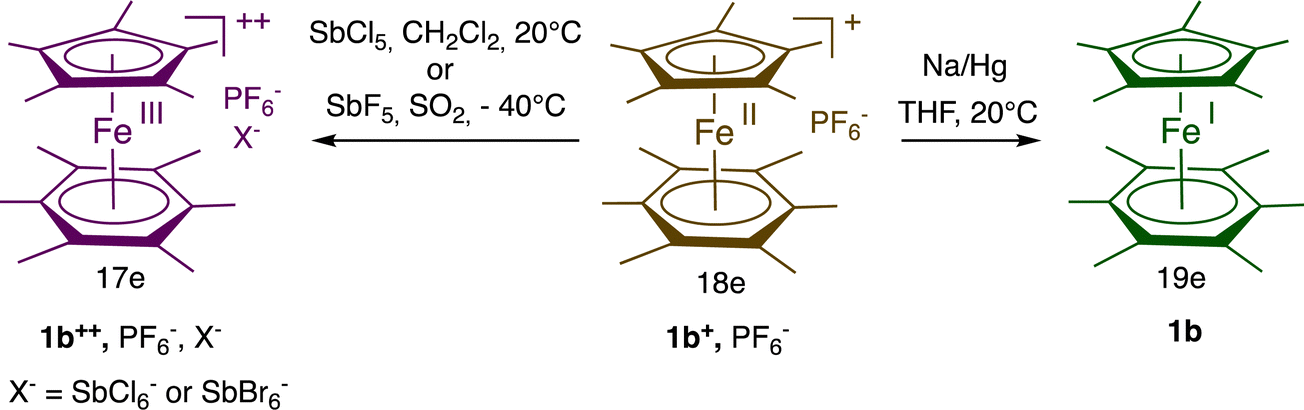 | ||
| Scheme 3 Synthesis of the stable, fully methylated mixed sandwich Fe complexes with 17 and 19 valence electrons [d5, Fe(III) resp. d7, Fe(I)] that are both redox catalysts and electrocatalysts. | ||
As shown early on by Wilkinson,4 ferrocene is easily oxidized to ferricenium salts upon reaction with I2, FeCl3 or AgNO3, and the resulting ferricenium salts are easily reduced back to ferrocene by reaction with, for instance, an acidic aqueous solution of TiCl3 or thiosulfate. The oxidizing property of ferricenium has been extensively applied to organometallic synthesis.33–35 An example of catalytic redox application was shown in the combination of ferricenium with the organometallic pre-catalyst [W(CO)3(NCMe)3], active in terminal alkyne polymerization.36,37 This type of alkyne polymerization proceeds according to the Chauvin–Katz metathesis mechanism,38,39 whereby the active catalyst precursor is a W-vinylidene species rearranging to a tungstacyclobutene intermediate. The role of ferricenium therein is to accelerate, by an ETC-catalyzed reaction, the introduction of the alkyne ligand rearranging to the vinylidene ligand (Scheme 4).40
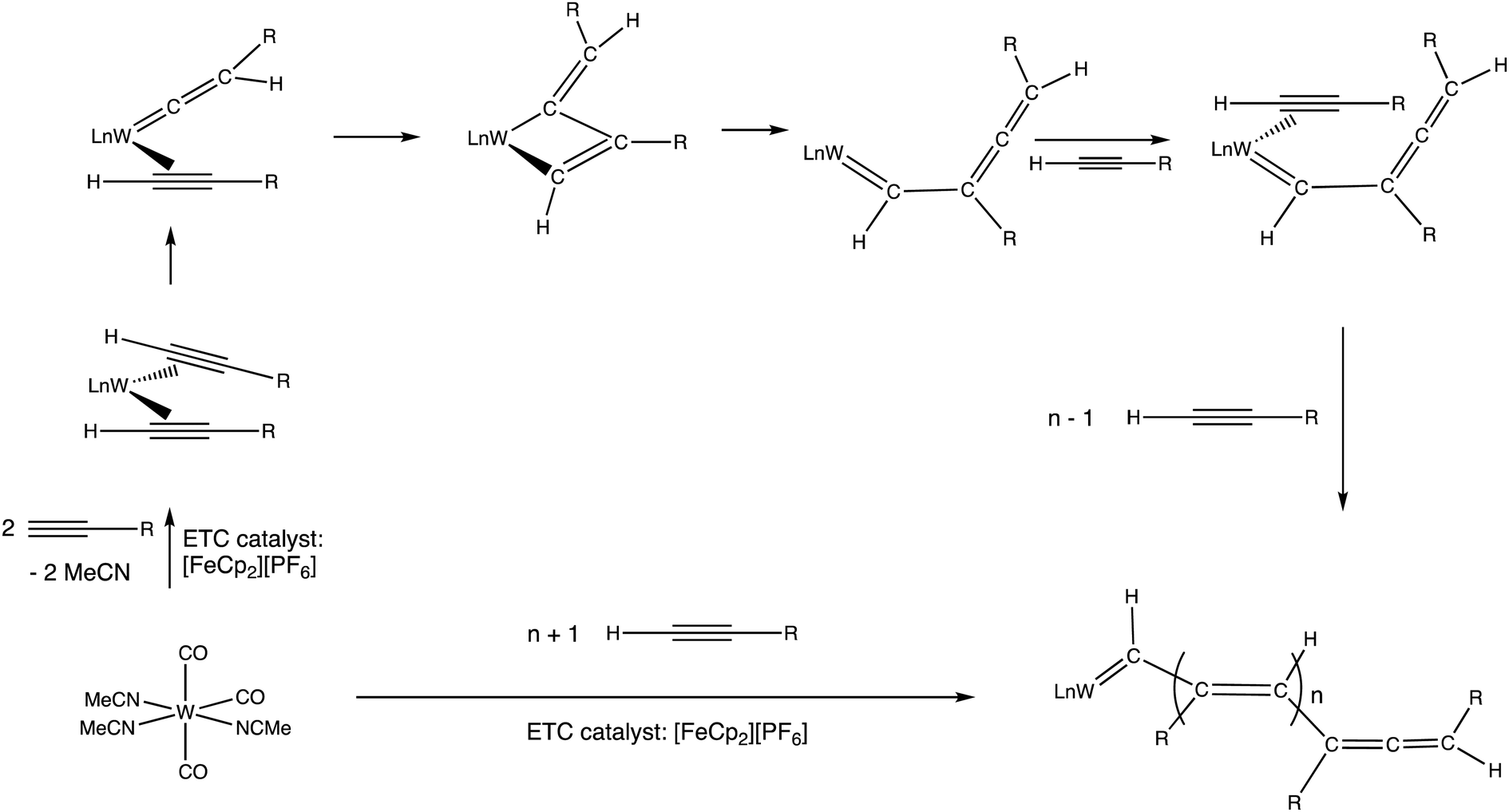 | ||
| Scheme 4 Polymerization of terminal alkyne catalyzed by a W(0) pre-catalyst according to the Chauvin–Katz metathesis mechanism. Addition of a catalytic amount of [FeCp2][PF6] vs. the W(0) catalyst considerably accelerates the reaction (from 100 °C, hours, in the absence of [FeCp2][PF6] to r.t. within a few minutes. Exchange of MeCN by the alkyne in the 18-electron W(0) complex is slow, but it becomes very fast at the 17-electron level. Following this exchange, the ETC cross redox step with the starting pre-catalyst is also fast due to its exergonicity, because the alkyne is a π-acceptor, contrary to MeCN.36 | ||
In summary, the FeI sandwich complexes are a rich family of electron-reservoir molecules, with a variety of substituents on the ligand rings, that undergo a variety of useful stoichiometric and catalytic electron-transfer processes in organic solvents (1a, 1b, etc.) or in water (for instance 1c and many others) summarized in Fig. 2. On the oxidation side, similar processes are accessible with the 17-electron ferricenium salts or, if insufficiently strong oxidant, the stronger 17-electron 1b2+ salts.
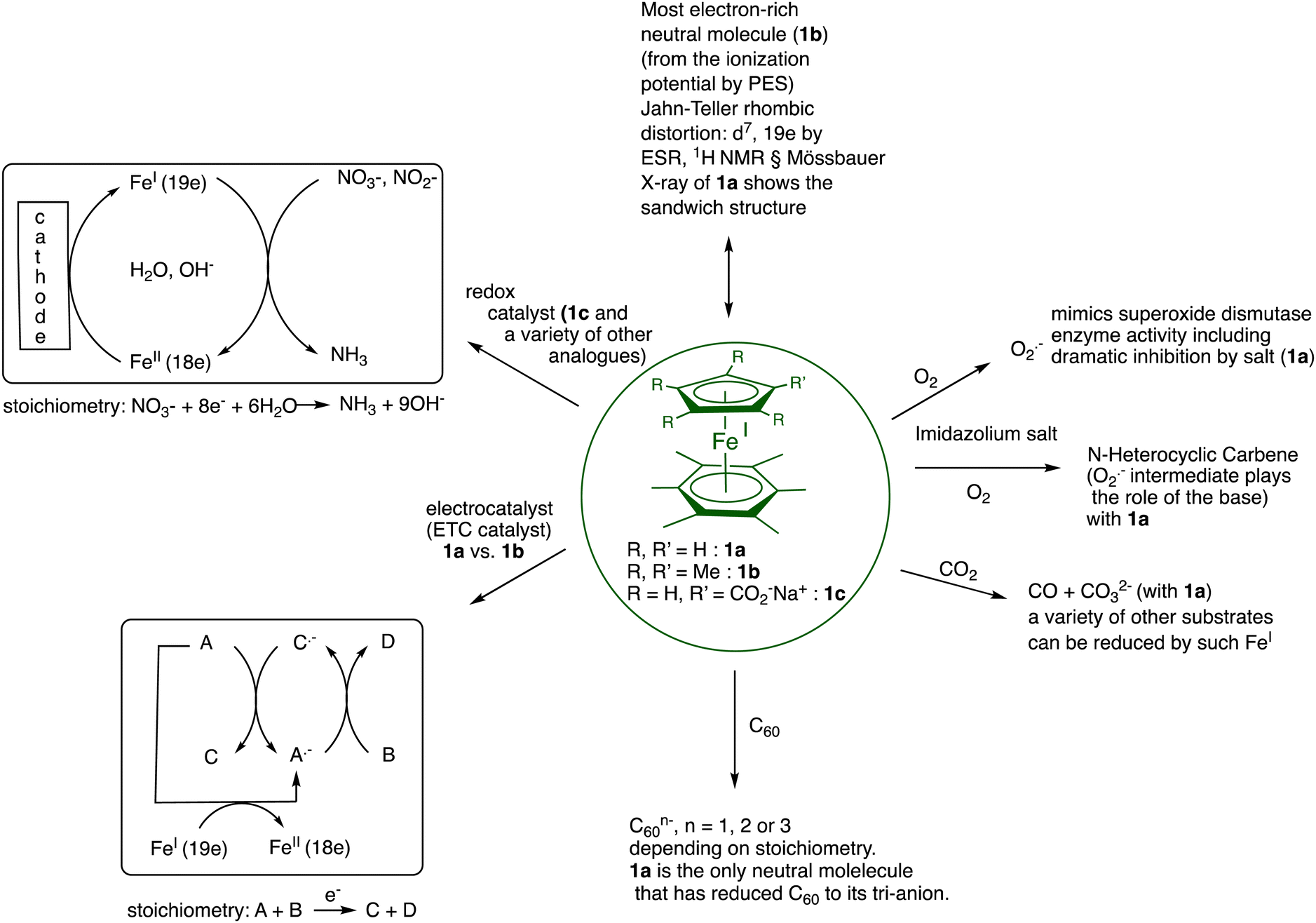 | ||
| Fig. 2 Illustration of the representative properties and applications of the family of 19-electron FeI complexes 1a, 1b and 1c as electron reservoirs. For the proton reservoir properties of the oxidized FeII form of these Fe sandwich complexes, see Section III. | ||
III. From organometallic chemistry to macromolecular chemistry: the advent of dendrimers with an original bursting reaction
The dendrimer field has been pioneered in the early 1980s mostly by Tomalia with PAMAM dendrimers41 and by Newkome with arborols,42 and in France, the Caminade–Majoral group, in particular, has been extremely active therein.43 Our story with dendrimers started when we discovered and reported in 1979 the bursting reaction of the complexes [FeCp(η6-C6Me6)][PF6].9 These one-pot multiple C–C branching reactions, extended to any number of methyl groups on the arene ligand, simply proceed under ambient conditions in the presence of an organic halide and a base such as t-BuOK or KOH in THF or DME, respectively. Deprotonation by the base proceeds first; then, nucleophilic substitution of the halide group occurs in the organic halide by the nucleophilic benzylic carbon that was previously deprotonated. Generally, for arenes bearing methyl substituents that do not have neighbors such as in mesitylene, this deprotonation–nucleophilic reaction sequence proceeds three times on each benzylic carbon,44 but it does so only twice for methyl groups that have only one methyl neighbor such as in o-xylene or durene,45 and only once (at least for short reaction times) for methyl groups that have two methyl neighbors such as in hexamethylbenzene.46,47 Organic halides that undergo such reactions include methyl iodide, benzyl bromide (including para-substituted dervatives) and allyl bromide. Poly-allyl branching was very favorable for further functionalization of the terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, and the 12-electron CpFe+ could be recycled upon visible-light photolysis in the presence of mesitylene (Scheme 5).
C bond, and the 12-electron CpFe+ could be recycled upon visible-light photolysis in the presence of mesitylene (Scheme 5).
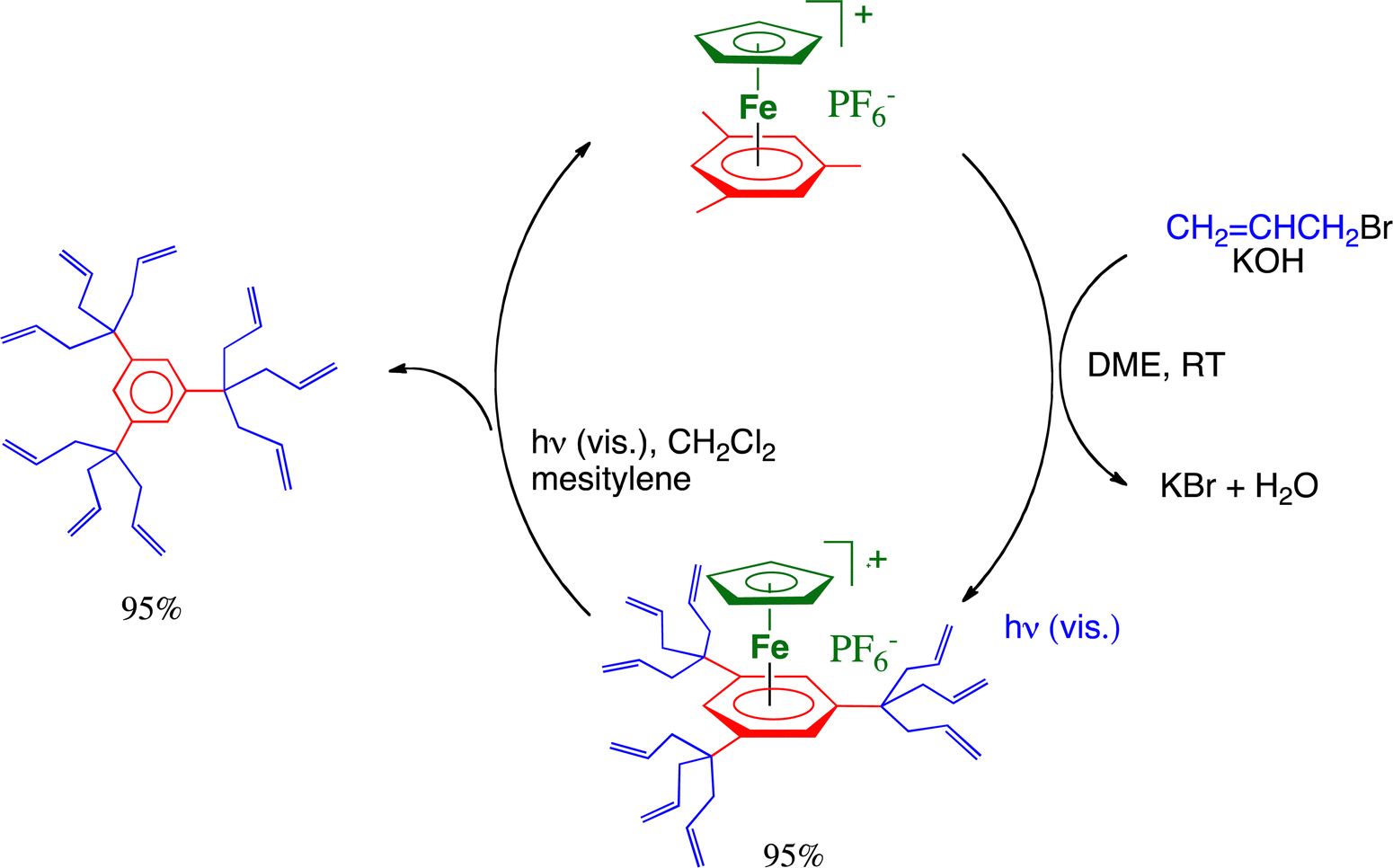 | ||
| Scheme 5 CpFe+-induced nona-allylation of mesitylene. The cationic charge of the Fe(II) complex activates benzylic deprotonation by the base, followed by nucleophilic substitution of Br− in CH2CHCH2Br, this sequence proceeding 9 times in situ under ambient conditions. The resulting nona-allylated complex is then photolyzed by visible light in CH2Cl2 in the presence of mesitylene to produce the iron-free nona-allylated arene, which also regenerates the starting sandwich complex of mesitylene. | ||
The nona-allyl structure served as core for dendrimer construction upon three-fold multiplication of the dendrimer branches at each generation.48 For this purpose, a triallylated phenol dendron was synthesized using the same CpFe+-induced triallylation strategy.49 For the divergent dendrimer growth, the construction using such a 1 → 3 branching, first initiated by Newkome with arborols,50 allows reaching much faster a high number of dendrimer tethers than the classic dendrimer double-branching construction. The dendrimer syntheses were followed by 1H, 13C and 29Si NMR indicating that they were clean on the NMR accuracy till the 9th generation (theoretical number of 177 407 terminal branches), which was confirmed by monodispersity measured by SEC (PDI = 1.00 or 1.01). The globular shape observed by HRTEM for the 9th-generation dendrimer was 16.7 nm. In AFM, a linear size progression of the layer thickness on graphitic HOPG support was observed till the 5th generation with a size of 10 nm, but from the 6th to the 9th generation, the thickness growed faster, reaching 25 nm, which presumably corresponded to a double dendrimer layer (Scheme 6 and Fig. 3).
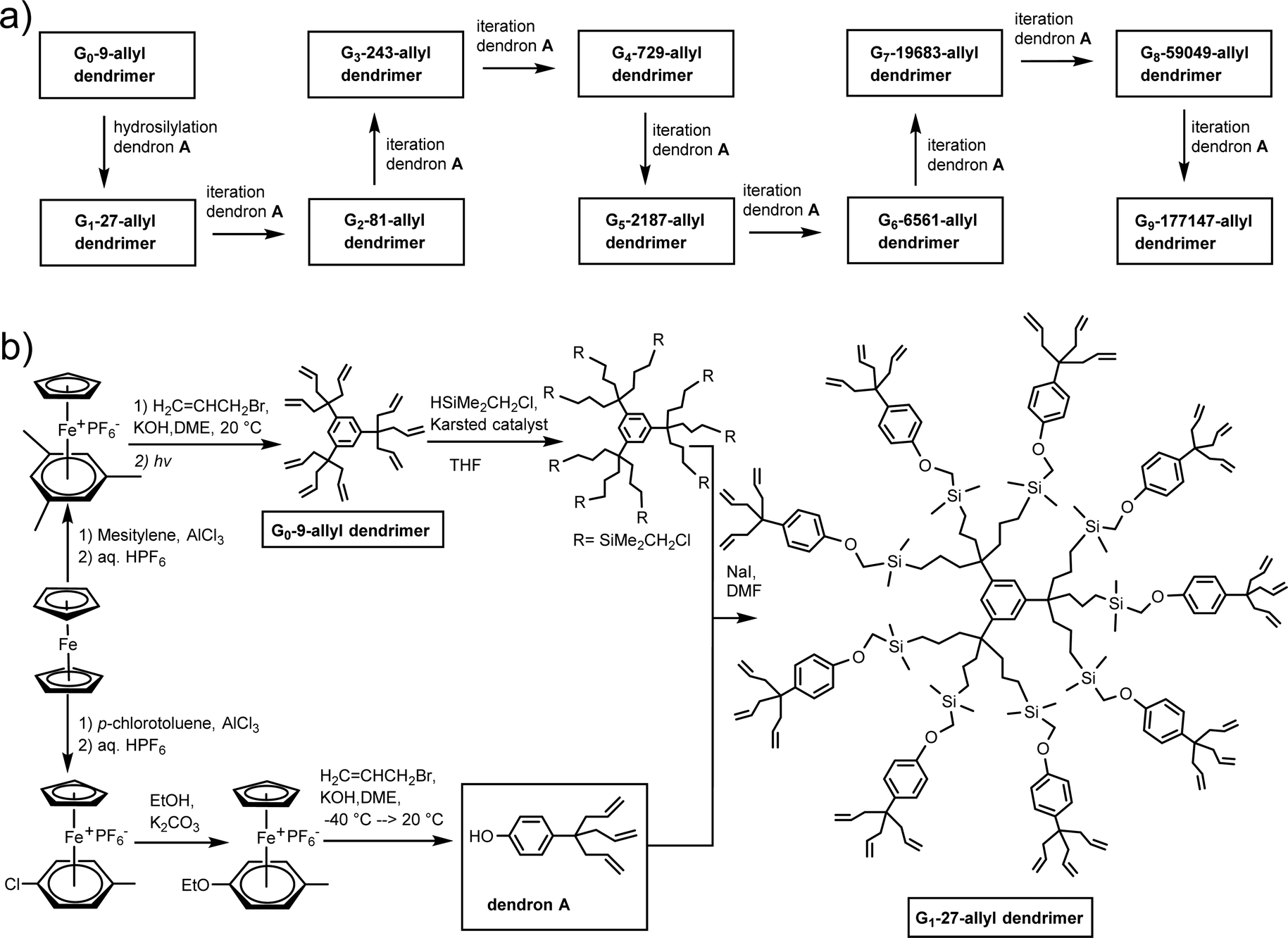 | ||
| Scheme 6 CpFe+-induced arene activation for the synthesis of the G0-9-allyl dendritic core and dendron A towards the multiple-generation divergent construction of large poly-allyl-terminated dendrimers following 1 → 3 branching until the 9th generation. | ||
 | ||
| Fig. 3 Molecular modeling of the dendritic size progression from G0 (9-allyl) to G3 (243-allyl). | ||
Defects, that are inherent to the divergent dendrimer construction, were characterized in the MALDI TOF mass spectrum of the 81-allyl dendrimer; although the molecular peak was largely dominant, a small peak was observed corresponding to the lack of one dendron. The characterization data show that the total number of branches is superior to 105 at the 9th generation level. Although de Gennes predicted that PAMAM dendrimer construction was limited due to steric limit at the dendrimer periphery,51 we believe that, on the contrary, our construction was not limited by the periphery bulk, because the lack of hydrogen bond at the termini allowed terminal branches to backfold toward the dendrimer core to avoid the bulky periphery. In this way, the construction is only limited by the overall dendrimer volume.
