
A dual-targeting fluorescent probe for simultaneous and discriminative visualization of lipid droplets and endoplasmic reticulum†
Fangfang
Meng
ac,
Junyi
He
a,
Jie
Niu
b,
Yawen
Li
a,
Peng
Gao
*ac and
Xiaoqiang
Yu
 *b
*b
aDepartment of Pathology, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, China. E-mail: gaopeng@sdu.edu.cn
bCenter of Bio & Micro/Nano Functional Materials, State Key Laboratory of Crystal Materials, Shandong University, Jinan, Shandong, China. E-mail: yuxq@sdu.edu.cn
cKey Laboratory for Experimental Teratology of Ministry of Education, Department of Pathology, School of Basic Medical Sciences, Shandong University, Jinan, Shandong, China
First published on 13th October 2022
Abstract
A single fluorescent probe (SF-probe) that can simultaneously and discriminatively visualize two organelles is a powerful tool to investigate their interaction in cellular processes. However, it is still challenging to develop this unique type of fluorescent probe due to the lack of a feasible design strategy. Herein, we proposed a dual-targeting group strategy to construct SF-probes by integrating two different organelle-targeting groups into the same fluorophore. A versatile fluorophore and two nonintrusive organelle-targeting groups are elements of this strategy. In view of only a few SF-probes having been developed for the simultaneous and discriminative imaging of lipid droplets (LDs) and endoplasmic reticulum (ER), as a proof of concept, a SF-probe, LDER, was designed and synthesized by introducing an LD targeting group and an ER targeting group onto the 1,8-naphthalimide fluorophore. Owing to the specific structure of the fluorophore, both targeting groups at two terminals of 1,8-naphthalimide can fully play their respective roles without mutual interference. Furthermore, the ability of the two groups to target their respective targets is comparable, enabling LDER to bind LDs and ER evenly. Meanwhile, LDER is very susceptible to polarity, which is advantageous for the discriminative imaging of LDs and ER. In addition, the interaction between LDs and ER was investigated.
Introduction
Organelles, as fundamental subunits in eukaryotic cells, play indispensable roles in complex biological processes.1–3 They not only have unique functionalities but also cooperate with each other to form organelle interaction networks.4–6 For example, lipid droplets (LDs) are actively involved in regulating lipid metabolism and cellular homeostasis.7 Endoplasmic reticulum (ER) has significant roles in protein synthesis, secretion, and signal transduction.8 Lysosomes are responsible for endocytic, autophagic, and phagocytic pathways.9 Furthermore, LDs and ER cooperate with each other to complete complicated biological procedures.10,11 Particularly, it is widely believed that LDs derive from ER.12,13 At the same time, LDs are degraded through lysosomes, which is known as lipophagy.14,15 To visualize two organelles and their associated interaction, a strategy of utilizing two individual organelle trackers has often been employed. However, the unsatisfying spectral cross-talk, increased cytotoxicity and tedious operating procedures limit their application in living cells.16 Therefore, it is of great significance to develop single fluorescent probes (SF-probes) to simultaneously and discriminatively visualize two organelles and their associated interaction. However, such a fluorescent probe or feasible design strategy has rarely been reported.Generally, an organelle-targeting fluorescent probe contains two components: a fluorophore and a targeting group.17 It should be noted that many organelles have a special targeting group18,19 (Scheme 1A). For example, LDs contain a hydrophobic lipid core covered by a phospholipid-monolayer membrane, thus probes with lipophilic groups such as alkylamino have been used to image LDs.20 ER is the largest membrane organelle in eukaryotic cells, and is where the ATP-sensitive K+ channel is mainly localized, thus methyl sulphonamide binding to the sulfonylurea receptor in the K+ channel can be used to construct ER probes.21 Mitochondria are organelles with a negative membrane potential of −180 mV, therefore Triphenylphosphonium (TPP+) with a cationic unit has been applied to target mitochondria.22 Lysosomes have an acidic environment, thus morpholine being weakly alkaline has often been used to design lysosomal probes.23 These key facts provide the possibility that a SF-probe for simultaneous and discriminative visualization of two organelles can be developed by exquisitely incorporating two different organelle-targeting groups into the same fluorophore (Scheme 1B). Obviously, the elements of this strategy should include: (1) a fluorophore ring that can be substituted at two terminals by two organelle-targeting groups. This will decrease the steric hindrance of the two organelle-targeting groups, making full use of their respective roles without mutual interference. (2) Organelle-targeting groups that have balanced targeting abilities towards two organelles. In this way, the probe can be evenly distributed among these two organelles. (3) High specificity of the probe. The constructed probe should be of high specificity towards these two organelles without interacting with others.
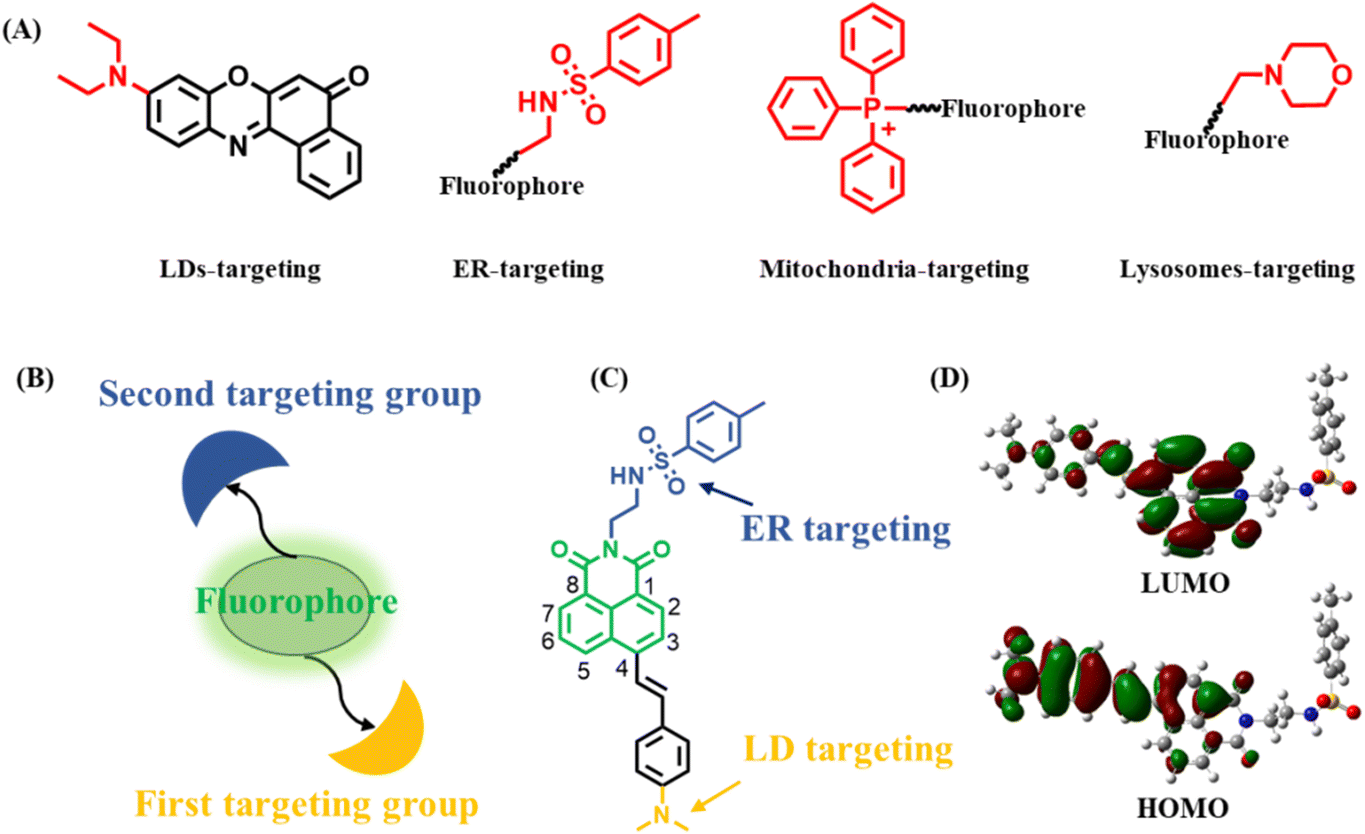 | ||
| Scheme 1 (A) Various targeting units used to design organelle-targeting fluorescent probes; (B) potential dual-targeting model; (C) structure of the probe LDER; (D) the frontier orbitals of LDER. | ||
