
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of the critical chain length for macromolecular crystallization using structurally flexible polyketones†
Yuki Ide a,
Yumehiro Manabeb,
Yuya Inabab,
Yusuke Kinoshitaa,
Jenny Pirilloa,
Yuh Hijikata*a,
Tomoki Yonedab,
Kilingaru I. Shivakumara,
Saki Tanakac,
Hitoshi Asakawac and
Yasuhide Inokuma*ab
a,
Yumehiro Manabeb,
Yuya Inabab,
Yusuke Kinoshitaa,
Jenny Pirilloa,
Yuh Hijikata*a,
Tomoki Yonedab,
Kilingaru I. Shivakumara,
Saki Tanakac,
Hitoshi Asakawac and
Yasuhide Inokuma*ab
aInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Kita 21, Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan. E-mail: inokuma@eng.hokudai.ac.jp; hijikata@icredd.hokudai.ac.jp
bDivision of Applied Chemistry, Faculty of Engineering, Hokkaido University, Kita 13, Nishi 8, Kita-ku, Sapporo, Hokkaido 060-8628, Japan
cNanomaterials Research Institute (NanoMaRi), Graduate School of Natural Science and Technology, and Nano Life Science Institute (WPI-NanoLSI), Kanazawa University, Kanazawa 920-1192, Japan
First published on 21st July 2022
Abstract
Critical chain length that divides small molecule crystallization from macromolecular crystallization is an important index in macro-organic chemistry to predict chain-length dependent properties of oligomers and polymers. However, extensive research on crystallization behavior of individual oligomers has been inhibited by difficulties in their synthesis and crystallization. Here, we report on the determination of critical chain length of macromolecular crystallization for structurally flexible polyketones consisting of 3,3-dimethylpentane-2,4-dione. Discrete polyketone oligomers were synthesized via stepwise elongation up to 20-mer. Powder and single crystal X-ray diffraction showed that the critical chain length for polyketones existed at an unexpectedly short chain length, 5-mer. While shorter oligomers adopted unique conformations and packing structures in the solid state, higher oligomers longer than 4-mer produced helical conformations and similar crystal packing. The critical chain length helped with understanding the inexplicable changes in melting point in the shorter chain length region resulting from chain conformations and packing styles.
Introduction
There has been growing interest in molecularly defined oligomers that combine the molecular precision of organic synthesis with the properties of macromolecules.1–8 This emerging research area is termed macro-organic chemistry. Interestingly, recent developments, such as the iterative exponential growth (IEG) strategy, facilitate an efficient synthetic access to discrete oligomers.9,10 Over the past few decades, a wide variety of discrete oligomers including oligo((meth)acrylate)s,11,12 oligo(ethylene imine)s,13–15 oligo(caprolactone)s,16–18 oligo(siloxane)s,19–22 oligo(thiophene)s,23–25 and oligo(peptide)s26–29 have been investigated. These oligomers aid in discovering length-specific properties, for example, aggregation, folding, host–guest interactions, and light absorption, which are otherwise hidden in polydisperse polymers. Such properties are not always proportional to chain length, but an abrupt change in a property can be sometimes observed at a certain chain length.30–34Crystallization behavior can often show discontinuous changes as the chain length of an oligomer is increased.35–38 In general, shorter oligomers show length-specific lattice cells, conformations (small molecule crystallization), and packing structures, whereas longer ones often crystallize virtually in a similar fashion to one another (macromolecular crystallization).