Activation of exo-cyclic C–H groups of arene ligands in the complexes [FeCp(arene)][PF6−] was useful to build more complex structures such as cyclophane, showing examples of the synthetic power of this activation type.52 Since this family of complexes are directly synthesized by reaction of ferrocene with the arene, this illustrates once more the synthetic potential of ferrocene.53 The CpFe+ activating group is not the only one able to activate the exocyclic C–H bonds in proton reservoir complexes,54 but in principle any cationic moiety in robust 18-electron complexes should be able to operate in a similar manner. In particular, this was the case in pentamethyl- and decamethylcobalticenium and rhodicenium PF6− salts. In such cases, more space is available around the permethylated Cp* ligand (Cp*C5Me5) for double branching than around the C6Me6 ligand. Thus, double methylation or allylation of each exocyclic methyl group was easy, and [CoCpCp*][PF6−] yielded the chiral complexes [CoCp(C5{CHMe2}5)][PF6−] and [CoCp(C5{CH{CH2CHCH2}2)}5][PF6−]. These complexes result from double branching at each exo-cyclic carbon upon reaction with excess KOH (or t-BuOK) and MeI or allyl bromide, respectively. At ambient temperature, they possess a single directionality (planar chirality) of the remaining C–H bonds with full rotation only allowed (from 1H NMR) at high temperature.55,56 With the decamethylcobalticenium PF6− salt, the reaction could not go to the end of the deprotonation–methylation sequences due to the limited space between the two rings restricting the space for the incoming groups. On the other hand, with decamethyl rhodicenium, this reaction readily gave the deca(isopropyl) rhodium complex, the only deca(isopropyl) metallocene known, because the distance between the two rings is sufficient to allow space for all these groups.57
IV. Mixed valences, molecular electronics and molecular batteries
The late transition-metal sandwich structures afford extensive possibilities in terms of oxidation states and electron and proton transfers involving stable 17 and 19 electron complexes.58 In the latter, such odd-electron non-sandwich structures, even stabilized ones, often have close energy levels and therefore involve fluxionality between these two valence states.59,60 A consequence is that these sandwich and related structures provide rich mixed and average valences with controllable electronic communication between two or several metals. An early example was that of the 35-electron biferrocenium salts disclosed in 1970,61 shortly following classification describing the various types of mixed-valence complexes62,63 and the fine analysis by Taube of the understanding of the energetic requirements of intermetallic electron-transfer processes.64 Optical properties of the biferrocenium salts indicate, due to the structural identity of both sandwiches in the neutral states, class II mixed-valence 35-electron complexes according to the Robin-Day classification.63 An example is found in the X-ray crystal structure of decamethylbiferrocenium for which the iron-ligand bond distances are distinct in the two sandwiches, indicating localization of the mixed valence in an FeII–FeIII complex.65 This is not the case when two CpFe(arene) units are linked through the Cp ring. The five oxidation states between the 36-electron and the 40-electron species are connected by 4 reversible cyclic voltammetry waves all separated by 0.5 V as a result of the electrostatic interactions, in a situation resembling that encountered in nanoclusters.66 In this series of sandwich complexes the infrared absorption and Mössbauer spectroscopy data showed electronic delocalization, both metals being in the 1.5 oxidation state in a class-III mixed (average) valence system.67,68 The other extreme with the two valence states of a fulvalene mixed-valence complex is that of the 37-electron complex Fc-CpFe(I)C6Me6 (Fc = ferrocenyl) that is clearly a localized Fe(I)–Fe(II) class-I mixed valence complex in the Robin-Day classification. Thus, the fulvalene bridge is extremely flexible concerning electronic communication between the two metals to which the two Cp parts of fulvalene are bound, this communication being directed by the nature of other, non-communicating (ancillary) ligands (Fig. 4).33,38,65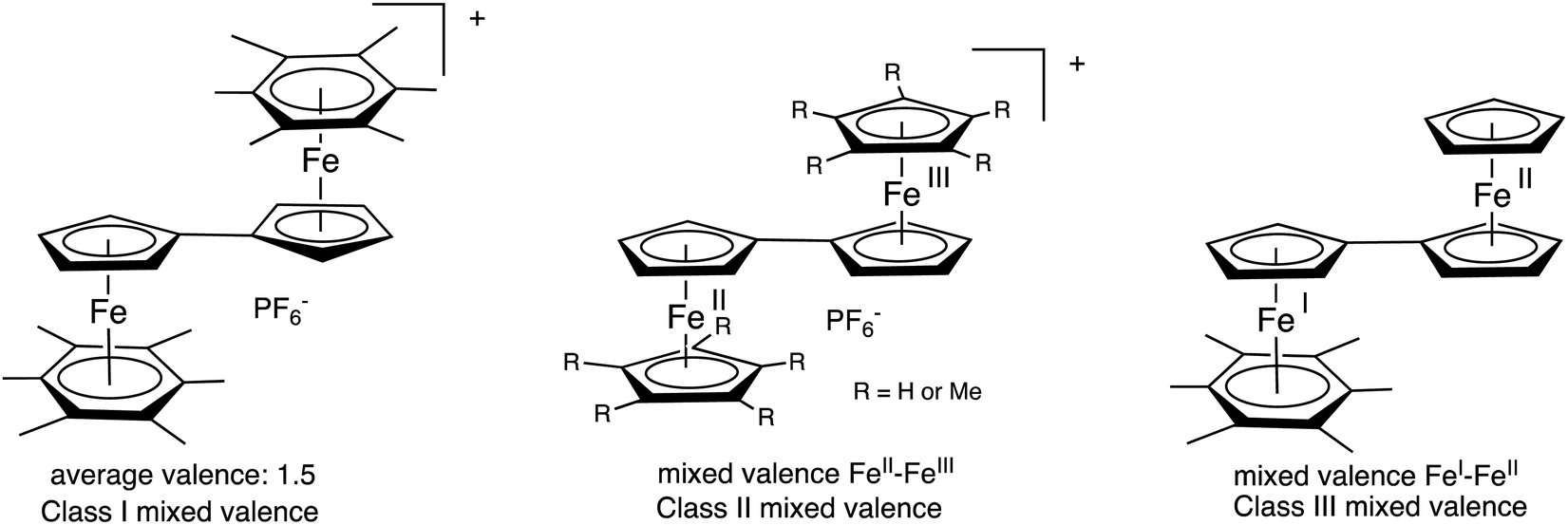 | ||
| Fig. 4 Ancillary ligand-dependent electronic communication between the two metals and mixed valence type in fulvalene diiron complexes [Fe2(η10,μ2-C10H8) Ln]p+, Ln = C5R5 (R = H or Me) or C6Me6, p = 0 or 1.65 | ||
The reaction of 19-electron,24 and also 38-electron complexes is very sensitive to the medium. For instance, if the ancillary ligand is benzene, the reaction of the 38-electron complex [FeI2(η10,μ2-10H8)(η6-C6H6)2] with CO is highly dependent on the presence of a salt. Indeed, the presence of a salt such as NaPF6 in THF completely changes the fate of the reaction,18 the salt-induced intramolecular electron transfer yielding the complex [FeII(η10,μ1-C10H8) (η6-C6H6)] instead of the carbonyl-containing dimer obtained in its absence (Scheme 7). Salt-induced reactions are known to proceed upon double ion exchange between two ion pairs in molecular chemistry.18 In the present case, they control intramolecular electron transfer across the fulvalene bridge.69
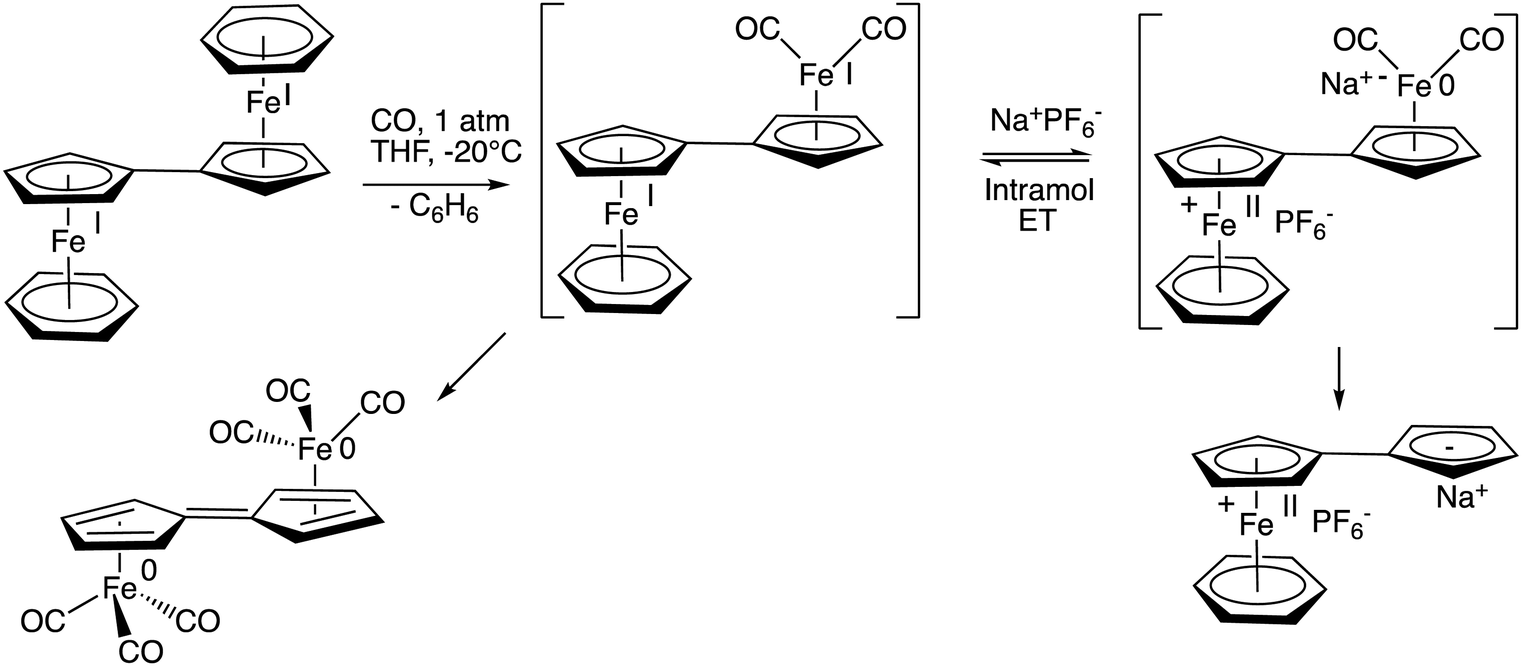 | ||
| Scheme 7 Salt-induced intramolecular electron transfer between the two Fe centers across the bridging fulvalene ligand. | ||
When two CpFe(arene) units are linked by a single bond through the Cp ligands, like in Scheme 7, the cyclovoltammogram shows a single-ET cascade (arene = C6H6 or C6Me6), because the 38 e− Fe(I) complex is thermally stable for arene = C6Me6. So, this shows that, like in cluster chemistry, there is no significant reorganization along the ET cascade. On the other hand, when the two CpFe(arene) units are linked through the arene ligand in diphenyl complexes, the electronic interaction between these two virtual 19-electron iron centers is so strong that it is quenched by intra-ligand coupling into a non-planar bis-cyclohexadienylidene ligand in which the cyclohexadienyl part of each ring is bonded in a ferrocene-like pentahapto coordination mode to iron, as shown by the X-ray crystal structure of the neutral blue complex in Scheme 8 (R = CH3). Formally, the two incoming electrons are found in the double bond of the link, and the result is a structure isolobal to biferrocene. The driving force to reach the 18-electron, ferrocene-type structure overtakes the requirement of diphenyl aromaticity. In the parent (Cp) complex, the energy involved in the intramolecular coupling overtakes the electrostatic energy involved in the second single-electron reduction, and the cyclovoltammogram shows a single 2-electron wave. With the C5Me5 analogue, this reorganization energy is a little less than the electrostatic factor, so that two close one-electron CV waves are observed, which also allows synthesizing the green average-valence 37-electron complex by comproportionation (Scheme 8). If the diphenyl group is kept rigid, however, such as in phenanthrene, triphenylene or pyrene, the cyclohexadienylydene formation is not possible.70,71
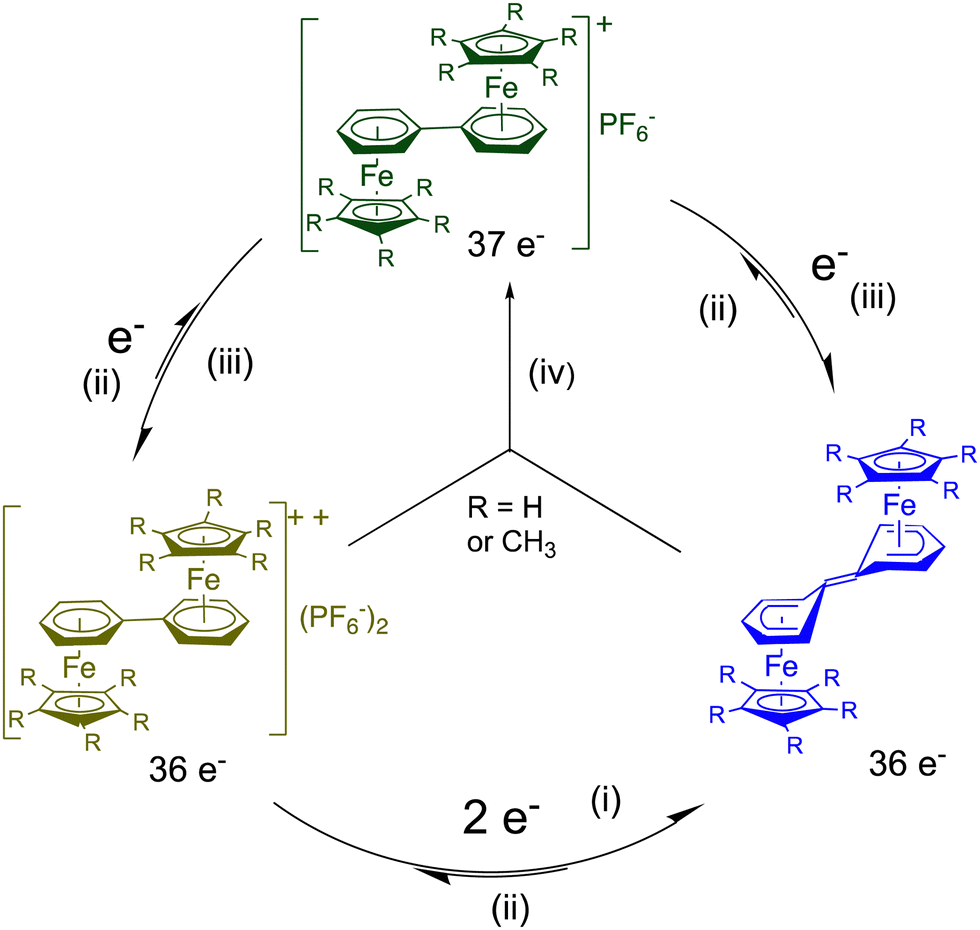 | ||
| Scheme 8 Electron transfer (ET) processes of diphenyl diiron complexes of biphenyl in THF˙ (i) LiAlH4, −80 °C; (ii) O2, NaPF6; (iii) Na/Hg, 20 °C; (iv) comproportionation, 20 °C.70 as shown by the X-ray crystal structure of the blue complex (R = CH3). | ||
Hexa(ferrocenylethynyl)benzene, synthesized by a Negishi coupling reaction between C6Br6 and ferrocenyl–ethynyl zinc chloride, FcCCZnCl, in the presence of the catalyst [Pd(PPh3)4] in refluxing THF–toluene, and methyl-substituted ferrocenyl analogues, showed interesting electrostatic and frustration effects by cyclic voltammetry (CV). With the supporting electrolyte N(n-Bu)4PF6 in CH2Cl2 and a platinum anode, a single six-electron CV wave was observed for hexa(ferrocenylethynyl)benzene as well as for 1,3,5-tris(ferrocenylethynyl)benzene, but, upon using N(n-Bu4)BArF4, (ArF = 3,5-C6H3-(CF3)2,), three different two-electron reversible CV waves were observed for both compounds (Fig. 5).72
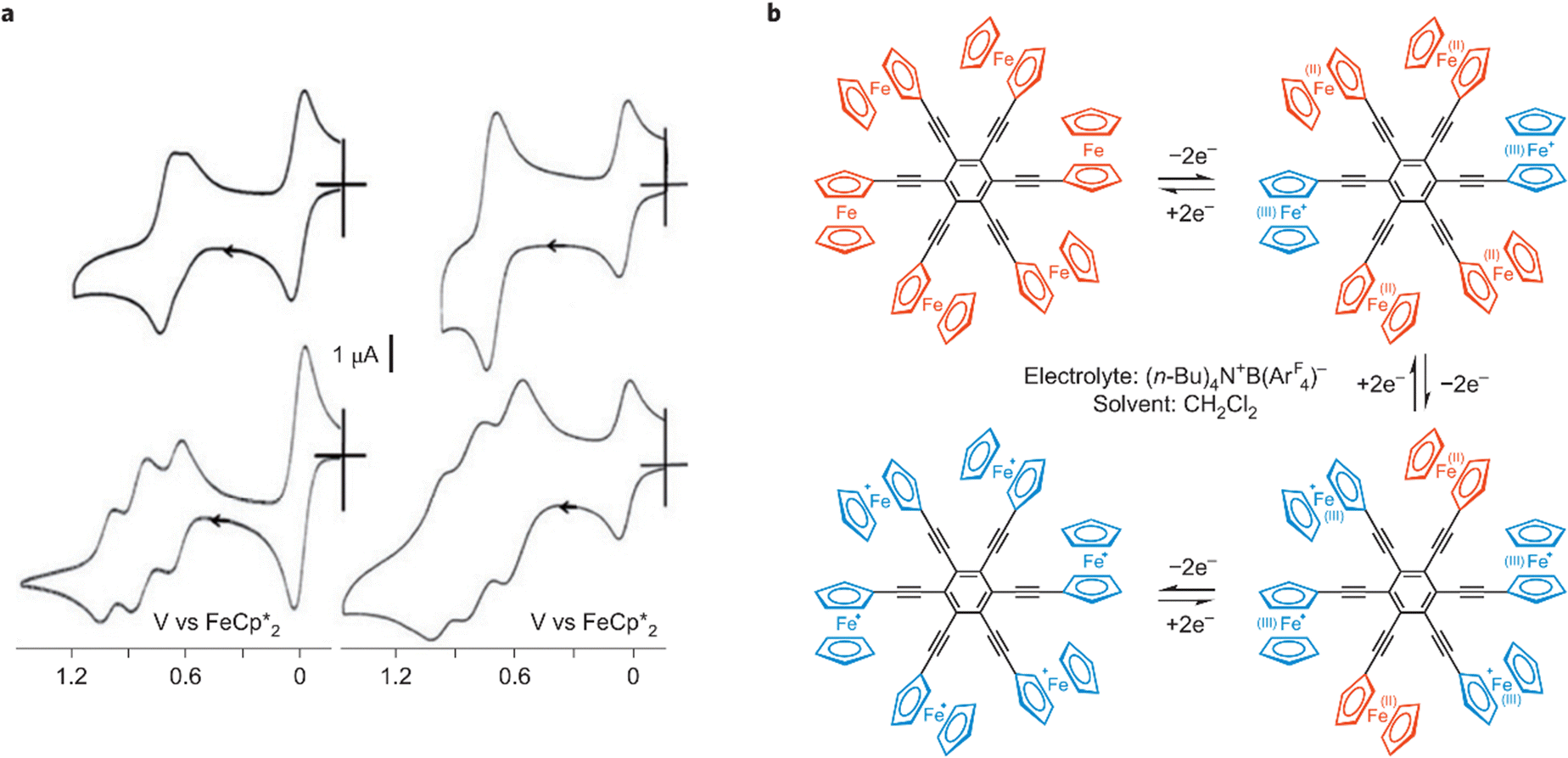 | ||
| Fig. 5 Electron transfers in hexa(ferrocenylethynyl)benzene governed (similarly to 1,3,5-tris (ferrocenylethynyl)benzene) by electrostatic and conformational frustration effects in the polycationic oxidized forms. (a) Compared cyclic voltammograms of 1,3,5-tris(ferrocenylethynyl)benzene (left) and hexakis-(ferrocenylethynyl)benzene (right) in CH2Cl2 with [N(n-Bu)4][PF6] (top) and [N(n-Bu)4][BArF4] (bottom, ArF = 3,5-C6H3(CF3)2. (b) Mechanism of the oxidation of hexakis-(ferrocenylethynyl)benzene in the presence of [N(n-Bu)4][BArF4] 0.1 N with ArF = 3,5-C6H3-(CF3)2, in CH2Cl2, a cascade of three two-electron oxidation steps shown in a with the CV (right, bottom). The ferrocenylethynyl groups are represented in red, whereas the ferriceniumethynyl groups are in blue; reproduced with permission from ref. 73 Copyright 2011 American Chemical Society. | ||
