
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Progress in electrode and electrolyte materials: path to all-solid-state Li-ion batteries
Sanjeev K.
Sharma
*a,
Gaurav
Sharma
b,
Anurag
Gaur
c,
Anil
Arya
d,
Fateme Sadat
Mirsafi
e,
Reza
Abolhassani
 e,
Horst-Günter
Rubahn
e,
Jong-Sung
Yu
f and
Yogendra Kumar
Mishra
*e
e,
Horst-Günter
Rubahn
e,
Jong-Sung
Yu
f and
Yogendra Kumar
Mishra
*e
aDepartment of Physics, CCS University, Meerut Campus, Meerut, Uttar Pradesh 250004, India. E-mail: sksharma18@ccsuniversity.ac.in
bDepartment of Basic Sciences, S.V.P. University of Agriculture & Technology, Modipuram, Meerut, Uttar Pradesh 250110, India
cDepartment of Physics, J.C. Bose University of Science & Technology (YMCA), Faridabad – 121006, Haryana, India
dDepartment of Physics, Kurukshetra University, Kurukshetra – 136119, Haryana, India
eMads Clausen Institute, NanoSYD, University of Southern Denmark, Alsion 2, 6400, Sønderborg, Denmark. E-mail: mishra@mci.sdu.dk
fDepartment of Energy Science and Engineering, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu, 42988, Republic of Korea
First published on 20th June 2022
Abstract
This review presents a brief scenario regarding the development of cathodes, anodes, and electrolytes for next-generation Li-ion batteries (LIBs) and supercapacitors for future energy technologies. The specific capacity and power density are two prime requirements for energy storage devices, which are mainly decided by the microstructure and composition of electrodes. Electrolyte, which is the highway for ions between electrodes, plays a crucial role in developing advanced batteries. Miniaturized electrode-based LIBs with high energy storage densities are a smart approach toward huge future energy demands, where nanomaterials play a crucial role. The ultra-large surface of nanostructure-based electrodes offers improved electrochemical performance per unit electrode area and/or material mass. Porous nanostructured material-based electrodes/electrolytes provide fast and shortened transportation pathways for carriers, facilitating improved reaction kinetics. This review presents the fabrication and electrochemical performances of different nanomaterial-based LIBs, including their critical challenges such as thermal runaway and dendrite growth. An overview of all-solid-state Li-ion batteries (ASSLIB), with the potential to bridge the gap between the laboratory and market, is presented. Finally, the status, challenges, and outlook for enhancing the performance of cathodes, anodes, electrolytes, and their integration in ASSLIB are briefly covered for the attention of the wider functional and energy material communities.
1. Introduction to energy systems
Currently, society depends on fossil fuels to generate electricity, drive vehicles, and power industries using internal combustion engines.1 Battery-driven energy has found a realistic way to liberate society,2–5 where green energy can be generated from natural resources such as solar energy, hydro power, wind-turbines, and wave upthrust.6,7 After harvesting energy, it needs to be stored it in an efficient system for long-lasting and maximum intercalation-deintercalation. According to the literature, energy storage systems have existed since ancient times. The first energy storage system was introduced by Agastya Rishi (Sages in Ancient Indian civilization) approximately ∼5000 BC.8 In 1780 AD, Luigi Galvani performed a unique electricity experiment on a frog, called the frog legs experiment.9 Later, based on the concept of the frog legs experiment, an Italian physicist, Sir Alessandro Volta, used metallic elements instead of a biological cell or living entities to produce electricity. Then, he investigated a way to store the produced energy, which was called a battery.10 In continuation of the investigation of energy storage devices, Edison invented the nickel-iron battery in 1968, producing a rechargeable system having nickel oxide-hydroxide positive plates and negative iron plates, with potassium hydroxide as the electrolyte.11 Thus, the development of energy storage systems dates back to the study by Agastya Sanhita, resulting in high energy density for the application of ASSLIB in HEV and smart/mobile devices.1.1 Solid-state electrolyte/batteries
The major challenges associated with electric-drive vehicles include their cost and performance, especially regarding their batteries, which are responsible for a large portion of the cost of these vehicles.12–14 The main thrust as the foundation for Li-ion batteries (LIBs) occurred after the oil crisis in the 1970s. Subsequently, the research community focused on developing the fossil-fuel-free natural energy harvesting and energy storage sectors to fulfill energy requirements globally.15,16 Accordingly, a suitable candidate with potential for energy storage was the LIB, which is a green energy storage system with a no-memory loss effect.17–20 The operative mechanism of LIBs is a simple principle similar to that of other storage systems, i.e., moving electrons from one region (anode) to another (cathode), which generates an electrical current. LIBs are considered safer compared to conventional batteries such as lead-acid and CdS, which exhibit several issues including the emission of toxic gases and overheating during charging and discharging.All-solid-state batteries (ASSBs) have attracted significant attention for application in future technologies due to their safety and high energy densities. However, many ASSBs are limited by their Coulombic efficiency, poor power performance, and short cycling life due to the high resistance at the interfaces in ASSBs. Banerjee et al.21 explored suitable materials that can serve as SEs for the fabrication of ASSBs, namely, materials with high ionic conductivity (σLi+ > 0.1 mS cm−1, which are commonly referred to as “superionic conductors”, but possess lower electronic conductivity (σe < 10−7 mS cm−1). These materials include polymer-, oxide-, and sulfide-based electrolytes. Before elaborating on LIBs, solid-state electrolytes (SSE) enable the utilization of Li metal anodes, which are considered the most promising anodes for next-generation rechargeable batteries due to their ultrahigh theoretical specific capacity of 3860 mA h g−1 and lowest negative electrochemical potential (−3.04 V versus the standard hydrogen electrode). However, in conventional organic electrolytes, lithium metal suffers from an unstable solid-state interphase, dendrite penetration, and pulverization issues. The state-of-the-art batteries possessing SSEs have been reviewed by Xu et al.22 to guarantee the development of next-generation battery systems with improved energy density and high safety. Numerous compounds including oxides, sulfides, and polymer ionic conductors have been developed and several achievements comparable to liquid electrolytes have been obtained. Materials possessing high theoretical capacities, such as lithium, sulfur, and lithium intercalation compounds, have also been introduced in the “solid family.”
The novel class of fast lithium ion-conducting metal oxides with the chemical composition Li5La3M2O12 (M = Nb and Ta) possess a garnet-related structure. Among the investigated compounds with garnet-related structures, Li6BaLa2Ta2O12 exhibit the highest Li+ ion conductivity of 4 × 10−5 S cm−1 at 22 °C with an activation energy of 0.40 eV; however, its bulk and total conductivity at room temperature are not sufficiently high to develop an ideal all-solid-state lithium ion rechargeable battery. Murugan et al.23 reported that due to the high lithium ion conductivity, good thermal and chemical stability against reactions with prospective electrode materials, environmental benignity, availability of its starting materials, low cost, and ease of preparation and densification of Li7La3Zr2O12 make it a promising solid electrolyte for all-solid-state lithium ion rechargeable batteries (ASSLIBs).
Lithium garnet (i.e., Li6.4La3Zr1.4Ta0.6O12, LLZTO) particle-based composite membranes and Li-salt-free polyethylene oxides (PEOs) as SSE were reported by Zhang et al.,24 which were crucial for the enhancement in the conductivity of the membranes containing 40 nm LLZTO particles due to the difference in the specific surface area and related to the percolation effect. Compared to the conventional PEO doped with lithium salt, the insulating PEO in PEO:LLZTO membrane electrolyte was conductive to the suppression of lithium dendrite growth because it hindered the current flow. The PEO:LLZTO membrane electrolyte exhibited a conductivity of 2.1 × 10−4 S cm−1 at 30 °C and 5.6 × 10−4 S cm−1 at 60 °C, and consequently the solid-state LiFePO4/PEO:LLZTO/Li and LiFe0.15Mn0.85PO4/PEO:LLZTO/Li cells delivered energy densities of 345 W h kg−1 (662 W h L−1) and 405 W h kg−1 (700 W h L−1, without the package weight or volume) with a good rate capability and cycling performance. The combination of nano-scale Li-ion-conducting particles and an insulating polymer provided a promising solution to produce powerful SSEs for high-performance solid-state lithium batteries (SSLBs). Thus, a polymer with improved stability is available, and consequently SSLBs can be constructed with enlarged voltage cathodes such as LiNi0.5Mn1.5O4 to further increase the energy density.
Zhang et al.25 reported that Li6.75La3Zr1.75Ta0.25O12 (LLZTO) ceramics could trigger structural modification in the poly(vinylidene fluoride) (PVDF) polymer electrolyte, which was prepared using the conventional solution-casting method. The LLZTO-modified PVDF membrane was shown to be a promising electrolyte material for use in ASSLIBs with significantly enhanced performances (a high ionic conductivity of about 5 × 10−4 S cm−1 at 25 °C, high mechanical strength, and good thermal stability). Furthermore, the LiCoO2|PVDF/LLZTO-CPE|Li cell presented a satisfactory rate capability and cycling stability at room temperature, which showed that PVDF/LLZTO-CPE has great potential to be used as the electrolyte in SSLBs.
An electrical current is created in LIBs because of the chemical reactions and movement of ions inside these batteries. Electrons move in the cell from the anode to cathode, which are located at the opposite ends of the cell. Significant progress has been achieved in the development of rechargeable LIBs since their introduction in the early 1990s, which are an integral part of all portable electronics and popular for powering hybrid vehicles.26,27 They can be recharged by using appropriate adaptors for several cycles (500–1000 cycles). The electrolytes inside these batteries also play an important role in the migration of ions from the anode to cathode, and vice versa. In the case of solid electrolytes, they can simultaneously act as a separator between the anode and cathode.28
In the modern digital era, among the various energy storage systems, LIBs represent the most popular rechargeable batteries for use in portable electronic devices such as mobile phones and laptops due to their long cycle life and high specific capacity. LIBs can also be formed into many shapes, making them ideal for use in the essential products of laptops, tablets, and cell phones. LIBs are widely used in these devices because of their rechargeability and negligible memory effect. Owing to their long cycle life and high capacity, LIBs are considered suitable for next-generation advanced mobile electronic devices (flexible and transparent devices), electric vehicles (EVs), hybrid electric vehicles (HEVs), and renewable energy storage applications.29Fig. 1 shows the increasing demand of LIBs since 2015 and will continue to rise sharply over the next few decades.
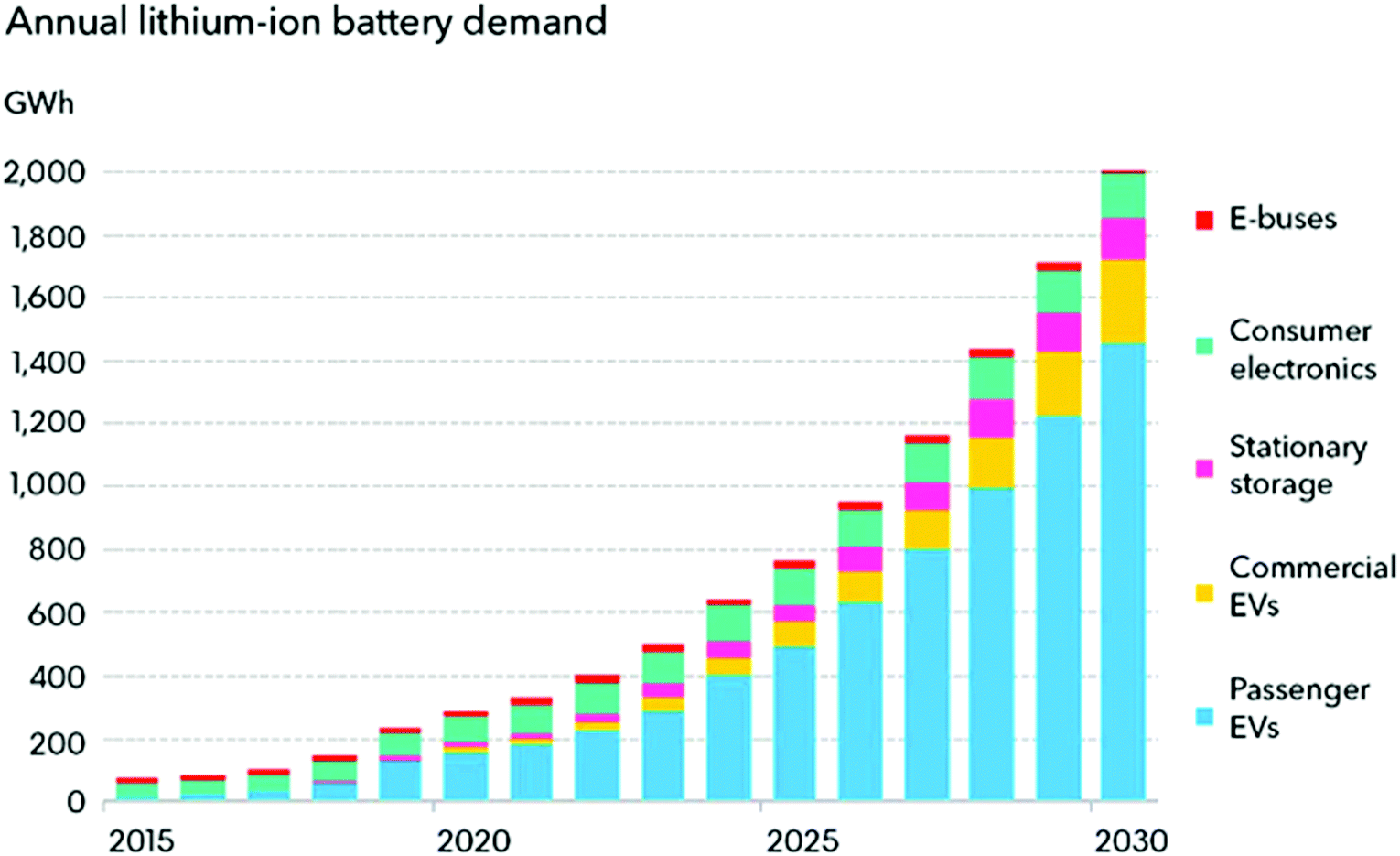 | ||
| Fig. 1 Li-ion battery demand forecast (https://about.bnef.com/electric-vehicle-outlook/) | ||
The greatest demand for electric passenger/commercial vehicles is expected to occur in the near future with stationary storage systems. However, the relatively low charge/discharge rates and safety concerns of these systems have limited their use in applied applications requiring both high-power density and high capacity for EVs and HEVs. Thus, the major scientific challenge associated with ASSLIBs is enhancing their power density, cycle life, recyclability, and safety concerns.30–32 The energy obtained from different sources can be stored in LIBs, and subsequently used according to the demand. Fig. 2(a) presents an overview of the energy production and storage from various energy sectors, including the functions of LIBs. To determine suitable candidates for the fabrication of LIBs, active, hybrid, and Si-based nanostructured materials must be tested to fulfill requirements of reversible capacity, good ionic and electrical conductivity, long cycle life, high lithium diffusion rate in the active material, and conclusively low cost and eco-compatibility.
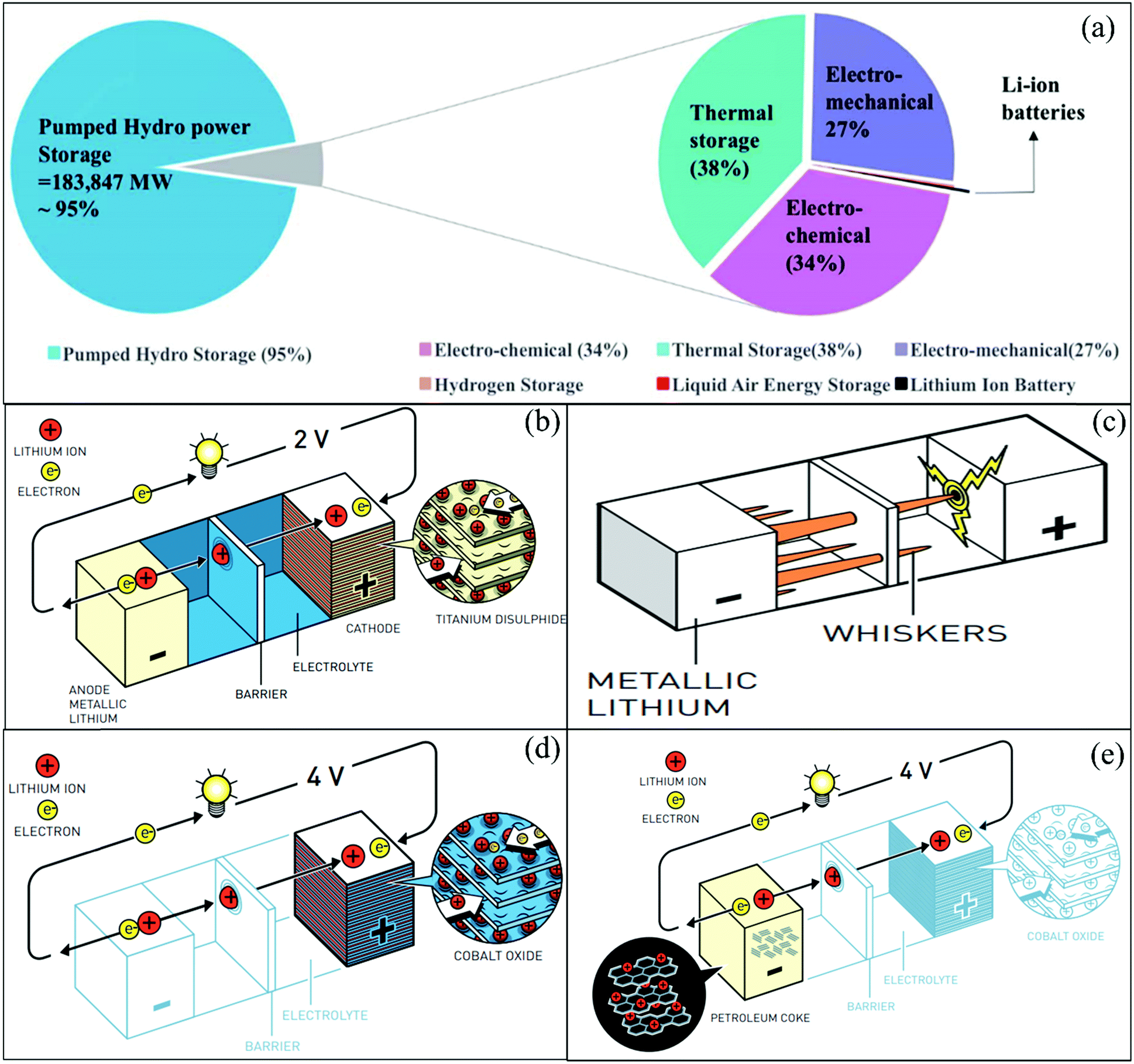 | ||
| Fig. 2 (a) Contribution of worldwide energy storage projects to grid applications.30 (b) Lithium-based battery using LixTiS2 as the cathode. (c) Formation of lithium whiskers/dendrites potentially leading to short-circuiting. (d) Lithium-based battery using LixCoO2 as the cathode. (e) Ion transfer cell lithium-ion battery configuration. (©Johan Jarnestad/Royal Swedish Academy of Sciences33). | ||
Currently, LIBs are the dominant power source for mobile phones, laptops, and numerous other portable electronic devices. Also, they have been increasingly used in electric vehicles (EVs) and flexible/mobile electronics since their commercialization. The Sony Corporation (1991) commercialized the first modern LIB, which held twice the energy density and was almost 10-times cheaper than the existing Li batteries.34 The Li-battery was first introduced by the American chemist Gilbert Newton Lewis (G. N. Lewis) in 1912,35 while the first lithium battery was invented in the 1970s, and the first attempts to develop rechargeable batteries were made in the 1980s by Bell Labs.36
Lithium (Li) is one of the lightest metals with the highest electrochemical potential, which can provide the largest specific energy density. It requires careful and systematic handling, where the development of breakthrough technologies based on new anodes, cathodes, and non-aqueous electrolytes can enable a steady improvement in high-energy lithium battery systems. Although LIBs are expensive, they have never experienced the memory issues that affect other battery technologies. LIBs exhibit a slightly lower energy density than lithium metal batteries; however, LIBs are safer than Li batteries and provide certain precautions during charging and discharging. Also, the LIB is a low-maintenance battery, and no schedule is required for cycling to prolong its lifetime. Compared to nickel–cadmium (Ni–Cd) batteries, the self-discharge of LIBs is less than half and is well-fitted for modern fuel gauge applications.
Based on the enormous success achieved from the laboratory to daily life, the discovery of lithium-ion batteries led to the 2019 Nobel Prize in Chemistry. The three key contributors to the development of LIBs were John B. Goodenough, M. Stanley Whittingham, and Akira Yoshino. Stanley Whittingham focused on developing titanium disulfide (TiS2)-based cathodes for LIBs, which possess space for ion intercalation, as shown in Fig. 2b. The metallic lithium was used as an anode for lithium batteries, which easily provides electrons. The battery had a very high energy density and a voltage of 2 V.37 The discovery of this battery was a big announcement at that time, but the formation of lithium whiskers/dendrites potentially led to short-circuiting in the battery (Fig. 2c). Goodenough investigated this failure and proposed the use of transition metal oxide-based cathodes instead of TiS2. His group discovered that lithium-cobalt oxide (LiCoO2) is a suitable cathode material, which was stable during cell operation (Fig. 2d). They successfully increased the voltage to 4 V.38,39 Then, another scientist interested in the development of lightweight LIBs was Akira Yoshino from the Asahi Kasei Corporation, Japan. He used LiCoO2 as a cathode and tried different carbon materials as the anode. Finally, petroleum coke was used as the anode and a full battery was developed (Fig. 2e), which demonstrated a high capacity and voltage.40
1.2 Principle of LIBs
An LIB consists of four components including a positive electrode (cathode), a negative electrode (anode), a separator (to separate electrodes), and electrolyte for the movement of ions through chemical reactions. The existing LIBs use LiCoO2 as the cathode and graphite as the anode. The standard electrolyte is liquid LiPF6, soaked by the separator between electrodes. For the cathode, Al is used as a current collector, and for the anode, Cu is used as a current collector (Fig. 3). During charging, the cathode releases Li+ (Li-ions), which moves through the electrolyte and gets accommodated in the anode (means energy stored). Meanwhile, electrons flow through the external circuit and the separator blocks the flow of electrons due to its insulating nature and provides easy access for ions. In the discharging process, Li+ ions migrate back to the cathode via the electrolyte and release the energy stored inside the battery. The performance of LIBs strongly depends on the utilized electrode materials, which is mainly decided by various parameters, such as, the physical and chemical properties, microstructure and composition of the material. Fig. 4(a) shows some important parameters (specific capacity, coulombic efficiency, power density, capacity retention, voltage stability and conductivity, toxicity and safety issues) that demonstrate the potential of electrode materials for use in LIBs. The voltage stability window of the cell is examined by evaluating the molecular properties of its materials via quantum chemical characterization of their highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). The relative energies of the electrolyte window (Eg) and the electrochemical potentials of the electrode, μA and μC, with no electrode/electrolyte reactions are shown in Fig. 4b. The difference between the HOMO and LUMO of a liquid electrolyte or the bottom of the conduction band and the top of the valence band of the solid electrolyte decides the electrochemical stability window (ESW) (Fig. 4b and c).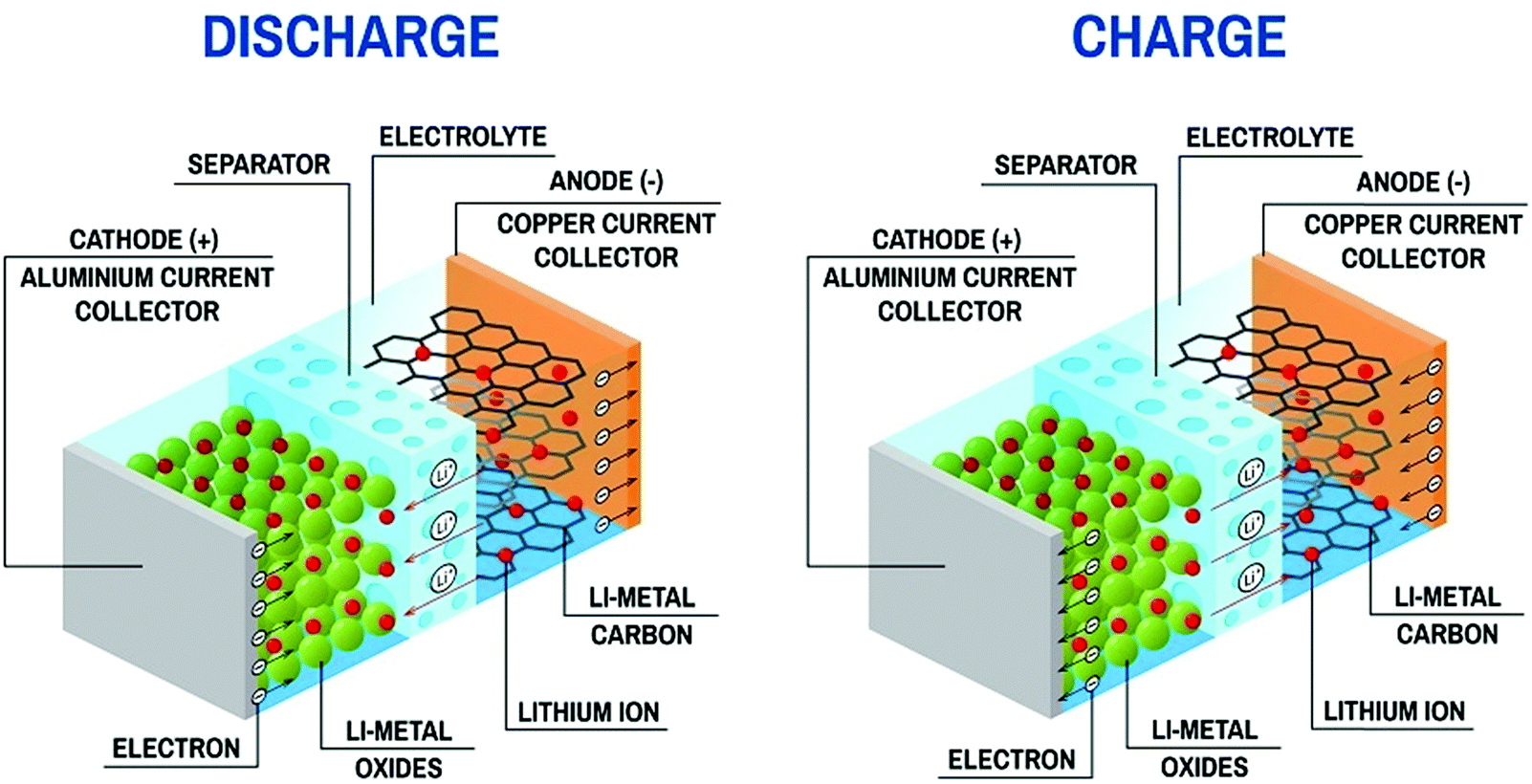 | ||
| Fig. 3 Charge–discharge process in Li-ion battery (Image Credit: https://sivVector/Shutterstock.com). | ||
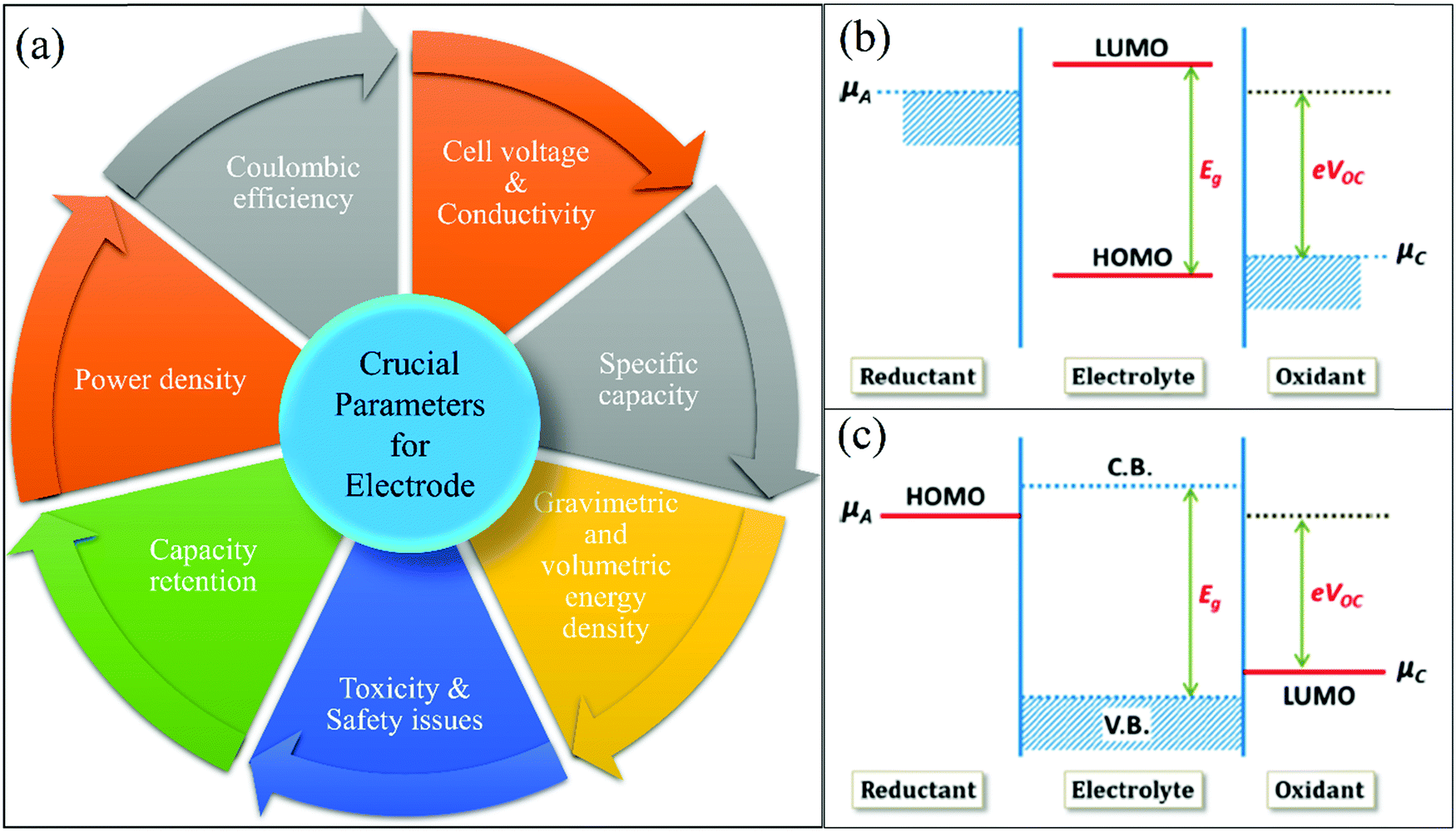 | ||
| Fig. 4 (a) Performance parameters of electrodes: specific capacity, coulombic efficiency, power density, capacity retention, voltage stability and conductivity, toxicity and safety issues. (b) Liquid electrolyte with solid electrodes (relative energies of the electrolyte window (Eg) and the electrochemical potentials of the electrode, μA and μC with no electrode/electrolyte reaction). (c) Solid electrolyte with liquid or gaseous reactants.41 (Reproduced with permission from the American Chemical Society41). | ||
To achieve stability, the ESW must be larger than the open circuit energy (Voc = (μA − μC)/e) (difference in Li chemical potential in each electrode).41–44Table 1 summarizes the critical cell performance parameters that need to be examined before commencing large scale commercial production.45,46 Another significant task in the case of battery technology is to carefully identify the failure mechanism (physical/electrical/mechanical/chemical). Thus, the failure mode, mechanism, and effect analysis (FMMEA) methodology has been adopted to discover the failure mechanism, as shown in Table 2. The FMMEA methodology provides details on the cell components, mode, and the cause of failure.47Table 2 also summarises the anode and cathode active materials and the current collectors, separator for Li-ions, electrolyte salts (organics solvents), and the terminals.
| Parameter | Measuring unit | Measuring formula | Information |
|---|---|---|---|
| Operating voltage | Volts (V) | Instrumental | Energy density and safety |
| Current density | mA g−1 | Instrumental | For testing rate capabilities |
| Theoretical capacity | mA h g−1 |
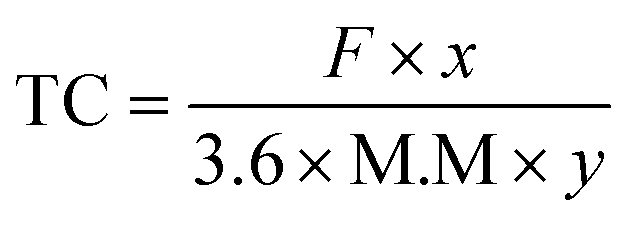
|
Lithium-ion storage capability |
| Gravimetric capacity | mA h g−1 |
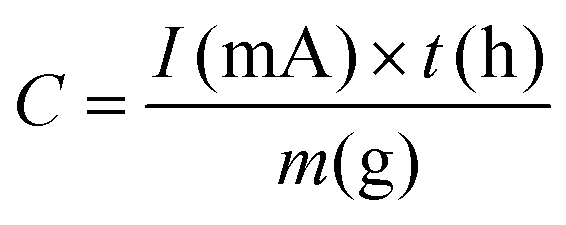
|
Li+ storage capability measured per unit mass |
| Areal capacity | mA h cm−2 |
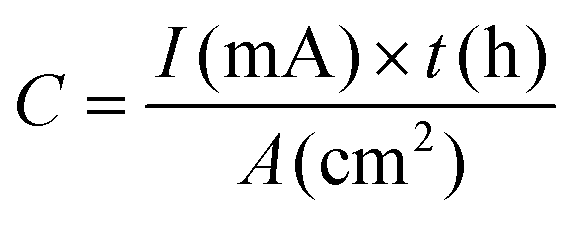
|
Li+ storage capability measured per unit area |
| Volumetric capacity | mA h cm−2 |
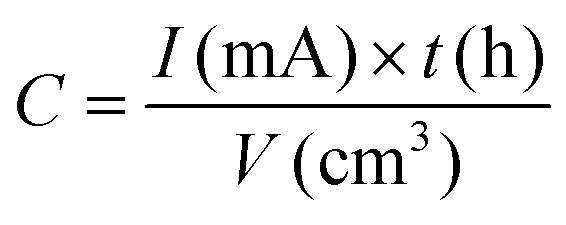
|
Li+ storage capability measured per unit volume |
| Specific energy density | W h g−1 or W h cm−2 or W h cm−3 | E = C × V | How much energy can be extracted |
| Power density | W g−1 or W cm−2 or W cm−3 | P = I × V | How fast the energy can be extracted |
| C rate | h−1 |
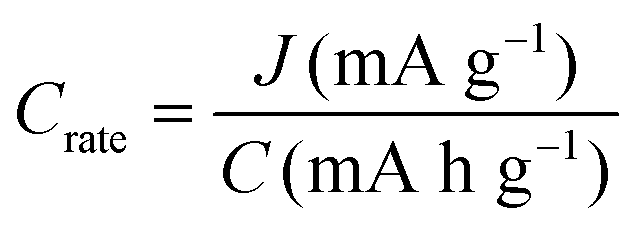
|
Rate of charging/discharging |
| Coulombic efficiency | N/A |
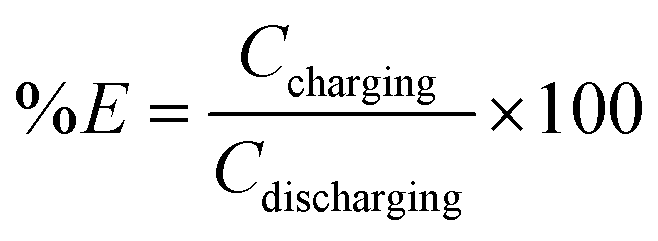
|
Reversible capacity |
| State of charge (SOC) | NA | SOC = remaining capacity/rated capacity | Remaining capacity of a battery |
| Depth of discharge (DOD) | NA | DOD = 1−SOC | Percentage of the total discharge battery capacity |
| Cycle life | NA | — | Number of discharge–charge cycles handled by a battery at a specific DOD |
| Calendar life | NA | — | The expected life span of the battery under storage or periodic cycling conditions. |
| Battery component | Potential failure mode (s) | Potential failure mechanism (s) | Mechanism type | Observed effect | Potential failure causes | Likelihood of occurrence | Severity of occurrence | Ease of detection |
|---|---|---|---|---|---|---|---|---|
| Anode (active material) | Thickening of solid electrolyte interphase layer | Chemical reduction reaction and deposition | Wear out | Increased charge transfer resistance, reduction of capacity, reduction of power | Chemical side reactions between lithium, electrode, and solvent | High | Low | High |
| Particle fracture | Mechanical stress | Over stress | Reduction of capacity, reduction of power | Intercalation stress | Moderate | Low | Low | |
| Reduced electrode porosity | Mechanical degradation | Wear out | Increased diffusion resistance, reduction of capacity, reduction of power | Dimensional changes in electrode | Moderate | Low | Low | |
| Lithium plating and dendrite growth on anode surface | Chemical reaction | Wear out | Can cause a short circuit if dendrites puncture the separator | Charging the battery at low temperatures or high rates | Low | High | Low | |
| Anode (current collector) | Free copper particles or copper plating | Chemical corrosion reaction and dissolution | Wear out | Increased resistance, reduction of power, reduction of current density | Over-discharge of the battery | Low | High | Low |
| Cathode (active material) | Thickening of solid electrolyte interphase layer | Chemical reduction reaction and deposition | Wear out | Increased charge transfer resistance, reduction of capacity, reduction of power | Chemical side reactions between lithium, electrode, and solvent | High | Low | High |
| Particle fracture | Mechanical stress | Over stress | Reduction of capacity, reduction of power | Intercalation stress | Moderate | Low | Low | |
| Reduced electrode porosity | Mechanical degradation | Wear out | Increased diffusion resistance, reduction of capacity, reduction of power | Dimensional changes in electrode | Moderate | Low | Low | |
| Gas generation and bloating of cell casing | Thermally driven electrode decomposition | Over stress | Reduction of capacity | Overcharge of the battery or short circuit | Low | High | Low | |
| Cathode (current collector) | Pitting corrosion of aluminum | Chemical corrosion reaction | Wear out | Increased resistance, reduction of power, reduction of current density | Overcharge of the battery | Low | Moderate | Low |
| Separator | Hole in separator | Mechanical damage | Over stress | High heat generation due to Joule heating, bloating of cell casing, drastic voltage reduction | Dendrite formation, external crushing of cell | Low | High | Moderate |
| Closing of separator pores | Thermally-induced melting of separator | Over stress | Inability to charge or discharge the battery | High internal cell temperature | Low | High | High | |
| Lithium ions | Reduction in lithium ions, thickening of solid electrolyte inter-phase layer | Electrolyte reduction and solid product formation | Wear out | Reduction of capacity | Chemical side reactions between lithium, electrodes, and solvent | High | Low | High |
| Electrolyte salt | Decrease in lithium salt concentration | Chemical reduction reaction and deposition | Wear out | Increased diffusion resistance | Chemical side reactions between lithium, electrodes, and solvent | Low | High | Low |
| Organic solvents | Gas generation and bloating of cell casing | Chemical decomposition of solvent | Over stress | Increased diffusion resistance, and may lead to thermal runaway | High external temperature, overcharging of the cell | Low | High | Low |
| Thickening of solid electrolyte interphase layer | Chemical reduction reaction and deposition | Wear out | Increased charge transfer resistance, reduction of capacity, reduction of power | Chemical side reactions between lithium, electrodes, and solvent | High | Low | High | |
| Terminals | External corrosive path between positive and negative leads | Chemical corrosion reaction | Wear out | High heat generation due to Joule heating, bloating of cell casing, drastic voltage reduction | Inadvertent shorting of the terminals | Low | High | Moderate |
| Solder cracking | Thermal fatigue mechanical vibration fatigue | Wear out | Loss of conductivity between battery and host device | Circuit disconnect | Low | Moderate | High | |
| Casing | Internal short circuit between anode and cathode | Mechanical stress | Over stress | High heat generation due to Joule heating, bloating of cell casing, drastic voltage reduction | External load on cell | Low | High | Moderate |
Before preparing the electrode materials, it is crucial to select the parent material based on parameters such as cost-effectiveness, non-toxicity, abundance, and safety. The low cost of the raw material and the preparation conditions (NTP) will lead to an overall cost that is within the affordable limit of the consumer market. The non-toxic nature of electrode materials is the primary requirement for the safe use of the device. The electrode materials should be abundant in nature to be commercialized and able to balance the supply according to the demand from the consumer sector. The most feasible elements from the periodic table are transition elements, and thus extensive studies have been performed based on their composites with various phases and structures (Fig. 5). It should be noted that parameters such as the ionic radius (helps in stabilizing oxidation states), ionization energy (decides oxidizing and reduction power), and electronegativity (helps in altering the redox potential) of elements need to be considered before the preparation of electrode materials.48Table 3 describes the anode and cathode electrodes, separator, etc., including their deficiencies and remedies.