Critical chain length,30–34 which is the division point between these crystallization types (Fig. 1), receives considerable interest because crystal packing and solid-state conformations can alter the physical properties of macromolecules, such as their melting point. However, research into oligomer crystallization is unexpectedly difficult from the viewpoint of precise synthesis and crystallization. In the case of discrete oligo(ethylene glycol)s, some of the most investigated materials, only a limited number of crystal structures have been reported, although recent syntheses have provided precise oligomers up to 64-mer.39 Moreover, incorporation of solvents and metal ions during crystallization hampers any direct comparison of their crystal structures and packing styles. Hence, extensive research of oligomer crystallization has been limited to conformationally rigid oligomers such as oligo(thiophene)s.40,41
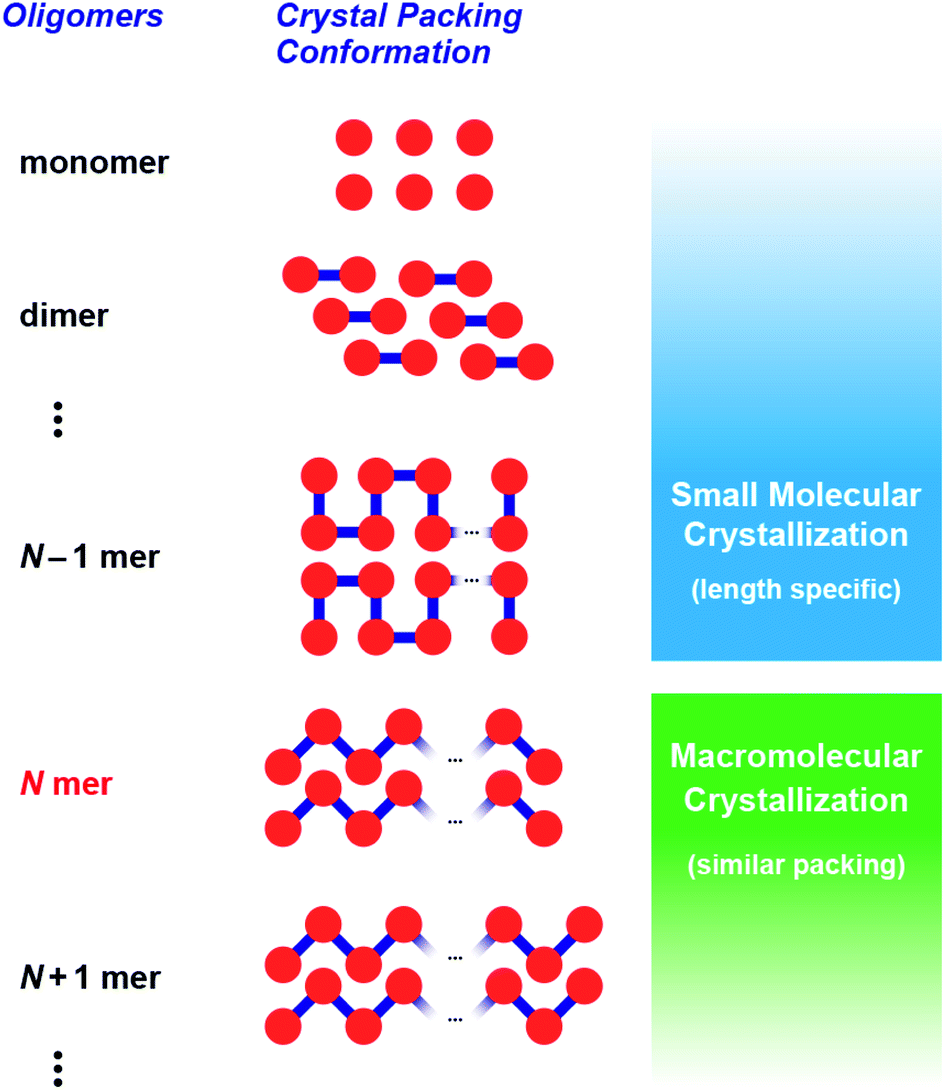 | ||
| Fig. 1 Critical chain length, N, as a boundary between small molecule-like crystallization and macromolecular crystallization. | ||
This situation prompted us to study the critical chain length of crystallization for structurally flexible oligomers and polymers. In this work, we focused on aliphatic polyketones consisting of 3,3-dimethylpentane-2,4-dione (1) as the monomer.42–46 Recently, these polyketones have received attention as lithium ion conducting materials47 and as structurally flexible synthetic precursors for generating metal ion adsorbents,48 π-conjugated chromophores,49–52 and anion-binding hosts.53 Furthermore, these polyketones have advantages in the stepwise synthesis of discrete oligomers. Namely, terminal-selective formation of enol silyl ether and subsequent Ag2O-mediated coupling reaction of oligomers allow accurate elongation of the chain length. Herein, we report on the synthesis and crystallization behavior of discrete oligo(3,3-dimethylpentane-2,4-dione)s up to icosamer (20-mer) to determine the critical chain length of macromolecular crystallization. Single crystal and powder X-ray diffraction analyses clearly showed that macromolecular crystallization behavior appears at the pentamer (N = 5 in Fig. 1). While shorter oligomers (dimer to tetramer) exhibited unique crystalline lattices, longer oligomers (N ≥ 5) commonly adopted helical conformations and similar packing styles. Our findings provide reliable evidence that macromolecular crystallization of flexible oligomers occurs at an unexpectedly short chain length.
Results and discussion