Since the CV of 1,3- and1,4-bis(ferrocenylethynyl) benzene showed only one wave whatever the supporting electrolyte, it was concluded that intramolecular electronic influence among the various redox groups was not observable, because it was too weak, but CV wave splitting observed with the tris-and hexa (ferrocenylethynyl) derivatives was due to more significant electrostatic effects. In addition, the striking contrast between the behavior of the 1,3- and 1,3,5- substituted derivatives indicated that the introduction of a 3rd substituent in meta position introduced a problem. This was taken into account by the fact that, if disubstituted derivatives could adopt an anti conformation of the two ferricenium groups about the benzene plane minimizing the electrostatic repulsion between the two positive charges, such relative anti conformation was no longer possible upon introduction of a 3rd substituent in meta position, involving stereo-electrostatic frustration and the splitting of the three ferrocenyl waves. In the hexasubstituted compound, each group of two substituents in para substitution is equivalent, which induces the observation of 3 CV waves, as in the 1,3,5-trisubstituted derivative.72–74 In large ferrocene-terminated dendrimers, these electrostatic effects become two weak to be observable, because these groups are far from one another.74
Electronic communication between two iron centers was established through a meta di-ethynylaryl group, providing sufficient electronic delocalization via this organic bridge for the stabilization of class-II mixed valency with the two iron centers.75 On this basis, hexanuclear and dendritic architectures were synthesized including the class-II mixed-valence system (Scheme 9 and Fig. 6).76
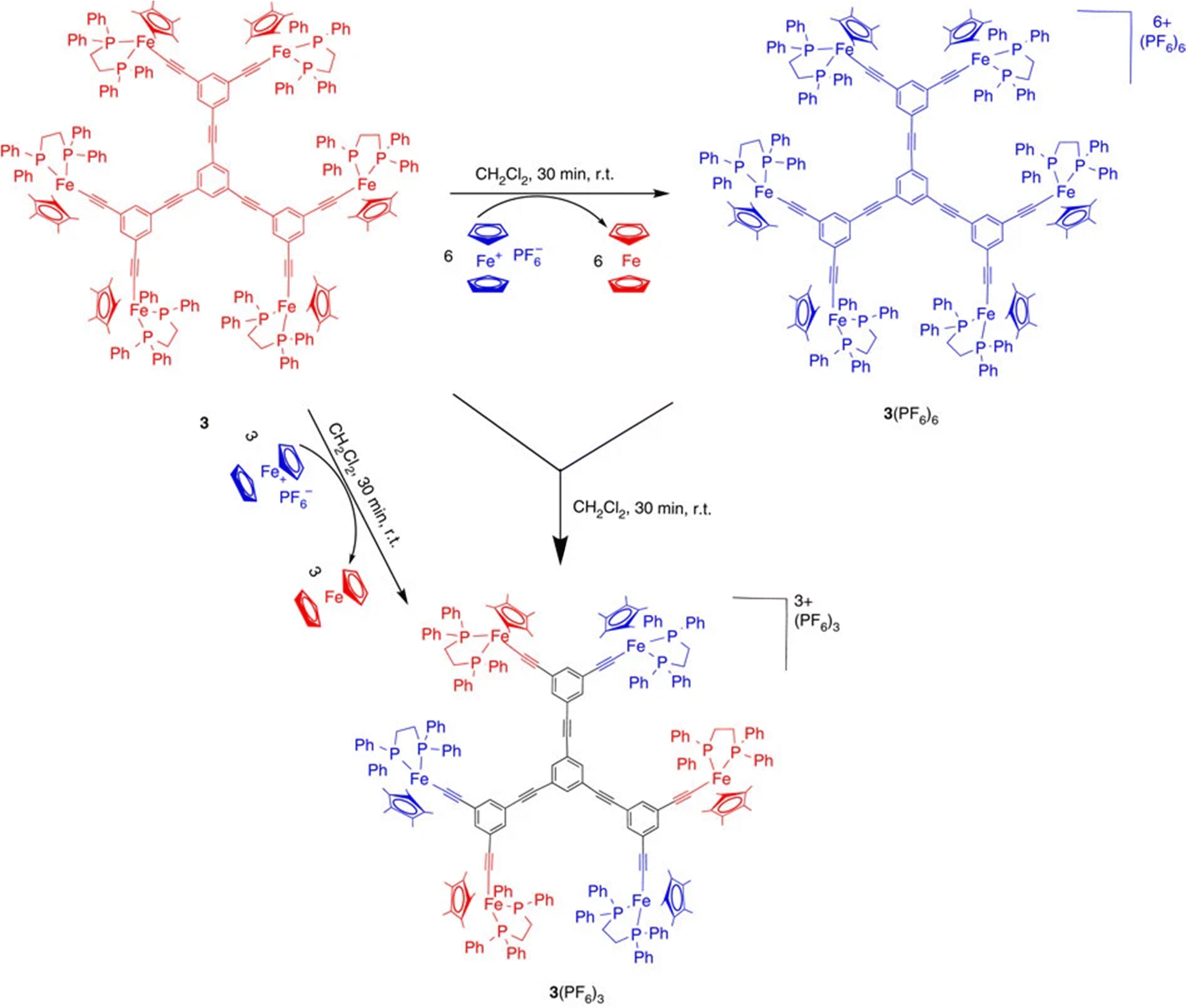 | ||
| Scheme 9 Syntheses using the 17-electron ferricenium complex of both the Fe(III) and class-II mixed-valent Fe(II)–Fe(III) hexanuclear complexes. The latter can also be obtained by quantitative comproportionation between the Fe(II) and Fe(III) hexanuclear complexes. Various dendrimers containing the same mixed-valent Fe(II)–Fe(III) periphery were obtained in the same way (see Fig. 6). Blue represents Fe(II), and red, Fe(III). Reproduced with permission from ref.76 Copyright 2014 Springer Nature. | ||
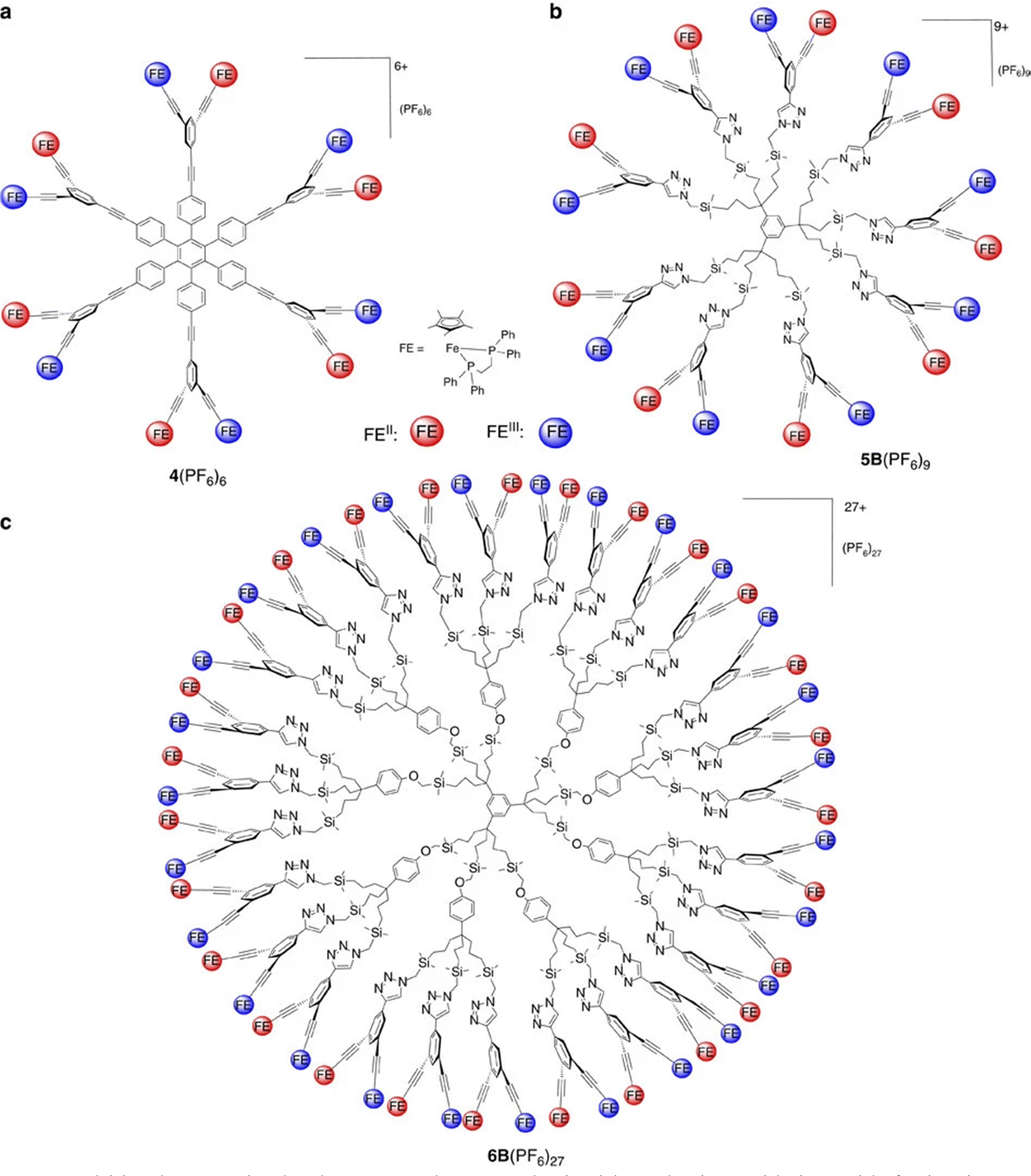 | ||
| Fig. 6 Dendritic class-II mixed valence complexes synthesized by selective oxidation with ferricenium or comproportionation as shown in Scheme 9. Reproduced with permission from ref. 76 Copyright 2016 Springer Nature. | ||
Metallopolymers containing ferrocene77,78 and cobaltocene79 or [CpFe(C6Me6]+ units (or two or more of these metallocenes)79 could be a means to target new molecular batteries.80–86 Whereas ferrocene polymers have been used for some time as redox flow batteries (RFBs) because of their adequate redox potential as a catholyte (3.44 V vs. Li/Li+),80 cobaltocenium salts have been introduced later as anolytes (1.3 V difference between the Cp2Fe+/0 and Cp2Co+/0 redox potentials).83 Very recently, polyferrocenes and polycobaltocenes have efficiently been used for the first time as catholytes and anolytes, respectively in graphene oxide composites for new (RFBs). Ideal Nerstian behavior was observed up to >1.2 C cm−2, high coulombic efficiency at ultrafast rates (200 A g−1). Charge was carried by the anion ClO4− upon using Li ClO4 (Scheme 10).84
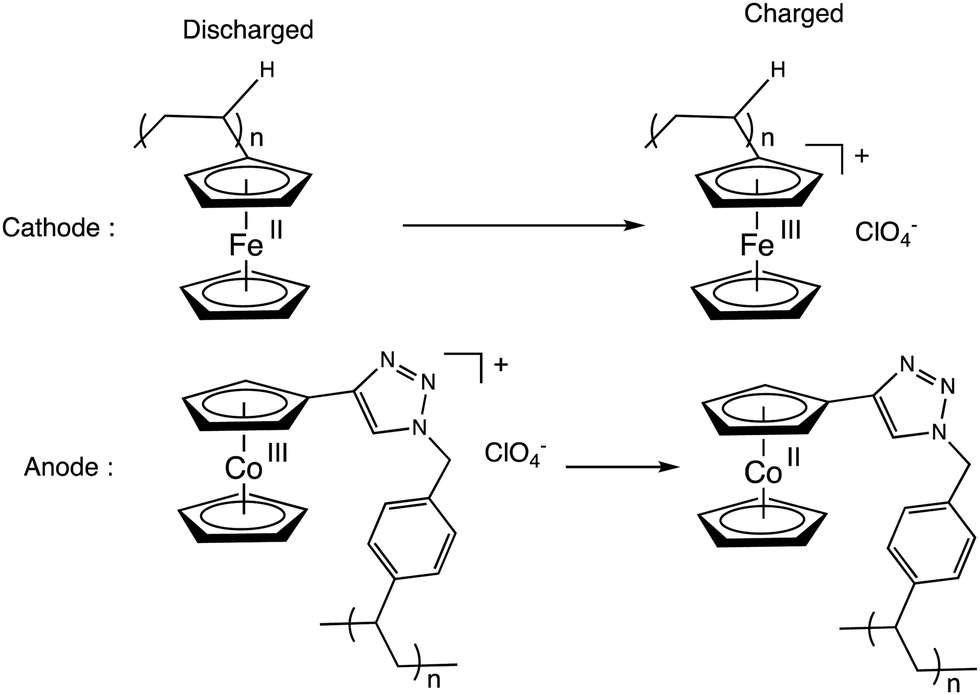 | ||
| Scheme 10 Electrode reactions in a recent RFBs based on ferrocene polymer as catholyte and cobaltocenium salt as anolyte (driving force: 1.3 V). Electrons are transported by ClO4− anions. | ||
The advantage of such RFBs is that electron self-exchange is fast with late metallocenes due to the fact that the metal–ligand bonds undergo only very small bond change between monocations and neutral forms (cf introduction); also, good ion percolation is obtained upon molecular self-assembly. Upon oxidation, metallocene materials undergo size increase (breathing) due to the anion income, but this breathing is reciprocal between ferrocene and cobaltocene, cancelling the pressure built up in the closed cell that would otherwise be damaging. As a result, excellent cycling was obtained (vide supra).84,85 Other recent studies involved charge transfer in metallocene (FeCp2 and CoCp2) intercalated MoS2 and WS2 and showed that the metallocene redox potentials were much dependent on the speciation and concentration of the cations in the electrolyte.86 Large dendrimers also represent a viable architecture for the design of molecular batteries with metallocene-terminated dendrimers, i.e. either ferrocenyl or pentamethylferrocenyl-terminated dendrimers (Fig. 7)87,88 on one hand and metallodendrimers terminated by cobalticenium89 or [CpFe(C6Me6]+ salts90 on the other hand, given the equivalence of the redox centers at the dendrimer periphery.74
 | ||
| Fig. 7 Planar representation of an 81-ferrocene dendrimer, with equivalent terminal groups, that can serve as battery catholyte. | ||
The equivalence of the many ferrocenyl redox sites at the dendrimer periphery for 9 generations of ferrocenyl and pentamethylferrocenyl-terminated dendrimers is shown by cyclic voltammetry (CV). Only one CV wave was observed, showing that the electrostatic factor is extremely weak, making the different redox potentials of these sites appearing equivalent. The CV waves were chemically reversible showing the stability of both Fe(II) and Fe(III) forms and electrochemically reversible showing fast rotation of the dendrimer bringing in turn all the redox centers near the electrode for fast electron transfer and tunneling from a ferrocene terminus to the next at the periphery.
Reversible molecular breathing between the 18-electron and the stable 17-electron oxidized forms of the metallodendrimers was measured by Atomic Force Microscopy (AFM) and Electronic Force Microscopy (EFM) showing that the dendrimer size increases for the fifth generation by 50% upon oxidation and returns to the initial size upon reduction of the ferricenium form back to Fe(II) (Fig. 8).
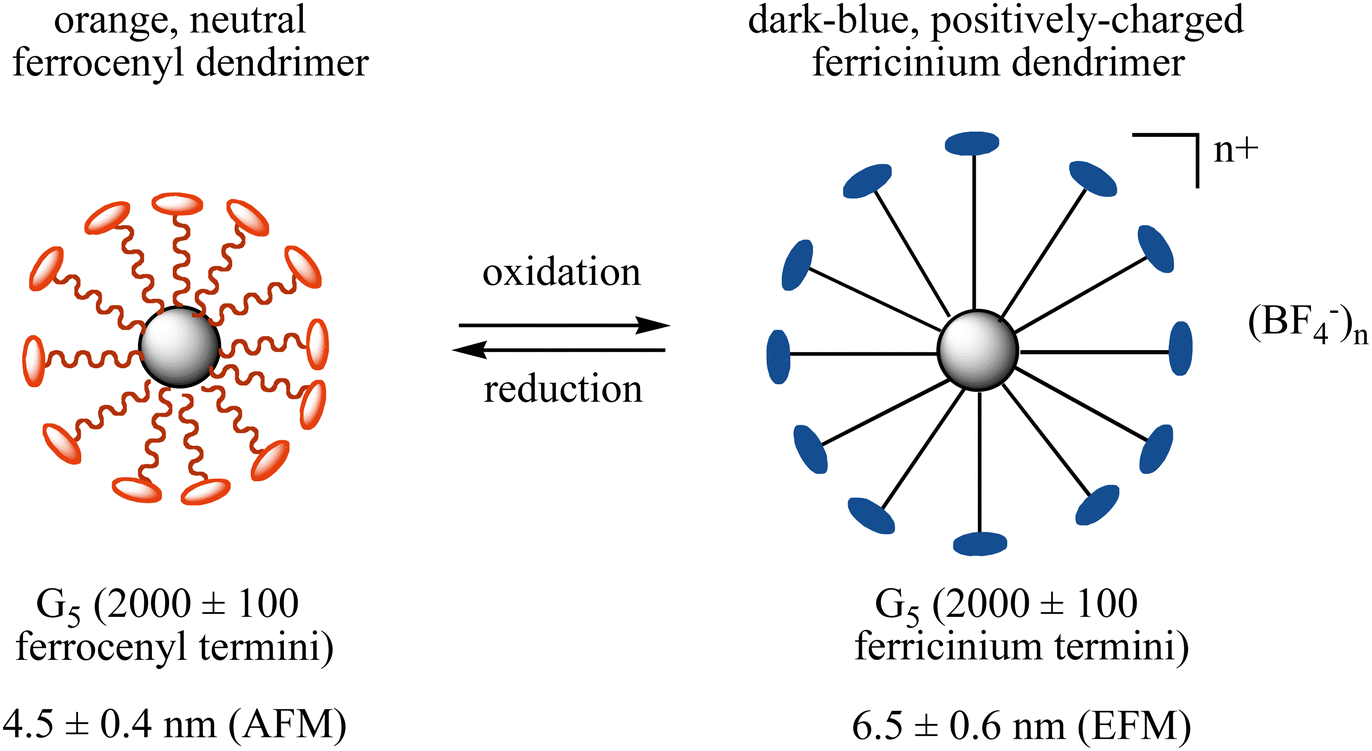 | ||
| Fig. 8 Redox breathing of a 5th generation ferrocenyl-terminated dendrimer constructed according to Scheme 6 followed by the reaction of G5-CH2I with Fc(CH2)11O-p-C6H4OH in the presence of K2CO3 in DMF for 2 d at 80 °C.87 | ||
Evidence for metal-sandwich-terminated dendrimers as electron-reservoir systems was also illustrated by single-electron transfer of each Fe(I) sandwich-terminated branch to C60 yielding a dendrimer with 64 positively charged Fe(II) sandwich complex, each of them electrostatically bonded to C60˙− (Fig. 9).
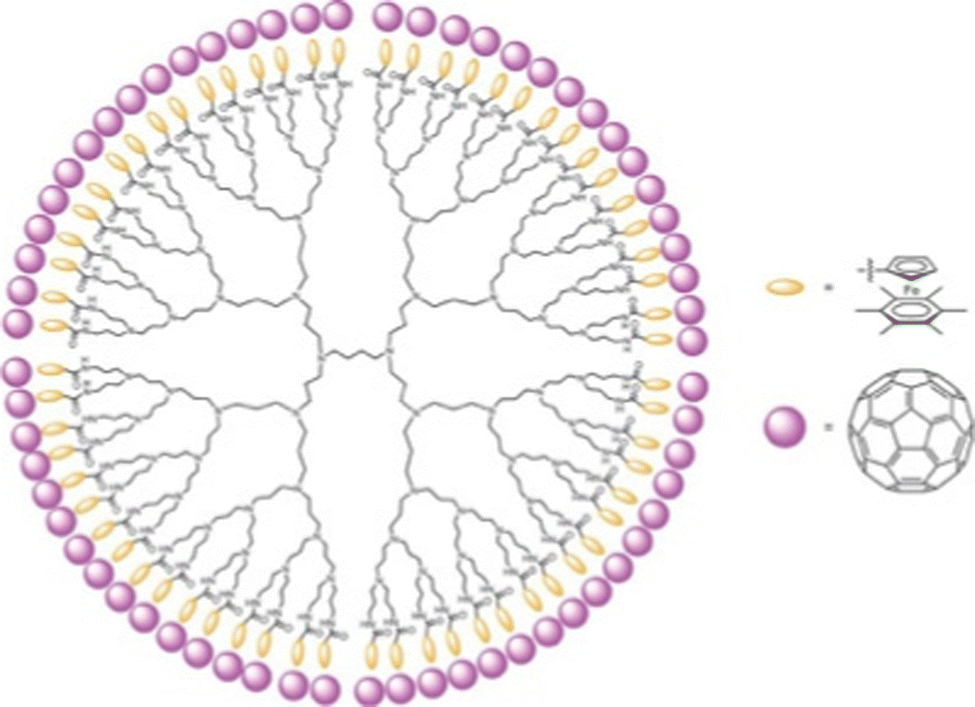 | ||
| Fig. 9 Dendri-64[–NHC(O)–CpFe(II)+C6Me6,C60˙−] (with Dendri = diaminobutane (DAB)-core G5 dendrimer) from the reaction between Dendri-64[–NHC(O)–CpFe(I)C6Me6 with 64 equiv. C60. EPR: g = 2.00093, ΔH = 3.12 G. Reproduced with permission from ref. 74. Copyright 2012 Springer Nature. | ||
V. Dendritic effect with supramolecular redox sensors of biologically important anions
Anion sensing is a major area, because anions are ubiquitous throughout biological systems (DNA, ATP, enzyme substrates and co-factors), playing a major role in medicine, catalysis, and environment (phosphate-containing fertilizers, carcinogenesis of nitrate metabolites, pertechnetate from nuclear fuel). Anion recognition and sensing are relevant to supramolecular chemistry, because they involve weak interaction with suitable hosts.91 This field has been well covered by Beer and Gale in their well-known review; the Beer group also designed families of endoreceptors linked to late transition-metal metallocenes for redox sensing.92Molecular recognition and sensing based on metallocene-containing polymers and materials are an extended research area, mostly involving ferrocenyl compounds, that has been reviewed.93 The apta-sensing strategy has recently been developed with ferrocene-labelled aptamers involving the hairpin DNA (hDNA) and linear single-stranded DNA (ssDNA) allowing simultaneous detection of multiple mycotoxins.94 For instance, aptamer-target recognition enabled Fc-ssDNA to be captured at the electrode surface via nucleic acid hybridization. Such electrochemical sensor was assessed bioaccumulated amount of microcystin (MC)-LR in the liver and meat of fish. Herewith, the aptamer-based strategy was utilized to develop an electrochemical MC-LR assay toward application in MC-LR-related aquatic product safety studies.95 Ferrocene-based electrochemical sensor studies are presently numerous, in particular toward electrochemical microRNAs96–99 detection.