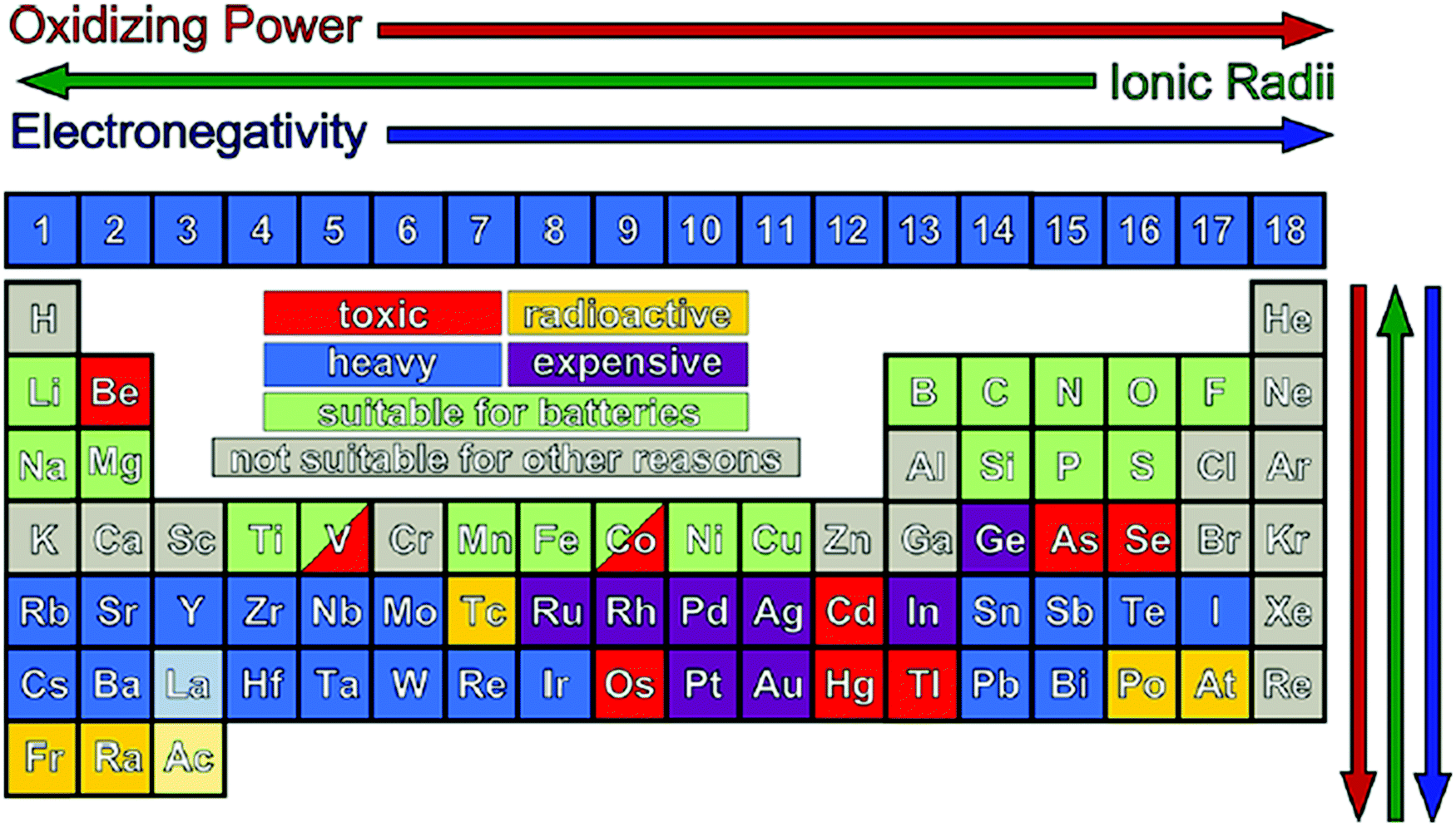 | ||
| Fig. 5 Periodic table is available to design new electrode materials. The colored squares are excluded due to either their high cost, low availability, toxicity, or radioactivity. This slightly restricts the elements available for the design of new materials but can be useful in guiding synthetic methods. Despite their toxicity, some transition metals, such as V and Co, are still actively investigated.48 Reproduced with permission from the American Chemical Society.48 | ||
| Location of deficiency | Deficiencies | Possible remedies |
|---|---|---|
| Carbonaceous anode (negative electrode) | Low capacity density (A h l−1) | Replace carbon with an improved alloy anode that allows high coulombic efficiency, good power capability, low irreversible capacity, and low cost with little or no loss of specific capacity or cell voltage |
| Negative electrode–electrolyte interface | Low coulombic efficiency with alloy anodes caused by solid electrolyte interphase (SEI) growth on the first cycle and continuing with cycling | Improved coatings, functional binders, and/or electrolyte additives to protect the interface during large volume changes |
| Positive electrode (lithiated transition metal oxide or phosphate) | Low specific capacity (A h kg−1) and charging voltage limited | Replace with new cathode material that allows high coulombic efficiency, good power capability, low irreversible capacity, and lower cost with little or no loss of capacity density or cell voltage |
| Positive electrode–electrolyte interface | Low coulombic efficiency at higher voltage limiting specific capacity and cycle life and causing increased cell impedance with cycling | Improve coating of cathode material, binders, and/or electrolyte additives that can prevent impedance increase with cycling, dissolution of transition metal ions |
| Separator | Penetration with conductive particles or lithium dendrites | Improved coatings of separators that do not impede ion flux, salt diffusion, or fluid flow, but can improve penetration strength or combine chemically with lithium dendrites |
| Metal collectors | Solid metal foils add to cost and take away from energy as they are inert in the system, yet must be thick enough to provide adequate electrical and thermal conductance | Perforated or expanded metal collectors are in common use for primary lithium batteries and secondary aqueous batteries, but have not been engineered for lithium-ion |
Crabtree pointed out the next breakthroughs expected at the other end of the battery for making better anodes. The anode stores lithium ions when the battery is charged and sends them to the cathode as the battery releases power. Japanese electronics giant Sony introduced carbon anodes to replace the troublesome lithium metal anodes in the early 1990s. These batteries were losing their performance, making necessary to restore it. Currently, one of the major problems is the graphite anodes developed after the lithium metal anodes, which eliminate lithium ions from the batteries, but they returning to the anode during charging. This leads to the formation of tree-like dendrite structures instead of a coating on the anode surface.
Materials scientist Nitash Balsara, University of California, Berkeley, explained that carbon anodes can accept lithium ions at a given rate. “If you try to send lithium (through the battery) too fast (while charging), the lithium doesn’t go into the graphite, it sticks on the outside. It becomes a safety hazard.” Small-size batteries can easily grow dendrites across the electrolyte and contact the opposite pole, as demonstrated by Goodenough. Permeable membranes, e.g., separators, are generally used to prevent the contact between electrodes, and thus stop short circuits, while allowing the flow of ions from the electrolyte. Nevertheless, this process involves a high risk of breaking the dendrite structures, leading to pore blockage. This often hampers the ion migration to the separator, thereby impacting the lifetime of the battery.
2. Electrodes for Li-ion batteries
2.1 Cathode materials of lithium-ion batteries (LIBs)
Cho et al.53 synthesized a high-performance LiCoO2 cathode via the sol–gel coating of Al2O3 on the LiCoO2 particle surface, followed by heat treatment at 600 °C for 3 h. The Al2O3-coated LiCoO2 cathode showed no decrease in its original specific capacity of 174 mA h g−1 compared to the pristine cathode (vs. lithium metal) together with excellent capacity retention (97% of its initial capacity) between 4.4 and 2.75 V (after 50 cycles). This enhancement in capacity retention has been attributed to the improvement in the structural stability of LiCoO2 during cycling owing to the presence of Al atoms on the electrode surface.
Li et al.54 examined the effects of halogen doping on the structural stability, electronic state, electrode potential, and Li diffusion behavior of LiCoO2 systems via density functional theory (DFT) calculations. It was observed that fluorine, chlorine, and bromine substitution of oxygen species suppresses the lattice changes upon Li de-intercalation. In contrast, an enhancement in structural stability, electronic conductivity, and Li mobility was confirmed from the intercalation-deintercalation studies. Chen et al.55 reported the synthesis of LiCoO2 (LCO) cathodes coated with a gel polymer Li-ion conductor layer, P(VDF-HFP)/LiTFSI (PHL) via the solution-casting technique at low temperature. The coated LCO cathode (thickness = 3 μm) exhibited 88.4% capacity retention of its original capacity (184.3 mA h g−1) after nearly 200 cycles in the range of 3.0–4.6 V. This is higher than that of the uncoated cathode, which showed only 80.4% of its original capacity (171.5 mA h g−1). This enhancement was attributed to the compact nature of the PHL layer, which forms a highly continuous surface coverage and penetrates the bulk of LCO. It also prevents side reactions between the charged LCO surface and electrolyte, leading to enhanced structural stability in LCO. Xie et al.56 reported the synthesis of an LiCoO2 cathode by modifying it with chemically inert and ionically conductive LiAlO2 interfacial layers. This conductive layer provides a path for the diffusion of lithium and also prevents interfacial reactions, as evidenced by Raman and impedance spectroscopy investigations. A capacity value close to 200 mA h g−1 was achieved for the LiCoO2 electrodes with commercial-level loading densities, cycled at the cut-off potential of 4.6 V versus Li+/Li for 50 stable cycles. This represents a 40% capacity gain with respect to the values obtained for the commercial samples cycled at the cut-off potential of 4.2 V versus Li+/Li.
The surface-engineered core–shell-like material demonstrated an excellent retention rate and cycling stability compared to pristine LMO. This enhancement was due to the increase in intrinsic conductivity and easy electrolyte access. For the full cell, the core–shell material exhibited 70% capacity retention, whereas the pristine material retained only 53% capacity after 1000 cycles at 0.1 A g−1. Abbas et al.61 examined the electrochemical performance of silver-modified LiMn2O4 cathode materials and the influence of the calcination atmosphere (vacuum and air). It was concluded that ∼3 wt% Ag coating is effective to minimize the drawbacks of the spinel LiMn2O4 (Mn dissolution and cycling instability). The Ag/LMO(v) electrode showed high capacity retention and good cyclability at the C/2 rate. Lee et al.62 synthesized a novel LiMn2O4 heterostructure with epitaxially grown layered (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) surface phase. No defect was observed at the interface between the host spinel and layered surface phase, which provided an efficient path for ionic and electronic mobilities. The heterostructure LiMn2O4 phase exhibited a discharge capacity of 123 mA h g−1 and retained 85% of its initial capacity after 100 cycles (at 60 °C). Zhu et al.63 reported the synthesis of Al2O3- and PPy-coated LiMn2O4 (PPy/Al2O3/LMO) via the sol–gel method, followed by oxidative chemical polymerization. The discharge capacity of PPy/Al2O3/LMO was reaching a value of 121.73 mA h g−1 at a rate of 1C. A high retention of around 95.81% was observed even after 100 charge/discharge cycles.
m) surface phase. No defect was observed at the interface between the host spinel and layered surface phase, which provided an efficient path for ionic and electronic mobilities. The heterostructure LiMn2O4 phase exhibited a discharge capacity of 123 mA h g−1 and retained 85% of its initial capacity after 100 cycles (at 60 °C). Zhu et al.63 reported the synthesis of Al2O3- and PPy-coated LiMn2O4 (PPy/Al2O3/LMO) via the sol–gel method, followed by oxidative chemical polymerization. The discharge capacity of PPy/Al2O3/LMO was reaching a value of 121.73 mA h g−1 at a rate of 1C. A high retention of around 95.81% was observed even after 100 charge/discharge cycles.
LiFePO4 belongs to the olivine family of lithium ortho-phosphates and has an orthorhombic lattice structure (space group Pnma),64,67 with the lattice parameters of a = 10.33 Å, b = 6.01 Å, c = 4.69 Å and V = 291.2 A3. Its structure consists of corner-shared FeO6 octahedra and edge-shared LiO6 octahedra, running parallel to the b-axis, which are linked by PO4 tetrahedra. In this structure, the Fe atoms occupy the octahedral (4c) sites (dark shading), the P atoms occupy the tetrahedral (4c) sites (light shading), and the Li ions (small circles) occupy octahedral (4a) sites.68
Li et al.69 prepared LiFePO4/graphite composites, which demonstrated a high reversible capacity (160 mA h g−1 under 0.2C), ultrahigh rate capability (107 mA h g−1 under 60C), and outstanding cycle performance (>95% reversible capacity retention over 2000 cycles). The high volumetric energy density of 427 W h L−1 under 60C was achieved. Wang et al.70 synthesized lithium iron phosphate (LFP) with Y–F co-doping. It was observed that the electronic conductivity increased upon doping with F owing to the rearrangement of the PO43+ electron cloud. Doping of Y reduced the space resistance of Li-ion owing to the introduction of Li+ vacancies. The XRD analysis confirmed that Y and F doping led to weakening of the Li–O bond and widening of the lithium-ion diffusion tunnel. The prepared cathode showed a discharge-specific capacity of 135.8 mA h g−1 at 10C and a discharge-specific capacity of 148.6 mA h g−1 without attenuation after 700 cycles at 5C.
Hsieh et al.71 examined the effect of the carbon layer on the cell performance of LiFePO4 (LFP). The carbon content on the surface of LFP powder was tuned via the addition of glucose. The moderate carbon layer-coated cathode exhibited a discharge capacity of ∼161.5 mA h g−1 at 0.1C and ∼99.6 mA h g−1 at 10C. However, at a higher content of glucose, a slow diffusion rate (DLi) and high equivalent series resistance (RES) were observed due to the formation of inter-grain LFP aggregates. The highest specific energy and specific power densities were observed to be 400 W h kg−1 and 1200 W kg−1, respectively. Motivated by the theoretical calculation, Gao et al.72 reported the synthesis of an Ru-doped LiFe1–xRuxPO4/C cathode through the sol–gel method. The sample designated as LFP-1 (x = 0.01) delivered excellent specific capacities of 162.6 and 110.6 mA h g−1 under 0.1 and 10C conditions, respectively. The capacity retention was 95.6% after 300 cycles at 5C. Liu et al.73 prepared Li1−XNaXFePO4 (X = 0, 0.01 or 0.05) composite cathode materials via the simple solvothermal method. The Li0.99Na0.01FePO4 cathode showed an excellent rate capacity (86.7% after 500 cycles at 10C) and cycle stability.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn:Co = 1/3:1/3:1/3), and thus the raw material cost is lower than the cobalt-based batteries.74,75
:Mn:Co = 1/3:1/3:1/3), and thus the raw material cost is lower than the cobalt-based batteries.74,75
Ren et al.76 prepared a lithium nickel manganese cobalt oxide (NMC) cathode, which was designated as LiNi0.33Mn0.33Co0.33O2 (NMC-111). This showed a superior electrochemical performance compared to the commercial NMC-111 (c-NMC), with discharge capacities of 138 and 131 mA h g−1 at high current rates of 20 and 30C, respectively. Even at both room temperature and at 50 °C, the cyclic stability was better. Reissing et al.77 investigated the combination of Zr as a common dopant in commercial materials with effective Li2WO4 and WO3 coatings in nickel-cobalt-manganese (NCM)‖graphite cells. It was concluded that the Zr4+ dopant diffused to the surface during annealing, improving the electrochemical performance compared to the samples without additional coatings. The pristine NCM‖graphite cell displayed an initial discharge capacity of 180 mA h g−1 at 0.1C and 173 mA h g−1 at 0.33C in the cell voltage window of 2.8–4.2 V, while its end of life was reached after approximately 343 cycles with an average Coulombic efficiency (CE) of 99.8%. The WO3-coated sample showed a similar initial discharge capacity and enhanced the life cycle up to >700 cycles.
Cao et al.80 reported the synthesis of an LiNi0.88Co0.09Al0.03O2 cathode via solvothermal and co-precipitation method. The discharge capacity of the solvothermal prepared cathode was observed to be 154.6 mA h g−1 at 55 °C after 100 cycles with the capacity retention of 75.93%. In contrast, the cathode prepared via the co-precipitation method delivered only 130.3 mA h g−1 after 100 cycles, with a capacity retention of 63.31%. Xiao et al.81 reported the synthesis of an LiNi0.88Co0.09Al0.03O2 cathode with the addition of trimethyl borate (TMB) in the commercial electrolyte, which enhanced the interfacial stability. The LiNi0.88Co0.09Al0.03O2 electrode with 10% TMB-containing electrolyte could achieve a capacity retention of up to 82% after 300 cycles at 1C rate (1C = 200 mA h g−1). Zhang et al.82 reported the synthesis of a high-nickel-content NCA (LiNi0.88Co0.09Al0.03) cathode material with a non-spherical morphology, which showed a good cycle performance (at both 25 °C and 45 °C), and also enhanced structural stability with suppressed phase transition from H2 to H3. The capacity retention of the pouch-type cells with non-spherical NCA as the cathode was greater than 91% after 1000 cycles.
| Material | LiFePO4 | LiMn2O4 | LiCoO2 | Li(NixCoyMnz)O2 | LiNi0.8Co0.15Al0.05O2 |
|---|---|---|---|---|---|
| Average voltage (V) | 3.4 | 3.8 | 3.7 | 3.6 | 3.6 |
| Specific capacity (mA h g−1) | 130–140 | 100–120 | 135–150 | 160–220 | 180–200 |
| Cycle number | 2000–5000 | 500–2000 | 500–1000 | 800–2000 | 800–2000 |
| Safety performance | Excellent | Good | Poor | Poor | Poor |
| Price (thousand $ ton−1) | 7.4–14.7 | 4.4–8.8 | 50–57 | 22–29 | 26–35 |
The limitation of traditional cathodes (layered, spinel, and olivine) has limited the capacity mismatch with that of silicon anodes (∼1000 mA h g−1). Thus, to fill this gap, Li-rich oxide (LRO) materials have emerged as a potential alternative to replace future cathodes due to their high theoretical capacity (∼300 mA h g−1) and high specific energy (∼900 W h kg−1). For LROs, the Li/TM ratio is greater than 1 (0 < x < 1), which is commonly referred to as Li1+xTM1−xO2 (e.g., Li2MnO3 and Li2RuO3).89,90 The research on LRO materials started in the early 1960s with the development of Li2SnO3 and Li2MnO3.91,92 Two remarkable developments were made by Thackeray's group,93 who synthesized Li1.09Mn0.91O2 (1991), and Dahn's group, who synthesized Li[NixLi(1/3−2x/3)Mn(2/3−x/3)]O294 in 2001. Two types of Li-rich materials are (i) layered lithium-rich oxides (LLROs), which exhibit a high working potential, low cost, and desirable cyclic stability, and (ii) cation disordered lithium-rich oxides (DLROs), which exhibit structural stability, high specific capacity, and poor cycling stability. Three major challenges with the precision use of LROs can be summarized as follows: (1) they regulate the oxygen loss/oxygen vacancy, where the excessive oxygen vacancies may shrink the cell volume, introduce stacking faults, cation mixing, and an undesirable new phase, which collectively decrease the electrochemical performance.95–97 (2) To obtain the deep insights into the bulk and surface structural evolutions during cycling, the structure transformation in LRO for the initial and final process showed a lower coulombic efficiency and interface breakdown.96 (3) To understand the relationship between the structure/charge-transfer mechanisms and electrochemical performance of LROs, they can be analysed using some advanced techniques (X-ray diffraction, neutron diffraction, X-ray and neutron pair distribution function (xPDF and nPDF), EXAFS, Raman spectroscopy, and Mössbauer spectroscopy). The performance of Li-rich cathode materials can be improved by five strategies, as follows: (i) elemental doping, (ii) controlling the morphology (iii) tuning the structure, (iv) optimisation of composition, and (v) electrolyte additives.98 Juan et al.99 prepared sulphur doped Li-rich cathode materials (LNMOS) via a co-precipitation method. The XRD-analysis confirmed the presence of the sulfur (S) dopant, which reduced the mixing degree of cations in the LNMOS and increased the ordered arrangement of the layered structure. The S-doped lithium-rich material released a higher initial efficiency of 96.06% (87.63% for LNMO), a specific capacity of 293.3 mA h g−1 (243.3 mA h g−1 for LNMO), and better cycling stability and rate performance (a capacity of 117 mA h g−1, maintained at a current density of 5C). Cui et al.99 prepared the stable O2-structured Li1.2Ni0.13Co0.13Mn0.54O2 (O2-LR-NCM) cathode, which demonstrated a high coulombic efficiency (CE) >99.82% and high reversible capacity of 278 mA h g−1. After 100 cycles, 83.3% capacity was retained by this optimised cathode material.
| Anode materials | Advantages | Disadvantages |
|---|---|---|
| Carbon | High electronic conductivity | Low specific capacity |
| Nice hierarchical structure | Low rate capacity | |
| Abundant and low-cost resources | Safety issues | |
| Alloys | High specific capacity (400–2300 mA h g−1) | Low electronic conductivity |
| Good security | Large volume change (100%) | |
| Transition metal oxides | High specific capacity (600–1000 mA h g−1) | Low coulombic efficiency |
| Nice stability | Large potential hysteresis | |
| Silicon | Highest specific capacity (3579 mA h g−1) | Large volume change (300%) |
| Rich, low-cost, clean resources |
Recently, Eftekhari et al.103 categorized anode materials in four classes based on the particular voltage range of their operation. Firstly, for low-voltage materials (group IV and V elements), the majority of the delithiation capacity can be achieved under 1.0 V versus Li/Li+, while secondly for mid-voltage materials (transition metal oxides and chalcogenides), the majority of the delithiation capacity can be achieved in the range of 1.0–2.0 V. The third type, i.e., high-voltage operating materials, the majority of the delithiation capacity occurs over 2.0 V. The fourth category covers nanostructured and mixed valence-based material, where their potential window varies in the range of 0–3.0 V and includes a wide variety of materials with nanostructured and mixed valences (Fig. 6).
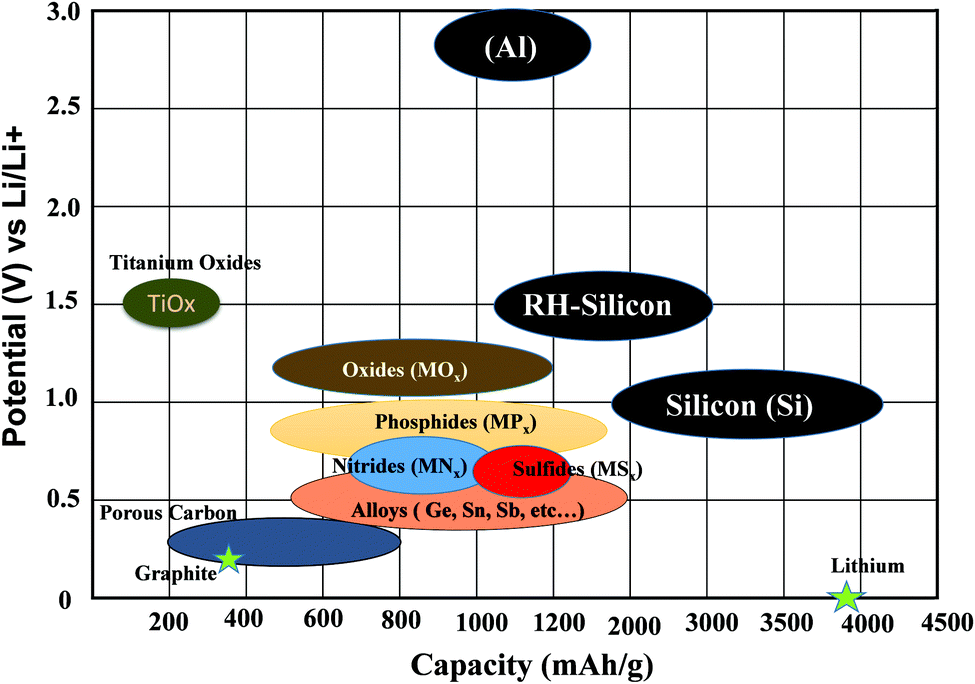 | ||
| Fig. 6 Comparison chart for potential versus specific capacity of various anode materials. | ||
| S. no. | Materials | Current (mA g−1) | Voltage (V) | Capacity (mA h g−1) | Scanning rate (mV s−1) | Coulombic efficiency (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Al | 0.01–1.2 | 890 | 10 | 104 | ||
| Al nanorods | 458 | 1.35 | 324 | 20 | 91.5 | 105 | |
| 2 | Al nanorod | 700 (0.5C) | 0.01–3 | 1243 | 0.1 | 106 | |
| 3 | Al nanorod | 700 | 0.01–3 | 1293 | 91 | 107 | |
| 4 | Fe2Al5 | 0.01–3 | 485 | 1 | 30 | 108 | |
| 5 | Al–Sn Composite | 0.1 mA cm−2 | 0.05–1.25 | 972.8 | 0.1–0.5 | 81 | 109 |
| 6 | Al–Sn composite | 0.1–04 mA cm−2 | 0.05–1.25 | 752 | 0.1–0.5 | 83 | 110 |
| 7 | Al–Fe3O4–rGO | C/10–10C | 0.005–3 | 717.4 | 99.17 | 111 | |
| 8 | Al foil | 182 | −0.02 to +0.55 | 967 | 92.6 | 112 | |
| 9 | Si–Al | 0.05 mA cm−2 | 0.005–2 | 3348 | 93.6 | 113 | |
| 10 | CNS/Si/Al2O3 | 1 A g−1 | 0.01–1 | 1560 | 1 | 84.8 | 114 |
| 11 | Al nanorod | 1C | 0.01–1 | 977 | 1 | 115 | |
| 12 | Si–Al thin film | 0.5C | 0–1.5 | 2257.8 | 0.05 | 85.9 | 116 |
However, the major problem in utilizing Si and Si-based anodes is their poor conductivity and very large volume change (about 310% for Li4Si) during the lithium alloying/dealloying (or lithiation/delithiation) process, which cause mechanical failure of the active material. Thus, it results in pulverization and poor cycle performance of the electrode.113 The specific capacity, coulombic efficiency, and scanning rates are summarized in Table 8.132–180
| S. no. | Materials | Current (mA g−1) | Voltage (V) | Capacity (mA h g−1) | Scanning rate (mV s−1) | Coulombic efficiency (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | a-C/Si | 100 A cm−2 (C/2) | 0.02–1.2 | 2500 | — | 99.5 | 132 |
| 2 | Si/C | 15–60C | 0–1.5 | 3107 | — | 83 | 133 |
| 3 | a-Si/SiOx/Cr/C | 100 | 0–1.5 | 810 | — | 99.2 | 134 |
| 4 | a-SiG | 100–140 | 0.01–1.5 | 2858 | — | 92.5 | 135 |
| 5 | a-Si thin film | 0.025C | 0–1.5 | 3134 | 0.05 | 87.1 | 136 |
| 6 | 3D-Si/C nanowire | 0.4 A g−1 | 0.1–2 | 2300 | — | 91 | 137 |
| 7 | Buddle-Si nanorod | 410 | 0–2 | 2411 | — | 94 | 138 |
| 8 | C–Si | 200 | 0–1.5 | 1280 | — | 99 | 139 |
| 9 | Si/SiO2/C | 100 | 0.01–1.5 | 786 | 0.5 | — | 140 |
| 10 | Si | 0.4 mA cm−2 | 0–1 | 3900 | 0.5 | 90 | 141 |
| 11 | Si/C | 100 | 0–1.5 | 781 | 0.05 | 61.8 | 142 |
| 12 | Si–C matrix | 0.2C | 0.01–3 | 2950 | — | 99.6 | 143 |
| 13 | Si NW | C/20 | 0.01–2 | 3193 | 1 | 90 | 144 |
| 14 | Hollow porous –SiO2 | 100 | 0–3 | 919 | 0.1 | 73 | 145 |
| 15 | Si NW-C | C/10 | 0.01–2 | 2000 | — | 96 | 146 |
| 16 | Li–Si alloy | 50 | 0.01–1.5 | 1000 | — | 93 | 147 |
| 17 | Mesoporous SiO2 | 100 | 0.01–1 | 3000 | — | — | 148 |
| 18 | Mesoporous Si | 0.1 A g−1 | 0.05–1.5 | 750 | — | 99.7 | 149 |
| 19 | P–Si NP | 0.1–1.5C | 0–3 | 2113 | — | 61.1 | 150 |
| 20 | Si spheres | C/20 | 0.01–2 | 3105 | 0.2 | 100 | 151 |
| 21 | Nano-Si | 2 A g−1 | 0.01–1 | 1024 | 0.02 | 99.1 | 152 |
| 22 | Si/PANI | 2 A g−1 | 0.01–1.5 | 766.6 | 0.1 | 72.5 | 153 |
| 23 | Si/CNT | 100 | 0.02–1.2 | 2050 | — | 80.3 | 154 |
| 24 | Mesoporous Si | 200 | 0.01–3 | 1038 | — | 98.4 | 155 |
| 25 | Si–O–C | 100 | 0.01–2 | 753.4 | 0.1 | 66.9 | 156 |
| 26 | Si/PANI | 100 | 0.01–1.5 | 840 | 0.5 | 56 | 157 |
| 27 | Si pomegranate | C/20 | 0.01–1 | 2350 | — | 99.87 | 158 |
| 28 | Si/S–C | 100 | 0.01–1.5 | 1947 | — | 76.1 | 159 |
| 29 | Si NWs | C/10 | 0.01–1 | 2000 | — | 160 | |
| 30 | Si N/SiOxNy | 0.2C | 0.01–2 | 2131 | — | 96 | 161 |
| 31 | Si–Ni3.5Sn4 | C/50 | 0.07–2 | 240 | 0.1 | 67 | 162 |
| 32 | Si/Ge DLNT | 0.2C | 0.01–2 | 1746.1 | — | 88.5 | 163 |
| 33 | Si/po-C/C | 0.2 A g−1 | 0.01–1.5 | 900 | — | — | 164 |
| 34 | Si/PCNF | 0.1 A g−1 | 0.01–1.5 | 2071 | 0.2 | 71.7 | 165 |
| 35 | Si NP-PANI | 1.0 A g−1 | 0.01–1 | 1600 | 0.1 | 99.8 | 166 |
| 36 | Si NW-C | 0.05C | 0.01–2 | 3701.8 | 0.5 | 83.2 | 167 |
| 37 | TiSi2 nanonets/Si | 0.6 A g−1 (0.2C) | 0.15–2 | 2700 | — | 97.5 | 168 |
| 38 | Si-CNT | 0.8 A g−1 (C/5) | 0.01–1 | 1200 | 43 | 169 | |
| 39 | Si/rGO-P | 50 | 0–3 | 1261 | 0.1 | 66 | 170 |
| 40 | Si–M C | 100 | 0.01–2 | 1220.9 | 0.1 | 56 | 171 |
| 41 | Si–Mn/rGO | 100 | 0.01–3.25 | 600 | 0.1 | 97.6 | 172 |
| 42 | Si/Cu–Al–Fe | 120 | 0–1.5 | 836 | — | 80.5 | 173 |
| 43 | SC-Si/G | 1.0 A g−1 | 0.005–1 | 1611 | 0.1 | 76.9 | 174 |
| 44 | SiO2 | C/2 | 0–3 | 1266 | 0.1 | 98.5 | 175 |
| 45 | Si-SWNT–Cu | 0.1C | 0.005–3 | 2221 | — | 74 | 176 |
| 46 | Si-CNT | C/5 | 0.01–1 | 494 | — | 98 | 177 |
| 47 | Tobacco Mosaic Virus (TMV)-Si | 1C | 0–1.5 | 3343 | — | 100 | 178 |
| 48 | Si Np–rGO | 0.5 A g−1 | 0.005–1.5 | 956.7 | — | 82.8 | 179 |
| 49 | PS@C | 100 | 0.05–2 | 1980 | 82 | 180 | |
| 50 | rGO-porous Si | 100 | 0.01–1.5 | 815 | 0.5 | 96.4 | 181 |
| S. no. | Materials | Current (mA g−1) | Voltage (V) | Capacity (mA h g−1) | Scanning rate (mV s−1) | Coulombic efficiency (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | CNT | 0.5C | 0–3 | 446 | 95 | 182 | |
| 2 | Co3O4/CoO/graphene | 21.12 | 0.005–3 | 1153.81 | 0.1 | 76 | 183 |
| 3 | Graphene nanosheet | 0.2 mA cm−2 | 0.01–3.5 | 672 | — | — | 184 |
| 4 | Fe2O3@MWCNTs | 100 | 0–3 | 515 | 0.1 | 95 | 185 |
| 5 | Fe2O3/graphene | 50 | 0.001–3 | 1069 | 0.1 | 77.2 | 186 |
| 6 | Fe2O3/Fe3C–graphene | 0.17C | 0.01–3 | 1118 | 0.5 | 187 | |
| 7 | Fe2O3–graphene | 0.1C | 0.05–3 | 1074.9 | 0.1 | 65 | 188 |
| 8 | Co3O4/graphene | 50 | 0.01–3 | 935 | 1 | 98 | 189 |
| 9 | Graphene NR/SnO2 | 100 | 0.01–2.5 | 1130 | 0.5 | 98.3 | 190 |
| 10 | G-CNT-Fe | 100 | 0–3 | 1024 | 0.05 | 99 | 191 |
| 11 | G/C–Si | 300 | 0.02–1.2 | 902 | 57.3 | 192 | |
| 12 | G-CoS2 | 100 | 0.01–3 | 800 | 0.05 | 98 | 193 |
| 13 | G–M-SnO2 | 100 | 0.005–3 | 1354 | 0.1 | 98 | 194 |
| 14 | GO/G/CNT | 0.5C | 0.01–3 | 1172.5 | 0.1 | 58 | 195 |
| 15 | MnO/RGO | 0.16 A g−1 | 0.01–3 | 855 | 0.1 | 69.7 | 196 |
| 16 | CNT-Si | C/10 | 0.01–1 | 1711 | — | 98 | 197 |
| 17 | C-Graphite | 0.1–3C | 0–2.5 | 358 | — | 81 | 198 |
| 18 | C/Si | 500 | 0.02–1.5 | 1018 | — | 98 | 199 |
| 19 | Nitrogen-doped-graphene | 100 | 0.01–3 | 2132 | 0.1 | 99.2 | 200 |
| 20 | nC-pSiMPs | C/4 | 0.01–1 | 1500 | — | 78 | 201 |
| 21 | p-CNTs@CFO | 0.1 A g−1 | 0.01–3 | 1077 | — | 202 | |
| 22 | Porous-G–C | 100 | 0–3 | 722 | 0.5 | 98 | 203 |
| 23 | N–C spheres | 50 | 0.005–3 | 816 | 0.2 | — | 204 |
| 24 | rGO/BN | 100 | 0–3 | 278 | 0.1 | 100 | 205 |
| 25 | Li2S-G | 0.1C | 0–3.5 | 791 | 0.1 | — | 206 |
The pulverization and disintegration of the active materials from the current collector increase by Li intercalation/deintercalation, leading to the formation of an unstable solid-electrolyte interphase (SEI) and severe capacity fading. Besides, one of the major obstacles of Sn-based anodes is the poor electrical conductivity of SnO2 nanostructures, which hinders the reaction with Li during the discharge. Several methods, such as, nano-scaled structure, doping, and core–shell structures, have been reported to overcome these limitations.209Table 10 highlights the tin-based anode electrodes for charging and discharging parameters.210–215
| S. no. | Materials | Current (mA g−1) | Voltage (V) | Capacity (mA h g−1) | Scanning rate (mV s−1) | Coulombic efficiency (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | SnO2/N–G | 0.5 A g−1 | 0.005–3 | 1352 | 0.1 | 97 | 210 |
| 2 | Sn/Cu6Sn5 thin film | 100 μA cm−2 | 0–1.5 | 1127 | 0.5 | 92 | 211 |
| 3 | SnO2/Co3O4/rGO | 100 | 0.01–3 | 1038 | 0.1 | 66.8 | 212 |
| 4 | SnO2/graphene | 100 | 0.01–3 | 2213 | 0.1 | 66.74 | 213 |
| 5 | C/Sn | 200 | 0–3 | 1300 | 0.2 | 100 | 214 |
| 6 | SnO2/C | 0.5C | 0.01–2 | 908 | 0.5 | 98 | 215 |
LiNi0.5Co0.2Mn0.3O2 (NCM523) is an interesting cathode due to its high discharge capacity and good cyclic stability. However, its safety issue and poor thermal stability are recognized as major drawbacks. Recently, Chen et al.219 examined the performance of NCM523 by modifying the surface of the electrode with Li3PO4 to eliminate the performance degradation. Fig. 7(a) shows the modification approach and role of coating in ion migration. The XRD analysis suggests that the coating did not affect the structure of bare NCM523. The FE-SEM and EDS analysis confirmed the formation of a core–shell structure and encapsulation of the bare cathode. The impedance analysis after 200 cycles showed the lower charge transfer resistance value for the coated NCM523 (156.5 Ω) compared to that of the bare NCM523 (340.1 Ω). Compared to the pristine cathode (DLi+ = 7.29 × 10−13 cm2 s−1), the diffusion coefficient was enhanced after the coating (DLi+ = 1.43 × 10−12 cm2 s−1). The discharge capacity, rate capability, and coulombic efficiency improved for the coated electrode (NCM523). The coating of the Li3PO4 layer acted as a carpet for Li-ion movement (Fig. 7a).