Discrete polyketone oligomers, from pentamer 5 to decamer 10, were synthesized by a stepwise chain length elongation strategy using shorter oligomers 2–4 42,50 as the starting materials (Scheme 1). Pentamer 5 was obtained by a cross-coupling reaction between silyl enolates of monomer 1-Si and tetramer 4-Si in 25% yield after separation by recycling gel permeation chromatography (GPC). Oxidative homo-coupling of mono-silylated trimer 3-Si gave discrete hexamer 6 in 32% yield. Although the cross-coupling reaction between mono-silylated dimer 2-Si and bis-silylated trimer 3-Si2 gave rise to a mixture of 2, 3, and pentamer 5, along with the desired heptamer 7, recycling GPC separation allowed isolation of pure 7 in 4% yield. Nonamer 9 was also prepared in 7% isolated yield by a cross-coupling reaction between 3-Si and 3-Si2. Treatment of pentamer 5 with chlorotrimethylsilane (2.00 equiv.), NaI, and triethylamine furnished mono-silylated 5-Si in 36% yield which was homo-coupled into decamer 10 in 24% yield. Furthermore, an oligomerization reaction of bis-silylated tetramer 4-Si2 enabled isolation of octamer 8, dodecamer 11, hexadecamer 12, and icosamer 13 in 9, 7, 5, and 3% yields, respectively (Scheme 1).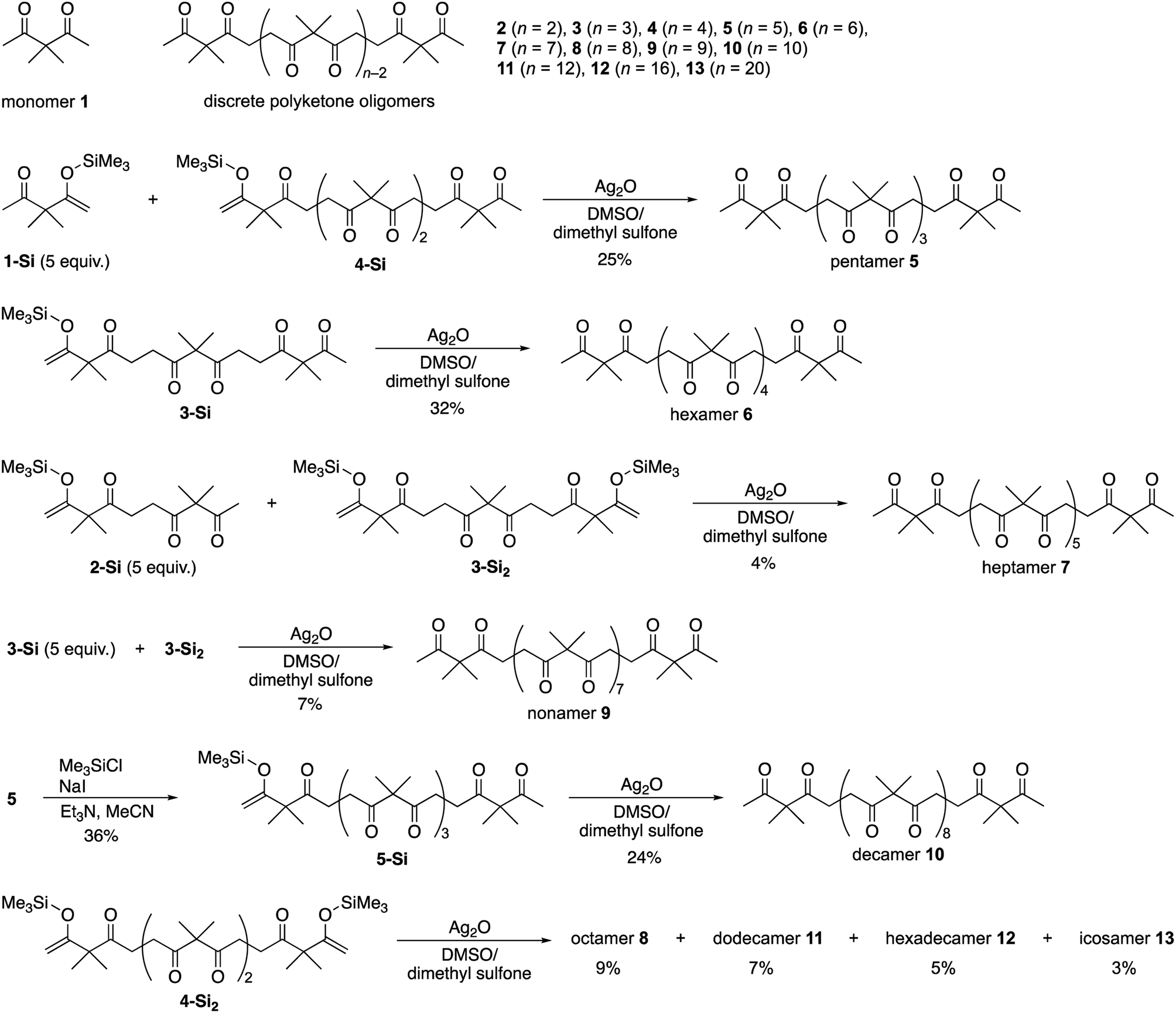 | ||
| Scheme 1 Synthesis of discrete polyketone oligomers 5–13. | ||
Matrix assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry and GPC-high performance liquid chromatography (HPLC) analysis unambiguously confirmed the monodisperse character of oligomers 2–13. MALDI-TOF mass spectra of each oligomer showed monocationic peaks assignable to sodium adducts [M + Na]+ at m/z = 277.3, 403.3, 529.3, 655.4, 781.4, 907.5, 1033.6, 1159.6, 1285.7, 1537.9, 2043.1, and 2547.5 for 2–13, respectively (Fig. S1 in the ESI†). GPC-HPLC chromatograms exhibited a monomodal peak for each oligomer whose retention time gradually decreased as the number of monomer units increased (Fig. S2†). Even for icosamer 13, contamination by any higher homologues was not detected in the HPLC chromatogram, which indicates >98% purity of the sample. 1H NMR spectroscopy of discrete oligomers 2–13 showed nearly identical signal patterns, as represented by icosamer 13; terminal acetyl groups resonated at 2.15 ppm as a sharp singlet, and rather broad signals for internal ethylene and dimethylmethylene groups appeared at 2.85–2.61 and 1.44–1.34 ppm, respectively.
Powder X-ray diffraction (PXRD) analysis of polyketone oligomers 2–13 clearly showed a discontinuous shift in their crystallization behavior (Fig. 2a). While monomer 1 was liquid at room temperature, oligomers 2–13 were obtained as crystalline solids by recrystallization from chloroform, except for trimer 3, which was crystallized via slow cooling of the melt. 1H NMR analysis confirmed that all the solid samples 2–13 were obtained as their pure forms without incorporation of solvent for crystallization. Dimer 2, trimer 3, tetramer 4, and pentamer 5 exhibited unique PXRD patterns that completely differ from each other. Longer oligomers 6–13 showed PXRD patterns similar to those of pentamer 5, indicating the critical chain length of N = 5. For shorter oligomers 2–4, the PXRD patterns matched with the simulated spectra generated from previously reported single crystal X-ray structures in which dimer 2, trimer 3 and tetramer 4 adopted U-shaped, linear, and S-shaped conformations in the space groups C2/c, P21/c, and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , respectively (Fig. 2b).42,50
, respectively (Fig. 2b).42,50
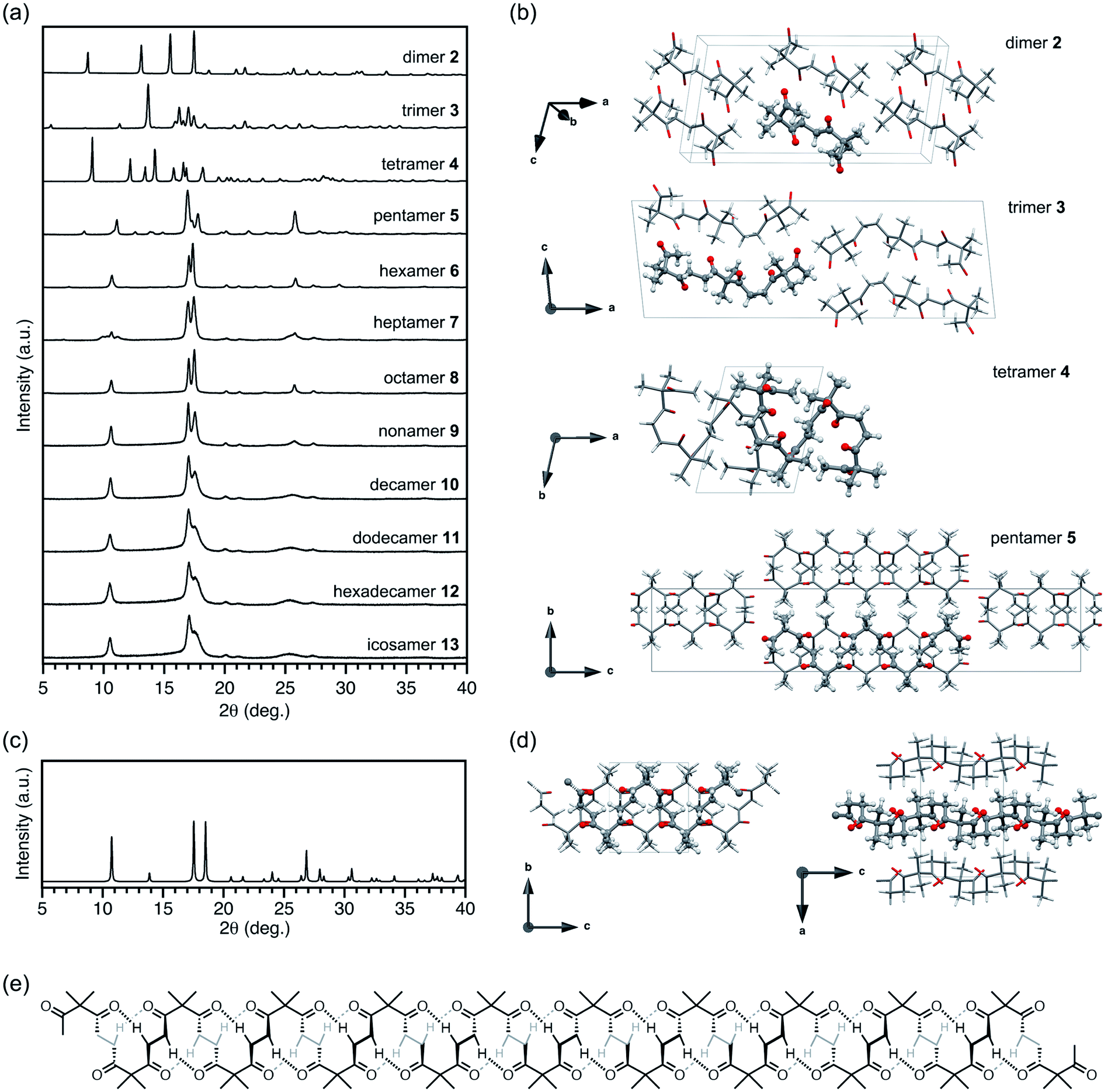 | ||
