
The in situ formation of defective CoOOH catalysts from semi-oxidized Co for alkaline oxygen evolution reaction†
Lan
Wang
a,
Ya
Pan
b,
Dan
Wu
b,
Xiaokang
Liu
b,
Linlin
Cao
 b,
Wei
Zhang
a,
Honglei
Chen
a,
Tong
Liu
b,
Dong
Liu
b,
Tao
Chen
c,
Tao
Ding
b,
Yi
Wang
d,
Chenglong
Ding
a,
Caitao
Kang
a,
Changli
Li
*a,
Jingfu
He
*a and
Tao
Yao
b
b,
Wei
Zhang
a,
Honglei
Chen
a,
Tong
Liu
b,
Dong
Liu
b,
Tao
Chen
c,
Tao
Ding
b,
Yi
Wang
d,
Chenglong
Ding
a,
Caitao
Kang
a,
Changli
Li
*a,
Jingfu
He
*a and
Tao
Yao
b
aSchool of Materials, Sun Yat-sen University, Guangzhou 510275, Guangdong, P. R. China. E-mail: lichli5@mail.sysu.edu.cn; hejf27@mail.sysu.edu.cn
bNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei 230029, Anhui, P. R. China
cSchool of National Defense Science and Technology, Southwest University of Science and Technology, Mianyang 621010, Sichuan, P. R. China
dReactor Operation and Application Research Sub-Institute, Nuclear Power Institute of China, Chengdu 610041, Sichuan, P. R. China
First published on 21st July 2022
Abstract
Electrochemical water splitting based on Co oxide electrocatalysts provides a promising means for renewable hydrogen production from water. Identifying the structure evolution and real-time active structure of Co-based electrocatalysts under operational conditions is crucial for understanding the OER mechanism and rational design of efficient catalysts. Here, we systematically synthesized three types of Co-based precatalysts, including Co/graphene oxide (GO), Co–CoO/GO and Co3O4/GO, to clarify the critical effects of the precatalyst structure for OER. The electrochemical test results confirm that the Co–CoO/GO precatalyst shows a lower overpotential compared to Co/GO and Co3O4/GO precatalysts and commercial RuO2/C. A thorough in situ analysis by XAFS revealed that the Co–CoO/GO precatalysts are oxidized and self-assembled into CoOOH during water oxidation and possess a much higher O vacancy density compared with the CoOOH derived from Co/GO, while the Co3O4/GO precatalyst maintains its structure during operation. This strategy of in situ creation of defects in CoOOH provides a guideline for the rational design of future catalysis systems.
 Jingfu He | Jingfu He is currently an Associate Professor at the School of Materials, Sun Yat-sen University. He received his PhD in 2012 from the University of Science and Technology of China. His current research interests focus on the development of advanced functional materials for the storage of renewable energy, including electrochemical CO2 reduction, water splitting and organic synthesis. He has also committed to developing advanced in situ/operando synchrotron radiation experimental techniques and their applications in energy storage and reaction mechanisms. |
1. Introduction
Clean fuels synthesized from renewable energy have been regarded as an alternative source to fossil fuels due to their high energy density and low greenhouse gas emission.1–5 Electrocatalytic water splitting has emerged as an attractive approach for producing clean and renewable hydrogen with high purity.6–8 However, the overall efficiency of water splitting is severely limited by the sluggish oxygen evolution reaction (OER) that occurs at the anode, which needs a large overpotential due to the complex four-electron reaction mechanism. Efforts in this area are mainly focused on developing highly efficient and stable electrocatalysts capable of mediating the OER at lower overpotential and the acquisition of mechanistic insights into the catalytic process.9–12 Besides the noble metal-based catalysts such as IrO2 and RuO2,13 many earth-abundant, low cost, transition metal-based (e.g., Co, Ni, and Fe) electrocatalysts are also OER active and highly stable in an alkaline environment.6 Among these earth-abundant electrocatalysts, Co-based compounds such as spinel Co3O4, CoO, MCo2O4, CoM2O4, perovskite-type MCoO3 (M, metal) and amorphous CoOx have been proved to exhibit desirable activity in alkaline electrolyte and attract significant attention.14–18 However, the lack of understanding about the complex relationship between catalyst composition and OER activity hinders further optimization of Co-based OER catalysts.Notably, cobalt-based oxides usually undergo a structural change during the OER. The formation of CoOOH through oxidation of cobalt ions to higher valence states under OER conditions is crucial since the newly generated CoOOH is considered the active species for the OER.19–25 This transformation process from a Co-based precatalyst to CoOOH is strongly related to its initial structure, which might be a key reason for the observed different OER activities of different Co-based compounds.26–28 For instance, based on operando XAS and other electrochemical techniques, it has been disclosed that for spinel Co3O4, the Co2+ sites act as active sites that release electrons under applied bias with a close affinity to oxygen to form CoOOH, while the Co3+ sites are relatively stable and thus limit its catalytic activity.29 To overcome the partial conversion of Co3O4 to CoOOH, a Co(OH)2 layer was prepared on Co3O4 to improve the catalytic activity through a facile chemical transformation process from Co(OH)2 to CoOOH.30,31 Despite these studies, our understanding of the direct relationship between the initial structure and activation results is still shallow, and many phenomena cannot be explained in depth. For instance, recent research found that some unique treatments for the initial Co-based precatalyst could effectively enhance the final performance. CoO nanostructures with abundant oxygen vacancies and heterogeneous Co/CoO electrocatalysts could both boost the catalytic activity and durability of the OER compared with standard CoO or Co.32–34 To fully understand the in situ activation of Co-based compounds, detailed experimental work should be conducted to illustrate the atomic-level dynamics of their local coordination environment during the reaction by advanced instrumental techniques such as EXAFS and XANES.35
Herein, we synthesized different phases of cobalt-based precatalysts (Co/GO, Co–CoO/GO and Co3O4/GO) by annealing the CoOx precursors in H2, Ar and air. The sample annealed in Ar composed of Co and CoO mixture phases shows higher alkaline OER activity than Co/GO and Co3O4/GO precatalysts, with an overpotential of 372 and 433 mV reaching a current density of 50 and 100 mA cm−2. XRD and XPS after the OER test demonstrate that Co/GO and Co–CoO/GO are converted into Co (oxy)hydroxide, while the Co3O4/GO structure is robust against oxidation. The in situ XAFS results further showed critical information that the oxidation of the Co sample resulted in the formation of CoOOH with a low density of defects during the OER, while the oxidation of the Co and CoO mixture during the OER resulted in a defective CoOxOHy structure. This research provides a potential solution for the defect engineering of OER catalysts.