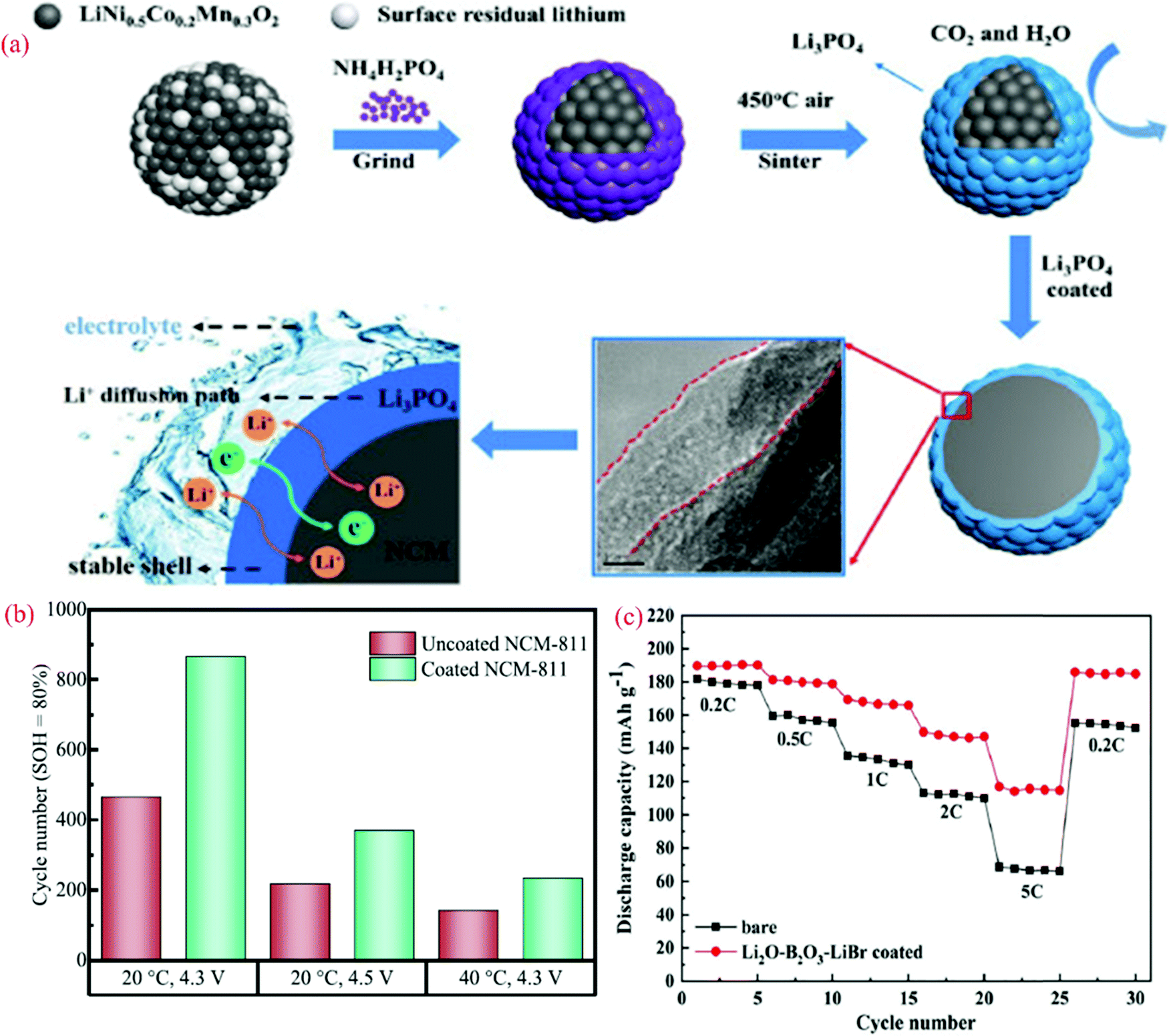 | ||
| Fig. 7 (a) Schematic diagram of the experimental process.219 (b) Achieved cycle numbers for different cycling conditions at an SOH of 80%. The calculation of the SOH is based on the capacity of the fifth cycle and (c) rate performance for uncoated NCM 523 and Li2O–B2O3–LiBr-coated NCM523. Reproduced with permission from Elsevier.219 | ||
The Ni0.8Co0.1Mn0.1O2 (NCM811) cathode is another promising electrode for batteries due to its high capacity. However, its poor thermal stability and tendency to react with moisture pose some challenges that need to be resolved. Becker et al.220 examined the electrochemical performance of NCM811 with a coating of Li2WO4via the sol–gel method. Li2WO4 was chosen due to its high Li+ conductivity, non-toxic nature, and desirable thermal stability.221 The increased thermal stability of the coated electrode was analyzed using XRD patterns. A comparison of the capacity retention for the coated and uncoated NCM811 is shown in Fig. 7(b), corresponding to different cyclic conditions (temperature and voltage). The state of health (SOH) of the cell is the ratio of discharge capacity of the actual cycle to the discharge capacity of the fifth cycle at 0.5C. For a SOH of about 80%, the coated electrode-based cell demonstrated an improved performance.
The NCM523 (LiNi0.5Co0.2Mn0.3O2) cathode is used for battery fabrication due to its low toxicity, cost-effectiveness, good safety, and high capacity.222 However, this type of electrode exhibits one drawback, it restricts the cyclic stability due to corrosion issues with the electrolyte. To eliminate the above-mentioned issue, Wang et al.223 recently examined an Li2O–B2O3–LiBr-coated (∼10 nm) electrode to improve the electrochemical performance of NCM523. The discharge capacity of the Li2O–B2O3–LiBr-coated coated electrode increased from 181.8 to 189.7 mA h g−1, and the coulombic efficiency was enhanced from 94.5% to 97.7%. Fig. 7(c) shows a comparison of the data for the rate capability of the bare and coated NCM523 electrode, where the discharge capacity was observed to be higher for the coated electrodes at all C-rates. Superior cyclic stability was also detected for the coated electrode, which is attributed to the elimination of side reactions and corrosion issues with the electrolyte. The capacity retention was observed to be 87.7% for the coated electrode, which was 29.8% higher than the uncoated electrode up to 100 cycles.
Gan et al.224 reported the preparation of an NCM811 cathode via modification with a coating of WO3. The modified layer thickness was around 10 to 15 nm, which was also confirmed via XRD and XPS analysis. The oxygen peak in the XPS survey of the modified electrode was detected, which was weaker than that of the uncoated NCM811 electrode, suggesting the presence of more reactive oxygen on uncoated NCM811. The presence of more reactive oxygen may provoke side reactions between the electrode and electrolyte. The negligible change in electrochemical performance was investigated in the voltage range of 2.8–4.3 V. Although no improvement was observed in the capacity, the rate capability and reversible capacity drastically improved. The capacity retention was 87.7% for the coated electrode, which was 29.8% higher than that of uncoated electrode for up to 100 cycles.
Therefore, it can be concluded that the coating or surface-modification of electrodes is a promising approach to tune the electrode surface chemistry. The notable features lead to (i) enhanced thermal stability, (ii) improved rate capability and capacity retention, (iii) prevention of side reactions and electrode corrosion due to the electrolyte, and (iv) suppressed capacity fading during long cycle run. Some of the important materials for electrode modification are suggested to be, for example, carbon (C), TiO2, ZnO, CuO, ZrO2, CeO2, Al2O3, and MgO. Ionic conducting materials such as PEDOT, LLTO (Li0.125La0.625TiO3), Li3PO4, Li3VO4, and LiAlO2 are also auspicious materials for the enhancement of electronic conductivity on the electrode surface. Some important modifications of (coating) materials and their comparative performances (capacity, voltage, capacity retention, and coulombic efficiency) are summarized in Table 11.218–220,223–242
| Coating | Host | Voltage (V) | Capacity (mA h g−1) | Capacity retention | Efficiency (%) | Ref. |
|---|---|---|---|---|---|---|
| Li3PO4 | LiNi0.5Co0.2Mn0.3O2 | 2.7–4.3 | 186.36 (184.36 for pristine) | 83% after 200 cycles (68.5 for pristine) | 86.06% (83.26% for pristine) | 219 |
| Li2WO4 | NCM 811 (LiNi0.8Co0.1Mn0.1O2) | 2.5–4.3 | 192 (188 for pristine) | 80% after 765 cycles (pristine after 465 cycles) | 220 | |
| Li2O–B2O3–LiBr | NCM523 (LiNi0.5Co0.2Mn0.3O2) | 2.5–4.5 | 116.9 at 5C (68.7 for pristine) | 87.7% after 100 cycles (29.8% for pristine) | 97.7% | 223 |
| LiVPO4F hybrid | LiNi0.8Co0.1Mn0.1O2 | 2.8–4.3 | 214.9 (208.1 for pristine) | 95.93 (91.68 for pristine) | 90.7 (85.5 for pristine) | 224 |
| LLTO | NCA (LiNi0.8Co0.15Al0.05O2) | 2.0–4.4 | 135 at C/20 (125 for pristine) | 99% capacity after 10 cycles (85% for pristine) | — | 218 |
| Al2O3 | LiNi0.8Co0.1Mn0.1O2 (NCM811) | 2.8–4.3 | 199.2 (201.7 for pristine) | 99.61, after 100th cycle | 88.02 (after 1st) | 225 |
| ZrO2 | 198.7 (201.7 for pristine) | 99.66, after 100th cycle | 88.04 (after 1st) | |||
| LBO (Li2O–B2O3–LiBr) | 204.3 (201.7 for pristine) | 99.78% after 100th cycle | 88.83 (after 1st cycle) | |||
| Li2O–B2O3–LiBr | Li1+xMn2O4 | 3–4.2 | 122.5 (12.5 for pristine) | 93% after 20 cycles (15.14% for pristine) | — | 226 |
| ZrO2 | LiMn2O4 | 3.0–4.3 | 118.8 (124.4 for pristine) | 90.1% after 400 cycles (at 55 °C) | 96.7 | 227 |
| CeO2 | Li1.2Ni0.2Mn0.16O2 | 2.0–4.8 | 270 at 0.1C (235 for pristine) | 78.5% after 200 cycles | 80.54 | 228 |
| Li2O–B2O3–LiBr | LiNi0.8Co0.15Al0.05O2 | 3.0–4.3 | 181 (175 for pristine) | 94.2% after 100 cycles | 91 | 229 |
| Ta and | LiNi0.6Co0.2Mn0.2O2O2 | 1.88–3.88 | 115 at 34 mA g−1 (52.7 for pristine) | 91% after 30 cycles (75% for pristine) | 95.9 (87.4 for pristine) | 230 |
| W | 93.3 (52.7 for pristine) | 82% after 30 cycles (75% for pristine) | 92.6 (87.4 for pristine) | |||
| Carbon | NCM811 (LiNi0.8Co0.1Mn0.1O2) | 3.0–4.3 | 188.6 (192.8 for pristine) | 87.8% after 80 cycles (74.3% for pristine) | — | 231 |
| Li5AlO4 | NCM811 (LiNi0.8Co0.1Mn0.1O2) | 2.8–4.3 | 147.61 (127.86 for pristine) | 89.15% after 100 cycles (75.06% for pristine) | — | 232 |
| Li3PO4 | LiNi0.8Co0.1Mn0.1O2 | 3.0–4.3 | 192.4 (186.0 for pristine) | 86.7% after 250 cycles (85.2% for pristine) | 86.4 (83.9 for pristine) | 233 |
| Li2CuO2 | LiNi0.33Co0.33Mn0.33O2 | 2.4–4.2 | 192 (182 for pristine) | 69% after 100 cycles (30% for pristine) | 99 after 30 cycle (93 for uncoated) | 234 |
| Co3O4 and LiMn2O4 layer | LiNi0.8Co0.1Mn0.1O2 | 2.7–4.5 | 203.9 at 0.1C (202.9 for pristine) | 91.4% after 100 cycles (73.5% for pristine) | 89.1 after 1st cycle (87.8 after pristine) | 235 |
| Ti3C2(OH)2 | LiNi0.6Co0.2Mn0.2O2O2 | 3.0–4.3 | 124.5 | 86.4% at 0.5C after 200 cycles (71.2% for pristine) | — | 236 |
| Nickel catalyzed graphitized carbon | LiFe1−xNixPO4 | 2.8–4.0 | 181.9 at 0.1C (143.3 for pristine) | 95.6% at 1C after 500 cycles | > 99 after 200 cycles | 237 |
| Li–Nb–O shell | Li1.2Ni0.13Co0.13Mn0.54O2 | 2.0–4.6 | 219.5 (212.3 for pristine) | 96.44% capacity retention after 100 cycles (83.09% for pristine) | 92.59% | 238 |
| N-Doped carbon-coated | LiNi0.6Co0.2Mn0.2O2O2 | 3.0–4.5 | 199.4 at 0.2C (202.4 for pristine) | 82.7% after 100 cycles (61.1% for pristine) | — | 239 |
| Nano-WO3 | LiNi0.5Co0.2Mn0.3O2O2 | 3.0–4.5 V | 138 (at 25 °C) and 170.9 (at 50 °C) (107.8 at 25 °C, and 143.9 at 50 °C for pristine) | 80.80% after 200 cycles | 87.39 after 1st cycle (83.19 for pristine) | 240 |
| LiBO2 | LiNi0.6Co0.2Mn0.2O2O2 | 2.8–4.3 | 123 after cycling at 0.5C after 150 cycles (94 for pristine) | 84.3% during 150 cycles at 0.5C (68.3% for pristine) | 93.7 (90.6 for pristine) | 241 |
| LFP | LiNi0.82Co0.12Mn0.06O2 | 3.0–4.2 | 165.3 after 500 cycles at 1C (130.7 for pristine) | 91.65% after 500 cycles (70.65% for pristine) | — | 242 |
Recently, Zheng et al.244 prepared the Li1.2Mn0.54Ni0.13Co0.13O2 (LMNCO) cathode with gadolinium-doped ceria (GDC) via doping using a wet-chemical deposition approach. The XRD pattern suggested that the structure of LMNCO was not changed by the GDC dopant concentration. The increased interlayer spacing of LMNCO indicated the successful doping of Ce+3/Ce+4 or Gd+3 without changing the structural properties. The investigated electrochemical performance was found to be superior for the LMNCO-GDC3 electrode with discharge capacities of 267.5 mA h g−1 and 255.2 mA h g−1 for the pristine electrode with the charging rate of 0.1C at different intervals. Therefore, it was concluded that the GDC-based modified electrode reduced the charge transfer resistance and promoted Li+ migration. After 100 cycles, the capacity retention of LMNCo-GDC3 was 92.9%, which was higher than that of the pristine electrode (75.3% at 0.5C). Fig. 8(a) shows the capacity fading mechanism in the pristine LMNCO. The electrolyte reacts with the electrode and side reactions occur. HF is released from the decomposition of the LiPF6 salt and reacts with the electrode, which leads to the formation of LiF, resulting in the reduction of Li ions. In contrast, the direct contact between the electrode and electrolyte was restricted in the modified electrode of LMNCO with GDC (Fig. 8b). Overall, this minimized the electrode dissolution tendency and loss of the active material.
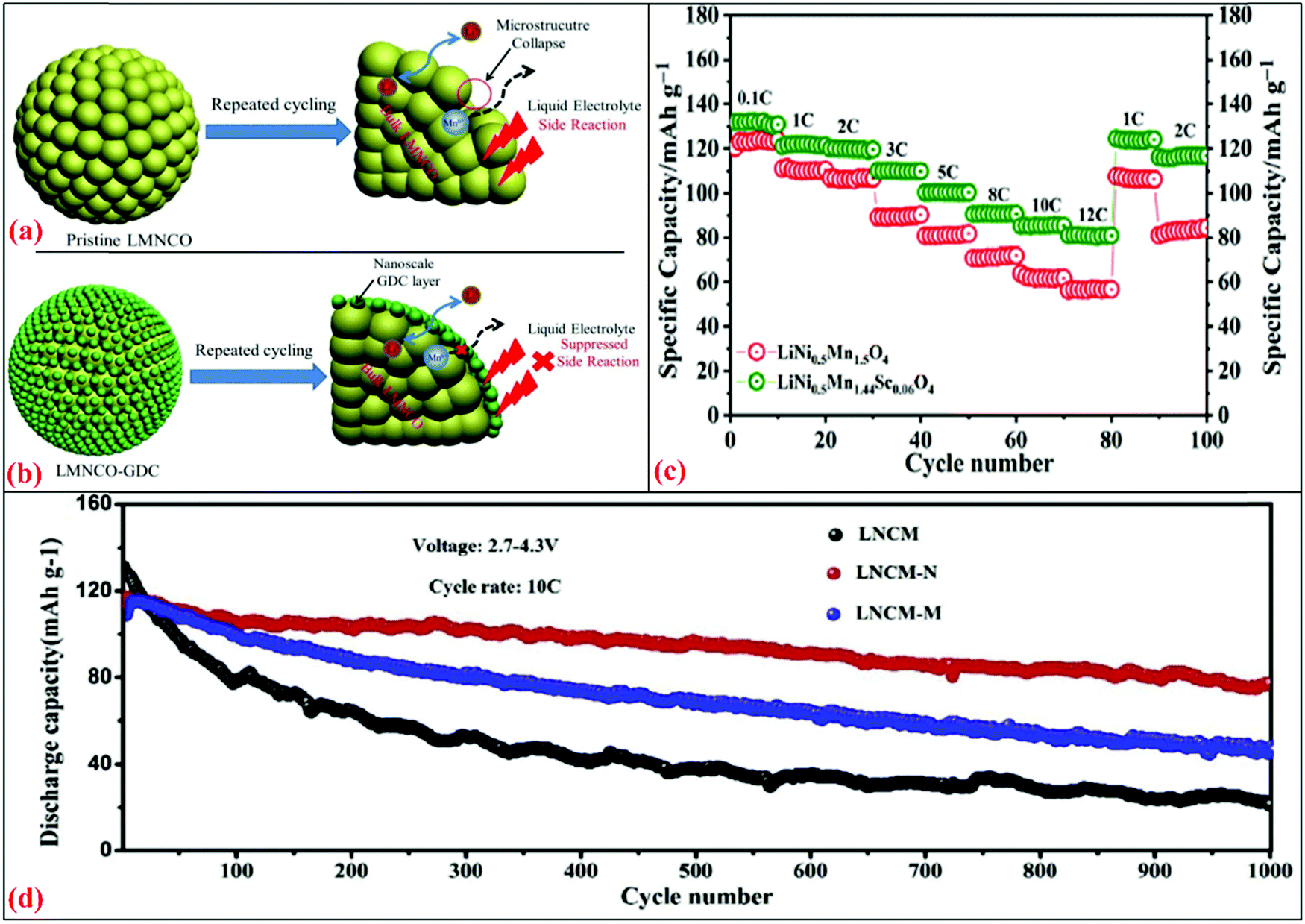 | ||
| Fig. 8 Schematic illustrations of (a) pristine LMNCO interfacial side reactions with liquid electrolyte after repeated cycling244 and (b) GDC coating layer acts as a protection layer to suppress the unwanted interfacial side reactions after repeated cycling.245 (c) Rate capability for LNMO and LNMSO cathode for 0.1–12C. (d) Cycling performances for LNCM, LNCM-N, LNCM-M at C-rate of 10C.246 Reproduced with permissions from Elsevier.244–246 | ||
Another environment-friendly material, LiNi0.5Mn1.5O4 (LNMO), has attracted attention from the energy storage community due to its spinal structure, high operating voltage (4.7 V versus Li) and rate capability. LNMO has a theoretical specific energy of ∼650 W h kg−1 and observed to be superior in comparison to other cathodes.247,248 The cation ordering in LNMO can be tuned by the annealing parameters, which disorder the spinel. To prepare disordered spinels, Bhuvaneswari et al.245 prepared Sc-doped LNMO (LNMSO) via the solution combustion method. Its XRD pattern confirmed the formation of a disordered spinel structure with the Fdm space group. The IR spectra also supported this, evidencing the absence of an ordered spinel structure (P4332). The first discharge capacity for LNMSO was 131 mA h g−1 with a coulombic efficiency of 88%, while the undoped LNMO demonstrated the first discharge capacity of 123 mA h g−1 with a coulombic efficiency of 81% at 0.1C. Even after 1000 cycles, LMNSO exhibited the capacity of 102 mA h g−1 (capacity retention = 98%), which was higher than that of LMNO (79 mA h g−1 with the capacity retention of 90% at 5C). Fig. 8(c) shows the rate capability in the range of 0.1C to 12C. The LMNSO electrode demonstrated a superior performance to the bare LMNO at all current rates. Even at a high C-rate, the capacity retention for LMNSO was observed to be 61%, which was higher than that of LMNO (45%). This enhancement in specific capacity and rate capability was attributed to the creation of disorder in Ni/Mn by Sc-doping, which facilitated faster Li diffusion. Higher DLi+ (10−10–10−14 cm2 s−1) values were obtained for LNMSO compared to LNMO (10−10–10−15 cm2 s−1), suggesting the fast cation diffusion regarding Sc concentration.
The atomic substitution of foreign elements is an effective strategy to improve the thermal stability and the rate capability of the NCM cathode. Li et al.246 examined the effect of Al3+ doping in the Ni-rich LiNi0.8Co0.1Mn0.09Al0.01O2 cathode via a continuous co-precipitation method. The samples were designated as LNCM-N (LiNi0.8Co0.1Mn0.09Al0.01O2), LNCM-M (LiNi0.8Co0.1Mn0.09Al0.01O2), and NCM811 (LiNi0.8Co0.1Mn0.1O2). XRD evidenced the shift to the 003 reflection, which was the highest. This suggests that the incorporation of Al3+ was the highest for LNCM-N. The FE-SEM image indicated the uniform distribution of Al3+. Compared to the Ni2+ on Li slabs of LNCM-M and LNCM, the XPS analysis indicated that the presence of a lower Ni2+ content on the Li slabs for LNCM-N. The lower Ni2+ content was favorable for the faster cation (Li+) intercalation/deintercalation, which showed the highest capacity in the electrochemical analysis. LNCM-N demonstrated a higher discharge capacity (at 1C) of about 126 mA h g−1 (capacity retention = 78.92%) after 200 cycles compared to LNCM-M (90 mA h g−1; capacity retention = 59.69%) and LNCM (83 mA h g−1; capacity retention = 48.62%). Fig. 8(d) shows the performance of the three cells for 1000 cycles at 10C. It shows the highest discharge capacity for LNCM-N (with the efficiency of >98% and capacity retention = 70%) compared to LNCM-M and PNCM. The diffusion coefficients of LNCM, LNCM-M, and LNCM-N were investigated to be 3.62 × 10−14 cm2 s−1, 6.51 × 10−14 cm2 s−1, and 9.77 × 10−14 cm2 s−1, respectively. The high value of the Li+-diffusion coefficient and low impedance (evidenced by the XPS) led to better cyclic stability and enhanced capacity.
Cation substitution is an attractive strategy to tune electrodes, which reduces the cation mixing to promote the structural integrity and increase the Coulombic efficiency of the electrode. This approach was used by Huang et al.,249 who investigated the effect of partial Mn substitution with Mg by preparing LiNi0.6Co0.2Mn0.2−xMgxO2via the hydroxide co-precipitation method. Mg reduced the cation mixing, preventing the structural collapse of the Li layer, i.e., stable pillar effect. The electrochemical performance was evaluated between 2.8–4.3 V at RT. The initial discharge capacity at 0.1C was 180.94 mA h g−1 for the pristine electrode and 186.23 mA h g−1 for x = 0.01. The capacity retention for the Mg-doped electrode was 91.04% for x = 0.03, which is higher than that of the pristine electrode (81.34%) for 100 cycles. This was attributed to the decreased cation mixing, which reduced the barrier for Li migration and enhanced the structural stability owing to strong Mg-O bonding. The effect of doping elements and metallic species on the specific capacity, capacity retention, Coulombic efficiency, and scanning rates is summarized in Table 12.239,244–246,249–261
| Dopant | Host | Voltage (V) | Capacity (mA h g−1) | Capacity retention | Efficiency (%) | Ref. |
|---|---|---|---|---|---|---|
| Gadolinium-doped ceria (GDC) | Li1.2Mn0.54Ni0.13Co0.13O2 | 2.0–4.8 | 267.5 at 0.1C (255.2 for pristine) | 92.9% after 100 cycles at 0.5C (75.3% for pristine) | 83.3 (73.7 for pristine) | 244 |
| Sc | LiNi0.5Mn1.5O4 | 3.5–4.9 | 131 (123 for undoped) | 94% after 300 cycles at 1C | 88 after 1st cycle (81 for un-doped) | 245 |
| Al3+ | LiNi0.8Co0.1Mn0.1O2 | 2.7–4.3 | 126 at 1C (83 for undoped) | 78.92% at 1C rate after 200 cycles, 70.0% at 10C rate after 1000 cycles. | 98% | 246 |
| Mg | LiNi0.6Co0.2Mn0.2O2 | 2.8–4.3 | 186.23 at 0.1C (180.94 for pristine) | 91.04% after 100 cycles (81.34%) for pristine) | 89.16 for first cycle (87.03 for pristine) | 249 |
| Mn | LiNi0.85Co0.10Al0.05−x | 3.0–4.3 | 171.4 (156.5 for pristine) | — | 88.6 (79.4 for pristine) | 250 |
| Ti | 179.6 (156.5 for pristine) | — | 81.7(79.4 for pristine) | |||
| F | Li1.15Ni0.275Ru0.575O2 | 2.2–4.3 | 103 (94 for pristine) | 42.2% (43.0% for undoped) | — | 251 |
| Ti | LiNi0.8Co0.1Mn0.1O2 | 2.8–4.3 | 165.02 (147.41 for undoped) | 77.01% after 150th at 1C | 82.5 after 1st cycle (79.3 for pristine) | 252 |
| Nb5+ | LiV3O8 nanorods | 1.8–4.0 | 401 at 0.1C | 99.7% after 500 cycles | >90 | 253 |
| N | LiNi0.6Co0.12Mn0.22 | 3.0–4.5 | 156.6 at 5C (129.2 for undoped) | 82.7% at 1C after 100th cycles (61.1% for undoped) | — | 239 |
| 1D Nb | LiNi1/3Co1/3Mn1/3O2 | 2.7–4.3 | 118.7 at 5C (109.6 for undoped) | 83.3% capacity retention after 200 cycles at 5C | 92.3 at 0.1C | 254 |
| V | LiMnPO4 | 2.2–5.0 | 126 at 0.2C | 74.4 after 50 cycles | 94% | 255 |
| W | LiNi0.90Co0.05Mn0.05O2 | 2.7–4.4 | 235 | 89% of after 500 cycles (60% for pristine) | — | 256 |
| Nb | Li1.2(Ni0.13Co0.13Mn0.54)1−xNbxO2 | 2.0–4.8 | 287.5 (234.2 for pristine) | 98.50% after 300 cycles (86.68% for pristine) | — | 257 |
| ZrO2 | LiNi0.92Co0.08O2 | 2.8–4.3 | 207.2 at 0.2C (201.3 for pristine) | 82.90% after 100 cycle (59.01% for pristine) | 84.74 after 1st cycle | 258 |
| Mg | NCM811 (LiNi0.8Co0.1Mn0.1O2) | 3.0–4.5 | 226.5 at 0.1C (208 for pristine) | 81% over 350 cycles at 0.5C (67% for pristine) | — | 259 |
| Mn | LiFePO4 | 3–4.4 | 45.7 at 0.05C after 1st cycle (43.8 for pristine) | 84% after 100 cycles at 0.5C | 94% after 100 cycles at 0.5C | 260 |
| W | Li4Ti5O12/brookite | 1.0–3.0 | ∼205 at 0.1 A g−1 at −20 °C (∼53 for pristine LTO) | 96% after 1000 cycles at 1 A g−1 | ∼100% | 261 |
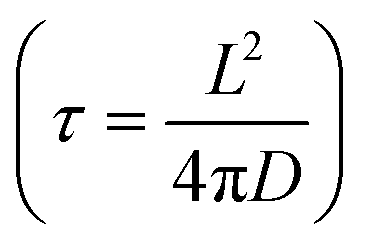 , where L is the particle size, D is the diffusion coefficient, and τ is the characteristic time.262 The synthetic method determines the morphology and the structure. Some of the important synthetic methods of nanoparticles include sol–gel, solid-state precipitation, hydrothermal, and solvothermal. The important parameters that are key to achieving a desirable morphology include the growth temperature, stirring time, sintering time, pressure, pH, and calcination temperature. Another important approach is the formation of core–shell microstructures, where the core and shell are tailored to achieve a balanced and optimum performance electrode. For the formation of good core–shell microstructures, the structural mismatch between the core and the shell must be eliminated or reduced to obtain the desired stability during the cell operation. The capacity and capacity retention of microstructure-based electrodes (nanoplate, rectangular prism, nanorods, hexagonal nanorods, nanowires, triaxial nanowires, nanotubes, and chain-like nanowires) are summarized in Table 13.263–270
, where L is the particle size, D is the diffusion coefficient, and τ is the characteristic time.262 The synthetic method determines the morphology and the structure. Some of the important synthetic methods of nanoparticles include sol–gel, solid-state precipitation, hydrothermal, and solvothermal. The important parameters that are key to achieving a desirable morphology include the growth temperature, stirring time, sintering time, pressure, pH, and calcination temperature. Another important approach is the formation of core–shell microstructures, where the core and shell are tailored to achieve a balanced and optimum performance electrode. For the formation of good core–shell microstructures, the structural mismatch between the core and the shell must be eliminated or reduced to obtain the desired stability during the cell operation. The capacity and capacity retention of microstructure-based electrodes (nanoplate, rectangular prism, nanorods, hexagonal nanorods, nanowires, triaxial nanowires, nanotubes, and chain-like nanowires) are summarized in Table 13.263–270
| Morphology | Material | Voltage (V) | Capacity (mA h g−1) | Capacity retention | Ref. |
|---|---|---|---|---|---|
| Nanoplate, rectangular prism nanorod | LiFePO4 | 2.4–4.2 | 163.8 at 0.2C | — | 263 |
| Nanoplate, hexagonal prism nanorod | 144.4 at 0.2C | — | |||
| Nanowire | LiFePO4/C | 2.5–4.1 | 155 at 1C rate | 98% after 100 cycles at 20C rate | 264 |
| Nanowire | LiFePO4 | 2–4.2 | 110 at a current rate of 30C | 86% after 1000 cycles at a current rate of 10C. | 265 |
| Triaxial nanowire | LiFePO4 | 2–4.5 | 130 at 0.1 A g−1 | — | 266 |
| Nanowire | LiCoO2 | 3.0–4.3 | 126 at 1 mA g−1 | 80% after 100 cycles | 267 |
| Nanotube | LiCoO2 | 3–4.3 | 185 at 10 mA g−1 | 89% after 100 cycles | 268 |
| Nanotube | LiNi0.8Co0.2O2 | 205 at 10 mA g−1 | 71% after 100 cycles | ||
| Nanotube | LiMn2O4 | 138 at 10 mA g−1 | 69% after 100 cycles | ||
| Chain like nanowire | LiCoO2 | 3.0–4.2 | 103 at 10C | 90% after 50 cycles | 269 |
| Aligned slanted nanowires | LiCoO2 | 3.0–4.2 | 97.3 at 0.1C | 89% after 150 cycles, 73% after 400 cycles | 270 |
Jiang et al.272 prepared an LiNi0.9Co0.05Mn0.025Mg0.025O2 electrode via the sol–gel method and investigated the effect of calcination temperature and time on its electrochemical performance. The XRD analysis evidenced a decrease in cation mixing with respect to an increase in temperature (650 °C to 800 °C). The microscopic images showed the growth of nanoparticles with an increase in temperature, which increased the size of the nanoparticles from the nanometer to micrometer range. The specific capacity of the electrode was 128.4, 201.0, and 180.5 mA h g−1 after the first cycle and 121.4, 199.6, and 170.0 mA h g−1 after the 10th cycle regarding the calcination temperature of 650 °C, 700 °C, and 750 °C, respectively. The specific capacity decreased at high temperatures due to the large particle size (as evidenced from SEM), which reduces the diffusion distance. The SEM analysis confirmed that the morphology changed with agglomeration at the calcination time of 18 h, together with a good hexagonal structure. The specific capacity changed to 144.0, 187.6, and 132.1 mA h g−1 after the fifth cycle and 106.3, 172.3, and 105.1 mA h g−1 at 1C with the calcination time of 6 h, 12 h, 18 h, respectively. The reduction in specific capacity at a high calcination time is attributed to the agglomeration tendency of particles, as evidenced by SEM. Table 14 summarizes the different methods, materials prepared, and performance parameters.80,271–289
| Method | Precursors | Voltage (V) | Capacity (mA h g−1) | Capacity retention (%) | Ref. |
|---|---|---|---|---|---|
| Sol–gel | LiNi0.8Co0.2O2 | 3.0–4.3 | 143 after 100 cycles at 0.2C | 77.3 | 271 |
| Solid-state | 158 after 100 cycles at 0.2C | 85.4 | |||
| Sol–gel (calcination temp = 650 °C) | LiNi0.9Co0.05Mn0.025Mg0.025O2 | 2.8–4.3 | 128.4 after 1st cycle | 121.4 after 10 cycles | 272 |
| Sol–gel (calcination temp = 700 °C) | 201.0 after 1st cycle | 199.6 after 10 cycles | |||
| Sol–gel (calcination temp = 750 °C) | 180.5 after 1st cycle | 170.0 after 10 cycles | |||
| Sol–gel (calcination time = 6 h) | LiNi0.9Co0.05Mn0.025Mg0.025O2 | 2.8–4.3 | 144.0 (at 1C) | 106.3 after 50 cycles | |
| Sol–gel (calcination temp = 12 h) | 187.6 (at 1C) | 172.3 after 50 cycles | |||
| Sol–gel (calcination temp = 18 h) | 132.1 (at 1C) | 105.1 after 50 cycles | |||
| Sol–gel | LiNi0.33Mn0.33Co0.33O2 (NMC111) | 3.0–4.6 | 157 | — | 273 |
| Thermal destruction | 147 | — | |||
| Solid-phase | 172 at 1C/0.5C | 95.2 after 10 cycles | |||
| Hydrothermal | Li2FeTiO4 | 1.5–4.8 | 153.8 at C/10 | — | 274 |
| Template-free hydrothermal | Li2FeSiO4 hollow sphere | 1.5–4.8 | 152 at 0.05C | 110 after 100 cycles at 0.1C | 275 |
| Hydrothermal | LiMn2O4 | 3.2–4.35 | 121 at a current density of 1/10C | 111 mA h g−1 at 1/2C after 40th cycle | 276 |
| Urea-based hydrothermal | LiNi0.5Co0.2 Mn0.3 O2 | 3.0–4.3 | 158.6 after 1st cycle at 20 mA h g−1 | 92.6 after 50 cycles | 277 |
| 3.0–4.6 | 200 after 1st cycle at 20 mA h g−1 | 79.4 after 50 cycles | |||
| Hydrothermal | LiFePO4 | 2.0–4.5 | 167 at 0.1C after 1st cycle | 98 after 30 cycles | 278 |
| Microwave synthesis | LiFePO4/Graphene | 2.7–4.2 | 166.3 at 0.1C after 1st cycle | 99.5 after 10th cycle | 279 |
| Microwave-assisted hydrothermal | LiFePO4 | 2.5–4.2 | 152.1 at 0.1C after 1st cycle | ∼95 after 40th cycle | 280 |
| Microwave synthesis | LiFePO4/C | 2.5–4.0 | 150 at 0.1C) | — | 281 |
| Microwave synthesis | LiMn1.5Ni0.5 O4 Spinel | 3.5–4.8 | 130 at the 25th cycle | 100 between 10th and 50th cycle | 282 |
| Microwave-assisted solvothermal | Li2MnSiO4 | 2.0–4.5 | 250 at C/10 | — | 283 |
| Spray drying | LiNi0.5Mn1.5O4 | 3.0–4.8 | 134 mA h g−1 | 95 at 3.5C | 284 |
| Modified co-precipitation | Al-Doped LiNi0.5Co0.2Mn0.3O2 | 3.0–4.3 | 159.7 mA h g−1 at 0.5C for 1st cycle | 86.56 after 100 cycles | 285 |
| Co-precipitation | LiNi0.5Mn1.5O4 (half-cell) | 3.5–4.95 | 141 mA h g−1 at 1C after 200 cycles | 94 over 200 cycles at 1C | 286 |
| LiNi0.5Mn1.5O4 (full cell) | 141 mA h g−1 at 1C after 200 cycles | 92.4 over 200 cycles at 1C | |||
| LiNi0.5Mn1.5O4 (full cell at 55 °C | 133.2 mA h g−1 at 1C after 200 cycles | 93.3 over 100 cycles at 1C | |||
| Solvothermal | LiNi0.88Co0.09Al0.03O2 | 3.0–4.3 | 210.7 mA h g−1 at 0.1 after 1st cycle | 75.93 after 100 cycles at 55 °C (0.1C) | 80 |
| Co-precipitation | 203.2 mA h g−1 at 0.1 after 1st cycle | 63.31 after 100 cycles at 55 °C (0.1C) | |||
| Antisolvent precipitation | LiBO2 coated LiNi0.5Co0.2Mn0.3O2 | 2.5–4.6 | 200 mA h g−1 at 0.1C | 78.5 after 100th cycle | 287 |
| Template method | LiNi0.5Mn1.5O4 | 3.5–5.0 | ∼129 mA h g−1 at 1C | 96.6 after 100 cycles at 1C | 288 |
| Chloride co-precipitation | LiNi0.8Co0.1Mn0.1O2 | 2.7–4.3 | 184 mA h g−1 at 0.1C | 169 mA h g−1 after 30 cycles. | 289 |
3. Electrolytes for Li-ion batteries
Since the first breakthrough in LIBs by John Goodenough and their commercialization by the Sony Corp. in 1991, these batteries have attracted global attention owing to their high energy density. Prof. J. B. Goodenough said that “the cost, safety, energy density, rates of charge/discharge and cycle life are critical for battery-driven cars to be more widely adopted”. During battery operation, the simultaneous movement of ions and electrons occurs. Ions flow through the electrolyte, while electrons are generated at the anode (negative electrode) and flow towards the cathode (positive electrode) via an external circuit. The electrode accommodates charge storage in the stacking layers, while the electrolyte acts as a carpet for the ions. The capacity of the battery depends on the rate of Li+ migration to and from via the spacer between electrodes. Despite the effective role of the electrode in the stability and safety of batteries, the electrolyte is also a key component and must be chosen carefully because of its dual role. A separator is placed between electrodes, which prevents short-circuit and provides a medium for ion migration.290,291 The electrolyte, together with the separator, must fulfill essential requirement. Table 15 provides a glimpse into the important electrolyte parameters for developing safe and efficient LIB separators.292| Parameter | Requirement |
|---|---|
| Chemical and electrochemical stability | Stable for an extended period |
| Wettability | Wet out quickly and completely |
| Mechanical property | >1000 kg cm−1 (98.06 MPa) |
| Thickness | 20–25 μm |
| Pore size | <1 μm |
| Porosity | 40–60% |
| Permeability (Gurley) | <0.025 s μm−1 |
| Dimensional stability | No curl up and lay flat |
| Thermal stability | <5% shrinkage after 60 min at 90 °C |
| Shutdown | Effectively shut down the battery at elevated temperatures |
The existing batteries are based on a liquid electrolyte; however, it threatens the safety of batteries due to the possibility of cell explosion as a result of side reactions. The leakage of the electrolyte may degrade the electrodes, causing capacity fading due to the loss of the active materials. This leads to the loss of sufficient ion storage sites in the electrode, low capacity, and energy density, resulting in a degradation of the battery performance. During the rapid charging–discharging process, the dendrites can grow at the anode and pass through the liquid electrolyte easily, which short-circuit the battery and explosion may also occur.