It is well known that LDs and ER are two organelles with similar polarity but different functions and there are multiple interactions between them,24 so their discriminative visualization is extremely valuable yet challenging. Also, as mentioned above, SF-probes capable of discriminative imaging of LDs and ER have rarely been reported.16,25,26 Therefore, as a proof-of-concept, we firstly applied this strategy to construct a SF-probe for simultaneous and discriminative visualization of LDs and ER. In view of the versatility, easy availability and substitution potential at the 3, 4, 5, 6 or N-imide position of 1,8-naphthalimide, it was selected as the basic building block.27–29 Dimethylamino and methyl sulphonamide moieties served as the LD- and ER-targeting groups owing to their similar targeting ability towards LDs and ER.30,31 By incorporating dimethylamino and methyl sulphonamide moieties at the 4-position and N-imide position of the 1,8-naphthalimide fluorophore, the probe LDER was developed (Scheme 1C). On the one hand, the two targeting groups were placed at opposite ends of 1,8-naphthalimide, which ensured that there was no interference caused by each other. On the other hand, these groups have the same ability to target their respective organelles, thus the probe can target LDs and ER evenly. Delightfully, as a strong electron donor, the dimethylamino group enhanced the intramolecular charge transfer (ICT) character of the fluorophore, resulting in a polarity sensitivity of this probe. The experimental results showed that the probe LDER could precisely and evenly target LDs and ER simultaneously and discriminately visualize them by their morphological differences. Meanwhile, LDER displayed high sensitivity to slight variations in polarity, enabling discriminative visualization of LDs and ER with different fluorescence. In addition, the dynamics of LDs and ER and their interaction in living cells were also investigated using LDER. The results demonstrated that this strategy has great potential in the design of SF-probes for simultaneous and discriminative visualization of two organelles.
Results and discussion
Synthesis and characterization of LDER
The designed fluorescent probe LDER was synthesized according to the synthesis route shown in Scheme S1 (ESI†), and the synthetic procedures are included in the ESI.† The chemical structure of LDER was characterized using 1H NMR, 13C NMR and HRMS spectra and they are presented in the ESI.†To confirm the ICT property of the probe, the frontier orbitals of LDER were calculated by Gaussian 09 software (Scheme 1D). The highest occupied molecular orbital (HOMO) of LDER is mainly distributed on the dimethylamino moiety, while the lowest unoccupied molecular orbital (LUMO) is mainly distributed on the 1,8-naphthalimide moiety. This result demonstrated that LDER may undergo a charge transfer process from dimethylamino to the 1,8-naphthalimide moiety owing to its typical D–π–A structural feature. Probes with such D–π–A structures are sensitive to solvent polarity, whereby their photophysical properties, particularly their emission behaviors, change upon varying the polarity. Thus, we think that the probe LDER has the potential to show polarity-sensitive emission behavior.
Optical properties of LDER
The absorption and emission spectra of LDER in various solvents were first recorded. As displayed in Fig. 1A, the absorption of LDER was in the range of 400–550 nm, and peaked at around 470 nm in the solvents with different polarity. In addition, the absorption spectra showed a slight red shift as the solvent polarity increased (Table S1, ESI†). On the other hand, under excitation at 470 nm, the fluorescence spectra of LDER displayed an obvious solvation effect (Fig. 1B and Table S1, ESI†). LDER showed an emission peak at 586 nm in toluene with low polarity. In contrast, in a high-polarity solvent, such as DMSO, LDER displayed a red-shifted emission peak at 646 nm. Simultaneously, a pronounced decrease in fluorescence intensity and fluorescence quantum yield with solvent polarity enhancement was observed. These results demonstrated that the emission of LDER was sensitive to solvent polarity.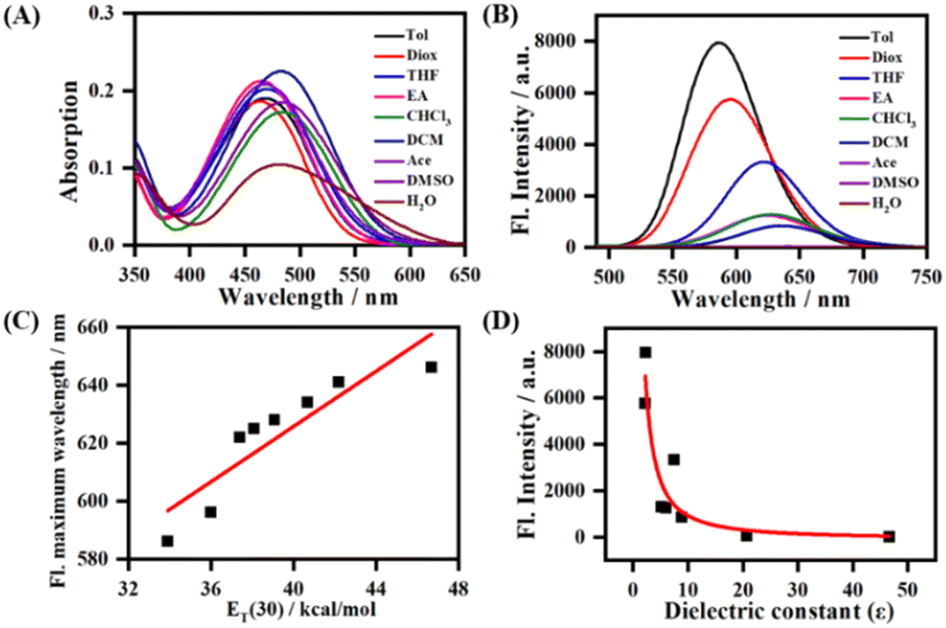 | ||
| Fig. 1 The absorption (A) and fluorescence (B) spectra of LDER in different polarity solvents; (C) plot of the maximum wavelength of LDER with ET(30); (D) fluorescence intensity change of LDERversus dielectric constant (ε). Concentration = 10 μM. Solvents: Tol: toluene; Diox: 1,4-dioxane; THF: tetrahydrofuran; EA: ethyl acetate; CHCl3: trichloromethane; DCM: dichloromethane; Ace: acetone; DMSO: dimethyl sulfoxide; H2O: water. | ||
To evaluate the polarity-sensitive emission behavior of LDER, the change in maximum emission wavelength with the solvent polarity parameter ET(30) value, a solvent polarity parameter that indicates solute–solvent interactions, was plotted.32 As ET(30) increased, the maximum emission wavelength of LDER increased linearly (Fig. 1C); such a good relationship indicated that LDER has a twisted ICT (TICT) character.33–35 The fluorescence intensity of LDER with dielectric constant (ε) of various media was also correlated, as shown in Fig. 1D. The fluorescence intensity varies inversely with the solvent dielectric constant (ε), which implies the polarity sensitivity of LDER.
