
Enhanced blue TADF in a D–A–D type naphthyridine derivative with an asymmetric carbazole-donor motif†
Dovydas
Banevičius
 a,
Gediminas
Kreiza
a,
Rokas
Klioštoraitis
a,
Saulius
Juršėnas
a,
Tomas
Javorskis
b,
Vytenis
Vaitkevičius
b,
Edvinas
Orentas
b and
Karolis
Kazlauskas
*a
a,
Gediminas
Kreiza
a,
Rokas
Klioštoraitis
a,
Saulius
Juršėnas
a,
Tomas
Javorskis
b,
Vytenis
Vaitkevičius
b,
Edvinas
Orentas
b and
Karolis
Kazlauskas
*a
aInstitute of Photonics and Nanotechnology, Vilnius University, Saulėtekio av. 3, Vilnius LT-10257, Lithuania. E-mail: karolis.kazlauskas@ff.vu.lt
bDepartment of Organic Chemistry, Vilnius University, Faculty of Chemistry and Geosciences, Naugarduko 24, Vilnius LT-03225, Lithuania
First published on 29th December 2021
Abstract
Efficient triplet-to-singlet conversion in conventional donor–acceptor TADF compounds relies on charge-transfer (CT) and locally-excited (LE) triplet state mixing as this provides the required spin–orbit coupling. In this work, an asymmetric carbazole-donor motif is shown to facilitate the coupling and spin–flip process in a naphthyridine-acceptor based TADF emitter due to the enhanced LE character of the lowest triplet state. This was revealed from the delayed fluorescence measurements, which clearly signified a doubled RISC rate (up to 1.33 × 106 s−1) and a shortened delayed fluorescence lifetime (down to 4.4 μs) in an asymmetric compound compared to those of symmetric or singly-carbazole-substituted compounds. The promising features of the asymmetric blue TADF emitter were demonstrated to result in high efficiency OLEDs (external quantum efficiency of 21%) with a reduced efficiency roll-off.
1. Introduction
Effective utilization of triplet excitons is of crucial importance for the development of highly efficient organic light emitting diodes (OLEDs) with a reduced efficiency roll-off.1–3 Purely organic emitters expressing a thermally activated delayed fluorescence (TADF) phenomenon are particularly promising for harvesting the dark triplets through their conversion to emissive singlet states.4 Thermally-assisted reverse intersystem crossing (RISC) enables the achievement of up to 100% internal quantum efficiency in TADF OLEDs via smart engineering of donor–acceptor (D–A) based emitter structures possessing small singlet–triplet energy splitting (ΔEST).1,5A high RISC rate promoting efficient delayed fluorescence is among the key features required for the realization of efficient TADF-based OLEDs.2 One of the best adopted strategies to achieve high kRISC is to minimize the energy gap between the lowest excited singlet and triplet states. Low ΔEST values can be attained in the compounds expressing a strong charge-transfer (CT) character,6i.e. typically, in D–A or D–A–D type compounds with large dihedral angles between the D and A fragments and, thus, large spatial separation of the highest occupied (HOMO) and lowest unoccupied molecular orbitals (LUMO).7 Phenoxazine- or acridan-based donors routinely employed in the synthesis of TADF emitters indeed demonstrate large dihedral angles (∼70–90°) compared to an acceptor.8–13 However, even though this strategy effectively reduces ΔEST, it subsequently decreases the radiative decay rate (kr) and, moreover, causes both the lowest triplet and singlet excited states to have a strong CT character. This adversely affects spin–orbit coupling, since, in accordance with El-Sayed's rule, spin conversion between pure CT states is forbidden.14 Therefore, such material design may not guarantee fast RISC. To address this issue, the involvement of a third locally excited (LE) triplet state is required.14–17 Another alternative to facilitate triplet-to-singlet conversion is to design compounds possessing a triplet state of stronger LE character.
The latter can be achieved by constructing compounds with a less twisted molecular geometry, e.g., by employing carbazole donor units. In fact, carbazoles are highly attractive for utilization in optoelectronic devices due to their superior stability and good charge transport properties.18,19 However, unsubstituted carbazole has weaker electron donating properties and forms smaller dihedral angles (∼40–50°) with an acceptor unless, e.g., steric hindrance groups such as methyl moieties are introduced at the 1st and/or 8th position.20–23 Besides, the compounds with asymmetric D–A–D* architecture were found to facilitate the formation of the mixed CT and LE states, thereby delivering efficient TADF.24,25
Aiming to further reveal the potential of asymmetric donor units in the structure of TADF emitters, we designed a series of carbazole–naphthyridine derivatives bearing D–A, D–A–D* and D–A–D structures. Here, 1,8-naphthyridine, which previously demonstrated promising results in applications in blue emitting OLEDs, was employed as the electron acceptor (A).20,26 Meanwhile, 3,5-di-tert-butylcarbazole (D*) and sterically demanding 3,3′,6,6′-tetra-tert-butyl-9H-1,9′-bicarbazole (D) were employed to create asymmetric donors. Although the synthesis of 3,3′,6,6′-tetra-tert-butyl-9H-1,9′-bicarbazole was recently reported,27 its applicability in optoelectronic devices remains unexplored. The obtained photophysical properties enabled disclosing of the key advantage of the asymmetric carbazole donors employed, i.e., enhanced triplet-to-singlet spin conversion due to the stronger LE character of the lowest triplet state. This resulted in a doubled RISC rate and a shortened delayed fluorescence lifetime. The endowed features of the asymmetric TADF emitter were demonstrated to result in high efficiency OLEDs (external quantum efficiency of 21%) with a reduced efficiency roll-off.