Unwanted chemical reactions between the liquid electrolyte and electrode lead to the release of gases inside, and pressure build-up occurs when the battery fails to accommodate the volume changes. The other main requirements for advanced batteries are the lower cost and weight. The current LIBs use both an electrolyte and separator, which affect their cost and weight. Therefore, these are some serious issues in the current battery systems that need to be resolved. They can be fixed by replacing the liquid electrolyte with solid electrolytes. Solid-state electrolytes (SSEs) are the main components in ASSLBs.293,294 The recent progress in inorganic SSE systems mainly including oxide SSEs, sulfide SSEs, and halide SSEs has been reported by many researchers.295–298 The ionic conductivity of the typical inorganic solid-state electrolyte is required ∼10−3 S cm−1 at room temperature, which is very close to the ionic conductivity level of liquid electrolytes.299 The poor performance was observed due to high interfacial impedance caused by the instability between the sulfide solid electrolyte and oxide cathode during the charge–discharge process. To overcome the interfacial impedance, the LiNbO3-coated NCM811 cathode was reported to exhibit significantly improved electrochemical performances at 35 °C and 60 °C in contrast to the bare cathode. Especially at 60 °C, the LiNbO3-coated NCM811 cathode displayed a discharge capacity of 203 mA h g−1 at 0.1C and a rate performance of 136.8 mA h g−1 at 5C, which are much higher than that for the reported oxide electrodes in ASSLIBs using sulfide solid electrolyte.300
The fundamental benefits of solid electrolytes are as follows: (i) better thermal and mechanical stability, (ii) better cell packaging with high pressure, (iii) no possibility of side reactions due to solid nature, (iv) better interfacial contact and prevention of dendrite growth, (v) low cost due to dual role (electrolyte and separator), and (vi) higher safety and broad temperature range of operation.291,301 For an ideal electrolyte, high ionic and negligible electronic conductivity are favorable.302 Ionic conductivity is linked to the number of free cations and electrolyte viscosity.303 Thus, a high number of free charge carriers and low viscosity of the electrolyte are specific requirements. Furthermore, the voltage stability, thermal stability, and mechanical stability determine the overall safety of the battery operation. Fig. 9 shows the characteristic parameters of the electrolyte (crystallinity, packaging, ion transport number, interfacial contacts, broad temperature range, voltage stability window, conductivity, salt dissociations, inert towards electrodes, and glass transition temperature) that influence the ion dynamics, capacity, and energy density of the battery.
 | ||
| Fig. 9 Characteristic properties of crystallinity, packaging, ion transport numbers, interfacial contacts, broad temperature range, voltage stability window, conductivity, salt dissociations, inert towards electrodes, and glass transition temperature for electrolytes of LIBs. | ||
For the synthesis of new polymer electrolytes, the involved salts play a crucial role. The parameters of the salt are the main deciding factors for the performance of electrodes, and thus it should be carefully selected. The ion dynamics are linked with the anion size, anion mobility, molecular weight, ion conductivity, donor number, thermal stability, toxicity, dissociation constant, and lattice energy of the salt.304Fig. 10 shows the possible structures of some of the dominant lithium salts in the R&D sector, and their key properties, that is, ionic conductivity, molecular weight, ion mobility, dissociation constant, and donor numbers are compared. Table 16 summarises the lithium salts according to their anion size and main characteristics (ionic conductivity, molecular weight, ion mobility, dissociation constant, and donor numbers), which affects the electrolyte conductivity.305
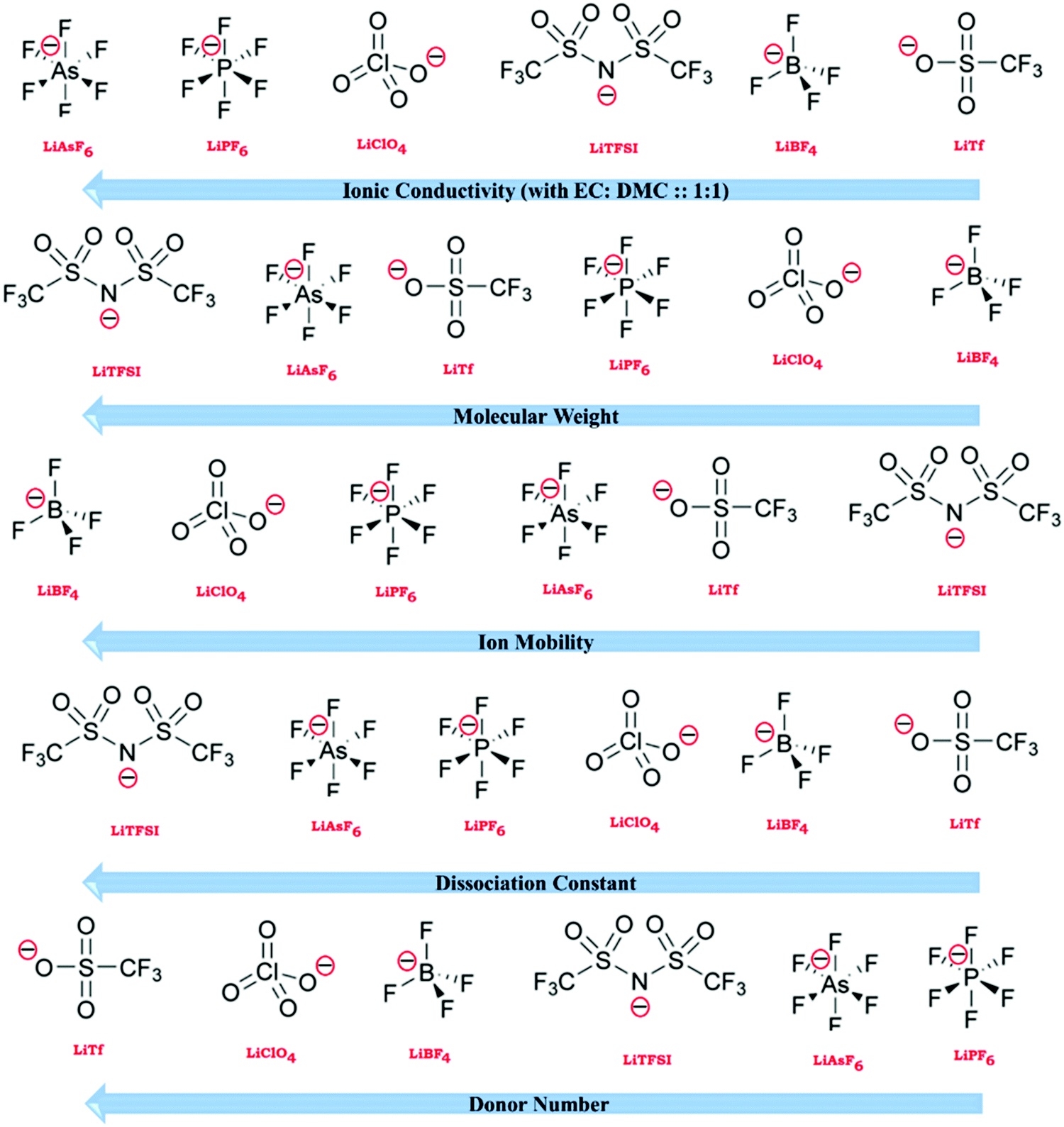 | ||
| Fig. 10 Comparison of the different characteristics of lithium salt (ionic conductivity, molecular weight, ion mobility, dissociation constant, and donor numbers), reproduced with permission from Springer.305 | ||
| Lithium salt | Anion | Main characteristics |
|---|---|---|
| LiClO4 |
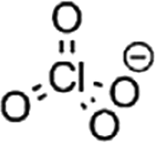
|
• Broad electrochemical stability window |
| • Low solubility in commonly used carbonate-type solvents | ||
| LiBF4 |
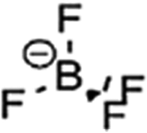
|
• Broad electrochemical stability window |
| • Low solubility in commonly used carbonate-type solvents | ||
| LiPF6 |
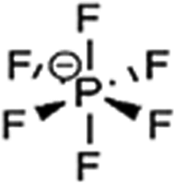
|
• High ionic conductivity, favors SEI formation, passivates Al substrate at the cathode side |
| • Decomposes in the presence of moisture and reacts with electrolytes at elevated temperatures, resulting in the formation of HF | ||
| LiFSI |
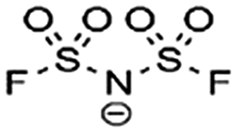
|
• Higher ionic conductivity compared to LiTFSI and high electrochemical stability |
| • Unable to form passivation layers on Al current collectors (in the presence of LiCl) But purified LiCl free salt passivates Al collectors | ||
| LiBETI |
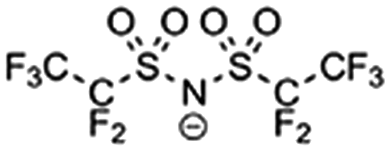
|
• High solubility and high ionic conductivity and high electrochemical stability |
| • Unable to form passivation layers on Al current collectors | ||
| LiBOB |
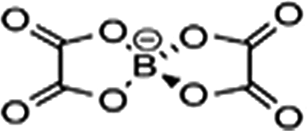
|
• High electrochemical stability and long-term stability |
| • Form highly resistive SEI-films (low conductivity in comparison to LiPF6 and LiTFSI) | ||
| LiDFOB |
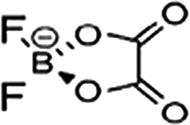
|
• High electrochemical stability and cycling behavior, Able to form a passivation layer on Al current collectors |
| • Lower solubility in carbonate type solvents compared to LiTFSI and LiPF6, but higher than LiBOB | ||
| LiTFSI |
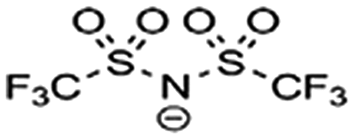
|
• High solubility and high ionic conductivity and high electrochemical stability |
| • Unable to form a passivation layer on Al current collectors (Al-degradation and corrosion) |
4. Major challenges associated with battery safety
The electrolyte used in commercial LIBs is an organic electrolyte, which threatens the safety of the battery during charging–discharging. Because of the poor safety of batteries with organic electrolytes, a high performance cannot be achieved (long cycle life and high-power density), where safety is one of the prime requirements for battery manufacturers. Due to major safety concerns, many challenges have been considered in the energy storage sector. Two important reasons that threaten the safety of LIBs are thermal runaway and dendrite growth, which both can cause battery explosion due to fire and short-circuit.307 Therefore, liquid electrolytes should be replaced with solid electrolytes for the safe operation of batteries, particularly for portable electronic devices and electric vehicles. Many accessories and precautions have been considered as a priority in the production of batteries to prevent heat generation and short-circuiting. Nevertheless, a feasible and optimized solution can enhance their safety by eliminating the inherent issues faced by the electrode, electrolyte, and interfacial layers.4.1. Thermal runaway issues
Thermal runaway (TR) indicates uncontrolled reactions inside the battery. Thermal runaway is initiated when the heat generated inside the battery cannot be compensated by heat loss to the environment. Because of the heat generated during operation, the battery catches fire and may explode, which can be avoided by replacing the liquid electrolyte with solid electrolytes. In brief, TR disturbs the physical and chemical properties of the materials used in batteries. Various fire incidents associated with LIBs have been reported in portable electronic products and electric vehicles worldwide. When they were analyzed in detail, four common reasons were observed, as follows: (i) overheating and overcharging, (ii) short-circuit due to self-ignition or mechanical damage, (iii) failure of the battery due to handling management, and (iv) pressure build-up due to the release of gas after degradation of the electrolyte.308,309Fig. 11 shows the main causes of LIB damage or battery explosions due to reasons such as deformation, separator tearing, dendrite growth, and increase in temperature.310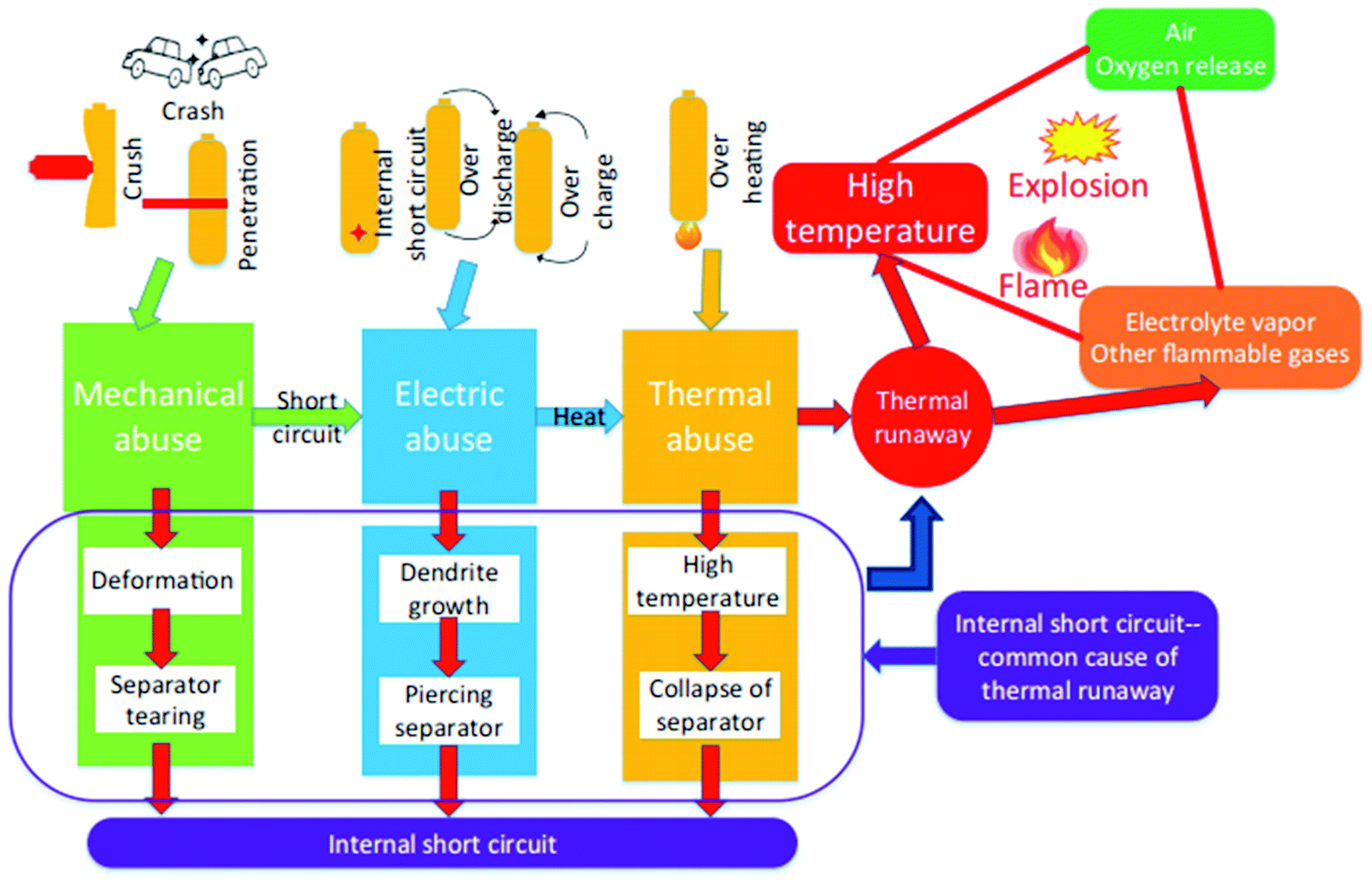 | ||
| Fig. 11 Schematic illustration of LIB fire accidents. | ||
Fig. 12(a) shows the role of temperature and its effect on the battery operation for safety concerns. With an increase in the temperature of the battery due to overcharging, overheating, or external impact during its operation, the active material of the electrolyte starts decomposing. This activity occurs in various side reactions of complex reaction mechanisms, which damage the battery. The cathode, anode, and electrolyte reactions lead to the decomposition of the SEI layer and electrolyte breakdown occurs with the release of harmful species (LiPF6 → LiF + PF5). The release of oxygen, heat, and dendrite formation lead to thermal runaway.311
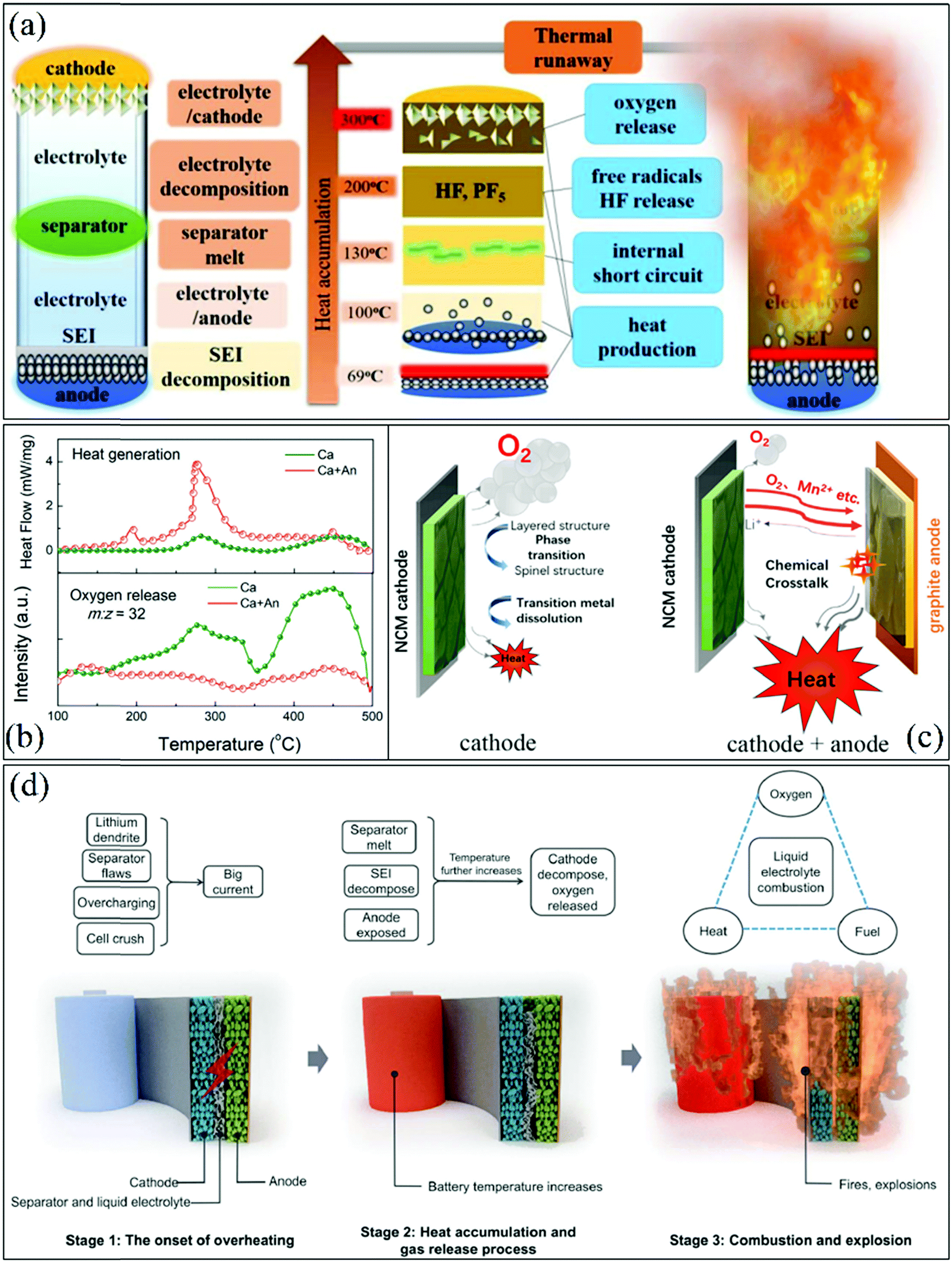 | ||
| Fig. 12 (a) Schematic of thermal runaway stages of lithium-ion batteries: chemical crosstalk between the charged cathode and anode. (b) Charged cathode alone exhibits a strong oxygen release peak, while the mixture of cathode/anode releases virtually no oxygen but has sharp heat generation enhancement at the same temperature range. (c) Illustration of proposed chemical crosstalk process between the cathode and anode. (d) Three stages for the thermal runaway process. Stage 1: The onset of overheating. The batteries change from a normal to an abnormal state and the internal temperature starts to increase. Stage 2: Heat accumulation and gas release process. The internal temperature quickly rises and the battery undergoes exothermal reactions. Stage 3: Combustion and explosion. The flammable electrolyte combusts, leading to fires and explosions.309 Reproduced with permission from AAAS Science.309 | ||
Although internal short-circuiting is one of reasons for TR, sometimes TR may occur without short-circuiting. The safety of the battery depends not only on the individual electrodes and electrolyte, but also on the overall properties of the cell components. The chemical cross-talk between the cathode and anode may lead to the TR mechanism. Recently, Liu et al.312 studied in detail large pouch cell batteries, where a polyethylene terephthalate (PET)-based ceramic separator was used to prevent short-circuiting of the battery. It was observed from DSC, heat generation, and the MS oxygen gas (m/z = 32) characterization versus temperature plot that the phase transition is linked to the generation of heat and release of oxygen. This released oxygen (at 276 °C) can diffuse through the separator and react with the reactive anode.
Fig. 12(b) shows the absence of any oxygen peak, which indicates that the anode consumed it, as also evidenced by the weight loss comparison. The cathode showed a larger (2.8%) weight loss than the cathode/anode mixture (0.7%). The exothermic reactions were larger for the cathode/anode mixture (770 J g−1) than the individual cathode (108 J g−1). This chemical cross-talk between the cathode and anode is shown in Fig. 12(c). The individual cathode releases oxygen, which leads to the initial generation of heat. During the device operation (charging–discharging), the heat is generated rapidly, and oxygen reaction leads to TR. The authors also confirmed that the TR could not be stopped by purging with liquid nitrogen. The liquid nitrogen failed to stop the TR because of oxygen was supplied from inside the cathode of the battery. Therefore, a solution to overcome this issue seems to be improving the battery thermal management system. The safety of the device needs to be the priority together with its performance.
Fig. 12(d) shows the battery explosion due to a thermal runaway during operation.309 In stage-1, the main reasons for initial heat generation are battery crash, dendrite growth, overcharging, and internal short-circuiting. In stage-2, the battery temperature increases due to the accumulation of heat, which results in the decomposition of SEI; hence it releases gases from electrode–electrolyte reactions where the separator starts melting. The melting of the separator leads to the short-circuiting of the battery, causing the breakdown of the active materials. In stage-3, the liquid electrolyte is present in the battery, resulting in the explosion of the battery or permanent shutdown. Simultaneously, the battery releases some gases due to internal pressure, which is a favorable condition for explosions. The electrolyte is a component that separates the electrodes, which prevents their interaction and plays an effective role in preventing the short-circuiting of the battery. The most feasible alternative electrolyte that has potential to eliminate the threat of thermal runaway is the solid electrolyte. The solid nature of electrolytes will automatically enhance the safety and prevent side reactions and there is no tendency to leak electrolytes.313
4.2 Dendrite growth: challenges and remedies
Together with the TR threat, the dendrite growth affects the safety of the battery, which diminishes the cyclic stability and restricts the operation or lifetime of the battery. Dendrites are generally rigid tree-like structures with needle-like projections (called whiskers) that grow at the anode. The growth of dendrite structures at the anode penetrates through the separator and reaches the cathode during the cell operation. Therefore, the specific capacity deteriorates and causes short-circuiting of the battery, and finally damages the device and shortens its life span. When increasing the miniaturization and compactness of devices, the growth of dendrite structures is one of the significant threats that need to be eliminated for developing high energy density and long life in the battery.Recently, Zhao et al.314 proposed the concept of ions redistribution to suppress the dendrite growth. The separator is an insulating layer through which Li-ions migrate/penetrate. These Li-ions accumulate on the anode surface through the pores of the separator. In the absence of distributed ions, the anode surface was faced with the separator skeleton. The Li-ion redistributor method is regulated to avoid the accumulation of ions on the anode electrode, where dendrite growth formation occurs (Fig. 13iA and B). A commercially available separator, named polypropylene (PP), was coated on Al-doped LLZTO (Al-doped Li6.75La3Zr1.75Ta0.25O12), which regulated the ion diffusion owing to the presence of inherent 3D conduction channels. The coated electrode enhanced the mechanical strength and suppressed the formation of dendrites even with liquid electrolytes (Fig. 13ii). Considering the safety issue of batteries, Zhao et al.315 proposed a flexible anion-immobilized ceramic–polymer composite electrolyte, that is, polyethylene oxide (PEO) and lithium bis(trifluoromethylsulfonyl)imide (LiTFSI), for the application of Al-doped LLZTO. Fig. 13(ii(A) and (B)) demonstrates a potential electrolyte that quenched the formation of dendrite structures, which is attributed to the rigid nature of uniform ion distribution (due to the effective immobilization of anions). The demonstrated electrolyte was stable up to 5.5 V and used to fabricate batteries, achieving a specific capacity of 150 mA h g−1. The internal health of the battery can provide a hint about the battery explosion, which can be prevented. D’innocenzo et al.316 developed a smart battery by changing the separator with a bifunctional separator (polymer–metal (Cu)–polymer triple layer configuration). This separator physically isolated the electrodes and reduced the voltage (Fig. 13iii-a and b). However, this type of separator did not stop the growth of dendrites, where the growth of dendrites increase with the time of cell operation, finally reaching the cathode.
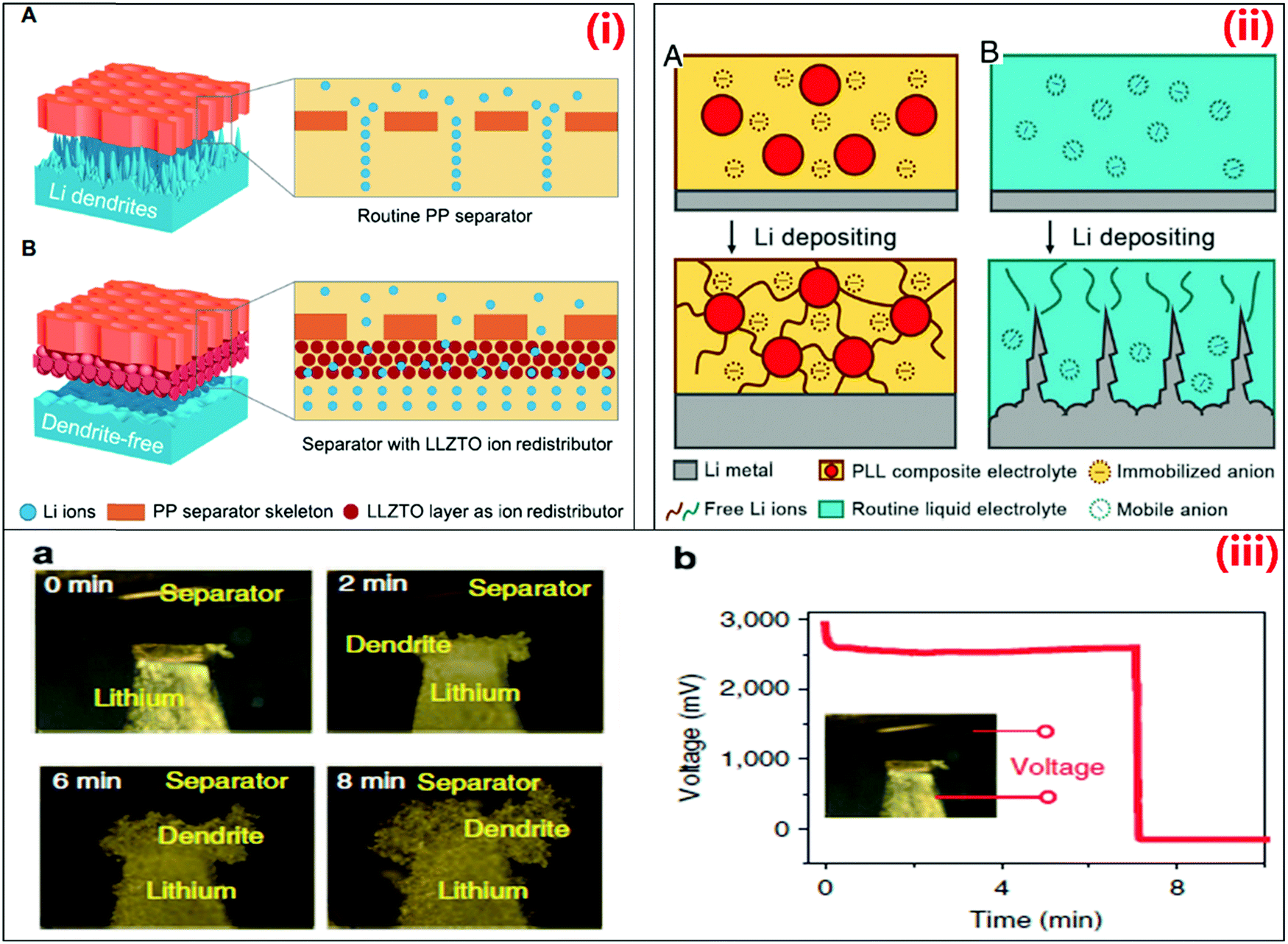 | ||
| Fig. 13 (i) Schematic illustration of the electrochemical deposition behaviors of the Li metal anodes using (A) routine PP separator and (B) composite separator with the LLZTO layer as an ion redistributor for uniform Li-ion distribution. (ii) Schematic of the electrochemical deposition behavior of the Li metal anode with (A) PLL solid electrolyte with immobilized anions and (B) routine liquid electrolyte with mobile anions.315 (iii) In situ observation of dendrite growth on lithium electrode. (a) Lithium anode and separator-wrapped lithium counter electrode with copper conductive layer facing the lithium anode housed in a glass cell for in situ optical microscopy observation. During charging of the cell, non-uniform deposition of lithium on the lithium electrode leads to mossy dendrite formation and growth on the surface. (b) Voltage profile of the device. The lithium dendrites contact the conductive copper layer on the separator within about 6–8 min, giving rise to a 3 V drop in VCu-Li, given that the potential difference between copper and lithium is dissipated on contact.316 Reproduced with permission from Nature Publishing Group.316 | ||
To suppress the growth of dendrites on the Li-metal anode, generally the coating of a polymer and ceramic on the anode is performed, where the representative investigations are presented in Fig. 14(a) and (b). This strategy enabled the control of dendrite growth, but the low ionic conductivity of the polymer and poor interfacial contact remain a considerable drawback to the overall cell performance.317 Thus, it becomes essential to prepare a suitable layer that provides faster ion diffusion and restricts the growth of dendrites. Xu et al.318 prepared an artificial protective layer (APL) based on PVDF-HFP and LiF on the Li metal anode to suppress the growth of dendrites. Fig. 14(c) shows the uniform Li deposition on the soft polymer matrix. A full cell was fabricated using LFP as the cathode, APL-modified Li as the anode, and carbonate electrolyte of 1.0 M lithium hexafluorophosphate (LiPF6)-ethylene carbonate/diethyl carbonate (v/v = 1:1). Initially, both cells (modified and unmodified Li anode) showed a capacity of 150.6 mA h g−1 with a Coulombic efficiency of >99%. After 50 cycles, the APL-modified cell demonstrated good cyclic stability up to 250 cycles with 80% capacity retention (Fig. 14d and e). In brief, the modified Li anode-based cells exhibited a 2.5-times longer cycle life than the unmodified anode. This approach may be very useful for the liquid electrolyte and a solid electrolyte, which can be adopted for the future design of Li-ion batteries.
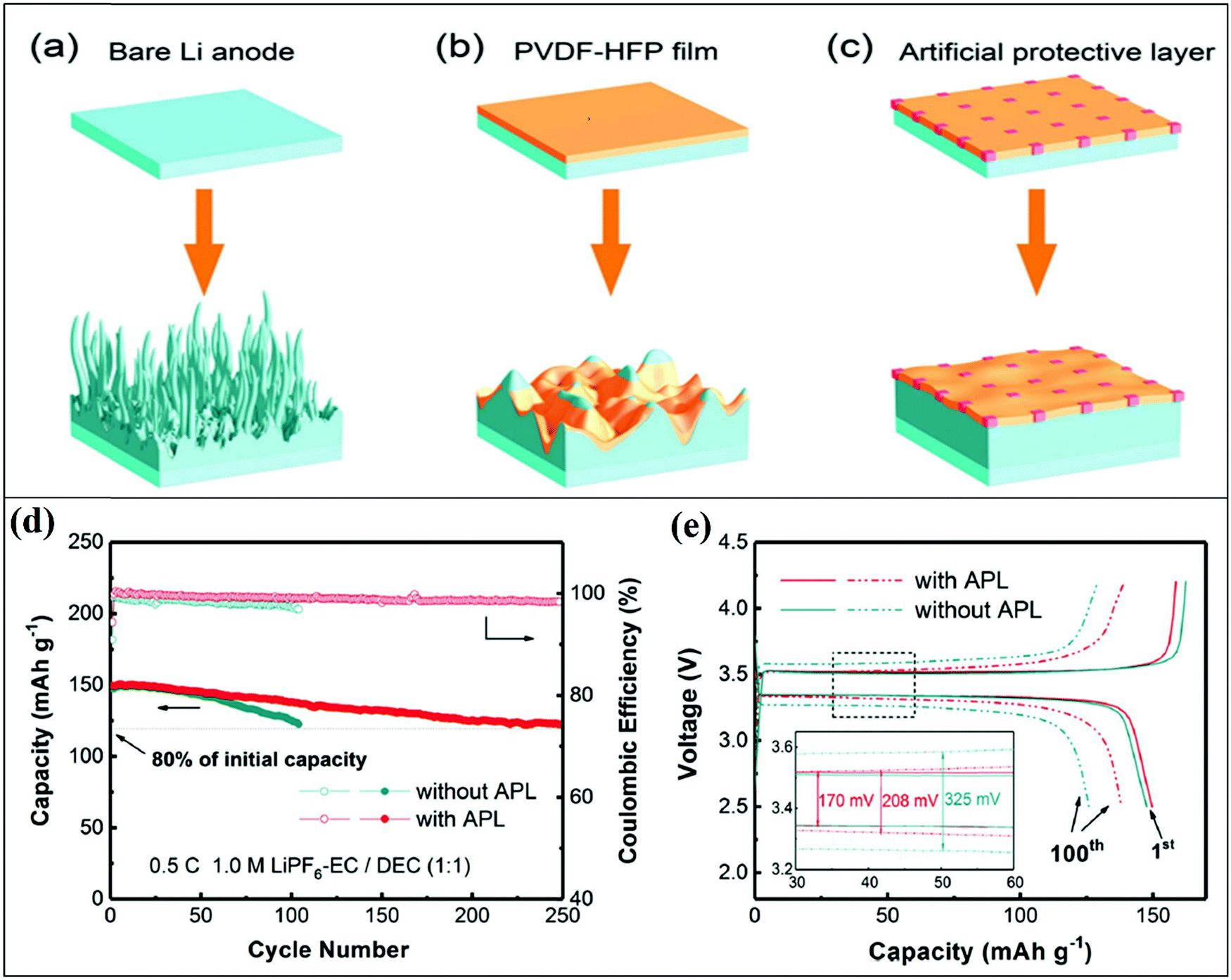 | ||
| Fig. 14 Schematic illustrations of Li deposition: (a) without protection, lithium metal dendrites, and dead. Li forms after cycling. (b) Pure PVDF-HFP layer with poor mechanical modulus, where interfacial fluctuation with dendrites piercing the PVDF-HFP layer occurs after cycling. (c) With APL composed of organic PVDF-HFP and inorganic LiF, which is conformal and mechanically strong to suppress Li dendrite penetration and stabilize Li metal surface, and electrochemical performance of Li|LFP cells and morphology of cycled Li metal anodes. (d) Long-term cycling performance at 0.5C. (e) Galvanostatic charge–discharge profiles at the 1st and 100th cycle.318 Reproduced with permission from Wiley-VCH.318 | ||
Besides the above-discussed strategies, some other strategies to moderate electrolytes include additive electrolyte, nanostructured electrolyte, solid electrolyte, and membrane modification.101 However, although these strategies effectively suppress dendrite growth, there are some negative effects that restrict their use on a large scale. Thus, the best strategy may be the optimization of these factors to develop efficient and long life span batteries. Table 17 shows a comparison of the proposed methods to suppress Li dendrite growth. It seems that the use of solid electrolytes has the potential to suppress the dendrite issue, whereas the limit of ionic conductivity is one of the major drawbacks. Accordingly, the scientific community has focused on enhancing the ionic conductivity of solid electrolytes and developing commercial liquid electrolytes. In the next section, we explore the possible use of solid electrolytes for Li-ion batteries and how this has opened new doors for developing high-power density and long life span innovative batteries, i.e., all-solid-state Li-ion batteries (ASSLIBs).
| Advantage | Disadvantage | |
|---|---|---|
| Electrolyte additive | (a) Facile operation | (a) Poor mechanical strength to suppress dendrite growth |
| (b) Forming a thin and highly conductive SEI | (b) Poor long-term stability during cycling | |
| Super-concentrated | High Coulombic efficiency and cycling stability | (a) High price |
| Electrolyte | (b) Limited rate performance | |
| Nanostructured electrolyte | High ionic conductivity | Complicated fabricating process |
| Solid-state electrolyte | (a) Without electrolyte leak | Low ionic conductivity |
| (b) Suppressing dendrite growth | ||
| Structured anode | Suppressing Li dendrite growth | Low Coulombic efficiency |
| Membrane | (a) Stopping dendrites to the cathode | Less effect on the dendrite |
| Modification | (b) Detecting the dendrite growth | Nucleation and growth |
4.3 Electrode/electrolyte interface (EEI) engineering
LIBs have various components that play a crucial role in deciding their performance. One of these crucial LIB components is recognized to be the electrode/electrolyte interface (EEI). The EEI is formed due to the decomposition of electrolytes and the electrode–electrolyte interactions, where (i) the interface generated on the negative electrode is the “solid electrolyte interphase” (SEI) and (ii) the interface generated on the positive electrode is the “cathode electrolyte interphase” (CEI). Generally, the effect of the cathode interface is smaller compared to that of the SEI. Two important challenges linked with the EEI are interfacial contact and chemical compatibility. In the EEI region, various phenomena occur such as charge transfer reactions, electrolyte decomposition, and electrode (cathode and anode) degradation. Thus, it becomes important to modify or tune the EEI for achieving optimum battery performances (high energy density, long cycle life, etc.). The EEI can be altered by varying the synthesis methods and engineering of the material. The quality of the EEI gives an idea about the safety and operation of the battery. An optimum EEI can be formed via three approaches, as follows: (i) minimizing the phase in the fabrication of the battery, (ii) enhancing the contact area by reducing the particle size and mixing the electrode and electrolyte, and (iii) addition of a buffer layer to enhance the chemical compatibility.320 The solid electrolyte interface (SEI) is an insulating film that covers the electrode surface to hinder the side reactions. Some key characteristics of the SEI film are, as follows: (1) high ionic conductance for ease of Li migration via the SEI, (2) stable morphology and chemical structure, (3) robust binding properties with active substances, (4) superior mechanical performance buffering volume expansion, and (5) superior electrochemical and thermal stability.321Fig. 15 displays the strategy to tune the interface in Li-ion battery by altering its structure. | ||
| Fig. 15 Schematic diagram of surface-interface modification strategies and classification in organic liquid electrolytes for LIBs and map of this review (Fig. 2 from ref. 321). | ||
Various fabrication techniques can be used to modify the surface of electrodes by creating a high-quality artificial buffer layer on the surface of SSE or/and electrode materials. The techniques classified based on this approach are (i) top-down approaches, including magnetron sputtering, spark plasma sintering, electron-beam evaporation, and pulsed laser deposition and (ii) bottom-up approaches, including sol–gel-derived synthesis, atomic layer deposition, chemical vapor deposition, and electrochemical-assisted synthesis.