To further study the sensitivity of LDER towards polarity, the absorption and emission spectra of the probe in mixed solvents with different polarity were recorded (Fig. 2). In mixtures of 1,4-dioxane and ethyl acetate (EA) with similarly low polarity, LDER exhibited tiny variations in absorption (Fig. 2A). As the EA content increased, the maximum emission wavelength of LDER red-shifted. Simultaneously, the fluorescence intensity decreased, showing a clear function of the solvent ratio (Fig. 2B). Such spectral changes are consistent with the TICT character. Furthermore, the absorption and emission of LDER were also recorded in a binary solvent system of toluene–DMSO and 1,4-dioxane–H2O. As the polar solvent (DMSO or H2O) content increased, a pronounced decrease in the fluorescence intensity was observed (Fig. 2D and F). These results indicated that the emission of the probe LDER was highly sensitive to polarity, which provided the potential for in situ imaging of organelles with polarity differences in living cells.
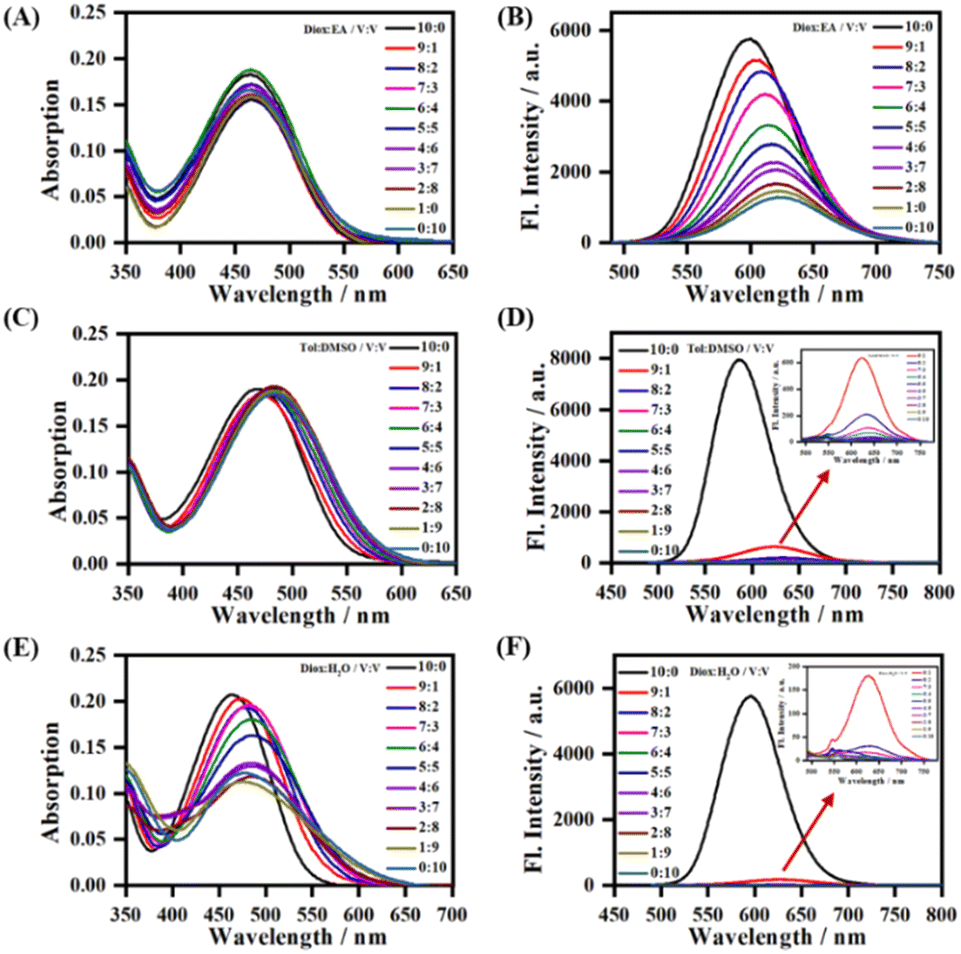 | ||
| Fig. 2 The absorption (A, C and E) and fluorescence (B, D and F) spectra of LDER in 1,4-dioxane–EA, toluene–DMSO and 1,4-dioxane–H2O mixtures with different volume ratios; inset: the enlarged part indicated by the red arrow. Concentration = 10 μM. | ||
In addition to polarity, the effects of different pH and viscosity on the probe LDER were studied. As shown in Fig. S1A (ESI†), in a wide pH range from 2 to 12, the fluorescence intensity did not change. Meanwhile, the fluorescence intensity remained stable at various viscosity levels (Fig. S1B, ESI†). These results indicated that the probe LDER was only sensitive to polarity.
Precise subcellular localization of LDER in living cells
Before exploring the application of LDER in living cell imaging, the cytotoxicity of the probe was evaluated. As shown in Fig. S2 (ESI†), LDER displayed no significant toxicity within the concentration range of 0–20 μM. This result indicated that LDER was favorable for bioimaging applications.Based on our design strategy, the probe LDER could simultaneously image LDs and ER in living cells. To verify the cellular localization of LDER, fluorescence imaging in living SiHa cells was carried out. As shown in Fig. 3A, spherical morphologies with bright green fluorescence and reticulate morphologies with weak green fluorescence were detected in the cytoplasm. Moreover, the two signals were not located in the same area, which indicated that LDER can stain two subcellular structures. We expected that the bright fluorescence was derived from LDs. There are two reasons for this: first, from the magnified images (Fig. 3B), the bright fluorescence from LDER appeared as spherical “droplets”. Some droplets are visible as individuals (yellow arrows), whereas some aggregate into large droplets (red arrows). Although the distribution, morphology and size of these droplets stained by LDER were different, they all well overlapped with the globular organelles in the DIC image. It is generally accepted that LDs present as globular spots in DIC owing to their high refractive index in relation to other subcellular structures.16,36 Thus, the bright dots shown in Fig. 3 should be LDs. Second, BODIPY 493/503, a commercially available probe for LD targeting, was used to co-stain with LDER. As shown in Fig. S3 (ESI†), the strong green fluorescence from LDER overlapped well with the red fluorescence from BODIPY 493/503, and the co-localization coefficient was around 0.86. This result demonstrated that the bright spherical dots stained by LDER were indeed LDs. The above results indicated that the probe LDER could stain both LDs and another subcellular structure.