2. Results and discussion
Three cobalt-based precatalysts with different oxidation states were prepared by high-pressure hydrothermal treatment of the Co precursor (Co(NO3)2·6H2O), followed by H2, Ar, or air annealing ambiance, respectively, to obtain the corresponding products (Fig. 1a). The X-ray diffraction (XRD) patterns were first recorded to explore the influence of the annealing atmosphere on the crystal structure. It can be seen in Fig. 1b that the annealed samples exhibit several distinct peaks, indicating their high crystallinity. The XRD pattern of the H2 treated sample is in accordance with the standard metal Co (red line, PDF# 15-0806) and this sample is named Co/GO. The phase of the air treated sample is identical to that of Co3O4 without an impurity peak (brown line, PDF# 43-1003) and is named Co3O4/GO. Impressively, the diffraction peaks of metallic Co and CoO (blue line, PDF# 43-1004) can be observed in the Ar treated sample, and this sample is named Co–CoO/GO. X-ray absorption spectra further revealed the local atomic structure of Co-based precatalysts (Fig. 1c, S1 and S2†). The Fourier transform (FT) k3-weighted extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structure spectroscopy (XANES) of Co/GO and Co3O4/GO are identical to those of Co foil and Co3O4, respectively (Fig. 1c and S1†). Notably, the absorption edge of Co–CoO/GO lies between that of the Co foil and CoO, implying that the valence state of Co in Co–CoO/GO is between 0 and +2 (Fig. S1†). The FT curve of Co–CoO/GO in Fig. 1c reveals two dominant peaks at 2–3 Å that are assigned to the Co–Co coordination of Co metal and CoO. This is another piece of evidence that ties in closely with the XRD results that metallic Co and CoO simultaneously exist in the Ar annealed sample. Moreover, the FT curve-fitting analysis of Co–CoO/GO by including one Co–O and two Co–Co coordinations gives the best fitting quality with the Co–O (R = 2.07 Å and N = 3.61) path plus the short (R = 2.51 Å and N = 5.44) and long (R = 3.03 Å and N = 5.04) Co–Co bonds (Table S1 and Fig. S2†). We use Linear combination fitting (LCF) method to determine the sample composition considering the contribution of the standard spectra of Co and CoO in Co K-edge XANES.36 And the metallic Co and CoO in Co–CoO/GO are calculated to be 53.3% and 46.7% by LCF, which is approximately one-to-one (Fig. S3†).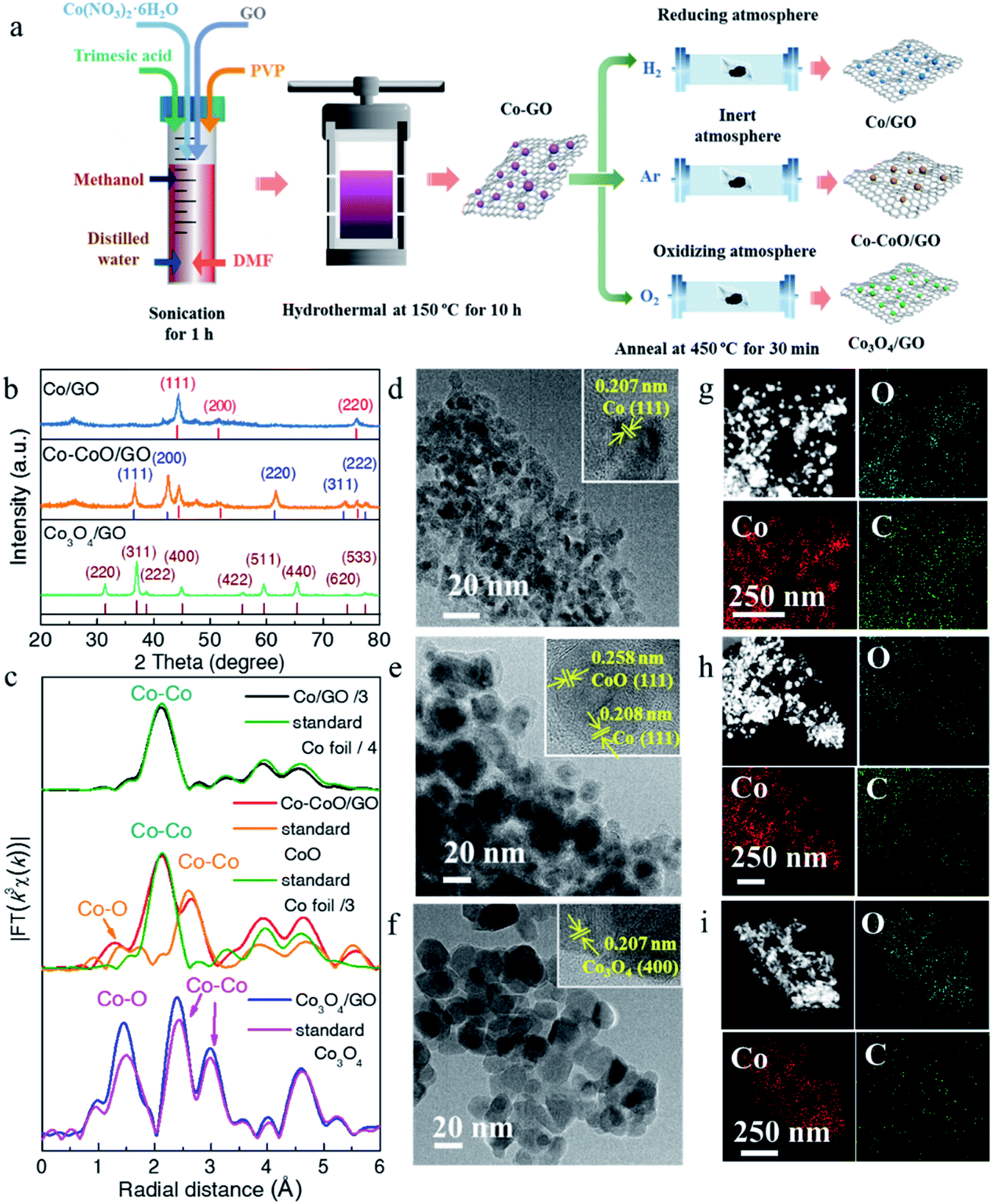 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of Co/GO, Co–CoO/GO and Co3O4/GO. (b) XRD patterns of Co/GO, Co–CoO/GO and Co3O4/GO. (c) The corresponding k3-weighted FT spectra of Co/GO, Co–CoO/GO and Co3O4/GO. The TEM images of Co/GO (d), Co–CoO/GO (e) and Co3O4/GO (f), and the insets display the HRTEM images of Co-based catalysts. EDS mapping images of Co/GO (g), Co–CoO/GO (h) and Co3O4/GO (i). | ||