Fig. 16(i) displays a typical schematic illustration of the SSB components and the interfacial challenges. Fig. 16(ii) shows the three types of interfaces (interphases), as follows: (i) intrinsically stable interface, where the solid-state electrolyte (SSE) is nonreactive with the Li metal and a distinct two-dimensional interface is formed (Fig. 16(ii)a) and (ii) solid-electrolyte interphase (SEI), with poor electron conductivity and desirable Li-ion conductivity (Fig. 16(ii)b). A perfectly stable interphase due to a self-limiting reaction between the SEI and Li effectively blocks electron transport. (iii) Mixed-conducting interphase, where the electronic conductivity is higher than the SSE (Fig. 16(ii)c).322
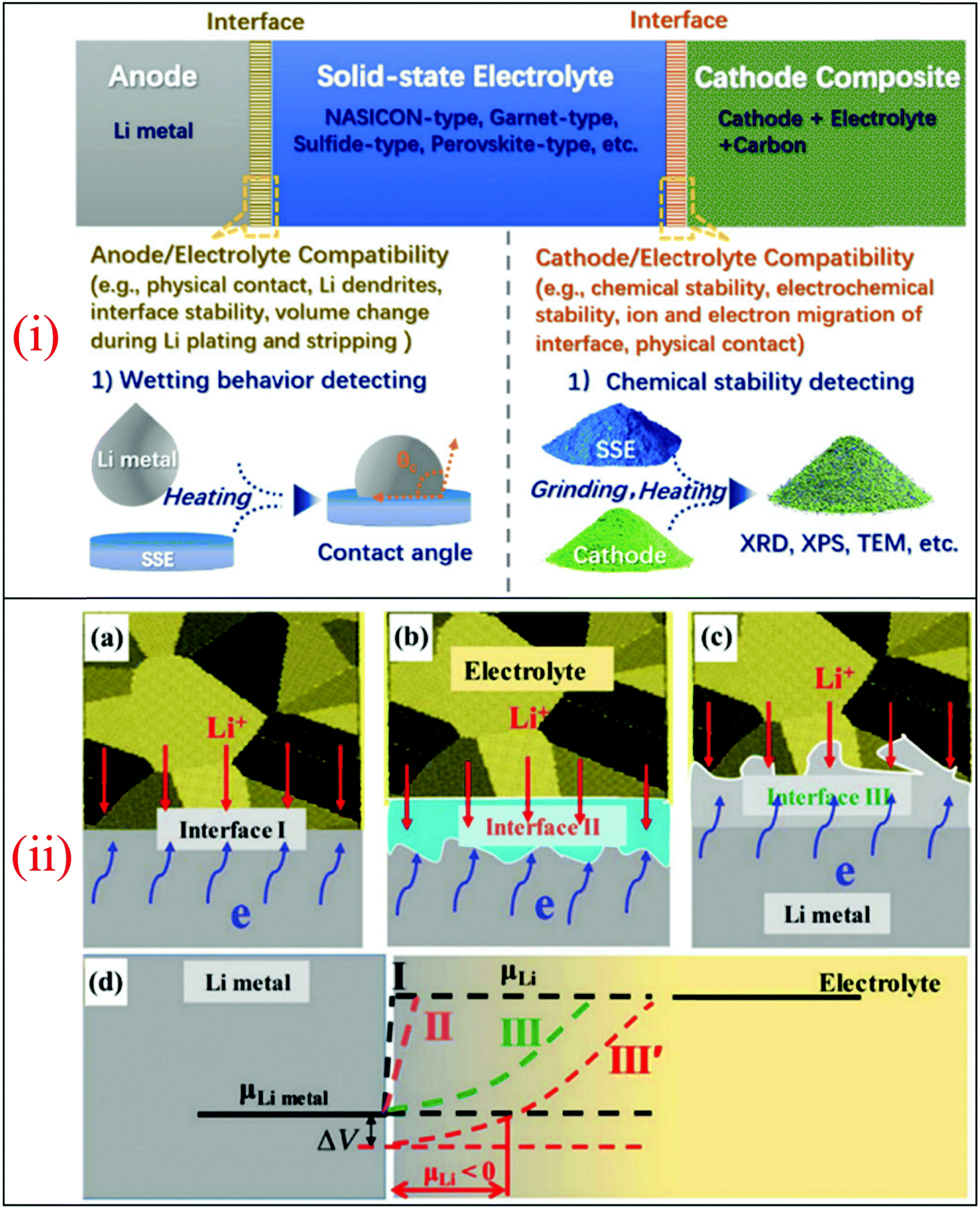 | ||
| Fig. 16 (i) Summary of compatibility problems associated with interfaces in SSBs (Fig. 1 from ref. 322). (ii) Formation of different types of Li/SSE interphases. (a) Intrinsically stable interface between the Li metal and the SSE. (b) Metastable solid-electrolyte interphase between the Li metal and the SSE. (c) Reactive mixed-conducting interphase between the Li metal and the SSE. (d) Corresponding Li potentials between the Li metal and different SSEs.322 | ||
Fig. 16(ii)d shows the potential drop from the SSE potential to Li metal at the interfaces for the first two types of SSEs, whereas the partial potential region of the third type of interphase drops below the potential of Li deposition. This drop indicates the growth of Li dendrites in the third type of interphase. The growth of dendrites is also attributed to the overpotentials during Li plating and the high electronic conductivity of SSEs. The high conductivity reduces the potential in the electron-conductive interphase (III’ curve). By introducing artificial buffer layers (ABLs), nonreactive/reactive interface with an Li-stable interphase can be created.323 The formation of an interface via this approach (between solid electrolyte and electrodes) provides enhanced chemical/electrochemical stabilities. By adding a compound with a special structure in the electrolyte, the properties of SEI can be tuned. Hogstrom et al. reported an increase in the irreversible capacity with the addition of an organic film-forming additive, propargyl methanesulfonate (PMS), and LiPF6 in EC/DEC electrolyte. This was attributed to the better thickness of the SEI film.324
Zheng et al.325 used tris(pentafluorophenyl)borane (TPFPB), a boron-based anion receptor, to decrease the side products on the cathode surface. The high coordination ability and high oxygen solubility of TPFPB restricted the electrolyte decomposition and enhanced the stability of the electrode–electrolyte interface. The surface chemistry of the electrode determines the SEI, which can be monitored by coating the electrode surface to form a stable structure.326 Initially, mostly metal oxides (Al2O3, TiO2, and Co3O4) are used as a coating material, which play two key roles, as follows: (i) preventing electrode decomposition by reacting with the acid species present in the electrolyte and (ii) preventing direct contact between the two electrodes.327–329
The metal oxide coating affects the conductivity, and thus an alternative strategy is to use a lithium-ion conductive-material such as Li3PO4330 and Li0.1B0.967PO4 (LBPO).331 Another strategy is to deposit a thin film via physical vapor deposition (PVD), chemical vapor deposition (CVD), molecular beam epitaxy (MBE), spin coating, and atomic layer deposition (ALD).332,333 Coating carbon on the electrode surface has also been shown to be efficient due to three actions, as follows: (i) enhancing the electrochemical performance, (ii) hindering the agglomeration of the electrode material for optimum use of the sites in the active material, and (iii) acting as a buffer layer to relieve the stress due to volume changes.334,335
5. Overview of all-solid-state Li-ion batteries (ASSLIBs)
The optimization of the architecture is one of the key points for all-solid-state Li-ion batteries (ASSLIBs). ASSLIBs have an inherent property for safety owing to the absence of flammable electrolytes. Further, the energy density and cycle stability are superior to the commercial LIBs.336 Heavy packaging makes batteries safer for commercial applications; however, it decreases their energy density. Fig. 17(i) and (ii)(a,b) show the key differences between the commercial LIBs and ASSLIBs in terms of their architecture. In ASSLIBs, a separator is not required, thus automatically opening the door for device miniaturization and weight/price reduction. The use of solid electrolytes eliminates all the barriers faced by commercial batteries such as dendrite growth (cause short-circuit), poor thermal stability, and poor safety. Another attractive feature of ASSLIBs is that they can be used as an anode with solid electrolytes. In commercial batteries, the liquid electrolyte interconnects all battery cell components with a parallel connection inside the cell stack (Fig. 17(ii)(c)). However, in all-solid-state batteries (ASSBs), the electrolyte is confined inside the galvanic cells and they enable bipolar stacking with single cells connected in series by a lithium-ion isolating layer.337 The bipolar stacking decreases the number of current collectors and increases the voltage of the battery cell (Fig. 17(ii)(d)). Also, the absence of a flammable electrolyte solvent in ASSBs eliminates the need for any type of cooling elements, as shown in Fig. 17(i).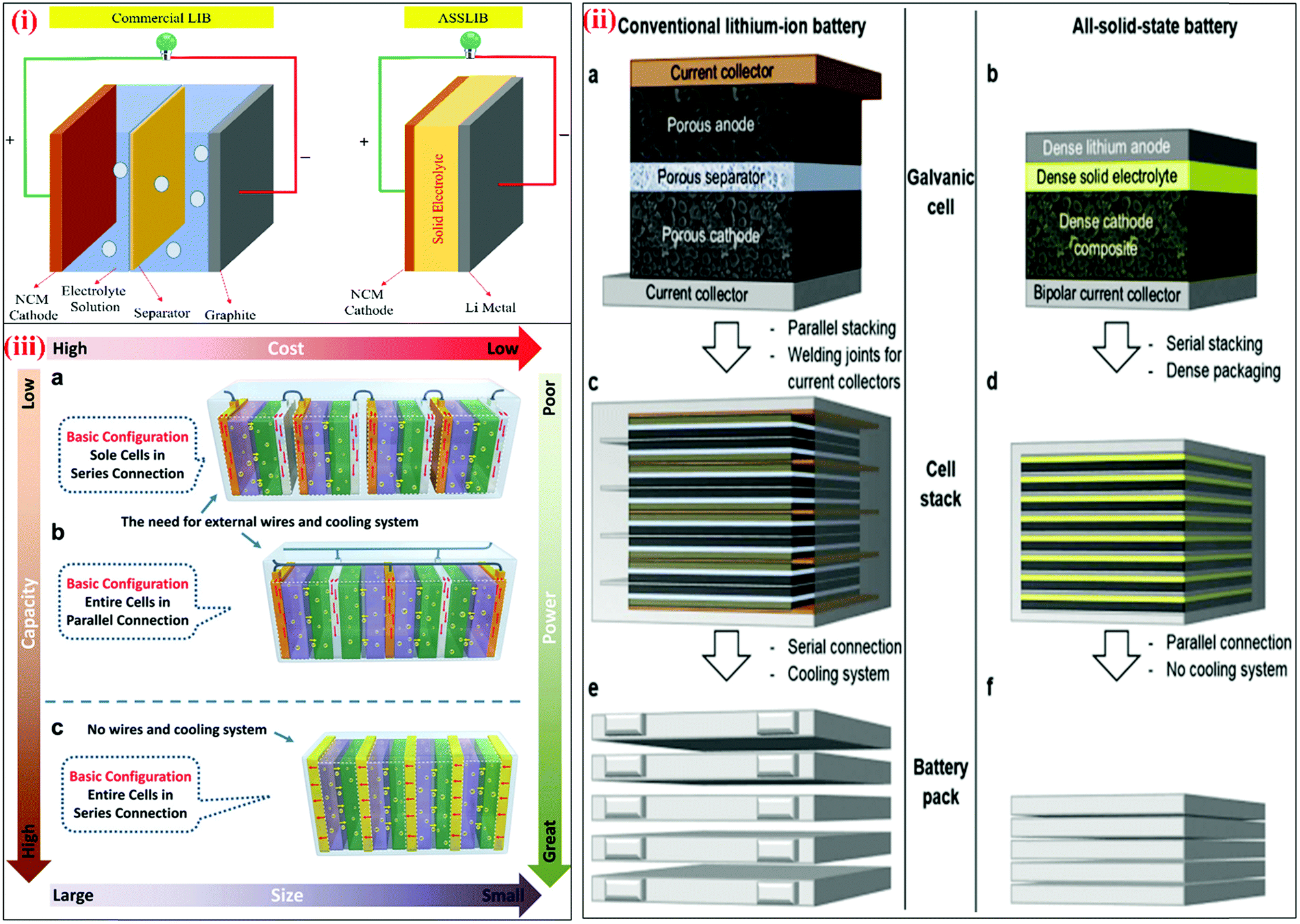 | ||
| Fig. 17 (i) Architectural comparison of commercial LIB and all-solid-state LIB. (ii) Comparison of conventional lithium-ion battery and all-solid-state lithium battery at the cell, stack, and pack levels with potentials for increased energy density. (iii) Schematic illustration of the stack configuration in rechargeable batteries: (a) SEs, (b) MEs, and (c) BEs. The direction and intensity of the red arrow represent the discharging current.337 Reproduced with permission from Wiley-VCH.337 | ||
The commercial advancement of fabricated LIBs depends on the optimization of their performance parameters. The electrochemical performance of the battery is indicated in terms of internal resistance, specific capacity, efficiency, capacity retention, and open-circuit voltage. Additionally, the measurement conditions (environment temperature and state of charge/discharge) need to be considered during electrochemical testing. Depending on the energy density demand, the capacity, material loading, thickness, and electrolyte uptake can be tuned. Table 18A summarizes the various performance parameters (top: Ionic conductivity, mechanical strength, interfacial functionality, safety, advantages and disadvantage and bottom: liquid, gel, SSP, and ceramic electrolytes) and information that can be extracted from any LIB, that is, measuring unit and formula and information (Table 18B).
| Electrolyte | Ionic conductivity | Mechanical strength | Price | Interfacial functionality | Safety |
|---|---|---|---|---|---|
| Liquid | 1 | 4 (needs separator) | 3 | 1 | 4 |
| Gel | 2 | 3 | 2 | 2 | 3 |
| Polymer | 4 | 2 | 1 | 3 | 2 |
| Ceramic | 3 | 1 | 4 | 4 | 1 |
| Classification | Advantages | Disadvantages |
|---|---|---|
| Liquid | High ionic conductivity | Poor thermal stability |
| Low interfacial impedance | Severe LiPS shuttling | |
| Gel | High ionic conductivity | Poor thermal stability |
| Low interfacial impedance | Low mechanical strength | |
| Suppressing LiPS shuttling | ||
| Solid-state polymer | Low interfacial impedance | Low ionic conductivity |
| Suppressing LiPS shuttling | Low mechanical strength | |
| Good thermal stability | ||
| Ceramic | High ionic conductivity | High interfacial impedance |
| Excellent thermal stability | Poor processability | |
| Preventing LiPS shuttling | ||
| Suppressing Li dendrite growth | ||
| Polymer/ceramic composite | Low interfacial impedance | Low ionic conductivity |
| Good thermal stability | ||
| Suppressing LiPS shuttling | ||
| Suppressing Li dendrite growth |
| Parameters | Measuring unit | Measuring formula | Information |
|---|---|---|---|
| Operating voltage | Volts (V) | Instrumental | Energy density and safety |
| Current density | mA g−1 | Instrumental | For testing rate capabilities |
| Theoretical capacity | mA h g−1 |
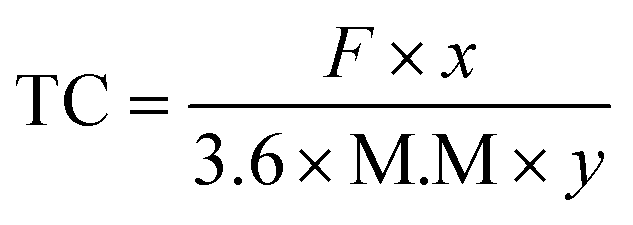
|
Lithium ion storage capability |
| Gravimetric capacity | mA h g−1 |
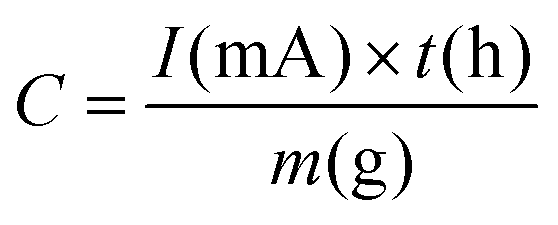
|
Li+ storage capability measured per unit mass |
| Areal capacity | mA h cm−2 |
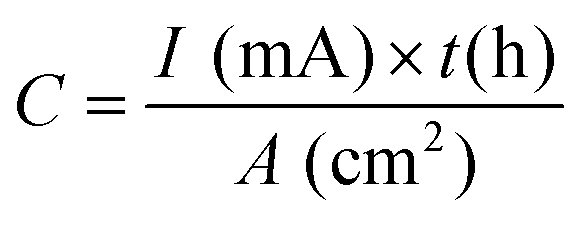
|
Li+ storage capability measured per unit area |
| Volumetric capacity | mA h cm−2 |
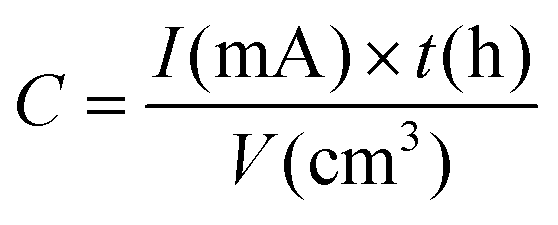
|
Li+ storage capability measured per unit volume |
| Specific energy | W h g−1 or W h cm−2 or W h cm−3 | E = C × V | How much energy can be extracted |
| Power density | W g−1 or W cm−2 or W cm−3 | P = I × V | How fast the energy can be extracted |
| C rate | h−1 |
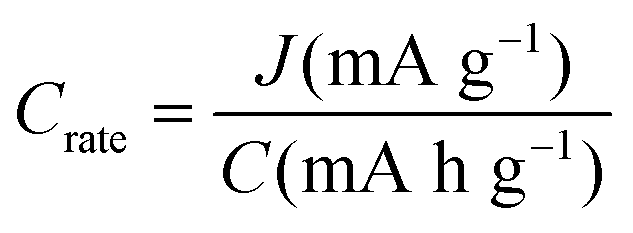
|
Rate of charging/discharging |
| Coulombic efficiency | — |
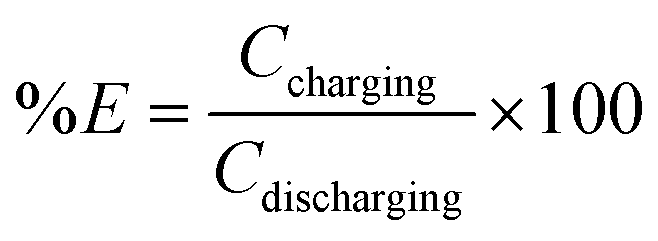
|
Reversible capacity |
| State of health | — |
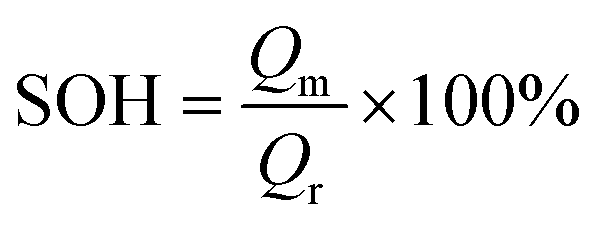
|
Q r—rated capacity and Qm—current maximum available capacity of the battery |
| If SOH < 80%, battery need to be replaced | |||
| Internal resistance | — |
|
R—internal resistance under the current state; Re—internal resistance of the battery when it reaches the end of life; and Rn—internal resistance of the new battery. |
Using the existing materials, cell optimization (single electrodes, SEs and monopolar electrodes, MEs) can enhance the specific energy from 80 W h kg−1 to 200 W h kg−1.341 The cell configuration allows the encapsulation of a greater amount of active materials, which significantly enhances the capacity and reduces the cost and size. Nowadays, bipolar electrodes (BEs) are also gaining attention, as shown in Fig. 17(iii)(c). Here, the cathode and anode slurries are separately coated on both sides of the substrate. This substrate allows the smooth migration of electrons with a lower ohmic resistance and homogeneous current distribution. The BEs demonstrated a superior specific energy, specific power, capacity, and voltage in contrast to SEs and MEs.342 Overall, it can be concluded that ASSLIBs are safer and reliable compared to all commercial LIBs.
Three possible configurations are discussed, as follows: (i) all three components (cathode, anode, and electrolyte) in solid form, (ii) liquid/polymer-based cathode together with solid electrolyte and anode, and (iii) cell with cathode and the separator only (here anode formation occurs after the first charge). The lithium-polymer differentiates itself from conventional battery systems in terms of the type of electrolytes used. In the original design back in the 1970s, a dry solid polymer electrolyte was used. This electrolyte resembles a plastic-like film, which does not conduct electricity but allows ion exchange (electrically charged atoms or groups of atoms). The polymer electrolyte replaces the traditional porous separator soaked in the electrolyte. Therefore, different types of electrolytes have been used. The solid electrolyte will provide only advantages, such as smaller size and higher energy density.305,343,344 To achieve the characteristic parameter of polymer electrolytes, a different strategy was carried out by using a different polymer. Fig. 18 summarizes different architectures of polymer electrolytes that hold potential to replace the existing liquid electrolyte for developing an efficient and safe battery.
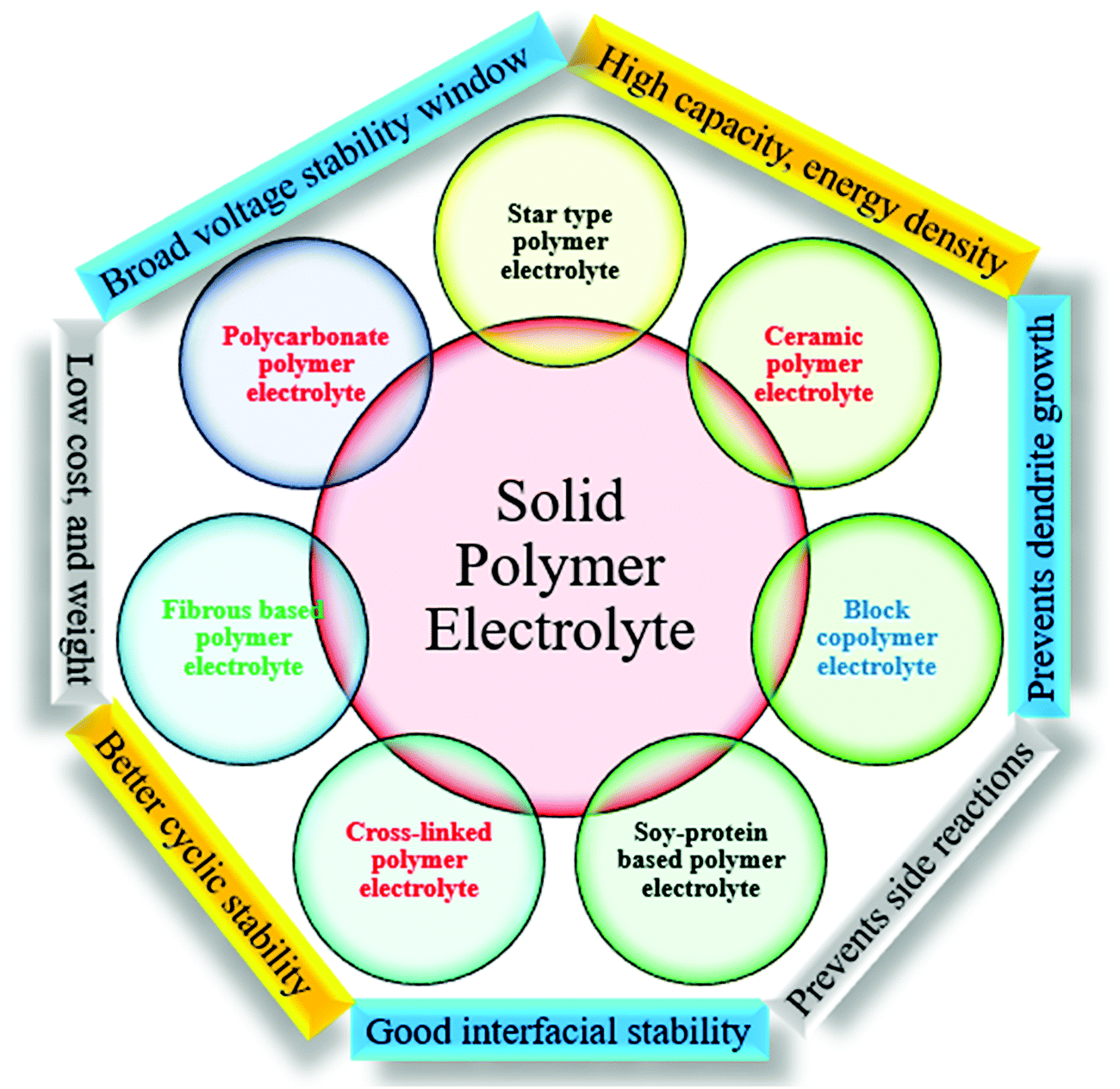 | ||
| Fig. 18 Different types of solid polymer-based electrolytes and their corresponding performances. | ||
Ceramic–polymer electrolytes are prepared by adding nanoparticles to the polymer salt matrix. The addition of nanoparticles enhances the conductivity and thermal and mechanical properties owing to the Lewis-acid-based interaction of the surface groups of the nanofiller with the polymer and salt. The oxygen in the surface groups (–OH) of the nanofiller also provides additional conducting sites for cation (Li+) migration.345–347 Another important and unique architecture block is the copolymer electrolyte, which is comprised of covalently bound polymers. It improves the electrical properties and stability compared to the individual polymer.348,349 Polycarbonate (PC)-based solid polymer electrolytes are another category of electrolytes that have attracted significant attention. Given that amorphous content is desirable for fast ion conduction in polymer electrolytes, PC-based electrolytes have a high amorphous content, good chain flexibility, and a high dielectric constant. Overall, they can enhance the cation transport number and broad voltage stability window.350,351
The ionic conductivity, voltage stability window, and ion transference number are three crucial parameters to enhance the overall cell performance. The ion transport is favored by the presence of amorphous content and segmental motion of the polymer chain. To promote faster ion migration, the crystallinity needs to be minimized (for faster segmental motion). The polymer must have a low glass transition temperature to achieve conductivity comparable to liquid electrolytes (10−4 S cm−1). Ionic conductivity is directly linked to several free-charge carriers participating in the conduction. Thus, the salt dissociation in polymer electrolytes needs to be improved by the addition of nanoparticles (NPs). The surface interactions among the polymer, salt, and NPs alter the environment of the ions and ionic conductivity. For polymer electrolytes, both cations and anions are mobile, where the mobility of cations is lower (than anions) due to their migration via the coordinating sites of the polymer matrix. The ratio of migrating Li+ to all the migrating ions including anions in the electrolyte is defined as the Li-ion transference number (LITN). For the optimum performance of the cell, the LITN must be high, which in the ideal case is 1. A high cation transference number also eliminates the issue of concentration polarization. By restricting the migration of anions via the addition of nanofillers, the cation migration can be improved.352 High ionic conductivity is crucial to achieve high charge/discharge rates. Another important parameter is the voltage stability window of the electrolytes, which limits the charge and discharge characteristics of the electrode materials in a particular voltage range. For this voltage window, the polymer electrolytes must be stable to achieve the optimum performance. The polymer electrolyte must be thermally stable and should not show any signs of shape/volume change, shrinkage, or melting.
Cross-linking is an effective strategy to prepare novel polymer structures, offering an enhancement in the mechanical, electrical, and voltage stability properties. The physical and topological properties are further improved by using a new architecture based on star polymers, where this is attributed to the presence of outer spheres of arms, which enhance the ion mobility and conductivity. The presence of various branching points interrupts the polymer re-crystallization tendency and enhances the ion migration owing to their high flexibility. High surface area fiber-based polymer membranes are also being investigated, which are better than linear polymers. They provide faster ion migration, a broad voltage window, and good interfacial contact.353,354 A new approach is using bio-based polymers such as soy-protein (SP). The ammonium group on SP interacts with the polymer chains and disrupts the crystallization tendency, which enhances the ion dynamics.355,356Table 19 compares the properties of different types of electrolytes.
| Electrolyte | Cathode | Anode | ESW (V) | Cation transport number (t+) | Electrolyte conductivity (S cm−1) | Specific capacity (mA h g−1) | Capacity retention | Efficiency | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| POSS/LiTFSI/(P(EO-co-PO)) | LFP | Li | 5.1 | 0.62 | 1.1 × 10−4 at 25 °C | 160 (at 25 °C) at 0.1C | 75% at 0.3C after 100 cycles | ∼100% | 357 |
| PLFY | LFP | Li | 4.99 | — | 3.23 × 10−4 at 25 °C and 8.58 × 10−4 at 45 °C | 160.1 after 200 cycles at 0.4C | 99.1% after 200 cycles | — | 358 |
| PEO–LiClO4-g-C3N4 | LFP | Li | 4.8 | 0.37 | 1.76 × 10−5 at 25 °C | 161.2 (at 1C) | 81% after 200 cycles at 1C at 80 °C | 99.7% | 359 |
| Functional gradient SPE | LFP | Li | 5.3 | 0.62 | 2.45 × 10−4 at 25 °C | 163.2 (at 0.1C) | 110.5% mA h g−1 after 500 cycles at 1C | 95.3% | 360 |
| Garnet Si–Li7La3Zr2O12 | NCM811 | Li | — | — | 6.68 × 10−4 at 25 °C | 137 after 100 cycles | 69.5% after 100 cycles | 99.0% at 50 °C | 361 |
| Garnet Si–Li7La3Zr2O12 | LFP | Li | 128.8 after 100 cycles | 91.8% after 100 cycles | 98.5% after 100 cycles | ||||
| PSF-PEO/LiTFSI/SN | LFP | Li | 4.2 | — | 1.6 × 10−4 (at RT) 1.14 × 10−3 (at 80 °C) | 152 (at C/3), ∼125 (after 30th cycle) | — | — | 362 |
| P(VDF-HFP)–(PE-PM-PVH) | LFP | Li | ∼5 | 0.72 | 0.81 × 10−3 | 152.7, 149.6 (after 100th cycle) | 98% | 99% | 363 |
| TEOS: PSZ | LCO | Graphite | — | — | 1.04 ± 0.05 × 10−3 | 134 | 93% (after 100th cycle) | — | 364 |
| PAN/PEO/PDMA | LFP | Li | 4.5 | 0.58 | 0.67 × 10−3 (30 °C) | 154 (at 0.1C) | — | — | 365 |
| PVDF-co-HFP/oligomeric ionic liquids | LFP | Li | 4.5 | — | 0.12 × 10−3 (RT) | 152 (at 0.1C) | 99% (after 100th cycle) | 366 | |
| PPC/LiTFSI/LAGP | LFP | Li | 4.5 | 0.75 | 1 × 10−4 | 138.3 at 0.1C | 97.1% after 100 cycles | — | 367 |
| PEO/LiTFSI | LATP-Coated LCO | Li | 4.5 | — | — | 177 at 0.1C | 93% after 50 cycles | — | 368 |
| Nanofiber-reinforced polymer electrolyte | LFP | Li | 4.5 | — | 1 × 10−4 | 159 at 70 °C | — | >99% | 369 |
| LSTHF, PVDF | LFP | Li | 4.8 | 0.50 | 0.53 × 10−3 at 23 °C, 0.89 × 10−3 at 70 °C | ∼134 at 1C | 100% after 150th cycle at 1C | > 98% | 370 |
| PEO–LiTFSI/PI-g-PEO nanofiber | LFP | Li | 5 | 0.45 | 1.0 × 10−4 at 40 °C | 140 at 0.05C | 120 mA h g−1 after 50th cycle | 99% at 0.05C | 371 |
| PVDF/LLTO–PEO/PVDF | LCO | Li | 5 | 0.67/0.70 | ∼3.01 × 10−3 | 144 (at 1C) | 91.8% (after 100th cycle) | — | 372 |
| β-Type PS4/Li3PS4 | LFP | Li | 5.1 (in situ) 4.9 (mechanical-mixing | 0.33 (in situ) 0.28 (mechanical-mixing | 8.01 × 10−4 (in situ) 6.98 × 10−4 (mechanical-mixing 60 °C) | 153 (at 0.1C) | 86.1% (after 100th cycle) 85.9% (after 325th cycle) | — | 373 |
| PEO/LLZTO | LFP | Li | 5 | — | 1.17 × 10−4 (at 30 °C) 1.58 × 10−3 (at 80 °C) | 149.1 (at 0.1C), 139.1 (at 0.1C; after 100th cycle) | — | 100% (after 50th cycle) | 374 |
| PEOBK-POSS | LFP | Li | 4.3 | — | 0.16 × 10−3 (30 °C), 0.7 × 10−3 (60 °C) | 146.5, 144.5 (after 100th cycle) (at 0.2C) | — | 99%, 99.7% (after 100th cycle) | 375 |
| PEO/LiTFSI/LLZO | LFP | Li | 5.7 | 0.207 (at 60 °C) | 5.5 × 10−4 (30 °C) | 150.1 (after 3 cycles); 149.5 (after 50 cycles), 121 (after 100 cycles) | 93.2% (after 1st), 89 (after 100th cycle) | 98.9% | 376 |
| PEO-PPC-LiTFSI-LLTO | LFP | Li | 5.1 | 0.227 | 5.66 × 10−5 (at 25 °C), 5.7 × 10−4 (at 80 °C) | 135 (at 0.5C), 130 (after 100th cycle) | 96% | 100% | 377 |
| PEO–LiTFSI/g-C3N4 | LFP | Li | 4.7 | 0.56 | 1.7 × 10−5 (at 30 °C) | 161.3, 155 (after 150th Cy.) | — | 99.5% | 378 |
| BCP with PS and Jeffamine | LFP | Li | 5.8 | 0.08 (at 70 °C) | 5.6 × 10−4 (at 70 °C), 7.9 × 10−5 (at 40 °C) | 140 (after 1st cycle) | — | 100% (after 30th)cycle | 379 |
| PAEC/LiTFSI | V2O5 | Li | — | — | 2 × 10−7 (at 25 °C) | 11 μA h cm−2 (areal capacity) | — | — | 380 |
| PEC/LiFSI | LFP | Li | 5 | 0.5 | 2.5 × 10−5 (at 30 °C) | 120–130 (at C/10) | — | — | 381 |
| Multi block copolymer (SI) | NCM | Li | 4.8/4.9 | 1 | 3.2 × 10−4 | 150 (at C/20) | 86% | 99.5% (after 230 cycles at C/5) | 382 |
| PCPU/PCDL/HDI/DEG/LiTFSI | LFP | Li | 4.5 (80 °C) | 0.45 (at 80 °C) | 2.2 × 10−6 (at 25 °C), 1.58 × 10−5 (at 60 °C) 1.12 × 10−4 (at 80 °C) | 128 (at 0.2C | 9% (after 100th cycle) | 100% | 383 |
| 127 (after 100th cycle) | 91% (after 600th cycle) | ||||||||
| WPU/PEG/HDI | LFP | Li | 4.8 V (60 °C) | — | 7.3 × 10−4 (at 60 °C) | 151 (at 0.1C), | 97% (after 50th cycle) | — | 384 |
| 2.2 × 10−3 (at 80 °C) | |||||||||
| Polypoly(ε-caprolactone) (PCL) | LFP | Li | — | 0.66 (at 60 °C), 0.62 (at 40 °C) | 4.1 × 10−5 (at 25 °C) | 150 (at C/50), | — | 100% | 385 |
| OV-POSS/PEGMEM | LFP | Li | 5.31 (SCP5.1) 5.04 (LSP5.1) | 0.35 (SCP5.1) 0.19 (LSP5.1) | 1.13 × 10−4 (SCP5.1), 5.63 × 10−5 (LCP5.1) (at 25 °C). | 163.8, 147.8 (after 100th cycle) | 90.2% | 100% | |
| PEGDMA550 (Li-SPE550-Li) | LFP | Li | 5.4 | 0.30 | 2.82 × 10−5 (at 20 °C) | 137.7, 130.5 (after 150th cycle) | 95% | 97% (after 1st cycle), 98% (after 150th cycle) | 386 |
| (HBPS-(PTFEMA-b-PPEGMA)27)/LiTFSI | LFP | Li | 4.9 | 0.26 | 2.36 × 10−5 (at 25 °C), 4.1 × 10−4 (at 80 °C) | 139, 147 (after 5th Cy.) (at 0.1C, 60 °C) | — | 100% (after 100th cycle) | 387 |
| PEGMA/DLC-((PS)23)6/LiTFSI | LFP | Li | 5.1 (30 °C) | 0.37 | 1.94 × 10−4 (at 30 °C), | 139 (at 0.1C), 130 (after 50th cycle) (at 60 °C) | — | 100% (after 50th cycle) (60 °C), 97% (80 °C) | 388 |
| PEO-LiTFSI/LLTO nanofiber | LFP | Li | 4.5 | 0.33 | 1.8 × 10−4 (at RT) | 80 (at 0.3C), 25 °C | — | 90–100% | 389 |
| PVDF-HFP/LiTFSI/LLZO nanofibre | LFP | Li | 5.2 | — | 9.5 × 10−4 (at 20 °C) | 140 (at 0.2C) | 93% (after 150th cycle Cy. at 0.5C) | 99.9% | 390 |
| PEOEC/LiClO4/OA-POSS | V2O5 | Li | 5.0 | — | 3.74 × 10−5 (at 30 °C), 3.26 × 10−4 (at 60 °C) | 280 | ∼100% (after 30 cycles) | — | 391 |
| PAN/SiO2 (MA-SiO2)/TEGDA | NCM | Graphite | — | — | 1.1 × 10−3 (non-porous), 1.8 × 10−3 (mesoporous) | 179.5, 157.9 (after 300 cycles) | 88.0% (for mesoporous) | — | 353 |
| PEGDA/DVB | LFP | Li | 5 | 0.23 | 1.4 × 10−4 | 123, 138 (after 20 cycles) | — | — | 392 |
| PEG/LiTFSI/RTIL | NMC | Li | 4.8 | — | 4 × 10−4 (at 25 °C), 1.45 × 10−3 (at 65 °C). | 118 (at C/10) | — | 99% | 393 |
| PEO-TEGDMA-TEGDME | LFP | Li | 5.38 | 0.56 | 2.7 × 10−4 (at 24 °C) | 160 (at 0.05C) | 98.8% (after 100 cycles (at 0.1C) | — | 393 |
| PEO/acryl-HBP/PEGDME | LFP-C | Li4Ti5O12 | 4.5 (30 °C) | 0.33 (at 30 °C) | 1.24 × 10−4 (at 20 °C), 1.97 × 10−3 (at 80 °C) for benzoyl peroxide radical initiator | 140 (at C/10) | — | 96% | 394 |
| PEO/acryl-HBP/PEGDME | LFP-C/Al (Full cell) | Cu/LTO | 4.5 (30 °C) | 0.33 (at 30 °C) | 2.44 × 10−4 (at 25 °C), 3.22 × 10−3 (at 80 °C). For EB radiation | 42 (at C/10) | 80% (after 340th cycle) | — | 394, 395 |
| PEO; UHMWPEO-LiClO4/core–shell protein@TiO2 NW | LCO (65 °C) | Li | 5.4 | 0.62 & 0.41 (PEO only) | 1.1 × 10−4 (at RT), 2 × 10−3 (at 80 °C) | 135 (at 0.2C) | 94.7% (after 70 cycles) | 98.6% | |
| PEO-LiTFSI/MXene (Ti3C2Tx) | LFP (60 °C) | Li | 5.2 | 0.18 | 2.2 × 10−5 (at 28 °C), 0.69 × 10−3 (at 60 °C) | 150 (at C/10) | 91.4% (after 100th cycle) | >97% (after 100th cycle) | 396 |
| PEO–LiClO4–lepidolite | LFP | Li | 6 | 0.72 | 1.39 × 10−6 (at RT), 1.23 × 10−4 (at 60 °C) | 120 (at 0.15C) | — | 100% | 397 |
| PEO–LiTFSI/vermiculite clay sheets | LFP | Li | 5.35 (25 °C), 5.0 (100 °C) | 0.246 (at 25 °C), 0.497 (at 90 °C) | 2.9 × 10−5 (at 25 °C), 1.2 × 10−3 (at 60 °C), 3.1 × 10−3 (at 100 °C) | 159.9 (at 0.1C) | — | — | 398 |
| PEO–LiTFSI/Vertically aligned vermiculite sheets | LFP | Li | — | 0.47 (at RT) | 1.89 × 10−4 (at 25 °C) | 167 (at 0.1C) | 82% (after 200th cycle) | — | 399 |
| PEO–LiTFSI-PAGP | LFP | Li | 5.0 | — | 1.6 × 10−5 | 100 (at 0.1C) | — | >99.5% | 400 |
| POSS-ILs | LFP | Li | 5.0 | — | 8.0 × 10−4 (at 22 °C) | 136.3 | — | — | 401 |
| 2.0 × 10−3 (at 62 °C) | |||||||||
| PEO–LiX–LAO | LFP | Li | 5.2 | — | 1.36 × 10−5 (at 30 °C) | 153.1 | — | 97% | 402 |
| PEO/LiBOB/LLZTO | LFP | Li | ∼5.0 | 0.57 | 165.9 | — | 403 | ||
| PEO–Zn (BEH) | LFP | Li | 4.2 | 0.5 (at 60 °C) | 1.1 × 10−5 (at 30 °C), 2 × 10−4 (at 60 °C) | 125, after 100th cycle at 0.1C | 84% after 100th cycle | — | 404 |
| Li6PS5Cl/poly(ethylene oxide) | NCM | Li | — | — | 1 × 10−3 | 110.2 (at 60 °C) | 91% over 200 cycles at 0.05C (at 30 °C) | — | 405 |
| LLZTO/PEO | LFP | Li | 5.5 | 0.41 | 1.22 × 10−4 (at 30 °C) | 151.1 (after 200 cycles) at 0.5C | 98% after 200 cycles at 0.5C | 99.5% | 406 |
| PVDF–PAN–ESFMs | LCO | Li | 5.1 | — | 7.8 × 10−3 (at 25 °C) | 120.4 (at 0.1C | 93% (after 150th cycle) | — | 407 |
The binder also plays an important role in the cell performance and its stability. Generally, polyvinylidene fluoride (PVDF) is used as an insulating binder. The cell performance can be amplified by replacing it with a conducting material such as lithium polyacrylate (PAALi) as a binder, which is solid up to 200 °C. Recently, He et al.408 fabricated ASSLIBs using LNMO as the cathode, RuO2 as the anode, and an ionic conductive thermosetting material (PAALi) as the binder. Fig. 19(a) shows the full-cell ASSLIB with the structure of LNMO/LAGP/RuO2. The NASICON-structured Li1.5Al0.5Ge1.5 (PO4)3 (LAGP) was used as a solid-state electrolyte. The ionic conductivity of the ASSLIB was determined to be 1.03 × 10−4 S cm−1. Fig. 19(b) shows the discharge capacity and efficiency for PAALi-based cells. The discharge capacity of the ASSLIB was 87.5 mA h g−1 at 0.2C (at 23.8 °C) for 120 cycles and 146 mA h g−1 at 0.5C and 50 °C for 43 cycles. The enhancement in the specific capacity is attributed to the decrease in resistance for Li-ion transportation with PAALi as a binder.