| Fig. 2 (a) PXRD patterns of discrete oligomers 2–13. (b) Conformations and packing structures of 2–5 obtained by SCXRD analysis. (c) Simulated PXRD pattern of the energy minimized crystal structure of infinite polyketones. (d) Energy minimized packing structure of infinite polyketone crystals. (e) Chemical structure of icosamer 13 in the helical conformation. | ||
Single crystals of 5 were obtained by vapor diffusion of cyclohexene as a poor solvent into a 1,2-dichloroethane solution of 5. However, single crystal X-ray diffraction (SCXRD) analysis of the as-obtained crystals gave a highly disordered structure along the oligomer chain with remarkably large R1 values (>0.20). As observed for some oligomer crystals having other repeating units,14,54–57 such a disordering can be attributable to aperiodic ordering of oligomer chains to give pseudo infinite chain structures. To improve the diffraction data quality, we examined crystal annealing at various temperatures. Eventually, a reasonable crystal structure of 5 was solved in the orthorhombic space group Pna21 using a crystal annealed at 50 °C for 3.5 days. The simulated PXRD pattern of 5 from a single crystal structure matched with the observation in Fig. 2 (see also Fig. S8†). Interestingly, a helical conformation in which two monomer units are incorporated per turn was observed for 5. In the centrosymmetric unit cell, each of the two left- and right-handed helices existed to form a racemic crystal. All the helical chains were oriented along the c-axis. An alternating alignment of left- and right-handed helices was found along the a-axis, whereas strip-shaped assemblies consisting of helices with the same handedness were oriented along the b-axis (Fig. S3†). Given the single crystal X-ray structure, the characteristic PXRD signals of 5 at 2θ = 11.2 and 17.0° in Fig. 2a were assigned to the (011) and (200) reflection planes, respectively. The 200 diffraction (d spacing = 5.22 Å) corresponded to the distance between two neighboring helices of different handedness, which is virtually independent of the chain length of the helices.
Single crystals of 6 were formed by vapor diffusion of cyclohexene into a 1,2-dichloroethane solution. As the best result of many attempts, a reasonable structure was obtained with a crystal annealed at 50 °C for 2 days. Hexamer 6 also showed a helical conformation resembling that of pentamer 5 (Fig. S4†). The simulated PXRD pattern of 6 was in good agreement with observations (Fig. S9†). Again, an intense PXRD peak at 2θ = 17.2° (d spacing = 5.16 Å) was assigned to the distance between two helices in different handedness.
Although SCXRD analysis of 7–13 was unsuccessful, we performed theoretical calculations to discuss their solid-state structures from the observed PXRD patterns. Considering the similarity of the PXRD patterns of 5–13, a packing model of infinite polyketone chains in a helical conformation was constructed based on the single crystal X-ray structure of 6. The packing structure was then refined under periodic boundary conditions with cell relaxation by the self-consistent-charge density-functional tight binding method (see the ESI†).58 In the optimized structure, a pentamer 5-like helical conformation was maintained for infinite polymers (Fig. 2d). Short H⋯O distances between carbonyl oxygen atoms and α-protons indicated that the helical conformations of polyketones were stabilized by multiple intramolecular hydrogen bonds (Fig. 2e).