The morphology and atomic structure of Co-based precatalysts were further investigated by transmission electron microscopy (TEM) and high resolution TEM (HRTEM). As shown in Fig. 1d–f, the mean sizes of Co-based nanoparticles are 7, 16 and 19 nm for Co/GO, Co–CoO/GO and Co3O4/GO, respectively, in agreement with the calculated results based on the Scherrer formula from the XRD results, which show that the sizes of the corresponding samples were about 8, 14 and 21 nm (Fig. S4†). The HRTEM image (Fig. 1d) of Co/GO shows the interplanar spacing was measured to be 0.207 nm, corresponding to the Co(111) plane. For Co–CoO/GO, the HRTEM image in Fig. 1e displays distinct lattice fringes of 0.208 nm and 0.258 nm corresponding to the (111) plane of the Co crystal and (111) plane of the CoO crystal, suggesting the Co and CoO are in simultaneous coexistence on the surface. The HRTEM image of Co3O4/GO shown in Fig. 1f well agrees with the XRD and XAFS results that Co3O4/GO samples match well with Co3O4 phase. The composition analysis of the annealed samples was confirmed by energy dispersive spectroscopy (EDS) mapping, showing the homogeneous dispersion of Co, O and C throughout the entire sample (Fig. 1g–i).
To further investigate the effect of precatalyst oxidation states on catalytic activity, the alkaline OER activities were evaluated with catalyst-modified glassy carbon electrodes (GCEs) and commercial RuO2/C for comparison. In the OER polarization curves (Fig. 2a), Co–CoO/GO exhibits the best OER activity, requiring the lowest overpotential of 372 mV and 433 mV to achieve a current density of 50 and 100 mA cm−2, slightly outperforming RuO2/C (394 and 464 mV to 50 and 100 mA cm−2). In these OER polarization curves, the anodic oxidation peaks of Co/GO, Co–CoO/GO and Co3O4/GO gradually shift to higher potentials, where the anodic peaks at 1.15 and 1.24 V vs. RHE are attributed to the oxidation of Co2+ to Co3+, and the peak at 1.47 V vs. RHE is usually assigned to the oxidation of Co3+ to Co4+.29 These results indicate that the valence states of the Co element increase during the OER. To reflect the intrinsic activity, the Tafel slopes were obtained from a cathodic polarization scan.37 The lowest Tafel slope of 89.5 mV dec−1 is obtained for Co–CoO/GO (Fig. 2b), smaller than that for RuO2 (93.8 mV dec−1), Co3O4/GO (99.8 mV dec−1) and Co/GO (139.2 mV dec−1), which indicates that Co–CoO/GO can show faster reaction kinetics.