Following Beer group’ sensor studies using metallocene endo-receptors,92 on the other hand, ferrocene-terminated dendrimers were designed as anion exo-receptors, i.e. a functional group was installed next to the ferrocenyl branch termini so that its supramolecular interactions with some anions, in particular oxo anions, influence the ferrocenyl redox potential during electrochemical titration. First successful attempts were obtained with the amidoferrocenyl-terminated dendrimers. Interestingly, it was found that the supramolecular interaction effect on the amidoferrocenyl redox potential was all the greater as the dendrimers of the same family contained a larger number of branches. This is illustrated in Fig. 10 representing a tris(amidoferrocenyl) dendron (3-Fc) and two dendrimers with 9- and 18 branches respectively (9-Fc resp. 18-Fc). In addition, a reference linear compound (1-Fc), [Fe(C5H5)(C5H4CONHCH2CH2OPh)], 1-Fc,was also used for comparison. All these amidoferrocene derivatives (10−3 M) were titrated in an electrochemical cell by n-Bu4N+HSO4− in CH2Cl2 containing n-Bu4N+BF4− (0.1 M). Fig. 10 shows that 1-Fc undergoes a redox potential shift of only about 10 mV at the equivalence point, whereas 3-Fc is shifted by 30 mV, 9-Fc by 65 mV, and 18-Fc by 130 mV (Fig. 10 and 11).100
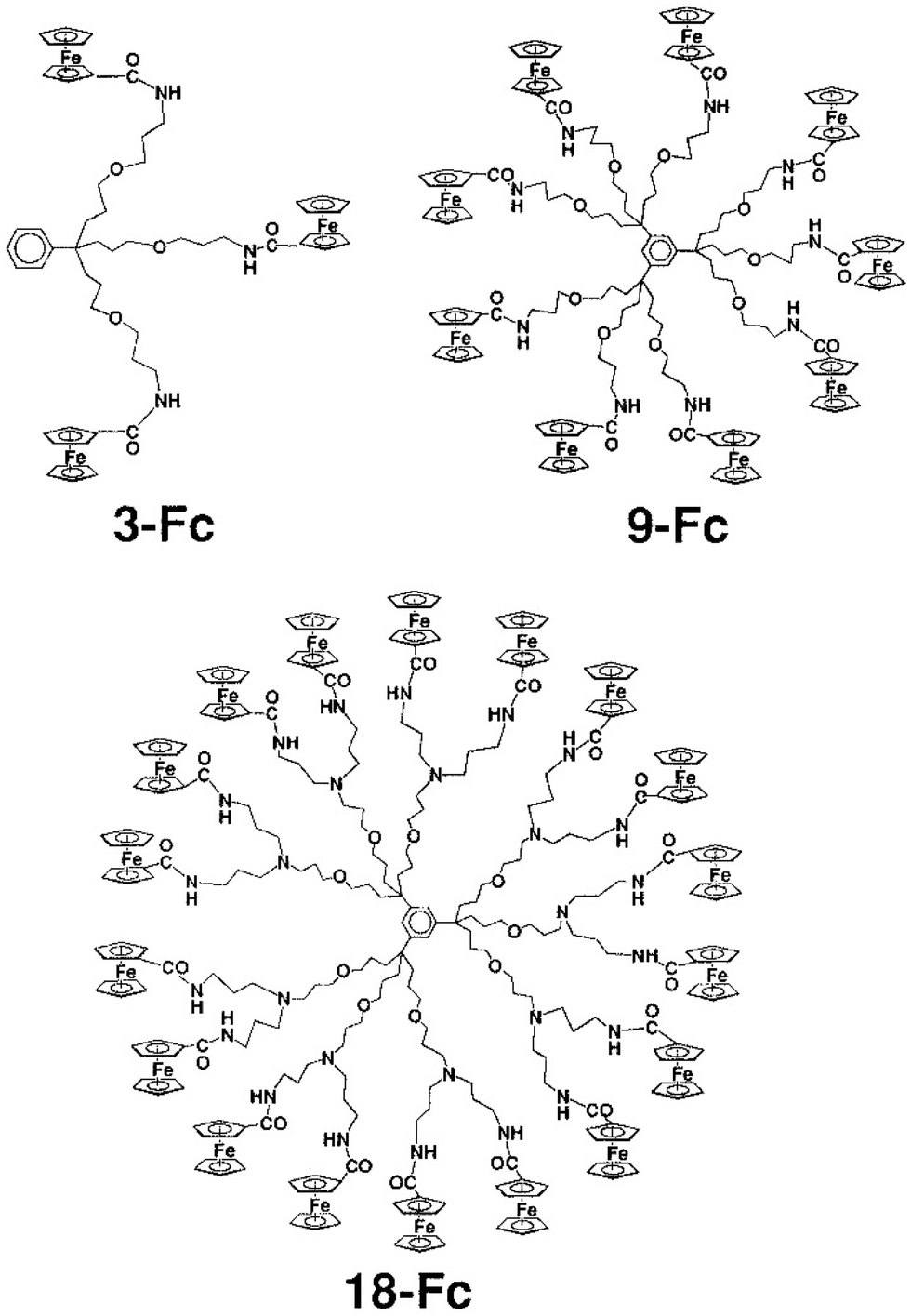 | ||
| Fig. 10 Metallodendrimers with 3 (3-Fc), 9 (9-Fc) and 27 (27-Fc) terminal amidoferrocenyl groups that recognize oxo-anions using cyclic voltammetry, CV (see Fig. 11). Reproduced with permission from ref. 100. Copyright 1997 American Chemical Society. | ||
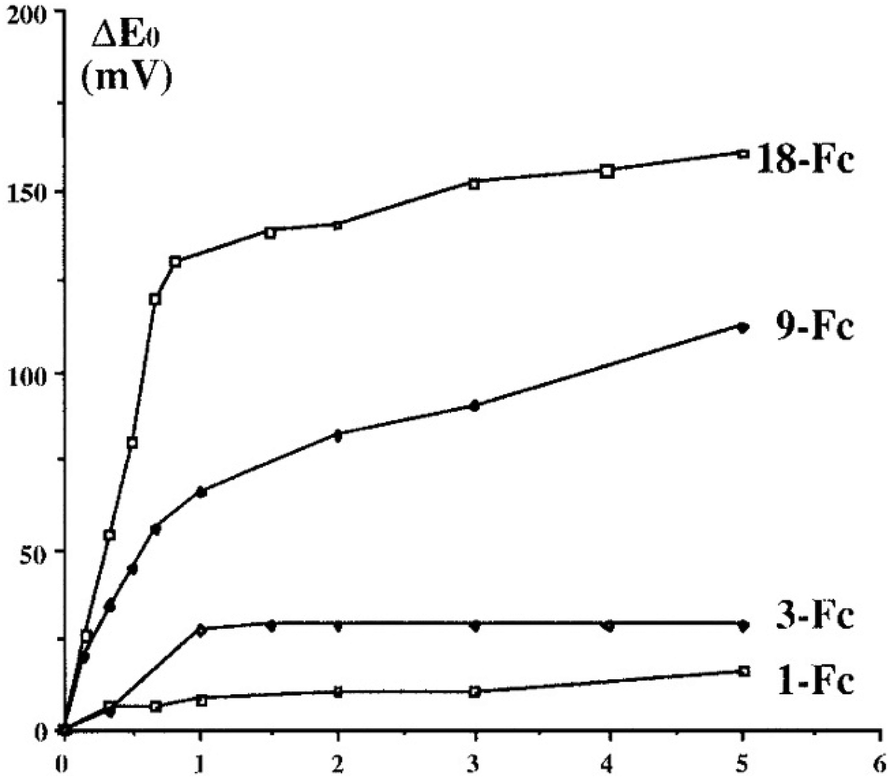 | ||
| Fig. 11 Dendritic effect for redox sensing of oxoanions: example of HSO4−. Number of equiv. of n-Bu4N+HSO4− added per branch. Titration of 1-Fc (1-Fc) [Fe(C5H5)(C5H4CONHCH2CH2OPh)], 3-Fc, 9-Fc, and 18-Fc (see Fig. 10) by n-Bu4N+HSO4− monitored by CV. Concentrations in FcDs were 0.001 M, CH2Cl2, n-Bu4N+BF4− (0.1 M), 20 °C, reference electrode: SCE, auxiliary and working electrodes: Pt, scan rate: 100 mV s−1. Reproduced with permission from ref. 100. Copyright 1997 American Chemical Society. | ||
There is thus a strongly positive dendritic effect. The magnitude of the redox potential shift varied as follows with the nature of the anion: H2PO4− > HSO4− > Cl− > NO3−. The chemical reversibility of the redox site is even strengthened upon using the permethylated ligand C5Me5 (Cp*) in the ferrocenyl group,101,102 which is also useful for ferrocene-based dendritic batteries.80,87,88 The reasons for these redox potential shifts are (i) the H-bonding interaction between the amido group and the anion, (ii) the electrostatic attraction between the positively charged ferricenium form in the course of electrochemical anodic scanning and the anion and (iii) the dendritic confinement effect provoked by the congestion between branches offering only a narrow channel to the anion for interaction that is all the more marked as the dendrimer generation is higher. Note that the two first effects are very small in a linear compound, and it is the third endo- or exo-receptor effect that very much increases the two first factors (Fig. 12).
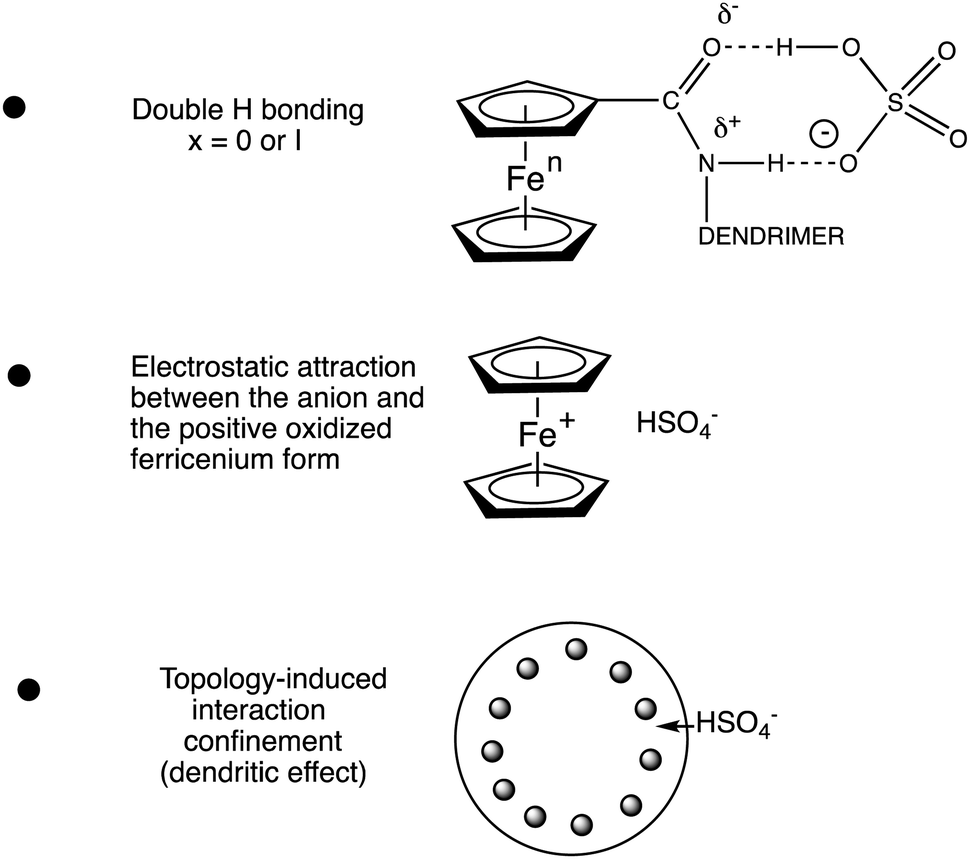 | ||
| Fig. 12 The three effects responsible for the redox recognition of anions. | ||
The binding constant between the dendritic host and the anion is much larger in the cationic ferricenium form (K+) than in the neutral ferrocene form (K0) due to the additional electrostatic interaction in the cationic form. The constant ratio is accessible from the difference between the redox potentials observed without interaction with the anion (beginning of titration) and that with full interaction (end of titration) as follows, K0 being then reached by 1H NMR (Scheme 11):103
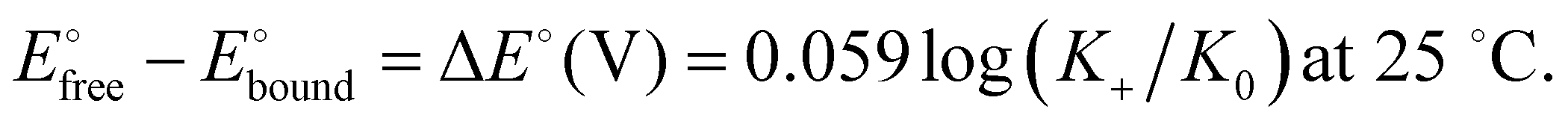
 | ||
Scheme 11 Square scheme showing the association constant K0 between the ferrocenyl dendrimer and the anion and that between the oxidized dendrimer and the anion (K+) as a function of the redox potentials of the ferrocenyl dendrimer alone before titration (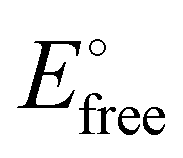 ) and in the presence of the anion at the end of titration ( ) and in the presence of the anion at the end of titration (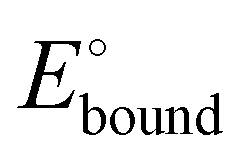 ). If K0 is large (strong association of A− with the ferrocenyl dendrimer), the second cyclovoltammetry wave appears during the titration. If K0 is small (weak association), the unique wave progressively shifts during the titration upon addition of the anion salt. ). If K0 is large (strong association of A− with the ferrocenyl dendrimer), the second cyclovoltammetry wave appears during the titration. If K0 is small (weak association), the unique wave progressively shifts during the titration upon addition of the anion salt. | ||
A variety of redox sites, functional links and receptors have been designed.92–110 Redox sites of the [FeCp(arene] family are also excellent choice, because the FeII/I redox system if often chemically and electrochemically reversible in this series. For instance, dendrimers terminated with [FeCp*(η6-C6H5NH-)] are excellent redox sensors for Cl− and Br−.106 Oxo anions deserve special attention, because genetic materials contain phosphate derived anions. Gold NP-centered amidoferrocenyl-terminated stars showed excellent oxo-anion sensing properties,108,109 and gold NP-centered silylferrocenyl-terminated dendrimers were selective ATP2− sensors (Fig. 13).109
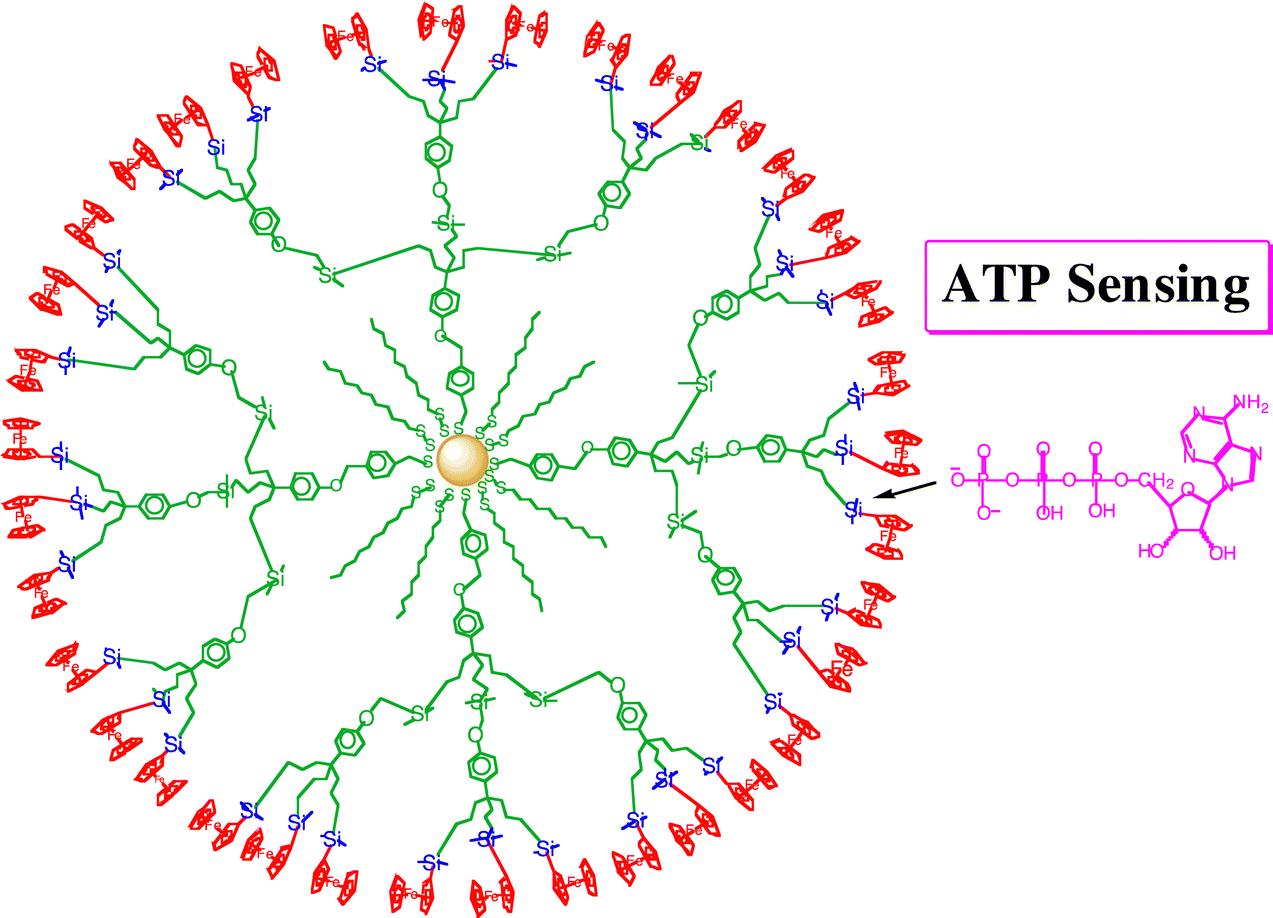 | ||
| Fig. 13 ATP anion redox sensing by gold NP-centered silylferrocenyl-terminated dendrimers (planar representation). The oxygen ATP atoms interact with the electrophilic Si atoms. | ||
Even dendrimers that are not assembled with covalent bonds, but supramolecularly assembled with a core and H-bonded dendrons are efficient and selective for oxo-anion sensing (Fig. 9).110 The dendritic topology with its dendritic effect represents an advantage over polymers or co-polymers, however, for ATP2− sensing with the same amido linker (Fig. 14 and 15).109 These macromolecular sensors are easily adsorbed on Pt electrodes upon scanning around the potential of the redox site, and the modified Pt electrode sensors are easily washed from substrate after sensing toward the next run.
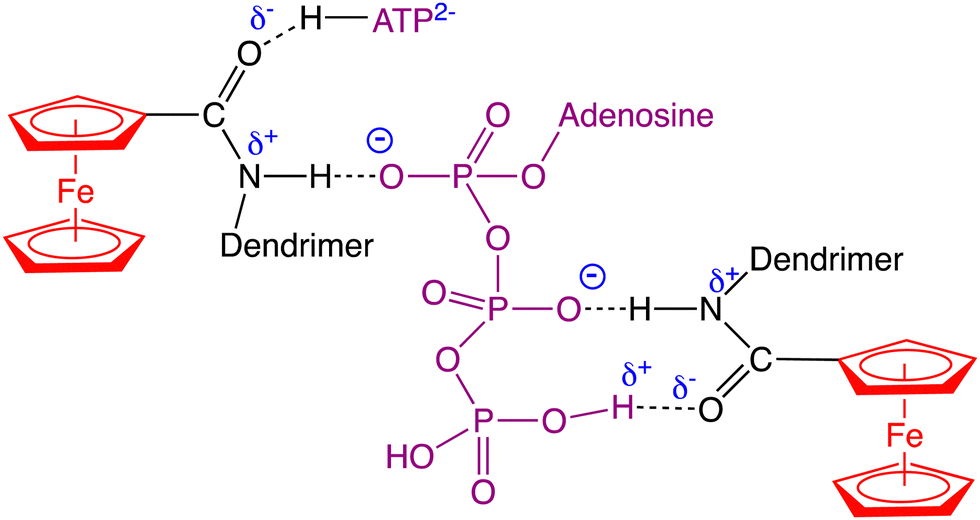 | ||
| Fig. 14 Supramolecular (H-bonding) interactions between ATP2− with amidoferrocenyl termini of dendrimers allowing to sense ATP2−. | ||
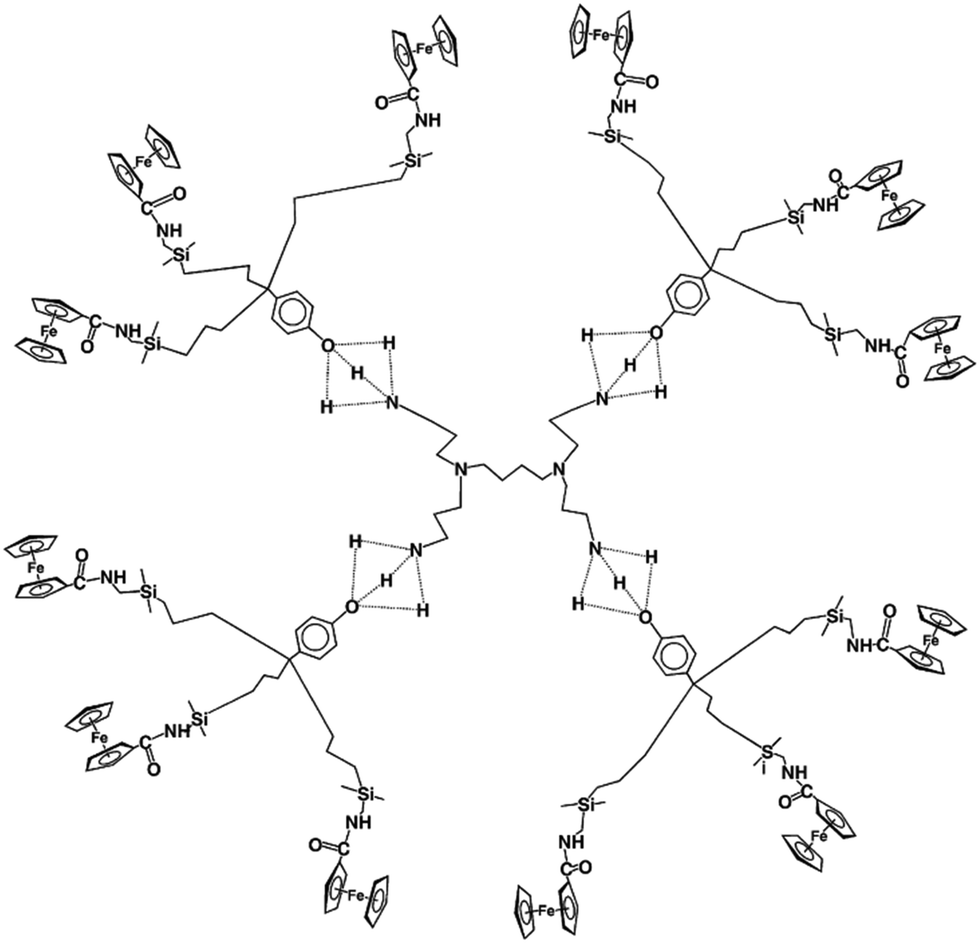 | ||
| Fig. 15 Arbitrary representation of the reversible hydrogen bonding between G1-DAB-dend-(NH2)4 and a triamidoferrocenyl dendron shown by the concentration-dependent average location of the (broad) NH2 + OH signal in 1H NMR between 2.4 and 4.1 ppm vs. TMS in CDCl3. | ||
Another useful link between the redox site and the receptor for redox sensing application in dendrimers111–119 and functionalized gold120 and palladium nanoparticles121 is the 1,2,3-triazole function formed by Cu(I) azide alkyne catalyzed (CuAAC) click reaction111–119 either between azido-terminated dendrimers and alkynyl-ferrocene111–121 or between alkynyl-terminated dendrimers and azido ferrocene122,123 (Scheme 12).