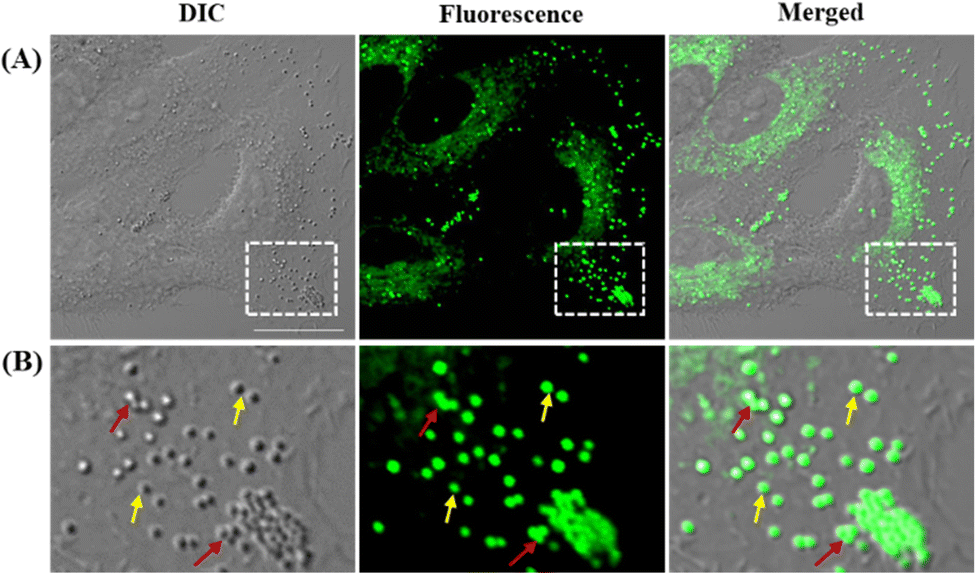 | ||
| Fig. 3 (A) Fluorescence images of living SiHa cells stained with 5 μM LDER; (B) enlarged fluorescence images of the regions of interest (ROIs) indicated by the white rectangles in (A). λex = 473 nm, λem = 550–650 nm; Scale bar = 20 μm. | ||
To reveal the specific localization of LDER in the other subcellular region, co-localization experiments were carried out. A co-localization experiment was first performed with a commercially available ER probe, ER-Tracker Blue. As displayed in Fig. 4, we could see that the weak green fluorescence from LDER overlapped well with the blue fluorescence of ER-Tracker Blue. To further analyse the imaging results, we randomly magnified three small parts of the merged image. As shown in the enlarged images in Fig. 4 and Fig. S4 (ESI†), the co-localization coefficients were determined to be 0.89, 0.87 and 0.90, respectively. This result indicated that the reticulate area stained by LDER was ER.
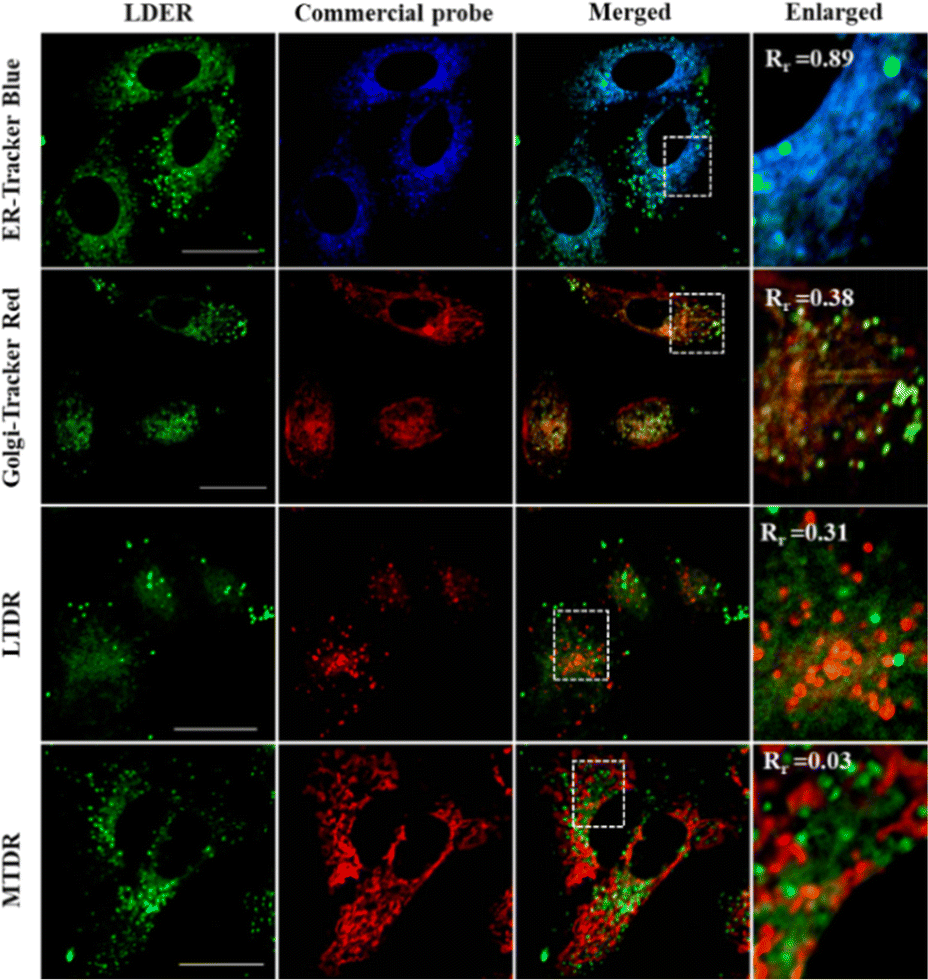 | ||
| Fig. 4 Fluorescence images of LDER co-stained with various commercial probes (ER-Tracker Blue, Golgi-Tracker Red, LTDR and MTDR) in living SiHa cells. LDER: λex = 473 nm, λem = 560–650 nm; ER-Tracker Blue: λex = 405 nm, λem = 450–500 nm; Golgi-Tracker Red: λex = 543 nm, λem = 600–700 nm; LTDR: λex = 633 nm, λem = 650–750 nm; MTDR: λex = 633 nm, λem = 650–750 nm. Scale bar = 20 μm. | ||
Besides this, co-localization experiments of LDER and other organelle markers (Golgi-Tracker Red, lysosomal deep red fluorescence marker (LTDR) and mitochondrial deep red fluorescence marker (MTDR)) were also carried out. As shown in Fig. 4 and Fig. S4 (ESI†), the overlap of LDER and Golgi-Tracker Red, LTDR and MTDR was not high, and the co-localization coefficients were 0.43, 0.47 and 0.03 on average, respectively. Taken together, the results show that the probe LDER can simultaneously image LDs and ER, which was consistent with our design.