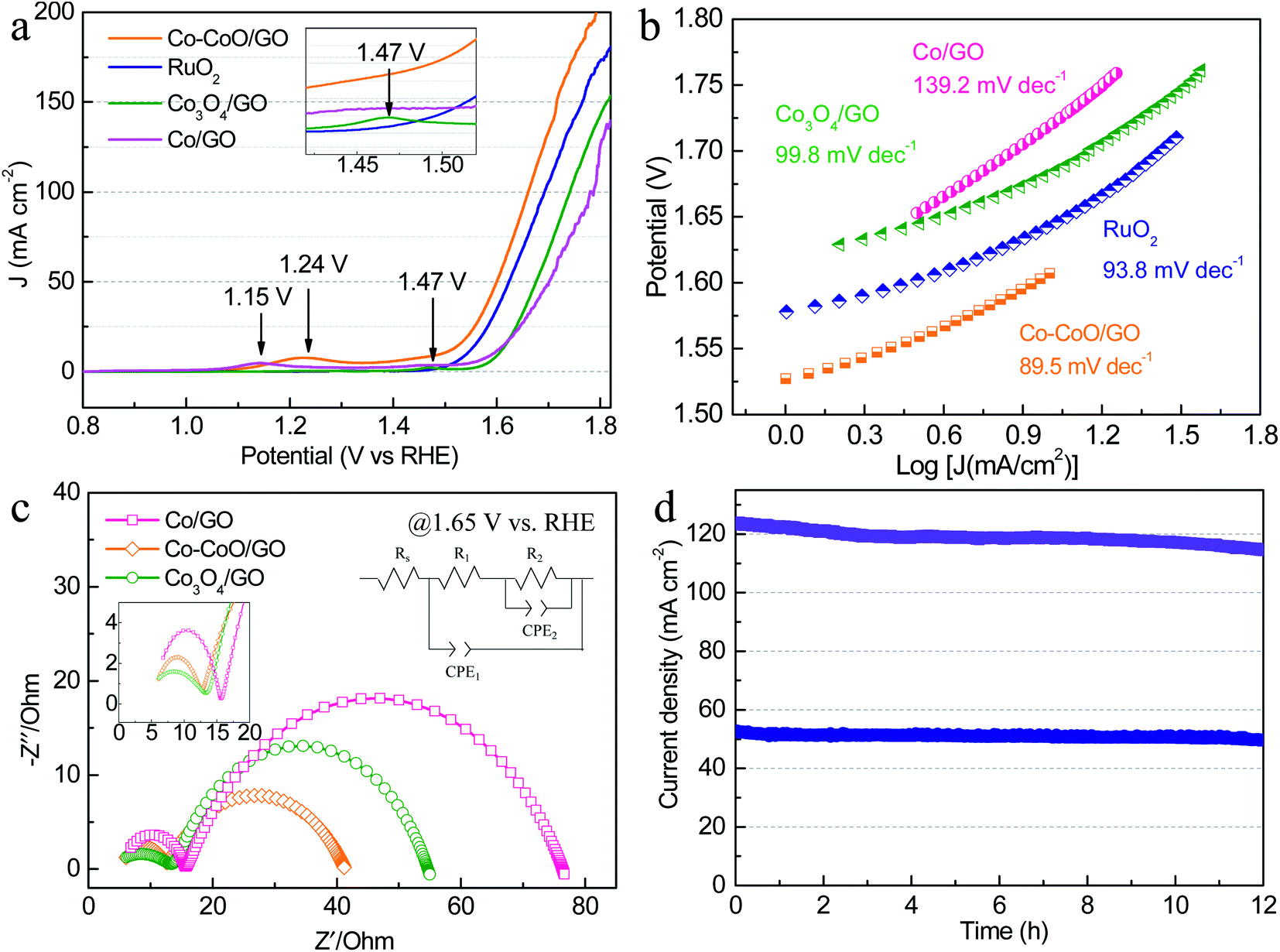 | ||
| Fig. 2 (a) LSV curves and (b) corresponding Tafel slopes of Co/GO, Co–CoO/GO, Co3O4/GO and commercial RuO2 in 1 M KOH. (c) EIS plots of Co/GO, Co–CoO/GO and Co3O4/GO at an overpotential of 420 mV. (d) The long-term amperometric (i–t) stability of Co–CoO/GO was measured at 1.63 and 1.71 V vs. RHE for 12 h. | ||
Fig. 2c shows the electrochemical impedance spectroscopy (EIS) results of cobalt-based precatalysts. The EIS data are characterized by two semicircles and an equivalent circuit used to fit the impedance data is shown in Fig. 2c. The high frequency semi-circle (CPE1-R1) can be viewed as a total charge transfer resistance for the combined steps of the OER, while the low frequency semi-circle (CPE2-R2) is related to the rate of production of one or more surface intermediates.38–40 As can be seen, Co–CoO/GO shows a charge transfer resistance (R1) of ∼7 Ω at 1.65 V vs. RHE, which is smaller than that of Co/GO and Co3O4/GO (∼11 Ω). Moreover, the Co–CoO/GO sample shows the lowest R2 (29 Ω) compared with Co/GO (61 Ω) and Co3O4/GO (41 Ω), indicating that the intermediate is more readily formed on Co–CoO/GO electrocatalysts. In addition, the electrochemical double-layer capacitance of Co–CoO/GO was measured to be 16.9 mF cm−2, higher than that of Co/GO (6.4 mF cm−2) and Co3O4/GO (1.5 mF cm−2), suggesting that Co–CoO/GO has a larger number of active sites (Fig. S5†). More importantly, the stability of Co–CoO/GO was assessed by current–time (i–t) measurements for 12 h (Fig. 2d). It can be seen that the current density of ∼50 mA cm−2 without apparent degradation and that of ∼120 mA cm−2 has ∼7% loss over a 12 hour operation period, demonstrating excellent long-time durability at high current density.