2. Experimental section
2.1. Sample preparation and instrumentation
Absorption spectra of the investigated carbazole–naphthyridine-based TADF compounds were recorded in toluene solutions (10−5 M) using a Lambda 950 UV-vis-NIR spectrophotometer (PerkinElmer). Bis[2-(diphenylphosphino)phenyl]ether oxide (DPEPO) films doped with 20 wt% of the compounds were prepared by spin-coating chloroform solutions (10 mg mL−1) of DPEPO and compound mixtures on quartz substrates at 2000 rpm. A back-thinned PMA-12 CCD spectrometer (Hamamatsu) was employed to record steady state fluorescence (FL) spectra by exciting samples with a xenon lamp coupled to a monochromator. Toluene solutions were degassed by the freeze–pump–thaw method (5 cycles) prior to FL measurements. FL quantum yields (ΦFL) of toluene solutions were evaluated by using the comparative method with quinine sulphate (in 0.1 M H2SO4) serving as the reference,28 whereas ΦFL of the films was estimated by using an integrating sphere (Sphere Optics). FL transients were measured using a nanosecond YAG:Nd3+ laser NT 242 equipped with an optical parametric oscillator (Ekspla, excitation wavelength 340 nm, pulse duration 5 ns, and repetition rate 1 kHz) for sample excitation and a time-gated intensified CCD camera iStar DH340T (Andor) mounted on a spectrograph SR-303i (Shamrock). A closed-cycle helium cryostat 204N (Cryo Industries) was used for low temperature measurements.The electrochemical behavior of the synthesized compounds was analyzed by cyclic voltammetry (CV) measurements performed in a three-electrode cell using ferrocene as an internal reference. A Pt/Ti-wire was employed as the counter-electrode, and Ag/AgCl and glassy-carbon electrodes served as the reference and working electrodes, respectively. All measurements were performed in nitrogen-degassed 1.0 mM dimethyl formamide solutions of compounds using 0.1 M [n-Bu4N]BF4 as the supporting electrolyte.
All materials and reagents were purchased from available commercial suppliers unless stated otherwise.
Detailed compound synthesis, identification and X-ray crystallographic data as well as experimental details are provided in the ESI.†
2.2. OLED fabrication and characterization
Organic light emitting devices were fabricated using patterned ITO/glass substrates (Kintec). The anode ITO layer was 125 nm thick with a sheet resistance of 15–20 Ohm per square. Prior to device fabrication, the substrates were cleaned in an ultrasonic bath filled with detergent solution (Hellmanex II) at 50 °C for 15 minutes. Substrates were then rinsed well with running distilled water and sonicated consecutively in acetone and isopropyl alcohol baths for 15 minutes. Afterwards, the substrates were immersed into boiling-hot distilled water and dried using a blow-gun. Eventually, the substrates were treated with oxygen plasma for 7 minutes. The clean substrates were transferred into a nitrogen-filled glove box with an integrated vacuum evaporation system for deposition of OLED stacks. During stack deposition, the temperature of organic sources was controlled for the deposition rate to remain in the range of 0.7–1 A s−1. The doped emission layer was deposited by co-evaporation from two organic sources with independently controlled deposition rates. The doping level was defined from the ratio of these deposition rates. To form cathode layers, the stacks were transferred into a metal deposition chamber without breaking the vacuum. An ultrathin layer of LiF (∼0.8 nm) was first deposited at a rate of 0.2 A s−1. Thereafter, aluminum was deposited on top at a rate of 1.5 A s−1. The active area of the devices