Discriminative imaging of LDs and ER in living cells
As described above, the probe LDER achieved the simultaneous visualization of LDs and ER in living SiHa cells, and could discriminate between them by morphological differences. Since LDER exhibited polarity-sensitive emission behavior and LDs and ER have different polarity, we suspected that the probe LDER could also discriminate between LDs and ER with different fluorescence.As shown in Fig. 5A, LDs and ER could be clearly seen in the LDER-stained SiHa cells, and these zones exhibited very different fluorescence intensities. According to the in situ fluorescence spectra, the LD- and ER-like regions were different (Fig. 5B). The emissive peak of LDER in the LDs was located at 580 nm, while it was at 600 nm in ER. Relative to ER, the LDs constitute more hydrophobic microenvironments, and these zones displayed high fluorescence intensity that was similar to those of LDER in solvents with low polarity. In contrast, the less hydrophobic microenvironments of ER translated into a weak fluorescence intensity for LDER, which was similar to its fluorescence in high polarity solvents. This result was in good accordance with the photophysical data recorded in solution (Fig. 1B).
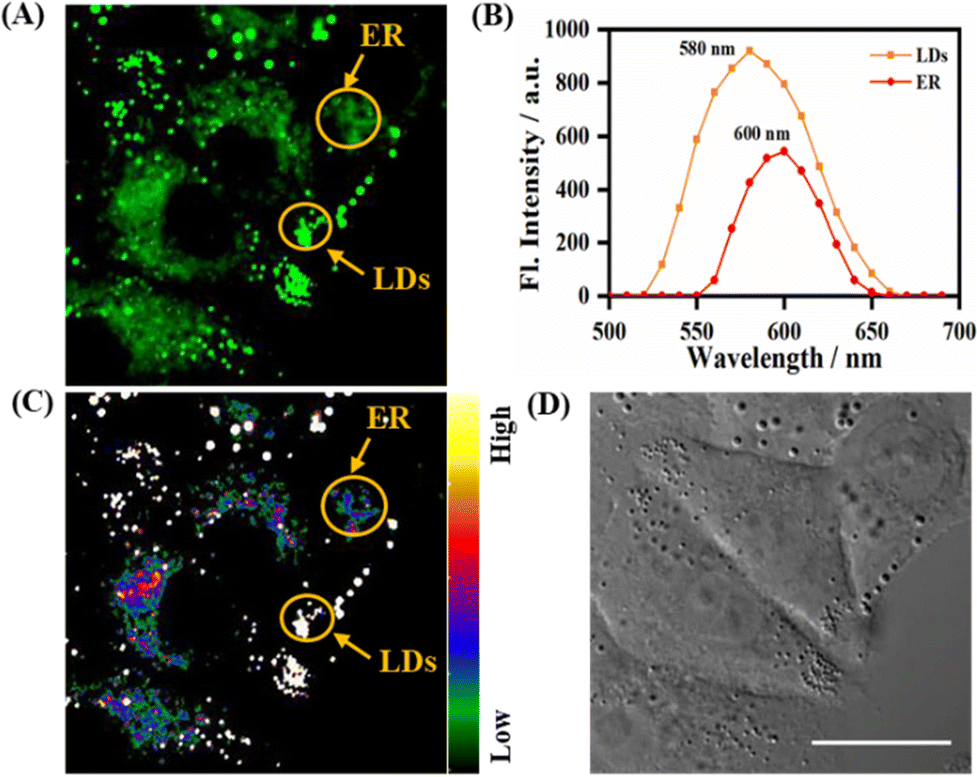 | ||
| Fig. 5 (A) Fluorescence images of LDER in living SiHa cells; (B) in situ emission spectra of LDs and ER in (A); (C) the relative fluorescence image of (A), with a rainbow of color representing the fluorescence intensity; (D) DIC image. Scale bar = 20 μm. | ||
In order to intuitively discriminate between LDs and ER, the conversion of different fluorescence intensities to different colors was achieved by automatic calculation by the microscopy software. With the aid of a rainbow of color, we could easily discern LDs and ER, as the LDs exhibited a higher emission intensity than the ER (Fig. 5C). The high fluorescence intensity of the LDs was mainly due to their low polarity, while the low fluorescence intensity of the ER was due to its high polarity. These differences demonstrated that the polarity-sensitive probe LDER may be used to discriminate between LDs and ER by fluorescence intensity.
To verify the universality of the staining pattern, fluorescence images were recorded in Human Cervical Carcinoma cells (HeLa cells) and Human Breast Cancer cells (MCF-7 cells) (Fig. S5, ESI†). It can be easily seen that LDER emitted different fluorescence in LDs and ER in these cell lines. The above results confirmed that the staining pattern of LDER is universal across different cell lines, and it can be used as a universal tool for the simultaneous and discriminative imaging of LDs and ER.