To understand the influence of the initial oxidation state of Co-based precatalysts on the OER activity, we measured the structure of Co-based electrocatalysts after OER tests. XRD patterns in Fig. 3a reveal that after the OER Co/GO and Co–CoO/GO exhibit the same diffraction peaks corresponding well to the different lattice planes of CoOOH (pink line, PDF# 73-1213) and Co(OH)2 (green line, PDF# 02-0925). Considering that there is a reversible transformation between Co(OH)2 and CoOOH, it is not sure from the XRD test which structure is responsible for the activity of alkaline OER.19,41,42 Co3O4/GO after OER conditioning still retains the crystallinity of Co3O4 (brown line, PDF# 43-1003), consistent with the reported result.20 The TEM results in Fig. S6–S8† show that the samples after the OER still exhibit clear lattice fringes, where the measured lattice fringe spacing of Co/GO and Co–CoO/GO is consistent with the crystallographic plane of Co(OH)2 and the CoOOH phase, and Co3O4/GO matches well with the Co3O4 phase. Notably, the full width at half maximum (FWHM) of Co/GO and Co–CoO/GO after the OER is much smaller than that before the OER, corresponding to a larger particle size of 18 and 31 nm (Fig. 3b–d). These structure characterization studies suggest that Co/GO and Co–CoO/GO precatalysts are totally transformed into cobalt (oxy)hydroxide with a layered structure during the OER, and small particles are assembled into larger particles.
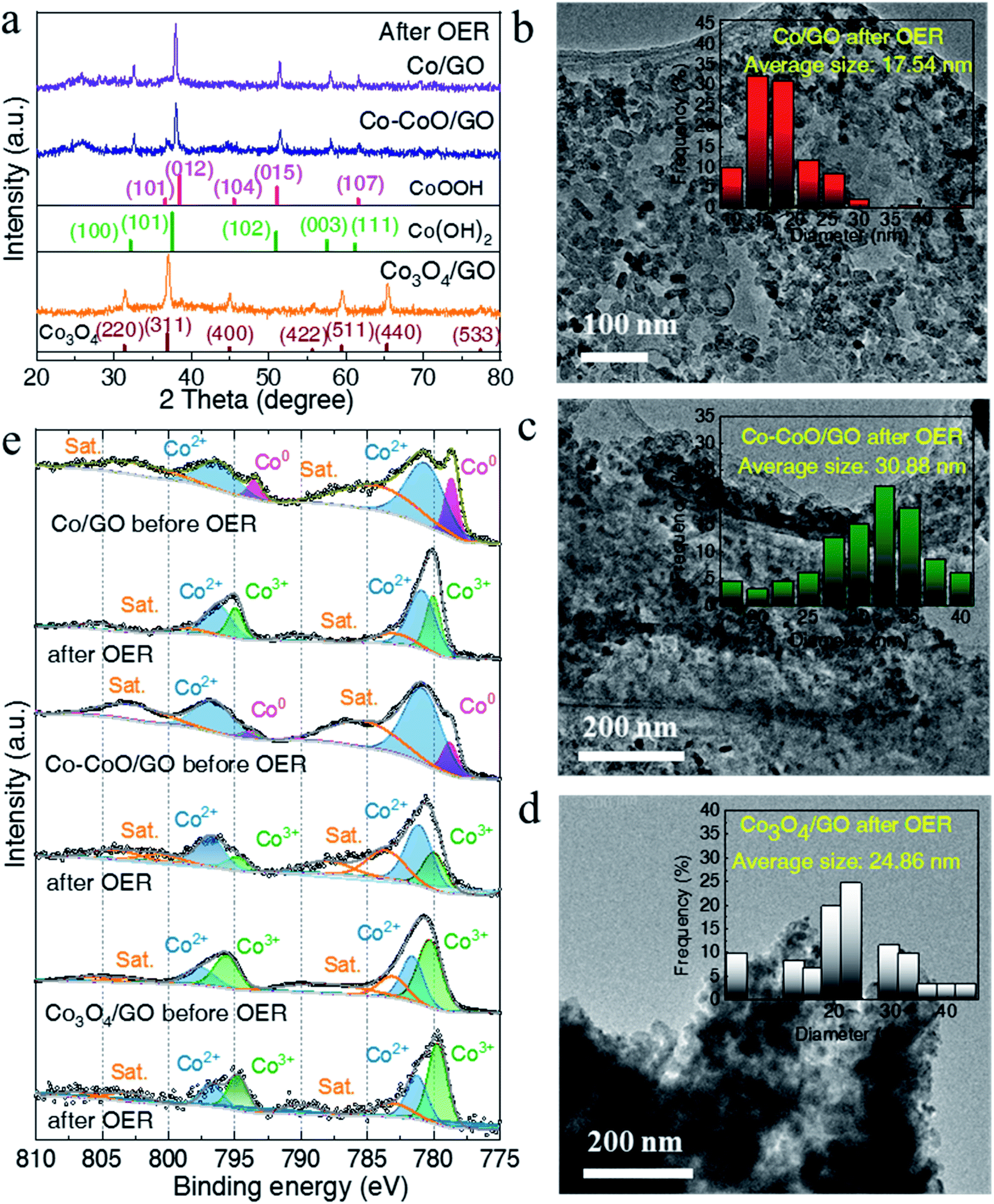 | ||
| Fig. 3 (a) XRD patterns of Co/GO, Co–CoO/GO and Co3O4/GO after OER tests. The TEM images of Co/GO (b), Co–CoO/GO (c) and Co3O4/GO (d) after OER tests. The inset is the corresponding size distribution. (e) Co 2p XPS spectra of Co/GO, Co–CoO/GO and Co3O4/GO before and after OER tests. | ||
Near-surface composition and element valences before and after the OER were characterized by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 3e, the fitting of the Co 2p XPS spectra of Co/GO and Co–CoO/GO shows the two peaks located at ca. 778.5 and 793.4 eV, well consistent with Co metal particles, suggesting that metallic Co exists on the surface of Co/GO and Co–CoO/GO.43 The peaks at ca. 780.6 and 796.1 eV indexed to Co2+ 2p can be observed in Co–CoO/GO that originates from CoO. After the OER, the peak attributed to Co2+ 2p remains and the new peaks at ca. 779.8 and 794.7 eV assigned to Co3+ 2p emerge. Taking XRD analysis into account, the Co2+ and Co3+ come from Co(OH)2 and CoOOH, separately. It is noteworthy that before and after the OER, the peaks attributed to Co3+ and Co2+ of Co3O4/GO remain, and no other Co valence can be found, indicating that Co3O4/GO keeps the same valence.