Simulated PXRD patterns calculated from the optimized packing structures essentially matched with the observations for oligomers 7–13. Intense peaks at 2θ = 10.8, 17.5, and 18.5° (d spacing = 8.19, 5.07 and 4.80 Å, respectively) were commonly observed for 7–13. Remarkably, the former two peaks, which are rather sharp compared with the other signals, were assignable to intermolecular distances between two helices in the same and different handedness, respectively. These results strongly suggested that the longer oligomers 7–13 also adopt helical conformations and follow packing structures similar to those of 5 and 6.
Small-angle X-ray scattering (SAXS) analysis also supported the helical conformations for longer oligomers, and the chain length dependence of long range periodic structures. While tetramer 4 did not show any observable diffraction signal in the range of 2θ < 4.0°, octamer 8 exhibited a signal at 2θ = 2.6° (d spacing: 3.4 nm). Assuming a helical structure, this long-periodic structure was assignable to the end-to-end distance of helical 8 (3.1 nm) (Fig. S16†). The SAXS signals shifted to a smaller angle region as the chain length increased: 2θ = 1.7, 1.4, and 1.1° (d spacing: 5.2, 6.3, and 8.0 nm) for dodecamer 11, hexadecamer 12, and icosamer 13, respectively (Fig. S10†). The periodic distance of the longest oligomer 13 showed good agreement with the chain length (7.5 nm) in its helical conformation (Fig. 2e and S16†).
Such a long-range periodic structure was also observed by atomic force microscopy (AFM) measurements. A 0.05 mM chloroform solution of octamer 8 (5 μL) was drop-cast onto a highly oriented pyrolytic graphite (HOPG) surface, and the solvent was slowly evaporated over 30 s. In the obtained AFM images, crystalline domains were observed, and each domain showed a striped texture with an average distance of 3.4 nm (Fig. 3). The observed periodic structure was in line with the distance between neighboring helices of 8 indicated by SAXS and PXRD analyses.
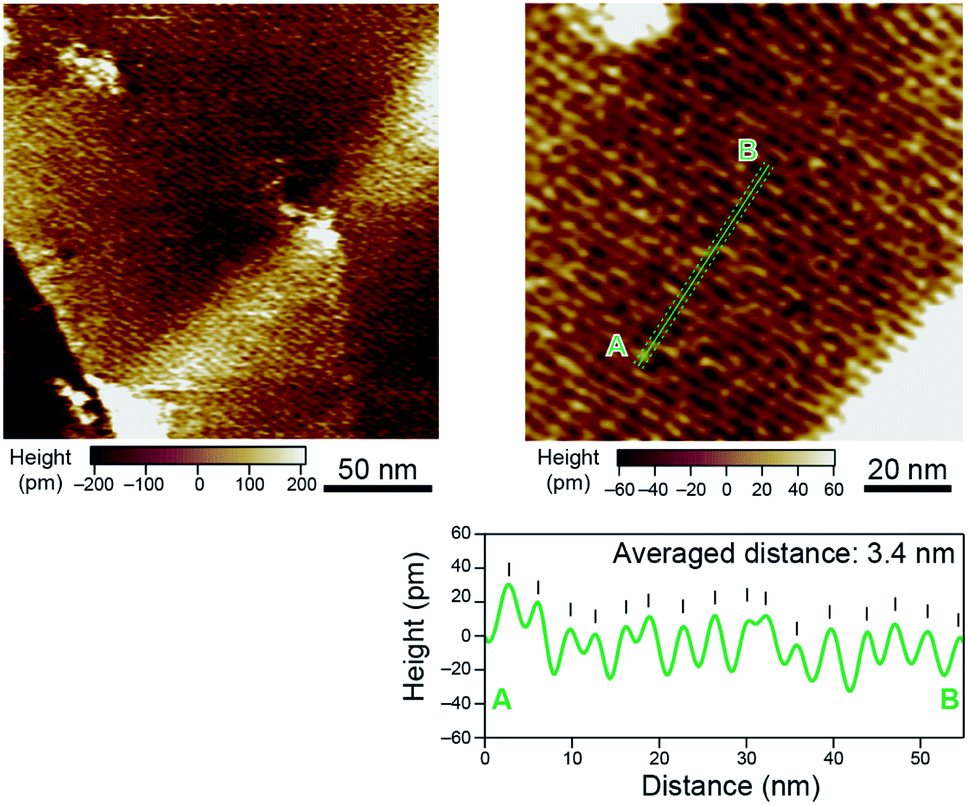 | ||
| Fig. 3 AFM images of octamer 8 on HOPG in ultra-pure water. | ||
A helical conformation and packing structures similar to those of discrete oligomers 5–13 were also suggested for polydisperse polymers in the solid state. Assuming the packing structure of infinite chains (Fig. 2d), the two characteristic PXRD peaks at 2θ ∼ 11° and 18° that correspond to helix–helix distances are basically independent of chain length. We thus conducted PXRD analysis of polyketone mixtures obtained from a homo-coupling reaction of bis-silylated tetramer 4-Si2. High-molecular weight components (N ≥ 20) were collected from the reaction mixture by recycling GPC. Crystalline powder of the polydisperse polymer was prepared by slow evaporation of the solvent from its chloroform solution over 1 day. As expected, the PXRD pattern of the polydisperse sample exhibited rather sharp peaks at 10.5 and 17.1°, which strongly suggested that longer oligomers tend to adopt helical conformations and packing structures similar to discrete 5 even in polydisperse forms (Fig. S13†).