was determined using the anode and cathode deposition shadow masks. Before exposing to air, the devices were transferred from the vacuum back into the nitrogen atmosphere and encapsulated using a UV-curable epoxy resin LP655 (Delo) and a cover glass on top.The performance of the fabricated devices was tested by measuring the current–voltage–luminance characteristics as well as external quantum efficiency using a calibrated integrating sphere (Labsphere) coupled with a photonic multichannel analyzer PMA-11 (Hamamatsu) and a source-meter 2610A (Keithley).
3. Results and discussion
3.1. Synthesis and DFT calculations
The synthesis of all 1,8-naphthyridine derivatives used in this study is outlined in Scheme 1. The branched N–C biscarbazole 2 (DCz) was obtained from carbazole 1 (Cz) in 82% yield by using the C–H activation method, previously reported by Patureau et al.27 The subsequent SNAr substitution reaction in 2,7-dichloro-1,8-naphthyridine 3 (ND) selectively furnished the monochloro derivative 4 in excellent yield. The latter intermediate was further elaborated into non-symmetric fully carbazole substituted naphthyridine DCz-ND-Cz or the reduced derivative DCz-ND. The last compound in the series, symmetric derivative DCz-ND-DCz, was also derived from intermediate 4 using the anion of 2 as the nucleophile. Synthesis and identification of the compounds are reported in detail in the ESI.† The structures were also unambiguously proved by X-ray diffraction studies (see Table S1 for crystallographic information, ESI†, and CCDC 2113723, 2113744 and 2113780).†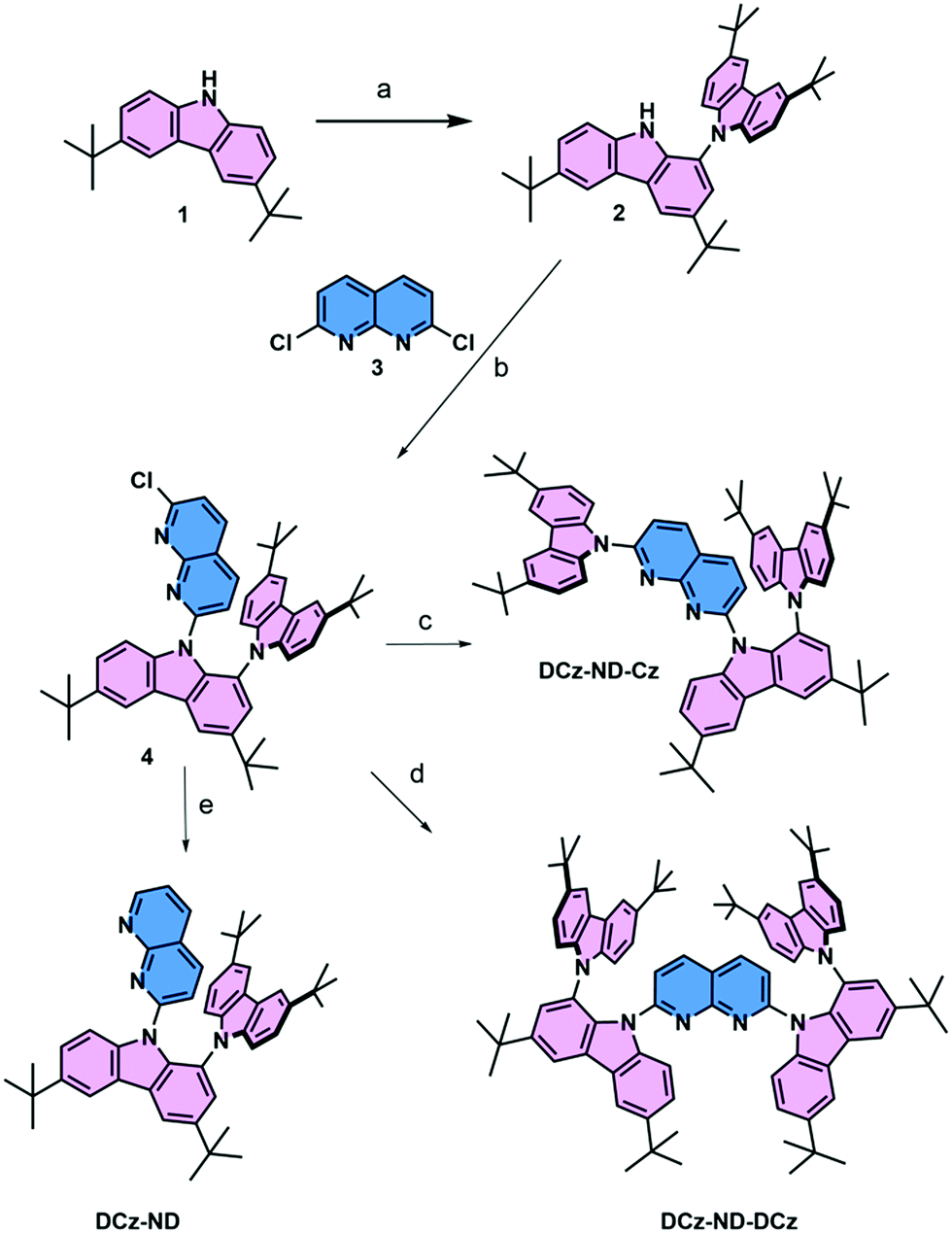 | ||
| Scheme 1 Reagents and conditions: (a) [Ru(p-cymene)Cl2]2, dppf, Cu(OAc)2, PhCl, 1,1,2,2-tetrachloroethylene, AcOH, O2, 150 °C, 82%; (b) 2, 3, n-BuLi, THF, 80 °C, 86%; (c) 3,5-di-tert-butylcarbazole, NaH, THF, 80 °C, 88%; (d) NaH, THF, 40%; and (e) Pd(OAc)2, Bu4NBr, DMF, iPrOH, 115 °C, 36%. | ||
To gain insight into the structure–property relationship of the investigated compounds, their ground state geometries were optimized under vacuum by using density functional theory (DFT) with the B3LYP functional at the 6-31G(d) basis set level implemented in the Gaussian 09W software.29 Although this functional tends to underestimate the energies of CT states, it was employed due to its cost-effectiveness and with a main goal to understand the nature of the S0 → Sn and S0 → Tn transitions. Time dependent DFT at the same basis set level was employed to calculate ground to excited state transition energies (E) and oscillator strengths (f) and to obtain the HOMO and LUMO. Calculated data are summarized in Table 1 and Fig. 1. A similar twisted ground state geometry was obtained for all the investigated naphthyridines regarding their DCz and ND motifs. The dihedral angle between the carbazole units in DCz was higher (67°) than that formed between DCz and ND groups (47–50°). Meanwhile, a significantly smaller dihedral angle of 36° was observed between unsubstituted Cz and ND in DCz-ND-Cz. Substantial separation of the HOMO and LUMO was ensured by the good electron donating properties of the twisted DCz groups, i.e. the HOMO was mainly localized on DCz groups, whereas the LUMO was localized on the ND acceptor. This implied a rather weak oscillator strength for the S0 → S1 transition, yet allowed anticipating relatively small ΔEST required for TADF emitters. It is noteworthy that a strong oscillator strength for the S0 → S2 transition (f = 0.3564) was estimated exclusively for the DCz-ND-Cz compound bearing a less twisted single Cz unit, resulting in a more pronounced LE character of this transition.