Visualization of the changes of LDs and ER during starvation and oleic acid treatment
Since LDER can simultaneously and discriminatively visualize LDs and ER, the changes in LDs and ER in reaction to different stimuli were studied. As dynamic lipid storage depots, LDs are highly responsive to the cellular metabolic state. During starvation, LD-associated neutral lipid lipases mediate the breakdown of LDs, leading to a decrease of LDs.37 Living SiHa cells were starved in nutrient-free medium for different times and then incubated with LDER to visualize the dynamic changes of LDs and ER. As shown in Fig. 6A, in the untreated cells, LDER emitted strong fluorescence in LDs and weak fluorescence in ER. After the cells were starved, the fluorescence of LDER in LDs decreased obviously, indicating the consumption of LDs during starvation. Meanwhile, no obvious changes of the fluorescence in ER were observed. This result suggested that the number of LDs decreased sharply with starvation time.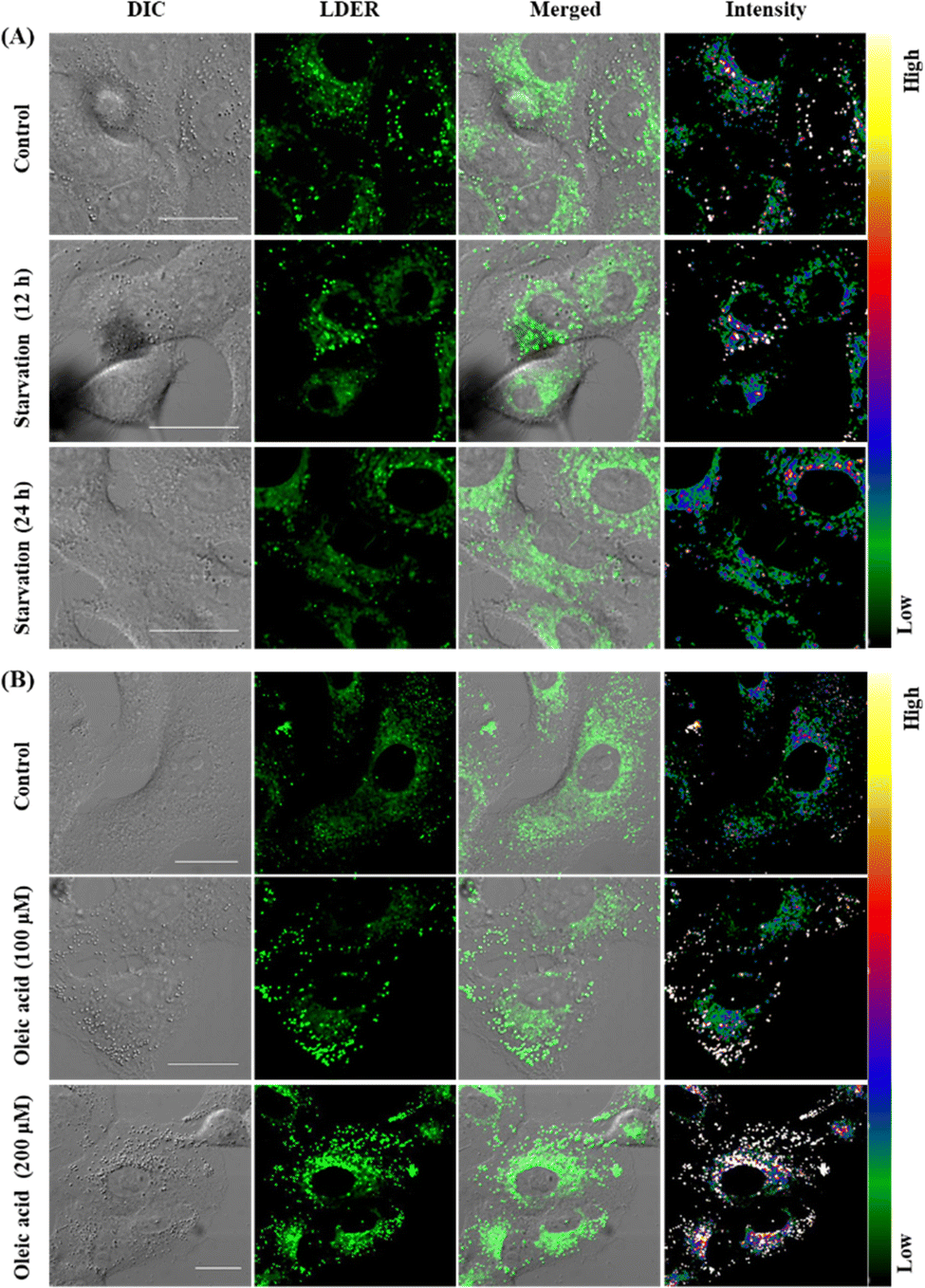 | ||
| Fig. 6 (A) Fluorescence images of SiHa cells incubated in nutrient-free medium for different times, followed by staining with LDER (5 μM); (B) fluorescence images of SiHa cells stained with LDER (5 μM) after the cells were pretreated with different concentrations of oleic acid for 4 h. The rainbow of color represents the fluorescence intensity. Scale bar = 20 μm. | ||
Oleic acid (OA) is known to stimulate the production of LDs in cells and thus we examined the dynamic changes of LDs and ER in SiHa cells before and after addition of OA.38 After staining with LDER, the LDs and ER in the control group could be seen with strong and weak fluorescence, respectively (Fig. 6B). The LDs were comparatively small and dispersed in the cytoplasm. Upon treatment with OA, the emission of the LDs enhanced evidently, and the number and size of the LDs increased as well. This result indicated that LDER could monitor OA-stimulated LD production. These results verified that LDER can be used for imaging the dynamic changes of LDs and ER during various biological procedures within living cells.
Visualization of the interaction between LDs and ER
LDs and ER are dynamic organelles and their interaction is closely related to cellular metabolism.39–41 To visualize their interaction, the following experiments were carried out.Photostability is an important factor for a probe used for real-time tracking of the dynamics of targets. Therefore, the photostability of LDER was investigated. As shown in Fig. S6 (ESI†), under continuous laser irradiation at 473 nm, the fluorescence of LDER in cells showed almost no loss (Fig. S6A, ESI†). Compared to its original fluorescence intensity, it still retained a fluorescence intensity of 92% in LDs and 74% in ER (Fig. S6B, ESI†). This implied that LDER has high stability and can be utilized for real-time monitoring of LD- and ER-related dynamics.
Encouraged by this, the concurrent movement of LDs and ER was tracked in real-time with LDER. The position, distribution and dynamics of LDs and ER are clearly shown in Fig. 7. It demonstrates that both the LDs and ER have a characteristic distribution and dispersion pattern in the cells, and the dynamic LDs and ER move distinctly over the course of a long observation period (60 min). To display the movements of the LDs and ER at different times more intuitively, four different pseudocolors were adopted. From the combined images at different time points, we could see the dynamics of the LDs and ER. In addition, the fluorescence intensity of the LDs and ER in the observed area did not show any obvious decrease during the imaging time. Specifically, the LDs revealed near spatial and temporal contacts with ER, suggesting interaction between LDs and ER.