To probe the active structure of Co-based catalysts in the working environment, in situ XAFS was used to collect the Co K-edge spectra of Co–CoO/GO, Co/GO and Co3O4/GO at different applied OER potentials (Fig. 4). The structure of Co3O4/GO is unchanged, where the curve is characterized by three iconic peaks of standard Co3O4 at 1.45, 2.45 and 3.03 Å, consistent with the XRD and TEM results. It is clear that dramatic structural changes have already happened for Co–CoO/GO and Co/GO at 1.1 V, where the anodic oxidation peaks first emerge (Fig. 4a). The FT k3-weighted spectra confirm that the Co valence of Co/GO and Co–CoO/GO are oxidized at 1.1 V vs. RHE before the OER process and form a CoOOH structure. These samples show two dominant peaks at approximately 1.48 and 2.45 Å that can be assigned to the Co–O and Co–Co coordination of CoOOH. Moreover, the Co–O coordination of around 1.68 Å and Co–Co coordination of around 2.79 Å corresponding to Co(OH)2 are absent in the spectra. A Co–Co characteristic peak of Co foil at 2.05 Å is found in Co–CoO/GO, indicating that a part of metallic Co exists in Co–CoO/GO, and Co is incompletely oxidized. The intensity of the white-line peak (Fig. S9a†), corresponding to a Co 1s–3d transition, is lowest in Co–CoO/GO, which indicates that the Co oxidation state is lower in Co–CoO/GO compared with that in Co/GO.
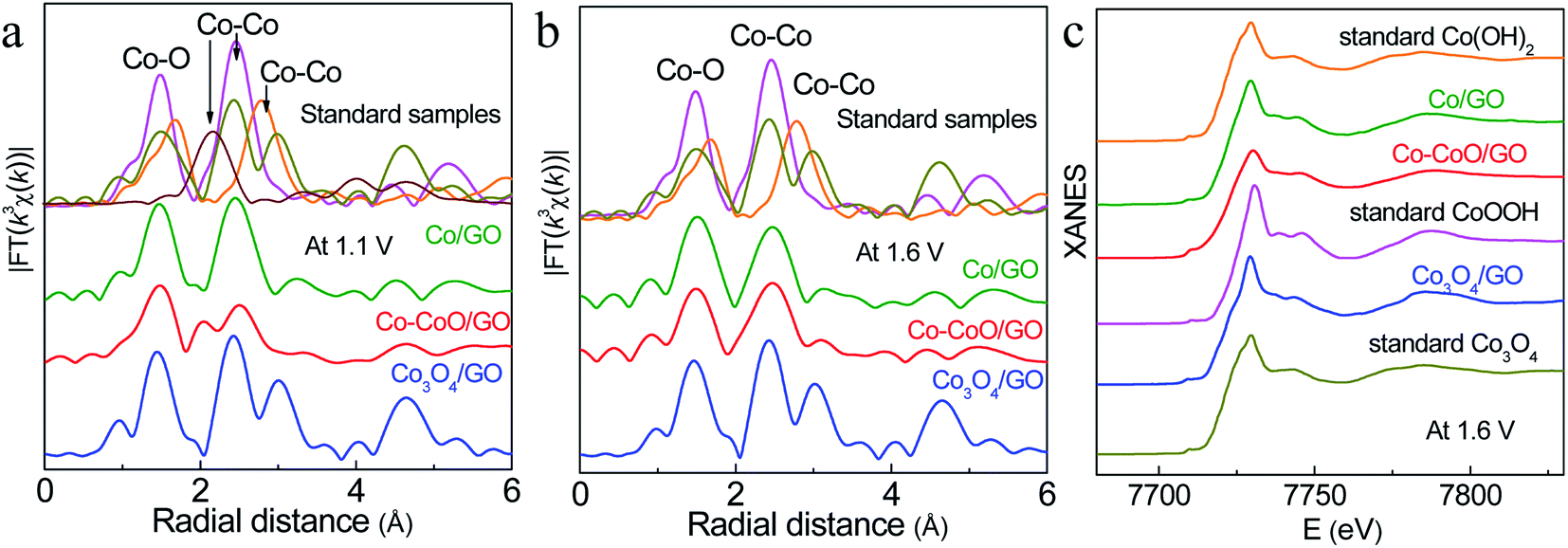 | ||
| Fig. 4 In situ k 3-weighted FT spectra of Co–CoO/GO, Co/GO and Co3O4/GO at 1.1 V (a) and 1.6 V (b). In situ Co K-edge XANES spectra of Co–CoO/GO, Co/GO and Co3O4/GO at 1.6 V (c). The pink, orange, dark yellow and wine red lines represent the standard samples CoOOH, Co(OH)2, Co3O4 and Co foil, respectively. | ||
Raising the potential to 1.6 V, the peak of metallic Co at 2.05 Å disappears in Co–CoO/GO, suggesting that Co–CoO/GO is also completely oxidized to CoOOH under working conditions (Fig. 4b, c and S10†). It can be observed that the intensity of the Co–O first coordination shell of Co–CoO/GO is lower than that of the Co/GO sample, and this difference is described by curve-fitting analysis in detail (Table S1 and Fig. S11†). The EXAFS fitting of standard CoOOH gives six Co–O bonds (R = 1.91 Å and N = 6). Compared with standard CoOOH, the formed CoOOH by direct oxidation of Co/GO at 1.6 V is more similar to standard CoOOH, and the self-assembled CoOOH formed from Co–CoO/GO at 1.6 V has defects in Co–O coordination (R = 1.89 Å and N = 5.24). These defects can also be found in the lower intensity of oscillation of Co–CoO/GO relative to standard CoOOH as shown in Fig. S10.† The oxygen vacancies are reported to influence the surface reconstruction of Co-based catalysts and can also modulate the electron configuration of Co cations and optimize the energy of the adsorption/desorption behavior of the oxygenated intermediates, such as OH− and O*, thus contributing to fast OER dynamics.44–51
Based on these experimental results, we propose a hypothesis for the activation mechanism of precatalysts with different Co oxidation states. Co3O4 with a high oxidation state is structurally stable and does not undergo overall structural changes in an OER environment. Metallic Co and semi-oxidized Co can be completely oxidized to CoOOH in an OER environment. The oxidation of metallic Co occurs at a lower potential, and the particles will assemble with each other during the oxidation process to form CoOOH with a larger size and fewer defects. The Co in Co–CoO is relatively difficult to oxidize, so a higher potential is required. Because the oxidation of Co and CoO does not occur simultaneously, the assembly process of the particles does not occur uniformly, resulting in a large number of O vacancies. The oxygen vacancies are reported to greatly facilitate the bonding of oxygen species on the catalyst surface and promote the OER activity of Co-based catalysts.