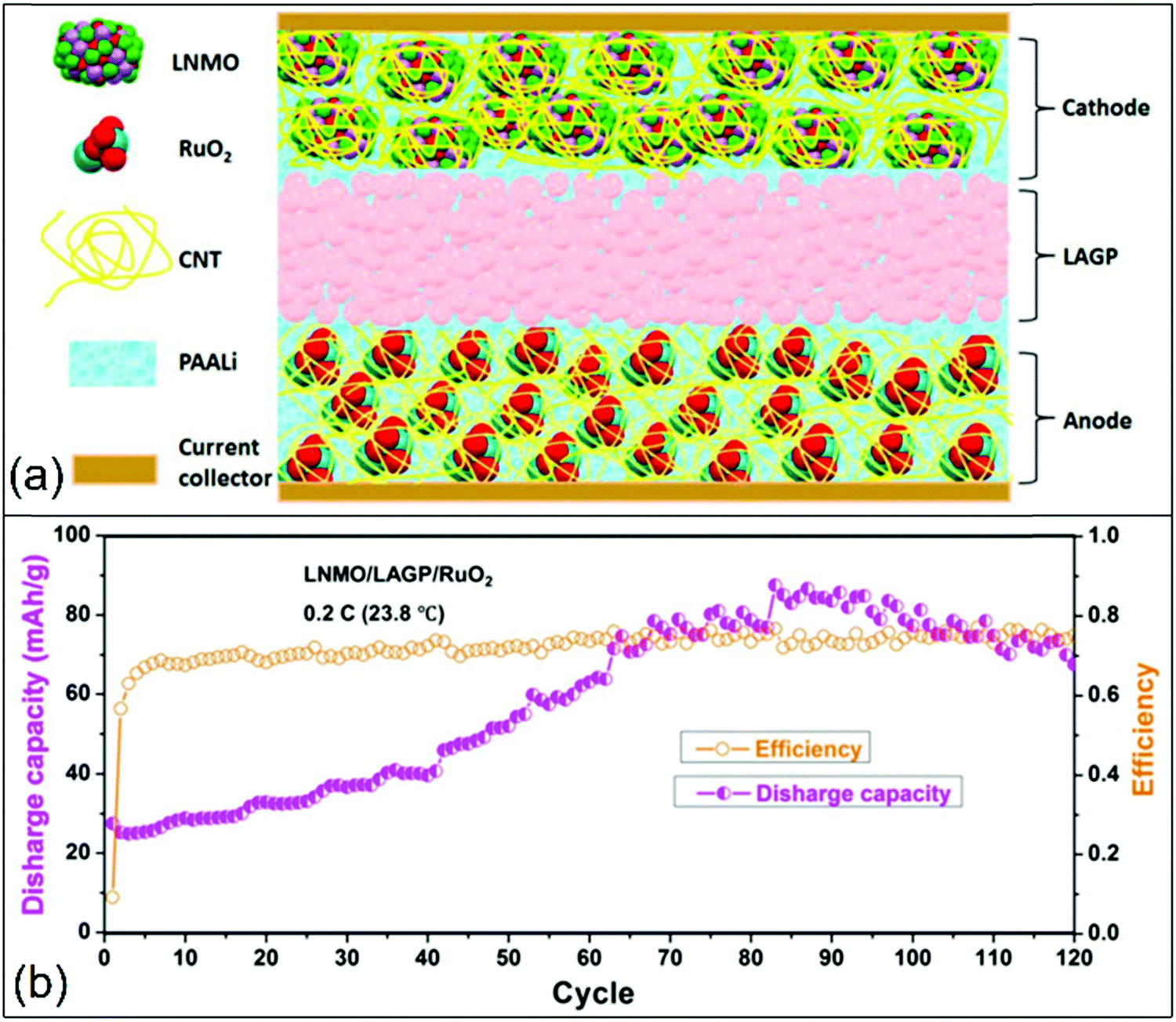 | ||
| Fig. 19 (a) Schematic illustration of the full lithium-ion solid-state battery using LNMO/CNT/PAALi as the cathode, LAGP as the solid electrolyte, and RuO2/CNT/PAALi as the anode, where both sides were sputtered with gold as current collectors. (b) Discharge capacity and efficiency of LNMO/LAGP/RuO2 at room temperature at 0.2C using PAALi as a binder.408 Reproduced with permission from The Royal Society of Chemistry, UK.408 | ||
Another attractive candidate as a solid-state electrolyte is NASICON-structure Li1.3Al0.3Ti1.7(PO4)3 (LATP). LATP has high ionic conductivity (1 mS cm−1) and better stability in water and air, which is attributed to the P-O bonding in its structure.409,410 The only drawback restricting its use is interfacial issues, which result in poor contact, side reactions, and formation of dendrites.411,412 The formation of a mixed ionic/electronic conducting interphase (MCI) was observed because the reduction of Ti4+ to Ti3+ led to the formation of dendrites. Interface engineering can eliminate these problems. Here, an interfacial layer with low electronic conductivity and high ionic conductivity was introduced to minimize the interfacial resistance and prevent side reactions.
PEO and polyacrylonitrile (PAN) were used by Liang et al.,413 which led to an improvement in mechanical stability and better performance. PEO enhanced the Li-ion migration and reduced the interfacial resistance; however, at high temperatures (80–100 °C) it cannot suppress the growth of dendrites. To overcome the above-mentioned issue, Jin et al.414 prepared a composite polymer electrolyte with LATP. Fig. 20(a) shows the interface evolution mechanism without modifying the interface microstructure. The growth of the dendrite structure occurred due to the formation of an MCI.
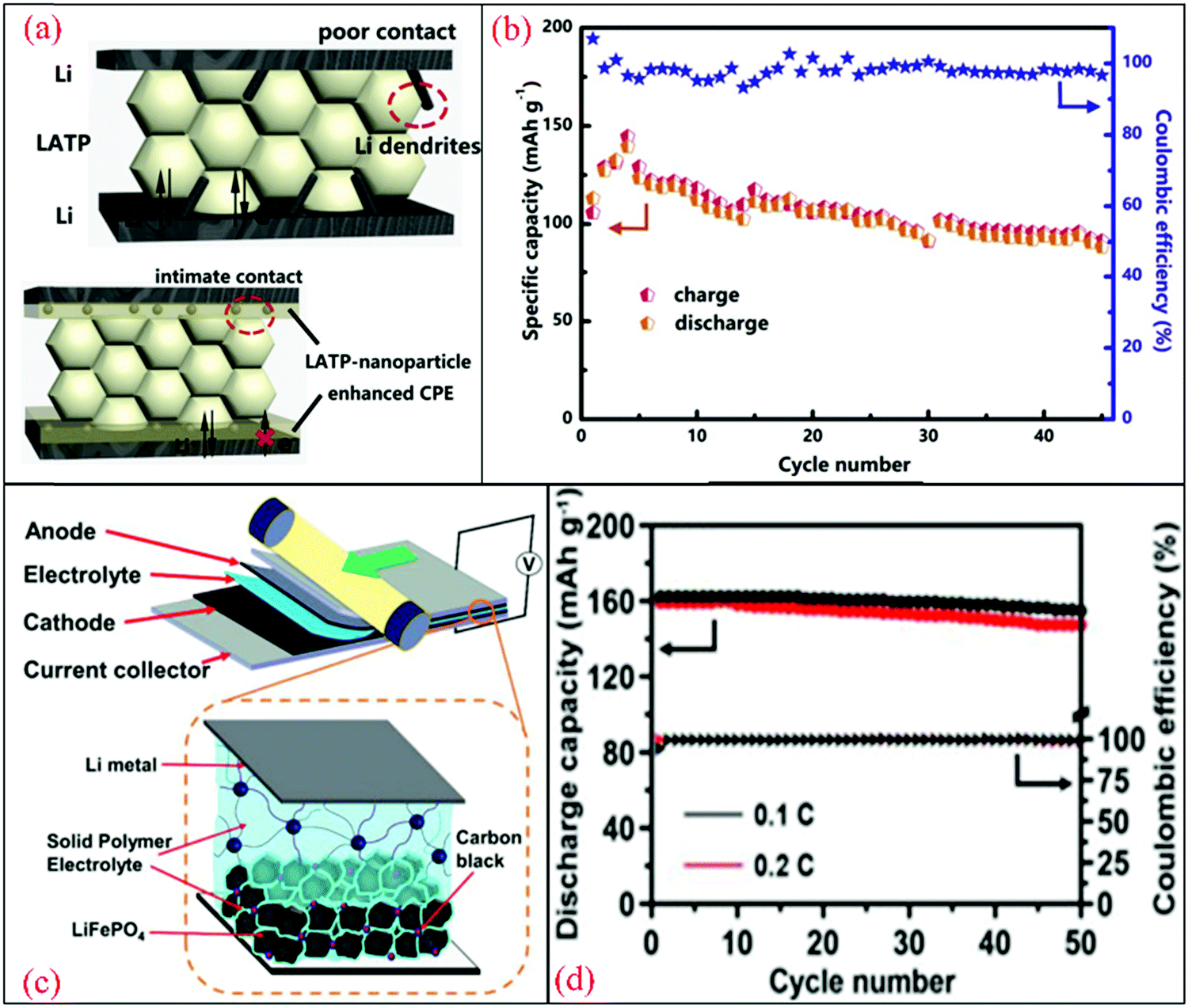 | ||
| Fig. 20 Schematic illustration of the interface evolution between LATP and Li. (a) Without interface modification and after introducing LATP nanoparticles, which enhanced the CPE interfacial layer at LATP/Li interface. (b) Long-term cycling performance and coulombic efficiency of the solid-state LFP/LATP/CPE/Li cell at 0.05C, 80 °C.414 (c) Schematic of the roll-to-roll assembly of an Li|N-SPE|LiFePO4 battery using a free-standing N-SPE film. (d) Variations in the capacity and Coulombic efficiency in the charge–discharge cycling at 0.1 and 0.2C-rates. The charge–discharge operated between 2.5 and 4.0 V, and the C rates were defined based on the theoretical capacity of LiFePO4 (170 mA h g−1). Reproduced with permission from Elsevier.414 | ||
With the introduction of an LATP layer, no MCI formation occurs, which is attributed to the elimination of side reactions, facilitating the fast migration of Li+. The suppression of dendrites is because of the high shear modulus (81–115 GPa). The ionic conductivity of LATP-CPE was 4.6 × 10−4 S cm−1 at 20 °C and 4 × 10−3 S cm−1 at 80 °C. This enhancement originated from the suppression of the crystalline phase and improvement in salt dissociation after the addition of LATP. The voltage window was close to 4.6 V. A solid-state battery (LFP/LATP/CPE/Li) was fabricated and its electrochemical performance was examined. The initial specific discharge capacity of the cell was 113.1 mA h g−1, which reached 139.5 mA h g−1 after the 4th cycle. The specific capacity was reduced to 91.3 mA h g−1 after 45 cycles, with a capacity retention of 80.7% and a Coulombic efficiency of >96% (Fig. 20b). PEO has been used as an electrolyte in batteries. However, the semi-crystalline nature of PEO hinders the desirable ionic conductivity. Thus, various strategies have been adopted to suppress the crystalline content of PEO. Some of the best-performance electrolytes are polymer blends, cross-linked polymers, co-polymers, block copolymers, networked solid polymer electrolyte (N-SPE), and polymers. The N-SPE is an attractive electrolyte due to the formation of a 3D radial network. It provides easy access to cation migration, which is associated with the segmental motion of polymer chains.
Recently, Hsu et al.357 prepared a network of solid polymer electrolyte (N-SPE), which is a cage-like polyhedral oligomeric silsesquioxane (POSS), serving as the hub of a network of poly(ethylene oxide-co-polypropylene oxide) (P(EO-co-PO)) with lithium bis(trifluoromethane sulfonyl)imide (LiTFSI). N-SPE demonstrated the highest conductivity of ∼1.1 × 10−4 S cm−1 at room temperature (25 °C) with an activation energy of 0.037 eV, which had a lower activation energy than PEO (0.5 eV). The lower activation energy and high ionic conductivity of 3D POSS are attributed to its perfect polymer networking. The voltage stability window for the electrolyte was 5.4 V with a cation transport number of 0.62. Table 19 demonstrates the comparison of the electrode, electrolyte, and cell performance parameters for ASSLIBs.
A solid-state battery was assembled with the configuration of Li|N-SPE|LiFePO4, as shown in Fig. 20(c). The N-SPE electrolyte was used as a sandwiched structure between two electrodes. It demonstrated a specific capacity of 160 mA h g−1 at the rate of 0.1C. Fig. 20(d) shows the cycling performance of the cell at 0.1C and 0.2C, where ∼100% coulombic efficiency with good capacity retention (95% at 0.1C and 92% at 0.2C) was observed. After 100 cycles with a high current rate (0.3C), the cell showed a capacity retention of 75% and Coulombic efficiency of 100%. The enhanced performance of the ASSLIB is attributed to the greater Li+ migration and good contact formation. Compared to the individual polymers, the blending of polymers is an effective strategy to suppress the crystallinity of PEO, facilitating higher electrical and mechanical properties. Recently, Bai et al.358 prepared a polymer-ceramic hybrid electrolyte via the blending of PEO/PVDF and nanofiller Li6.4La3Zr1.4Ta0.6O12 (LLZTO).
Fig. 21(a) shows the detailed preparation process of solid composite electrolyte-based PEO/PVDF/LLZTO/LiTFSI systems. The high tensile strength (5.21 MPa) and large strain (1888%) of the electrolyte film were achieved due to the reduction in crystallinity of the hybrid structure. The highest ionic conductivity was 3.23 × 10−4 S cm−1 at 25 °C and 8.58 × 10−4 S cm−1 at 45 °C with an activation energy of 0.32 eV. The enhancement in conductivity, reduction in crystallinity, and improved salt dissociation were attributed to the formation of additional conducting pathways with LLZTO due to the increased dielectric constant of PEO. Therefore, ASSLIBs were fabricated using LiFePO4 as a cathode, Li metal as an anode, and the hybrid electrolyte.
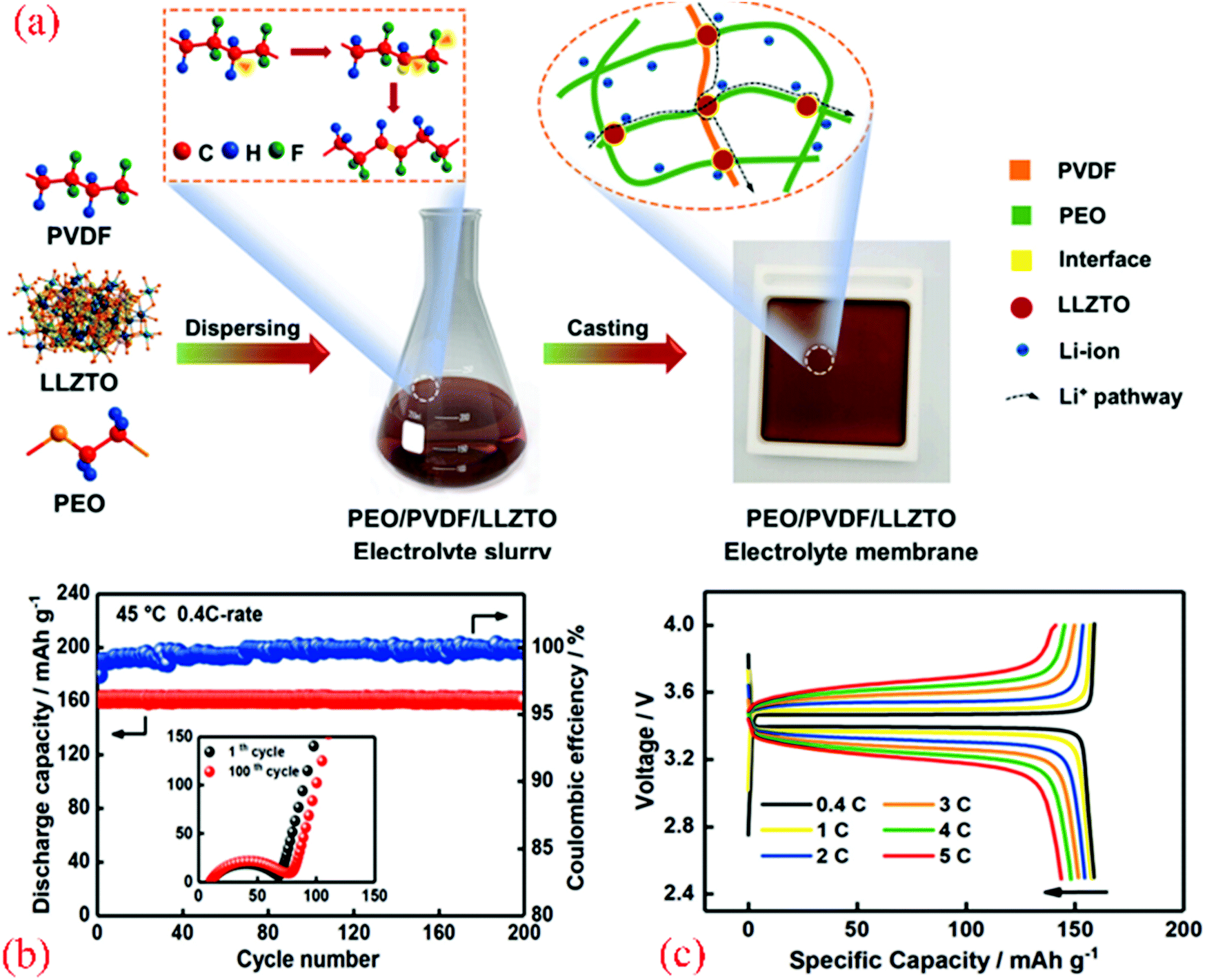 | ||
| Fig. 21 (a) Fabrication process of the PLFF (PEO/PVDF/LLZTO/LiTFSI) solid composite electrolyte. (b) Cycling performances at 0.4C and 45 °C and the impedance of LiFePO4/Li cell cycling with PLFY. (c) Different rates of galvanostatic charge–discharge profiles of LiFePO4/PLFY/Li cells under 45 °C.358 Reproduced with permission from Elsevier.358 | ||
Fig. 21(b) shows the cyclic performance of the fabricated LiFePO4/Li cell at 0.4C (45 °C). The cell demonstrated a high discharge capacity of 160.1 mA h g−1 and 99.1% capacity retention after 200 cycles. The smooth charge–discharge curves and low migration of discharge plateau with C-rate variation indicate better interfacial stability (Fig. 21c). This confirmed the better electrochemical performance, where the ASSLIB operated for 1000 h without short-circuit. Thus, it can be concluded from the above-detailed discussion on the different components of LIBs that the solid-state battery is the ultimate goal, which has the potential to eliminate the existing problems of liquid electrolytes. Fig. 22 provides a glimpse into the advantages and challenges of solid-state LIBs compared to liquid LIBs.
 | ||
| Fig. 22 Advantages and challenges in solid-state batteries (LIBs). [https://www.futurebridge.com/blog/solid-state-batteries/]. | ||
6. Improving performance of Si-based anode for LIBs
Significant efforts have been made to improve the performances of Si-based anode for LIBs. To overcome the volume expansion during the intercalation of electrochemical reactions, the inorganic/organic/Si nanocomposite anode of LIBs should be accommodated with the optimized microstructures. The hybrid nanostructured materials exhibit a genuine prospect to significantly impact the electrochemical performance of Si anodes. Therefore, the intercalation/deintercalation rates can be enhanced using Si-NP-based nanocomposites. The significance of nano-sized Si (n-Si) for the battery performance was demonstrated by the LIB innovative researchers.8,14,4157. Limitations
The traditional LIBs are manufactured using liquid electrolytes, which cause toxic emissions and flammable accidents, which can be overcome by using solid electrolytes. In this case, LIBs can be manufactured without the use of a separator free with the implementation of solid-state electrolytes (SSE). ASSLIBs are manufactured as separator-free batteries, which automatically open the door for device miniaturization. Therefore, the weight/price can be controlled with the replacement of SSEs. The standard lifespan of LIBs is about three years (500 to 1000 cycles), where after this prescribed period, they usually do not show power backup and stability and seem to be worthless. The use of SSEs removes all the barriers faced by the commercial manufacturing of lithium ion-based batteries such as dendrite growth (cause short-circuit), pitiable thermal stability and safety concerns for portability and use.The safety, cycle lifetime, and power density of LIBs cannot be easily controlled and tackled within the wide range of operational temperature conditions. Thus, a protection circuit must be included to maintain the voltage and current within safe limits together with the temperature sealing layers. The aging effect of LIBs is also a serious concern when these batteries are not in use and stored in a cool place. It significantly reduces the charge over time. There are transportation restrictions for the shipment of larger quantities, which may be troubling for regulatory controls. These restrictions do not apply to personal carry-on batteries. Also, their manufacturing cost is about 40% higher than the nickel–cadmium batteries, even though the materials, metals, and chemicals are changing continuously.
8. Challenges
Over the past few decades, the progress in the development of materials for energy storage/conversion devices, especially Li-ion batteries (LIBs) has been satisfactory. However, although LIBs have been commercialized successfully with the use of liquid electrolytes, the scientific community has focused on alternative electrolytes to fulfill the dream of all-solid-state batteries (ASSBs). All three components of LIBs (cathode, anode, and electrolyte) need to be developed or innovated simultaneously to optimize the electrochemical performance of the cell. Accordingly, there are still several challenges related to LIBs that need to be resolved. A few of the major challenges are described, as follows: (i) disposing of damaged LIBs from mobile electronics, (ii) developing the large-scale production of LIBs containing advanced electrodes and electrolytes for HEVs, (iii) fulfilling the required power density, and cyclability and (iv) recycling of automotive LIBs. Compared to lead-acid batteries, the recycling/disposal of LIBs are one of the most complicated issues, which have not been established to date, and thus further research is necessary for the recycling of the electrodes. The market for LIBs is driven by increasing the demand for mobile electronics, computers, and portable devices. The remarkable growth of mobile and robotic electronic systems has demonstrated that we need to improve the engineering and manufacturing process of Si-based nanocomposite electrodes. In response to the current demands of modern society and emerging ecological concerns, low-cost and environmentally friendly energy storage systems are required. Hence, the rapid R & D in energy storage systems should be aware of the market requirement. The performance of LIBs depends directly on the properties of their electrode materials and microstructure/compositions. Innovative materials chemistry demonstrates the advancement in energy storage mechanisms for LIBs. Some new strategies must be developed for stabilizing the cathode and anode to achieve the optimum performance in terms of durability. The contact between the current collector and active material needs to be examined for enhancing the charge transport via full use of the active material. The solid electrolyte interface needs to be improved to enhance the rate capability of the full cell. Therefore, no single modification strategy is suitable to achieve the optimum performance and the combination of various strategies will be more efficient in tuning the properties of materials. Cobalt is an important raw material for batteries, considering that the search for alternative green electrodes for the next generation of ASSBs is a top priority.The electrolyte is another crucial component of ASSLIBs. Solid polymer electrolytes have the potential to become a future electrolyte. Although significant efforts have been devoted by researchers to achieve better ionic conductivity, there is still a need to improve the ionic conductivity before commercialization. Different strategies have been adopted to design single-ion conductors, as follows, (i) addition of nanoparticles, (ii) optimizing polymer chain movement, (iii) and designing new polymer backbones. The role of additives is to enhance the salt dissociation and provide additional conducting sites for cation migration. The surface groups of nanoparticles minimize the overall conducting path length for cations. However, in the development of solid polymer electrolytes (SPE), the ion transport mechanism needs to be investigated deeply. It can be understood from this review that the key approach is to adopt SPE, that is the fabrication of flexible energy storage devices to broaden the application range and durability. In this case, the key properties of SPEs such as thermal/chemical/electrochemical/mechanical stability need to be examined. Furthermore, to maintain the energy density of ASSBs, these stability parameters play an important role. Further investigation needs to be focused on the performance of flexible batteries under different conditions such as bending, stretching, and reshaping. Nanostructured materials have attracted the great interest in recent years because of their unique mechanical, electrical, optical properties and have the maximum surface area. To create a roadmap for nanomaterial-based electrodes for energy storage systems, the synthesis and manipulation of nanostructured materials need to be optimized for high-power density and long cycle ability.
The compatibility between electrodes and electrolyte needs to be enhanced for the fabrication of efficient ASSBs. Given that the internal resistance needs to be minimized for faster ion conduction from one electrode to another electrode, the interfacial stability between the components needs to be examined in detail together with electrochemical analysis. To meet the requirement of practical applications of LIBs, the optimization of the electrodes and the electrolyte is very important for the performance and durability. The aging effect of batteries and self-discharge are also a topic of research but hardly studied in the literature. To moderate the aging effect, some advanced characterization techniques should be used for the optimization of electrode materials such as neutron diffraction, small-angle X-ray scattering, and in situ tools for understanding of material behavior. Usually, all energy devices are operated at room temperature. Therefore, the current R&D of energy storage systems should be focused on low/high temperatures operation to achieve an efficient energy density and the memory effect. For the commercialization of LIBs globally, the universal testing parameters should be established for the comparison of data from various research groups/industries. Thus, the performance environment of ASSBs needs to consider the optimum feasibility worldwide. For the commercialization of LIBs, the cell architecture plays an important role in the performance and durability. The scientific community has devoted their efforts to the development of smart energy devices, which are superior to traditional devices. Thus, artificial intelligence (AI) and deep learning may be adopted for developing smart devices, which can optimize their performance themselves, strengthening the application of smart devices.
9. Future outlook
The recent development of novel negative electrodes (anodes) for lithium-ion batteries (LIBs) has focused on silicon-based nanostructured composites or hybrid materials. Silicon is one of the highest specific capacity anode materials, which can replace the standard C-based electrodes (carbon has a specific capacity of 372 mA h g−1). Although the pure form of Si is not available in the Earth's crust or the environment, nanostructured Si can be obtained via synthetic methods or the biogenic synthesis. Nanostructured Si can be synthesized economically from biomass waste (biogenic silicon), which has the highest theoretical specific capacity (4200 mA h g−1). For the setup of a materials database (cathode, anode, and electrolyte) for proper identification, research and development have been carried out to reduce the cost and processing time, resulting in efficient device development. To achieve a core-level understanding of the ion transport in the electrode/electrolyte, simulation tools can be used for the predetermination/demonstration before the experiment. To overcome the traditional instrument failure, advanced characterization techniques (cryo-electron microscopy) should be used to gain insights into materials and the interfaces in batteries. The utility and performance of ASSLIBs can be enhanced especially for HEVs and next-generation portable electronics due to the unique characteristics of materials and applications. Therefore, research needs to be carried out toward the development of solid electrolytes that exhibit high conductivity even at sub-zero temperatures. The progress and performance of ASSLIBs depend on their components and compatibility. To resolve the existing environmental and safety concerns, efficient ASSBs are future devices that have potential to promote growth in various sectors (from automobiles to the digital market).Author contributions
Conceptualization: SKS, GS, AG, AA, YKM; data curation: SKS, GS, AG H-GR; formal analysis: SKS, GS, AG, AA, FSM, RA, J-SY, YKM H-GR; funding acquisition: SKS, YKM; investigation: SKS, GS, AA, AG, YKM; methodology: SKS, AG, AA, YKM; project administration: SKS, AG, YKM; supervision: SKS, AG, H-GR, YKM; writing, reviewing & editing: SKS, GS, AG, AA, FSM, RA, J-SY, YKM H-GR.List of abbreviations
| ASSLIB | All-solid-state Li-ion batteries |
| LIBs | Li-ion batteries |
| EVs | Electric vehicles |
| HEVs | Hybrid electric vehicles |
| TiS2 | Titanium disulfide |
| LiCoO2 | Lithium-cobalt oxide |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| ESW | Electrochemical stability window |
| FMMEA | Failure mode, mechanism, and effect analysis |
| Li-NMC | Lithium-manganese-cobalt-oxide |
| NCA | Lithium nickel cobalt aluminum oxide batteries |
| ANL | Argonne national laboratory |
| a-C | Amorphous carbon |
| CNTs | Carbon nanotubes |
| SEI | Solid-electrolyte interphase |
| PVDF | Polyvinylidene fluoride |
| PAALi | Lithium polyacrylate |
| MCI | Mixed ionic/electronic conducting interphase |
| PAN | Polyacrylonitrile |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Prof. Sanjeev K. Sharma acknowledges the Government of Uttar Pradesh, India for sanctioned the project under UP-CST Research & Development programme (108/2021/2585/Sattar-4-2021-4(28)/2021/20). Danish authors acknowledge the funding by Interreg Deutschland-Denmark with money from the European Regional Development Fund, project number 096-1.1-18 (Access and Acceleration) and from the ESS lighthouse on hard materials in 3D, SOLID, funded by the Danish Agency for Science and Higher Education, grant number 8144-00002B.References
- J. Xiao, et al., Recent progress of emerging cathode materials for sodium ion batteries, Mater. Chem. Front., 2021, 5(10), 3735–3764 RSC.
- X. Shen, et al., Advanced electrode materials in lithium batteries: Retrospect and prospect, Energy Mater. Adv., 2021, 2021, 1205324 Search PubMed.
- C. Cao, Green Innovation in China: China's Wind Power Industry and the Global Transition to a Low Carbon Economy, JSTOR, 2014 Search PubMed.
- J. Tian, Z. Chen and Y. Zhao, Review on modeling for chemo-mechanical behavior at interfaces of all-solid-state lithium-ion batteries and beyond, ACS Omega, 2022, 7(8), 6455–6462 CrossRef CAS PubMed.
- N. Kamaya, et al., A lithium superionic conductor, Nat. Mater., 2011, 10(9), 682–686 CrossRef CAS PubMed.
- J. C. R. Kumar and M. A. Majid, Renewable energy for sustainable development in India: current status, future prospects, challenges, employment, and investment opportunities, Energy, Sustainability Soc., 2020, 10(1), 2 CrossRef.
- F. Porté-Agel, M. Bastankhah and S. Shamsoddin, Wind-Turbine and Wind-Farm Flows: A Review, Boundary-Layer Meteorol., 2020, 174(1), 1–59 CrossRef PubMed.
- A. Dutta, et al., A comprehensive review on batteries and supercapacitors: development and challenges since their inception, Energy Storage, 2022, e339 Search PubMed.
- A. P. Wickens, A history of the brain: from stone age surgery to modern neuroscience, Psychology Press, 2014 Search PubMed.
- M. Piccolino, Animal electricity and the birth of electrophysiology: the legacy of Luigi Galvani, Brain Res. Bull., 1998, 46(5), 381–407 CrossRef CAS PubMed.
- M. Krepelková Evolution of batteries: From experiments to everyday usage, 21th International Student Conference on Electrical Engineering. 2017.
- J. Park, et al., Challenges, laser processing and electrochemical characteristics on application of ultra-thick electrode for high-energy lithium-ion battery., J. Power Sources, 2021, 482, 228948 CrossRef CAS.
- L. Gijs, Parafilieën en parafiele stoornissen., Leerboek Seksuologie, 2018, 453–478 Search PubMed.
- A. Masias, J. Marcicki and W. A. Paxton, Opportunities and challenges of lithium ion batteries in automotive applications, ACS Energy Lett., 2021, 6(2), 621–630 CrossRef CAS.
- Y. Liu, et al., Current and future lithium-ion battery manufacturing., iScience, 2021, 24(4), 102332 CrossRef CAS PubMed.
- X. Zhang, et al., A review on thermal management of lithium-ion batteries for electric vehicles, Energy, 2022, 238, 121652 CrossRef CAS.
- Y. Liu, et al., Nanostructured strategies towards boosting organic lithium-ion batteries, J. Energy Chem., 2021, 54, 179–193 CrossRef.
- M. Rahimi, Lithium-Ion Batteries: Latest Advances and Prospects, Multidisciplinary Digital Publishing Institute, 2021, p. 8 Search PubMed.
- M. Yoshio and H. Noguchi, Lithium-Ion Batteries, ed E. M. Yoshio, R. J. Brodd and A. Kozawa, New York, Springer Science and Business Media, 2009 Search PubMed.
- Y. Wu, et al., Progress in thermal stability of all-solid-state-Li-ion-batteries, InfoMat, 2021, 3(8), 827–853 CrossRef CAS.
- A. Banerjee, et al., Interfaces and interphases in all-solid-state batteries with inorganic solid electrolytes, Chem. Rev., 2020, 120(14), 6878–6933 CrossRef CAS PubMed.
- L. Xu, et al., Toward the Scale-Up of Solid-State Lithium Metal Batteries: The Gaps between Lab-Level Cells and Practical Large-Format Batteries, Adv. Energy Mater., 2021, 11(4), 2002360 CrossRef CAS.
- R. Murugan, V. Thangadurai and W. Weppner, Fast lithium ion conduction in garnet-type Li7La3Zr2O12., Angew. Chem., Int. Ed., 2007, 46(41), 7778–7781 CrossRef CAS PubMed.
- J. Zhang, et al., Flexible and ion-conducting membrane electrolytes for solid-state lithium batteries: Dispersion of garnet nanoparticles in insulating polyethylene oxide., Nano Energy, 2016, 28, 447–454 CrossRef CAS.
- X. Zhang, et al., Synergistic coupling between Li6.75La3Zr1.75Ta0.25O12 and poly(vinylidene fluoride) induces high ionic conductivity, mechanical strength, and thermal stability of solid composite electrolytes, J. Am. Chem. Soc., 2017, 139(39), 13779–13785 CrossRef CAS PubMed.
- J. Besenhard, J. Yang and M. Winter, Will advanced lithium-alloy anodes have a chance in lithium-ion batteries?, J. Power Sources, 1997, 68(1), 87–90 CrossRef CAS.
- K. Ozawa, Lithium-ion rechargeable batteries with LiCoO2 and carbon electrodes: the LiCoO2/C system., Solid State Ionics, 1994, 69(3–4), 212–221 CrossRef CAS.
- J. C.-Y. Jung, P.-C. Sui and J. Zhang, A review of recycling spent lithium-ion battery cathode materials using hydrometallurgical treatments, J. Energy Storage, 2021, 35, 102217 CrossRef.
- S. Goriparti, et al., Review on recent progress of nanostructured anode materials for Li-ion batteries., J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- N. Mohamed and N. Allam, Recent advances in the design of cathode materials for Li-ion batteries., RSC Adv., 2020, 10, 21662–21685 RSC.
- J. Zhang, L. Huang and X. Gu, Failure mechanism of solid-state electrolyte Li10GeP2S12 in a moist atmosphere: a first-principles study, Mater. Adv., 2022, 3(7), 3143–3150 RSC.
- L. Li, Y. Deng and G. Chen, Status and prospect of garnet/polymer solid composite electrolytes for all-solid-state lithium batteries., J. Energy Chem., 2020, 50, 154–177 CrossRef.
- M. S. Whittingham and A. Yoshino, Lithium-Ion Batteries, 2019 Search PubMed.
- W. V. Schalkwijk and B. Scrosati, Advances in lithium ion batteries introduction, in Advances in lithium-ion batteries, Springer, 2002, pp. 1–5 Search PubMed.
- M. Calvin and G. T. Seaborg, The college of chemistry in the GN Lewis Era: 1912-1946, ACS Publications, 1984 Search PubMed.
- R. J. Brodd, A. Kozawa and M. Yoshio, Lithium-Ion Batteries: Science and Technologies, Springer, 2009 Search PubMed.
- M. S. Whittingham, Electrical energy storage and intercalation chemistry, Science, 1976, 192(4244), 1126–1127 CrossRef CAS PubMed.
- A. Morachevskii, Lithium–Tellurium System: Thermodynamic and Electrochemical Studies and Prospects for Use in Chemical Current Sources., Russ. J. Appl. Chem., 2020, 93(3), 313–324 CrossRef CAS.
- K. Mizushima, et al., LixCoO2 (0< x ⩽ 1): A new cathode material for batteries of high energy density, Solid State Ionics, 1981, 3, 171–174 CrossRef.
- A. Yoshino, The birth of the lithium-ion battery, Angew. Chem., Int. Ed., 2012, 51(24), 5798–5800 CrossRef CAS PubMed.
