
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen activation and cleavage by a multimetallic uranium complex†
Megan
Keener
 a,
Farzaneh
Fadaei-Tirani
a,
Rosario
Scopelliti
a,
Ivica
Zivkovic
b and
Marinella
Mazzanti
*a
a,
Farzaneh
Fadaei-Tirani
a,
Rosario
Scopelliti
a,
Ivica
Zivkovic
b and
Marinella
Mazzanti
*a
aInstitut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), 1015, Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bLaboratory for Quantum Magnetism, Institute of Physics, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
First published on 22nd June 2022
Abstract
Multimetallic-multielectron cooperativity plays a key role in the metal-mediated cleavage of N2 to nitrides (N3−). In particular, low-valent uranium complexes coupled with strong alkali metal reducing agents can lead to N2 cleavage, but often, it is ambiguous how many electrons are transferred from the uranium centers to cleave N2. Herein, we designed new dinuclear uranium nitride complexes presenting a combination of electronically diverse ancillary ligands to promote the multielectron transformation of N2. Two heteroleptic diuranium nitride complexes, [K{UIV(OSi(OtBu)3)(N(SiMe3)2)2}2(μ-N)] (1) and [Cs{UIV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] (3-Cs), containing different combinations of OSi(OtBu)3 and N(SiMe3)2 ancillary ligands, were synthesized. We found that both complexes could be reduced to their U(III)/U(IV) analogues, and the complex, [K2{UIV/III(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] (6-K), could be further reduced to a putative U(III)/U(III) species that is capable of promoting the 4e− reduction of N2, yielding the N24−complex [K3{UV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)(μ-η2:η2-N2)], 7. Parallel N2 reduction pathways were also identified, leading to the isolation of N2 cleavage products, [K3{UVI(OSi(OtBu)3)2(N(SiMe3)2)(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N)}(μ-N)2{UV(OSi(OtBu)3)2(N(SiMe3)2)}]2, 8, and [K4{(OSi(OtBu)3)2UV)(N)}(μ-NH)(μ-κ2:C,N-CH2SiMe2NSiMe3)-{UV(OSi(OtBu)3)2][K(N(SiMe3)2]2, 9. These complexes provide the first example of N2 cleavage to nitride by a uranium complex in the absence of reducing alkali metals.
N)}(μ-N)2{UV(OSi(OtBu)3)2(N(SiMe3)2)}]2, 8, and [K4{(OSi(OtBu)3)2UV)(N)}(μ-NH)(μ-κ2:C,N-CH2SiMe2NSiMe3)-{UV(OSi(OtBu)3)2][K(N(SiMe3)2]2, 9. These complexes provide the first example of N2 cleavage to nitride by a uranium complex in the absence of reducing alkali metals.
Introduction
The search for catalysts capable of converting dinitrogen into ammonia, or higher-value organic products under mild low-energy conditions, continues to attract intensive research on dinitrogen binding and cleavage by molecular compounds of d and f block elements.1 Uranium nitrides were shown to be highly active catalysts in the conversion of dinitrogen (N2) to ammonia (NH3), before equally active iron oxides were adopted as catalysts in the industrial production of NH3.2 This has generated significant interest towards the N2 chemistry of uranium,1a,b,1d,3a–l leading to the discovery of complexes capable of stoichiometrically converting N2 to NH3,1b,d,3j and in one system, catalytically converting N2 to amine (6.4 equiv.).1d However, the reaction of dinitrogen with well characterized molecular U(III) complexes, in most cases resulted in reversible binding,3b–f or in the 2e− reduction to N22−, with electrons being provided by two 1e− transfers from two mononuclear complexes.3a,g In contrast, the few reported cases of 4e− reduction of N2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1d or N2 cleavage to nitride,3d,k,3l usually required a combination of U(III) or U(IV) complexes and highly reducing alkali ions (K or Cs), leaving ambiguity on the number of electrons transferred by the uranium centers.
1d or N2 cleavage to nitride,3d,k,3l usually required a combination of U(III) or U(IV) complexes and highly reducing alkali ions (K or Cs), leaving ambiguity on the number of electrons transferred by the uranium centers.
Recently, we identified a molecular dinuclear nitride U(III) complex (complex C prepared from the reduction of the U(IV)/U(IV) analogue A, Scheme 1) that effects the 4e− reduction of N2 to N24− (complex D), which can be further functionalized by protons (H+) or CO, without addition of highly reducing alkali ions. In this dinuclear system, each uranium center is capable of transferring two electrons to N2.1b
 | ||
| Scheme 1 Reactivity of previously reported uranium nitride complexes A, B, C, E, F, and G with reducing agents and N2. | ||
Although multielectron transfer reactions are not common in uranium chemistry, reports of metal based two-electron transfer reactions have been rapidly increasing.4,5
In contrast, redox reactivity involving more than two electrons has been so far limited to complexes of uranium with redox-active ligands.6 However, our recent report of the first unambiguous example of metal-based 4e− transfer by a U(II) complex,7 suggests that 3e− transfer reactions may also be possible. Specifically, the 3e− transfer in dinuclear U(III) complexes is a pre-requisite to affect N2 cleavage without addition of reducing agents.
We reasoned that dinuclear U(III) nitride complexes presenting ligands more electron-donating than the siloxide ligand, (OSi(OtBu)3), would be able to transfer a high number of electrons to dinitrogen. We recently identified the bulky silylamide ligand (N(SiMe3)2) as well-suited for determining ligand effects on the reactivity and magnetic properties of nitride-bridged uranium complexes, due to the more electron-donating character and a slightly higher steric demand.3g
Inspired by the original report by Fortier and Hayton8 of the synthesis of the nitride bridged diuranium(IV) complex, [Na(dme)3][((N(SiMe3)2)2U(μ-N)(μ-κ2-C,N-CH2SiMe2NSiMe3)-U(N(SiMe3)2)2], we previously identified routes to homoleptic anionic, [NBu4{UIV(N(SiMe3)2)3}2(μ-N)], (G), and cationic, [{UIV(N(SiMe3)2)2(THF)}2(μ-N)] (E), U(IV)/U(IV) nitride bridged complexes of the N(SiMe3)2 ligand (Scheme 1).9 However, reduction of these complexes to their U(III)/U(III) analogue revealed impossible due to the very electron-rich character of the U centers. Herein, we have developed synthetic routes to the heteroleptic complexes, [K{UIV(OSi(OtBu)3)(N(SiMe3)2)2}2(μ-N)] (1) and [Cs{UIV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] (3-Cs), containing different combinations of OSi(OtBu)3 and N(SiMe3)2 ligands. Both complexes could be reduced to the U(III)/U(IV) analogue and complex, [K2{UIV/III(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] (6-K), could be further reduced to a highly reactive putative U(III)/U(III) species that is able to promote the 4e− reduction of N2. This reactivity provides the third example of 4e− reduction of N2 by a dinuclear U(III) complex. Here, parallel N2 reduction pathways were also identified, leading to the isolation of N2 cleavage products, providing the first example of N2 cleavage to nitride by a uranium complex in the absence of reducing alkali metals.
Results and discussion
Synthesis of heteroleptic U(IV) nitride complexes
In order to probe ligand effects on N2 binding, we sought to synthesize a series of heteroleptic uranium complexes with OSi(OtBu)3 and N(SiMe3)2 ancillary ligands in various ratios.First, we attempted to incorporate a higher number of OSi(OtBu)3 ligands within the previously reported mixed-ligand nitride system, [{UIV((N(SiMe3)2)3}[{UIV(N(SiMe3)2)2(OSi(OtBu)3)}(μ-N)][Na(DME)3] (I),9a by ligand protonolysis with 3.0 equiv. of HOSi(OtBu)3 (see ESI†). Analysis of the reaction mixture by 1H NMR spectroscopy indicated unreacted starting materials (Fig. S1†), suggesting that protonolysis of the U–N(SiMe3)2 bonds by HOSi(OtBu)3 cannot occur in these U(IV) systems.