| Compound | E S0→S1 (eV) | f S0→S1 | E S0→S2 (eV) | f S0→S2 | E S0→T1 (eV) | E S0→T2 (eV) | ΔES1–T1 (eV) |
|---|---|---|---|---|---|---|---|
| DCz-ND | 2.920 | 0.0098 | 3.165 | 0.0747 | 2.778 | 3.017 | 0.142 |
| DCz-ND-Cz | 2.822 | 0.0156 | 3.004 | 0.3564 | 2.662 | 2.735 | 0.160 |
| DCz-ND-DCz | 2.922 | 0.0025 | 2.926 | 0.0325 | 2.790 | 2.798 | 0.132 |
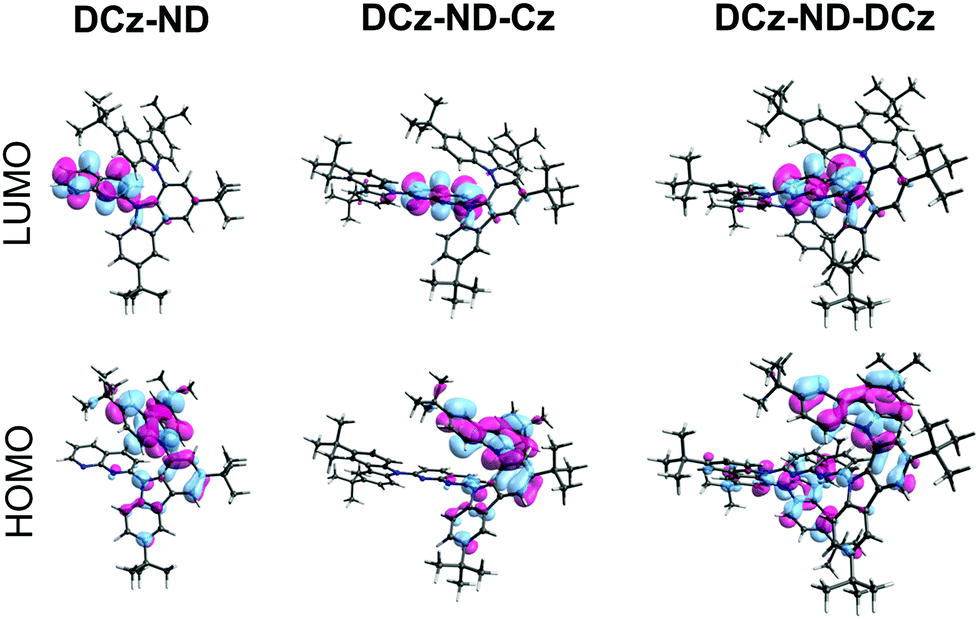 | ||
| Fig. 1 HOMO and LUMO of the investigated naphthyridine compounds obtained by DFT B3LYP/6-31G(d) calculations. | ||
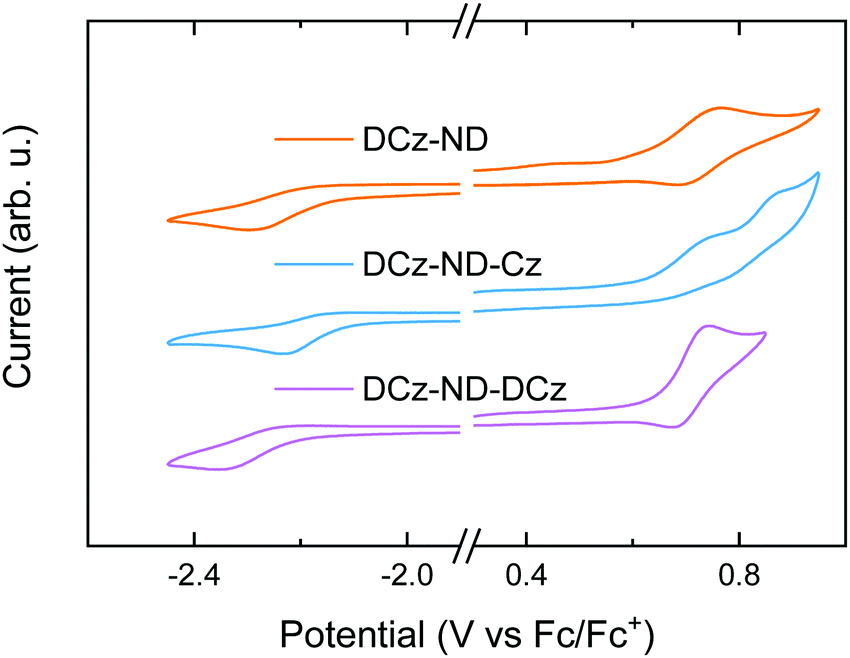 | ||
| Fig. 2 CV traces of the naphthyridine compounds in DMF under nitrogen with 0.1 M [n-Bu4N]BF4 as the supporting electrolyte. Scan rate = 50 mV s−1; compound concentration = 1 mM. | ||
To analyze the electrochemical behavior of the synthesized compounds, CV measurements were performed (see Fig. 2). Reversible oxidation was demonstrated by DCz-ND and DCz-ND-DCz, whereas quasi-reversible oxidation of carbazole units was observed for DCz-ND-Cz. The corresponding reduction processes were found to be irreversible for all the studied naphthyridines. The HOMO and LUMO energies were calculated using the relation EHOMO/LUMO = −(Eoxonset/Eredonset + 4.8), where Eoxonset and Eredonset are the onset oxidation and reduction potentials, respectively (see Table 2 for summarized data). Similar HOMO (−5.4) and LUMO (−2.6 eV) energies were determined for all the compounds, which were found to be favorable for both electron and hole injection in the emitting layer of an OLED.
| Compound | E oxonset (V) | E redonset (V) | HOMO (eV) | LUMO (eV) |
|---|---|---|---|---|
| DCz-ND | 0.61 | −2.16 | −5.41 | −2.64 |
| DCz-ND-Cz | 0.62 | −2.12 | −5.42 | −2.68 |
| DCz-ND-DCz | 0.64 | −2.22 | −5.44 | −2.58 |
3.2. Photophysical properties
Absorption and fluorescence (FL) spectra of the synthesized naphthyridine compounds in dilute toluene solutions are shown in Fig. 3. The detailed photophysical properties are summarized in Table 3. All the derivatives demonstrated sharp absorption peaks at 346 nm with well-resolved vibronic replicas typical for LE transitions associated with carbazole units. The main differences between compounds were observed at longer wavelengths of the absorption spectra. DCz-ND demonstrated a broad and structureless absorption band typical for the CT transition, whereas DCz-ND-Cz was distinguished by an intense and vibronically-modulated band at 401 nm typical for the LE transition.30,31 The latter was most likely determined by a smaller dihedral angle between the single Cz donor unit and the ND acceptor. Meanwhile, DCz-ND-DCz showed a weaker, yet still structured absorption shoulder with a maximum at 377 nm.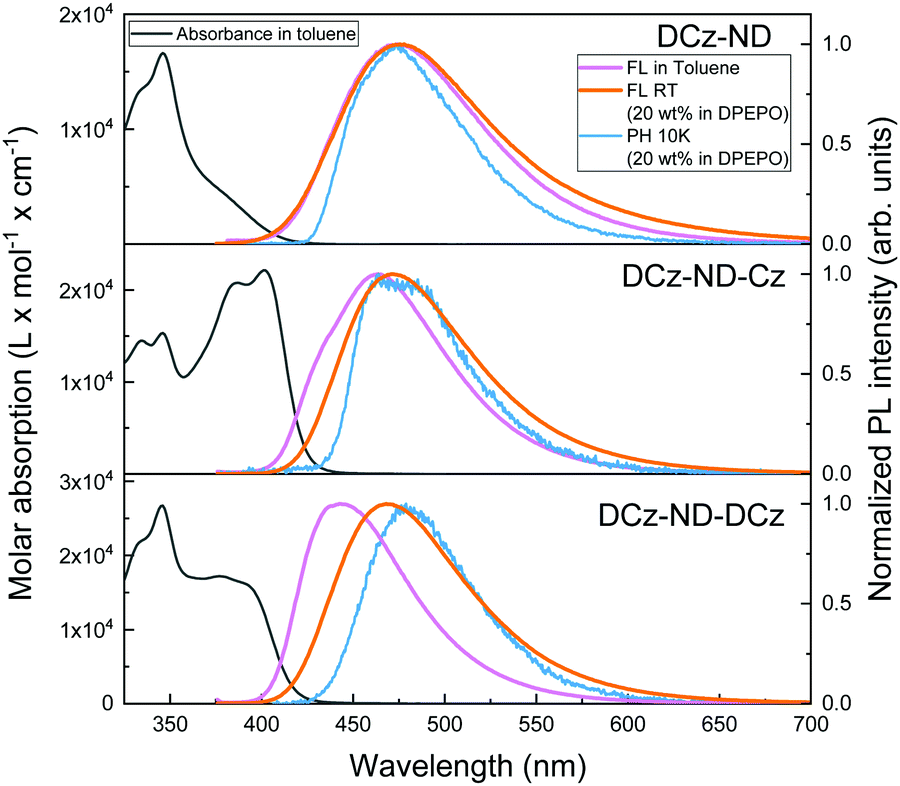 | ||
| Fig. 3 Absorption and fluorescence (1 × 10−5 mol L−1 in toluene), room-temperature fluorescence (20 wt% in DPEPO) and phosphorescence (20 wt% in DPEPO at 10 K, delay time 100 μs) spectra of the carbazole–naphthyridine compounds DCz-ND, DCz-ND-Cz and DCz-ND-DCz. | ||
| Compound | Form | λ FL,max (nm) | Φ FL | τ PF (ns) | τ DF (μs) | k r (106 s−1) | k RISC (106 s−1) | k ISC (107 s−1) | k nr,T (106 s−1) | S1c (eV) | T1c (eV) | ΔESTc (eV) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a In an oxygen-free environment, DF/PF ratios in parentheses determined from the integrals in FL transients. b Estimated from knr,T = kRISC/ΦRISC − kRISC.32 c Determined from the onsets of PH and FL spectra. | ||||||||||||
| DCz-ND | DPEPO | 475 | 0.46(2.3) | 11.0 | 6.0 | 12.5 | 0.45 | 7.8 | 0.104 | 3.01 | 2.89 | 0.12 |
| Toluene | 474 | 0.19(0.03) | 18.8 | 0.24 | 8.6 | 0.91 | 4.5 | 4.091 | ||||