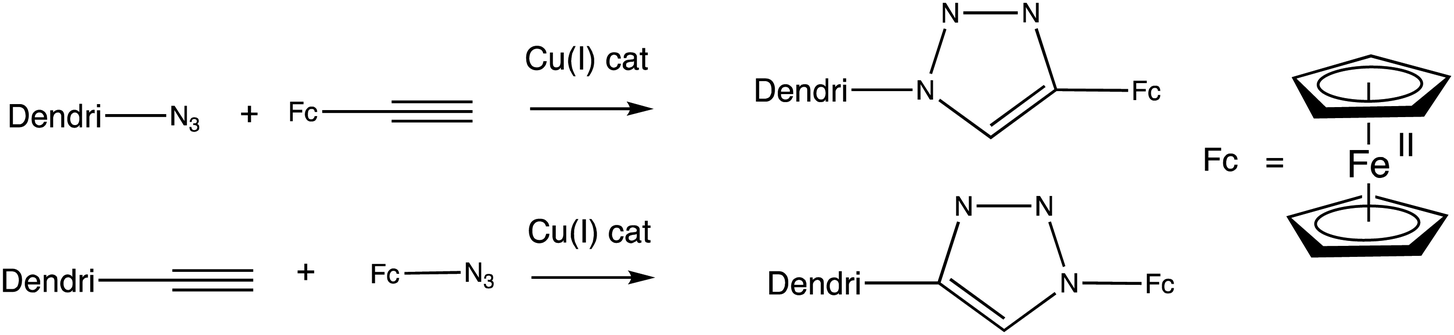 | ||
| Scheme 12 Two ways to branch a redox site with a 1,2,3-triazole to a dendritic receptor. | ||
Ferrocenyl (or cobaltocenyl) triazolyl dendrimers are good sensors for both oxo-anions with supramolecular interactions connected via the triazole N atoms and for transition metal cations through binding to the triazole as ligand.107–109 The single CV wave observed for all the ferrocenyl groups facilitates sensing, as their number was determined using the Bard–Anson equation using the redox system [FeCp*2+/0] as a reference.
For the oxo anions (n-Bu4N)(H2PO4) or (n-Bu4N)2(ATP), the new redox wave is located at a less positive potential than the initial wave, because the dendrimer–oxo anion assembly is easier to oxidize than the dendrimer alone with the anion donating electron density to the redox system. For transition metal cations, the new CV wave appears upon titration at a more positive potential than the initial wave, because the cation-dendrimer assembly is more difficult to oxidize than the initial redox center due to electron-releasing coordination of the triazole ligand to the added metal cation. This molecular redox recognition is all the larger (difference in redox potential upon ion salt addition) as the dendrimer generation is higher, in accord with the positive dendritic effect in redox sensing.
A remarkable case is that of the click triazolylbiferrocenyl-terminated dendrimers that can recognize both oxo-anion from the terminal ferrocenyl groups oxidized first because they are more electron-rich than the inner ferrocenyl groups, and metal cations through coordination to the triazolferrocenyl group, a sensor system that can be installed onto a Pt electrode to modify it upon scanning (Fig. 16 and 17).109
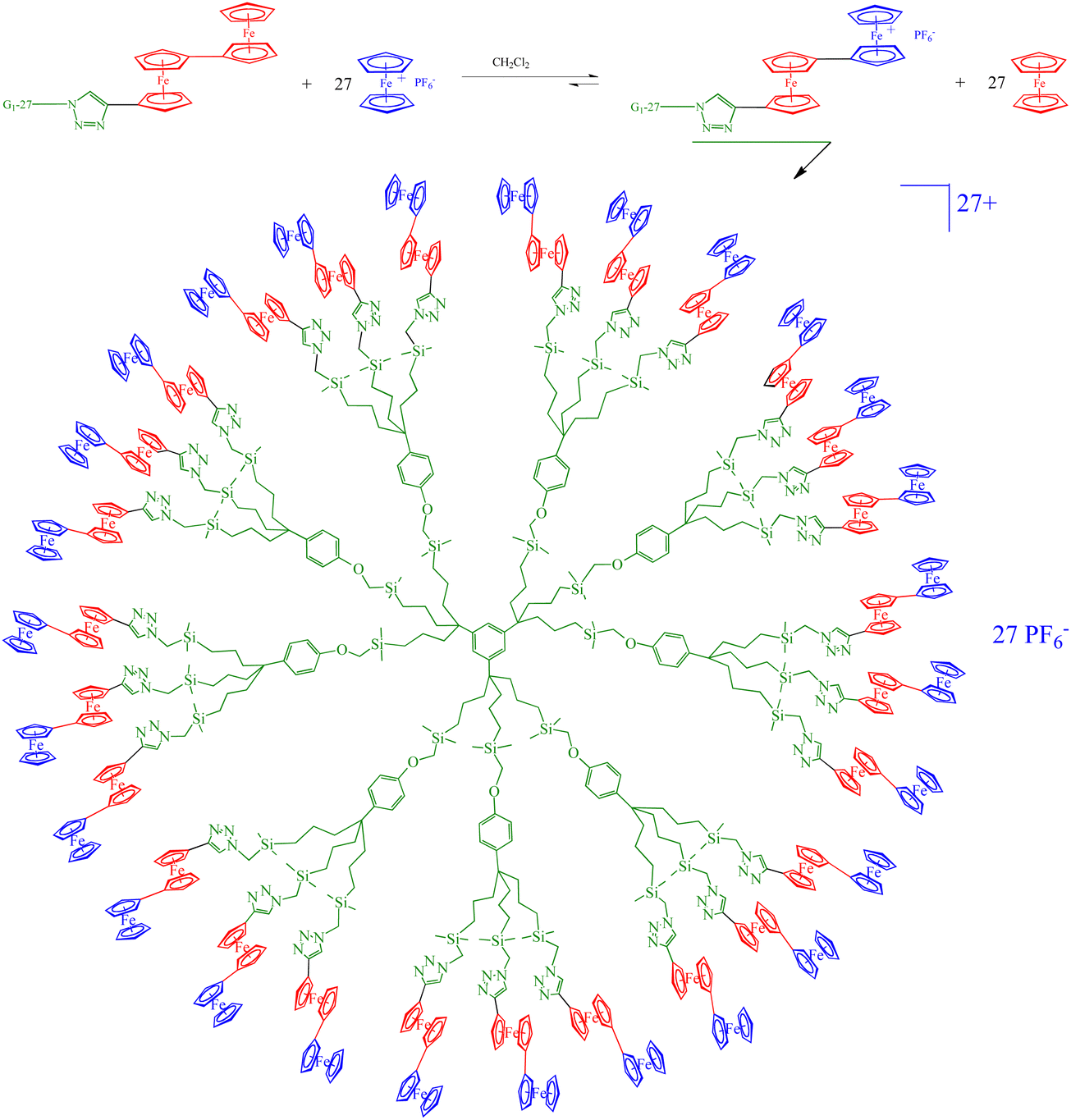 | ||
| Fig. 16 Selective oxidation of the outer ferrocenyl groups (blue) in the 27-biferrocenyl dendrimer. | ||
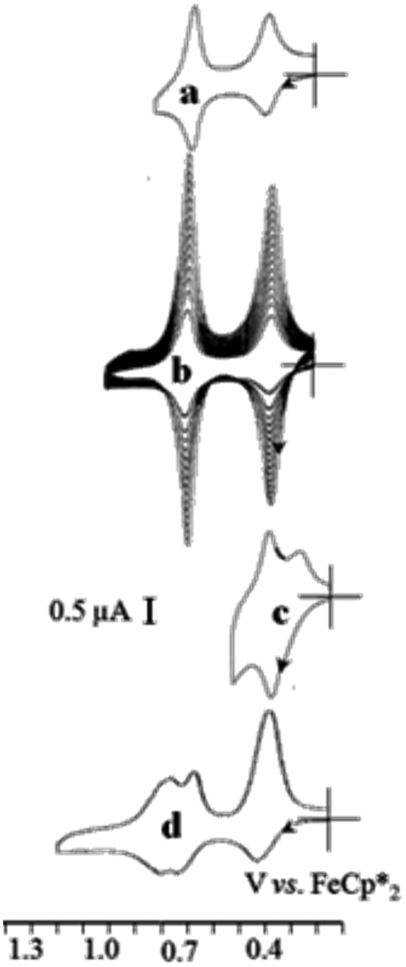 | ||
| Fig. 17 Selective roles of the inner and outer ferrocenyl redox centers of the 81-biferrocenyl dendrimer in the redox recognition of ATP2− and Pd2+. Cyclic voltammograms of the G2-81-biferrocenyl click dendrimer, (a) in CH2Cl2, [n-Bu4N][PF6] 0.1M; (b) progressive adsorption upon scanning around the potential of the biFc area; (c) splitting of the outer-Fc CV wave at 0.4V upon addition of [ATP][n-Bu4N]2; (d) addition of Pd(OAc)2 provoking the splitting of the inner-Fc wave at 0.7 V. Reproduced with permission from ref. 117. Copyright 2010 Wiley-VCH. | ||
VI. Dendrimers and ferrocene-containing macromolecular devices in biomedicine
Ferrocene and dendrimers have long been the subjects of focuses due to their biomedical properties. Since the finding in 1971 of the properties of ferrocenone (o-sodium carboxylate benzoyl ferrocene, Fig. 18, left, the only ferrocenyl drug so far approved for clinical use) against anemia124 and the first discovery and investigation in 1978 of ferrocene derivatives as anti-cancer drugs,125 ferrocene-containing substrates have constantly been investigated for their anti-cancer and other biomedical properties.126–129 The other most advanced drug in clinical trials, ferroquine (Fig. 18, middle), is in phase IIb of clinical trials for its anti-malarial activity in vivo, in combination with artefenomel (a fast-killing antiparasitic trioxolanel).128 A good example of ferrocene-containing antitumoral drug is the ferrocifen family (Fig. 18, bottom, right) for which the ferrocenyl group replaces the phenyl group of the anticancer drug tamoxiphen.129 The strength of all the ferrocene-containing drug candidates is their ability to produce reactive oxygen species (ROS) such as OH˙ near cancer cells where H2O2 is present, using ferrocene redox chemistry in Fenton-type reactions (Fig. 18(a)).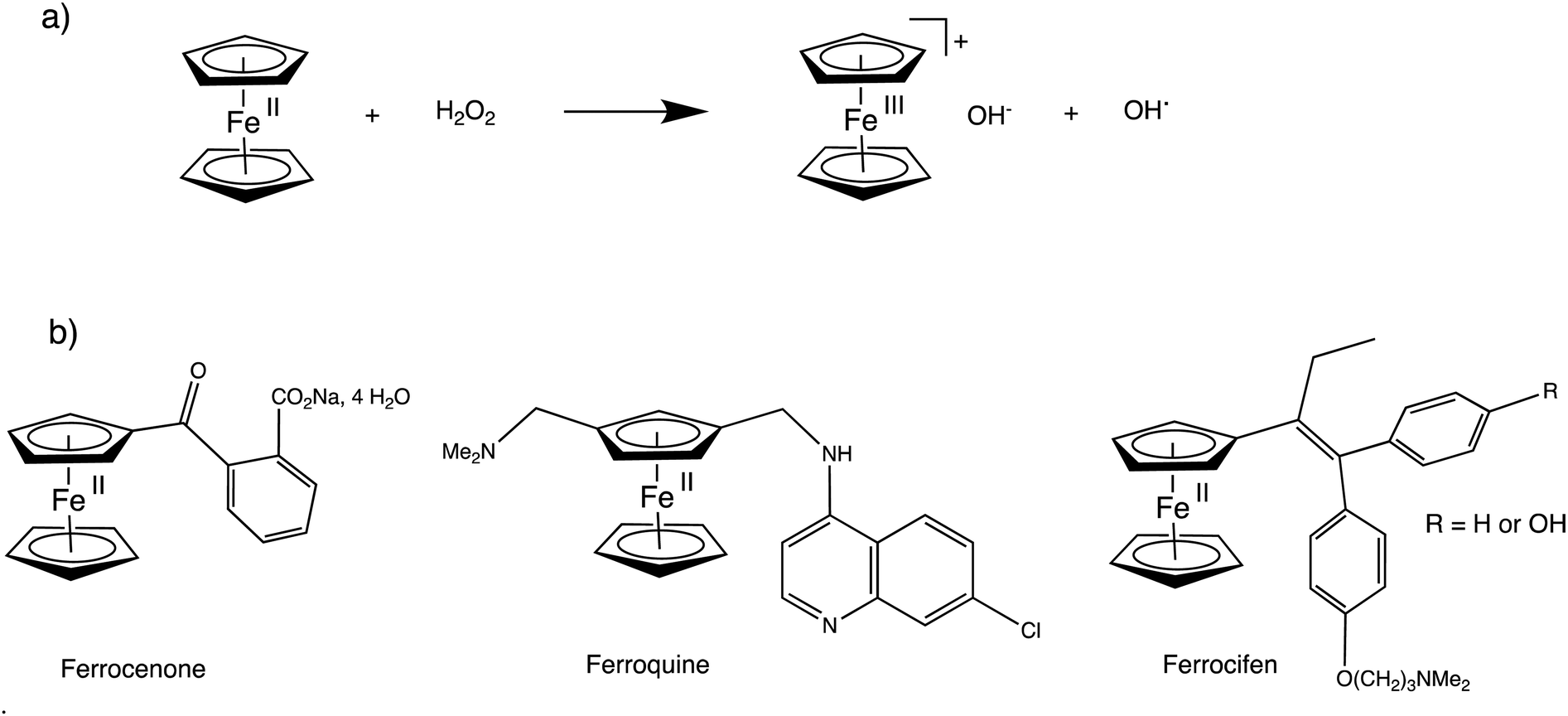 | ||
| Fig. 18 (a) Fenton-type reaction of ferrocene partly responsible for ferrocene drug activity upon generating OH˙ radicals at the vicinity of the sick cells; (b) main types of ferrocene-containing drugs. | ||
Therefore, a 80 nm-size nanozyme Co-ferrocene metal–organic framework combining glucose oxidase was reported to function as enzymatic/Fenton catalytic platform generating gluconic acid and H2O2 resulting in excellent Fenton effect for the generation of highly toxic OH˙ radicals remarkably increasing tumor treatment.130
The incorporation of metal-containing artificial analogues into DNA strands has been reported to control the size and the functions of noncanonical self-assemblies of multifunctional DNA nanostructures, assembled from long DNA building blocks, and named DNA nanoflowers. The introduction of a ferrocene (Fc) base allowed size controllability and self-degradability of Sgc8-NFs-Fc with H2O2via Fenton's reaction.131 Amphiphilic polymer micelles formed by self-assembling arachidonic acid and azobenzene linked to Fc was shown to be an effective cell carrier and expected to develop in sensitizing ferroptosis (programmed cell death pathway) and anti-tumor.132 An Ir(III) complex liganded to a ferrocene-modified diphosphine that localizes in lysosomes also showed ferroptosis effective for cancer immunotherapy.133
Dendritic macromolecules have long been another area of interest for a wide variety of biomedical applications,134–139 because of their capacity of drug encapsulation.129,140 An example from our group is docetaxel (Fig. 19), commercialized as taxoter (from the taxoid antineoplastic family), and discovered in 1989 by Pierre Pottier and his group at the ICSN institute of Gif-sur-Yvette, near Paris, upon hemi-synthesis from 10-deacetylbaccatin-III extracted from the leaves of the European yew (Taxus baccata).141 It is registered in the updated Model List of Essential Medicines by the World Organization of Health (WOH).
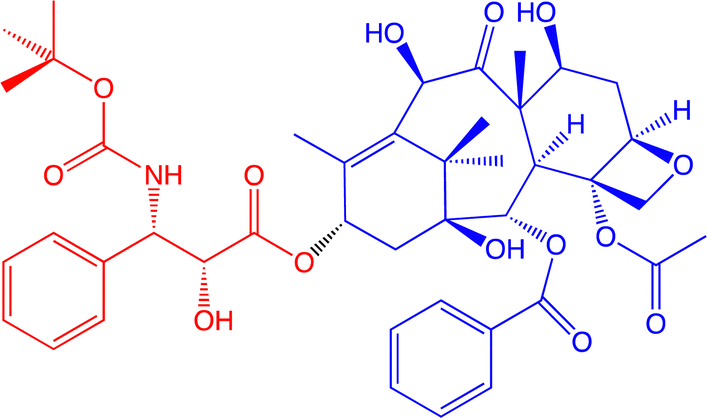 | ||
| Fig. 19 Structure of docetaxel (formulated as taxoter). The red part was added upon hemi-synthesis from 10-deacetylbaccatin-III (blue part) extracted from the European yew leaves.141 | ||
Docetaxel if very efficient against breast, lung and metastatic prostate cancers by stabilizing the microtubules upon inhibition of their depolymerization by stable bond formation with its cellular receptor, tubuline, which generates the blockade of mitosis. Docetaxel nanotechnology in anti-cancer therapy is essential towards the vectorization of the drug in order to optimize its efficiency and minimize side effects.141,142 Therefore, docetaxel, that is hydrophobic, insoluble in water, was solubilized upon encapsulation into dendrimer-like PEGylated gold nanoparticles, which allowed recording its 1H NMR spectrum in D2O (Fig. 20). This encapsulation of docetaxel was utilized for its vectorization to cancer cells, which was achieved with positive in vitro results.143
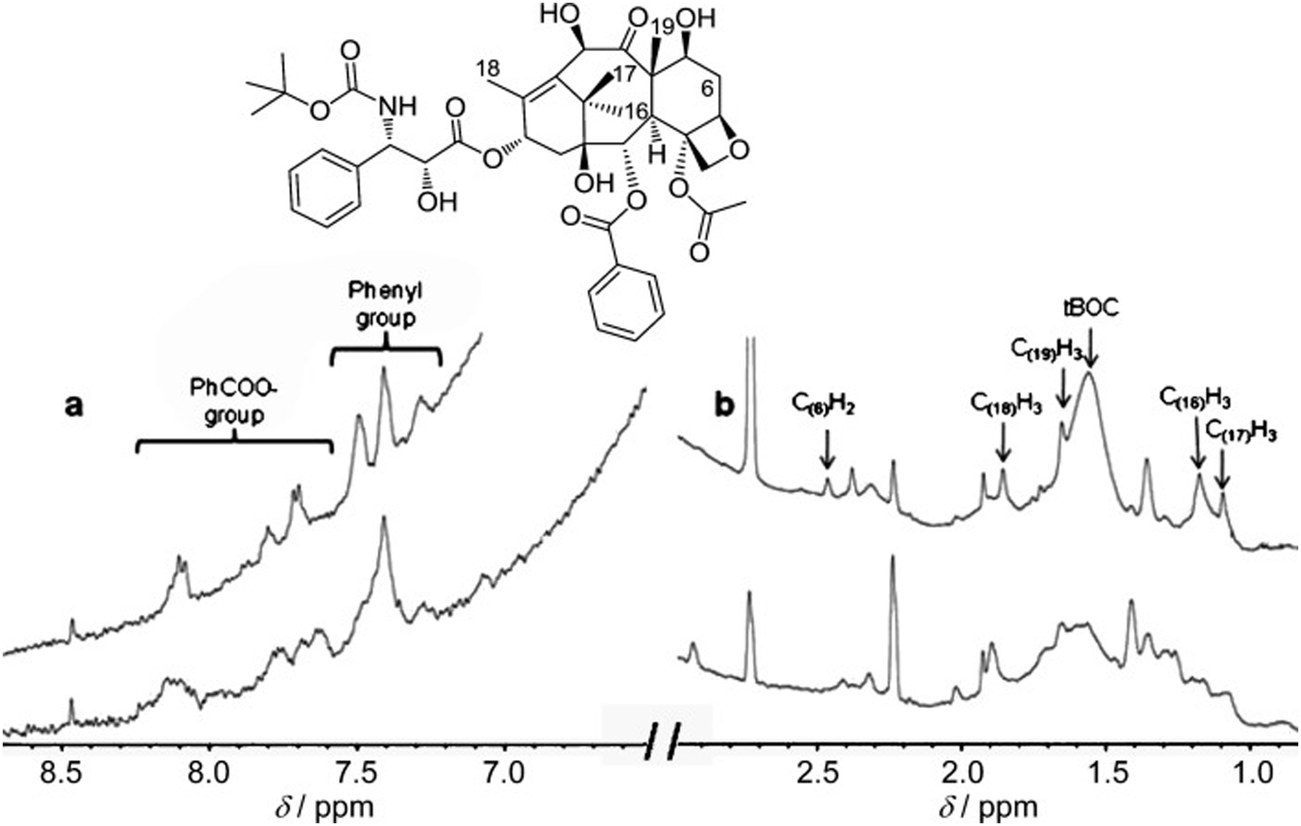 | ||
| Fig. 20 1H NMR spectroscopic (600 MHz) analysis (a) >6 ppm and (b) <3 ppm of AuNP-encapsulated docetaxel (9.5 molecules per Au atom) in H2O. Upper traces: docetaxel only; lower traces: docetaxel-AuNPs. Signal assignments were carried out by comparison with the spectrum in DMSO. Note that the solubility of docetaxel alone in H2O is by far too low to record a spectrum in this solvent without AuNPs. Reproduced with permission from ref. 143. Copyright 2011 Wiley-VCH. | ||
As opposed to the long-known aggregation-caused quenching (ACQ) of light emission in the condensed phase due to decay via non-radiative pathways of aggregate excited states,144 Tang discovered in 2001 the phenomenon of aggregation-induced emission (AIE) that he assigned to restriction of intramolecular rotation (RIR) inhibiting non-radiative decay.145 Tang extended the concept to collective intra- and inter-chain n → π* interactions of heteroatoms and to various other types of macromolecules.146–148 Along this line and following the emission properties of dendrimers, Tomalia later proposed that, similar to RIR, restricted intramolecular mobility (RIM) in dendrimer structure due to aggregation of numerous non-emissive, electron rich, heteroatomic groups was a source of AIE named non-traditional intrinsic luminescence (NTIL).149,150 Merging dendrimer and ferrocene properties towards theranostic applications was a recent target of our group in collaboration with the Ornelas group, particularly because dendrimers containing some functions can undergo AIE. Indeed, triazolylferrocenyl dendrimers of the family of that shown in Fig. 7 self-assembled into nanovesicles and vesosomes in water (Fig. 21) and presented green NTIL fluorescence and cytotoxicity allowing the development of new theranostic systems.151
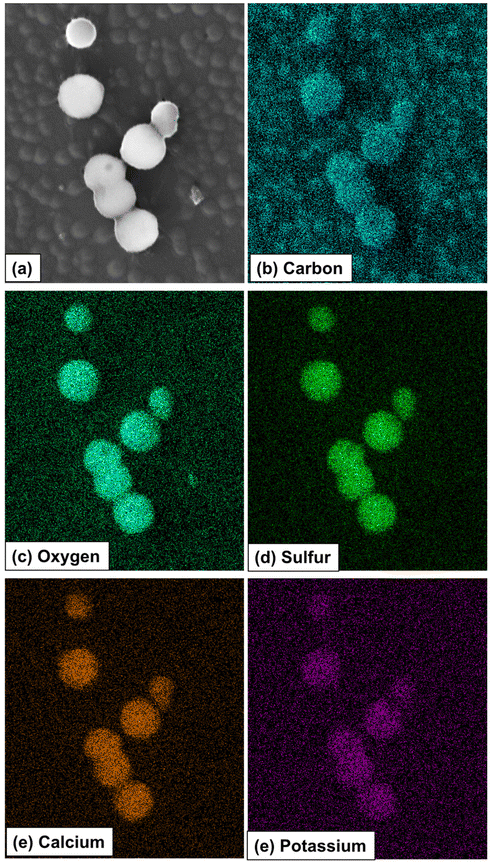 | ||
| Fig. 21 SEM/EDS elemental mapping profiles of the vesosomes resulting from self-assembling triazolylferrocenyl dendrimers in water. Reproduced from ref. 151. Copyright 2021 American Chemical Society. | ||
Several generations of our triazolyferrocene-terminated dendrimers of the family shown in Fig. 7 contain numerous ether, silyl and triazolyl heteroatoms and therefore could lead to such “hybridized assemblies of single quantum state-like clouds of delocalizable electrons”.150 The location of large ferrocene groups at the dendrimer peripheny contributes to restricting intradendritic motion responsible for enhancement of emission.