Therefore, we next investigated the addition of KOSi(OtBu)3 (2.0 equiv. total) to the cationic, all-N(SiMe3)2 complex, [{UIV(N(SiMe3)2)2(THF)}2(μ-N)], E. Addition of 2.0 equiv. of KOSi(OtBu)3 to a solution of E in THF at room temperature, resulted in the immediate formation of a pink–brown solution and precipitation of a white solid. Large orange crystals of complex [K{UIV(OSi(OtBu)3)(N(SiMe3)2)2}2(μ-N)] (1) (Scheme 2, Fig. 1a), were obtained from a concentrated hexane solution at −40 °C in 82% yield. The 1H NMR spectrum of isolated complex 1 in d8-toluene displays two resonances at δ −0.10 and −3.81 ppm, corresponding to the OSi(OtBu)3 and N(SiMe3)2 signals, respectively (Fig. S8†). The solid-state molecular structure of complex 1 was determined by X-ray diffraction studies, and shows the presence of an ion pair consisting of one K cation and the [{UIV(OSi(OtBu)3)(N(SiMe3)2)2}2(μ-N)] anion. The two uranium(IV) ions in 1 are bridged by a nitride (N3−), and each are bound by one OSi(OtBu)3 and two N(SiMe3)2 ligands, resulting in a mixed-ligand framework. The K cation binds to the bridging nitride ligand (2.953(3)Å) and to four oxygens of the two OSi(OtBu)3 ligands. The U–N–U bond angle (162.5(1)°) and distances (2.071(2), 2.077(2)Å; Table 1) are consistent with those found for the previously reported all-OSi(OtBu)3 ([{UIV(OSi(OtBu)3)3}2(μ-N)][Cs] (A, Scheme 1); (170.2(3)°; 2.058(5)Å),4c and the all-N(SiMe3)2 complexes, E (168.97(14)°; 2.055(3)Å)9b and G (179.0(1)°; 2.076(5)Å).9a
 | ||
| Scheme 2 Synthesis of uranium heteroleptic complexes 1, 3-Cs, and 3b-Cs. | ||
 | ||
| Fig. 1 Molecular structures of (a) [K{UIV(OSi(OtBu)3)(N(SiMe3)2)2}2(μ-N)], 1, and (b) [Cs{UIV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)], 3-Cs, with thermal ellipsoids at drawn at the 50% probability level. Hydrogen atoms and methyl groups on the OSi(OtBu)3 ligands have been omitted for clarity. | ||
| Complex | 1 | 3-Cs | 3b-Cs | 4 | 5 | 6-K | 6-Cs |
|---|---|---|---|---|---|---|---|
| U–Nnitride | 2.071(2), 2.077(2) | 2.064(3), 2.074(3) | — | U2: 1.995(4), U1: 2.210(4) | — | U2: 2.024(11), U1: 2.187(12) | U2: 2.043(6), U1: 2.124(6) |
| Nnitride–M (M = K or Cs) | K1: 2.953(3) | Cs1: 3.086(3) | — | K1: 2.864(5) | — | K1: 2.967(11) K2: 3.256(11) | Cs1: 3.260(6) Cs2: 3.477(6) |
| U–NHimido | — | — | 2.167(8), 2.198(8) | — | U1: 2.184(11),U1′: 2.355(12) | — | — |
| U–N–U | 162.51(13) | 156.91(17) | — | 163.9(3) | — | 151.6(6) | 166.0(3) |
| U–NH–U | — | — | 119.7(4) | — | 119.5(6) | — | — |
Next, we investigated the synthesis of a heteroleptic complex with a different combination of OSi(OtBu)3 (four) and N(SiMe3)2 (two) ligands, to probe the effect of ancillary ligands in N2 binding and activation.
We pursued the synthesis of the targeted nitride complex by reacting the heteroleptic precursor “[UIII(OSi(OtBu)3)2(N(SiMe3)2)]” with CsN3, a method that had proven successful for the synthesis of the all-OSi(OtBu)3 complex, A,4c and other homoleptic nitride bridged U(IV) compounds.9a,10 First, we synthesized the monomeric heteroleptic complex, [UIII(OSi(OtBu)3)2I(THF)3],11 by reaction of [UI3(THF)3.5] with 2.0 equiv. KOSi(OtBu)3, and then further incorporated a N(SiMe3)2 ligand by salt metathesis to afford the heteroleptic complex “[UIII(OSi(OtBu)3)2(N(SiMe3)2)]” (Scheme S1†).
Isolation of the heteroleptic complex “[UIII(OSi(OtBu)3)2(N(SiMe3)2)]” in analytically pure form proved unsuccessful, leading to ligand scrambling products (see ESI† for details), and therefore further reactivity studies were carried out using the in situ generated heteroleptic U(III) complex.
Addition of 0.5 equiv. CsN3 to an Et2O solution of in situ generated heteroleptic complex “[UIII(OSi(OtBu)3)2(N(SiMe3)2)]” at −40 °C for four days, resulted in the formation of a light brown–pink solution. Analysis of the reaction mixture by 1H NMR spectroscopy in d8-toluene showed disappearance of the U(III) precursor and appearance of new resonances (Fig. S7†). Light brown crystals of the heteroleptic complex, [Cs{UIV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] (3-Cs) (Scheme 2, Fig. 1b), were obtained from a concentrated Et2O solution at −40 °C in 56% yield. The solid-state molecular structure of complex 3-Cs was determined by X-ray diffraction studies, and shows the presence of an ion pair consisting of one Cs cation and the [{UIV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] anion. The two uranium(IV) ions in 3-Cs are bridged by a nitride (N3−), and each ion is bound by two OSi(OtBu)3 and one N(SiMe3)2 ligand, resulting in a mixed-ligand framework. This altogether differs from complex 1 in the OSi(OtBu)3/N(SiMe3)2 ligand ratio (e.g. 2:4 in 1vs. 4:2 in 3-Cs). The Cs cation binds the bridging nitride (3.063(3)Å) and four oxygens from two OSi(OtBu)3 ligands. The U–N–U bond angle (156.9(2)°) is slightly smaller than 1, but has similar U–N bond distances (2.064(3), 2.074(3)Å; Table 1).
Complex 3-Cs is stable in toluene at −40 °C for one month and in THF at −80 °C for up to 8 hours. However, the product begins to decompose in THF after 1 hour at −40 °C, and is fully consumed after 2 hours at room temperature with a solution color change from pink to yellow (Fig. S14†). From this mixture, yellow crystals were isolated at −40 °C in concentrated n-hexane and characterized by XRD analysis as a diuranium imido cyclometallated complex, [Cs{(OSi(OtBu)3)3UIV}(μ-NH)(μ-κ2:C,N-CH2SiMe2NSiMe3)-{UIV(N(SiMe3)2)(OSi(OtBu)3)] (3b-Cs) in 89% yield (Scheme 2, Fig. 2).
 | ||
| Fig. 2 Molecular structure of complex, [Cs{(OSi(OtBu)3)3UIV}(μ-NH)(μ-κ2:C,N-CH2SiMe2NSiMe3)-{UIV(N(SiMe3)2)(OSi(OtBu)3)], 3b-Cs, with thermal ellipsoids at drawn at the 50% probability level. Hydrogen atoms and methyl groups on the OSi(OtBu)3 ligands have been omitted for clarity. | ||
The solid-state molecular structure of 3b-Cs suggests that decomposition involves 1,2-addition of a C–H bond of N(SiMe3)2 across the uranium–nitride bond. This is accompanied by an intramolecular ligand rearrangement, resulting in three OSi(OtBu)3 ligands binding one U(IV) ion, whereas, one OSi(OtBu)3, one N(SiMe3)2, and a μ-κ2-N,C-CH2SiMe2NSiMe3 ligand binds the second U(IV). The two uranium(IV) ions in 3b-Cs are bridged by two dianionic ligands, namely, an imido (NH2−) with a U–N–U bond angle (119.7(4)°) significantly bent compared to 3-Cs, and a μ-κ2-N,C-CH2SiMe2NSiMe3 ligand.