The critical chain length (N = 5) for crystal packing and conformations of polyketones is helpful for understanding the dependency of melting point on chain length. Differential scanning calorimetry (DSC) analysis of discrete polyketones 2–13 exhibited melting peaks at 87, 45, 104, 91, 119, 131, 139, 140, 142, 148, 157, and 161 °C, respectively (Fig. 4 and S14†). While no apparent regularity between peak shift and chain length was seen for dimer 2 to tetramer 4, the melting point did monotonically increase from pentamer 5 to icosamer 13. The irregular changes in melting point for 2–4 were accounted for by the differences in crystal packings. On the other hand, given the similarities in conformations and packing styles for 5–13, the monotonic increase of melting point for longer oligomers (N ≥ 5) can be explained by the increased number of monomer units.
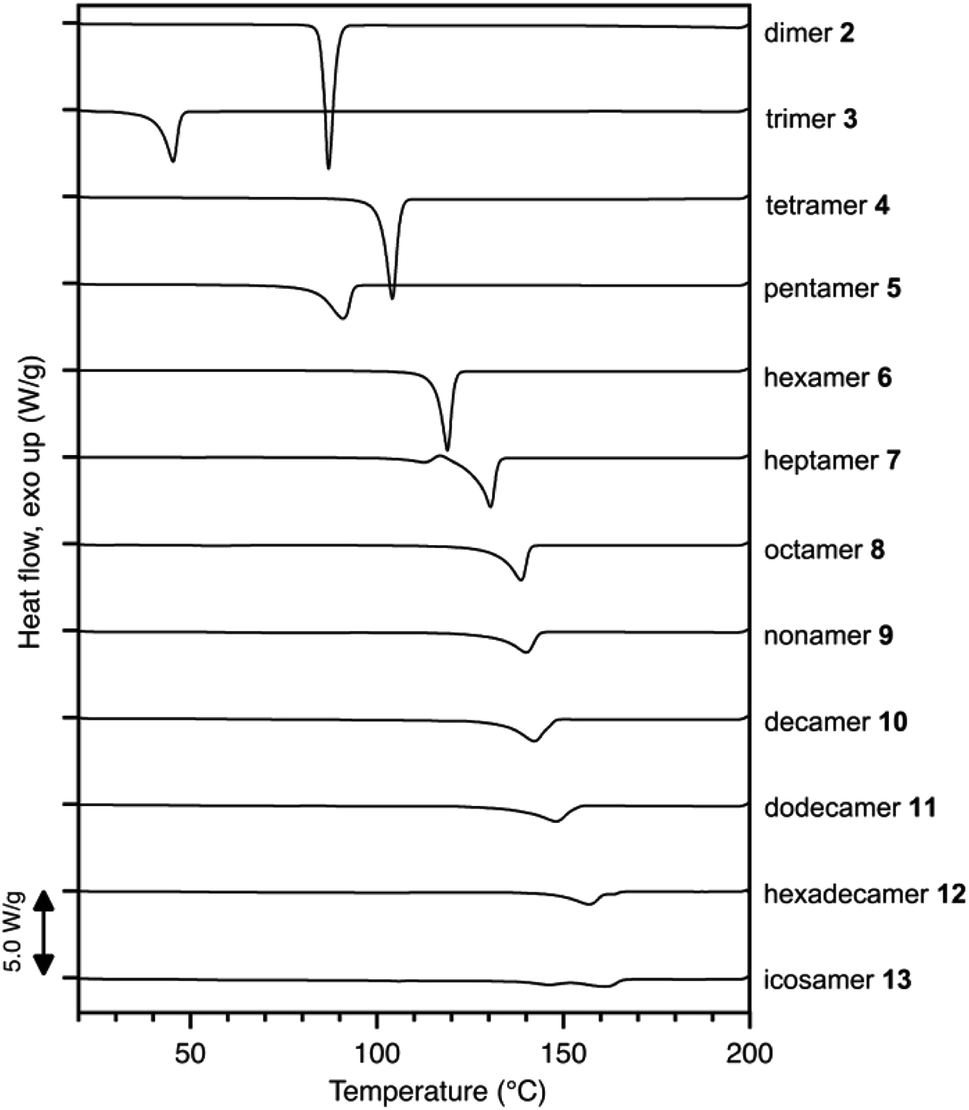 | ||
| Fig. 4 DSC traces (1st heating run) of discrete oligomers 2–13 at 10 °C min−1. | ||
Conclusions
In summary, we have extensively studied the conformations and crystal packings of structurally flexible polyketone oligomers. PXRD and SCXRD analyses clearly showed that the critical chain length, which divides small molecule-like crystallization from macromolecular crystallization, exists at an unexpectedly short chain length (N = 5). Longer oligomers 5–13 and even polydisperse polymers commonly adopted helical conformations and similar packing styles. Theoretical calculations reasonably explained that the helical structure was constructed via multiple intramolecular hydrogen bonds. Furthermore, determination of the critical chain length for macromolecular crystallization helped with understanding the irregularity of changes in melting point for oligomers in terms of conformations and packing structures. Our research has provided a rare example of critical chain length determination with respect to crystallization, which is beneficial for investigating other physical properties in macro-organic chemistry. Similar approaches for existing polymers would aid in finding and understanding hidden properties and functions of macromolecules.Data availability
The ESI contains synthetic procedures, mass spectra, HPLC chromatograms, computational details, crystallographic data, and NMR spectra.†Author contributions
Y. Inokuma designed the study and supervised the project. Y. Ide, Y. M., Y. Inaba, Y. K., T. Y, and K. I. S. performed experimental work. J. P. and Y. H. carried out computational analyses. S. T. and H. A. performed AFM studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by a JSPS Grant-in-Aid for Challenging Research (Exploratory) (No. 20K21214), Scientific Research (B) (No. 22H02058), JST FOREST Program (No. JPMJFR211H) and the Asahi Glass Foundation, where Y. Inokuma is the principal investigator. The Institute for Chemical Reaction Design and Discovery (ICReDD) was established by the World Premier International Research Initiative (WPI), MEXT, Japan. Y. Ide is grateful for a Grant-in-Aid for Early-Career Scientists grant (No. 21K14597) and Scientific Research on Innovative Areas “Soft Crystals”. Y. H. is grateful for a JSPS Grant-in-Aid for Challenging Research (Exploratory) (No. 21K18970). Y. M. and Y. Inaba would like to appreciate the Grant-in-Aid for JSPS Research Fellow (No. 21J11228 and No. 21J20973, respectively).Notes and references
- B. van Genabeek, B. A. G. Lamers, C. J. Hawker, E. W. Meijer, W. R. Gutekunst and B. V. K. J. Schmidt, J. Polym. Sci., 2021, 59, 373–403 CrossRef CAS.