3. Conclusions
In summary, Co-based precatalysts with different oxidation states are synthesized by annealing CoOx precursors in H2, Ar and air. From the electrochemical OER test results, it is demonstrated that the Co–CoO/GO precatalyst shows a lower overpotential compared to Co/GO and Co3O4/GO precatalysts and commercial RuO2/C. The XRD and XPS results after OER tests suggest that Co3O4/GO retains its initial structure, while Co/GO and Co–CoO/GO samples are oxidized and transformed into layered structures of CoOxOHy. In situ analysis by XAFS further reveals that the Co–CoO/GO precatalysts are oxidized and self-assembled into CoOOH during water oxidation and possess a much higher O vacancy density compared with the CoOOH derived from Co/GO. This work provides atomic-level insights into the structural evolution of different cobalt oxides during the OER process.Author contributions
J. H., C. L. and T. Y. conceived this project. L. W., X. L., W. Z., H. C., T. L., D. L., T. C., T. D., C. D and C. K. performed the catalyst synthesis and characterization, XAFS and electrochemical experiments. L. W., Y. P., W. D., L. C., and Y. W. analyzed the experimental data. The manuscript was written by J. H., C. Li. and W. L. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22002191, 12105287, 22002147, 22179125 and 12025505), the Natural Science Foundation of Guangdong Province (No. 2022A1515010928 and 2022A1515012661), Fundamental Research Funds for the Central Universities, University of Science and Technology of China (No. KY2310000020) and Sun Yat-sen University (No. 22qntd0206). We thank NSRL, BSRF and SSRF for the synchrotron beam time.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci., 2006, 103, 15729 CrossRef CAS PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337 RSC.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347 CrossRef CAS PubMed.
- C. L. Li, Q. Cao, F. Z. Wang, Y. Q. Xiao, Y. B. Li, J. J. Delaunay and H. W. Zhu, Chem. Soc. Rev., 2018, 47, 1029 Search PubMed.
- X. Li, X. Hao, A. Abudula and G. Guan, J. Mater. Chem. A, 2016, 4, 11973 RSC.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- Y. Zhou and H. J. Fan, ACS Mater. Lett., 2020, 3, 136 CrossRef.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215 CrossRef CAS PubMed.
- L. Li, P. Wang, Q. Shao and X. Huang, Chem. Soc. Rev., 2020, 49, 3072 RSC.
- J. B. Gerken, J. G. McAlpin and J. Y. C. Chen, J. Am. Chem. Soc., 2011, 133, 14431 CrossRef CAS PubMed.
- C. Zhu, Q. Shi, S. Feng, D. Du and Y. Lin, ACS Energy Lett., 2018, 3, 1713 CrossRef CAS.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501 CrossRef CAS PubMed.
- N. Danilovic, R. Subbaraman, K. C. Chang, S. H. Chang, Y. J. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y. T. Kim and D. Myers, J. Phys. Chem. Lett., 2014, 5, 2474 CrossRef CAS PubMed.
- S. Ye, Y. Zhang, W. Xiong, T. Xu, P. Liao, P. Zhang, X. Ren, C. He, L. Zheng and X. Ouyang, Nanoscale, 2020, 12, 11079 RSC.
- R. Zhang, Y. C. Zhang, L. Pan, G. Q. Shen, N. Mahmood, Y. H. Ma, Y. Shi, W. Jia, L. Wang and X. Zhang, ACS Catal., 2018, 8, 3803 CrossRef CAS.
- H. M. Amin and H. Baltruschat, Phys. Chem. Chem. Phys., 2017, 19, 25527 RSC.
- L. Cao, Y. Cao, X. Liu, Q. Luo, W. Liu, W. Zhang, X. Mou, T. Yao and S. Wei, J. Mater. Chem. A, 2018, 6, 15684 RSC.
- K. Dai, N. Zhang, L. Zhang, L. Yin and B. Zhang, Chem. Eng. J., 2021, 414, 128804 CrossRef CAS.
- Y. C. Liu, J. A. Koza and J. A. Switzer, Electrochim. Acta, 2014, 140, 359–365 CrossRef CAS.
- B. Arno, T. E. Jones, M. M. Elias, T. Detre, C. Petko, G. Manuel, R. Tobias, D. Holger and S. Peter, Nat. Catal., 2018, 1, 711 CrossRef.