The U–NH bond distances (2.167(8), 2.198(8)Å; Table 1) are significantly longer than the parent nitride complex 3-Cs (156.19(17); 2.064(3), 2.074(3)Å). The 1,2-addition of a C–H bond of a N(SiMe3)2 ligand across the uranium–nitride has been previously demonstrated in a U(IV)/U(III) imido cyclometalate complex, [((Me3Si)2N)2U(THF)(μ-NH)(μ-κ2:C,N-CH2SiMe2NSiMe3)-U(N(SiMe3)2)(THF)].9b However, this type of 1,2-addition reactivity remains rare,12 as silylmethyl metalation in U–N(SiMe3)2 chemistry typically occurs with concomitant loss of one N(SiMe3)2 ligand to yield HN(SiMe3)2.8,9,13 Conversely, C–H bond activation was not observed for complex 1, suggesting that the higher number of OSi(OtBu)3 ligands found in complex 3-Cs, compared to 1 (four vs. two), leads to an increased nucleophilicity of the nitride moiety. Higher nucleophilicity of the bridging nitride had already been observed for the all-OSi(OtBu)3 complex, A, compared to the all-N(SiMe3)2 complex, G.9a,14
Synthesis and characterization of heteroleptic U(III)/U(IV) and U(III)/(III) nitrides
With heteroleptic nitride complexes 1 and 3-Cs in hand, we next explored their reduction chemistry to prepare new, low-valent, mixed-ligand uranium–nitrides and to investigate their ability to bind and reduce N2.Addition of 10.0 equiv. of KC8 to a solution of 1 in d8-THF at −80 °C led to the appearance of new resonances and consumption of 1 as seen by 1H NMR spectroscopy (Fig. S31†). Dark black prism crystals of [K{UIII/IV(OSi(OtBu)3)(N(SiMe3)2)2}2(μ-N)][K(THF)6] (4) (Scheme 3, Fig. 3), were obtained from a THF/hexane mixture both at −80 and −40 °C (78% yield), and characterized by X-ray diffraction studies. The solid-state molecular structure of 4 shows the presence of an ion pair consisting of two K cations and the [{UIII/IV(OSi(OtBu)3)(N(SiMe3)2)2}2(μ-N)] anion. The two uranium(III)/(IV) ions are bridged by a nitride (N3−) with a U–N–U bond angle (163.9(3)°) and U–N bond distances (U2: 1.995(4), U1: 2.210(4)Å; Table 1) consistent with a localized U(IV) and U(III), respectively. The U1–Namide and U1–Osiloxide bond distances for the U(III) ion (2.415(5)Å, 2.429(5)Å and 2.274(4)Å, respectively) are elongated compared to the U(IV) ion, where the U2–Namide and U2–Osiloxide bond distances (2.398(6)Å, 2.374(5)Å and 2.253(4)Å, respectively), are comparable to that of the parent U(IV) compound 1 (U–Namide, 2.319(2)–2.344(2)Å; U–Osiloxide, 2.216(2)Å, 2.221(2)Å), overall consistent with a localized U(IV)/U(III) motif. One K cation is outer sphere, and is bound by six THF molecules, whereas the second K binds the bridging nitride (2.864(5)Å), and four oxygens of the two OSi(OtBu)3 ligands. Remarkably, the structure of complex 4 indicates that the reduction proceeds without ligand scrambling reactions in this heteroleptic complex.
 | ||
| Scheme 3 Reactivity of complexes (a) 1 and 4 and (b) 3-Cs, 6-K, 6-Cs, X, and 7. The –OtBu and –(SiMe3)2 substituents on complexes 6-K, 6-Cs, 7, 8, and 9 were omitted for clarity. | ||
 | ||
| Fig. 3 Molecular structure of complex, [K{UIII/IV(OSi(OtBu)3)(N(SiMe3)2)2}2(μ-N)][K(THF)6], 4, with thermal ellipsoids at drawn at the 50% probability level. Hydrogen atoms, methyl groups on the OSi(OtBu)3 ligands, and the [K(THF)6] cation have been omitted for clarity. | ||
The isolation of complex 4 from the reduction of 1 with excess KC8, suggests that further reduction to the U(III)/U(III) analogue is not accessible in this ligand environment. This is comparable with the results of the previously reported reduction of complex E, which also led to the U(III)/U(IV) nitride, [{UIII/IV((N(SiMe3)2)2(THF)}2(μ-N)] (F, Scheme 1).9b This also shows that replacement of two N(SiMe3)2 ligands in the all-N(SiMe3)2 complex, [UIV(N(SiMe3)2)3}2(μ-N)] (G), by two OSi(OtBu)3 ligands, allows for the reduction of one uranium center, yielding the U(III)/U(IV) complex, which was unattainable in the reduction of the all-amide complex, G (Scheme 1). So far, nitride–bridged diuranium(III) complexes have only been obtained by reduction of the all-OSi(OtBu)3 analogue, complex A.4c The addition of two OSi(OtBu)3 ligands to complex E also results in a higher stability of the U(III)/U(IV) complex 4, when compared to the previously reported U(III)/U(IV) nitride (F) – which readily undergoes 1,2 addition of the C–H bond of a N(SiMe3)2 ligand across the uranium–nitride at −40 °C.9b Whereas, complex 4 is relatively stable over 24 hours in THF at −40 °C (Fig. S18†). The increased stability of 4 is likely due to the presence of the K+ cation capping the nitride.
Notably, when the K cation is removed from the core by addition of 2.0 equiv. of 2.2.2-cryptand to 4 in d8-THF at −40 °C, 1,2-addition of the C–H bond across the nitride occurs readily. Analysis of the reaction mixture by 1H NMR spectroscopy shows the consumption of 4 and the appearance of [K(2.2.2-cryptand)] (Fig. S32†). Dark black crystals of the U(III)/U(IV) cyclometalated imido complex, [((Me3Si)2N)2UIV(OSi(OtBu)3)(μ-NH)(μ-κ2:C,N-CH2SiMe2NSiMe3)-UIII(N(SiMe3)2)(OSi(OtBu)3)][{K(2.2.2-cryptand)}2] (5) (Scheme 3, Fig. S61†), were obtained from a THF/hexane mixture at −40 °C in 74% yield, and characterized by X-ray diffraction studies (see ESI†).
Next, we investigated the reduction of complex 3-Cs. Addition of 1.0 or 10.0 equiv. of KC8 to solutions of 3-Cs in d8-THF at −40 °C, led to significant differences in reactivity. Notably, the reaction with 1.0 equiv. of KC8 led to a brown–red solution and the appearance of new resonances and unreacted 3-Cs (a, Fig. S35†). Crystals of [K2{UIV/III(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] (6-K) (Scheme 3) were isolated from a concentrated n-hexane solution at −40 °C and characterized by XRD analysis. Attempts to isolate analytically pure 6-K from this pathway proved unsuccessful due to the contaminant precipitation of 3-Cs.
Instead, isolation of 6-K could be carried out by reacting 10.0 equiv. of KC8 with 3-Cs in Et2O at −40 °C. Analysis of the reaction mixture by 1H NMR spectroscopy in d8-toluene indicated full consumption of 3-Cs and formation of 6-K (Fig. S36†), which could be isolated from a concentrated hexane solution at −40 °C in 72% yield. Complex 6-K is stable in toluene at −40 °C for one month, whereas in THF, the product begins to decompose at −40 °C after 24 hours. The solid state molecular structure of complex 6-K shows the presence of an ion pair consisting of two K cations and the [{UIV/III(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] anion (Fig. 4a). The two uranium(III)/(IV) ions are bridged by a nitride (N3−), and are bound by two OSi(OtBu)3 and one N(SiMe3)2 ligand. The K cations bind to the bridging nitride (2.967(11), 3.256(11)Å) and the four OSi(OtBu)3 ligands. The U–N–U bond angle (151.6(6)°) is slightly bent, but has similar U–N bond distances (U2: 2.024(12), U1: 2.187(12)Å; Table 1) found in complex 4 (163.9(3)°; 1.995(4)Å, 2.210(4)Å)), indicating localized U(IV)/U(III) centers.