- R. Aksakal, C. Mertens, M. Soete, N. Badi and F. Du. Prez, Adv. Sci., 2021, 8, 2004038 CrossRef CAS PubMed.
- M. Nerantzaki and J.-F. Lutz, Macromol. Chem. Phys., 2022, 223, 2100368 CrossRef CAS.
- S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1600711 CrossRef PubMed.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- S. Binauld, D. Damiron, L. A. Connal, C. J. Hawker and E. Drockenmuller, Macromol. Rapid Commun., 2011, 32, 147–168 CrossRef CAS PubMed.
- O. O. Mykhaylyk, C. M. Fernyhough, M. Okura, J. P. A. Fairclough, A. J. Ryan and R. Graham, Eur. Polym. J., 2011, 47, 447–464 CrossRef CAS.
- D. Zhou, M. Xu, Z. Ma, Z. Gan, R. Tan, S. Wang, Z. Zhang and X.-H. Dong, J. Am. Chem. Soc., 2021, 143, 18744–18754 CrossRef CAS PubMed.
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS PubMed.
- Y. Jiang, M. R. Golder, H. V.-T. Nguyen, Y. Wang, M. Zhong, J. C. Barnes, D. J. C. Ehrlich and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 9369–9372 CrossRef CAS PubMed.
- J. J. Haven, J. De Neve, A. C. Villavicencio and T. Junkers, Polym. Chem., 2019, 10, 6540–6544 RSC.
- J. Lawrence, S.-H. Lee, A. Abdilla, M. D. Nothling, J. M. Ren, A. S. Knight, C. Fleischmann, Y. Li, A. S. Abrams, B. V. K. J. Schmidt, M. C. Hawker, L. A. Connal, A. J. McGrath, P. G. Clark, W. R. Gutekunst and C. J. Hawker, J. Am. Chem. Soc., 2016, 138, 6306–6310 CrossRef CAS PubMed.
- M. Landa, M. Kotera, J.-S. Remy and N. Badi, Eur. Polym. J., 2016, 84, 338–344 CrossRef CAS.
- A. C. French, A. L. Thompson and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 1248–1252 CrossRef CAS PubMed.
- K. Abe, M. Koide and E. Tsuchida, Polym. J., 1977, 9, 73–78 CrossRef CAS.
- J. E. Báez, K. J. Shea, P. R. Dennison, A. Obregón-Herrera and J. Bonilla-Cruz, Polym. Chem., 2020, 11, 4228–4236 RSC.
- K. Takizawa, C. Tang and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 1718–1726 CrossRef CAS PubMed.
- E. Ihara, M. Tanabe, Y. Nakayama, A. Nakamura and H. Yasuda, Macromol. Chem. Phys., 1999, 200, 758–762 CrossRef CAS.
- B. A. G. Lamers, B. F. M. de Waal and E. W. Meijer, J. Polym. Sci., 2021, 59, 1142–1150 CrossRef CAS.
- K. Matsumoto, J. Jpn. Pet. Inst., 2021, 64, 307–316 CrossRef CAS.
- B. A. G. Lamers, A. Herdlitschka, T. Schnitzer, M. F. J. Mabesoone, S. M. C. Schoenmakers, B. F. M. de Waal, A. R. A. Palmans, H. Wennemers and E. W. Meijer, J. Am. Chem. Soc., 2021, 143, 4032–4042 CrossRef CAS PubMed.
- B. van Genabeek, B. F. M. de Waal, M. M. J. Gosens, L. M. Pitet, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 4210–4218 CrossRef CAS PubMed.
- J. T. Liu, H. Hase, S. Taylor, I. Salzmann and P. Forgione, Angew. Chem., Int. Ed., 2020, 59, 7146–7153 CrossRef CAS PubMed.
- M. Scheuble, M. Goll and S. Ludwigs, Macromol. Rapid Commun., 2015, 36, 115–137 CrossRef CAS PubMed.
- A. Mishra, C.-Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS.
- Z. Li, B. Cai, W. Yang and C.-L. Chen, Chem. Rev., 2021, 121, 14031–14087 CrossRef CAS.
- S. Lou, X. Wang, Z. Yu and L. Shi, Adv. Sci., 2019, 6, 1802043 CrossRef PubMed.
- S. Dobitz, M. R. Aronoff and H. Wennemers, Acc. Chem. Res., 2017, 50, 2420–2428 CrossRef CAS PubMed.
- A. Altmayer-Henzien, V. Declerck, J. Farjon, D. Merlet, R. Guillot and D. J. Aitken, Angew. Chem., Int. Ed., 2015, 54, 10807–10810 CrossRef CAS PubMed.
- M. T. Stone, J. M. Heemstra and J. S. Moore, Acc. Chem. Res., 2006, 39, 11–20 CrossRef CAS PubMed.
- J. M. Ren, J. Lawrence, A. S. Knight, A. Abdilla, R. B. Zerdan, A. E. Levi, B. Oschmann, W. R. Gutekunst, S.-H. Lee, Y. Li, A. J. McGrath, C. M. Bates, G. G. Qiao and C. J. Hawker, J. Am. Chem. Soc., 2018, 140, 1945–1951 CrossRef CAS PubMed.