- J. B. Goodenough and K.-S. Park, The Li-ion rechargeable battery: a perspective, J. Am. Chem. Soc., 2013, 135(4), 1167–1176 CrossRef CAS PubMed.
- H. Maeshima, et al., Quantitative evaluation of electrochemical potential windows of electrolytes for electric double-layer capacitors using ab initio calculations, J. Electrochem. Soc., 2010, 157(6), A696 CrossRef CAS.
- M. D. Halls and K. Tasaki, High-throughput quantum chemistry and virtual screening for lithium ion battery electrolyte additives, J. Power Sources, 2010, 195(5), 1472–1478 CrossRef CAS.
- K. Wang, et al., A cost-effective and humidity-tolerant chloride solid electrolyte for lithium batteries, Nat. Commun., 2021, 12(1), 4410 CrossRef CAS PubMed.
- U. Gulzar, et al., Next-generation textiles: from embedded supercapacitors to lithium ion batteries, J. Mater. Chem. A, 2016, 4(43), 16771–16800 RSC.
- K. Young, et al., Electric vehicle battery technologies, Electric vehicle integration into modern power networks, Springer, 2013, pp. 15–56 Search PubMed.
- C. Hendricks, et al., A failure modes, mechanisms, and effects analysis (FMMEA) of lithium-ion batteries, J. Power Sources, 2015, 297, 113–120 CrossRef CAS.
- B. C. Melot and J.-M. Tarascon, Design and preparation of materials for advanced electrochemical storage, Acc. Chem. Res., 2013, 46(5), 1226–1238 CrossRef CAS PubMed.
- X. Lin, et al., High temperature electrical energy storage: advances, challenges, and frontiers, Chem. Soc. Rev., 2016, 45(21), 5848–5887 RSC.
- Y. Lyu, et al., An Overview on the Advances of LiCoO2 Cathodes for Lithium-Ion Batteries. Advanced Energy, Materials, 2021, 11(2), 2000982 CAS.
- C. Daniel, et al. Cathode materials review, AIP Conference Proceedings, American Institute of Physics, 2014.
- K. Mizushima, et al., LixCoO2 (0 < x < −1): A new cathode material for batteries of high energy density, Mater. Res. Bull., 1980, 15(6), 783–789 CrossRef CAS.
- J. Cho, Y. J. Kim and B. Park, Novel LiCoO2 cathode material with Al2O3 coating for a Li ion cell, Chem. Mater., 2000, 12(12), 3788–3791 CrossRef CAS.
- G. Li, et al., Halogen-doping in LiCoO2 cathode materials for Li-ion batteries: insights from ab initio calculations, RSC Adv., 2015, 5(130), 107326–107332 RSC.
- H. Chen, et al., Direct surface coating of high voltage LiCoO2 cathode with P (VDF-HFP) based gel polymer electrolyte, RSC Adv., 2020, 10(41), 24533–24541 RSC.
- J. Xie, et al., Engineering the surface of LiCoO2 electrodes using atomic layer deposition for stable high-voltage lithium ion batteries, Nano Res., 2017, 10(11), 3754–3764 CrossRef CAS.
- M. Thackeray, P. Hohnson, L. A. de Picciono, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1984, 19, 179 CrossRef CAS.
- S. A. A. Wahab, et al., A study on optical properties of zinc silicate glass-ceramics as a host for green phosphor, J. Appl. Sci., 2020, 10(14), 4938 CrossRef.
- S. G. Booth, et al., Perspectives for next generation lithium-ion battery cathode materials, APL Mater., 2021, 9(10), 109201 CrossRef CAS.
- V. Selvamani, et al., High-performance spinel LiMn2O4@carbon core–shell cathode materials for Li-ion batteries, Sustainable Energy Fuels, 2019, 3(8), 1988–1994 RSC.
- S. M. Abbas, et al., Ag-modified LiMn2O4 cathode for lithium-ion batteries: coating functionalization, Energies, 2020, 13(19), 5194 CrossRef CAS.
- M.-J. Lee, et al., High performance LiMn2O4 cathode materials grown with epitaxial layered nanostructure for Li-ion batteries, Nano Lett., 2014, 14(2), 993–999 CrossRef CAS PubMed.
- L. Zhu, et al., Polypyrrole/Al2O3/LiMn2O4 cathode for enhanced storage of Li ions, Electrochem. Commun., 2021, 124, 106951 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, Phospho-olivines as positive-electrode materials for rechargeable lithium batteries, J. Electrochem. Soc., 1997, 144(4), 1188 CrossRef CAS.
- D. Ganguly and S. Ramaprabhu, Facile synthesis and electrochemical properties of α-Fe2O3 nanoparticles/etched carbon nanotube composites as anode for lithium-ion batteries, Mater. Chem. Phys., 2021, 267, 124664 CrossRef CAS.
- A. Eftekhari, LiFePO4/C nanocomposites for lithium-ion batteries, J. Power Sources, 2017, 343, 395–411 CrossRef CAS.
- J. Li, et al., Lithium ion conductivity in single crystal LiFePO4, Solid State Ionics, 2008, 179(35–36), 2016–2019 CrossRef CAS.
- A. Andersson and J. O. Thomas, The source of first-cycle capacity loss in LiFePO4, J. Power Sources, 2001, 97, 498–502 CrossRef.
- F. Li, et al., Graphite-Embedded Lithium Iron Phosphate for High-Power–Energy Cathodes, Nano Lett., 2021, 21(6), 2572–2579 CrossRef CAS PubMed.
- H. Wang, et al., Y–F co-doping behavior of LiFePO4/C nanocomposites for high-rate lithium-ion batteries, New J. Chem., 2021, 45(12), 5695–5703 RSC.
- C.-T. Hsieh, et al., Preparation of lithium iron phosphate cathode materials with different carbon contents using glucose additive for Li-ion batteries, J. Taiwan Inst. Chem. Eng., 2014, 45(4), 1501–1508 CrossRef CAS.
- Y. Gao, et al., Effect of Ru Doping on the Properties of LiFePO4/C Cathode Materials for Lithium-Ion Batteries, ACS Omega, 2021, 6(22), 14122–14129 CrossRef CAS PubMed.
- Y. Liu, et al., Effect of Na+ in situ doping on LiFePO4/C cathode material for lithium-ion batteries, Prog. Nat. Sci.: Mater. Int., 2021, 31(1), 14–18 CrossRef CAS.
- S. Dou, Review and prospect of layered lithium nickel manganese oxide as cathode materials for Li-ion batteries, J. Solid State Electrochem., 2013, 17(4), 911–926 CrossRef CAS.
- D. Pant and T. Dolker, Green and facile method for the recovery of spent Lithium Nickel Manganese Cobalt Oxide (NMC) based Lithium ion batteries, Waste Manage., 2017, 60, 689–695 CrossRef CAS PubMed.
- D. Ren, et al., Ultrahigh rate performance of a robust lithium nickel manganese cobalt oxide cathode with preferentially orientated Li-diffusing channels, ACS Appl. Mater. Interfaces, 2019, 11(44), 41178–41187 CrossRef CAS PubMed.
- F. Reissig, et al., Synergistic Effects of Surface Coating and Bulk Doping in Ni-Rich Lithium Nickel Cobalt Manganese Oxide Cathode Materials for High-Energy Lithium Ion Batteries, ChemSusChem, 2022, 15(4), e202102220 CAS.
- S. U. Muzayanha, et al., A fast metals recovery method for the synthesis of lithium nickel cobalt aluminum oxide material from cathode waste, Metals, 2019, 9(5), 615 CrossRef CAS.
- D. Wong, et al., Impact of high rate discharge on the aging of lithium nickel cobalt aluminum oxide batteries, J. Power Sources, 2015, 280, 363–372 CrossRef CAS.
- G. Cao, et al., Synthesis of a fine LiNi0.88Co0.09Al0.03O2 cathode material for lithium-ion batteries via a solvothermal route and its improved high-temperature cyclic performance, RSC Adv., 2020, 10(17), 9917–9923 RSC.
- Z.-X. Xiao, et al., Enabling LiNi0.88Co0.09Al0.03O2 Cathode Materials with Stable Interface by Modifying Electrolyte with Trimethyl Borate, ACS Sustainable Chem. Eng., 2021, 9(4), 1958–1968 CrossRef CAS.
- H. Zhang, et al., Synthesis of non-spherical LiNi0.88Co0.09Al0.03O2 cathode material for lithium-ion batteries, Energy Fuels, 2020, 34(7), 9002–9010 CrossRef CAS.
- O. Bohnke, C. Bohnke and J. Fourquet, Mechanism of ionic conduction and electrochemical intercalation of lithium into the perovskite lanthanum lithium titanate, Solid State Ionics, 1996, 91(1–2), 21–31 CrossRef CAS.
- F. Zhu, et al., Atomically Intimate Solid Electrolyte/Electrode Contact Capable of Surviving Long-Term Cycling with Repeated Phase Transitions, Nano Lett., 2022, 22, 3457–3464 CrossRef CAS PubMed.
- X. Chen and Y. Tian, Review of Graphene in Cathode Materials for Lithium-Ion Batteries, Energy Fuels, 2021, 35(5), 3572–3580 CrossRef CAS.
- H. Krüger, et al. Novel Tailored 3D Carbon Nanotube Cathodes for Effective Trapping of Polysulfides in Lithium Sulfur Batteries, ECS Meeting Abstracts, IOP Publishing, 2019.
- S. K. Sharma, et al., Synthesis of silica and carbon-based nanomaterials from rice husk ash by ambient fiery and furnace sweltering using a chemical method, Appl. Surf. Sci. Adv., 2022, 8, 100225 CrossRef.
- G. Suciu and A. Pasat, Challenges and opportunities for batteries of electric vehicles, 2017 10th International Symposium on Advanced Topics in Electrical Engineering (ATEE), IEEE, 2017.
- W. Zuo, et al., Li-rich cathodes for rechargeable Li-based batteries: reaction mechanisms and advanced characterization techniques, Energy Environ. Sci., 2020, 13(12), 4450–4497 RSC.
- B. Çetin, Z. Camtakan and N. Yuca, Synthesis and characterization of li-rich cathode material for lithium ion batteries, Mater. Lett., 2020, 273, 127927 CrossRef.
- G. Lang, Strukturvergleiche an ternären und quarternären Oxiden, Z. Anorg. Allg. Chem., 1966, 348(5–6), 246–256 CrossRef CAS.
- M. Jansen and R. Hoppe, Zur Kenntnis der NaCl-Strukturfamilie: Neue Untersuchungen an Li2MnO3, Z. Anorg. Allg. Chem., 1973, 397(3), 279–289 CrossRef CAS.
- M. Rossouw and M. Thackeray, Lithium manganese oxides from Li2MnO3 for rechargeable lithium battery applications, Mater. Res. Bull., 1991, 26(6), 463–473 CrossRef CAS.
- Z. Lu, D. MacNeil and J. Dahn, Layered cathode materials Li [NixLi(1/3−2x/3)Mn(2/3−x/3)]O2 for lithium-ion batteries., Electrochem. Solid-State Lett., 2001, 4(11), A191 CrossRef CAS.
- X. Li, et al., Direct Visualization of the Reversible O2−/O− Redox Process in Li-Rich Cathode Materials., Adv. Mater., 2018, 30(14), 1705197 CrossRef PubMed.
- C. R. Fell, et al., Correlation between oxygen vacancy, microstrain, and cation distribution in lithium-excess layered oxides during the first electrochemical cycle, Chem. Mater., 2013, 25(9), 1621–1629 CrossRef CAS.
- Y. Okamoto, Ambivalent effect of oxygen vacancies on Li2MnO3: a first-principles study, J. Electrochem. Soc., 2011, 159(2), A152 CrossRef.
- S. Zhao, et al., Towards high-energy-density lithium-ion batteries: Strategies for developing high-capacity lithium-rich cathode materials, Energy Storage Mater., 2021, 34, 716–734 CrossRef.
- J. An, et al., Insights into the stable layered structure of a Li-rich cathode material for lithium-ion batteries, J. Mater. Chem. A, 2017, 5(37), 19738–19744 RSC.
- S. Xiao, et al., Practical strategies for enhanced performance of anode materials in Na+/K+-ion batteries, J. Mater. Chem. A, 2021, 9(12), 7317–7335 RSC.
- W. Xiao, et al., Stable all-solid-state battery enabled with Li6.25PS5.25Cl0.75 as fast ion-conducting electrolyte, J. Energy Chem., 2021, 53, 147–154 CrossRef.
- A. Ja’farawy, et al., A Review: The Development of SiO2/C Anode Materials for Lithium-Ion Batteries, J. Electron. Mater., 2021, 50(12), 6667–6687 CrossRef.
- A. Eftekhari, Low voltage anode materials for lithium-ion batteries, Energy Storage Mater., 2017, 7, 157–180 CrossRef.
- Y. Hamon, et al., Aluminum negative electrode in lithium ion batteries, J. Power Sources, 2001, 97, 185–187 CrossRef.
- C. Li, et al., Metallic aluminum nanorods: synthesis via vapor-deposition and applications in Al/air batteries, Chem. Mater., 2007, 19(24), 5812–5814 CrossRef CAS.
- M. Au, et al., Free standing aluminum nanostructures as anodes for Li-ion rechargeable batteries, J. Power Sources, 2010, 195(10), 3333–3337 CrossRef CAS.
- S. Sharma, et al., Al nanorod thin films as anode electrode for Li ion rechargeable batteries, Electrochim. Acta, 2013, 87, 872–879 CrossRef CAS.
- M. Lindsay, G. Wang and H. Liu, Al-based anode materials for Li-ion batteries, J. Power Sources, 2003, 119, 84–87 CrossRef.
- R. Hu, et al., Investigation of immiscible alloy system of Al–Sn thin films as anodes for lithium ion batteries, Electrochem. Commun., 2008, 10(7), 1109–1112 CrossRef CAS.
- R. Hu, et al., Microstructure and electrochemical performance of thin film anodes for lithium ion batteries in immiscible Al–Sn system, J. Power Sources, 2009, 188(1), 268–273 CrossRef CAS.
- Q.-H. Wu, et al., An alumina-coated Fe3O4-reduced graphene oxide composite electrode as a stable anode for lithium-ion battery, Electrochim. Acta, 2015, 156, 147–153 CrossRef CAS.
- S. Kuksenko, Aluminum foil as anode material of lithium-ion batteries: Effect of electrolyte compositions on cycling parameters, Russ. J. Electrochem., 2013, 49(1), 67–75 CrossRef CAS.
- P. Liu, et al., Fabrication and characterization of porous Si-Al films anode with different macroporous substrates for lithium-ion batteries, J. Solid State Electrochem., 2014, 18(7), 1799–1806 CrossRef CAS.
- B. Li, et al., Hollow carbon nanospheres/silicon/alumina core–shell film as an anode for lithium-ion batteries, Sci. Rep., 2015, 5(1), 1–9 Search PubMed.
- E. Sugawara and H. Nikaido, Properties of AdeABC and AdeIJK efflux systems of Acinetobacter baumannii compared with those of the AcrAB-TolC system of Escherichia coli, Antimicrob. Agents Chemother., 2014, 58(12), 7250–7257 CrossRef PubMed.
- L. Chen, et al., Si–Al thin film anode material with superior cycle performance and rate capability for lithium ion batteries, Electrochim. Acta, 2008, 53(28), 8149–8153 CrossRef CAS.
- S. K. Sharma, et al., Review on Se-and S-doped hydrogenated amorphous silicon thin films, Indian J. Pure Appl. Phys., 2015, 52(5), 293–313 Search PubMed.
- J. Wu, et al., A review of PEM fuel cell durability: Degradation mechanisms and mitigation strategies, J. Power Sources, 2008, 184, 104–119 CrossRef CAS.
- S. Sharma, J. Baveja and R. Mehra, Study of thermal equilibration in selenium-and sulphur-doped a-Si: H, Int. J. Electron., 2003, 90(7), 423–431 CrossRef CAS.
- S. Sharma, et al., Optical properties of Se or S-doped hydrogenated amorphous silicon thin films with annealing temperature and dopant concentration, J. Alloys Compd., 2011, 509(7), 3338–3342 CrossRef CAS.
- J. W. Silverstone, et al., On-chip quantum interference between silicon photon-pair sources, Nat. Photonics, 2014, 8(2), 104–108 CrossRef CAS.
- S. Durán, et al., Silicon-nanowire based attachment of silicon chips for mouse embryo labelling, Lab Chip, 2015, 15(6), 1508–1514 RSC.
- X. Li, et al., Mesoporous silicon sponge as an anti-pulverization structure for high-performance lithium-ion battery anodes, Nat. Commun., 2014, 5(1), 1–7 Search PubMed.
- G. Sharma, et al., Biomass as a sustainable resource for value-added modern materials: a review, Biofuels, Bioprod. Biorefin., 2020, 14(3), 673–695 CrossRef CAS.
- C. Erk, et al., Toward silicon anodes for next-generation lithium ion batteries: a comparative performance study of various polymer binders and silicon nanopowders, ACS Appl. Mater. Interfaces, 2013, 5(15), 7299–7307 CrossRef CAS PubMed.
- B. Liang, Y. Liu and Y. Xu, Silicon-based materials as high capacity anodes for next generation lithium ion batteries, J. Power Sources, 2014, 267, 469–490 CrossRef CAS.
- D. M. Piper, et al., Stable silicon-ionic liquid interface for next-generation lithium-ion batteries, Nat. Commun., 2015, 6(1), 1–10 Search PubMed.
- D. S. Jung, et al., Recycling rice husks for high-capacity lithium battery anodes, Proc. Natl. Acad. Sci., 2013, 110(30), 12229–12234 CrossRef CAS PubMed.
- D. P. Wong, et al., Binder-free rice husk-based silicon–graphene composite as energy efficient Li-ion battery anodes, J. Mater. Chem. A, 2014, 2(33), 13437–13441 RSC.
- N. Liu, et al., Rice husks as a sustainable source of nanostructured silicon for high performance Li-ion battery anodes, Sci. Rep., 2013, 3(1), 1–7 Search PubMed.
- A. Xing, et al., Mesoporous silicon engineered by the reduction of biosilica from rice husk as a high-performance anode for lithium-ion batteries, RSC Adv., 2013, 3(26), 10145–10149 RSC.
- M. K. Datta, et al., Amorphous silicon–carbon based nano-scale thin film anode materials for lithium ion batteries, Electrochim. Acta, 2011, 56(13), 4717–4723 CrossRef CAS.
- J. Wang, et al., Amorphous silicon/carbon multilayer thin films as the anode for high rate rechargeable Li-ion batteries, Mater. Lett., 2013, 97, 37–39 CrossRef CAS.
- M. Li, et al., Amorphous-silicon@ silicon oxide/chromium/carbon as an anode for lithium-ion batteries with excellent cyclic stability, Electrochim. Acta, 2015, 164, 163–170 CrossRef CAS.
- M. Ko, et al., Elastic a-silicon nanoparticle backboned graphene hybrid as a self-compacting anode for high-rate lithium ion batteries, ACS Nano, 2014, 8(8), 8591–8599 CrossRef CAS PubMed.
- L. Chen, et al., An amorphous Si thin film anode with high capacity and long cycling life for lithium ion batteries, J. Appl. Electrochem., 2009, 39(8), 1157–1162 CrossRef CAS.
- W. Wang, et al., Binder-free three-dimensional silicon/carbon nanowire networks for high performance lithium-ion battery anodes, Nano Energy, 2013, 2(5), 943–950 CrossRef CAS.
- J. S. Kim, et al., Bundle-type silicon nanorod anodes produced by electroless etching using silver ions and their electrochemical characteristics in lithium ion cells, Int. J. Hydrogen Energy, 2014, 39(36), 21420–21428 CrossRef CAS.
- J. Guo, X. Chen and C. Wang, Carbon scaffold structured silicon anodes for lithium-ion batteries, J. Mater. Chem., 2010, 20(24), 5035–5040 RSC.
- H.-C. Tao, et al., Double-walled core–shell structured Si@ SiO2@ C nanocomposite as anode for lithium-ion batteries, Ionics, 2014, 20(11), 1547–1552 CrossRef CAS.
- S. Ivanov, et al., Electrochemical performance of nanoporous Si as anode for lithium ion batteries in alkyl carbonate and ionic liquid-based electrolytes, J. Appl. Electrochem., 2014, 44(1), 159–168 CrossRef CAS.
- J. Wang, et al., Electrochemical stability of optimized Si/C composites anode for lithium-ion batteries, Ionics, 2015, 21(2), 579–585 CrossRef.
- B. Liu, et al., Hierarchical silicon nanowires-carbon textiles matrix as a binder-free anode for high-performance advanced lithium-ion batteries, Sci. Rep., 2013, 3(1), 1–7 Search PubMed.
- C. K. Chan, et al., High-performance lithium battery anodes using silicon nanowires, Nat. Nanotechnol., 2008, 3(1), 31–35 CrossRef CAS PubMed.
- N. Yan, et al., Hollow porous SiO2 nanocubes towards high-performance anodes for lithium-ion batteries, Sci. Rep., 2013, 3(1), 1–6 Search PubMed.
- T. D. Bogart, et al., Lithium ion battery peformance of silicon nanowires with carbon skin, ACS Nano, 2014, 8(1), 915–922 CrossRef CAS.
- S. Iwamura, et al., Li-rich Li-Si alloy as a lithium-containing negative electrode material towards high energy lithium-ion batteries, Sci. Rep., 2015, 5(1), 1–8 Search PubMed.
- D. Qiu, et al., Mesoporous silicon microspheres fabricated via in situ magnesiothermic reduction of silicon oxide as a high-performance anode material for lithium–ion batteries, J. Solid State Electrochem., 2015, 19(3), 935–939 CrossRef CAS.
- X. H. Liu, et al., Size-dependent fracture of silicon nanoparticles during lithiation, ACS Nano, 2012, 6(2), 1522–1531 CrossRef CAS.
- C.-y Jung, et al., Effects of phosphorous incorporation on the microstructure of Si nanoparticles as an anode material for lithium-ion battery, Thin Solid Films, 2015, 587, 142–149 CrossRef CAS.
- W. Wang, et al., Monodisperse porous silicon spheres as anode materials for lithium ion batteries, Sci. Rep., 2015, 5(1), 1–6 CAS.
- Z. Favors, et al., Towards scalable binderless electrodes: carbon coated silicon nanofiber paper via Mg reduction of electrospun SiO2 nanofibers, Sci. Rep., 2015, 5(1), 1–7 Search PubMed.
- M. Feng, et al., Nano-silicon/polyaniline composites with an enhanced reversible capacity as anode materials for lithium ion batteries, J. Solid State Electrochem., 2015, 19(6), 1773–1782 CrossRef CAS.
- H. Anodes, Nanostructured hybrid silicon/carbon, ACS Nano, 2010, 4, 2233–2241 CrossRef PubMed.
- Y. Zhou, et al., Novel mesoporous silicon nanorod as an anode material for lithium ion batteries, Electrochim. Acta, 2014, 127, 252–258 CrossRef CAS.
- W. Xiao, et al., Novel silicon–oxygen–carbon composite with excellent cycling steady performance as anode for lithium-ion batteries, Ionics, 2015, 21(8), 2149–2153 CrossRef CAS.
- H.-C. Tao, et al., Polyaniline encapsulated silicon nanocomposite as high-performance anode materials for lithium ion batteries, J. Solid State Electrochem., 2014, 18(7), 1989–1994 CrossRef CAS.
- N. Liu, et al., A pomegranate-inspired nanoscale design for large-volume-change lithium battery anodes, Nat. Nanotechnol., 2014, 9(3), 187–192 CrossRef CAS PubMed.
- L. Yue, et al., Porous Si coated with S-doped carbon as anode material for lithium ion batteries, J. Solid State Electrochem., 2013, 17(4), 961–968 CrossRef CAS.
- N. Liu, et al., Prelithiated silicon nanowires as an anode for lithium ion batteries, ACS Nano, 2011, 5(8), 6487–6493 CrossRef CAS.
- T. Song, Y. Jeon and U. Paik, Si nanotubes array sheathed with SiN/SiOxNy layer as an anode material for lithium ion batteries, J. Electroceram., 2014, 32(1), 66–71 CrossRef CAS.
- Z. Edfouf, et al., Nanostructured Ni3.5Sn4 intermetallic compound: An efficient buffering material for Si-containing composite anodes in lithium ion batteries, Electrochim. Acta, 2013, 89, 365–371 CrossRef CAS.
- T. Song, et al., Si/Ge double-layered nanotube array as a lithium ion battery anode, ACS Nano, 2012, 6(1), 303–309 CrossRef CAS PubMed.
- S. Li, et al., Silicon/carbon composite microspheres with hierarchical core–shell structure as anode for lithium ion batteries, Electrochem. Commun., 2014, 49, 98–102 CrossRef CAS.
- M.-S. Wang, et al., Highly uniform silicon nanoparticle/porous carbon nanofiber hybrids towards free-standing high-performance anodes for lithium-ion batteries, Carbon, 2015, 82, 337–345 CrossRef CAS.
- H. Wu, et al., Stable Li-ion battery anodes by in-situ polymerization of conducting hydrogel to conformally coat silicon nanoparticles, Nat. Commun., 2013, 4(1), 1–6 Search PubMed.
- H. Chen, et al., Silicon nanowires with and without carbon coating as anode materials for lithium-ion batteries, J. Solid State Electrochem., 2010, 14(10), 1829–1834 CrossRef CAS.
- J. Xie, et al., Comparing one-and two-dimensional heteronanostructures as silicon-based lithium ion battery anode materials, ACS Nano, 2011, 5(11), 9225–9231 CrossRef CAS PubMed.
- L. Hu, et al., Silicon-conductive nanopaper for Li-ion batteries, Nano Energy, 2013, 2(1), 138–145 CrossRef CAS.
- H. Tang, et al., Self-assembly silicon/porous reduced graphene oxide composite film as a binder-free and flexible anode for lithium-ion batteries, Electrochim. Acta, 2015, 156, 86–93 CrossRef CAS.
- A. R. Park, et al., Si–Mn/Reduced graphene oxide nanocomposite anodes with enhanced capacity and stability for lithium-ion batteries, ACS Appl. Mater. Interfaces, 2014, 6(3), 1702–1708 CrossRef CAS PubMed.
- Y. Yang, et al., ATF-2/CREB/IRF-3-targeted anti-inflammatory activity of Korean red ginseng water extract, J. Ethnopharmacol., 2014, 154(1), 218–228 CrossRef CAS PubMed.
- X. Zhou, et al., Spin-coated silicon nanoparticle/graphene electrode as a binder-free anode for high-performance lithium-ion batteries, Nano Res., 2012, 5(12), 845–853 CrossRef CAS.
- Z. Favors, et al., Stable cycling of SiO2 nanotubes as high-performance anodes for lithium-ion batteries, Sci. Rep., 2014, 4(1), 1–7 Search PubMed.
- J. Rong, et al., Tandem structure of porous silicon film on single-walled carbon nanotube macrofilms for lithium-ion battery applications, ACS Nano, 2010, 4(8), 4683–4690 CrossRef CAS PubMed.
- K. Evanoff, et al., Ultra strong silicon-coated carbon nanotube nonwoven fabric as a multifunctional lithium-ion battery anode, ACS Nano, 2012, 6(11), 9837–9845 CrossRef CAS PubMed.
- X. Chen, et al., Virus-enabled silicon anode for lithium-ion batteries, ACS Nano, 2010, 4(9), 5366–5372 CrossRef CAS PubMed.
- K. H. Kim, et al., Complete magnesiothermic reduction reaction of vertically aligned mesoporous silica channels to form pure silicon nanoparticles, Sci. Rep., 2015, 5(1), 1–7 Search PubMed.
- J. S. Kim, et al., Amorphous carbon-coated prickle-like silicon of micro and nano hybrid anode materials for lithium-ion batteries, Solid State Ionics, 2014, 260, 36–42 CrossRef CAS.
- H.-C. Tao, et al., Reduced graphene oxide/porous Si composite as anode for high-performance lithium ion batteries, Ionics, 2015, 21(3), 617–622 CrossRef CAS.
- D. Sui, et al., A Comprehensive Review of Graphene-Based Anode Materials for Lithium-ion Capacitors, Chemistry, 2021, 3(4), 1215–1246 CrossRef CAS.
- S. Yoon, et al., Carbon nanotube film anodes for flexible lithium ion batteries, J. Power Sources, 2015, 279, 495–501 CrossRef CAS.
- A. K. Rai, et al., Partially reduced Co3O4/graphene nanocomposite as an anode material for secondary lithium ion battery, Electrochim. Acta, 2013, 100, 63–71 CrossRef CAS.
- P. Guo, H. Song and X. Chen, Electrochemical performance of graphene nanosheets as anode material for lithium-ion batteries, Electrochem. Commun., 2009, 11(6), 1320–1324 CrossRef CAS.
- N. Yan, et al., Fe2O3 nanoparticles wrapped in multi-walled carbon nanotubes with enhanced lithium storage capability, Sci. Rep., 2013, 3(1), 1–6 CrossRef.
- W. Xiao, et al., Fe2O3 particles enwrapped by graphene with excellent cyclability and rate capability as anode materials for lithium ion batteries, Appl. Surf. Sci., 2013, 266, 148–154 CrossRef CAS.
- Y. Yang, et al., Three-dimensional nanoporous Fe2O3/Fe3C-graphene heterogeneous thin films for lithium-ion batteries, ACS Nano, 2014, 8(4), 3939–3946 CrossRef CAS PubMed.
- J. Kan and Y. Wang, Large and fast reversible Li-ion storages in Fe2O3-graphene sheet-on-sheet sandwich-like nanocomposites, Sci. Rep., 2013, 3(1), 1–10 Search PubMed.
- Z.-S. Wu, et al., Graphene anchored with Co3O4 nanoparticles as anode of lithium ion batteries with enhanced reversible capacity and cyclic performance, ACS Nano, 2010, 4(6), 3187–3194 CrossRef CAS PubMed.
- J. Lin, et al., Graphene nanoribbon and nanostructured SnO2 composite anodes for lithium ion batteries, ACS Nano, 2013, 7(7), 6001–6006 CrossRef CAS PubMed.
- S.-H. Lee, et al., Graphene–nanotube–iron hierarchical nanostructure as lithium ion battery anode, ACS Nano, 2013, 7(5), 4242–4251 CrossRef CAS PubMed.
- M. Zhou, et al., Graphene/carbon-coated Si nanoparticle hybrids as high-performance anode materials for Li-ion batteries, ACS Appl. Mater. Interfaces, 2013, 5(8), 3449–3455 CrossRef CAS PubMed.
- J. Guo, et al., Graphene-encapsulated cobalt sulfides nanocages with excellent anode performances for lithium ion batteries, Electrochim. Acta, 2015, 167, 32–38 CrossRef CAS.
- X. Liu, et al., Gram-scale synthesis of graphene-mesoporous SnO2 composite as anode for lithium-ion batteries, Electrochim. Acta, 2015, 152, 178–186 CrossRef CAS.
- J. Zhang, et al., High-capacity graphene oxide/graphite/carbon nanotube composites for use in Li-ion battery anodes, Carbon, 2014, 74, 153–162 CrossRef CAS.
- B.-K. Zou, et al., Hydrothermally enhanced MnO/reduced graphite oxide composite anode materials for high performance lithium-ion batteries, Electrochim. Acta, 2015, 167, 25–31 CrossRef CAS.
- L.-F. Cui, et al., Light-weight free-standing carbon nanotube-silicon films for anodes of lithium ion batteries, ACS Nano, 2010, 4(7), 3671–3678 CrossRef CAS PubMed.
- R. Yuge, et al., High-rate charge/discharge properties of Li-ion battery using carbon-coated composites of graphites, vapor grown carbon fibers, and carbon nanohorns, J. Power Sources, 2014, 266, 471–474 CrossRef CAS.
- Y. Xu, Y. Zhu and C. Wang, Mesoporous carbon/silicon composite anodes with enhanced performance for lithium-ion batteries, J. Mater. Chem. A, 2014, 2(25), 9751–9757 RSC.
- F. Zheng, Y. Yang and Q. Chen, High lithium anodic performance of highly nitrogen-doped porous carbon prepared from a metal-organic framework, Nat. Commun., 2014, 5(1), 1–10 Search PubMed.
- Z. Lu, et al., Nonfilling carbon coating of porous silicon micrometer-sized particles for high-performance lithium battery anodes, ACS Nano, 2015, 9(3), 2540–2547 CrossRef CAS PubMed.
- L. Wang, et al., Porous carbon nanotubes decorated with nanosized cobalt ferrite as anode materials for high-performance lithium-ion batteries, J. Power Sources, 2015, 283, 289–299 CrossRef CAS.
- L. Chen, et al., Porous graphitic carbon nanosheets as a high-rate anode material for lithium-ion batteries, ACS Appl. Mater. Interfaces, 2013, 5(19), 9537–9545 CrossRef CAS PubMed.
- T. Chen, et al., Porous nitrogen-doped carbon microspheres as anode materials for lithium ion batteries, Dalton Trans., 2014, 43(40), 14931–14935 RSC.
- H. Li, et al., Reduced graphene oxide/boron nitride composite film as a novel binder-free anode for lithium ion batteries with enhanced performances, Electrochim. Acta, 2015, 166, 197–205 CrossRef CAS.
- J. He, et al., Highly-flexible 3D Li2S/graphene cathode for high-performance lithium sulfur batteries, J. Power Sources, 2016, 327, 474–480 CrossRef CAS.
- A. R. Kamali and D. J. Fray, Tin-based materials as advanced anode materials for lithium ion batteries: a review, Rev. Adv. Mater. Sci., 2011, 27(1), 14–24 CAS.
- F. Xin and M. S. Whittingham, Challenges and development of tin-based anode with high volumetric capacity for Li-ion batteries, Electrochem. Energy Rev., 2020, 3(4), 643–655 CrossRef CAS.
- M. Mousavi, et al., Antimony doped SnO2 nanowire@ C core–shell structure as a high-performance anode material for lithium-ion battery, Nanotechnology, 2021, 32(28), 285403 CrossRef CAS PubMed.
- X. Zhou, L. J. Wan and Y. G. Guo, Binding SnO2 nanocrystals in nitrogen-doped graphene sheets as anode materials for lithium-ion batteries, Adv. Mater., 2013, 25(15), 2152–2157 CrossRef CAS PubMed.
- R. Hu, M. Zeng and M. Zhu, Cyclic durable high-capacity Sn/Cu6Sn5 composite thin film anodes for lithium ion batteries prepared by electron-beam evaporation deposition, Electrochim. Acta, 2009, 54(10), 2843–2850 CrossRef CAS.
- Y. Wang, et al., Designed hybrid nanostructure with catalytic effect: beyond the theoretical capacity of SnO2 anode material for lithium ion batteries, Sci. Rep., 2015, 5(1), 1–8 CAS.
- L. Liu, et al., Superior cycle performance and high reversible capacity of SnO2/graphene composite as an anode material for lithium-ion batteries, Sci. Rep., 2015, 5(1), 1–10 CAS.
- Y. Xu, J. Guo and C. Wang, Sponge-like porous carbon/tin composite anode materials for lithium ion batteries, J. Mater. Chem., 2012, 22(19), 9562–9567 RSC.
- L. Ding, et al., Ultrasmall SnO2 nanocrystals: hot-bubbling synthesis, encapsulation in carbon layers and applications in high capacity Li-ion storage, Sci. Rep., 2014, 4(1), 1–8 Search PubMed.
- C. Li, et al., Cathode materials modified by surface coating for lithium ion batteries, Electrochim. Acta, 2006, 51(19), 3872–3883 CrossRef CAS.
- S.-T. Myung, K. Amine and Y.-K. Sun, Surface modification of cathode materials from nano-to microscale for rechargeable lithium-ion batteries, J. Mater. Chem., 2010, 20(34), 7074–7095 RSC.
- D. Qian, et al., Lithium lanthanum titanium oxides: a fast ionic conductive coating for lithium-ion battery cathodes, Chem. Mater., 2012, 24(14), 2744–2751 CrossRef CAS.
- H. Chen, et al., Construction of core–shell Li3PO4@ LiNi0·5Co0·2Mn0·3O2 cathode with improved cycling stability for lithium ion batteries, Electrochim. Acta, 2020, 344, 136142 CrossRef CAS.
- D. Becker, et al., Surface modification of Ni-rich LiNi0.8Co0.1Mn0.1O2 cathode material by tungsten oxide coating for improved electrochemical performance in lithium-ion batteries, ACS Appl. Mater. Interfaces, 2019, 11(20), 18404–18414 CrossRef CAS PubMed.
- T. Aida, et al., Ammonium tungstate modified Li-rich Li1+xNi0.35Co0.35Mn0.30O2 to improve rate capability and productivity of lithium-ion batteries, J. Solid State Electrochem., 2017, 21(7), 2047–2054 CrossRef CAS.
- K. Wu, et al., Effect of precursor and synthesis temperature on the structural and electrochemical properties of Li (Ni0.5Co0.2Mn0.3)O2, Electrochim. Acta, 2012, 75, 393–398 CrossRef CAS.
- L. Wang and Y. H. Hu, Surface modification of LiNi0.5Co0.2Mn0.3O2 cathode materials with Li2O–B2O3–LiBr for lithium-ion batteries, Int. J. Energy Res., 2019, 43(9), 4644–4651 CrossRef CAS.
- Z. Gan, et al., Surface modification of LiNi0.8Co0.1Mn0.1O2 by WO3 as a cathode material for LIB, Appl. Surf. Sci., 2019, 481, 1228–1238 CrossRef CAS.
- H. Zhang, J. Xu and J. Zhang, Surface-coated LiNi0.8Co0.1Mn0.1O2 (NCM811) cathode materials by Al2O3, ZrO2, and Li2O-2B2O3 thin-layers for improving the performance of lithium ion batteries, Front. Mater., 2019, 309 CrossRef CAS.
- H. W. Chan, J. G. Duh and S. R. Sheen, Surface treatment of the lithium boron oxide coated LiMn2O4 cathode material in Li-ion battery, Key Engineering Materials, Trans Tech Publ, 2005 Search PubMed.
- G. Li, et al., One-time sintering process to synthesize ZrO2-coated LiMn2O4 materials for lithium-ion batteries, RSC Adv., 2018, 8(30), 16753–16761 RSC.
- J. Wu, et al., Nanocoating of Ce-tannic acid metal-organic coordination complex: surface modification of layered Li1.2Mn0.6Ni0.2O2 by CeO2 coating for lithium-ion batteries, Ionics, 2019, 25(7), 3031–3040 CrossRef CAS.
- S. N. Lim, et al., Enhanced elevated-temperature performance of Li (Ni0.8Co0.15Al0.05) O2 electrodes coated with Li2O-2B2O3 glass, Electrochim. Acta, 2014, 136, 1–9 CrossRef CAS.
- C. B. Lim and Y. J. Park, Precursor-based surface modification of cathodes using Ta and W for sulfide-based all-solid-state batteries, Sci. Rep., 2020, 10(1), 1–12 CrossRef PubMed.
- S.-J. Sim, et al., Use of carbon coating on LiNi0.8Co0.1Mn0.1O2 cathode material for enhanced performances of lithium-ion batteries, Sci. Rep., 2020, 10(1), 1–9 CrossRef PubMed.
- J. Zhou, et al., In situ formed Li5AlO4-coated LiNi0·8Co0·1Mn0·1O2 cathode material assisted by hydrocarbonate with improved electrochemical performance for lithium-ion batteries, Electrochim. Acta, 2020, 353, 136541 CrossRef CAS.