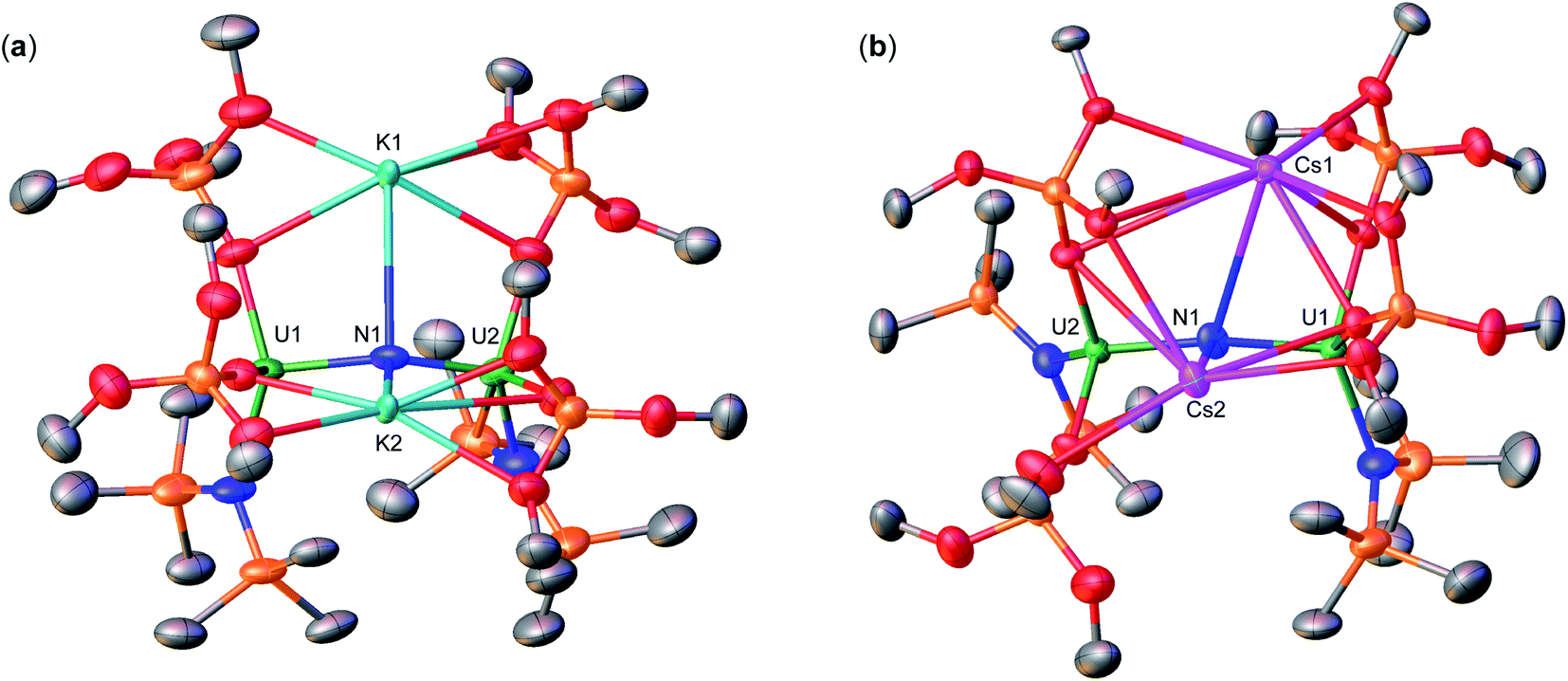 | ||
| Fig. 4 Molecular structures of (a) [K2{UIV/III(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)], 6-K, and (b) [Cs2{UIII/IV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)], 6-Cs, with thermal ellipsoids at drawn at the 50% probability level. Hydrogen atoms and methyl groups on the OSi(OtBu)3 ligands have been omitted for clarity. | ||
Alternatively, when the reduction of 3-Cs is carried out in the presence of 10.0 equiv. KC8 in THF at −40 °C, a dark purple solution is obtained. Analysis of the reaction mixture by variable temperature 1H NMR spectroscopy indicated full consumption of complex 3-Cs, and formation of new resonances. (Fig. S39 and S42†). The same species was also obtained by reduction of isolated 6-K with KC8 in THF (Fig. S43†). This suggests that further reduction of 6-K yields a putative U(III)/U(III) nitride, “[K3{UIII(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)]” (X), analogous to the one previously observed in homoleptic nitride systems (C; Scheme 1).1b,15 Attempts to isolate single crystals or analytically pure material from the reaction mixture proved unsuccessful, most likely due to the highly reactive nature of the formed species, which results in the formation of 6-K in toluene solution at −40 °C after 24 hours (Fig. S41†). These results indicate that depending on the nature of the solvent (Et2O or THF), reduction with excess KC8 can either lead to the formation of a U(III)/U(IV) or U(III)/U(III) species, highlighting the importance in tuning the reaction conditions in order to reach low-valent species.
Next, to probe the role of the cation in the synthesis and stability of the proposed heteroleptic U(III)/U(III) species, X, we also performed the reduction of 3-Cs with 10.0 equiv. CsC8 in THF at −40 °C. In comparison to the KC8 reactivity, this reaction instead resulted in a red–brown solution (Fig. S37†). Light orange crystals suitable for XRD analysis were obtained from a concentrated hexane solution at −40 °C in 78% yield, and characterized as the Cs analogue of 6-K, [Cs2{UIII/IV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] (6-Cs) (Scheme 3, Fig. 4b). The solid state molecular structure of complex 6-Cs shows the presence of an ion pair consisting of two Cs cations and the [{UIV/III(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] anion. The two uranium(III)/(IV) ions are bridged by a nitride (N3−) moiety, and are bound by two OSi(OtBu)3 and one N(SiMe3)2 ligand.
The Cs cations bind to the bridging nitride (3.260(6), 3.477(6)Å) and the four OSi(OtBu)3 ligands. The U–N–U bond angle (166.0(3)°) and distances (U2: 2.043(6), U1: 2.124(6)Å) are consistent with complex 6-K, indicating a localized U(IV)/U(III) structure, and the previously reported complex, [Cs2{UIII/IV(OSi(OtBu)3)3}2(μ-N)] (B; where M = Cs, Scheme 1) (169.1(7)°; 2.081(12)–2.099(12)Å).15 These results indicate that with excess CsC8, we cannot produce the analogous Cs3U(III)–N–U(III) species. This contrasts the previous studies for the reduction of the all-OSi(OtBu)3 analogue, [Cs{UIII(OSi(OtBu)3)3}2(μ-N)] (A), with KC8 and CsC8, where in both systems, the respective K3U(III)–N–U(III) and Cs3U(III)–N–U(III) species were obtained. The unreactive nature of the U(III)–N–U(IV) species, with excess CsC8, can be explained by a predicted, more negative reduction potential in comparison to 6-K, due to the replacement of the K cation by Cs in the second coordination sphere. Notably, the stronger K–O bonds (vs. Cs–O bonds) in the secondary coordination sphere help drive the reaction to the U(III)/U(III) state. However, the redox potentials for these species could not be obtained due to the rapid decomposition in THF solution at room temperature (3-Cs → 3b-Cs).