- L. Yang, H. Qin, Z. Dong, T. Wang, G. Wang and L. Jiao, Small, 2021, 17, 12102027 Search PubMed.
- N. Yao, G. Wang, H. Jia, J. Yin, H. Cong, S. Chen and W. Luo, Angew. Chem., Int. Ed., 2022, 61, e202117178 CAS.
- M. Kim, B. Lee, H. Ju, S. W. Lee and J. Kim, Adv. Mater., 2019, 31, 1901977 CrossRef PubMed.
- Y. Zhang, C. Wu, H. Jiang, Y. Lin, H. Liu, Q. He, S. Chen, T. Duan and L. Song, Adv. Mater., 2018, 30, 1707522 CrossRef PubMed.
- R. Q. Gao, M. Deng, Q. Yan, Z. X. Fang, L. C. Li, H. Y. Shen and Z. F. Chen, Small Methods, 2021, 5, 2100834 CrossRef CAS PubMed.
- P. Guo, J. Wu, X. B. Li, J. Luo, W. M. Lau, H. Liu, X. L. Sun and L. M. Liu, Nano Energy, 2018, 47, 96 CrossRef CAS.
- K. Fan, H. Zou, L. Duan and L. Sun, Adv. Energy Mater., 2020, 10, 1903571 CrossRef CAS.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6, 1502313 CrossRef.
- H. Y. Wang, S. F. Hung, H. Y. Chen, T. S. Chan and B. Liu, J. Am. Chem. Soc., 2015, 138, 36 CrossRef PubMed.
- M. Favaro, J. Yang, S. Nappini, E. Magnano, F. M. Toma, E. J. Crumlin, J. Yano and I. D. Sharp, J. Am. Chem. Soc., 2017, 139, 8960 CrossRef CAS PubMed.
- T. Ling, D. Y. Yan, Y. Jiao, H. Wang, Y. Zheng, X. Zheng, J. Mao, X. W. Du, Z. Hu and M. Jaroniec, Nat. Commun., 2016, 7, 12876 CrossRef CAS PubMed.
- D. Guo, F. Chen, W. Zhang and R. Cao, Sci. Bull., 2017, 62, 626 CrossRef CAS.
- Z. Liang, Z. Huang, H. Yuan, Z. Yang, C. Zhang, Y. Xu, W. Zhang, H. Zheng and R. Cao, Chem. Sci., 2018, 9, 34 Search PubMed.
- X. Fan, Y. Fan, X. Zhang, L. Tang and J. Guo, J. Alloy. Compd., 2021, 877, 160279 CrossRef CAS.
- D. Cao, D. Liu, S. Chen, O. A. Moses, X. Chen, W. Xu, C. Wu, L. Zheng, S. Chu, H. Jiang, C. Wang, B. Ge, X. Wu, J. Zhang and L. Song, Energy Environ. Sci., 2021, 14, 906 RSC.
- Y. S. Shen, S. L. Wang, S. T. Huang, Y. M. Tzou and J. H. Huang, J. Hazard. Mater., 2010, 179, 160 CrossRef CAS PubMed.
- X. Sun, X. X. Zhang, Y. L. Li, Y. Z. Xu, H. Su, W. Che, J. F. He, H. Zhang, M. H. Liu, W. L. Zhou, W. R. Cheng and Q. H. Liu, Small Methods, 2021, 5, 2100573 CrossRef CAS PubMed.
- L. Bai, D. A. Harrington and B. E. Conway, Electrochim. Acta, 1987, 32, 1713 CrossRef CAS.
- R. L. Doyle and M. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 5224 RSC.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248 CrossRef CAS PubMed.
- S. Liu, X. Tan, X. Zheng, S. Liang and H. Zhang, Ionics, 2020, 26, 3531 CrossRef CAS.
- Y. Lu, D. Q. Fan, Z. P. Chen, W. P. Xiao, C. C. Cao and X. F. Yang, Sci. Bull., 2019, 65, 460 CrossRef.
- X. Ren, T. Wu, Y. Sun, Y. Li and Z. J. Xu, Nat. Commun., 2021, 12, 2608 CrossRef CAS PubMed.
- S. Wang, Y. Xu, R. Fu, H. Zhu and Y. Zhao, Nano-Micro Lett., 2019, 11, 76 CrossRef CAS PubMed.
- Z. Xiao, Y. C. Huang, C. L. Dong, C. Xie and S. Wang, J. Am. Chem. Soc., 2020, 142, 12087 CrossRef CAS PubMed.
- Y. Zhu, L. Zhang, B. Zhao, H. Chen, X. Liu, R. Zhao, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Funct. Mater., 2019, 29, 1901783 CrossRef.
- G. Zhuang, Y. Chen, Z. Zhuang, Y. Yu and J. Yu, Sci. China Mater., 2020, 63, 2089 CrossRef CAS.
- A. Badreldin, A. E. Abusrafa and A. Abdel-Wahab, ChemSusChem, 2021, 14, 10 CrossRef CAS PubMed.
- L. Yan, B. Zhang, Z. Liu and J. Zhu, Chem. Eng. J., 2020, 405, 126198 CrossRef.
- Y. Yan, C. Y. Liu, H. W. Jian, X. Cheng, T. Hu, D. Wang, L. Shang, G. Chen, P. Schaaf, X. Y. Wang, E. J. Kan and T. R. Zhang, Adv. Funct. Mater., 2021, 31, 2009610 CrossRef CAS.
- X. Zhang, Y. Zhao, Y. Zhao, R. Shi, G. Waterhouse and T. Zhang, Adv. Energy Mater., 2019, 9, 1900881 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02989h |
| This journal is © The Royal Society of Chemistry 2022 |