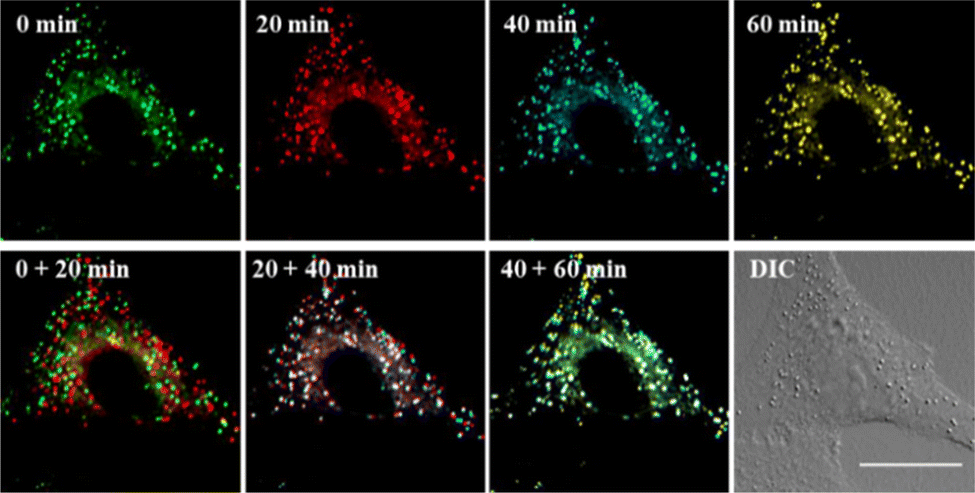 | ||
| Fig. 7 Fluorescence images of living SiHa cells stained with LDER. Different pseudocolors are used to represent the fluorescence images at times of 0, 20, 40, and 60 min. Merged images at two different times, DIC image obtained at 0 min. Scale bar = 20 μm. | ||
Visualization of the generation of LDs from ER
It is widely thought that the spatiotemporal generation of LDs occurs from ER. Here, we used LDER to intuitively visualize the emergence of LDs from the ER. First, SiHa cells were starved to force them to consume LDs, incubated with LDER, and then treated with OA to stimulate LD formation. Subsequently, the dynamics of the LDs and ER were observed on the same area of the dish. As illustrated in Fig. 8A, several globular LDs and large amounts of reticular ER were seen in the OA-untreated cells (0 min). After treatment with OA, more globular LDs appeared in the ER regions, indicating the formation of LDs in ER. With prolonged OA incubation time, the number and size of LDs increased, and they aggregated to form enlarged LDs. From the magnified images of the ROIs of the white rectangles (Fig. 8B), no green LDs (the position of the yellow arrow) could be seen in the beginning. As time went on, a green LD dot appeared near the ER region, and grew during the first 30 min. The above results demonstrated that LDER can monitor LD production from the ER. | ||
| Fig. 8 (A) In situ monitoring of LD formation in OA-treated SiHa cells stained with LDER. (B) Enlarged fluorescence images of the ROIs indicated by the white rectangles in (A). Scale bar = 20 μm. | ||
Conclusions
In summary, we proposed a dual-targeting group strategy for construction of a SF-probe to simultaneously and discriminatively visualize two types of organelles by incorporating two different organelle-targeting groups into the same fluorophore. As a proof of concept, a 1,8-naphthalimide-based fluorescent probe (LDER) was precisely designed. The probe LDER was capable of imaging LDs and ER simultaneously. We found that the fluorescence intensity of LDER was sensitive to polarity, which guaranteed its usability in the discrimination of LDs and ER. Additionally, LDER could visualize the dynamics of LDs and ER upon either starvation stress or nutrient-rich oleic acid exposure. Notably, the interaction between LDs and ER was investigated with LDER, which revealed that the LDs are formed in ER regions. We expect that this strategy can serve as a novel strategy to design SF-probes for imaging different organelles and uncovering interactions between them.Experimental section
Materials and instrumentation
All of the chemicals were purchased from commercial suppliers. 1H NMR and 13C NMR spectra were recorded by 400 MHz Bruker Avance NMR spectrometers. The mass spectra were recorded by an Agilent Technologies 6510 Q-TOF LC/MS system. The absorbance and fluorescence spectra were recorded on a HITACHI U-2910 spectrophotometer and HITACHI F-2700 spectrofluorimeter, respectively. Confocal fluorescence imaging was performed using an Olympus FV-1200 confocal laser scanning microscope.Synthesis of LDER
The syntheses of compound 1, compound 2 and compound 3 are described in the supporting information (Scheme S1, ESI†). Compound 3 (1 mmol; 470 mg), N,N-dimethyl-4-vinylaniline (1.5 mmol; 220 mg), Pd(OAc)2 (0.1 mmol; 25 mg) and (o-tolyl)3P (0.2 mmol; 60 mg) were dissolved in NEt3/DMF (3 mL/15 mL). The mixture was reacted at 100 °C for 48 h under N2; after cooling to room temperature, the product was poured into water. The resulting precipitate was purified using ethanol to yield LDER as a red powder (54%). 1H NMR (400 MHz, DMSO-d6) δ 8.98 (d, J = 8 Hz, 1H), 8.48 (d, J = 8 Hz, 1H), 8.39 (d, J = 8 Hz, 1H), 8.18 (d, J = 8 Hz, 1H), 7.93 (d, J = 16 Hz, 1H), 7.87 (t, J = 8 Hz, 1H), 7.76–7.70 (m, 3H), 7.60–7.53 (m, 3H), 7.21 (d, J = 8 Hz, 2H), 6.77 (d, J = 8 Hz, 2H), 4.12 (t, J = 8 Hz, 2H), 3.11(t, J = 8 Hz, 2H), 3.00 (s, 6H), 2.25 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 164.04, 163.70, 151.17, 142.90, 142.41, 138.09, 136.16, 131.18, 131.15, 128.77, 124.84, 122.53, 117.88, 112.42, 40.40, 21.35. HRMS (ESI+): m/z calcd for C31H30N3O4S+ 540.1952; found: [M + H]+ 540.1952.Theoretical calculation
The geometrical structure and frontier orbitals of LDER were obtained using the Gaussian 09 program package. All geometry optimizations were carried out using the B3LYP functional basis set.Cell culture and fluorescence imaging
SiHa cells are Human Cervical Carcinoma cells, which are the commonly used cells in fluorescence imaging research. So, we picked SiHa cells as the cellular model. SiHa cells were incubated in H-DMEM (Dulbecco's Modified Eagle's Medium, High Glucose) supplemented with 10% FBS (fetal bovine serum) and 1% penicillin and streptomycin, and cultured in a 5% CO2 incubator at 37 °C. The probe LDER was dissolved in DMSO at a concentration of 5 mM.For living cell staining experiments, SiHa cells were incubated with 5 μM LDER for 30 min. After rinsing with PBS, SiHa cells were imaged using an FV-1200.
For co-localization experiments, SiHa cells were incubated with 5 μM LDER for 30 min, then incubated with various commercial probes (BODIPY 493/503, ER-Tracker Blue, Golgi-Tracker Red, Lyso-Tracker Deep Red (LTDR) and MitoTracker Deep Red (MTDR)) for another 10 min. Cells were rinsed before imaging with an FV-1200.
The calculation was performed using the software of FV10-ASW 4.2 Viewer based on the intensity analysis function of the confocal microscope. This function enables the conversion of fluorescence intensity to color. As a result, the fluorescence intensity change can be presented as a change in color. Based on an image consisting of different fluorescence intensities, a new image with different colors could be automatically obtained. The color legend represents the values of fluorescence intensity. With the aid of the rainbow of color, the fluorescence intensity in different areas can be easily visualized.
Starvation-induced experiments
SiHa cells were incubated in culture medium without nutrition for different times, then stained with LDER (5 μM, 30 min). After washing with PBS, cells were imaged with an FV-1200.Oleic acid treatment experiments
SiHa cells were pretreated with different concentrations of oleic acid (0, 100, and 200 μM) for 4 h, and then washed with PBS. After staining with LDER (5 μM, 30 min), cells were imaged with an FV-1200.Dynamic monitoring of LDs and ER
SiHa cells were incubated with LDER (5 μM, 30 min) and then washed with PBS. Using an FV-1200, fluorescence images were taken every 20 min for a total of 60 min.Dynamic monitoring of LD formation
SiHa cells were starved for 12 h, then cultured in H-DMEM, stained with LDER, and treated with 400 μM oleic acid. Fluorescence images were obtained at different time points using an FV-1200.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52150222, 52073163, and 51773111), Natural Science Foundation of Shandong Province, China (ZR2017ZC0227), and Major Science and Technology Project of Zhejiang Provincial Department of Science and Technology (2020C03030).References
- C. P. Satori, M. M. Henderson, E. A. Krautkramer, V. Kostal, M. D. Distefano and E. A. Arriaga, Chem. Rev., 2013, 113, 2733–2811 CrossRef CAS PubMed.