Reactivity of heteroleptic nitrides
Now with the U(III)/U(IV) complexes 4, 6-K, and 6-Cs in hand, we sought to probe their reactivity toward N2 activation and cleavage. However, in all systems, the U(III)/U(IV) precursors showed no reactivity with 1 atm N2 in toluene or THF solutions, resulting in unreacted starting materials as evidenced by 1H NMR studies (see ESI†).In contrast, addition of 1 atm N2 to the in situ generated U(III)/U(III) complex X, in toluene solution at −100 °C, led to an unambiguous color change from dark purple to light brown. Analysis of the reaction mixture by 1H NMR spectroscopy at variable temperature displays new resonances and the consumption of complex X (Fig. S48†). Dark brown crystals suitable for XRD analysis were obtained from the toluene reaction mixture at −40 °C after 24 hours, and characterized as the N24− complex, [K3{UV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)(μ-η2:η2-N2)] (7) in 56% yield (Scheme 3, Fig. 5). Complex 7 shows the presence of an ion pair consisting of three K cations and the [{UV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)(μ-η2:η2-N2)] anion. The two uranium(V) ions are bridged by a nitride (N3−) and a side-on-bound hydrazido (N24−) moiety, and each U(V) is bound by two OSi(OtBu)3 and one N(SiMe3)2 ancillary ligand. All K cations in complex 7 are inner-sphere, bound by the nitride, N24− moiety, and the four OSi(OtBu)3 ligands. One K cation, binds to the bridging nitride (N1) and four oxygens of two OSi(OtBu)3 ligands. The other two K atoms bind the N24− moiety, where K2 is bound to both nitrogen atoms, and K3 interacts with only one. Due to the mixed-ligand nature, the four OSi(OtBu)3 ligands support all three K cations. This contrasts the previously reported all-OSi(OtBu)3 U(V)/U(V) nitride–hydrazido complex (D, Scheme 1), in which the three K cations are bound to six OSi(OtBu)3 ligands and one K is bound to the N24− moiety, while two K atoms are bound to the bridging nitride.1b The U–Nnitride bond distances (2.101(15), 2.137(13)Å) (Table 2) are consistent with the previously reported complex D (2.069(2)Å),1b and U(V)/U(V) bis-nitride complexes (2.022(5), 2.101(6)Å).16 The U2(μ-η2:η2-N2) moiety features U–N bond distances which range between (2.178(15)–2.318(15)Å), consistent with U–N single bonds.17 The value of the NN bond length (1.54(2)Å) corresponds to a highly-activated N2, that has undergone a 4e− reduction to a hydrazido moiety (N24−), and is elongated compared to previously reported U–(N2)0 (1.109(7)–1.120(14)Å) and U–(N2)2− (1.124(12)–1.232(10) Å) moieties.3b,3c,3e,3f,3g Additionally, this value is comparable to the observed N–N bond length in complex D (1.521(18)Å).1b
 | ||
| Fig. 5 Molecular structure of [K3{UV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)(μ-η2:η2-N2)], 7, with thermal ellipsoids at drawn at the 50% probability level. Hydrogen atoms and methyl groups on the OSi(OtBu)3 ligands have been omitted for clarity. | ||
N)}(μ-N)2{UV(OSi(OtBu)3)2(N(SiMe3)2)}]2 (8), and [K4{(OSi(OtBu)3)2UV)(N)}(μ-NH)(μ-κ2:C,N-CH2SiMe2NSiMe3)-{UV(OSi(OtBu)3)2][K(N(SiMe3)2]2} (9)
| Complex | 7 | 8 | 9 |
|---|---|---|---|
| U–N24−–U | 1.54(2) | — | — |
| U–Nnitride | 2.101(15), 2.137(13) | 1.922(4)–2.311(4) | 1.916(13), 2.323(13) |
| U–NHimido | — | — | 2.218(14), 2.318(14) |
| UNterminal |
— | 1.784(4) | 1.847(13) |
To date, only one other example of a dinuclear U(III)/U(III) complex capable of reducing N2 to N24− has been reported (D),1b the homoleptic-OSi(OtBu)3 analogue of X.
Next, variable-temperature magnetic and EPR data were measured on isolated complex 7 in order to confirm the presence of two U(V) (5f1) ions, consistent with the 4e− reduction of N2. The magnetic susceptibility (χM) of complex 7 shows a paramagnetic behaviour with two magnetically independent U ions (purple, Fig. S65a†). This is in contrast to the previously reported complex D, which displays antiferromagnetic coupling at 50 K. The solid-state magnetic moment of 7 at 250 K was found to be 1.76 μB per uranium, (data were collected between 2–250 K to avoid thermal decomposition of the product). This value is lower than the theoretical value expected for a free U(V) ion (2.54 μB; 2F5/2 ground term),18 however it is in the range of other reported bimetallic U(V) systems (1.35–3.32 μB).4b,19a,b,19c–e,10a The μeff for complex 7 shows a steady decrease when the temperature is lowered, reaching 1.23 μB at 50 K, and then more rapidly to reach a value of 0.32 μB at 2 K (blue, Fig. S65a†). Although this temperature profile is typical for U(V), the value at 2 K is rather low. However, this behavior has been previously observed for the dimeric U(V) bridging bis–oxo complex, [{((AdArO)3N)U}2(μ-O)2],20 in which they report a similar χM plot and absence of antiferromagnetic coupling, altogether combined with a low μeff value at 2 K (0.36 μB per U). This was attributed to a potential antiferromagnetic interaction that was not observable, leading to the unexpected low temperature value. Additionally, the X-band (9.4 GHz) EPR spectrum of complex 7 shows an intense signal at 6 K that was fit to an axial set of signals (g1 = 2.740; g2 = 2.008; g3 = 1.797), further supporting the U(V) oxidation state (Fig. S64†).4a,4f,19b,21
Interestingly, separation and crystallization of the supernatant from four independent reactions of complex X with N2, after removal of complex 7, led to the identification of two N24−-cleavage products, characterized by XRD analysis as a formal U(VI)/U(V) tris-nitride complex, [K3{UVI(OSi(OtBu)3)2(N(SiMe3)2)(N)}(μ-N)2{UV(OSi(OtBu)3)2(N(SiMe3)2)}]2 (8), and a U(V)/U(V) bis-nitride imido cyclometallated product, [K4{((OSi(OtBu)3)2UV)(N)}(μ-NH)(μ-κ2:C,N-CH2SiMe2NSiMe3)-{UV(OSi(OtBu)3)2][K(N(SiMe3)2]2 (9) (Fig. 6). Over four trials, complex 8 was isolated once, while complex 9 was isolated on three occasions. Each crystallization of the supernatant always led to a mixture of species, in which the only other product that could be crystallographically identified was the K analogue of complex 3-Cs, [K{UIV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] (3-K) (Fig. S62†). Overall preventing the further characterization of these N2 cleavage products. However, these crystal structures provide important insight into N2 cleavage and reactivity.