VII. Olefin metathesis reactions with dendrimers and application to soft materials
The discovery of efficient olefin metathesis polymerization catalysts has been a considerable breakthrough towards new functional macromolecules.152–154 In our group, for instance, an early example consisted in adapting such a catalyst at the periphery of several generations of poly(propylene imine) (so-called DAB) dendrimers, then polymerize norbornene using these dendritic Ring-Opening Metathesis Polymerization (ROMP) catalysts, which produced giant DAB-dendrimer-cored macromolecules (Scheme 13). Remarkably, the polymerization rates were higher for the dendrimers than with a monomer propylene imine-derived ruthenium catalyst, possibly because steric congestion among the ruthenium ligands of the catalyst, but the rates decreased as the dendrimer generation increased. Indeed, with catalysts located at dendrimer peripheries, we usually observed a negative dendritic effect (i.e. decreased rates as the dendrimer generation increased) due to steric bulk inhibiting substrate approach to the catalytic center, which was opposite to redox sensors for which the dendritic effect was always positive.155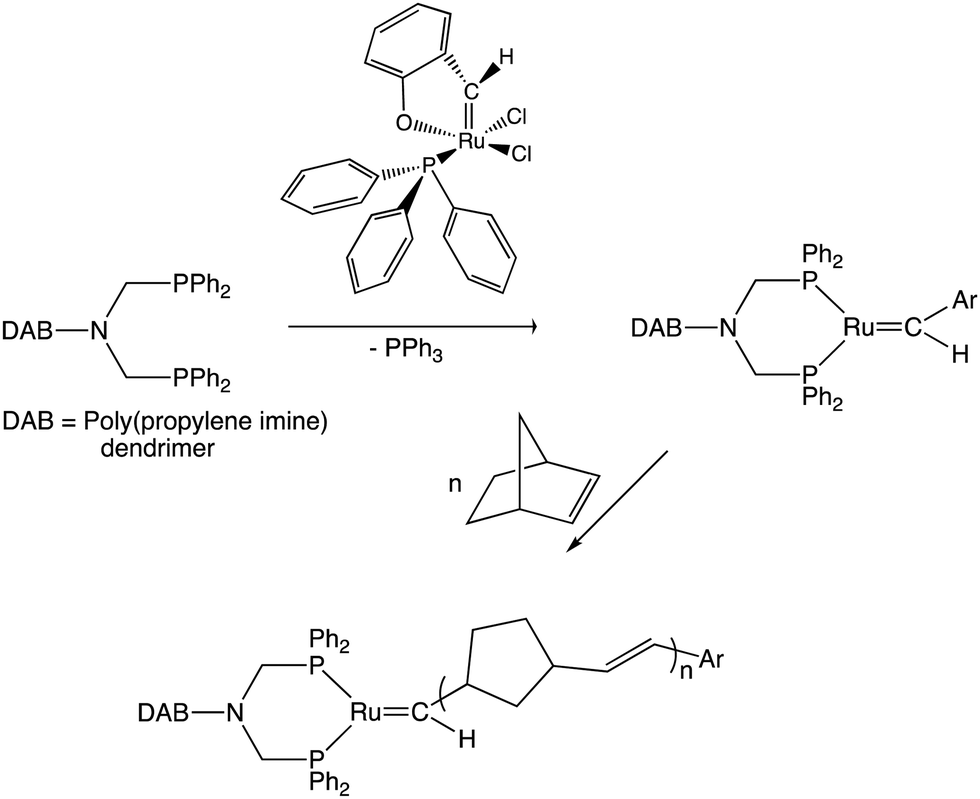 | ||
| Scheme 13 ROMP of norbornene (n = 100) at the periphery of 3 generations (G1, G2 and G3) of DAB dendrimers producing giant dendritic-cored star-shaped macromolecules using a ruthenium benzylidene catalyst. In the dendrimers of this scheme, the Ru benzylidene ligand is simplified as CHAr and the chloro ligands are omitted for clarity. The dendritic DAB-cored phosphines were accessible by reactions of the first, second and third generation DAB dendrimers with P(Cl)Ph2. | ||
The nona-allyl dendrimer core obtained in Scheme 6 from mesitylene was the subject of remarkably selective olefin metathesis reactions catalyzed by Grubb's second-generation catalyst, [Ru(CHPh) (NHC)Cl2]]. Ring Closing Metathesis (RCM) of two of the three allyl group of each triallyl tripod was fast, giving a cyclopentenyl substituent. On the other end, the reaction of the third allyl group of each tripod was much slower and depended of the reaction conditions (Scheme 14).156,157
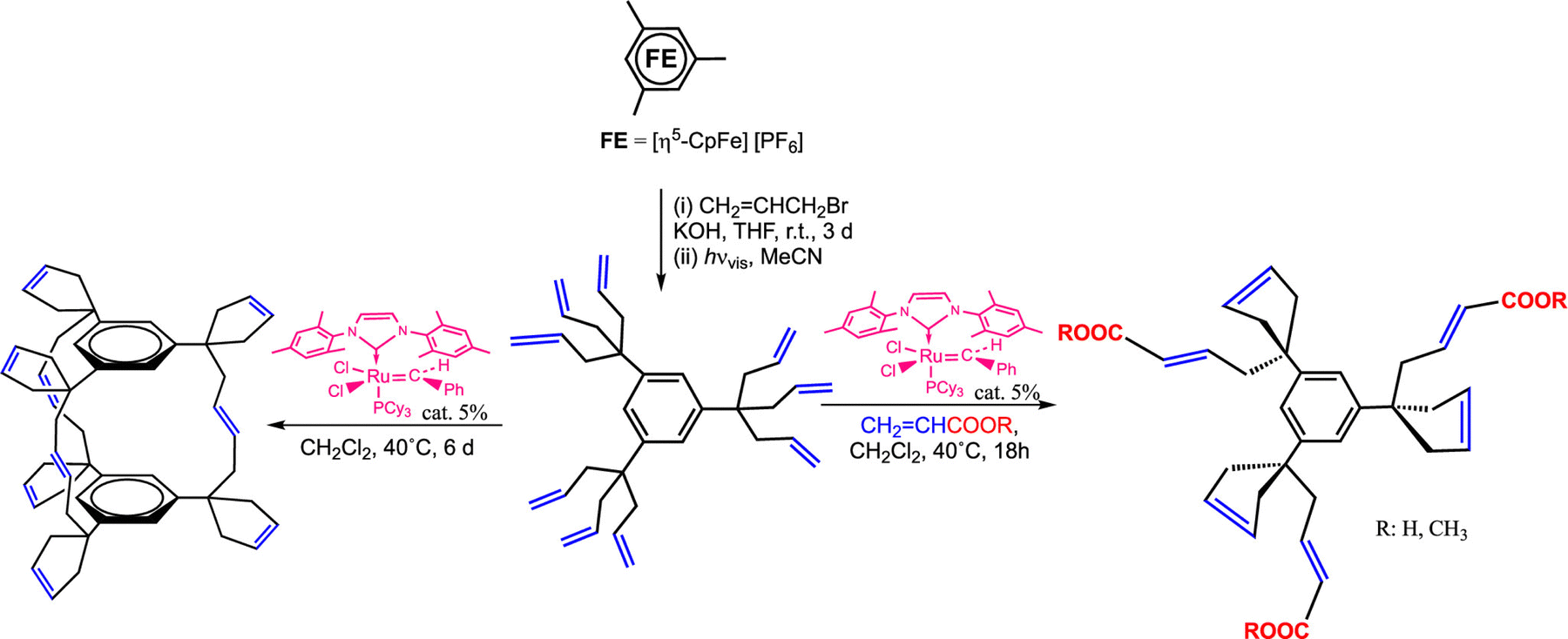 | ||
| Scheme 14 Single-pot CpFe+-induced nona-allylation of mesitylene followed by olefin metathesis reaction of the 9 terminal double bonds catalyzed by the 2nd-generation Ru Grubbs catalyst: cyclophane cage synthesis (left) or cyclization of two tethers of each tripod along with functionalization of the 3rd one in the presence of acrylic acid or acrylate (right). Adapted with permission of ref. 140. Copyright 2008 American Chemical Society. | ||
In the presence of an acrylic ester, cross metathesis with this functional olefin allowed substrate functionalization, but, in its absence, slow triple cross metathesis with another molecule yielded a single capsule compound as the thermodynamic end-product assembling two arene moieties. Many intermediate oligomers were observed by MALDI TOF mass spectrometry as kinetic products in this latter reaction. Lengthening the tethers before metathesis highlighted the interest of the cross metathesis for the functionalization of olefin-terminated dendrimers with acrylic acid and esters, as illustrated in Scheme 15.157
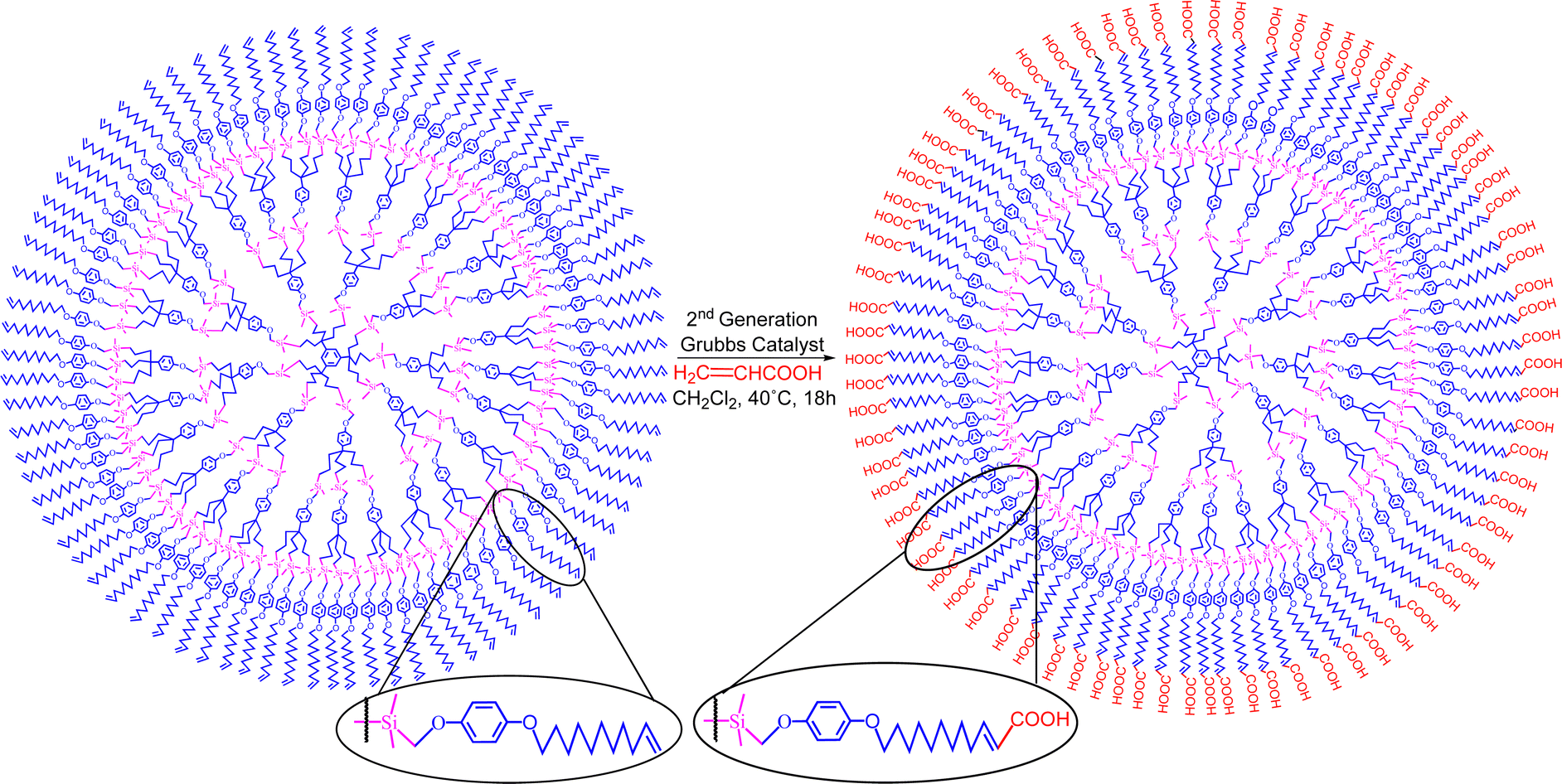 | ||
| Scheme 15 Selective functionalization of olefin-terminated dendrimers by cross metathesis. Reproduced with permission from ref. 157. Copyright 2008 American Chemical Society. | ||
Another useful aspect of ferrocene or cobalticenium-containing macromolecules concerns metallohydrogels. Supramolecular gels are soft materials composed of small gelator molecule assembled into supramolecular networks, with space filled by solvent. Some gel materials are able to respond to various stimuli including with redox groups such as ferrocene, making them attractive drug delivery vehicles and as matrices for tissue regeneration.158,159 Immobilization in the hydrogel of glucose oxidase (GOx) modified with ferrocene produces an electrically contacted enzyme electrode that stimulates the bioelectrocatalyzed oxidation of glucose. Upon incorporation of a load into the hydrogel, the electrocatalyzed oxidation of glucose and the accompanying acidification allow controlling the release of the load.160 Hydrogels that contained dendronic and polymeric ferrocene ensembles synthesized using the ROMP technique with the same Grubbs-II catalyst in macromolecular di-block assemblies allowed access to self-healing materials for biomedical applications including redox-stimuli-responsive drug delivery systems.161 Examples of the encapsulation/release of substrates include rhodamine, benzocain and the anti-cancer drug doxorubicin upon redox switch with suitable redox reagents of the ferrocene unit in metallopolymer micelles (Fig. 22).161–164
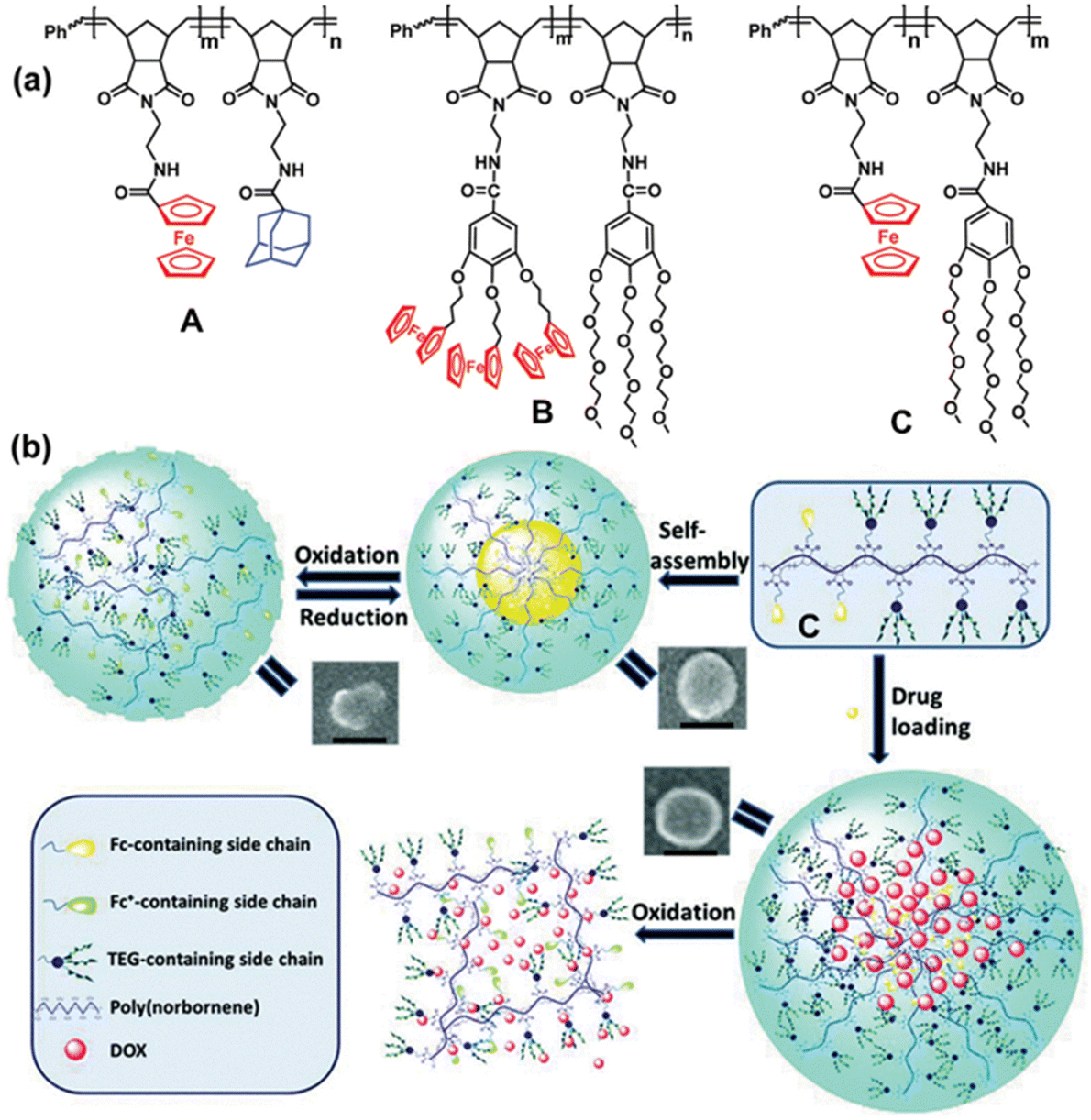 | ||
| Fig. 22 (a) ROMP-synthesized metallopoymers A, B and C containing ferrocenes. A forms an amphiphile upon β-cyclodextrin addition, and B and C are amphiphiles that self-assembles in redox-responsive micelle (b) Redox-controlled micelles of C for the encapsulation/release of drugs upon redox switch of the ferrocene units; SEM images are shown (scale bar: 100 nm). Reproduced with permission from ref. 163. Copyright 2019 Royal Society of Chemistry. | ||
Supramolecular soft-templating mesoporous materials have been reported to allow regulating uniform pore structure with designed dimensions hosting situ synthesis of nano-objects toward applications for separation, sensing, catalysis, energy conversion and storage, photonics, solar cells, photo- and electrochromism, microelectronics, bio-oriented devices.165,166 Therefore, a newly designed electrochemically assisted self-assembly method provides a fast and versatile way to generate highly ordered mesopore channels and creates a platform for the development of functionalized oriented films carrying functional groups such as ferrocene and cobaltocene with electron hoping between close redox sites as the charge transfer mechanism.165
Electro-responsive threadlike micelles were formed using new cationic gemini surfactants (Fc)11-n11(Fc) with redox-active ferrocenyl groups in the gemini alkyl chains and showed to form a class of highly viscous smart soft materials.167 The new gelator, stigmasteryl glycinate ferrocenoylamide (SGF) utilizes plant sterols as raw materials and has multi-stimulus response performance with gelation behavior including lamellar aggregation pattern showing excellent responsiveness to redox stimuli.168
VIII. Dendritic catalysis, unimolecular micellar effect, auto-catalysis, homeopathic catalysis, and nanozymes
In section VII, cross metathesis was shown to be useful as a dendrimer functionalization strategy. Other efficient dendrimer functionalization methods involved, for instance ETC reactions26 using the Fe(I) sandwich 1a as initiator, as shown in Scheme 2. When dendrimers were decorated with 32 or 64 CH2PPh2 groups, ETC exchange of a carbonyl ligand of the well-known cluster [Ru3(CO)12] by each of the dendritic phosphine using 1% mol equiv. of this initiator, 1a, was very clean at ambient temperature as shown by 31P NMR and provided multi cluster-functionalized dendrimers.169,170 A dendrimer with nona-peroxophospho-tungstate {[PO4{WO(O2)2}4]3−} tri(hexyl) ammonium salts as termini synthesized by functionalization of the nona-allyl arene proved to be a mild, efficient and fully recyclable metallodendritic catalyst for the epoxidation of olefins by H2O2 in water/CDCl3 and for the oxidation of thioanisole to sulfone.171 In this later case, as in many others in the literature for a variety of reactions,172 the dendrimer acts as a support allowing catalyst recycling. Dendritic unimolecular micelles such as that synthesized according to Scheme 16 served as catalyst templates for various catalytic reactions in water or aqueous media including olefin metathesis reactions,172–174 click chemistry,174–176 carbon–carbon coupling of Miyaura–Suzuki or Sonogashira type177–179 and redox reactions including nitrophenol reduction179–182 by NaBH4183 or aerobic benzylic alcohol oxidation.184 Click dendrimers were indeed major candidates for the mild stabilization of efficient gold and palladium nanocatalysts and nanosensors,185,186 although efficient gold nanocatalysts185–187 were also synthesized by simple Au(III) reduction with NaBH4.188 This dendrimer synthesis of Scheme 16 itself is autocatalytic,175 since conducting the reaction in the presence of 1% of the final product more than doubled the reaction yield, and the conversion yield was already 46% after 1.5 h instead of 2% in absence of this addition. The reason of the autocatalysis is that the dendrimer served as a template for the substrates. These micellar-type reactions needed only a small amount (such as 0.1 mol%) of the dendritic micelle42 (that was fully recyclable) and involved only a few ppm of nanocatalyst (and even less than 1 ppm for some Miyaura–Suzuki reactions). With triazolyl-ferrocene terminated click dendrimers, these reactions required, at ambient temperature, all the less ppm Pd catalyst as the concentration was lower, which led to the concept of “homeopathic catalysis”. This phenomenon, known in de Vries’ work for Heck reactions at 130 °C,189 was taken into account by tiny catalytic cluster loss from the mother nanoparticle that recombined all the less easily as the nanoparticle concentration was lower.190–192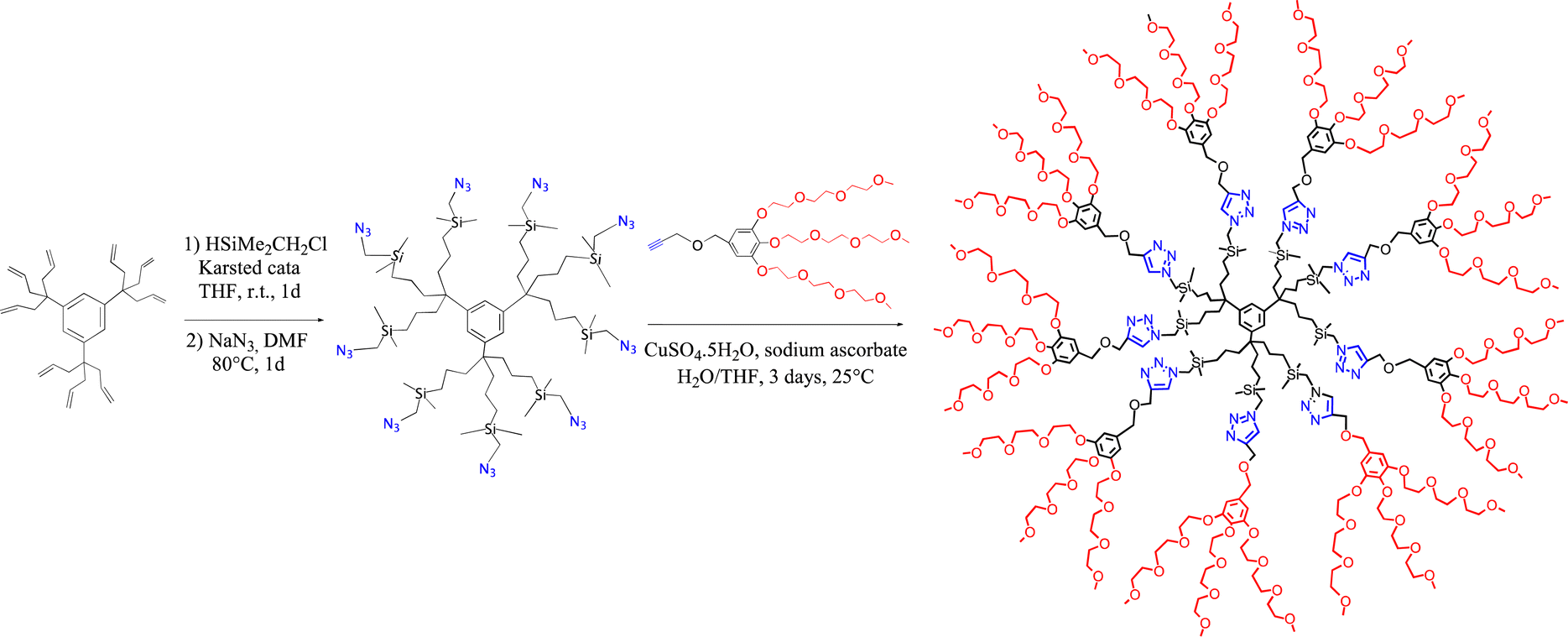 | ||
| Scheme 16 Synthesis of a dendritic unimolecular micelle as template for multiple catalytic uses (molecular catalysis with Grubbs II catalyst, metal ion click catalysis with Cu(I), and nanoparticle catalysis). | ||
The localization and shape of nanoparticles encapsulated in dendrimers also play key roles on their catalytic activity, depending on the reaction involved.193,194 In most of these reactions, especially micellar ones, the dendrimer-nanocatalyst assembly behaves as an enzyme, i.e. it represents a nanozyme, a known biochemical concept.195,196 For instance, with Cu(I)tren-centered dendrimer terminated with 18 or 54 tri(ethyleneglycol) branches, kinetic studies with phenyl acetylene and benzyl azide showed even faster reactions than with non-dendritic Cu(I)-(PhCH2)6tren, a trend similar to that known with enzymes.197 Click-dendrimer-Pd nanoparticle assemblies also behaved as enzyme mimics in the colorimetric H2O2 detection upon catalytic o-phenylene diamine oxidation.198
IX. Hydrogen production
Hydrogen (H2) is crucial for sustainable energy of the future.199 Therefore, the necessity of safe H2 carriers to minimize risks and insure H2 storage and transportation has undergone considerable research.200 Boranes and related derivatives201 are hydrogen rich, and their hydrolysis under ambient conditions, providing H2 in the presence of various transition metal-based nanocatalysts, has drawn the attention of chemists,201–203 with particular emphasis first on NaBH4, then on ammonia–borane, NH3BH3.203 If these two derivatives are the most investigated ones, research has been extended to others in the liquid phase such as hydrazine borane, silanes, formic acid and other hydrocarbons.203,204 Our group has studied a variety of these hydrogen carriers essentially using, as nanocatalyst stabilizers, either click dendrimers205–208 or MOFs of the ZIF-8 type,209–216 because both of these families, beside their steric effects, contain nitrogen atoms that mildly stabilize nanoparticle catalysts for optimized reactions and provide synergy in catalysis. In these reactions, one hydrogen atoms of H2 formed is provided by the hydrogen carrier, and the other from water, which is shown by labeling experiments.215 Kinetic isotope effects and DFT calculations have provided evidence that water activation is the rate-determining step, facilitated by hydrogen bonding with the hydridic atom coming from the carrier at the nanocatalyst surface.195 In the particular case of the nanocatalyzed hydrolysis of bis-boric acid, {B2(OH)4}, however, both hydrogen atoms of H2 formed are coming from water, the side product being boric acid, B(OH)3.207,217 The H2 generation reaction rates can be switched on and off for instance by pH control,212,215,217 or be boosted by visible-light irradiation, either upon including a semiconductor in the nanocatalyst composite (Mott–Schottky junction at the metal–semiconductor interface),211 or more efficiently utilizing the Ag or Au plasmon207–209,215–219 or a combination of both the semiconductor and plasmonic effects.211 The studies of these nanocatalysts bring about considerable knowledge on how the key nanocatalyst hydride are formed and how they reductively eliminate to give H2. In particular, the introduction of negative charges onto the nanocatalyst by anion coordination or plasmonic hot electron transfer has been shown to boost H2 formation during these reactions, which also is of interest for other energy-providing nanocatalyzed or electrocatalyzed reactions. This detailed mechanistic information disclosed here, confirmed by DFT studies, is also useful for the design of energy-related catalytic devices.220–223X. Conclusion and outlook