- W. Zhang, et al., Ni-rich LiNi0·8Co0·1Mn0·1O2 coated with Li-ion conductive Li3PO4 as competitive cathodes for high-energy-density lithium ion batteries, Electrochim. Acta, 2020, 340, 135871 CrossRef CAS.
- S. K. Singh, D. Dutta and R. K. Singh, Enhanced structural and cycling stability of Li2CuO2-coated LiNi0.33Mn0.33Co0.33O2 cathode with flexible ionic liquid-based gel polymer electrolyte for lithium polymer batteries, Electrochim. Acta, 2020, 343, 136122 CrossRef CAS.
- X. Chen, et al., A highly stabilized single crystalline nickel-rich LiNi0.8Co0.1Mn0.1O2 cathode through a novel surface spinel-phase modification, Electrochim. Acta, 2020, 341, 136075 CrossRef CAS.
- L. Xu, et al., Ti3C2 (OH) 2 coated Li (Ni0.6Co0.2Mn0.2) O2 cathode material with enhanced electrochemical properties for lithium ion battery, Electrochim. Acta, 2018, 289, 120–130 CrossRef CAS.
- Z. Yan, et al., Nickel catalyzed graphitized carbon coated LiFe1-xNixPO4 composites as cathode material for high-performance lithium-ion batteries, Electrochim. Acta, 2020, 353, 136565 CrossRef CAS.
- Y. Chen, et al., Surface dual-shell construction enhances the electrochemical performances of Li1· 2Ni0· 13Co0· 13Mn0· 54O2 cathode materials, Electrochim. Acta, 2020, 341, 136082 CrossRef CAS.
- X. Chen, et al., Nitrogen-doped carbon coated LiNi0.6Co0.2Mn0.2O2 cathode with enhanced electrochemical performance for Li-Ion batteries, Electrochim. Acta, 2018, 284, 526–533 CrossRef CAS.
- Y. He, et al., Enhancement of the high-voltage electrochemical performance of an LiNi0.5Co0.2Mn0.3O2 cathode via WO3 coating, Appl. Surf. Sci., 2020, 508, 145259 CrossRef CAS.
- X.-D. Zhang, et al., An effective LiBO2 coating to ameliorate the cathode/electrolyte interfacial issues of LiNi0.6Co0.2Mn0.2O2 in solid-state Li batteries, J. Power Sources, 2019, 426, 242–249 CrossRef CAS.
- Z. Zhong, et al., Nano LiFePO4 coated Ni rich composite as cathode for lithium ion batteries with high thermal ability and excellent cycling performance, J. Power Sources, 2020, 464, 228235 CrossRef CAS.
- H. H. Sun, et al., Transition metal-doped Ni-rich layered cathode materials for durable Li-ion batteries, Nat. Commun., 2021, 12(1), 1–11 CrossRef.
- F. Zheng, et al., Nanoscale gadolinium doped ceria (GDC) surface modification of Li-rich layered oxide as a high performance cathode material for lithium ion batteries, Chem. Eng. J., 2018, 334, 497–507 CrossRef CAS.
- S. Bhuvaneswari, et al., Sc-doping induced cation-disorder in LiNi0.5Mn1.5O4 spinel leading to improved electrochemical performance as cathode in lithium ion batteries, Electrochim. Acta, 2019, 327, 135008 CrossRef CAS.
- Y.-C. Li, et al., Construction of homogeneously Al3+ doped Ni rich Ni-Co-Mn cathode with high stable cycling performance and storage stability via scalable continuous precipitation, Electrochim. Acta, 2018, 291, 84–94 CrossRef CAS.
- K. Ariyoshi, et al., Effect of Primary Particle Size upon Polarization and Cycling Stability of 5-V Lithium Insertion Material of Li [Ni1/2Mn3/2]O4, J. Electrochem. Soc., 2011, 158(3), A281 CrossRef CAS.
- E.-S. Lee, et al., Influence of cation ordering and lattice distortion on the charge–discharge behavior of LiMn1.5Ni0.5O4 spinel between 5.0 and 2.0 V, Chem. Mater., 2012, 24(18), 3610–3620 CrossRef CAS.
- Z. Huang, et al., Structural and electrochemical properties of Mg-doped nickel based cathode materials LiNi0.6Co0.2Mn0.2−xMgxO2 for lithium ion batteries, RSC Adv., 2015, 5(108), 88773–88779 RSC.
- D. Y. Wan, et al., Effect of metal (Mn, Ti) doping on NCA cathode materials for lithium ion batteries, J. Nanomater., 2018, 2018, 8082502 CrossRef.
- S. Choi, et al., F-doped Li1.15Ni0.275Ru0.575O2 cathode materials with long cycle life and improved rate performance, Electrochim. Acta, 2019, 326, 135015 CrossRef CAS.
- D. Zhang, et al., Effect of Ti ion doping on electrochemical performance of Ni-rich LiNi0.8Co0.1Mn0.1O2 cathode material, Electrochim. Acta, 2019, 328, 135086 CrossRef CAS.
- L. Wang, et al., Nb5+ doped LiV3O8 nanorods with extraordinary rate performance and cycling stability as cathodes for lithium-ion batteries, Electrochim. Acta, 2018, 284, 366–375 CrossRef CAS.
- C. Lv, et al., 1D Nb-doped LiNi1/3Co1/3Mn1/3O2 nanostructures as excellent cathodes for Li-ion battery, Electrochim. Acta, 2019, 297, 258–266 CrossRef CAS.
- F. Vásquez and J. Calderón, Vanadium doping of LiMnPO4 cathode material: correlation between changes in the material lattice and the enhancement of the electrochemical performance, Electrochim. Acta, 2019, 325, 134930 CrossRef.
- G.-T. Park, et al., Tungsten doping for stabilization of Li [Ni0.90Co0.05Mn0.05] O2 cathode for Li-ion battery at high voltage, J. Power Sources, 2019, 442, 227242 CrossRef CAS.
- S. Dong, et al., Understanding electrochemical performance improvement with Nb doping in lithium-rich manganese-based cathode materials, J. Power Sources, 2020, 462, 228185 CrossRef CAS.
- X. Yang, et al., Bifunctional nano-ZrO2 modification of LiNi0·92Co0.08O2 cathode enabling high-energy density lithium ion batteries, J. Power Sources, 2019, 438, 226978 CrossRef CAS.
- X. Liu, et al., Stabilizing the high-voltage cycle performance of LiNi0.8Co0.1Mn0.1O2 cathode material by Mg doping, J. Power Sources, 2019, 438, 227017 CrossRef CAS.
- H. Lee, et al., Carbon-free Mn-doped LiFePO4 cathode for highly transparent thin-film batteries, J. Power Sources, 2019, 434, 226713 CrossRef CAS.
- Z. Pu, et al., Preparation of W-doped hierarchical porous Li4Ti5O12/brookite nanocomposites for high rate lithium ion batteries at −20 °C, J. Power Sources, 2019, 437, 226890 CrossRef CAS.
- X.-L. Wu, et al., LiFePO4 nanoparticles embedded in a nanoporous carbon matrix: superior cathode material for electrochemical energy-storage devices, Adv. Mater., 2009, 21(25–26), 2710–2714 CrossRef CAS.
- Z. Ma, et al., Tunable morphology synthesis of LiFePO4 nanoparticles as cathode materials for lithium ion batteries, ACS Appl. Mater. Interfaces, 2014, 6(12), 9236–9244 CrossRef CAS PubMed.
- C. Li, et al., Preparation and characterization LiFePO4/C nanowires and their improved performance for lithium-ion batteries, Ionics, 2015, 21(9), 2465–2469 CrossRef CAS.
- L. Peng, et al., Self-assembled LiFePO4 nanowires with high rate capability for Li-ion batteries, Chem. Commun., 2014, 50(67), 9569–9572 RSC.
- E. Hosono, et al., Synthesis of triaxial LiFePO4 nanowire with a VGCF core column and a carbon shell through the electrospinning method, ACS Appl. Mater. Interfaces, 2010, 2(1), 212–218 CrossRef CAS PubMed.
- X. Xiao, et al., Facile synthesis of LiCoO2 nanowires with high electrochemical performance, Nano Res., 2012, 5(1), 27–32 CrossRef CAS.
- X. Li, et al., Template-synthesized LiCoO2, LiMn2O4, and LiNi0.8Co0.2O2 nanotubes as the cathode materials of lithium ion batteries, J. Phys. Chem. B, 2005, 109(29), 14017–14024 CrossRef CAS.
- H. Xia, et al., Facile synthesis of chain-like LiCoO2 nanowire arrays as three-dimensional cathode for microbatteries, NPG Asia Mater., 2014, 6(9), e126 CrossRef CAS.
- M. Yoon, et al., All-solid-state thin film battery based on well-aligned slanted LiCoO2 nanowires fabricated by glancing angle deposition, Appl. Surf. Sci., 2017, 412, 537–544 CrossRef CAS.
- Z.-L. Gong, et al., Effects of preparation methods of LiNi0.8Co0.2O2 cathode materials on their morphology and electrochemical performance, J. Power Sources, 2004, 136(1), 139–144 CrossRef CAS.
- D. Jiang, et al., Preparation and characterization of layered LiNi0.9Co0.05Mn0.025Mg0.025O2 cathode material by a sol–gel method for lithium-ion batteries, RSC Adv., 2015, 5(51), 40779–40784 RSC.
- V. Voronov, et al., Effect of the preparation method of the cathode material LiNi0.33Mn0.33Co0.33O2 on the electrochemical characteristics of a lithium ion cell, Russ. J. Inorg. Chem., 2016, 61(9), 1153–1159 CrossRef CAS.
- P. Hou, et al., Optimize hydrothermal synthesis and electrochemical performance of Li2FeTiO4 composite cathode materials by using orthogonal experimental design method, Ionics, 2020, 26(4), 1657–1662 CrossRef CAS.
- Y. Xu, et al., Template-free hydrothermal synthesis of Li 2 FeSiO4 hollow spheres as cathode materials for lithium-ion batteries, J. Mater. Chem. A, 2014, 2(32), 12982–12990 RSC.
- H. Wu, et al., One-step synthesis LiMn2O4 cathode by a hydrothermal method, J. Power Sources, 2006, 161(2), 1260–1263 CrossRef CAS.
- Y. Shi, et al., Urea-based hydrothermal synthesis of LiNi0.5Co0.2Mn0.3O2 cathode material for Li-ion battery, J. Power Sources, 2018, 394, 114–121 CrossRef CAS.
- B. Jin and H.-B. Gu, Preparation and characterization of LiFePO4 cathode materials by hydrothermal method, Solid State Ionics, 2008, 178(37–38), 1907–1914 CrossRef CAS.
- T.-Y. Huang, et al., Supramolecular photothermal nanomedicine mediated distant tumor inhibition via PD-1 and TIM-3 blockage, Front. Chem., 2020, 8, 1 CrossRef CAS PubMed.
- L. Zhang and H. Liang, Rapid synthesis of LiFePO4 nanoparticles by microwave-assisted hydrothermal method, Russ. J. Electrochem., 2013, 49(5), 492–495 CrossRef CAS.
- X.-F. Guo, H. Zhan and Y.-H. Zhou, Rapid synthesis of LiFePO4/C composite by microwave method, Solid State Ionics, 2009, 180(4–5), 386–391 CrossRef CAS.
- C. J. Jafta, et al., Microwave-assisted synthesis of high-voltage nanostructured LiMn1.5Ni0.5O4 spinel: tuning the Mn3+ content and electrochemical performance, ACS Appl. Mater. Interfaces, 2013, 5(15), 7592–7598 CrossRef CAS.
- M. Kuezma, S. Devaraj and P. Balaya, Li2MnSiO4 obtained by microwave assisted solvothermal method: electrochemical and surface studies, J. Mater. Chem., 2012, 22(39), 21279–21284 RSC.
- D. Li, et al., Electrochemical characteristics of LiNi0.5Mn1.5O4 prepared by spray drying and post-annealing, Electrochim. Acta, 2007, 52(5), 1919–1924 CrossRef CAS.
- X. Zhao, G. Liang and D. Lin, Synthesis and characterization of Al-substituted LiNi0.5Co0.2Mn0.3O2 cathode materials by a modified co-precipitation method, RSC Adv., 2017, 7(60), 37588–37595 RSC.
- Y. Shen, et al., A convenient co-precipitation method to prepare high performance LiNi0.5Mn1.5O4 cathode for lithium ion batteries, Mater. Chem. Phys., 2020, 240, 122137 CrossRef CAS.
- S. Hashigami, et al., Improvement of Cycleability and Rate-Capability of LiNi0.5Co0.2Mn0.3O2 Cathode Materials Coated with Lithium Boron Oxide by an Antisolvent Precipitation Method, ChemistrySelect, 2019, 4(29), 8676–8681 CrossRef CAS.
- G. Liu, et al., A facile template method to synthesize significantly improved LiNi0.5Mn1.5O4 using corn stalk as a bio-template, Electrochim. Acta, 2014, 141, 141–148 CrossRef CAS.
- Z. Tan, et al., One-dimensional Hierarchical Porous Layered Oxide LiNi0.8Co0.1Mn0.1O2 Cathode for Lithium-ion Batteries via Self-template Interstitial Co-precipitation Method, Chem. Lett., 2021, 50, 1385–1387 CrossRef CAS.
- A. Arya and A. Sharma, Polymer electrolytes for lithium ion batteries: a critical study, Ionics, 2017, 23(3), 497–540 CrossRef CAS.
- A. Arya and A. L. Sharma, Structural, microstructural and electrochemical properties of dispersed-type polymer nanocomposite films, J. Phys. D: Appl. Phys., 2018, 51(4), 045504 CrossRef.
- H. Lee, et al. , Energy Environ. Sci., 2014, 7(12), 3857–3886 RSC.
- F. Hippauf, et al., Overcoming binder limitations of sheet-type solid-state cathodes using a solvent-free dry-film approach, Energy Storage Mater., 2019, 21, 390–398 CrossRef.
- X. Li, et al., LiNbO3-coated LiNi0.8Co0.1Mn0.1O2 cathode with high discharge capacity and rate performance for all-solid-state lithium battery, J. Energy Chem., 2020, 40, 39–45 CrossRef.
- K. Kisu, et al., Microstructural analyses of all-solid-state Li–S batteries using LiBH4-based solid electrolyte for prolonged cycle performance, J. Energy Chem., 2020, 50, 424–429 CrossRef.
- Y. Lu, et al., Highly stable garnet solid electrolyte based Li-S battery with modified anodic and cathodic interfaces, Energy Storage Mater., 2018, 15, 282–290 CrossRef.
- Y. Lu, et al., The carrier transition from Li atoms to Li vacancies in solid-state lithium alloy anodes, Sci. Adv., 2021, 7(38), eabi5520 CrossRef CAS PubMed.
- L. Yong-Gun, et al., High-energy long-cycling all-solid-state lithium metal batteries enabled by silver–carbon composite anodes, Nat. Energy, 2020, 5(4), 299–308 CrossRef.
- T. Yu, et al., Progress and perspectives on typical inorganic solid-state electrolytes, J. Alloys Compd., 2021, 885, 161013 CrossRef CAS.
- X. Li, et al., LiNbO3-coated LiNi0.8Co0.1Mn0.1O2 cathode with high discharge capacity and rate performance for all-solid-state lithium battery, J. Energy Chem., 2020, 40, 39–45 CrossRef.
- A. Arya and A. L. Sharma, Electrolyte for energy storage/conversion (Li+, Na+, Mg2+) devices based on PVC and their associated polymer: a comprehensive review, J. Solid State Electrochem., 2019, 23(4), 997–1059 CrossRef CAS.
- L. Zhou, et al., Solvent-engineered design of argyrodite Li6PS5X (X = Cl, Br, I) solid electrolytes with high ionic conductivity, ACS Energy Lett., 2018, 4(1), 265–270 CrossRef.
- N. Bansal, B. C. Mohanty and K. Singh, Designing composition tuned glasses with enhanced properties for use as substrate in Cu2ZnSnS4 based thin film solar cells, J. Alloys Compd., 2020, 819, 152984 CrossRef CAS.
- R. Younesi, et al. , Energy Environ. Sci., 2015, 8, 1905–1922 RSC.
- A. Arya and A. Sharma, A glimpse on all-solid-state Li-ion battery (ASSLIB) performance based on novel solid polymer electrolytes: a topical review, J. Mater. Sci., 2020, 55(15), 6242–6304 CrossRef CAS.
- M. Grünebaum, et al., Synthesis and electrochemistry of polymer based electrolytes for lithium batteries, Prog. Solid State Chem., 2014, 42(4), 85–105 CrossRef.
- L. Chen, et al., Enhancing interfacial stability in solid-state lithium batteries with polymer/garnet solid electrolyte and composite cathode framework, J. Energy Chem., 2021, 52, 210–217 CrossRef.
- L. Kong, et al., Li-ion battery fire hazards and safety strategies, Energies, 2018, 11(9), 2191 CrossRef.
- K. Liu, et al., Materials for lithium-ion battery safety, Sci. Adv., 2018, 4(6), eaas9820 CrossRef.
- Q. Wang, et al., A review of lithium ion battery failure mechanisms and fire prevention strategies, Prog. Energy Combust. Sci., 2019, 73, 95–131 CrossRef.
- Q. Wang, et al., Progress of enhancing the safety of lithium ion battery from the electrolyte aspect, Nano Energy, 2019, 55, 93–114 CrossRef CAS.
- X. Liu, et al., Thermal runaway of lithium-ion batteries without internal short circuit, Joule, 2018, 2(10), 2047–2064 CrossRef CAS.
- A. Petronico, et al., Solid–Liquid Lithium Electrolyte Nanocomposites Derived from Porous Molecular Cages, J. Am. Chem. Soc., 2018, 140(24), 7504–7509 CrossRef CAS PubMed.
- S. N. Willner, et al., Adaptation required to preserve future high-end river flood risk at present levels, Sci. Adv., 2018, 4(1), eaao1914 CrossRef.
- C.-Z. Zhao, et al., An anion-immobilized composite electrolyte for dendrite-free lithium metal anodes, Proc. Natl. Acad. Sci., 2017, 114(42), 11069–11074 CrossRef CAS.
- V. D’innocenzo, et al., Excitons versus free charges in organo-lead tri-halide perovskites, Nat. Commun., 2014, 5(1), 1–6 Search PubMed.
- B. Zhu, et al., Poly(dimethylsiloxane) thin film as a stable interfacial layer for high-performance lithium-metal battery anodes, Adv. Mater., 2017, 29(2), 1603755 CrossRef.
- R. Xu, et al., Artificial soft–rigid protective layer for dendrite-free lithium metal anode, Adv. Funct. Mater., 2018, 28(8), 1705838 CrossRef.
- X.-B. Cheng, et al., Toward safe lithium metal anode in rechargeable batteries: a review, Chem. Rev., 2017, 117(15), 10403–10473 CrossRef CAS PubMed.
- Y. Lu, et al., Electrolyte and interface engineering for solid-state sodium batteries, Joule, 2018, 2(9), 1747–1770 CrossRef CAS.
- W. Guo, et al., Surface and Interface Modification of Electrode Materials for Lithium-Ion Batteries With Organic Liquid Electrolyte, Front. Energy Res., 2020, 170 CrossRef.
- M. Du, et al., Recent advances in the interface engineering of solid-state Li-ion batteries with artificial buffer layers: challenges, materials, construction, and characterization, Energy Environ. Sci., 2019, 12(6), 1780–1804 RSC.
- H. Xu, et al., Li3N-modified garnet electrolyte for all-solid-state lithium metal batteries operated at 40 °C, Nano Lett., 2018, 18(11), 7414–7418 CrossRef CAS PubMed.
- K. Ciosek Högström, et al., The influence of PMS-additive on the electrode/electrolyte interfaces in LiFePO4/graphite Li-ion batteries, J. Phys. Chem. C, 2013, 117(45), 23476–23486 CrossRef.
- J. Zheng, et al., Interface modifications by anion receptors for high energy lithium ion batteries, J. Power Sources, 2014, 250, 313–318 CrossRef CAS.
- F. Zhao, et al., Vapor-assisted synthesis of Al2O3-coated LiCoO2 for high-voltage lithium ion batteries, Electrochim. Acta, 2015, 174, 384–390 CrossRef CAS.
- J. W. Kim, et al., Surface chemistry of LiNi0.5Mn1.5O4 particles coated by Al2O3 using atomic layer deposition for lithium-ion batteries, J. Power Sources, 2015, 274, 1254–1262 CrossRef CAS.
- S. Tao, et al., Nanoscale TiO2 membrane coating spinel LiNi0.5Mn1.5O4 cathode material for advanced lithium-ion batteries, J. Alloys Compd., 2017, 705, 413–419 CrossRef CAS.
- M.-M. Deng, et al., Enhancing the electrochemical performances of LiNi0.5Mn1.5O4 by Co3O4 surface coating, J. Alloys Compd., 2018, 762, 163–170 CrossRef CAS.
- S.-X. Zhao, et al., Improving rate performance of LiFePO4 cathode materials by hybrid coating of nano-Li3PO4 and carbon, J. Alloys Compd., 2013, 566, 206–211 CrossRef CAS.
- X. Yang, et al., Modification of LiNi0.5Mn1.5O4 high potential cathode from the inner lattice to the outer surface with Cr 3 + -doping and Li + -conductor coating, J. Mater. Chem. A, 2014, 2(27), 10359–10364 RSC.
- R. L. Puurunen, Surface chemistry of atomic layer deposition: A case study for the trimethylaluminum/water process, J. Appl. Phys., 2005, 97(12), 9 CrossRef.
- J. Elam, et al., Conformal coating on ultrahigh-aspect-ratio nanopores of anodic alumina by atomic layer deposition, Chem. Mater., 2003, 15(18), 3507–3517 CrossRef CAS.
- H. Huo, et al., Investigating the Structure of an Active Material–Carbon Interface in the Monoclinic Li3V2 (PO4) 3/C Composite Cathode, ACS Appl. Energy Mater., 2019, 2(5), 3692–3702 CrossRef CAS.
- B. Wang, et al., Nitrogen-doped Li4Ti5O12 nanosheets with enhanced lithium storage properties, J. Power Sources, 2014, 266, 150–154 CrossRef CAS.
- S. Wang, et al., Lithium chlorides and bromides as promising solid-state chemistries for fast ion conductors with good electrochemical stability, Angew. Chem., Int. Ed., 2019, 58(24), 8039–8043 CrossRef CAS PubMed.
- H. S. Shin, et al., Multilayered, Bipolar, All-Solid-State Battery Enabled by a Perovskite-Based Biphasic Solid Electrolyte, ChemSusChem, 2018, 11(18), 3184–3190 CrossRef CAS PubMed.
- Z. Wen, C. Shen and Y. Lu, Air electrode for the lithium–air batteries: Materials and structure designs, ChemPlusChem, 2015, 80(2), 270–287 CrossRef CAS.
- V. Etacheri, et al., Challenges in the development of advanced Li-ion batteries: a review, Energy Environ. Sci., 2011, 4(9), 3243–3262 RSC.
- L. Yao, et al., A Review of Lithium-Ion Battery State of Health Estimation and Prediction Methods, World Electr. Veh. J., 2021, 12(3), 113 CrossRef.
- H. Li, Practical evaluation of Li-ion batteries, Joule, 2019, 3(4), 911–914 CrossRef CAS.
- R. M. LaFollette and D. N. Bennion, Design Fundamentals of High Power Density, Pulsed Discharge, Lead-Acid Batteries: II. Modeling, J. Electrochem. Soc., 1990, 137(12), 3701 CrossRef CAS.
- K. Kerman, et al., Practical challenges hindering the development of solid state Li ion batteries, J. Electrochem. Soc., 2017, 164(7), A1731 CrossRef CAS.
- F. Lu, et al., A High-Performance Li–B–H Electrolyte for All-Solid-State Li Batteries, Adv. Funct. Mater., 2019, 29(15), 1809219 CrossRef.
- K. Vignarooban, et al., Effect of TiO2 nano-filler and EC plasticizer on electrical and thermal properties of poly(ethylene oxide)(PEO) based solid polymer electrolytes, Solid State Ionics, 2014, 266, 25–28 CrossRef CAS.
- M. R. Johan and L. B. Fen, Combined effect of CuO nanofillers and DBP plasticizer on ionic conductivity enhancement in the solid polymer electrolyte PEO–LiCF3SO3, Ionics, 2010, 16(4), 335–338 CrossRef CAS.
- S. N. Banitaba, et al., Evaluating the electrochemical properties of PEO-based nanofibrous electrolytes incorporated with TiO2 nanofiller applicable in lithium-ion batteries, Polym. Adv. Technol., 2019, 30(5), 1234–1242 CrossRef CAS.
- Q. Xiao, et al., Macroporous polymer electrolytes based on PVDF/PEO-b-PMMA block copolymer blends for rechargeable lithium ion battery, J. Membr. Sci., 2009, 334(1–2), 117–122 CrossRef CAS.
- A. Bergfelt, et al., D8-poly(methyl methacrylate)-poly [(oligo ethylene glycol) methyl ether methacrylate] tri-block-copolymer electrolytes: Morphology, conductivity and battery performance, Polymer, 2017, 131, 234–242 CrossRef CAS.
- M. Ebadi, et al., Restricted ion transport by plasticizing side chains in polycarbonate-based solid electrolytes, Macromolecules, 2020, 53(3), 764–774 CrossRef CAS PubMed.
- B. Sun, et al., Polycarbonate-based solid polymer electrolytes for Li-ion batteries, Solid State Ionics, 2014, 262, 738–742 CrossRef CAS.
- H. Zhang, et al., Single lithium-ion conducting solid polymer electrolytes: advances and perspectives, Chem. Soc. Rev., 2017, 46(3), 797–815 RSC.
- O. Garcia-Calvo, et al., Cross-linked solid polymer electrolyte for all-solid-state rechargeable lithium batteries, Electrochim. Acta, 2016, 220, 587–594 CrossRef.
- M. S. Grewal, M. Tanaka and H. Kawakami, Free-standing polydimethylsiloxane-based cross-linked network solid polymer electrolytes for future lithium ion battery applications, Electrochim. Acta, 2019, 307, 148–156 CrossRef CAS.
- M. Zhu, et al., High performance and biodegradable skeleton material based on soy protein isolate for gel polymer electrolyte, ACS Sustainable Chem. Eng., 2016, 4(9), 4498–4505 CrossRef CAS.
- X. Fu, et al., Building ion-conduction highways in polymeric electrolytes by manipulating protein configuration, ACS Appl. Mater. Interfaces, 2018, 10(5), 4726–4736 CrossRef CAS PubMed.
- S.-T. Hsu, et al., Free-standing polymer electrolyte for all-solid-state lithium batteries operated at room temperature, J. Power Sources, 2020, 449, 227518 CrossRef CAS.
- C. Bai, et al., Poly(ethylene oxide)/Poly(vinylidene fluoride)/Li6.4La3Zr1.4Ta0.6O12 composite electrolyte with a stable interface for high performance solid state lithium metal batteries, J. Power Sources, 2020, 472, 228461 CrossRef CAS.
- J. Yang, et al., High-performance solid composite polymer electrolyte for all solid-state lithium battery through facile microstructure regulation, Front. Chem., 2019, 7, 388 CrossRef CAS PubMed.
- J. Liu, et al., A functional-gradient-structured ultrahigh modulus solid polymer electrolyte for all-solid-state lithium metal batteries, J. Mater. Chem. A, 2019, 7(42), 24477–24485 RSC.
- X. Ma, et al., Garnet Si–Li7La3Zr2O12 electrolyte with a durable, low resistance interface layer for all-solid-state lithium metal batteries, J. Power Sources, 2020, 453, 227881 CrossRef CAS.
- Q. Lu, et al., A novel solid composite polymer electrolyte based on poly(ethylene oxide) segmented polysulfone copolymers for rechargeable lithium batteries, J. Membr. Sci., 2013, 425, 105–112 CrossRef.
- J. Shi, Y. Yang and H. Shao, Co-polymerization and blending based PEO/PMMA/P (VDF-HFP) gel polymer electrolyte for rechargeable lithium metal batteries, J. Membr. Sci., 2018, 547, 1–10 CrossRef CAS.
- S. A. Smith, B. P. Williams and Y. L. Joo, Effect of polymer and ceramic morphology on the material and electrochemical properties of electrospun PAN/polymer derived ceramic composite nanofiber membranes for lithium ion battery separators, J. Membr. Sci., 2017, 526, 315–322 CrossRef CAS.
- C.-H. Tsao and P.-L. Kuo, Poly(dimethylsiloxane) hybrid gel polymer electrolytes of a porous structure for lithium ion battery, J. Membr. Sci., 2015, 489, 36–42 CrossRef CAS.
- P.-L. Kuo, et al., A new strategy for preparing oligomeric ionic liquid gel polymer electrolytes for high-performance and nonflammable lithium ion batteries, J. Membr. Sci., 2016, 499, 462–469 CrossRef CAS.
- Y. Li, et al., Ambient temperature solid-state Li-battery based on high-salt-concentrated solid polymeric electrolyte, J. Power Sources, 2018, 397, 95–101 CrossRef CAS.
- Q. Yang, et al., Surface-protected LiCoO2 with ultrathin solid oxide electrolyte film for high-voltage lithium ion batteries and lithium polymer batteries, J. Power Sources, 2018, 388, 65–70 CrossRef CAS.
- I. Aldalur, et al., Nanofiber-reinforced polymer electrolytes toward room temperature solid-state lithium batteries, J. Power Sources, 2020, 448, 227424 CrossRef CAS.
- Z. Dai, et al., Highly conductive and nonflammable composite polymer electrolytes for rechargeable quasi-solid-state Li-metal batteries, J. Power Sources, 2020, 464, 228182 CrossRef CAS.
- T. Watanabe, et al., Development of all-solid-state battery based on lithium ion conductive polymer nanofiber framework, J. Power Sources, 2019, 423, 255–262 CrossRef CAS.
- H. Li, et al., A sandwich structure polymer/polymer-ceramics/polymer gel electrolytes for the safe, stable cycling of lithium metal batteries, J. Membr. Sci., 2018, 555, 169–176 CrossRef CAS.
- K. Homma, et al., Crystal structure and phase transitions of the lithium ionic conductor Li3PS4, Solid State Ionics, 2011, 182(1), 53–58 CrossRef CAS.
- S. Chen, et al., In-situ preparation of poly(ethylene oxide)/Li3PS4 hybrid polymer electrolyte with good nanofiller distribution for rechargeable solid-state lithium batteries, J. Power Sources, 2018, 387, 72–80 CrossRef CAS.
- K. K. Fu, et al., Flexible, solid-state, ion-conducting membrane with 3D garnet nanofiber networks for lithium batteries, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(26), 7094–7099 CrossRef CAS PubMed.
- F. Chen, et al., Solid polymer electrolytes incorporating cubic Li7La3Zr2O12 for all-solid-state lithium rechargeable batteries, Electrochim. Acta, 2017, 258, 1106–1114 CrossRef CAS.
- L. Zhu, et al., High-performance solid PEO/PPC/LLTO-nanowires polymer composite electrolyte for solid-state lithium battery, Int. J. Energy Res., 2019, 43(9), 4854–4866 CrossRef CAS.
- Z. Sun, et al., gC 3 N 4 nanosheets enhanced solid polymer electrolytes with excellent electrochemical performance, mechanical properties, and thermal stability, J. Mater. Chem. A, 2019, 7(18), 11069–11076 RSC.
- I. Aldalur, et al., Self-standing highly conductive solid electrolytes based on block copolymers for rechargeable all-solid-state lithium-metal batteries, Batteries Supercaps, 2018, 1(4), 149–159 CrossRef CAS.
- J. Mindemark, et al., Allyl ethers as combined plasticizing and crosslinkable side groups in polycarbonate-based polymer electrolytes for solid-state Li batteries, J. Polym. Sci., Part A: Polym. Chem., 2016, 54(14), 2128–2135 CrossRef CAS.
- K. Kimura, M. Yajima and Y. Tominaga, A highly-concentrated poly(ethylene carbonate)-based electrolyte for all-solid-state Li battery working at room temperature, Electrochem. Commun., 2016, 66, 46–48 CrossRef CAS.
- H.-D. Nguyen, et al., Nanostructured multi-block copolymer single-ion conductors for safer high-performance lithium batteries, Energy Environ. Sci., 2018, 11(11), 3298–3309 RSC.
- J. Bao, et al., Polycarbonate-based polyurethane as a polymer electrolyte matrix for all-solid-state lithium batteries, J. Power Sources, 2018, 389, 84–92 CrossRef CAS.
- B. Cong, et al., Polyethylene glycol-based waterborne polyurethane as solid polymer electrolyte for all-solid-state lithium ion batteries, Mater. Des., 2018, 142, 221–228 CrossRef CAS.
- J. Mindemark, et al., High-performance solid polymer electrolytes for lithium batteries operational at ambient temperature, J. Power Sources, 2015, 298, 166–170 CrossRef CAS.
- Z. Xiao, et al., PEO-based electrolytes blended with star polymers with precisely imprinted polymeric pseudo-crown ether cavities for alkali metal ion batteries, J. Membr. Sci., 2019, 576, 182–189 CrossRef CAS.
- H. Xu, et al., A new fluorine-containing star-branched polymer as electrolyte for all-solid-state lithium-ion batteries, Polymer, 2018, 146, 249–255 CrossRef CAS.
- S. Wang, et al., Six-arm star polymer based on discotic liquid crystal as high performance all-solid-state polymer electrolyte for lithium-ion batteries, J. Power Sources, 2018, 395, 137–147 CrossRef CAS.
- X. Wang, et al., Lithium-salt-rich PEO/Li0.3La0.557TiO3 interpenetrating composite electrolyte with three-dimensional ceramic nano-backbone for all-solid-state lithium-ion batteries, ACS Appl. Mater. Interfaces, 2018, 10(29), 24791–24798 CrossRef CAS PubMed.
- Y. Li, et al., Li7La3Zr2O12 ceramic nanofiber-incorporated composite polymer electrolytes for lithium metal batteries, J. Mater. Chem. A, 2019, 7(7), 3391–3398 RSC.
- S.-J. Kwon, et al., Preparation of organic/inorganic hybrid semi-interpenetrating network polymer electrolytes based on poly(ethylene oxide-co-ethylene carbonate) for all-solid-state lithium batteries at elevated temperatures, Polymer, 2014, 55(12), 2799–2808 CrossRef CAS.
- V. Chaudoy, et al., Cross-linked polymer electrolytes for Li-based batteries: from solid to gel electrolytes, Ind. Eng. Chem. Res., 2016, 55(37), 9925–9933 CrossRef CAS.
- Y. Zhang, et al., Cross-linking network based on Poly(ethylene oxide): Solid polymer electrolyte for room temperature lithium battery, J. Power Sources, 2019, 420, 63–72 CrossRef CAS.
- T. Sakakibara, et al., Cross-linked polymer electrolyte and its application to lithium polymer battery, Electrochim. Acta, 2019, 296, 1018–1026 CrossRef CAS.
- X. Fu, et al., Core–shell hybrid nanowires with protein enabling fast ion conduction for high-performance composite polymer electrolytes, Small, 2018, 14(49), 1803564 CrossRef PubMed.
- Q. Pan, et al., 2D MXene-containing polymer electrolytes for all-solid-state lithium metal batteries, Nanoscale Adv., 2019, 1(1), 395–402 RSC.
- B. Wang, et al., A 2D layered natural ore as a novel solid-state electrolyte. ACS Applied Energy, Materials, 2019, 2(8), 5909–5916 CAS.
- W. Tang, et al., Simultaneously enhancing the thermal stability, mechanical modulus, and electrochemical performance of solid polymer electrolytes by incorporating 2D sheets. Advanced Energy, Materials, 2018, 8(24), 1800866 Search PubMed.
- W. Tang, et al., High-performance solid polymer electrolytes filled with vertically aligned 2D materials, Adv. Funct. Mater., 2019, 29(16), 1900648 CrossRef.
- G. Piana, et al., PEO/LAGP hybrid solid polymer electrolytes for ambient temperature lithium batteries by solvent-free,“one pot” preparation, J. Energy Storage, 2019, 26, 100947 CrossRef.
- J. Fu, et al., A novel room temperature POSS ionic liquid-based solid polymer electrolyte, J. Mater. Sci., 2018, 53(11), 8420–8435 CrossRef CAS.
- N. Zhang, et al., Composite solid electrolyte PEO/SN/LiAlO2 for a solid-state lithium battery, J. Mater. Sci., 2019, 54(13), 9603–9612 CrossRef CAS.
- H.-L. Guo, et al., A new type of composite electrolyte with high performance for room-temperature solid-state lithium battery, J. Mater. Sci., 2019, 54(6), 4874–4883 CrossRef CAS.
- Z. Zeng, et al., Zinc bis (2-ethylhexanoate), a homogeneous and bifunctional additive, to improve conductivity and lithium deposition for poly(ethylene oxide) based all-solid-state lithium metal battery, J. Power Sources, 2020, 451, 227730 CrossRef CAS.
- J. Zhang, et al., Poly(ethylene oxide) reinforced Li6PS5Cl composite solid electrolyte for all-solid-state lithium battery: Enhanced electrochemical performance, mechanical property and interfacial stability, J. Power Sources, 2019, 412, 78–85 CrossRef CAS.
- W. Zha, et al., High-performance Li6.4La3Zr1.4Ta0.6O12/Poly(ethylene oxide)/Succinonitrile composite electrolyte for solid-state lithium batteries, J. Power Sources, 2018, 397, 87–94 CrossRef CAS.
- A. I. Gopalan, et al., Development of electrospun PVdF–PAN membrane-based polymer electrolytes for lithium batteries, J. Membr. Sci., 2008, 325(2), 683–690 CrossRef CAS.
- L. He, et al., A new approach for synthesizing bulk-type all-solid-state lithium-ion batteries, J. Mater. Chem. A, 2019, 7(16), 9748–9760 RSC.
- F. Ma, et al., Preparation and evaluation of high lithium ion conductivity Li1.3Al0.3Ti1.7(PO4)3 solid electrolyte obtained using a new solution method, Solid State Ionics, 2016, 295, 7–12 CrossRef CAS.
- S. D. Jackman and R. A. Cutler, Effect of microcracking on ionic conductivity in LATP, J. Power Sources, 2012, 218, 65–72 CrossRef CAS.
- Y. Li, et al., Mastering the interface for advanced all-solid-state lithium rechargeable batteries, Proc. Natl. Acad. Sci., 2016, 113(47), 13313–13317 CrossRef CAS PubMed.
- G. Wang, et al., High performance lithium-ion and lithium–sulfur batteries using prelithiated phosphorus/carbon composite anode, Energy Storage Mater., 2020, 24, 147–152 CrossRef.
- J.-Y. Liang, et al., Engineering janus interfaces of ceramic electrolyte via distinct functional polymers for stable high-voltage Li-metal batteries, J. Am. Chem. Soc., 2019, 141(23), 9165–9169 CrossRef PubMed.
- Y. Jin, et al., Interface engineering of Li1.3Al0.3Ti1.7 (PO4) 3 ceramic electrolyte via multifunctional interfacial layer for all-solid-state lithium batteries, J. Power Sources, 2020, 460, 228125 CrossRef CAS.
- B. Akkinepally, et al., Temperature effect and kinetics, LiZr2(PO4)3 and Li1.2Al0.2Zr1.8(PO4)3 and electrochemical properties for rechargeable ion batteries, Int. J. Energy Res., 2022 DOI:10.1002/er.8129.
| This journal is © The Royal Society of Chemistry 2022 |