 | ||
| Fig. 6 Molecular structures of (a) [K3{UVI(OSi(OtBu)3)2(N(SiMe3)2)(N)}(μ-N)2{UV(OSi(OtBu)3)2(N(SiMe3)2)}]2, 8, and (b) [K4{(OSi(OtBu)3)2UV)(N)}(μ-NH)(μ-κ2:C,N-CH2SiMe2NSiMe3)-{UV(OSi(OtBu)3)2][K(N(SiMe3)2]2, 9, with thermal ellipsoids at drawn at the 50% probability level. Hydrogen atoms and methyl groups on the OSi(OtBu)3 ligands have been omitted, and the KN(SiMe3)2 ligands for (b) have been depicted as wire frames for clarity. | ||
The solid-state molecular structure of complex 8 shows the presence of a symmetric tetrameric structure, comprising of two dimers in a diamond-shaped core, with two K atoms at the apical positions, capping the terminal and bridging nitride moieties (Fig. S63†). Due to the imposed crystal symmetry, identical bond metrics are found for each dimeric unit. U1 is bound by two OSi(OtBu)3 and one N(SiMe3)2 ligands. Conversely, U2 is bound by one OSi(OtBu)3, one N(SiMe3)2, and a terminal nitride, indicating that loss of one OSi(OtBu)3 ligand has occurred, most likely as KOSi(OtBu)3. The U1 and U2 ions are bridged by two nitride (N3−) ligands, where N2 is coordinated to two K atoms (K1, K2), bound through a combination of the OSi(OtBu)3 and N(SiMe3)2 ancillary ligands. The bridging U–N bond distances are asymmetric, featuring a combination of short (U1–N1: 1.922(4), U2–N2: 1.927(3)Å) and elongated (U1–N2: 2.253(4), U2–N1: 2.311(4)Å) bond distances (Table 2). This is consistent with the presence of UN multiple bonds and singly bound U–N, respectively, as previously observed in U(VI) bis-nitride bridged complexes.22
Two nitrides (N2, N3), bind the U2 ion in a slightly bent trans configuration (167.59(17)°), with different bond distances for U2–N3 and U2–N2 (1.784(4) and 1.927(3)Å). These parameters can be compared to those of the two previously reported linear trans-bis-nitride U(VI) complexes. In the complex, [K6{(OSi(OtBu)3)2UIV}3-{(OSi(OtBu)3)2UVI}(μ4-N)3(μ3-N)(μ3-O)2], the NUcenN bond distances are also slightly asymmetric (1.963(8) and 1.902(8) Å) and the bond angle is linear (179.07(11)°).3k In contrast, in the complex [(NH3)8UIV-(μ-N)-UVI(NH3)5-(μ-N)-UIV(NH3)8]8+, the NUcenN bond distances are identical (1.853(3) and 1.854(3)Å) and the bond angle is linear (179.07(11)°) probably due to the absence of coordinating cations.23 The terminal nitride has a bond distance of 1.784(4)Å, consistent with the bond distances found in the only two previously reported terminal U(VI) nitrides (1.769(2), 1.799(7)Å)24 and shorter than those found in alkali-capped (1.846(3)–1.929(6))25 and borane-capped U(V) nitrides (1.880(4), 1.916(4)Å).26
The solid-state molecular structure of complex 9 displays the presence of a dimeric imido cyclometallated anion, where the two uranium ions are bridged by three anionic ligands, namely a nitride (N3−), an imido (NH2−), and a μ-κ2-N,C-CH2SiMe2NSiMe3 ligand. The presence of an imido group, arising from the 1,2 addition of a C–H bond of the N(SiMe3)2 ligand across the U–N–U bond, was confirmed by 1H NMR spectroscopy of the reaction mixture, showing resonances of the imido and methylene protons at δ 627.91 ppm and δ −138.25 and −153.55 ppm, respectively. In complex 9, one U-bound N(SiMe3)2 ligand is lost, and two KN(SiMe3)2 are bound in the second coordination sphere. Two nitrides bind the U2 ion in a trans configuration (170.0(6)°) and the U(V) bond distances for the terminal and bridging nitride are 1.847(13)Å and 1.916(13)Å, respectively. The bis-imido motif R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UVN–R, an analogue of the UO22+ cation, has been previously observed by Boncella and coworkers.27 However, such an analogous bis-nitride motif, NUN, has not been previously observed for U(V). The terminal nitride (1.847(13)Å) is elongated compared to the terminal K-capped U(VI) nitride in complex 8 (1.784(4)Å; Table 2), and is comparable to that of the previously reported alkali-capped24 and borane-capped U(V) nitrides.26 The bridging U–N–U (U1–N1: 2.323(13), U2–N1: 1.916(13)Å) and U–NH–U (U1–N2: 2.218(14), U2–N2: 2.318(14)Å)14 bond distances are consistent with the presence of a U–N single and a short UN multiple bond (similar to complex 8), and a bridging imido, respectively. Complexes 8 and 9 provide the first example of N2 cleavage promoted by a low-valent U(III) complex without addition of alkali reducing agents. We recently reported N2 cleavage in the all-OSi(OtBu)3 diuranium(V) oxide bridged complex, [K2{[UV(OSi(OtBu)3)3]2(μ-O)(μ-η2:η2-N2)}], to yield a tetranitride cluster.3k However, this reactivity was only observed upon reduction with KC8.
UVN–R, an analogue of the UO22+ cation, has been previously observed by Boncella and coworkers.27 However, such an analogous bis-nitride motif, NUN, has not been previously observed for U(V). The terminal nitride (1.847(13)Å) is elongated compared to the terminal K-capped U(VI) nitride in complex 8 (1.784(4)Å; Table 2), and is comparable to that of the previously reported alkali-capped24 and borane-capped U(V) nitrides.26 The bridging U–N–U (U1–N1: 2.323(13), U2–N1: 1.916(13)Å) and U–NH–U (U1–N2: 2.218(14), U2–N2: 2.318(14)Å)14 bond distances are consistent with the presence of a U–N single and a short UN multiple bond (similar to complex 8), and a bridging imido, respectively. Complexes 8 and 9 provide the first example of N2 cleavage promoted by a low-valent U(III) complex without addition of alkali reducing agents. We recently reported N2 cleavage in the all-OSi(OtBu)3 diuranium(V) oxide bridged complex, [K2{[UV(OSi(OtBu)3)3]2(μ-O)(μ-η2:η2-N2)}], to yield a tetranitride cluster.3k However, this reactivity was only observed upon reduction with KC8.
Once isolated, the diuranium(V)–N24− complex 7 is stable in toluene solution at −40 °C for one month, indicating that the formation of the nitride complexes, 8 and 9, must arise from parallel redox pathways. The formal oxidation state of complex 8, indicates that its formation requires the 1e− reduction of complex 7 by an external reducing agent. This would result in the overall 2e− reduction and cleavage of the N24− single bond, where an additional 1e− is provided by the U(V)/U(VI) oxidation.
Overall, complex 8 is the product of an unprecedented case of 3e− transfer from a U(III) center. Whereas the formation of complex 9 most likely involves a 2e− reduction of the N24− moiety by an external reducing agent. Formation of these unusual species suggests a high redox flexibility of the uranium centers in the mixed-ligand environment, provided by the combination of OSi(OtBu)3 and N(SiMe3)3 ligands.
A potential source for the additional 1 and 2e− required to form complexes 8 and 9, respectively, is the U(III)/U(III) nitride complex, X. One could envision that the reduction of complex 7, promoted by the highly reducing U(III)/U(III) complex X, occurs in parallel with the binding of N2.
To confirm such a pathway, we sought out the reduction of 7 with the U(III)/U(III) complex X. Addition of 1.0 equiv. (2e−) of in situ generated complex X in d8-toluene to 7 at −80 °C, resulted in an unambiguous colour change from dark purple to brown with precipitation of orange crystals. Analysis of the reaction mixture by variable temperature 1H NMR spectroscopy, led to the complete consumption of X and formation of new resonances, including the imido (NH; δ 627.91 ppm) and methylene protons (δ −138.25 and −153.55 ppm), attributed to complex 9 (Fig. S50†). Quantitative 1H NMR studies of the reaction mixture demonstrated that complex 9 is formed in 51% yield (Fig. S51†). Crystallization of the reaction mixture from a concentrated toluene solution at −40 °C led to a mixture of pale green, orange, and colorless crystals. The pale green and orange crystals were identifiable as complexes 9 and 3-K, respectively. This indicates that complex 7 undergoes a 2e− reduction promoted by the oxidation of the U(III)/U(III) complex to U(IV)/U(IV). Attempts to isolate analytically pure 9 from this route proved unsuccessful due to the contaminant precipitation of 3-K.
In order to probe the reactivity of the U2(N24−) moiety in complex 7, we next investigated the reactivity with acid (H+). Addition of HCl in Et2O to isolated complex 7, led to the formation of a broad, ill-defined triplet at δ 7.41 ppm in the 1H NMR spectrum, possibly consistent with 14NH4Cl (Fig. S57a†). However, we have previously shown that uranium bridging nitride complexes react readily with strong acids to form NH3.14 Therefore, to determine if the NH4Cl was derived from the (N3−) and (N24−) ligands, the analogous experiment with 15N2-labelled complex, 7-14/15N, was also carried out. HCl in Et2O addition to 7-14/15N led to a comparable spectrum as 7 (Fig. S57b†), making the assignment to 15NH4Cl or 14NH4Cl ambiguous. Broad resonances are not common in NH4Cl quantification experiments, and are most likely due to our ligand system. Indeed, addition of HCl in Et2O to neat KN(SiMe3)2, also led to a broad singlet at δ 7.35 ppm in the 1H NMR spectrum (Fig. S55†), directly overlapping with the resonance for 14/15NH4Cl at δ 7.41 ppm. Altogether, this rendered NH3 quantification difficult, yet comparable yields were obtained for the nitride precursor 3-Cs and complexes 7 and 7-14/15N (see ESI†), suggesting that protonation of the N24− does not result in NH3 formation.
However, addition of HCl in Et2O to the reaction mixture obtained after reacting complex X and 7, led to the formation of a broad, ill-defined signal at δ 7.41 ppm in the 1H NMR spectrum assigned to 14NH4Cl (Fig. S59†). Although ammonia quantification is difficult in this system, the number of equivalents of NH3 increased by about ∼1.7, compared to the amount obtained from the addition of acid to isolated complex 7.
This indicates that complex 7 undergoes a 2e− reduction and N24− cleavage to a tris-nitride complex, promoted via the U(III)/U(III) reducing agent, X.