| DCz-ND-Cz | DPEPO | 471 | 0.74(5.1) | 6.3 | 4.4 | 19.1 | 1.33 | 14.0 | 0.067 | 2.95 | 2.82 | 0.13 |
| Toluene | 463 | 0.58(0.69) | 13.5 | 2.0 | 25.5 | 0.52 | 4.9 | 0.315 | ||||
| DCz-ND-DCz | DPEPO | 468 | 0.72(2.9) | 9.9 | 7.0 | 19.3 | 0.49 | 8.2 | 0.049 | 2.98 | 2.85 | 0.13 |
| Toluene | 443 | 0.33(0.12) | 8.2 | 1.5 | 34.1 | 0.12 | 8.2 | 0.637 | ||||
Broad FL spectra of DCz-ND toluene solution that peaked at 474 nm confirmed the CT character of the lowest excited state governed by large separation of the HOMO and LUMO localized on a twisted DCz donor against the ND acceptor. Symmetric compound DCz-ND-DCz showed narrower and blue-shifted FL with maximum at 443 nm indicating suppressed relaxation of the excited state geometry due to a more rigid molecular structure. Interestingly, FL spectra of its asymmetric analogue DCz-ND-Cz were somewhat structured and possessed both a shoulder at higher energies and a broad red-shifted CT-like band at 463 nm. This could be explained by the scenario where FL is visible simultaneously from the LE and CT states. In fact, the existence of those close-lying states was also predicted by DFT calculations. Aiming to further disclose the effect of the asymmetric donor motif, solvatochromic measurements of the compounds were carried out. The corresponding discussion based on the Lippert–Mataga formalism is provided in the ESI.†
To ensure efficient energy transfer from the host to guest molecules, while avoiding FL quenching, 20 wt% doping concentration of the naphthyridine emitters in DPEPO was selected. No distinct absorption bands associated with the formation of H- or J-aggregates were observed (see Fig. S9 in the ESI†). However, the absorption spectra were redshifted by 30–40 meV from those observed in solutions for all the investigated naphthyridines suggesting the presence of enhanced intermolecular interactions, which is expected at such doping ratios. The FL spectrum of DCz-ND dispersed in DPEPO was very similar to that observed in toluene solution. Conversely, DCz-ND-Cz, and, particularly, DCz-ND-DCz in DPEPO exhibited broad and red-shifted FL (λmax = 471 nm and 468 nm, respectively) indicating more effective stabilization of the CT state due to higher environment polarity as compared to its solution. Since the redshift of absorption spectra occurred for all the compounds, yet the difference between the FL spectra of the toluene solutions and 20 wt% DPEPO films was most pronounced for DCz-ND-DCz, we tend to associate it with different excited state relaxation dynamics. Moreover, the absorption spectra of DPEPO films were broadened and less structured than those of solutions suggesting the presence of static conformational disorder in the films.
DPEPO films were further employed for low temperature measurements, which enabled determination of promising ΔEST values being in the range of 0.12–0.13 eV for all the investigated naphthyridine derivatives. Toluene solutions of DCz-ND, DCz-ND-Cz and DCz-ND-DCz demonstrated moderate ΦFL values of 0.19, 0.58 and 0.33, respectively (see Table 3). Furthermore, the compounds demonstrated oxygen-sensitive FL in solutions with distinguishable prompt FL (PF) and delayed FL (DF) components in FL transients indicating TADF properties (Fig. 4a). τPF was determined to be in the nanosecond time-domain – 11.0 ns, 6.3 ns and 9.9 ns for DCz-ND, DCz-ND-Cz and DCz-ND-DCz, respectively. A microsecond DF was observed for toluene solutions of DCz-ND-Cz and DCz-ND-DCz with τDF values of 2.0 μs and 1.5 μs, respectively, meanwhile DCz-ND showed a very weak contribution of DF (ΦDF/ΦPF = 0.03) with a τDF of 0.24 μs.