- D. Zhou, M. Xu, J. Li, R. Tan, Z. Ma and X.-H. Dong, Macromolecules, 2021, 54, 4827–4833 CrossRef CAS.
- D. Zhou, M. Xu, R. Tan, Y. Sun, Z. Ma, J. Li and X.-H. Dong, Polymer, 2021, 225, 123746 CrossRef CAS.
- C. Zhang, D. S. Kim, J. Lawrence, C. J. Hawker and A. K. Whittaker, ACS Macro Lett., 2018, 7, 921–926 CrossRef CAS PubMed.
- K. Hatada, T. Kitayama, K. Ute and T. Nishiura, Macromol. Rapid Commun., 2004, 25, 1447–1477 CrossRef CAS.
- K. Hatada, T. Kitayama, K. Ute and T. Nishiura, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 416–431 CrossRef CAS.
- J. Li, R. Xia, H. Xu, J. Yang, X. Zhang, J. Kougo, H. Lei, S. Dai, H. Huang, G. Zhang, F. Cen, Y. Jiang, S. Aya and M. Huang, J. Am. Chem. Soc., 2021, 143, 17857–17861 CrossRef CAS PubMed.
- B. Oschmann, J. Lawrence, M. W. Schulze, J. M. Ren, A. Anastasaki, Y. Luo, M. D. Nothling, C. W. Pester, K. T. Delaney, L. A. Connal, A. J. McGrath, P. G. Clark, C. M. Bates and C. J. Hawker, ACS Macro Lett., 2017, 6, 668–673 CrossRef CAS PubMed.
- H. Zhang, X. Li, Q. Shi, Y. Li, G. Xia, L. Chen, Z. Yang and Z.-X. Jiang, Angew. Chem., Int. Ed., 2015, 54, 3763–3767 CrossRef CAS PubMed.
- Y. Kim, H. Park, A. Abdilla, H. Yun, J. Han, G. E. Stein, C. J. Hawker and B. J. Kim, Chem. Mater., 2020, 32, 3597–3607 CrossRef CAS.
- F. P. V. Koch, M. Heeney and P. Smith, J. Am. Chem. Soc., 2013, 135, 13699–13709 CrossRef CAS PubMed.
- M. Uesaka, Y. Saito, S. Yoshioka, Y. Domoto, M. Fujita and Y. Inokuma, Commun. Chem., 2018, 1, 23 CrossRef.
- Y. Inokuma, T. Yoneda, Y. Ide and S. Yoshioka, Chem. Commun., 2020, 56, 9079–9093 RSC.
- Y. Inokuma, J. Synth. Org. Chem. Jpn., 2019, 77, 1078–1085 CrossRef CAS.
- Y. Inokuma and Y. Inaba, Bull. Chem. Soc. Jpn., 2021, 94, 2187–2194 CrossRef CAS.
- P. Sarkar, Y. Inaba, H. Shirakura, T. Yoneda and Y. Inokuma, Org. Biomol. Chem., 2020, 18, 3297–3302 RSC.
- T. Eriksson, A. Mace, Y. Manabe, M. Yoshizawa-Fujita, Y. Inokuma, D. Brandell and J. Mindemark, J. Electrochem. Soc., 2020, 167, 070537 CrossRef.
- H. Shirakura, Y. Hijikata, J. Pirillo, T. Yoneda, Y. Manabe, M. Murugavel, Y. Ide and Y. Inokuma, Eur. J. Inorg. Chem., 2021, 1705–1708 CrossRef CAS.
- Y. Saito, M. Higuchi, S. Yoshioka, H. Senboku and Y. Inokuma, Chem. Commun., 2018, 54, 6788–6791 RSC.
- Y. Manabe, M. Uesaka, T. Yoneda and Y. Inokuma, J. Org. Chem., 2019, 84, 9957–9964 CrossRef CAS PubMed.
- Y. Inaba, T. Yoneda, Y. Kitagawa, K. Miyata, Y. Hasegawa and Y. Inokuma, Chem. Commun., 2020, 56, 348–351 RSC.
- H. Shirakura, Y. Manabe, C. Kasai, Y. Inaba, M. Tsurui, Y. Kitagawa, Y. Hasegawa, T. Yoneda, Y. Ide and Y. Inokuma, Eur. J. Org. Chem., 2021, 4345–4349 CrossRef CAS.
- Y. Inaba, Y. Nomata, Y. Ide, J. Pirillo, Y. Hijikata, T. Yoneda, A. Osuka, J. L. Sessler and Y. Inokuma, J. Am. Chem. Soc., 2021, 143, 12355–12360 CrossRef CAS PubMed.
- P. Teng, M. Zheng, D. C. Cerrato, Y. Shi, M. Zhou, S. Xue, W. Jiang, L. Wojtas, L.-J. Ming, Y. Hu and J. Cai, Commun. Chem., 2021, 4, 58 CrossRef CAS.
- P. Teng, G. M. Gray, M. Zheng, S. Singh, X. Li, L. Wojtas, A. van der Vaart and J. Cai, Angew. Chem., Int. Ed., 2019, 58, 7778–7782 CrossRef CAS PubMed.
- F. Sue, P. Teng, A. Peguero-Tejada, M. Wang, N. Ma, T. Odom, M. Zhou, E. Gjonaj, L. Wojtas, A. van der Vaart and J. Cai, Angew. Chem., Int. Ed., 2018, 57, 9916–9920 CrossRef PubMed.
- D. J. Williams, H. M. Colquhoun and C. A. O'Mahoney, J. Chem. Soc. Chem. Commun., 1994, 1643–1644 RSC.
- In the initial model, helical chains were oriented along the c-axis. Alternating assemblies of left- and right-handed helices were aligned along the a-axis in analogy with the crystal structure of 6.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2173799 and 2173800. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03083g |
| This journal is © The Royal Society of Chemistry 2022 |