Conclusions
Herein, we have identified rational routes for the synthesis of heteroleptic nitride bridged diuranium(IV) complexes containing OSi(OtBu)3 and N(SiMe3)3 ligands in varying ratios. The differences in the electronic and steric properties of these ligands allow for the tuning of redox properties and reactivity of these diuranium complexes. The reactivity of the heteroleptic complexes [K{UIV(OSi(OtBu)3)(N(SiMe3)2)2}2(μ-N)] (1) and [Cs{UIV(OSi(OtBu)3)2(N(SiMe3)2)}2(μ-N)] (3-Cs), differs significantly from the previously reported all-OSi(OtBu)3 and all-N(SiMe3)2 analogues. Notably, the reduction of one of the U(IV) centers can be performed for 1 and 3-Cs to yield the nitride bridged U(III)/U(IV) complexes, 4, 6-K, and 6-Cs, while reduction of the anionic all-amide complex G was unattainable, probably due the higher electron-rich character of the N(SiMe3)3 ligand compared to OSi(OtBu)3. The presence of two additional OSi(OtBu)3 ligands in the U(III)/U(IV) complex 4, compared to the cationic all-amide complex E, results in an increased stability towards the 1,2 addition of the C–H bond of a N(SiMe3)2 ligand across the uranium–nitride,9b most likely due to the siloxide-bound K cation capping the nitride. Removal of the K cation by addition of 2.2.2-cryptand, increases nitride reactivity towards C–H activation, affording the imido cyclometallated complex 5. The U(III)/U(IV) complexes 4 and 6-Cs cannot be reduced further, most likely due to the very electron-rich character of the U. However, reduction of the second U(IV) center is possible for complex 6-K, but is highly dependent upon the reduction reaction conditions. The putative U(III)/U(III) heteroleptic nitride complex X, is significantly more reactive than the only other example of the all-OSi(OtBu)3 U(III)/U(III) nitride complex, A. Similar to complex A, X reacts with N2 and promotes the 4e− reduction to yield the U(V)/U(V)–N24− complex 7. N2 cleavage products were also identified from the reaction of X and N2, and were crystallographically characterized. The tris–nitride complex 8 is generated through the overall 6e− cleavage of the bound N2 moiety, promoted by a 5e− transfer from the two U(III) centers to form a U(V)/U(VI) complex, with an additional 1e− arising from an external source. In complex 9, cleavage of N2 has also occurred to yield a U(V)/U(V) nitride, with 2e− arising from an external source. We found that addition of 7 to X results in the formation of complexes 9 and 3-K, indicating that the diuranium(III) precursor, X, provides 1 or 2e− to further reduce the bound N24− moiety in 7. These results demonstrate that by tuning the ancillary ligands, the reducing power and the redox flexibility of the uranium center can be altered leading to the 3e− transfer from U(III) to N2. This work has demonstrated the first example of N2 cleavage by a uranium complex in the absence of alkali metals. Further tuning of the ancillary ligands in diuranium nitride complexes, should allow better control of these transformations.Data availability
All data associted were included in the supporting information.Author contributions
M. K. carried out the synthetic experiments and analyzed the experimental data. F. F.-T. and R. S. carried out the X-ray single crystal structure analyses. I. V. collected the variable-temperature magnetic data. M. M. originated the central idea, coordinated the work, and analyzed the experimental data. The manuscript was written through contributions of all authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge support from the Swiss National Science Foundation grant number 200021_178793, and the Ecole Polytechnique Fédérale de Lausanne (EPFL). We thank Dr Andrzej Sienkiewicz for EPR data collection.References
- (a) M. D. Walter, Adv. Organomet. Chem., 2016, 65, 261–377 CrossRef CAS; (b) M. Falcone, L. Chatelain, R. Scopelliti, I. Zivkovic and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS PubMed; (c) Y. Roux, C. Duboc and M. Gennari, Chemphyschem, 2017, 18, 2606–2617 CrossRef CAS PubMed; (d) P. L. Arnold, T. Ochiai, F. Y. T. Lam, R. P. Kelly, M. L. Seymour and L. Maron, Nat. Chem., 2020, 12, 654–659 CrossRef CAS; (e) M. Reiners, D. Baabe, K. Munster, M. K. Zaretzke, M. Freytag, P. G. Jones, Y. Coppel, S. Bontemps, I. del Rosal, L. Maron and M. D. Walter, Nat. Chem., 2020, 12, 740–746 CrossRef PubMed; (f) S. J. K. Forrest, B. Schluschass, E. Y. Yuzik-Klimova and S. Schneider, Chem. Rev., 2021, 121, 6522–6587 CrossRef CAS; (g) F. Masero, M. A. Perrin, S. Dey and V. Mougel, Chem.–Eur. J., 2021, 27, 3892–3928 CrossRef CAS PubMed.
- (a) F. Haber, Ammonia German Patent, DE 229126 Pat., 1909; (b) F. Haber, Angew. Chem., 1914, 27, 473–477 CrossRef CAS.
- (a) A. L. Odom, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 1998, 120, 5836–5837 CrossRef CAS; (b) P. Roussel and P. Scott, J. Am. Chem. Soc., 1998, 120, 1070–1071 CrossRef CAS; (c) F. G. N. Cloke and P. B. Hitchcock, J. Am. Chem. Soc., 2002, 124, 9352–9353 CrossRef CAS PubMed; (d) I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed. Engl., 2002, 41, 3433–3436 CrossRef CAS; (e) W. J. Evans, S. A. Kozimor and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 14264–14265 CrossRef CAS; (f) S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed; (g) S. M. Mansell, J. H. Farnaby, A. I. Germeroth and P. L. Arnold, Organometallics, 2013, 32, 4214–4222 CrossRef CAS; (h) M. Falcone, L. Barluzzi, J. Andrez, F. F. Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS PubMed; (i) E. Lu, B. E. Atkinson, A. J. Wooles, J. T. Boronski, L. R. Doyle, F. Tuna, J. D. Cryer, P. J. Cobb, I. J. Vitorica-Yrezabal, G. F. S. Whitehead, N. Kaltsoyannis and S. T. Liddle, Nat. Chem., 2019, 11, 806–811 CrossRef CAS PubMed; (j) X. Q. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Q. Zhu, J. Am. Chem. Soc., 2020, 142, 15004–15011 CrossRef CAS PubMed; (k) N. Jori, L. Barluzzi, I. Douair, L. Maron, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2021, 143, 11225–11234 CrossRef CAS PubMed; (l) P. L. Wang, I. Douair, Y. Zhao, S. Wang, J. Zhu, L. Maron and C. Q. Zhu, Angew. Chem., Int. Ed. Engl., 2021, 60, 473–479 CrossRef CAS PubMed.
- (a) S. Fortier, N. Kaltsoyannis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 14224–14227 CrossRef CAS; (b) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS PubMed; (c) C. Camp, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed; (d) T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC; (e) C. Camp, M. A. Antunes, G. Garcia, I. Ciofini, I. C. Santos, J. Pecaut, M. Almeida, J. Marcalo and M. Mazzanti, Chem. Sci., 2014, 5, 841–846 RSC; (f) O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS PubMed; (g) D. P. Halter, F. W. Heinemann, J. Bachmann and K. Meyer, Nature, 2016, 530, 317–321 CrossRef CAS PubMed; (h) N. Tsoureas, A. F. R. Kilpatrick, C. J. Inman and F. G. N. Cloke, Chem. Sci., 2016, 7, 4624–4632 RSC; (i) B. M. Gardner, C. E. Kefalidis, E. Lu, D. Patel, E. J. L. McInnes, F. Tuna, A. J. Wooles, L. Maron and S. T. Liddle, Nat. Commun., 2017, 8, 1898 CrossRef PubMed; (j) N. S. Settineri, A. A. Shiau and J. Arnold, Chem. Commun., 2018, 54, 10913–10916 RSC.