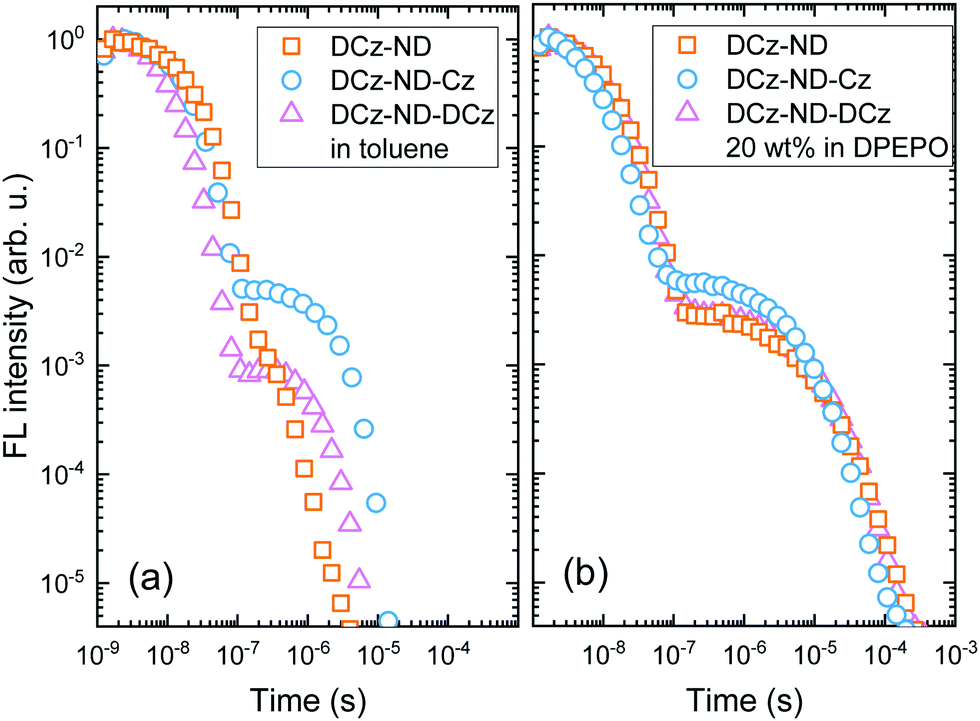 | ||
| Fig. 4 FL transients of the compounds DCz-ND, DCz-ND-Cz and DCz-ND-DCz in degassed toluene (a) and DPEPO at 20 wt% (b). | ||
The ΦFL values were significantly enhanced for the naphthyridine compounds dispersed in a rigid DPEPO host at 20 wt% suggesting effective suppression of non-radiative decay processes occurring in solutions. The ΦFL of DCz-ND reached 0.46, whereas almost twice as high ΦFL values of 0.74 and 0.72 were estimated for DCz-ND-Cz and DCz-ND-DCz, respectively. The enhancement of ΦFL in DPEPO was followed by the enlarged contribution of DF components in FL transients and prolonged τDF (see Table 3 and Fig. 4b), which were determined to be 6.0 μs for DCz-ND and 7.0 μs for the symmetric derivative DCz-ND-DCz. The prolonged tails of FL transients in DPEPO with respect to those in solutions supported the presence of conformational disorder. Interestingly, the asymmetric D–A–D* structure-based derivative DCz-ND-Cz expressed a shorter DF lifetime in the DPEPO host (τDF = 4.4 μs), which is rather promising for application in TADF OLEDs.
Data obtained from ΦFL and FL transient measurements were used to calculate reverse intersystem crossing rates (kRISC) using eqn (1):33
 | (1) |
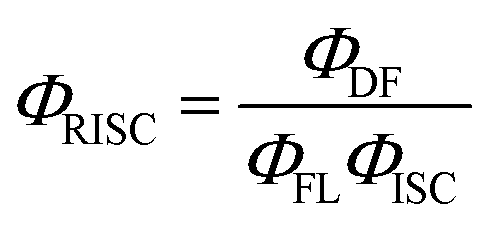 | (2) |
Here ΦISC was calculated in a simplified manner according to the previously described procedures (ΦISC = 1 − ΦPF).32 Regardless of the similar ΔEST values obtained for the doped DPEPO films, the DCz-ND-Cz compound demonstrated a more than two-fold higher RISC rate (kRISC = 1.33 × 106 s−1) than those observed for DCz-ND (kRISC = 4.5 × 105 s−1) and DCz-ND-DCz (kRISC = 4.9 × 105 s−1). The higher RISC rate of asymmetrically donor-substituted DCz-ND-Cz can be justified by the stronger LE character of the T1 state, which is expected to facilitate triplet-to-singlet spin conversion due to the improved spin–orbit coupling with the S1 state of CT character according to El-Sayed's rule.14,34 Indeed, the phosphorescence spectrum of DCz-ND-Cz with more pronounced vibronic features supports the LE character of the lowest triplet state. Even though, all the studied compounds demonstrated rather short DF lifetimes (<10 μs), asymmetric DCz-ND-Cz additionally expressed a combination of fast RISC and relatively high radiative decay rate (kr = 1.91 × 107 s−1), which is advantageous for the application of this compound in OLEDs as a TADF emitter.
It is also worth noting that τDF in toluene solutions is mainly determined by strong variations in knr,T among the investigated compounds. For instance, DCz-ND-Cz expressing the lowest knr,T has the longest τDF. Meanwhile embedding the compounds in a rigid DPEPO host significantly reduces and equalizes knr,T making τDF reflect more the changes in kRISC. Hence, τDF becomes the shortest for DCz-ND-Cz in DPEPO featuring the highest kRISC.
3.3. OLED properties
The naphthyridine-based TADF emitters with symmetric and asymmetric donor motifs were tested and compared in fabricated OLED devices. Except the different emitters DCz-ND, DCz-ND-Cz and DCz-ND-DCz employed, the devices otherwise featured the same configuration and the same emitter doping level (20 wt%) in the emissive layer. The main OLED characteristics are presented in Fig. 5a–d, while the key parameters are summarized in Table 4. OLEDs were fabricated using the following device architecture: ITO (125 nm)/TAPC (30 nm)/TCTA (5 nm)/emitter (20 wt%): DPEPO (20 nm)/DPEPO (5 nm)/TmPyPB (50 nm)/LiF (0.8 nm)/Al (100 nm). Here, TAPC (1,1-bis[(di-4-tolylamino)phenyl]cyclohexane) and TCTA (tris(4-carbazoyl-9-ylphenyl)amine) acted as the hole injection and transport layers, while LiF and TmPyPB (1,3,5-tris(3-pyridyl-3-phenyl)benzene) layers were employed for electron injection and transport. DPEPO with a high triplet energy (3.0 eV) served a dual purpose, i.e., it was used as the host for the naphthyridine-based TADF emitters and as a thin triplet exciton confining layer. We note that the same host was also employed in the evaluation of the photophysical properties of the emissive layers.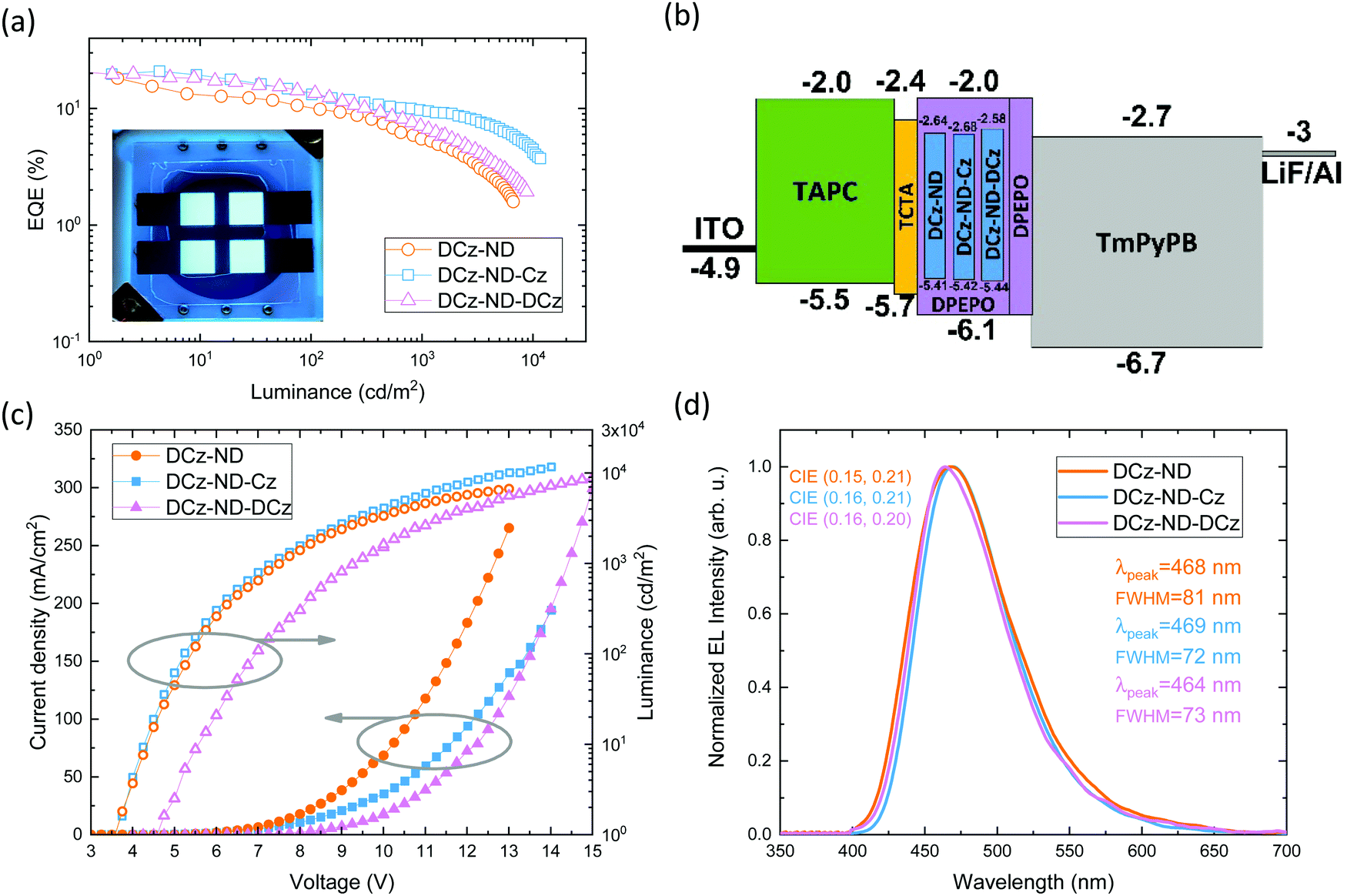 | ||
| Fig. 5 Main characteristics of naphthyridine-based TADF-OLEDs: (a) EQE vs. luminance with a picture of a working DCz-ND-Cz device in the inset; (b) energy level diagram; (c) current density and luminance vs. applied voltage; and (d) EL spectra at 100 cd m−2. | ||
| Device | V on (V) | EQEb (%) | L max (cd m−2) | CEmaxd (cd A−1) | LEmaxe (lm W−1) | λ max (nm) | FWHM (nm) | CIE1931f (x, y) |
|---|---|---|---|---|---|---|---|---|
| a Turn-on voltage at 1 cd m−2. b Maximum EQE/EQE at 100 cd m−2/EQE at 1000 cd m−2. c Maximum brightness. d Maximum current efficiency. e Maximum luminous efficiency. f Commission Internationale de L’Eclairage colour coordinates (x, y). | ||||||||
| DCz-ND | 3.75 | 18.1/10.0/5.4 | 6666 | 30.8 | 25.8 | 469 | 81 | (0.15, 0.21) |
| DCz-ND-Cz | 3.75 | 20.8/13.2/9.6 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
36.2 | 30.3 | 468 | 72 | (0.16, 0.21) |
| DCz-ND-DCz | 4.50 | 20.8/13.6/6.9 | 8816 | 28.8 | 19.0 | 464 | 73 | (0.16, 0.20) |
Devices utilizing DCz-ND and DCz-ND-Cz emitters demonstrated a turn-on voltage of 3.75 V, while the DCz-ND-DCz based OLED showed a somewhat higher turn-on voltage of 4.5 V as well as higher operating voltages. This can be associated with the bulky DCz units, which shield the naphthyridine acceptor, resulting in poor electron transport and overall imbalance of electron and hole currents in the device. All the fabricated devices displayed blue electroluminescence peaking at 468, 469 and 464 nm for DCz-ND, DCz-ND-Cz, DCz-ND-DCz devices, respectively. While the DCz-ND based device demonstrated EL spectra with a FWHM of 81 nm, the other two devices exhibited narrower emission with FWHM of 72 and 73 nm. Commission Internationale de L’Eclairage CIE 1931 color coordinates of DCz-ND, DCz-ND-Cz, and DCz-ND-DCz devices were estimated to be (0.15, 0.21), (0.16, 0.21), and (0.16, 0.20), respectively.
OLEDs with DCz-ND-Cz or DCz-ND-DCz emitters exhibited the same maximum external quantum efficiency (EQE) of 20.8%, whereas the DCz-ND based device demonstrated a somewhat lower maximum EQE of 18.1%. The EQE dependence vs. luminance can be justified by the delayed FL lifetime and RISC rate of the respective emitters. Since the emitter DCz-ND-Cz expresses the shortest delayed FL lifetime as well as the highest RISC rate, this emitter-based device demonstrates a relatively low efficiency roll-off. On the other hand, the DCz-ND emitter based device suffers from the highest EQE roll-off due to the longest living triplet species and the slowest RISC rate. EQE losses of DCz-ND, DCz-ND-Cz and DCz-ND-DCz based devices compared to their maximum efficiency are 45, 36 and 35%, respectively, at a luminance of 100 cd m−2 and 70, 53 and 67%, respectively, at 1000 cd m−2. The brightness of the best performing OLED based on DCz-ND-Cz maxed out at 11600 cd m−2.