- H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115–2126 CrossRef CAS PubMed.
- W. Xu, Z. Zeng, J. H. Jiang, Y. T. Chang and L. Yuan, Angew. Chem., Int. Ed., 2016, 55, 13658–13699 CrossRef CAS PubMed.
- A. M. Valm, S. Cohen, W. R. Legant, J. Melunis, U. Hershberg, E. Wait, A. R. Cohen, M. W. Davidson, E. Betzig and J. Lippincott-Schwartz, Nature, 2017, 546, 162–167 CrossRef CAS PubMed.
- Q. Ba and G. Yang, Front. Biol., 2017, 12, 7–18 CrossRef CAS.
- Y. Tu, L. Zhao, D. D. Billadeau and D. Jia, Front. Cell Dev. Biol., 2020, 8, 163 CrossRef PubMed.
- R. V. Farese, Jr. and T. C. Walther, Cell, 2009, 139, 855–860 CrossRef PubMed.
- S. Chen, P. Novick and S. Ferro-Novick, Curr. Opin. Cell Biol., 2013, 25, 428–433 CrossRef CAS PubMed.
- B. Turk and V. Turk, J. Biol. Chem., 2009, 284, 21783–21787 CrossRef CAS PubMed.
- M. J. Phillips and G. K. Voeltz, Nat. Rev. Mol. Cell Biol., 2016, 17, 69–82 CrossRef CAS PubMed.
- F. Wilfling, J. T. Haas, T. C. Walther and R. V. Farese, Jr., Curr. Opin. Cell Biol., 2014, 29, 39–45 CrossRef CAS PubMed.
- D. Li, Y. G. Zhao, D. Li, H. Zhao, J. Huang, G. Miao, D. Feng, P. Liu, D. Li and H. Zhang, Cell Rep., 2019, 27, 343–358 CrossRef CAS PubMed.
- S. Martin and R. G. Parton, Nat. Rev. Mol. Cell Biol., 2006, 7, 373–378 CrossRef CAS PubMed.
- K. Liu and M. J. Czaja, Cell Death Differ., 2013, 20, 3–11 CrossRef CAS PubMed.
- H. Dong and M. J. Czaja, Trends Endocrinol. Metab., 2011, 22, 234–240 CrossRef CAS PubMed.
- L. Guo, M. Tian, Z. Zhang, Q. Lu, Z. Liu, G. Niu and X. Yu, J. Am. Chem. Soc., 2021, 143, 3169–3179 CrossRef CAS PubMed.
- I. Johnson, Histochem. J., 1998, 30, 123–140 CrossRef CAS PubMed.
- N.-E. Choi, J.-Y. Lee, E.-C. Park, J.-H. Lee and J. Lee, Molecules, 2021, 26, 217 CrossRef CAS PubMed.
- P. Gao, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 6035–6071 RSC.
- T. K. Fam, A. S. Klymchenko and M. Collot, Materials, 2018, 11, 1768 CrossRef PubMed.
- D. Singh, D. Rajput and S. Kanvah, Chem. Commun., 2022, 58, 2413–2429 RSC.
- R. A. Smith, R. C. Hartley and M. P. Murphy, Antioxid. Redox Signaling, 2011, 15, 3021–3038 CrossRef CAS PubMed.
- Q. Wan, S. Chen, W. Shi, L. Li and H. Ma, Angew. Chem., Int. Ed., 2014, 53, 10916–10920 CrossRef CAS PubMed.
- D. I. Danylchuk, P. H. Jouard and A. S. Klymchenko, J. Am. Chem. Soc., 2021, 143, 912–924 CrossRef CAS PubMed.
- Z. Zhang, B. Dong and M. Tian, Sens. Actuators B Chem., 2021, 343, 130168 CrossRef CAS.
- Z. Zhang, Z. Gou, B. Dong and M. Tian, Sens. Actuators B Chem., 2022, 355, 131349 CrossRef CAS.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- C. Geraghty, C. Wynne and R. B. P. Elmes, Coord. Chem. Rev., 2021, 437, 213713 CrossRef CAS.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC.
- F. Meng, J. Niu, H. Zhang, R. Yang, Q. Lu, Y. Yu, Z. Liu, G. Niu and X. Yu, Sens. Actuators B Chem., 2021, 329, 129148 CrossRef CAS.
- H. Xiao, P. Li, X. Hu, X. Shi, W. Zhang and B. Tang, Chem. Sci., 2016, 7, 6153–6159 RSC.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- C. W. Song, U. Tamima, Y. J. Reo, M. Dai, S. Sarkar and K. H. Ahn, Dyes Pigm., 2019, 171, 107718 CrossRef CAS.
- M. Jiang, X. Gu, J. W. Y. Lam, Y. Zhang, R. T. K. Kwok, K. S. Wong and B. Z. Tang, Chem. Sci., 2017, 8, 5440–5446 RSC.
- R. Miao, J. Li, C. Wang, X. Jiang, Y. Gao, X. Liu, D. Wang, X. Li, X. Liu and Y. Fang, Adv. Sci., 2022, 9, 2104609 CrossRef CAS PubMed.
- L. Guo, M. Tian, R. Feng, G. Zhang, R. Zhang, X. Li, Z. Liu, X. He, J. Z. Sun and X. Yu, ACS Appl. Mater. Interfaces, 2018, 10, 10706–10717 CrossRef CAS PubMed.
- X. Zheng, W. Zhu, F. Ni, H. Ai and C. Yang, Sens. Actuators, B, 2018, 255, 3148–3154 CrossRef CAS.
- Y. Guo, T. C. Walther, M. Rao, N. Stuurman, G. Goshima, K. Terayama, J. S. Wong, R. D. Vale, P. Walter and R. V. Farese, Nature, 2008, 453, 657–661 CrossRef CAS PubMed.
- H. Xu, H. Zhang, G. Liu, L. Kong, X. Zhu, X. Tian, Z. Zhang, R. Zhang, Z. Wu, Y. Tian and H. Zhou, Anal. Chem., 2019, 91, 977–982 CrossRef CAS PubMed.
- E. Zhao, C. Kam, M.-Y. Wu, E. Wang, J. Wang, B. Z. Tang and S. Chen, CCS Chem., 2022, 4, 515–525 CrossRef CAS.
- S. Zang, X. Kong, J. Cui, S. Su, W. Shu, J. Jing and X. Zhang, J. Mater. Chem. B, 2020, 8, 2660–2665 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and additional spectra. See DOI: https://doi.org/10.1039/d2tb01536f |
| This journal is © The Royal Society of Chemistry 2022 |