The simplest chemical events, electron transfer and proton transfer, have been utilized with late transition metal sandwiches whose robustness was eventually reinforced by sandwich ring permethylation. The considerable acidity increase of these juxtacyclic methyl or alkyl groups compared to isostructural neutral compounds has allowed us to establish in the late 1970s a novel and powerful method of multiple C–C bond formation that led to the formation of molecular stars and dendrimers. It is also the choice of such robust cationic 18-electron complexes that afforded easily synthesizing very electron-rich 19-electron complexes, yet at redox potential far less negative than isolobal ferrocene anion that cannot be synthesized. The designed 19-electron and 17-electron iron complexes, besides oroginal electronic properties including mixed and average valencies and application to battery materials, are very useful stoichiometric redox reagents and also possess redox catalyst and electrocatalyst properties allowing many redox transformations of a variety of substrates.The resulting dendrimer structure with 1 → 3 branching led us to the design of the first click metallodendrimers, i.e. with metals either attached to the termini of the dendritic tethers or as nanoparticules coordinated to the triazole link. This specifically mild triazole coordination to nanoparticles rendered these nanocatalysts extremely efficient for a broad range of key reactions including C–C or C–N bond formation in substrates as well as fast and efficient H2 generation from hydrogen-rich precursors, sometimes accelerated by visible light illumination, for instance when the alloy contains a plasmonic metal besides the substrate-activating metal.
Focusing on electron and proton transfer processes obviously involved a biological context and applications, as already noticed at an early time by the easy generation and demonstration of great reactivity of superoxide radical anion and the total inhibition of its reactions in the model system. Redox catalysis of reduction of nitrogen oxide and other nitro derivatives catalyzed by Fe(I) or nanoparticles is a method of choice of their destruction from the environment. Olefin metathesis reactions are a useful complement to the dendritic structure in molecular engineering towards both redox sensing and drug delivery, for which the redox aspect of the metal sandwich compounds plays an essential part. The supramolecular aspect of these ensembles plays a key role in their properties and functions, as exemplified by the in vitro toxicity of dendritic structures in docetaxel delivery against prostate cancer and theranostic application of fluorescent vesosomes produced upon metallodendrimers assembly.
In the future, such sophistication of molecular and inorganic devices and their interplay should be essential, in particular in the design of both optimized nanocatalysts for energy conversion and “magic bullets” for drug delivery.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Contributions and enlightening and enthusiastic discussions with the numerous colleagues and students cited in the references, and financial support from the University of Bordeaux, the Centre National de la Recherche Scientifique (CNRS), L’Oréal Recherche, the Institut Universitaire de France, the Agence National pour la Recherche (ANR), the Nanosolution European FP7 program, the China Scolarship Council (CSC) and 111 Project of China are gratefully acknowledged.References
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- I. Langmuir, Science, 1921, 54, 59–67 CrossRef CAS PubMed.
- R. B. Woodward, M. Rosenblum and M. C. Whiting, J. Am. Chem. Soc., 1952, 74, 3458–3459 CrossRef CAS.
- G. Wilkinson, J. Organomet. Chem., 1975, 100, 273–278 CrossRef CAS.
- M. L. H. Green and G. Parkin, J. Chem. Ed., 2014, 91, 807–816 Search PubMed.
- P. Stepnicka, Dalton Trans., 2022, 51, 8085–8102 RSC.
- M. C. Baird, J. Organomet. Chem., 2014, 751, 50–54 CrossRef CAS.
- A. N. Nesmeyanov, N. A. Volkenau and I. N. Bolesova, Tetrahedron Lett., 1963, 4, 1725–1729 CrossRef.
- D. Astruc, J.-R. Hamon, G. Althoff, E. Román, P. Batail, P. Michaud, J.-P. Mariot, F. Varret and D. Cozak, J. Am. Chem. Soc., 1979, 101, 5445–5447 CrossRef CAS.
- J.-R. Hamon, D. Astruc and P. Michaud, J. Am. Chem. Soc., 1981, 103, 758–766 CrossRef CAS.
- M. V. Rajasekharan, S. Giezynski, J. H. Ammeter, N. Oswald, J.-R. Hamon, P. Michaud and D. Astruc, J. Am. Chem. Soc., 1982, 104, 2400–2407 CrossRef CAS.
- D. Astruc, Acc. Chem. Res., 1986, 19, 377–383 CrossRef CAS.
- D. Astruc, E. Román, J.-R. Hamon and P. Batail, J. Am. Chem. Soc., 1979, 101, 2240–2242 CrossRef CAS.
- D. Astruc, J.-R. Hamon, E. Román and P. Michaud, J. Am. Chem. Soc., 1981, 103, 7502–7514 CrossRef CAS.
- J.-R. Hamon, D. Astruc, E. Román, P. Batail and J. J. Mayerle, J. Am. Chem. Soc., 1981, 103, 2431–2433 CrossRef CAS.
- J.-R. Hamon and D. Astruc, J. Am. Chem. Soc., 1983, 105, 5951–5953 CrossRef CAS.
- T. Finkel and N. J. Holbrook, Nature, 2000, 408, 239–247 CrossRef CAS PubMed.
- A. Loupy, B. Tchoubar and D. Astruc, Chem. Rev., 1992, 92, 1141–1165 CrossRef CAS.
- C. Bossard, S. Rigaut, D. Astruc, M.-H. Delville, G. Félix, A. Février-Bouvier, J. Amiell, S. Flandrois and P. Delhaès, J. Chem. Soc., Chem. Commun., 1993, 333–334 RSC.
- D. Méry, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2006, 128, 5602–5603 CrossRef PubMed.
- F. Fu, A. Dedieu, W. Wang, T. Chen, Y. Song, E. Fouquet, J.-R. Hamon, M. Zhu and D. Astruc, Chem. Commun., 2019, 55, 10277–10280 RSC.
- S. Rigaut, M.-H. Delville and D. Astruc, J. Am. Chem. Soc., 1997, 119, 11132–11133 CrossRef CAS.
- S. Rigaut, M.-H. Delville, J. Losada and D. Astruc, Inorg. Chim. Acta, 2002, 334, 225–242 CrossRef CAS.
- J. Ruiz, M. Lacoste and D. Astruc, J. Am. Chem. Soc., 1990, 112, 5471–5483 CrossRef CAS.
- D. S. Brown, M.-H. Delville, R. Boese, K. P. C. Vollhardt and D. Astruc, Angew. Chem., Int. Ed. Engl., 1994, 33, 661–663 CrossRef.
- D. Astruc, Angew. Chem., Int. Ed. Engl., 1988, 27, 643–660 CrossRef.
- H. Taube, Chem. Rev., 1952, 50, 69–126 CrossRef CAS.
- J. Ruiz and D. Astruc, C. R. Seances Acad. Sci., Ser. C, 1998, 21–27 CrossRef CAS.
- I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters and L. Phillips, J. Phys. Chem. B, 1999, 103, 6713–6722 CrossRef CAS.
- C. A. P. Goodwin, M. J. Giansiracusa, S. M. Greer, H. M. Nicholas, P. Evans, M. Vonci, S. Hill, N. F. Chilton and D. P. Mills, Nat. Chem., 2021, 13, 243–248 CrossRef CAS PubMed.
- H. A. Trujillo, C. Casado, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 1999, 121, 5674–5686 CrossRef CAS.
- J. Ruiz, F. Ogliaro, J.-Y. Saillard, J.-F. Halet, F. Varret and D. Astruc, J. Am. Chem. Soc., 1998, 120, 11693–11705 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- H. Nishihara, Adv. Inorg. Chem., 2002, 53, 41–86 CrossRef CAS.
- D. Astruc, Electron Transfer and Radical Processes, Transition-Metal Chemistry. Part I: Theories, Techniques, Electronic Structures and Molecular Electronics. Part II: Mechanisms, Catalysis and Applications to Synthesis, VCH, New York, 1995, p. 630, ISBN 1-56081-566-3; foreword: Henry Taube Search PubMed.
- M.-H. Desbois and D. Astruc, New J. Chem., 1989, 13, 595–600 CAS.
- D. Astruc, Acc. Chem. Res., 1991, 24, 36–42 CrossRef CAS.
- J. L. Hérisson and Y. Chauvin, Makromol. Chem., 1971, 141, 161–176 CrossRef.
- T. J. Katz, Adv. Organomet. Chem., 1978, 16, 283–317 CrossRef.
- D. Astruc, Organometallic Chemistry and Catalysis, Springer, Berlin, 2007, p. 608, ISBN 978-3-540-46128-9. French edition: Chimie organométallique et catalyse. EDP Sciences, Les Ullis, 2013 Search PubMed.
- D. A. Tomalia, A. M. Naylor and W. A. Goddard, Angew. Chem., Int. Ed. Engl., 1990, 29, 138–175 CrossRef.
- G. R. Newkome, E. F. He and C. N. Moorefield, Chem. Rev., 1999, 99, 1689–1746 CrossRef CAS PubMed.
- A. M. Caminade and J. P. Majoral, in Dendrimers, ed. F. Vögtle, Top. Curr. Chem., 1998, vol. 197, pp. 79–124 Search PubMed.
- F. Moulines, L. Djakovitch, R. Boese, B. Gloaguen, W. Thiel, J.-L. Fillaut, M.-H. Delville and D. Astruc, Angew. Chem., Int. Ed. Engl., 1993, 32, 1075–1077 CrossRef.
- F. Moulines, B. Gloaguen and D. Astruc, Angew. Chem., Int. Ed. Engl., 1992, 31, 458–460 CrossRef.
- J.-R. Hamon, J.-Y. Saillard, A. Le Beuze, M. J. McGlinchey and D. Astruc, J. Am. Chem. Soc., 1982, 104, 7549–7555 CrossRef CAS.
- F. Moulines and D. Astruc, Angew. Chem., Int. Ed. Engl., 1988, 27, 1347–1349 CrossRef.
- J. Ruiz, G. Lafuente, S. Marcen, C. Ornelas, S. Lazare, E. Cloutet, J.-C. Blais and D. Astruc, J. Am. Chem. Soc., 2003, 125, 7250–7257 CrossRef CAS PubMed.
- V. Sartor, L. Djakovitch, J.-L. Fillaut, F. Moulines, F. Neveu, V. Marvaud, J. Guittard, J.-C. Blais and D. Astruc, J. Am. Chem. Soc., 1999, 121, 2929–2930 CrossRef CAS.
- G. R. Newkome and C. Shreiner, Chem. Rev., 2010, 110, 6338–6442 CrossRef CAS PubMed.
- P.-G. De Gennes and H. Hervet, J. Phys. Lett., 1983, 44, L351–L360 CrossRef.
- V. Martinez, J.-C. Blais and D. Astruc, Angew. Chem., Int. Ed., 2003, 42, 4366–4369 CrossRef CAS PubMed.
- D. Catheline and D. Astruc, J. Organomet. Chem., 1984, 272, 417–426 CrossRef CAS.
- D. Astruc, New J. Chem., 1992, 16, 305–328 CAS.
- B. Gloaguen and D. Astruc, J. Am. Chem. Soc., 1990, 112, 4607–4609 CrossRef CAS.
- D. Buchholz, B. Gloaguen, J.-L. Fillaut, M. Cotrait and D. Astruc, Chem. – Eur. J., 1995, 1, 374–381 CrossRef CAS.
- D. Buchholz and D. Astruc, Angew. Chem., Int. Ed. Engl., 1994, 33, 1637–1639 CrossRef.
- D. Astruc, Acc. Chem. Res., 2000, 33, 287–298 CrossRef CAS PubMed.
- M.-H. Desbois and D. Astruc, Angew. Chem., Int. Ed. Engl., 1989, 28, 460–461 CrossRef.
- M.-H. Delville, S. Mross, D. Astruc, J. Linares, F. Varret, H. Rabaa, A. Le Beuze, J.-Y. Saillard, R. D. Culp, D. A. Atwood and A. H. Cowley, J. Am. Chem. Soc., 1996, 118, 4133–4147 CrossRef.
- D. O. Cowan, C. LeVanda, J. Park and F. Kaufmann, Acc. Chem. Res., 1973, 6, 1–7 CrossRef CAS.
- G. C. Allen and N. S. Hush, Prog. Inorg. Chem., 1967, 8, 357–389 CAS.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1968, 10, 247–422 CrossRef.
- C. Creuz and H. Taube, J. Am. Chem. Soc., 1969, 91, 3988–3989 CrossRef.
- D. Astruc, Acc. Chem. Res., 1997, 30, 383–391 CrossRef CAS.
- D. Astruc, Chem. – Eur. J., 2021, 27, 16291–16308 CrossRef CAS PubMed.
- M.-H. Desbois, D. Astruc, J. Guillin, J.-P. Mariot and F. Varret, J. Am. Chem. Soc., 1985, 107, 5280–5282 CrossRef CAS.
- M.-H. Desbois, D. Astruc, J. Guillin, F. Varret, A. X. Trautwein and G. Villeneuve, J. Am. Chem. Soc., 1989, 111, 5800–5809 CrossRef CAS.
- M.-H. Delville-Desbois, M. Lacoste and D. Astruc, J. Am. Chem. Soc., 1992, 114, 8310–8311 CrossRef.
- M. Lacoste, F. Varret, L. Toupet and D. Astruc, J. Am. Chem. Soc., 1987, 109, 6504–6506 CrossRef CAS.
- M. Lacoste, H. Rabaa, D. Astruc, N. Ardoin, F. Varret, J.-Y. Saillard and A. Le Beuze, J. Am. Chem. Soc., 1990, 112, 9548–9557 CrossRef CAS.
- A. K. Diallo, J.-C. Daran, F. Varret, J. Ruiz and D. Astruc, Angew. Chem., Int. Ed., 2009, 48, 3141–3145 CrossRef CAS PubMed.
- A. K. Diallo, C. Absalon, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2011, 133, 629–641 CrossRef CAS PubMed.
- D. Astruc, Nat. Chem., 2012, 4, 255–267 CrossRef CAS PubMed.
- F. Paul and C. Lapinte, Coord. Chem. Rev., 1998, 178–180, 431–509 CrossRef CAS.
- Y. Wang, L. Salmon, J. Ruiz and D. Astruc, Nat. Commun., 2014, 5, 3489 CrossRef PubMed.