4. Conclusions
In summary, a series of blue-emitting donor–acceptor TADF compounds possessing D–A, D–A–D* and D–A–D architecture was designed to elucidate the potential of an asymmetric donor motif in thermally-assisted triplet upconversion. In the study, 1,8-naphthyridine was employed as an acceptor (A), whereas tert-butylcarbazole (D*) and sterically demanding branched N–C biscarbazole (D) served as the donors. The assessed photophysical properties of the carbazole–naphthyridine derivatives revealed the advantage of the asymmetric donor motif in terms of close-lying CT and LE states with more pronounced LE character, which were also predicted by DFT calculations. The stronger mixing of the different molecular orbital possessing states ensured a 2-fold increased RISC rate (up to 1.33 × 106 s−1) and a shortened delayed fluorescence lifetime (down to 4.4 μs) in the asymmetric donor compound DCz-ND-Cz compared to those of symmetric DCz-ND-DCz or singly-carbazole-substituted DCz-ND compounds. OLEDs employing asymmetric DCz-ND-Cz as the TADF emitter demonstrated a high external quantum efficiency (21%) with a reduced efficiency roll-off promoting this asymmetric donor motif for exploitation in TADF-based light emitting devices.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The research was funded by the European Social Fund (project No. 09.3.3-LMT-K-712-01-0084) under grant agreement with the Research Council of Lithuania (LMTLT).References
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- X. Cai, X. Li, G. Xie, Z. He, K. Gao, K. Liu, D. Chen, Y. Cao and S. J. Su, Chem. Sci., 2016, 7, 4264–4275 RSC.
- B. Li, Z. Li, X. Wei, F. Guo, Y. Wang, L. Zhao and Y. Zhang, J. Mater. Chem. C, 2019, 7, 9966–9974 RSC.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- T. Chen, L. Zheng, J. Yuan, Z. An, R. Chen, Y. Tao, H. Li, X. Xie and W. Huang, Sci. Rep., 2015, 5, 10923 CrossRef PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- W. L. Tsai, M. H. Huang, W. K. Lee, Y. J. Hsu, K. C. Pan, Y. H. Huang, H. C. Ting, M. Sarma, Y. Y. Ho, H. C. Hu, C. C. Chen, M. T. Lee, K. T. Wong and C. C. Wu, Chem. Commun., 2015, 51, 13662–13665 RSC.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Y. Mei, D. Liu, J. Li, H. Li and W. Wei, J. Mater. Chem. C, 2021, 9, 5885–5892 RSC.
- R. Braveenth, H. Lee, S. Kim, K. Raagulan, S. Kim, J. H. Kwon and K. Y. Chai, J. Mater. Chem. C, 2019, 7, 7672–7680 RSC.
- M. Han, Y. Chen, Y. Xie, F. Zhang, X. Li, A. Huang, Y. Fan, Y. Fan, Y. Gong, Q. Peng, Q. Li, D. Ma and Z. Li, Cell Rep. Phys. Sci., 2020, 1, 100252 CrossRef.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- C. M. Marian, J. Phys. Chem. C, 2016, 120, 3715–3721 CrossRef CAS.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- D. Berenis, G. Kreiza, S. Juršėnas, E. Kamarauskas, V. Ruibys, O. Bobrovas, P. Adomėnas and K. Kazlauskas, Dyes Pigm., 2020, 182, 108579 CrossRef CAS.
- B. Wex and B. R. Kaafarani, J. Mater. Chem. C, 2017, 5, 8622–8653 RSC.
- N. Blouin and M. Leclerc, Acc. Chem. Res., 2008, 41, 1110–1119 CrossRef CAS PubMed.
- G. Kreiza, D. Banevičius, J. Jovaišaitė, S. Juršėnas, T. Javorskis, V. Vaitkevičius, E. Orentas and K. Kazlauskas, J. Mater. Chem. C, 2020, 8, 8560–8566 RSC.
- L. Cui, H. Nomura, Y. Geng, J. U. Kim, H. Nakanotani and C. Adachi, Angew. Chem., Int. Ed., 2017, 56, 1571–1575 CrossRef CAS PubMed.
- G. Kreiza, D. Berenis, D. Banevičius, S. Juršėnas, T. Javorskis, E. Orentas and K. Kazlauskas, Chem. Eng. J., 2021, 412, 128574 CrossRef CAS.
- J. U. Kim, I. S. Park, C. Y. Chan, M. Tanaka, Y. Tsuchiya, H. Nakanotani and C. Adachi, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- Z. Yang, Z. Mao, C. Xu, X. Chen, J. Zhao, Z. Yang, Y. Zhang, W. Wu, S. Jiao, Y. Liu, M. P. Aldred and Z. Chi, Chem. Sci., 2019, 10, 8129–8134 RSC.
- W. Wei, Z. Yang, X. Chen, T. Liu, Z. Mao, J. Zhao and Z. Chi, J. Mater. Chem. C, 2020, 8, 3663–3668 RSC.
- Y.-F. Shen, W.-L. Zhao, H.-Y. Lu, Y.-F. Wang, D.-W. Zhang, M. Li and C.-F. Chen, Dyes Pigm., 2020, 178, 108324 CrossRef CAS.
- A. W. Jones, C. K. Rank, Y. Becker, C. Malchau, I. Funes-Ardoiz, F. Maseras and F. W. Patureau, Chem. – Eur. J., 2018, 24, 15178–15184 CrossRef CAS PubMed.
- M. J. Adams, J. G. Highfield and G. F. Kirkbright, Anal. Chem., 1977, 49, 1850–1852 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, 2016.
- X. He, T. Shan, X. Tang, Y. Gao, J. Li, B. Yang and P. Lu, J. Mater. Chem. C, 2016, 4, 10205–10208 RSC.
- U. Subuddhi, S. Haldar, S. Sankararaman and A. K. Mishra, Photochem. Photobiol. Sci., 2006, 5, 459–466 CrossRef CAS PubMed.
- G. Kreiza, D. Banevičius, J. Jovaišaitė, K. Maleckaitė, D. Gudeika, D. Volyniuk, J. V. Gražulevičius, S. Juršėnas and K. Kazlauskas, J. Mater. Chem. C, 2019, 7, 11522–11531 RSC.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2113723, 2113744 and 2113780. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc05061c |
| This journal is © The Royal Society of Chemistry 2022 |