- (a) D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448–9460 CrossRef CAS; (b) J. L. Kiplinger, D. E. Morris, B. L. Scott and C. J. Burns, Chem. Commun., 2002, 30–31 RSC; (c) J. L. Brown, S. Fortier, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2013, 135, 5352–5355 CrossRef CAS PubMed; (d) A. J. Lewis, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 13185–13192 CrossRef CAS PubMed; (e) E. L. Lu, O. J. Cooper, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed. Engl., 2014, 53, 6696–6700 CrossRef CAS PubMed; (f) N. T. Rice, K. McCabe, J. Bacsa, L. Maron and H. S. La Pierre, J. Am. Chem. Soc., 2020, 142, 7368–7373 CrossRef CAS; (g) R. J. Ward, P. Rungthanaphatsophon, I. del Rosal, S. P. Kelley, L. Maron and J. R. Walensky, Chem. Sci., 2020, 11, 5830–5835 RSC.
- (a) J. M. Manriquez, P. J. Fagan, T. J. Marks, S. H. Vollmer, C. S. Day and V. W. Day, J. Am. Chem. Soc., 1979, 101, 5075–5078 CrossRef CAS; (b) P. J. Fagan, J. M. Manriquez, T. J. Marks, C. S. Day, S. H. Vollmer and V. W. Day, Organometallics, 1982, 1, 170–180 CrossRef CAS; (c) P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108–6109 CrossRef CAS; (d) W. J. Evans, S. A. Kozimor and J. W. Ziller, Chem. Commun., 2005, 4681–4683 RSC; (e) G. F. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriedsen and R. A. Andersen, Organometallics, 2005, 24, 4251–4264 CrossRef CAS; (f) W. J. Evans, K. A. Miller, S. A. Kozimor, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2007, 26, 3568–3576 CrossRef CAS; (g) W. J. Evans, E. Montalvo, S. A. Kozimor and K. A. Miller, J. Am. Chem. Soc., 2008, 130, 12258–12259 CrossRef CAS; (h) C. Camp, V. Mougel, P. Horeglad, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2010, 132, 17374–17377 CrossRef CAS PubMed; (i) D. P. Cladis, J. J. Kiernicki, P. E. Fanwick and S. C. Bart, Chem. Commun., 2013, 49, 4169–4171 RSC; (j) N. H. Anderson, S. O. Odoh, Y. Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919–926 CrossRef CAS PubMed; (k) J. J. Kiernicki, P. E. Fanwick and S. C. Bart, Chem. Commun., 2014, 50, 8189–8192 RSC; (l) P. L. Diaconescu and C. C. Cummins, Dalton Trans., 2015, 44, 2676–2683 RSC; (m) J. J. Kiernicki, R. F. Higgins, S. J. Kraft, M. Zeller, M. P. Shores and S. C. Bart, Inorg. Chem., 2016, 55, 11854–11866 CrossRef CAS; (n) L. Zhang, C. C. Zhang, G. H. Hou, G. F. Zi and M. D. Walter, Organometallics, 2017, 36, 1179–1187 CrossRef CAS; (o) P. Rungthanaphatsophon, C. L. Barnes, S. P. Kelley and J. R. Walensky, Dalton Trans., 2018, 47, 8189–8192 RSC; (p) N. Jori, M. Falcone, R. Scopelliti and M. Mazzanti, Organometallics, 2020, 39, 1590–1601 CrossRef CAS.
- D. K. Modder, C. T. Palumbo, I. Douair, R. Scopelliti, L. Maron and M. Mazzanti, Chem. Sci., 2021, 12, 6153–6158 RSC.
- S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS.
- (a) C. T. Palumbo, L. Barluzzi, R. Scopelliti, I. Zivkovic, A. Fabrizio, C. Corminboeuf and M. Mazzanti, Chem. Sci., 2019, 10, 8840–8849 RSC; (b) C. T. Palumbo, R. Scopelliti, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 3149–3157 CrossRef CAS PubMed.
- (a) A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS PubMed; (b) J. Z. Du, D. M. King, L. Chatelain, E. L. Lu, F. Tuna, E. J. L. McInnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738–3745 RSC.
- L. Barluzzi, PhD, EPFL, 2021 Search PubMed.
- K. C. Mullane, H. Ryu, T. Cheisson, L. N. Grant, J. Y. Park, B. C. Manor, P. J. Carroll, M. H. Baik, D. J. Mindiola and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 11335–11340 CrossRef CAS PubMed.
- (a) S. J. Simpson, H. W. Turner and R. A. Andersen, J. Am. Chem. Soc., 1979, 101, 7728–7729 CrossRef CAS; (b) S. J. Simpson, H. W. Turner and R. A. Andersen, Inorg. Chem., 1981, 20, 2991–2995 CrossRef CAS; (c) C. R. Graves, E. J. Schelter, T. Cantat, B. L. Scott and J. L. Kiplinger, Organometallics, 2008, 27, 5371–5378 CrossRef CAS; (d) B. M. Gardner, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2009, 131, 10388–10389 CrossRef CAS PubMed.
- M. Keener, R. Scopelliti and M. Mazzanti, Chem. Sci., 2021, 12, 12610–12618 RSC.
- L. Chatelain, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2016, 138, 1784–1787 CrossRef CAS PubMed.
- L. Barluzzi, L. Chatelain, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, Chem. Sci., 2019, 10, 3543–3555 RSC.
- L. P. Spencer, E. J. Schelter, P. Yang, R. L. Gdula, B. L. Scott, J. D. Thompson, J. L. Kiplinger, E. R. Batista and J. M. Boncella, Angew. Chem., Int. Ed. Engl., 2009, 48, 3795–3798 CrossRef CAS PubMed.
- D. R. Kindra and W. J. Evans, Chem. Rev., 2014, 114, 8865–8882 CrossRef CAS PubMed.
- (a) R. K. Rosen, R. A. Andersen and N. M. Edelstein, J. Am. Chem. Soc., 1990, 112, 4588–4590 CrossRef CAS; (b) S. C. Bart, C. Anthon, F. W. Heinemann, E. Bill, N. M. Edelstein and K. Meyer, J. Am. Chem. Soc., 2008, 130, 12536–12546 CrossRef CAS PubMed; (c) C. Camp, V. Mougel, J. Pecaut, L. Maron and M. Mazzanti, Chem.–Eur. J., 2013, 19, 17528–17540 CrossRef CAS PubMed; (d) D. Patel, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2013, 42, 5224–5227 RSC; (e) A. C. Schmidt, F. W. Heinemann, W. W. Lukens and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed.
- O. P. Lam, F. W. Heinemann and K. Meyer, Chem. Sci., 2011, 2, 1538–1547 RSC.
- J. L. Brown, S. Fortier, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2013, 135, 5352–5355 CrossRef CAS PubMed.
- L. Barluzzi, F. C. Hsueh, R. Scopelliti, B. E. Atkinson, N. Kaltsoyannis and M. Mazzanti, Chem. Sci., 2021, 12, 8096–8104 RSC.
- S. S. Rudel, H. L. Deubner, M. Muller, A. J. Karttunen and F. Kraus, Nat. Chem., 2020, 12, 962–967 CrossRef CAS PubMed.
- (a) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482–488 CrossRef CAS PubMed; (b) L. Barluzzi, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 19047–19051 CrossRef CAS PubMed.
- D. M. King, P. A. Cleaves, A. J. Wooles, B. M. Gardner, N. F. Chilton, F. Tuna, W. Lewis, E. J. L. McInnes and S. T. Liddle, Nat. Commun., 2016, 7, 13773 CrossRef CAS PubMed.
- (a) A. R. Fox and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 5716–5717 CrossRef CAS PubMed; (b) M. A. Boreen, G. D. Rao, D. G. Villarreal, F. A. Watt, R. D. Britt, S. Hohloch and J. Arnold, Chem. Commun., 2020, 56, 4535–4538 RSC.
- R. E. Jilek, L. P. Spencer, R. A. Lewis, B. L. Scott, T. W. Hayton and J. M. Boncella, J. Am. Chem. Soc., 2012, 134, 9876–9878 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC [2174705, 2174938, 2101093, 2141646, 2174960, 2174751, 2174754, 2141660, 2141333, 2174752, 2142105 and 2174753]. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02997a |
| This journal is © The Royal Society of Chemistry 2022 |