- R. L. N. Hailes, A. M. Oliver, J. Gwyther, G. R. Whytell and I. Manners, Chem. Soc. Rev., 2016, 45, 5358–5407 RSC.
- C. Deraedt, A. Rapakousiou, Y. Wang, L. Salmon, M. Bousquet and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 8445–8449 CrossRef CAS PubMed.
- H. Gu, R. Ciganda, P. Castel, S. Moya, R. Hernandez, J. Ruiz and D. Astruc, Angew. Chem., Int. Ed., 2018, 57, 2204–2208 CrossRef CAS PubMed.
- P. Roland, E. Schoeter, O. Nolte, G. R. Newkome, M. D. Hager and U. S. Schubert, Prog. Mater. Sci., 2022, 125, 101474 Search PubMed.
- K. S. Park, S. B. Schougaard and J. B. Goodenough, Adv. Mater., 2007, 19, 848–851 CrossRef CAS.
- S. M. Beladi-Mousavi, S. Sadaf, L. Walder, M. Gallei, C. Rüttiger, S. Eigler and C. E. Halbig, Adv. Energy Mater., 2016, 6, 1600108 CrossRef.
- B. Hwang, M.-S. Park and K. Kim, ChemSusChem, 2015, 8, 310–314 CrossRef CAS PubMed.
- S. M. Beladi-Messavi, S. Sadaf, A. K. Hennecke, J. Klein, A. M. Mahmood, C. Ruttiger, M. Gallei, F. Fu, E. Fouquet, J. Ruiz, D. Astruc and L. Walder, Angew. Chem., Int. Ed., 2021, 60, 13554–13558 CrossRef PubMed.
- X. S. M. Beladi-Messavi and L. Walder, Polymer, 2022, 245, 124658 CrossRef.
- D. Y. Kuo, P. S. Price, S. Raugei and B. M. Cossairt, J. Phys. Chem. C, 2022, 126, 13994–14002 CrossRef CAS.
- C. Ornelas, J. Ruiz, C. Belin and D. Astruc, J. Am. Chem. Soc., 2009, 131, 590–601 CrossRef CAS PubMed.
- D. Astruc, C. Ornelas and J. Ruiz, Chem. – Eur. J., 2009, 15, 8936–8944 CrossRef CAS PubMed.
- C. Ornelas, J. Ruiz and D. Astruc, Organometallics, 2009, 28, 2716–2723 CrossRef CAS.
- R. Djeda, C. Ornelas, J. Ruiz and D. Astruc, Inorg. Chem., 2010, 49, 6085–6101 CrossRef CAS PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS PubMed.
- R. L. Sun, L. Wang, H. J. Yu, Zain-ul-Abdin, Y. S. Chen, J. Huang and R. B. Tong, Organometallics, 2014, 33, 4560–4573 CrossRef CAS.
- C. X. Zhu, D. Liu, Y. Y. Li, S. Ma, M. Wang and T. Y. Yu, Biosens. Bioelectron., 2021, 174, 112654 CrossRef CAS PubMed.
- H. Y. Li, Q. Li, S. X. Zhao, X. M. Wang and F. Li, J. Agric. Food Chem., 2022, 70, 680–686 CrossRef CAS PubMed.
- X. Y. Hua, H. L. Wang, Y. Q. Wen, X. J. Zhang and L. Su, Sens. Actuators, B, 2022, 372, 132610 CrossRef CAS.
- S. N. Feng, Y. Xue, J. S. Huang and X. R. Yang, Anal. Chem., 2022, 94, 16945–16952 CrossRef CAS PubMed.
- R. J. Zeng, J. H. Xu, T. K. Liang, M. J. Li and D. P. Tang, ACS Sens., 2023, 8, 317–325 CrossRef CAS PubMed.
- M. Y. Wang, W. J. Jing, L. J. Wang, L. P. Jia, R. N. Ma, W. Zhang, L. Shang, X. J. Li, Q. W. Xu and H. S. Wang, Biosens. Bioelectron., 2023, 226, 115116 CrossRef CAS PubMed.
- C. Valério, J.-L. Fillaut, J. Ruiz, J. Guittard, J.-C. Blais and D. Astruc, J. Am. Chem. Soc., 1997, 119, 2588–2589 CrossRef.
- S. Nlate, J. Ruiz, J.-C. Blais and D. Astruc, Chem. Commun., 2000, 417–418 RSC.
- M.-C. Daniel, J. Ruiz, J.-C. Blais, N. Daro and D. Astruc, Chem. – Eur. J., 2003, 9, 4371–4379 CrossRef CAS PubMed.
- S. R. Miller, D. A. Gustowski, Z.-H. Chen, G. W. Gokel, L. Echegoyen and A. E. Kaifer, Anal. Chem., 1988, 60, 2021–20924 CrossRef CAS.
- C. M. Casado, I. Cuadrado, M. Moran, B. Alonso, B. Garcia, B. Gonzales and J. Losada, Coord. Chem. Rev., 1999, 185–186, 53–79 CrossRef CAS.
- C. M. Cardona, S. Mendoza and A. E. Kaifer, Chem. Soc. Rev., 2000, 29, 37–42 RSC.
- C. Valério, E. Alonso, J. Ruiz, J.-C. Blais and D. Astruc, Angew. Chem., Int. Ed., 1999, 38, 1747–1751 CrossRef.
- M. Yamada and H. Nishihara, C. R. Chimie, 2001, 6, 919–934 CrossRef.
- A. Labande, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2002, 124, 1782–1789 CrossRef CAS PubMed.
- M.-C. Daniel, J. Ruiz, S. Nlate, J.-C. Blais and D. Astruc, J. Am. Chem. Soc., 2003, 125, 2617–2628 CrossRef CAS PubMed.
- M.-C. Daniel, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2003, 125, 1150–1151 CrossRef CAS PubMed.
- H. Gu, A. Rapakousiou, P. Castel, N. Guidolin, N. Pinaud, J. Ruiz and D. Astruc, Organometallics, 2014, 33, 4323–4335 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- C. R. Bercer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef PubMed.
- C. Ornelas, J. Ruiz, E. Cloutet, S. Alves and D. Astruc, Angew. Chem., Int. Ed., 2007, 46, 872–877 CrossRef CAS PubMed.
- C. Ornelas, L. Salmon, J. Ruiz and D. Astruc, Chem. – Eur. J., 2008, 14, 50–64 CrossRef CAS PubMed.
- R. Djeda, A. Rapakousiou, L. Liang, N. Guidolin, J. Ruiz and D. Astruc, Angew. Chem., Int. Ed., 2010, 49, 8152–8156 CrossRef CAS PubMed.
- S. Badèche, J.-C. Daran, J. Ruiz and D. Astruc, Inorg. Chem., 2008, 47, 4903–4908 CrossRef PubMed.
- D. Astruc, C. Ornelas and J. Ruiz, J. Inorg. Organomet. Polym. Mater., 2008, 18, 4–17 CrossRef CAS.
- E. Boisselier, L. Salmon, J. Ruiz and D. Astruc, Chem. Commun., 2008, 5788–5790 RSC.
- C. Ornelas, L. Salmon, J. Ruiz and D. Astruc, Chem. Commun., 2007, 4946–4948 RSC.
- J. Camponovo, J. Ruiz, E. Cloutet and D. Astruc, Chem. – Eur. J., 2009, 15, 2990–3002 CrossRef CAS PubMed.
- H.-W. Marx, F. Moulines, T. Wagner and D. Astruc, Angew. Chem., Int. Ed. Engl., 1996, 35, 1701–1704 CrossRef CAS.
- A. N. Nesmeyanov, L. G. Bogomolova and V. Viltcheskaya, US Pat., 119 356, 1971 Search PubMed.
- V. J. Fiorina, R. J. Dubois and S. Brynes, J. Med. Chem., 1978, 21, 393–395 CrossRef CAS PubMed.
- C. Biot, G. Glorian, L. A. Maciejewski and J. Brocard, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS PubMed.
- M. Patra and G. Gasser, Nat. Rev. Chem., 2017, 1, 0066 CrossRef CAS.
- C. Ornelas, New J. Chem., 2011, 35, 973–1985 RSC.
- G. Jaouen, A. Vessieres and S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817 RSC.
- C. Fang, Z. Deng, G. D. Cao, Q. Chu, Y. L. Wu, X. Li, X. S. Peng and G. R. Han, Adv. Funct. Mater., 2020, 30, 1910085 CrossRef CAS.
- L. L. Zhang, R. Abdullah, X. X. Hu, H. R. Bai, H. H. Fan, L. He, H. Liang, J. M. Zou, Y. L. Liu and Y. Sun, J. Am. Chem. Soc., 2019, 141, 4282–4290 CrossRef CAS PubMed.
- X. Liu, Z. F. Wu, C. J. Guo, H. M. Guo, Y. G. Su, Q. Chen, C. G. Sun, Q. M. Liu, D. Q. Chen and H. J. Mu, Drug Delivery, 2022, 29, 138–148 CrossRef CAS PubMed.
- W. J. Wang, Y. Y. Ling, Y. M. Zhong, Z. Y. Li, C. P. Tan and Z. W. Mao, Angew. Chem., Int. Ed., 2022, 61, e202115247 CAS.
- D. Astruc, C. R. Seances Acad. Sci., Ser. B, 1996, 322, 757–766 CAS.
- C. C. Lee, J. A. MacKay, J. M. J. Fréchet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS PubMed.
- J. Khandare, M. Calderon, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- R. M. Kannan, E. Nance, S. Kannan and D. A. Tomalia, J. Intern. Med., 2014, 276, 579–617 CrossRef CAS PubMed.
- A.-M. Caminade and C.-O. Turrin, J. Mater. Chem. B, 2014, 2, 4055–4066 RSC.
- E. Boisselier, A. K. Diallo, L. Salmon, C. Ornelas, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2010, 132, 2729–2742 CrossRef CAS PubMed.
- E. Boisselier, L. Liang, M. Dalko-Csiba, J. Ruiz and D. Astruc, Chem. – Eur. J., 2010, 16, 6056–6068 CrossRef CAS PubMed.
- D. Guénard, F. Guéritte-Voegelein and P. Pottier, Acc. Chem. Res., 1993, 26, 160–167 CrossRef.
- P. Zhao and D. Astruc, ChemMedChem, 2012, 7, 952–972 CrossRef CAS PubMed.
- A. François, A. Laroche, N. Pinaud, L. Salmon, J. Ruiz, J. Robert and D. Astruc, ChemMedChem, 2011, 6, 2003–2008 CrossRef PubMed.
- T. Förster and K. Kasper, Z. Phys. Chem., 1954, 1, 275–277 CrossRef.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Y. Wang, H. Li, D. Wang and B. Z. Tang, Adv. Funct. Mater., 2021, 31, 2006952 CrossRef CAS.
- X. Zhou, W. Luo, H. Nie, L. Xu, R. Hu, Z. Zhao, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 4775–4779 RSC.
- M. Studzian, L. Pułaski, D. A. Tomalia and B. Klajnert-Maculewicz, J. Phys. Chem. C, 2019, 123, 18007–18016 CrossRef CAS.
- D. A. Tomalia, B. Klajnert-Maculewicz, K. A. M. Johnson, H. F. Brinkman, A. Janaszewska and D. M. Hedstrand, Prog. Polym. Sci., 2019, 90, 35–117 CrossRef CAS.
- G. Perli, Qi Wang, C. B. Braga, D. L. Bertuzzi, L. A. Fontana, M. C. P. Soares, J. Ruiz, J. D. Megiatto, D. Astruc and C. Ornelas, J. Am. Chem. Soc., 2021, 143, 12948–12954 CrossRef CAS PubMed.
- Y. Chauvin, Angew. Chem., Int. Ed., 2006, 45, 3740–3747 CrossRef PubMed.
- R. R. Schrock, Acc. Chem. Res., 1990, 23, 158–165 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- S. Gatard, S. Nlate, E. Cloutet, G. Bravic, J.-C. Blais and D. Astruc, Angew. Chem., Int. Ed., 2003, 42, 452–456 CrossRef CAS PubMed.
- C. Ornelas, D. Méry, J. Ruiz, J.-C. Blais, E. Cloutet and D. Astruc, Angew. Chem., Int. Ed., 2005, 44, 7399–7404 CrossRef CAS PubMed.
- C. Ornelas, D. Méry, E. Cloutet, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2008, 130, 1495–1506 CrossRef CAS PubMed.
- N. Falcone and H. B. Kratz, Chem. – Eur. J., 2018, 54, 14316–14328 CrossRef PubMed.
- M. F. Ni, N. Zhang, W. Xia, X. Wu, C. H. Yao, X. Liu, X. Y. Hu, C. Lin and L. Y. Wang, J. Am. Chem. Soc., 2016, 138, 6643–6649 CrossRef CAS PubMed.
- M. Fadeev, Y. Ouyang, G. Davidson-Rosenfeld and I. Wilner, J. Electroanal. Chem., 2021, 904, 115926 CrossRef.
- H. Gu, S. Mu, G. Qiu, X. Liu, L. Zhang, Y. Yuan and D. Astruc, Coord. Chem. Rev., 2018, 364, 51–85 CrossRef CAS.
- H. B. Gu, R. Ciganda, P. Castel, A. Vax, D. Gregurec, J. Irigoyen, S. Moya, L. Salmon, P. X. Zhao, J. Ruiz, R. Hernandez and D. Astruc, Chem. - Eur. J., 2015, 21, 18177–18186 CrossRef CAS PubMed.
- G. R. Qiu, X. Liu, B. R. Wang, H. B. Gu and W. X. Wang, Polym. Chem., 2019, 10, 2527–2539 RSC.
- X. Liu, A. Rapakousiou, C. Deraedt, R. Ciganda, Y. Wang, J. Ruiz and D. Astruc, Chem. Commun., 2020, 56, 11374–11385 RSC.
- A. Walcarius, Acc. Chem. Res., 2021, 54, 3563–3575 CrossRef CAS PubMed.
- L. Liang, F. Qin, S. Wang, J. Wu, R. Li, Z. Wang, M. Ren, D. Liu, D. Wang and D. Astruc, Coord. Chem. Rev., 2023, 478, 214998 CrossRef CAS.
- J. Sugai, N. Saito, Y. Takahashi and Y. Kondo, Colloids Surf., 2019, 572, 197–202 CrossRef CAS.
- J. C. Wang, M. Chen, H. T. Zhao, H. Zhang, H. Zhang and X. Yang, Colloids Surf., 2020, 39, 100321 CAS.
- E. Alonso and D. Astruc, J. Am. Chem. Soc., 2000, 122, 3222–3223 CrossRef CAS.
- D. Astruc, K. Heuze, S. Gatard, D. Méry, S. Nlate and L. Plault, Adv. Synth. Catal., 2005, 347, 329–338 CrossRef CAS.
- L. Plault, A. Hauseler, S. Nlate, D. Astruc, S. Gatard and R. Neumann, Angew. Chem., Int. Ed., 2004, 43, 2924–2928 CrossRef CAS PubMed.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS PubMed.
- A. Diallo, E. Boisselier and D. Astruc, Chem. – Eur. J., 2010, 16, 11832–11835 CrossRef CAS PubMed.
- D. Astruc, L. Liang, A. Rapakousiou and J. Ruiz, Acc. Chem. Res., 2012, 45, 630–640 CrossRef CAS PubMed.
- C. Deraedt, N. Pinaud and D. Astruc, J. Am. Chem. Soc., 2014, 136, 12092–12098 CrossRef CAS PubMed.
- C. Wang, D. Wang, S. Yu, T. Cornilleau, J. Ruiz, L. Salmon and D. Astruc, ACS Catal., 2016, 6, 5424–5431 CrossRef CAS.
- F. Liu, X. Liu, D. Astruc and H. Gu, J. Colloid Interface Sci., 2019, 533, 161–170 CrossRef CAS PubMed.
- W. Wang, E. S. Chamkina, E. G. Cal, D. Di Silvio, M. Martínez Moro, S. Moya, J.-R. Hamon, D. Astruc and Z. B. Shifrina, Dalton Trans., 2021, 50, 11852–11860 RSC.
- C. Deraedt, L. Salmon and D. Astruc, Adv. Synth. Catal., 2014, 356, 2525–2538 CrossRef CAS.
- D. Wang, C. Deraedt, L. Salmon, C. Labrugère, L. Etienne, J. Ruiz and D. Astruc, Chem. – Eur. J., 2015, 21, 1508–1519 CrossRef CAS PubMed.
- C. Deraedt, L. Salmon and D. Astruc, Adv. Synth. Catal., 2014, 356, 2525–2538 CrossRef CAS.
- C. Wang, R. Ciganda, L. Salmon, D. Gregurec, J. Irigoyen, S. Moya, J. Ruiz and D. Astruc, Angew. Chem., Int. Ed., 2016, 55, 3091–3095 CrossRef CAS PubMed.
- P. Michaud, D. Astruc and J. H. Ammeter, J. Am. Chem. Soc., 1982, 104, 3755–3757 CrossRef CAS.
- D. Wang, C. Deraedt, L. Salmon, C. Labrugère, L. Etienne, J. Ruiz and D. Astruc, Chem. – Eur. J., 2015, 21, 6501–6510 CrossRef CAS PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- D. Astruc, F. Lu and J. Ruiz, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- N. Li, P. Zhao and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 1756–1789 CrossRef CAS PubMed.
- C. Deraedt, L. Salmon, S. Gatard, R. Ciganda, R. Hernandez, J. Ruiz and D. Astruc, Chem. Commun., 2014, 50, 14194–14196 RSC.
- J. G. A. de Vries, Dalton Trans., 2006, 421–429 RSC.
- A. K. Diallo, C. Ornelas, L. Salmon, J. Ruiz and D. Astruc, Angew. Chem., Int. Ed., 2007, 46, 8644–8648 CrossRef CAS PubMed.
- D. Astruc, Tetrahedron Asym., 2010, 21, 1041–1054 CrossRef CAS.
- C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494–503 CrossRef CAS PubMed.
- X. Liu, D. Gregurec, J. Irigoyen, A. Martinez, S. Moya, R. Ciganda, P. Hermange, J. Ruiz and D. Astruc, Nat. Commun., 2016, 7, 13152 CrossRef CAS PubMed.
- A. Rapakousiou, C. Deraedt, H. Gu, L. Salmon, C. Belin, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2014, 136, 13995–13998 CrossRef CAS PubMed.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060 RSC.
- Y. Huang, J. Ren and X. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS PubMed.
- L. Liang, J. Ruiz and D. Astruc, Adv. Synth. Catal., 2011, 353, 3434–3450 CrossRef CAS.
- Y. Liu, R. Pereira Lopes, T. Lüdtke, D. Di Silvio, S. Moya, J.-R. Hamon and D. Astruc, Inorg. Chem. Front., 2021, 8, 3301–3307 RSC.
- M. L. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Renewable Sustainable Energy Rev., 2021, 146, 111180 CrossRef.
- T. He, P. Pachfule, H. Wu, Q. Xu and P. Chen, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed.
- Z. Huang, S. Wang, R. D. Dewhurst, N. V. Ignat’ev, M. Finze and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 8800–8816 CrossRef CAS PubMed.
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2021, 50, 3437–3484 RSC.
- M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698 RSC.
- Q. Wang, F. Fu, S. Yang, M. Martinez Moro, M. de los A. Ramirez, S. Moya, L. Salmon, J. Ruiz and D. Astruc, ACS Catal., 2019, 9, 1110–1119 CrossRef CAS.
- N. Kang, R. Djeda, Q. Wang, F. Y. Fu, J. Ruiz, J.-L. Pozzo and D. Astruc, ChemCatChem, 2019, 11, 2341–2349 CrossRef CAS.
- Q. Zhao, N. Kang, M. Martinez Moro, E. Guisasola Cal, S. Moya, E. Coy, L. Salmon, X. Liu and D. Astruc, ACS Appl. Energy Mater., 2022, 5, 3834–3844 CrossRef CAS.
- N. Kang, Q. Wang, R. Djeda, W. Wang, F. Fu, M. M. Moro, M. de los A. Ramirez, S. Moya, E. Coy, L. Salmon, J.-L. Pozzo and D. Astruc, ACS Appl. Mater. Interfaces, 2022, 12, 53816–53826 CrossRef PubMed.
- N. Kang, X. Wei, R. Shen, B. J. Li, E. Guisasola Cal, S. Moya, L. Salmon, C. Wang, E. Coy, M. Berlande, J.-L. Pozzo and D. Astruc, Appl. Catal., B, 2023, 320, 121957 CrossRef CAS.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. D. Huang, R. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- S. Zhang, M. Li, L. Li, F. Dushimimana, J. Zhao, S. Wang, J. Han, X. Zhu, X. Liu, Q. Ge and H. Wang, ACS Catal., 2020, 10, 14903–14915 CrossRef CAS.
- C. Wang, J. Tuninetti, Z. Wang, C. Zhang, R. Ciganda, L. Salmon, S. Moya, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2017, 139, 11610–11615 CrossRef CAS PubMed.
- F. Fu, C. Wang, Q. Wang, A. M. Martinez-Villacorta, A. Escobar, H. Chong, X. Wang, S. Moya, L. Salmon, E. Fouquet, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2018, 140, 10034–10042 CrossRef CAS PubMed.
- C. Wang, X. Liu, Y. Wu and D. Astruc, J. Mater. Chem. A, 2022, 10, 17614–17623 RSC.
- N. Kang, R. Shen, B. Li, F. Fu, B. Espuche, S. Moya, L. Salmon, J.-L. Pozzo and D. Astruc, J. Mater. Chem. A, 2023, 11, 5245–5256 RSC.
- C. Wang, Q. Wang, F. Fu and D. Astruc, Acc. Chem. Res., 2020, 10, 2483–2493 CrossRef PubMed.
- Q. Zhao, X. Liu and D. Astruc, Eur. J. Inorg. Chem., 2023, e202300024 CrossRef CAS.
- W. Huang, F. Xu, D.-S. Li, D. Astruc and X. Liu, Carbon Energy, 2023, 5, e269 CAS.
- N. Kang, C. Wang and D. Astruc, Chemistry, 2023, 5, 886–899 CrossRef.
- K. Yamamoto, T. Imaoka, M. Tanabe and T. Kambe, Chem. Rev., 2020, 120, 1397–1437 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- W. J. Wang, J. Ruiz, C. Ornelas and J.-R. Hamon, ACS Catal., 2023, 13, 1574–1586 CrossRef CAS.
Footnote |
| † This article is dedicated to my distinguished friend and colleague Professor Henri Bouas-Laurent on the occasion of his 90th birthday. |
| This journal is © The Royal Society of Chemistry 2023 |